the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Nov 2020
| 12 Nov 2020
Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates
R. Anthony Cox
Markus Ammann
Hartmut Herrmann
Michael E. Jenkin
V. Faye McNeill
Abdelwahid Mellouki
Jürgen Troe
Timothy J. Wallington
This article, the seventh in the series, presents kinetic and photochemical data sheets evaluated by the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation. It covers an extension of the gas-phase and photochemical reactions related to Criegee intermediates previously published in Atmospheric Chemistry and Physics (ACP) in 2006 and implemented on the IUPAC website up to 2020. The article consists of an introduction, description of laboratory measurements, a discussion of rate coefficients for reactions of O3 with alkenes producing Criegee intermediates, rate coefficients of unimolecular and bimolecular reactions and photochemical data for reactions of Criegee intermediates, and an overview of the atmospheric chemistry of Criegee intermediates. Summary tables of the recommended kinetic and mechanistic parameters for the evaluated reactions are provided. Data sheets summarizing information upon which the recommendations are based are given in two files, provided as a Supplement to this article.
- Article
(1341 KB) - Full-text XML
-
Supplement
(3476 KB) - BibTeX
- EndNote





Laboratory kinetic and mechanistic studies of the reactions of alkenes with ozone (O3) have established that “Criegee intermediates” (CIs) produced from these reactions are potentially important oxidants in atmospheric chemistry (e.g. Calvert et al., 2000; Johnson and Marston, 2008; Taatjes et al., 2014). This followed the suggestion by Cox and Penkett (1971, 1972) that the rapid oxidation of SO2 in the presence of reacting mixtures of O3 and alkenes in air was caused by production of a reactive intermediate, namely the peroxidic zwitterion, R1R2C=OO−, proposed by Rudolf Criegee, based on studies of the liquid-phase ozonolysis of alkenes (e.g. Criegee et al., 1954).
This has led to extensive study of the mechanisms of O3 + alkene reactions, and of the chemistry of the CIs formed. It is well established that the reaction proceeds by initial cyclo-addition of O3 across the C=C double bond in an alkene to form an energy-rich primary ozonide (POZ), which rapidly decomposes to form either of two sets of CI and carbonyl compound, as shown in Fig. 1. The reaction is exothermic, leading to an excess of energy (200–250 kJ mol−1) distributed between these reaction products. Some of this excess energy is deposited as internal energy in the nascent CIs, which can promote unimolecular decomposition or which can be lost by collisional energy transfer to other gas molecules, leading to formation of stabilized Criegee intermediates (sCIs), which can themselves react with other atmospheric trace species. The impact of the ozonolysis reaction on atmospheric oxidation chemistry is therefore influenced by the relative importance of prompt CI decomposition vs. formation of sCI. The recognition of the important distinction between the chemically activated CI, formed promptly in excited state, and the thermally equilibrated sCI has led to extensive experimental efforts to determine the yield of sCI (Y) formed from the ozonolysis of a variety of alkenes, as discussed further in Sect. 3. It is also well established that the decomposition of both CI and sCI leads to the formation of hydroxyl (HO) radicals and other radical products. Due to its important role in initiating the oxidation of organics (including alkenes), the formation of HO radicals has received particular attention, and this is discussed further in Sect. 4.
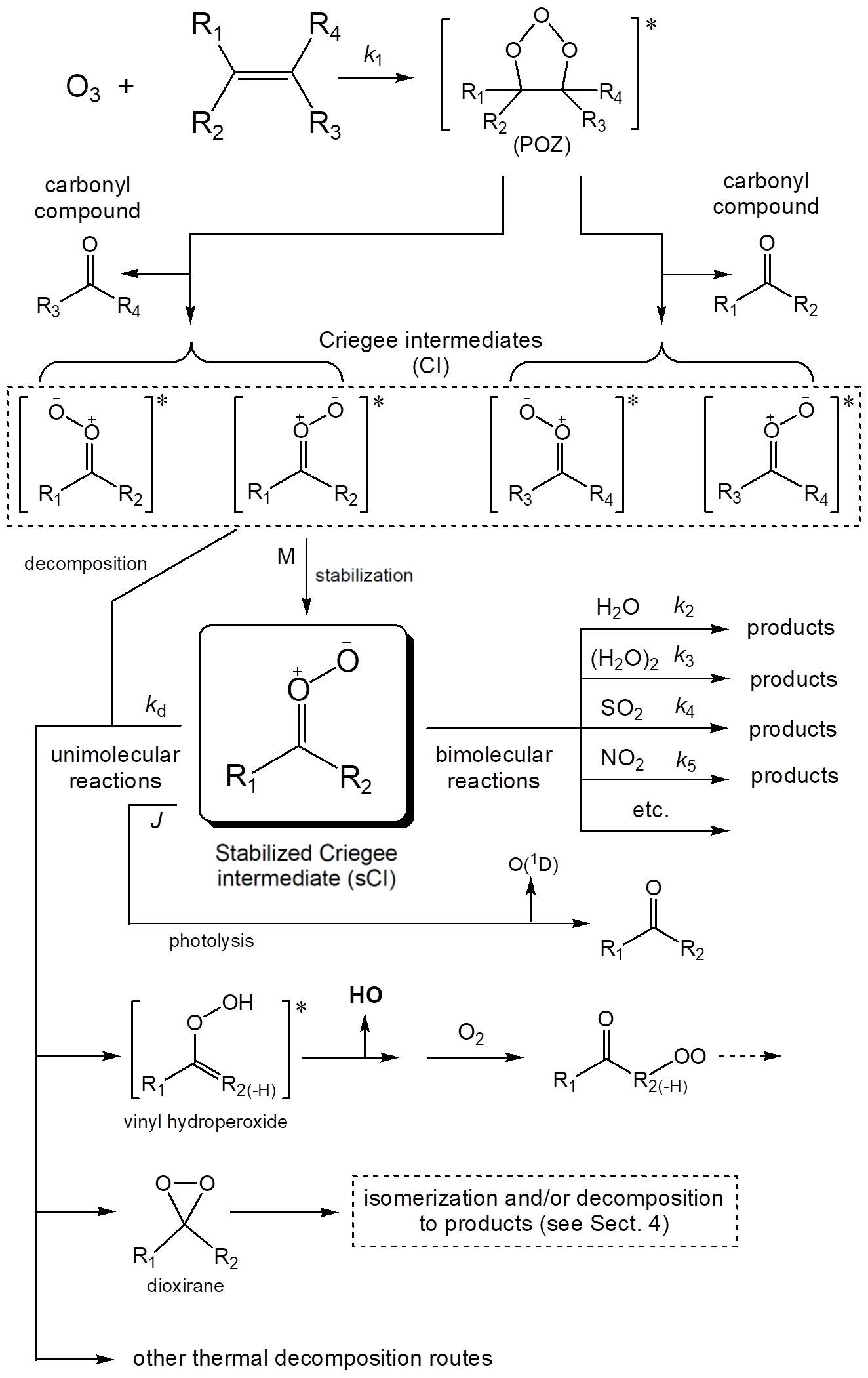
Figure 1Mechanism of the reaction of ozone with alkenes, showing formation of Criegee intermediates (CIs). The general types of reaction available for stabilized Criegee intermediates (sCIs) are also illustrated for one example. The substituents R1 to R4 can be either H atoms or organic groups, although the illustrated sCI/CI vinyl hydroperoxide route is unavailable if R2 = H. Note that in the case of endocyclic C=C bonds in cycloalkenes, the initially formed carbonyl and CI moieties are substituents of the same organic product.
The mechanism in Fig. 1 shows that the CIs (and sCIs) formed from the ozonolysis of a simple alkene can each be formed as either of two stereo-isomers, with different orientations of the outer O atom relative to the substituent groups. This potentially has an important impact on the chemical pathways available, and their relative rates. The stereo-isomers have generally been distinguished using the terms syn- and anti-, to specify the orientation of the outer O atom relative to a particular substituent; although the use of the IUPAC Z- and E- nomenclature is becoming increasingly adopted (e.g. Vereecken et al., 2017). As shown in Fig. 1, the ozonolysis of a simple alkene containing four different substituents therefore produces four distinct CIs, with this number being systematically reduced in symmetrical alkenes (because the products of the two POZ fragmentation pathways are the same) or in alkenes possessing two identical substituents on the same carbon atom (because the stereo-isomerism in the CI is removed). However, the number of different CI isomers can also be increased if the alkene contains alkenyl substituents, as is the case for the CIs formed from the ozonolysis of dienes such as the C4 species derived from isoprene because of additional stereo-isomerism in the substituent group(s).
Based on current understanding of the mechanism of alkene ozonolysis (as illustrated in Fig. 1), the steady-state concentration of a given stabilized Criegee intermediate, [sCIi], maintained by a balance between production and loss, is described by Eq. (1):
where k1j is the rate coefficient for the reaction of O3 with alkenej and Yij is the yield of sCIi from that reaction. The numerator of Eq. (1) therefore quantifies the source term for formation of sCIi from all relevant alkenes, and the denominator quantifies the sum of the rates of the unimolecular and individual bimolecular loss processes for sCIi, with the example contributors to the summation being based on the processes shown in Fig. 1. This illustrates that knowledge of the yields, rate coefficients and products for the component reactions is important for quantitative description of the chemical pathways controlling the atmospheric chemistry and impact of the given sCIi. It also shows the importance of establishing how these parameters vary from one sCI to another, e.g. the structural dependence of the rate coefficients for their unimolecular and bimolecular reactions.
Although rate constants have been determined accurately for a large number of O3 + alkene reactions, using both direct and relative rate techniques, all kinetic data reported for sCI reactions prior to 2012 were based on indirect relative rate techniques. Many of these data were previously evaluated by the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation (Atkinson et al., 2006). Since 2012, many new rate coefficients for sCI reactions have been reported in direct kinetic studies, providing a wealth of data for the elementary reaction kinetics and spectroscopy of sCI reactions. The current evaluation therefore addresses these reactions, substantially extending the scope of our former evaluation published in ACP in 2006. This includes a major extension of the scope of the evaluation to include rate coefficients of elementary reactions of selected sCIs, which have provided a better understanding of the atmospheric impact of sCI chemistry.
In this review we summarize the results of this evaluation activity, presenting in turn the recommended kinetic data for the key reactions in the above mechanism, using data for those species which are representative of the chemistry of the terrestrial atmosphere. The rate coefficients for O3 + alkene initiation reactions are presented and discussed in Sect. 2, with reference to a series of detailed data sheets which are provided in Supplement A. Information on the sCI and HO radical yields from the ozone + alkene reactions is presented in Sects. 3 and 4, with additional discussion once again provided in the corresponding data sheets in Supplement A. The data sheets therefore each include summary information on the kinetics studies of the given reaction and provide an overview of mechanistic information and product yields where available.
The spectroscopy and kinetics recommendations for the sCI reactions are presented and discussed in Sects. 5 and 6. These include data for bimolecular and unimolecular reactions of selected sCIs of particular atmospheric relevance for which direct kinetic data have been reported, namely CH2OO, Z- and E- CH3CHOO, (CH3)2COO and E-(CH=CH2)(CH3)COO, and we also provide some discussion of the complete set of C4 intermediates formed from isoprene (see Fig. 2 for sCI structures). These are predicted to be among the most important sCIs in tropospheric chemistry (Vereecken et al., 2017), and can also act as a systematic set of template species for representing the fates of some larger and more complex sCIs. Detailed data sheets for the sCI reactions are provided in Supplement B, providing supporting summary information and discussion. Finally, the recommended kinetics parameters are used to evaluate the relative importance of the different fates of the sCIs under representative atmospheric conditions in Sect. 7, and an overview of the impact of Criegee intermediates in atmospheric oxidation chemistry is given in Sect. 8.
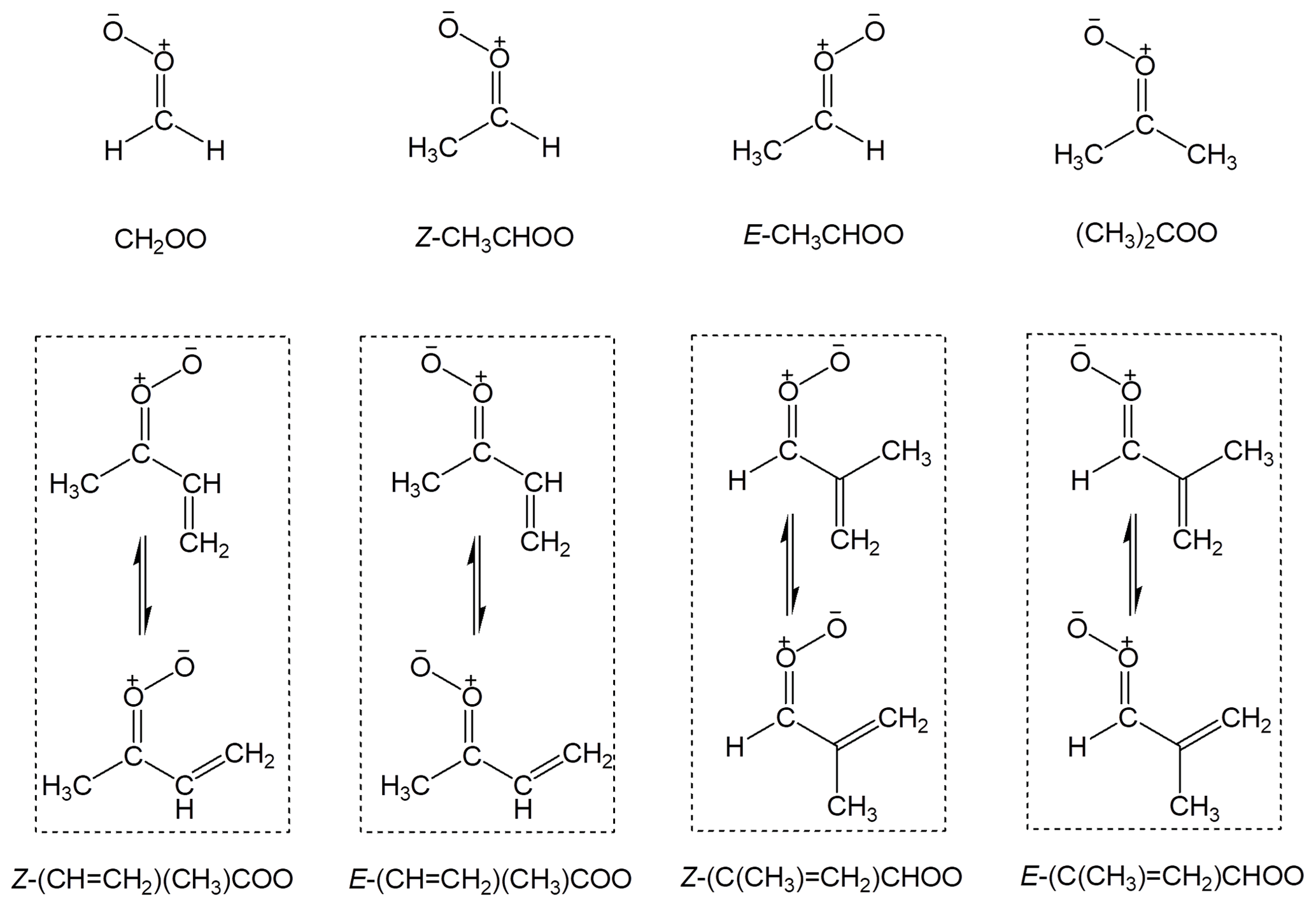
Figure 2Structures of the stabilized Criegee intermediates considered in the present evaluation, and the nomenclature assigned. In the cases of the di-substituted (isoprene-derived) C4 intermediates, the Z and E notations specify the orientation of the first named substituent (which has the higher Cahn–Ingold–Prelog priority) relative to the CI moiety. The displayed rotamers of the C4 intermediates are calculated to be in near equilibrium under atmospheric conditions (Vereecken et al., 2017) and are assumed to act as a single species in each case.
