the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Feb 2024
| 19 Feb 2024
Oxygenated organic molecules produced by low-NOx photooxidation of aromatic compounds: contributions to secondary organic aerosol and steric hindrance
Xi Cheng
Yong Jie Li
Yan Zheng
Keren Liao
Theodore K. Koenig
Yanli Ge
Chunxiang Ye
Xinghua Qiu
Oxygenated organic molecules (OOMs) produced by the oxidation of aromatic compounds are key components of secondary organic aerosol (SOA) in urban environments. The steric effects of substitutions and rings and the role of key reaction pathways in altering the OOM distributions remain unclear because of the lack of systematic multi-precursor study over a wide range of OH exposure. In this study, we conducted flow-tube experiments and used the nitrate adduct time-of-flight chemical ionization mass spectrometer (-TOF-CIMS) to measure the OOMs produced by the photooxidation of six key aromatic precursors under low-NOx conditions. For single aromatic precursors, the detected OOM peak clusters show an oxygen atom difference of one or two, indicating the involvement of multi-step auto-oxidation and alkoxy radical pathways. Multi-generation OH oxidation is needed to explain the diverse hydrogen numbers in the observed formulae. In particular, for double-ring precursors at higher OH exposure, multi-generation OH oxidation may have significantly enriched the dimer formulae. The results suggest that methyl substitutions in precursor lead to less fragmented OOM products, while the double-ring structure corresponds to less efficient formation of closed-shell monomeric and dimeric products, both highlighting significant steric effects of precursor molecular structure on the OOM formation. Naphthalene-derived OOMs however have lower volatilities and greater SOA contributions than the other-type of OOMs, which may be more important in initial particle growth. Overall, the OOMs identified by the -TOF-CIMS may have contributed up to 30.0 % of the measured SOA mass, suggesting significant mass contributions of less oxygenated, undetected semi-volatile products. Our results highlight the key roles of progressive OH oxidation, methyl substitution and ring structure in the OOM formation from aromatic precursors, which need to be considered in future model developments to improve the model performance for organic aerosol.
- Article
(3832 KB) - Full-text XML
-
Supplement
(1060 KB) - BibTeX
- EndNote




Oxidation of volatile organic compounds (VOCs) leads to the formation of ozone and secondary organic aerosols (SOAs) (Ziemann and Atkinson, 2012). Light and heavy aromatic VOCs are important SOA precursors that can be emitted from various anthropogenic sources such as transportation, solvent use, wood burning, coal burning and cooking (Henze et al., 2008; Pye and Pouliot, 2012). There are many chamber studies on the SOA formation from aromatic VOCs (Li et al., 2016a, b; Lambe et al., 2011; Ng et al., 2007; Chan et al., 2009; Kautzman et al., 2010). The derived SOA yields vary by precursors and NOx levels, owing to the difference in forming condensable products, i.e., oxygenated organic molecules (OOMs). Characterization of these OOM species is however limited and challenging. Most atmospheric chemical transport models (CTMs) still use SOA-yield-based parameterizations, which have difficulties in reproducing the concentration and the variability of organic aerosol in urban areas (Tsigaridis et al., 2014).
Recent developments in time-of-flight chemical ionization mass spectrometry provide new insights into the formation of OOMs and their contribution to SOA (Bianchi et al., 2019). Wang et al. (2017) indicate that the intramolecular H shift of peroxy radicals (RO2) from alkyl benzenes can compete with bimolecular reactions under atmospheric conditions, thereby substantially increasing the product oxygen numbers via auto-oxidation. Laboratory studies show further evidence for the occurrence of both multi-generation OH oxidation and multi-step auto-oxidation in the aromatic oxidation to increase the oxidation state of the products (Garmash et al., 2020; Cheng et al., 2021b). Additionally, Berndt et al. (2018b) suggest high formation rates of accretion products from self- and cross-reactions of RO2 radicals for aromatic precursors, resulting in great carbon and oxygen number increments that lower the product volatility. The three mechanisms, i.e., multi-generation OH oxidation, multi-step auto-oxidation and accretion reactions, play different roles under different oxidation conditions. Moreover, aromatic precursors differ in substituents and ring numbers, which may cause steric effects in the OOM formation. Cheng et al. (2021b) suggest that for benzene and toluene, the multi-generation OH oxidation likely proceeds more favorably by H abstraction than OH addition, and the dimer formation is unfavorable at high OH exposures. Other laboratory studies indicate that under similar OH exposures, the oxidation of isopropylbenzene, ethylbenzene and toluene likely forms more oxidized RO2 radicals than the oxidation of benzene does (Wang et al., 2017). The formation of dimeric products from the oxidation of trimethylbenzene seemed to be more significant for meta-substituents than for ortho-substituents (Y. Wang et al., 2020). However, there is still a lack of multi-precursor studies that compare the OOM formation systematically under a wide range of OH exposure. A better understanding of the precursor steric effects and the OOM potential of forming SOA is needed to assist future developments of detailed model representation of SOA formation.
Herein, the formation of OOMs from six aromatic VOCs (i.e., benzene, toluene, m-xylene, 1,3,5-trimethylbenzene, naphthalene and 1-methylnaphthalene) is investigated in flow-tube experiments under low-NOx conditions, aiming at gaining insights into the OH-initiated chemistry and the low-NOx representation of suburban and downwind environments. The OOMs are characterized using a nitrate adduct time-of-flight chemical ionization mass spectrometer (-TOF-CIMS). The experiments cover a wide range of OH exposures that are equivalent to approximately 1–19 d, assuming a mean OH concentration of 1.5×106 molecules cm−3. The influences of precursor structure and OH exposure on the formation of OOMs and the contributions of the identified OOMs to SOA are investigated.
2.1 Experimental procedures
Experiments were conducted in a 13.3 L Aerodyne oxidation flow reactor (OFR) with a mean residence time of 95 s under low-NOx conditions. The operation of the OFR has been described previously by Cheng et al. (2021b). The OFR was operated in an OFR254-5 mode, with 254 nm lights on and 5 ppm of externally generated O3 introduced into the OFR (Lambe et al., 2017; Peng et al., 2018). The OH radicals are generated via
The relative humidity (RH) inside the OFR was maintained at 23.4 %–29.4 %. Concentrations of OH radicals were varied by ramping the voltage of the UV lamps in the OFR, for which each lamp voltage represents one experimental condition. All experiments herein were conducted without injection of seed particles and NOx. We estimate a background NOx level of <1 ppb throughout the experiments. In total, 27 experiments were performed for the six aromatic precursors. Table 1 lists the experimental conditions and the key measured and derived quantities, including those for the oxidation of benzene and toluene that have been discussed in our previous study (Cheng et al., 2021b). The concentrations of OH and HO2 radicals were estimated using a photochemical box model (PAMchem) (Lambe et al., 2017; Cheng et al., 2021b). The results suggest that the first step of oxidation of the aromatic VOCs was dominated by the OH radicals rather than by O3 because of much higher reaction rate constants for the former than the latter (Lambe et al., 2017; Peng et al., 2016). The reaction rates of early-generation oxidation products that contain double bounds with O3 are also likely slower than those with OH radicals (Molteni et al., 2018; Y. Wang et al., 2020). Therefore, we believe that the oxidation chemistry was prevalently initiated by OH radicals in our experiments. The modeled HO2-to-OH concentration ratio is about 2–18, while the rate constant for RO2+HO2 is typically an order of magnitude smaller than that for RO2+OH (Peng and Jimenez, 2020, and references therein). Figure S1 in the Supplement shows the calculated contributions of various pathways to the RO2 loss in our experiments for a given set of rate constants. Both the RO2+HO2 and RO2+OH channels are important for RO2 loss under conditions of our experiments considering the uncertainties of the estimations and the variations of reaction constants. The RO2 isomerization rate coefficients are highly structure dependent, ranging from ∼ 10−3 to ∼ 10 s−1 (per second) for aromatic RO2 (Wang et al., 2017; Praske et al., 2018). Therefore, the fast RO2+HO2 reactions may limit some RO2 auto-oxidation in our experiments.
