the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Mar 2018
| 20 Mar 2018
Secondary sulfate is internally mixed with sea spray aerosol and organic aerosol in the winter Arctic
Rachel M. Kirpes
Amy L. Bondy
Daniel Bonanno
Ryan C. Moffet
Bingbing Wang
Alexander Laskin
Few measurements of aerosol chemical composition have been made during the winter–spring transition (following polar sunrise) to constrain Arctic aerosol–cloud–climate feedbacks. Herein, we report the first measurements of individual particle chemical composition near Utqiaġvik (Barrow), Alaska, in winter (seven sample days in January and February 2014). Individual particles were analyzed by computer-controlled scanning electron microscopy with energy dispersive X-ray spectroscopy (CCSEM-EDX, 24 847 particles), Raman microspectroscopy (300 particles), and scanning transmission X-ray microscopy with near-edge X-ray absorption fine structure spectroscopy (STXM-NEXAFS, 290 particles). Sea spray aerosol (SSA) was observed in all samples, with fresh and aged SSA comprising 99 %, by number, of 2.5–7.5 µm diameter particles, 65–95 % from 0.5–2.5 µm, and 50–60 % from 0.1–0.5 µm, indicating SSA is the dominant contributor to accumulation and coarse-mode aerosol during the winter. The aged SSA particles were characterized by reduced chlorine content with 94 %, by number, internally mixed with secondary sulfate (39 %, by number, internally mixed with both nitrate and sulfate), indicative of multiphase aging reactions during transport. There was a large number fraction (40 % of 1.0–4.0 µm diameter particles) of aged SSA during periods when particles were transported from near Prudhoe Bay, consistent with pollutant emissions from the oil fields participating in atmospheric processing of aerosol particles. Organic carbon and sulfate particles were observed in all samples and comprised 40–50 %, by number, of 0.1–0.4 µm diameter particles, indicative of Arctic haze influence. Soot was internally mixed with organic and sulfate components. All sulfate was mixed with organic carbon or SSA particles. Therefore, aerosol sources in the Alaskan Arctic and resulting aerosol chemical mixing states need to be considered when predicting aerosol climate effects, particularly cloud formation, in the winter Arctic.
- Article
(1028 KB) - Full-text XML
-
Supplement
(221 KB) - BibTeX
- EndNote





The Arctic region is experiencing warming at a greater rate than elsewhere on Earth (Pachauri et al., 2014) and undergoing substantial transformations, including rapid loss of sea ice (Overland and Wang, 2013). This is leading to increased aerosol emissions, resulting in changes to atmospheric aerosol budgets and associated climate feedbacks (Struthers et al., 2011). Characterizing the chemical composition and morphology of individual Arctic aerosol particles is important for understanding the influence of local and transported aerosols on climate (Leck et al., 2002; Leck and Svensson, 2015), which remains one of the largest uncertainties in radiative forcing (Boucher et al., 2013). Aerosol mixing state, the distribution of chemical species across an aerosol population and within each individual particle, determines particle reactivity, hygroscopicity, cloud activation efficiency, and optical properties (Prather et al., 2008; Ault and Axson, 2017). However, the few studies that have used single-particle analysis techniques to characterize the chemical mixing state of the full aerosol population have been limited to Svalbard (Weinbruch et al., 2012; Hara et al., 2003; Geng et al., 2010; Chi et al., 2015; Moroni et al., 2015, 2017; Young et al., 2016), the summertime Canadian archipelago (Köllner et al., 2017), the summertime central Arctic (Hamacher-Barth et al., 2016; Sierau et al., 2014), and the Alaskan Arctic during spring (Brock et al., 2011; Parungo et al., 1990; Parungo et al., 1993) and summer (Gunsch et al., 2017). Evaluating aerosol impacts on climate across the Arctic region is of particular importance given rapid changes in aerosol sources. Therefore, there is an urgent need to study the chemical composition of individual Arctic aerosol particles.
Aerosol influences on cloud formation and cloud–climate feedbacks in the Arctic are highly uncertain during winter, when there is little direct solar radiation and longwave radiative forcing dominates (Holland and Bitz, 2003; Letterly et al., 2016; Pithan and Mauritsen, 2014; Garrett and Zhao, 2006). Few studies have characterized Arctic aerosols, particularly those that may act as cloud condensation nuclei (CCN) and ice-nucleating particles (INP), during this period. Most studies in the winter–spring have focused on the components of Arctic haze, long-range transported pollution from the midlatitudes present in the Arctic after polar sunrise, including non-sea-salt sulfate, soot, organics, and metals (e.g., Sturges and Barrie, 1988; Norman et al., 1999; Sirois and Barrie, 1999; Quinn et al., 2002; Polissar et al., 1999; Hara et al., 2002b; Fisher et al., 2011). Notably, particulate sulfate concentrations in the Alaskan Arctic during haze season are 0.1–0.4 µg m−3 on average, and much higher than average nitrate concentrations of 0.01–0.03 µg m−3 (Quinn et al., 2007). Sea spray aerosol (SSA) has also been identified as a significant contributor to the winter–spring aerosol budget by mass (10–30 %) in the Canadian Arctic (Sirois and Barrie, 1999; Norman et al., 1999; Quinn et al., 2002) and by number (55–85 %) in the Norwegian Arctic (Weinbruch et al., 2012). SSA are efficient CCN (Collins et al., 2013; Quinn et al., 2014) and can act as INP (DeMott et al., 2016), resulting in complex sea ice–aerosol–cloud interactions in the Arctic (Browse et al., 2014). Gaseous sulfuric acid or sulfur dioxide associated with Arctic haze has been shown to react with SSA, resulting in sulfate formation and internally mixed SSA–sulfate particles (Hara et al., 2002a, 2003). While less commonly observed in the Arctic, reactions between gaseous HNO3 or N2O5 and SSA can also form mixed SSA–nitrate particles (Hara et al., 1999). These multiphase reactions result in chlorine (HCl, ClNO2, Cl2) liberation from SSA, contributing to atmospheric halogen chemistry (Sturges and Barrie, 1988; Barrie and Barrie, 1990; Hara et al., 2002c, a). Given changing marine emissions coupled with transported pollution, it is important to understand aerosol chemical composition and heterogeneous processing to determine impacts on climate in the winter Arctic.
To improve our understanding of Arctic aerosol chemical mixing state under the changing radiation and sea ice conditions during the winter–spring transition (following polar sunrise), atmospheric particles were collected near Utqiaġvik (Barrow), Alaska, during January and February 2014. Scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX), Raman microspectroscopy, and scanning transmission X-ray microscopy with near-edge X-ray absorption fine structure spectroscopy (STXM-NEXAFS) were utilized to characterize individual particle chemical composition and mixing state. To our knowledge, these are the first measurements of individual particle chemical composition in the Alaskan Arctic during winter. The relative contributions of regional Arctic haze and SSA on the aerosol budget during this winter–spring transition were examined, and the mixing states of individual aerosol particles were evaluated to examine atmospheric aging by multiphase reactions forming sulfate and nitrate.
Atmospheric particle sampling was conducted from 23 to 28 January and 24 to 28 February 2014 near Utqiaġvik (Barrow), Alaska at a tundra field site (71.28∘ N, 156.64∘ W) located ∼ 5 km inland from the Arctic Ocean. Ozone concentrations and meteorological data, including wind speed, wind direction, and solar radiation, were obtained from the NOAA Barrow Observatory (71.32∘ N, 156.61∘ W), located 5 km to the northeast of the sampling site and separated only by flat tundra. Atmospheric particles were collected using a rotating micro-orifice uniform deposition impactor (MOUDI, MSP Corp., model 110) sampling at 30 LPM through a 10 µm cut-point cyclone (URG-2000-30EA) located ∼ 2 m above the snow surface. The 50 % particle collection efficiency size cuts for the six MOUDI stages used were 3.2, 1.8, 1.0, 0.56, 0.32, and 0.18 µm aerodynamic diameter (Da). Particles were impacted on transmission electron microscopy (TEM) grids (Carbon Type-B film copper grids, Ted Pella, Inc.) and silicon substrates (Ted Pella, Inc.) for SEM analysis, and quartz substrates (Ted Pella, Inc.) for Raman microspectroscopy analysis. Particle samples were stored frozen prior to analysis to keep near the ambient temperature at collection. Samples selected for analysis were collected for ∼ 24 h on 24–25 January (10:15–10:00 AKST) and 27–28 January (11:00–10:30 AKST), ∼ 18 h on 26 January (11:00–17:15 AKST), ∼ 12 h during 26 February daytime (09:00–19:30 AKST), 26 February nighttime (19:45–08:30 AKST), 27 February daytime (09:00–19:30 AKST), and 27 February nighttime (20:00–07:30 AKST). These time periods were characterized by wind directions of 75–225∘ such that the town of Utqiaġvik was not upwind during sampling. Polar sunrise occurred at Utqiaġvik on 22 January 2014.
