the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Dec 2021
| 06 Dec 2021
Synergetic effects of NH3 and NOx on the production and optical absorption of secondary organic aerosol formation from toluene photooxidation
Shijie Liu
Dandan Huang
Yiqian Wang
Si Zhang
Xiaodi Liu
Can Wu
Wei Du
NH3 is the most important alkaline gas in the atmosphere and one of the key species affecting the behaviors of atmospheric aerosols. However, the impact of NH3 on secondary organic aerosol (SOA) formation remains poorly understood, especially the dynamic evolution of chemical compositions in the SOA formation process. In this study, a series of chamber experiments were performed to probe the individual and common effects of NH3 and NOx on toluene SOA formation through OH photooxidation. The chemical compositions of toluene SOA were characterized using the Aerodyne high-resolution time-of-flight aerosol mass spectrometer (AMS). The SOA yield increased from 28.1 % in the absence of NH3 to 34.7 % in the presence of NH3 but decreased to 19.5 % in the presence of NOx. However, the highest SOA yield of 42.7 % and the lowest carbon oxidation state (OSC) occurred in the presence of both NH3 and NOx, indicating that the higher-volatility products that formed in the presence of NOx could partition into the particle phase when NH3 was added. This resulted in a synergetic effect on SOA formation when NH3 and NOx co-existed. The heterogeneous reaction was the main pathway by which NH3 participated in SOA formation in the photooxidation process. The synergetic effect of NH3 and NOx was also observed in SOA optical absorption. A peak at 280 nm, which is characteristic of organonitrogen imidazole compounds, was observed in the presence of NH3, and its intensity increased when NOx was added into the chamber. This work improves our understanding of how the synergistic interactions between NH3 and NOx influence SOA formation and offers new insights into mitigating haze pollution.
- Article
(492 KB) - Full-text XML
-
Supplement
(1051 KB) - BibTeX
- EndNote





Secondary organic aerosols (SOAs) are an important component of atmospheric particulate matter (Moise et al., 2015; Liu et al., 2017) and can significantly affect atmospheric visibility, air quality, and human health (Paciga et al., 2014; Yang et al., 2016; Liu et al., 2017). Optical properties of SOA have been directly and indirectly linked to their effects on the climate (Laskin et al., 2015; Xie et al., 2017; Peng et al., 2020). Because of the complexity of their chemical components, oxidation processes, and environmental factors, SOA formation mechanisms are very complex, and the current understanding of SOA formation is incomplete. This limited understanding hampers the ability of models to predict the magnitudes, dynamics, and distributions of atmospheric aerosols from particulate and precursor emissions (Ortiz-Montalvo et al., 2014). In the past decades, although our understanding of SOA formation mechanisms has been constantly improving, there is still a gap between the simulated SOA concentration in large-scale atmospheric models and field observations (Volkamer et al., 2006; Yang et al., 2018).
Ammonia (NH3) is the most abundant and ubiquitous alkaline inorganic gas in the atmosphere and one of the critical factors influencing SOA formation (Wang et al., 2016, 2018b; Chen et al., 2019). Some studies have noted that the presence of NH3 can contribute to the formation of more aerosol mass through photooxidation (Na et al., 2007; Li et al., 2018). Na et al. (2007) observed that aerosol yields in the α-pinene–ozone oxidation system increased by 8 % when NH3 was added. Li et al. (2018) reported that the presence of NH3 in the aromatic hydrocarbon photooxidation system increased aerosol size growth potentials (by 7 %–108 %) and resulted in enhanced SOA formation. Qi et al. (2020) found that the concentration and average diameter of SOA showed an immediate and rapid increase after adding NH3. Furthermore, the acid–base reactions between NH3 NH and the carboxyl groups in SOA molecules might enhance SOA formation (Qi et al., 2020; Liu et al., 2015). The condensable ammonium salts formed from the reaction between NH3 and organic acids reduce the volatility of the organic acids by several orders of magnitude (Paciga et al., 2014), and they act as particle-phase organics that further promote SOA formation (Na et al., 2007; Huang et al., 2012; Chen et al., 2019; Qi et al., 2020; Wu et al., 2020). In addition, carbonyls can undergo nucleophilic attack by NH3 through the Maillard reactions and form the corresponding iminium intermediates (Noziere et al., 2009; Laskin et al., 2015; Liu et al., 2015). The iminium intermediates can continue to react with carbonyls, which activates further transformations such as the formation of heterocyclic compounds and oligomerization reactions and form condensation (oligomeric) products with more stable secondary imines (Schiff bases) (Laskin et al., 2014). Both Noziere et al. (2009) and Ortiz-Montalvo et al. (2014) reported that NH3 is an efficient catalyst for reactions with carbonyl compounds to form nitrogen-containing organic aerosols (NOAs). The reaction between carbonyl and NH3 can significantly decrease the volatility of oxidation products, which further increases the yield of SOA (Lee et al., 2013; Zhang et al., 2015a; Qi et al., 2020). Babar et al. (2017) found that the substantial formation of secondary imines in the presence of NH3 was responsible for the higher α-pinene SOA yields. However, not all studies have shown that the presence of NH3 increases SOA yields. One study observed that NH3 suppressed SOA formation under certain ozonation conditions (Ma et al., 2018b). Furthermore, the consumption of NH3 by Criegee intermediates was reported to decrease the secondary ozonide yield and thus affect SOA formation.
Nitrogen oxides (NOx = NO + NO2), which are mainly emitted from the combustion of fossil fuels, have received significant attention due to their effects on the photooxidation process of volatile organic compounds (VOCs) and SOA formation (Surratt et al., 2006; Ng et al., 2007b; Draper et al., 2015; Berkemeier et al., 2016; Sarrafzadeh et al., 2016; Zhao et al., 2018). A clear increase at first and then a decrease in the SOA yield was found with increasing NOx concentration from the laboratory experiments with both anthropogenic (trimethylbenzene) and biogenic (β-pinene) VOCs (Sarrafzadeh et al., 2016; Yang et al., 2020). The competitive chemistry of organic peroxyl radicals (RO2) with hydroperoxyl radicals (HO2) and NO was responsible for the variability in SOA formation (Ng et al., 2007a; Xu et al., 2014; Jiang et al., 2020). RO2 mainly reacts with HO2 under low-NOx conditions to form oxidation products with lower volatility, which may enable it to partition into the particle phase and contribute to the SOA mass (Ng et al., 2007a). While the RO2 + NO reaction is dominant in high-NOx conditions, the increase in volatile products formed through fragmentation was responsible for the decrease in SOA yield with increasing NOx (Zhao et al., 2018; Liu et al., 2019a; Xu et al., 2020). In addition, the increase in OH concentration formed through the NO + HO2 → O2 + OH reaction at low-NOx conditions and a suppressing effect of NOx on OH formation under high-NOx conditions were partly responsible for the first increasing and then decreasing trend of SOA yield with NOx concentration (Sarrafzadeh et al., 2016; Bates et al., 2021).
In the last decade, atmospheric pollutants in China have changed significantly in their concentrations and composition (Wang et al., 2015; Xia et al., 2016) with the emissions of SO2 and NOx decreased by 75 % from 2007–2015 and 10 % from 2011–2015, respectively (de Foy et al., 2016; Vu et al., 2019; Wang et al., 2020). However, owing to the lack of regulation regarding NH3 emissions, NH3 emissions increased by ∼ 30 % from 2008–2016 over the North China Plain (Liu et al., 2018). As has been pointed out in previous research, the promoting effect of NH3 on the formation of SOA may counteract the decreases in aerosol formation due to reductions in SO2 and NOx (Zhang et al., 2021a). Indeed, field observation and model simulation have pointed out that the simultaneous control of NH3 emissions in conjunction with SO2 emission is more effective in reducing PM2.5 than the process without NH3 emissions control, and PM2.5 concentration can be more effectively reduced if NH3 emission is decreased as much as that of SO2 (Erisman and Schaap, 2004). Hence, the mechanism by which NH3 affects SOA formation has attracted more and more attention (Wang et al., 2018a; Ge et al., 2019; Zhang et al., 2021b). However, previous studies have not paid sufficient attention to the joint impacts of NH3 and NOx on the formation of SOA and its corresponding optical properties. Due to the lack of real-time detection methods for SOA chemical composition, the dynamic characteristics of how NH3 participates in SOA formation via photooxidation have not been extensively studied.
Toluene is one of the most abundant aromatic VOCs in the urban atmosphere, which is also an important source of brown carbon (Laskin et al., 2010; Ma et al., 2018a). The effects of NH3 and NOx on SOA formation through the toluene photooxidation process were investigated in this study. The chemical composition of toluene SOA was characterized online with an aerosol mass spectrometer, and the characteristics of SOA chemical composition under different conditions were further explored by applying a positive matrix factorization (PMF) analysis. The optical properties of toluene SOA particles were determined based on a UV–Vis spectrum analysis. Possible mechanisms of the effects of both NH3 and NOx on SOA formation were discussed. The results will help us to better understand SOA formation mechanisms under complex pollution conditions with elevated NH3 and NOx concentrations in an urban atmospheric environment.
2.1 Photooxidation chamber experiments
All toluene photooxidation experiments were performed in a 4 m3 chamber. The chamber has been described in detail in our previous study (Liu et al., 2021). Briefly, the chamber was constructed with a 0.08 mm thick Teflon FEP film. Fifty UV-B lamps (TUV36W, Philips) with peak wavelengths of 254 nm were set up around the chamber and used as the light source to drive OH radical formation through hydrogen peroxide (H2O2) photolysis. Mirror-surfaced stainless steel was used as the interior wall of the enclosure to maximize and homogenize the interior light intensity. All experiments were performed at room temperature (293–298 K), and atmospheric pressure was maintained in the chamber at all times.