Table 1Summary of experimental parameters and measured and derived quantities for aromatic (CxHy) oxidation in the OFR under low-NOx conditions.
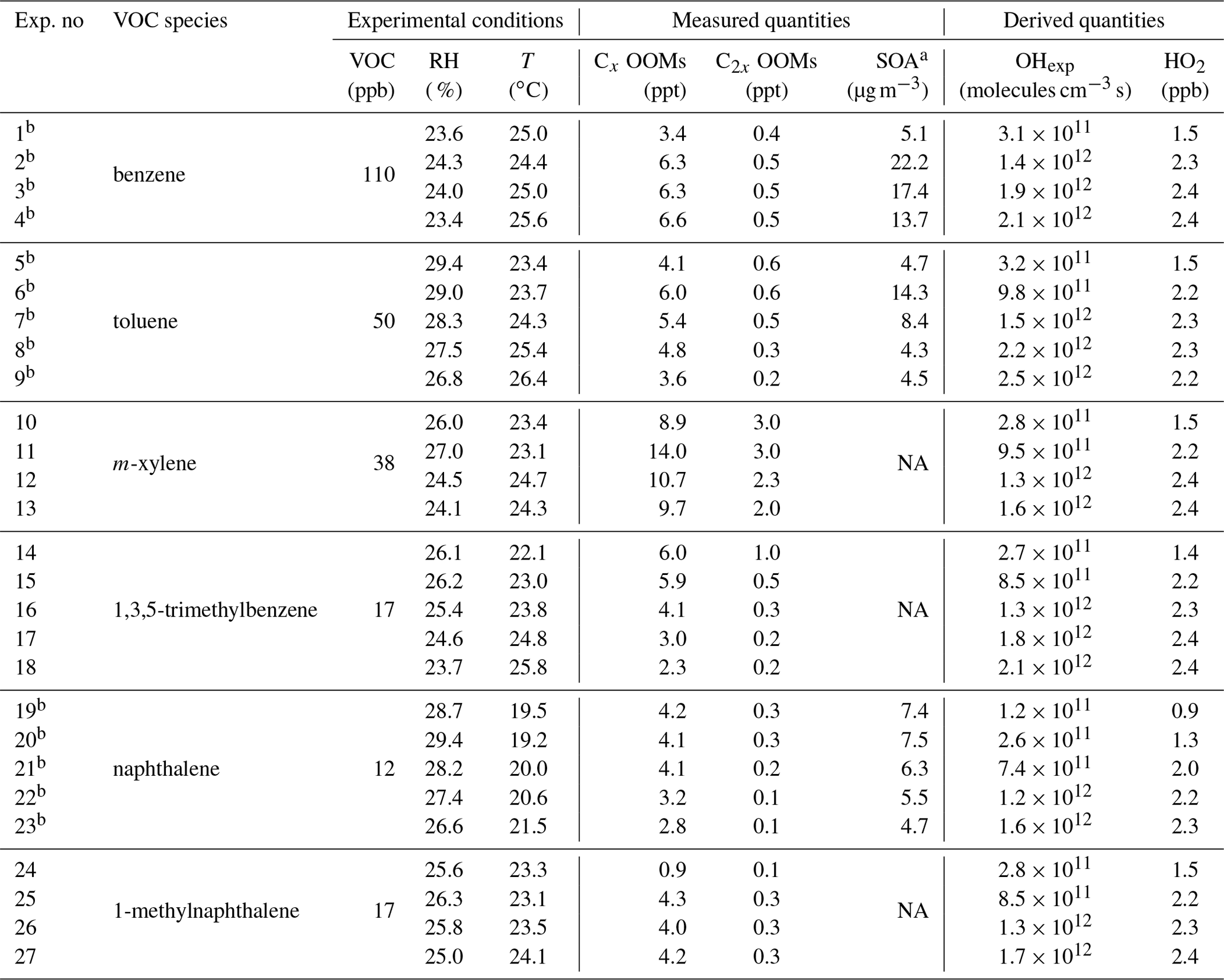
a Not corrected by particle wall losses.
b Experiments that have LTOF-SP-AMS measurements.
NA: not available.
2.2 Measurements
The concentration of O3 was monitored with an UV photometric analyzer (2B Technologies, 202). Concentrations of aromatic VOCs were measured by an IONICON proton transfer reaction quadrupole interface time-of-flight mass spectrometer (PTR-QiTOF). Calibration of the PTR-QiTOF was conducted using gas standards (Spectra Gases, ∼ 1 ppm) at different concentration levels. The measurement uncertainty is about 15 % for calibrated species (Huang et al., 2019). Particle size distributions were measured by a scanning mobility particle sizer (SMPS; TSI, 3938). The SOA mass concentrations were measured by an Aerodyne long time-of-flight soot particle aerosol mass spectrometer (LTOF-SP-AMS). Calibrations of ionization efficiency (IE) and relative IE (RIE) followed standard procedures using pure ammonium nitrate (NH4NO3) and ammonium sulfate ((NH4)2SO4) (Zheng et al., 2020). A collection efficiency (CE) value of 1 was applied to the LTOF-SP-AMS data herein, which leads to a good agreement between the AMS and SMPS results. The oxygen-to-carbon (O:C) and hydrogen-to-carbon (H:C) ratios of SOA were estimated by the method introduced by Aiken et al. (2008). Because the measured ratios of (i.e., around 0.33) and (i.e., 1.1–1.2) for chamber aromatic SOA were similar to the default ratios of 0.225 and 1, respectively (Chhabra et al., 2011; Nakao et al., 2013), we did not update these ratios in the calculation of elemental ratios (Chen et al., 2011).
