the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Sep 2021
| 09 Sep 2021
Role of Criegee intermediates in the formation of sulfuric acid at a Mediterranean (Cape Corsica) site under influence of biogenic emissions
Alexandre Kukui
Michel Chartier
Jinhe Wang
Sébastien Dusanter
Stéphane Sauvage
Vincent Michoud
Nadine Locoge
Valérie Gros
Thierry Bourrianne
Karine Sellegri
Jean-Marc Pichon
Reaction of stabilized Criegee intermediates (SCIs) with SO2 was proposed as an additional pathway of gaseous sulfuric acid (H2SO4) formation in the atmosphere, supplementary to the conventional mechanism of H2SO4 production by oxidation of SO2 in reaction with OH radicals. However, because of a large uncertainty in mechanism and rate coefficients for the atmospheric formation and loss reactions of different SCIs, the importance of this additional source is not well established. In this work, we present an estimation of the role of SCIs in H2SO4 formation at a western Mediterranean (Cape Corsica) remote site, where comprehensive field observations including gas-phase H2SO4, OH radicals, SO2, volatile organic compounds (VOCs) and aerosol size distribution measurements were performed in July–August 2013 as a part of the project ChArMEx (Chemistry-Aerosols Mediterranean Experiment). The measurement site was under strong influence of local emissions of biogenic volatile organic compounds, including monoterpenes and isoprene generating SCIs in reactions with ozone, and, hence, presenting an additional source of H2SO4 via SO2 oxidation by the SCIs. Assuming the validity of a steady state between H2SO4 production and its loss by condensation on existing aerosol particles with a unity accommodation coefficient, about 90 % of the H2SO4 formation during the day could be explained by the reaction of SO2 with OH. During the night the oxidation of SO2 by OH radicals was found to contribute only about 10 % to the H2SO4 formation. The accuracy of the derived values for the contribution of OH + SO2 reaction to the H2SO4 formation is limited mostly by a large, at present factor of 2, uncertainty in the OH + SO2 reaction rate coefficient. The contribution of the SO2 oxidation by SCIs to the H2SO4 formation was evaluated using available measurements of unsaturated VOCs and steady-state SCI concentrations estimated by adopting rate coefficients for SCI reactions based on structure–activity relationships (SARs). The estimated concentration of the sum of SCIs was in the range of (1–3) × 103 molec. cm−3. During the day the reaction of SCIs with SO2 was found to account for about 10 % and during the night for about 40 % of the H2SO4 production, closing the H2SO4 budget during the day but leaving unexplained about 50 % of the H2SO4 formation during the night. Despite large uncertainties in used kinetic parameters, these results indicate that the SO2 oxidation by SCIs may represent an important H2SO4 source in VOC-rich environments, especially during nighttime.
- Article
(6245 KB) - Full-text XML
-
Supplement
(2366 KB) - BibTeX
- EndNote





Sulfuric acid, H2SO4, is an important atmospheric component identified as playing a key role in formation of secondary atmospheric aerosol through new particle formation processes (Dunne et al., 2016; Paasonen et al., 2010; Sipilä et al., 2010; Weber et al., 1997). H2SO4 is considered to be a major precursor of newly formed atmospheric nucleation-mode particles and may play a significant role in their subsequent growth (Boy et al., 2005; Smith et al., 2005; Zhang et al., 2012). It is therefore important to understand well the atmospheric mechanisms determining the H2SO4 concentrations in different atmospheric environments.
Until recently it was generally accepted that the dominant atmospheric source of H2SO4 is the reaction of OH radicals with SO2 (Reaction R1) presenting a rate-limiting step leading in the troposphere to fast production of H2SO4 in the presence of water vapour and oxygen via Reactions (R2)–(R3) (Finlayson-Pitts and Pitts, 2000). It was assumed that H2SO4 atmospheric concentrations are determined predominantly by this source and the loss of sulfuric acid on the surface of existing particles, with the loss rate depending on the efficiency of H2SO4 uptake.
Another possible atmospheric source of H2SO4 via oxidation of SO2 by stabilized Criegee intermediates (SCIs), compounds formed by ozonolysis of unsaturated organic compounds, was suggested by Cox and Penkett (1971) and discussed first in view of its atmospheric importance by Calvert and Stockwell (1983). For a long time the reactions of SO2 with SCIs were considered to be too slow to represent an important atmospheric source of H2SO4, until in the more recent study of Welz et al. (2012) a rate constant of (3.9±0.7) × 10−11 cm−3 molec.−1 s−1 was derived for the reaction of SO2 with the simplest SCI, formaldehyde oxide (CH2OO), which is significantly larger than previous estimates of around cm−3 molec.−1 s−1 for the reactions of SO2 with CH2OO (Hatakeyama et al., 1986) and for the reactions of SO2 with other SCIs (Johnson and Marston, 2008). The importance of this additional source of H2SO4, which is still under discussion, depends on the atmospheric SCI concentrations and the kinetics and mechanisms of the SCI reactions with SO2.
Criegee intermediates (CIs), also known as carbonyl oxides, (R1)(R2)COO, with R1 and R2 representing different substituents, are produced via ozonolysis of alkenes by cycloaddition of an ozone molecule on a double bond forming a primary ozonide (POZ), a highly energized compound containing an O–O–O group. Subsequent rapid cleavage of either of the O–O bonds leads to the formation of a carbonyl compound and chemically activated CIs, which can undergo either prompt dissociation or thermal stabilization, leading to the formation of the SCIs (Criegee, 1975; Criegee and Wenner, 1949; Donahue et al., 2011; Johnson and Marston, 2008; Vereecken and Francisco, 2012; Vereecken et al., 2012).
Atmospheric concentrations of SCIs depend on their production rates by the ozonolysis of alkenes and their loss rates, predominantly via unimolecular decomposition and reactions with water monomers and dimers (Vereecken et al., 2017). However, the kinetic parameters and the reaction mechanisms of these processes are not well known.
The SCI production rate is determined by the ozonolysis reaction rate constants and corresponding speciated yields of different SCIs. The speciated SCI yield depends on a relative yield of two different CIs formed by decomposition of the POZ and a yield of SCI produced by the CI collisional stabilization depending on the CI structure and its energy content. For most of atmospherically relevant alkenes, the total SCI yields were not studied directly, while for those for which multiple studies are available there is in many cases a large data scatter. The data on the speciated SCI yields are available only for a few of alkenes (see e.g. Vereecken et al., 2017, and references therein).
For the loss of SCIs via unimolecular decomposition and reactions with water monomers and dimers, the results of experimental and theoretical studies show that the corresponding rate coefficients may vary by orders of magnitude depending on the SCI substituents and conformers (see e.g. Vereecken et al., 2017, and references therein). In recent years experimental studies of these reactions with direct detection and generation of specific SCIs were performed for several among the simplest of them, such as formaldehyde oxide (CH2OO) (Chao et al., 2015; Lewis et al., 2015; Sheps et al., 2017; Smith et al., 2015; Stone et al., 2018), acetaldehyde oxide (CH3CHOO) (Li et al., 2020; Lin et al., 2016; Sheps et al., 2014) and acetone oxide ((CH3)2COO) (Chhantyal-Pun et al., 2017; Fang et al., 2017; Huang et al., 2015; Lester and Klippenstein, 2018; Smith et al., 2016). Very recently the decomposition rate and an estimation of the rate coefficients for the reaction with water vapour were obtained for the first time in direct kinetic studies for syn methyl vinyl ketone oxide (syn-MVK-oxide) and anti methacrolein oxide (anti-MACR-oxide), four-carbon unsaturated Criegee intermediates derived from the ozonolysis of isoprene (Barber et al., 2018; Caravan et al., 2020; Lin et al., 2021). For other large SCIs, only estimations based on theoretical and indirect studies are available for their reactions with water vapour and their thermal decomposition.
The result of Welz et al. (2012) about the fast reaction of SO2 with formaldehyde oxide was extended in later direct and indirect studies, where similarly fast reactions with SO2 were confirmed for CH3CHOO, (CH3)2COO, Z-nopinone oxide (product of β-pinene ozonolysis), syn-MVK-oxide and anti-MACR-oxide (Ahrens et al., 2014; Caravan et al., 2020; Lin et al., 2021; Vereecken et al., 2017, and references therein), with rate coefficients in the range of (3–16) × 10−11 cm3 molec.−1 s−1. Reactions of other SCIs with SO2 were suggested to be similarly fast on the basis of theoretical results (Kurtén et al., 2011).
Theoretical analysis suggests that the reaction of SCI with SO2 proceeds via a barrierless cycloaddition of SO2 to SCI forming a sulfur-bearing secondary ozonide (SOZ) which can either be stabilized or decompose to form SO3 or other products (Kurtén et al., 2011; Kuwata et al., 2015; Vereecken et al., 2012). For smaller SCIs, e.g. formaldehyde and acetone oxides, the theory predicts negligible SOZ stabilization and about unity yield of SO3 (Kuwata et al., 2015). These results are supported by experimental studies for CH2OO (Berndt et al., 2014a; Wang et al., 2018) and (CH3)2OO, CH3CHOO (Berndt et al., 2014b). For larger SCIs with expected longer SOZ lifetimes, the SO3 yield may depend on the SOZ fate in the atmosphere with respect to its decomposition or further reactions, e.g. with H2O (Kuwata et al., 2015; Vereecken et al., 2012), although a large yield of sulfur trioxide exceeding 80 % was observed by Ahrens et al. (2014) for the large SCIs formed during the ozonolysis of β-pinene.
Estimations based on the available or evaluated kinetic parameters show that the atmospheric SCI concentrations vary by orders of magnitude depending on conditions specific to different environments, such as the concentrations and composition of alkenes, ozone concentration or humidity. Using chemistry-transport global modelling, the highest SCI concentrations of the order of 104–105 molec. cm−3 were inferred for the regions with the highest isoprene and terpene emissions, e.g. above the tropical forest (Chhantyal-Pun et al., 2019; Khan et al., 2018; Newland et al., 2018; Vereecken et al., 2017; Novelli et al., 2017). Estimated using steady-state calculations, the concentration of SCI ranges from 2.3 × 103 molec. cm−3 at a rural site to 5.5 × 104 molec. cm−3 in an urban polluted environment (Vereecken et al., 2017). The estimated contribution of SCI to H2SO4 formation is also highly variable: about 7 % in rural environments and up to 70 % over tropical regions (Vereecken et al., 2017). At the global scale, the contribution of SCI to SO2 oxidation was estimated to be negligible, contributing less than 1 % (Newland et al., 2018). The uncertainty associated with the predicted SCI concentrations was estimated to be 1 order of magnitude (Vereecken et al., 2017) due to poorly defined SCI formation and loss rates. Even a higher uncertainty may be expected for the estimated contribution of SCIs to SO2 oxidation considering not well-defined reaction rate coefficients for the reaction of different SCIs with SO2.
The adequacy of the mechanism treating the SO2 oxidation by OH as a predominant source of the atmospheric H2SO4 was tested in a number of field campaigns where simultaneous measurements of OH and H2SO4 were conducted (Table S1 in the Supplement). A selected ion chemical ionization mass spectrometry (CIMS) technique for simultaneous measurements of OH and H2SO4 was first introduced by Eisele and Tanner (1993), and since then it has been used in a number of field measurements in different environments. In these studies the measurements of OH, H2SO4, SO2 and aerosol surface area were used to compare the rate of H2SO4 production in Reaction (R1) and the rate of H2SO4 loss on aerosol particles assuming a steady-state condition between these processes. In a number of measurement campaigns the H2SO4 budget was found to be closed using the uptake coefficient of unity corresponding to the upper limit of the H2SO4 loss rate on existing particles. This was observed in different environments including remote marine (Weber et al., 1997), forested rural (Birmili et al., 2000; Boy et al., 2013) and forested remote sites (Eisele and Tanner, 1993; Weber et al., 1997). However, in other field studies conducted in various environments, the H2SO4 condensation sink calculated using an uptake coefficient of unity was found to significantly exceed its formation rate via SO2 oxidation by OH, indicating either an H2SO4 uptake efficiency lower than unity or the presence of sources of H2SO4 other than Reaction (R1) (Bardouki et al., 2003; Berresheim et al., 2002, 2014; Boy et al., 2013; Jefferson et al., 1998; Mauldin III et al., 2012; Petäjä et al., 2009). Several additional H2SO4 gas-phase sources were suggested, such as the oxidation of DMS or DMDS in remote coastal environments, proceeding with SO3 formation (Berresheim et al., 2002, 2014; Jefferson et al., 1998) or SO2 oxidation by SCIs in the boreal forest and in moderately polluted environments (Boy et al., 2013; Kim et al., 2015; Mauldin et al., 2012). A heterogeneous formation of gas-phase H2SO4 via the catalytic oxidation of SO2 on the surface of black carbon aerosols has also recently been shown to be important under polluted conditions (Yao et al., 2020).