Computer-controlled SEM (CCSEM) analysis of individual atmospheric particles was completed using a FEI Quanta environmental SEM with a field emission gun operating at 20 keV with a high-angle annular dark field (HAADF) detector (Laskin et al., 2006, 2012). An EDX spectrometer (EDAX, Inc.) collected X-ray spectra from elements with atomic numbers higher than Be (Z=4). A total of 24 847 individual particles, typically ∼ 1000 per substrate, were analyzed by CCSEM-EDX. A size distribution showing the number of particles analyzed by CCSEM-EDX is shown in Fig. S1 in the Supplement. Morphological data, including projected area diameter (Dpa) and perimeter, were collected for each particle, in addition to the relative abundance of the following elements quantified from the EDX spectra: C, N, O, Na, Mg, Al, Si, P, S, Cl, K, Ca, and Fe. Individual particle data were analyzed using K-means clustering of the EDX spectra (Ault et al., 2012; Shen et al., 2016; Axson et al., 2016). K-means cluster analysis resulted in 50 clusters, which were then grouped into five particle classes (fresh SSA, partially aged SSA, organic + sulfate aerosol, fly ash aerosol, and mineral dust aerosol), based on comparisons of cluster EDX spectra with particle classes identified in previous studies. Prior ambient aerosol CCSEM-EDX studies have established EDX spectral signatures for fresh and aged SSA (Ault et al., 2013a; Hara et al., 2002c, 2003), organic + sulfate aerosol (Moffet et al., 2010b; Laskin et al., 2006; Allen et al., 2015), fly ash (Ault et al., 2012), and mineral dust (Coz et al., 2009; Sobanska et al., 2003; Axson et al., 2016; Creamean et al., 2016).
Individual particles from two MOUDI stages (1.0–1.8 and 0.56–1.0 µm aerodynamic diameter size ranges) for each of the seven samples were also analyzed by Raman microspectroscopy using a Horiba Scientific Labram HR Evolution spectrometer coupled with a confocal optical microscope (100× Olympus objective, 0.9 numerical aperture) equipped with a Nd : YAG laser source (50 mW, 532 nm) and CCD detector. A 600 groove mm−1 diffraction grating was used, yielding spectral resolution of 1.8 cm−1. The laser power was adjusted between 25 and 100 % by varying a neutral density filter to prevent damage to the sample. Raman spectra were obtained over the 500–4000 cm−1 range for ∼ 300 particles. Spectra were compared with prior Raman studies of nascent and reacted sea spray aerosol (Ault et al., 2013c, 2014).
Beamline 5.3.2 on the Advanced Light Source at Lawrence Berkeley National Laboratory (Berkeley, CA) was used for STXM-NEXAFS analysis over the carbon K edge (280–320 eV), as previously described by Moffet et al. (2010a). Briefly, X-rays from the synchrotron were energy-selected using a monochromator, focused on the sample, and raster scanned across a selected area. The sample was rescanned at closely spaced X-ray energies to complete a spectral image stack. After the X-ray spectra were converted to optical density using the Beer–Lambert law, STXM-NEXAFS maps were generated to show the distribution of organic carbon, soot, and inorganic components in individual aerosol particles, based on the X-ray absorptions at 288.5, 285.4, and 283 eV, respectively. From the 26 February nighttime sample (0.10–0.18 µm Da), 290 particles were analyzed for detection of organic carbon. Dpa was measured by CCSEM-EDX, Raman, and STXM-NEXAFS; therefore, it is the parameter reported for all data herein. Dpa is often larger than geometric diameter due to particle deformation upon impaction (Sobanska et al., 2014; Hinds, 2012; O'Brien et al., 2014), indicating that particle size reported here is an upper bound and could represent smaller diameter in the atmosphere.
3.1 Chemical composition and size distribution of observed particle types
Five individual particle classes, including fresh SSA, partially aged SSA, organic + sulfate particles, fly ash, and mineral dust particles, were identified from the CCSEM-EDX data (Fig. 1). SSA (both fresh and partially aged) and organic (with and without sulfate) particles were the most commonly observed types, indicating that mixing of sulfate with SSA and organic aerosol may be significant in the winter Arctic. Fresh and partially aged SSA comprised 99 %, by number, of the observed supermicron particles (1.0–7.5 µm Dpa) (Fig. 2). Across the submicron size range (0.1–1.0 µm Dpa), the majority of particles were also SSA (50–75 %, by number) (Fig. 2). The prevalence of SSA particles, even in the winter, may be a result of changing conditions in the Arctic, with previous work showing local SSA influence in Utqiaġvik, Alaska, from nearby sea ice leads, even during winter (May et al., 2016). Organic particles (with and without sulfate) were also a significant fraction (25–50 %, by number) of submicron particles. Only a limited fraction of particles (∼ 1 % by number across the entire size range) were classified as fly ash or mineral dust, characterized by silicon and oxygen, with trace amounts of aluminum, sodium, and iron (Coz et al., 2009; Sobanska et al., 2003).
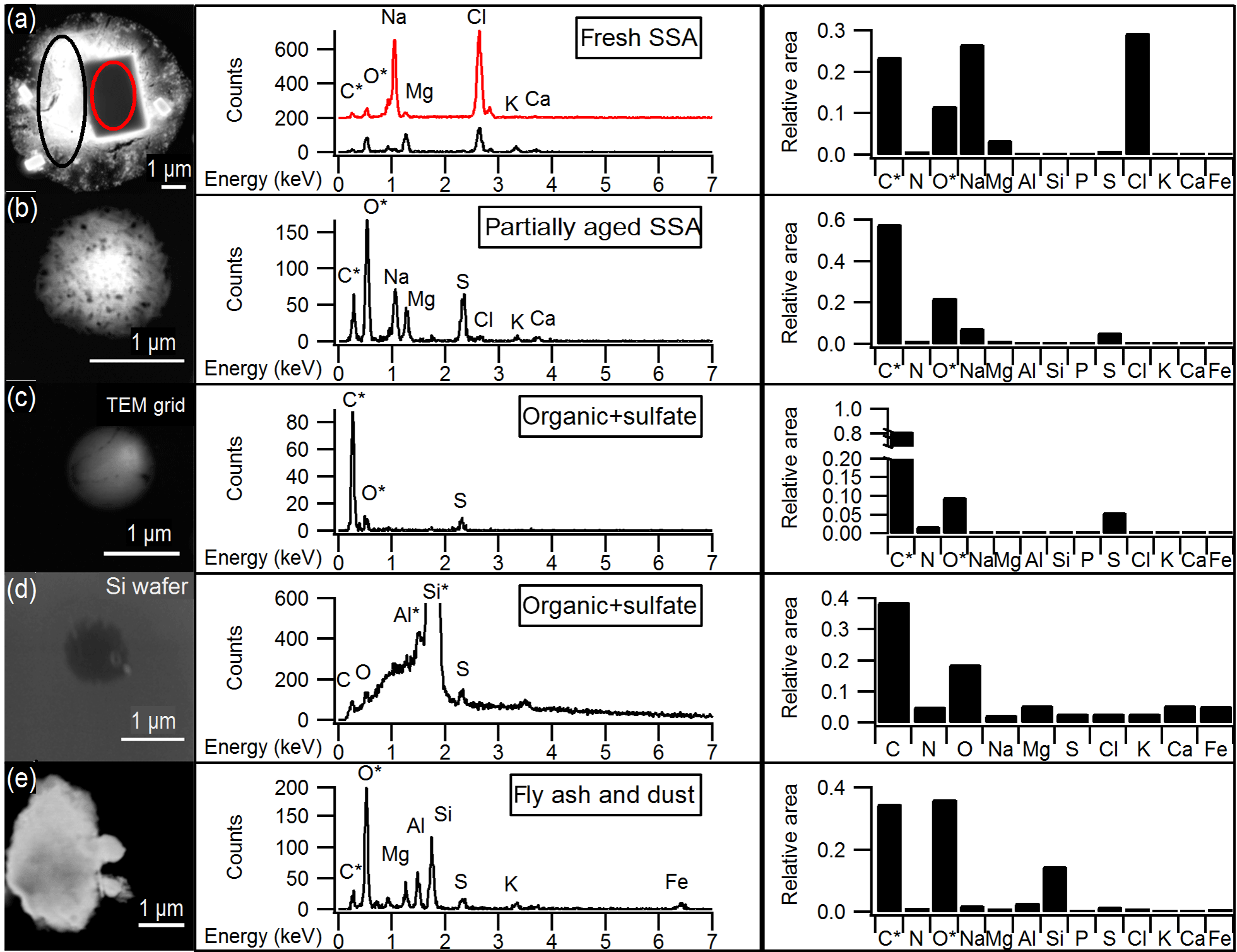
Figure 1Representative SEM images and EDX spectra of individual particles corresponding to the main particle types observed by CCSEM-EDX, and the average EDX spectrum for each particle type. Average spectra show the relative peak areas of all elements analyzed by CCSEM-EDX. (a) Fresh SSA particle comprised of sodium chloride core (red) and magnesium chloride shell (black). The spectrum for the core is offset for clarity. (b) Partially aged SSA particle containing sodium and more sulfur than chlorine. (c) Organic + sulfate particle. (d) Organic + sulfate particle on silicon substrate. (e) Aluminum- and silicon-containing dust particle. *Carbon and oxygen peaks include some signal from TEM grid substrate background for particles (a), (b), (c), and (e). Aluminum and silicon peaks are due to sample holder and silicon substrate background, respectively, for particle (d).