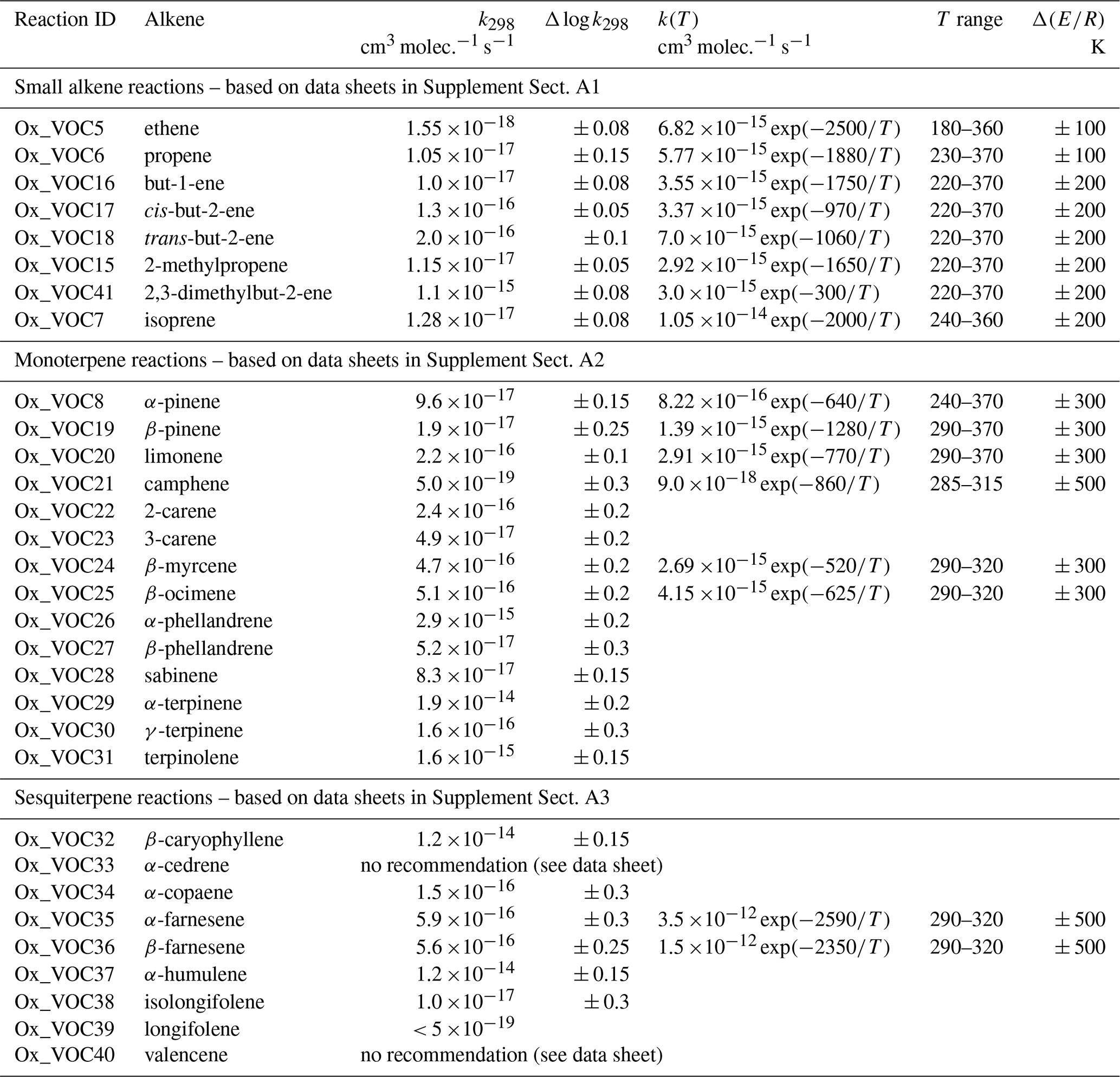
The present evaluation considers the reactions of O3 with 31 alkenes, including small (C1 to C4) alkenes, isoprene, monoterpenes and sesquiterpenes. This represents a substantial increase on those considered in our previous evaluation (Atkinson et al., 2006), which were limited to ethene, propene, isoprene and α-pinene. The reactions are listed in Table 1, along with the associated recommended rate coefficients. A detailed data sheet for each reaction is also provided in Supplement A. As discussed in detail previously (e.g. Calvert et al., 2000, 2015), the data indicate that the rate coefficients are highly sensitive to alkene structure, and depend on the degree of alkyl substitution of the unsaturated bond(s), on steric effects and on ring-strain effects in cyclic compounds. The lower tropospheric lifetimes of the alkenes, with respect to reaction with 20 ppb O3, therefore cover several orders of magnitude, ranging from as short as 2–3 min for reactive species such as α-terpinene, β-caryophyllene and α-humulene, to about 7 weeks or longer for camphene and longifolene. For the simple alkenes, the lifetimes range from about 30 min for the fully substituted 2,3-dimethylbut-2-ene to about 2 weeks for ethene. With the exception of the least reactive compounds, removal by ozonolysis is expected to make a contribution for all the evaluated alkenes under lower tropospheric conditions and is generally the dominant fate for those with rate coefficients in excess of about 10−15 cm3 molec.−1 s−1.
Table 1Summary of recommended rate coefficients for reactions of O3 with alkenes.
As described in Sect. 1 and Fig. 1, the chemically activated CIs formed from the O3 + alkene reactions may either decompose promptly or lose energy by collisions with other molecules to form stabilized Criegee intermediates (sCIs). The sCIs have the potential to undergo reactions with other atmospheric trace gases, leading to their oxidation and formation of characteristic products. Thus, it is important to quantify the yield (Y) of each sCI from each relevant precursor alkene if the impact of alkene ozonolysis and Criegee chemistry on oxidation processes is to be correctly represented in atmospheric mechanisms, or to allow the local steady-state concentration of the sCIs to be estimated by Eq. (1).
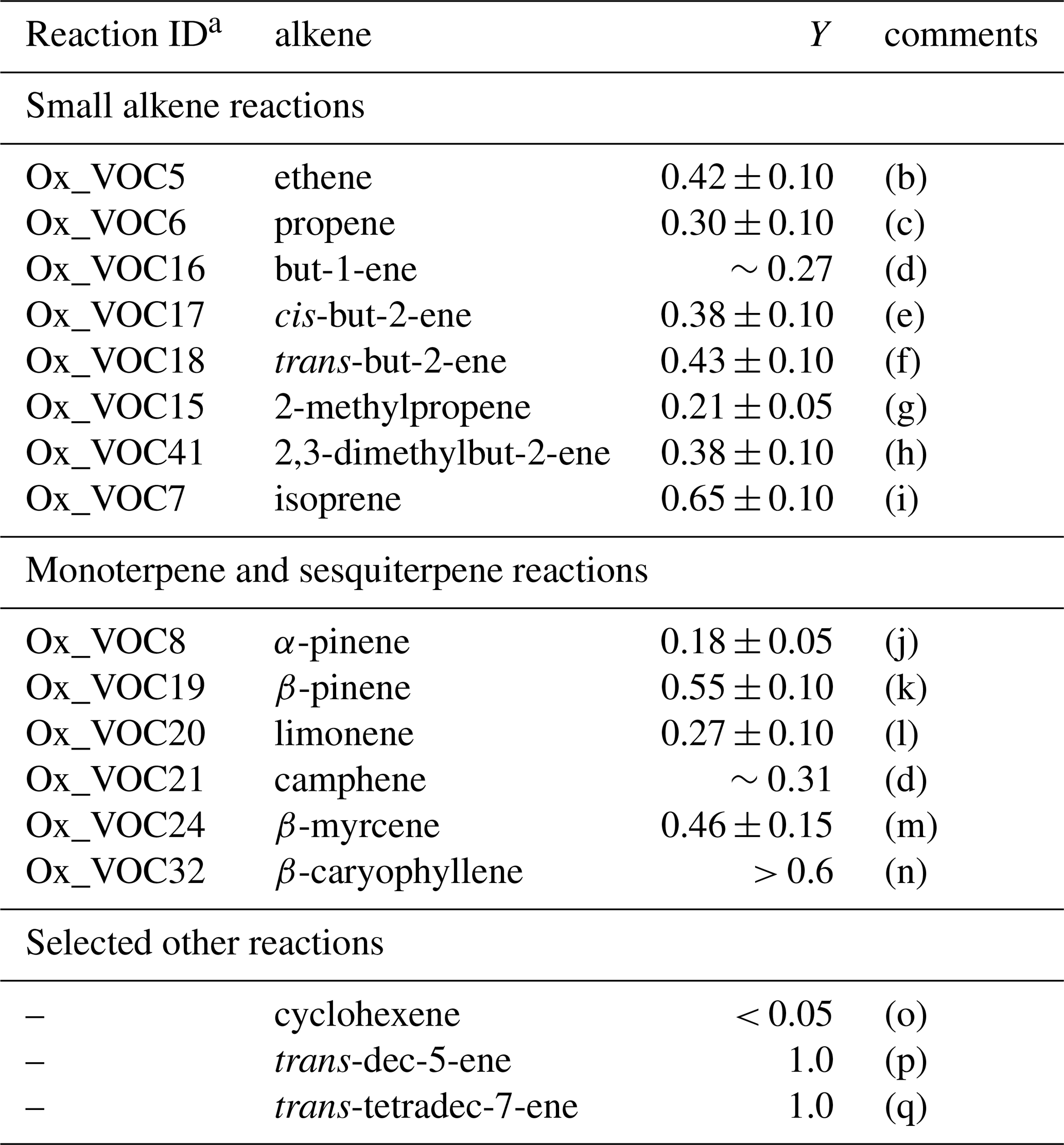
There have been extensive experimental efforts to determine Y for a variety of O3 + alkene systems. The yields have generally been expressed as a fraction of the molar amount of O3 reacted, and have been determined by reaction of the sCIs formed with an appropriate added scavenger reagent (e.g. SO2, H2O, HCHO, HCOOH, CF3C(O)CF3). The yield is determined either from quantitative analysis of a characteristic product of the sCI + scavenger reaction, or through measurement of the loss of the scavenger. The reported values of Y are therefore indirect measurements that have generally quantified the total yield of sCIs formed in a given alkene + O3 system, with little or no information on the contributions of the component sCI species for asymmetric alkenes being reported. Table 2 gives a summary of the recommended values of Y for the evaluated O3 + alkene reactions at 298 K and 1 bar, with additional details provided in the corresponding reaction data sheets in Supplement A. Values for selected other alkenes (trans-dec-5-ene, trans-tetradec-7-ene and cyclohexene) are also given to help illustrate structural variations in Y, as discussed further below.
Table 2Summary of recommended total sCI yields (Y) from O3 + alkene reactions at 298 K and 1 bar.
a See corresponding data sheets in Supplement A for further information. b Based on
Su et al. (1980), Kan et al. (1981), Hatekayama et al. (1984, 1986), Horie
and Moortgat (1991), Neeb et al. (1996, 1998), Horie et al. (1999), Hasson et al.
(2001a), Alam et al. (2011), Newland et al. (2015a, 2020). c Based on Hatekayama et
al. (1984), Horie and Moortgat (1991) and Newland et al. (2020). d Based on Hasson
et al. (2001b). e Based on Newland et al. (2015a). f Based on Berndt et al. (2014),
Newland et al. (2015a), and Hakala and Donahue (2018). Pressure dependence measurements suggest Y falls to ∼ 0.25 at 50 Torr (Hakala and Donahue, 2018).
g Based on Hatekayama et al. (1986) and Newland et al. (2020). h Based on Berndt et al. (2014), Newland et al. (2015a, 2020), and Hakala and Donahue (2016). Pressure dependence measurements suggest Y falls to 0.12–0.15 at zero pressure (e.g. Hakala
and Donahue, 2016; Campos-Pineda and Zhang, 2017). i Based on Sipilä et al. (2014), Newland et al. (2015b) and Nguyen et al. (2016), as also discussed further in data
sheet CGI_21 (Supplement B). j Based on Drozd and Donahue (2011), Sipilä et al.
(2014) and Newland et al. (2018). Approximately linear pressure dependence observed
by Drozd and Donahue (2011), with Y≈ 0.05 at 110 Torr. k Based on Winterhalter et al. (2000) and Newland et al. (2018) with support from theoretical study of Nguyen et al. (2009a). l Based on Sipilä et al. (2014) and Newland et al. (2018). m Based on
Newland et al. (2020). n Based on Winterhalter et al. (2009) with support from
theoretical study of Nguyen et al. (2009b). o Based on Hatekayama et al. (1984),
Drozd and Donahue (2011), who observed no stabilization at 550–640 Torr, and Campos-Pineda and Zhang (2018), who observed no stabilization at 10–20 Torr.
p Based on Drozd and Donahue (2011). Full stabilization observed at pressures above ∼ 400 Torr, with Y falling at lower pressures to ∼ 0.6 at 70 Torr. q Based on Hakala
and Donahue (2018). Pressure dependence measurements suggest Y falls to ∼ 0.35 at 50 Torr.
As indicated in Sect. 1, the stabilization of the promptly formed chemically activated CIs requires loss of internal energy through collisional energy transfer to other gas molecules. As a result, studies of a number of O3 + alkene systems have shown that Y depends on pressure (e.g. Hatekayama et al., 1986; Drodz and Donahue, 2011; Hakala and Donahue, 2016, 2018; Campos-Pineda and Zhang, 2017) but with significant residual values at zero pressure. This indicates that a limiting yield of sCIs (i.e. with internal energy below the threshold required for decomposition or isomerization) is typically formed directly from decomposition of the primary ozonide, POZ (although this is not illustrated in Fig. 1 for simplicity), with additional sCI formation resulting from collisional stabilization of the chemically activated CIs.
The value of Y is also expected to vary systematically with alkene structure, and the reported data display some logical structural trends that are consistent with theoretical treatments. These can be rationalized in terms of the nascent internal energy in the Criegee intermediate and how this is influenced by partitioning of the excess energy into either translational modes (i.e. recoil), internal energy of the carbonyl co-product, or into non-reactive vibrational or rotational modes within the Criegee intermediate (e.g. Choung et al., 2004; Drozd et al., 2011). The value of Y is therefore expected to increase with alkene size along a homologous series, as has been confirmed for a set of trans- symmetric alkenes between C4 and C14 by Hakala and Donahue (2018). As a result, the values at 298 K and 1 bar for large acyclic alkenes (e.g. Y=1.0 for trans-dec-5-ene and trans-tetradec-7-ene) tend to be larger than those for the smaller acyclic alkenes, as listed in Table 2. Similarly, those for large cycloalkenes with endocylic double bonds (e.g. Y > 0.6 for the C15 β-caryophyllene) tend to be larger than those for smaller species such as the C10 α-pinene (Y = 0.18) and the C6 cyclohexene (Y < 0.05). It is also clear that the values for cycloalkenes with endocylic double bonds are systematically lower than those for similarly sized acyclic alkenes (or cycloalkenes with exocylic double bonds). This can be rationalized in terms of the excess energy being confined within a single product possessing both Criegee and carbonyl functionalities for the cycloalkenes with endocylic double bonds; whereas it can be dissipated into translational modes, and the internal energy of the carbonyl co-product, for acyclic alkenes (and for cycloalkenes with exocylic double bonds).
It is also recognized that the indirect yield measurements are often subject to significant uncertainties and variability between different measurement methods (e.g. see Hakala and Donahue, 2016), such that systematic trends can be masked. Another important consideration is that exceptionally rapid unimolecular decomposition and isomerization reactions are predicted to be available for some sCIs, e.g. some of the C4 species formed from O3 + isoprene (Vereecken et al., 2017), as is discussed further in Sects. 4 and 7. In these cases it is possible that the bimolecular reactions with scavengers are unable to compete with the fast unimolecular processes, and consequently the reported yields for sCI formation may be underestimated.
It has been established for several decades that the reactions of O3 with alkenes lead to the formation of HO radicals (e.g. Finlayson et al., 1972; Donahue et al., 1998), and considerable attention has been given to quantifying HO radical yields for many O3 + alkene systems. In most of the reported studies, the yields have been determined indirectly by reaction of the HO formed with an appropriate added scavenger reagent (e.g. cyclohexane, 1,3,5-trimethylbenzene, butan-2-ol). The yield is then determined either from quantitative analysis of the yield of a characteristic product of the HO + scavenger reaction or through measurement of the loss of the scavenger. However, direct detection methods (particularly laser-induced fluorescence) have also been used to quantify HO yields in a number of studies (e.g. Donahue et al., 1998; Siese et al., 2001; Kroll et al., 2001a, b; Malkin et al., 2010; Alam et al., 2013), providing unequivocal identification of HO as a product of O3 + alkene reactions, and a means of confirming the validity and interpretation of the indirect methods (e.g. Malkin et al., 2010). Table 3 gives a summary of the recommended HO radical yields for the evaluated O3 + alkene reactions at 298 K and 1 bar, with additional details provided in the corresponding reaction data sheets in Supplement A.
Table 3Summary of recommended HO yields for reactions of O3 with alkenes at 298 K and 1 bar.
a See corresponding data sheets in Supplement A for further information. b Based on Atkinson et al. (1992), Paulson et al. (1999), Rickard et al. (1999), Mihelcic et
al. (1999), Fenske et al. (2000) and Alam et al. (2011). Comparable pressure-
independent yield (0.14) reported by Kroll et al. (2001a) over pressure range
13–80 mbar. c Based on Atkinson and Aschmann (1993), Neeb and Moortgat (1999), Paulson et al. (1999), Rickard et al. (1999), Aschmann et al. (2003), Qi et al. (2009)
and Alam et al. (2013). d Based on Atkinson and Aschmann (1993), Paulson et al.