The OOMs were measured by the Aerodyne -TOF-CIMS in the form of deprotonated ions, clusters with a nitrate monomer or clusters with a nitrate dimer. This study only presents clusters that have one nitrate ion. The oxidation of benzene and toluene has been discussed in our previous study for a fitted mass-to-charge ratio () range of 150 to 450 Thomson (Th) (Cheng et al., 2021b). In this study, we refitted the spectra to up to 650 Th, resulting in slightly different molar yields (Table S1 in the Supplement). Although our work and other studies report similar calibration factors for the range of 100–450 Th for a high-resolution time-of-flight (HTOF) -CIMS (Ehn et al., 2014; Jokinen et al., 2012; Kürten et al., 2012), it does not guarantee a flat transmission for a wider range of m/z. The transmission correction is instrument-specific and strongly depends on the chosen settings of the instrument (Heinritzi et al., 2016). Tian et al. (2023) and Zheng et al. (2023) show a relatively smooth transmission curve for 450 Th, with an enhancement factor of 1–1.2. We used a uniform calibration factor of 1.23×1010 molecules cm−3, with additional sampling-line vapor loss correction of 26 % for all OOMs in this study, with considerations of the transmission uncertainty in the result interpretation. This may underestimate the sensitivity of large molecules by a factor of 2 if we consider an average transmission efficiency of ∼ 0.5 for 450–650 Th, suggested by Tian et al. (2023). The background signals of individual OOMs were determined from measurements made without the injection of VOC precursors. The instrument operation and data analysis have been described in detail in our previous studies (Cheng et al., 2021a, b).
2.3 Dimer formation rates
Dimeric products may be formed by the accretion reactions between RO2 and R′O2 radicals. At steady state, the formation rate of dimers is represented as follows:
where kR,i (cm3 molecule−1 s−1) is the apparent rate constants of accretion reactions (i≥1), and kloss (s−1) represents the loss rate of OOMs (0.04–0.1 s−1) (Sect. S1 of the Supplement). We use Eq. (1) to provide rough constraints to the formation rate constants of detected dimeric products, which is similar to the approach used by Molteni et al. (2019). There are 46, 54 and 76 assigned dimeric products from benzene, toluene and naphthalene experiments, respectively, from the -TOF-CIMS measurements of OOM molecular formulae. However, for each precursor, only five to eight dimers have sufficient combinations of above-detection-limit signals of possible RO2 and R′O2 to solve kR,i using a non-negative multi-linear regression method (sklearn from Python 3). Only the kR,i values that are greater than zero but less than cm3 molecule−1 s−1 (considering the collision limit) with correlation coefficients (r) of > 0.5 are considered reasonable rate constants and are listed in Table 2. Theoretical and laboratory studies indicate that the production rate constant of ROOR′ can reach 10−10 cm3 molecules−1 s−1 (Molteni et al., 2019; Hasan et al., 2020; Perakyla et al., 2023). The OOMs that form slowly would not be detectable by the -TOF-CIMS because of the fast vapor losses at the SOA loading level at the exit of OFR. The detected dimeric products (e.g., C14H18O14) showed rapid increases to stable concentrations with time series similar with their parent RO2 (e.g., C7H9O10), indicating a fast production of these dimers. The PAMchem model simulations indicate minor changes of the ROOR′ concentrations when the reaction rate constant of reaches above cm3 molecules−1 s−1 (Fig. S2). We therefore think the steady-state assumption may stand for the few dimers that are listed in Tables S2–S7.
Table 2Rate coefficients k for benzene- and naphthalene-derived dimer and comparison with data from other studies. R-squared values (R2) are shown to verify the fitting results.
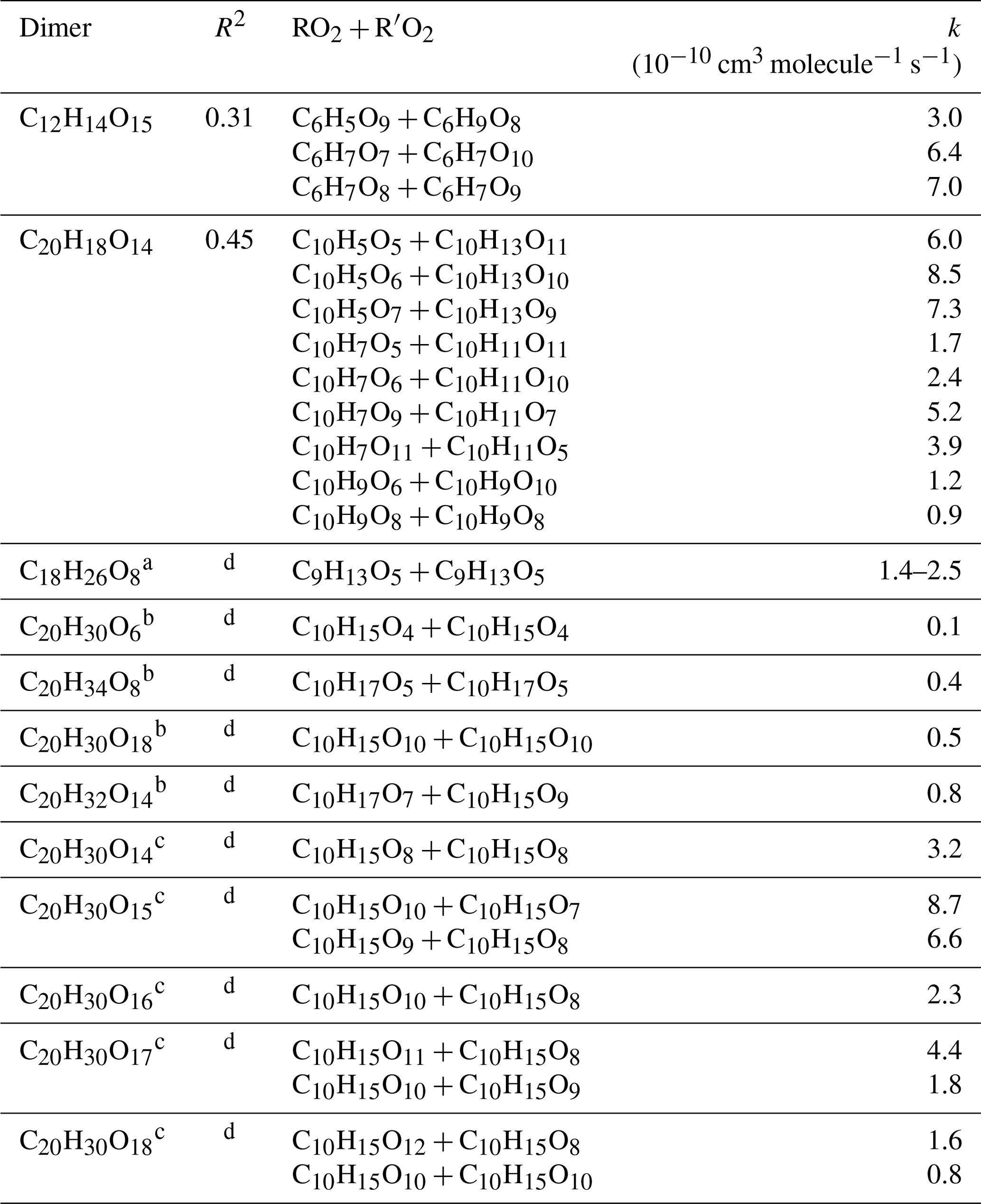
a From experiments with 1,3.5-trimethylbenzene addition (Berndt et al., 2018b).
b From experiments with α-pinene addition (Berndt et al., 2018a).
c From experiments with α-pinene addition (Molteni et al., 2019).
d Not provided in the literature.