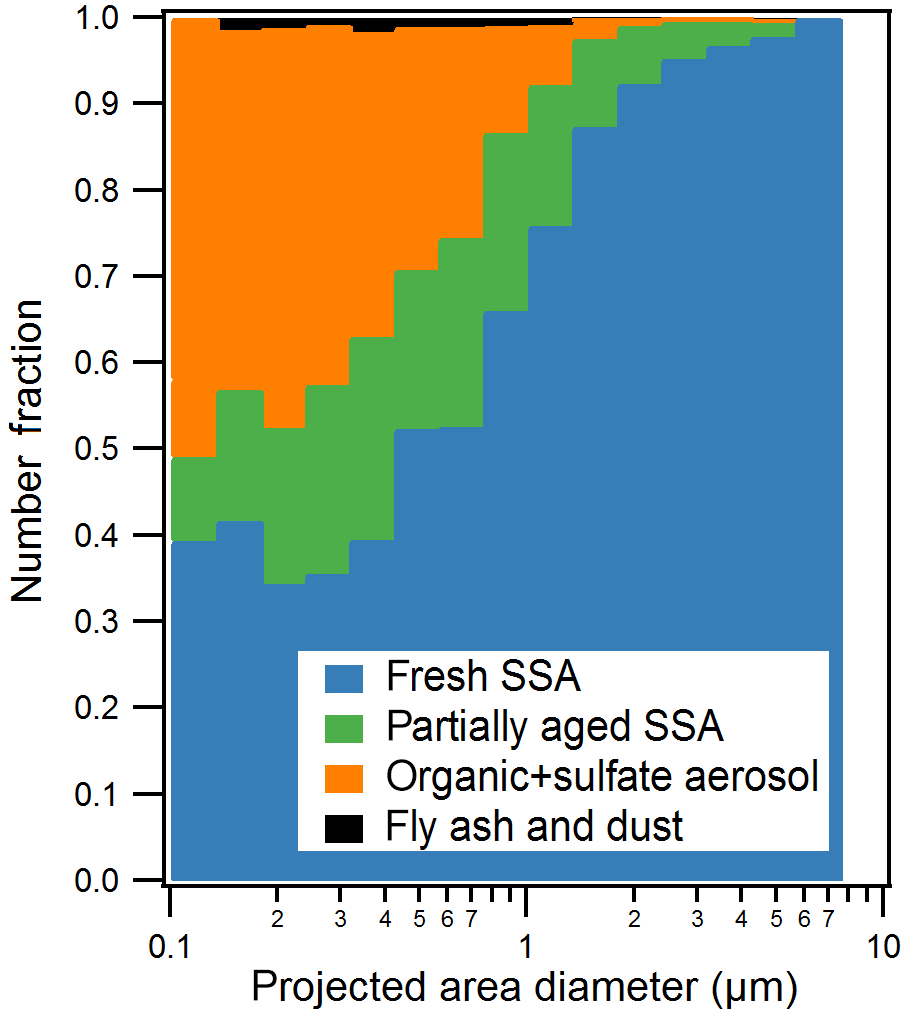
Figure 2Size-resolved CCSEM-EDX number fraction distributions of observed particle types for all samples. Particles were sorted into 16 bins (logarithmic) from 0.1 to 10.0 µm projected area diameter (8 bins per decade). Organic + sulfate class includes a small fraction of internally mixed soot.
Table 1Size-resolved number fractions of individual fresh SSA, partially aged SSA, and organic + sulfate particles containing Cl, S, and N, in addition to average atomic (mole) ratios of Cl ∕ Na, S ∕ Na, and N ∕ Na for individual fresh and partially aged SSA.
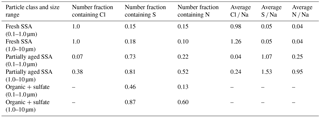
Particles classified as fresh SSA, based on grouping by chemical composition by K-means analysis, contained sodium, magnesium, sulfur, and chlorine in similar mole ratios (Table 1) to those found in seawater (Cl ∕ Na = 1.2, Mg ∕ Na = 0.11, S ∕ Na = 0.06) (Quinn et al., 2015; Pilson, 2013), indicating these particles had not undergone chemical aging processes during atmospheric transport. Some SSA particles were observed with a sodium chloride core and magnesium chloride outer coating (Fig. 1), which is likely due to the particle undergoing efflorescence after collection (Ault et al., 2013b); this morphology has been previously observed for Arctic SSA particles (Chi et al., 2015). The partially aged SSA particles contained sulfur and/or nitrogen and were characterized by Cl ∕ (Na + 0.5 Mg) ratios of less than 1 (Laskin et al., 2012). This indicates that multiphase reactions had occurred, releasing chlorine-containing trace gases, primarily hydrochloric acid (Laskin et al., 2002, 2003; Gard et al., 1998), and resulting in the formation of sulfate and nitrate in the particles. SSA chemical mixing state information is further discussed in Sect. 3.2. SSA aging was observed for few 1.0–7.5 µm particles (7 %, by number, aged SSA and 90 % fresh SSA), with a greater fraction of submicron 0.1–1.0 µm SSA particles having undergone aging (18 %, by number, aged SSA and 42 % fresh SSA) (Fig. 2). Compared to supermicron particles, submicron particles have longer atmospheric lifetimes, a smaller Cl reservoir, and greater surface area to volume ratios, which are conducive to increased atmospheric processing (Hara et al., 2002a; Leck et al., 2002; Williams et al., 2002; Ault et al., 2014). While concentrations of sulfur- and nitrogen-containing gases are lower in the Arctic winter compared to the peak of spring haze season, allowing for SSA particles to remain chemically fresh further from the emission point, aged SSA particles have also been observed during winter at Svalbard (Hara et al., 1999, 2002a). Overall, fresh and aged SSA were significant contributors to the winter Arctic aerosol budget (Figs. 2 and S2). This observation is consistent with studies of annual Arctic aerosol trends that have shown a large influence of SSA in the winter by mass: constituting up to 40 % of supermicron mass at Barrow (Quinn et al., 2002) and 60–90 % of 0.5–10 µm particles, by number, for winter samples at Svalbard (Weinbruch et al., 2012).
Organic particles, classified by K-means analysis, were characterized by spherical morphology and carbon and oxygen in the single-particle EDX spectra. Since there is background C and O EDX signal from the TEM grid substrate film, the contribution of C and O to this particle class was confirmed by CCSEM-EDX analysis of 110 particles that had been collected simultaneously on silicon substrates that do not have these interferences. Figure 1 shows the representative EDX spectra of organic particles analyzed on TEM grids and silicon substrates for comparison. Sulfur was present in 47 %, by number, of organic particles, at levels of at least 2 % atomic content in the EDX spectrum; therefore, these organic particles will be discussed together as an organic + sulfate particle class (Laskin et al., 2006; Moffet et al., 2010b). Example organic + sulfate particles are shown in Fig. 1c and d. Organic + sulfate particles were primarily observed in the submicron size range (Fig. 2). Overall, 40–50 % of the particles 0.1–0.5 µm in diameter and 15–25 % of the 0.5–1.0 µm particles, by number, were classified as organic + sulfate (Fig. 2). The detailed chemical mixing states of these organic + sulfate particles will be discussed in Sect. 3.3. The presence of a large number fraction of submicron organic + sulfate particles is consistent with previous winter–spring Arctic studies, which have observed organic particles contributing up to 30 % of submicron aerosol by mass and greater than 80 %, by number, at Barrow (Shaw et al., 2010; Hiranuma et al., 2013) and greater than 80 %, by number, of 0.1–0.5 µm (aerodynamic diameter) particles at Svalbard (Weinbruch et al., 2012). Internal mixing of organic and sulfate aerosol has previously been observed in the Arctic winter–spring at Svalbard, with most 0.2–2.0 µm (aerodynamic diameter) organic particles containing sulfate (Hara et al., 2002b). Internally mixed organic + sulfate aerosol is now being observed across the Arctic during the winter, highlighting the importance of considering sulfate mixing states during this period.
3.2 Internal mixing of SSA with sulfate and nitrate
Raman microspectroscopic analysis of individual aged SSA particles confirmed that the sulfur and nitrogen detected by EDX in SSA were in the forms of sulfate and nitrate, respectively, based on the presence of sharp peaks corresponding to characteristic symmetric stretches at ∼ 1000 cm−1 for νs(SO and ∼ 1050 cm−1 for νs(NO (Fig. 3) (Ault et al., 2014; Deng et al., 2014; Eom et al., 2016). In addition, these particles were characterized by broad peaks in the 3000–3500 cm−1 range (Fig. 3), corresponding to O–H stretching, likely due to particle-phase water (Ault et al., 2014), confirmed by the frequency of the νs(NO mode at ∼ 1050 cm−1. Raman C–H stretching peaks in the 2800–3000 cm−1 range indicated that organic compounds were present in both fresh and aged SSA (Ault et al., 2013c; Baustian et al., 2012; Eom et al., 2016); the organic functional groups present will be discussed further in a future publication.