In this work, we present an evaluation of the role of SCIs in H2SO4 production at a remote site on Cape Corsica near the northern tip of Corsica (Ersa station, western Mediterranean). In July–early August 2013, comprehensive field observations including gas-phase (OH radicals, H2SO4, VOCs, NOx, SO2, others) and aerosol-size distribution measurements were conducted at this site in the framework of the SAFMED (Secondary Aerosol Formation in the Mediterranean) campaign as part of the summer 2013 experimental effort of project ChArMEx (Chemistry-Aerosols Mediterranean Experiment). During the field campaign, the site was strongly influenced by local emissions of biogenic volatile organic compounds, including isoprene and terpenes, forming different SCIs in reactions with ozone and, hence, potentially representing an additional source of H2SO4 via reactions of SCIs with SO2. We use the OH, H2SO4 and SO2 measurements to estimate an upper limit for the contribution of H2SO4 sources other than Reaction (R1). Using available measurements of unsaturated VOCs and adopting rate coefficients for SCI reactions based on structure–activity relationships (SARs) from Vereecken et al. (2017), we estimate steady-state SCI concentrations. These SCI concentrations are used for the estimation of the rate of H2SO4 formation in the reactions of SCIs with SO2 and its comparison with OH + SO2 source resulting from the OH and H2SO4 measurements.
2.1 Field site
Measurements were performed at the Ersa site from 18 July to 5 August 2013 during the ChArMEx/SAFFMED field campaign (Dulac et al., 2021). The Ersa station (42.969∘ N, 9.380∘ E) is located at Cape Corsica on the northern edge of Corsica (Michoud et al., 2017; Zannoni et al., 2017). It is situated at an altitude of 533 m above sea level on the top of a hill dominating the northern part of the cape. On its eastern, northern and western sides it is a few kilometres away from the coast and has a direct view of the sea. The measurement site is isolated by a mountain range from the closest large city, Bastia, situated about 30 km south of the site. The site is surrounded by widespread vegetation such as scrubland typical of the Mediterranean areas, responsible for biogenic VOC emissions (Debevec et al., 2021; Zannoni et al., 2015).
2.2 Experimental methods
2.2.1 OH and H2SO4 measurements
Concentrations of OH radicals and H2SO4, as well as total peroxy radicals (HO2 + RO2, not discussed here), were measured using chemical ionization mass spectrometry (CIMS) (Berresheim et al., 2000; Eisele and Tanner, 1991). A detailed description of the instrument is presented elsewhere (Kukui et al., 2008, 2012). Here we briefly present the measurement technique and essential details about the setup and performance of the instrument during the ChArMEx/SAFMED campaign. A detailed description of the calibration system used during the campaign, which was not presented before, is given in Sect. S3 in the Supplement.
OH was detected by conversion of the sampled OH with isotopically labelled 34SO2 to form HSO4 in a chemical conversion reactor (CCR) in the presence of ambient water vapour and oxygen. The isotopically labelled HSO4 and ambient H2SO4 were detected by mass spectrometry as H34SO and H32SO product ions. The product ions were produced by chemical ionization with a NO reagent ion in an ion-molecule reactor (IMR) following the CCR. The reagent ions were generated in a corona NO2/air discharge ion source (CD). A scheme of the reactor is presented in Fig. S2 in the Supplement.
Ambient air was sampled at a volumetric flow rate of 10 SLM (standard litre per minute), creating turbulent flow in the chemical conversion region of the reactor. The turbulent flow conditions minimize possible influence of wind speed on the measurements and ensure fast mixing of reactants. The reactants used for the chemical conversion (34SO2 for OH conversion into sulfuric acid and NO for peroxy radical conversion into OH and their subsequent detection as OH) and the radical quencher (NO2) are introduced into the reactor through a set of injectors. NO2 used as a scavenger removes not only the OH radicals, but also peroxy radicals, converting them into HO2NO2 and RO2NO2 peroxy nitrates. Switching the reactant flows between the different injectors allows measurements in four different modes: the background mode, two different OH radical measurement modes and the peroxy radical measurement mode (Fig. S1). The two OH measurement modes differ by the times used for the chemical conversion, 4 and 20 ms. The ratio of the signals with the short and long conversion times may be used as an indicator of an artificial OH formation in the reactor (Kukui et al., 2008).
Measurements were performed by monitoring the peak intensities at (NO), (H32SO), and (H34SO) with the CIMS, respectively, denoted I62, I97, and I99 hereafter. Every measurement of OH was derived from 1 min of an OH ion signal count and two 30 s background ion signal counts before and after the OH signal measurement. Peroxy radicals were measured at the end of the OH detection sequence by switching on the NO flow to the corresponding injector for a duration of 2 min. To avoid any possible influence of traces of NO on the OH measurements, a time delay of 6 min was imposed after switching off the NO flow and before starting the next OH measurement sequence in order to ensure flushing of the CCR. The OH and the H2SO4 data were averaged, resulting either in a sequence of three points with a step of 7 min separated by a time gap of 15 min or yielding a sequence with a time step of about 90 min. The latter was used to match the time resolution of the VOC measurements (Table S4).
The concentrations of OH and H2SO4 were derived from the measured ratios of the H32SO, H34SO and NO ion peak intensities, and : , where R corresponds to OH or H2SO4, IR is a combination of and ratios corresponding to OH or H2SO4 accounting for isotopic composition of SO2 used for the chemical conversion (99 % isotopic enrichment of 34S, Eurisotop, Cambridge Isotope Laboratories, Inc.) and sulfur isotope natural abundance (32S (95.02 %) and 34S (4.21 %); Hoefs, 2018), and CR is a calibration coefficient determined in calibration measurements by production of OH or H2SO4 in a turbulent flow reactor using photolysis of water vapour at 184.9 nm and quantified by chemical actinometry using photolysis of N2O (Faloona et al., 2004). A detailed description of the calibration system with definitions of CR and IR is given in Sect. S3.
The overall accuracy of the calibration coefficients was estimated taking into account uncertainties of all parameters used for calculation of the radical concentrations in the photolysis reactor and the precision of the measurements of the ratios and . The overall estimated calibration uncertainty (1σ) was 30 % for OH, 32 % for H2SO4 and 8 % for a ratio of [H2SO4] to [OH], (Table S3). Accounting for the calibration uncertainties and the measurement precision, the overall 1σ uncertainty of the 14 min-averaged measurements of OH, H2SO4 and the ratio of [H2SO4] [OH] was estimated to be around 32 %, 34 % and 16 % during the daytime and 42 %, 44 % and 40 % during the nighttime, respectively. During the ChArMEx/SAFMED campaign the observed level of the OH background signal was significantly higher than the typical OH background found during calibration or field measurements in air with low VOC concentrations (see Fig. 7 and the discussion in Sect. 4.4). Accordingly, the lower limits of detection at a signal-to-noise ratio of 2 and a 15 min integration time were 2×105 molec. cm−3 for H2SO4 and 5×105 and 2×105 molec. cm−3 for OH daytime and nighttime measurements, respectively.
During the ChArMEx/SAFMED campaign the instrument was installed in a dedicated container with the CCR fixed to the roof of the container via an interface cap covered with a PTFE sheet. The sampling aperture of the reactor (3 mm diameter) was positioned 50 cm above the roof and about 3 m above the ground.
To avoid possible contamination of ambient air by the SO2, NO and NO2 reactants added to the CCR, a trap was set up at the pump exhaust by using two 100 L cylinders containing zeolites. The cylinders were refilled several times during measurements. A flexible exhaust tube of 30 m length was always placed downwind from the container.
2.2.2 Complementary measurements
The aerosol particle size distribution was measured using a scanning mobility particle sizer (SMPS TSI 3080, associated with a CPC TSI 3010) in the range from 10.9 to 495.8 nm and with an aerodynamic particle sizer (APS, TSI 3321) in the range from 542 nm to 19.48 µm. As the SMPS measurements were made with dehydrated particles, the particle diameters were corrected to ambient humidity using a particle growth factor (GF) of 1.5 at 90 % determined with a volatility hygroscopic–tandem differential mobility analyser (VH-TDMA) (Villani et al., 2008). The dependence of the GF on relative humidity (RH) was calculated using the one-parameter approximation from Rissler et al. (2006). Estimated uncertainties of measured particle number densities and GF-corrected particle diameters were 10 % and 15 %, respectively.
SO2 concentration was measured by UV fluorescence (Thermo Environmental Instruments (TEI), model 43C-TLE) with an estimated accuracy of 20 %, a lower detection limit of 0.05 ppb and a time resolution of 5 min.
Ozone concentration was measured by means of a CraNOx II (Eco Physics) NOx and O3 monitor with an estimated accuracy of 10 %.
A detailed description of VOC measurements during the ChArMEx/SAFMED campaign is given in Michoud et al. (2017). The measurements of 23 unsaturated VOCs, including alkenes, aldehydes, ketones, isoprene and monoterpenes, were used in this work for estimation of SCI concentrations. Employed measurement techniques and concentration ranges for measured unsaturated VOCs are given in Table S4 together with associated time resolution, limit of detection and uncertainties. The data were averaged or interpolated with a time step of 90 min.
Wind speed and direction, relative humidity, temperature and photolysis rates were also measured throughout the campaign.
2.3 Estimation of H2SO4 steady-state concentrations
Concentrations of H2SO4 produced via SO2 oxidation by OH and sum of SCIs, H2SO and H2SO, respectively, were calculated assuming validity of a steady state between the H2SO4 production and its loss (see discussion in Sect. 4.1),
Here [OH] and [SO2] are measured concentrations, [SCIi] are estimated concentrations of speciated SCIs, and CS, a condensation sink, is a rate of H2SO4 loss by its condensation on aerosol particles which is assumed to be the predominant H2SO4 loss process, while the dry deposition on macroscopic surfaces is neglected considering a long associated lifetime estimated of about 1 d using a typical deposition velocity of 1 cm s−1 (Seinfeld and Pandis, 2016) and a boundary-layer depth of 1 km. Considering the CS to be a major sulfuric acid loss process, the median value of H2SO4 lifetime was 2.7 min (2.2–3.7 min interquartile range, Figs. 1, 2). The rate coefficient for the reaction of OH with SO2, cm3 molec.−1 s−1 (at 760 torr and 298 K) was taken from the IUPAC 2004 recommendation (Atkinson et al., 2004) (see Sect. 4.2). Production of H2SO is calculated as a sum of contributions from different speciated SCIi reacting with SO2 with rate coefficients (Table S5). It is assumed here that the H2SO4 yield in reaction of SCIs with SO2 is a unity for all SCIs, giving an upper limit for the contribution to H2SO4 formation of accounted SCIs.
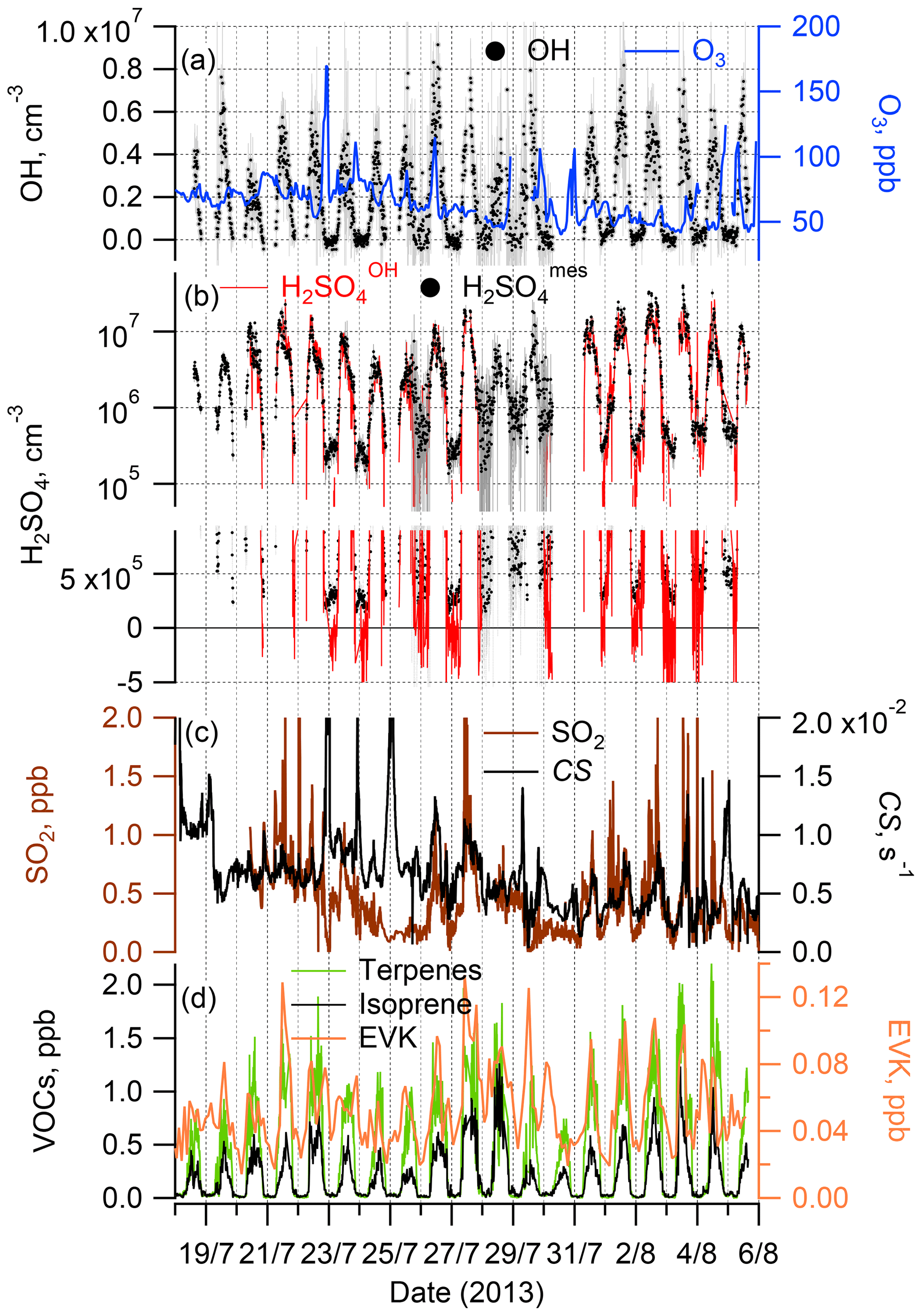
Figure 1Time series of the observations during the ChArMEx summer 2013 campaign: OH radicals and O3 (a); sulfuric acid observed, H2SO, and calculated assuming only an SO2+OH source, H2SO (see Sect. 2.3) (b); SO2 and condensation sink (CS) (c); total monoterpenes and isoprene (left axis) and EVK (right axis) (d). For clarity, a comparison of the measured H2SO and calculated H2SO concentrations is presented in (b) using two plots, with logarithmic and linear scales on the y axis.