Before each experiment, the chamber was flushed with zero air for at least 18 h, after which the concentration of particles was less than 1 cm−3. Zero air was generated by a zero air supply (111-D3N, Thermo Scientific™, USA). The flow rate of zero air was controlled at 20 L min−1 by a mass flow controller (D088C/ZM, Beijing Sevenstar Electronics Co.) during the process of chamber reaction bag inflating. The relative humidity (RH) of zero air was about 20 %. For each experiment, measured amounts of toluene (Sigma-Aldrich, analytically pure) and H2O2 solution (Sigma-Aldrich, 30 wt % in H2O) were injected into a Teflon bulb with microsyringes. Zero air was passed through the injection tube to make sure all the liquids had evaporated to the gas phase and were blown into the chamber. Toluene concentration was measured with a proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS, Ionicon Analytik, Austria). The evolution of toluene concentration for different experiments was shown in Fig. S1. The OH concentration in the chamber was calculated based on the first-order decay of toluene concentration. There was no obvious difference in OH concentrations in the different NOx and NH3 levels (Fig. S2). The average OH concentration over the entire reaction period was 5.87×107 molec. cm−3. NOx (Air Liquid Shanghai, 510 ppm NO2 in N2) and/or NH3 (Air Liquid Shanghai, 502 ppm NH3 in N2) were introduced directly into the chamber to reach the required concentrations. For experiments with NOx, although only NO2 was introduced into the chamber before photooxidation, NO could be formed through NO2 photolysis under the UV light irradiation, so NO always co-existed with NO2 in the photooxidation system (Zhao et al., 2018). NOx concentration was measured online by the NO–NO2–NOx analyzer (model 42C, Thermo Electron Corporation, USA). Each experiment was performed without seed aerosols. After all the reactants were added, the chamber stood undisturbed for 10 min without turning on the light to ensure that the reactant gases in the chamber were evenly mixed. The photooxidation started when the UV light was turn on.
Table 1Summary of experimental conditions in this study.

a The concentration of NH3 is estimated by the amount of NH3 added and the volume of the smog chamber. b SOA concentration and yield were calculated after taking into account the wall loss. c The reported SOA mass concentrations were the peak values after the wall-loss correction.
The experimental conditions for the toluene photooxidation are listed in Table 1. In our work, the OH and toluene concentrations were higher than those of urban conditions. The purpose of the high OH and toluene concentrations is to obtain enough particle production samples for offline collections and accurate measurements. The toluene concentrations remained stable under the different experimental conditions, the variations in toluene-derived SOA mass concentration and yield were only affected by the different NO2 and/or NH3 concentrations in this study. Toluene was studied here as the representative of total aromatic VOCs in the urban atmosphere, thus its concentration used in this study was much higher than that in the real atmosphere. However, the concentration ratio of toluene to OH in this study is similar to that under real atmospheric conditions (Prinn et al., 1995; Zou et al., 2015).
2.2 Particle concentration measurements
For each experiment, a scanning mobility particle sizer (SMPS) was used to record the particle size distribution and volume concentration of the toluene-derived SOA. The SMPS was composed of a differential mobility analyzer (DMA model 3081, TSI Inc., USA) and a condensation particle counter (CPC model 3776, TSI Inc., USA), which were used for screening particles with specific electrical mobility diameter (from 14.1 to 736.5 nm) and for counting the number of the selected particles, respectively. The sheath gas velocity was 3 L min−1, and the sample gas velocity was 0.3 L min−1. The size scan was repeated every 5 min. During each scan cycle, the scan time was 240 s, and the particle sizes ranged from 13.6 to 726.5 nm. A density of 1.4 g m−3, which was measured by Ng et al. (2007b), was used for the calculation of toluene SOA mass concentration from the particle volume concentration (Ng et al., 2007b).
2.3 Chemical characterization
In this study, the toluene SOA chemical compositions were characterized with an online high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc., USA). The sample flow passed through a Nafion dryer, and the RH of the sample gas was reduced to below 20 % before entering the AMS. In the injection port, an aerodynamic lens focused particles with a vacuum aerodynamic diameter below 1 µm into a narrow beam. Particles impacted a flash vaporizer (600 ∘C) at the rear of the sizing region under high vacuum (∼ 10−7 Torr) and were subsequently ionized by electron impact ionization (70 eV). Then, the positively charged ions entered the ToF section and were separated. V-mode () was used in the AMS ToF section to achieve the high signal-to-noise ratio. The separated ion fragments were analyzed by a mass spectrometer with scans from 1 to 300 . The composition-dependent collection efficiency (CE) was applied to the data based on the methods established by Middlebrook et al. (2012). For mass concentration calculations, 1.1, 1.2, and 1.4 were applied as the default relative ionization efficiency (RIE) values of nitrate, sulfate, and organic compounds, respectively. The standard AMS data analysis software SQUIRREL 1.63B, coupled with PIKA 1.23B in Igor Pro (WaveMetrics, Inc., Portland, Oregon), which was retrieved from http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/ (last access: 26 November 2021), was used for the analysis of elemental ratios and the ion speciated compositions of toluene SOA in the chamber. Note that the elemental ratios (i.e., , , and ) and mass-to-carbon ratio () were all calculated using the Aiken-Ambient method for comparability with previous studies (Aiken et al., 2008). In order to further explore the changes in SOA chemical composition, a PMF of the high-resolution mass spectra was performed to determine the different organic aerosol (OA) factors during the toluene photooxidation process. We preformed the PMF analysis in the same way as Zhang et al. (2011), the details of which are provided in the Supplement.
2.4 Absorption measurements
The changes in absorption spectra and the absorbance of the toluene-derived SOA under different conditions were determined using a UV spectrophotometer (UV-3600, Shimadzu, Japan) with a 1 cm cuvette. The SOA was collected from a 3 m3 sample gas onto the 46.2 mm PTFE filter (Whatman™, UK). The collected SOA sample was dissolved in 5 mL of methanol (HPLC grade, > 99.8 %) with 30 min of sonication. As reported by Chen and Bond (2010), > 92 % of SOA is extractable by organic solvents (e.g., methanol), which means that almost all organic matter was extracted in this study. The filter extracts were filtered through 0.2 µm PTFE syringe filters to remove suspended insoluble particles. Before detection of the optical absorbance, a cuvette filled with pure methanol was scanned as a blank to provide a spectral background. The absorption was detected over the range of 200 to 800 nm with a resolution of 0.5 nm−1. The light absorption coefficient of the particles at a specific wavelength λ (Absλ, Mm−1) was calculated according to Eq. (1):
where A700 is the background value of light absorption intensity, calculated as the average value of light absorption intensity from 695–705 nm to reduce the limits of error in measurement; Vl and Va are the volumes of methanol with dissolved particles and sampled air, respectively; and L is the optical path length. Because Absλ was strongly dependent on the amount of SOA, all Absλ results were normalized based on the SOA mass collected on the filter. The normalized result was defined as the mass absorption coefficient (MAC, m2 g−1), calculated using Eq. (2):
where M (µg m−3) represents the concentration of methanol-soluble organic carbon.
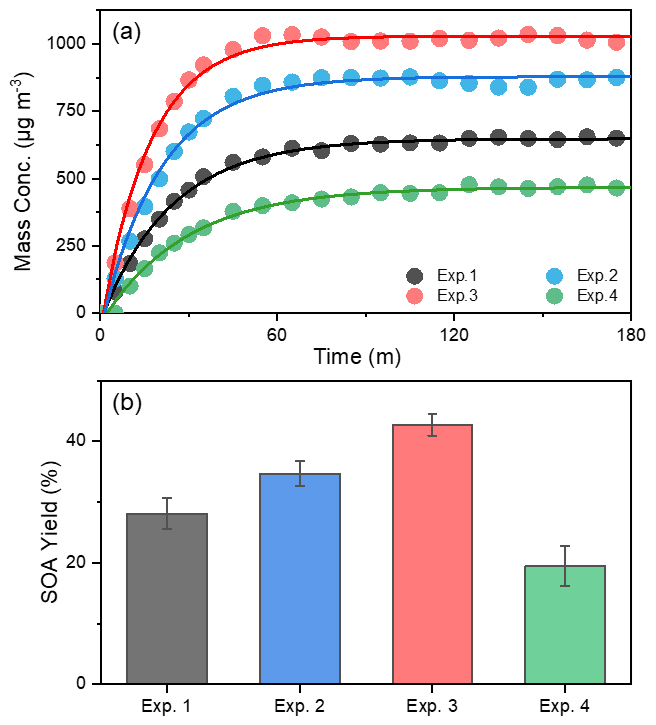
Figure 1The evolution of mass concentration (a) and yield (b) of toluene-derived SOA in different experiments. All the mass concentrations were wall-loss corrected. The error bars were calculated by the fluctuation of measured SOA concentration after the UV light was turn off at the end of each experiment.