2.4 Contribution of OOMs to SOA
The contributions of the detected OOMs to SOA were only estimated for benzene, toluene and naphthalene experiments, for which both LTOF-SP-AMS and SMPS measurements are available. We consider both the irreversible and equilibrium-type condensation. The former was represented by the net condensation flux of the observed OOMs, which can be described using an aerosol growth model (Tröstl et al., 2016). The latter was represented by the particulate fraction estimated by the partitioning theory and the vapor pressure of OOMs (Pankow, 1994). The calculation details are provided in Sect. S1. Aromatic OOMs consist of abundant hydroxyl (–OH) and carboxylic (=O) groups but less hydroperoxide (–OOH) groups compared to monoterpene OOMs (M. Wang et al., 2020). We therefore applied the empirical volatility parameterization that was used in previous naphthalene studies to estimate the saturation concentrations (C∗) of the detected OOMs (Epstein et al., 2010; Donahue et al., 2011). The OOMs were grouped into volatility bins based on the volatility-basis-set (VBS) method (Donahue et al., 2006). They were classified as ultralow-volatility organic compounds (ULVOCs; ), extremely low volatility organic compounds (ELVOCs; ), low-volatility organic compounds (LVOCs; ), semi-volatile organic compounds (SVOCs; ) and intermediate-volatility organic compounds (IVOCs; ) (Bianchi et al., 2019; Schervish and Donahue, 2020). In this study, SOA contributions from OOMs with C∗ of ≤0.01 µg m−3 were calculated by the aerosol growth model, and those from OOMs with µg m−3 were calculated by the partitioning method.
3.1 Product distributions
The general oxidation chemistry for OH-initiated pathways of aromatic VOCs is described in Sect. S2. Figure 1 shows the OOM product distributions for the oxidation of the six aromatic precursors under similar OH exposures of approximately (1.2–1.5) ×1012 molecules cm−3 s. For a given precursor of CxHy, we name the detected OOMs as monomeric products (C<x and Cx series) and dimeric products (C<2x and C2x series). Monomeric products contribute to the majority of the detected OOM signals for the six aromatic precursors studied herein, while the dimeric products contribute to up to 18 % of the OOM signals. The highest dimeric signal fraction is observed for m-xylene OOMs. Overall, ion peaks in the range of 200–650 Th show in clusters with progressions and sequences that preserve the precursor carbon structure. The lower ends of the peak clusters of monomeric products for benzene, toluene, m-xylene and 1,3,5-trimethylbenzene are shifted by progressive differences of 14 Th (–CH2). The products from naphthalene and 1-methylnaphthalene also show a similar –CH2 progression. The progression is doubled for dimeric products (28 Th, –(CH2)2). Moreover, there are series of clusters with peak sequences that differ by one and two oxygen atoms in both of monomeric and dimeric products, respectively, in each mass spectrum. The peak clusters then differ by hydrogen atom numbers for each oxygen atom addition. The general shifting of peak abundance to products with one to four more hydrogen (i.e., to the right side) is consistent with the importance of OH addition in the oxidation of aromatic precursors. By contrast, dimeric clusters show abundant distributions in the hydrogen-deficient values (i.e., on the left side), suggesting a significant H abstraction process before the accretion reactions occur.
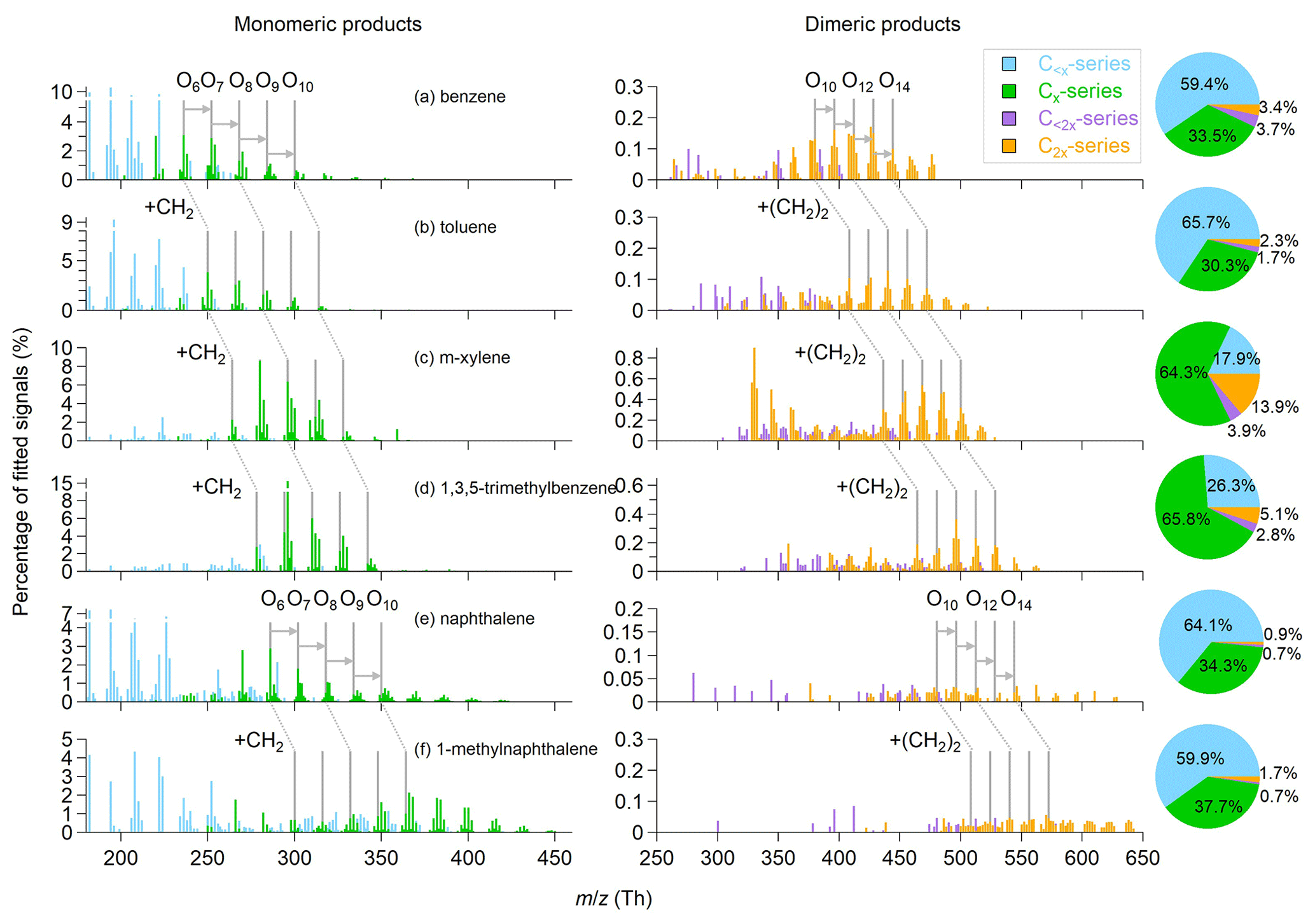
Figure 1Mass spectra and fractions of different categories of gaseous OOMs observed in the oxidation of (a) benzene, (b) toluene, (c) m-xylene, (d) 1,3,5-trimethylbenzene, (e) naphthalene and (f) 1-methylnaphthalene, corresponding to experiment nos. 2, 7, 12, 16, 22 and 26 in Table 1, respectively.