Figure 2Median diel profiles of calculated [H2SO4] produced in OH + SO2 (H2SO), observed [H2SO4], [OH] and photolysis rate J(O1D) (a), CS and [SO2] (b), [O3] and concentrations of selected VOCs (c), and relative humidity (RH) and temperature (T) (d). Shaded areas represent the 25th/75th percentiles.
The CS was calculated using measured particle size distributions and number concentrations by calculating diffusional flux to the aerosol particles assuming an accommodation coefficient of unity (Hanson, 2005) and using a Fuchs–Sutugin transition correction (Jefferson et al., 1998; Seinfeld and Pandis, 2016). A diffusion coefficient of H2SO4 was estimated using its dependence on relative humidity from Hanson and Eisele (2000), giving 0.077 cm2 s−1 at an RH of 60.6 % (median value). Without considering an uncertainty in the accommodation coefficient, the accuracy of the calculated CS of 20 % was assessed, accounting for the uncertainties in particle measurements given in Sect. 2.2.2 and an uncertainty in the H2SO4 diffusion coefficient of 5 %.
Concentrations of speciated SCIs, [SCIi], were calculated assuming steady-state conditions considering their production by ozonolysis of the measured unsaturated VOCs and their loss by thermal decomposition and in reactions with water vapour (with H2O and (H2O)2):
where X denotes a specific VOC, is a rate coefficient for the reaction of X with O3, is a yield of stabilized SCIi from CIi, is a specific yield of CIi in the reaction X+O3, and Ki, , and are rate coefficients for the thermal decomposition, the reaction with H2O, and the reaction with water dimer for the specific SCIi, respectively. Concentration of (H2O)2 was calculated using an equilibrium constant for water dimer formation and its temperature dependence from Ruscic (2013).
The rate coefficients and the yields and for 36 SCIs derived from the ozonolysis of 23 measured VOCs are presented in Tables S4 and S5. Apart from a few rate coefficients and yields available from more recent experimental and theoretical studies, most of the parameters in Eqs. (2) and (3) are taken from recommendations of Vereecken et al. (2017), which are based either on an analysis of available experimental data or derived from theory-based structure–activity relationships (SARs). For the 36 SCIs listed in Tables S4 and S5, we have adopted the SCI naming convention from Vereecken et al. (2017). Using this approach, uncertainty in the SCI concentrations is estimated in Vereecken et al. (2017) to be of an order of magnitude, mainly due to the uncertainties in CI speciation and the SCI decay rates.
3.1 Observed data
Time series and median diel profiles of observed concentrations of OH, H2SO4, SO2, O3, selected VOCs and a calculated condensation sink of H2SO4 are presented in Fig. 1 (time series) and Fig. 2 (median diel cycles, also including J(O1D), T, RH, and CS data). The nighttime OH and H2SO4 measurements on 26 and 28–30 July were influenced by a strong fog event deteriorating the accuracy of the corresponding OH and H2SO4 data.
OH and H2SO4 concentrations reached around midday, with maximum values of 4.3 × 106 and 8.5 × 106 molec. cm−3 (mean values for daytime hours from 11:00 to 13:00 local time (= GMT+2 h)), respectively, with nighttime concentrations around 1 × 105 and 5 × 105 molec. cm−3 for OH and H2SO4, respectively, close to the detection limit.
Comparing the present observations (Fig. 2) with previous measurements in the Mediterranean region, the mean peak OH concentration during the noon hours was close to the peak OH levels observed during the CYPHEX campaign in the summer of 2014 in Cyprus in the eastern Mediterranean, 5.8 × 106 molec. cm−3 (Mallik et al., 2018), where O3 concentrations and J(O1D) peak levels were similar to those observed during ChArMEx (J(O1D) was measured during ChArMEx but not yet published). The somewhat lower ChArMEx OH noon concentrations compared to CYPHEX are consistent with higher OH reactivity observed during ChArMEx (Zannoni et al., 2017), although the direct comparison of radical chemistry at these two sites is not straightforward considering that, among other differences between these sites, biogenic VOCs at the Cyprus site, e.g. isoprene and monoterpenes, were 3–5 times lower compared to ChArMEx. About 4 times higher OH peak concentrations, 2.1 × 107 molec. cm−3, were observed during the MINOS campaign in the summer of 2001 in Crete (central Mediterranean), with similar O3 and J(O1D) observed levels (Berresheim et al., 2003). This difference is difficult to explain based on the available data (Mallik et al., 2018).
The observed ChArMEx H2SO4 concentrations were about 2 times higher than the observed OH concentrations. For other sites the observed ratios [H2SO4] [OH] were in the range from 1 to 9, with only one example when this ratio was less than unity (Table S1). The ratio [H2SO4] [OH] depends on the SO2 concentration and condensation sink, correlating with the aerosol particle surface area concentration. The condensation sink and [SO2] values during ChArMEx, with median values of 6.1 × 10−3 s−1 and 0.44 ppb, respectively, were typical of clean remote continental or coastal environments (Table S1).
The unsaturated VOCs observed during ChArMEx showed strong diel variation from about 0.4 ppb during the night to about 2 ppb at noon for the concentration of the sum of the VOCs (Figs. 1, 2 and S9 and Table S4). The major contribution during the day (from 07:00 to 20:00) was from biogenic VOCs (isoprene (22 % on average), β-pinene (14 %), α-pinene (9 %), α-terpinene (7 %), methyl vinyl ketone (MVK) (5 %) and methacrolein (MACR) (5 %)), with emission rates correlating with temperature and solar radiation (Kesselmeier and Staudt, 1999). At nighttime, the unsaturated VOCs were represented mostly by compounds of mixed biogenic and anthropogenic origin: ethene (31 % on average), acrolein (14 %) and ethyl vinyl ketone (9 %). A significant nighttime contribution was also found from isoprene (9 %) and its first-generation oxidation products MVK (6 %) and MACR (6 %).
3.2 Comparison of observed H2SO4 with sulfuric acid produced from OH + SO2 (H2SO)
Time series of the H2SO4 produced in the reaction of OH with SO2 and H2SO, calculated according to Eq. (1), and of the observed H2SO4 are presented in Figs. 1b and 2b showing apparently good agreement between them during the day and an underestimation of the sulfuric acid concentration by H2SO during the night. The inverse variance weighted mean value of the ratio of [H2SO4]OH [H2SO4] for the data presented in Fig. 1b is 0.86±0.04 during the day (07:00–20:00) and 0.09±0.02 during the night (20:00–07:00), respectively (Table 1). Uncertainties in the ratio [H2SO4]OH [H2SO4] were estimated by accounting for the uncertainty in the measured ratio of [OH] [H2SO4] (see Sect. 2.2.1), in the SO2 measurements (20 %) and in the CS calculations (20 %) without considering uncertainties in the reaction rate coefficient k1 and the H2SO4 uptake coefficient (see Sect. 4.2 and 4.3).
Table 1Comparison of observed [H2SO4] with calculations assuming H2SO4 formation via oxidation of SO2 by OH and SCIs. [OH] and [H2SO4] are observed concentrations, and [H2SO4]OH and [H2SO4]SCI are calculated H2SO4 produced by oxidation of SO2 by OH and SCI, respectively. Values in square brackets correspond to [H2SO4]OH concentrations calculated with a rate coefficient for the reaction of OH with SO2 from Medeiros et al. (2018) (see discussion in Sect. 4.2).
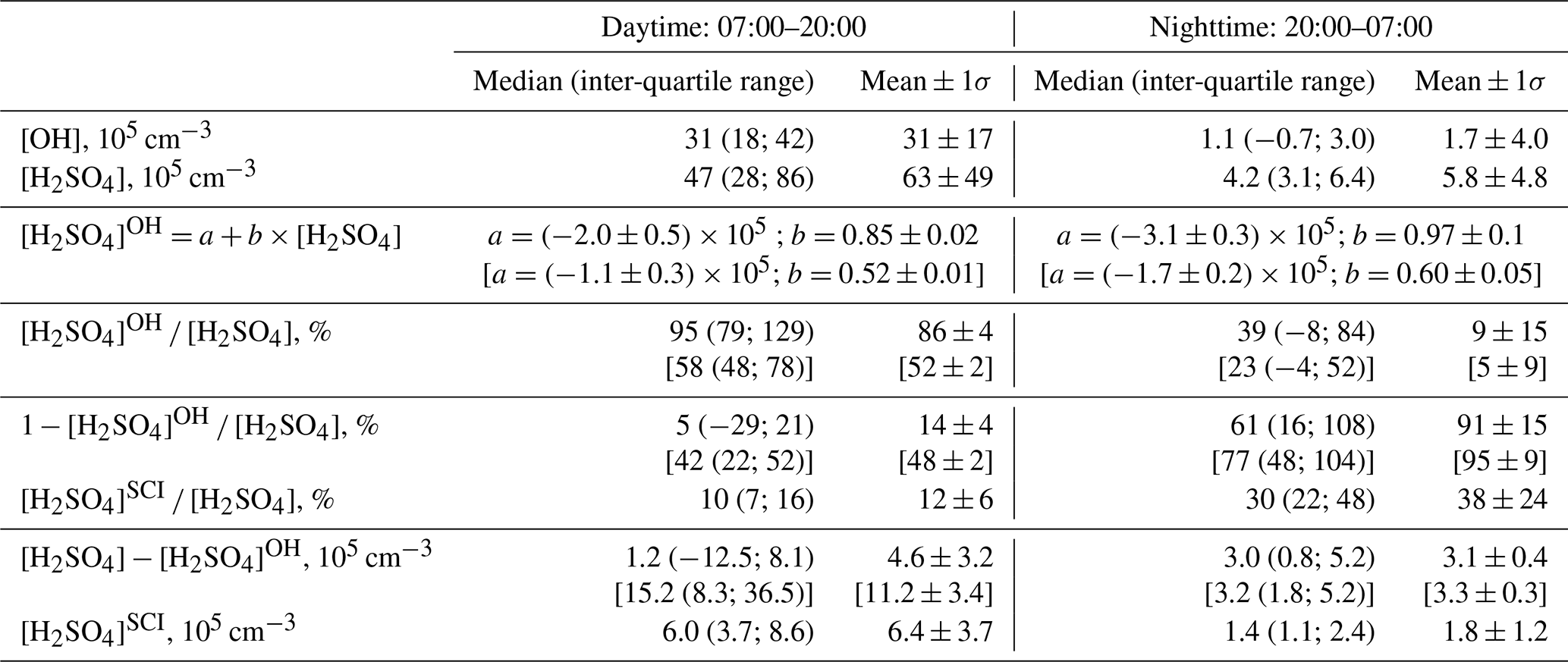
Considering the correlation of the calculated sulfuric acid concentration [H2SO4]OH with the measured [H2SO4] shown in Fig. 3, we find that the linear regression using a bivariate fit procedure accounting for the measurement errors of both H2SO4 and H2SO (York et al., 2004) results in slopes of 0.85 ± 0.02 and 0.97 ± 0.1 for the daytime and nighttime, respectively. The linear regression yields significant intercepts of () × 105 molec. cm−3 during the day and of () × 105 molec. cm−3 during the night. The close-to-unity slopes and the negative intercepts may be interpreted as a presence of an additional source contributing to H2SO4 on an average level of several of 105 molec. cm−3. During the night this source is important compared with the observed nighttime average [H2SO4] of (5.8±4.8) × 105 molec. cm−3. During the night the estimation of the contribution from missing sources derived from the linear fit is in qualitative agreement with the mean ratio of [H2SO4]OH [H2SO4] of 0.09, indicating that the SO2 + OH source explains only about 10 % of the H2SO4 formation. During the daytime, the reaction of SO2 with the OH source explains around 90 % of the H2SO4 formation (Table 1).