3.1 SOA formation
In order to investigate the effect of NH3 and NOx on SOA formation from toluene photooxidation, a control test was carried out. SOA yield (Y) is defined as HC, where ΔM0 is the produced organic aerosol mass concentration (µg m−3), and ΔHC is the mass concentration of reacted toluene (µg m−3). The evolution of SOA mass concentrations and SOA yield at different conditions during the photooxidation process are shown in Fig. 1. Recent experiments showed that the wall loss of organic vapors to the Teflon walls should not be ignored (Zhang et al., 2014, 2015b) and represented a major challenge in investigating SOA formation with environmental chambers (Krechmer et al., 2020). The formation of SOA in laboratory chambers may be substantially suppressed due to losses of SOA-forming vapors to chamber walls, but this effect on SOA formation has not yet been quantitatively established. However, the particle wall-loss rates were detected at the end of the chamber experiment after the UV lamps were turned off, and the mass concentration was corrected in the same way as by Jiang et al. (2020) and Pathak et al. (2007). After the wall-loss correction, the particle mass concentration was almost constant; the different wall-loss effect caused by gaseous oxidation products formed in the different experiment conditions was considered remedied. There was a noticeable increase in the SOA mass concentration in the presence of NH3, which was consistent with previous studies (Qi et al., 2020; Chu et al., 2016). The mass concentration of SOA increased from 637±14.6 µg m−3 without NH3 to a maximum of 867±12.7 µg m−3 with 200 ppb NH3. Previous studies attributed the enhancement of SOA to the formation of NOA from acid–base reactions between NH3 NH and carboxyl groups or Maillard reactions of NH3 NH with carbonyl functional groups (Noziere et al., 2009; Ortiz-Montalvo et al., 2014; Liu et al., 2015; Qi et al., 2020). In contrast, SOA concentrations were lower in the presence of NOx, and the maximum mass concentration of toluene SOA was only 452±18.9 µg m−3 with 63 ppb of initial NOx. The branching of RO2 loss among different pathways has an important influence on the product distribution and SOA formation. The fate of RO2 mainly depends on the concentrations of NOx (Zhao et al., 2018; Liu et al., 2019a; Xu et al., 2020). Several studies have shown that, instead of RO2 reacting with RO2 HO2, NO would react with RO2 to form the RO intermediate and produces more oxidation products with higher volatilities through fragmentation in the presence of NOx (Zhao et al., 2018; Liu et al., 2019a; Xu et al., 2020). Highly volatile compounds cannot readily partition into the particle phase, so this substantially suppresses the formation of SOA.
The NOx and NH3 had opposite effects on toluene SOA formation in this study. Interestingly, however, the highest SOA mass concentration (1020±10.6 µg m−3) occurred in the presence of both NOx and NH3, which was nearly 1.6 times higher than that observed in the absence of NOx or NH3. Although inorganic aerosol (i.e., NH4NO3) was formed from the interaction of NH3 and NOx in the chamber, the upper limit of the inorganic matter only accounted for 6.6 % of the total mass of particulate matter (Table S1) in the NH3 + NOx experiment. Therefore, NH4NO3 was not the main cause of the increase in particulate matter; the co-occurrence of NH3 and NOx had a synergistic effect on the SOA formation, because their combined effect on SOA formation was greater than the sum of their separate effects. Qi et al. (2020) found that the promotion of NH3 on toluene SOA formation was more obviously under high NOx concentration; SOA yield increased 3.7 % and 4.6 % for 70 and 160 ppb initial NOx concentration, respectively, when 200 ppb NH3 was added into the chamber. Li et al. (2018) showed that the presence NH3 can promote the particle size growth of SOA; at the same time, this particle growth rate was higher under low VOC NOx (or high-NOx) conditions. All in all, the joint effect of multiple environmental factors on SOA formation is not the simple summation of the influences of various factors on SOA formation. This may at least partly explain why predictions of SOA concentrations in large-scale atmospheric models, which typically describe SOA formation from data derived from chamber experiments, are frequently lower than field observations (Volkamer et al., 2006).
3.2 SOA chemical composition
The traditional SOA formation mechanism is based on the chemical compositions obtained through offline detection of the chemical composition of SOA (Jang et al., 2002; Liu et al., 2019a; Xu et al., 2020). SOA is continually evolving in the atmosphere, and the aging process of SOA co-occurs with its formation process, resulting in the transformation of SOA chemical composition continuously proceeding during the photooxidation process. Therefore, the AMS was used for online measurement of the SOA chemical composition and how the chemical composition evolved in the photooxidation process will be discussed in the following section.
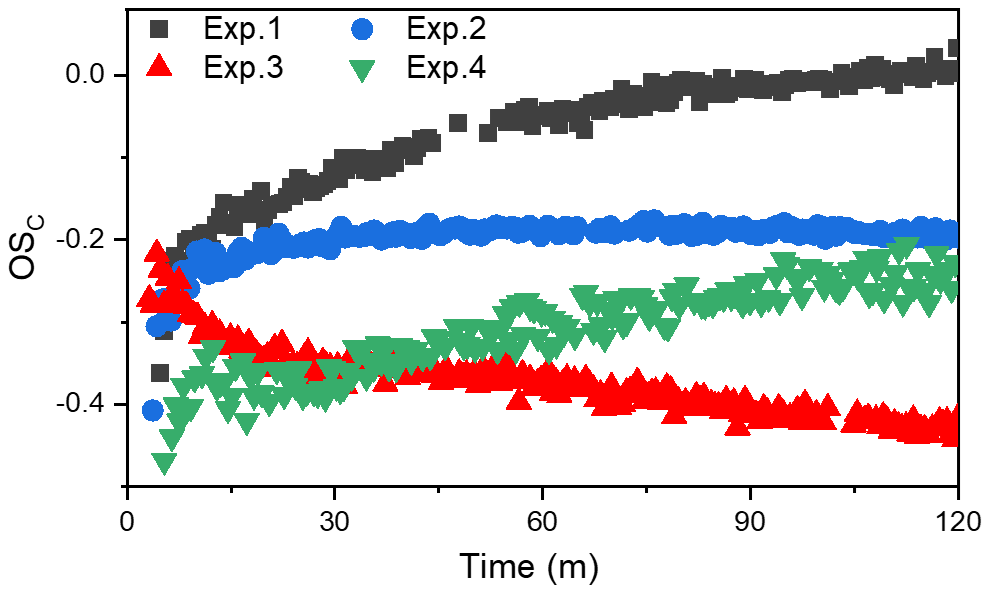
Chemical composition of SOA is very complex. The average carbon oxidation state (OSC) has been shown to be an ideal conceptual framework to describe changes in the degree of oxidation undergone by SOA (Kroll et al., 2011) and has been widely applied in field and laboratory studies (Chen et al., 2018; Mandariya et al., 2019). Average OSC calculation is shown in the Supplement. Figure 2 shows the changes in the OSC of toluene SOA formed in different experiments. Notably, toluene SOA OSC values were in the range between −0.5 and 0, which is consistent with that of semi-volatile oxygenated organic aerosols (SV-OOAs). However, the different OSC values and the change trends observed for the toluene SOA formed in different conditions (with and without NH3 NOx) in Fig. 2 indicated that there was a photooxidation mechanism active during SOA formation, which ultimately changed the SOA chemical compositions.
The OSC increased over time for all SOAs that were formed in the absence of NH3. There are several possible reasons for the increasing trend of OSC values. Firstly, a dynamic equilibrium of semi-volatile vapors may have been achieved between the particle phase and gas phase during the earlier toluene oxidation process. The increase in SOA led to a reduction in the concentration of gas-phase semi-volatile organic products. A decreasing concentration of gas-phase semi-volatile organic compound products would suppress their transformation from gas phase to particulate phase. More lower-volatility gas-phase oxidation products with higher OSC values would then be shifted to the particle phase, which would be responsible for the continuing increase in SOA and its OSC. Secondly, the formed SOA could have further been oxidized by OH radicals through heterogeneous reactions (Kourtchev et al., 2015; Liu et al., 2019b). This could be the main reason for the increase in the OSC when the SOA concentration was no longer increasing. Finally, as pointed out by Malecha and Nizkorodov (2016), even if there was no OH radical in the chamber, the photodegradation of SOA can produce small oxygenated volatile organic compounds (e.g., acetaldehyde OS, and acetone OS) under UV light irradiation. The photoproduction of OVOCs (oxygenated VOCs) from SOA had a lower OSC value than that of SOA. Although the loss of SOA through photodegradation is small, the OSC value of SOA still had increased to a certain extent (Malecha and Nizkorodov, 2016).
The fact that additional photochemical processing results in the dynamic evolution of the OSC over time has been demonstrated in both field and laboratory experiments (Jimenez et al., 2009). The atmospheric oxidation of OA tends towards higher OSC regardless of the original OA source (Herndon et al., 2008). However, when NH3 was present, the OSC of total SOA was almost unchanged for the whole photooxidation period. Carboxyl and carbonyl are the main oxygen-containing functional groups responsible for the toluene photooxidation production (Ji et al., 2017). An organic ammonium salt with four H atoms can offset an increase in OSC value caused by the formation of organic acids or carboxy group with two O atoms through acid–base reactions (Kuwata and Martin, 2012; Liu et al., 2015). In addition, NH3 NH may react with carbonyl functional groups through Maillard reactions, consuming the oxygen atom in the carbonyl group and leading to the formation of species with covalent carbon-nitrogen bonds (Lee et al., 2013; Zhang et al., 2015a; Qi et al., 2020). Xu et al. (2018) showed that imidazole compounds (OSC ) generated through heterogeneous reaction between NH3 and carbonyl compounds might contribute to the decrease in the OSC of SOA. It is clear that an increase in OSC caused by the formation of oxygen-containing functional groups (carboxyl, carbonyl, etc.) would be counteracted through acid–base reactions or Maillard reactions in the presence of NH3. After 60 min of UV light irradiation, there was no more SOA formation; however, the OSC did decrease slightly in Exp. 2 and 3, illustrating that the NH3 could continue to react with SOA through heterogeneous processes. Huang et al. (2016) also pointed out that the portion of semi-volatile products with low OSC formed at the later stage of photooxidation also contributed to the decreased OSC.