(1999), Fenske et al. (2000) and Alam et al. (2013). e Based on Atkinson and
Aschmann (1993), McGill et al. (1999), Orzechowska and Paulson (2002) and Alam
et al. (2013). f Based on Atkinson and Aschmann (1993), McGill et al. (1999), Orzechowska and Paulson (2002), Hasson et al. (2003) and Alam et al. (2013).
g Based on Atkinson and Aschmann (1993), Neeb and Moortgat (1999), Paulson et al. (1999), Rickard et al. (1999) and Alam et al. (2013). h Based on Chew and
Atkinson (1996), Rickard et al. (1999), Fenske et al. (2000), Siese et al. (2001), Orzechowska and Paulson (2002), Aschmann et al. (2003) and Alam et al. (2013).
i Based on Aschmann et al. (1996), Paulson et al. (1998), Neeb and Moortgat (1999), Malkin et al. (2010), Nguyen et al. (2016) and Ren et al. (2017). j Based on Atkinson
et al. (1992), Chew and Atkinson (1996), Paulson et al. (1998), Rickard et al. (1999),
Siese et al. (2001), Aschmann et al. (2002), Berndt et al. (2003), Presto and Donahue (2004) and Forester and Wells (2011). k Based on Atkinson et al. (1992) and Rickard
et al. (1999). l Based on Aschmann et al. (2002), Herrmann et al. (2010) and Forester
and Wells (2011). m Based on Atkinson et al. (1992). n Based on Aschmann et al.
(2002). o Based on Herrmann et al. (2010). p Based on Aschmann et al. (2002) and Herrmann et al. (2010). q Based on Aschmann et al. (2002) and Herrmann et al.
(2010). r Based on Shu and Atkinson (1994), Winterhalter et al. (2009) and Jenkin et al. (2012). s Based on Shu and Atkinson (1994) and Yao et al. (2014). Substantially
lower yield, 0.090 ± 0.016, reported in the presence sCI scavengers, CH3C(O)OH or
SO2, by Yao et al. (2014). t Based on Shu and Atkinson (1994). u Based on Shu and Atkinson (1994) and Beck et al. (2011).
The formation of HO radicals can result from both prompt unimolecular decomposition of chemically activated CIs and decomposition of thermally equilibrated sCIs over longer timescales (e.g. as demonstrated in the time-resolved pressure dependence measurements of Kroll et al., 2001c). The most important mechanism forming HO is generally accepted to proceed by a 1,4 H shift isomerization to an excited vinyl hydroperoxide intermediate, which decomposes to form HO and a vinoxy or β-oxo alkyl radical; and this has been characterized for a variety of Criegee intermediates in theoretical studies (e.g. Vereecken et al., 2017). Using Z-CH3CHOO as an example, the mechanism proceeds as shown below.
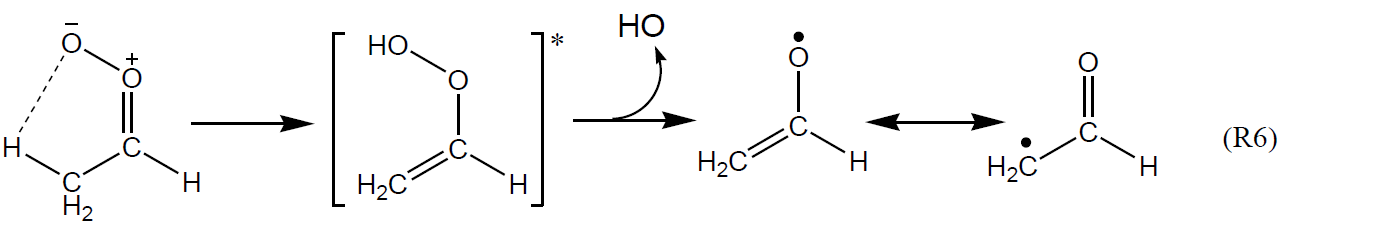
First proposed for (CH3)2COO by Niki et al. (1987), this mechanism is therefore potentially available for all di-substituted and Z- mono-substituted Criegee intermediates that possess a β-hydrogen atom but is unavailable for CH2OO and E- mono-substituted Criegee intermediates (e.g. E-CH3CHOO), where the outer O atom of the CI moiety is not directed towards an organic group. In the cases where Reaction (R6) is available, it is believed to be the dominant decomposition route for small (e.g. methyl- and ethyl-substituted) Criegee intermediates. The reported variation of HO yields with structure for simple small alkenes (e.g. as shown in Table 3) can therefore be broadly rationalized in terms of the combination of Criegee intermediates formed, and whether or not Reaction (R6) is available; and this has provided the basis of simple structure–activity relationships (SARs) for HO yields from O3 + alkene reactions (e.g. Rickard et al., 1999). Thus, those for fully methyl-substituted alkenes (e.g. 0.93 for 2,3-dimethyl-but-2-ene) tend to be systematically higher than those for partially methyl-substituted alkenes (e.g. 0.33 for cis-but-2-ene and 0.60 for trans-but-2-ene), which in turn are higher than that for the unsubstituted ethene (0.17).
Another key route involves initial rearrangement (1,3 ring-closure) to form a dioxirane intermediate (see Fig. 1). This provides an alternative unimolecular decomposition pathway for CH2OO and E- mono-substituted Criegee intermediates (e.g. E-CH3CHOO) and is also calculated to be competitive for some Z- mono-substituted Criegee intermediates possessing oxygenated substituents (Vereecken et al., 2017). In the former case, it is likely to be significant only for the chemically activated CI, [CH2OO]∗, because unimolecular loss of the thermally equilibrated sCI, CH2OO, is observed and calculated to be slow (see Sect. 7). The dioxirane intermediate isomerizes to form “hot” formic acid, [HC(O)OH]∗, which can fragment via a number of pathways:
Reaction (R7a) can therefore account for the small HO yield (0.17) resulting from the O3 + ethene reaction (Table 3), and the set of product channels can also rationalize the observed formation of HO2 (from the reactions of O2 with H and HCO), CO and CO2 (see data sheet OX_VOC5 in Supplement A). In the cases of E- mono-substituted Criegee intermediates (e.g. E-CH3CHOO), isomerization via a dioxirane intermediate is again expected to be significant for chemically activated CIs and is also calculated to occur for thermally equilibrated sCIs (e.g. Vereecken et al., 2017). For sCIs, however, it is in competition with some particularly rapid bimolecular reactions under atmospheric conditions (see Sects. 7 and 8) and may therefore be of limited importance. In the case of the chemically activated [E-CH3CHOO]∗, the dioxirane intermediate isomerizes to form “hot” acetic acid, [CH3C(O)OH]∗, and the following pathways can rationalize the observed formation of HO2, CO2, CH2=CO (ketene), CH4 and CH3OH from the reactions of O3 with propene and cis- and trans-but-2-ene (see data sheets OX_VOC6 , OX_VOC17 and OX_VOC18 in Supplement Sect. A1).
In principle, formation of HO (and CH3CO, or CH3 and CO) may also occur by a route analogous to Reaction (R7a). The possible contribution of the corresponding channel more generally for E- mono-substituted CIs is poorly characterized, although it is generally accepted to be only a minor source of HO compared with the 1,4 H shift isomerization route for the Z- conformers. The dioxirane route has also been reported to lead to the formation of stabilized acid, ester or lactone products, particularly in larger O3 + alkene systems (e.g. Hakola et al., 1994; Griesbaum et al., 1998; Winterhalter et al., 2009; Nguyen et al., 2009a, b).
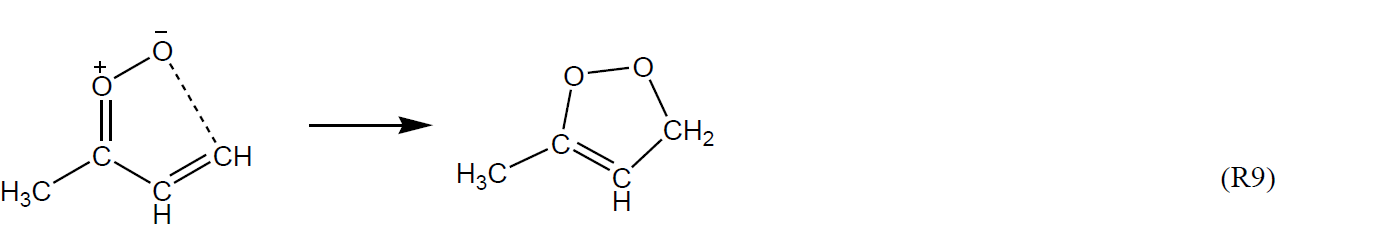
A number of other unimolecular reactions are available for Criegee intermediates possessing larger organic substituents, particularly those that are unsaturated (e.g. see Vereecken et al., 2017). For example, very rapid 1,5 ring-closure reactions are expected to dominate for Z-α,β-unsaturated sCIs, such as Z-(CH=CH2)(CH3)COO and Z-(C(CH3)=CH2)CHOO formed from O3 + isoprene, e.g. for Z-(CH=CH2)(CH3)COO.
In the cases of carbonyl-substituted sCIs, such as those formed from cycloalkenes with endocyclic double bonds, the potential for (bicyclic) ring-closure to form intramolecular secondary ozonides is well established.
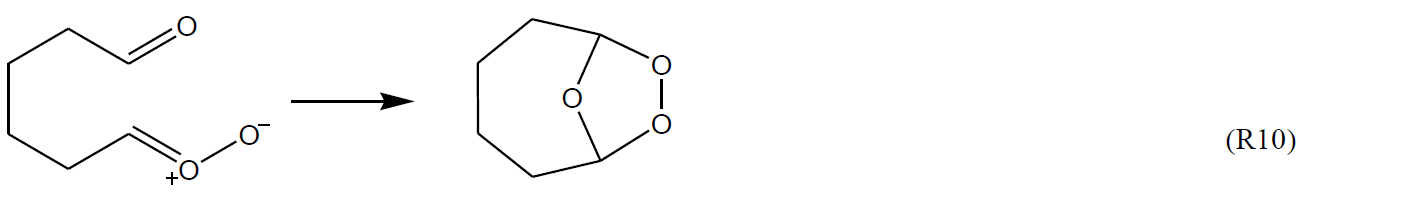
These reactions have been characterized in a number of theoretical studies (e.g. Chuong et al., 2004; Nguyen et al., 2009b; Mackenzie-Rae et al., 2016; Vereecken et al., 2017; Long et al., 2019), with experimental evidence for their formation also reported (e.g. Winterhalter et al., 2009; Beck et al., 2011). In practice, however, these reactions are only observed (and calculated) to be significant for larger systems (e.g. sesquiterpene ozonolysis), where the Criegee intermediates are formed significantly stabilized, and the ring closure reaction does not result in prohibitive ring strain. Where these criteria are met, they are predicted to be rapid reactions that can compete with, or dominate over, other decomposition routes, and this is one factor contributing to the low HO yields reported for some sesquiterpenes (e.g. β-caryophyllene and α-humulene).
Assessment of the photolysis rates and product channels for sCIs requires quantitative data for the absorption cross sections and quantum yields for individual sCI species at atmospheric, actinic wavelengths, mainly in the UV and visible. The development of methods for creating specific sCIs in defined concentrations has allowed experimental investigation of their spectroscopy and structure; see the review by Osborn and Taatjes (2015), for example. Moreover, advances in theoretical methods have also provided insight into the spectra and structure of sCIs, and quantum calculations have given further details of reaction mechanisms and product channels of sCI photolysis (e.g. Samanta et al., 2014).
The spectroscopic studies of sCIs have led to the recognition that they have a single ground electronic state, whose dominant configuration is that of a zwitterion, and this is reflected in the large dipole moment of these species (Chhantyal-Pun et al., 2017a). The observed spectra of the C1–C3 sCIs exhibit strong and broad absorptions centred in the near UV, with maximum cross sections of the order of 10−17 cm2 molec.−1. These features, and their detailed rovibrational structures, are consistent with transitions, i.e. intense transitions analogous to the familiar UV spectrum of O3 in the Hartley–Huggins bands. Photodissociation of CH2OO from this excitation is reported to proceed with a quantum yield of unity (e.g. Ting et al., 2014), producing HCHO and either O(3P) or O(1D). Production of O(1D) has been reported to be dominant (e.g. Li et al., 2015) and exclusive in the long wavelength tail (λ≥364 nm; Vansco et al., 2017).
5.1 Conformers
As described in Sect. 1 and Fig. 1, Criegee intermediates with dissimilar substituents can exist as two possible conformers, denoted E- and Z-, which differ in the orientation of the outer O atom relative to the substituent groups. The simplest examples are E- and Z-CH3CHOO (acetaldehyde oxide, see Fig. 2), for which conformer dependence has been demonstrated experimentally and theoretically in its spectra and its reaction rates and pathways. Calculations place Z-CH3CHOO about 15 kJ mol−1 lower in energy than E-CH3CHOO (Kuwata et al., 2010), reflecting the zwitterionic character of the Criegee intermediate structure. Calculated energies of the E- and Z- conformers of CH3CHOO are consistent with the spectral shift of λmax(Z-) < λmax(E-) shown in Fig. 3. This has enabled conformer-specific reactions to be investigated, using direct observation of the kinetics and products of the two conformers. The barrier to interconversion is substantial (∼ 160 kJ mol−1), and consequently E- and Z-CH3CHOO act as distinct chemical species at atmospheric temperatures. The absence of rotation is an important indicator of the zwitterionic character of the intermediate, as originally proposed by Criegee et al. (1954).
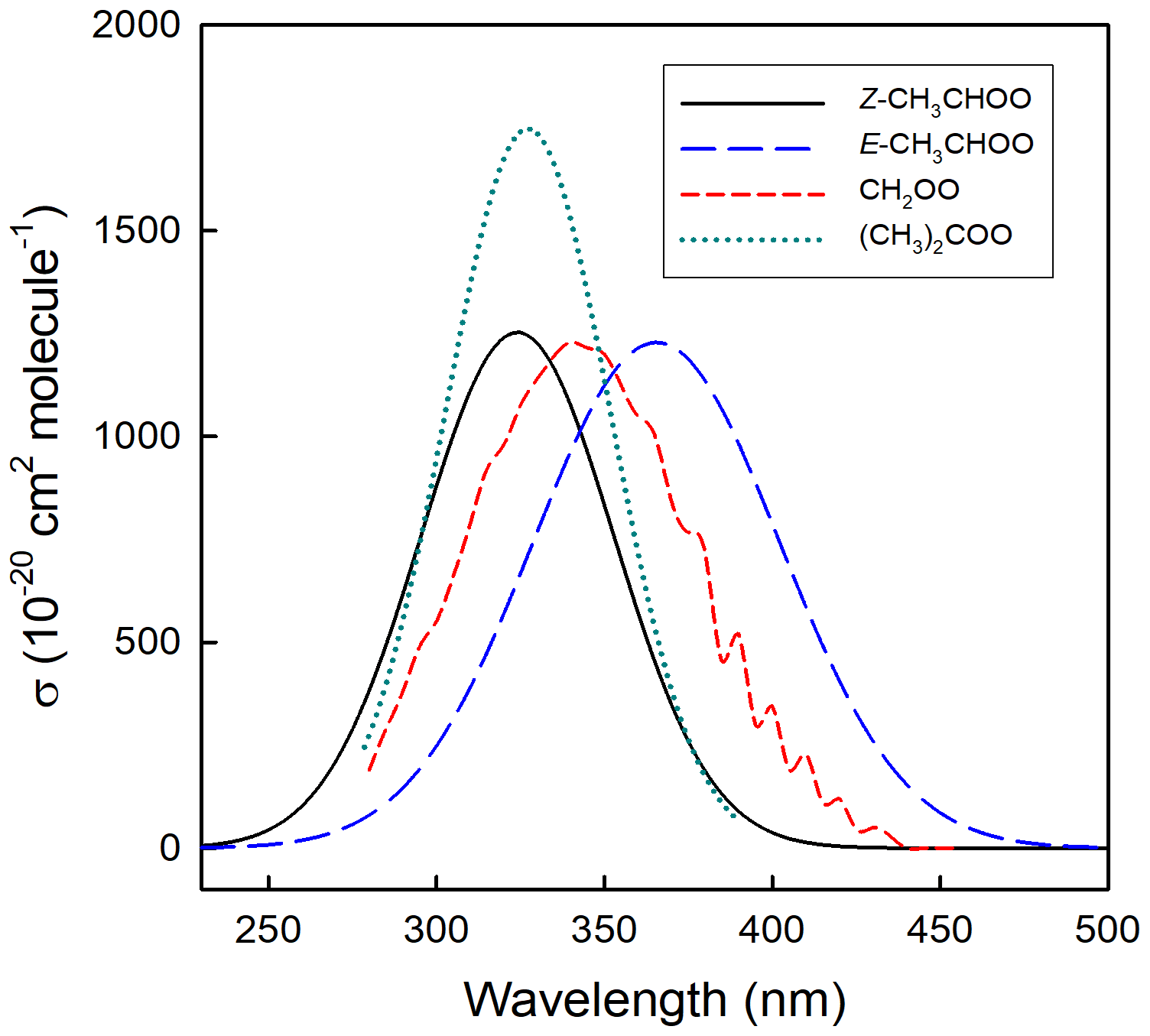
5.2 UV spectra of stabilized Criegee intermediates
In experimental studies of UV-visible spectra, the series of C1–C3 sCIs have mainly been formed by photolysis of the corresponding di-iodoalkane (via C-I bond fission), followed by the reaction of the iodoalkyl radical with O2, e.g. in the case of CH2OO (e.g. Beames et al., 2012; Sheps, 2013).