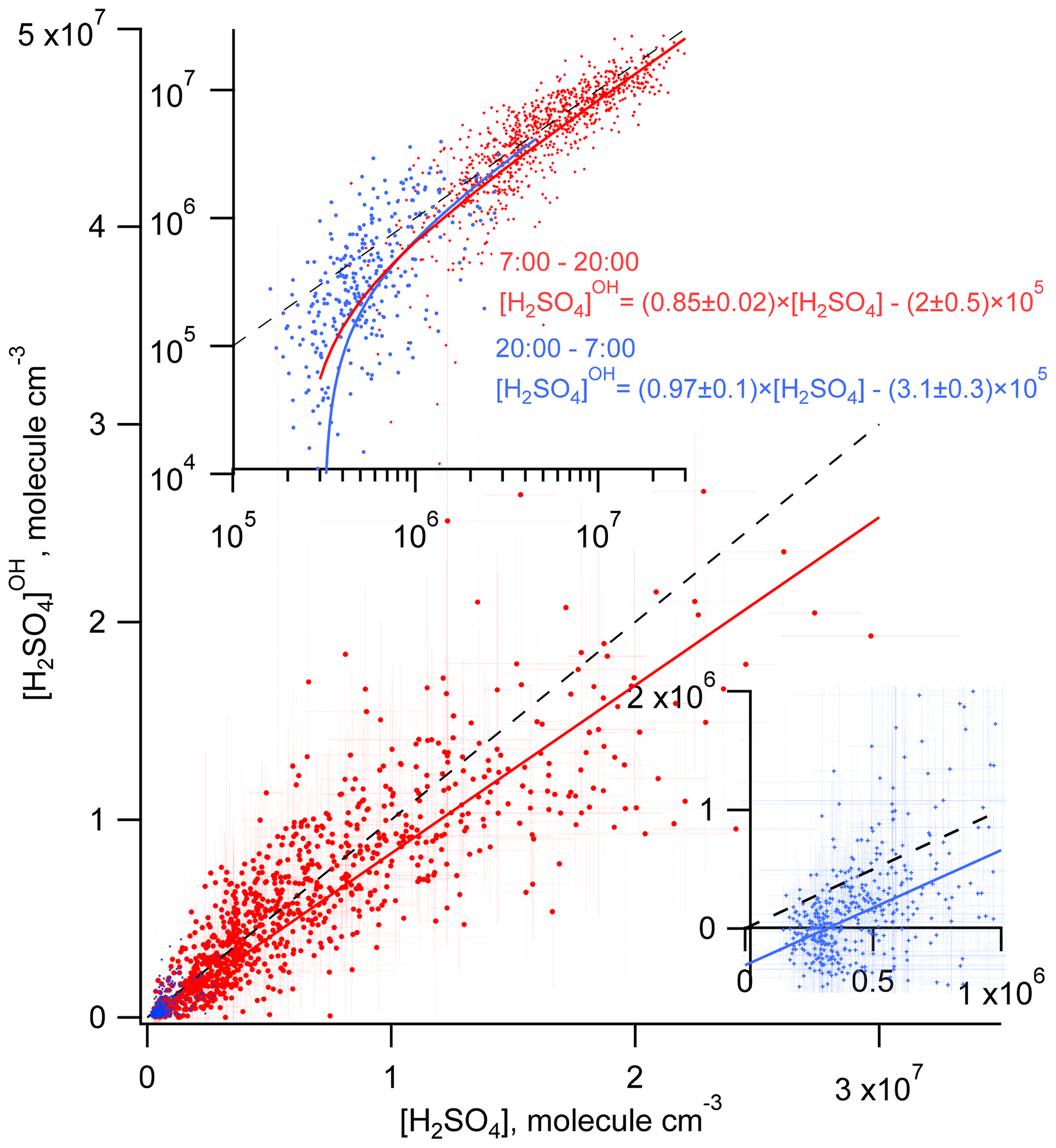
Figure 3Comparison of the measured H2SO4 and the H2SO calculated accounting only for the OH + SO2 source during the day (red) and during the night (blue). Solid lines correspond to linear regression fitting accounting for both X and Y measurement uncertainties. Dashed lines represent 1 : 1 ratios. Inserts, log plot (upper) and linear plot (lower right) are added for clearer presentation of the nighttime data.
3.3 Comparison of observed H2SO4 with sulfuric acid produced from SCIs + SO2 (H2SO)
Estimated according to Eqs. (2) and (3), mean diel profiles of the sulfuric acid H2SO produced in the reactions of SO2 with SCIs generated by the ozonolysis of the measured unsaturated VOCs, excluding α-terpinene, are presented in Fig. 4a. The calculated contribution from α-terpinene alone is up to 6 times larger than observed [H2SO4] (Fig. 4b). It is not clear whether the reason for this large overestimation can be related to erroneous α-terpinene measurements and/or to incorrect kinetic parameters used for the calculation of H2SO4 production from α-terpinene. The observed α-terpinene daytime concentrations were similar to the concentrations of α-pinene and β-pinene. Accounting for about 100 times faster α-terpinene consumption in reactions with OH and O3 compared to other terpenes (Atkinson et al., 2006; IUPAC, 2020), that would imply about a 100 times larger α-terpinene emission rate at the measurement site, which is unlikely considering observed compositions of monoterpene emissions of biogenic origin (Geron et al., 2000). Being well outside of the uncertainty of the H2SO4 measurements, the contribution from α-terpinene was therefore excluded from consideration in this work.

Figure 4Diel profiles of the mean calculated H2SO4 concentrations produced from ozonolysis of different VOCs: (a) filled regions correspond to [H2SO4]SCI produced by ozonolysis of different VOCs; (b) profiles of ratios of [H2SO4]SCI from different VOCs to the measured [H2SO4]. Black dotted line in (b) corresponds to [H2SO4]SCI produced by the ozonolysis of α-terpinene (right axis).
The calculated concentration of the sum of H2SO reaches a maximum of about 7×105 molec. cm−3 around midday and goes down to about 1.5×105 molec. cm−3 during the night. The largest estimated contribution to the H2SO is from α-pinene, limonene and isoprene during the day and from MVK and EVK at nighttime (Fig. 4).
The largest contribution to the calculated sum of SCIs was from the syn form of acetaldehyde oxide, Z-CH3CHOO (2), E-MVK-oxide (10) from isoprene, oxo-substituted E-(C(O)R)CHOO (31) (with R = H, CH3, and C2H5 for the SCI from acrolein, MVK and EVK, respectively), and phenyl-substituted carbonyl oxide PhCHOO (35), from styrene and Z-pinonaldehyde-K-oxide (13) from α-pinene (the SCIs numbering given in parentheses corresponds to the SCI numbering in Tables S4 and S5.
The nighttime and daytime mean concentrations of the sums of all SCIs were about 103 and 3×103 molec. cm−3, respectively. Z-pinonaldehyde-K-oxide (13) (from α-pinene) and E-(C(O)R)CHOO (31) (from MVK, EVK and acrolein) were also the major SCI producing H2SO4. In addition, a comparable contribution to H2SO4 formation was from Z-(CHRaRb)(CH3)COO (21) (from limonene ozonolysis) and E-MVK-oxide (10) (from isoprene ozonolysis). Detailed information is given in Fig. S10.
Comparing the calculated [H2SO4]SCI with the missing H2SO4 source derived from the difference of the measured [H2SO4] and calculated [H2SO4]OH, Fig. 5a shows that, compared to the difference of [H2SO4] − [H2SO4]OH representing the missing H2SO4 source, the [H2SO4]SCI is lower during the night and higher during the day, with a difference of several 105 molec. cm−3 (Table 1). The large variance of the average [H2SO4] − [H2SO4]OH difference presented in Table 1 is due to the large scattering of these values derived either from a small difference of large OH and H2SO4 signals or from the measurements of OH and H2SO4 concentrations close to their lower detection limits as well as due to a stochastic day-to-day natural variation.
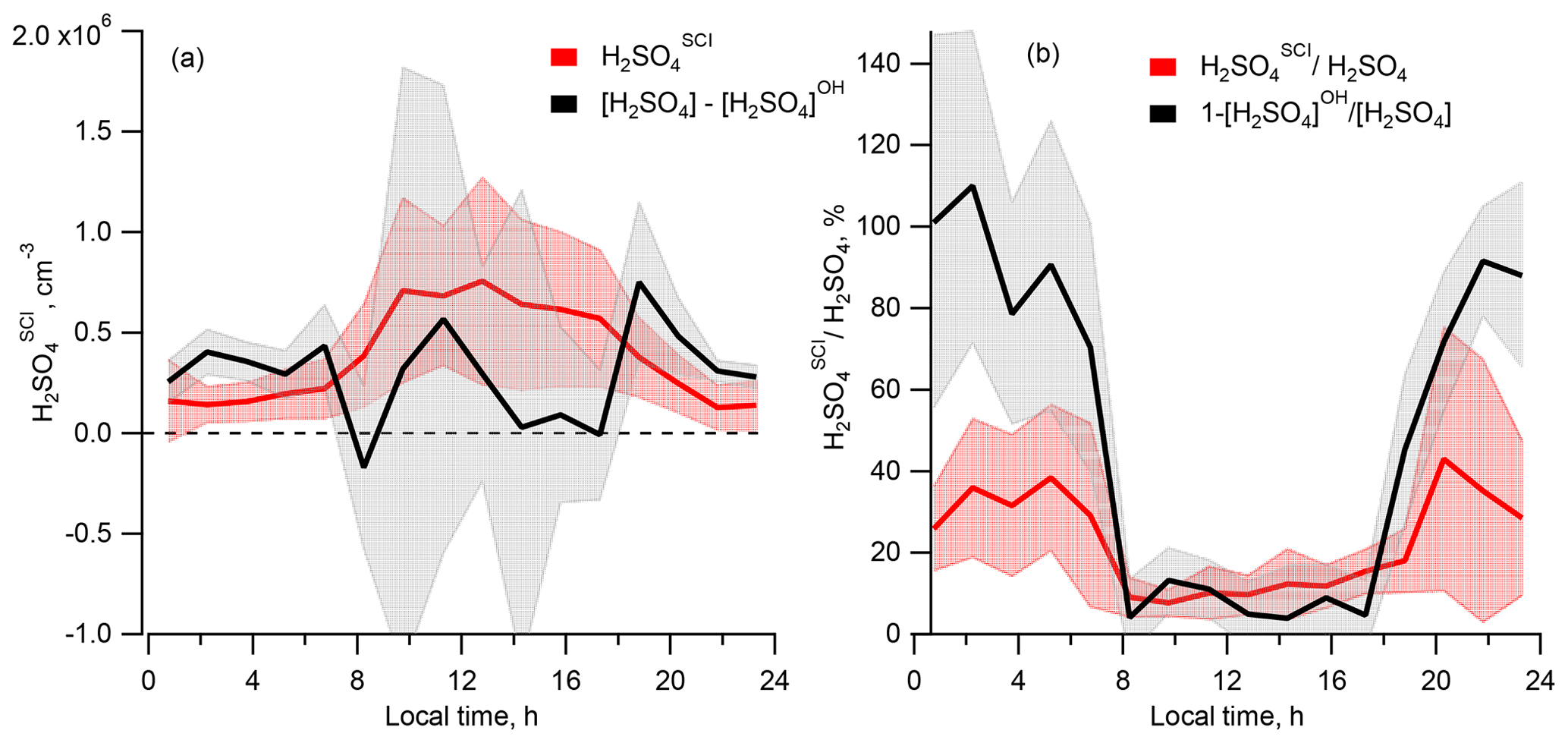
Figure 5Comparison of the sulfuric acid produced by oxidation of SO2 by SCIs, H2SO, and the missing source of H2SO4 derived from the difference of the measured [H2SO4] and [H2SO4]OH. (a) Diel profiles of the mean [H2SO4]SCI (red) and [H2SO4] − [H2SO4]OH (black). (b) Diel profiles of the mean of the relative contributions [H2SO4]SCI [H2SO4] (red) and 1 − [H2SO4]OH [H2SO4] (black). Shaded areas correspond to ±1σ standard deviation.
Shown in Fig. 4b, the calculated dial profile of the relative H2SO to H2SO4 contribution resembles the dial profile of a missing H2SO4 source derived from the relative concentration difference of H2SO4 and H2SO, i.e. 1 − [H2SO4]OH [H2SO4]. Both profiles exhibit a shallow well from about 08:00 to 18:00 local time (UTC+2) during the day, with a higher relative contribution during the night. According to the [H2SO4]SCI calculations, the SCIs contribution is about 3 times higher during the night, constituting (38±24) % compared to (12±6) % during daytime. The calculated [H2SO4]SCI contribution is significantly lower than the nighttime missing source of (91±2) % and is in good agreement with the missing (14±4) % during the day (Table 1). The large unexplained formation of H2SO4 during the night can be related to the large uncertainty in the calculated SCI concentrations and the rates of H2SO4 production from their reactions with SO2 (Vereecken et al., 2017) as well as with an unaccounted-for contribution from the ozonolysis of some unsaturated compounds not measured during the campaign. The latter explanation is supported by significant daytime and nighttime missing OH reactivity of about 50 %, observed by Zannoni et al. (2017) during ChArMEx using the same VOC data as in the present work. The main unaccounted species were suggested to be reactive biogenic VOCs, including sesquiterpenes, oxygenated terpenes and their oxidation products. Ozonolysis of these compounds could be an additional unaccounted-for source of SCI in the present work.
4.1 Validity of H2SO4 steady-state conditions
Estimated concentrations of H2SO4 produced in reactions of SO2 with OH and with SCIs are based on the assumption of a steady state between production and loss pathways of H2SO4. All variations of the parameters influencing the H2SO4 concentration in the air masses incoming to the measurement site are assumed to occur on a timescale that is longer than the lifetime of H2SO4 estimated on the basis of the local aerosol measurements. These conditions are not obviously fulfilled at the Ersa site because the air masses arriving at the island may become influenced by local biogenic emissions, possibly leading to varying concentrations of these compounds along the trajectory from the coast to the measurement location (due to non-uniformly distributed emissions, development of vertical profiles of VOCs and other reasons). As the production rate of the SCIs is proportional to the corresponding concentrations of the unsaturated compounds, the variation of VOCs may induce variations in the SCI concentrations. The concentration of OH could also be affected by varying VOC concentrations considering that the lifetime of OH at the measurement site was about 0.2 s, with half of the OH reactivity explained by the local biogenic emissions (Zannoni et al., 2017).