The C<x products are typically fragmented products formed from the decomposition of alkoxy (RO) radicals (Li and Wang, 2014). Methyl substitution may prevent the efficient formation of phenoxy radicals, thereby lowering the abundance of fragmented products (Schwantes et al., 2017). Indeed, we observed high signal fractions of C<x products (59.4 %–65.7 %) in OOMs produced by the oxidation of benzene, toluene, naphthalene and 1-methylnaphthalene that have low methyl-to-aryl carbon ratios (CMe : CAr<0.2), whereas the signal fractions of C<x products produced by the oxidation of m-xylene and 1,3,5-trimethylbenzene (CMe : CAr >0.2) are much lower (17.9 %–26.3 %). Tables S2–S7 list the formulae and the relative signals of major OOMs for the experiments presented in Fig. 1. For the oxidation of benzene, toluene, naphthalene and 1-methylnaphthalene, C3H4O5 and C4H4O5 are the main common C<x products. These molecules are likely second- or third-generation products formed via the fragmentation of phenoxy radicals with rich unsaturated aryl carbon moieties (Schwantes et al., 2017). C5H6O6 and C8H10O7 are the most abundant C<x products for the oxidation of m-xylene and 1,3,5-trimethylbenzene, respectively. C5H6O6 is likely a ring-scission product (e.g., carboxylic acid), while C8H10O7 may be a fragmented ring-retaining product formed by the dealkylation pathway (Zaytsev et al., 2019; Mehra et al., 2020).
The Cx products with odd H numbers in their formulae for single-precursor oxidation systems are plausibly open-shell products (i.e., mainly RO2) (Zhao et al., 2018), and the Cx products with even H numbers are normally closed-shell products. Central in the formation of OOMs from aromatic VOCs, the bicyclic peroxy radicals (BPRs) contain five oxygen numbers (CxHy+1O5), which are formed by two steps of O2 addition after OH addition (Birdsall et al., 2010). As shown in Table S8, the signal fractions of CxHy+1O7 and CxHy+1O9 (0.1 %–1.0 %) are greater than those of CxHy+1O5( 0.05 %–0.4 %), explained by aggressive multiple steps of auto-oxidation. These odd-oxygen RO2 radicals are the results of the predominant OH-addition plus sequential O2 incorporation (auto-oxidation). Besides, even-oxygen RO2 radicals are observed, with CxHy+1O6 and CxHy+1O8 (0.1 %–1.1 %) being the most abundant. Their formation might involve the RO pathway (Orlando et al., 2003; Cheng et al., 2021b). In the RO pathway, the RO radicals undergo an intermolecular H shift (isomerization) instead of decomposition, which forms C-centered alkyl radicals that are ready for further auto-oxidation. For example, the BPR (CxHy+1O5) can react with HO2 to form the RO radical CxHy+1O4. The newly formed RO radical then isomerizes to become a hydroxylated alkyl radical and results in a new RO2 radical (CxHy+1O6) via O2 addition. The most abundant closed-shell Cx products are C6H6O6 (3.1 %), C7H8O6 (3.8 %), C8H10O7 (8.6 %), C9H14O7 (17.5 %), C10H8O6 (2.9 %) and C11H12O10 (2.1 %) for the oxidation of benzene, toluene, m-xylene, 1,3,5-trimethylbenzene, naphthalene and 1-methylnaphthalene, respectively. We obtained average O:C ratios of 0.9–1.1 for closed-shell OOM products formed from monocyclic precursors and 0.7–0.8 for those formed from double-ring precursors. The lower O:C ratios for the latter are probably because the second aromatic ring prohibits extensive auto-oxidation (Molteni et al., 2018).
Among the dimeric products, the C2x products are much more abundant than the C<2x ones in terms of both number and amount (Tables S2–S7). The observed C2x dimers have odd and even oxygen numbers in their formulae, suggesting the occurrence of both the cross- and self-reactions of RO2 radicals. For example, C18H26O12 is a product from the 1,3,5-trimethylbenzene oxidation, which may be formed via self-reactions of C9H13O7 or cross-reactions of other even-oxygen RO2 radicals. Dimers that are formed from BPR or RO2 with further auto-oxidation are expected to contain at least eight oxygen atoms. Interestingly, the C2x products with four to seven oxygen numbers are abundant, suggesting the occurrence of accretion reaction between BPR and less oxygenated RO2 radicals. These C2x products show a greater abundance for the oxidation of m-xylene (Fig. 1c) than for those of other precursors, indicating more stable accretion reactions for the less oxygenated RO2 radicals derived from m-xylene due to stereoselectivity. Consistently, Y. Wang et al. (2020) identified more abundant dimeric products for the meta-substitution aromatic. Moreover, different numbers of hydrogen atoms in the dimer formulae suggest different formation routes. For monocyclic aromatic precursors, the main C2x products are C2xH2y+2O10−14, likely formed by the accretion reactions of two CxHy+1Oz radicals (Table S8). These CxHy+1Oz products are usually RO2 radicals formed from multi-step auto-oxidation with one step of OH addition. Unlike the monocyclic precursors, the main C2x products produced by the oxidation of naphthalene and 1-methylnaphthalene are C2xH2yO10−14, C2xH2y+2O10−14 and C2xH2y+4O10−14, showing more diverse H number distributions. This result suggests more efficient multi-generation OH oxidation (e.g., adding one H atom via OH addition vs. losing one H atom via H abstraction) before accretion reactions occur for double-ring aromatic precursors than for single-ring precursors.
The apparent molar yields of the OOMs detected by the -TOF-CIMS at the exit of OFR were calculated as , where kloss is the loss rate constant of OOMs, and k1 is the VOC–OH reaction rate constant. The AMS and SMPS particle measurements are only available for benzene, toluene and naphthalene, for which we can correct the vapor loss of OOMs to the particle phase and the OFR wall. Figure 2 shows the OOM yields under different OH exposures. The yields range from 0.07 % to 2.05 %, among which the yields of naphthalene-derived OOMs are greater than those of the other two monocyclic aromatic precursors. This is consistent with the result from Molteni et al. (2018), who obtained a higher OOM yield for naphthalene (1.8 %) than for benzene (0.2 %) and toluene (0.1 %). As the OH exposure increases, the OOM yields for benzene and toluene first increase and then decrease after the equivalent photochemical age exceeds 7 d. The initial increase in OOM yields is consistent with the results presented by Garmash et al. (2020) for the oxidation of benzene under low OH exposure. The later decrease in the apparent OOM yields under high OH exposure suggests the production of more fragmented products that remain undetected by the -TOF-CIMS. For the oxidation of naphthalene, we find increasing OOM yields under high OH exposure. Multi-generation OH oxidation may occur more efficiently for precursors with two aromatic rings than the monocyclic precursors, thereby forming more of the detectable OOMs by the -TOF-CIMS even under high OH exposure. The parameters in the yield calculation are associated with various kinds of uncertainties. We used a uniform calibration factor to determine the OOM concentrations which may underestimate the sensitivity of large molecules. Given an average transmission efficiency of ∼ 0.5 for 450–650 Th, as suggested by Tian et al. (2023), the underestimations of OOM yields (about 0.01 %–0.03 %) are minor. The main bias in yields is perhaps associated with kloss, which is most sensitive to the condensation sink (Palm et al., 2016). It is worth noting that the yield calculation herein assumes that the production rate is equal to the loss rate of OOMs, which may represent only the lower limit of the actual yield if particles are still growing when exiting the OFR.