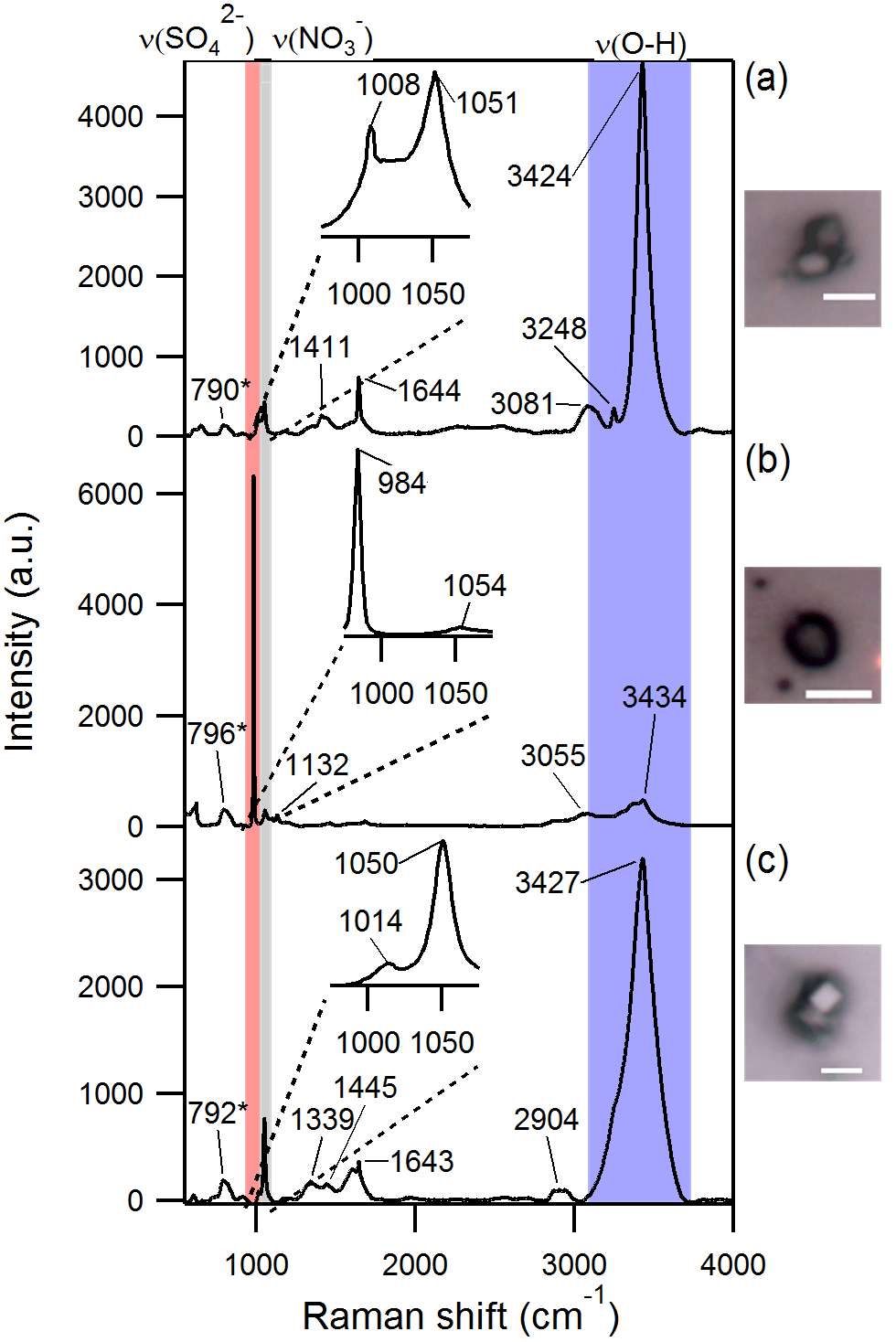
Figure 3Optical images and Raman spectra of three representative SSA particles containing nitrate and/or sulfate and hydroxyl groups. A total of ∼ 300 individual particles were analyzed by Raman microspectroscopy. * The 790–796 cm−1 peak is due to quartz substrate background. Scale bar for all images is 5 µm.
Based on the CCSEM-EDX analysis, SSA aging by sulfur species (e.g., sulfuric acid) was more prevalent than aging by nitrogen species (e.g., nitric acid) in the submicron size range, consistent with previous measurements of SSA during Arctic haze periods in the Norwegian Arctic (Hara et al., 2002c). A total of 73 % of partially aged SSA, by number, in the 0.1–1.0 µm size range contained secondary sulfate. This was determined by a S ∕ Na ratio at least 25 % greater than the seawater mole ratio 0.06 (Pilson, 2013), with these particles having an average S ∕ Na ratio of 1.07 (Table 1). In comparison, only 22 % of 0.1–1.0 µm particles contained nitrate (Table 1). The diffusion-limited uptake of SO2 in submicron particles is favored over the thermodynamically controlled uptake of HNO3, resulting in a preference for sulfate in submicron aged SSA (Liu et al., 2007; Zhuang et al., 1999; Kerminen et al., 1998). However, sulfate was also more prevalent than nitrate in supermicron SSA (Table 1), where kinetically favorable uptake of HNO3 would be expected to dominate, suggesting that higher concentrations of H2SO4, compared to HNO3, and aqueous-phase sulfate formation influenced particle aging. The prevalence of SSA aging by sulfur species near Utqiaġvik is consistent with the appearance of springtime Arctic haze, as 30 % of submicron particle mass corresponds to sulfate during haze season (January to May) (Quinn et al., 2007, 2002). Sulfate mass concentrations peak in winter–spring near Utqiaġvik, while methanesulfonic acid mass is greatest in the summer and has not been observed during winter months (Quinn et al., 2007). Therefore, the prevalence of mixed SSA–sulfate suggests that reactions with sulfuric acid from Arctic haze are an important source of SSA sulfate (Hara et al., 2002a; Barrie and Barrie, 1990). SSA aging through sulfate addition was likely also due to influence from Prudhoe Bay SO2 emissions (Peters et al., 2011; Gunsch et al., 2017), discussed further in Sect. 3.4.2.
3.3 Organic particle mixing states
Organic particles and internally mixed organic + sulfate particles composed a significant number fraction of submicron particles, which is consistent with the presence of organic aerosol, sulfuric acid, and ammonium sulfate in Arctic haze (Hara et al., 2002b; Hirdman et al., 2010). STXM-NEXAFS indicated the presence of organic carbon in these particles, based on X-ray absorption at 288.5 eV, characteristic of carboxylic acids (Moffet et al., 2010a). Additionally, STXM-NEXAFS analysis confirmed that organic and inorganic (likely sulfate, based on sulfur detected during CCSEM-EDX analyses) components were internally mixed within individual particles (Fig. 4), with particles showing an internal mix of both inorganic-dominant (> 50 %) and organic-dominant regions. The pre- and post-edge ratio of inorganic to organic components also indicated that most analyzed particles contained both inorganic and organic species (Fig. 4b). Raman analysis confirmed sulfur was present in the form of sulfate. Nitrogen (nitrate, according to Raman analysis) was also present in 15 % of 0.1–1.0 µm organic + sulfate particles, by number.
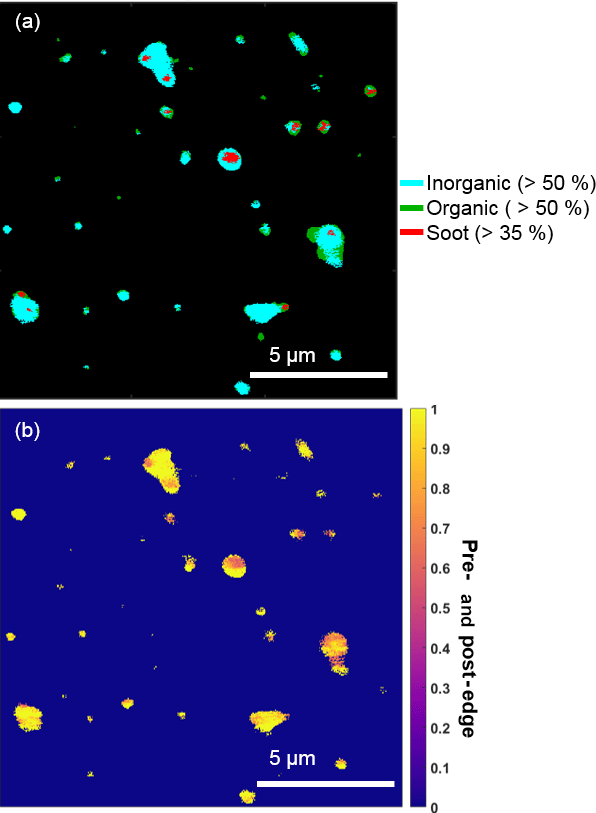
Figure 4Representative STXM-NEXAFS map from 26 February nighttime showing (a) the distributions of inorganic dominant (blue, > 50 % by mass), organic carbon dominant (green, > 50 % by mass), and soot (red, sp2 > 35 %) and (b) the ratio of inorganic (pre-) and organic (post-edge) components between populations of individual particles sampled during a period with a high fraction of organic + sulfate particles.
Chemical mixing state analysis determined that a small fraction of particles classified as organic + sulfate (7 % of this particle class, by number) by CCSEM-EDX were primarily carbon-containing particles with less than 5 % oxygen and sulfur. For the 26 February nighttime sample analyzed by STXM-NEXAFS, elevated levels of sp2 carbon, indicative of soot, were observed in some particles (Fig. 4) (Moffet et al., 2010a). These small soot particles observed by STXM-NEXAFS were likely members of the “primarily carbon” group identified by CCSEM-EDX and were internally mixed with organic carbon and inorganic species (likely sulfate, based on sulfur detected during CCSEM-EDX analyses). Therefore, these particles were included in the organic + sulfate class. Externally mixed soot particles, comprised solely of elemental carbon with no organic or sulfate component, were not observed in any sample, indicating that all soot was internally mixed with organic + sulfate particles. Soot present in Arctic haze (Quinn et al., 2007; Law and Stohl, 2007) has previously been observed to be internally mixed with sulfate for winter–spring Arctic aerosol, with soot-sulfate particles contributing ∼ 10–20 % of observed particles sampled (< 2.0 µm), by number, at Svalbard (Hara et al., 2003).