The steady-state conditions were hardly disturbed by variations of the concentrations of SO2 and O3, species of non-local origin with lifetimes longer than the lifetime of H2SO4. Concerning the H2SO4 condensation sink, a variation in the aerosol particle number concentration and size distribution could be present. Recent studies evidenced that highly oxygenated molecules (HOMs) from the ozonolysis of monoterpenes may initiate new particle formation and its fast initial growth with the growth rates of tens of nanometres per hour (Stolzenburg et al., 2018; Tröstl et al., 2016). However, these variations are still slow compared to the H2SO4 lifetime of several minutes.
The timescales for the variations of VOCs, SCI, OH and CS may depend on the distribution of the biogenic emissions, wind speed, conditions of turbulent mixing and others. Without information about all these details, we can only estimate the maximum time for such variations, corresponding to the time of air mass presence over the land. To fulfill the steady-state conditions, this presence time has to be at least longer than the lifetime of H2SO4. The prevailing wind directions were from the north-east and, predominantly, from the south-west. The south-westerly direction corresponds to the shortest presence times because of the shortest distance to the coastline and the highest wind speeds corresponding to this wind direction (Figs. S11 and S12). On average, the presence time over the land, tpr, significantly exceeded the H2SO4 lifetime, , with the median value of the ratio of being 7.7 (4.3–17.9 interquartile range). To test the influence of the presence time on the calculated H2SO4 budget, the dependencies between the relative difference 1 − [H2SO4]OH [H2SO4] and the relative contribution [H2SO4]SCI [H2SO4] were analysed with filtered data using criteria tpr > 1 and tpr > 10. No significant difference was found compared with the unfiltered data, as is also illustrated in Fig. 6, showing no apparent correlation between the relative contribution of the OH + SO2 source and the ratio of . Similarly, no apparent effect was found by filtering the data according to the wind speed and the wind direction (not presented). Finally, although relatively short timescale spatial variability of OH or SCI concentrations around the measurement site cannot be excluded, an apparent independence of the presence time or the wind speed supports the validity of the assumption used here regarding H2SO4 steady-state conditions.
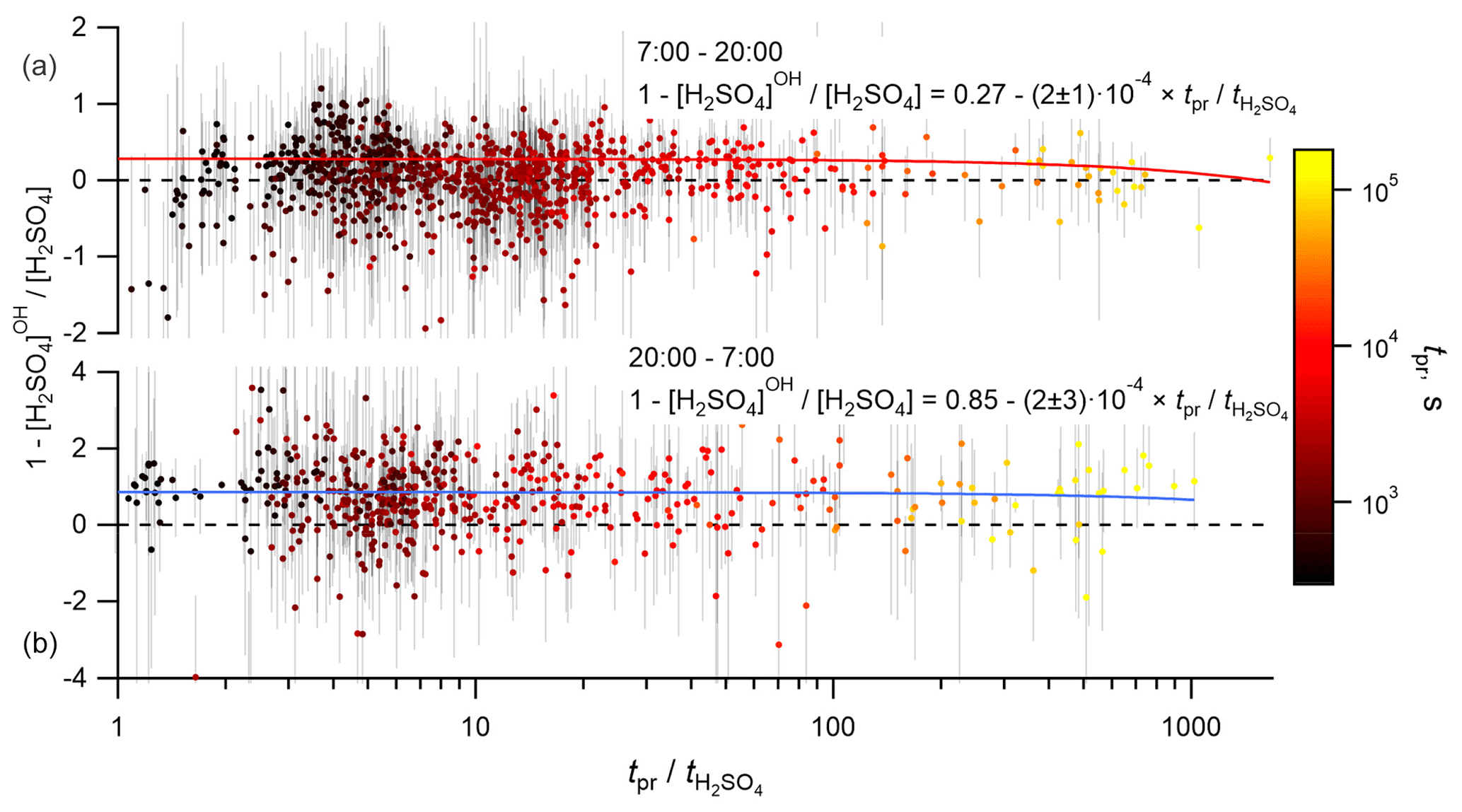
Figure 6Dependence of the normalized difference of the H2SO4 concentrations observed and produced from SO2 + OH, 1 − [H2SO4]OH [H2SO4], on the ratio of presence time over the land to the lifetime of H2SO4, , coloured by the presence time over the land, tpr. Panels (a) and (b) correspond to the daytime and nighttime data, respectively. Solid lines correspond to linear regression fits.
4.2 OH + SO2 reaction rate constant
In this work the rate coefficient for the reaction of OH with SO2, cm3 molec.−1 s−1 (at 760 torr and 298 K), was taken from the last published IUPAC recommendation (Atkinson et al., 2004). The two latest JPL evaluations recommend an approximately 20 % larger rate constant of cm3 molec.−1 s−1 (Burkholder et al., 2015, 2019). Using the rate constant from the JPL evaluations in this work would result in about a 20 % larger H2SO4 production rate via OH + SO2. This would yield a closed H2SO4 budget with only an OH + SO2 source during the day and about 2 times lower contribution from a missing source during the night.
In a recent study of Blitz et al. (2017a, b), a significantly lower rate constant of cm3 molec.−1 s−1 was derived from experiments with vibrationally excited OH (v=1, 2, 3) + SO2 and using the master equation analysis of the pressure and temperature dependence of their own and some other experimental OH + SO2 reaction rate constants. An even lower rate constant of cm3 molec.−1 s−1 was derived by Medeiros et al. (2018) by applying more detailed master equation analysis of experimental data from Blitz et al. (2017a, b) and some other data. These recent results have not been confirmed by other studies. Also, they have been discussed but not recommended by the latest JPL evaluation (Burkholder et al., 2019).
Using the lower rate constant from Medeiros et al. (2018) in our study would result in about 2 times reduced H2SO4 production by oxidation of SO2 by OH and would invoke either significantly larger contribution from an additional H2SO4 source or a lower H2SO4 uptake coefficient of about 0.5 instead of unity. As shown in Table 1, the reaction of OH with SO2 would explain only about 50 % and 5 % of the observed H2SO4 production during the day and during the night, respectively.
In previous field studies of the H2SO4 budget listed in Table S1, the OH + SO2 production rate of H2SO4 was calculated using a k1 rate coefficient in the range of (8.5–12) × 10−13 cm3 molec.−1 s−1. Reanalysis of these data using the lower k1 would lead to a conclusion that the H2SO4 budget was never observed to be closed with the uptake coefficient close to unity. On the other hand, employing the lower k1 from Blitz et al. (2017a, b) in model studies results in a significantly larger relative contribution of SCI to the H2SO4 formation. For example, the SCI contribution of 7 % in a rural environment and of 70 % in tropical regions, which were estimated assuming the lower k1 by Vereecken et al. (2017), would be reduced, respectively, to negligible and to about 30 % if the larger k1 from IUPAC or JPL were used.
4.3 H2SO4 loss
The mass accommodation coefficient of unity used in this work was measured by Hanson (2005) for the H2SO4 uptake on 5–20 nm diameter particles composed of water and sulfuric acid. The efficient uptake of H2SO4 is supported by other studies where the accommodation coefficients of about 0.7 were determined for the uptake on liquid sulfuric acid (Pöschl et al., 1998) and on ammonium sulfate and sodium chloride particles (Jefferson et al., 1997). Lower mass accommodation coefficients in the range of 0.2–0.3 were determined for the uptake on hydrocarbon-coated particles (Jefferson et al., 1997), suggesting that the uptake coefficient may depend on aerosol composition. Considering the measurement uncertainty, the results obtained in this work are consistent with the accommodation coefficient in the range from about 0.8 to 1. At lower uptake values the OH + SO2 source would significantly override the calculated H2SO4 loss during the day. At the same time, the apparently missing H2SO4 source during the night can be explained by a lower uptake coefficient, down to about 0.5.
Another possible loss mechanism of H2SO4 can be via collisions of sulfuric acid molecules leading to new particle formation (NPF) in the atmosphere. For some atmospheric conditions, like in the presence of high concentrations of base atmospheric components, e.g. ammonia and amines (Almeida et al., 2013), stabilizing the H2SO4 dimer and larger clusters, the nucleation may proceed at a collisionally limited rate corresponding to an effective bimolecular H2SO4 loss rate coefficient of about cm3 molec.−1 s−1 (Kürten et al., 2014). Such conditions might have been encountered in highly polluted industrial and urban environments, regions influenced by strong agricultural emissions, and chamber experiments (Kürten et al., 2016, 2018; Yao et al., 2018). Removal of H2SO4 with this rate constant would significantly contribute to its loss during ChArMEx, increasing it by about a factor of 2 for the largest observed H2SO4 concentrations. During the ChArMEx/SAFMED experiment, the H2SO4 loss rate assuming sulfuric acid-initiated kinetic particle formation can be estimated from several episodes of the NPF observed during the campaign (on days from 29 July to 2 August; Berland et al., 2017). Figure S13 shows an example of NPF observed around the midday of 31 July. For this day the profiles of H2SO4 and N10–15 are similar, with a time delay of about 2 h for the maximum of the size distribution of 15 nm. This allows us to assume that the observed NPF on this day could be related to the evolution of the H2SO4 concentration. The growth rate (GR) and 12 nm particle apparent formation rate from the SMPS measurements are 3.1 nm h−1 and 8.2 × 10−3 cm−3 s−1, respectively. GR was calculated using a maximum-concentration method (Kulmala et al., 2012). Using the approach of Kerminen and Kulmala (2002) and Sihto et al. (2006) to derive the rate of formation of the critical cluster of size 1 nm, J1, we obtain around 1 cm−3 s−1 at H2SO4 concentration of around 107 molec. cm−3. Assuming a kinetically limited nucleation mechanism in which the critical cluster contains two sulfuric acid molecules, this corresponds to the bimolecular rate constant of for the removal of H2SO4, which would correspond to a negligible H2SO4 loss compared to the condensation on existing particles. Similar rates, several orders of magnitude below the collision limited rate, were found in other diverse continental and marine atmospheric environments (Kuang et al., 2008). In addition, the GR estimated from the time delay between an H2SO4 profile and a corresponding increase in N10–15 particles is about 2 times faster than derived from the particle size growth rate. Using this latter GR would result in an even slower particle formation. Finally, analysis of particles' origins and their chemical composition at the measurement site made with ATOMF MS indicates that even if some traces of amines were present, they were of remote origin and not present at the measurement site (Arndt et al., 2017).
4.4 SCI interference with OH and H2SO4 measurements
High concentrations of SO2 used in the chemical conversion reactor of CIMS instruments for the conversion of OH into H2SO4 may lead to an interference with OH and H2SO4 measurements due to an artificial generation of H2SO4 in reactions of SCIs with SO2 inside the reactor. When the reactant used as a scavenger of OH does not react quickly with SCIs, e.g. using propane, the contribution of the artificially formed H2SO4 is the same for the OH measurements in the background (BG) and OH signal + background modes (OH + BG) if the SO2 injection position rests unchanged for the measurements in both modes. In this case the OH signal derived from the difference of the OH + BG and BG signals is free from the artificial H2SO4 formation. However, the presence of an additional SO2 oxidant not efficiently removed by an OH scavenger may lead to a significant increase in the BG level, as was observed in previous field measurements (Berresheim et al., 2014; Mauldin et al., 2012).