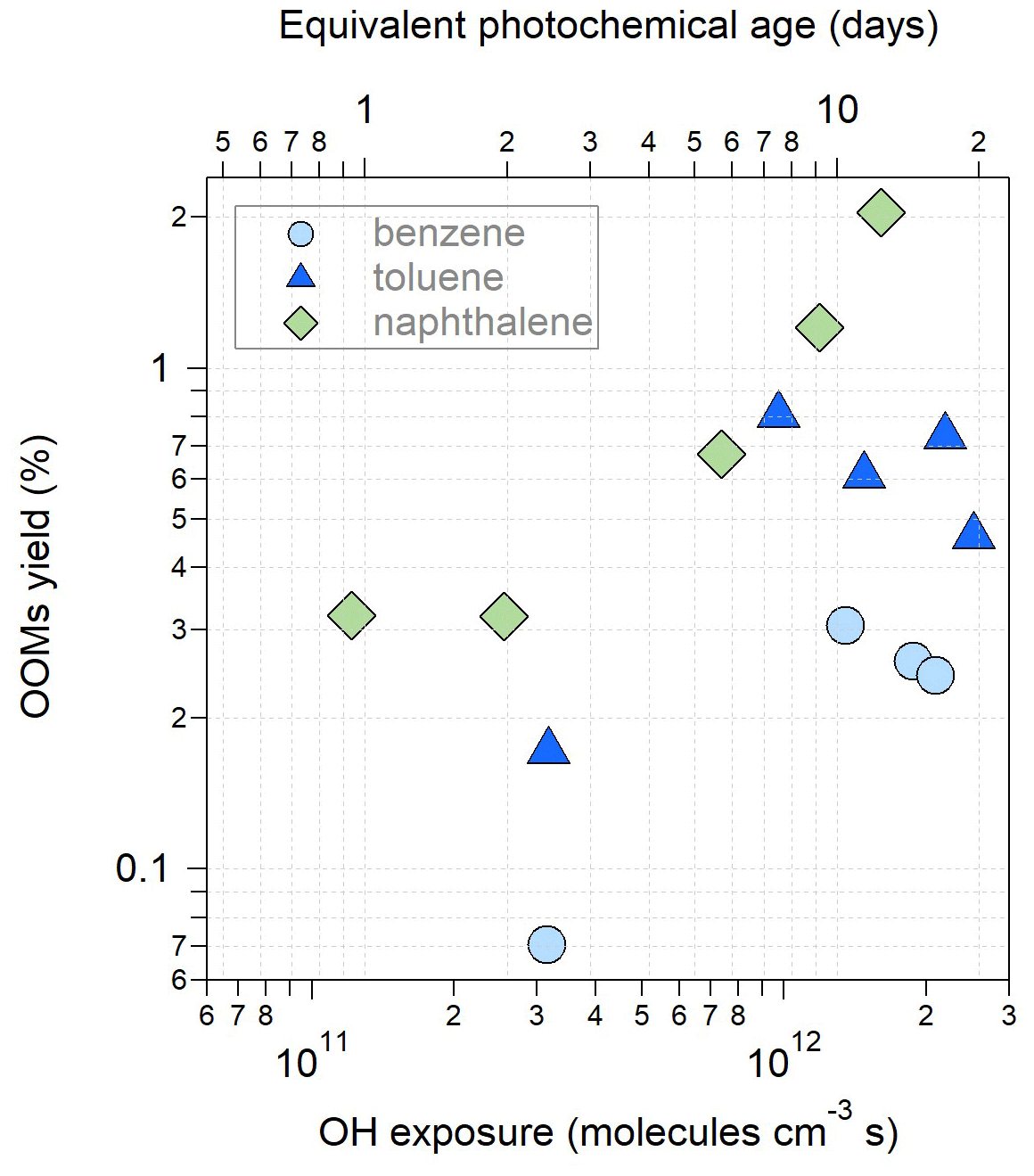
Figure 2Yields of OOMs produced from photooxidation of benzene, toluene, and naphthalene in OFR as a function of OH exposure. Equivalent photochemical ages are referred to a mean OH concentration of 1.5×106 molecules cm−3.
3.2 Formation of Cx and C2x OOMs
For all precursors, the concentrations of closed-shell Cx products are much greater than those of open-shell products (i.e., mainly RO2) (Fig. S3). Under low-NOx conditions, the closed-shell Cx products are formed primarily from the RO2+HO2 pathways rather than the reaction of RO2+RO2. This is due to the much higher HO2 concentrations (0.9–2.4 ppb) than the RO2 concentrations (0.1–2.2 ppt) (Table 1), which hints that the formation of closed-shell Cx products is limited by the RO2 availability. Indeed, good correlations between closed-shell Cx products and RO2 are observed (Fig. 3). The slopes are generally lower for double-ring precursors than for monocyclic precursors, indicating a steric hindrance of double rings on the Cx product formation.
