the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Dec 2023
| 08 Dec 2023
Direct formation of HONO through aqueous-phase photolysis of organic nitrates
Juan Miguel González-Sánchez
Miquel Huix-Rotllant
Nicolas Brun
Julien Morin
Carine Demelas
Amandine Durand
Sylvain Ravier
Jean-Louis Clément
Anne Monod
Organic nitrates (RONO2) are secondary compounds whose fate is closely related to the transport and removal of NOx in the atmosphere. Despite their ubiquitous presence in submicron aerosols, the photochemistry of RONO2 has only been investigated in the gas phase, leaving their reactivity in condensed phases poorly explored. This work aims to address this gap by investigating, for the first time, the reaction products and the mechanisms of aqueous-phase photolysis of four RONO2 (i.e., isopropyl nitrate, isobutyl nitrate, α-nitrooxy acetone, and 1-nitrooxy-2-propanol). The results show that the reactivity of RONO2 in the aqueous phase differs significantly from that in the gas phase. In contrast to the gas phase, where RONO2 release NOx upon photolysis, the aqueous-phase photolysis of RONO2 leads primarily to the direct formation of nitrous acid (HONO or HNO2), which was confirmed by quantum chemistry calculations. Hence, the aqueous-phase photolysis of RONO2 represents both a NOx sink and a source of atmospheric nitrous acid, a significant precursor of ⋅ OH and ⋅ NO. These secondary radicals (⋅ OH and ⋅ NO) are efficiently trapped in the aqueous phase, leading to the formation of HNO3 and functionalized RONO2. This reactivity can thus potentially contribute to the aging of secondary organic aerosol (SOA) and serves as an additional source of aqueous-phase SOA.
- Article
(2856 KB) - Full-text XML
-
Supplement
(1159 KB) - BibTeX
- EndNote





Organic nitrates (RONO2) are secondary compounds, formed through NOx+ volatile organic compound reactions, which play an essential role in the transport and removal of NOx in the atmosphere. These compounds can have long lifetimes, lasting from a few hours to several days, which allow them to travel to remote regions (Shepson, 1999). During their long-range transport, they can undergo reactions such as gas-phase photolysis and/or ⋅ OH oxidation, which can release NOx back into the atmosphere. As a result, organic nitrates are responsible for a more uniform distribution of NOx and are thus indirectly responsible for the transport of other pollutants such as O3 and secondary organic aerosol (SOA; Perring et al., 2013).
Furthermore, RONO2 also participate in NOx removal from the atmosphere, which can occur either through its deposition to the Earth's surface or by transformation into a less reactive chemical compound, such as nitric acid (Hu et al., 2011; Nguyen et al., 2015). Therefore, its atmospheric reactivity and fate must be considered to accurately predict pollution transport on a regional scale. This is especially important for world regions experiencing decreasing NOx levels, such as Europe and North America, where the relative importance of RONO2 in NOx transport and removal is growing due to the increase in the overall transformation of NOx into RONO2 (Romer Present et al., 2020).
RONO2 are not only present in the gas phase, as some of them have low volatility and can partition into condensed phases. As a result, RONO2 account for a significant fraction of submicron organic aerosol, ranging from 5 % to 77 % (Kiendler-Scharr et al., 2016; Ng et al., 2017). The reactivity of RONO2 in condensed phases may differ from that in the gas phase and may affect the role of RONO2 as NOx reservoirs. For instance, it is well established that the hydrolysis of tertiary and allylic RONO2 serves as a fast and permanent sink of NOx in the atmosphere, as the nitrate group is transformed into nitric acid (Darer et al., 2011; Rindelaub et al., 2015; Hu et al., 2011). However, only a small fraction of RONO2 (between 9 % and 34 % for α- and β-pinene-related RONO2) undergo hydrolysis (Takeuchi and Ng, 2018; Wang et al., 2021). Other aqueous-phase reactions are thus to be considered: photolysis and ⋅ OH oxidation. Our previous studies have emphasized the significance of aqueous-phase reactivity for atmospherically relevant RONO2, such as isoprene and terpene nitrates, with intermediate to high water solubilities (González-Sánchez et al., 2021, 2023a). Under typical cloud/fog conditions (liquid water content (LWC) of 0.35 g m−3), the aqueous-phase photoreactivity can act as a major sink (> 50 %) for water-soluble RONO2 (KH>105 M atm−1), while at a very low LWC (3 ⋅ 10−5 g m−3), it can serve as a major sink for very highly water-soluble RONO2 (KH > 109 M atm−1). Nevertheless, the fate of the nitrate group during these processes is still unknown, and it is uncertain whether this reactivity acts as a NOx sink or as an additional transport mechanism.
This work intends to address these questions for the aqueous-phase photolysis of RONO2. While aqueous-phase photolysis is a minor sink when compared with ⋅ OH oxidation (González-Sánchez et al., 2023a), the investigation of the photolysis mechanisms is of high importance as it is a first step towards the fundamental understanding of photooxidation pathways of RONO2.
To explore the aqueous-phase photolysis of RONO2, the fate of four molecules (i.e., isopropyl nitrate, isobutyl nitrate, α-nitrooxy acetone, and 1-nitrooxy-2-propanol) was experimentally investigated. These RONO2 served as proxies to understand the fate of the nitrate group. The two alkyl nitrates are simple molecules, simplifying the comprehension of mechanisms related to the nitrate group's reactivity. Furthermore, the other two investigated RONO2 are polyfunctional; they combine highly relevant functional groups (hydroxy and carbonyl groups) with the nitrate group, allowing for the assessment of their influence on the reactivity. The aqueous-phase photolysis primary and secondary reaction products were identified and quantified, and the fate of the nitrate group was elucidated with support from theoretical calculations. The atmospheric implications of these findings are discussed.
2.1 Experimental setup
The aqueous-phase photolysis experiments were conducted using the experimental setup previously described in detail by González-Sánchez et al. (2023a). Briefly, a 450 cm3 double-wall Pyrex aqueous-phase photoreactor filled with 400 mL of aqueous solution and covered by a quartz lid was used. It was equipped with four apertures for reagent injections and sampling and an optical IDS dissolved oxygen sensor FDO® 925 (WTW), which included temperature monitoring. The reactor was thermostated at 298 K and continuously stirred. Irradiation was provided by an arc light source (LOT Quantum Design) equipped with a 1000 W arc xenon lamp. Wavelengths below 290 nm were removed using an ASTM 892 AM1.5 standard filter (see lamp spectra in Fig. S1 in the Supplement along with the liquid-phase absorption cross-sections of the investigated RONO2) (González-Sánchez et al., 2023). A constant distance of 18.4 cm between the lamp and the water surface was carefully maintained in all experiments.
2.2 Photolysis experiments
Prior to each photolysis experiment, the photoreactor was filled with Milli-Q water, and RONO2 were added. The solution was stirred for 30 min in the dark to ensure complete dilution of the RONO2. Meanwhile, the lamp was turned on for 10 min to stabilize the light beam. The first aliquot was sampled when the reactor was placed under the light beam, marking the reaction time as 0. Photolysis reactions were performed for 4 to 7 h at 298.0±0.2 K. The specific experimental conditions of all photolysis experiments are appended to Table 1.
During the reaction, aliquots were regularly sampled for offline analyses. The pH of the reaction mixture was measured using a 9110DJWP pH probe (Thermo Scientific). Ultrahigh-performance liquid chromatography ultraviolet (UHPLC-UV) detector analyses were performed to monitor the decay of RONO2 or to identify and quantify carbonyl compounds after 2,4-dinitrophenylhydrazine (DNPH) derivatization. High-performance ionic chromatography conductivity detector (HPIC-CD) analyses were conducted to quantify HNO2, HNO3, and organic acids. At the end of the reaction, the remaining volume was used to perform liquid–liquid extraction and gas chromatograph–mass spectrometer (GC–MS) analyses to identify the formed oxidized RONO2. In experiment 1, the headspace of the reactor was monitored with a NOx analyzer to investigate the possible formation of these compounds.
Table 1Initial conditions and analytical instruments used during the photolysis experiments of four individual RONO2.
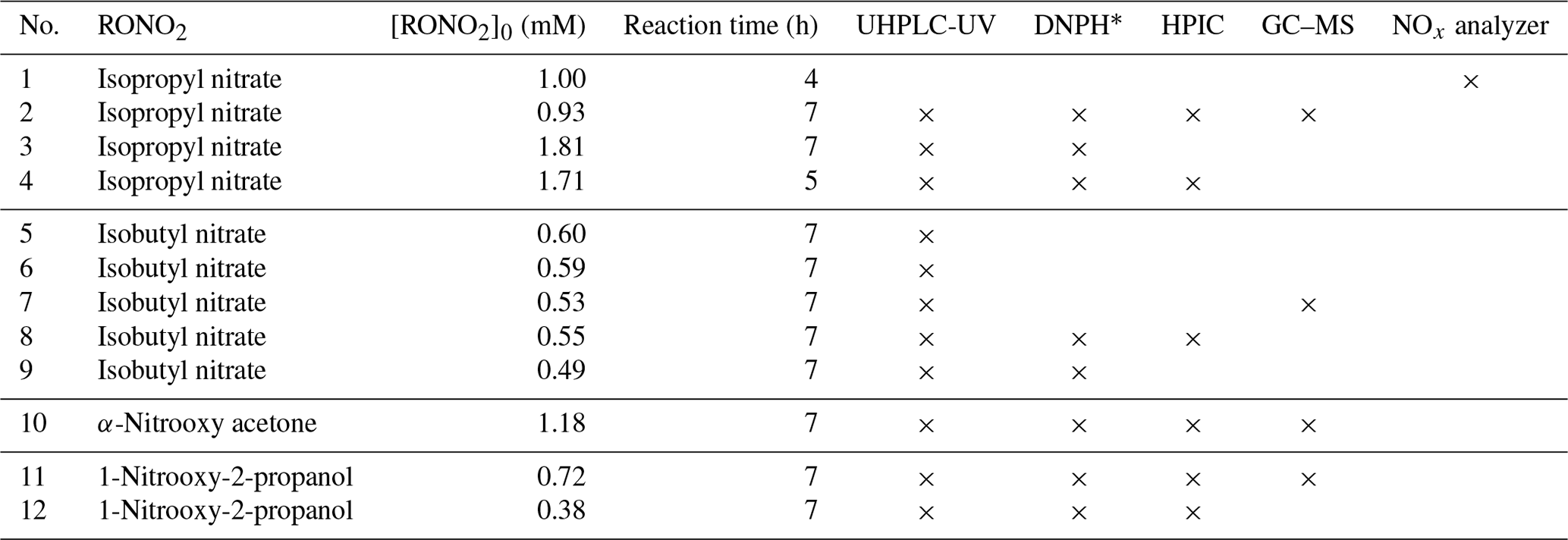
∗ UHPLC-UV analyses after DNPH derivatization of the sample were used to identify and quantify carbonyl compounds. All experiments were performed at 298.0±0.2 K.
2.3 Analytical instruments
2.3.1 UHPLC-UV
An ultrahigh-performance liquid chromatography (UHPLC) ultraviolet (UV) detector (Thermo Scientific Accela) equipped with a Hypersil Gold C18 column (50×2.1 mm) with a particle size of 1.9 µm and an injection loop of 5 µL was used to quantify carbonyl compounds after 2,4-dinitrophenylhydrazine (DNPH) derivatization and RONO2.