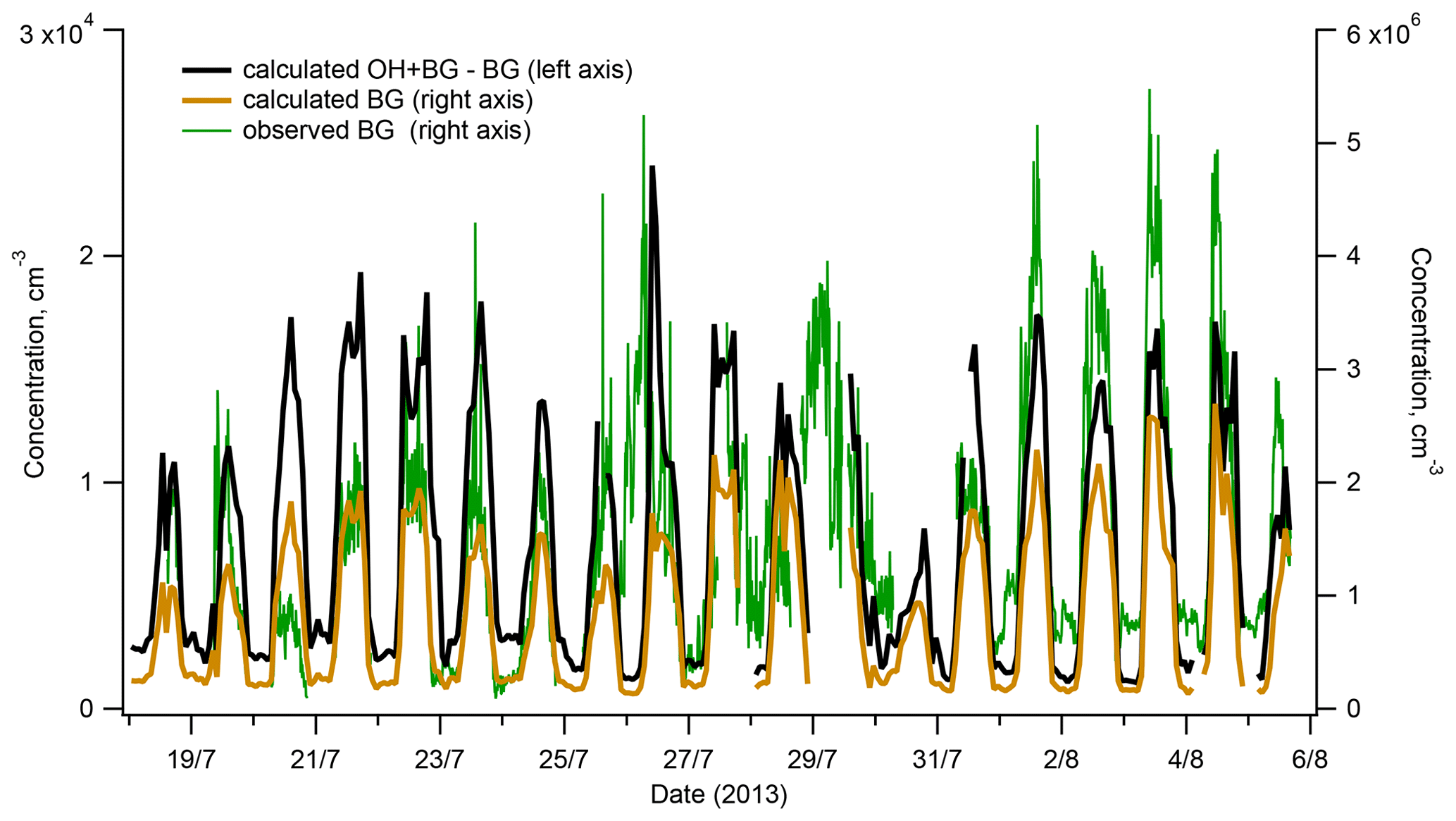
Figure 7Interference with OH measurements from the H2SO4 produced in the reactor by reactions of SO2 with SCIs: calculated contribution to measured OH (red line, left axis) and comparison of the observed background (BG) (blue line, right axis) and the BG calculated assuming its origin from the reactions of SCIs with SO2 inside the reactor (black line, right axis).
In the instrument used in this study the OH is scavenged with NO2. The rate constant for the reaction of NO2 with CH2OO, CH3CHOO and (CH3)2COO stabilized CIs was found to be of about cm3 molec.−1 s−1 (Chhantyal-Pun et al., 2017; Stone et al., 2014; Taatjes et al., 2013), which is about 10–100 times smaller than typical rate constants of the reactions of SCIs with SO2 (Table S5). As the ratio of [NO2] [34SO2] in the reactor was about 100, the SCIs in the reactor may react with similar rates with both SO2 and NO2 contributing to the BG signal. In addition, as the NO2 and SO2 injection positions are different for the BG and OH + BG modes (see Fig. S1 for details), the OH signal derived from the OH + BG and the BG difference may also be influenced by the artificial HSO4 production in reactions of SCIs with SO2.
As presented in Fig. 7, the BG signal observed during ChArMEx showed a diel profile similar to that of VOCs or OH (Figs. 1 and 2) with maximum at noon and minimum during the night. The peak noon and nighttime BG signals reached levels corresponding to OH concentrations of about 5×106 and 1×106 molec. cm−3, respectively. These BG levels are significantly higher than values of about (1–5) × 105 molec. cm−3 typically observed with the present instrument during calibration with zero air or during measurements in clean air with low VOCs concentrations, e.g. in Antarctica (Kukui et al., 2014).
To examine whether the observed high BG levels can be explained by the SO2 reaction with SCIs, the H2SO4 concentration produced in the reactor in reactions of the sum of SCIs with SO2, [HSO4], was calculated for BG and OH+BG modes using Eq. (4), which is similar to Eq. (2) but neglects the H2SO4 losses in the reactor:
where the index Rj corresponds to different parts of the reactor, and are the concentrations and is the bulk flow time in the corresponding parts of the reactor (see also Fig. S1). The concentrations of SCIs inside the reactor generated by ozonolysis of the measured unsaturated VOCs were calculated similarly to Eq. (3) but with accounting for the additional SCI loss in reactions with SO2 and NO2 injected into the reactor. The concentrations of VOCs, O3 and H2O inside the reactor were calculated using the ambient measurements with accounting for a dilution inside the reactor.
The concentration of O3 in the IMR was corrected by accounting for ozone generated in the corona discharge ion source and added to the IMR with the flow of primary ions. Amount of O3 generated by the ion source depends on the ion source operating configuration, i.e. flow rates of injected mixtures, composition of the mixtures and potentials of the ion source electrodes. The concentration of O3 in the IMR was not monitored during the campaign, but according to later checks this concentration was of 2±1 ppm and this value was used for the estimation of [HSO4].
As shown in Fig. 7, the calculated and the observed BGs exhibit similar variability and correspond to comparable concentration levels allowing us to suggest that the observed elevated BG levels were related to the SO2 oxidation by SCIs in the reactor. This hypothesis is supported by results of later experiments on the ozonolysis of terpenes conducted in an environmental chamber where a strong dependence of the observed BG level on the SCI production rate was confirmed, as shown in Fig. S14 for the ozonolysis of α-pinene.
Concerning a possible interference of the SO2 + SCI reaction with the OH measurements, Fig. 7 shows that during the campaign, this interference was negligible because the difference of [HSO4] produced in the OH + BG and the BG modes corresponded to OH concentration lower than 2 × 104 molec. cm−3, about 10 times lower than the OH lower detection limit. Notably, this contribution is independent of the [O3] in the IMR, influencing equally the OH + BG and BG measurements.
The formation of H2SO4 was observed at the Ersa station in northern Corsica, a site influenced by local emissions of biogenic VOCs. The H2SO4 concentration reached 107 molec. cm−3 at midday and was around 5 × 105 molec. cm−3 during the night. Based on the OH, H2SO4, SO2 and particle number density measurements and assuming validity of a steady state between H2SO4 production and its loss by condensation on existing aerosol particles with a unity accommodation coefficient, we have found that the contribution of the SO2 + OH reaction accounts for (86±4) % and only for (9±2) % of the H2SO4 production during the day and night, respectively. The given accuracy of these values has been estimated without accounting for the large uncertainty in the OH + SO2 reaction rate coefficient, which results in a larger uncertainty in the contribution to H2SO4 formation from the OH + SO2 source, from about a factor of 1.5 of its overestimation to about 20 % of its underestimation.
Estimating the H2SO4 production from the SO2 oxidation by SCIs, we conclude that despite the low calculated SCI concentrations ((1–3) × 103 molec. cm−3 for the sum of SCIs), this source may explain about 10 % of the H2SO4 formation during the day and represents a major source of H2SO4 accounting for about 40 % of its formation during the night. The sum of the H2SO4 production rates via SO2 + OH and SO2 + SCIs correspond to a closure of the H2SO4 budget during the day, but seem to underestimate by 50 % the H2SO4 production during the night, with the latter being possibly related to uncertainties in the used in this work kinetic parameters, an unaccounted contribution from the ozonolysis of some unsaturated compounds not measured during the campaign, as well as to some yet unidentified H2SO4 production mechanisms during nighttime.
Both the daytime and nighttime results of this study indicate that the oxidation of SO2 by SCIs may be an important source of H2SO4 in VOC-rich environments, especially during nighttime.
Data are available upon request from the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-13333-2021-supplement.
AK designed the CIMS instrument, performed the OH and H2SO4 measurements, analysed the data and wrote the paper. MC participated in the design of the new calibration system. All the other coauthors participated in data collection. All the coauthors participated in paper discussion.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors would like to thank Thierry Vincent, Stephane Chevrier and Gilles Chalumeau from LPC2E for logistical help in preparation and during the campaign, Karine Sartelet from CEREA for managing the SAFMED project, Jean Sciare from LSCE (now at CyI) for managing the campaign site and providing meteorology data, Eric Hamonou and François Dulac from LSCE for organizing the ChArMEx campaign and for managing and coordinating the ChArMEx project, and François Dulac for editorial corrections and suggestions.
This research received funding from French National Research Agency (ANR) project SAFMED (grant no. ANR-12-BS06-0013). This work is part of the ChArMEx project supported by ADEME, CEA, CNRS-INSU and Météo-France through the multidisciplinary programme MISTRALS (Mediterranean Integrated Studies aT Regional And Local Scales). The station at Ersa was partly supported by the CORSiCA project funded by the Collectivité Territoriale de Corse (CTC) through the Fonds Européen de Développement Régional of the European Operational Program 2007–2013 and the Contrat de Plan Etat-Région. This project was also supported by the CaPPA project (Chemical and Physical Properties of the Atmosphere), funded by the French National Research Agency (ANR) through the PIA (Programme d'Investissement d'Avenir) under contract ANR-11-LABX-0005-01, and by the Regional Council Nord-Pas de Calais and the European Funds for Regional Economic Development (FEDER). This work was also supported by the LABEX VOLTAIRE ANR-10-LABX-100-01 (2011–2020) and PIVOTS projects provided by the Région Centre–Val de Loire (ARD 2020 programme and CPER 2015–2020).
This paper was edited by Ivan Kourtchev and reviewed by four anonymous referees.
Ahrens, J., Carlsson, P. T. M., Hertl, N., Olzmann, M., Pfeifle, M., Wolf, J. L., and Zeuch, T.: Infrared detection of Criegee intermediates formed during the ozonolysis of β-pinene and their reactivity towards sulfur dioxide, Angew. Chem.-Int. Ed., 53, 715–719, https://doi.org/10.1002/anie.201307327, 2014.
Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J.: Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Arndt, J., Sciare, J., Mallet, M., Roberts, G. C., Marchand, N., Sartelet, K., Sellegri, K., Dulac, F., Healy, R. M., and Wenger, J. C.: Sources and mixing state of summertime background aerosol in the north-western Mediterranean basin, Atmos. Chem. Phys., 17, 6975–7001, https://doi.org/10.5194/acp-17-6975-2017, 2017.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Barber, V. P., Pandit, S., Green, A. M., Trongsiriwat, N., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Four-Carbon Criegee Intermediate from Isoprene Ozonolysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production, J. Am. Chem. Soc., 140, 10866–10880, https://doi.org/10.1021/jacs.8b06010, 2018.
Bardouki, H., Berresheim, H., Vrekoussis, M., Sciare, J., Kouvarakis, G., Oikonomou, K., Schneider, J., and Mihalopoulos, N.: Gaseous (DMS, MSA, SO2, H2SO4 and DMSO) and particulate (sulfate and methanesulfonate) sulfur species over the northeastern coast of Crete, Atmos. Chem. Phys., 3, 1871–1886, https://doi.org/10.5194/acp-3-1871-2003, 2003.
Berland, K., Rose, C., Pey, J., Culot, A., Freney, E., Kalivitis, N., Kouvarakis, G., Cerro, J. C., Mallet, M., Sartelet, K., Beckmann, M., Bourriane, T., Roberts, G., Marchand, N., Mihalopoulos, N., and Sellegri, K.: Spatial extent of new particle formation events over the Mediterranean Basin from multiple ground-based and airborne measurements, Atmos. Chem. Phys., 17, 9567–9583, https://doi.org/10.5194/acp-17-9567-2017, 2017.
Berndt, T., Voigtländer, J., Stratmann, F., Junninen, H., Mauldin, R. L., Sipilä, M., Kulmala, M., and Herrmann, H.: Competing atmospheric reactions of CH2OO with SO2 and water vapour, Phys. Chem. Chem. Phys., 16, 19130, https://doi.org/10.1039/C4CP02345E, 2014a.