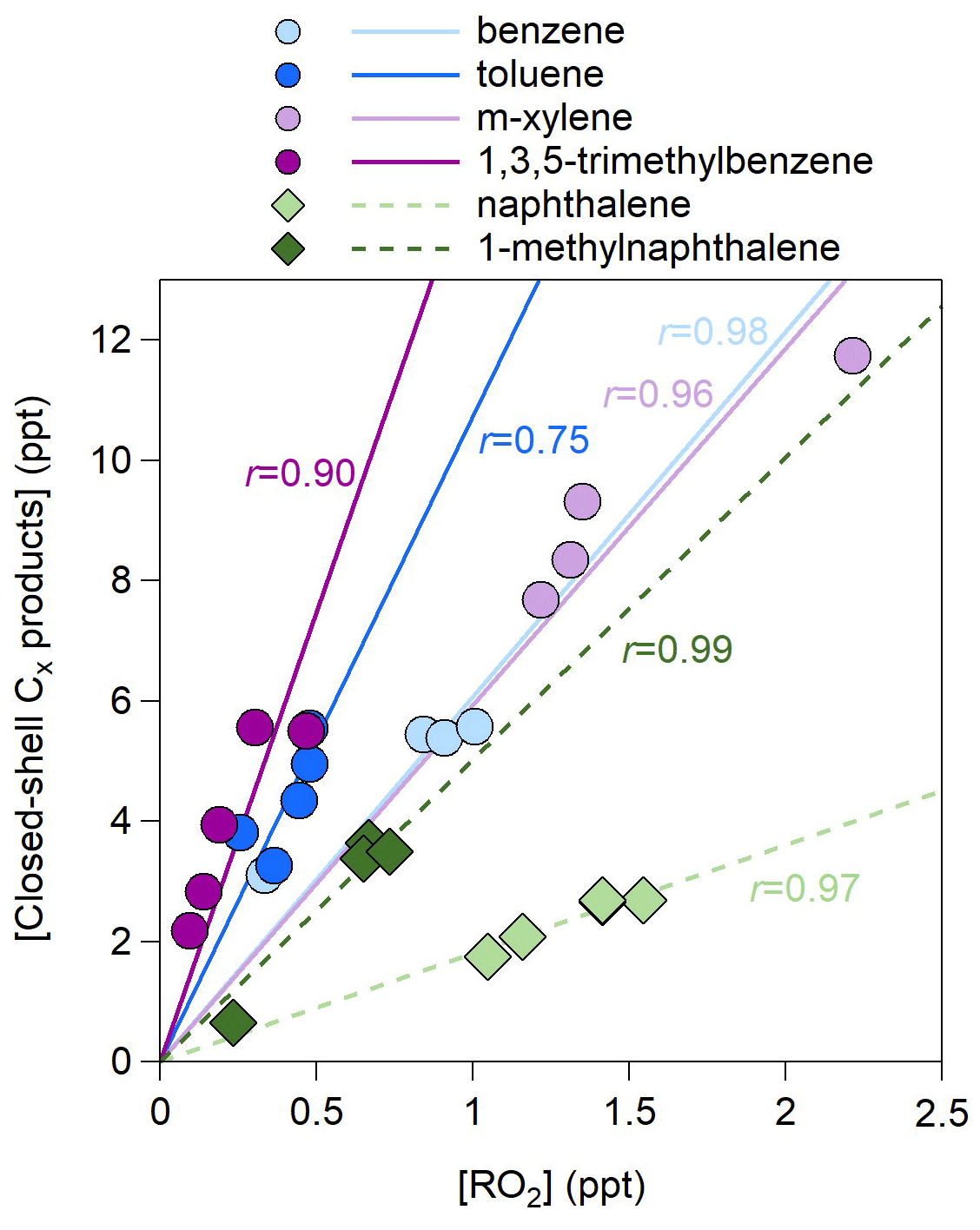
Figure 3Scatter plot of the concentrations of closed-shell Cx products and those of open-shell Cx products (RO2) for the 27 experiments listed in Table 1. The r values represent the Pearson correlation coefficients.
For a given precursor of CxHy, further auto-oxidation of BPRs (CxHy+1O5) may lead to the formation of Cx products with hydrogen numbers of y or y+2, whereas multi-generation OH reactions (i.e., a second or third OH attack) would lead to the formation of Cx products with hydrogen numbers of y−2 (H abstraction) or (OH addition) (Garmash et al., 2020; Cheng et al., 2021b). Figure 4 shows the signal fractions of closed-shell Cx products that have different hydrogen numbers at different OH exposure. The signal fractions of products with y−2 hydrogen numbers increase with increasing OH exposure, suggesting that more H abstraction may have occurred at higher OH exposure. Consistently, the signal fractions of Cx products with hydrogen numbers decrease as the OH exposure increases, suggesting less OH addition at higher OH exposure. H abstraction has been found to be involved in the formation of the majority of monoterpene OOMs (Shen et al., 2022). Moreover, the signal fractions of products with y−2 and hydrogen numbers are relatively higher in naphthalene- and 1-methylnaphthalene-derived OOMs than in other types of OOMs, highlighting the importance of multi-generation OH oxidation for double-ring aromatic precursors. Additionally, for naphthalene and 1-methylnaphthalene, both of the products with y+4 (15.9 %–27.8 %) and y+6 (3.4 %–10.9 %) hydrogen numbers have high signal fractions, whereas for monocyclic aromatic OOMs, the products with y+4 (9.7 %–37.9 %) hydrogen numbers are the main products (Tables S2–S7 and Fig. S4). The presence of abundant Cx products with hydrogen numbers of y+6 from double-ring precursors suggests a significant occurrence of third OH addition in early-generation products that still possess a high degree of unsaturation.
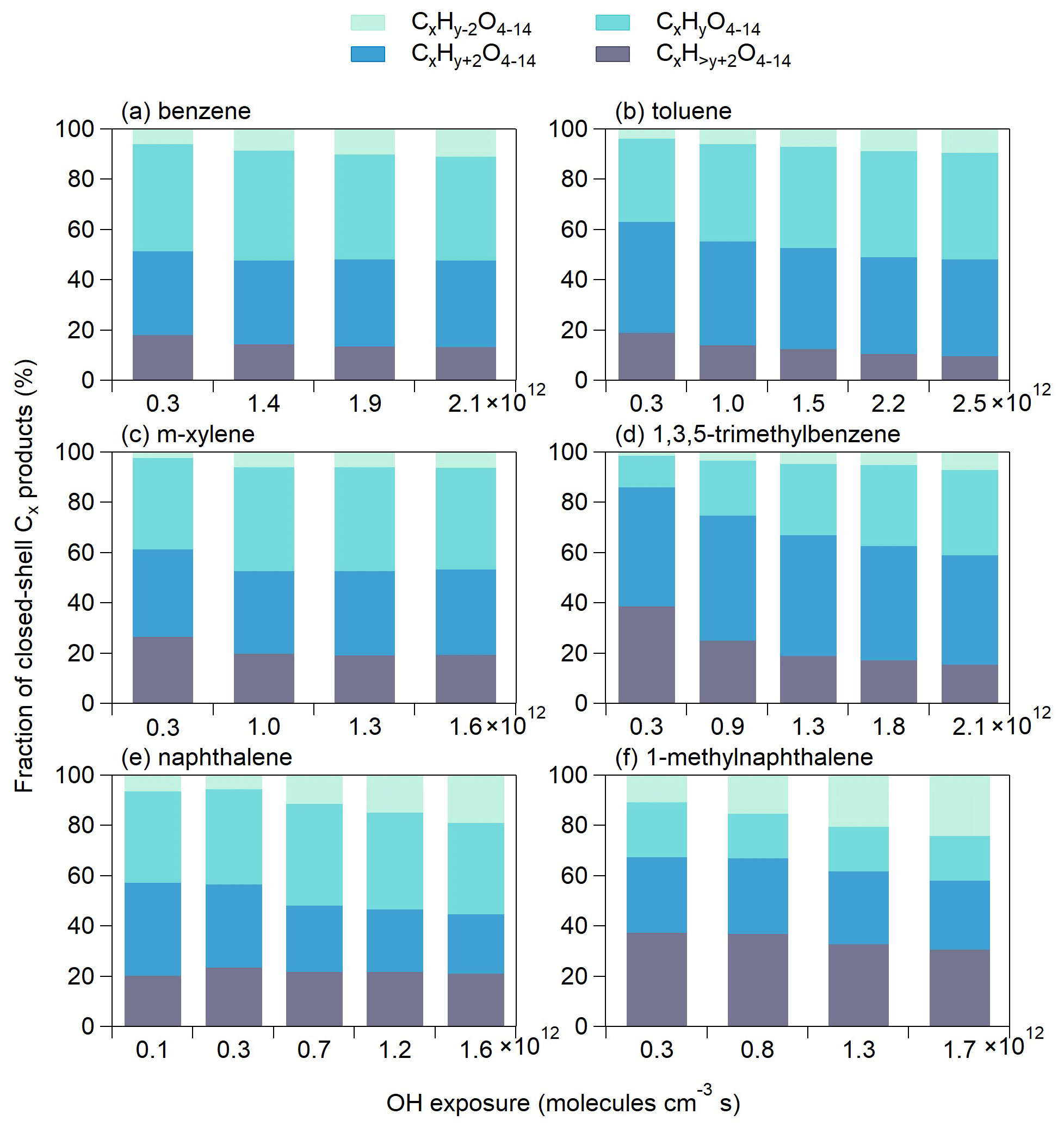
Figure 4Fractions of different series of closed-shell Cx products formed from the oxidation of (a) benzene, (b) toluene, (c) m-xylene, (d) 1,3,5-trimethylbenzene, (e) naphthalene and (f) 1-methylnaphthalene at different OH exposure. x and y represent the numbers of carbon and hydrogen atoms of the aromatic precursor (CxHy).