The OSC of the toluene SOA formed with NOx was lower than that formed in the absence of NOx, no matter whether NH3 was present in the chamber or not. This indicated that an increased NOx concentration benefits the formation of high volatility oxidation products with lower OSC values (Kroll et al., 2011; Jimenez et al., 2009). However, the relationships between OSC and SOA mass concentration with and without NH3 were opposite to each other. Predictably, the SOA formation mechanism in the presence of NOx is different from that with NOx + NH3. In the absence of NH3, the RO intermediate, which is easily fragmented to produce relatively high-volatility compounds, was the dominant product of the NOx = RO2 reaction (Zhao et al., 2018; Liu et al., 2019a; Xu et al., 2020). Highly volatile compounds cannot readily partition into the particle phase, which subsequently results in a lower SOA yield in the presence of NOx (Yang et al., 2020). Thereby, both OSC and the SOA mass concentration were lower when the 60 ppb of NOx that was added into the chamber. However, when both NOx and NH3 were present, the toluene-derived SOA had the lowest OSC value, but the highest mass concentration. This result suggested that although NOx promotes the formation of higher-volatility compounds, these higher-volatility compounds (e.g., glyoxal) can react with NH3 and partition into the particle phase, which could contribute to the increase in SOA formation. Huffman et al. (2009) observed that aerosol volatility was inversely correlated with the extent of oxidation of OA components. The low value of OSC in the presence of NOx indicated that NOx would promote the formation of the relatively high-volatility compounds. However, the lower OSC value in the presence of NH3 indicated that the high-volatility compounds would promote partitioning into the particle phase when reacting with NH3.
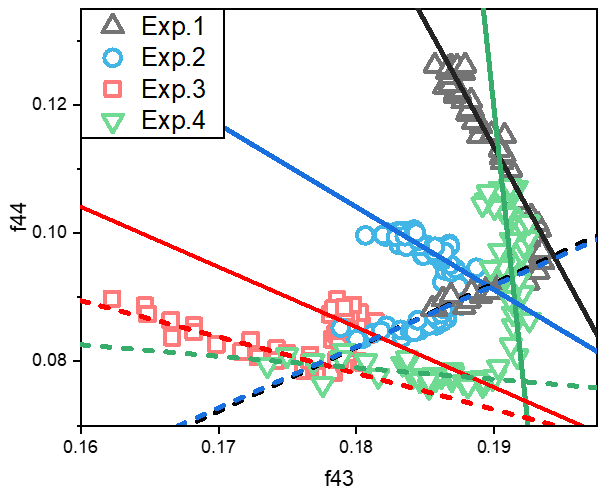
Figure 3The relationship between total organic signals at 43 (f43) vs. 44 (f44) from SOA data during the photooxidation process. The f43 vs. f44 plots exhibited inflection points during the photooxidation process. The dashed lines indicate the trends of f43 vs. f44 for the SOA formation stage (before the inflection point), and the solid lines are for the stable stage.
Fragments derived from the AMS data have also been widely used to explore the bulk compositions and properties of SOA (Ng et al., 2010, 2017). The 43 (f43) frequency was dominated by ion C2H3O+, which is the tracer for organic compounds with alcohol and carbonyl functional groups (Alfarra et al., 2006). Meanwhile, the 44 (f44) signal was dominated by CO ions, which is the tracer for organic compounds with carboxyl functional groups and an indicator of highly oxygenated organic aerosols (Ng et al., 2010). Here, we use the approach of Ng et al. (2010) by plotting the fractions of the total organic signal at 43 vs. 44 (f43 vs. f44). The change of f43 vs. f44, which has an inflection point during the photooxidation process, is shown in Figs. 3 and S3. In our study, the change before the inflection point was defined as the formation stage, and the linear fit of f43 vs. f44 for the formation stage is shown by the dashed lines. The change in f43 vs. f44 after the inflection point was defined as the stable stage, and the linear fit of f43 vs. f44 in this stage is shown by the solid lines. The formation and stable stages of the f43 vs. f44 relationship during the experiment are discussed separately here.
In the stable stage, the increase in f44 and decrease in f43 with increasing OH exposure indicated that the carbonyl groups in toluene SOA were oxidized to carboxyl groups by the aging process. For the experiments without NH3 and NOx, the slope ratio of f43 vs. f44 was −3.9. When there was 60 ppb initial NOx, the f43 was almost stable, while the f44 increased with the oxidation process. There was a lower slope ratio of f43 vs. f44, indicating that organic compounds with more alcohol and carbonyl functional groups had formed in the presence of NOx. But for the experiments with 200 ppb initial NH3, the slope ratios of f43 vs. f44 were only −1.1 and −1.3 in the presence and absence of NOx, respectively. According to the above results, we can see that more carbonyl groups are consumed as carboxyl groups are formed in the presence of NH3. The carbonyls can be oxidized to organic acids (Kawamura and Bikkina, 2016), but unreacted carbonyl can be nucleophilically attacked by NH3 NH to form nitrogen-containing heterocyclic compounds, e.g., imidazole (Grace et al., 2019; Lian et al., 2020). Meanwhile, the peak f44 value decreased from 0.13 to 0.10 when NH3 was added into the chamber. This suggested that the heterogeneous reaction of NH3 NH could promote the consumption of particle-phase carbonyl groups (Xu et al., 2018) and must inhibit the formation of carboxyl groups in the SOA aging process. According to the changing trend of SOA concentration over time, the photooxidation process was divided into formation stage and stable stage. As shown in Fig. 1, the first half hour of photooxidation when SOA concentration increased linearly with time was defined as SOA formation stage. After 60 min of photooxidation, SOA concentration was not changed with reaction time, and it was defined as stable stage. The differences in spectra of toluene SOA in the formation stage and stable stage are shown in Fig. 4. A lower signal intensity variation in CO in the presence of NH3 also illustrated that NH3 would inhibit heterogeneous reactions that form carboxyl groups.
In the formation stage, the slope ratios of f43 vs. f44 were almost the same for both experiments without NOx. It can thus be seen that the presence or absence of NH3 does not affect the change trend of f43 vs. f44 in the SOA formation stage. Therefore, the gas-phase homogeneous reaction of NH3 on SOA formation is not important. Clearly, the particle-phase heterogeneous reaction was the main reaction pathway by which NH3 participated in the photooxidation process and toluene SOA formation. However, negative correlations were observed between f43 and f44 in the presence of NOx. Based on this, we concluded that NOx not only affects the SOA formation through the particle-phase heterogeneous reactions but also through gas-phase homogeneous reactions.
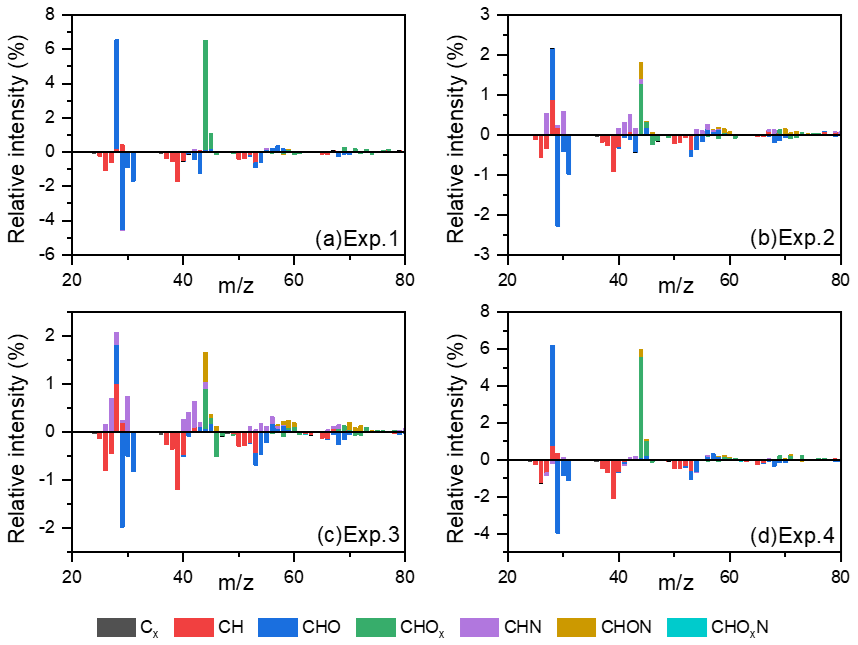
Figure 4The differential spectra of toluene SOA in the formation and stable stages. Data were taken and analyzed at a high resolution but were summarized to a unit mass resolution for display. Only minimal N-containing fragments could be observed in Exp. 1 without added NH3 and NOx. These N-containing fragments could be attributed to the background NH3 and NOx in the chamber or systematic errors from AMS.
3.3 PMF results
A temporal evaluation of the toluene SOA chemical composition during photooxidation is vital to the analysis of the NOA formation mechanism in the presence of NH3 and/or NOx. Therefore, this study further compared the chemical properties of the SOA generated under different experimental conditions by applying a PMF analysis to the HR-ToF-AMS data (Chen et al., 2019). A summary of the PMF results is presented in Figs. S4–S7. For the toluene OH-photooxidation experiments with NH3 and/or NOx present, two factors were identified from the PMF analysis in the same way as in Chen et al. (2019). The , , and values of these two factors are shown in Fig. 5. The factor with higher values was defined as high-nitrogen OA (Hi-NOA). In contrast, the factor with lower values was defined as low-nitrogen OA (Lo-NOA). Figure 6 exhibits the evolution of Hi-NOA and Lo-NOA during the photooxidation process as resolved from the PMF analysis of different initial NOx NH3 concentrations. While similar evolutionary trends were observed under different conditions, the relative intensities and the chemical compositions of these two factors in each experiment were not consistent.