3.4 Influence of marine- and Prudhoe Bay-influenced air masses on particle composition
There was no clear dependence or trend with wind speed or month (January vs. February) for SSA S ∕ Na or Cl ∕ Na ratios, with average wind speeds ranging from 5–12 m s−1 for the selected sampling periods, but some variability in particle composition between samples could be attributed to the influence of different air masses. Though all samples experienced some degree of Arctic Ocean air mass influence due to the sampling location and prevailing wind direction from the north over the Beaufort Sea to the sampling site, using NOAA HYSPLIT 48 h backward air mass trajectory analysis (Rolph, 2016), two main air mass source regions (Arctic Ocean and Prudhoe Bay influence) were determined for the seven analyzed sample periods. Most notably, the 26 February daytime sample was influenced by air from the north and east over the Arctic Ocean within the boundary layer for the 6–7 h prior to arrival at the sampling site, whereas the 27 January sample had prolonged surface influence (18 h) along the air mass trajectory from the east to the southeast, during which the air mass passed over Prudhoe Bay, the third largest oilfield in North America (U.S. Energy Information Administration, 2015) (Fig. 5). Prudhoe Bay influence was determined by HYSPLIT trajectories that passed within 1∘ (∼ 50 km) of the Prudhoe Bay emissions box, described in Kolesar et al. (2017) as the area significantly influenced by combustion emissions from the oilfields. The air mass trajectories for the remaining samples (24 January, 26 January day, 26 February night, 27 February day, 27 February night) fell in between the two regions (Arctic Ocean and Prudhoe Bay influence).

Figure 5Size-resolved number fractions of observed particle types (CCSEM-EDX), for example sample periods influenced by (a) the Arctic Ocean (26 February day, 4490 particles) and (b) Prudhoe Bay (27 January, 1475 particles). Air mass influence is shown for (c) 26 February daytime and (d) 27 January as determined by NOAA HYSPLIT 48 h backward air mass trajectories. Both ensemble (dotted line) and single representative trajectories are shown. Color scale indicates air mass altitude, and markers are placed at 6 h intervals. Red line shows extent of Prudhoe Bay emissions influence box (Kolesar et al., 2017). Yellow diamond indicates sampling site near Utqiaġvik.
Comparison of particle type contributions as a function of size for the representative Arctic Ocean-influenced (26 February day) and Prudhoe Bay-influenced (27 January) samples are shown in Fig. 5 (with results of additional samples shown in Fig. S2). The Arctic Ocean-influenced sample was characterized by a large fraction (95 %, by number) of fresh SSA in the 1.0–7.5 µm size range. In comparison, the Prudhoe Bay-influenced sample was characterized by 55 % fresh SSA and 40–45 % partially aged SSA, by number, in the supermicron range. This is indicative of multiphase reactions between SSA and gaseous emissions from combustion at the oilfields (e.g., SO2, NOx) (Jaffe et al., 1991; Peters et al., 2011; Gunsch et al., 2017), contributing to a greater number fraction of aged SSA during Prudhoe Bay-influenced periods. The Prudhoe Bay-influenced sample also had a greater number fraction of organic + sulfate particles in the 0.1–0.5 µm range (60–70 %) compared to the Arctic Ocean-influenced sample (40–50 %). Given that organic + sulfate particles were a significant fraction of submicron particles in all samples, including ocean-influenced periods, these samples were likely influenced by long-range transported pollution from the midlatitudes, consistent with regional background haze (Quinn et al., 2007). However, it is likely that gas-particle partitioning of oxidation products from and multiphase reactions of Prudhoe Bay oilfield combustion emissions, including volatile organic compounds and SO2 (Peters et al., 2011; Jaffe et al., 1991; Gunsch et al., 2017), also results in the formation of organic + sulfate particles, including particles internally mixed with soot (Sect. 3.3), contributing to the increased number fraction of organic + sulfate particles observed during Prudhoe Bay-influenced periods.
For atmospheric particles collected in January and February 2014 near Utqiaġvik, Alaska, SSA was observed to be the most prevalent particle type, composing 50–75 and 99 %, by number, of particles in the 0.1–1.0 and 1.0–7.5 µm projected area diameter ranges, respectively. Internal mixing of sulfate and nitrate with SSA particles was observed in all samples, regardless of air mass influence, suggesting prevalent regional pollution, such as Arctic haze influence, for secondary inorganic aerosol formation. Prudhoe Bay-influenced air masses were characterized by higher number fractions of partially aged SSA, however, suggesting that oilfield emissions also contribute significantly to multiphase reactions with SSA. Most global and regional climate models assume that Arctic haze components (sulfate, organic aerosol, black carbon) and natural aerosols are externally mixed and do not predict climate impacts of internally mixed species (Eckhardt et al., 2015; Alterskjaer et al., 2010; Korhonen et al., 2008). However, no externally mixed sulfate or sulfuric acid particles were observed during January or February sampling in Utqiaġvik, Alaska; all sulfate was internally mixed with organic aerosol particles or with SSA. Internal mixing of SSA and sulfate reduces CCN efficiencies compared to externally mixed sulfate aerosol or SSA, as sodium sulfate is less hygroscopic than sodium chloride or sulfuric acid (Gong and Barrie, 2003; Petters and Kreidenweis, 2007). The prevalence of SSA internally mixed with sulfate should be considered in the interpretation of elevated sulfate concentrations in the winter–spring Arctic atmosphere (Sturges and Barrie, 1988; Sirois and Barrie, 1999; Hara et al., 2002a).
While SSA comprised 50–60 % of 0.1–0.5 µm particles, by number, organic + sulfate particles made up 40–50 %, by number, in this particle diameter range and were present in similar number fractions in all samples, suggesting the importance of Arctic haze as a source of submicron particles in January and February in Utqiaġvik, Alaska. Internal mixing of sulfate and nitrate with organic aerosol is consistent with previous single-particle measurements at Svalbard, where organic aerosol mixed with sulfate and nitrate was observed to be the dominant particle type in the submicron size range in the winter and spring (Weinbruch et al., 2012). Weinbruch et al. (2012) also observed soot particles internally mixed with organics, sulfate, and nitrate, consistent with the small fraction of internally mixed organic + sulfate and soot particles (∼ 2–3 % of total observed particles, by number) observed in this study. The internal mixing of sulfate with organic aerosol is important to consider in climate predictions, as the CCN activity of internally mixed organic + sulfate aerosol is reduced relative to externally mixed sulfate, due to the lower hygroscopicity of the organic fraction (Wang et al., 2015; Petters and Petters, 2016). Continuing oil and gas development in the Arctic region will influence both SSA and organic aerosol composition (Peters et al., 2011), as well as mixing state, due to secondary inorganic aerosol formation.
Data are available by contacting the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-3937-2018-supplement.
RMK, KAP, and APA prepared the manuscript and led data interpretation. KAP collected the samples. RMK analyzed the samples, with assistance from ALB. DB and RM conducted the STXM-NEXAFS analysis. AL and BW assisted with CCSEM-EDX analysis. APA provided guidance with CCSEM-EDX and Raman microspectroscopy analysis.
The authors declare that they have no conflict of interest.
CCSEM-EDX analyses were performed at the Environmental Molecular
Sciences Laboratory (EMSL), a national scientific user facility located at
the Pacific Northwest National Laboratory (PNNL) and sponsored by the Office
of Biological and Environmental Research of the US Department of Energy
(DOE). PNNL is operated for DOE by Battelle Memorial Institute under
contract no. DE-AC06-76RL0 1830. Travel funds to PNNL and Alaska were
provided by the University of Michigan College of Literature, Science, and
the Arts and Department of Chemistry. Additional travel funds and logistics
support for sampling in Alaska were provided by the National Science
Foundation (PLR-1107695). Ryan C. Moffet acknowledges funding by US DOE's
Atmospheric System Research Program, BER under grant DE-SC0008643. The
STXM-NEXAFS particle analysis was performed at beamlines 5.3.2 at the
Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory. The
work at the ALS was supported by the Director, Office of Science, Office of
Basic Energy Sciences, of the US DOE under contract DE-AC02-05CH11231.
Bingbing Wang acknowledges the support by Chinese Fundamental Research Funds for the
Central Universities (no. 20720160111) and the Recruitment Program of Global
Youth Experts of China. Rachel M. Kirpes received funding in part from a University
of Michigan Davis Graduate Fellowship. Meteorological data were obtained
from the NOAA Earth System Research Laboratory Barrow Observatory. The
authors gratefully acknowledge the NOAA Air Resources Laboratory for the
provision of the HYSPLIT transport and dispersion model and READY website
(http://www.ready.noaa.gov) (Rolph, 2016) used in this publication.
Edited by: Annmarie Carlton
Reviewed by: two anonymous referees
Allen, H. M., Draper, D. C., Ayres, B. R., Ault, A., Bondy, A., Takahama, S., Modini, R. L., Baumann, K., Edgerton, E., Knote, C., Laskin, A., Wang, B., and Fry, J. L.: Influence of crustal dust and sea spray supermicron particle concentrations and acidity on inorganic NO aerosol during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 15, 10669–10685, https://doi.org/10.5194/acp-15-10669-2015, 2015.
Alterskjaer, K., Kristjansson, J. E., and Hoose, C.: Do anthropogenic aerosols enhance or suppress the surface cloud forcing in the Arctic?, J. Geophys. Res.-Atmos., 115, DS22204, https://doi.org/10.1029/2010jd014015, 2010.
Ault, A. P. and Axson, J. L.: Atmospheric Aerosol Chemistry: Spectroscopic and Microscopic Advances, Anal. Chem., 89, 430–452, https://doi.org/10.1021/acs.analchem.6b04670, 2017.