The formation of CH2I (and subsequently CH2OO) from the photolysis of CH2IBr (via C-Br bond fission) has also recently been reported (Peltola et al., 2020), suggesting that bromo-iodoalkanes may also be used more widely as sCI precursors. Absolute cross sections at specific wavelengths have been derived by monitoring the laser UV-induced depletion of the sCIs, monitored, for example, by mass spectrometry or by time resolved UV-absorption spectroscopy. The experimental data reveal some discrepancies regarding the shapes, structure, and intensities of the spectra determined using transient absorption spectroscopy, compared with laser-induced depletion techniques, determined under molecular beam conditions. This discrepancy has been attributed to the much lower temperatures reached in the molecular beams, compared to measurements at ambient temperature, but lack of detailed data on the temperature dependence of the cross sections over the required range precludes firm conclusions from being drawn. The evaluation of the spectral data and detailed discussion of the reported studies are given for the C1–C3 sCIs in the data sheets in Supplement Sect. B5. The recommendations for the maximum absorption cross sections are given in Table 4, and the spectra are presented in Fig. 3. Using these data, representative lower tropospheric photolysis removal rates in the range 0.4–1.4 s−1 can be calculated for a solar zenith angle of 30∘ at the surface, based on the actinic flux estimates of Madronich, presented by Finlayson-Pitts and Pitts (2000). This indicates that loss by photolysis is likely only a minor or negligible loss process for sCIs in the lower atmosphere, compared with their collective removal by the thermal reactions discussed in the following section.
Table 4Summary of the recommended spectral data for C1–C3 sCIs.

a See corresponding data sheets in Supplement Sect. B5 for further information. b ϕ is the photodissociation quantum yield. c Data sheet for CH3CH2CHOO included for completeness, although thermal reactions of this sCI are not included in the current evaluation
Novel methods for the production of the C4 isoprene-derived sCIs have also been reported (Barber et al., 2018; Vansco et al., 2019), with Z- and E-(CH=CH2)(CH3)COO formed from the photolysis of 1,3-di-iodobut-2-ene and Z- and E-(C(CH3)=CH2)CHOO formed from the photolysis of 1,3-di-iodo-2-methylprop-1-ene, both in the presence of O2. This has allowed characterization of the UV-visible spectra of the unsaturated C4 sCIs (Vansco et al., 2018, 2019), which are reported to be broader and shifted to longer wavelengths compared with those of the simple C1–C3 sCIs as a result of the conjugation of the vinyl and carbonyl oxide groups (see the detailed discussion in the data sheets in Supplement Sect. B5).
6.1 Measurements of absolute rate coefficients for reactions of sCIs
In recent years, numerous direct studies of the elementary reaction kinetics of sCIs have been reported. This has been made possible by two developments. First, the discovery of a novel fast photochemical source of sCIs, from the reactions of iodo-alkyl radicals with O2, has allowed generation of specific sCIs, following the photolysis of the corresponding di-iodoalkane (and recently, bromo-iodoalkene), as shown above in Sect. 5 for the example of CH2OO (Reactions R11 and R12). Second, direct time-resolved detection and measurements of sCI concentrations have been achieved using spectroscopic methods involving both tunable vacuum UV multiplexed photoionization mass spectrometry (MPIMS) and UV or IR absorption.
The first breakthrough in these developments for direct studies of sCI kinetics came from the work of Taatjes and co-workers, who used MPIMS to monitor the time-resolved decay of CH2OO in the presence of bimolecular reaction partners such as SO2, NO and NO2 (Welz et al., 2012; Taatjes et al., 2012). Subsequently it was shown that this technique for detection and production could be equally well applied to kinetics studies of the larger Criegee intermediates (e.g. Taatjes et al., 2013; Chhantyal-Pun et al., 2017b), so that structural effects on the basic oxidation rates and mechanisms could be explored directly.
The tunable light sources needed for MPIMS are not readily available for conventional laboratory rate constant measurements. The characterization of the strong absorption spectrum of CH2OO in the mid UV (Sheps, 2013) offered a second, more flexible and sensitive (but less specific) detection method for following sCI kinetics, which has the advantage of the ability to monitor sCI kinetics at up to 1 bar pressure, appropriate for lower atmospheric conditions. The UV absorption method is also applicable to the larger Criegee intermediates, produced from the same source chemistry (e.g. Sheps et al., 2014; Huang et al., 2015), and can provide kinetic–spectroscopic distinction of the Z- and E- conformers, where applicable.
The lower atmosphere contains many alkenes from both natural and anthropogenic sources, which react with O3 to form Criegee intermediates with a wide variety of structures. Earlier work on ozone–alkene reactions gave little clue on the structural dependence of sCI reactivity with trace gases such as SO2, H2O, NO2 and organics, or of their unimolecular decomposition rates. Direct kinetic studies have provided new information on the reaction rate constants and mechanisms of C1–C3 sCIs formed from ozonolysis of simple alkenes. As indicated in Sect. 5.2, methods for the production and spectroscopic characterization of more complex isoprene-derived species are emerging (e.g. Barber et al., 2018; Vansco et al., 2018, 2019), and these have provided the basis for their direct kinetics study (Caravan et al., 2020). However, direct experimental determinations of rate coefficients have not yet been reported for larger complex species (> C4) derived, for example, from monoterpenes and sesquiterpenes.
6.2 Evaluation of rate coefficients for bimolecular reactions
As noted in Sect. 1, most of the information on the kinetics of sCI reactions up to 2006 was based on data obtained using indirect relative rate techniques. These were evaluated by the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation and published in ACP in 2006 (Atkinson et al., 2006). Since 2012, the direct kinetics studies described above have provided a wealth of new data on the elementary reaction kinetics and spectroscopy of sCIs. This has stimulated further competitive rate studies using static and slow-flow experiments in chambers to generate sCIs from O3 + alkene reactions under atmospheric pressure and temperature conditions. In this evaluation, recommended rate coefficients are generally based on the results of direct kinetic studies of the sCIs, derived from di-iodoalkane precursors as described above (Sects. 5.2 and 6.1). However, the results of relative rate studies are also used to assess information on the kinetics and to check for consistency of the kinetic data for C1–C3 sCIs, when they are produced by alkene ozonolysis. Table 5 provides a summary of the preferred values of bimolecular reaction rate coefficients, with additional details given in the corresponding reaction data sheets in Supplement B. As indicated in Sect. 1, the evaluation focuses on classes of reaction that are of particular significance in tropospheric chemistry. Where data are available, these include reactions with SO2, NO2, H2O, (H2O)2, CH3CHO, CH3C(O)CH3, CF3C(O)CF3, HC(O)OH, CH3C(O)OH and CF3C(O)OH for the set of sCIs, with some additional reactions also considered for CH2OO. It is noted that data have been reported for some other classes of reaction (e.g. with alkenes and alcohols) that are uncompetitive under tropospheric conditions and some relative rate data are also available for large sCIs not considered here, e.g. as summarized in the compilation reported by Khan et al. (2018).
Table 5Summary of recommended rate coefficients for gas-phase bimolecular reactions of sCIs.
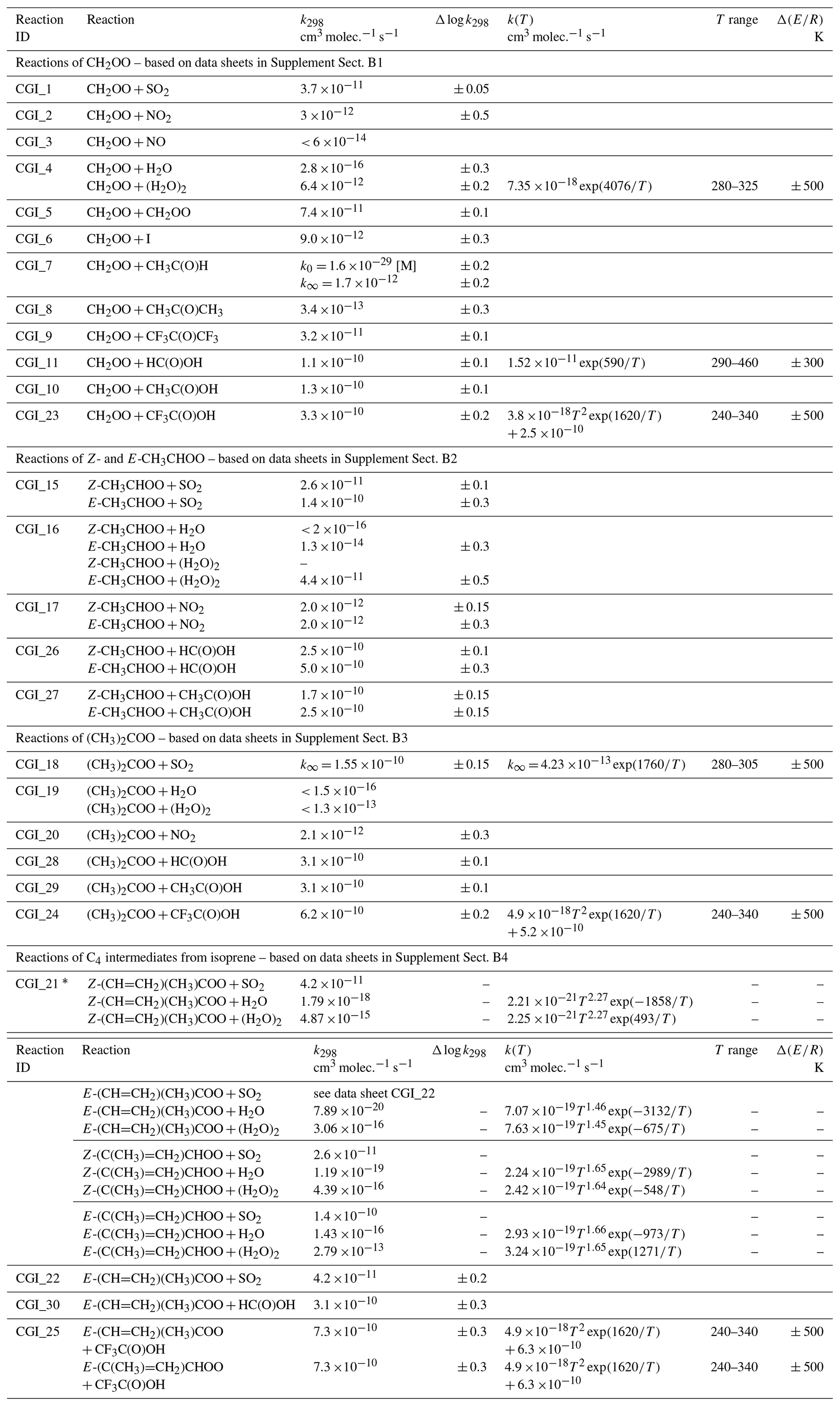
∗ Rate coefficients for SO2 reactions with Z-(CH=CH2)(CH3)COO, Z-(C(CH3)=CH2)CHOO and E-(C(CH3)=CH2)CHOO are inferred from those for
E-(CH=CH2)(CH3)COO, Z-CH3CHOO and E-CH3CHOO, respectively. Temperature-dependent rate coefficients for H2O and (H2O)2 reactions are adopted from the
theoretical/ SAR methods reported by Vereecken et al. (2017), as presented in Supplement Tables 35 and 40 of that paper. Individual parameters are not currently assigned
uncertainties, but performance of ensemble of reactions (also including sCI decomposition reactions) was tested against reported O3 + isoprene product observations (see data
sheet CGI_21, Supplement B).
With the exception of the reactions of E-(CH=CH2)(CH3)COO with SO2 and HC(O)OH (Caravan et al., 2020), there are currently no direct kinetics determinations for reactions of the C4 sCIs derived from isoprene. In these cases, the recommendations are either inferred from those for the simpler C2 and C3 species and E-(CH=CH2)(CH3)COO or adopted from reported theoretical studies (Vereecken et al., 2017; Chhantyal-Pun et al., 2017a). The performance of the ensemble of bimolecular reactions with SO2, H2O and (H2O)2 (and unimolecular decomposition reactions) has been checked for consistency, using the results of published chamber and slow-flow experiments (Sipilä et al., 2014; Newland et al., 2015b; Nguyen et al., 2016), as described in detail in data sheet CGI_21 (Supplement B). The results provide some confidence in these recommendations for use in practical applications, although the data also support some tolerance in the parameter values applied. Measurements of speciated sCI yields, and further direct kinetics studies of the rate coefficients and product channels for the reactions of the C4 sCI isomers are therefore required to allow these recommendations to be confirmed or refined.
6.3 Evaluation of rate coefficients for unimolecular decomposition
Table 6 shows a summary of preferred values of unimolecular decomposition rate coefficients, kd, given in the corresponding data sheets in Supplement B. The evaluations are based on both consideration of direct time-resolved measurements, and those reported in relative rate experiments, where kd is determined relative to the loss rate of sCI via a well-defined competing bimolecular reaction. In the former case, kd is determined from observation of the decay kinetics of the Criegee intermediates themselves (or of a marker species) in the absence of a second reagent or by extrapolation of the observed first-order removal rate vs. reagent concentration plots to zero. Although unimolecular decomposition can make a major (or even the dominant) contribution to such limiting first-order removal rates, other processes also need to be taken into account (e.g. wall loss, reaction with impurity gases or diffusive loss from the monitoring probe area), as is usually discussed in the individual studies. In some cases, therefore, direct kinetics studies can only provide upper limit estimates of unimolecular decomposition rates for sCIs, particularly when the decomposition rate is slow. Relative rate determinations can also be influenced by background loss processes for the sCI (e.g. reaction with impurity gases or products), and reported rate coefficient ratios need to be placed on an absolute basis using the rate coefficient for the competing, reference bimolecular reaction, which itself has an associated uncertainty. In the present evaluations, the reference (bimolecular) rate coefficients are all based on the preferred values given in Table 5.
Table 6Summary of recommended rate coefficients for gas-phase unimolecular reactions of sCIs.
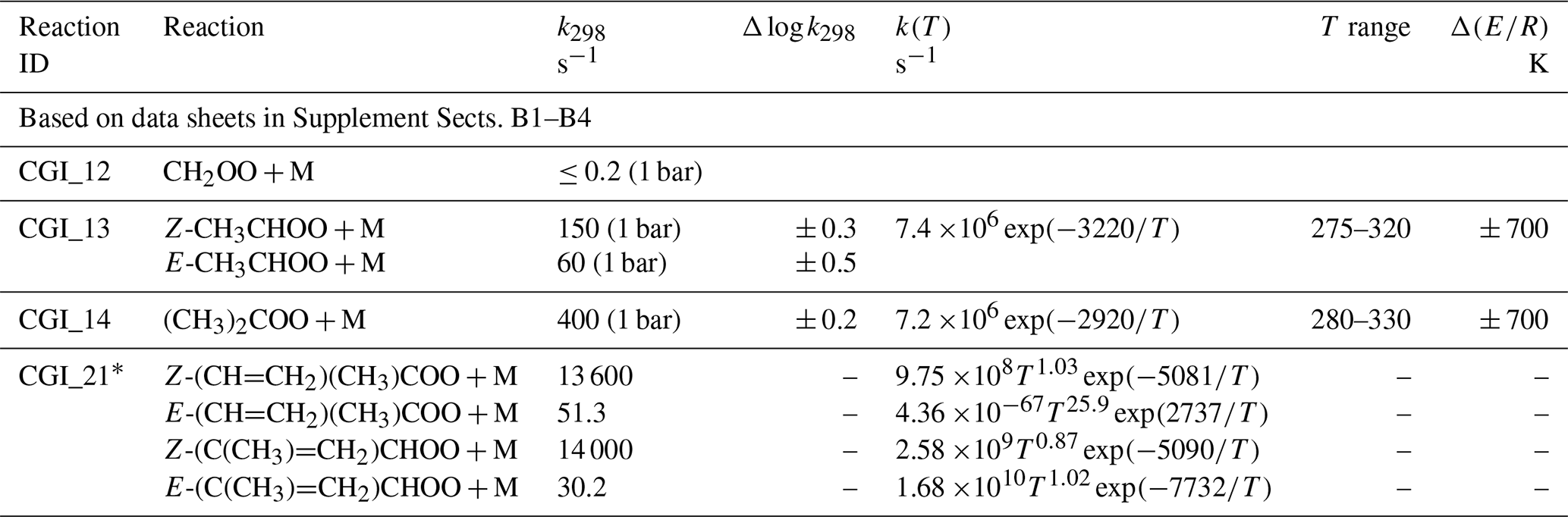
∗ Temperature-dependent rate coefficients adopted from the theoretical and SAR methods reported by Vereecken et al. (2017), as presented in Supplement Table 31 of that paper (note that the exponent of the pre-exponential factor changed from 9 to 8 in the case of Z-(CH=CH2)(CH3)COO, for consistency with 298 K rate coefficient reported by Vereecken et al., 2017). Individual parameters are not currently assigned uncertainties, but performance of the ensemble of reactions (also including sCI reactions with SO2, H2O and (H2O)2) was tested against reported O3 + isoprene product observations (see data sheet CGI_21, Supplement B).