Berndt, T., Jokinen, T., Sipila, M., Mauldin, R. L., Herrmann, H., Stratmann, F., Junninen, H., and Kulmala, M.: H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 89, 603–612, https://doi.org/10.1016/j.atmosenv.2014.02.062, 2014b.
Berresheim, H., Elste, T., Plass-Dülmer, C., Eisele, F. L., and Tanner, D. J.: Chemical ionization mass spectrometer for long-term measurements of atmospheric OH and H2SO4, Int. J. Mass Spectrom., 202, 91–109, https://doi.org/10.1016/S1387-3806(00)00233-5, 2000.
Berresheim, H., Elste, T., Tremmel, H. G., Allen, A. G., Hansson, H., Rosman, K., Maso, M. D., Mäkelä, J. M., Kulmala, M., and O'Dowd, C. D.: Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res., 107, 1–12, https://doi.org/10.1029/2000JD000229, 2002.
Berresheim, H., Plass-Dülmer, C., Elste, T., Mihalopoulos, N., and Rohrer, F.: OH in the coastal boundary layer of Crete during MINOS: Measurements and relationship with ozone photolysis, Atmos. Chem. Phys., 3, 639–649, https://doi.org/10.5194/acp-3-639-2003, 2003.
Berresheim, H., Adam, M., Monahan, C., O'Dowd, C., Plane, J. M. C., Bohn, B., and Rohrer, F.: Missing SO2 oxidant in the coastal atmosphere? – observations from high-resolution measurements of OH and atmospheric sulfur compounds, Atmos. Chem. Phys., 14, 12209–12223, https://doi.org/10.5194/acp-14-12209-2014, 2014.
Birmili, W., Wiedensohler, A., Plass-Dülmer, C., and Berresheim, H.: Evolution of Newly Formed Aerosol Particles in the Continental Boundary Layer: A Case Study Including OH and H2SO4 Measurements, Geophys. Res. Lett., 27, 2205–2208, 2000.
Blitz, M. A., Salter, R. J., Heard, D. E., and Seakins, P. W.: An Experimental and Master Equation Study of the Kinetics of OH/OD + SO2: The Limiting High-Pressure Rate Coefficients, J. Phys. Chem. A, 121, 3184–3191, https://doi.org/10.1021/acs.jpca.7b01295, 2017a.
Blitz, M. A., Salter, R. J., Heard, D. E., and Seakins, P. W.: An Experimental Study of the Kinetics of OH/OD (v=1, 2, 3) + SO2: The Limiting High-Pressure Rate Coefficients as a Function of Temperature, J. Phys. Chem. A, 121, 3175–3183, https://doi.org/10.1021/acs.jpca.7b01294, 2017b.
Boy, M., Kulmala, M., Ruuskanen, T. M., Pihlatie, M., Reissell, A., Aalto, P. P., Keronen, P., Dal Maso, M., Hellen, H., Hakola, H., Jansson, R., Hanke, M., and Arnold, F.: Sulphuric acid closure and contribution to nucleation mode particle growth, Atmos. Chem. Phys., 5, 863–878, https://doi.org/10.5194/acp-5-863-2005, 2005.
Boy, M., Mogensen, D., Smolander, S., Zhou, L., Nieminen, T., Paasonen, P., Plass-Dülmer, C., Sipilä, M., Petäjä, T., Mauldin, L., Berresheim, H., and Kulmala, M.: Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 13, 3865–3879, https://doi.org/10.5194/acp-13-3865-2013, 2013.
Burkholder, J. B., Sander, S. P., Abbat, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov/ (last access: 1 March 2021), 2015.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov/ (last access: 1 March 2021), 2019.
Calvert, J. G. and Stockwell, W. R.: Acid generation in the troposphere by gas-phase chemistry, Environ. Sci. Technol., 17, 428A–443A, https://doi.org/10.1021/es00115a727, 1983.
Caravan, R. L., Vansco, M. F., Au, K., Khan, M. A. H., Li, Y.-L., Winiberg, F. A. F., Zuraski, K., Lin, Y.-H., Chao, W., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Lin, J. J.-M., Shallcross, D. E., Sheps, L., Klippenstein, S. J., Taatjes, C. A., and Lester, M. I.: Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate, P. Natl. Acad. Sci. USA, 117, 9733–9740, https://doi.org/10.1073/pnas.1916711117, 2020.
Chao, W., Hsieh, J.-T., Chang, C.-H., and Lin, J. J.-M.: Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 347, 751–754, https://doi.org/10.1126/science.1261549, 2015.
Chhantyal-Pun, R., Welz, O., Savee, J. D., Eskola, A. J., Lee, E. P. F., Blacker, L., Hill, H. R., Ashcroft, M., Khan, M. A. H., Lloyd-Jones, G. C., Evans, L., Rotavera, B., Huang, H., Osborn, D. L., Mok, D. K. W., Dyke, J. M., Shallcross, D. E., Percival, C. J., Orr-Ewing, A. J., and Taatjes, C. A.: Direct Measurements of Unimolecular and Bimolecular Reaction Kinetics of the Criegee Intermediate (CH3)2COO, J. Phys. Chem. A, 121, 4–15, https://doi.org/10.1021/acs.jpca.6b07810, 2017.
Chhantyal-Pun, R., Khan, M. A. H., Martin, R., Zachhuber, N., Buras, Z. J., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Direct Kinetic and Atmospheric Modeling Studies of Criegee Intermediate Reactions with Acetone, ACS Earth Sp. Chem., 3, 2363–2371, https://doi.org/10.1021/acsearthspacechem.9b00213, 2019.
Cox, R. A. and Penkett, S. A.: Oxidation of Atmospheric SO2 by Products of the Ozone-Olefin Reaction, Nature, 230, 321–322, 1971.
Criegee, R.: Mechanism of Ozonolysis, Angew. Chem. Int. Edit., 14, 745–752, 1975.
Criegee, R. and Wenner, G.: Die Ozonisierung des 9,10-Oktalins, Liebigs Ann. Chem., 564, 9–15, https://doi.org/10.1002/jlac.19495640103, 1949.
Debevec, C., Sauvage, S., Gros, V., Salameh, T., Sciare, J., Dulac, F., and Locoge, N.: Seasonal variation and origins of volatile organic compounds observed during 2 years at a western Mediterranean remote background site (Ersa, Cape Corsica), Atmos. Chem. Phys., 21, 1449–1484, https://doi.org/10.5194/acp-21-1449-2021, 2021.
Donahue, N. M., Drozd, G. T., Epstein, S. A., Presto, A. A., and Kroll, J. H.: Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 13, 10848, https://doi.org/10.1039/c0cp02564j, 2011.
Dulac, F., Hamonou, E., Sauvage, S., and Debevec, C.: Introduction to the volume 1 of Atmospheric Chemistry in the Mediterranean and to the ChArMEx experimental effort, in Atmospheric Chemistry in the Mediterranean – Vol. 1, Background Information and Pollutants Distribution, Springer, in press, 2021.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Makhmutov, V., Mann, G., Mathot, S., Merikanto, J., Miettinen, P., Nenes, A., Onnela, A., Rap, A., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipilä, M., Smith, J. N., Stozkhov, Y., Tomé, A., Tröstl, J., Wagner, P. E., Wimmer, D., Winkler, P. M., Worsnop, D. R., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1124, 2016.
Eisele, F. L. and Tanner, D. J.: Ion-Assisted Tropospheric OH Measurements, J. Geophys. Res., 96, 9295–9308, 1991.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimetes of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 98, 9001–9010, 1993.
Faloona, I. C., Tan, D., Lesher, R. L., Hazen, N. L., Frame, C. L., Simpas, J. B., Harder, H., Martinez, M., Di Carlo, P., Ren, X., and Brune, W. H.: A Laser-induced Fluorescence Instrument for Detecting Tropospheric OH and HO2: Characteristics and Calibration, J. Atmos. Chem., 47, 139–167, https://doi.org/10.1023/B:JOCH.0000021036.53185.0e, 2004.
Fang, Y., Barber, V. P., Klippenstein, S. J., McCoy, A. B., and Lester, M. I.: Tunneling effects in the unimolecular decay of (CH3)2COO Criegee intermediates to OH radical products, J. Chem. Phys., 146, 134307, https://doi.org/10.1063/1.4979297, 2017.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 2000.
Geron, C., Rasmussen, R., Arnts, R. R., and Guenther, A.: A review and synthesis of monoterpene speciation from forests in the United States, Atmos. Environ., 34, 1761–1781, https://doi.org/10.1016/S1352-2310(99)00364-7, 2000.
Hanson, D. R.: Mass Accommodation of H2SO4 and CH3SO3H on Water – Sulfuric Acid Solutions from 6 % to 97 % RH, J. Phys. Chem. A, 109, 6919–6927, 2005.
Hanson, D. R. and Eisele, F.: Diffusion of H2SO4 in Humidified Nitrogen: Hydrated H2SO4, J. Phys. Chem. A, 104, 1715–1719, https://doi.org/10.1021/jp993622j, 2000.
Hatakeyama, S., Kobayashi, H., Lin, Z. Y., Takagi, H., and Akimoto, H.: Mechanism for the reaction of peroxymethylene with sulfur dioxide, J. Phys. Chem., 90, 4131–4135, https://doi.org/10.1021/j100408a059, 1986.
Hoefs, J.: Stable isotope geochemistry, 8th Edn., Springer, available at: https://www.springer.com/gp/book/9783319785264 (last access: 7 September 2021), 2018.
Huang, H.-L., Chao, W., and Lin, J. J.-M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, available at: http://iupac.pole-ether.fr, last access: April 2020.
Jefferson, A., Eisele, F. L., Ziemann, P. J., Weber, R. J., Marti, J. J., and Mcmurry, P. H.: Measurements of the H2SO4 mass accommodation coefficient onto polydisperse aerosol, J. Geophys. Res.-Atmos., 102, 19021–19028, 1997.
Jefferson, A., Tanner, D. J., Eisele, F. L., and Berresheim, H.: Sources and sinks of H2SO4 in the remote Antarctic marine boundary layer, J. Geophys. Res.-Atmos., 103, 1639–1645, https://doi.org/10.1029/97JD01212, 1998.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/b704260b, 2008.
Kerminen, V.-M. and Kulmala, M.: Analytical formulae connecting the “real” and the “apparent” nucleation rate and the nuclei number concentration for atmospheric nucleation events, J. Aerosol Sci., 33, 609–622, 2002.
Kesselmeier, J. and Staudt, M.: Biogenic Volatile Organic Compounds (VOC): An Overview on Emission, Physiology and Ecology, J. Atmos. Chem., 33, 23–88, 1999.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci. Process. Impacts, 20, 437–453, https://doi.org/10.1039/C7EM00585G, 2018.
Kim, S., Guenther, A., Lefer, B., Flynn, J., Griffin, R., Rutter, A. P., Gong, L., and Cevik, B. K.: Potential role of stabilized Criegee radicals in sulfuric acid production in a high biogenic VOC environment, Environ. Sci. Technol., 49, 3383–3391, https://doi.org/10.1021/es505793t, 2015.
Kuang, C., Mcmurry, P. H., Mccormick, A. V., and Eisele, F. L.: Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations, J. Geophys. Res., 113, D10209, https://doi.org/10.1029/2007JD009253, 2008.
Kukui, A., Ancellet, G., and Le Bras, G.: Chemical ionisation mass spectrometer for measurements of OH and Peroxy radical concentrations in moderately polluted atmospheres, J. Atmos. Chem., 61, 133–154, https://doi.org/10.1007/s10874-009-9130-9, 2008.
Kukui, A., Legrand, M., Ancellet, G., Gros, V., Bekki, S., Loisil, R., and Preunkert, S.: Measurements of OH and RO2 radicals at the coastal Antarctic site of Dumont d'Urville (East Antarctica) in summer 2010–2011, J. Geophys. Res., 117, D12310, https://doi.org/10.1029/2012JD017614, 2012.
Kukui, A., Legrand, M., Preunkert, S., Frey, M. M., Loisil, R., Gil Roca, J., Jourdain, B., King, M. D., France, J. L., and Ancellet, G.: Measurements of OH and RO2 radicals at Dome C, East Antarctica, Atmos. Chem. Phys., 14, 12373–12392, https://doi.org/10.5194/acp-14-12373-2014, 2014.
Kulmala, M., Petäjä, T., Nieminen, T., Sipilä, M., Manninen, H. E., Lehtipalo, K., Dal Maso, M., Aalto, P. P., Junninen, H., Paasonen, P., Riipinen, I., Lehtinen, K. E. J., Laaksonen, A., and Kerminen, V.-M.: Measurement of the nucleation of atmospheric aerosol particles, Nat. Protoc., 7, 1651–1667, https://doi.org/10.1038/nprot.2012.091, 2012.
Kürten, A., Jokinen, T., Simon, M., Sipilä, M., Sarnela, N., Junninen, H., Adamov, A., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Hutterli, M., Kangasluoma, J., Kirkby, J., Laaksonen, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schobesberger, S., Seinfeld, J. H., Steiner, G., Tomé, A., Tröstl, J., Winkler, P. M., Williamson, C., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K. S., Kulmala, M., Worsnop, D. R., and Curtius, J.: Neutral molecular cluster formation of sulfuric acid – dimethylamine observed in real time under atmospheric conditions, P. Natl. Acad. Sci. USA, 111, 15019–15024, https://doi.org/10.1073/pnas.1404853111, 2014.