Ault, A. P., Peters, T. M., Sawvel, E. J., Casuccio, G. S., Willis, R. D., Norris, G. A., and Grassian, V. H.: Single-particle SEM-EDX analysis of iron-containing coarse particulate matter in an urban environment: sources and distribution of iron within Cleveland, Ohio, Environ. Sci. Technol., 46, 4331–4339, https://doi.org/10.1021/es204006k, 2012.
Ault, A. P., Guasco, T. L., Ryder, O. S., Baltrusaitis, J., Cuadra-Rodriguez, L. A., Collins, D. B., Ruppel, M. J., Bertram, T. H., Prather, K. A., and Grassian, V. H.: Inside versus Outside: Ion Redistribution in Nitric Acid Reacted Sea Spray Aerosol Particles as Determined by Single Particle Analysis, J. Am. Chem. Soc., 135, 14528–14531, https://doi.org/10.1021/ja407117x, 2013a.
Ault, A. P., Moffet, R. C., Baltruaitis, J., Collins, D. B., Ruppel, M. J., Cuadra-Rodriguez, L. A., Zhao, D., Guasco, T. L., Ebben, C. J., Geiger, F. M., Bertram, T. H., Prather, K. A., and Grassian, V. H.: Size-Dependent Changes in Sea Spray Aerosol Composition and Properties with Different Seawater Conditions, Environ. Sci. Technol., 47, 5603–5612, https://doi.org/10.1021/es400416g, 2013b.
Ault, A. P., Zhao, D., Ebben, C. J., Tauber, M. J., Geiger, F. M., Prather, K. A., and Grassian, V. H.: Raman microspectroscopy and vibrational sum frequency generation spectroscopy as probes of the bulk and surface compositions of size-resolved sea spray aerosol particles, Phys. Chem. Chem. Phys., 15, 6206–6214, 2013c.
Ault, A. P., Guasco, T. L., Baltrusaitis, J., Ryder, O. S., Trueblood, J. V., Collins, D. B., Ruppel, M. J., Cuadra-Rodriguez, L. A., Prather, K. A., and Grassian, V. H.: Heterogeneous Reactivity of Nitric Acid with Nascent Sea Spray Aerosol: Large Differences Observed between and within Individual Particles, J. Phys. Chem. Lett., 5, 2493–2500, https://doi.org/10.1021/jz5008802, 2014.
Axson, J. L., Shen, H., Bondy, A. L., Landry, C. C., Welz, J., Creamean, J. M., and Ault, A. P.: Transported Mineral Dust Deposition Case Study at a Hydrologically Sensitive Mountain Site: Size and Composition Shifts in Ambient Aerosol and Snowpack, Aerosol Air Qual. Res., 16, 555–567, 2016.
Barrie, L. and Barrie, M.: Chemical components of lower tropospheric aerosols in the high Arctic: Six years of observations, J. Atmos. Chem., 11, 211–226, https://doi.org/10.1007/BF00118349, 1990.
Baustian, K. J., Cziczo, D. J., Wise, M. E., Pratt, K. A., Kulkarni, G., Hallar, A. G., and Tolbert, M. A.: Importance of aerosol composition, mixing state, and morphology for heterogeneous ice nucleation: A combined field and laboratory approach, J. Geophys. Res.-Atmos., 117, D06217, https://doi.org/10.1029/2011jd016784, 2012.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., and Lohmann, U.: Clouds and Aerosols, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 571–657, 2013.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Browse, J., Carslaw, K. S., Mann, G. W., Birch, C. E., Arnold, S. R., and Leck, C.: The complex response of Arctic aerosol to sea-ice retreat, Atmos. Chem. Phys., 14, 7543–7557, https://doi.org/10.5194/acp-14-7543-2014, 2014.
Chi, J. W., Li, W. J., Zhang, D. Z., Zhang, J. C., Lin, Y. T., Shen, X. J., Sun, J. Y., Chen, J. M., Zhang, X. Y., Zhang, Y. M., and Wang, W. X.: Sea salt aerosols as a reactive surface for inorganic and organic acidic gases in the Arctic troposphere, Atmos. Chem. Phys., 15, 11341–11353, https://doi.org/10.5194/acp-15-11341-2015, 2015.
Collins, D. B., Ault, A. P., Moffet, R. C., Ruppel, M. J., Cuadra-Rodriguez, L. A., Guasco, T. L., Corrigan, C. E., Pedler, B. E., Azam, F., Aluwihare, L. I., Bertram, T. H., Roberts, G. C., Grassian, V. H., and Prather, K. A.: Impact of marine biogeochemistry on the chemical mixing state and cloud forming ability of nascent sea spray aerosol, J. Geophys. Res.-Atmos., 118, 8553–8565, https://doi.org/10.1002/jgrd.50598, 2013.
Coz, E., Gomez-Moreno, F. J., Pujadas, M., Casuccio, G. S., Lersch, T. L., and Artinano, B.: Individual particle characteristics of North African dust under different long-range transport scenarios, Atmos. Environ., 43, 1850–1863, https://doi.org/10.1016/j.atmosenv.2008.12.045, 2009.
Creamean, J. M., Axson, J. L., Bondy, A. L., Craig, R. L., May, N. W., Shen, H., Weber, M. H., Pratt, K. A., and Ault, A. P.: Changes in precipitating snow chemistry with location and elevation in the California Sierra Nevada, J. Geophys. Res.-Atmos., 121, 2015JD024700, https://doi.org/10.1002/2015JD024700, 2016.
DeMott, P. J., Hill, T. C. J., McCluskey, C. S., Prather, K. A., Collins, D. B., Sullivan, R. C., Ruppel, M. J., Mason, R. H., Irish, V. E., Lee, T., Hwang, C. Y., Rhee, T. S., Snider, J. R., McMeeking, G. R., Dhaniyala, S., Lewis, E. R., Wentzell, J. J. B., Abbatt, J., Lee, C., Sultana, C. M., Ault, A. P., Axson, J. L., Diaz Martinez, M., Venero, I., Santos-Figueroa, G., Stokes, M. D., Deane, G. B., Mayol-Bracero, O. L., Grassian, V. H., Bertram, T. H., Bertram, A. K., Moffett, B. F., and Franc, G. D.: Sea spray aerosol as a unique source of ice nucleating particles, P. Natl. Acad. Sci. USA, 113, 5797–5803, https://doi.org/10.1073/pnas.1514034112, 2016.
Deng, C. H., Brooks, S. D., Vidaurre, G., and Thornton, D. C. O.: Using Raman Microspectroscopy to Determine Chemical Composition and Mixing State of Airborne Marine Aerosols over the Pacific Ocean, Aerosol Sci. Technol., 48, 193–206, https://doi.org/10.1080/02786826.2013.867297, 2014.
Eckhardt, S., Quennehen, B., Olivié, D. J. L., Berntsen, T. K., Cherian, R., Christensen, J. H., Collins, W., Crepinsek, S., Daskalakis, N., Flanner, M., Herber, A., Heyes, C., Hodnebrog, Ø., Huang, L., Kanakidou, M., Klimont, Z., Langner, J., Law, K. S., Lund, M. T., Mahmood, R., Massling, A., Myriokefalitakis, S., Nielsen, I. E., Nøjgaard, J. K., Quaas, J., Quinn, P. K., Raut, J.-C., Rumbold, S. T., Schulz, M., Sharma, S., Skeie, R. B., Skov, H., Uttal, T., von Salzen, K., and Stohl, A.: Current model capabilities for simulating black carbon and sulfate concentrations in the Arctic atmosphere: a multi-model evaluation using a comprehensive measurement data set, Atmos. Chem. Phys., 15, 9413–9433, https://doi.org/10.5194/acp-15-9413-2015, 2015.
Eom, H.-J., Gupta, D., Cho, H.-R., Hwang, H. J., Hur, S. D., Gim, Y., and Ro, C.-U.: Single-particle investigation of summertime and wintertime Antarctic sea spray aerosols using low-Z particle EPMA, Raman microspectrometry, and ATR-FTIR imaging techniques, Atmos. Chem. Phys., 16, 13823–13836, https://doi.org/10.5194/acp-16-13823-2016, 2016.
Fisher, J. A., Jacob, D. J., Wang, Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate-ammonium aerosol in the Arctic in winter-spring, Atmos. Environ., 45, 7301–7318, https://doi.org/10.1016/j.atmosenv.2011.08.030, 2011.
Gard, E. E., Kleeman, M. J., Gross, D. S., Hughes, L. S., Allen, J. O., Morrical, B. D., Fergenson, D. P., Dienes, T., M, E. G., Johnson, R. J., Cass, G. R., and Prather, K. A.: Direct observation of heterogeneous chemistry in the atmosphere, Science, 279, 1184–1187, https://doi.org/10.1126/science.279.5354.1184, 1998.
Garrett, T. J. and Zhao, C.: Increased Arctic cloud longwave emissivity associated with pollution from mid-latitudes, Natur, 440, 787, https://doi.org/10.1038/nature04636, 2006.
Geng, H., Ryu, J., Jung, H.-J., Chung, H., Ahn, K.-H., and Ro, C.-U.: Single-Particle Characterization of Summertime Arctic Aerosols Collected at Ny-Alesund, Svalbard, Environ. Sci. Technol., 44, 2348–2353, https://doi.org/10.1021/es903268j, 2010.
Gong, S. L. and Barrie, L. A.: Simulating the impact of sea salt on global nss sulphate aerosols, J. Geophys. Res.-Atmos., 108, 4516, https://doi.org/10.1029/2002JD003181, 2003.