As indicated above in Sect. 6.2, the recommendations for the unimolecular decomposition of C4 sCIs derived from isoprene ozonolysis are adopted from the theoretical study of Vereecken et al. (2017). These recommendations have been assessed, along with the recommended bimolecular rate coefficients for reactions with SO2, H2O and (H2O)2, using the results of published chamber and slow-flow experiments (Sipilä et al., 2014; Newland et al., 2015b; Nguyen et al., 2016), as described in detail in data sheet CGI_21 (Supplement B).
In addition to the progressive increase in the availability of experimental data, there have been substantial advances in the theoretical treatment of structure and reaction kinetics of sCIs in the gas phase (e.g. Olzmann et al., 1997; Zhang et al., 2002; Anglada et al., 2002, 2011; Ryzhkov and Ariya, 2004; Kuwata et al., 2010; Vereecken et al., 2012, 2017; Liu et al., 2014; Anglada and Solé, 2016; Long et al., 2016; Vereecken and Nguyen, 2017; Chhantyal-Pun et al., 2017a; Stephenson and Lester, 2020). Theoretical studies provided particular guidance prior to the advances in sCI production and detection methods for direct kinetics measurements, as described above. However, the body of experimental information now available for a series of sCIs allows the results of theoretical studies to be validated and for the methods to be refined, optimized and extended. This has included a comparison of experimental and theoretical unimolecular decay rates of a number of infra-red activated sCIs for a range of excitation energies (e.g. Fang et al., 2016, 2017; Barber et al., 2018).
Table 7Rate coefficients (k) and representative lower tropospheric first-order loss rates (kI) at 298 K for sCI bimolecular reactions with SO2, H2O and (H2O)2, and unimolecular decomposition. The present recommendations (IUPAC) are compared with calculated values using the theory-based SAR developed by Vereecken et al. (2017) (except where noted).
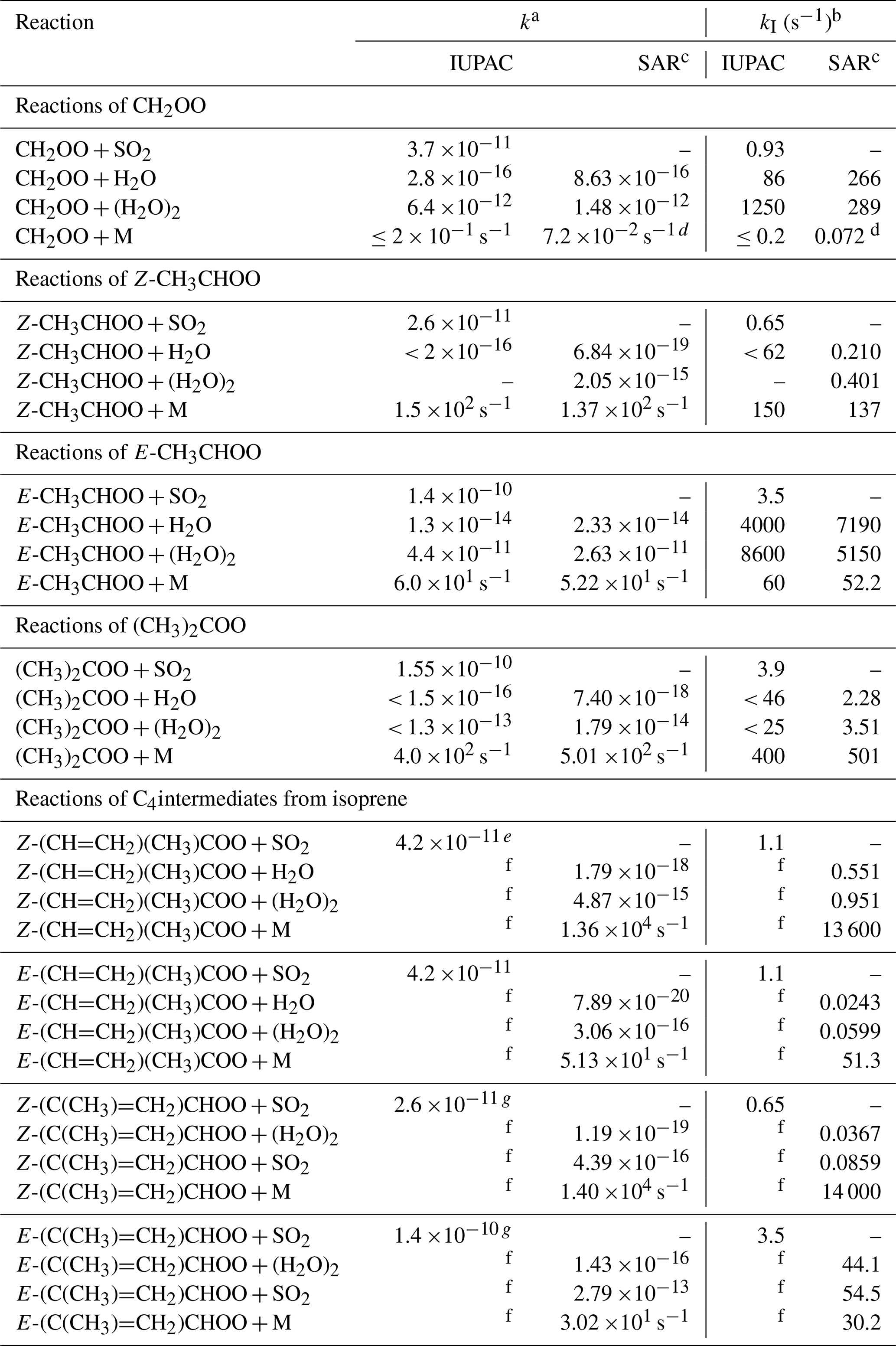
a Units of k are cm3 molec.−1 s−1, unless otherwise stated. b kI at 298 K and 1 bar determined for 40 % relative humidity ([H2O] = 3.08 ×1017 molec. cm−3 and [(H2O)2] = 1.96 ×1014 molec. cm−3), and for [SO2] = 2.5 ×1010 molec. cm−3 (∼ 1 ppbv). c Except where noted, based on high-pressure limiting T-dependence parameters given in Tables 31, 35 and 40 of Vereecken et al. (2017). d Based on the value at 1 bar reported by Long et al. (2016). e Value inferred from that for E-(CH=CH2)(CH3)COO. f IUPAC k value is adopted from Vereecken et al. (2017), and each given entry is therefore identical to the SAR value shown. g Values for Z- and E-(C(CH3)=CH2)CHOO inferred from those for Z- and E-CH3CHOO, respectively.
In this section, the recommended rate coefficients for unimolecular decomposition and bimolecular reactions of the C1–C3 sCIs with H2O and (H2O)2 at 298 K and atmospheric pressure are compared with those derived from theoretical calculations (see Table 7). With the exception of the unimolecular decomposition of CH2OO, the calculated values are based on the high-pressure limiting values reported in the comprehensive study of Vereecken et al. (2017), which presented theory-based structure-activity relationships (SARs) for 98 atmospherically relevant classes of sCI. Those for unimolecular decomposition reactions were derived from consideration of 14 reaction types (e.g. including the 1,4 H shift isomerization, 1,3 ring-closure and 1,5 ring-closure reactions discussed and illustrated in Sect. 4) for a benchmark series of sCIs containing key substituents, comprising a set of about 170 calculated rate coefficients. Those for the bimolecular reactions with H2O and (H2O)2 were based on fitting theory-derived reactivity trends to a set of literature data, which included rate coefficients available at the time for the same set of C1–C3 sCIs considered in the present evaluation. As a result, the absolute scaling of the theory-based SAR values cannot be considered to be entirely independent of our experimentally based evaluations for the H2O and (H2O)2 reactions, although comparison of the (considerable) reactivity variation across the series of sCIs is valid.
The comparisons shown in Table 7 demonstrate that the theory-based SAR rate coefficients reported by Vereecken et al. (2017) show a good level of consistency with our recommended rate coefficients for the reactions of the C1–C3 sCIs. Both sets of parameters display a similar structural dependence across the series of sCIs. The rate coefficients for the bimolecular reactions with H2O and (H2O)2 agree to within about a factor of about four (which is well within the combined uncertainties), where direct comparison is possible (i.e. for CH2OO and E-CH3CHOO), and where only an upper limit recommendation is possible in the present work (i.e. for Z-CH3CHOO and (CH3)2COO), the SAR rate coefficient is fully compatible with that recommendation. This indicates a consistent structure-reactivity variation across the series, with systematically higher reactivities for the reactions of CH2OO and E-CH3CHOO with both H2O and (H2O)2.
The unimolecular decomposition parameters recommended in the present study for E- and Z-CH3CHOO and (CH3)2COO are in very good agreement with the theory-based SAR values; being consistent with dominant 1,3 ring-closure to form a dioxirane intermediate for E-CH3CHOO, and dominant 1,4 H shift isomerization to form vinyl hydroperoxide intermediates for Z-CH3CHOO and (CH3)2COO. In the case of CH2OO, the upper limit rate coefficient at 298 K and atmospheric pressure recommended in the present work is consistent with the value calculated by Long et al. (2016), which is about a factor of four lower than the calculated high-pressure limiting value (see data sheet CGI_12). Both the recommended and calculated values are sufficiently slow for unimolecular decomposition to be uncompetitive under tropospheric conditions.
Also shown in Table 7 are representative lower tropospheric first-order loss rates (kI) (at 298 K and atmospheric pressure) calculated for the same series of reactions using both sets of rate parameters; and for the bimolecular reactions with SO2, based on the rate coefficients recommended here. The representative rates for the bimolecular reactions assume a mid-range relative humidity of 40 % (corresponding to [H2O] ≈ 3 ×1017 molec. cm−3 and [(H2O)2] ≈ 2 ×1014 molec. cm−3), and an SO2 concentration of 2.5 ×1010 molec. cm−3 (about 1 ppbv), broadly typical of urban background air. Although these concentrations are only representative, the associated values of kI nevertheless give a clear indication that unimolecular decomposition is the dominant loss route for Z-CH3CHOO and (CH3)2COO, consistent with a widespread role for 1,4 H shift isomerization for Z- monoalkyl-substituted and dialkyl-substituted sCIs possessing a β-hydrogen atom. In contrast, bimolecular reactions with H2O and/or (H2O)2 are the dominant loss routes for CH2OO and E-CH3CHOO, and likely most other E- monoalkyl-substituted sCIs. The values of kI also give an indication that loss due to reaction with SO2 generally only makes a minor contribution to sCI removal, away from the immediate vicinity of SO2 sources, such that their ambient concentration is mainly controlled by either decomposition or reaction with H2O and/or (H2O)2.
The match between the experimentally based recommendations presented here and those derived from the theory-based SARs for this set of sCIs, gives some confidence that the SAR rate coefficients of Vereecken et al. (2017) provide a very reasonable basis for representing the structural dependence of the kinetic parameters for unimolecular decomposition and bimolecular reactions with H2O and (H2O)2. In view of this, our corresponding recommendations for the more complex C4 isoprene-derived species (E- and Z-(CH=CH2)(CH3)COO and E- and Z-(C(CH3)=CH2)CHOO) are currently adopted from Vereecken et al. (2017), as evaluated and discussed in detail in data sheet CGI_21 (Supplement B). The 298 K rate coefficients and representative first-order loss rates (kI) are also shown for these C4 sCIs in Table 7. The values of kI clearly demonstrate that bimolecular reactions cannot compete with the very rapid unimolecular decomposition of Z-(CH=CH2)(CH3)COO and Z-(C(CH3)=CH2)CHOO (via 1,5 ring-closure) under atmospheric conditions (and indeed most reported experimental conditions) and that decomposition of Z-(CH=CH2)(CH3)COO (via 1,4 H shift isomerization) is also likely to be its major loss route, by virtue of its slow bimolecular reactions with H2O and (H2O)2. In the case of E-(C(CH3)=CH2)CHOO, however, loss via unimolecular decomposition and bimolecular reactions with H2O and (H2O)2 are predicted to occur at comparable rates, and experimental confirmation of the rate coefficients would be of particular value for this species.
The kinetics and mechanistic information for sCI reactions recommended in the present evaluation provides the basis for representing the associated impact of alkene ozonolysis in atmospheric chemical mechanisms. The significance of sCIs as atmospheric oxidants can be discussed in terms of Eq. (1), which defines the local balance between production and loss of sCIs, and hence their steady-state concentrations. As described in more detail in Supplement C, the parameters recommended in this evaluation (supplemented by data from other sources) have been used to calculate surface production rates, loss rates and steady-state concentrations of a series of sCIs for average ambient conditions representative of rural background, suburban background and urban kerbside (urban traffic) locations in the south-eastern UK in both winter and summer. The calculations make use of measured or inferred concentrations of a series of C1–C6 alkenes, isoprene, α-pinene, limonene, O3, NO2, SO2 and HC(O)OH, in conjunction with H2O and (H2O)2 concentrations based on modelled temperature and relative humidity data typical of the region. In this section, the key results are summarized and placed in context by comparison with reported results calculated for other locations, and in global modelling studies. Although the calculations presented here aim to take account of the most important production and loss routes, the estimates are inevitably subject to potential omissions and uncertainties in the sources and sinks of the sCIs (as discussed further in Sect. C4), in addition to uncertainties associated with the kinetic parameters and sCI yields, as discussed in earlier sections and in the data sheets in Supplements A and B.
8.1 sCI production rates
The formation of a series of 28 C1–C10 sCIs from the speciation of 19 precursor alkenes was represented in the present calculations (see Supplement C for full details). The core set of C1–C4 sCIs specifically considered in the present evaluation collectively makes an important contribution to the total production rate at each of the three locations; the respective winter and summer contributions lying in the ranges 88 %–91 % and 45 %–86 % (see Table 8). The speciation of this core set of sCIs is also presented in Fig. 4 for the example of the rural background location (Chilbolton Observatory). The formation of E- and Z-CH3CHOO is most significant because they are formed from propene and all the alk-2-enes in the applied speciation. They are also favoured because reaction with O3 is a major (and sometimes the dominant) removal route for alk-2-enes (and other internal alkenes) because of their particularly rapid reactions with O3 (see Table 1). CH2OO also makes a notable contribution to the totals because it is formed from all the alk-1-enes and isoprene. The higher alkyl-substituted sCIs make systematically lower contributions, primarily because their precursor alkenes have systematically lower abundances (see Table 8).
Table 8Summary of results for the representative ambient calculations (see Sect. 8 and Supplement C for further details).