For the toluene SOA formed under NH3 conditions, both Lo-NOA and Hi-NOA had similar values, which were fully oxygenated with an average of 0.74±0.04 (Fig. 5a). These values were comparable to the low-volatility oxygenated organic aerosols (LV-OOAs) with an value ranging from 0.6 to 1 (Jimenez et al., 2009). The main difference between these two OA sources was the ratio. The ratio of Hi-NOA ( = 0.032) was about 3 times higher than that of Lo-NOA ( = 0.010) (Fig. 5a). The evolution of these two OA sources during the photooxidation process is shown in Fig. 6a. The components of toluene SOA were mostly Lo-NOA during the initial phase of SOA formation, but Hi-NOA toluene SOA started forming after 1 min and continued to increase. The Lo-NOA reached the maximum mass concentration after 30 min of the photooxidation, and then decreased. Such a declining trend of Lo-NOA at longer reaction times reflected the conversion of Lo-NOA into something else in the particle phase. As the Lo-NOA decreased, the mass concentration of Hi-NOA gradually increased. Thus, the Hi-NOA should be derived from the heterogeneous reaction of Lo-NOA with NH3 NH. At the same time, it was proved that the formation pathway of Hi-NOA was not through reaction of NH3 with later-generation gas-phase products in the homogeneous gas phase. With the gradual replacement of Lo-NOA by Hi-NOA, the ratio of [Hi-NOA] [Lo-NOA] stabilized at 5–6.

Figure 5The , , and values of Hi-NOA and Lo-NOA for each experiment. (a) Exp. 2 with 200 ppb NH3, (b) Exp. 3 with 200 ppb NH3 and 62 ppb NO2, and (c) Exp. 4 with 63 ppb NO2.
For the toluene SOA formed under NOx conditions, there was not a large difference between the ratios of Hi-NOA ( = 0.019) and Lo-NOA ( = 0.014) (Fig. 5c). At the end of the NOx experiment, the ratio of [Hi-NOA] [Lo-NOA] was only 3:2 (Fig. 6c). It follows that the contribution of the heterogeneous NOx reaction to the ratio of toluene SOA was not obvious. Therefore, the formation of NOA in the presence of NOx mainly occurred through gas-phase homogeneous reactions, which was consistent with the results in Sect. 3.3.
The changing trend of with time in the presence of NH3 was different to that with NOx present. The evolution trends of the of SOA in different experiments are shown in Fig. 7. In the presence of NH3, the value gradually increased throughout the photooxidation process. The increased value in the photooxidation process was attributed to the heterogeneous NH3 reaction with SOA. But in the presence of NOx, the increased rapidly to its maximum value where it was stable for the rest of the reaction. This could mean that the heterogeneous reaction of toluene SOA with NOx to form NOA was not as important as the gas-phase homogeneous reaction.
When both NOx and NH3 were added into the chamber, the ratios of Hi-NOA and Lo-NOA were 0.062 and 0.029, respectively (Fig. 5b). The ratio of Hi-NOA, which was comparable to the recently isolated nitrogen-enriched OA value (0.053) observed by Sun et al. (2011), was much higher than that observed in the experiments with only NH3 or NOx. It was even higher than the sum of the ratios from both Exp. 2 with NH3 and Exp. 4 with NOx. In order to calculate the relative contributions of NH3 and NOx to , it was assumed that the effects of NH3 or NOx on the ratio in the Hi-NOA and Lo-NOA factors did not change among different experimental conditions. For Lo-NOA, the contributions of NH3 and NOx to the value were 0.0126 and 0.0164, and their relative intensities were 43 % and 57 %, respectively. For Hi-NOA, the contributions of NH3 and NOx to the values were 0.0404 and 0.0216, and their relative intensities were 65 % and 35 %, respectively. For the experiment with both NH3 and NOx, the contribution of NH3 to was higher by 26 %, and the contribution of NOx to was higher by 17 % compared to the experiments with the single pollutants. The co-existence of NH3 and NOx further enhanced the value of toluene SOA, indicating that a synergetic interaction between NH3 and NOx further enhanced organic nitrogen formation.

Figure 6The evolution of high-nitrogen OA (Hi-NOA) and low-nitrogen OA (Lo-NOA) during the photooxidation process under different NOx NH3 concentrations. Hi-NOA and Lo-NOA were not consistent among experiments. (a) Exp. 2 with 200 ppb NH3, (b) Exp. 3 with 200 ppb NH3 and 62 ppb NO2, and (c) Exp. 4 with 63 ppb NO2.
3.4 Optical absorption
The optical characteristics of toluene SOA formed from different NH3 and NOx conditions were investigated. The MAC of toluene-derived SOA detected over the range of 230–600 nm is displayed in Fig. 8. Over the entire UV detection range, an increase in light absorption was observed when the toluene SOA formed in the presence of NOx or NH3.
By looking at Fig. 8 in detail, one can see that the MAC of toluene SOA formed with (red line) and without (black line) NH3 overlapped at 250 nm, but when the UV wavelength exceeded 250 nm the MAC of the toluene SOA formed in the presence of NH3 was higher. The red line reflects an obvious characteristic absorption peak at 270–280 nm, which was mainly due to the absorption of the electronic transitions. The imidazole compounds were formed through the Maillard reactions between NH3 NH with carbonyl functional groups (Zhang et al., 2015a). The C=N double bonds in the organonitrogen imidazole compounds can act as effective chromophores since both and transitions are chromatically active (Nguyen et al., 2013). The UV–Vis spectra of imine and pyrrole show broad bands at 270 nm (NIST, 2020), which was consistent with the UV absorption peak of the band observed here. According to the AMS results, carbonyl was the main functional group of toluene SOA. The emergence of absorption peaks at 270–280 nm demonstrated that some organonitrogen imidazole compounds (e.g., imines and pyrrole) were formed through the heterogeneous reaction of toluene with NH3. Meanwhile, the high-molecular-weight nitrogen-containing organic species might have formed through Maillard reactions in the particle phase (Wang et al., 2010). This was also a reason for the increase in SOA mass concentration in the presence of NH3.
The green line in Fig. 8 represents the MAC value of toluene-derived SOA in the presence of NOx, which was also higher than the black line (control) throughout the UV detection range. When compared with the red line, the green line had no obvious characteristic peak at 280 nm, but it had higher absorbance in the range between 240 and 280 nm. This indicated that both NOx and NH3 increased the absorbance of toluene SOA, while the chromophores generated from the reactions between toluene-derived SOA with NH3 or NOx did not behave in the same way.
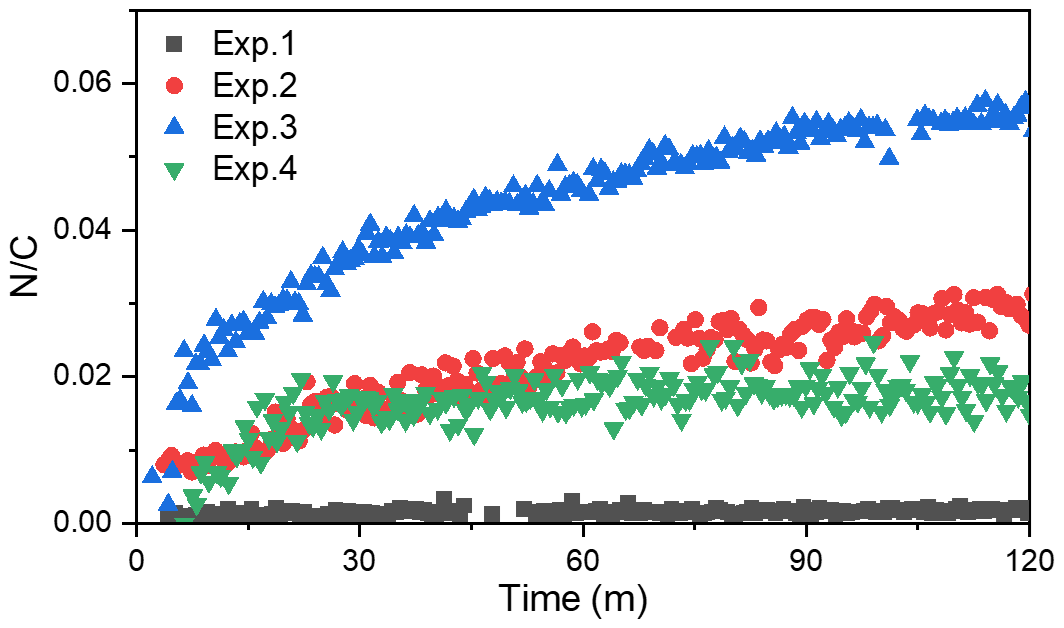
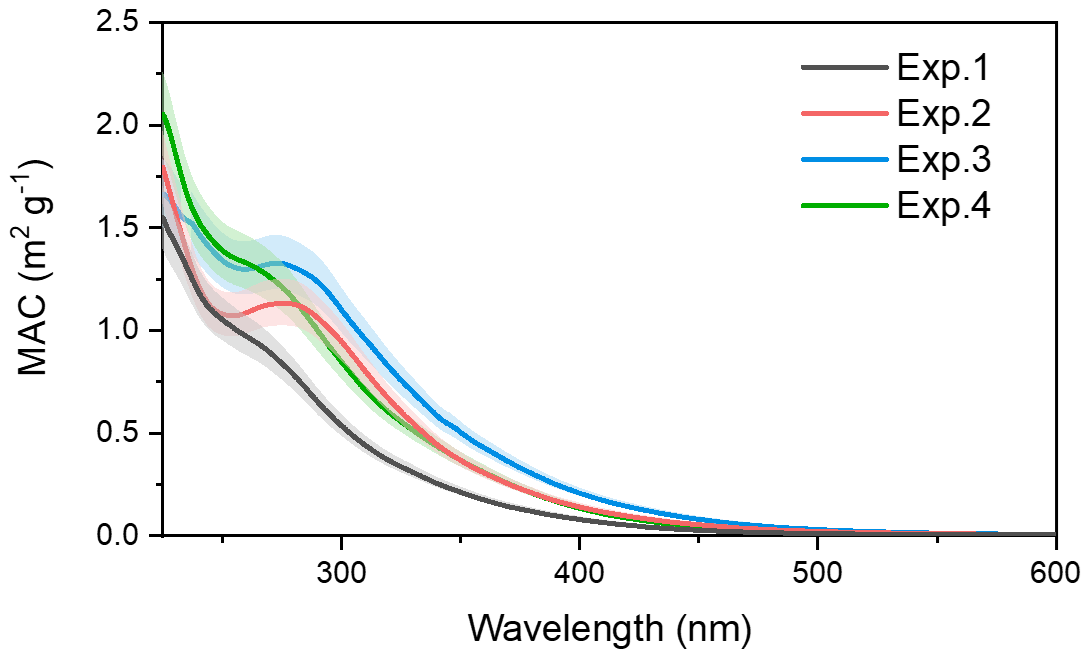
Figure 8The MAC over the range of 230–600 nm for the toluene SOA formed under different experiment conditions.