Gunsch, M. J., Kirpes, R. M., Kolesar, K. R., Barrett, T. E., China, S., Sheesley, R. J., Laskin, A., Wiedensohler, A., Tuch, T., and Pratt, K. A.: Contributions of transported Prudhoe Bay oil field emissions to the aerosol population in Utqiagvik, Alaska, Atmos. Chem. Phys., 17, 10879–10892, https://doi.org/10.5194/acp-17-10879-2017, 2017.
Hamacher-Barth, E., Leck, C., and Jansson, K.: Size-resolved morphological properties of the high Arctic summer aerosol during ASCOS-2008, Atmos. Chem. Phys., 16, 6577–6593, https://doi.org/10.5194/acp-16-6577-2016, 2016.
Hara, K., Osada, K., Hayashi, M., Matsunaga, K., Shibata, T., Iwasaka, Y., and Furuya, K.: Fractionation of inorganic nitrates in winter Arctic troposphere: Coarse aerosol particles containing inorganic nitrates, J. Geophys. Res.-Atmos., 104, 23671–23679, https://doi.org/10.1029/1999jd900348, 1999.
Hara, K., Osada, K., Matsunaga, K., Iwasaka, Y., Shibata, T., and Furuya, K.: Atmospheric inorganic chlorine and bromine species in Arctic boundary layer of the winter/spring, J. Geophys. Res.-Atmos., 107, 4361, https://doi.org/10.1029/2001jd001008, 2002a.
Hara, K., Osada, K., Matsunaga, K., Sakai, T., Iwasaka, Y., and Furuya, K.: Concentration trends and mixing states of particulate oxalate in Arctic boundary layer in winter/spring, J. Geophys. Res.-Atmos., 107, 4361, https://doi.org/10.1029/2001jd001584, 2002b.
Hara, K., Osada, K., Nishita, C., Yamagata, S., Yamanocuhi, T., Herber, A., Matsunaga, K., Iwasaka, Y., Nagatani, M., and Nakata, H.: Vertical variations of sea-salt modification in the boundary layer of spring Arctic during the ASTAR 2000 campaign, Tellus B, 54, 361–376, https://doi.org/10.1034/j.1600-0889.2002.201253.x, 2002c.
Hara, K., Yamagata, S., Yamanouchi, T., Sato, K., Herber, A., Iwasaka, Y., Nagatami, M., and Nakata, H.: Mixing states of individual aerosol particles in spring Arctic troposphere during ASTAR 2000 campaign, J. Geophys. Res., 108, 4209, https://doi.org/10.1029/2002JD002513, 2003.
Hinds, W. C.: Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles, Wiley, 504 pp., 2012.
Hiranuma, N., Brooks, S. D., Moffet, R. C., Glen, A., Laskin, A., Gilles, M. K., Liu, P., Macdonald, A. M., Strapp, J. W., and McFarquhar, G. M.: Chemical characterization of individual particles and residuals of cloud droplets and ice crystals collected on board research aircraft in the ISDAC 2008 study, J. Geophys. Res.-Atmos., 118, 6564–6579, https://doi.org/10.1002/jgrd.50484, 2013.
Hirdman, D., Burkhart, J. F., Sodemann, H., Eckhardt, S., Jefferson, A., Quinn, P. K., Sharma, S., Ström, J., and Stohl, A.: Long-term trends of black carbon and sulphate aerosol in the Arctic: changes in atmospheric transport and source region emissions, Atmos. Chem. Phys., 10, 9351–9368, https://doi.org/10.5194/acp-10-9351-2010, 2010.
Holland, M. M. and Bitz, C. M.: Polar amplification of climate change in coupled models, Clim. Dynam., 21, 221–232, https://doi.org/10.1007/s00382-003-0332-6, 2003.
Jaffe, D. A., Honrath, R. E., Herring, J. A., Li, S. M., and Kahl, J. D.: Measurements of Nitrogen-Oxides at Barrow, Alaska during Spring – Evidence for Regional and Northern Hemispheric Sources of Pollution, J. Geophys. Res.-Atmos., 96, 7395-7405, https://doi.org/10.1029/91jd00065, 1991.
Kerminen, V. M., Teinila, K., Hillamo, R., and Pakkanen, T.: Substitution of chloride in sea-salt particles by inorganic and organic anions, J. Aerosol Sci., 29, 929–942, https://doi.org/10.1016/S0021-8502(98)00002-0, 1998.
Kolesar, K. R., Cellini, J., Peterson, P. K., Jefferson, A., Tuch, T., Birmili, W., Wiedensohler, A., and Pratt, K. A.: Effect of Prudhoe Bay emissions on atmospheric aerosol growth events observed in Utqiagvik (Barrow), Alaska, Atmos. Environ., 152, 146–155, https://doi.org/10.1016/j.atmosenv.2016.12.019, 2017.
Köllner, F., Schneider, J., Willis, M. D., Klimach, T., Helleis, F., Bozem, H., Kunkel, D., Hoor, P., Burkart, J., Leaitch, W. R., Aliabadi, A. A., Abbatt, J. P. D., Herber, A. B., and Borrmann, S.: Particulate trimethylamine in the summertime Canadian high Arctic lower troposphere, Atmos. Chem. Phys., 17, 13747–13766, https://doi.org/10.5194/acp-17-13747-2017, 2017.
Korhonen, H., Carslaw, K. S., Spracklen, D. V., Ridley, D. A., and Strom, J.: A global model study of processes controlling aerosol size distributions in the Arctic spring and summer, J. Geophys. Res.-Atmos., 113, D08211, https://doi.org/10.1029/2007jd009114, 2008.
Laskin, A., Iedema, M. J., and Cowin, J. P.: Quantitative time-resolved monitoring of nitrate formation in sea salt particles using a CCSEM/EDX single particle analysis, Environ. Sci. Technol., 36, 4948–4955, https://doi.org/10.1021/es020551k, 2002.
Laskin, A., Gaspar, D. J., Wang, W. H., Hunt, S. W., Cowin, J. P., Colson, S. D., and Finlayson-Pitts, B. J.: Reactions at interfaces as a source of sulfate formation in sea-salt particles, Science, 301, 340–344, https://doi.org/10.1126/science.1085374, 2003.
Laskin, A., Cowin, J. P., and Iedema, M. J.: Analysis of individual environmental particles using modern methods of electron microscopy and X-ray microanalysis, J. Electron. Spectrosc. Relat. Phenom., 150, 260–274, https://doi.org/10.1016/j.elspec.2005.06.008, 2006.
Laskin, A., Moffet, R. C., Gilles, M. K., Fast, J. D., Zaveri, R. A., Wang, B., Nigge, P., and Shutthanandan, J.: Tropospheric chemistry of internally mixed sea salt and organic particles: Surprising reactivity of NaCl with weak organic acids, J. Geophys. Res.-Atmos., 117, D15302, https://doi.org/10.1029/2012jd017743, 2012.
Law, K. S. and Stohl, A.: Arctic Air Pollution: Origins and Impacts, Science, 315, 1537–1540, https://doi.org/10.1126/science.1137695, 2007.
Leck, C. and Svensson, E.: Importance of aerosol composition and mixing state for cloud droplet activation over the Arctic pack ice in summer, Atmos. Chem. Phys., 15, 2545–2568, https://doi.org/10.5194/acp-15-2545-2015, 2015.
Leck, C., Norman, M., Bigg, E. K., and Hillamo, R.: Chemical composition and sources of the high Arctic aerosol relevant for cloud formation, J. Geophys. Res.-Atmos., 107, 4135, https://doi.org/10.1029/2001JD001463, 2002.
Letterly, A., Key, J., and Liu, Y.: The influence of winter cloud on summer sea ice in the Arctic, 1983–2013, J. Geophys. Res.-Atmos., 121, 2178–2187, https://doi.org/10.1002/2015JD024316, 2016.
Liu, Y., Cain, J. P., Wang, H., and Laskin, A.: Kinetic study of heterogeneous reaction of deliquesced NaCl particles with gaseous HNO3 using particle-on-substrate stagnation flow reactor approach, J. Phys. Chem. A, 111, 10026–10043, https://doi.org/10.1021/jp072005p, 2007.
May, N. W., Quinn, P. K., McNamara, S. M., and Pratt, K. A.: Multiyear study of the dependence of sea salt aerosol on wind speed and sea ice conditions in the coastal Arctic, J. Geophys. Res.-Atmos., 121, 9208–9219, 10.1002/2016jd025273, 2016.
Moffet, R. C., Henn, T., Laskin, A., and Gilles, M. K.: Automated chemical analysis of internally mixed aerosol particles using X-ray spectromicroscopy at the carbon K-edge, Anal. Chem., 82, 7906–7914, https://doi.org/10.1021/ac1012909, 2010a.
Moffet, R. C., Henn, T. R., Tivanski, A. V., Hopkins, R. J., Desyaterik, Y., Kilcoyne, A. L. D., Tyliszczak, T., Fast, J., Barnard, J., Shutthanandan, V., Cliff, S. S., Perry, K. D., Laskin, A., and Gilles, M. K.: Microscopic characterization of carbonaceous aerosol particle aging in the outflow from Mexico City, Atmos. Chem. Phys., 10, 961–976, https://doi.org/10.5194/acp-10-961-2010, 2010b.
Moroni, B., Becagli, S., Bolzacchini, E., Busetto, M., Cappelletti, D., Crocchianti, S., Ferrero, L., Frosini, D., Lanconelli, C., Lupi, A., Maturilli, M., Mazzola, M., Perrone, M. G., Sangiorgi, G., Traversi, R., Udisti, R., Viola, A., and Vitale, V.: Vertical profiles and chemical properties of aerosol particles upon Ny-Ålesund (Svalbard Islands), Adv. Meteor., 2015, 292081, https://doi.org/10.1155/2015/292081, 2015.