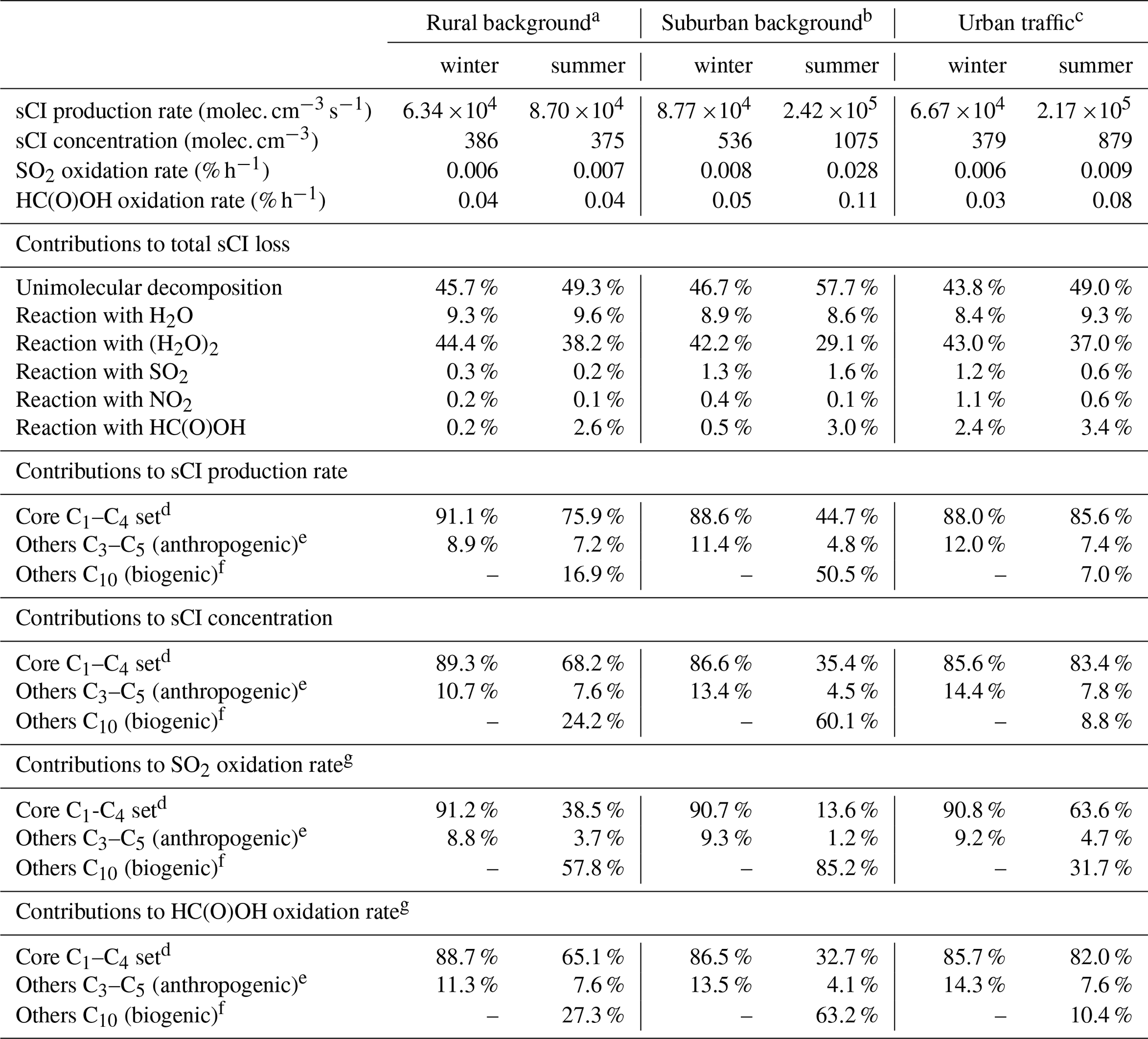
a Based on data for the Chilbolton Observatory site (51.149617, −1.438228). b Based on data for the London Eltham site (51.452580, 0.070766). c Based on data for the London Marylebone Road site (51.522530, −0.154611). d The core C1-C4 set comprises the sCIs shown in Fig. 2, for which evaluated rate parameters have been presented in Sect. 6. e Comprises E- and Z-C2H5CHOO, E- and Z-n-C3H7CHOO, E- and Z-i-C3H7CHOO, E- and Z-n-C4H9CHOO, E- and Z-(C2H5)(CH3)COO, and E- and Z-(CH=CH2)CHOO formed from the C4-C6 alkenes considered (see Supplement C). f Comprises eight pinonaldehyde oxide and limononaldehyde oxide isomers formed from the monoterpenes α-pinene and limonene (see Supplement C). g Specifies the contribution to the total SO2 or HC(O)OH oxidation rate due to reaction with sCIs.
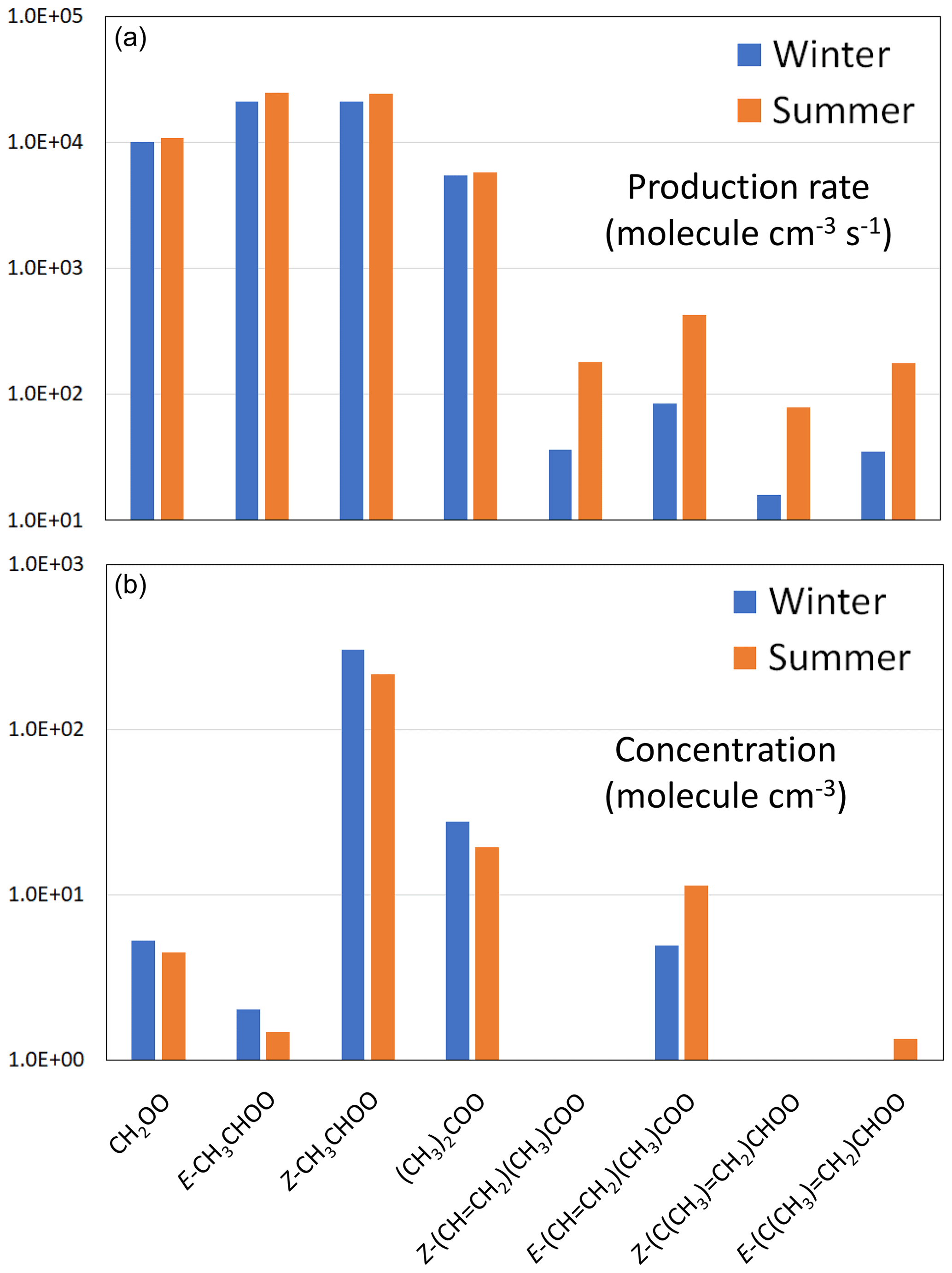
Figure 4Production rates (a) and steady-state concentrations (b) for the core set of C1–C4 sCIs for representative rural background conditions in the south-eastern UK. Note that the information is presented on log scales, with cut-offs of 10 molec. cm−3 s−1 and 1 molec. cm−3, respectively. The total sCI production rates and concentrations are given in Table 8. Results for an extended series of 28 sCIs, and for suburban and urban traffic conditions, are presented in Supplement C.
As also shown in Table 8, sCIs formed from biogenic hydrocarbons logically make an increased and important collective contribution under the summer conditions. This is particularly the case at the suburban background location (London Eltham), which has a mixture of trees and other vegetation in close proximity to the measurement site. The monoterpene-derived (α-pinene and limonene) sCIs are calculated to be particularly significant because ozonolysis generally makes a major contribution to the removal of endocyclic alkenes, by virtue of their particularly rapid reactions with O3 (see Table 1). In contrast, the reaction of O3 with isoprene is comparatively slow, its dominant removal reaction being with HO radicals. Therefore, the production rates of the isoprene-derived sCIs are approaching an order of magnitude lower than those of the monoterpene-derived sCIs under the conditions represented here. However, isoprene-derived sCIs have been shown to make more important contributions globally, particularly in specific regions such as the Amazonian rainforest (e.g. Vereecken et al., 2017; Khan et al., 2018), as a result of the dominant contribution of isoprene to global biogenic VOC emissions.
8.2 sCI loss rates
Based on the information presented and discussed in Sects. 6 and 7, sCI removal by unimolecular decomposition and bimolecular reactions with H2O, (H2O)2, NO2, SO2 and HC(O)OH was taken into account. Table 8 and Fig. 5 present information on the speciated and total first-order loss rates of the sCIs, and the contributions made by the series of removal reactions considered. Consistent with the analysis presented in Sect. 7, the results confirm that the major loss routes for most of the sCIs are either thermal decomposition or reaction with (H2O)2, supplemented by reaction with H2O. As a result, these reaction classes dominate total sCI removal under all conditions, with reaction with (H2O)2 and H2O being slightly more important in the winter, and thermal decomposition being slightly more important in the summer, based on the average of the three sites. As also indicated in Sect. 7, thermal decomposition tends to dominate the removal of Z- mono-substituted and di-substituted sCIs, with reaction with (H2O)2 and H2O dominating the removal of CH2OO and E- mono-substituted sCIs.
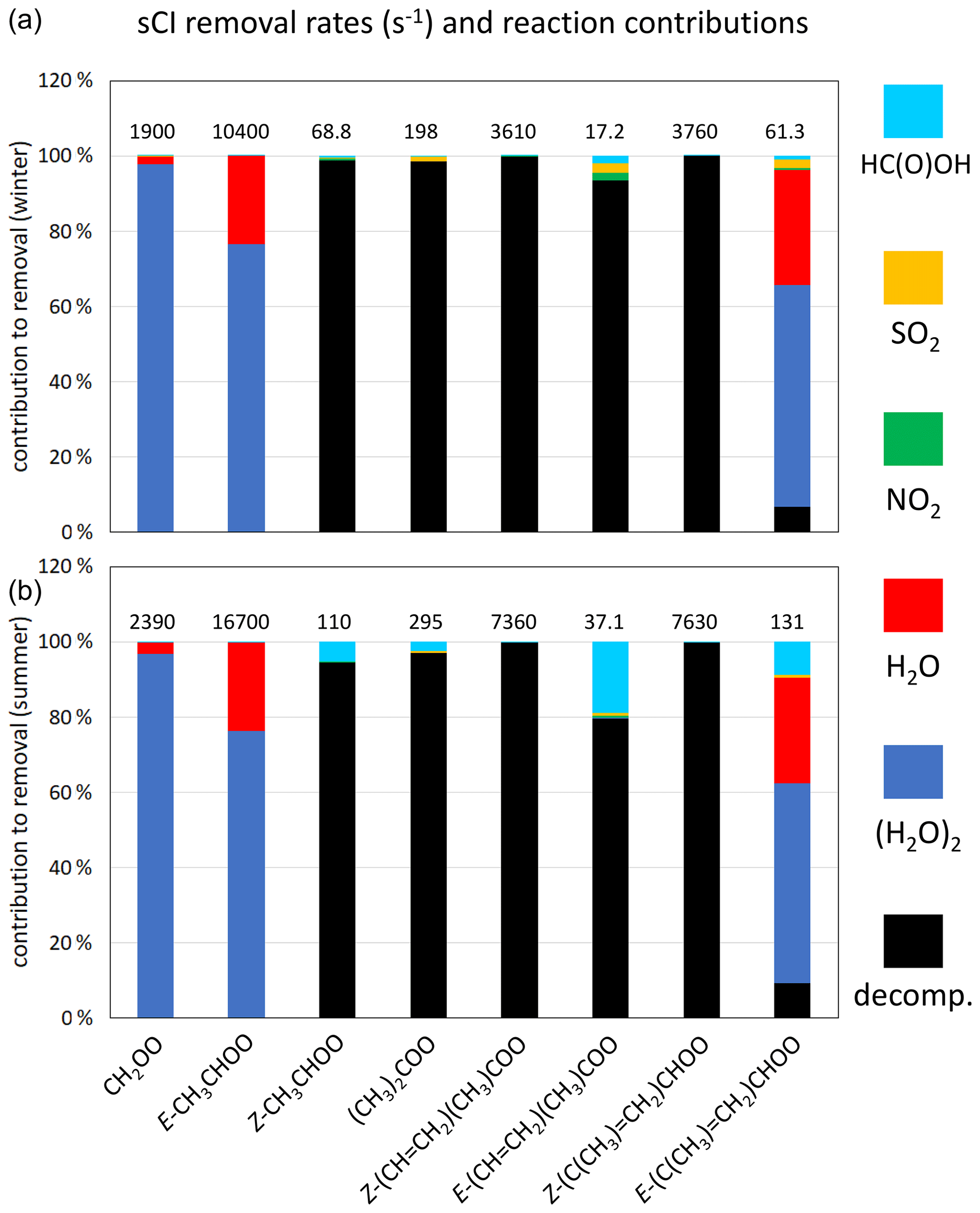
Figure 5Contributions of the unimolecular and bimolecular removal routes for the core set of C1–C4 sCIs under winter conditions (a) and summer conditions (b) for representative rural background conditions in the south-eastern UK. The data label shows the total removal rate (in s−1) for the given sCI. Results for an extended series of sCIs, and for suburban and urban traffic conditions, are presented in Supplement C.
The total first-order loss rates for the individual sCIs lie in an approximate range of 20 to 20 000 s−1 for the full series of considered conditions (see Supplement C). Those toward the low end of the range generally correspond to sCIs for which the dominant removal route is 1,4 H atom migration, occurring at only a modest rate (e.g. in the cases of Z-CH3CHOO and E-(CH=CH2)(CH3)COO), particularly under winter conditions, and for which the reactions with (H2O)2 and H2O are very slow. Those at the high end of the range generally correspond to E- monosubstituted sCIs (e.g. E-CH3CHOO) for which the dominant removal reactions with (H2O)2 and H2O are very fast. The associated loss rates for selected sCIs in the former category are sufficiently slow for removal by reaction with HC(O)OH and SO2 (and to a lesser extent, reaction with NO2) to make a notable contribution (e.g. E-(CH=CH2)(CH3)COO in Fig. 5). In collective terms, however, these classes of reaction each make only a small (< 4 %) contribution to total sCI removal under the series of conditions considered here, as shown in Table 8.
8.3 Steady-state concentrations and speciation
The total first-order sCI loss rates indicated above correspond to individual sCI lifetimes lying in the range 50 µs to 50 ms, confirming that calculation of their concentrations using the steady-state approximation, described by Eq. (1), is valid. The resultant calculated steady-state concentrations of the core set of C1–C4 sCIs for the rural background conditions are shown in Fig. 4, with the total sCI concentrations for the three scenarios given in Table 8. The totals calculated for the rural background conditions (386 and 375 molec. cm−3 for winter and summer, respectively) are broadly consistent with the low annual average values simulated for the UK in the global modelling calculations of Vereecken et al. (2017), and the concentrations calculated for the series of locations and conditions (up to about 1100 molec. cm−3) are comparable with those reported by Khan et al. (2018), based on similar UK calculations to those reported here.
The distributions of sCIs are generally dominated by a limited number of individual species and show similarities to those reported elsewhere for locations with significant anthropogenic VOC emissions (e.g. Vereecken et al., 2017). Z-CH3CHOO is the most abundant sCI for all the considered scenarios, accounting for 75 %–79 % of the totals for winter conditions, and 25 %–77 % of the totals for summer conditions. This results from the combination of its high production rate (see Fig. 4) and its relatively slow removal rate (see Fig. 5), as discussed above. Its lowest contribution occurs for summer conditions at the suburban location, when the sCIs derived from α-pinene and limonene (and to a lesser extent, isoprene) are collectively dominant (67 %), as discussed in more detail in Supplement C. Given the relatively low biogenic hydrocarbon emission rates in the UK, this result for a suburban location in London gives a strong indication that biogenic hydrocarbon-derived sCIs will dominate the global concentrations, as clearly demonstrated in the modelling studies presented by Vereecken et al. (2017) and Khan et al. (2018). Those studies reported important and widespread global contributions from sCIs derived from both isoprene and monoterpenes, although they report substantially different sCI concentrations. In the work of Vereecken et al. (2017), the rapid unimolecular decomposition rates calculated for many of the of sCIs (as also adopted in the present work) strongly suppress the simulated concentrations compared with those reported by Khan et al. (2018). The resultant annual average sCI concentration at the surface maximizes at 7 ×103 molec. cm−3 over the Amazon basin but is generally less than 2 ×103 molec. cm−3 over most of the globe (Vereecken et al., 2017). This further emphasizes the need for direct kinetics studies of a structurally diverse series of isoprene and terpene-derived sCIs to help validate and refine the rate coefficients calculated in theoretical studies.