Kürten, A., Bergen, A., Heinritzi, M., Leiminger, M., Lorenz, V., Piel, F., Simon, M., Sitals, R., Wagner, A. C. and Curtius, J.: Observation of new particle formation and measurement of sulfuric acid, ammonia, amines and highly oxidized organic molecules at a rural site in central Germany, Atmos. Chem. Phys., 16, 12793–12813, https://doi.org/10.5194/acp-16-12793-2016, 2016.
Kürten, A., Li, C., Bianchi, F., Curtius, J., Dias, A., Donahue, N. M., Duplissy, J., Flagan, R. C., Hakala, J., Jokinen, T., Kirkby, J., Kulmala, M., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Onnela, A., Rissanen, M. P., Simon, M., Sipilä, M., Stozhkov, Y., Tröstl, J., Ye, P., and McMurry, P. H.: New particle formation in the sulfuric acid–dimethylamine–water system: reevaluation of CLOUD chamber measurements and comparison to an aerosol nucleation and growth model, Atmos. Chem. Phys., 18, 845–863, https://doi.org/10.5194/acp-18-845-2018, 2018.
Kurtén, T., Lane, J. R., Jørgensen, S., and Kjaergaard, H. G.: A Computational Study of the Oxidation of SO2 to SO3 by Gas-Phase Organic Oxidants, J. Phys. Chem. A, 115, 8669–8681, https://doi.org/10.1021/jp203907d, 2011.
Kuwata, K. T., Guinn, E. J., Hermes, M. R., Fernandez, J. A., Mathison, J. M., and Huang, K.: A Computational Re-examination of the Criegee Intermediate-Sulfur Dioxide Reaction, J. Phys. Chem. A, 119, 10316–10335, https://doi.org/10.1021/acs.jpca.5b06565, 2015.
Lester, M. I. and Klippenstein, S. J.: Unimolecular Decay of Criegee Intermediates to OH Radical Products: Prompt and Thermal Decay Processes, Acc. Chem. Res., 51, 978–985, https://doi.org/10.1021/acs.accounts.8b00077, 2018.
Lewis, T. R., Blitz, M. A., Heard, D. E., and Seakins, P. W.: Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 17, 4859–4863, https://doi.org/10.1039/C4CP04750H, 2015.
Li, Y.-L., Kuo, M.-T., and Lin, J. J.-M.: Unimolecular decomposition rates of a methylsubstituted Criegee intermediate syn-CH3CHOO, RSC Adv., 10, 8518–8524, https://doi.org/10.1039/D0RA01406K, 2020.
Lin, L.-C., Chao, W., Chang, C.-H., Takahashi, K., and Lin, J. J.-M.: Temperature dependence of the reaction of anti-CH3CHOO with water vapor, Phys. Chem. Chem. Phys., 18, 28189–28197, https://doi.org/10.1039/C6CP05171E, 2016.
Lin, Y.-H., Yin, C., Takahashi, K., and Lin, J.-M.: Surprisingly long lifetime of methacrolein oxide, an isoprene derived Criegee intermediate, under humid conditions, Commun. Chem., 4, https://doi.org/10.1038/s42004-021-00451-z, 2021.
Mallik, C., Tomsche, L., Bourtsoukidis, E., Crowley, J. N., Derstroff, B., Fischer, H., Hafermann, S., Hüser, I., Javed, U., Keßel, S., Lelieveld, J., Martinez, M., Meusel, H., Novelli, A., Phillips, G. J., Pozzer, A., Reiffs, A., Sander, R., Taraborrelli, D., Sauvage, C., Schuladen, J., Su, H., Williams, J., and Harder, H.: Oxidation processes in the eastern Mediterranean atmosphere: evidence from the modelling of HOx measurements over Cyprus, Atmos. Chem. Phys., 18, 10825–10847, https://doi.org/10.5194/acp-18-10825-2018, 2018.
Mauldin III, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V.-M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
Medeiros, D. J., Blitz, M. A., and Seakins, P. W.: Exploring the features on the OH + SO2 potential energy surface using theory and testing its accuracy by comparison to experimental data, Phys. Chem. Chem. Phys., 20, 8984–8990, https://doi.org/10.1039/C8CP00091C, 2018.
Michoud, V., Sciare, J., Sauvage, S., Dusanter, S., Léonardis, T., Gros, V., Kalogridis, C., Zannoni, N., Féron, A., Petit, J.-E., Crenn, V., Baisnée, D., Sarda-Estève, R., Bonnaire, N., Marchand, N., DeWitt, H. L., Pey, J., Colomb, A., Gheusi, F., Szidat, S., Stavroulas, I., Borbon, A., and Locoge, N.: Organic carbon at a remote site of the western Mediterranean Basin: sources and chemistry during the ChArMEx SOP2 field experiment, Atmos. Chem. Phys., 17, 8837–8865, https://doi.org/10.5194/acp-17-8837-2017, 2017.
Newland, M. J., Rickard, A. R., Sherwen, T., Evans, M. J., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling, Atmos. Chem. Phys., 18, 6095–6120, https://doi.org/10.5194/acp-18-6095-2018, 2018.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Paasonen, P., Nieminen, T., Asmi, E., Manninen, H. E., Petäjä, T., Plass-Dülmer, C., Flentje, H., Birmili, W., Wiedensohler, A., Hõrrak, U., Metzger, A., Hamed, A., Laaksonen, A., Facchini, M. C., Kerminen, V.-M., and Kulmala, M.: On the roles of sulphuric acid and low-volatility organic vapours in the initial steps of atmospheric new particle formation, Atmos. Chem. Phys., 10, 11223–11242, https://doi.org/10.5194/acp-10-11223-2010, 2010.
Petäjä, T., Mauldin, III, R. L., Kosciuch, E., McGrath, J., Nieminen, T., Paasonen, P., Boy, M., Adamov, A., Kotiaho, T., and Kulmala, M.: Sulfuric acid and OH concentrations in a boreal forest site, Atmos. Chem. Phys., 9, 7435–7448, https://doi.org/10.5194/acp-9-7435-2009, 2009.
Pöschl, U., Canagaratna, M., Jayne, J. T., Molina, L. T., Worsnop, D. R., Kolb, C. E., and Molina, M. J.: Mass Accommodation Coefficient of H2SO4 Vapor on Aqueous Sulfuric Acid Surfaces and Gaseous Diffusion Coefficient of H2SO4 in N2/H2O, J. Phys. Chem. A, 102, 10082–10089, https://doi.org/10.1021/jp982809s, 1998.
Rissler, J., Vestin, A., Swietlicki, E., Fisch, G., Zhou, J., Artaxo, P., and Andreae, M. O.: Size distribution and hygroscopic properties of aerosol particles from dry-season biomass burning in Amazonia, Atmos. Chem. Phys., 6, 471–491, https://doi.org/10.5194/acp-6-471-2006, 2006.
Ruscic, B.: Active Thermochemical Tables: Water and Water Dimer, J. Phys. Chem. A, 117, 11940–11953, https://doi.org/10.1021/jp403197t, 2013.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, Inc., Hoboken, New Jersey, 2016.
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 16, 26701–26706, https://doi.org/10.1039/C4CP04408H, 2014.
Sheps, L., Rotavera, B., Eskola, A. J., Osborn, D. L., Taatjes, C. A., Au, K., Shallcross, D. E., Khan, M. A. H., and Percival, C. J.: The Reaction of Criegee Intermediate CH2OO with Water Dimer: Primary Products and Atmospheric Impact, Phys. Chem. Chem. Phys., 19, 21970–21979, https://doi.org/10.1039/C7CP03265J, 2017.
Sihto, S.-L., Kulmala, M., Kerminen, V.-M., Dal Maso, M., Petäjä, T., Riipinen, I., Korhonen, H., Arnold, F., Janson, R., Boy, M., Laaksonen, A., and Lehtinen, K. E. J.: Atmospheric sulphuric acid and aerosol formation: implications from atmospheric measurements for nucleation and early growth mechanisms, Atmos. Chem. Phys., 6, 4079–4091, https://doi.org/10.5194/acp-6-4079-2006, 2006.
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L., Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M.: The Role of Sulfuric Acid in Atmospheric Nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010.
Smith, J. N., Moore, K. F., Eisele, F. L., Voisin, D., Ghimire, A. K., Sakurai, H., and McMurry, P. H.: Chemical composition of atmospheric nanoparticles during nucleation events in Atlanta, J. Geophys. Res., 110, D22S03, https://doi.org/10.1029/2005JD005912, 2005.
Smith, M. C., Chang, C. H., Chao, W., Lin, L. C., Takahashi, K., Boering, K. A., and Lin, J. J. M.: Strong negative temperature dependence of the simplest criegee intermediate CH2OO reaction with water dimer, J. Phys. Chem. Lett., 6, 2708–2713, https://doi.org/10.1021/acs.jpclett.5b01109, 2015.
Smith, M. C., Chao, W., Takahashi, K., Boering, K. A., and Lin, J. J. M.: Unimolecular Decomposition Rate of the Criegee Intermediate (CH3)2COO Measured Directly with UV Absorption Spectroscopy, J. Phys. Chem. A, 120, 4789–4798, https://doi.org/10.1021/acs.jpca.5b12124, 2016.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Buenrostro Mazon, S., Chen, D., Dias, A., Draper, D. C., Duplissy, J., El Haddad, I., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Stone, D., Blitz, M., Daubney, L., Howes, N. U. M., and Seakins, P.: Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 16, 1139–1149, https://doi.org/10.1039/C3CP54391A, 2014.
Stone, D., Au, K., Sime, S., Medeiros, D. J., Blitz, M., Seakins, P. W., and Sheps, L.: Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and, Phys. Chem. Chem. Phys., 20, 24940–24954, https://doi.org/10.1039/C8CP05332D, 2018.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 340, 177–180, https://doi.org/10.1126/science.1234689, 2013.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A., Breitenlechner, M., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527, https://doi.org/10.1038/nature18271, 2016.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259, https://doi.org/10.1039/c2cs35070j, 2012.
Vereecken, L., Harder, H., and Novelli, A.: The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 14, 14682, https://doi.org/10.1039/c2cp42300f, 2012.
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, https://doi.org/10.1039/C7CP05541B, 2017.
Villani, P., Picard, D., Michaud, V., Laj, P., and Wiedensohler, A.: Design and Validation of a Volatility Hygroscopic Tandem Differential Mobility Analyzer (VH-TDMA) to Characterize the Relationships Between the Thermal and Hygroscopic Properties of Atmospheric Aerosol Particles, Aerosol Sci. Tech., 42, 729–741, https://doi.org/10.1080/02786820802255668, 2008.
Wang, Y.-Y., Dash, M. R., Chung, C.-Y., and Lee, Y.-P.: Detection of transient infrared absorption of SO3 and 1,3,2-dioxathietane-2,2-dioxide [cyc-(CH2)O(SO2)O] in the reaction CH2OO + SO2, J. Chem. Phys., 148, 064301, https://doi.org/10.1063/1.5019205, 2018.
Weber, R. J., Marti, J. J., Mcmurry, P. H., Eisele, F. L., Tanner, D. J., and Jefferson, A.: Measurements of new particle formation and ultrafine particle growth rates at a clean continental site, J. Geophys. Res., 102, 4375–4385, 1997.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2, Science, 335, 204–207, https://doi.org/10.1126/science.1213229, 2012.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Paasonen, P., Sipilä, M., Wang, M., Wang, X., and Xiao, S.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 361, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
Yao, L., Fan, X., Yan, C., Kurtén, T., Daellenbach, K. R., Li, C., Wang, Y., Guo, Y., Dada, L., Rissanen, M. P., Cai, J., Tham, Y. J., Zha, Q., Zhang, S., Du, W., Yu, M., Zheng, F., Zhou, Y., Kontkanen, J., Chan, T., Shen, J., Kujansuu, J. T., Kangasluoma, J., Jiang, J., Wang, L., Worsnop, D. R., Petäjä, T., Kerminen, V.-M., Liu, Y., Chu, B., He, H., Kulmala, M., and Bianchi, F.: Unprecedented Ambient Sulfur Trioxide (SO3) Detection: Possible Formation Mechanism and Atmospheric Implications, Environ. Sci. Technol. Lett., 7, 809–818, https://doi.org/10.1021/acs.estlett.0c00615, 2020.
York, D., Evensen, N. M., Martínez, M. L., and De Basabe Delgado, J.: Unified equations for the slope, intercept, and standard errors of the best straight line, Am. J. Phys., 72, 367–375, https://doi.org/10.1119/1.1632486, 2004.
Zannoni, N., Dusanter, S., Gros, V., Sarda Esteve, R., Michoud, V., Sinha, V., Locoge, N., and Bonsang, B.: Intercomparison of two comparative reactivity method instruments inf the Mediterranean basin during summer 2013, Atmos. Meas. Tech., 8, 3851–3865, https://doi.org/10.5194/amt-8-3851-2015, 2015.
Zannoni, N., Gros, V., Sarda Esteve, R., Kalogridis, C., Michoud, V., Dusanter, S., Sauvage, S., Locoge, N., Colomb, A., and Bonsang, B.: Summertime OH reactivity from a receptor coastal site in the Mediterranean Basin, Atmos. Chem. Phys., 17, 12645–12658, https://doi.org/10.5194/acp-17-12645-2017, 2017.
Zhang, R., Khalizov, A., Wang, L., Hu, M., and Xu, W.: Nucleation and Growth of Nanoparticles in the Atmosphere, Chem. Rev., 112, 1957–2011, https://doi.org/10.1021/cr2001756, 2012.