Moroni, B., Cappelletti, D., Crocchianti, S., Becagli, S., Caiazzo, L., Traversi, R., Udisti, R., Mazzola, M., Markowicz, K., Ritter, C., and Zielinski, T.: Morphochemical characteristics and mixing state of long range transported wildfire particles at Ny-Ålesund (Svalbard Islands), Atmos. Environ., 156, 135–145, https://doi.org/10.1016/j.atmosenv.2017.02.037, 2017.
Norman, A., Barrie, L., Toom-Sauntry, D., Sirois, A., Krouse, H., Li, S., and Sharma, S.: Sources of aerosol sulphate at Alert: Apportionment using stable isotopes, J. Geophys. Res.-Atmos., 104, 11619–11631, https://doi.org/10.1029/1999JD900078, 1999.
O'Brien, R. E., Neu, A., Epstein, S. A., MacMillan, A. C., Wang, B., Kelly, S. T., Nizkorodov, S. A., Laskin, A., Moffet, R. C., and Gilles, M. K.: Physical properties of ambient and laboratory-generated secondary organic aerosol, Geophys. Res. Lett., 41, 4347–4353, https://doi.org/10.1002/2014GL060219, 2014.
Overland, J. E. and Wang, M.: When will the summer Arctic be nearly sea ice free?, Geophys. Res. Lett., 40, 2097–2101, https://doi.org/10.1002/grl.50316, 2013.
Pachauri, R. K., Allen, M., Barros, V., Broome, J., Cramer, W., Christ, R., Church, J., Clarke, L., Dahe, Q., and Dasgupta, P.: Climate Change 2014: Synthesis Report, Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, 2014.
Parungo, F., Nagamoto, C., Herbert, G., Harris, J., Schnell, R., Sheridan, P., and Zhang, N.: Individual particle analyses of Arctic aerosol samples collected during AGASP-III, Atmos. Environ., 27, 2825–2837, 1993.
Parungo, F. P., Nagamoto, C. T., Sheridan, P. J., and Schnell, R. C.: Aerosol characteristics of Arctic haze sampled during AGASP-II, Atmos. Environ., 24A, 937–949, 1990.
Peters, G. P., Nilssen, T. B., Lindholt, L., Eide, M. S., Glomsrød, S., Eide, L. I., and Fuglestvedt, J. S.: Future emissions from shipping and petroleum activities in the Arctic, Atmos. Chem. Phys., 11, 5305–5320, https://doi.org/10.5194/acp-11-5305-2011, 2011.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Petters, S. S. and Petters, M. D.: Surfactant effect on cloud condensation nuclei for two-component internally mixed aerosols, J. Geophys. Res.-Atmos., 121, 1878–1895, https://doi.org/10.1002/2015JD024090, 2016.
Pilson, M. E. Q.: An Introduction to the Chemistry of the Sea, Cambridge University Press, 533 pp., 2013.
Pithan, F. and Mauritsen, T.: Arctic amplification dominated by temperature feedbacks in contemporary climate models, Nat. Geosci., 7, 181–184, https://doi.org/10.1038/ngeo2071, 2014.
Polissar, A., Hopke, P., Paatero, P., Kaufmann, Y., Hall, D., Bodhaine, B., Dutton, E., and Harris, J.: The aerosol at Barrow, Alaska: long-term trends and source locations, Atmos. Environ., 33, 2441–2458, https://doi.org/10.1016/S1352-2310(98)00423-3, 1999.
Prather, K. A., Hatch, C. D., and Grassian, V. H.: Analysis of atmospheric aerosols, Annu. Rev. Anal. Chem., 1, 485–514, https://doi.org/10.1146/annurev.anchem.1.031207.113030, 2008.
Quinn, P. K., Miller, T. L., Bates, T. S., Ogren, J. A., Andrews, E., and Shaw, G. E.: A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res.-Atmos., 107, 4130, https://doi.org/10.1029/2001jd001248, 2002.
Quinn, P. K., Shaw, G., Andrews, E., Dutton, E. G., Ruoho-Airola, T., and Gong, S. L.: Arctic haze: current trends and knowledge gaps, Tellus B, 59, 99–114, https://doi.org/10.1111/j.1600-0889.2006.00238.x, 2007.
Quinn, P. K., Bates, T. S., Schulz, K. S., Coffman, D. J., Frossard, A. A., Russell, L. M., Keene, W. C., and Kieber, D. J.: Contribution of sea surface carbon pool to organic matter enrichment in sea spray aerosol, Nat. Geosci., 7, 228–232, https://doi.org/10.1038/NGEO2092, 2014.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and related properties of freshly emitted sea spray aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr500713g, 2015.
Rolph, G. D.: Real-time Environmental Applications and Display sYstem (READY) Website (https://ready.arl.noaa.gov/HYSPLIT.php, last access: 12 February 2017), 2016.
Shaw, P. M., Russell, L. M., Jefferson, A., and Quinn, P. K.: Arctic organic aerosol measurements show particles from mixed combustion in spring haze and from frost flowers in winter, Geophys. Res. Lett., 37, L10803, https://doi.org/10.1029/2010gl042831, 2010.
Shen, H., Peters, T. M., Casuccio, G. S., Lersch, T. L., West, R. R., Kumar, A., Kumar, N., and Ault, A. P.: Elevated Concentrations of Lead in Particulate Matter on the Neighborhood-Scale in Delhi, India As Determined by Single Particle Analysis, Environ. Sci. Technol., 50, 4961–4970, https://doi.org/10.1021/acs.est.5b06202, 2016.
Sierau, B., Chang, R. Y.-W., Leck, C., Paatero, J., and Lohmann, U.: Single-particle characterization of the high-Arctic summertime aerosol, Atmos. Chem. Phys., 14, 7409–7430, https://doi.org/10.5194/acp-14-7409-2014, 2014.
Sirois, A. and Barrie, L. A.: Arctic lower tropospheric aerosol trends and composition at Alert, Canada: 1980–1995, J. Geophys. Res.-Atmos., 104, 11599–11618, https://doi.org/10.1029/1999JD900077, 1999.
Sobanska, S., Coeur, C., Maenhaut, W., and Adams, F.: SEM-EDX characterisation of tropospheric aerosols in the Negev desert (Israel), J. Atmos. Chem., 44, 299–322, https://doi.org/10.1023/a:1022969302107, 2003.
Sobanska, S., Falgayrac, G., Rimetz-Planchon, J., Perdrix, E., Bremard, C., and Barbillat, J.: Resolving the internal structure of individual atmospheric aerosol particle by the combination of Atomic Force Microscopy, ESEM-EDX, Raman and ToF-SIMS imaging, Microchem. J., 114, 89–98, https://doi.org/10.1016/j.microc.2013.12.007, 2014.
Struthers, H., Ekman, A. M. L., Glantz, P., Iversen, T., Kirkevåg, A., Mårtensson, E. M., Seland, Ø., and Nilsson, E. D.: The effect of sea ice loss on sea salt aerosol concentrations and the radiative balance in the Arctic, Atmos. Chem. Phys., 11, 3459–3477, https://doi.org/10.5194/acp-11-3459-2011, 2011.
Sturges, W. and Barrie, L.: Chlorine, bromine and iodine in Arctic aerosols, Atmos. Environ., 22, 1179–1194, https://doi.org/10.1016/0004-6981(88)90349-6, 1988.
U.S. Energy Information Administration: I. S. A.: Top 100 U.S. Oil and Gas Fields, U.S. Department of Energy, Washington, D.C., 2015.
Wang, Z., Su, H., Wang, X., Ma, N., Wiedensohler, A., Pöschl, U., and Cheng, Y.: Scanning supersaturation condensation particle counter applied as a nano-CCN counter for size-resolved analysis of the hygroscopicity and chemical composition of nanoparticles, Atmos. Meas. Tech., 8, 2161–2172, https://doi.org/10.5194/amt-8-2161-2015, 2015.
Weinbruch, S., Wiesemann, D., Ebert, M., Schutze, K., Kallenborn, R., and Strom, J.: Chemical composition and sources of aerosol particles at Zeppelin Mountain (Ny Alesund, Svalbard): An electron microscopy study, Atmos. Environ., 49, 142–150, https://doi.org/10.1016/j.atmosenv.2011.12.008, 2012.
Williams, J., de Reus, M., Krejci, R., Fischer, H., and Ström, J.: Application of the variability-size relationship to atmospheric aerosol studies: estimating aerosol lifetimes and ages, Atmos. Chem. Phys., 2, 133–145, https://doi.org/10.5194/acp-2-133-2002, 2002.
Young, G., Jones, H. M., Darbyshire, E., Baustian, K. J., McQuaid, J. B., Bower, K. N., Connolly, P. J., Gallagher, M. W., and Choularton, T. W.: Size-segregated compositional analysis of aerosol particles collected in the European Arctic during the ACCACIA campaign, Atmos. Chem. Phys., 16, 4063–4079, https://doi.org/10.5194/acp-16-4063-2016, 2016.
Zhuang, H., Chan, C. K., Fang, M., and Wexler, A. S.: Formation of nitrate and non-sea-salt sulfate on coarse particles, Atmos. Environ., 33, 4223–4233, https://doi.org/10.1016/S1352-2310(99)00186-7, 1999.