8.4 Oxidation of SO2 and organic acids
The speciated sCI distributions have also been used to calculate the associated SO2 oxidation rates. As shown in Table 8, the total oxidation rates are calculated to be between 0.006 % h−1 and 0.028 % h−1. They broadly follow the simulated trend in total sCI concentrations but also reflect that the rate coefficient values for individual species span almost an order of magnitude. These oxidation rates can be compared with a reference SO2 oxidation rate of about 0.3 % h−1 for reaction with HO radicals at a concentration of 106 molec. cm−3. This comparison is therefore consistent with the < 10 % annual average contribution to gas-phase SO2 oxidation for the UK, reported in the global modelling calculations of Vereecken et al. (2017) and Khan et al. (2018). The more widespread potential role of biogenic hydrocarbon derived sCIs in global SO2 oxidation has also been considered in those modelling studies. Although the results possess some similarities, in terms of the relative regional variation, their role is much more limited in the Vereecken et al. (2017) calculations because of the high calculated decomposition rates applied to many of the sCIs and the resultant suppression of sCI concentrations commented on above. Nevertheless, annual average contributions of up to about 70 % were still simulated for the terrestrial equatorial belt.
The largest bimolecular rate coefficients for sCI reactions that have been measured experimentally are those for reactions with organic acids such as HC(O)OH, CH3C(O)OH and CF3C(O)OH (see Table 5). HC(O)OH and CH3C(O)OH are present in the troposphere in significant concentrations (e.g. Andreae et al., 1988; Millet et al., 2015; Bannan et al., 2017) due in part to their formation in the photochemical oxidation or ozonolysis of many VOCs, from both anthropogenic and natural sources. As discussed above and shown more widely in the calculations of Vereecken et al. (2017), the reactions with organic acids can make small but significant contributions to sCI removal in some regions. The total sCI concentrations calculated here suggest associated oxidation rates of (0.04–0.11) % h−1 for HC(O)OH and similar rates for CH3C(O)OH. This can be compared with reference oxidation rates of about 0.16 % h−1 and 0.25 % h−1 for the reaction of HC(O)OH and CH3C(O)OH with HO radicals at a concentration of 106 molec. cm−3. This indicates that reaction with sCIs makes an important contribution to the oxidation of these acids under the conditions considered here, with oxidation rates comparable to those via HO reaction calculated for equatorial regions in the global modelling study of Vereecken et al. (2017). It is noted that the reactions of larger sCIs and organic acids (e.g. derived from the ozonolysis of monoterpenes and sesquiterpenes) potentially form highly oxidized, low-volatility products (hydroperoxyl-esters), which may play a role in secondary organic aerosol (SOA) formation (e.g. Tobias and Ziemann, 2001; Chhantyal-Pun et al., 2018).
As shown in Table 5, the reactions of sCIs with CF3C(O)OH are particularly rapid. The total sCI concentrations calculated here suggest an associated oxidation rate of (0.1–0.3) % h−1 for CF3C(O)OH, compared with 0.25 % h−1 for its reaction with HO radicals at a concentration of 106 molec. cm−3. This demonstrates the potential importance of sCIs as gas-phase oxidants for CF3C(O)OH (and other perfluoro-carboxylic acids, CnF2n+1C(O)OH) over land masses. However, it is noted that the reaction of the resultant hydroperoxyl-fluoroester products with HO radicals probably reforms the perfluoro-carboxylic acids on a timescale of 1–2 d (Taatjes et al., 2019).
Current understanding of the atmospheric chemistry of sCIs therefore supports the original hypothesis of Cox and Penkett (1971, 1972) regarding their potential importance as atmospheric oxidants, identified from chamber measurements of SO2 oxidation associated with alkene ozonolysis, and the observed effect of relative humidity on the oxidation rates. Considerable progress has since been made in the understanding of the kinetics and mechanisms of alkene ozonolysis, resulting from both experimental and theoretical studies, with particular advances since the pioneering work of Taatjes and co-workers less than a decade ago (e.g. Welz et al., 2012; Taatjes et al., 2013) in the detection of sCIs and direct measurements of the kinetics of their reactions. However, significant uncertainties remain in some aspects of mechanistic understanding, including measurements of the yields of sCIs and their speciation in asymmetric alkene systems. The current evaluation has focused primarily on those sCIs for which direct kinetics measurements are available (i.e. CH2OO, E-CH3CHOO, Z-CH3CHOO and (CH3)2COO), with some consideration also given to the C4 intermediates formed from isoprene. Whilst these represent an important subset of atmospheric sCIs, it is recognized that an enormous variety of sCIs are generated, with particularly important global contributions from those generated from the ozonolysis of monoterpenes and sesquiterpenes. The concurrent progress in the theoretical treatment of the structure and reaction kinetics of sCIs in the gas phase has allowed the development of theory-based structure–activity relationships (SARs) (Vereecken et al., 2017), which provide a basis for representing the reactions of structurally complex sCIs in atmospheric mechanisms. As a result, there is a need for direct kinetics studies of a structurally diverse series of isoprene and terpene-derived sCIs to help validate and refine the rate coefficients calculated in theoretical studies. In addition, the continued development of sensitive detection methods for sCIs (e.g. Berndt et al., 2017) that may eventually allow sCI concentrations to be measured in the field would be valuable for evaluation and testing of the representation of sCI sources and sinks in atmospheric models.
All relevant data and supporting information have been provided in the Supplement. The detailed reaction data sheets are also available at the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation website: http://iupac.pole-ether.fr/ (IUPAC, 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-13497-2020-supplement.
All authors defined the scope of the work. RAC, MEJ and TJW developed and drafted the data sheets and manuscript. All authors reviewed, refined, and revised the data sheets, and all sections of the manuscript and Supplement.
The authors declare that they have no conflict of interest.
We thank Cathy Boonne (CNRS-IPSL) for developing and maintaining the Task Group website.
This research has been supported by the International Union of Pure and Applied Chemistry, IUPAC (project nos. 2015-038-1-100 and 2017-024-1-100); the Swiss National Science Foundation (grant no. 169176); the Department of Chemistry, University of Cambridge; the Centre National de la Recherche Scientifique-Institut National des Sciences de l'Univers (CNRS-INSU); and the Orléans University and Observatoire des Sciences de l'Univers en région Centre (OSUC).
The article processing charges for this open-access
publication were covered by the Max Planck Society.
This paper was edited by Sergey A. Nizkorodov and reviewed by Craig Taatjes and one anonymous referee.
Alam, M. S., Camredon, M., Rickard, A. R., Carr, T., Wyche, K. P., Hornsby, K. E., Monks, P. S., and Bloss, W. J.: Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 13, 11002–11015, 2011.
Alam, M. S., Rickard, A. R., Camredon, M., Wyche, K. P., Carr, T., Hornsby, K. E., Monks, P. S., and Bloss, W. J.: Radical product yields from the ozonolysis of short chain alkenes under atmospheric boundary layer conditions, J. Phys. Chem., 117, 12468–12483, 2013.
Andreae, M. O., Talbot, R. W., Andreae, T. W., and Harriss, R. C.: Formic and acetic-acid over the central Amazon region, Brazil .1. Dry season, J. Geophys. Res.-Atmos., 93, 1616–1624, 1988.
Anglada, J. M. and Solé, A.: Impact of water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 18, 17698–17712, 2016.
Anglada, J. M., Aplincourt, P., Bofill, J. M., and Cremer, D.: Atmospheric formation of OH radicals and H2O2 from alkene ozonolysis under humid conditions, Chem. Phys. Chem., 3, 215–221, 2002.
Anglada, J. M., González, J., and Torrent-Sucarrat, M.: Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 13, 13034–13045, 2011.
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with methacrolein and methyl vinyl ketone, Atmos. Environ., 30, 2939–2943, 1996.
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with a series of terpenes, Atmos. Environ., 36, 4347–4355, 2002.
Aschmann, S. M., Tuazon, E. C., Arey, J., and Atkinson, R.: Products of the gas-phase reaction of O3 with cyclohexene, J. Phys. Chem. A, 107, 2247–2255, 2003.
Atkinson, R. and Aschmann, S. M.: OH radical production from the gas-phase reactions of O3 with a series of alkenes under atmospheric conditions, Environ. Sci. Technol., 27, 1357–1363, 1993.
Atkinson, R., Aschmann, S. M., Arey, J., and Shorees, B.: Formation of OH radicals in the gas phase reactions of O3 with a series of terpenes, J. Geophys. Res., 97, 6065–6073, 1992.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bannan, T. J., Booth, A. M., Le Breton, M., Bacak, A., Muller, J. B. A., Leather, K. E., Khan, M. A. H., Lee, J. D., Dunmore, R. E., Hopkins, J. R., Fleming, Z. L., Sheps, L., Taatjes, C. A., Shallcross, D. E., and Percival, C. J.: Seasonality of formic acid (HCOOH) in London during the ClearfLo campaign, J. Geophys. Res.-Atmos., 122, 12488–12498, 2017.
Barber, V. P., Pandit, S., Green, A. M., Trongsiriwat, N., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Four-carbon Criegee intermediate from isoprene ozonolysis: Methyl vinyl ketone oxide synthesis, infrared spectrum, and OH production, J. Am. Chem. Soc., 140, 10866–10880, https://doi.org/10.1021/jacs.8b06010, 2018.
Beames, J. M., Liu, F., Lu. L., and Lester, M. I.: Ultraviolet spectrum and photochemistry of the simplest Criegee intermediate CH2OO, J. Am. Chem. Soc., 134, 20045–20048, https://doi.org/10.1021/ja310603j, 2012.
Beck, M., Winterhalter, R., Herrmann, F., and Moortgat, G. K.: The gas-phase ozonolysis of α-humulene, Phys. Chem. Chem. Phys., 13, 10970–11001, 2011.
Berndt, T., Böge, O., and Stratmann, F.: Gas-phase ozonolysis of alpha-pinene: gaseous products and particle formation, Atmos. Environ., 37, 3933–3945, 2003.
Berndt, T., Jokinen, T., Sipilä. M., Mauldin, R. L., Herrmann, H., Stratmann, F., Junninen, H., and Kulmala, M.: H2SO4 formation from the gas-phase reaction of stabilized Criegee intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 89, 603–612, 2014.
Berndt, T., Herrmann, H., and Kurteìn, T.: Direct probing of Criegee intermediates from gas-phase ozonolysis using chemical ionization mass spectrometry, J. Am. Chem. Soc., 139, 13387–13392, 2017.
Calvert, J. G., Mellouki, A., Orlando, J. J., Pilling, M. J., and Wallington, T. J.: The mechanisms of atmospheric oxidation of the alkenes, Oxford University Press, Oxford, UK, ISBN 978-0-19513177-2, 2000.
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The mechanisms of reactions influencing atmospheric ozone, Oxford University Press, Oxford, UK, ISBN 978-0-19023302-0, 2015.
Campos-Pineda, M. and Zhang, J.: Low-pressure yields of stabilized Criegee intermediates CH3CHOO and (CH3)2COO in ozonolysis of trans-2-butene and 2,3-dimethyl-2-butene, Chem. Phys. Lett., 683, 647–652, 2017.
Campos-Pineda, M. and Zhang, J.: Product yields of stabilized Criegee intermediates in the ozonolysis reactions of cis-2-butene, 2-methyl-2-butene, cyclopentene, and cyclohexene, Sci. China Chem., 61, 850–856, https://doi.org/10.1007/s11426-017-9229-0, 2018.
Caravan, R. L., Vansco, M. F., Au, K., Khan, M. A. H., Li, Y.-L. Winiberg, F. A. F., Zuraski, K., Lin, Y.-H., Chao, W., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Lin, J.-J. M., Shallcross, D. E., Sheps, L., Klippenstein, S. J., Taatjes, C. A., and Lester, M. I.: Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate, P. Nat. Acad. Sci. USA, 117, 9733–9740, https://doi.org/10.1073/pnas.1916711117, 2020.
Chhantyal-Pun, R., McGillen, M. R., Beames, J. M., Khan, M. A. H., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Temperature-dependence of the rates of reaction of trifluoroacetic acid with Criegee intermediates Angew. Chem. Int. Ed., 56, 9044–9047, 2017a.
Chhantyal-Pun, R., Welz, O., Savee J. D., Eskola, A. J., Lee, E. P. F., Blacker, L., Hill, H. R., Ashcroft, M., Khan, M. A. H., Lloyd-Jones, G. C., Evans, L., Rotavera, B., Rotavera H., Osborn, D. L., Mok, D. K. W., Dyke, J. M., Shallcross, D. E., Percival, C. J., Orr-Ewing, A. J., and Taatjes, C. A.: Direct measurements of unimolecular and bimolecular reaction kinetics of the Criegee intermediate (CH3)2COO, J. Phys. Chem. A, 121, 4–15, https://doi.org/10.1021/acs.jpca.6b07810, 2017b.
Chhantyal-Pun, R., Rotavera, B., McGillen, M. R., Khan, M. A. H., Eskola, A. J., Caravan, R. L., Blacker, L., Tew, D. P., Osborn, D. L., Percival, C. J., Shallcross, D. E., and Orr-Ewing A. J.: Criegee intermediate reactions with carboxylic acids: a potential source of secondary organic aerosol in the atmosphere, ACS Earth Space Chem., 2, 833–842, 2018.
Chew, A. A. and Atkinson, R.: OH radical formation yields from the gas-phase reactions of O3 with alkenes and monoterpenes, J. Geophys. Res., 101, 28649–28653, 1996.
Chuong, B., Zhang, J., and Donahue, N. M.: Cycloalkene ozonolysis: collisionally mediated mechanistic branching, J. Am. Chem. Soc., 126, 12363–12373, 2004.
Cox, R. A. and Penkett, S. A.: Oxidation of atmospheric SO2 by products of the ozone–olefin reaction, Nature, 230, 321–322, 1971.
Cox, R. A. and Penkett, S. A.: Aerosol formation from sulphur dioxide in the presence of ozone and olefinic hydrocarbons, J. Chem. Soc., Faraday Trans. 1, 1735–1753, 1972.
Criegee, R., Blust, G., and Zinke, H.: Eine neuartige synthese von ozoniden, Chem. Ber., 87,766–768, 1954.
Donahue, N. M., Kroll, J. H., Anderson, J. G., and Demerjian, K. L.: Direct observation of OH production from the ozonolysis of olefins, Geophys. Res. Lett., 25, 59–62, https://doi.org/10.1029/97GL53560, 1998.
Drozd, G. T. and Donahue, N. M.: Pressure dependence of stabilized Criegee intermediate formation from a sequence of alkenes, J. Phys. Chem. A, 115, 4381–4387, 2011.
Drozd, G. T., Kroll, J., and Donahue, N. M.: 2,3-Dimethyl-2-butene (TME) ozonolysis: pressure dependence of stabilized Criegee intermediates and evidence of stabilized vinyl hydroperoxides, J. Phys. Chem. A, 115, 161–166, 2011.
Fang, Y., Liu, F., Barber, V. P., Klippenstein, S. J., McCoy, A. B., and Lester, M. I.: Deep tunneling in the unimolecular decay of CH3CHOO Criegee intermediates to OH radical products, J. Chem. Phys. 145, 234308, https://doi.org/10.1063/1.4972015, 2016.
Fang, Y., Liu, F., Barber, V. P., Klippenstein, S. J., McCoy, A. B., and Lester, M. I.: Tunneling effects in the unimolecular decay of (CH3)2COO Criegee intermediates to OH radical products, J. Chem. Phys. 146, 134307, https://doi.org/10.1063/1.4979297, 2017.
Fenske, J. D., Hasson, A. S., Paulson, S. E., Kuwata, K. T., Ho, A., and Houk, K. N.: The pressure dependence of the OH radical yield from ozone-alkene reactions, J. Phys. Chem. A, 104, 7821–7833, 2000.
Finlayson, B. J., Pitts, J. N., and Akimoto, H.: Production of vibrationally excited OH in chemiluminescent ozone-olefin reactions, Chem. Phys. Lett., 12, 495–498, 1972.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the upper and lower atmosphere: theory, experiments, and applications, Academic Press, https://doi.org/10.1016/B978-012257060-5/50010-1, 2000.
Forester, C. D. and Wells, J. R.: Hydroxyl radical yields from reactions of terpene mixtures with ozone, Indoor Air, 21, 400–409, 2011.
Griesbaum, K., Miclaus, V., and Jung, I. C.: Isolation of ozonides from gas-phase ozonolyses of terpenes, Environ. Sci. Technol., 32, 647–649, 1998.
Hakala, J. P. and Donahue, N. M.: Pressure-dependent Criegee intermediate stabilization from alkene ozonolysis, J. Phys. Chem. A, 120, 2173–2178, 2016.
Hakala, J. P. and Donahue, N. M.: Pressure stabilization of Criegee intermediates formed from symmetric trans-alkene ozonolysis, J. Phys. Chem. A, 122, 9426–9434, 2018.
Hakola, H., Arey, J., Aschmann, S. M., and Atkinson, R.: Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes, J. Atmos. Chem., 18, 75–102, 1994.
