the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Jul 2019
| 31 Jul 2019
A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol
Kelvin H. Bates
Daniel J. Jacob
Atmospheric oxidation of isoprene, the most abundantly emitted non-methane hydrocarbon, affects the abundances of ozone (O3), the hydroxyl radical (OH), nitrogen oxide radicals (NOx), carbon monoxide (CO), oxygenated and nitrated organic compounds, and secondary organic aerosol (SOA). We analyze these effects in box models and in the global GEOS-Chem chemical transport model using the new reduced Caltech isoprene mechanism (RCIM) condensed from a recently developed explicit isoprene oxidation mechanism. We find many similarities with previous global models of isoprene chemistry along with a number of important differences. Proper accounting of the isomer distribution of peroxy radicals following the addition of OH and O2 to isoprene influences the subsequent distribution of products, decreasing in particular the yield of methacrolein and increasing the capacity of intramolecular hydrogen shifts to promptly regenerate OH. Hydrogen shift reactions throughout the mechanism lead to increased OH recycling, resulting in less depletion of OH under low-NO conditions than in previous mechanisms. Higher organonitrate yields and faster tertiary nitrate hydrolysis lead to more efficient NOx removal by isoprene and conversion to inorganic nitrate. Only 20 % of isoprene-derived organonitrates (excluding peroxyacyl nitrates) are chemically recycled to NOx. The global yield of formaldehyde from isoprene is 22 % per carbon and less sensitive to NO than in previous mechanisms. The global molar yield of glyoxal is 2 %, much lower than in previous mechanisms because of deposition and aerosol uptake of glyoxal precursors. Global production of isoprene SOA is about one-third from each of the following: isoprene epoxydiols (IEPOX), organonitrates, and tetrafunctional compounds. We find a SOA yield from isoprene of 13 % per carbon, much higher than commonly assumed in models and likely offset by SOA chemical loss. We use the results of our simulations to further condense RCIM into a mini Caltech isoprene mechanism (Mini-CIM) for less expensive implementation in atmospheric models, with a total size (108 species, 345 reactions) comparable to currently used mechanisms.
- Article
(10344 KB) - Full-text XML
-
Supplement
(44983 KB) - BibTeX
- EndNote





Isoprene (2-methyl-1,3-butadiene), the dominant hydrocarbon emitted to the atmosphere by plants, plays a central role in tropospheric chemistry. Its global emission is estimated to be ∼500 Tg a−1, comparable to that of methane (Guenther et al., 2012). Its atmospheric lifetime is only ∼1 h against oxidation by the hydroxyl radical (OH), the main tropospheric oxidant (τOH=1.1 h for [OH] molecules cm−3 at T=298 K). The high reactivity of isoprene and the subsequent cascade of oxidation products have important implications for tropospheric ozone (Squire et al., 2015), the hydroxyl radical (Lelieveld et al., 2008), the nitrogen cycle (Paulot et al., 2013), and secondary organic aerosol (SOA) (Carlton et al., 2009). The persistence of long-lived oxidation products extends isoprene's influence to regional and global scales (Kanakidou et al., 2005; Paulot et al., 2012).
Proper representation of isoprene chemistry is of critical importance for global models of atmospheric chemistry, but the mechanism is complicated and models often use outdated information. Wennberg et al. (2018) presented a detailed review of current knowledge and compiled a comprehensive mechanism. This mechanism is far too complex for implementation in atmospheric models, but Wennberg et al. (2018) also compiled a reduced version suitable for the range of conditions found in the atmosphere. We examine here its implications for the range of effects of isoprene on atmospheric chemistry.
The isoprene oxidation cascade varies considerably depending on local atmospheric conditions. Different branches in the chemical mechanism develop depending on the reactions of the peroxy radicals (RO2) produced in the initial and subsequent oxidation steps. Reaction with NO produces ozone and organic nitrates, and reactions of acylperoxy radicals with NO2 produce peroxyacyl nitrates (PANs). Reactions with HO2 are typically OH-consuming via hydroperoxide formation. Intramolecular hydrogen-shift reactions (H-shift reactions) tend to propagate radical chains and regenerate OH. These different branches of RO2 chemistry also produce a large and differing ensemble of oxygenated multifunctional compounds, some of which have low volatility and/or aqueous-phase chemistry leading to SOA formation.
Isoprene oxidation mechanisms in atmospheric models have evolved considerably over the past decades. Early mechanisms focused on high-NO conditions representative of polluted regions and the role of isoprene in driving the production of ozone and organic nitrates (Lloyd et al., 1983; Brewer et al., 1984; Trainer et al., 1987; Madronich and Calvert, 1990). These model studies led to a number of chamber experiments to test and improve the mechanisms (Atkinson et al., 1989; Tuazon and Atkinson, 1990; Paulson et al., 1992; Paulson and Seinfeld, 1992; Grosjean et al., 1993; Miyoshi et al., 1994; Kwok and Atkinson, 1995; Stevens et al., 1999; Ruppert and Becker, 2000) along with field observations of isoprene chemistry (Biesenthal and Shepson, 1997; Starn et al., 1998; Roberts et al., 1998; Wiedinmyer et al., 2001). Improved understanding of the chemistry under low-NO conditions and growing interest in formation of organic aerosol led to the development of increasingly complex mechanisms (Carter, 1996; Stockwell et al., 1997; Pöschl et al., 2000; Geiger et al., 2003; Aumont et al., 2005). The regularly updated Master Chemical Mechanism (MCM) presents a nearly explicit compilation of isoprene chemistry (Jenkin et al., 1997, 2015; Saunders et al., 2003). Various versions of these mechanisms have been incorporated into atmospheric models (Fan and Zhang, 2004; Pfister et al., 2008; Taraborrelli et al., 2009; Archibald et al., 2010; Mao et al., 2013; Squire et al., 2015; Chan Miller et al., 2017; Müller et al., 2019), with no coalescence toward a unified mechanism across models.
The effect of isoprene on OH concentrations has elicited much controversy. Early mechanisms exhibited near-complete titration of OH by isoprene under low-NO conditions (Jacob and Wofsy, 1988). However, this was contradicted in the early 2000s by observations of elevated OH concentrations in tropical forests (Carslaw et al., 2001; Lelieveld et al., 2008; Martinez et al., 2010; Pugh et al., 2010; Whalley et al., 2011; Stone et al., 2011). Models attempted to correct for this behavior by invoking hypothetical OH-recycling mechanisms in their low-NO oxidation schemes (Butler et al., 2008; Taraborrelli et al., 2009; Kubistin et al., 2010). These were later replaced with mechanistic OH-recycling pathways, including isoprene epoxydiol (IEPOX) formation (Paulot et al., 2009b), radical propagation in reactions of HO2 with acylperoxy radicals (Hasson et al., 2004; Jenkin et al., 2007; Dillon and Crowley, 2008), and H-shift isomerizations of the initial isoprene-hydroxy-peroxy radicals (Peeters et al., 2009). Incorporation of these mechanistic OH-recycling pathways into models showed that the latter pathway is most important for sustaining elevated OH concentrations under low-NO conditions (Archibald et al., 2010), and subsequent studies have identified and characterized additional OH-regenerating H-shift reactions throughout the isoprene oxidation mechanism (Peeters and Muller, 2010; Crounse et al., 2012, 2013; Peeters et al., 2014; Jørgensen et al., 2016; Wang et al., 2018; Møller et al., 2019).
Another focus of interest has been the role of isoprene as a sink for NOx (≡ NO+NO2) through organonitrate formation and the subsequent fates of these organonitrates (von Kuhlmann et al., 2004; Horowitz et al., 2007; Wu et al., 2007; Paulot et al., 2012). Differences between models in organonitrate yields and recycling have large effects on simulated ozone (Fiore et al., 2012; Xie et al., 2013; Müller et al., 2014). Recent work has established that hydrolysis and deposition of isoprene-derived organonitrates can be a dominant NOx loss process in some environments (Romer et al., 2016; Fisher et al., 2016).
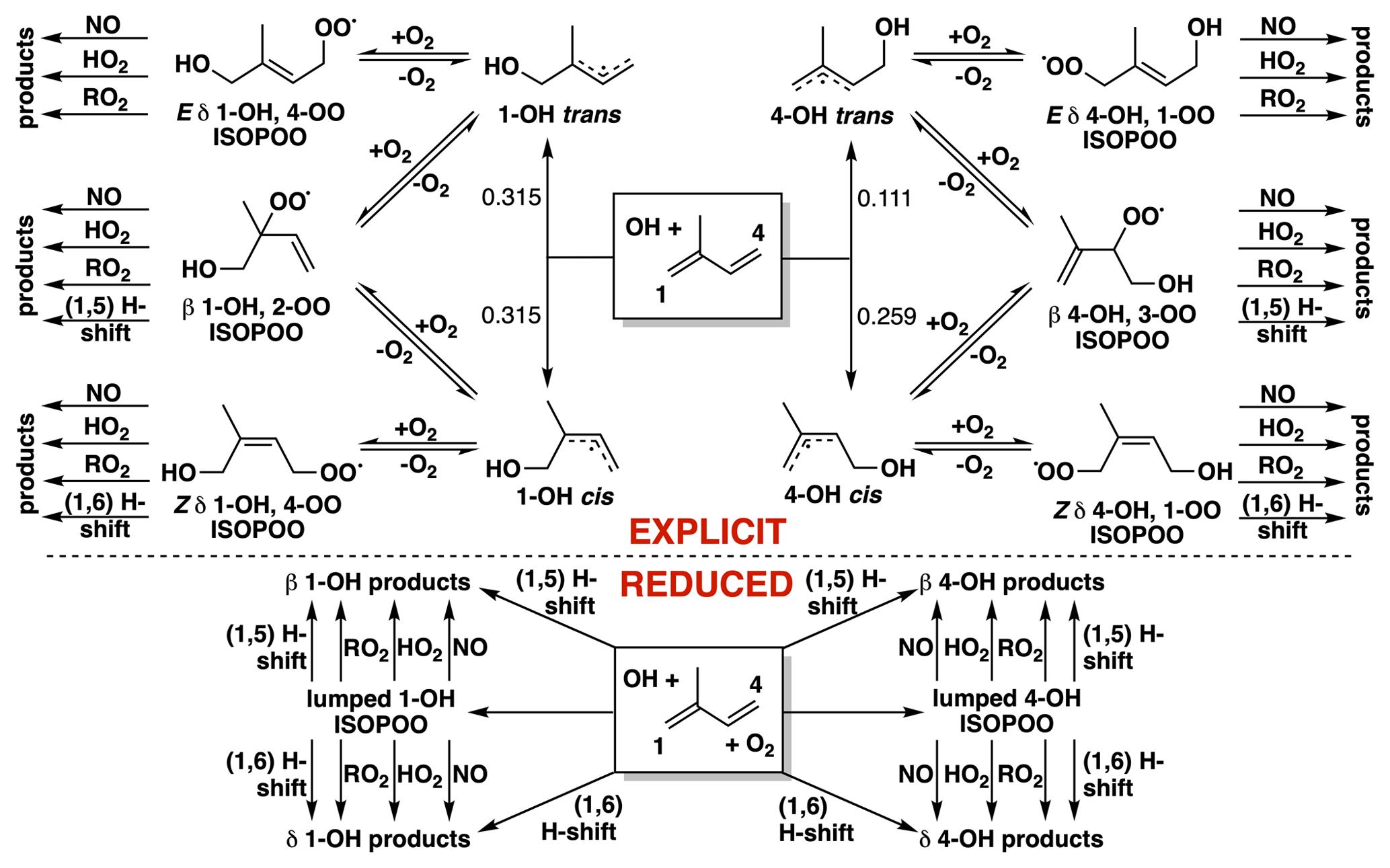
Figure 1Fate of the allylic and peroxy radicals produced from the reaction of isoprene with OH in the presence of O2. The explicit Wennberg et al. (2018) scheme is on top, and the reduced Caltech isoprene mechanism (RCIM) scheme is on bottom.
The role of isoprene as a SOA precursor has received increasing interest following evidence from field studies of C5 compounds in ambient particles (Claeys et al., 2004; Edney et al., 2005; Kleindienst et al., 2007), which led to experimental work measuring SOA yields from isoprene oxidation (Kroll et al., 2005, 2006; Surratt et al., 2006). Further investigations of the isoprene oxidation mechanism identified specific SOA precursors such as IEPOX (Paulot et al., 2009b; Nguyen et al., 2014), oxidation products of methacryloyl peroxynitrate (MPAN; Nguyen et al., 2015a), and highly functionalized compounds with low volatility (Krechmer et al., 2015; D'Ambro et al., 2017). These SOA formation pathways are now commonly implemented in models (Marais et al., 2016; Stadtler et al., 2018).
Here we implement the condensed version of the Wennberg et al. (2018) comprehensive mechanism, which we call the reduced Caltech isoprene mechanism (RCIM), in three types of atmospheric models – a fixed-radical box model, a diurnal steady-state box model, and the global GEOS-Chem chemical transport model. RCIM introduces a number of components not currently included in the models, as described in Sect. 2. We use it to investigate the effects of isoprene chemistry on hydrogen oxide radicals (HOx ≡ OH + peroxy radicals), NOx, and ozone. We also investigate the fate of the isoprene carbon, including the yields of oxygenated organic products, CO, CO2, and SOA. In the process we compare RCIM to previous isoprene oxidation mechanisms, including MCM v3.3.1, the current GEOS-Chem standard mechanism (v11-02c), and others in the literature. Finally, we use the results of our simulations to further simplify the isoprene mechanism for computational savings in model applications.
2.1 Chemical mechanism
RCIM is version 4.1 of the “Reduced-plus” mechanism found in the Wennberg et al. (2018) mechanism repository (https://doi.org/10.7907/Z9S75DHB, Bates and Wennberg, 2017). It includes the oxidation of isoprene by OH, ozone, and NO3 and condenses the ensuing oxidation cascade for the practical range of atmospheric conditions. The mechanism includes 148 species and 412 reactions, representing the complete isoprene oxidation cascade, in contrast to the 385 species and 810 reactions in the Wennberg et al. (2018) explicit mechanism, which did not seek to provide loss processes for compounds without experimental constraints and therefore did not represent a complete oxidation cascade (i.e., full conversion to CO2). RCIM was compiled concurrently with the full explicit mechanism and was designed to keep product yields of known compounds the same, with minimal simplifications beyond lumping of isomeric compounds with similar reaction pathways and removal of especially minor (<1 % yield) pathways. Under atmospheric conditions, early-generation compound yields and mixing ratios in simulations with RCIM therefore closely track those of the full explicit mechanism. Major deviations occur only for later-generation compounds for which minimal experimental evidence exists to constrain reactive pathways and for the proposed products of these reactions. For such compounds, the authors applied a self-consistent set of assumptions (Sect. 2 in Wennberg et al., 2018) based on extrapolation from similar compounds and structure–activity relationships. While these assumptions were grounded in experimental evidence, they necessarily include high levels of uncertainty, which are discussed in greater detail in Sect. 8 of Wennberg et al. (2018).
The reader is directed to Wennberg et al. (2018) for a detailed description of RCIM. Most importantly, the mechanism treats the initial system of allylic and peroxy radicals formed following the addition of OH to isoprene dynamically, as shown in Fig. 1. Older mechanisms implicitly used fixed distributions of isoprene-hydroxy-peroxy (ISOPOO) radicals, often derived from experiments performed under high-NO conditions. Addition of O2 to allylic radicals under ambient conditions is in fact a reversible process, resulting in a dynamic system with differing initial (kinetic) and equilibrium radical distributions, as first postulated by Peeters et al. (2009) and demonstrated experimentally by Teng et al., 2017. In RCIM, we simplify this 10-species, 69-reaction radical system to two species, as shown in the bottom panel of Fig. 1 and described in Wennberg et al. (2018). The implications of this novel treatment of the isoprene-hydroxy-peroxy radical system are manifest throughout this paper but are discussed specifically in Sect. 3.3.
Additional aspects of RCIM relative to older mechanisms include new products and decreased C5-hydroperoxy-aldehyde (HPALD) yields following the 1,6-H shifts of the Z-δ-OH-peroxy radicals shown in Fig. 1 (Teng et al., 2017); more intramolecular H shifts, including rapid peroxy-hydroperoxy shifts (Jørgensen et al., 2016; Møller et al., 2019), resulting in higher OH recycling under low-NO conditions; new parameterizations of nitrate yields from RO2 + NO reactions, including pressure and temperature dependence; explicit treatment of highly functionalized products such as C5-dihydroxy-dihydroperoxides and other tetrafunctionalized C5 compounds; and more detailed chemistry following the reactions of isoprene with NO3 (Schwantes et al., 2015) and ozone (Nguyen et al., 2016).
In this paper we compare RCIM to MCM v3.3.1 (Jenkin et al., 2015) and to the current standard GEOS-Chem mechanism implemented in version 11-02c. MCM v3.3.1 (subsequently MCM for short) treats the chemistry of isoprene more explicitly than RCIM or the v11-02c mechanism. It includes 602 species and 1926 reactions. The GEOS-Chem v11-02c chemical mechanism includes 106 species and 335 reactions and is primarily derived from chamber experiments separating the high- and low-NO oxidation schemes of isoprene (Mao et al., 2013; Paulot et al., 2009a, b) with targeted updates to nitrate yields, peroxy radical H-shift chemistry, and IEPOX chemistry (Bates et al., 2014, 2016; Travis et al., 2016; Fisher et al., 2016; Marais et al., 2016; Chan Miller et al., 2017). The GEOS-Chem mechanism lumps the initial isoprene peroxy radicals into a single ISOPOO species.
Neither RCIM nor MCM includes the loss of organic products by deposition or SOA formation. We exclude these processes from box model simulations so as to isolate the effects of gas phase chemistry. For global GEOS-Chem simulations with RCIM, we extend the existing deposition (Nguyen et al., 2015b; Travis et al., 2016) and SOA formation (Marais et al., 2016) parameterizations from GEOS-Chem v11-02c to analogous species in the mechanism. All C5 epoxides are thus treated identically to IEPOX, tetrafunctional C5 nitrates are treated identically to isoprene dihydroxy-dinitrates, and tetrafunctional C5 non-nitrates are treated identically to the “LVOC” low-volatility species that represents this group of SOA precursors in GEOS-Chem v11-02c. The one major change we implement relative to the standard v11-02c mechanism is to increase the aerosol uptake rate for tertiary nitrates by a factor of 10, following evidence that the hydrolysis of these nitrates proceeds rapidly in the atmosphere (Darer et al., 2011; Hu et al., 2011; Rindelaub et al., 2014; Xu et al., 2015; Rindelaub et al., 2016).
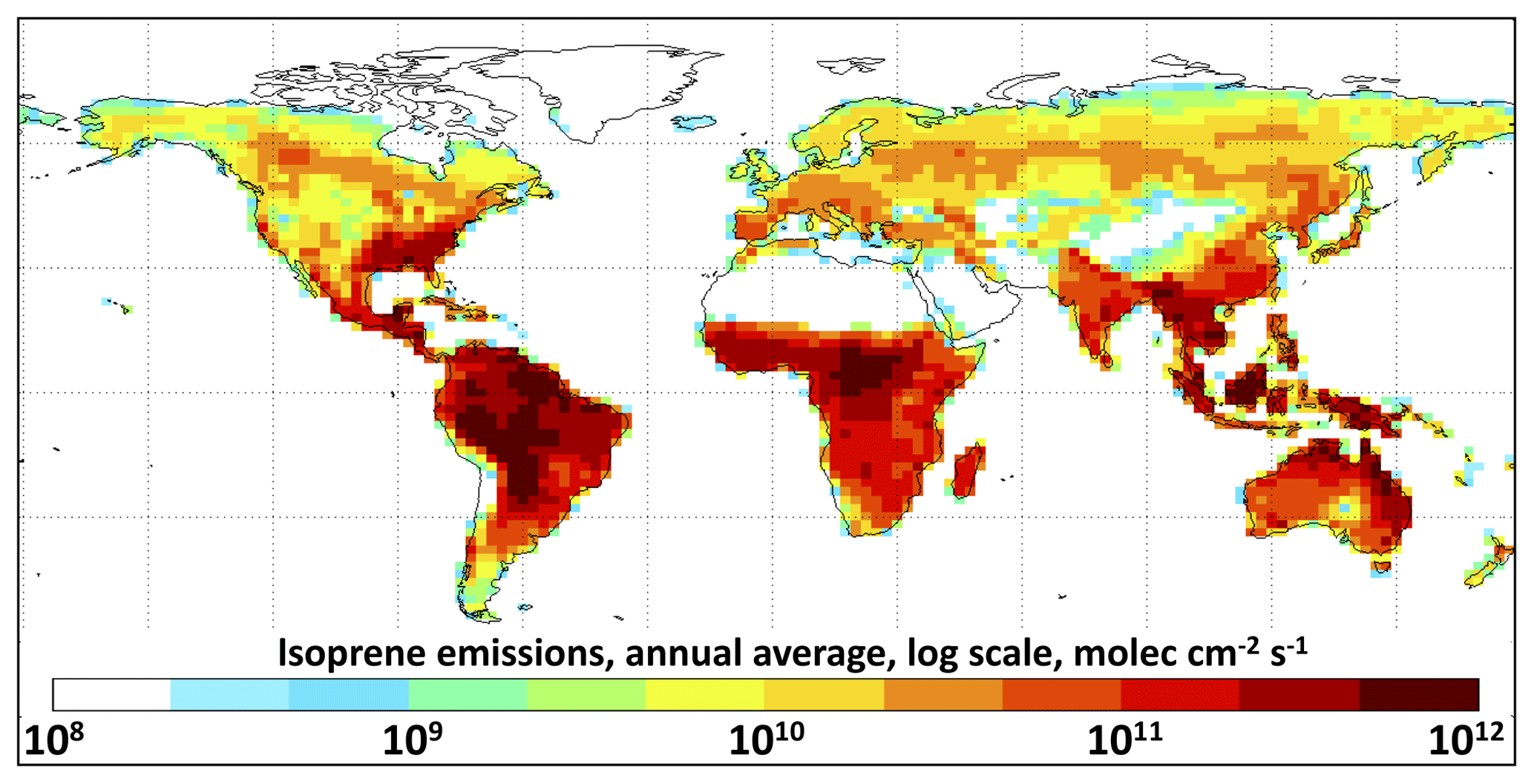
Figure 2Annually averaged MEGAN v2.1 isoprene emissions for July 2014–June 2015 as implemented in GEOS-Chem at horizontal resolution.
2.2 Simulations
All models use the same fast Rosenbrock kinetic solver implemented with the Kinetic PreProcessor tool (KPP; Damian et al., 2002; Daescu et al., 2003; Sandu et al., 2003).
Table 1Branching ratios (%) of isoprene oxidation pathwaysa.
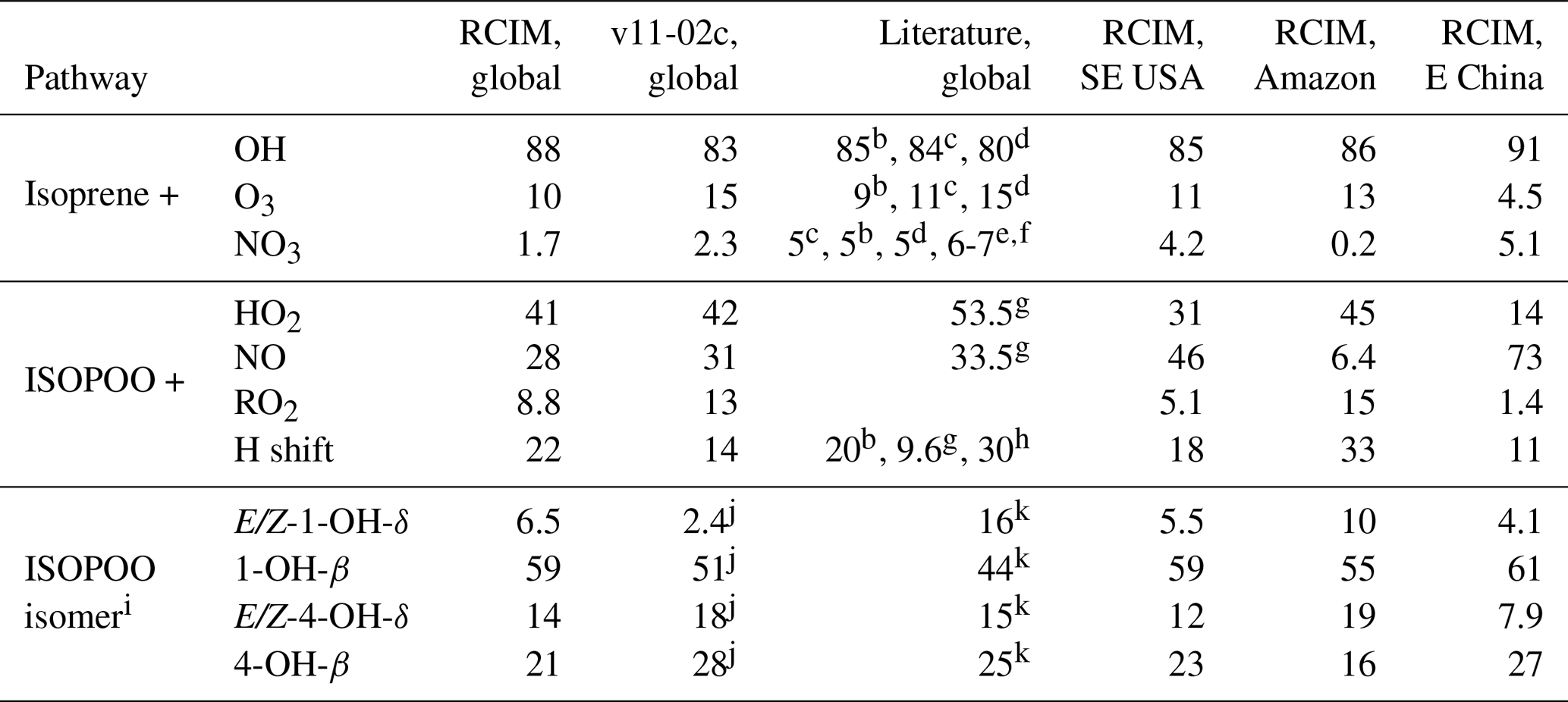
a Annual totals. Percentage values from the reduced Caltech isoprene mechanism (RCIM) implemented in GEOS-Chem are compared to the standard GEOS-Chem v11-02c mechanism and to literature values. Regional domains are defined in the text – SE USA is the southeastern United States. b Müller et al. (2019). cTaraborrelli et al. (2009). d Pfister et al. (2008). e Horowitz et al. (2007). f Ng et al. (2008). g Crounse et al. (2011). h Peeters et al. (2014). i Values represent the reacted fractions, which govern product formation. j Inferred from product distribution. k Paulot et al. (2009a).
Fixed-radical box modeling. In order to quantify product yields per unit of isoprene oxidized by OH under a given set of atmospheric conditions, a series of box model simulations are conducted with fixed concentrations of NO and HO2. This method serves to remove most nonlinearities and feedbacks inherent in the isoprene oxidation mechanism, so as to isolate the effects of the radicals on the oxidation pathways, and provides a quantitative reference of organic product yields from isoprene oxidation under fixed ambient conditions. The model is initialized with 1 ppbv of isoprene and run until complete conversion to CO2. (In the absence of deposition or SOA formation, all isoprene carbon is eventually converted to CO2.) The branching pathways of isoprene oxidation products are computed assuming 0.1 pptv OH, an NO2∕NO molar ratio of 5, no ozone or NO3, a temperature of 298.15 K, and solar radiation for clear-sky equatorial midday with an ozone column of 350 DU (Dobson units). Product yields are calculated by dividing the total molar production over the entirety of the simulation of each compound of interest by the amount of isoprene oxidized. Additional sensitivity simulations with differing temperature and photolysis settings, along with simulations investigating isoprene + NO3 chemistry, can be found in Sects. S2 and S4 of the Supplement, respectively.
Diurnal steady-state box modeling. Additional box model simulations are run with variable radical concentrations and diurnal cycles of temperature, sunlight, and isoprene emissions. These simulations follow the setup and conditions of Jenkin et al. (2015) for MCM to facilitate comparison. The box model simulates a tropical continental boundary layer with isoprene and NO emissions, ventilated by the background free troposphere with a fixed exchange rate constant corresponding to a ventilation timescale of 1 d. The free tropospheric background includes 1.8 ppmv CH4, 100 ppbv CO, 20 ppbv ozone, 300 pptv formaldehyde, and 1 % H2O. Isoprene emissions vary diurnally with both temperature and sunlight, as parameterized by Guenther et al. (1995), for an average daytime emission rate of 7.6×106 molecules cm−3 s−1 (Eerdekens et al., 2009). NO emissions are constant for a given simulation and are varied between simulations to diagnose the sensitivity of the isoprene oxidation cascade to NOx; results are presented as a function of the daytime NOx concentration. Photolysis rates follow the diurnal pattern for clear sky at the Equator, with an ozone column of 350 DU. Temperature follows a sinusoidal diurnal pattern with an amplitude of 4 ∘C, peaking at 13:00 local time. Simulations are initialized for 7 d, after which concentrations from the 8th day (daytime averages between 06:00 and 18:00) are used in the results reported below.
Global modeling. We incorporate RCIM into the GEOS-Chem global 3-D model (http://geos-chem.org, last access: 26 July 2019). GEOS-Chem is driven by assimilated meteorological observations from the NASA Goddard Earth Observing System – Fast Processing (GEOS-FP) – of the NASA Global Modeling and Assimilation Office (GMAO). We use UCX version 11-02c as a base, including both tropospheric and stratospheric chemistry (Eastham et al., 2014) and with tropospheric methane fixed on the basis of observations. Emissions use the standard HEMCO configuration in v11-02c (Keller et al., 2014), including isoprene emissions from the MEGAN v2.1 inventory (Guenther et al., 2012) as implemented into GEOS-Chem by Hu et al. (2015) and scaled uniformly to 535 Tg a−1 (Guenther et al., 2012). Annual isoprene emissions are shown in Fig. 2. We conduct simulations for 1 year (July2014–June 2015), following 1.5 years of initialization starting in January 2013. Baseline simulations are conducted at horizontal resolution with 72 vertical levels, and additional sensitivity simulations are conducted at horizontal resolution. We find no significant differences in results between the two resolutions, consistent with a previous GEOS-Chem investigation of the effects of model resolution on isoprene chemistry (Yu et al., 2016). For regional-scale results, we use the outputs of horizontal resolution simulations at 0–1 km altitude and average over 81.25–93.75∘ W, 31–39∘ N, for the southeastern United States, 53.75–76.25∘ W, 11∘ S–3∘ N, for the Amazon Basin, and 111.25–121.25∘ E, 23–41∘ N, for eastern China.
3.1 Initial oxidant branching
Figure 3a and d show the contributions of OH, ozone, and NO3 to the overall oxidation of isoprene using RCIM in diurnal steady-state box model simulations and in GEOS-Chem. On a global scale, we find that 88 % of isoprene is oxidized by OH, 10 % by ozone, and 1.7 % by NO3. These global averages mask some spatial variability, as shown in Fig. 4 and Table 1; for example, NO3 oxidation contributes up to 5 % of isoprene loss locally in the southeastern United States, and oxidation by ozone contributes up to 15 % over tropical forests.
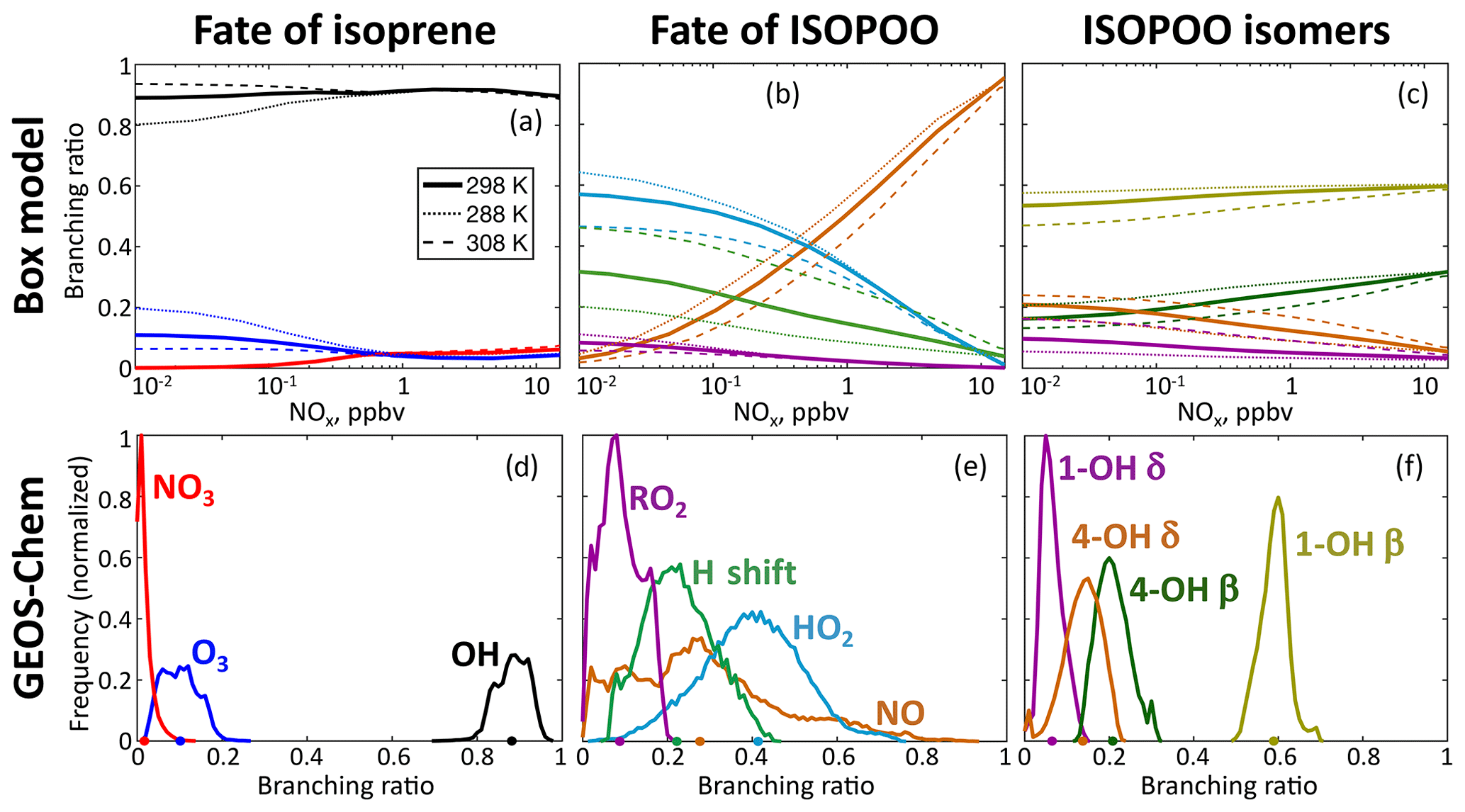
Figure 3Fate of isoprene-hydroxy-peroxy radicals (ISOPOO) in the reduced Caltech isoprene mechanism (RCIM). The figure shows isoprene oxidation pathway branching ratios in a diurnal steady-state box model for clear-sky equatorial conditions (a, b, c) as a function of daytime mean NOx concentration and temperature, and the global spatial frequency distribution of annual mean branching ratios in GEOS-Chem (d, e, f) at horizontal resolution. The GEOS-Chem frequency distributions are weighted by the amount of isoprene reacting in each grid box. Dots on the x axis indicate the global annual total reacting via each pathway. The distributions of ISOPOO isomers are weighted by their subsequent reactivity.
Although the ozone and NO3 oxidation pathways represent relatively minor contributions to global isoprene oxidation, they can be important for the global budgets of certain compounds. For example, a sensitivity GEOS-Chem simulation without isoprene ozonolysis results in a 51 % global mean decrease in formic acid production from isoprene and a 25 % decrease in hydroxymethyl-methyl-α-lactone (HMML), a product of methacrolein oxidation and a SOA precursor (Nguyen et al., 2015a). Similarly, a GEOS-Chem simulation without isoprene + NO3 results in a 39 % decrease in isoprene-derived C2+ organonitrate concentrations. Additional results from simulations excluding the ozone and NO3 pathways can be found in Table S1.
RCIM results in a higher contribution of isoprene + OH to the total oxidation of isoprene than past estimates, as shown in Table 1, and lower contributions from ozone and NO3. We ascribe this change primarily to increased OH recycling relative to older mechanisms (see Sect. 4). This further explains the temperature dependence of the OH pathway contribution in the diurnal steady-state box model (Fig. 3a), which is also stronger than in past mechanisms as a result of the temperature dependence of OH-recycling H-shift reactions. Nighttime oxidation by NO3 is particularly lower than previously reported in the literature (5 %–7 % globally; Table 1), which largely reflects the amount of isoprene remaining at sunset. More efficient recycling of OH in RCIM results in less isoprene at sunset.
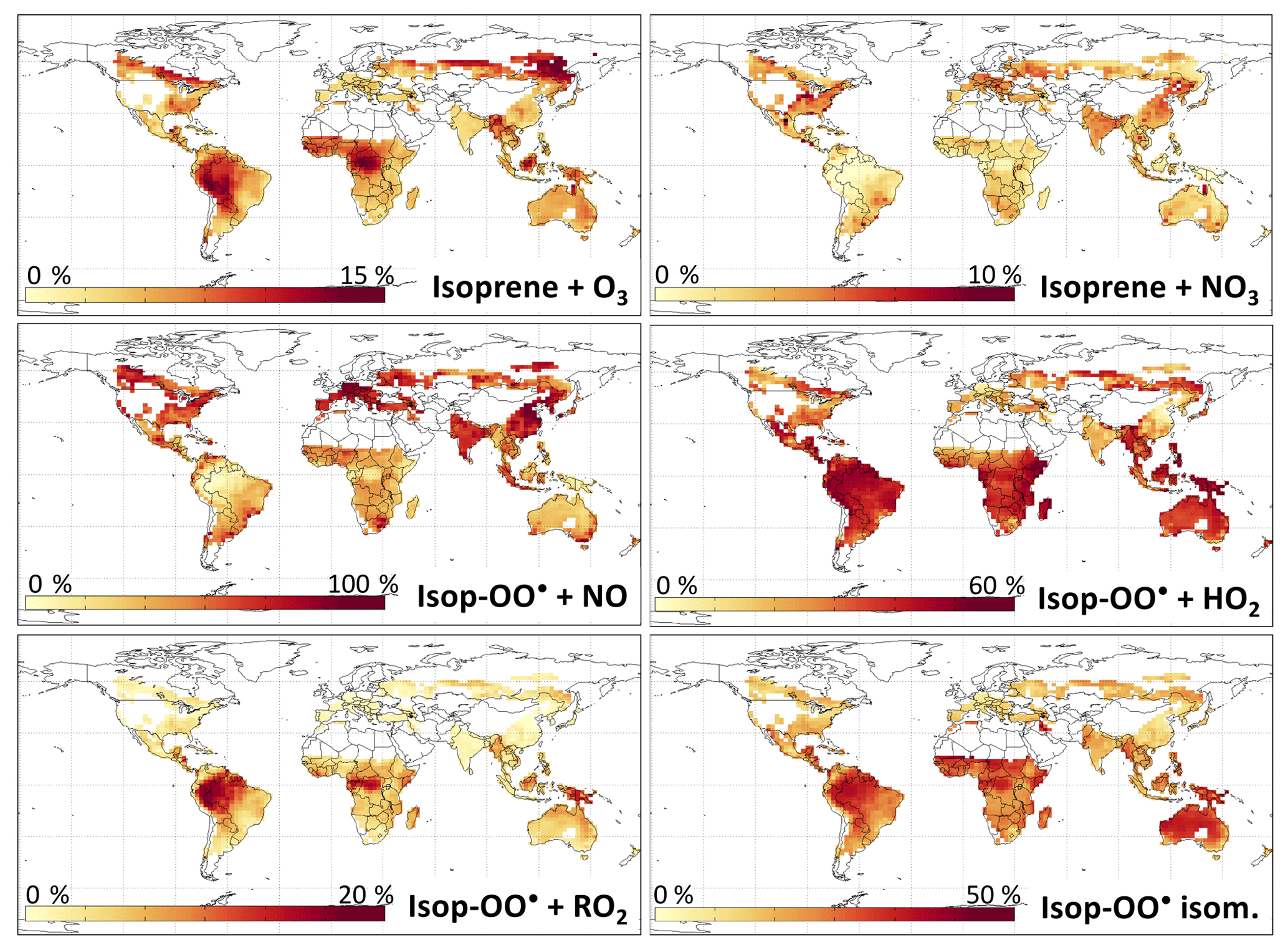
Figure 4Percent of isoprene reacting with O3 and NO3, and percent of the products from the reaction of isoprene with OH (ISOPOO radicals) reacting via each pathway. Values are annual averages from the reduced Caltech isoprene mechanism (RCIM) as implemented in GEOS-Chem and for the bottom 1 km of the troposphere. Grid boxes with an average isoprene oxidation rate of molecules cm−3 s−1 are excluded. Note the different scales for each panel.
3.2 Fate of ISOPOO
The ISOPOO radicals produced following the reaction of isoprene with OH and addition of O2 (Fig. 1) represent an important branching point in the isoprene oxidation cascade. The relative contributions of ISOPOO's reactions with NO, HO2, and RO2 and via unimolecular H shifts largely set the chemical outcomes of the oxidation mechanism, including ozone formation, OH titration vs. recycling, and SOA production.
Figure 3b shows the relative contributions of each pathway as a function of NOx and temperature in diurnal steady-state box model simulations. At a mean temperature of 25 ∘C, reaction with NO dominates the ISOPOO fate at NOx >500 pptv and reaction with HO2 dominates at lower NOx, while H shifts account for up to 30 % at low NOx. H-shift rates are strongly temperature-dependent (Teng et al., 2017) and reach a comparable importance to reaction with HO2 at 35 ∘C.
The global contributions of each pathway as computed by GEOS-Chem are summarized in Table 1. While reaction with HO2 represents the dominant fate of ISOPOO in the atmosphere, reaction with NO and H shifts contribute major portions of the global total. We estimate a 50 % larger flux through the H-shift pathway than in the GEOS-Chem v11-02c mechanism; this contributes to the higher OH recycling in RCIM (Sect. 4.1). Our estimated fraction of ISOPOO undergoing H shifts is 25 % lower than that of Peeters et al. (2014), which used faster 1,6-H-shift rates, and similar to the recent estimate of Müller et al. (2019).
Figure 4 illustrates the global distribution of the ISOPOO fate. The NO pathway dominates in polluted regions of northern midlatitudes, contributing up to 50 % of ISOPOO reactivity in the southeastern United States and 90 % in eastern China. In remote tropical forests, where low NOx and HOx lead to peroxy radical lifetimes in excess of 100 s, the H-shift pathway can account for up to 45 % of the ISOPOO fate, and reaction with RO2 can contribute up to 20 %.
3.3 ISOPOO isomer distributions
Figure 3c and f show the fractional contributions of the ISOPOO isomers to total ISOPOO reactivity in the diurnal steady-state and global simulations. The δ-ISOPOO isomers are largely responsible for HOx recycling via rapid unimolecular H shifts, while the two β isomers lead to different subsequent product formation. Methyl vinyl ketone (MVK) is the major product of β-1-OH-ISOPOO + NO or RO2, while methacrolein (MACR) is the main product from the equivalent β-4-OH-ISOPOO reactions. MACR leads to SOA formation via the production of HMML (Nguyen et al., 2015a), while MVK does not.
The δ isomers comprise a higher fraction of total ISOPOO reactivity under low-NO conditions (up to 30 % at 10 pptv NOx and a mean diurnal temperature of 25 ∘C) due to the importance of the rapid 1,6-H shifts of the Z-δ isomers. Within the ISOPOO pool derived from OH addition to isoprene at C4 (comprising 37 % of the total; see numbering in Fig. 1), the δ isomer contribution is even higher (up to 57 %) due to the more rapid 1,6-H shift of Z-δ-4-OH-ISOPOO. The δ isomers account for <10 % of ISOPOO reactivity at NO >10 ppbv. At even higher concentrations of NO, bimolecular reactivity can be high enough to trap the ISOPOO isomer distribution at its kinetic limit, leading to much higher δ isomer abundances. Here RCIM deviates substantially from the explicit mechanism of Wennberg et al. (2018) and MCM (see Fig. S14). These conditions are not of general atmospheric relevance but may occur in chamber experiments; for such applications, we recommend the use of a mechanism that resolves the full system of allylic and peroxy radicals (Fig. 1).
Table 1 and Fig. 3f show the isomers' contributions to total ISOPOO reactivity from GEOS-Chem simulations. We find that the δ isomers contribute 21 % of the total ISOPOO reactivity in a global annual average. This contribution increases in areas with high ISOPOO H-shift fractions (e.g., to 30 % in the Amazon) and decreases in areas with higher NO (e.g., to 17 % in the southeastern United States).
In the GEOS-Chem v11-02c and older mechanisms, which treat the ISOPOO system as a single species, H shifts deplete the whole ISOPOO pool rather than preferentially depleting the 4-OH-ISOPOO radicals. As a result, the fraction of ISOPOO that goes on to react bimolecularly, which should be enriched in 1-OH-ISOPOO, is instead assigned the same initial ratio it would have been assigned without H shifts. This leads to far higher contributions from the 4-OH pathway – up to 58 % over the Amazon in the v11-02c mechanism.
The most prominent effect of this change is in the unique subsequent products of the 1-OH and 4-OH systems. By preferentially depleting the 4-OH-ISOPOO pool, H shifts predominantly remove the potential for formation of MACR and the secondary β-hydroxynitrate, while the much slower 1,6-H shift of 1-OH Z-δ-ISOPOO has a smaller effect on potential MVK and tertiary β-hydroxynitrate formation from the 1-OH-ISOPOO system. In RCIM, increasing H-shift contributions thus increase the MVK ∕ MACR and tertiary ∕ secondary nitrate ratios, while in GEOS-Chem v11-02c and older mechanisms these ratios are unaffected by H-shift chemistry. Global simulations with the single-radical ISOPOO representation of Mao et al. (2013) and with the fixed ISOPOO distribution of Paulot et al. (2009a) (while leaving the rest of the chemistry unchanged from RCIM) result in 21 % and 18 % decreases in tropospheric production of MVK relative to RCIM, respectively, and 24 % and 10 % increases in tropospheric production of MACR, approximately doubling the MVK ∕ MACR ratio under low-NO conditions. The dynamic ISOPOO system also results in 25 % higher tropospheric production of the tertiary β-hydroxynitrate in RCIM relative to the single-radical and fixed-distribution ISOPOO representations of Mao et al. (2013) and Paulot et al. (2009a). The rapid hydrolysis of this tertiary nitrate in turn leads to more efficient NOx removal by isoprene nitrates (see Sect. 5.2). Table S1 shows additional results of GEOS-Chem simulations with fixed-distribution and single-radical ISOPOO chemistry.
4.1 Effects on HOx radicals
Figure 5 shows the effects of isoprene on OH and HO2 concentrations in diurnal steady-state box model simulations with the RCIM, MCM, and v11-02c mechanisms. OH is depleted under low-NO conditions by direct reactions with isoprene and its oxidation products. The effect reverses under high-NOx conditions when these reactions compete with the reaction of NO2 with OH that is the dominant HOx sink. Isoprene chemistry enhances HO2 concentrations under all conditions because of photolysis of formaldehyde and other carbonyls producing HOx radicals, particularly under high-NOx conditions by competing with the NO2 + OH reaction. OH depletion from isoprene oxidation under low-NO conditions is strongly temperature-dependent in RCIM and less pronounced than in previous mechanisms. In diurnal steady-state simulations at <100 pptv NOx, we find that an increase in mean daytime temperature of 10 ∘C causes up to a doubling in OH concentrations and that RCIM sustains OH concentrations twice as high as MCM and 3 times higher than GEOS-Chem v11-02c.
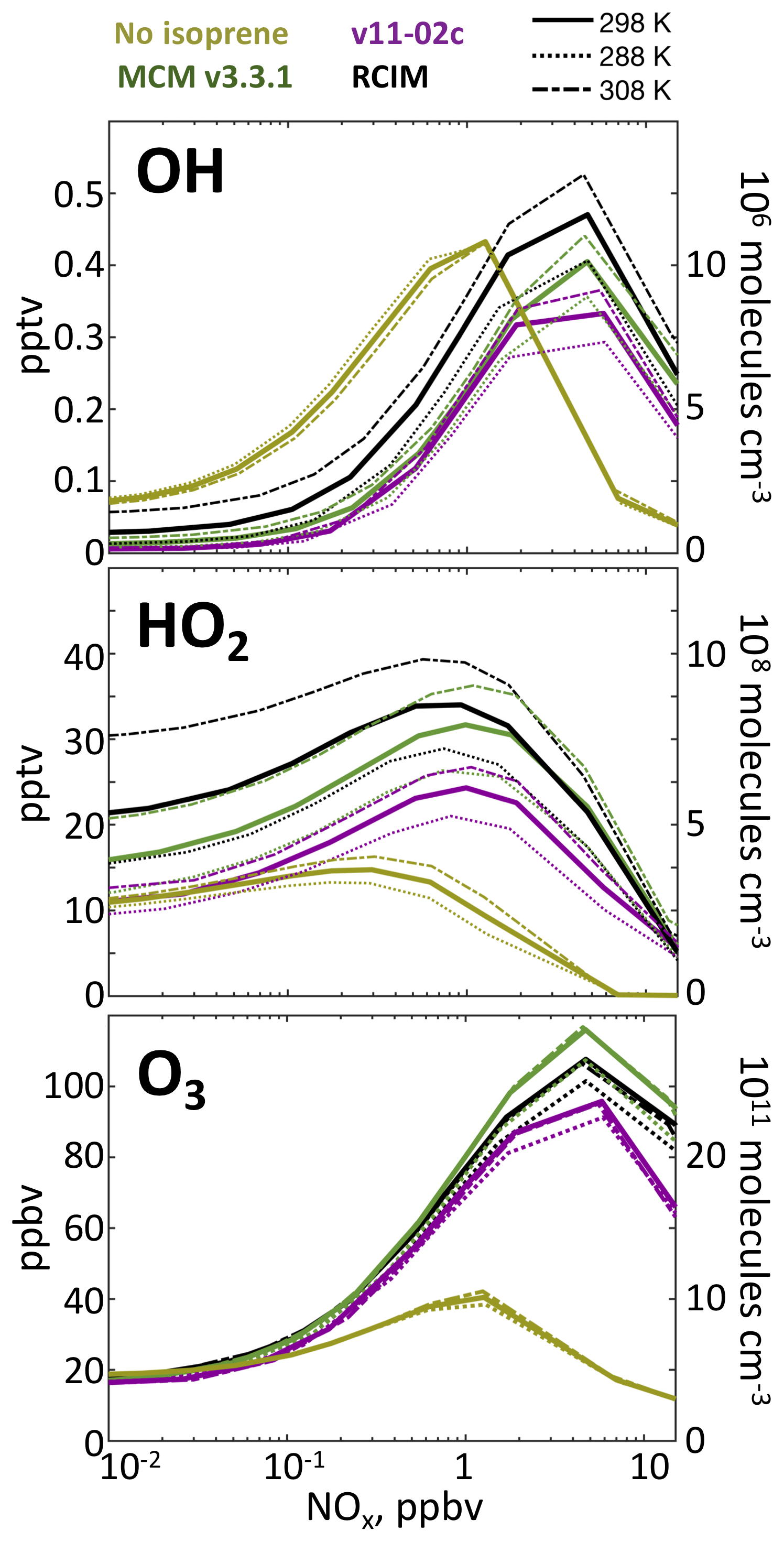
Figure 5Effects of isoprene oxidation on OH, HO2, and ozone concentrations in a diurnal steady-state box model for clear-sky equatorial conditions as a function of NOx and temperature. The RCIM, MCM, and GEOS-Chem v11-02c mechanisms are compared.
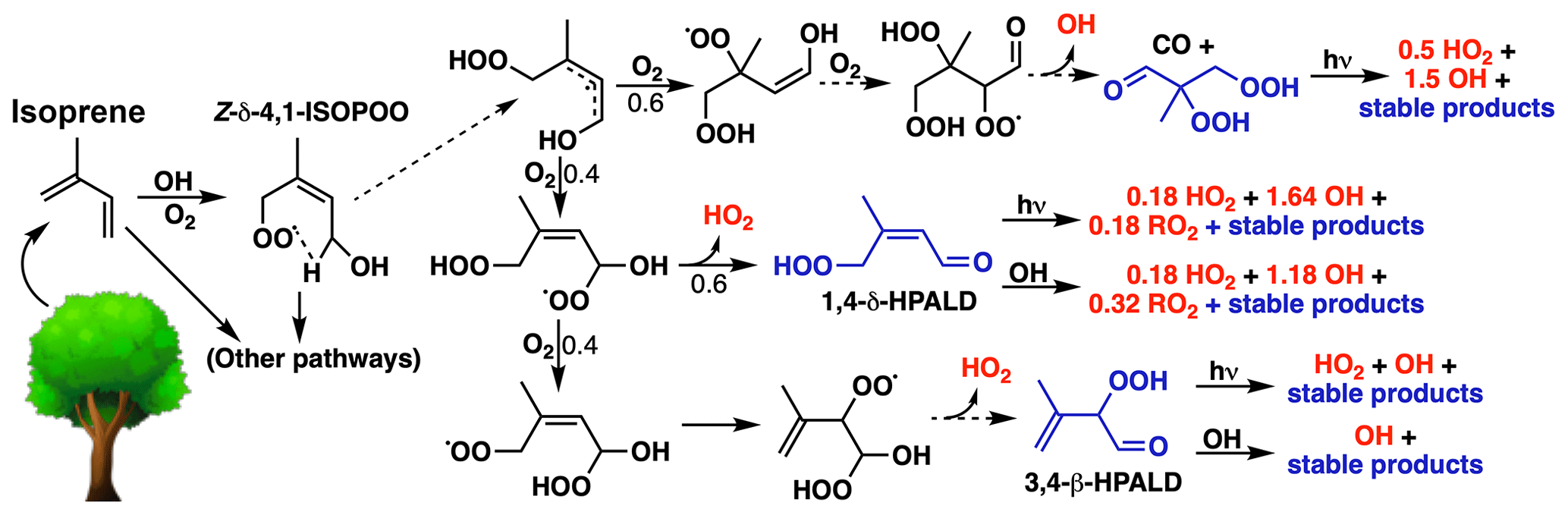
Figure 6HOx production from H-shift chemistry in the reduced Caltech isoprene mechanism (RCIM). The figure shows HOx-generating pathways following the 1,6-H shift of Z-δ-1,4-ISOPOO. H-shift reactions are shown as dashed arrows, non-radical (closed-shell) products are shown in blue, and HOx production is shown in red. In RCIM, C4-dihydroperoxy-aldehydes (top right) are assumed to photolyze rapidly, resulting in a first-generation HOx recycling yield of 2.2 (1.5 OH + 0.7 HO2) produced per ISOPOO H shift.
Differences with MCM and GEOS-Chem v11-02c are due to updated H-shift chemistry in RCIM, which efficiently recycles HOx, as shown in Fig. 6. The initial H shift of the Z-δ-4-OH-ISOPOO radical (the dominant ISOPOO H-shift pathway) is highly temperature-dependent and regenerates one equivalent of HOx (0.6 OH + 0.4 HO2) concurrently with the first generation of non-radical products. In RCIM, the C4-dihydroperoxy-carbonyl compounds (top right of Fig. 6) produced in this reaction are assumed to rapidly photolyze as postulated in Wennberg et al. (2018), which produces an additional 1.2 HOx equivalents, for a total HOx regeneration of 2.2 equivalents (1.5 OH + 0.7 HO2) from the 1,6-H shifts of Z-δ-ISOPOO isomers. Reaction with OH could possibly provide a competitive loss pathway for the C4-dihydroperoxy-carbonyl compounds, which would result in lower net HOx production. An upper limit of 3.0 equivalents of HOx production (2.2 OH + 0.75 HO2 + 0.04 RO2) can be achieved in the second oxidative generation if photolysis is also the dominant fate of the HPALDs that make up the remaining 40 % of the stable products. Such a regeneration of HOx is necessary to reconcile models and measurements in low-NO conditions (Fuchs et al., 2013; Kaser et al., 2015; Feiner et al., 2016; Kaiser et al., 2016; Mao et al., 2018).
Table 2Percent changes in tropospheric concentrations due to isoprene.a
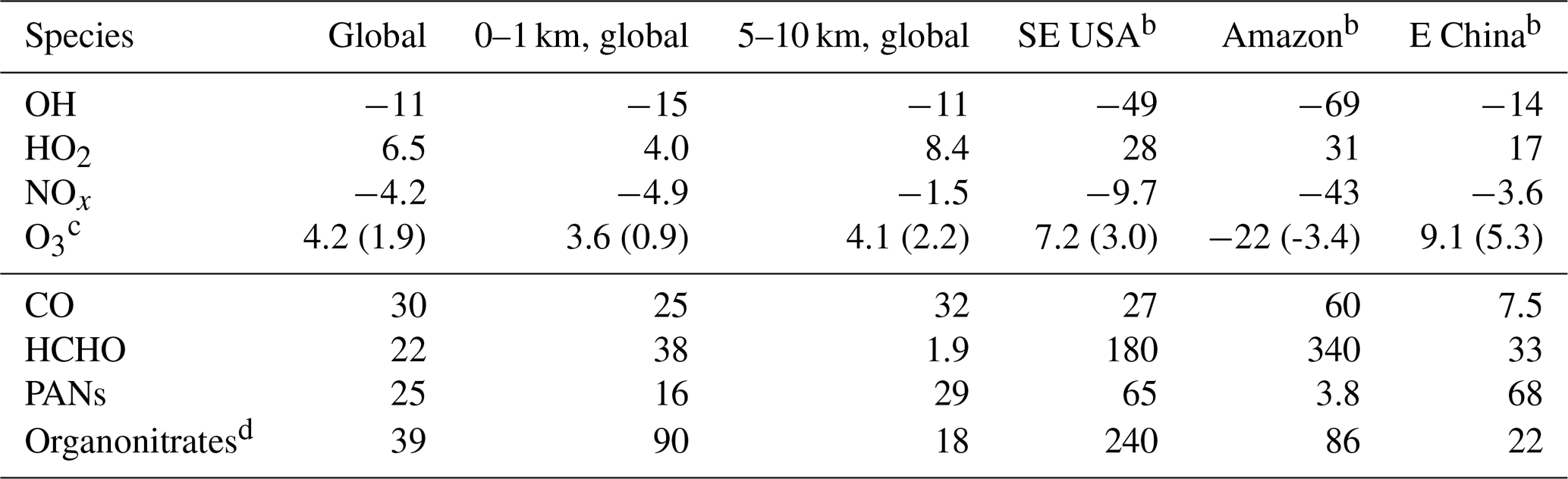
a Annual mean differences between GEOS-Chem simulations with and without isoprene emissions. Isoprene chemistry uses RCIM. b Regional results are for 0–1 km altitude (see Sect. 2.2 for precise geographic definitions) – SE USA is the southeastern United States. c Numbers in parentheses are annual mean absolute changes in parts per billion volume. d Not including PANs.
Figure 7 and Table 2 show the effects of isoprene oxidation as simulated in GEOS-Chem. The global annual mean tropospheric concentration of OH decreases by 11 %, and that of HO2 increases by 6.5 %. OH decreases are largest in tropical continental boundary layers, but diffuse effects extend throughout the global troposphere due to the influence of longer-lived isoprene oxidation products, in particular CO. Thus the globally integrated effects in the upper troposphere (5–10 km) are comparable to those in the boundary layer (0–1 km). We calculate that isoprene chemistry causes a 12 % increase in the tropospheric lifetime of methane with respect to oxidation by OH, thus enhancing the climatological effects of an already-potent greenhouse gas.
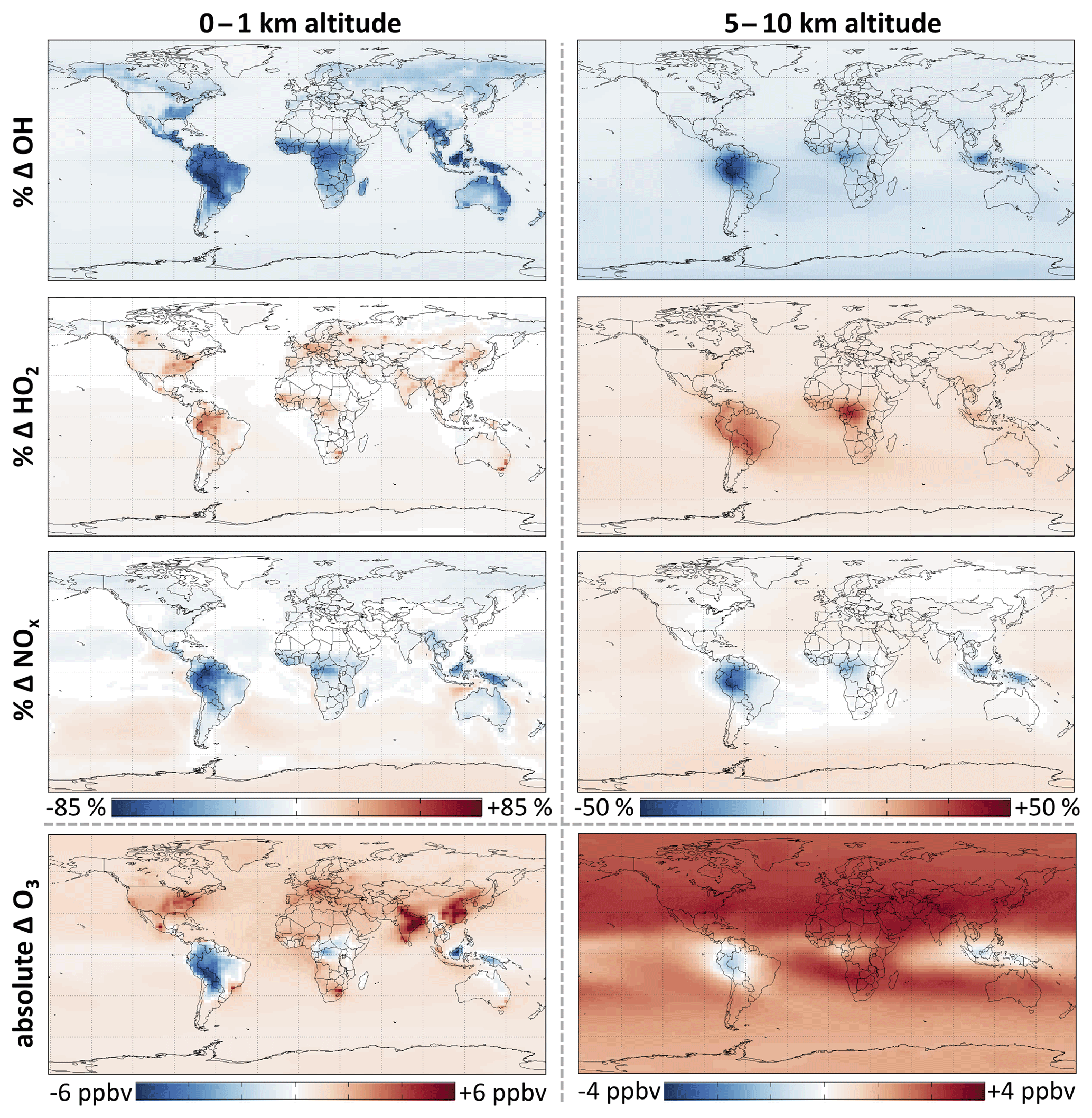
Figure 7Effects of RCIM isoprene chemistry on OH, HO2, NOx, and ozone concentrations. The figure shows annual mean differences in GEOS-Chem simulations with versus without isoprene emissions.
As in the diurnal steady-state simulations, the titration of OH by isoprene oxidation under low-NO conditions is substantially weaker in global simulations with RCIM than with the GEOS-Chem v11-02c mechanism. Whereas isoprene oxidation in the v11-02c mechanism causes reductions in annual mean OH of ∼90 % over the Amazon and Congo basins, RCIM exhibits only ∼70 % reductions. RCIM increases the simulated annual mean OH concentration over the Amazon by +170 % relative to GEOS-Chem v11-02c, and that of HO2 by +30 %, both in better agreement with field observations in the region (Barkley et al., 2011). Again, this change is largely due to increased HOx production from the H shifts of ISOPOO in RCIM. For additional comparison to the MCM and GEOS-Chem mechanisms, see Sect. S5.2.
4.2 Effects on NOx
Table 2 and Fig. 7 summarize the effects of RCIM isoprene chemistry on tropospheric NOx. These effects largely involve the formation and fate of PANs and other organonitrates and changes in the NOx lifetime due to changes in HOx. In a global annual average, isoprene chemistry depletes tropospheric NOx by 4.2 %. NOx depletion reaches 50 % in tropical continental regions where high VOC∕NOx ratios promote NOx titration by organonitrate formation. NOx increases by up to 10 % in remote regions such as the oceanic free troposphere due to release of NOx from transported PANs and other organonitrates. The fate of organonitrates including the fraction recycled as NOx will be discussed in more detail in Sect. 5.2.
The effects described above have been shown in past models (von Kuhlmann et al., 2004; Ito et al., 2009; Fischer et al., 2014; Jenkin et al., 2015) that calculated similar magnitudes for the overall contribution of isoprene to NOx and nitrate budgets. One significant difference that we find in comparison with the GEOS-Chem v11-02c mechanism is the composition of the organonitrate pool and its effects on NOx transport and removal. For example, due to higher formation of tertiary nitrates and their increased hydrolysis rate in RCIM, we estimate tropospheric NOx loss to hydrolysis of nitrates to be 4.9 TgN a−1 compared to only 1.8 TgN a−1 with the GEOS-Chem v11-02c mechanism. This increased NOx loss rate in RCIM is offset by smaller overall organonitrate production and a substantial reduction in the formation of MPAN in low-NO conditions. Thus, the two mechanisms simulate a nearly identical tropospheric NOx burden, but with distributional differences. For example, global simulations with RCIM result in a 17 % increase in annual mean surface NOx mixing ratios relative to the GEOS-Chem mechanism over the Amazon Basin (see Fig. S17 and Table S6), a region where surface NOx is typically underestimated in GEOS-Chem (Barkley et al., 2011; Liu et al., 2016).
4.3 Effects on ozone
The bottom panel of Fig. 5 shows the effect of isoprene oxidation on ozone in diurnal steady-state box model simulations. Isoprene has little effect under low-NO conditions but stimulates ozone production at higher NO due to increased peroxy radical concentrations, accelerating cycling of NO to NO2. At very high NO, ozone production becomes VOC-limited and the effect of isoprene becomes very large. These dependences are relatively invariant with temperature and similar to those seen in MCM and GEOS-Chem v11-02c.
Figure 7 and Table 2 summarize the effect of isoprene chemistry on ozone as simulated in GEOS-Chem. While isoprene oxidation decreases boundary layer ozone over the Amazon by 22 % (3.4 ppbv), mainly because of NOx depletion, it causes an overall increase in the annual average tropospheric ozone burden of 4.2 % (1.9 ppbv) and local increases of up to 6 ppbv in China, where ozone production is often VOC-limited (Jin and Holloway, 2015). The release of NOx from isoprene-derived organonitrates extends these effects to the free troposphere, with stronger ozone enhancement in the Northern Hemisphere, where NO is higher. These results are consistent with past studies diagnosing the influence of isoprene and its oxidation mechanism on ozone (Wang and Shallcross, 2000; von Kuhlmann et al., 2004; Squire et al., 2015) and investigating the effects of changing isoprene emissions (Sanderson et al., 2003; Wiedinmyer et al., 2006; Ganzeveld et al., 2010; Wu et al., 2012; Pacifico et al., 2012; Squire et al., 2014).
However, certain new aspects of RCIM cause slight distributional changes in the effects of isoprene on ozone relative to past mechanisms. RCIM's higher first-generation nitrate yields and faster tertiary nitrate hydrolysis relative to the GEOS-Chem v11-02c mechanism lead to reduced ozone formation from the ISOPOO + NO pathway. In the southeastern United States, where past mechanisms have tended to overestimate surface ozone (Murazaki and Hess, 2006; Yu et al., 2007, 2010; Lin et al., 2008; Fiore et al., 2009; Rasmussen et al., 2012; Travis et al., 2016), RCIM results in a 1.5 % decrease in annual mean boundary layer ozone relative to the GEOS-Chem v11-02c mechanism. Reduced NOx transport in RCIM and reduced MPAN formation under low-NO conditions also results in higher sustained ozone over the Amazon (+3 ppbv) and lower ozone in the remote Southern Hemisphere (−1.5 ppbv) than the GEOS-Chem v11-02c mechanism. For more detailed comparisons with MCM and GEOS-Chem v11-02c, see Sect. S5.2.
RCIM is carbon-conserving and can be used to track the fate of isoprene-derived carbon. Figure 8a shows global mean results for the fate of isoprene carbon in GEOS-Chem; 50 % of isoprene carbon is oxidized fully to CO2 in the gas phase, in good agreement with the value of 52 % calculated by Safieddine et al. (2017) for all non-methane VOCs, and 37 % is lost to wet and dry deposition of organic oxidation products before full conversion to CO2. This falls between previous estimates of 32 % (Müller et al., 2019) and 44 % (Safieddine et al., 2017) for all oxidized non-methane VOCs. The remaining 13 % forms isoprene SOA, which is assumed in GEOS-Chem to have no further chemical reactivity (Marais et al., 2016).
The following subsections describe the fate of isoprene-derived carbon and its organic products in greater detail: oxygenated gas-phase VOCs for which observations are available (Sect. 5.1), organonitrates (Sect. 5.2), and SOA precursors (Sect. 5.3). Figure 9 shows the annual average spatial distributions of important isoprene oxidation products in GEOS-Chem. Figure 10 shows molar yields of individual products from OH-initiated oxidation as a function of NO and HO2 in fixed-radical box model simulations. Figure 11 shows daytime mean concentrations of major classes of isoprene products as a function of NOx and temperature in diurnal steady-state simulations. Additional details on the organic products of isoprene oxidation are provided in the Supplement, including simulated global and regional molar yields (Table S3), the contributions of specific oxidation pathways to global production (Tables S1 and S2), sensitivities of yields to light and temperature (Sect. S2), molar yields from NO3-initiated oxidation (Sect. S4), and comparisons to MCM and GEOS-Chem v11-02c (Sect. S5).
5.1 Oxygenated VOCs and CO
CO. We find the molar yield of carbon monoxide from isoprene oxidation to be 190 % globally, or 38 % per carbon. Of the 50 % of isoprene carbon oxidized fully to CO2, 76 % proceeds via CO (Fig. 8a). The global CO source from isoprene oxidation is 415 Tg a−1, which in GEOS-Chem represents 17 % of the total atmospheric CO source (including 36 % from methane, 8 % from the oxidation of other VOCs, and 39 % from direct emissions). Isoprene oxidation generates up to 100 ppbv of CO locally over tropical forests. Our simulated global molar CO yield from isoprene is slightly higher than the range of 100 %–170 % from previous estimates (Miyoshi et al., 1994; Bergamaschi et al., 2000; Duncan et al., 2007; Pfister et al., 2008) but only a small increase from the 180 % molar yield with the GEOS-Chem v11-02c mechanism. Distributional changes from the GEOS-Chem v11-02c mechanism include 9 % higher CO concentrations over the Amazon (due to faster in situ isoprene oxidation from higher OH) and a more diffuse increase of ∼2 % in CO concentrations throughout the Southern Hemisphere (see Fig. S24), where GEOS-Chem tends to underestimate remote surface, column, and upper-tropospheric CO (Zeng et al., 2015; Huang et al., 2016; Fisher et al., 2017).
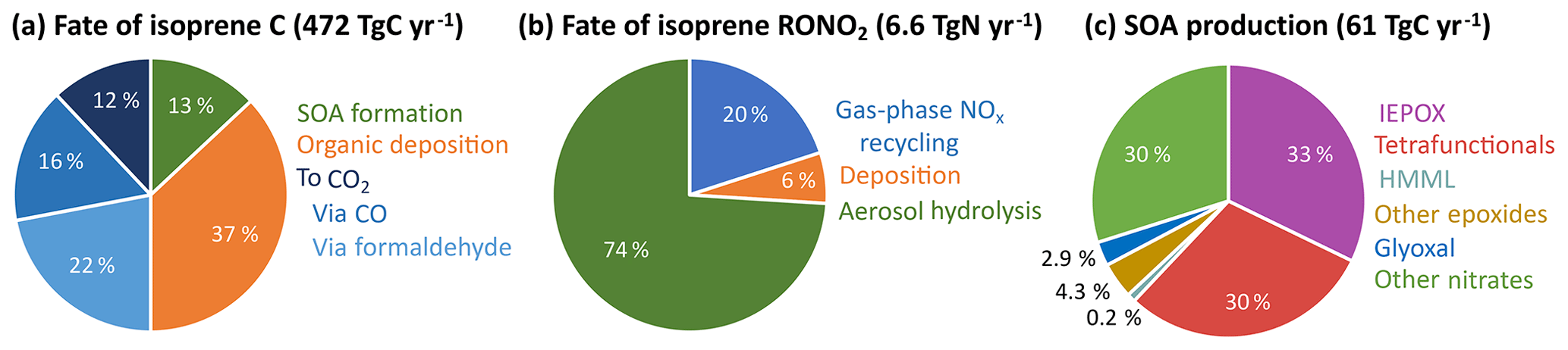
Figure 8Accounting in the reduced Caltech isoprene mechanism (RCIM) of (a) isoprene carbon, (b) the fate of isoprene-derived organonitrates (not including PANs), and (c) isoprene-derived SOA production. Values are global annual means from RCIM implemented in GEOS-Chem.
Formaldehyde. Formaldehyde is measurable by satellites and has been used in this manner to infer isoprene emissions based on estimated formaldehyde yields from isoprene (Palmer et al., 2003; Marais et al., 2012; Barkley et al., 2013; Kaiser et al., 2018). Marvin et al. (2017) and Wolfe et al. (2016) found from field observations that most current mechanisms underestimate formaldehyde yields from isoprene. Fixed-radical box model simulations with RCIM show 140 %–190 % molar yields (28 %–38 % per-carbon yields) of formaldehyde in the complete gas-phase oxidation of isoprene by OH under atmospherically relevant NO and HO2 concentrations, with increasing yields at higher NO. The variation with NO is less steep than in past mechanisms, exhibiting higher formaldehyde production under low-NO conditions, much of it being from the rapid (and unconstrained) photolysis of C4-dihydroperoxy-carbonyls produced by H-shift chemistry (top-right corner of Fig. 6), and lower production under high-NO conditions (see Figs. S20 and S24 in the Supplement). While this difference has regional impacts, e.g., increasing the mean annual boundary layer formaldehyde burden by 50 % over the Amazon from GEOS-Chem v11-02c, it does not substantially change the overall global molar yield of formaldehyde from isoprene, which we estimate to be 111 % (22 % per carbon, a 4 % increase from GEOS-Chem v11-02c). The yield is lower than in the box model simulations of Fig. 10 because of deposition and aerosol uptake of isoprene oxidation intermediates. We find that isoprene contributes 18 % of the global formaldehyde burden (Table 2), in line with previous estimates (Pfister et al., 2008).
Glyoxal. Glyoxal (C2H2O2) is also measured by satellites (Vrekoussis et al., 2009; Alvarado et al., 2014; Chan Miller et al., 2014), and different yields relative to formaldehyde can discriminate between emissions of different VOCs (Chan Miller et al., 2016). Past mechanisms have provided differing estimates on the isoprene oxidation pathways that produce the most glyoxal (Li et al., 2016), and comparisons with field measurements show that glyoxal production is higher under low-NO conditions than most mechanisms predict (Li et al., 2016; Chan Miller et al., 2017). Our diurnal steady-state box model simulations show that the RCIM glyoxal / formaldehyde ratio remains in the 2 %–3 % range over the ensemble of atmospheric conditions (see Fig. S21), in line with field observations for isoprene-dominated environments (Kaiser et al., 2015; Chan Miller et al., 2017).
RCIM yields of glyoxal from isoprene peak at 10 % under low-NO conditions (Fig. 10), while glyoxal yields in MCM are highest under high-NO conditions, and yields in the GEOS-Chem v11-02c mechanism are even higher than RCIM under low-NO conditions (Figs. S18–S19). Mechanistically, these differences primarily reflect changes in the contributions from two low-NO pathways in RCIM relative to MCM and v11-02c: the products of Z-δ-ISOPOO H shifts and the reactions of IEPOX-derived peroxy radicals with HO2. While both MCM and RCIM include moderate yields of glyoxal (largely via hydroperoxyethanal) from the C4-dihydroperoxy-carbonyl products of Z-δ-ISOPOO H shifts, GEOS-Chem v11-02c incorporates much higher second-generation glyoxal yields from these H-shift pathways (primarily via HPALD and dihydroperoxy-dicarbonyl compounds), consistent with field observations (Chan Miller et al., 2017). For the reactions of IEPOX-derived peroxy radicals with HO2, both RCIM and GEOS-Chem v11-02c include moderate yields of glyoxal presumed to form in the radical-propagating reaction channel (RO2+HO2 → ), as suggested in Bates et al. (2014) and implemented in Wennberg et al. (2018), while MCM includes no glyoxal formation under low-NO conditions from IEPOX-derived peroxy radicals. Both the atmospheric fates of C4-dihydroperoxy-carbonyl compounds and the radical-propagating channels of non-acyl RO2+HO2 reactions are poorly constrained (Wennberg et al., 2018), and the glyoxal yields from these pathways therefore remain uncertain.
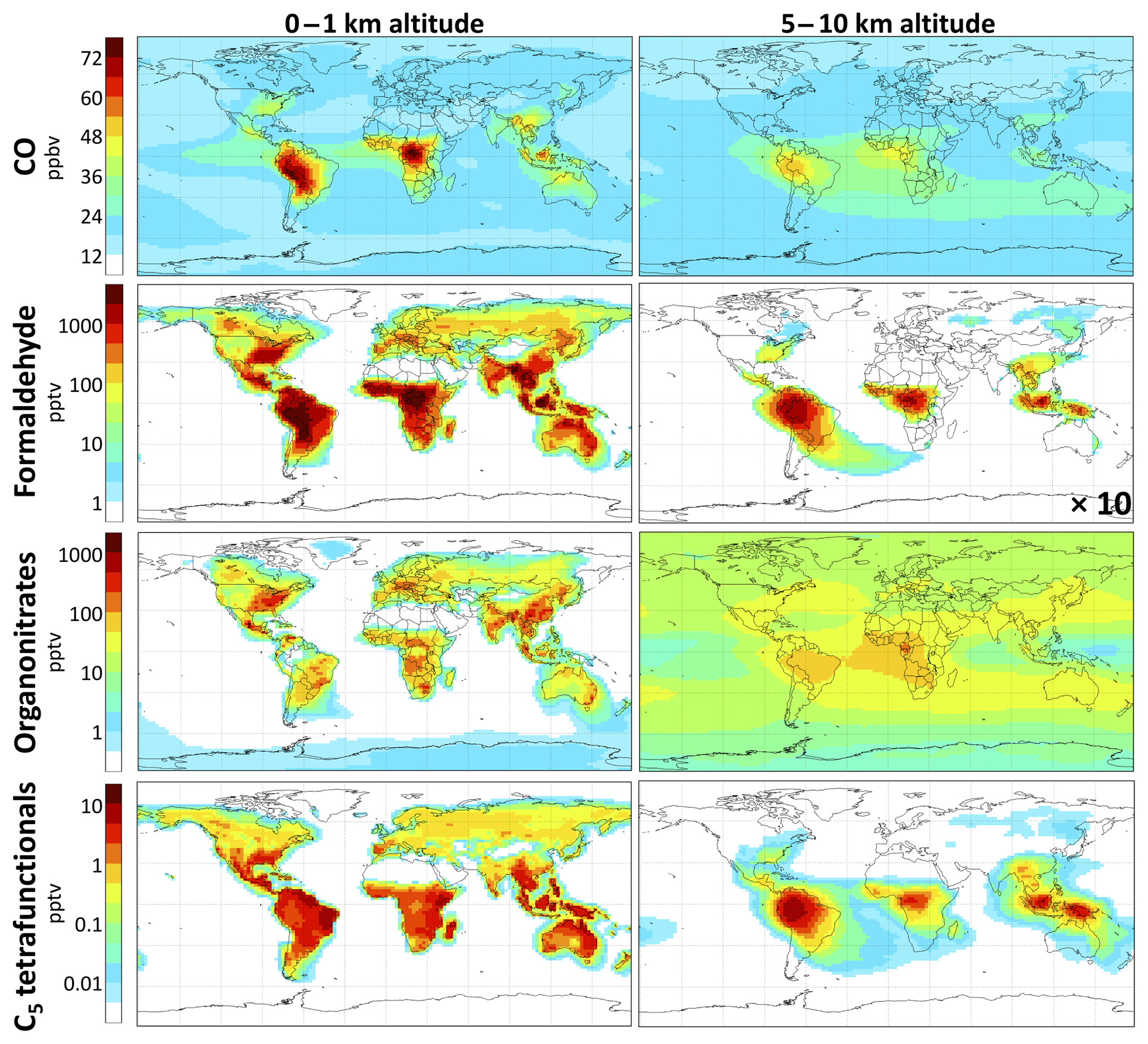
Figure 9Contributions of isoprene to concentrations of CO, formaldehyde, organonitrates (including peroxyacyl nitrates), and C5 tetrafunctional compounds using RCIM. Values are annual averages, calculated as the differences between GEOS-Chem simulations with and without isoprene emissions. Color scale is linear for CO and logarithmic for other species.
We find in GEOS-Chem that many glyoxal precursors (IEPOX, nitrates, and tetrafunctional C5 compounds) are lost to aerosol or deposition before they can react in the gas phase, depressing the glyoxal yield relative to the box model simulations where aerosol deposition effects are not included. This results in a global glyoxal yield from isoprene of 2 % in GEOS-Chem with RCIM, only half that reported recently by Müller et al. (2019) and even lower than in some past simulations (Fu et al., 2008; Myriokefalitakis et al., 2008; Taraborrelli et al., 2009). We find a reduction in global tropospheric glyoxal loading of 60 % relative to the GEOS-Chem v11-02c mechanism. However, Chan Miller et al. (2017) found good agreement between glyoxal simulated by GEOS-Chem v11-02c and aircraft observations in the southeastern United States. This suggests that RCIM may underestimate glyoxal yields from isoprene.
Organic acids. In GEOS-Chem simulations with RCIM, isoprene contributes 21 Tg a−1 of formic acid (a 5.8 % molar yield) and 25 Tg a−1 of acetic acid (a 5.5 % molar yield) globally. Over half of this formic acid comes from the initial reaction of isoprene with ozone, either directly from the stabilized C1 Criegee intermediate (Nguyen et al., 2016) or secondarily through the reaction of hydroxymethyl hydroperoxide with OH (Allen et al., 2018), while the rest is formed in the ozonolysis of MVK and MACR or the reactions of MVK-derived enols and nitrates with OH. Acetic acid is produced in the reactions of HO2 and RO2 with the acylperoxy radical, a fragmentation product from many oxidation pathways. These overall yields are similar to past estimates of isoprene's contribution to organic acid budgets (Millet et al., 2015; Müller et al., 2019).
Hydroperoxides. Organic hydroperoxides serve as a HOx reservoir in the gas phase, can contribute to the oxidation of SO2 to sulfate in aerosol and cloud water (Lind et al., 1987; Zhou and Lee, 1992), and are harmful to plants and human cells (Hewitt et al., 1990; Williams et al., 1983; Runge-Morris et al., 1989; Pöschl and Shiraiwa, 2015). We simulate an overall molar yield of hydroperoxides from isoprene in excess of 50 % globally (Table S3). As shown in Fig. 11, the majority of this production (67 %) consists of the first-generation hydroxy-hydroperoxide (ISOPOOH), with additional contributions from highly functionalized C4 and C5 compounds and from hydroperoxyacetone. Many of these are later-generation products of the initial ISOPOO + NO pathway either through subsequent H shifts or subsequent RO2+HO2 reactions; as a result, even in eastern China, where reaction with NO dominates the RO2 fate, the molar yield of hydroperoxides from isoprene oxidation still reaches 25 %.
MVK and MACR. MVK and MACR are formed in the first generation of isoprene oxidation via multiple pathways, including high production branching ratios from isoprene ozonolysis, H shifts of β-ISOPOO isomers, and the reactions of ISOPOO with NO and RO2. In the GEOS-Chem simulation with RCIM we find 28 % and 16 % global mean molar yields for MVK and MACR, respectively. This represents a pronounced decrease in the relative yield of MACR compared to past mechanisms, largely caused by the dynamic representation of the ISOPOO isomer distribution in RCIM and resulting titration of methacrolein-forming 4-OH-ISOPOO via rapid H shift (see Sect. 3.3). The decreased relative importance of isoprene ozonolysis in RCIM, which generates 26 % of global MACR, also contributes. This causes a sharp increase of up to 50 % from past mechanisms in the simulated MVK ∕ MACR ratio in diurnal steady-state simulations to a range of 1.6–2.6, depending on NOx (see Figs. S20 and S21), which is in better agreement with observations (Greenberg et al., 1999; Karl et al., 2009; Wolfe et al., 2016). Ratios of MVK + MACR to isoprene and MVK + MACR + ISOPOOH to isoprene, which are used as proxies for OH and photochemical age (Kuhn et al., 2007; Karl et al., 2009), remain considerably more consistent across mechanisms.
5.2 Organonitrates
RCIM includes important updates to the formation and fates of organonitrates through pressure- and temperature-dependent parameterizations of nitrate branching ratios and a new structure–activity relationship for calculating the formation of nitrates from multifunctional peroxy radicals without measured yields (Wennberg et al., 2018). We also implement faster particle-phase hydrolysis of the 1-OH,2-ONO2 isoprene hydroxynitrate (Darer et al., 2011; Hu et al., 2011; Rindelaub et al., 2014; Xu et al., 2015; Rindelaub et al., 2016), which decreases its ability to transport and recycle NOx. Finally, the dynamic representation of the ISOPOO isomers causes lower production of methacrolein-derived nitrates, including MPAN, and higher production of the hydrolysis-prone tertiary hydroxynitrate than previous mechanisms, as discussed in Sect. 3.3.
The effects of these updates are shown in Table 2 and Figs. 9–11. Isoprene is found to contribute 20 % of the tropospheric burden of peroxyacyl nitrates and 28 % of non-peroxyacyl nitrates, with higher contributions in the Southern Hemisphere and local contributions up to 80 % in regions of concurrent isoprene and NOx emissions. Among isoprene-derived PANs, peroxyacetyl nitrate (PAN) dominates; we estimate that 19 % of global PAN is derived from isoprene. This represents a considerably smaller fraction than in the v11-02c mechanism (44 %) and in Fischer et al. (2014) (37 %), due primarily to lower yields of precursors; methylglyoxal yields are reduced due to higher losses of intermediates to deposition and SOA, and acetaldehyde is not produced from isoprene in RCIM. We simulate PAN / MPAN ratios between 10 and 20, which are similar to MCM and to observed ratios (Roberts et al., 2002, 2007; Cleary et al., 2007; Jenkin et al., 2015). This is in contrast to the GEOS-Chem v11-02c mechanism, which found a large contribution from MPAN due to high methacrolein yields and a high formation rate taken from Lin et al. (2013) (see Fig. S23); the rate has since been revised down in GEOS-Chem v12.
GEOS-Chem with RCIM shows substantial daytime contributions from a number of non-PAN organonitrates (Fig. 11), including first-generation C5 hydroxynitrates as well as later-generation C4 nitrates, C5 tetrafunctionalized nitrates, and propanone nitrate, similar to those in GEOS-Chem v11-02c (Fisher et al., 2016) and MCM (see Sect. S5.3). We simulate higher yields of first-generation hydroxynitrates than GEOS-Chem v11-02c but lower later-generation yields, leading to an overall 14 % decrease in non-PAN organonitrate production in RCIM. Nitrates derived from NO3-initiated oxidation contribute substantially to nighttime burdens, but their relatively short lifetimes against photolysis and oxidation by OH mean that they are rapidly lost in the day; for more on isoprene-NO3 chemistry, see Sect. S4.
In global simulations, as described in Sect. 4.2, the rapid hydrolysis of tertiary nitrates is an important sink of NOx. Figure 8b shows the global fate of non-PAN isoprene-derived organonitrates in GEOS-Chem with RCIM. We find that only 20 % of these organonitrates recycle NOx via gas-phase oxidation, while 6 % undergo deposition and 74 % hydrolyze to inorganic nitrate. Organonitrate hydrolysis constitutes a NOx sink of 4.9 TgN a−1 globally, or 10 % of total NOx loss. Including organonitrate deposition and hydrogen abstraction from isoprene-derived VOCs by NO3 to form HNO3, the overall contribution of isoprene to global NOx loss reaches 15 %. The increased hydrolysis rate also causes a 14 % reduction in the production of C5 tetrafunctional compounds, which may contribute to SOA (see Table S2) because their organonitrate precursors are lost to hydrolysis.
Over the southeastern United States, where isoprene nitrate chemistry has been extensively observed (Lee et al., 2016; Romer et al., 2016), we simulate that loss to hydrolysis is the fate of 69 % of isoprene-derived nitrates annually, comprising 45 % of the total regional NOx sink, while deposition and gas-phase NOx recycling contribute 9 % and 22 %, respectively. Fisher et al. (2016) estimated a similar fraction of NOx recycling from isoprene nitrates of 23 % over the southeastern United States in summer, with a larger contribution from deposition (18 %) and a smaller fraction lost to hydrolysis (59 %). The average lifetime of isoprene-derived nitrates in the region is 3.6 h in RCIM, more consistent with the observational estimate of 2–4 h (Romer et al., 2016; Lee et al., 2016) than the simulated lifetimes of 0.48 d with GEOS-Chem v11-02c and 0.58 d in Horowitz et al. (2007).
5.3 SOA and its precursors
Figures 10–11 show the yields of major precursors of isoprene-derived SOA (iSOA) in RCIM, including IEPOX, highly functionalized C5 compounds, HMML, other epoxides, and organonitrates, all of which are discussed in greater detail below. We find a global isoprene-derived SOA (iSOA) production of 61 TgC a−1 (13 % yield per carbon; Fig. 8a) in GEOS-Chem using RCIM. IEPOX, tetrafunctional C5 compounds, and organonitrates each contribute ∼30 % to this total (Fig. 8c). If we simply consider the individual molecular weights of the iSOA precursors, we obtain a global iSOA source of 136 Tg SOA a−1, corresponding to a mass yield of 25 % from isoprene, and a ratio of organic mass to organic carbon (OM / OC) of 2.2 for iSOA, consistent with observations for highly oxidized SOA (Aiken et al., 2008). We assume this ratio in what follows, recognizing that subsequent aerosol-phase reactions not described here would modify it.
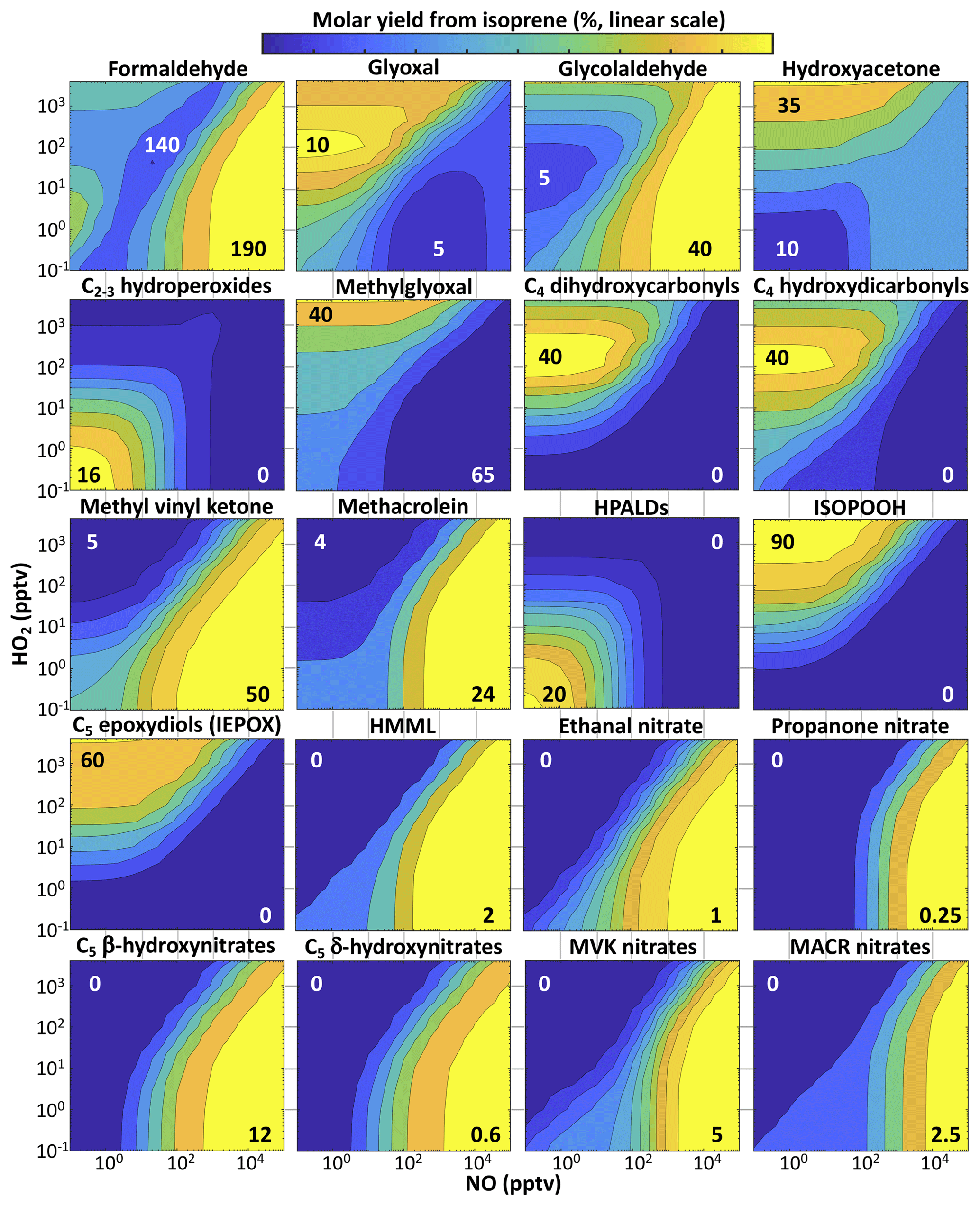
Figure 10Percent yields of organic products from isoprene + OH oxidation as a function of NO and HO2. Results are from fixed-radical box model simulations with RCIM, run at 25 ∘C for clear-sky equatorial radiation at solar noon and an ozone column of 350 DU. The fixed-radical box model does not account for deposition or aerosol uptake. Contours are evenly spaced on a linear scale between the minimum values (in white) and maximum values (in black) located on each plot. HPALDs ≡ C5 hydroperoxy-aldehydes. ISOPOOH: C5 hydroxy-hydroperoxides. HMML: hydroxymethyl-methyl-α-lactone. MVK: methyl vinyl ketone. MACR: methacrolein.
The 25 % mass yield of SOA from isoprene simulated with RCIM is considerably higher than values commonly used in global models, e.g., in the range of 0.9 %–6.8 % in the models discussed in Carlton et al. (2009). The standard GEOS-Chem model has two options for simulating iSOA: either a fixed mass yield of 3 % (Kim et al., 2015) or an explicit representation coupled to the gas-phase mechanism (Marais et al., 2016). Marais et al. (2016) find from that explicit representation a 3.3 % mass yield over the southeastern US in summer, with glyoxal and IEPOX dominating the formation in the high-NO and low-NO regimes, respectively. In contrast, RCIM has a low yield of glyoxal from isoprene, as discussed above, and far larger contributions from other iSOA precursors. The Marais et al. (2016) GEOS-Chem mechanism has limited representation of iSOA formation from tetrafunctional C5 compounds and organic nitrates.
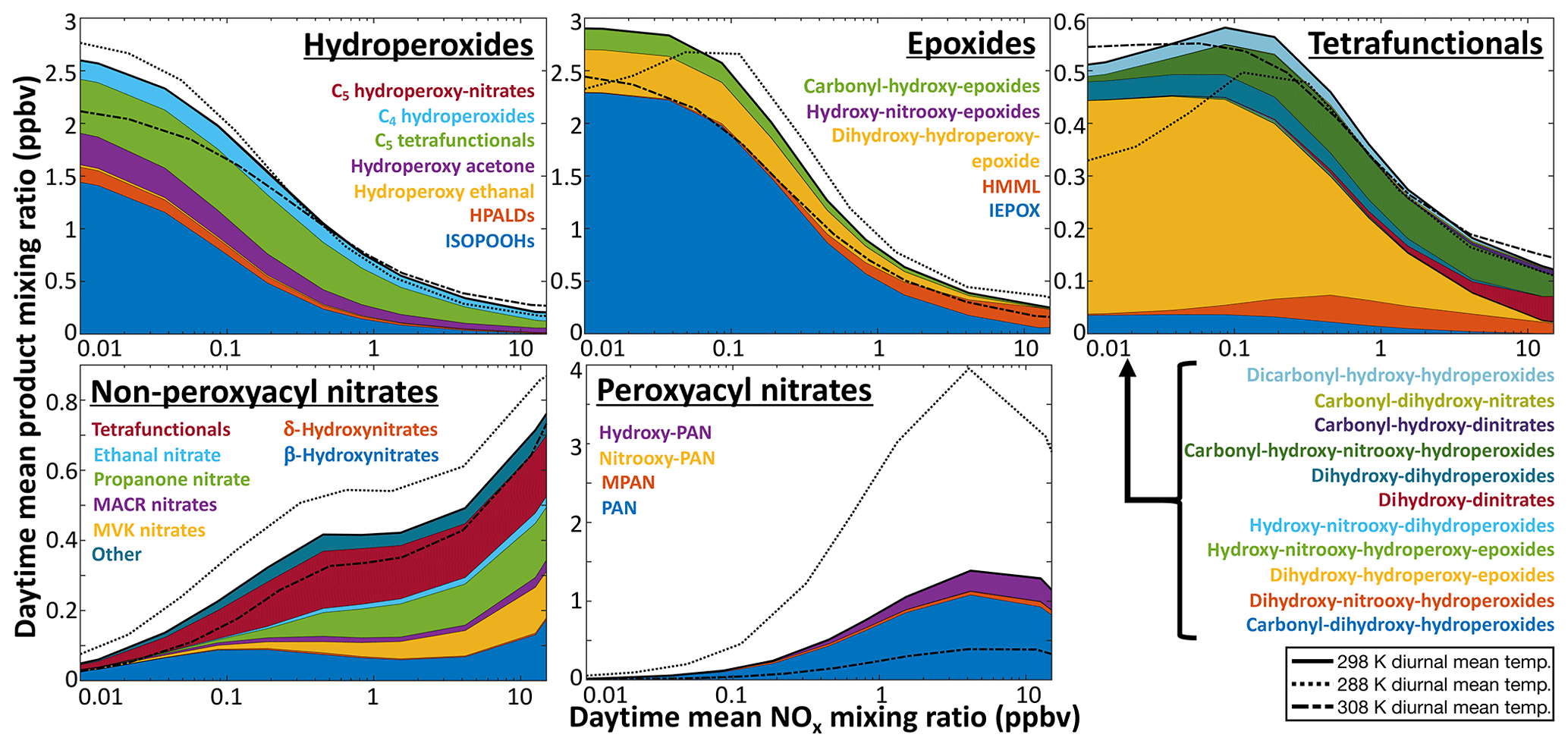
Figure 11Daytime average concentrations of isoprene oxidation products as a function of NOx. Results are from diurnal steady-state box model simulations for equatorial conditions using RCIM. Y axis scales vary between panels.
The ∼3 % mass yield of SOA from isoprene in GEOS-Chem was previously found to successfully account for organic aerosol observations over the US (Kim et al., 2015; Marais et al., 2016, 2017). A 25 % mass yield would lead to a severe overestimate. However, these and other SOA observations in isoprene-dominated environments tend to be in high-NO conditions, where yields from IEPOX and tetrafunctionals are low (Fig. 11). Organonitrates dominate iSOA formation in RCIM under high-NO conditions but hydrolyze rapidly in the aerosol phase, and the organic moiety could further react and volatilize.
Recent work suggests that increased SOA yields from isoprene may be appropriate in global simulations, and are likely to be partially balanced by iSOA chemical sinks in order to reconcile with SOA observations. Global models tend to underestimate the atmospheric burden of organic aerosol (Volkamer et al., 2006; de Gouw and Jimenez, 2009; Tsigaridis et al., 2014). Chamber studies isolating specific isoprene oxidation pathways have measured SOA mass yields in excess of 15 % (Liu et al., 2016; Schwantes et al., 2019), while top-down (Hallquist et al., 2009; Heald et al., 2010; Spracklen et al., 2011) and mass-balance (Goldstein and Galbally, 2007) assessments of global SOA production consistently arrive at higher estimates than the models. Stadtler et al. (2018) found better agreement with the top-down assessments by implementing a new isoprene mechanism (including the major tetrafunctional C5 compounds) into global simulations, resulting in a 33 % global iSOA mass yield (16 % per carbon). Hodzic et al. (2016) showed that discrepancies between observed SOA yields and modeled SOA budgets could be reconciled by incorporating increased rates of SOA loss to deposition, photolysis, and heterogeneous reactions, balanced by SOA sources 3.9 times higher than the GEOS-Chem standard model.
IEPOX. As the dominant contributor to iSOA worldwide (Marais et al., 2016; Stadtler et al., 2018), IEPOX is a second-generation oxidation product of isoprene via the ISOPOO + HO2 reaction pathway. IEPOX can form in high yields of up to 75 % from isoprene in HO2-dominated conditions (Fig. 10). In remote regions, these yields are strongly temperature-dependent due to competition from ISOPOO H-shift pathways. We estimate global IEPOX production to be 185 Tg a−1, or a 20 % molar yield from isoprene, similar to past estimates (Bates et al., 2014, 2016; St. Clair et al., 2015) and to the GEOS-Chem v11-02c mechanism (183 Tg a−1). This results in 38 Tg a−1 (20 TgC a−1) of iSOA formation from IEPOX, slightly lower than a recent estimate by Stadtler et al. (2018). While the uptake parameterization of Marais et al. (2016) used here varies with particle acidity and sulfate content as seen in chamber studies and field observations (Gaston et al., 2014; Nguyen et al., 2014; Liao et al., 2015), it does not include the known effects of organic coatings and aerosol-phase state (Riva et al., 2016; Zhang et al., 2018), which may also be important for the uptake of other precursors.
C5 tetrafunctional species. RCIM includes 11 distinct C5 tetrafunctional compounds with unique combinations of functional groups, each of which represents a variety of isomers. The global distribution of these compounds is shown in Fig. 9, while their simulated daytime concentrations in diurnal steady-state box models are shown in Fig. 11 as a function of NOx. C5 dihydroxy-hydroperoxy-epoxides (IDHPE), formed in H-shift reactions following the addition of OH to ISOPOOH (D'Ambro et al., 2017), are estimated to contribute the bulk of the tetrafunctional compounds globally (54 % of molar production) and under most NOx conditions. MCM and GEOS-Chem v11-02c predict similar yields of C5 tetrafunctional species, but the relative contributions of individual species vary substantially between mechanisms (see Figs. S22–S23). GEOS-Chem v11-02c only considered SOA formation from two such species (dihydroxy-dinitrates and LVOC produced in the reaction of ISOPOOH with OH), resulting in 4 Tg a−1 iSOA from C5 tetrafunctional compounds. Because the rates of gas-phase oxidation, deposition, and aerosol uptake for these compounds are all poorly constrained, their contribution to iSOA remains highly uncertain, and future studies will need to evaluate the volatilities, solubilities, and particle-phase reactivities of the individual tetrafunctional species.
While the individual yields of these compounds from isoprene may be small, their cumulative production (4.1 % molar yield from isoprene globally) and relatively low volatility make them potentially substantial contributors to iSOA, and aerosol formation has been observed from these pathways in chamber experiments (Krechmer et al., 2015; D'Ambro et al., 2017). Quantitative descriptions of their contribution to SOA remain uncertain, but we estimate a global source of 46 Tg a−1 (18 TgC a−1) of iSOA from C5 tetrafunctional species. IDHPE accounts for 51 % of this total, with dihydroxy-dihydroperoxides and dihydroxy-hydroperoxy-carbonyls contributing over 10 % each. This total carries high uncertainty, due both to the SOA uptake parameterization and the lack of constraints on other loss pathways of the C5 tetrafunctional compounds, but is similar to a recent estimate by Stadtler et al. (2018) and highlights the importance of further investigations of this iSOA formation pathway. Until such studies are performed, we recommend reducing the LVOC uptake coefficient applied to the tetrafunctional species by a factor of 10 in GEOS-Chem implementations to bring iSOA production from this pathway into line with previous model–measurement comparisons (Marais et al., 2016; Pai et al., 2019).
HMML. Hydroxymethyl-methyl-α-lactone, a product of methacrolein oxidation via MPAN, is considered to be a major iSOA precursor under high-NOx conditions (Nguyen et al., 2015a; Kjaergaard et al., 2012; Jiang et al., 2018). Its contribution to SOA is identified in ambient aerosol from its hydrolysis product, 2-methylglyceric acid (Edney et al., 2005; Szmigielski et al., 2007; Zhang et al., 2011). While HMML is better classified as a lactone, we include it with the epoxides in Fig. 11 and Tables S1–S3, as it is thought to react similarly in aerosol (Jiang et al., 2018). Our mechanism shows only minor yields of HMML under most conditions, up to a maximum of 2 % molar yield from isoprene at extremely high NO (Fig. 10), but it may contribute substantially to iSOA production locally; HMML production reaches 25 % that of IEPOX in the NOx-dominated conditions of eastern China (Table S3). RCIM results in similar production of HMML as in MCM, but a lower yield than in GEOS-Chem v11-02c, largely due to lower MACR production and MPAN formation rates (see Sect. 5.2); as a result, we estimate that HMML contributes only 0.18 Tg iSOA a−1 (0.11 TgC a−1) globally, much lower than the 1.7 Tg SOA a−1 predicted in the v11-02c mechanism. However, a recent chamber study comparing iSOA yields to RCIM showed an underprediction of iSOA formation from the HMML pathway, suggesting that this global estimate may be too low (Schwantes et al., 2019).
Other epoxides. A new element of RCIM is the introduction of additional organic epoxide products. These include IDHPE, discussed above, C5 carbonyl-hydroxy-epoxides (ICHE), produced from the reaction of IEPOX with OH and in the H shifts of Z-δ-ISOPOO radicals (Bates et al., 2014; Wennberg et al., 2018), and two varieties of C5 hydroxy-nitrooxy-epoxides, formed in the morning from the reactions of isoprene + NO3 products with OH (Schwantes et al., 2015). The contributions of these epoxides relative to IEPOX in diurnal steady-state simulations are shown in Fig. 11; we find that they can comprise up to 20 % of ambient epoxide concentrations under low-NO conditions. In global simulations, we find that non-IEPOX, non-IDHPE epoxides contribute 5.1 Tg a−1 (2.6 TgC a−1) of iSOA globally.
Nitrates. Multifunctional nitrates derived from both the ISOPOO + NO and isoprene + NO3 pathways are also known to contribute to iSOA (Ng et al., 2008; Lee et al., 2014; Schwantes et al., 2019). In GEOS-Chem, nitrate hydrolysis results in irreversible iSOA formation, and the higher organonitrate uptake and hydrolysis rates implemented in RCIM therefore result in a high iSOA formation from organonitrates of 49 Tg a−1 (21 TgC a−1); 6.9 Tg a−1 (3.0 TgC a−1) of this total comes from C5 tetrafunctional compounds and is already included in the amounts listed in that section above. Much of the rest comes from C5 difunctional compounds, which are expected to form alcohols (diols in the case of hydroxynitrates) following their particle-phase hydrolysis, many of which may be volatile enough to partition back to the gas phase. The organonitrate iSOA formation simulated in GEOS-Chem is therefore likely an upper limit.
Other compounds. Additional known iSOA precursors include glyoxal and methylglyoxal. As discussed previously, we find that RCIM leads to low glyoxal yields relative to previous mechanisms; this results in a small estimated contribution of glyoxal to global iSOA of 4.2 Tg a−1 (1.7 TgC a−1), 36 % lower than in GEOS-Chem v11-02c and 58 % lower than a recent estimate by Stadtler et al. (2018). In the southeastern United States, where Marais et al. (2016) found that glyoxal contributed about half as much iSOA as IEPOX, we instead find that production of iSOA from glyoxal is 10 % of that from IEPOX. Locally, however, glyoxal can still be an important contributor to iSOA; we find that it contributes 23 % of iSOA in eastern China. The production of methylglyoxal is much higher than that of glyoxal (20 % molar yield from isoprene globally), but due to its low SOA yield it contributes only 0.01 Tg iSOA a−1 (McNeill et al., 2012). Finally, RCIM predicts a large molar yield of semivolatile highly oxidized C4 compounds, including 51 Tg a−1 of dihydroxy-carbonyls, 84 Tg a−1 of hydroxy-dicarbonyls, and 56 Tg a−1 of hydroxy-hydroperoxy-carbonyls, which may also contribute to iSOA formation; as with many other elements of the isoprene SOA formation scheme, further study is required to better constrain this pathway.
We use the results of the simulations described above to implement further simplifications to RCIM and compile a mini isoprene mechanism (Mini-CIM) for use in chemical transport modeling, where computational cost is a concern. The speciation of highly functionalized isoprene oxidation products with low individual yields in RCIM goes beyond many measurement capabilities and the needs of most atmospheric model applications. We therefore combine and remove many such products, with an aim of maintaining the effects of isoprene on OH, NOx, ozone, SOA precursors, readily measured organic products, and organonitrates as shown in Sects. 3–5.
In Mini-CIM, we create lumped species from isoprene oxidation products that meet two criteria: (1) <0.1 % molar yield from isoprene globally and (2) <1 % molar yield from isoprene in each of the following: the Amazon, southeastern United States, and eastern China. These products are then lumped according to their number of carbon atoms (to conserve carbon) and similarity of lifetimes and functional groups. To maintain the effects of isoprene oxidation on NOx transport and removal, we prioritize lumping of functional groups by nitrate content. For example, all C5 dinitrate compounds are lumped into a single species, while C5 tetrafuntional mononitrates are lumped into two categories (those with and without an aldehyde, which substantially shortens the compounds' lifetimes). In addition to lumping species that meet the low-yield criteria, we remove peroxy radicals that have recently been shown to undergo rapid H shifts (Møller et al., 2019) and replace them with the products of those H shifts. We further combine five pairs of isomeric species that exceed the molar yield thresholds but have identical loss rates and are predominantly produced concurrently, which means that their lumping has no effect on the species' lifetimes and minimal effect on product distributions. A detailed list of the simplifications made in the Mini mechanism can be found in the Supporting Information along with a list of the excluded species and their global and regional molar yields (Table S7).
Global simulations with Mini-CIM exhibit only minimal differences from simulations with RCIM in the outcomes described in Sects. 3–5. Table S6 shows the effects of these simplifications on the simulated global and regional production and burden of tropospheric radicals, ozone, SOA, and organic products. The tropospheric methane lifetime increases by only 0.1 % from RCIM to Mini-CIM. Changes in annual average HOx, NOx, ozone, CO, and formaldehyde concentrations between the two mechanisms are all below 0.2 % globally, and regional differences are only minimally larger. Changes in PANs, epoxides, and SOA are below 0.5 % globally and regionally, while C2–C5 nitrates and hydroperoxides exhibit similarly small global changes but some regional differences of up to 4.2 % in areas with low absolute loadings.
Whereas RCIM originally compiled in Wennberg et al. (2018) includes 148 organic species and 412 reactions, new Mini-CIM contains 108 organic species involved in 345 reactions, which is comparable to the current mechanism in GEOS-Chem v11-02c (106 organic species involved in 335 reactions). We recommend the use of Mini-CIM in atmospheric models except when more detailed speciation of highly functionalized, low-yield isoprene oxidation products is required for model–measurement comparisons. A complete listing of the species and reactions in Mini-CIM can be found in KPP format in the online repository with the original mechanisms (https://doi.org/10.7907/Z9S75DHB).
We have presented a detailed analysis of the reduced Caltech isoprene mechanism (RCIM), a new isoprene oxidation mechanism based on a recently developed explicit scheme (Wennberg et al., 2018), to examine its atmospheric implications for HOx and NOx radicals, ozone, organic products, and secondary organic aerosol (SOA) formation. We used for that purpose a combination of box models and the GEOS-Chem global chemical transport model and compared RCIM to the explicit MCM v3.3.1 and to the previous v11-02c version of the GEOS-Chem isoprene mechanism.
RCIM estimates a higher fraction of isoprene reacting with OH globally (88 %) than past mechanisms. The resulting hydroxy-peroxy radicals (ISOPOO) react with HO2 (41 %), NO (28 %), and RO2 (9 %) or undergo H shifts to regenerate HOx (22 %). The dynamic system of ISOPOO isomers, and the differences in H-shift rates between isomers, has important consequences for subsequent product formation. We show that the depletion of 4-OH ISOPOO due to its rapid H shift leads to higher MVK ∕ MACR ratios, higher tertiary nitrate production, and lower MPAN production than is simulated by mechanisms that do not treat the 1-OH and 4-OH ISOPOO systems separately.
The global effects of isoprene chemistry on radical families and ozone are similar in RCIM to past mechanisms, with notable regional differences. We find that isoprene is responsible for an 11 % reduction in OH averaged over the troposphere, causing a 12 % increase in the tropospheric lifetime of methane. Depletion of OH under low-NO conditions is much less than in previous mechanisms because of HOx recycling from H-shift pathways. Isoprene oxidation results in a 6.5 % increase in mean tropospheric HO2 and a 4.2 % decrease in NOx. It increases tropospheric ozone by 1.9 ppbv globally but depresses ozone by up to 3.4 ppbv over tropical forests.
Mass conservation in RCIM enables a detailed accounting of the atmospheric fate of isoprene-derived carbon and the yields of oxidation products. We find globally that 50 % of isoprene is oxidized to CO2 in the gas phase, 76 % of which proceeds via CO, including 44 % via formaldehyde. Another 37 % of isoprene-derived carbon is lost to organic deposition, while 13 % forms SOA. For both formaldehyde and glyoxal, RCIM results in higher yields under low-NO conditions than previous mechanisms. However, deposition and aerosol uptake of isoprene oxidation intermediates greatly depress the glyoxal yield relative to previous mechanisms.
The largest changes in RCIM relative to previous mechanisms are for organonitrates and SOA. We find that isoprene contributes 20 % of the tropospheric burden of peroxyacyl nitrates and 28 % of non-peroxyacyl nitrates, lower than in previous mechanisms. The implementation of fast tertiary nitrate hydrolysis leads to a NOx sink of 4.9 TgN a−1 globally, or 10 % of total NOx loss. Only 20 % of isoprene-derived organonitrates (excluding peroxyacyl nitrates) chemically recycle NOx. We estimate the total global source of SOA from isoprene to be 61 TgC a−1 (136 Tg a−1), with approximately equal contributions from IEPOX, organonitrates, and highly functionalized C5 compounds. This 13 % SOA yield per carbon (25 % yield by mass) is much higher than in previous global models, due primarily to our inclusion of additional precursors, but is similar to a recent estimate by Stadtler et al. (2018). Such high yields imply that SOA produced from isoprene cannot be regarded as chemically inert and must further react in the aerosol phase to generate volatile products. This aerosol-phase chemistry is not yet included in RCIM and is a topic for further research.
Finally, we compiled a Mini-CIM mechanism that makes further simplifications to RCIM to decrease the computational burden of simulating isoprene chemistry. The Mini-CIM has 108 species and 345 reactions, comparable in size to previous mechanisms implemented in GEOS-Chem while remaining closely consistent with the original mechanism of Wennberg et al. (2018). Global simulations with the Mini-CIM exhibit minimal deviations from RCIM for atmospherically relevant applications.
The RCIM and Mini-CIM mechanisms used here are available online (https://doi.org/10.22002/d1.247, Bates and Wennberg, 2017) along with the KPP code for conducting box model simulations and the model output discussed in this paper. MCM (http://mcm.leeds.ac.uk/MCM/, Rickard and Young, 2019) and GEOS-Chem (http://geos-chem.org, International GEOS-Chem Community, 2019) are both available online for public use.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-9613-2019-supplement.
KHB designed and carried out the simulations described herein and prepared the paper with substantial assistance from DJJ.
The authors declare that they have no conflict of interest.
Kelvin H. Bates acknowledges the support of the Harvard University Center for the Environment and the National Oceanic and Atmospheric Administration's Climate and Global Change Fellowship programs.
This paper was edited by Ulrich Pöschl and reviewed by two anonymous referees.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008. a
Allen, H. M., Crounse, J. D., Bates, K. H., Teng, A. P., Krawiec-Thayer, M. P., Rivera-Rios, J. C., Keutsch, F. N., St. Clair, J. M., Hanisco, T. F., Möller, K. H., Kjaergaard, H. G., and Wennberg, P. O.: Kinetics and product yields of the OH initiated oxidation of hydroxymethyl hydroperoxide, J. Phys. Chem. A, 122, 6292–6302, https://doi.org/10.1021/acs.jpca.8b04577, 2018. a
Alvarado, L. M. A., Richter, A., Vrekoussis, M., Wittrock, F., Hilboll, A., Schreier, S. F., and Burrows, J. P.: An improved glyoxal retrieval from OMI measurements, Atmos. Meas. Tech., 7, 4133–4150, https://doi.org/10.5194/amt-7-4133-2014, 2014. a
Archibald, A. T., Cooke, M. C., Utembe, S. R., Shallcross, D. E., Derwent, R. G., and Jenkin, M. E.: Impacts of mechanistic changes on HOx formation and recycling in the oxidation of isoprene, Atmos. Chem. Phys., 10, 8097–8118, https://doi.org/10.5194/acp-10-8097-2010, 2010. a, b
Atkinson, R., Aschmann, S. M., Tuazon, E. C., Arey, J., and Zielinska, B.: Formation of 3-Methylfuran from the gas-phase reaction of OH radicals with isoprene and the rate constant for its reaction with the OH radical, Int. J. Chem. Kinet., 21, 593–604, https://doi.org/10.1002/kin.550210709, 1989. a
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005. a
Barkley, M. P., Palmer, P. I., Ganzeveld, L., Arneth, A., Hagberg, D., Karl, T., Guenther, A., Paulot, F., Wennberg, P. O., Mao, J., Kurosu, T. P., Chance, K., Müller, J.-F., De Smedt, I., Van Roozendael, M., Chen, D., Wang, Y., and Yantosca, R. M.: Can a “state of the art” chemistry transport model simulate Amazonian tropospheric chemistry?, J. Geophys. Res.-Atmos., 116, D16302, https://doi.org/10.1029/2011JD015893, 2011. a, b
Barkley, M. P., de Smedt, I., Van Roozendael, M., Kurosu, T. P., Chance, K., Arneth, A., Hagberg, D., Guenther, A., Paulot, F., Marais, E., and Mao, J.: Top-down isoprene emissions over tropical South America inferred from SCIAMACHY and OMI formaldehyde columns, J. Geophys. Res.-Atmos., 118, 6849–6868, https://doi.org/10.1002/jgrd.50552, 2013. a
Bates, K. H., Crounse, J. D., St Clair, J. M., Bennett, N. B., Nguyen, T. B., Seinfeld, J. H., Stoltz, B. M., and Wennberg, P. O.: Gas phase production and loss of isoprene epoxydiols, J. Phys. Chem. A, 118, 1237–46, https://doi.org/10.1021/jp4107958, 2014. a, b, c, d
Bates, K. H., Nguyen, T. B., Teng, A. P., Crounse, J. D., Kjaergaard, H. G., Stoltz, B. M., Seinfeld, J. H., and Wennberg, P. O.: Production and fate of C4 dihydroxycarbonyl compounds from isoprene oxidation, J. Phys. Chem. A, 120, 106–117, https://doi.org/10.1021/acs.jpca.5b10335, 2016. a, b
Bates, K. H. and Wennberg, P. O.: Isoprene Oxidation Model, https://doi.org/10.22002/d1.247, 2017. a, b
Bergamaschi, P., Hein, R., Heimann, M., and Crutzen, P. J.: Inverse modeling of the global CO cycle: 1. Inversion of CO mixing ratios, J. Geophys. Res.-Atmos., 105, 1909–1927, https://doi.org/10.1029/1999JD900818, 2000. a
Biesenthal, T. A. and Shepson, P. B.: Observations of anthropogenic inputs of the isoprene oxidation products methyl vinyl ketone and methacrolein to the atmosphere, Geophys. Res. Lett., 24, 1375–1378, https://doi.org/10.1029/97GL01337, 1997. a
Brewer, D. A., Ogliaruso, M. A., Augustsson, T. R., and Levine, J. S.: The oxidation of isoprene in the troposphere: Mechanism and model calculations, Atmos. Environ., 18, 2723–2744, https://doi.org/10.1016/0004-6981(84)90338-X, 1984. a
Butler, T. M., Taraborrelli, D., Brühl, C., Fischer, H., Harder, H., Martinez, M., Williams, J., Lawrence, M. G., and Lelieveld, J.: Improved simulation of isoprene oxidation chemistry with the ECHAM5/MESSy chemistry-climate model: lessons from the GABRIEL airborne field campaign, Atmos. Chem. Phys., 8, 4529–4546, https://doi.org/10.5194/acp-8-4529-2008, 2008. a
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009. a, b
Carslaw, N., Creasey, D., Harrison, D., Heard, D., Hunter, M., Jacobs, P., Jenkin, M., Lee, J., Lewis, A., Pilling, M., Saunders, S., and Seakins, P.: OH and HO2 radical chemistry in a forested region of north-western Greece, Atmos. Environ., 35, 4725–4737, https://doi.org/10.1016/S1352-2310(01)00089-9, 2001. a
Carter, W. P. L.: Condensed atmospheric photooxidation mechanisms for isoprene, Atmos. Environ., 30, 4275–4290, https://doi.org/10.1016/1352-2310(96)00088-X, 1996. a
Chan Miller, C., Gonzalez Abad, G., Wang, H., Liu, X., Kurosu, T., Jacob, D. J., and Chance, K.: Glyoxal retrieval from the Ozone Monitoring Instrument, Atmos. Meas. Tech., 7, 3891–3907, https://doi.org/10.5194/amt-7-3891-2014, 2014. a
Chan Miller, C., Jacob, D. J., González Abad, G., and Chance, K.: Hotspot of glyoxal over the Pearl River delta seen from the OMI satellite instrument: implications for emissions of aromatic hydrocarbons, Atmos. Chem. Phys., 16, 4631–4639, https://doi.org/10.5194/acp-16-4631-2016, 2016. a
Chan Miller, C., Jacob, D. J., Marais, E. A., Yu, K., Travis, K. R., Kim, P. S., Fisher, J. A., Zhu, L., Wolfe, G. M., Hanisco, T. F., Keutsch, F. N., Kaiser, J., Min, K.-E., Brown, S. S., Washenfelder, R. A., González Abad, G., and Chance, K.: Glyoxal yield from isoprene oxidation and relation to formaldehyde: chemical mechanism, constraints from SENEX aircraft observations, and interpretation of OMI satellite data, Atmos. Chem. Phys., 17, 8725–8738, https://doi.org/10.5194/acp-17-8725-2017, 2017. a, b, c, d, e, f
Claeys, M., Graham, B., Vas, G., Wang, W., Vermeylen, R., Pashynska, V., Cafmeyer, J., Guyon, P., Andreae, M. O., Artaxo, P., and Maenhaut, W.: Formation of secondary organic aerosols through photooxidation of isoprene, Science, 303, 1173–1176, https://doi.org/10.1126/science.1092805, 2004. a
Cleary, P. A., Wooldridge, P. J., Millet, D. B., McKay, M., Goldstein, A. H., and Cohen, R. C.: Observations of total peroxy nitrates and aldehydes: measurement interpretation and inference of OH radical concentrations, Atmos. Chem. Phys., 7, 1947–1960, https://doi.org/10.5194/acp-7-1947-2007, 2007. a
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011. a
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric fate of methacrolein. 1. Peroxy radical isomerization following adition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012. a
Crounse, J. D., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a
Daescu, D., Sandu, A., and Carmichael, G.: Direct and adjoint sensitivity analysis of chemical kinetic systems with KPP: II – validation and numerical experiments, Atmos. Environ., 37, 5097–5114, https://doi.org/10.1016/j.atmosenv.2003.08.020, 2003. a
D'Ambro, E. L., Moller, K. H., Lopez-Hilfiker, F. D., Schobesberger, S., Liu, J., Shilling, J. E., Lee, B. H., Kjaergaard, H. G., and Thornton, J. A.: Isomerization of second generation isoprene peroxy radicals: Epoxide formation and implications for secondary organic aerosol yields, Environ. Sci. Technol., 51, 4978–4987, https://doi.org/10.1021/acs.est.7b00460, 2017. a, b, c
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G.: The Kinetic PreProcessor (KPP) – a software environment for solving chemical kinetics, Comput. Chem. Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002. a
Darer, A. I., Cole-Filipiak, N. C., O’Connor, A. E., and Elrod, M. J.: Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011. a, b
de Gouw, J. and Jimenez, J. L.: Organic aerosols in the Earth's atmosphere, Environ. Sci. Technol., 43, 7614–7618, https://doi.org/10.1021/es9006004, 2009. a
Dillon, T. J. and Crowley, J. N.: Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals, Atmos. Chem. Phys., 8, 4877–4889, https://doi.org/10.5194/acp-8-4877-2008, 2008. a
Duncan, B. N., Logan, J. A., Bey, I., Megretskaia, I. A., Yantosca, R. M., Novelli, P. C., Jones, N. B., and Rinsland, C. P.: Global budget of CO, 1988–1997: Source estimates and validation with a global model, J. Geophys. Res.-Atmos., 112, D22301, https://doi.org/10.1029/2007JD008459, 2007. a
Eastham, S. D., Weisenstein, D. K., and Barrett, S. R.: Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem, Atmos. Environ., 89, 52–63, https://doi.org/10.1016/j.atmosenv.2014.02.001, 2014. a
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005. a, b
Eerdekens, G., Ganzeveld, L., Vilà-Guerau de Arellano, J., Klüpfel, T., Sinha, V., Yassaa, N., Williams, J., Harder, H., Kubistin, D., Martinez, M., and Lelieveld, J.: Flux estimates of isoprene, methanol and acetone from airborne PTR-MS measurements over the tropical rainforest during the GABRIEL 2005 campaign, Atmos. Chem. Phys., 9, 4207–4227, https://doi.org/10.5194/acp-9-4207-2009, 2009. a
Fan, J. and Zhang, R.: Atmospheric oxidation mechanism of isoprene, Environ. Chem., 1, 140–149, https://doi.org/10.1071/EN04045, 2004. a
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., DeGouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing atmospheric oxidation in an Alabama forest, J. Atmos. Sci., 73, 4699–4710, https://doi.org/10.1175/JAS-D-16-0044.1, 2016. a
Fiore, A. M., Dentener, F. J., Wild, O., Cuvelier, C., Schultz, M. G., Hess, P., Textor, C., Schulz, M., Doherty, R. M., Horowitz, L. W., MacKenzie, I. A., Sanderson, M. G., Shindell, D. T., Stevenson, D. S., Szopa, S., Van Dingenen, R., Zeng, G., Atherton, C., Bergmann, D., Bey, I., Carmichael, G., Collins, W. J., Duncan, B. N., Faluvegi, G., Folberth, G., Gauss, M., Gong, S., Hauglustaine, D., Holloway, T., Isaksen, I. S. A., Jacob, D. J., Jonson, J. E., Kaminski, J. W., Keating, T. J., Lupu, A., Marmer, E., Montanaro, V., Park, R. J., Pitari, G., Pringle, K. J., Pyle, J. A., Schroeder, S., Vivanco, M. G., Wind, P., Wojcik, G., Wu, S., and Zuber, A.: Multimodel estimates of intercontinental source-receptor relationships for ozone pollution, J. Geophys. Res.-Atmos., 114, D04301, https://doi.org/10.1029/2008JD010816, 2009. a
Fiore, A. M., Naik, V., Spracklen, D. V., Steiner, A., Unger, N., Prather, M., Bergmann, D., Cameron-Smith, P. J., Cionni, I., Collins, W. J., Dalsøren, S., Eyring, V., Folberth, G. A., Ginoux, P., Horowitz, L. W., Josse, B., Lamarque, J.-F., MacKenzie, I. A., Nagashima, T., O'Connor, F. M., Righi, M., Rumbold, S. T., Shindell, D. T., Skeie, R. B., Sudo, K., Szopa, S., Takemura, T., and Zeng, G.: Global air quality and climate, Chem. Soc. Rev., 41, 6663–6683, https://doi.org/10.1039/C2CS35095E, 2012. a
Fischer, E. V., Jacob, D. J., Yantosca, R. M., Sulprizio, M. P., Millet, D. B., Mao, J., Paulot, F., Singh, H. B., Roiger, A., Ries, L., Talbot, R. W., Dzepina, K., and Pandey Deolal, S.: Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 14, 2679–2698, https://doi.org/10.5194/acp-14-2679-2014, 2014. a, b
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016. a, b, c, d
Fisher, J. A., Murray, L. T., Jones, D. B. A., and Deutscher, N. M.: Improved method for linear carbon monoxide simulation and source attribution in atmospheric chemistry models illustrated using GEOS-Chem v9, Geosci. Model Dev., 10, 4129–4144, https://doi.org/10.5194/gmd-10-4129-2017, 2017. a
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.-Atmos., 113, D15303, https://doi.org/10.1029/2007JD009505, 2008. a
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Haseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013. a
Ganzeveld, L., Bouwman, L., Stehfest, E., van Vuuren, D. P., Eickhout, B., and Lelieveld, J.: Impact of future land use and land cover changes on atmospheric chemistry-climate interactions, J. Geophys. Res.-Atmos., 115, D23301, https://doi.org/10.1029/2010JD014041, 2010. a
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014. a
Geiger, H., Barnes, I., Bejan, I., Benter, T., and Spittler, M.: The tropospheric degradation of isoprene: An updated module for the regional atmospheric chemistry mechanism, Atmos. Environ., 37, 1503–1519, https://doi.org/10.1016/S1352-2310(02)01047-6, 2003. a
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the Earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007. a
Greenberg, J., Guenther, A., Zimmerman, P., Baugh, W., Geron, C., Davis, K., Helmig, D., and Klinger, L.: Tethered balloon measurements of biogenic VOCs in the atmospheric boundary layer, Atmos. Environ., 33, 855–867, https://doi.org/10.1016/S1352-2310(98)00302-1, 1999. a
Grosjean, D., Williams II, E. L., and Grosjean, E.: Atmospheric chemistry of isoprene and of its carbonyl products, Environ. Sci. Technol., 27, 830–840, https://doi.org/10.1021/es00042a004, 1993. a
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., McKay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995. a
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a, b, c
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009. a
Hasson, A. S., Tyndall, G. S., and Orlando, J. J.: A product yield study of the reaction of HO2 radicals with ethyl peroxy (C2H5O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals, J. Phys. Chem. A, 108, 5979–5989, https://doi.org/10.1021/jp048873t, 2004. a
Heald, C. L., Ridley, D. A., Kreidenweis, S. M., and Drury, E. E.: Satellite observations cap the atmospheric organic aerosol budget, Geophys. Res. Lett., L24808, 37, https://doi.org/10.1029/2010GL045095, 2010. a
Hewitt, C. N., Kok, G. L., and Fall, R.: Hydroperoxides in plants exposed to ozone mediate air pollution damage to alkene emitters, Nature, 344, 56–58, https://doi.org/10.1038/344056a0, 1990. a
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016. a
Horowitz, L. W., Fiore, A. M., Milly, G. P., Cohen, R. C., Perring, A., Wooldridge, P. J., Hess, P. G., Emmons, L. K., and Lamarque, J.-F.: Observational constraints on the chemistry of isoprene nitrates over the eastern United States, J. Geophys. Res.-Atmos., 112, D12S08, https://doi.org/10.1029/2006JD007747, 2007. a, b, c
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011. a, b
Hu, L., Millet, D. B., Baasandorj, M., Griffis, T. J., Turner, P., Helmig, D., Curtis, A. J., and Hueber, J.: Isoprene emissions and impacts over an ecological transition region in the U.S. Upper Midwest inferred from tall tower measurements, J. Geophys. Res.-Atmos., 120, 3553–3571, https://doi.org/10.1002/2014JD022732, 2015. a
Huang, L., Jiang, J. H., Murray, L. T., Damon, M. R., Su, H., and Livesey, N. J.: Evaluation of UTLS carbon monoxide simulations in GMI and GEOS-Chem chemical transport models using Aura MLS observations, Atmos. Chem. Phys., 16, 5641–5663, https://doi.org/10.5194/acp-16-5641-2016, 2016. a
International GEOS-Chem Community: GEOS-Chem, available at: http://www.geos-chem.org, last access: 26 July 2019. a
Ito, A., Sillman, S., and Penner, J. E.: Global chemical transport model study of ozone response to changes in chemical kinetics and biogenic volatile organic compounds emissions due to increasing temperatures: Sensitivities to isoprene nitrate chemistry and grid resolution, J. Geophys. Res.-Atmos., 114, D09301, https://doi.org/10.1029/2008jd011254, 2009. a
Jacob, D. J. and Wofsy, S. C.: Photochemistry of biogenic emissions over the Amazon forest, J. Geophys. Res.-Atmos., 93, 1477–1486, https://doi.org/10.1029/JD093iD02p01477, 1988. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jenkin, M. E., Hurley, M. D., and Wallington, T. J.: Investigation of the radical product channel of the CH3C(O)O2 + HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 9, 3149–3162, https://doi.org/10.1039/B702757E, 2007. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a, b, c, d, e
Jiang, K., Hill, D. R., and Elrod, M. J.: Assessing the potential for oligomer formation from the reactions of lactones in secondary organic aerosols, J. Phys. Chem. A, 122, 292–302, https://doi.org/10.1021/acs.jpca.7b10411, 2018. a, b
Jin, X. and Holloway, T.: Spatial and temporal variability of ozone sensitivity over China observed from the Ozone Monitoring Instrument, J. Geophys. Res.-Atmos., 120, 7229–7246, https://doi.org/10.1002/2015JD023250, 2015. a
Jørgensen, S., Knap, H. C., Otkjær, R. V., Jensen, A. M., Kjeldsen, M. L. H., Wennberg, P. O., and Kjaergaard, H. G.: Rapid hydrogen shift scrambling in hydroperoxy-substituted organic peroxy radicals, J. Phys. Chem. A, 120, 266–275, https://doi.org/10.1021/acs.jpca.5b06768, 2016. a, b
Kaiser, J., Wolfe, G. M., Min, K. E., Brown, S. S., Miller, C. C., Jacob, D. J., deGouw, J. A., Graus, M., Hanisco, T. F., Holloway, J., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Washenfelder, R. A., and Keutsch, F. N.: Reassessing the ratio of glyoxal to formaldehyde as an indicator of hydrocarbon precursor speciation, Atmos. Chem. Phys., 15, 7571–7583, https://doi.org/10.5194/acp-15-7571-2015, 2015. a
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016. a
Kaiser, J., Jacob, D. J., Zhu, L., Travis, K. R., Fisher, J. A., González Abad, G., Zhang, L., Zhang, X., Fried, A., Crounse, J. D., St. Clair, J. M., and Wisthaler, A.: High-resolution inversion of OMI formaldehyde columns to quantify isoprene emission on ecosystem-relevant scales: application to the southeast US, Atmos. Chem. Phys., 18, 5483–5497, https://doi.org/10.5194/acp-18-5483-2018, 2018. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a
Karl, T., Guenther, A., Turnipseed, A., Tyndall, G., Artaxo, P., and Martin, S.: Rapid formation of isoprene photo-oxidation products observed in Amazonia, Atmos. Chem. Phys., 9, 7753–7767, https://doi.org/10.5194/acp-9-7753-2009, 2009. a, b
Kaser, L., Karl, T., Yuan, B., Mauldin, R. L., Cantrell, C. A., Guenther, A. B., Patton, E. G., Weinheimer, A. J., Knote, C., Orlando, J., Emmons, L., Apel, E., Hornbrook, R., Shertz, S., Ullmann, K., Hall, S., Graus, M., Gouw, J., Zhou, X., and Ye, C.: Chemistry-turbulence interactions and mesoscale variability influence the cleansing efficiency of the atmosphere, Geophys. Res. Lett., 42, 10,894–10,903, https://doi.org/10.1002/2015GL066641, 2015. a
Keller, C. A., Long, M. S., Yantosca, R. M., Da Silva, A. M., Pawson, S., and Jacob, D. J.: HEMCO v1.0: a versatile, ESMF-compliant component for calculating emissions in atmospheric models, Geosci. Model Dev., 7, 1409–1417, https://doi.org/10.5194/gmd-7-1409-2014, 2014. a
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015. a, b
Kjaergaard, H. G., Knap, H. C., Ornso, K. B., Jorgensen, S., Crounse, J. D., Paulot, F., and Wennberg, P. O.: Atmospheric fate of methacrolein. 2. Formation of lactone and implications for organic aerosol production, J. Phys. Chem. A, 116, 5763–5768, https://doi.org/10.1021/jp210853h, 2012. a
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007. a
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., Clair, J. M. S., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation hydroxyhydroperoxide low-NO oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015. a, b
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation under high‐NOx conditions, Geophys. Res. Lett., 32, L18808, https://doi.org/10.1029/2005GL023637, 2005. a
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006. a
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010. a
Kuhn, U., Andreae, M. O., Ammann, C., Araújo, A. C., Brancaleoni, E., Ciccioli, P., Dindorf, T., Frattoni, M., Gatti, L. V., Ganzeveld, L., Kruijt, B., Lelieveld, J., Lloyd, J., Meixner, F. X., Nobre, A. D., Pöschl, U., Spirig, C., Stefani, P., Thielmann, A., Valentini, R., and Kesselmeier, J.: Isoprene and monoterpene fluxes from Central Amazonian rainforest inferred from tower-based and airborne measurements, and implications on the atmospheric chemistry and the local carbon budget, Atmos. Chem. Phys., 7, 2855–2879, https://doi.org/10.5194/acp-7-2855-2007, 2007. a
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995. a
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L. Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016. a, b
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On rates and mechanisms of OH and O3 reactions with isoprene-derived hydroxy nitrates, J. Phys. Chem. A, 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014. a
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008. a, b
Li, C., Balluz, L. S., Vaidyanathan, A., Wen, X.-J., Hao, Y., and Qualters, J. R.: Long-term exposure to ozone and life expectancy in the United States, 2002 to 2008, Medicine, 95, e2474, https://doi.org/10.1097/MD.0000000000002474, 2016. a, b
Liao, J., Froyd, K. D., Murphy, D. M., Keutsch, F. N., Yu, G., Wennberg, P. O., St. Clair, J. M., Crounse, J. D., Wisthaler, A., Mikoviny, T., Jimenez, J.-L., Campuzano-Jost, P., Day, D. A., Hu, W., Ryerson, T. B., Pollack, I. B., Peischl, J., Anderson, B. E., Ziemba, L. D., Blake, D. R., Meinardi, S., and Diskin, G.: Airborne measurements of organosulfates over the continental U.S., J. Geophys. Res.-Atmos., 120, 2990–3005, https://doi.org/10.1002/2014JD022378, 2015. a
Lin, J.-T., Youn, D., Liang, X.-Z., and Wuebbles, D. J.: Global model simulation of summertime U.S. ozone diurnal cycle and its sensitivity to PBL mixing, spatial resolution, and emissions, Atmos. Environ., 42, 8470–8483, https://doi.org/10.1016/j.atmosenv.2008.08.012, 2008. a
Lin, Y.-H., Zhang, H., Pye, H. O., Zhang, Z., Marth, W. J., Park, S., Arashiro, M., Cui, T., Budisulistiorini, S. H., Sexton, K. G., Vizuete, W., Xie, Y., Luecken, D. J., Piletic, I. R., Edney, E. O., Bartolotti, L. J., Gold, A., and Surratt, J. D.: Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, P. Natl. Acad. Sci. USA, 110, 6718–6723, https://doi.org/10.1073/pnas.1221150110, 2013. a
Lind, J. A., Lazrus, A. L., and Kok, G. L.: Aqueous phase oxidation of sulfur(IV) by hydrogen peroxide, methylhydroperoxide, and peroxyacetic acid, J. Geophys. Res.-Atmos., 92, 4171–4177, https://doi.org/10.1029/JD092iD04p04171, 1987. a
Liu, J., D’Ambro, E. L., Lee, B. H., Lopez-Hilfiker, F. D., Zaveri, R. A., Rivera-Rios, J. C., Keutsch, F. N., Iyer, S., Kurten, T., Zhang, Z., Gold, A., Surratt, J. D., Shilling, J. E., and Thornton, J. A.: Efficient isoprene secondary organic aerosol formation from a non-IEPOX pathway, Environ. Sci. Technol., 50, 9872–9880, https://doi.org/10.1021/acs.est.6b01872, 2016. a, b
Lloyd, A. C., Atkinson, R., Lurmann, F., and Nitta, B.: Modeling potential ozone impacts from natural hydrocarbons – I. Development and testing of a chemical mechanism for the NOx-air photooxidations of isoprene and alpha-pinene under ambient conditions, Atmos. Environ., 17, 1931–1950, https://doi.org/10.1016/0004-6981(83)90351-7, 1983. a
Madronich, S. and Calvert, J. G.: Permutation reactions of organic peroxy radicals in the troposphere, J. Geophys. Res., 95, 5697–5715, https://doi.org/10.1029/JD095iD05p05697, 1990. a
Mao, J., Paulot, F., Jacob, D. J., Cohen, R. C., Crounse, J. D., Wennberg, P. O., Keller, C. A., Hudman, R. C., Barkley, M. P., and Horowitz, L. W.: Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry, J. Geophys. Res., 118, 11256–11268, https://doi.org/10.1002/jgrd.50817, 2013. a, b, c, d
Mao, J., Carlton, A., Cohen, R. C., Brune, W. H., Brown, S. S., Wolfe, G. M., Jimenez, J. L., Pye, H. O. T., Lee Ng, N., Xu, L., McNeill, V. F., Tsigaridis, K., McDonald, B. C., Warneke, C., Guenther, A., Alvarado, M. J., de Gouw, J., Mickley, L. J., Leibensperger, E. M., Mathur, R., Nolte, C. G., Portmann, R. W., Unger, N., Tosca, M., and Horowitz, L. W.: Southeast Atmosphere Studies: learning from model-observation syntheses, Atmos. Chem. Phys., 18, 2615–2651, https://doi.org/10.5194/acp-18-2615-2018, 2018. a
Marais, E. A., Jacob, D. J., Kurosu, T. P., Chance, K., Murphy, J. G., Reeves, C., Mills, G., Casadio, S., Millet, D. B., Barkley, M. P., Paulot, F., and Mao, J.: Isoprene emissions in Africa inferred from OMI observations of formaldehyde columns, Atmos. Chem. Phys., 12, 6219–6235, https://doi.org/10.5194/acp-12-6219-2012, 2012. a
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016. a, b, c, d, e, f, g, h, i, j, k, l
Marais, E. A., Jacob, D. J., Turner, J. R., and Mickley, L. J.: Evidence of 1991–2013 decrease of biogenic secondary organic aerosol in response to SO2 emission controls, Environ. Res. Lett., 12, 054018, https://doi.org/10.1088/1748-9326/aa69c8, 2017. a
Martinez, M., Harder, H., Kubistin, D., Rudolf, M., Bozem, H., Eerdekens, G., Fischer, H., Klüpfel, T., Gurk, C., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: airborne measurements, Atmos. Chem. Phys., 10, 3759–3773, https://doi.org/10.5194/acp-10-3759-2010, 2010. a
Marvin, M. R., Wolfe, G. M., Salawitch, R. J., Canty, T. P., Roberts, S. J., Travis, K. R., Aikin, K. C., de Gouw, J. A., Graus, M., Hanisco, T. F., Holloway, J. S., Hüler, G., Kaiser, J., Keutsch, F. N., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Veres, P. R., and Warneke, C.: Impact of evolving isoprene mechanisms on simulated formaldehyde: An inter-comparison supported by in situ observations from SENEX, Atmos. Environ., 164, 325–336, https://doi.org/10.1016/j.atmosenv.2017.05.049, 2017. a
McNeill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: A modeling study, Environ. Sci. Technol., 46, 8075–8081, https://doi.org/10.1021/es3002986, 2012. a
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015. a
Miyoshi, A., Hatakeyama, S., and Washida, N.: OH radical-initiated photooxidation of isoprene: An estimate of global CO production, J. Geophys. Res.-Atmos., 99, 18779–18787, https://doi.org/10.1029/94JD01334, 1994. a, b
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The importance of peroxy radical hydrogen-shift reactions in atmospheric isoprene oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019. a, b, c
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014. a
Müller, J.-F., Stavrakou, T., and Peeters, J.: Chemistry and deposition in the Model of Atmospheric composition at Global and Regional scales using Inversion Techniques for Trace gas Emissions (MAGRITTE v1.1) – Part 1: Chemical mechanism, Geosci. Model Dev., 12, 2307–2356, https://doi.org/10.5194/gmd-12-2307-2019, 2019. a, b, c, d, e, f
Murazaki, K. and Hess, P.: How does climate change contribute to surface ozone change over the United States?, J. Geophys. Res.-Atmos., 111, D05301, https://doi.org/10.1029/2005JD005873, 2006. a
Myriokefalitakis, S., Vrekoussis, M., Tsigaridis, K., Wittrock, F., Richter, A., Brühl, C., Volkamer, R., Burrows, J. P., and Kanakidou, M.: The influence of natural and anthropogenic secondary sources on the glyoxal global distribution, Atmos. Chem. Phys., 8, 4965–4981, https://doi.org/10.5194/acp-8-4965-2008, 2008. a
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008. a, b
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014. a, b
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/C5CP02001H, 2015a. a, b, c, d
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015b. a
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/C6CP00053C, 2016. a, b
Pacifico, F., Folberth, G. A., Jones, C. D., Harrison, S. P., and Collins, W. J.: Sensitivity of biogenic isoprene emissions to past, present, and future environmental conditions and implications for atmospheric chemistry, J. Geophys. Res.-Atmos., 117, D22302, https://doi.org/10.1029/2012JD018276, 2012. a
Pai, S. J., Heald, C. L., Pierce, J. R., Farina, S. C., Marais, E. A., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Middlebrook, A. M., Coe, H., Shilling, J. E., Bahreini, R., Dingle, J. H., and Vu, K.: An evaluation of global organic aerosol schemes using airborne observations, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-331, in review, 2019. a
Palmer, P. I., Jacob, D. J., Fiore, A. M., Martin, R. V., Chance, K., and Kurosu, T. P.: Mapping isoprene emissions over North America using formaldehyde column observations from space, J. Geophys. Res.-Atmos., 108, 4180, https://doi.org/10.1029/2002JD002153, 2003. a
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, https://doi.org/10.5194/acp-9-1479-2009, 2009a. a, b, c, d
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kurten, A., St Clair, J., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009b. a, b, c
Paulot, F., Henze, D. K., and Wennberg, P. O.: Impact of the isoprene photochemical cascade on tropical ozone, Atmos. Chem. Phys., 12, 1307–1325, https://doi.org/10.5194/acp-12-1307-2012, 2012. a, b
Paulot, F., Jacob, D. J., and Henze, D. K.: Sources and processes contributing to nitrogen deposition: An adjoint model analysis applied to biodiversity hotspots worldwide, Environ. Sci. Technol., 47, 3226–3233, https://doi.org/10.1021/es3027727, 2013. a
Paulson, S. E. and Seinfeld, J. H.: Development and evaluation of a photooxidation mechanism for isoprene, J. Geophys. Res.-Atmos., 97, 20703–20715, https://doi.org/10.1029/92JD01914, 1992. a
Paulson, S. E., Flagan, R. C., and Seinfeld, J. H.: Atmospheric photooxidation of isoprene part I: The hydroxyl radical and ground state atomic oxygen reactions, Int. J. Chem. Kinet., 24, 79–101, https://doi.org/10.1002/kin.550240109, 1992. a
Peeters, J. and Muller, J. F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: Experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, https://doi.org/10.1039/c0cp00811g, 2010. a
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009. a, b
Peeters, J., Muller, J. F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014. a, b, c
Pfister, G. G., Emmons, L. K., Hess, P. G., Lamarque, J.-F., Orlando, J. J., Walters, S., Guenther, A., Palmer, P. I., and Lawrence, P. J.: Contribution of isoprene to chemical budgets: A model tracer study with the NCAR CTM MOZART-4, J. Geophys. Res., 113, D05308, https://doi.org/10.1029/2007JD008948, 2008. a, b, c, d
Pöschl, U. and Shiraiwa, M.: Multiphase chemistry at the atmosphere-biosphere interface influencing climate and public health in the anthropocene, Chem. Rev., 115, 4440–4475, https://doi.org/10.1021/cr500487s, 2015. a
Pöschl, U., von Kuhlmann, R., Poisson, N., and Crutzen, P. J.: Development and intercomparison of condensed isoprene oxidation mechanisms for global atmospheric modeling, J. Atmos. Chem., 37, 29–52, https://doi.org/10.1023/A:1006391009798, 2000. a
Pugh, T. A. M., MacKenzie, A. R., Hewitt, C. N., Langford, B., Edwards, P. M., Furneaux, K. L., Heard, D. E., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J., Mills, G., Misztal, P., Moller, S., Monks, P. S., and Whalley, L. K.: Simulating atmospheric composition over a South-East Asian tropical rainforest: performance of a chemistry box model, Atmos. Chem. Phys., 10, 279–298, https://doi.org/10.5194/acp-10-279-2010, 2010. a
Rasmussen, D., Fiore, A., Naik, V., Horowitz, L., McGinnis, S., and Schultz, M.: Surface ozone-temperature relationships in the eastern US: A monthly climatology for evaluating chemistry-climate models, Atmos. Environ., 47, 142–153, https://doi.org/10.1016/j.atmosenv.2011.11.021, 2012. a
Rickard, A. and Young, J.: The Master Chemical Mechanism Version 3.3.1, available at: http://mcm.leeds.ac.uk/MCM/, last access: 26 July 2019. a
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: Determination of α-pinene-derived organic nitrate yields: particle phase partitioning and hydrolysis, Atmos. Chem. Phys. Discuss., 14, 3301–3335, https://doi.org/10.5194/acpd-14-3301-2014, 2014. a, b
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016. a, b
Riva, M., Bell, D. M., Hansen, A.-M. K., Drozd, G. T., Zhang, Z., Gold, A., Imre, D., Surratt, J. D., Glasius, M., and Zelenyuk, A.: Effect of organic coatings, humidity and aerosol acidity on multiphase chemistry of isoprene epoxydiols, Environ. Sci. Technol., 50, 5580–5588, https://doi.org/10.1021/acs.est.5b06050, 2016. a
Roberts, J. M., Williams, J., Baumann, K., Buhr, M. P., Goldan, P. D., Holloway, J., Hübler, G., Kuster, W. C., McKeen, S. A., Ryerson, T. B., Trainer, M., Williams, E. J., Fehsenfeld, F. C., Bertman, S. B., Nouaime, G., Seaver, C., Grodzinsky, G., Rodgers, M., and Young, V. L.: Measurements of PAN, PPN, and MPAN made during the 1994 and 1995 Nashville Intensives of the Southern Oxidant Study: Implications for regional ozone production from biogenic hydrocarbons, J. Geophys. Res.-Atmos., 103, 22473–22490, https://doi.org/10.1029/98JD01637, 1998. a
Roberts, J. M., Flocke, F., Stroud, C. A., Hereid, D., Williams, E., Fehsenfeld, F., Brune, W., Martinez, M., and Harder, H.: Ground-based measurements of peroxycarboxylic nitric anhydrides (PANs) during the 1999 Southern Oxidants Study Nashville Intensive, J. Geophys. Res.-Atmos., 107, ACH 1-1–ACH 1-10, https://doi.org/10.1029/2001JD000947, 2002. a
Roberts, J. M., Marchewka, M., Bertman, S. B., Sommariva, R., Warneke, C., de Gouw, J., Kuster, W., Goldan, P., Williams, E., Lerner, B. M., Murphy, P., and Fehsenfeld, F. C.: Measurements of PANs during the New England Air Quality Study 2002, J. Geophys. Res.-Atmos., 112, D20306, https://doi.org/10.1029/2007JD008667, 2007. a
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016. a, b, c
Runge-Morris, M., Frank, P., and Novak, R. F.: Differential effects of organic hydroperoxides and hydrogen peroxide on proteolysis in human erythrocytes, Chem. Res. Toxicol., 2, 76–83, https://doi.org/10.1021/tx00008a002, 1989. a
Ruppert, L. and Becker, K.-H.: A product study of the OH radical-initiated oxidation of isoprene: formation of C5-unsaturated diols, Atmos. Environ., 34, 1529–1542, https://doi.org/10.1016/S1352-2310(99)00408-2, 2000. a
Safieddine, S. A., Heald, C. L., and Henderson, B. H.: The global nonmethane reactive organic carbon budget: A modeling perspective, Geophys. Res. Lett., 44, 3897–3906, https://doi.org/10.1002/2017GL072602, 2017. a, b
Sanderson, M. G., Jones, C. D., Collins, W. J., Johnson, C. E., and Derwent, R. G.: Effect of climate change on isoprene emissions and surface ozone levels, Geophys. Res. Lett., 30, 1936, https://doi.org/10.1029/2003GL017642, 2003. a
Sandu, A., Daescu, D., and Carmichael, G.: Direct and adjoint sensitivity analysis of chemical kinetic systems with KPP: I – theory and software tools, Atmos. Environ., 37, 5083–5096, https://doi.org/10.1016/j.atmosenv.2003.08.019, 2003. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., Clair, J. M. S., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 oxidation products from the RO2+HO2 pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015. a, b
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019. a, b, c
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011. a
Squire, O. J., Archibald, A. T., Abraham, N. L., Beerling, D. J., Hewitt, C. N., Lathière, J., Pike, R. C., Telford, P. J., and Pyle, J. A.: Influence of future climate and cropland expansion on isoprene emissions and tropospheric ozone, Atmos. Chem. Phys., 14, 1011–1024, https://doi.org/10.5194/acp-14-1011-2014, 2014. a
Squire, O. J., Archibald, A. T., Griffiths, P. T., Jenkin, M. E., Smith, D., and Pyle, J. A.: Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century, Atmos. Chem. Phys., 15, 5123–5143, https://doi.org/10.5194/acp-15-5123-2015, 2015. a, b, c
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jorgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and products of the reaction of the first-generation isoprene hydroxy hydroperoxide (ISOPOOH) with OH, J. Phys. Chem. A, 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2015. a
Stadtler, S., Kühn, T., Schröder, S., Taraborrelli, D., Schultz, M. G., and Kokkola, H.: Isoprene-derived secondary organic aerosol in the global aerosol–chemistry–climate model ECHAM6.3.0–HAM2.3–MOZ1.0, Geosci. Model Dev., 11, 3235–3260, https://doi.org/10.5194/gmd-11-3235-2018, 2018. a, b, c, d, e, f, g
Starn, T. K., Shepson, P. B., Bertman, S. B., Riemer, D. D., Zika, R. G., and Olszyna, K.: Nighttime isoprene chemistry at an urban-impacted forest site, J. Geophys. Res.-Atmos., 103, 22437–22447, https://doi.org/10.1029/98JD01201, 1998. a
Stevens, P., L'Esperance, D., Chuong, B., and Martin, G.: Measurements of the kinetics of the OH-initiated oxidation of isoprene: Radical propagation in the OH + isoprene + O2 + NO reaction system, Int. J. Chem. Kinet., 31, 637–643, https://doi.org/10.1002/(SICI)1097-4601(1999)31:9<637::AID-KIN5>3.0.CO;2-O, 1999. a
Stockwell, W. R., Kirchner, F., Kuhn, M., and Seefeld, S.: A new mechanism for regional atmospheric chemistry modeling, J. Geophys. Res.-Atmos., 102, 25847–25879, https://doi.org/10.1029/97JD00849, 1997. a
Stone, D., Evans, M. J., Edwards, P. M., Commane, R., Ingham, T., Rickard, A. R., Brookes, D. M., Hopkins, J., Leigh, R. J., Lewis, A. C., Monks, P. S., Oram, D., Reeves, C. E., Stewart, D., and Heard, D. E.: Isoprene oxidation mechanisms: measurements and modelling of OH and HO2 over a South-East Asian tropical rainforest during the OP3 field campaign, Atmos. Chem. Phys., 11, 6749–6771, https://doi.org/10.5194/acp-11-6749-2011, 2011. a
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene., J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006. a
Szmigielski, R., Surratt, J. D., Vermeylen, R., Szmigielska, K., Kroll, J. H., Ng, N. L., Murphy, S. M., Sorooshian, A., Seinfeld, J. H., and Claeys, M.: Characterization of 2-methylglyceric acid oligomers in secondary organic aerosol formed from the photooxidation of isoprene using trimethylsilylation and gas chromatography/ion trap mass spectrometry, J. Mass Spectrom., 42, 101–116, https://doi.org/10.1002/jms.1146, 2007. a
Taraborrelli, D., Lawrence, M. G., Butler, T. M., Sander, R., and Lelieveld, J.: Mainz Isoprene Mechanism 2 (MIM2): an isoprene oxidation mechanism for regional and global atmospheric modelling, Atmos. Chem. Phys., 9, 2751–2777, https://doi.org/10.5194/acp-9-2751-2009, 2009. a, b, c, d
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017. a, b, c
Trainer, M., Williams, E. J., Parrish, D. D., Buhr, M. P., Allwine, E. J., Westberg, H. H., Fehsenfeld, F. C., and Liu, S. C.: Models and observations of the impact of natural hydrocarbons on rural ozone, Nature, 329, 705–707, 1987. a
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016. a, b, c
Tsigaridis, K., Daskalakis, N., Kanakidou, M., Adams, P. J., Artaxo, P., Bahadur, R., Balkanski, Y., Bauer, S. E., Bellouin, N., Benedetti, A., Bergman, T., Berntsen, T. K., Beukes, J. P., Bian, H., Carslaw, K. S., Chin, M., Curci, G., Diehl, T., Easter, R. C., Ghan, S. J., Gong, S. L., Hodzic, A., Hoyle, C. R., Iversen, T., Jathar, S., Jimenez, J. L., Kaiser, J. W., Kirkevåg, A., Koch, D., Kokkola, H., Lee, Y. H., Lin, G., Liu, X., Luo, G., Ma, X., Mann, G. W., Mihalopoulos, N., Morcrette, J.-J., Müller, J.-F., Myhre, G., Myriokefalitakis, S., Ng, N. L., O'Donnell, D., Penner, J. E., Pozzoli, L., Pringle, K. J., Russell, L. M., Schulz, M., Sciare, J., Seland, Ø., Shindell, D. T., Sillman, S., Skeie, R. B., Spracklen, D., Stavrakou, T., Steenrod, S. D., Takemura, T., Tiitta, P., Tilmes, S., Tost, H., van Noije, T., van Zyl, P. G., von Salzen, K., Yu, F., Wang, Z., Wang, Z., Zaveri, R. A., Zhang, H., Zhang, K., Zhang, Q., and Zhang, X.: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 14, 10845–10895, https://doi.org/10.5194/acp-14-10845-2014, 2014. a
Tuazon, E. C. and Atkinson, R.: A product study of the gas-phase reaction of methacrolein with the OH radical in the presence of NOx, Int. J. Chem. Kinet., 22, 591–602, https://doi.org/10.1002/kin.550220604, 1990. a
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006. a
von Kuhlmann, R., Lawrence, M. G., Pöschl, U., and Crutzen, P. J.: Sensitivities in global scale modeling of isoprene, Atmos. Chem. Phys., 4, 1–17, https://doi.org/10.5194/acp-4-1-2004, 2004. a, b, c
Vrekoussis, M., Wittrock, F., Richter, A., and Burrows, J. P.: Temporal and spatial variability of glyoxal as observed from space, Atmos. Chem. Phys., 9, 4485–4504, https://doi.org/10.5194/acp-9-4485-2009, 2009. a
Wang, K. Y. and Shallcross, D. E.: Modelling terrestrial biogenic isoprene fluxes and their potential impact on global chemical species using a coupled LSM-CTM model, Atmos. Environ., 34, 2909–2925, https://doi.org/10.1016/S1352-2310(99)00525-7, 2000. a
Wang, S., Riva, M., Yan, C., Ehn, M., and Wang, L.: Primary formation of highly oxidized multifunctional products in the OH-initiated oxidation of isoprene: A combined theoretical and experimental study, Environ. Sci. Technol., 52, 12255–12264, https://doi.org/10.1021/acs.est.8b02783, 2018. a
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011. a
Wiedinmyer, C., Friedfeld, S., Baugh, W., Greenberg, J., Guenther, A., Fraser, M., and Allen, D.: Measurement and analysis of atmospheric concentrations of isoprene and its reaction products in central Texas, Atmos. Environ., 35, 1001–1013, https://doi.org/10.1016/S1352-2310(00)00406-4, 2001. a
Wiedinmyer, C., Tie, X., Guenther, A., Neilson, R., and Granier, C.: Future changes in biogenic isoprene emissions: how might they affect regional and global atmospheric chemistry?, Earth Interact., 10, 1–19, https://doi.org/10.1175/EI174.1, 2006. a
Williams, P., Calabrese, E. J., and Moore, G. S.: The effect of methyl oleate hydroperoxide, a possible toxic ozone intermediate, on human normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes, Ecotox. Environ. Safe., 7, 242–248, https://doi.org/10.1016/0147-6513(83)90069-6, 1983. a
Wolfe, G. M., Kaiser, J., Hanisco, T. F., Keutsch, F. N., de Gouw, J. A., Gilman, J. B., Graus, M., Hatch, C. D., Holloway, J., Horowitz, L. W., Lee, B. H., Lerner, B. M., Lopez-Hilifiker, F., Mao, J., Marvin, M. R., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Thornton, J. A., Veres, P. R., and Warneke, C.: Formaldehyde production from isoprene oxidation across NOx regimes, Atmos. Chem. Phys., 16, 2597–2610, https://doi.org/10.5194/acp-16-2597-2016, 2016. a, b
Wu, S., Mickley, L. J., Jacob, D. J., Logan, J. A., Yantosca, R. M., and Rind, D.: Why are there large differences between models in global budgets of tropospheric ozone?, J. Geophys. Res.-Atmos., 112, D05302, https://doi.org/10.1029/2006JD007801, 2007. a
Wu, S., Mickley, L. J., Kaplan, J. O., and Jacob, D. J.: Impacts of changes in land use and land cover on atmospheric chemistry and air quality over the 21st century, Atmos. Chem. Phys., 12, 1597–1609, https://doi.org/10.5194/acp-12-1597-2012, 2012. a
Xie, Y., Paulot, F., Carter, W. P. L., Nolte, C. G., Luecken, D. J., Hutzell, W. T., Wennberg, P. O., Cohen, R. C., and Pinder, R. W.: Understanding the impact of recent advances in isoprene photooxidation on simulations of regional air quality, Atmos. Chem. Phys., 13, 8439–8455, https://doi.org/10.5194/acp-13-8439-2013, 2013. a
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effect of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015. a, b
Yu, K., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Miller, C. C., Travis, K. R., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Cohen, R. C., Dibb, J. E., Fried, A., Mikoviny, T., Ryerson, T. B., Wennberg, P. O., and Wisthaler, A.: Sensitivity to grid resolution in the ability of a chemical transport model to simulate observed oxidant chemistry under high-isoprene conditions, Atmos. Chem. Phys., 16, 4369–4378, https://doi.org/10.5194/acp-16-4369-2016, 2016. a
Yu, S., Mathur, R., Schere, K., Kang, D., Pleim, J., and Otte, T. L.: A detailed evaluation of the Eta-CMAQ forecast model performance for O3, its related precursors, and meteorological parameters during the 2004 ICARTT study, J. Geophys. Res.-Atmos., 112, D12S14, https://doi.org/10.1029/2006JD007715, 2007. a
Yu, S., Mathur, R., Sarwar, G., Kang, D., Tong, D., Pouliot, G., and Pleim, J.: Eta-CMAQ air quality forecasts for O3 and related species using three different photochemical mechanisms (CB4, CB05, SAPRC-99): comparisons with measurements during the 2004 ICARTT study, Atmos. Chem. Phys., 10, 3001–3025, https://doi.org/10.5194/acp-10-3001-2010, 2010. a
Zeng, G., Williams, J. E., Fisher, J. A., Emmons, L. K., Jones, N. B., Morgenstern, O., Robinson, J., Smale, D., Paton-Walsh, C., and Griffith, D. W. T.: Multi-model simulation of CO and HCHO in the Southern Hemisphere: comparison with observations and impact of biogenic emissions, Atmos. Chem. Phys., 15, 7217–7245, https://doi.org/10.5194/acp-15-7217-2015, 2015. a
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J., and Kamens, R. M.: Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions, Atmos. Chem. Phys., 11, 6411–6424, https://doi.org/10.5194/acp-11-6411-2011, 2011. a
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the aerosol-phase state on secondary organic aerosol formation from the reactive uptake of isoprene-derived epoxydiols (IEPOX), Environ. Sci. Technol., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018. a
Zhou, X. and Lee, Y. N.: Aqueous solubility and reaction kinetics of hydroxymethyl hydroperoxide, J. Phys. Chem., 96, 265–272, https://doi.org/10.1021/j100180a051, 1992. a