Measurements of RONO2
A binary eluent of H2O and CH3CN was used for all analyses, with a flow rate of 400 µL min−1. Two gradients were used depending on the polarity of the compounds. For isopropyl nitrate and isobutyl nitrate, the gradient started at () and was gradually adjusted to () over 3 min; it was held at this proportion for 1 min and then set back to () within 10 s until the end of the run at 5 min. For more polar compounds, i.e., α-nitrooxy acetone and 1-nitrooxy-2-propanol, a similar gradient was employed, but the initial and final proportions were adjusted to () to optimize their retention times (rt's). All RONO2 were detected at their maximum absorbance wavelength at 200 nm (González-Sánchez et al., 2023a).
Calibration curves were linear (as determined by the Mandel test) between and mol L−1 with R2>0.9995. Aliquots with expected concentrations higher than mol L−1 were diluted before analyses. The retention times were 0.9, 1.2, 2.4, and 3.3 min for 1-nitrooxy-2-propanol, α-nitrooxy acetone, isopropyl nitrate, and isobutyl nitrate, respectively (Fig. S2). Limits of detection (LODs) were mol L−1 for isopropyl nitrate and mol L−1 for the three other compounds.
Measurements of carbonyl compounds
To derivatize the carbonyl compounds, 500 µL of the aqueous sample was mixed with 450 µL of 0.005 M DNPH and 50 µL of 0.1 M HCl. The mixture was allowed to react for 24 h to achieve high yields. A specific method was developed for separating and quantifying the formed hydrazones. The gradient, with a flow rate of 400 µL min−1, started from () for 1 min and was then gradually adjusted to () over 6 min; it was held at this proportion for 1 min and then set back to () within 10 s until the end of the run at 9 min.
The resulting hydrazones from formaldehyde, acetaldehyde, acetone, hydroxyacetone, and isobutyraldehyde were identified and quantified at 360 nm. External calibrations were performed to quantify carbonyl compounds with concentrations ranging from to M and R2>0.9995. Their retention times were 4.4, 5.1, 5.8, 3.8, and 6.8 min, respectively. LODs were 4.1, 2.1, 1.5, 4.2, and M, respectively.
2.3.2 HPIC-CD
The formation of HNO2 and HNO3 and organic acids, such as formic acid and acetic acid, was quantified using the Dionex ICS-3000 high-performance ionic chromatography (HPIC) system with a Dionex IonPac™ AG11-HC precolumn (4×50 mm) and a Dionex IonPac™ AS11-HC column (4×250 mm) coupled with a CD25 conductivity detector.
A binary eluent gradient method composed of H2O and NaOH 0.1 mol L−1 aqueous solution was optimized to separate the formed organic acids at relatively short retention times. At a flow rate of 1 mL min−1, the gradient started at 0.1 mol L−1 () for 10 min, then gradually went to () at 12 min, then went back to () within 1 min and was held at this proportion until the end of the analyses at 25 min. The injection volume was 200 µL, and a constant flow of H2SO4 0.05 M continuously passed through the suppressor at a flow rate of 3 mL min−1.
The retention times of acetate, formate, NO, and NO were 5.9, 7.2, 17.3, and 21.9 min, respectively. Calibration curves were optimized to obtain good linearity and low LODs (within the concentrations range expected). LODs were , , , and M for acetic acid, formic acid, NO, and NO, respectively.
2.3.3 GC–MS
The Clarus® 680 gas chromatograph (GC; PerkinElmer) equipped with the Elite-5MS capillary column (PerkinElmer) with a length of 30 m, diameter of 0.25 mm, and film thickness of 0.25 µm coupled with an AxION® iQT™ quadrupole time-of-flight mass spectrometer (MS; PerkinElmer) was used to qualitatively detect and identify oxidized RONO2 formed during the aqueous-phase photolysis experiments. RONO2 were extracted and preconcentrated from the remaining solution after the end of each photolysis experiment. The remaining solutions were stored at ∼ 4 ∘C for up to 48 h before the analyses.
A total of 100 mL of the remaining solution was extracted using 3×20 mL of dichloromethane in a separatory funnel. UHPLC-UV analyses of the aqueous phase before and after the extraction confirmed that all RONO2 efficiently partitioned to dichloromethane. The extracts were washed with 20 mL of Milli-Q water and were concentrated in a TurboVap II system (Biotage). The concentration workstation used a nitrogen flow at 11 psi and a water bath at 30 ∘C to evaporate dichloromethane until a 500 µL sample was obtained.
A total of 1 µL of the concentrated extract was then injected into the GC–MS. The carrier gas was helium at a flow rate of 1 mL min−1. A split of was used due to the high concentration of the compounds. The injector temperature was set to increase from 60 to 200 ∘C within 1 min to prevent thermolysis of RONO2. The following program was set in the oven: hold the temperature for 10 min at 30 ∘C, increase it to 300 ∘C at a rate of 15 ∘C min−1, and hold it for 10 min at 300 ∘C before the end of the analyses.
The analytes were detected with a time-of-flight mass spectrometer using electron impact ionization with an electron energy of 70 eV and an ion source temperature of 250 ∘C. The ion source was turned on 5–7 min after the analysis started in order to avoid the saturation of the source due to the solvent signal. The detector performed full scan measurements from to 300 amu. The mass-to-charge ratio of the ion NO (), specific to RONO2, was extracted to detect these compounds. Seven known RONO2 were analyzed by GC–MS to investigate their retention times and fragmentation patterns (Sect. S1 in the Supplement).
2.3.4 NOx analyzer in the reactor's headspace
The CLD 88 p Eco Physics NOx analyzer was used to determine if ⋅ NO and ⋅ NO2 were formed and partitioned into the gas-phase headspace of the solution during the photolysis of isopropyl nitrate. Indeed, both ⋅ NO and ⋅ NO2 are highly volatile compounds (K M atm−1 and K M atm−1, respectively; Sander, 2015), i.e., from 30 to 107 times more volatile than the investigated RONO2. Therefore, if any ⋅ NO or ⋅ NO2 was formed during the aqueous-phase photolysis, it would have partitioned to the reactor's headspace.
As the NOx analyzer monitored the headspace of the reactor, a specific experimental setup consisting of a hermetic 1 L three-neck round-bottom flask was used (Fig. S3). It was irradiated by the lamplight beam on its side. Note that since the reactor's headspace was also illuminated, photolysis of isopropyl nitrate could occur in the reactor's headspace. However, although isopropyl nitrate is highly volatile, most of the compound remained in the aqueous phase in the timescale of the experiments (only 3 % of isopropyl nitrate partitioned into the reactor's headspace after 7 h) (González-Sánchez et al., 2023a).
The NOx analyzer LOD is 0.1 ppbv for both ⋅ NO and ⋅ NO2. Considering the gas-phase dilutions performed downward of the reactor, ⋅ NOx could be detected, if formed, at concentrations higher than ∼ 2 ppbv using this setup. Although the CLD 88 p Eco Physics NOx analyzer uses a photolytic converter, interferences with the RONO2 were observed. A slight proportion (less than 0.6 %) of gas-phase isopropyl nitrate was detected as ⋅ NO2. Further details are given in Sect. S2.
In addition, a control experiment was performed to test the efficiency of the gas-phase ⋅ NO2 photolysis and conversion to ⋅ NO under our experimental conditions by bubbling gas-phase ⋅ NO2 into the reactor's aqueous phase and photolyzing it with the lamplight. The experimental setup is depicted in Fig. S4.
2.4 Molar yield determinations
The molar yields of the primary reaction products were determined by plotting their concentrations against Δ[RONO2], which represents the consumption of the parent organic nitrate (Eq. 1).
Since the reaction products were susceptible to undergoing photolysis over time, the yields were calculated for the initial aliquots, sampled during the first 1–2 h of reaction. The evaporation rate of some RONO2 could be non-negligible compared with photolysis (González-Sánchez et al., 2023a), and Δ[RONO2] was thus systematically corrected from evaporation using Eqs. (2)–(3):
where [RONO2]0 is the initial concentration and kexp the pseudo-first-order decay, determined by fitting the RONO2 concentrations ([RONO2]t) versus time (t) following Eq. (4).
The evaporation rate constant, kvap, was determined by control experiments reported in González-Sánchez et al. (2023a).
2.5 Theoretical calculations
Theoretical simulations of the photolysis reaction of isopropyl nitrate photolysis were performed in a model of aqueous solution and in the gas phase. To build the model in the gas phase, snapshots from 10 ps ab initio molecular dynamics (MD) dynamics were performed using a thermostat at 300 K. For the static calculations of minima, conical intersections, and transition states we used B3LYP/6–31G* for the ground-state calculations and TDDFT using the Tamm–Dancoff approximation for the excited-state calculations, with water treated as an implicit solvent via a polarizable continuum model (PCM). To perform the quantum dynamic simulations, we built a model of isopropyl nitrate in aqueous solution by first constructing a water box of 11.39 nm3 equilibrated using Amber99 TIP3P water force field parameters. Isopropyl nitrate was then soaked in the water box and re-equilibrated with the following protocol (see Sect. S3): (1) a canonical ensemble molecular mechanics (MM) MD at a fixed isopropyl geometry during 125 ps; (2) an isobaric–isothermal MM MD at a fixed isopropyl during 1 ns; and (3) an isobaric–isothermal B3LYP/6–31G*//Amber99 quantum mechanics/molecular mechanics (QM/MM) MD in period boundary conditions, relaxing the full system for 12 ps (Bonfrate et al., 2023). The snapshots were taken from the latter, discarding the first 2 ps. For each snapshot, a water droplet of 1 nm was extracted, including a spherical wall potential to avoid evaporation of water during the excited-state dynamics. In each snapshot (gas phase and aqueous solution), non-adiabatic excited-state molecular dynamics were operated using Tully's fewest-switch surface-hopping algorithm (Huix-Rotllant et al., 2023). The trajectories were started from the second excited state (S2). Excited states were computed using mixed-reference time-dependent density-functional theory, which can describe the multi-configuration character of wave functions during photolysis at the cost of a density-functional theory calculation (Lee et al., 2018; Huix-Rotllant et al., 2023).
2.6 Reagents
Chemicals were commercially available and were used as supplied: isopropyl nitrate (96 %, Sigma Aldrich), isobutyl nitrate (98 %, Sigma Aldrich), chloroacetone (95 %, Sigma Aldrich), AgNO3 (99 %, VWR Chemicals), KI (98 %, Sigma Aldrich), NaBH4 (98 %, Sigma Aldrich), 2,4-dinitrophenylhydrazine (≥ 99 %, Sigma Aldrich), formaldehyde DNPH (≥ 99 %, Sigma Aldrich), acetaldehyde DNPH (≥ 99 %, Sigma Aldrich), acetone DNPH (≥ 99 %, Sigma Aldrich), isobutyraldehyde (≥ 99 %, Sigma Aldrich), formaldehyde DNPH (≥ 99 %, Sigma Aldrich), hydroxy acetone (95 %, Alfa Aesar), acetic acid (≥ 99.7 %, Sigma Aldrich), formic acid (≥ 96 %, Sigma Aldrich), (34 %–37 %, TraceMetal Grade, Fisher Chemical), nitrite standard for IC (1000±4 mg L−1, Sigma Aldrich), nitrate standard for IC (1000±4 mg L−1, Sigma Aldrich), NaOH (46 %–51 %, analytical reagent grade, Fisher Chemical), and H2SO4 (95 %–98 %, Merck). Acetonitrile (Fisher Optima) and isopropanol (Honeywell) were LC/MS grade and were used as supplied. Acetone (Carlo Erba Reagents), dichloromethane (Fisher Chemical), and ether (Fisher Chemical) were HPLC grade. Tap water was purified with the Millipore Milli-Q system (18.2 MΩ cm and total organic carbon (TOC) < 2 ppb). Gases were used as supplied: synthetic air (Linde, > 99.999 stated purity), helium 5.0 (Linde), and ⋅ NO2 (2 ppm in He 5.0, Linde).
Non-commercial organic nitrates, i.e., α-nitrooxy acetone and 1-nitrooxy-2-propanol, were synthesized and purified. α-Nitrooxy acetone was synthesized by the nucleophilic substitution reaction of iodoacetone, which was synthesized previously from chloroacetone. The ketone group from α-nitrooxy acetone was reduced to produce 1-nitrooxy-2- propanol (see details in González-Sánchez et al., 2023a).
The results of the aqueous-phase photolysis of organic nitrates are presented in a stepwise manner. Since NOx compounds are the known major primary product formed in the gas-phase photolysis of RONO2, this process is examined first in Sect. 3.1, which describes the attempt to measure any formation and partitioning of NOx to the headspace of the reactor. Section 3.2, 3.3, and 3.4 present the identified reaction products in the aqueous phase, including HNO2, HNO3, carbonyls, organic acids, and oxidized RONO2, and their associated yields. All results are reported in Table S1. Finally, Sect. 4 provides a detailed discussion of the mechanisms involved, focusing on the fate of the nitrate group.
3.1 Absence of NOx in the reactor's headspace
Experiment 1 investigated isopropyl nitrate (1 mM) photolysis by analyzing the reactor's gas-phase headspace with a NOx analyzer (Fig. 1a). Prior to turning on the lamp, the ⋅ NO2 signal increased up to ∼ 150 ppb, corresponding to a fraction of gas-phase isopropyl nitrate that was photolyzed inside the NOx analyzer photolytic converter (see Sect. S2 for further details). Once the lamp was turned on (shown in shaded blue in Fig. 1a), the aqueous-phase photolysis of isopropyl nitrate started, but no ⋅ NO signal was detected, while the ⋅ NO2 signal peaked at 800 ppb within ∼ 10 min of photolysis. However, this signal did not correspond to ⋅ NO2, as demonstrated by the control experiment where ∼ 800 ppb of ⋅ NO2(g) was bubbled through the same volume of ultrapure water. When the lamp was turned on (shown in shaded blue in Fig. 1b), ⋅ NO2(g) was effectively photolyzed, directly forming ⋅ NO(g). In this experiment, barely any ⋅ NO2(g) partitioned to the aqueous phase (confirmed by the absence of aqueous-phase HNO2 or HNO3, measured by HPIC), and thus the photolysis of ⋅ NO2(g) exclusively occurred in the reactor's headspace. From this control experiment, it was concluded that if the measured ⋅ NO2 signal represented actual ⋅ NO2(g) directly formed in experiment 1, it would be photolyzed in the headspace of the photoreactor to produce measurable amounts of ⋅ NO(g).
Since no ⋅ NO(g) was observed when the lamp was turned on in experiment 1 (Fig. 1a), one can conclude that no substantial amounts of ⋅ NO2(g) were present in the system. The signal detected as ⋅ NO2(g) likely represented an interfering reagent. Nitrous acid (HONO or HNO2) cannot be this interfering reagent since the concentrations of the interference in the reactor decreased as the reaction progressed, while the measured HONO in the aqueous phase continuously increased. It likely corresponded to another volatile N-containing compound that was detected by the NOx analyzer as a ⋅ NO2 signal (as isopropyl nitrate does). Its signal could be higher than that observed for isopropyl nitrate if the compound presented less water solubility and/or if it decomposed more efficiently in the photocatalytic converter of the NOx analyzer. It is worth noting that the estimated interference for isopropyl nitrate is very low; it would represent 0.01 % if equilibrium was reached.
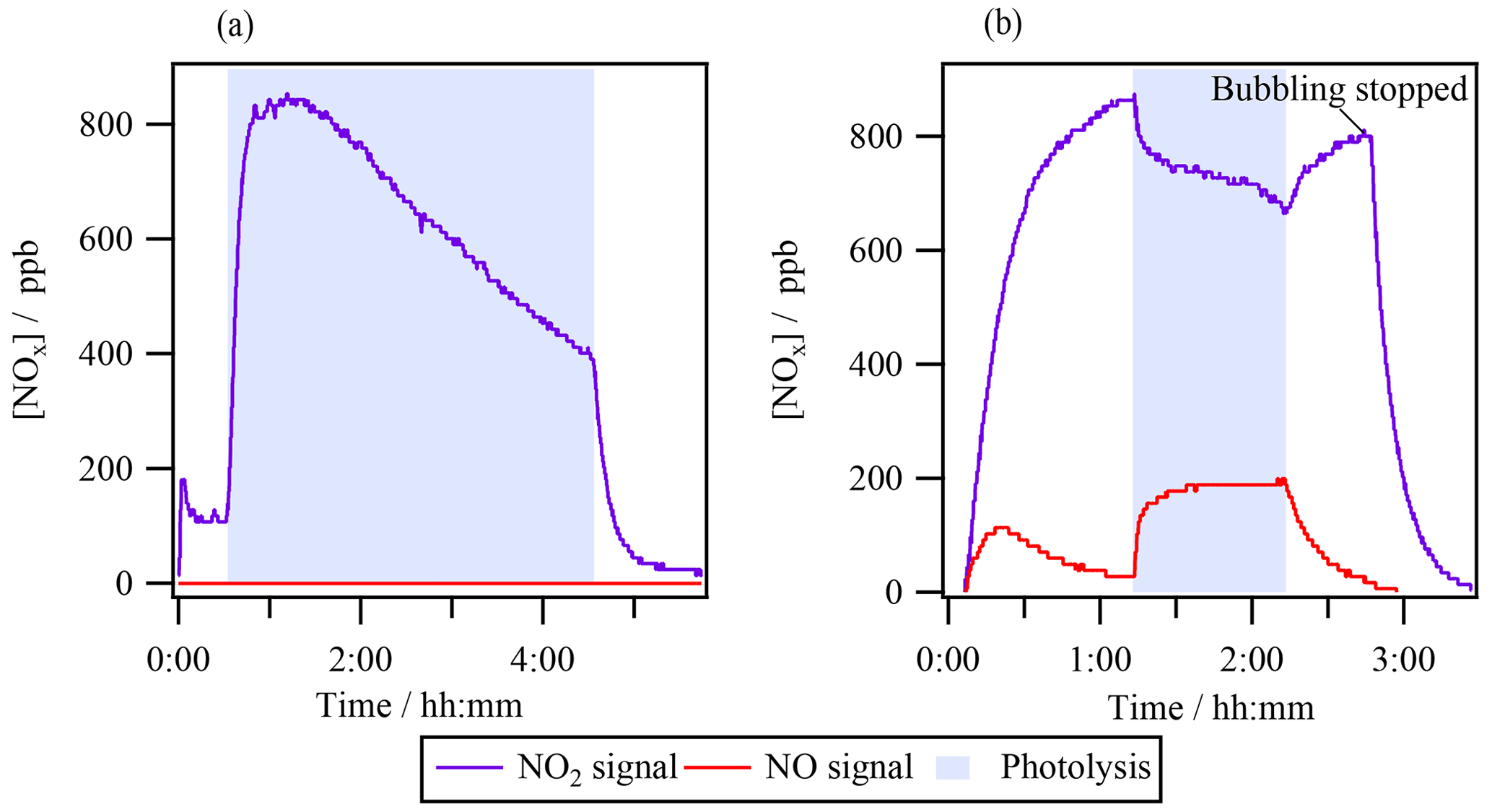
Figure 1Headspace time profiles of ⋅ NO(g) and ⋅ NO2(g) signals during (a) aqueous-phase photolysis of isopropyl nitrate with [RONO2]0 = 10−3 M (exp. 1 in Table 1) and (b) photolysis of ⋅ NO2(g) bubbled in water.
3.2 Formation of HNO2 and HNO3
HNO2 and HNO3 were formed during aqueous-phase photolysis of RONO2. Both compounds were detected as NO and NO using the HPIC-CD, but their formation as acids was inferred by the observed fast decrease in pH (Fig. S5) and was confirmed by theoretical calculations (see Sect. 4.1.1).
Figure 2 shows an example of HNO2 and HNO3 time profiles during the photolysis experiments of isopropyl nitrate (Fig. 2a), isobutyl nitrate (Fig. 2b), α-nitrooxy acetone (Fig. 2c), and 1-nitrooxy-2-propanol (Fig. 2d). HNO2 could not be quantified during the aqueous-phase photolysis of α-nitrooxy acetone due to its fast hydrolysis in the HPIC system that used high-pH eluents, where the molecule decomposes into lactate and NO (Brun et al., 2023).
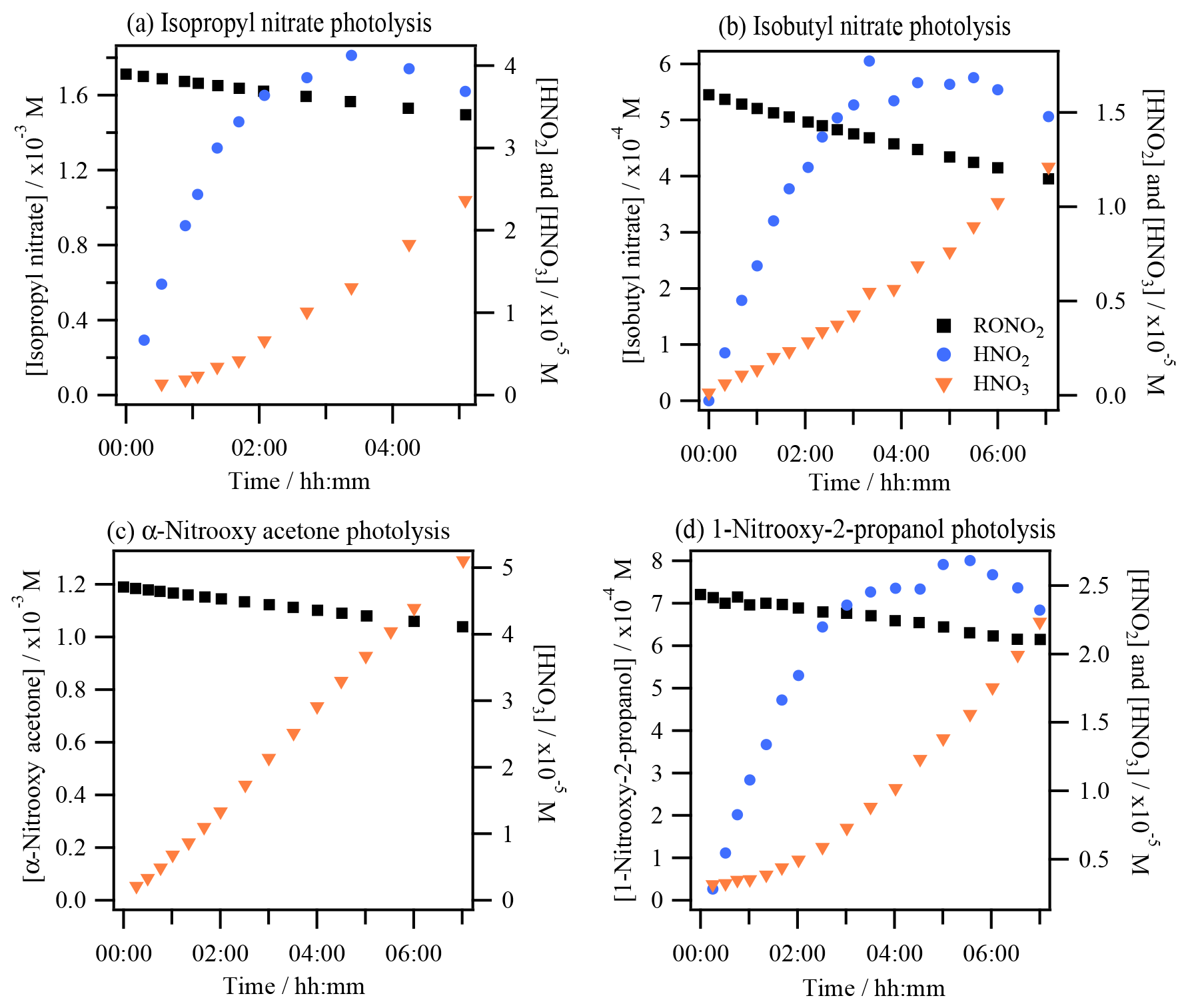
Figure 2Photolysis experiments of RONO2: time profiles of RONO2, HNO2, and HNO3 for (a) isopropyl nitrate (exp. 4), (b) isobutyl nitrate (exp. 8), (c) α-nitrooxy acetone (exp. 10), and (d) 1-nitrooxy-2-propanol (exp. 11). HNO2 and HNO3 were detected as NO and NO in the HPIC-CD.
The figure shows that HNO2 was efficiently formed as a primary product during all aqueous-phase photolysis reactions of RONO2. HNO2 formation slowed down over time due to its fast oxidation to HNO3, whose time profiles present exponential growth due to its secondary formation. Since this conversion is fast, HNO3 formation of the first aliquots has been included in the HNO2 primary yields, assuming that all HNO3 was formed via HNO2 oxidation. The detailed chemistry of HNO2 and HNO3 that validates this approach is discussed in Sect. 4.1.2. The HNO2 yields ranged from 40 % to 59 % for isopropyl nitrate (experiments 2 and 4) and from 59 % to 62 % for 1-nitrooxy-2-propanol (experiments 11 and 12), and they were 31±7 % for isobutyl nitrate (experiment 8) and higher than 28 % for α-nitrooxy acetone (experiment 10).
3.3 Formation of carbonyl compounds and organic acids
The formation of primary and secondary carbonyl compounds and organic acids was observed during the aqueous-phase photolysis of RONO2 (Fig. 3).
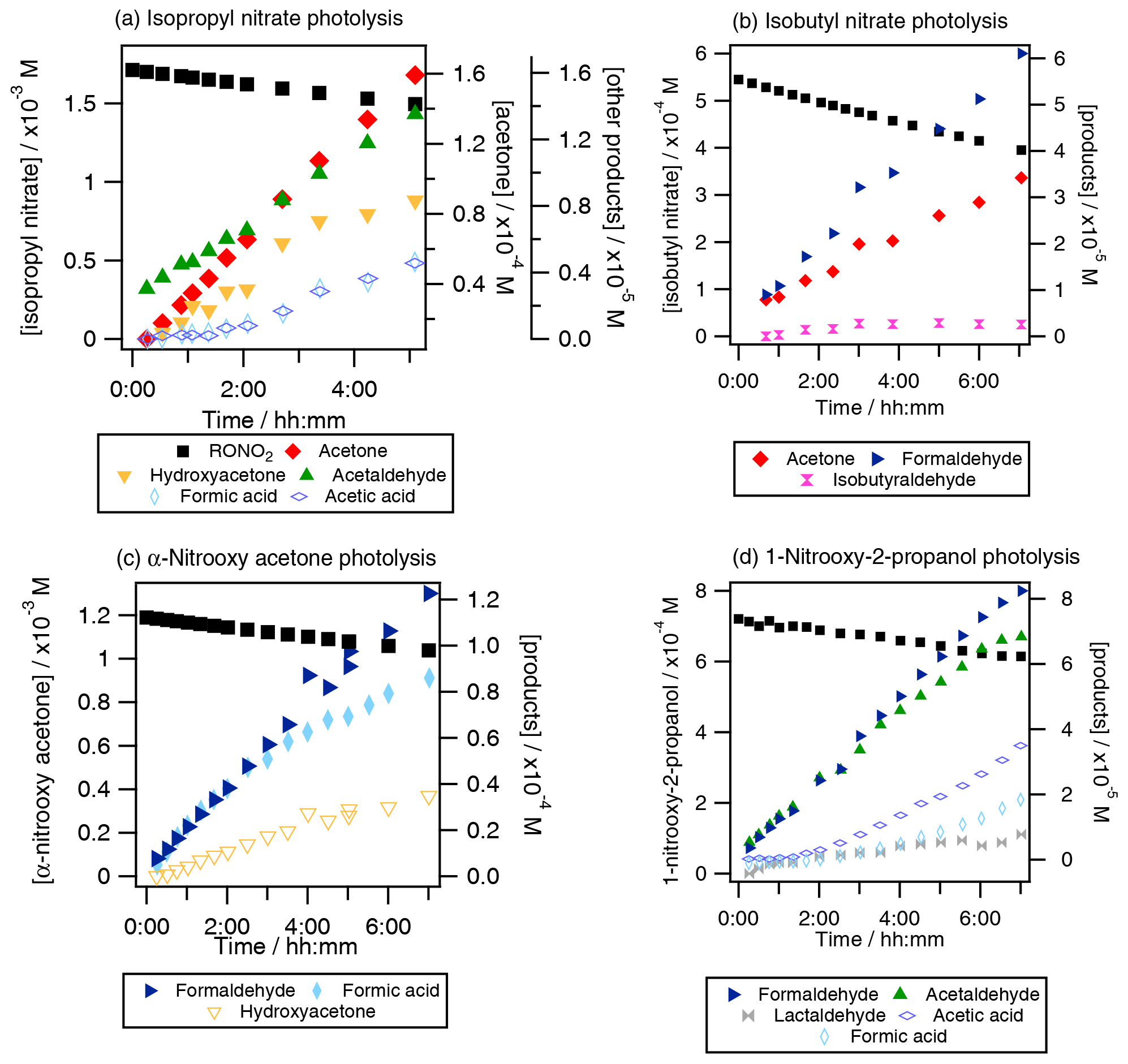
Figure 3Photolysis experiments of RONO2: time profiles of RONO2, carbonyl compounds and organic acids for (a) isopropyl nitrate (Exp. 4), (b) isobutyl nitrate (Exp. 8), (c) α-nitrooxy acetone (Exp. 10), and (d) 1-nitrooxy-2-propanol (Exp. 11). Plain markers are used for primary products.
For isopropyl nitrate (Fig. 3a), the main primary reaction product was acetone, with yields ranging from 32 % to 88 % (experiments 2–4), and acetaldehyde was formed primarily with lower yields (5 %). Additionally, hydroxyacetone, formic acid, and acetic acid were formed as secondary products. These compounds were likely formed via acetone photooxidation (Poulain et al., 2010). For isobutyl nitrate (Fig. 3b), formaldehyde and acetone were the main non-nitrogen-containing photolysis products (primary yields of 37 %–39 % and 20 %–32 %, respectively, experiments 8–9). Additionally, isobutyraldehyde was detected as a minor product (with a primary yield of 4 %–5 %). For α-nitrooxy acetone (Fig. 3c), formaldehyde and formic acid appeared as primary products, while hydroxyacetone and acetaldehyde were likely secondary products. The formation yields were found to be 96±5 % and 79±3 % for formic acid and formaldehyde, respectively. Other reaction products, such as acetic acid and methylglyoxal, were identified but not quantified due to interferences in the analyzers caused by hydrolysis of α-nitrooxy acetone or oligomerization of methylglyoxal (see Sect. S4). For 1-nitrooxy-2-propanol (Fig. 3d), formaldehyde and acetaldehyde were identified as the main primary reaction products with yields of 63 %–71 % and 50 %–70 %, respectively. Furthermore, lactaldehyde was detected as a primary product with a minor yield of 8 %–14 %. Formic acid and acetic acid were observed as secondary products, likely formed via the photooxidation of formaldehyde and acetaldehyde.
3.4 Secondary formation of oxidized RONO2
GC–MS analyses at the end of each reaction were performed to look for nitrogen-containing organic products. Figure 4a compares the gas chromatograms obtained for the sample analyzed after 7 h of photolysis with one obtained during a control experiment of isopropyl nitrate in the dark. In both chromatograms, (which corresponds to a NO fragment) was extracted to display chromatographic peaks related to compounds of RONO2. The figure shows the formation of at least five oxidized RONO2 (IP 1, IP 2, IP 3, IP 4, and IP 5), with IP 3 presenting an intensity of 1 order of magnitude higher than the others.
The observed compounds were less volatile than isopropyl nitrate (which had a retention time of 6 min, not shown in Fig. 4a) given their higher retention times and were thus probably oxidized species. The mass spectra of IP 1 to IP 5 confirm that all compounds were RONO2 with similar chemical structures as isopropyl nitrate (shown in the bottom right of Fig. 4b for comparison). Apart from the NO fragment, other fragments observed for isopropyl nitrate were detected. Fragments such as C3H () and C2H4ONO () were observed in IP 2, IP 3, and IP 5 (and also in IP 1 for ). Note that can also correspond to an oxygenated fragment (C2H3O+), but the resolution of 1 amu did not allow for separation from C3H fragments. Additionally, a specific fragment of a molecule of RONO2 bearing its nitrate group on a primary carbon atom (CH2ONO at ) was observed for IP 1, IP 3, IP 4, and IP 5. Since IP 3 and IP 5 combine this fragment with a fragment that is specific for the secondary nitrate group (C2H4ONO at ), these compounds might be dinitrates. This is the case for the most intense chromatographic peak (IP 3). IP 3 was thus assigned to the 1,2-propyl dinitrate molecule due to its mass spectra. Additionally, IP 2 was assigned to 2-nitrooxy-1-propanol due to the C3H7O+ and C2H5ONO fragments ( and , respectively). These identifications are consistent with the proposed mechanism (see Sect. 4.2). However, the absence of standards prevented the precise identification and quantification of these compounds.
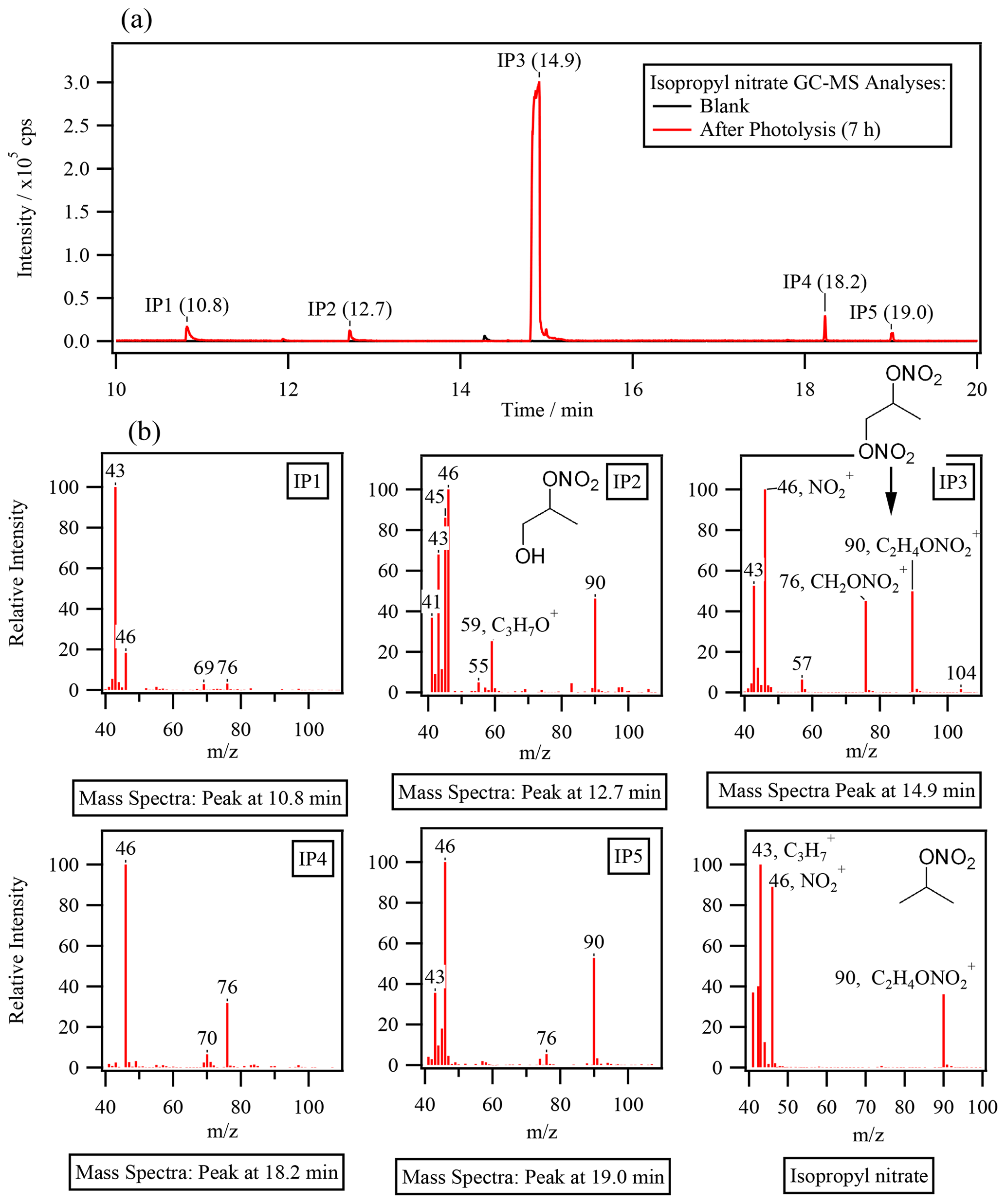
Figure 4Isopropyl nitrate photolysis. (a) Gas chromatogram (extracted for , NO of the end of the reaction of exp. 2 in Table 1 (after 7 h of photolysis) and the control experiment (isopropyl nitrate in water). (b) Mass spectra of the detected peaks.
Hints of the formation of an oxidized molecule of RONO2 were also observed in the non-derivatized UHPLC-UV analyses. An unidentified peak was detected at a retention time close to isopropyl nitrate (2.7 vs. 2.4 min). The peak presented similar UV absorption spectra to the standards of RONO2 (Fig. S6) and was thus assigned to be IP 3 (1,2-propyl dinitrate) due to its major concentrations. The compound was a secondary product since its occurrence started after 2 h of reaction. A rough estimation of its concentration was performed using average calibration curve parameters obtained for isopropyl nitrate, isobutyl nitrate, α-nitrooxy acetone, and 1-nitrooxy-2-propanol. Assuming that IP 3 was a dinitrate, it represented 9 % of the consumed nitrogen at the end of the reaction.
The secondary formation of oxidized RONO2 was also confirmed for isobutyl nitrate and 1-nitrooxy-2-propanol. For isobutyl nitrate, two unidentified peaks assigned to oxidized RONO2 (IB 1 and IB 2) were observed by UHPLC-UV. Both compounds present UV–Vis absorption spectra identical to isobutyl nitrate at lower retention times (1.6 min for IB 1 and 3.1 min for IB 2 vs. 3.4 min for isobutyl nitrate) related to a higher polarity of the molecules. Their time profiles show that both compounds were formed through secondary reactions (Fig. S7). GC–MS analyses (performed after preconcentration of the sample) allowed for the detection of up to nine oxidized RONO2. For 1-nitrooxy-2-propanol, four oxidized RONO2, including α-nitrooxy acetone, were observed by GC–MS. The chromatograms and mass spectra as well as comments on the identification of the formed molecules are presented in the Supplement (Sect. S5).
For α-nitrooxy acetone, no oxidized RONO2 were found in UHPLC-UV analyses or in GC–MS analyses.
4.1 N budget during aqueous-phase photolysis of RONO2
Gas-phase photolysis of RONO2 is known to induce homolytic rupture of the RO–NO2 bond, releasing ⋅ NO2 into the atmosphere with yields close to 100 % (Talukdar et al., 1997; Carbajo and Orr-Ewing, 2010). This reactivity turns RONO2 into NOx reservoirs and shifts pollution transportation from the local to the regional scale. Our results show that, in the aqueous phase, a primary formation of HNO2 (with yields ranging from 31 % to 62 %) is followed by a secondary formation of HNO3. Therefore, one of the main questions about the aqueous-phase photolysis of RONO2 is whether (or not) it can regenerate NOx that would partition to the gas phase.
To address this question, we explored the viability of two different chemical pathways that lead to NO HNO2 and NO HNO3 in the aqueous phase. The first explored pathway was the direct formation of ⋅ NO2,(aq) followed by its known aqueous reactivity (i.e., hydrolysis and reactivity towards other radicals). This pathway was rejected since ⋅ NO and ⋅ NO2 should be observable in the system in this scenario (see details in Sect. S6). The second explored pathway was the direct formation of HNO2 in the aqueous phase. This pathway was confirmed by theoretical calculations for isopropyl nitrate aqueous-phase photolysis. Herein, the Discussion section focuses on this pathway and the secondary chemistry of the photolysis products in our system. Finally, a conclusion is given with proposed mechanisms of aqueous-phase photolysis reactions of isopropyl nitrate, isobutyl nitrate, 1-nitrooxy-2-propanol, and α-nitrooxy acetone, including a detailed discussion for isopropyl nitrate.
4.1.1 Direct formation of HNO2 in the aqueous phase
Theoretical calculations were performed to evaluate if the direct formation of HNO2 is possible in the aqueous phase. The static calculations showed that the formation of HNO2 is thermodynamically favorable. Figure 5 represents the potential energy surfaces of a potential reaction pathway of isopropyl nitrate aqueous-phase photolysis, showing that it is indeed a possible reaction. Upon photon absorption, isopropyl nitrate is in the first excited state and can relax rapidly to the minimum of this state. From the excited state, it undergoes a non-radiative internal conversion back to the ground state through a degenerated point between the excited state and the ground state, a conical intersection. The conical intersection has a sloped topology. −ONO2 presents a pyramidal structure (instead of triangular) in the conical intersection. After reaching the conical intersection, the isopropyl nitrate accumulates all the photon energy as excess vibrational energy. Indeed, this energy is in principle sufficient to cross the large barrier to form HNO2 as a product. The transition state is a concerted RO–NO2 dissociation and a proton transfer, with an imaginary frequency of 1241.3 cm−1, which leads directly to the formation of acetone and HNO2 as final products.
To confirm this pathway, we performed excited-state non-adiabatic dynamics in an atomistic model of isopropyl nitrate in water using QM/MM methodology. The dynamics in such a model allow us not only to determine the reaction hypothesis described in Fig. 5, but also to estimate the timescales at which this reaction can happen. In Fig. 6, an example of a reactive trajectory in the excited state is depicted. Initially, the R–ONO2 is in a trigonal planar conformation. Once the photon is absorbed, the group displays a pyramidal conformation that allows a non-radiative conversion from the excited to the ground state via a conical intersection. This leads directly to the dissociation of ⋅ NO2, which diffuses towards water. This is at odds with the found transition state, which corresponded to a concerted ⋅ NO2 and proton transfer. Therefore, we can rather claim that the reaction happens in two steps. The interaction of ⋅ NO2 with water favors the 180 ∘ twist, in which nitrogen is pointing towards water molecules, thus favoring a conformation in which a proton transfer is favored, occurring in less than 1 ps. Despite the fact that this happens to be the main reaction channel, other reactions, in which direct formation of acetaldehyde or dissociation of HNO2 in ⋅ OH and ⋅ NO is observed, are possible. This is due to the excess vibrational energy of the photoproducts encapsulated in a water cavity with a diameter of around 0.7 nm, which prevents their diffusion. Still, in longer timescales the photoproducts will either react with water or dissipate the energy to the solvent.
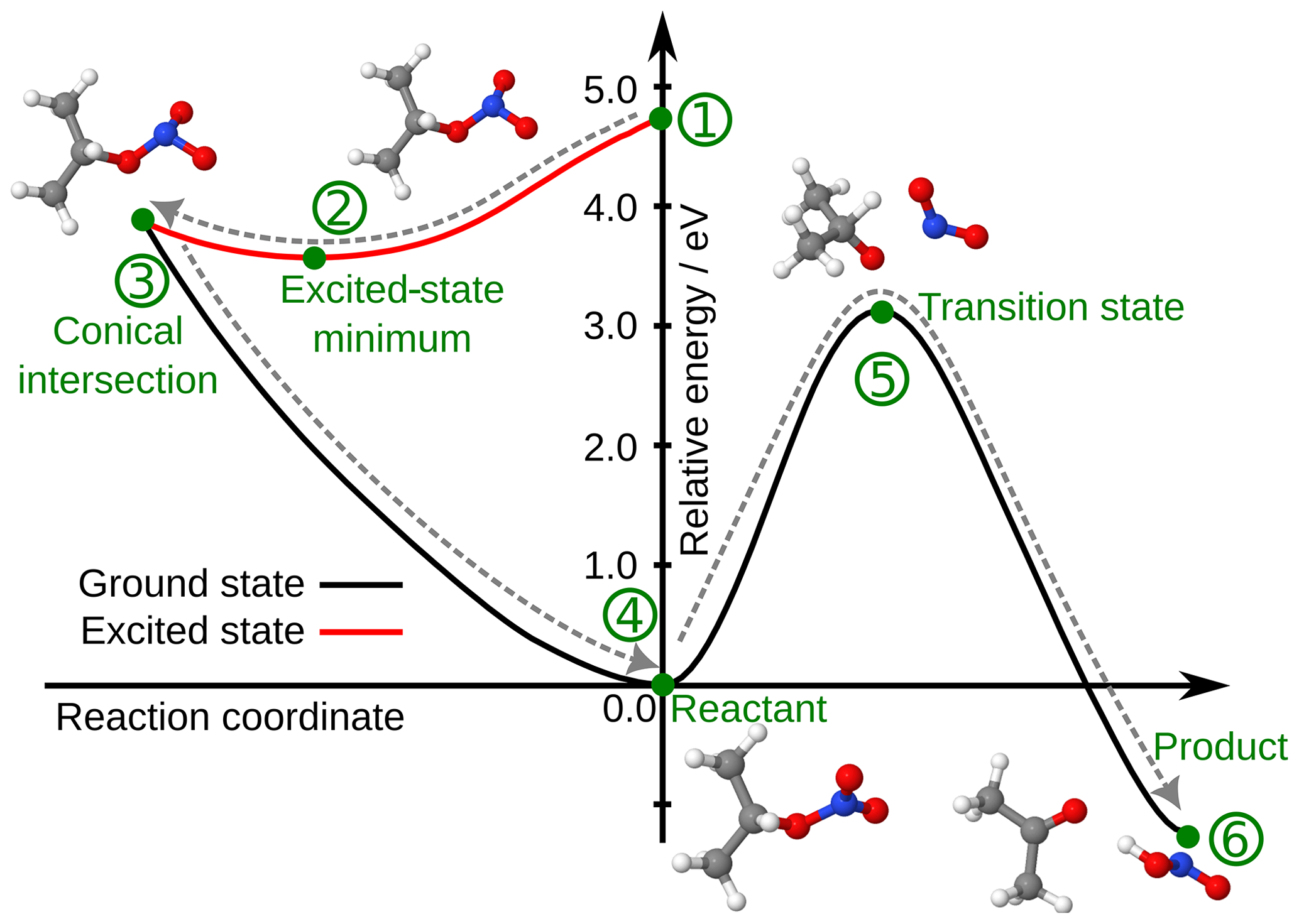
Figure 5Relative energy potential energy surface of isopropyl nitrate photolysis. The structures of the key points of the reaction, marked with green dots, are also depicted in the figure. The grey arrow shows the hypothetical reaction: (1) the molecule is in an excited state after photon absorption, traveling first to the minimum energy structure of the excited state (2) and accessing a conical intersection (3) through a barrierless surface. The molecule then goes back to the reactant (4) with excess vibrational energy, which allows it to cross a transition state (5) up to the products (6). The reactant's energy has been arbitrarily placed at 0.0 eV. Calculations were performed using a density functional theory (DFT)/time-dependent DFT (TDDFT) model in implicit solvent.
The gas-phase photolysis of isopropyl nitrate was also studied using the same type of dynamics. In the gas phase, the photochemical pathway through a conical intersection until the formation of the ⋅ NO2 radical happens in a similar manner to the aqueous solution. In the gas phase, however, the absence of a water cavity prevents the resulting fragments from reorienting and reacting. Indeed, similar to an explosion, the two resulting fragments ⋅ NO2 and RO⋅ diffuse in opposite directions before any proton transfer can occur between them. This explains the observed direct formation of ⋅ NO2,(g) (Talukdar et al., 1997). In contrast, in the cavity, collisions with the solvent are frequent, and thus the photolysis likely follows the pathway with the minimum-energy barrier, which leads to the formation of HNO2 and acetone.
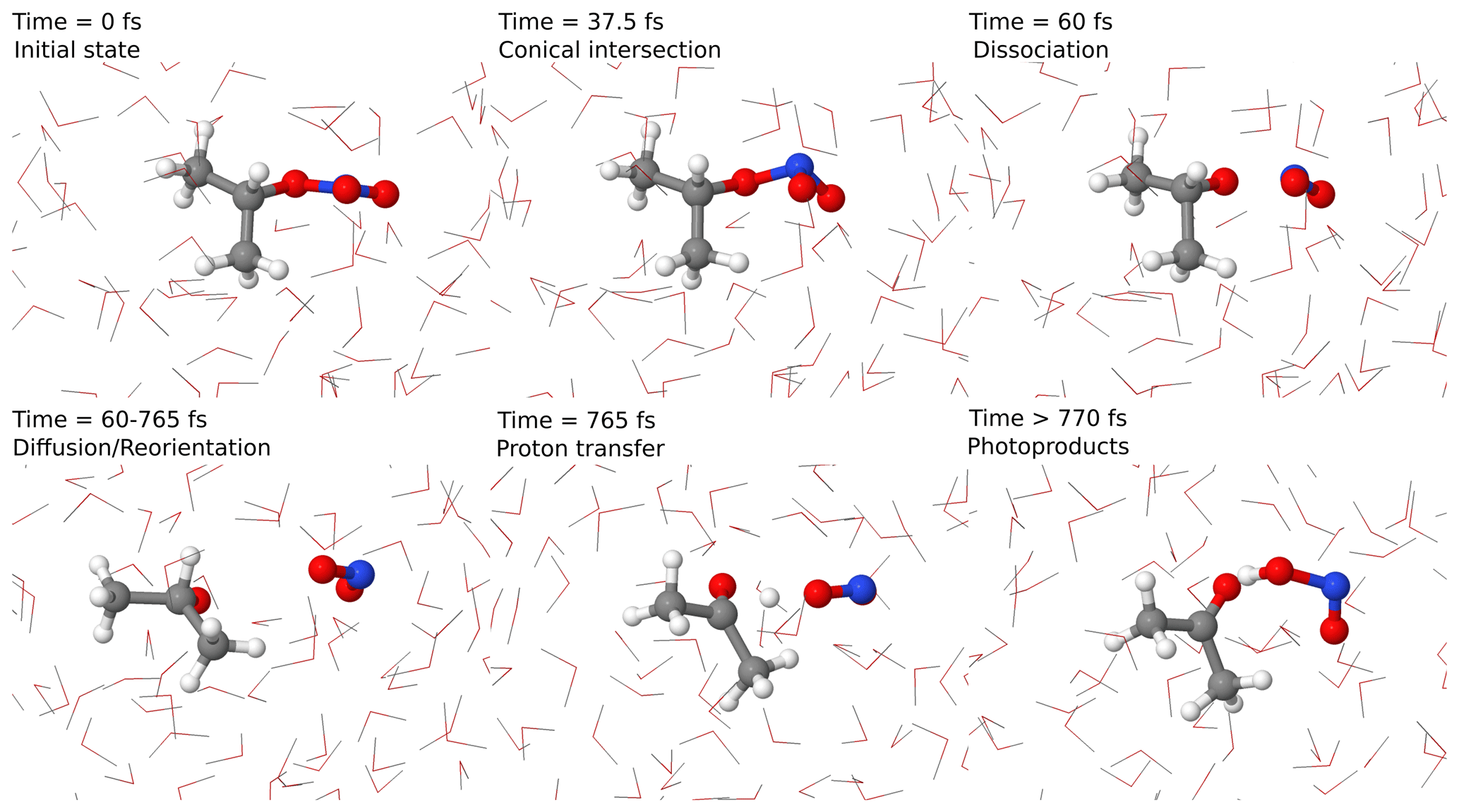
Figure 6Representative reactive trajectories for the formation of acetone and HNO2 with the main reaction steps depicted. Timescales are just indicative.
These calculations agree with our observations that the photolysis of isopropyl nitrate caused the direct concomitant formation of HNO2 and acetone as shown in Fig. 7. Furthermore, minor trajectories where the carbon backbone structure of isopropyl nitrate breaks, leading to the formation of acetaldehyde and other species, have been also experimentally observed since acetaldehyde was determined to be a primary product with low yields (∼ 4 %).
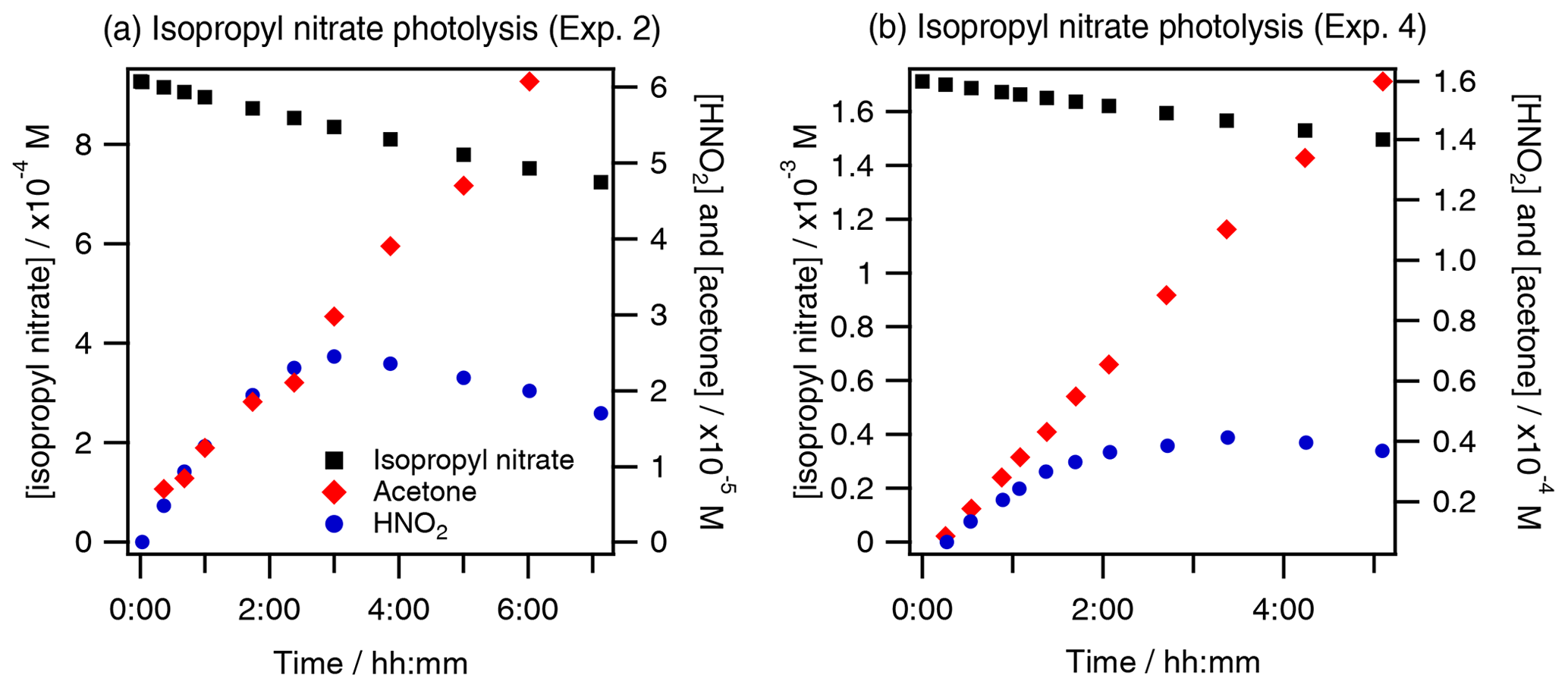
Figure 7Concomitant acetone and HNO2 formation during isopropyl nitrate photolysis at two different initial concentrations, (a) 0.57 mM (exp. 2) and (b) 1.42 mM (exp. 4).
4.1.2 Secondary chemistry of HNO2 in the aqueous phase
Once formed in the solution, HNO2 was highly reactive, as shown by its time profiles (in Fig. 7). It may disproportionate to yield ⋅ NO and ⋅ NO2 (R1).
However, this reaction is quite slow under our experimental conditions (rate constant of 28.6 M−1 s−1, Vione et al., 2004). Nevertheless, considering the lamp actinic flux, the photolysis/photooxidation of HNO2 was likely its major sink. The photolysis of HNO2 and NO is known to form ⋅ NO and a ⋅ OH radical (R2–4) (Mack and Bolton, 1999; Fischer and Warneck, 1996; Kim et al., 2014). HNO2 can also decompose due to the additional energy of the photolysis of RONO2.
Additionally, ⋅ OH radicals readily react with NO and HNO2 (rate constants of 1.0 × 1010 and 2.6 × 109 M−1 s−1, respectively) (Mack and Bolton, 1999; Kim et al., 2014).
HNO3 can then be formed through ⋅ NO2 hydrolysis (R7) (Finlayson-Pitts and Pitts, 2000).
Another pathway can be initiated by the reaction of ⋅ NO with HO2⋅ radicals to yield peroxynitrite (R8). The latter can isomerize into HNO3 NO (R9) or decompose, yielding ⋅ NO2 and ⋅ OH radicals (R10).
HO2⋅ radicals were likely formed by the photooxidation of organic compounds. Since ⋅ OH radicals were formed through HNO2 NO photolysis, they could attack the organic molecules present in the photoreactor (i.e., the RONO2, as no scavenger was used). Upon oxygen addition, the ⋅ OH attack yielded peroxy radicals. The formation of peroxy radicals was confirmed by the dissolved oxygen time profiles: during each photolysis experiment, dissolved [O2] underwent slight decay due to the reaction of alkyl radicals (R⋅) and oxygen (Fig. S8).
Peroxy radicals can readily react with ⋅ NO to form peroxynitrites (ROONO) that can isomerize to RONO2 (R11). Additionally, peroxy radicals react with ⋅ NO2, forming peroxynitrates (ROONO2) (Goldstein et al., 2004).
The rate constants observed for R11 and R12 range, respectively, from 2.8 × 109 to 3.5 × 109 M−1 s−1 (with R being (CH3)2CCH2− and CH3−) and from 0.7 to 1.5 × 109 M−1 s−1 (with R being (CH3)2CCH2−, CH3−, and c-C5H9−). Therefore, these reactions could readily occur under our experimental conditions but not in the first step, as they would be limited by the formation of ⋅ OH radicals to be initiated.
In our experiments, the formation of oxidized RONO2 during isopropyl nitrate and isobutyl nitrate photolysis was confirmed by GC–MS and UHPLC-UV analyses (Fig. 4 and Sect. S4). The possibility of forming oxidized RONO2 via the aforementioned reactions is consistent with the substantial number of compounds displaying the NO fragment found by GC–MS analyses (up to six compounds for isopropyl nitrate photolysis and up to eight for isobutyl nitrate). Nevertheless, ROONO2, if formed, was not detected due to its thermolysis during the analysis.
During isopropyl nitrate photolysis, the main formed oxidized organic nitrate (IP 3 in Fig. 4) was suspected to be a dinitrate (1,2-propyldinitrate) since its mass spectra conjugate mass fragments that correspond to both primary ( = 76, CH2ONO and secondary nitrate groups ( = 90, CH(ONO2)CH. The formation of this compound through secondary photochemistry of HNO2 NO agrees well with the observed secondary time profile of this product. An equivalent compound was observed during isobutyl nitrate photolysis (IB 6 in Fig. S4.1).
4.2 Proposed chemical mechanisms
4.2.1 Proposed mechanism of isopropyl nitrate aqueous-phase photolysis
Combining all the reactions mentioned in the Discussion, Fig. 8 proposes a complete mechanism of isopropyl nitrate aqueous-phase photolysis.
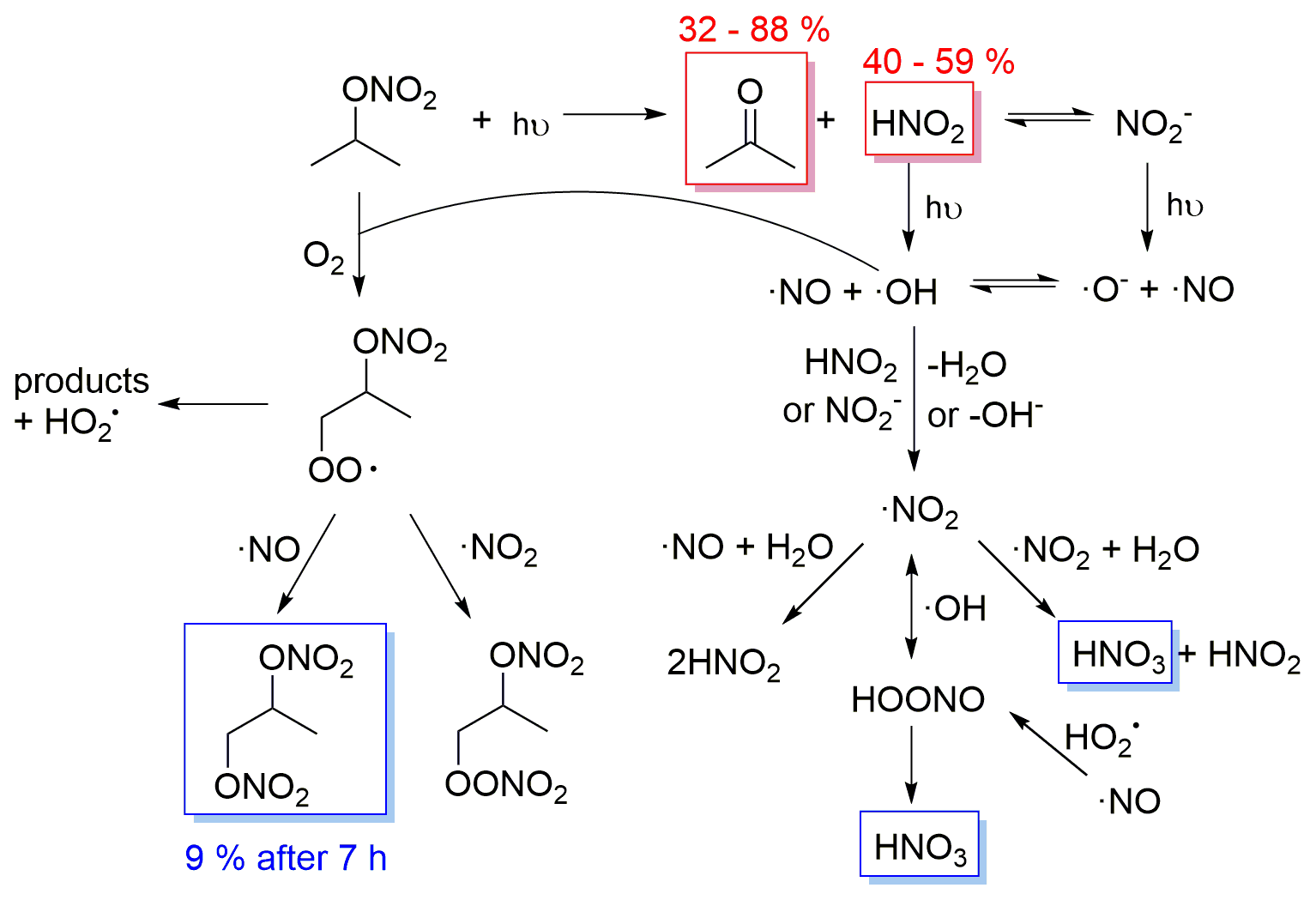
Figure 8Proposed mechanism of isopropyl nitrate photolysis in the aqueous phase. In red are the measured primary products with their molar yields. In blue are the measured secondary products.
Isopropyl nitrate photolyzes into acetone and nitrous acid. Nitrous acid undergoes equilibrium with nitrite in the aqueous phase. Both HNO2 and NO can undergo photolysis, yielding ⋅ NO and ⋅ OH radicals (R5 to 7). ⋅ OH radicals can react with isopropyl nitrate, yielding an alkyl radical that upon oxygen addition forms a peroxy radical. The peroxy radical can decompose into products (i.e., acetone, formic acid, acetic acid, hydroxy acetone, and acetaldehyde, which could also be issued from acetone photooxidation), or react with ⋅ NO or ⋅ NO2, to form a dinitrate or a peroxynitrate. The dinitrate likely corresponds to the compound detected by GC–MS (IP 3 in Fig. 4) and is formed secondarily, in agreement with the proposed mechanism. Additionally, IP 3 was estimated to account for 18 % of the reactive N at the end of the reaction, in agreement with the 20 % of isopropyl nitrate estimated to undergo ⋅ OH oxidation. Furthermore, HNO3 is formed through secondary reactions such as ⋅ NO2 hydrolysis (R1) or peroxynitrite isomerization (R11).
4.2.2 Proposed mechanism of isobutyl nitrate and 1-nitrooxy-2-propanol aqueous-phase photolysis
The primary formation of HNO2 was also observed during the photolysis of isobutyl nitrate and 1-nitrooxy-2-propanol in the aqueous phase (Fig. 2). The determined yields were 31 % and 59 %–62 % for isobutyl nitrate (Fig. 9a) and 1-nitrooxy-2-propanol (Fig. 9b), respectively. Although no DFT calculations were performed specifically for these molecules, they likely undergo a similar photolysis process to the one detailed for isopropyl nitrate, where an adjacent hydrogen atom is captured by the −NO2-leaving moiety (as shown in Fig. 6).
Nevertheless, the formation of carbonyl products concomitant to HNO2 was different from that expected from the main isopropyl nitrate mechanism. The corresponding carbonyl compounds were only observed in minor proportions: yields of 5 % isobutyraldehyde and 8 %–10 % lactaldehyde were obtained for isobutyl nitrate and 1-nitrooxy-2-propanol, respectively. The major carbonyl products were formed after the breakdown of the organic chain, probably due to the excess energy the molecules have after light absorption. This pathway has been observed during the isopropyl nitrate calculations, although as a minor pathway, leading to the formation of acetaldehyde. Figure S9 clearly shows that the carbonyl products formed concomitantly to HNO2 were acetone and formaldehyde (yields of 20 %–32 % and 37 %–39 %, respectively) during isobutyl nitrate photolysis and formaldehyde and acetaldehyde (yields of 63 %–71 % and 50 %–70 %, respectively) during 1-nitrooxy-2-propanol photolysis.
The proposed pathways for their photolysis are given in Fig. 9. Further studies should be conducted to understand the breakdown of the organic chain.
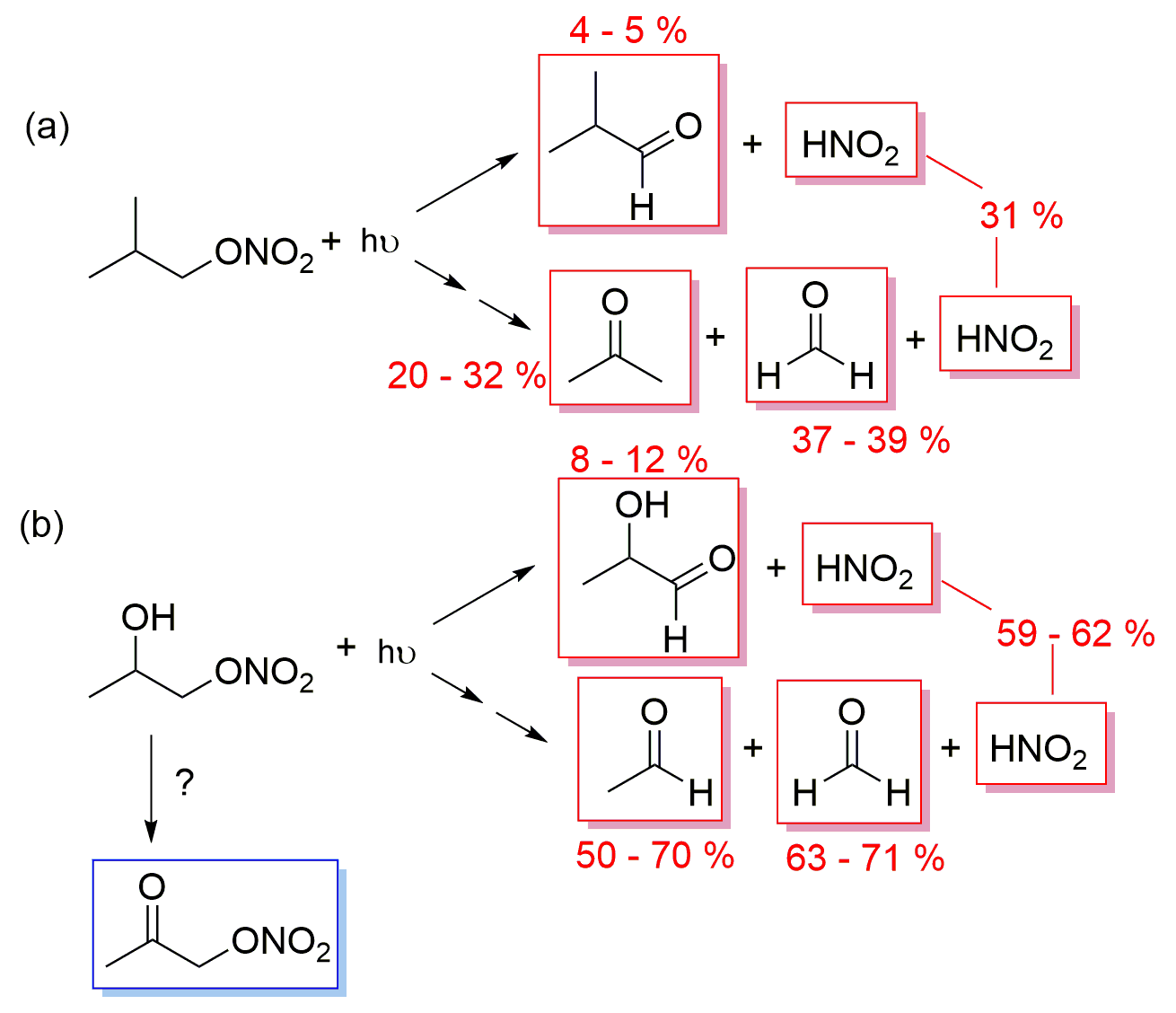
Figure 9Proposed mechanisms of aqueous-phase photolysis of (a) isobutyl nitrate and (b) 1-nitrooxy-2-propanol. In red are the measured primary products and in blue the detected secondary products.
During isobutyl nitrate photolysis, two oxidized RONO2 were observed; these compounds were likely formed through the ⋅ OH oxidation of isobutyl nitrate initiated by HNO2 NO aqueous-phase photochemistry (as it occurs in isopropyl nitrate photolysis). During 1-nitrooxy-2-propanol photolysis, the secondary formation of α-nitrooxy acetone was observed (Fig. 9b).
4.2.3 Proposed mechanism of α-nitrooxy acetone aqueous-phase photolysis
During α-nitrooxy acetone photolysis, NO could not be measured due to its base-catalyzed hydrolysis in the HPIC system (at a pH of 12, Brun et al., 2023), but NO was quantified and showed a secondary formation. Primary formation of HNO2 was therefore expected with a minimum yield of 28 %. The complete mechanism could not be elucidated for this molecule, since some of its primary products could not be quantified (i.e., methylglyoxal and acetic acid). Nevertheless, the mechanism appears to be similar to that of isobutyl nitrate and 1-nitrooxy-2-propanol. The identification of methylglyoxal indicates one pathway, leading to its direct formation along with HNO2 (homologous mechanism as in Fig. 5). However, the detection of significant quantities of formic acid and formaldehyde and the identification of acetic acid suggest that the molecule tends to break at different points (similar to isobutyl nitrate and 1-nitrooxy-2-propanol). Further analytical efforts or modeling should be conducted to clearly identify how the molecule undergoes rupture by photolysis.
This work investigated the fate of the nitrate group during the aqueous-phase photolysis of four species of RONO2: isopropyl nitrate, isobutyl nitrate, 1-nitrooxy-2-propanol, and α-nitrooxy acetone. Our findings suggest a completely different reactivity from the gas phase one. While RONO2 release NOx back into the atmosphere upon photolysis in the gas phase, HNO2 is directly formed in the aqueous phase.
HNO2 was detected as a primary reaction product along with others such as carbonyl compounds or organic acids. The direct formation of HNO2 through aqueous-phase photolysis was confirmed by DFT theoretical calculations and was supported by the absence of direct ⋅ NO2 formation due to solvent cage effects.
Therefore, aqueous-phase photolysis of RONO2 represents both a sink of NOx and a source of atmospheric HNO2 (or HONO). The latter is an important precursor of ⋅ OH and ⋅ NO radicals. During our experiments, these secondarily formed radicals were shown to be trapped in the aqueous phase, producing HNO3 and functionalized RONO2. In the atmosphere, this reactivity can potentially contribute to the sink of NOx, a source of ⋅ OH radicals in condensed phases, and an additional source of SOAaq. Aqueous-phase photolysis has been reported to be negligible in the sinks of RONO2 in the atmosphere due to the hindering effect of the solvent cage (González-Sánchez et al., 2023a). Nevertheless, the mechanisms of this reactivity might be relevant to more significant reactions such as the aqueous-phase ⋅ OH oxidation of RONO2 or potentially their heterogeneous photolysis. Therefore, further work should be done to better assess the role of RONO2 in NOx sink and transport in the formation of atmospheric HONO and SOA.
Data related to this article are available at https://doi.org/10.7910/DVN/USWU6V (González-Sánchez et al., 2023b). Data related to the theoretical calculations can be requested from Miquel Huix-Rotllant (miquel.huixrotllant@univ-amu.fr).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-15135-2023-supplement.
JMGS performed all experiments and treated all experimental data. MHR developed the quantum chemistry model and conducted the calculations. JMGS, NB, and CD developed the HPIC-CD method. JM built the NOx analyzer experimental setup. JMGS and SR developed the UHPLC-UV method. JMGS and AD developed the GC–MS method. JMGS and JLC performed the organic synthesis of RONO2. AM and JLC coordinated the work. JMGS, MHR, and AM wrote the article with inputs from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors thank Benjamin Chazeau and Baptiste Marques for their help and insightful discussions.
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Actions program (grant no. 713750). The project has been carried out with the financial support of the Regional Council of Provence-Alpes-Côte d'Azur and A*Midex (grant no. ANR-11-IDEX-0001-02), funded by the Investissements d'Avenir project, which is further funded by the French Government, and managed by the French National Research Agency (ANR). This study also received funding from the French CNRS-LEFE-CHAT (Programme National-Les Enveloppes Fluides et l'Environnement-Chimie Atmosphérique through the Multinitrates project) and from the ANR-PRCI program (grant no. ANR-18-CE92-0038-02) through the Paramount project.
This paper was edited by Eleanor Browne and reviewed by two anonymous referees.
Bonfrate, S., Ferré, N., and Huix-Rotllant, M.: An efficient electrostatic embedding QM/MM method using periodic boundary conditions based on particle-mesh Ewald sums and electrostatic potential fitted charge operators, J. Chem. Phys., 158, 21101, https://doi.org/10.1063/5.0133646, 2023.
Brun, N., González-Sánchez, J. M., Demelas, C., Clément, J.-L., and Monod, A.: A fast and efficient method for the analysis of α-dicarbonyl compounds in aqueous solutions: Development and application, Chemosphere, 137977, https://doi.org/10.1016/J.CHEMOSPHERE.2023.137977, 2023.
Carbajo, P. G. and Orr-Ewing, A. J.: NO2 quantum yields from ultraviolet photodissociation of methyl and isopropyl nitrate, Phys. Chem. Chem. Phys., 12, 6084–6091, https://doi.org/10.1039/c001425g, 2010.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the Upper and Lower Atmosphere, Academic P., Elsevier, https://doi.org/10.1016/b978-0-12-257060-5.x5000-x, 2000.
Fischer, M. and Warneck, P.: Photodecomposition of nitrite and undissociated nitrous acid in aqueous solution, J. Phys. Chem., 100, 18749–18756, https://doi.org/10.1021/jp961692+, 1996.
Goldstein, S., Lind, J., and Merenyi, G.: Reaction of Organic Peroxyl Radicals with NO2 and NO in Aqueous Solution: Intermediacy of Organic Peroxynitrate and Peroxynitrite Species, J. Phys. Chem. A, 108, 1719–1725, https://doi.org/10.1021/jp037431z, 2004.
González-Sánchez, J. M., Brun, N., Wu, J., Morin, J., Temime-Roussel, B., Ravier, S., Mouchel-Vallon, C., Clément, J.-L., and Monod, A.: On the importance of atmospheric loss of organic nitrates by aqueous-phase •OH oxidation, Atmos. Chem. Phys., 21, 4915–4937, https://doi.org/10.5194/acp-21-4915-2021, 2021.
González-Sánchez, J. M., Brun, N., Wu, J., Ravier, S., Clément, J.-L., and Monod, A.: On the importance of multiphase photolysis of organic nitrates on their global atmospheric removal, Atmos. Chem. Phys., 23, 5851–5866, https://doi.org/10.5194/acp-23-5851-2023, 2023a.
González-Sánchez, J. M., Huix-Rotllant, M., Brun, N., Morin, J., Demelas, C., Durand, A., Ravier, S., Clément, J.-L., and Monod, A.: Replication Data for: Direct formation of HONO through aqueous-phase photolysis of organic nitrates, V1, Harvard Dataverse [data set], https://doi.org/10.7910/DVN/USWU6V, 2023b.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Huix-Rotllant, M., Schwinn, K., Pomogaev, V., Farmani, M., Ferré, N., Lee, S., and Choi, C. H.: Photochemistry of Thymine in Solution and DNA Revealed by an Electrostatic Embedding QM/MM Combined with Mixed-Reference Spin-Flip TDDFT, J. Chem. Theory Comput., 19, 147–156, https://doi.org/10.1021/acs.jctc.2c01010, 2023.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kim, D. H., Lee, J., Ryu, J., Kim, K., and Choi, W.: Arsenite oxidation initiated by the uv photolysis of nitrite and nitrate, Environ. Sci. Technol., 48, 4030–4037, https://doi.org/10.1021/es500001q, 2014.
Lee, S., Filatov, M., Lee, S., and Choi, C. H.: Eliminating spin-contamination of spin-flip time dependent density functional theory within linear response formalism by the use of zeroth-order mixed-reference (MR) reduced density matrix, J. Chem. Phys., 149, 104101, https://doi.org/10.1063/1.5044202, 2018.
Mack, J. and Bolton, J. R.: Photochemistry of nitrite and nitrate in aqueous solution: a review, J. Photochem. Photobiol. A Chem., 128, 1–13, https://doi.org/10.1016/S1010-6030(99)00155-0, 1999.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Crounse, J. D., Teng, A. P., Clair, J. M. S., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Poulain, L., Katrib, Y., Isikli, E., Liu, Y., Wortham, H., Mirabel, P., Le Calvé, S., and Monod, A.: In-cloud multiphase behaviour of acetone in the troposphere: Gas uptake, Henry's law equilibrium and aqueous phase photooxidation, Chemosphere, 81, 312–320, https://doi.org/10.1016/j.chemosphere.2010.07.032, 2010.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Romer Present, P. S., Zare, A., and Cohen, R. C.: The changing role of organic nitrates in the removal and transport of NOx, Atmos. Chem. Phys., 20, 267–279, https://doi.org/10.5194/acp-20-267-2020, 2020.
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015.
Shepson, P. B.: Organic nitrates, Blackwell Publishing Ltd, Oxford, UK, 58–63 pp., https://doi.org/10.1358/dnp.1999.12.1.863615, 1999.
Takeuchi, M. and Ng, N. L.: Organic nitrates and secondary organic aerosol (SOA) formation from oxidation of biogenic volatile organic compounds, in: ACS Symposium Series, vol. 1299, American Chemical Society, 105–125, https://doi.org/10.1021/bk-2018-1299.ch006, 2018.
Talukdar, R. K., Burkholder, J. B., Hunter, M., Gilles, M. K., Roberts, J. M., and Ravishankara, A. R.: Atmospheric fate of several alkyl nitrates: Part 2. UV absorption cross-sections and photodissociation quantum yields, J. Chem. Soc. – Faraday Transactions, 93, 2797–2805, https://doi.org/10.1039/a701781b, 1997.
Vione, D., Belmondo, S., and Carnino, L.: A kinetic study of phenol nitration and nitrosation with nitrous acid in the dark, Environ. Chem. Lett., 2, 135–139, https://doi.org/10.1007/s10311-004-0088-1, 2004.
Wang, Y., Piletic, I. R., Takeuchi, M., Xu, T., France, S., and Ng, N. L.: Synthesis and Hydrolysis of Atmospherically Relevant Monoterpene-Derived Organic Nitrates, Environ. Sci. Technol., 55, 14595–14606, https://doi.org/10.1021/acs.est.1c05310, 2021.