Hasson, A. S., Orzechowska, G., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 1. Ethene, trans-2-butene, and 2,3-dimethyl-2-butene, J. Geophys. Res.-Atmos., 106, 34131–34142, 2001a.
Hasson, A. S., Ho, A. W., Kuwata, K. T., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 2. Asymmetric and biogenic alkenes, J. Geophys. Res.-Atmos., 106, 34143–34153, 2001b.
Hasson, A. S., Chung, M. Y., Kuwata, K. T., Converse, A. D., Krohn, D., and Paulson, S. E.: Reaction of Criegee intermediates with water vapor – An additional source of OH radicals in alkene ozonolysis?, J. Phys. Chem. A, 107, 6176–6182, 2003.
Hatakeyama, S., Kobayashi, H., and Akimoto, H.: Gas-phase oxidation of SO2 in the ozone-olefin reactions, J. Phys. Chem., 88, 4736–4739, 1984.
Hatakeyama, S., Kobayashi, H., Lin, Z.-Y., Takagi, H., and Akimoto, H.: Mechanism for the reaction of CH2OO with SO2, J. Phys. Chem., 90, 4131–4135, 1986.
Herrmann, F., Winterhalter, R., Moortgat, G. K., and Williams, J.: Hydroxyl radical (OH) yields from the ozonolysis of both double bonds for five monoterpenes, Atmos. Environ., 44, 3458–3464, 2010.
Horie, O. and Moortgat, G. K.: Decomposition pathways of the excited Criegee intermediates in the ozonolysis of simple alkenes, Atmos. Environ., 25A, 1881–1896, 1991.
Horie, O., Schafer, C., and Moortgat, G. K.: High reactivity of hexafluoro acetone toward Criegee intermediates in the gas-phase ozonolysis of simple alkenes, Int. J. Chem. Kinet., 31, 261–269, 1999.
Huang, H.-L., Chao, W., and Lin, J. J.-M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
IUPAC, Task Group on Atmospheric Chemical Kinetic Data Evaluation: Evaluated kinetic data, available at: http://iupac.pole-ether.fr/, last access: 22 September 2020.
Jenkin, M. E., Wyche, K. P., Evans, C. J., Carr, T., Monks, P. S., Alfarra, M. R., Barley, M. H., McFiggans, G. B., Young, J. C., and Rickard, A. R.: Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene, Atmos. Chem. Phys., 12, 5275–5308, https://doi.org/10.5194/acp-12-5275-2012, 2012.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, 2008.
Kan, C. S., Su, F., Calvert, J. G., and Shaw, J. H.: Mechanism of the ozone-ethene reaction in dilute N2 ∕ O2 mixtures near 1-atm Pressure, J. Phys. Chem., 85, 2359–2363, 1981.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci.-Proc. Imp., 20, 437–453, 2018.
Kroll, J. H., Hanisco, T. F, Donahue, N. M., Demerjian, K. L., and Anderson, J. G.: Accurate, direct measurements of OH yields from gas-phase ozone-alkene reactions using an in situ LIF Instrument, Geophys. Res. Lett., 28, 3863–3866, https://doi.org/10.1029/2001GL013406, 2001a.
Kroll, J. H., Clarke, J. S., Donahue, N. M., Anderson, J. G., and Demerjian, K. L.: Mechanism of HOx formation in the gas-phase ozone-alkene reaction. 1. Direct, pressure-dependent measurements of prompt OH yields, J. Phys. Chem. A, 105, 1554–1560, 2001b.
Kroll, J. H., Sahay, S. R., Anderson, J. G., Demerjian, K. L., and Donahue, N. M.: Mechanism of HOx formation in the gas-phase ozone-alkene reaction. 2. Prompt versus thermal dissociation of carbonyl oxides to form OH, J. Phys. Chem. A, 105, 4446–4457, 2001c.
Kuwata, K. T., Hermes, M. R., Carlson, M. J., and Zogg, C. K.: Computational studies of the isomerization and hydration reactions of acetaldehyde oxide and methyl vinyl carbonyl oxide, J. Phys. Chem. A, 114, 9192–9204, 2010.
Li, H., Fang, Y., Beames, J. M., and Lester, M. I.: Velocity map imaging of O-atom products from UV photodissociation of the CH2OO Criegee intermediate, J. Chem. Phys., 142, 214312, https://doi.org/10.1063/1.4921990, 2015.
Liu, F., Beames, J. M., Green, A. M., and Lester, M. I.: UV spectroscopic characterization of dimethyl- and ethyl-substituted carbonyl oxides, J. Phys. Chem. A, 118, 2298–2306, 2014.
Long, B., Bao, J. L., and Truhlar, D.G.: Atmospheric chemistry of Criegee intermediates: Unimolecular reactions and reactions with water, J. Am. Chem. Soc., 138, 14409–14422, https://doi.org/10.1021/jacs.6b08655, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Rapid unimolecular reaction of stabilized Criegee intermediates and implications for atmospheric chemistry, Nat. Commun., 10, 2003, https://doi.org/10.1038/s41467-019-09948-7, 2019.
Mackenzie-Rae, F. A., Karton, A., and Saunders, S. M.: Computational investigation into the gas-phase ozonolysis of the conjugated monoterpene alpha-phellandrene, Phys. Chem. Chem. Phys., 18, 27991–28002, 2016.
Malkin, T. L., Goddard, A., Heard, D. E., and Seakins, P. W.: Measurements of OH and HO2 yields from the gas phase ozonolysis of isoprene, Atmos. Chem. Phys., 10, 1441–1459, https://doi.org/10.5194/acp-10-1441-2010, 2010.
McGill, C. D., Rickard, A. R., Johnson, D., and Marston, G.: Product yields in the reactions of ozone with Z-but-2-ene, E-but-2-ene and 2-methylbut-2-ene, Chemosphere, 38, 1205–1212, 1999.
Mihelcic, D., Heitlinger, M., Kley, D., Musgen P., and Volz-Thomas, A.: Formation of hydroxyl and hydroperoxy radicals in the gas-phase ozonolysis of ethene, Chem. Phys. Lett., 301, 559–564, 1999.
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015.
Neeb, P. and Moortgat, G. K.: Formation of OH radicals in the gas-phase reaction of propene, isobutene, and isoprene with O3: Yields and mechanistic implications, J. Phys. Chem. A, 103, 9003–9012, 1999.
Neeb, P., Horie, O., and Moortgat, G. K.: Gas-phase ozonolysis of ethene in the presence of hydroxylic compounds, Int. J. Chem. Kinet., 28, 721–730, 1996.
Neeb, P., Horie, O., and Moortgat, G. K.: The ethene-ozone reaction in the gas phase, J. Phys. Chem. A, 102, 6778–6785, 1998.
Newland, M. J., Rickard, A. R., Alam, M. S., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions, Phys. Chem. Chem. Phys., 17, 4076–4088, 2015a.
Newland, M. J., Rickard, A. R., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide, Atmos. Chem. Phys., 15, 9521–9536, https://doi.org/10.5194/acp-15-9521-2015, 2015b.
Newland, M. J., Rickard, A. R., Sherwen, T., Evans, M. J., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling, Atmos. Chem. Phys., 18, 6095–6120, https://doi.org/10.5194/acp-18-6095-2018, 2018.
Newland, M. J., Nelson, B. S., Muñoz, A., Ródenas, M., Vera, T., Tárrega, J., and Rickard, A. R.: Trends in stabilisation of Criegee intermediates from alkene ozonolysis, Phys. Chem. Chem. Phys., 22, 13698–13706, https://doi.org/10.1039/D0CP00897D, 2020.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J, H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, 2016.
Nguyen, T. L., Peeters, J., and Vereecken, L.: Theoretical study of the gas-phase ozonolysis of β-pinene (C10H16), Phys. Chem. Chem. Phys., 11, 5643–5656, 2009a.
Nguyen, T. L., Winterhalter, R., Moortgat, G., Kanawati, B., Peeters, J., and Vereecken, L.: The gas-phase ozonolysis of β-caryophyllene (C15H24). Part II: a theoretical study, Phys. Chem. Chem. Phys., 11, 4173–4183, 2009b.
Niki, H., Maker, P. D., Savage, C. M., Breitenbach, L. P., and Hurley, M. D.: FTIR spectroscopic study of the mechanism for the gas-phase reaction between ozone and tetramethylethylene, J. Phys. Chem., 91, 941–946, 1987.
Olzmann, M., Kraka, E., Cremer, D., Gutbrod, R., and Andersson, S.: Energetics, kinetics, and product distributions of the reactions of ozone with ethene and 2,3-dimethyl-2-butene, J. Phys. Chem. A, 101, 9421–9429, 1997.
Orzechowska, G. and Paulson, S. E.: Production of OH radicals from the reactions of C4-C6 internal alkenes and styrenes with ozone in the gas phase, Atmos. Environ., 36, 571–581, 2002.
Osborn, D. L. and Taatjes, C. A.: The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 34, 309–360, 2015.
Paulson, S. E., Chung, M., Sen, A. D., and Orzechowska, G.: Measurement of OH radical formation from the reaction of ozone with several biogenic alkenes, J. Geophys. Res.-Atmos., 103, 25533–25539, 1998.
Paulson, S. E., Fenske, J. D., Sen, A. D., and Callahan, T. W.: A novel small-ratio relative-rate technique for measuring OH formation yields from the reactions of O3 with alkenes in the gas phase, and its application to the reactions of ethene and propene, J. Phys. Chem. A, 103, 2050–2059, 1999.
Peltola, J., Seal, P., Inkilä, A., and Eskola, A.: Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 22, 11797–11808, https://doi.org/10.1039/D0CP00302F, 2020.
Presto, A. A. and Donahue, N. M.: Ozonolysis fragment quenching by nitrate formation: The pressure dependence of prompt OH radical formation, J. Phys. Chem. A, 108, 9096–9104, 2004.
Qi, B., Yang, B., Wang, Z. Q., Yang, H. Y., and Liu, L.: Production of radicals in the ozonolysis of propene in air, Sci. China, Ser. B, 52, 356–361, 2009.
Ren, Y., Grosselin, B., Daële, V., and Mellouki, A.: Investigation of the reaction of ozone with isoprene, methacrolein and methyl vinyl ketone using the HELIOS chamber, Faraday Discuss., 200, 289–311, 2017.
Rickard, A. R., Johnson, D., McGill, C. D., and Marston, G.: OH yields in the gas-phase reactions of ozone with alkenes, J. Phys. Chem. A, 103, 7656–7664, 1999.
Ryzhkov, A. B. and Ariya, P. A.: A theoretical study of the reactions of parent and substituted Criegee intermediates with water and the water dimer, Phys. Chem. Chem. Phys., 6, 5042–5050, 2004.
Samanta, K., Beames, J. M., Lester M. I., and Subotnik, J. E.: Quantum dynamical investigation of the simplest Criegee intermediate CH2OO and its O–O photodissociation channels, J. Chem. Phys. 141, 134303, https://doi.org/10.1063/1.4894746, 2014.
Sheps, L.: Absolute ultraviolet absorption spectrum of a Criegee intermediate CH2OO, J. Phys. Chem. Lett., 4, 4201–4205, 2013.
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO Phys. Chem. Chem. Phys., 16, 26701–26706, 2014.
Shu, Y. and Atkinson, R.: Rate constants for the gas-phase reactions of O3 with a series of terpenes and OH radical formation from the O3 reactions with sesquiterpenes at 296 ± 2 K, Int. J. Chem. Kinet., 26, 1193–1205, 1994.
Siese, M., Becker, K. H., Brockmann, K. J., Geiger, H., Hofzumahaus, A., Holland, F., Mihelcic, D., and Wirtz, K.: Direct measurement of OH radicals from ozonolysis of selected alkenes: A EUPHORE simulation chamber study, Environ. Sci. Technol., 35, 4660–4667, 2001.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
Stephenson, T. A. and Lester, M. I.: Unimolecular decay dynamics of Criegee intermediates: Energy-resolved rates, thermal rates, and their atmospheric impact, Int. Rev. Phys. Chem., 39, 1–33, https://doi.org/10.1080/0144235X.2020.1688530, 2020.
Su, F., Calvert, J. G., and Shaw, J. H.: A FT-IR Spectroscopic study of the ozone-ethene reaction mechanism in O2-rich mixtures, J. Phys. Chem., 84, 239–246, 1980.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Osborn, D. L., Lee, E. P. F., Dyke, J. M., Mok, D. W. K., Shallcross, D. E., and Percival, C. J.: Direct measurement of Criegee intermediate (CH2OO) reactions with acetone, acetaldehyde, and hexafluoroacetone, Phys. Chem. Chem. Phys., 14, 10391–10400, 2012.
Taatjes, C. A., Welz, C. A.; Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO, Science, 340, 177–181, 2013.
Taatjes, C. A., Shallcross, D. E., and Percival, C. J.: Research frontiers in the chemistry of Criegee intermediates and tropospheric ozonolysis, Phys. Chem. Chem. Phys., 16, 1704–1718, https://doi.org/10.1039/c3cp52842a, 2014.
Taatjes, C. A., Khan, M. A. H., Eskola, A. J., Percival, C. J., Osborn, D. L., Wallington, T. J., and Shallcross, D. E.: Reaction with Criegee intermediates: An important atmospheric fate of perfluorocarboxylic acids, Environ. Sci. Technol., 53, 1245–1251, 2019.
Ting, W.-L, Chen, Y.-H., Chao, W., Smith, M. C., and Lin Jr., J.-M.: The UV absorption spectrum of the simplest Criegee intermediate CH2OO, Phys. Chem. Chem. Phys., 16, 10438–10443, 2014.
Tobias, H. J. and Ziemann, P. J.: Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids, and water with the C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene, J. Phys. Chem. A, 105, 6129–6135, 2001.
Vansco, M. F., Li, H., and Lester, M. I.: Prompt release of O1D products upon UV excitation of CH2OO Criegee intermediates, J. Chem. Phys. 147, 013907, https://doi.org/10.1063/1.4977987, 2017.
Vansco, M. F., Marchetti, B., and Lester, M. I.: Electronic spectroscopy of methyl vinyl ketone oxide: A four-carbon unsaturated Criegee intermediate from isoprene ozonolysis, J. Chem. Phys., 149, 244309, https://doi.org/10.1063/1.5064716, 2018.
Vansco, M. F., Marchetti, B. Trongsiriwat, N., Bhagde, T., Wang, G., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Synthesis, electronic spectroscopy, and photochemistry of methacrolein oxide: A four-carbon unsaturated Criegee intermediate from isoprene ozonolysis, J. Am. Chem. Soc., 141, 15058–15069, https://doi.org/10.1021/jacs.9b05193, 2019.
Vereecken, L. and Nguyen, H. M. T.: Theoretical study on the reaction of carbonyl oxide with nitrogen dioxide: CH2OO + NO2, Int. J. Chem. Kinet., 49, 752–760, 2017.
Vereecken, L., Harder, H., and Novelli, A.: The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 14, 14682–14695, https://doi.org/10.1039/C2CP42300F, 2012.
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, https://doi.org/10.1039/c7cp05541b, 2017.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, 2012.
Winterhalter, R., Neeb, P., Grossmann, D., Kolloff, A., Horie, O., and Moortgat, G. K.: Products and mechanism of the gas phase-reaction of ozone with β-pinene, J. Atmos. Chem., 35, 165–197, 2000.
Winterhalter, R., Herrmann, F., Kanawati, B., Nguyen, T. L., Peeters, J., Vereecken, L., and Moortgat, G. K.: The gas-phase ozonolysis of β-caryophyllene (C15H24). Part I: an experimental study, Phys. Chem. Chem. Phys., 11, 4152–4172, 2009.
Yao, L., Ma, Y., Wang, L., Zheng, J., Khalizov, A., Chen, M., Zhou, Y., Qi, L., and Cui, F.: Role of stabilized Criegee intermediate in secondary organic aerosol formation from the ozonolysis of α-cedrene, Atmos. Environ., 94, 448–457, 2014.
Zhang, D., Lei, W., and Zhang, R.: Mechanism of OH formation from ozonolysis of isoprene: kinetics and product yields, Chem. Phys. Lett., 358, 171–179, 2002.
- Abstract
- Introduction
- Rate coefficients of O3 + alkene reactions
- sCI yields from O3 + alkene reactions
- HO radical yields from O3 + alkene reactions
- Structure and spectroscopy of Criegee intermediates
- Rate coefficients for thermal reactions of sCIs
- Overall reactivity conclusions – comparison of experiment and theory
- Impact of Criegee intermediates in atmospheric oxidation chemistry
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Rate coefficients of O3 + alkene reactions
- sCI yields from O3 + alkene reactions
- HO radical yields from O3 + alkene reactions
- Structure and spectroscopy of Criegee intermediates
- Rate coefficients for thermal reactions of sCIs
- Overall reactivity conclusions – comparison of experiment and theory
- Impact of Criegee intermediates in atmospheric oxidation chemistry
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement