the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Oct 2019
| 14 Oct 2019
Secondary organic aerosol formation from the laboratory oxidation of biomass burning emissions
David H. Hagan
Matthew M. Coggon
Abigail R. Koss
Kanako Sekimoto
Joost de Gouw
Carsten Warneke
Christopher D. Cappa
In the original version of the Supplement, the authors had mistakenly published primary organic aerosol (POA) values that were not corrected for AMS collection efficiency. The error is limited to Table S1 and Figure S9b in the Supplement and does not impact any other figures or conclusions in the paper. In the attached version of the Supplement, the authors have edited Table S1 and Figure S9b to use the properly corrected POA values.
Biomass burning is an important source of aerosol and trace gases to the atmosphere, but how these emissions change chemically during their lifetimes is not fully understood. As part of the Fire Influence on Regional and Global Environments Experiment (FIREX 2016), we investigated the effect of photochemical aging on biomass burning organic aerosol (BBOA) with a focus on fuels from the western United States. Emissions were sampled into a small (150 L) environmental chamber and photochemically aged via the addition of ozone and irradiation by 254 nm light. While some fraction of species undergoes photolysis, the vast majority of aging occurs via reaction with OH radicals, with total OH exposures corresponding to the equivalent of up to 10 d of atmospheric oxidation. For all fuels burned, large and rapid changes are seen in the ensemble chemical composition of BBOA, as measured by an aerosol mass spectrometer (AMS). Secondary organic aerosol (SOA) formation is seen for all aging experiments and continues to grow with increasing OH exposure, but the magnitude of the SOA formation is highly variable between experiments. This variability can be explained well by a combination of differences in OH exposure and the total concentration of non-methane organic gases (NMOGs) in the chamber before oxidation, as measured by PTR-ToF-MS (r2 values from 0.64 to 0.83). From this relationship, we calculate the fraction of carbon from biomass burning NMOGs that is converted to SOA as a function of equivalent atmospheric aging time, with carbon yields ranging from 24±4 % after 6 h to 56±9 % after 4 d.
-
Please read the editorial note first before accessing the article.
-
Article
(1231 KB)
-
Supplement
(2141 KB)
-
Please read the editorial note first before accessing the article.
- Article
(1231 KB) - Full-text XML
-
Supplement
(2141 KB) - BibTeX
- EndNote




Biomass burning is a major source of particulate matter and trace gases to the atmosphere, and strongly affects global air quality and climate (Akagi et al., 2011; Bond et al., 2004; Liu et al., 2017). In fire-prone regions such as the western United States, the frequency and intensity of wildfires have increased over the past several decades due to fire management practices and climate change (Westerling et al., 2006), and this trend is expected to continue in the coming decades (Dennison et al., 2014; Spracklen et al., 2009). Emissions from fires have been the subject of intense study, but primary emissions alone do not determine the atmospheric impacts of biomass burning since smoke plumes can be transported thousands of kilometers and undergo dramatic chemical changes over their lifetimes in the atmosphere (Andreae et al., 1988; Cubison et al., 2011). In particular, biomass burning organic aerosol (BBOA) is subject to atmospheric aging processes that could significantly alter the climate- and health-relevant properties of biomass burning emissions (Hennigan et al., 2012; Vakkari et al., 2014). Such processes include oxidation of gas-phase compounds followed by partitioning to the particle phase, forming secondary organic aerosol (SOA); direct oxidation of molecules in the particle phase through heterogeneous reactions; and evaporation of particulate semivolatile molecules upon plume dilution, potentially followed by subsequent gas-phase oxidation. However, despite the potential importance of aging on biomass burning emissions, the effect of aging on BBOA composition and loading over multiday timescales is not well-constrained, and usually is not included in global chemical transport models (Shrivastava et al., 2017).
Field measurements provide strong evidence that the composition of BBOA changes significantly when photochemically aged. In aircraft measurements of biomass burning plumes, OA consistently becomes more oxidized downwind, relative to the source of emissions (Capes et al., 2008; Cubison et al., 2011; Forrister et al., 2015; Jolleys et al., 2015, 2012). Additionally, decreases in reactive tracers from biomass burning, such as levoglucosan, are observed after aging when compared to their contribution to fresh emissions (Cubison et al., 2011). Despite these consistencies, field measurements show mixed results with regards to whether or not there is an increase in net SOA downwind of fires. Net SOA formation is usually characterized by an OA enhancement ratio, defined as the ratio between fresh and aged ΔOA∕ΔCO measurements to account for plume dilution. Some studies show that little to no net secondary organic aerosol is formed over the course of several days of aging, or even that a loss of organic mass can occur (Akagi et al., 2012; Capes et al., 2008; Cubison et al., 2011; Hecobian et al., 2011; Jolleys et al., 2015, 2012; May et al., 2015). However, other studies show that significant OA enhancement can occur as well (DeCarlo et al., 2010; Vakkari et al., 2018; Yokelson et al., 2009).
Laboratory studies intended to constrain the effects of aging on biomass burning emissions have also had variable results. Consistent with field measurements, laboratory experiments in which emissions from open burning and wood stoves were photochemically aged found that BBOA became increasingly oxidized and tracers were depleted with increased aging time (Ahern et al., 2019; Bertrand et al., 2018; Cubison et al., 2011; Grieshop et al., 2009a; Hennigan et al., 2011; Ortega et al., 2013; Tkacik et al., 2017). Most laboratory experiments investigating the aging of biomass burning emissions find that significant amounts of SOA are formed in most, but not all, cases (Ahern et al., 2019; Bruns et al., 2016; Grieshop et al., 2009b; Hennigan et al., 2011; Ortega et al., 2013; Tiitta et al., 2016; Tkacik et al., 2017). Even under constrained laboratory experimental conditions, these studies show significant variability in SOA formation between burns of similar or even identical fuels. This variability is often attributed to differences in burning conditions (e.g., flaming and smoldering) (Hennigan et al., 2011) or the presence of unmeasured SOA precursors (Bruns et al., 2016; Grieshop et al., 2009b; Ortega et al., 2013), but predicting biomass burning SOA across fuel types and burning conditions has remained a challenge. Very recently, Ahern et al. (2019) showed that the detailed characterization of hundreds of compounds emitted from a given burn, coupled with estimated SOA yields from each, enables the prediction of SOA formation to within roughly a factor of 2. This approach establishes a clear link between the gas-phase emissions and SOA formation but relies critically on a comprehensive understanding of emission profiles, which may exhibit substantial burn-to-burn variability.
The high degree of variability in net OA observed from biomass burning studies leads to a large range of estimates of SOA from biomass burning, which span nearly 2 orders of magnitude (Shrivastava et al., 2017). The range of global estimates is thus essentially unconstrained, with some studies ranking biomass burning as an insignificant source of SOA and others ranking it as the major source of global SOA (Shrivastava et al., 2015, 2017). Understanding the evolution of biomass burning emissions is necessary to better evaluate the effects of biomass burning on air quality, human health, and climate. To this end, we describe the results from a set of laboratory aging experiments on a variety of fuels, employing an oxidation reactor coupled with real-time measurements of the composition of both the particle-phase and gas-phase emissions, to better constrain the effects of aging on biomass burning emissions.
2.1 Experimental setup and emissions sampling
Experiments were carried out as part of the Fire Influence on Regional and Global Environments Experiment (FIREX 2016) at the USDA Fire Sciences Laboratory (FSL) in Missoula, MT, USA, with the goal of better understanding the evolution of biomass burning emissions within a controlled environment. Experiments took place during the “stack burn” portion of FIREX, in which fuels were burned beneath the exhaust stack (1.6 m diameter, 17 m tall), and were well-mixed before being characterized at the top of the stack. Fuels burned were characteristic of the western US and included Engelmann spruce, lodgepole pine, subalpine fir, chamise, manzanita, Douglas fir, ponderosa pine, and sagebrush (Selimovic et al., 2018). For each of these fuels, components of each fuel (e.g., canopy, litter, duff) were burned individually to determine differences between components and in combination as they would in natural wildfires and prescribed burns. In addition to these fuels, several other fuel types were included, such as peat, dung, Excelsior (wood shavings), rice straw, loblolly pine, and bear grass. The weight of fuel used for each experiment was between 250 and 6000 g. Details of each sampled burn are given in the Supplement (Table S1); here we focus on results from 20 burns (out of 56 sampled total) for which aging experiments were carried out, the full analytical instrument suite (described in Sect. 2.2) collected data, and particle wall-loss rates were unaffected by UV irradiation (also discussed in Sect. 2.2).
Aging experiments were conducted in a 150 L PFA (perfluoroalkoxy) environmental chamber (the “mini-chamber”), an oxidation reactor of intermediate size between commonly used oxidation flow reactors (with volumes of <15 L) and large environmental chambers (>1000 L). The mini-chamber was located in the wind tunnel room in the FSL; smoke from the top of the stack was transported to the wind-tunnel room with a 30 m long community inlet (see Fig. S2 in the Supplement). In order to minimize interactions of the smoke with the walls of the inlet, the tubing had a large diameter (8 in. diameter aluminum ducting) and fast flow (inline fan at the ducting exhaust pulling at 0.33 m3 s−1, giving a transport time of <2 s). Smoke from the community inlet was subsampled from the center of the flow using an ejector diluter pressurized with clean air, then passed through 1 m of passivated stainless-steel tubing and a PM1 cyclone, and injected into the chamber. Comparisons of fresh emissions between direct measurements from the top of the stack and measurements in the mini-chamber indicated some loss of gases and particles along the community inlet and transfer line. However, these were relatively minor (<8 % per volatility bin on average) and, while they might affect SOA yields somewhat, are not expected to affect the overall results significantly (Fig. S3).
Prior to sampling, the chamber was flushed with clean air from a zero-air generator (Teledyne 701H) and humidified air (total 15 slpm, standard liters per minute) for approximately 45 min, leading to background particle concentrations 103–104 times smaller than the peak concentrations during filling. Relative humidity remained in the range of 25 %–40 % throughout the entire experiment. Emissions sampling was initiated at the beginning of the burn and continued until the fuel was completely burned or the chamber particle concentration substantially declined from its maximum concentration. Sampling generally lasted for a significant fraction of each fire (5–20 min sampling time, while fires burned for 5–40 min) and initial concentrations of species within the chamber were diluted by a factor of ∼7 relative to concentrations in the stack. Once sampling was stopped, the chamber was allowed to mix passively for 5–10 min while the fresh emissions were characterized. The chamber was operated in semi-batch mode, meaning that after sampling the chamber was continuously diluted with clean air while the smoke was oxidized and monitored. Chemical aging was initiated by OH, generated from both ozone photolysis and reaction of the resulting O(1D) with water vapor and the photolysis of other OH precursors present in the smoke (e.g., HONO). A mercury pen-ray lamp (Jelight model 600 ozone generator) was used to generate ozone (50–100 ppb in dilution air), which was added to the chamber starting just after the sampling period and continually added over the course of the experiment. The O3 concentration in the chamber was typically lower at the start of each experiment and higher toward the end, and ranged from 10 to 80 ppb throughout. OH oxidation was initiated by exposing the chamber to 254 nm UV light (one UVP, LLC. XX-40S bulb, for a chamber-averaged photon flux of ). Use of low-wavelength UV light can introduce non-OH chemistry (Peng et al., 2016); however, loss rates of common compounds from biomass burning such as toluene, phenol, and naphthalene agree well with predicted loss rates from OH reaction alone (average molec cm−3), indicating that for these compounds UV photolysis is negligible. However, photolysis can be competitive with OH reactions for compounds that undergo rapid photolytic degradation at 254 nm. Such species, which are characterized by a low ratio between their OH reaction rate constant to absorption cross section ( cm s−1) and high quantum yields, include conjugated carbonyls (e.g., furfural and benzaldehyde) (Coggon et al., 2019). These species will therefore undergo substantially more photolysis in the present experiments compared to atmospheric conditions; however, for the vast majority of compounds studied here, the dominant reactive loss (as in the atmosphere) is by reaction with OH (Coggon et al., 2019). Oxidation lasted 30–60 min, then the chamber was flushed with clean air before the next experiment. At the end of each day, the chamber was left to flush with clean air and with four UV lamps on overnight. Seeded blanks (with ammonium sulfate particles) were run and showed negligible OA formation, indicating that the cleaning procedure was successful. In addition to the aging experiments, control runs were conducted without UV light and used to characterize the evolution of the smoke in the absence of OH oxidation.
Particles and gases exiting the reactor were monitored with a suite of analytical instruments. Particle composition measurements were made with an aerosol mass spectrometer with a standard tungsten vaporizer (AMS, Aerodyne Research, Inc.), which measures the mass and composition of nonrefractory particles with diameters between 70 nm and 1 µm (DeCarlo et al., 2006). Black carbon mass was measured with a single-particle soot photometer (SP2, Droplet Measurement Technologies) and soot particle AMS (SP-AMS, Aerodyne Research Inc.; Onasch et al., 2012). Particle size distributions were measured with a scanning electrical mobility spectrometer (SEMS, Brechtel). All particle-phase measurements were made alternating between a two-stage thermal denuder (150 and 250 ∘C) and a room-temperature bypass line. From these measurements, particle organic mass fraction remaining (MFR) was calculated by comparing the thermally denuded particle organic mass to the mass after the bypass line. Non-methane organic gases (NMOGs) were measured with a proton-transfer-reaction time-of-flight mass spectrometer (NOAA PTR-ToF-MS) (Yuan et al., 2016); these measurements are described in detail elsewhere (Koss et al., 2018; Sekimoto et al., 2018), and the oxidation chemistry of NMOGs is described in a companion paper (Coggon et al., 2019). Auxiliary measurements of inorganic gases included O3 (2B Technologies, model 202), CO (Teledyne, model T300), and CO2 (LI-COR, LI-840A). Particle optical properties were monitored with a three-wavelength photoacoustic spectrometer (PASS-3), a two-wavelength cavity ring-down photoacoustic spectrometer (CRD-PAS), and a cavity attenuated phase shift spectrometer (CAPS). Optical measurements from these instruments are not used in the present study and instead are discussed in a separate publication (McClure et al., 2019).
2.2 Data analysis
Particle mass and composition data from the AMS were analyzed using the ToF-AMS analysis toolkits (Squirrel version 1.57I, Pika version 1.16I) using the “improved-ambient” method for calculating oxygen-to-carbon (O∕C) and hydrogen-to-carbon (H∕C) elemental ratios (Canagaratna et al., 2015). Mass concentrations must be corrected for changes in AMS collection efficiency (CE), as well as wall loss and dilution. This is often done in chamber experiments by normalizing measured SOA mass concentrations to an inert internal tracer. However, we were unable to find a suitable tracer in these experiments: sulfate changes as a result of oxidation of emitted SO2, black carbon (BC, when present in high concentrations) exhibited wall losses different from OA (as described below) and appeared not to be homogeneously mixed with the OA (McClure et al., 2019), and POA tracers (such as the ion, recently used by Ahern et al., 2019) are likely to be lost via heterogeneous oxidation at the high OH exposures examined here. Thus, corrections for CE, dilution, and particle wall loss were carried out individually, as described below.
CE and particle density were calculated by comparing AMS particle time-of-flight (PToF) and SEMS size distributions along with AMS organic mass and SEMS volume (Bahreini et al., 2005). This could only be done for the subset of data points with PToF and SEMS distributions that could be fit to lognormal functions, did not show significant particle nucleation, and had low SP-AMS rBC concentration (see below). An AMS CE correction was then applied to the entire data set by parameterizing the exponential relationship between AMS CE with the particle MFR (Fig. S4). An alternative calculation of OA mass using SEMS volume multiplied by OA density agrees with OA mass calculated using this MFR CE parameterization, and is shown in Fig. S5. Generally, POA has low organic MFR (indicating that it is relatively volatile) and particle MFR increases with oxidation, consistent with previous work (Hennigan et al., 2011). This indicates that particles are becoming less volatile and more likely to be (semi-)solid, and therefore likely to have a lower CE due to increased bounce off of the AMS vaporizer (Matthew et al., 2008; Virtanen et al., 2010). Calculated AMS collection efficiencies range from 0.35 to 0.64, with the average CE of fresh emissions equal to 0.54 and average CE of aged (i.e., end-of-oxidation) emissions equal to 0.40. We note that this CE value of 0.54 for fresh emissions is substantially lower than the value of 1 typically assumed in such experiments (Ahern et al., 2019; Hennigan et al., 2011; Heringa et al., 2011; Ortega et al., 2013; Tkacik et al., 2017).
Acetonitrile, an inert tracer species ( ) (Atkinson et al., 2006), was used to correct all data for chamber dilution. An exponential function was fitted to the decay of acetonitrile for each experiment (τdilution∼20 min). All gas- and particle-phase species concentrations are corrected for dilution using an experiment-specific dilution rate. Dilution rates calculated from the fitted decay of acetonitrile agree well with estimates based on chamber volume and flow rates; this is in contrast with dilution rates derived from CO, which is produced in the chamber during oxidation. Particle mass concentrations were also corrected for loss to the chamber walls by fitting an exponential equation to the particle organic mass concentration during a control experiment in which the UV lamps were not turned on (τwall=35 min; Fig. S6). Comparison of wall loss rates calculated between two dark experiments and size-dependent wall loss rates calculated from SEMS data yield similar results. Fires with the lowest initial AMS OA to SP-AMS BC (rBC) ratios () showed enhanced wall loss rates upon UV irradiation, likely due to photoionization of BC or polycyclic aromatic hydrocarbons present on the particle surface (Burtscher, 1992; Mohr et al., 1996), followed by increased electrostatic interactions with the chamber walls. This complicates the wall loss correction, and such low-OA∕rBC burns are excluded from this analysis (see Table S1). All particle concentrations presented, unless otherwise noted, are corrected for collection efficiency, dilution, and wall loss. Gases were corrected for dilution only; control runs showed that wall loss was not a major loss pathway for most primary intermediate volatility organic compounds (IVOCs; Fig. S7).
OH exposures in the chamber were estimated by measuring the decay of an OH tracer, deuterated n-butanol (D9, 98 %, Cambridge Isotope Laboratories), added at the beginning of each experiment (10 µL, 2 % in water). OH exposure was then calculated from the dilution-corrected concentration of deuterated butanol and its reaction rate coefficient with OH ( ) (Barmet et al., 2012). Differences in the initial concentration of OH precursors (e.g., O3 and HONO) in the chamber, OH sinks, and experiment duration resulted in variations in total OH exposure from experiment to experiment, with end-of-experiment OH exposures (calculated at the point when UV lamps are turned off) ranging from 1 to 10 d of atmospheric aging. Throughout this work, OH exposure is converted to equivalent atmospheric aging time (in days) by assuming an average atmospheric OH concentration of 1.5×106 molec cm−3.
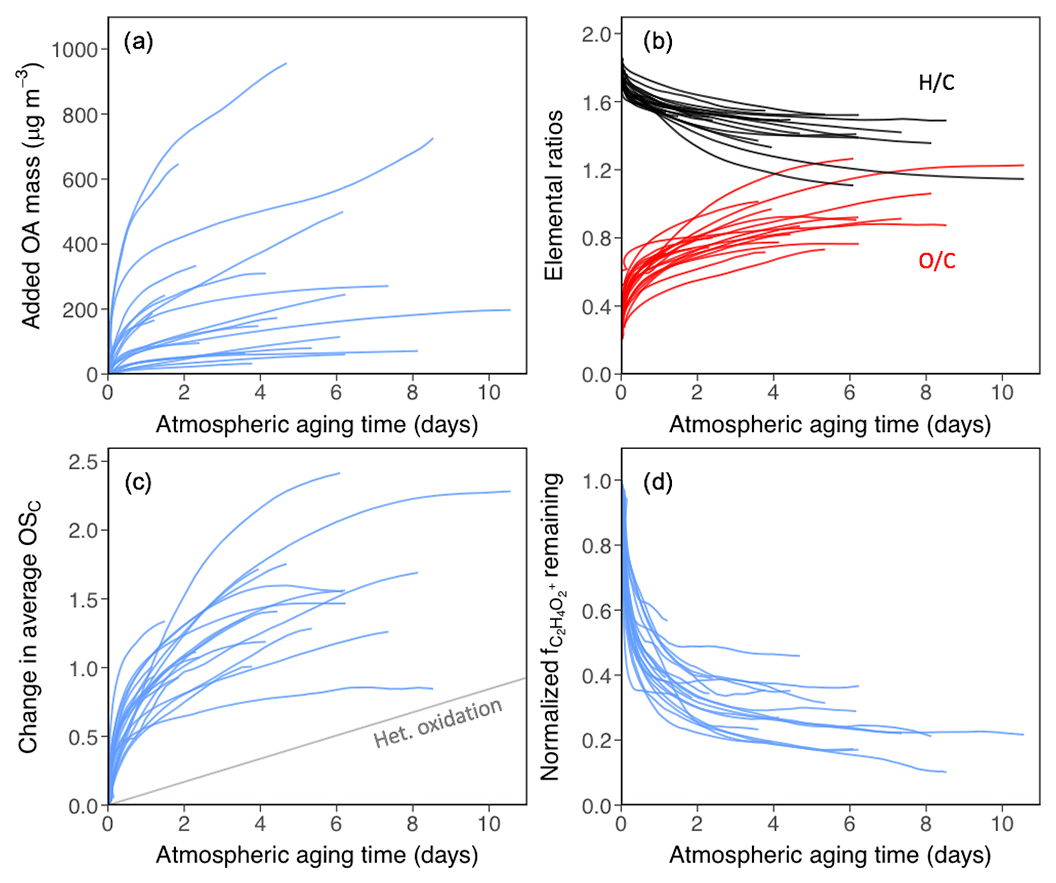
Figure 1Changes in OA mass and composition as a function of aging time, assuming an atmospheric [OH] of 1.5×106 molec cm−3. Each line represents a separate aging experiment. (a) Increase in OA mass with oxidation, showing variable SOA across all experiments. (b) Elemental ratios (red: O∕C, black: H∕C). (c) Change in OSC; reference line shows average change due to heterogeneous oxidation of laboratory flow tube experiments for comparison (Kroll et al., 2015). (d) Normalized fraction of AMS organic signal due to the fragment ion, a common primary biomass burning OA tracer indicative of levoglucosan and related compounds.
3.1 Loading and composition of fresh and aged biomass burning particles
The total initial aerosol mass in the chamber varied widely from experiment to experiment (Table S1), averaging 130±103 µg m−3 (mean±1σ) depending on the amount of fuel burned, fuel type, sampling time, and dilution prior to oxidation. For all experiments considered here, the organic fraction dominated the composition of the primary particle mass (as measured by the sum total of the AMS and SP2), with initial primary OA (POA) concentrations accounting for 70 %–99 % of the total aerosol mass. The initial fraction of black carbon (SP2 BC) mass also varied significantly (0 %–30 % of total aerosol mass) and was highly dependent on fuel type, with the highest BC mass fractions observed for chaparral and Excelsior fuels. Concentrations of non-BC inorganic components (nitrate, sulfate, ammonium, and chloride, all measured by the AMS) were variable but low for all experiments (≤8 % of total aerosol mass). After initiation of oxidation, organic aerosol loadings grew substantially for all experiments (Fig. 1a). The average mass loading of SOA formed (corrected OA mass at the end of the experiment minus the OA mass prior to OH oxidation) was 260±250 µg m−3. Alternatively, the amount of SOA at the end of experiment can be expressed as an OA enhancement ratio, defined as the final OA mass (secondary + primary) divided by the initial (primary) OA mass. The average OA enhancement ratio was 3.5±1.7. This is considerably higher than reported in previous studies (Ahern et al., 2019; Hennigan et al., 2011; Ortega et al., 2016; Tkacik et al., 2017) but once differences in OH exposure (as well as AMS CE) are taken into account these results are reasonably consistent with previous chamber studies and somewhat higher than previous flow tube experiments (Fig. S8). Discussion of the relationship between SOA formation and gas-phase composition will be discussed further in Sect. 3.2.
The chemical composition of the primary organic particulate matter varied substantially between experiments. The initial O∕C ranged from 0.20 to 0.60 (0.35±0.09) and initial H∕C ranged from 1.72 to 1.85 (1.77±0.04), indicating significant variation in POA elemental composition. Elemental ratios stayed constant in the chamber for each given experiment until the lights were turned on. Similarly, control experiments (with no oxidation) showed constant OA∕BC, O∕C, and H∕C ratios over the course of the run, indicating that OA mass and overall oxidation state of biomass burning POA is stable in the chamber without exposure to UV light, despite the potential for semivolatile compounds to partition to the gas phase and/or be lost to the chamber walls (Ahern et al., 2019; Grieshop et al., 2009b; Hennigan et al., 2011; May et al., 2013).
OH oxidation, initiated in the chamber by exposure to UV light, rapidly changes the composition of OA, as shown in Fig. 1b–d. Gas-phase chemistry in the mini-chamber is discussed in detail in a companion publication (Coggon et al., 2019). Despite differences in initial composition between experiments, OA in all experiments undergoes a large increase in O∕C and decrease in H∕C (Fig. 1b), and a corresponding increase in average carbon oxidation state (OSC, equal to 2 O∕C – H∕C) (Fig. 1c). OSC for all fuels increases with increasing OH exposure, with an average end-of-experiment increase in OSC of 1.33±0.50. Most of this change occurs during the initial period of oxidation (equivalent timescales of just 1–2 d); after this, changes in OSC still occur but over much longer timescales. Over longer timescales, when dilution is more significant and OA concentrations are lower, such OSC changes are likely to reflect the oxidation of longer-lived gases. Both condensation of SOA and heterogeneous oxidation (direct reactions between gas-phase oxidants and particle-phase organic molecules) can contribute to the observed increases in oxidation state. However, the initial rate of change for aging of biomass burning emissions (gases and particles combined) is much faster than the average rate of OSC change measured in laboratory heterogeneous OH-oxidation experiments (Kroll et al., 2015) (shown as the gray line in Fig. 1c), implying that condensation of oxidized secondary species is the main driver for the changes in composition observed.
Similar to the elemental ratios, the initial fraction of the primary organic signal from the AMS fragment ion () varies from burn to burn (mean 2.2±1.2 %). This fragment is a small (<6 %) contributor to the overall OA mass spectrum but is known to correlate with levoglucosan (and related molecules) and is commonly used as a tracer for biomass burning POA (Cubison et al., 2011). Figure 1d shows the evolution of , normalized to its value at the start of each experiment. This ion is known to correspond to semivolatile species (Grieshop et al., 2009b), and changes to (∼25 % loss) are observed even in the absence of oxidation; however, oxidation greatly enhances the rate and magnitude of its decrease. Under oxidation conditions, the contribution of this fragment to the total organic mass decreases dramatically over the course of 1–2 d of equivalent OH exposure, then stabilizes after that. This is in agreement with previous aircraft studies which show that, even in highly aged air masses, a small amount of this tracer remains elevated relative to the atmospheric background level (Cubison et al., 2011), potentially due to the contribution of other molecules to this tracer ion. Overall, the chemical changes observed here are likely dominated by the formation of SOA, although heterogeneous oxidation, dilution-driven evaporation, and wall loss may also contribute.
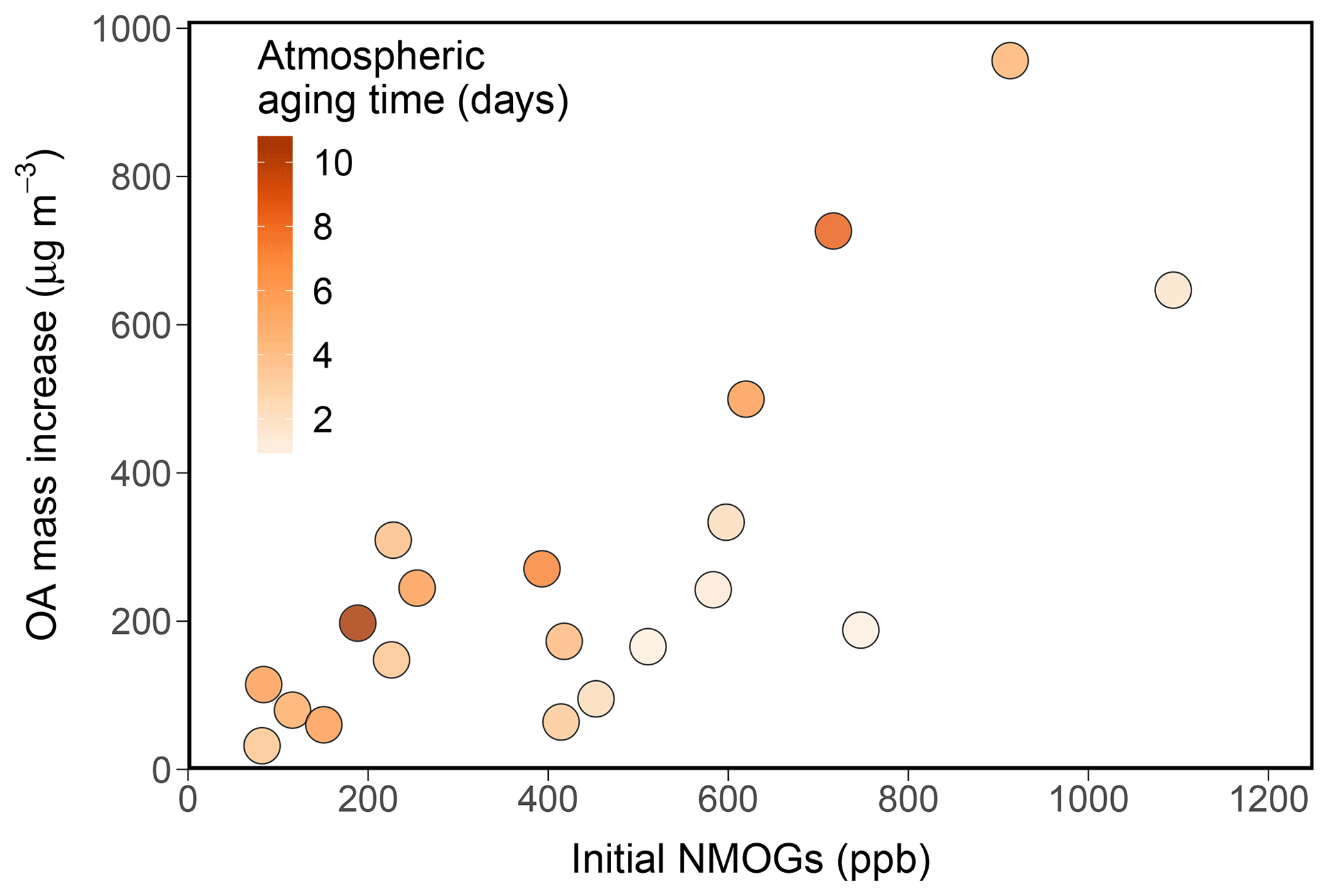
Figure 2End-of-experiment SOA formation vs. total NMOG concentration in the chamber prior to OH oxidation. Points are colored by the atmospheric equivalent aging time corresponding to the end of each experiment.
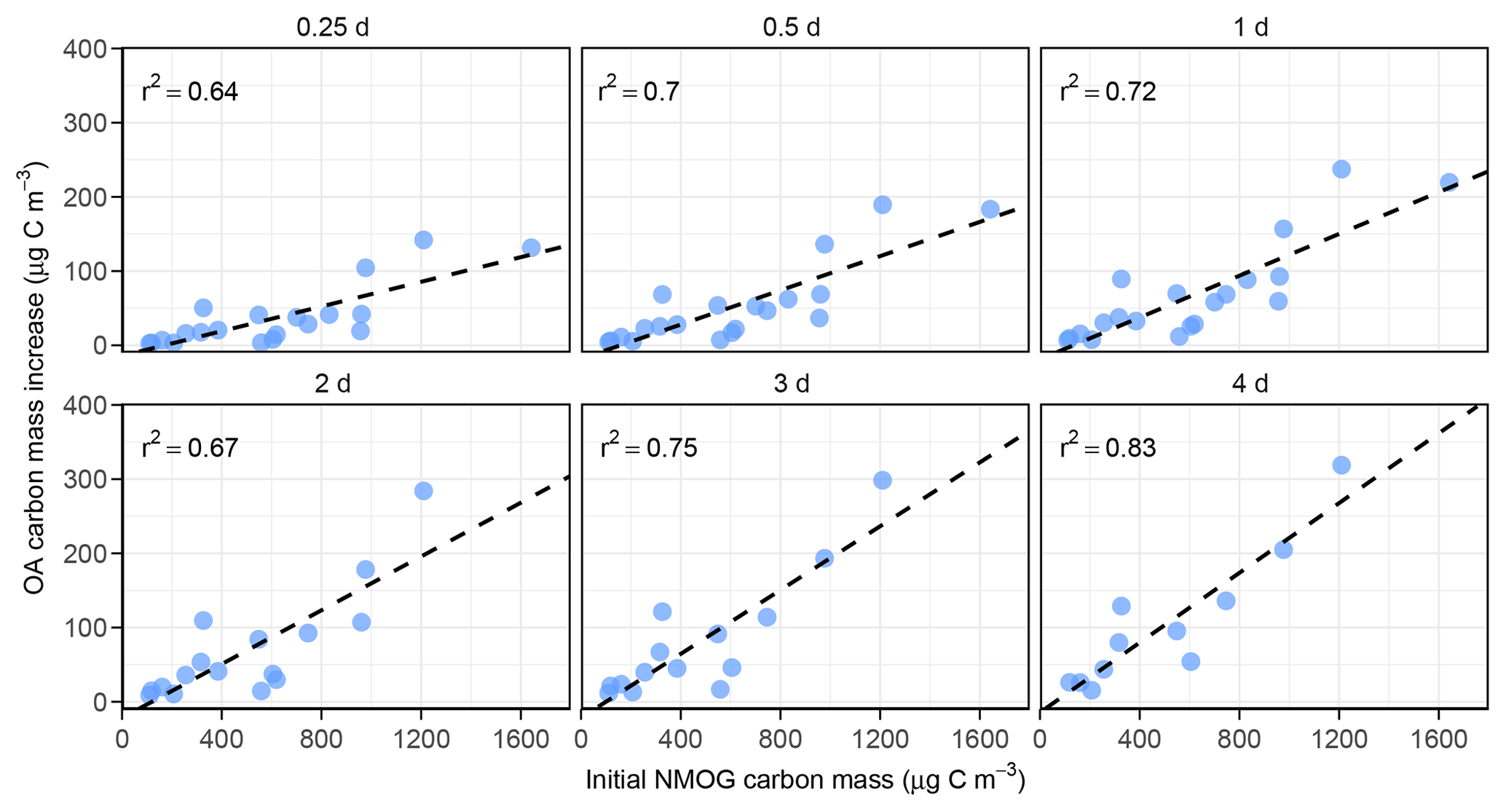
Figure 3OA carbon mass added vs. initial NMOG carbon mass from PTR-ToF-MS measurements at various OH exposures (0.25–4 d of equivalent atmospheric aging). All panels show correlation coefficients (r2) of 0.64 or higher, and the linear relationships at longer aging times show larger slopes (i.e., more SOA; see Fig. 6).
3.2 Secondary organic aerosol formation
All aging experiments show substantial SOA formation with OA mass continuing to increase with extended aging time (Fig. 1a). Consistent with previous studies, the correlations between OA mass enhancement ratio and various parameters that could affect SOA production (e.g., OH exposure, POA, monoterpenes, total NMOG concentration) are weak at best (Fig. S9). However, the absolute amount of SOA formed does appear to correlate with some of these. Figure 2 shows the relationship between SOA formed by the end of each experiment (µg m−3) and the initial concentration of total NMOGs in the chamber (ppb) measured by the PTR-ToF-MS. A positive correlation is seen between the two variables, but the correlation is not especially strong (r2=0.51). A confounding variable in this relationship is the difference in total OH exposure between experiments, as shown in the color scale. To account for the differences in OH exposure, Fig. 3 shows the same relationship between SOA and NMOG concentration but now compared at equal OH exposures (0.25, 0.5, 1, 2, 3, and 4 d of atmospheric aging) and with both axes converted to carbon concentration. Although there is still significant scatter, possibly due to differences in initial POA levels (Fig. S10), there is an improved correlation (r2=0.64–0.83) between SOA and NMOG carbon mass at each OH exposure. Panels in Fig. 3 contain different numbers of data points due to the differences in final OH exposure achieved in each experiment. As such, the r2 values are not strictly comparable but are given to show the correlation in each case. Additionally, the slopes of these plots exhibit a clear trend, increasing with OH exposure. This indicates that a greater fraction of carbon from NMOGs is converted to SOA as aging time increases, consistent with continual SOA formation over long aging timescales. Total PTR-ToF-MS measured NMOGs also correlates well with POA concentration; as such, POA shows a similarly strong relationship with SOA. As the goal of this work is to provide a more mechanistic understanding of the underlying chemistry, relationships between SOA and NMOGs are shown in the main text; relationships between POA and SOA are given in Fig. S11 for cases in which POA measurements may be more readily available.
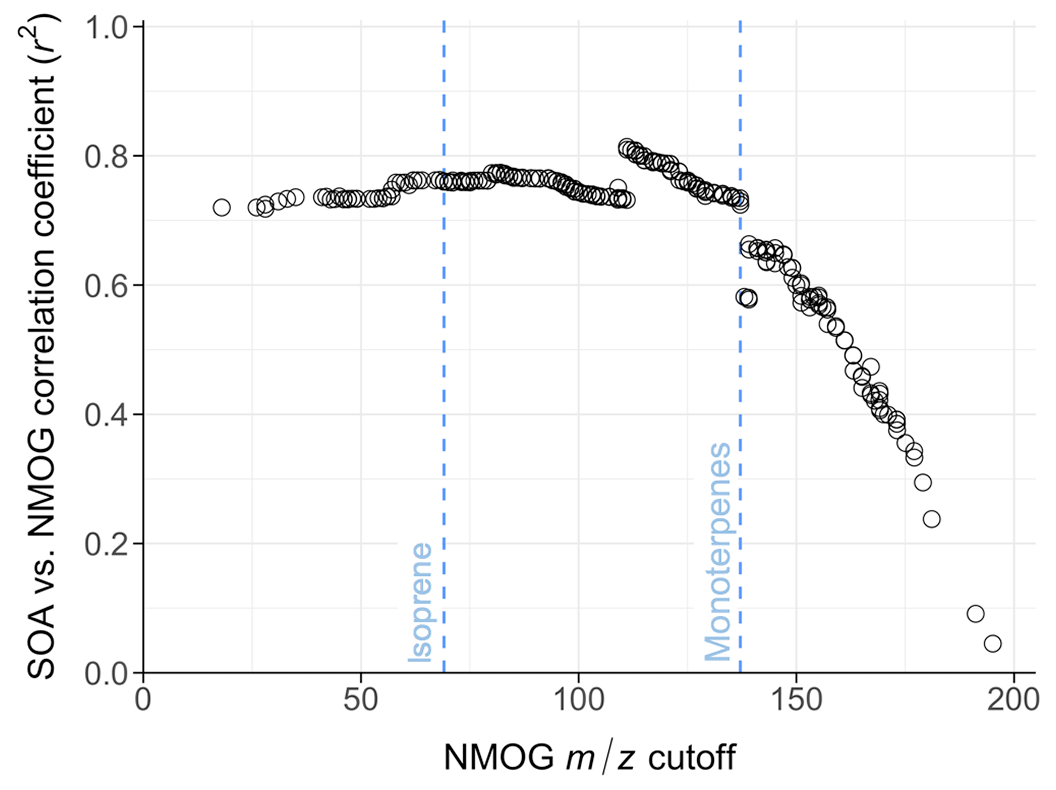
Figure 4Correlation coefficients (r2) between OA carbon mass added and the summed NMOG carbon mass above some ion mass (m∕z) cutoff. For example, the point labeled “isoprene” shows the correlation between SOA carbon mass and initial NMOG carbon mass for all measured ions with mass-to-charge ratio equal to or greater than that of isoprene (i.e., species of molecular weight 68 g mol−1 or higher). Initial NMOG carbon mass is calculated prior to oxidation and data points all correspond to r2 values at 1 d of atmospheric aging time. Correlation coefficients are high for all cutoff points below monoterpenes, then drop off due to loss of signal and the importance of smaller compounds to SOA formation.
The correlation between SOA formation and the initial chamber concentration of NMOGs is reasonable since NMOGs provide the carbon that drives SOA growth. However, the PTR-ToF-MS measures many compounds that likely do not contribute to SOA formation (e.g., small compounds such as methanol and acetonitrile). In addition to comparing SOA to the initial NMOG concentration, we can examine how SOA formation correlates with the concentration of NMOGs above some molecular weight cutoff. Figure 4 shows the correlation coefficient for the linear fit between SOA carbon mass and summed NMOG carbon mass at each molecular weight cutoff for 1 d of equivalent aging. While low-molecular-weight NMOGs are not expected to contribute to SOA mass, the correlation between SOA carbon mass and initial NMOG carbon mass does not improve substantially when these are excluded (left side of Fig. 4). In fact, the correlation between SOA is relatively insensitive to the m∕z cutoff point until only compounds with molecular weight greater than monoterpenes () are considered. After this point the correlation drops rapidly to zero, likely because the PTR-ToF-MS signal is very low in this mass range and/or compounds with 10 or fewer carbon atoms dominate SOA formation (or at least correlate with some unmeasured SOA-forming species). This suggests that the ratio of SOA precursors to the total concentration of measured NMOGs is relatively constant between experiments. The rapid step change after m∕z 137 might suggest the importance of monoterpene chemistry to SOA formation, consistent with Ahern et al. (2019); however, we do not observe strong correlations between SOA and monoterpenes alone, nor any other single standard SOA precursor (e.g., isoprene, benzene, phenol, naphthalene), nor any single volatility class measured by the PTR-ToF-MS (e.g., IVOCs). PTR-ToF-MS measurements taken directly from the FSL exhaust stack show that although the NMOG emissions are highly complex, much of the variability in emissions (∼85 %) can be described by just two emission factors (derived using positive matrix factorization or PMF): one high-temperature combustion factor and one low-temperature combustion factor (Sekimoto et al., 2018). Correlations between NMOGs and SOA when splitting total NMOGs by factor type are weak (Fig. S12), indicating that both factors include compounds that contribute to SOA formation.
Previous biomass burning aging experiments with both aerosol and NMOG measurements have not observed this relationship between SOA and total NMOGs (Ortega et al., 2013) or individual SOA precursors (Bruns et al., 2016; Grieshop et al., 2009b; Ortega et al., 2013; Tkacik et al., 2017). Most such studies identified only half or less of the NMOG signal and/or were limited to a small number of experiments (Grieshop et al., 2009b; Ortega et al., 2013; Tkacik et al., 2017); this could potentially explain why similar correlations between total volatile organic compounds (VOCs) and SOA have not been observed before. By contrast, the use of PTR-ToF-MS in the present study enables the measurement of ∼50 %–80 % of reactive gas-phase carbon from biomass burning, making it a good tool for characterizing the total organic gas-phase emissions from fires (Hatch et al., 2017). In this analysis, we have included both identified and calibrated PTR-ToF-MS ions (∼90 % of the signal, approximately 150 ions) as well as an additional 370 unidentified ions (Koss et al., 2018). This is roughly comparable to the number of compounds identified and quantified in recent speciated studies of biomass burning emissions (Hatch et al., 2015), which have been shown to enable reasonable bottom-up estimates of total SOA formation (Ahern et al., 2019). However, such speciated approaches require estimates of SOA yields from each precursor, which might be unknown or highly uncertain. As an alternative approach, the initial total NMOG concentration measured by the PTR-ToF-MS, in conjunction with OH exposure, provides a reasonable predictor for the amount of SOA formation without the need for speciated aerosol yields.
Recent work has pointed to the importance of non-traditional SOA precursors to SOA formation for residential wood combustion of a single fuel type (beech wood) (Bruns et al., 2016). These precursors include semivolatile and intermediate-volatility volatile organic compounds (S/IVOCs) such as phenols and naphthalenes (Bruns et al., 2016). However, the majority of gas-phase carbon observed in this study is in compounds with low carbon number (nC<7), with corresponding volatilities that are weighted towards volatile compounds (c0>107 µg m−3) rather than S/IVOCs (Fig. 5). This means that SOA from biomass burning is strongly influenced by the oxidation of relatively small, volatile species and not S/IVOCs, a result consistent with Ahern et al. (2019); alternatively, this could mean that the PTR-ToF-MS does not measure all important SOA precursors but measures compounds that are co-emitted and correlate well with them.
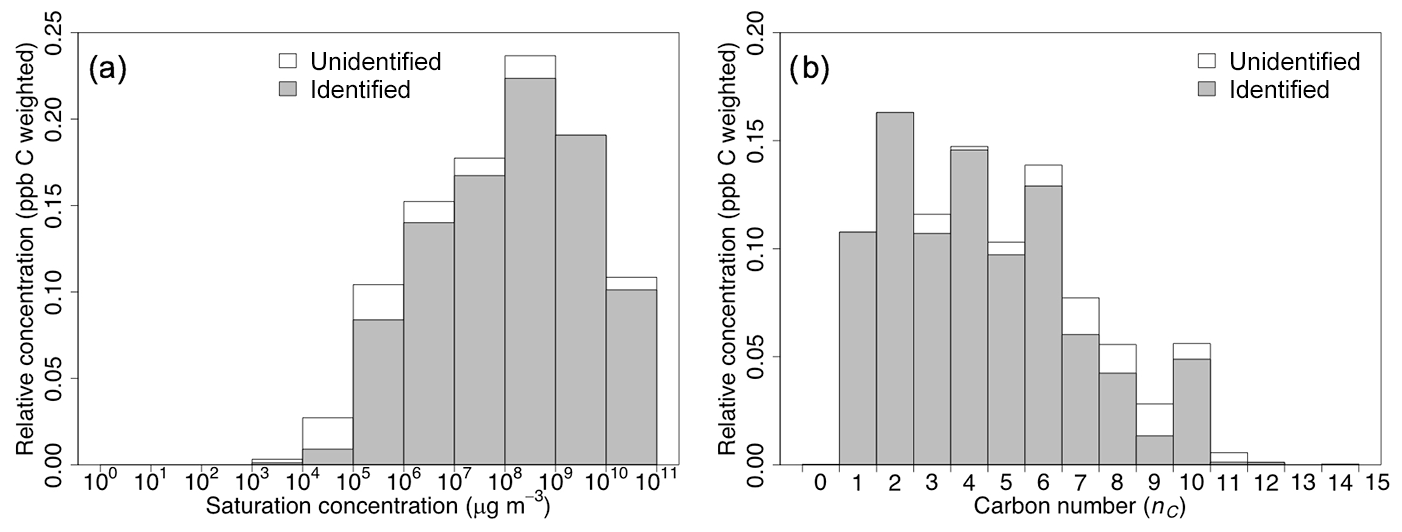
Figure 5Estimated saturation vapor concentration distribution (a) and carbon number (nC) distribution (b) for compounds (NMOGs) measured by the PTR-ToF-MS (Koss et al., 2018) in the chamber prior to oxidation, averaged over all burns. Distributions are separated into identified and unidentified ions and are weighted by parts-per-billion carbon (ppb C).
From each of the relationships between SOA carbon mass and initial NMOG carbon mass (linear fits in Fig. 3), an effective carbon yield can be calculated. Carbon yield is defined here as the SOA carbon formed divided by the total NMOG carbon reacted at each respective OH exposure. The amount of gas-phase carbon reacted (Δ[C]NMOG) is estimated from the initial concentration of PTR-ToF-MS-measured gas-phase organic carbon in the chamber before oxidation, experiment-specific dilution rates, and speciated OH reaction rates for identified compounds present in each fire (Koss et al., 2018). We are not able to directly measure the amount of NMOG reacted because of a number of complications: the dilution loss of NMOG (gases removed from the chamber before they can be reacted with OH), formation of secondary gas-phase products, and potential off-gassing of non-SOA forming low-molecular-weight NMOG from the chamber walls. Carbon yields as a function of atmospheric age are shown in Fig. 6 and range from 24±4 % at 0.25 d of equivalent atmospheric oxidation to 56±9 % after 4 d of equivalent atmospheric oxidation. Since the PTR-ToF-MS does not measure the true total NMOG concentration (e.g., not all alkanes and alkenes are measured), these carbon yields are likely to be upper bounds. The calculated yields use our best estimate of OA carbon mass, using the AMS CE correction described previously. The gray points in Fig. 6 show the carbon yields assuming a constant AMS CE equal to 1. In both cases, the amount of SOA formed increases with increasing aging time. Accounting for changes in collection efficiency that occur with aging leads to a factor of approximately 4 increase in yields; previous studies assuming a constant or initial AMS CE equal to 1 may thus significantly underestimate SOA formation (Ahern et al., 2019; Hennigan et al., 2011; Heringa et al., 2011; Ortega et al., 2013). While a recent review of biomass burning suggests that AMS CE is not a major factor in quantifying SOA from aged biomass burning emissions (Hodshire et al., 2019a), this work shows that, in fact, it may have a large effect and should be considered in future laboratory and field studies. Improved constraints on estimates of AMS collection efficiency are thus needed for improved estimates of SOA formation from biomass burning. Nonetheless, this carbon yield, combined with laboratory- or field-based estimates of NMOG emissions, provides a means for including SOA formation from biomass burning sources within chemical transport models.

Figure 6SOA carbon yield from aging of biomass burning emissions. Black points are carbon yields using our best estimate of OA carbon mass. Yields are calculated from the ratios of SOA carbon formed to NMOG carbon reacted (estimated from the average carbon-weighted OH rate coefficient for identified compounds and accounting for chamber dilution). Error bars are ±1σ in the slope of the linear fit between SOA and NMOG carbon mass. Gray triangles are estimated yields using AMS CE=1 for all OA, both before and after aging, which is a common assumption made in previous studies on the aging of biomass burning. See Supplement (Table S13) for tabulated yields.
The variability in findings from previous lab and field studies on the effect aging has on net SOA from biomass burning can be potentially explained by the effects of dilution on the evolution of BBOA mass. Some fraction of BBOA is semivolatile, and dilution (in chambers or ambient smoke plumes) will cause volatile OA components to partition from the particle phase to the gas phase (May et al., 2013). Recent modeling work has shown that even in plumes that show no net SOA formation, significant condensation of secondary organic mass may occur (Bian et al., 2017; Hodshire et al., 2019b), but net growth can be low (or even negligible) due to dilution-driven evaporation of OA. In ambient plumes, dilution drives semivolatile species from the particle phase to gas phase; although this causes a loss in OA mass, it also serves as a source of semivolatile organic compounds (SVOCs) that can condense back onto particles after oxidation, leading to little to no net change in OA. Related to this point, calculated net-OA values are also sensitive to the choice of starting point (t0). Initial dilution in chamber experiments may result in substantial POA evaporation, which can provide high concentrations of SVOCs that are efficiently converted to SOA upon oxidation. However, some laboratory experiments find a net loss of OA mass during aging (Hennigan et al., 2011; Ortega et al., 2013; Tkacik et al., 2017), which we do not observe in the present experiments. Instead we see SOA formation in all photooxidation studies and no evaporative loss during dilution-only experiments, results consistent with recent work by Ahern et al. (2019). The reason for this is unclear but could be due to some combination of differences in dilution and wall losses (gas and particle) in chambers and flow tubes, the method of AMS collection efficiency correction, or experimental conditions leading to different peroxy radical (RO2) chemistry. Small chambers such as the mini-chamber have lower initial dilution factors but much higher OH concentrations than large chambers, potentially favoring condensation from VOC oxidation over evaporation of particle mass. With the potential preference for condensation over evaporation, this present study may be effectively measuring SOA formation while excluding evaporation from the extensive dilution that occurs in biomass burning plumes; therefore, it is primarily accessing the “chemistry” component of OA evolution (Bian et al., 2017). Thus, the carbon yields shown in Fig. 6 need to be combined with a realistic treatment of BBOA partitioning for effective model inputs to describe BBOA evolution in the atmosphere.
We show that the OH-initiated aging of biomass burning emissions leads to significant changes in BBOA composition and loading. These changes are dependent on OH exposure and are especially large over the first few days after emission. Significant amounts of SOA are formed from all fuels studied here, but SOA formation is highly variable. Despite large differences in fuel type and burning conditions, much of this variability can be explained by differences in the initial total NMOG concentration and OH exposure. Correlations between SOA formation and the concentration of initial measured NMOGs in the chamber at given OH exposures are good, with r2 values between 0.64 and 0.83, and indicate SOA carbon yields between 24 % (after 6 h of equivalent atmospheric oxidation) to 56 % (after 4 d). Given total NMOG measurements from future field campaigns, the calculated SOA carbon yields can be used to estimate gross SOA formation from biomass burning in chemical transport models. However, these estimates would likely need to be used in conjunction with estimates of BBOA evaporation rates to calculate the net effect of aging on OA concentrations. Future work investigating the evolution of biomass burning emissions should attempt to further constrain the rates of BBOA evaporation and compare the relative rates of oxidation and dilution from field and laboratory studies. In addition to this, laboratory studies on a wider range of fuels (i.e., those found in areas other than the western US), and under a wider range of reaction conditions (i.e., different RO2 reaction regimes), will help improve the ability to predict the loadings, properties, and impacts of biomass burning emissions globally.
Data are available from the CSD NOAA archive at https://esrl.noaa.gov/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/ (NOAA, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-12797-2019-supplement.
Data were interpreted and the article was written by CYL and JHK. Mini-chamber construction and operation were done by CYL, DHH, and CDC. The AMS was operated and data were analyzed by CYL. PTR-ToF-MS was operated and data were analyzed by MMC, ARK, and KS. Experiments were conceived by JHK, CDC, and CW. All co-authors provided article feedback and comments.
The authors declare that they have no conflict of interest.
Christopher Y. Lim and Abigail R. Koss were supported by the NSF graduate research fellowship program. The authors would like to thank Timothy Onasch for support of the SP-AMS; Colette Heald for helpful comments; and Edward Fortner, Berk Knighton, Robert Yokelson, the entire FIREX science team, and Missoula Fire Sciences Laboratory staff for support during the project.
This research has been supported by the NOAA AC4 program (grant nos. NA16OAR4310112 and NA16OAR4310111).
This paper was edited by Ryan Sullivan and reviewed by two anonymous referees.
Ahern, A. T., Robinson, E. S., Tkacik, D. S., Saleh, R., Hatch, L. E., Barsanti, K. C., Stockwell, C. E., Yokelson, R. J., Presto, A. A., Robinson, A. L., Sullivan, R. C., and Donahue, N. M.: Production of Secondary Organic Aerosol During Aging of Biomass Burning Smoke From Fresh Fuels and Its Relationship to VOC Precursors, J. Geophys. Res.-Atmos., 124, 3583–3606, https://doi.org/10.1029/2018JD029068, 2019.
Akagi, S. K., Yokelson, R. J., Wiedinmyer, C., Alvarado, M. J., Reid, J. S., Karl, T., Crounse, J. D., and Wennberg, P. O.: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011, 2011.
Akagi, S. K., Craven, J. S., Taylor, J. W., McMeeking, G. R., Yokelson, R. J., Burling, I. R., Urbanski, S. P., Wold, C. E., Seinfeld, J. H., Coe, H., Alvarado, M. J., and Weise, D. R.: Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 12, 1397–1421, https://doi.org/10.5194/acp-12-1397-2012, 2012.
Andreae, M. O., Browell, E. V., Garstang, M., Gregory, G. L., Harriss, R. C., Hill, G. F., Jacob, D. J., Pereira, M. C., Sachse, G. W., Setzer, A. W., Dias, P. L. S., Talbot, R. W., Torres, A. L., and Wofsy, S. C.: Biomass-burning emissions and associated haze layers over Amazonia, J. Geophys. Res., 93, 1509–1527, https://doi.org/10.1029/JD093iD02p01509, 1988.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bahreini, R., Keywood, M. D., Ng, N. L., Varutbangkul, V., Gao, S., Flagan, R. C., Seinfeld, J. H., Worsnop, D. R., and Jimenez, J. L.: Measurements of secondary organic aerosol from oxidation of cycloalkenes, terpenes, and m-xylene using an Aerodyne aerosol mass spectrometer, Environ. Sci. Technol., 39, 5674–5688, https://doi.org/10.1021/es048061a, 2005.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018.
Bian, Q., Jathar, S. H., Kodros, J. K., Barsanti, K. C., Hatch, L. E., May, A. A., Kreidenweis, S. M., and Pierce, J. R.: Secondary organic aerosol formation in biomass-burning plumes: theoretical analysis of lab studies and ambient plumes, Atmos. Chem. Phys., 17, 5459–5475, https://doi.org/10.5194/acp-17-5459-2017, 2017.
Bond, T. C., Streets, D. G., Yarber, K. F., Nelson, S. M., Woo, J. H., and Klimont, Z.: A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res.-Atmos., 109, 1–43, https://doi.org/10.1029/2003JD003697, 2004.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep.-UK, 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Burtscher, H.: Measurement and characteristics of combustion aerosols with special consideration of photoelectric charging and charging by flame ions, J. Aerosol Sci., 23, 549–595, https://doi.org/10.1016/0021-8502(92)90026-R, 1992.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Capes, G., Johnson, B., McFiggans, G., Williams, P. I., Haywood, J., and Coe, H.: Aging of biomass burning aerosols over West Africa: Aircraft measurements of chemical composition, microphysical properties, and emission ratios, J. Geophys. Res.-Atmos., 113, 1–13, https://doi.org/10.1029/2008JD009845, 2008.
Coggon, M. M., Lim, C. Y., Koss, A. R., Sekimoto, K., Yuan, B., Gilman, J. B., Hagan, D. H., Selimovic, V., Zarzana, K., Brown, S. S., Roberts, J. M., Müller, M., Yokelson, R., Wisthaler, A., Krechmer, J. E., Jimenez, J. L., Cappa, C., Kroll, J., de Gouw, J., and Warneke, C.: OH-chemistry of non-methane organic gases (NMOG) emitted from laboratory and ambient biomass burning smoke: evaluating the influence of furans and oxygenated aromatics on ozone and secondary NMOG formation, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-516, in review, 2019.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
DeCarlo, P. F., Ulbrich, I. M., Crounse, J., de Foy, B., Dunlea, E. J., Aiken, A. C., Knapp, D., Weinheimer, A. J., Campos, T., Wennberg, P. O., and Jimenez, J. L.: Investigation of the sources and processing of organic aerosol over the Central Mexican Plateau from aircraft measurements during MILAGRO, Atmos. Chem. Phys., 10, 5257–5280, https://doi.org/10.5194/acp-10-5257-2010, 2010.
Dennison, P. E., Brewer, S. C., Arnold, J. D., and Moritz, M. A.: Large wildfire trends in the western United States, 1984–2011, Geophys. Res. Lett., 41, 2928–2933, https://doi.org/10.1002/2014GL059576, 2014.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015GL063897, 2015.
Grieshop, A. P., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 2: analysis of aerosol mass spectrometer data, Atmos. Chem. Phys., 9, 2227–2240, https://doi.org/10.5194/acp-9-2227-2009, 2009a.
Grieshop, A. P., Logue, J. M., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution, Atmos. Chem. Phys., 9, 1263–1277, https://doi.org/10.5194/acp-9-1263-2009, 2009b.
Hatch, L. E., Luo, W., Pankow, J. F., Yokelson, R. J., Stockwell, C. E., and Barsanti, K. C.: Identification and quantification of gaseous organic compounds emitted from biomass burning using two-dimensional gas chromatography–time-of-flight mass spectrometry, Atmos. Chem. Phys., 15, 1865–1899, https://doi.org/10.5194/acp-15-1865-2015, 2015.
Hatch, L. E., Yokelson, R. J., Stockwell, C. E., Veres, P. R., Simpson, I. J., Blake, D. R., Orlando, J. J., and Barsanti, K. C.: Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 17, 1471–1489, https://doi.org/10.5194/acp-17-1471-2017, 2017.
Hecobian, A., Liu, Z., Hennigan, C. J., Huey, L. G., Jimenez, J. L., Cubison, M. J., Vay, S., Diskin, G. S., Sachse, G. W., Wisthaler, A., Mikoviny, T., Weinheimer, A. J., Liao, J., Knapp, D. J., Wennberg, P. O., Kürten, A., Crounse, J. D., Clair, J. St., Wang, Y., and Weber, R. J.: Comparison of chemical characteristics of 495 biomass burning plumes intercepted by the NASA DC-8 aircraft during the ARCTAS/CARB-2008 field campaign, Atmos. Chem. Phys., 11, 13325–13337, https://doi.org/10.5194/acp-11-13325-2011, 2011.
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011.
Hennigan, C. J., Westervelt, D. M., Riipinen, I., Engelhart, G. J., Lee, T., Collett, J. L., Pandis, S. N., Adams, P. J., and Robinson, A. L.: New particle formation and growth in biomass burning plumes: An important source of cloud condensation nuclei, Geophys. Res. Lett., 39, 1–5, https://doi.org/10.1029/2012GL050930, 2012.
Heringa, M. F., DeCarlo, P. F., Chirico, R., Tritscher, T., Dommen, J., Weingartner, E., Richter, R., Wehrle, G., Prévôt, A. S. H., and Baltensperger, U.: Investigations of primary and secondary particulate matter of different wood combustion appliances with a high-resolution time-of-flight aerosol mass spectrometer, Atmos. Chem. Phys., 11, 5945–5957, https://doi.org/10.5194/acp-11-5945-2011, 2011.
Hodshire, A. L., Akherati, A., Alvarado, M. J., Brown-Steiner, B., Jathar, S. H., Jimenez, J. L., Kreidenweis, S. M., Lonsdale, C. R., Onasch, T. B., Ortega, A. M. and Pierce, J. R.: Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 53, 10007–10022, https://doi.org/10.1021/acs.est.9b02588, 2019a.
Hodshire, A. L., Bian, Q., Ramnarine, E., Lonsdale, C. R., Alvarado, M. J., Kreidenweis, S. M., Jathar, S. H., and Pierce, J. R.: More Than Emissions and Chemistry: Fire Size, Dilution, and Background Aerosol Also Greatly Influence Near-Field Biomass Burning Aerosol Aging, J. Geophys. Res.-Atmos., 124, 5589–5611, https://doi.org/10.1029/2018JD029674, 2019b.
Jolleys, M. D., Coe, H., McFiggans, G., Capes, G., Allan, J. D., Crosier, J., Williams, P. I., Allen, G., Bower, K. N., Jimenez, J. L., Russell, L. M., Grutter, M., and Baumgardner, D.: Characterizing the aging of biomass burning organic aerosol by use of mixing ratios: A meta-analysis of four regions, Environ. Sci. Technol., 46, 13093–13102, https://doi.org/10.1021/es302386v, 2012.
Jolleys, M. D., Coe, H., McFiggans, G., Taylor, J. W., O'Shea, S. J., Le Breton, M., Bauguitte, S. J.-B., Moller, S., Di Carlo, P., Aruffo, E., Palmer, P. I., Lee, J. D., Percival, C. J., and Gallagher, M. W.: Properties and evolution of biomass burning organic aerosol from Canadian boreal forest fires, Atmos. Chem. Phys., 15, 3077–3095, https://doi.org/10.5194/acp-15-3077-2015, 2015.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, J. Phys. Chem. A, 119, 10767–10783, 2015.
Liu, X., Huey, L. G., Yokelson, R. J., Selimovic, V., Simpson, I. J., Müller, M., Jimenez, J. L., Campuzano-Jost, P., Beyersdorf, A. J., Blake, D. R., Butterfield, Z., Choi, Y., Crounse, J. D., Day, D. A., Diskin, G. S., Dubey, M. K., Fortner, E., Hanisco, T. F., Hu, W., King, L. E., Kleinman, L., Meinardi, S., Mikoviny, T., Onasch, T. B., Palm, B. B., Peischl, J., Pollack, I. B., Ryerson, T. B., Sachse, G. W., Sedlacek, A. J., Shilling, J. E., Springston, S., St. Clair, J. M., Tanner, D. J., Teng, A. P., Wennberg, P. O., Wisthaler, A., and Wolfe, G. M.: Airborne measurements of western U.S. wildfire emissions: Comparison with prescribed burning and air quality implications, J. Geophys. Res., 122, 6108–6129, https://doi.org/10.1002/2016JD026315, 2017.
Matthew, B. M., Middlebrook, A. M., and Onasch, T. B.: Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols, Aerosol Sci. Technol., 42, 884–898, https://doi.org/10.1080/02786820802356797, 2008.
May, A. A., Levin, E. J. T., Hennigan, C. J., Riipinen, I., Lee, T., Collett, J. L., Jimenez, J. L., Kreidenweis, S. M., and Robinson, A. L.: Gas-particle partitioning of primary organic aerosol emissions: 3. Biomass burning, J. Geophys. Res.-Atmos., 118, 11327–11338, https://doi.org/10.1002/jgrd.50828, 2013.
May, A. A., Lee, T., McMeeking, G. R., Akagi, S., Sullivan, A. P., Urbanski, S., Yokelson, R. J., and Kreidenweis, S. M.: Observations and analysis of organic aerosol evolution in some prescribed fire smoke plumes, Atmos. Chem. Phys., 15, 6323–6335, https://doi.org/10.5194/acp-15-6323-2015, 2015.
McClure, C. D., Lim, C. Y., Hagan, D. H., Kroll, J. H., and Cappa, C. D.: Biomass-burning derived particles from a wide variety of fuels: Part 1: Properties of primary particles, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-707, in review, 2019.
Mohr, M., Matter, D., Burtscher, H., Mohr, M., and Burtscher, H.: Efficient multiple charging of diesel particles by photoemission, Aerosol Sci. Technol., 24, 14–20, https://doi.org/10.1080/02786829608965348, 1996.
NOAA: Fire Lab 2016 Data Download, available at: https://esrl.noaa.gov/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload, last access: 9 September 2019.
Onasch, T. B., Trimborn, A., Fortner, E. C., Jayne, J. T., Kok, G. L., Williams, L. R., Davidovits, P., and Worsnop, D. R.: Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application, Aerosol Sci. Technol., 46, 804–817, https://doi.org/10.1080/02786826.2012.663948, 2012.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Ortega, A. M., Hayes, P. L., Peng, Z., Palm, B. B., Hu, W., Day, D. A., Li, R., Cubison, M. J., Brune, W. H., Graus, M., Warneke, C., Gilman, J. B., Kuster, W. C., de Gouw, J., Gutiérrez-Montes, C., and Jimenez, J. L.: Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area, Atmos. Chem. Phys., 16, 7411–7433, https://doi.org/10.5194/acp-16-7411-2016, 2016.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Sekimoto, K., Koss, A. R., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Warneke, C., Yokelson, R. J., Roberts, J. M., and de Gouw, J.: High- and low-temperature pyrolysis profiles describe volatile organic compound emissions from western US wildfire fuels, Atmos. Chem. Phys., 18, 9263–9281, https://doi.org/10.5194/acp-18-9263-2018, 2018.
Selimovic, V., Yokelson, R. J., Warneke, C., Roberts, J. M., de Gouw, J., Reardon, J., and Griffith, D. W. T.: Aerosol optical properties and trace gas emissions by PAX and OP-FTIR for laboratory-simulated western US wildfires during FIREX, Atmos. Chem. Phys., 18, 2929–2948, https://doi.org/10.5194/acp-18-2929-2018, 2018.
Shrivastava, M., Easter, R. C., Liu, X., Zelenyuk, A., Singh, B., Zhang, K., Ma, P.-L., Chand, D., Ghan, S., Jimenez, J. L., Zhang, Q., Fast, J., Rasch, P. J., and Tiitta, P.: Global transformation and fate of SOA: Implications of low-volatility SOA and gas-phase fragmentation reactions, J. Geophys. Res.-Atmos., 120, 4169–4195, https://doi.org/10.1002/2014JD022563, 2015.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Spracklen, D. V., Mickley, L. J., Logan, J. A., Hudman, R. C., Yevich, R., Flannigan, M. D., and Westerling, A. L.: Impacts of climate change from 2000 to 2050 on wildfire activity and carbonaceous aerosol concentrations in the western United States, J. Geophys. Res.-Atmos., 114, 1–17, https://doi.org/10.1029/2008JD010966, 2009.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
Tkacik, D. S., Robinson, E. S., Ahern, A., Saleh, R., Stockwell, C., Veres, P., Simpson, I. J., Meinardi, S., Blake, D. R., Yokelson, R. J., Presto, A. A., Sullivan, R. C., Donahue, N. M., and Robinson, A. L.: A dual-chamber method for quantifying the effects of atmospheric perturbations on secondary organic aerosol formation from biomass burning emissions, J. Geophys. Res., 122, 6043–6058, https://doi.org/10.1002/2016JD025784, 2017.
Vakkari, V., Kerminen, V.-M., Beukes, J. P., Tiitta, P., van Zyl, P. G., Josipovic, M., Venter, A. D., Jaars, K., Worsnop, D. R., Kulmala, M., and Laakso, L.: Rapid changes in biomass burning aerosols by atmospheric oxidation, Geophys. Res. Lett., 41, 2644–2651, https://doi.org/10.1002/2014GL059396, 2014.
Vakkari, V., Beukes, J. P., Jaars, K., Josipovic, M., Venter, A. D., and Zyl, P. G. Van: Major secondary aerosol formation in southern African open biomass burning plumes, Nat. Geosci., 11, 580–583, https://doi.org/10.1038/s41561-018-0170-0, 2018.
Virtanen, A., Joutsensaari, J., Koop, T., Kannosto, J., Yli-Pirilä, P., Leskinen, J., Mäkelä, J. M., Holopainen, J. K., Pöschl, U., Kulmala, M., Worsnop, D. R., and Laaksonen, A.: An amorphous solid state of biogenic secondary organic aerosol particles, Nature, 467, 824–827, https://doi.org/10.1038/nature09455, 2010.
Westerling, A. L., Hidalgo, H. G., Cayan, D. R., and Swetnam, T. W.: Warming and earlier spring increase Western U.S. forest wildfire activity, Science, 313, 940–943, https://doi.org/10.1126/science.1128834, 2006.
Yokelson, R. J., Crounse, J. D., DeCarlo, P. F., Karl, T., Urbanski, S., Atlas, E., Campos, T., Shinozuka, Y., Kapustin, V., Clarke, A. D., Weinheimer, A., Knapp, D. J., Montzka, D. D., Holloway, J., Weibring, P., Flocke, F., Zheng, W., Toohey, D., Wennberg, P. O., Wiedinmyer, C., Mauldin, L., Fried, A., Richter, D., Walega, J., Jimenez, J. L., Adachi, K., Buseck, P. R., Hall, S. R., and Shetter, R.: Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 9, 5785–5812, https://doi.org/10.5194/acp-9-5785-2009, 2009.
Yuan, B., Koss, A., Warneke, C., Gilman, J. B., Lerner, B. M., Stark, H., and de Gouw, J. A.: A high-resolution time-of-flight chemical ionization mass spectrometer utilizing hydronium ions (H3O+ ToF-CIMS) for measurements of volatile organic compounds in the atmosphere, Atmos. Meas. Tech., 9, 2735–2752, https://doi.org/10.5194/amt-9-2735-2016, 2016.
Please read the editorial note first before accessing the article.
- Article
(1231 KB) - Full-text XML
-
Supplement
(2141 KB) - BibTeX
- EndNote