The blue line in Fig. 8 represents the absorbance of toluene SOA formed in the presence of both NOx and NH3. The MAC of toluene SOA formed in the presence of both NOx and NH3 was higher than the toluene SOA formed in the presence of either NH3 or NOx. There might have been a synergetic effect between NOx and NH3 on the absorbance of toluene SOA. Considering that the mass concentration of toluene SOA formed in the presence of both NH3 and NOx was the highest, as described in Sect. 3.1, the co-existence of NH3 and NOx may also result in the toluene SOA having stronger light absorption and atmospheric radiative forcing. We also noted a higher MAC value at 280 nm, which illustrated that the presence of NOx could promote the formation of imines and pyrrole in the photooxidation system of toluene with NH3.
Here we present the results of a study in which we characterized the mass concentrations, chemical compositions, and optical properties of SOA formed from the photooxidation of toluene under different NH3 and NOx concentrations. When compared with the control experiment, the SOA mass concentration data showed that the formation of toluene-derived SOA was enhanced in the presence of NH3, through acid–base reactions between carboxyl groups or Maillard reactions with carbonyls, but inhibited in the presence of NOx. Meanwhile, the mass concentration of toluene SOA formed in the presence of both NOx and NH3 was higher than those formed under either NH3 or NOx alone. This result indicated that there was a synergistic interaction between NH3 and NOx that further enhanced toluene-derived SOA formation. At the same time, the lowest OSC value was obtained when both NH3 and NOx were present. We concluded that highly volatile compounds, which were formed from toluene photooxidation in the presence of NOx, could react with NH3 to form products with lower volatilities and promoted the partitioning of these products into the particle phase.
Synergetic effects of NH3 and NOx on the formation of NOA and the optical properties of SOA were also observed in this study. The heterogeneous reaction was responsible for the formation of NOA in the presence of NH3. Meanwhile, an absorption peak at 270–280 nm, which is characteristic of imine and pyrrole, was observed. In contrast, the formation of NOA caused by NOx alone was mainly due to a gas-phase homogeneous reaction.
In the actual atmosphere, especially in Chinese urban atmosphere, NOx and NH3 abundantly co-exist. Therefore, the findings presented here clearly show that the synergetic effects of NOx and NH3 should not be neglected. In the meantime, our work provides a scientific basis for the consideration of synergistic emission reductions of NH3 and NOx under the compound pollution conditions, which will contribute to reducing the burden of aerosols in the atmosphere. It has to be noted that the concentration of reactants used for the experiments is much higher than that observed in polluted areas. Although the reactant concentrations including NH3 used in this work are much higher than those in the real urban environment, our results are applicable for the polluted urban atmosphere. In the urban atmosphere aromatic VOCs consist of numerous species, and their total concentration is much higher than a single species such as toluene. On the other hand, carboxylic acids and carbonyls in the urban polluted atmosphere can be produced from aromatics and many other species. Therefore, it is reasonable for our smog chamber experiments to use toluene as a single precursor with a concentration much higher than that in the real atmosphere. Although the mechanisms of SOA formed under high precursor concentrations is expected to be the same as that under low concentrations, the kinetics are probably different. Thus, the effect of NH3 and NOx on the photooxidation of toluene with lower concentrations should be checked in a future study.
The datasets are available at https://doi.org/10.6084/m9.figshare.16910953 (last access: 26 November 2021, Liu, 2021.).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-17759-2021-supplement.
SL and GW designed the experiment. SL, DH, and YW conducted the experiments. SL, DH, YW, and GW performed the data interpretation. SL and GW wrote the paper. YW, SZ, XL, CW, and WD contributed to the paper with useful scientific discussions or comments.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was financially supported by National Natural Science Foundation of China (Grant No. 42130704, 42005088); the China Postdoctoral Science Foundation (Grant No. 2019M661427); Fundamental Research Funds for the Central Universities, Director's Fund of Key Laboratory of Geographic Information Science (Ministry of Education), East China Normal University (Grant No. KLGIS2021C02); and ECNU Happiness Flower Program.
This paper was edited by Arthur Chan and reviewed by four anonymous referees.
Alfarra, M. R., Paulsen, D., Gysel, M., Garforth, A. A., Dommen, J., Prévôt, A. S. H., Worsnop, D. R., Baltensperger, U., and Coe, H.: A mass spectrometric study of secondary organic aerosols formed from the photooxidation of anthropogenic and biogenic precursors in a reaction chamber, Atmos. Chem. Phys., 6, 5279–5293, https://doi.org/10.5194/acp-6-5279-2006, 2006.
Babar, Z. B., Park, J.-H., and Lim, H.-J.: Influence of NH3 on secondary organic aerosols from the ozonolysis and photooxidation of α-pinene in a flow reactor, Atmos. Environ., 164, 71–84, https://doi.org/10.1016/j.atmosenv.2017.05.034, 2017.
Bates, K., Jacob, D., Li, K., Ivatt, P., Evans, M., Yan, Y., and Lin, J.: Development and evaluation of a new compact mechanism for aromatic oxidation in atmospheric models, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-605, in review, 2021.
Berkemeier, T., Ammann, M., Mentel, T. F., Poschl, U., and Shiraiwa, M.: Organic nitrate contribution to new particle formation and growth in secondary organic aerosols from α-pinene ozonolysis, Environ. Sci. Technol., 50, 6334–6342, https://doi.org/10.1021/acs.est.6b00961, 2016.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Chen, C. L., Li, L. J., Tang, P., and Cocker, D. R.: SOA formation from photooxidation of naphthalene and methylnaphthalenes with m-xylene and surrogate mixtures, Atmos. Environ., 180, 256–264, https://doi.org/10.1016/j.atmosenv.2018.02.051, 2018.
Chen, T., Liu, Y., Ma, Q., Chu, B., Zhang, P., Liu, C., Liu, J., and He, H.: Significant source of secondary aerosol: formation from gasoline evaporative emissions in the presence of SO2 and NH3, Atmos. Chem. Phys., 19, 8063–8081, https://doi.org/10.5194/acp-19-8063-2019, 2019.
Chu, B., Zhang, X., Liu, Y., He, H., Sun, Y., Jiang, J., Li, J., and Hao, J.: Synergetic formation of secondary inorganic and organic aerosol: effect of SO2 and NH3 on particle formation and growth, Atmos. Chem. Phys., 16, 14219–14230, https://doi.org/10.5194/acp-16-14219-2016, 2016.
de Foy, B., Lu, Z., and Streets, D. G.: Satellite NO2 retrievals suggest China has exceeded its NOx reduction goals from the twelfth Five-Year Plan, Sci. Rep., 6, 35912, https://doi.org/10.1038/srep35912, 2016.
Draper, D. C., Farmer, D. K., Desyaterik, Y., and Fry, J. L.: A qualitative comparison of secondary organic aerosol yields and composition from ozonolysis of monoterpenes at varying concentrations of NO2, Atmos. Chem. Phys., 15, 12267–12281, https://doi.org/10.5194/acp-15-12267-2015, 2015.
Erisman, J. W. and Schaap, M.: The need for ammonia abatement with respect to secondary PM reductions in Europe, Environ. Pollut., 129, 159–163, https://doi.org/10.1016/j.envpol.2003.08.042, 2004.
Ge, S., Wang, G., Zhang, S., Li, D., Xie, Y., Wu, C., Yuan, Q., Chen, J., and Zhang, H.: Abundant NH3 in China enhances atmospheric HONO production by promoting the heterogeneous reaction of SO2 with NO2, Environ. Sci. Technol., 53, 14339–14347, https://doi.org/10.1021/acs.est.9b04196, 2019.
Grace, D. N., Sharp, J. R., Holappa, R. E., Lugos, E. N., Sebold, M. B., Griffith, D. R., Hendrickson, H. P., and Galloway, M. M.: Heterocyclic product formation in aqueous brown carbon systems, ACS Earth Space Chem., 3, 2472–2481, https://doi.org/10.1021/acsearthspacechem.9b00235, 2019.
Herndon, S. C., Onasch, T. B., Wood, E. C., Kroll, J. H., Canagaratna, M. R., Jayne, J. T., Zavala, M. A., Knighton, W. B., Mazzoleni, C., Dubey, M. K., Ulbrich, I. M., Jimenez, J. L., Seila, R., de Gouw, J. A., de Foy, B., Fast, J., Molina, L. T., Kolb, C. E., and Worsnop, D. R.: Correlation of secondary organic aerosol with odd oxygen in Mexico City, Geophys. Res. Lett., 35, L15804, https://doi.org/10.1029/2008gl034058, 2008.
Huang, D. D., Zhang, X., Dalleska, N. F., Lignell, H., Coggon, M. M., Chan, C. M., Flagan, R. C., Seinfeld, J. H., and Chan, C. K.: A note on the effects of inorganic seed aerosol on the oxidation state of secondary organic aerosol-α-Pinene ozonolysis, J. Geophys. Res.-Atmos, 121, 12476–12483, https://doi.org/10.1002/2016jd025999, 2016.
Huang, Y., Lee, S. C., Ho, K. F., Ho, S. S. H., Cao, N. Y., Cheng, Y., and Gao, Y.: Effect of ammonia on ozone-initiated formation of indoor secondary products with emissions from cleaning products, Atmos. Environ., 59, 224–231, https://doi.org/10.1016/j.atmosenv.2012.04.059, 2012.
Huffman, J. A., Docherty, K. S., Mohr, C., Cubison, M. J., Ulbrich, I. M., Ziemann, P. J., Onasch, T. B., and Jimenez, J. L.: Chemically-resolved volatility measurements of organic aerosol fom different sources, Environ. Sci. Technol., 43, 5351–5357, https://doi.org/10.1021/es803539d, 2009.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Jiang, X. T., Lv, C., You, B., Liu, Z. Y., Wang, X. F., and Du, L.: Joint impact of atmospheric SO2 and NH3 on the formation of nanoparticles from photo-oxidation of a typical biomass burning compound, Environ. Sci.-Nano, 7, 2532–2545, https://doi.org/10.1039/d0en00520g, 2020.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kawamura, K. and Bikkina, S.: A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 170, 140–160, https://doi.org/10.1016/j.atmosres.2015.11.018, 2016.
Kourtchev, I., Doussin, J.-F., Giorio, C., Mahon, B., Wilson, E. M., Maurin, N., Pangui, E., Venables, D. S., Wenger, J. C., and Kalberer, M.: Molecular composition of fresh and aged secondary organic aerosol from a mixture of biogenic volatile compounds: a high-resolution mass spectrometry study, Atmos. Chem. Phys., 15, 5683–5695, https://doi.org/10.5194/acp-15-5683-2015, 2015.
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always lost but never forgotten: Gas-phase wall losses are important in all teflon environmental chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kuwata, M. and Martin, S. T.: Phase of atmospheric secondary organic material affects its reactivity, P. Natl. Acad. Sci. USA, 109, 17354–17359, https://doi.org/10.1073/pnas.1209071109, 2012.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of atmospheric brown carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Laskin, J., Laskin, A., Roach, P. J., Slysz, G. W., Anderson, G. A., Nizkorodov, S. A., Bones, D. L., and Nguyen, L. Q.: High-resolution desorption electrospray ionization mass spectrometry for chemical characterization of organic aerosols, Anal. Chem., 82, 2048–2058, https://doi.org/10.1021/ac902801f, 2010.
Laskin, J., Laskin, A., Nizkorodov, S. A., Roach, P., Eckert, P., Gilles, M. K., Wang, B., Lee, H. J., and Hu, Q.: Molecular selectivity of brown carbon chromophores, Environ. Sci. Technol., 48, 12047–12055, https://doi.org/10.1021/es503432r, 2014.
Lee, A. K., Zhao, R., Li, R., Liggio, J., Li, S. M., and Abbatt, J. P.: Formation of light absorbing organo-nitrogen species from evaporation of droplets containing glyoxal and ammonium sulfate, Environ. Sci. Technol., 47, 12819–12826, https://doi.org/10.1021/es402687w, 2013.
Li, K., Chen, L., White, S. J., Yu, H., Wu, X., Gao, X., Azzi, M., and Cen, K.: Smog chamber study of the role of NH3 in new particle formation from photo-oxidation of aromatic hydrocarbons, Sci. Total Environ., 619, 927–937, https://doi.org/10.1016/j.scitotenv.2017.11.180, 2018.
Lian, X., Zhang, G., Yang, Y., Lin, Q., Fu, Y., Jiang, F., Peng, L., Hu, X., Chen, D., Wang, X., Peng, P. a., Sheng, G., and Bi, X.: Evidence for the formation of imidazole from carbonyls and reduced nitrogen species at the individual particle level in the ambient atmosphere, Environ. Sci. Tech. Lett., 8, 9–15, https://doi.org/10.1021/acs.estlett.0c00722, 2021.
Liu, S.: Synergetic effect of NH3 and NOx on SOA [data set], available at: https://doi.org/10.6084/m9.figshare.16910953, last access: 26 November 2021.
Liu, M., Huang, X., Song, Y., Xu, T., Wang, S., Wu, Z., Hu, M., Zhang, L., Zhang, Q., Pan, Y., Liu, X., and Zhu, T.: Rapid SO2 emission reductions significantly increase tropospheric ammonia concentrations over the North China Plain, Atmos. Chem. Phys., 18, 17933–17943, https://doi.org/10.5194/acp-18-17933-2018, 2018.
Liu, S., Jia, L., Xu, Y., Tsona, N. T., Ge, S., and Du, L.: Photooxidation of cyclohexene in the presence of SO2: SOA yield and chemical composition, Atmos. Chem. Phys., 17, 13329–13343, https://doi.org/10.5194/acp-17-13329-2017, 2017.
Liu, S. J., Jiang, X. T., Tsona, N. T., Lv, C., and Du, L.: Effects of NOx, SO2 and RH on the SOA formation from cyclohexene photooxidation, Chemosphere, 216, 794–804, https://doi.org/10.1016/j.chemosphere.2018.10.180, 2019a.
Liu, S. J., Tsona, N. T., Zhang, Q., Jia, L., Xu, Y. F., and Du, L.: Influence of relative humidity on cyclohexene SOA formation from OH photooxidation, Chemosphere, 231, 478–486, https://doi.org/10.1016/j.chemosphere.2019.05.131, 2019b.
Liu, S. J., Wang, Y., Wang, G., Zhang, S., Li, D., Du, L., Wu, C., Du, W., and Ge, S.: Enhancing effect of NO2 on the formation of light-absorbing secondary organic aerosols from toluene photooxidation, Sci. Total Environ., 794, 148714, https://doi.org/10.1016/j.scitotenv.2021.148714, 2021.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Ma, P., Zhang, P., Shu, J., Yang, B., and Zhang, H.: Characterization of secondary organic aerosol from photo-oxidation of gasoline exhaust and specific sources of major components, Environ. Pollut., 232, 65–72, https://doi.org/10.1016/j.envpol.2017.09.018, 2018a.
Ma, Q., Lin, X., Yang, C., Long, B., Gai, Y., and Zhang, W.: The influences of ammonia on aerosol formation in the ozonolysis of styrene: Roles of Criegee intermediate reactions, R. Soc. Open Sci., 5, 172171, https://doi.org/10.1098/rsos.172171, 2018b.
Malecha, K. T., and Nizkorodov, S. A.: Photodegradation of secondary organic aerosol particles as a source of small, oxygenated volatile organic compounds, Environ. Sci. Technol., 50, 9990–9997, https://doi.org/10.1021/acs.est.6b02313, 2016.
Mandariya, A. K., Gupta, T., and Tripathi, S. N.: Effect of aqueous-phase processing on the formation and evolution of organic aerosol (OA) under different stages of fog life cycles, Atmos. Environ., 206, 60–71, https://doi.org/10.1016/j.atmosenv.2019.02.047, 2019.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Moise, T., Flores, J. M., and Rudich, Y.: Optical properties of secondary organic aerosols and their changes by chemical processes, Chem. Rev., 115, 4400–4439, https://doi.org/10.1021/cr5005259, 2015.
Na, K., Song, C., Switzer, C., and Cocker, D. R.: Effect of ammonia on secondary organic aerosol formation from α-pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, https://doi.org/10.1021/es061956y, 2007.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007a.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007b.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Brown carbon formation from ketoaldehydes of biogenic monoterpenest, Faraday Discuss., 165, 473–494, https://doi.org/10.1039/c3fd00036b, 2013.
NIST: NIST Chemistry WebBook Standard Reference Database 69, https://doi.org/10.18434/T4D303, 2020.
Noziere, B., Dziedzic, P., and Cordova, A.: Products and kinetics of the liquid-phase reaction of glyoxal catalyzed by ammonium ions (NH, J. Phys. Chem. A, 113, 231–237, https://doi.org/10.1021/jp8078293, 2009.
Ortiz-Montalvo, D. L., Hakkinen, S. A., Schwier, A. N., Lim, Y. B., McNeill, V. F., and Turpin, B. J.: Ammonium addition (and aerosol pH) has a dramatic impact on the volatility and yield of glyoxal secondary organic aerosol, Environ. Sci. Technol., 48, 255–262, https://doi.org/10.1021/es4035667, 2014.
Paciga, A. L., Riipinen, I., and Pandis, S. N.: Effect of ammonia on the volatility of organic diacids, Environ. Sci. Technol., 48, 13769–13775, https://doi.org/10.1021/es5037805, 2014.
Pathak, R. K., Stanier, C. O., Donahue, N. M., and Pandis, S. N.: Ozonolysis of alpha-pinene at atmospherically relevant concentrations: Temperature dependence of aerosol mass fractions (yields), J. Geophys. Res.-Atmos, 112, D03201, https://doi.org/10.1029/2006jd007436, 2007.
Peng, C., Yang, F., Tian, M., Shi, G., Li, L., Huang, R. J., Yao, X., Luo, B., Zhai, C., and Chen, Y.: Brown carbon aerosol in two megacities in the Sichuan Basin of southwestern China: Light absorption properties and implications, Sci. Total Environ., 719, 137483, https://doi.org/10.1016/j.scitotenv.2020.137483, 2020.
Prinn, R. G., Weiss, R. F., Miller, B. R., Huang, J., Alyea, F. N., Cunnold, D. M., Fraser, P. J., Hartley, D. E., and Simmonds, P. G.: Atmospheric trends and lifetime of CH3CCI3 and global OH concentrations, Science, 269, 187–192, https://doi.org/10.1126/science.269.5221.187, 1995.
Qi, X., Zhu, S., Zhu, C., Hu, J., Lou, S., Xu, L., Dong, J., and Cheng, P.: Smog chamber study of the effects of NOx and NH3 on the formation of secondary organic aerosols and optical properties from photo-oxidation of toluene, Sci. Total Environ., 727, 138632, https://doi.org/10.1016/j.scitotenv.2020.138632, 2020.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Sun, Y.-L., Zhang, Q., Schwab, J. J., Demerjian, K. L., Chen, W.-N., Bae, M.-S., Hung, H.-M., Hogrefe, O., Frank, B., Rattigan, O. V., and Lin, Y.-C.: Characterization of the sources and processes of organic and inorganic aerosols in New York city with a high-resolution time-of-flight aerosol mass apectrometer, Atmos. Chem. Phys., 11, 1581–1602, https://doi.org/10.5194/acp-11-1581-2011, 2011.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006gl026899, 2006.
Vu, T. V., Shi, Z., Cheng, J., Zhang, Q., He, K., Wang, S., and Harrison, R. M.: Assessing the impact of clean air action on air quality trends in Beijing using a machine learning technique, Atmos. Chem. Phys., 19, 11303–11314, https://doi.org/10.5194/acp-19-11303-2019, 2019.
Wang, G., Zhang, F., Peng, J., Duan, L., Ji, Y., Marrero-Ortiz, W., Wang, J., Li, J., Wu, C., Cao, C., Wang, Y., Zheng, J., Secrest, J., Li, Y., Wang, Y., Li, H., Li, N., and Zhang, R.: Particle acidity and sulfate production during severe haze events in China cannot be reliably inferred by assuming a mixture of inorganic salts, Atmos. Chem. Phys., 18, 10123–10132, https://doi.org/10.5194/acp-18-10123-2018, 2018a.
Wang, G. H., Zhang, R. Y., Gomez, M. E., Yang, L. X., Zamora, M. L., Hu, M., Lin, Y., Peng, J. F., Guo, S., Meng, J. J., Li, J. J., Cheng, C. L., Hu, T. F., Ren, Y. Q., Wang, Y. S., Gao, J., Cao, J. J., An, Z. S., Zhou, W. J., Li, G. H., Wang, J. Y., Tian, P. F., Marrero-Ortiz, W., Secrest, J., Du, Z. F., Zheng, J., Shang, D. J., Zeng, L. M., Shao, M., Wang, W. G., Huang, Y., Wang, Y., Zhu, Y. J., Li, Y. X., Hu, J. X., Pan, B., Cai, L., Cheng, Y. T., Ji, Y. M., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630-13635, https://doi.org/10.1073/pnas.1616540113, 2016.
Wang, R. Y., Ye, X. N., Liu, Y. X., Li, H. W., Yang, X., Chen, J. M., Gao, W., and Yin, Z.: Characteristics of atmospheric ammonia and its relationship with vehicle emissions in a megacity in China, Atmos. Environ., 182, 97–104, https://doi.org/10.1016/j.atmosenv.2018.03.047, 2018b.
Wang, S. W., Zhang, Q., Martin, R. V., Philip, S., Liu, F., Li, M., Jiang, X. T., and He, K. B.: Satellite measurements oversee China's sulfur dioxide emission reductions from coal-fired power plants, Environ. Res. Lett., 10, 114015, https://doi.org/10.1088/1748-9326/10/11/114015, 2015.
Wang, X., Gao, S., Yang, X., Chen, H., Chen, J., Zhuang, G., Surratt, J. D., Chan, M. N., and Seinfeld, J. H.: Evidence for high molecular weight nitrogen-containing organic salts in urban aerosols, Environ. Sci. Technol., 44, 4441–4446, https://doi.org/10.1021/es1001117, 2010.
Wang, Y., Chen, Y., Wu, Z., Shang, D., Bian, Y., Du, Z., Schmitt, S. H., Su, R., Gkatzelis, G. I., Schlag, P., Hohaus, T., Voliotis, A., Lu, K., Zeng, L., Zhao, C., Alfarra, M. R., McFiggans, G., Wiedensohler, A., Kiendler-Scharr, A., Zhang, Y., and Hu, M.: Mutual promotion between aerosol particle liquid water and particulate nitrate enhancement leads to severe nitrate-dominated particulate matter pollution and low visibility, Atmos. Chem. Phys., 20, 2161–2175, https://doi.org/10.5194/acp-20-2161-2020, 2020.
Wu, C., Zhang, S., Wang, G., Lv, S., Li, D., Liu, L., Li, J., Liu, S., Du, W., Meng, J., Qiao, L., Zhou, M., Huang, C., and Wang, H.: Efficient heterogeneous formation of ammonium nitrate on the saline mineral particle surface in the atmosphere of east asia during dust storm periods, Environ. Sci. Technol., 54, 15622–15630, https://doi.org/10.1021/acs.est.0c04544, 2020.
Xia, Y., Zhao, Y., and Nielsen, C. P.: Benefits of China's efforts in gaseous pollutant control indicated by the bottom-up emissions and satellite observations 2000-2014, Atmos. Environ., 136, 43–53, https://doi.org/10.1016/j.atmosenv.2016.04.013, 2016.
Xie, M., Chen, X., Hays, M. D., Lewandowski, M., Offenberg, J., Kleindienst, T. E., and Holder, A. L.: Light absorption of secondary organic aerosol: Composition and contribution of nitroaromatic compounds, Environ. Sci. Technol., 51, 11607–11616, https://doi.org/10.1021/acs.est.7b03263, 2017.
Xu, J., Huang, M. Q., Cai, S. Y., Liao, Y. M., Hu, C. J., Zhao, W. X., Gu, X. J., and Zhang, W. J.: Chemical composition and reaction mechanisms for aged p-xylene secondary organic aerosol in the presence of ammonia, J. Chin. Chem. Soc., 65, 578–590, https://doi.org/10.1002/jccs.201700249, 2018.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the volatility of secondary organic aerosol from isoprene photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Xu, L., Moller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Yang, W. Y., Li, J., Wang, M., Sun, Y. L., and Wang, Z. F.: A case study of investigating secondary organic aerosol formation pathways in beijing using an observation-based SOA box model, Aerosol Air Qual. Res., 18, 1606–1616, https://doi.org/10.4209/aaqr.2017.10.0415, 2018.
Yang, Y., Vance, M., Tou, F. Y., Tiwari, A., Liu, M., and Hochella, M. F.: Nanoparticles in road dust from impervious urban surfaces: Distribution, identification, and environmental implications, Environ. Sci.-Nano, 3, 534–544, https://doi.org/10.1039/c6en00056h, 2016.
Yang, Z., Tsona, N. T., Li, J., Wang, S., Xu, L., You, B., and Du, L.: Effects of NOx and SO2 on the secondary organic aerosol formation from the photooxidation of 1,3,5-trimethylbenzene: A new source of organosulfates, Environ. Pollut., 264, 114742, https://doi.org/10.1016/j.envpol.2020.114742, 2020.
Zhang, L., Wang, Y., Feng, C., Liang, S., Liu, Y., Du, H., and Jia, N.: Understanding the industrial NOx and SO2 pollutant emissions in China from sector linkage perspective, Sci. Total Environ., 770, 145242, https://doi.org/10.1016/j.scitotenv.2021.145242, 2021a.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of urban fine particulate matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015a.
Zhang, S., Li, D., Ge, S., Liu, S., Wu, C., Wang, Y., Chen, Y., Lv, S., Wang, F., Meng, J., and Wang, G.: Rapid sulfate formation from synergetic oxidation of SO2 by O3 and NO2 under ammonia-rich conditions: Implications for the explosive growth of atmospheric PM2.5 during haze events in China, Sci. Total Environ., 772, 144897, https://doi.org/10.1016/j.scitotenv.2020.144897, 2021b.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015b.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zou, Y., Deng, X. J., Zhu, D., Gong, D. C., Wang, H., Li, F., Tan, H. B., Deng, T., Mai, B. R., Liu, X. T., and Wang, B. G.: Characteristics of 1 year of observational data of VOCs, NOx and O3 at a suburban site in Guangzhou, China, Atmos. Chem. Phys., 15, 6625–6636, https://doi.org/10.5194/acp-15-6625-2015, 2015.