The concentrations of C2x products are several times lower than those of Cx products (Fig. S5). Their total concentrations vary by OH exposures but show different trends for different precursors. For benzene and 1-methylnaphthalene, the C2x concentrations increase for higher OH exposures, whereas for the other four precursors, the C2x concentrations decrease as the OH exposure increases. This highlights a large difference in the RO2 distribution and their functional groups for different precursors, calling for further investigations. The concentrations of C2x products correlate well with the square of the RO2 concentrations except for m-xylene (Fig. 5). The slopes are lower for naphthalene than for monocyclic precursors, again suggesting significant steric effects of double rings on the formation of C2x products (Tomaz et al., 2021). In addition, the slopes are greater for methyl substituted precursors than for non-substituted ones.
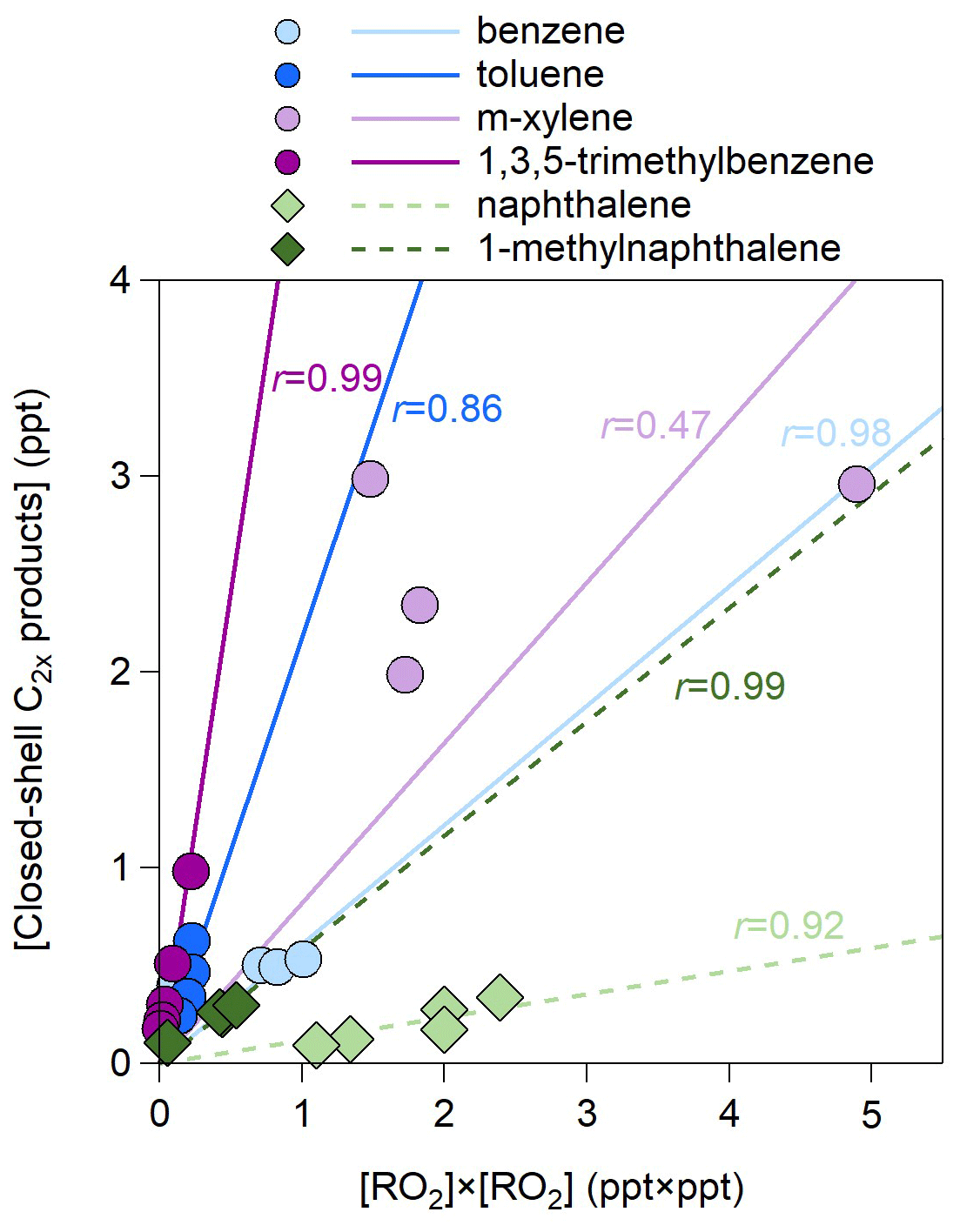
Figure 5Scatter plot of the concentrations of closed-shell C2x products and those of RO2× RO2 for the 27 experiments listed in Table 1. The r values represent the Pearson correlation coefficients.
Table 2 lists the constrained rate constants for benzene- and naphthalene-derived dimeric products with reasonable fittings (r>0.5). Our estimated rate coefficients for C2x products from the self- and cross-reactions of RO2 radicals span a range of 1 order of magnitude (i.e., 0.9–8.5 cm3 molecule−1 s−1). The rates are similar to those reported for α-pinene and 1,3,5-trimethylbenzene (i.e., 0.1–8.7 cm3 molecule−1 s−1) (Berndt et al., 2018a, b; Molteni et al., 2019). The estimated rate constants are cm3 molecules−1 s−1 for and cm3 molecules−1 s−1 for , suggesting that the accretion reaction rate is sensitive to precursor structures and RO2 functional groups. These RO2 radicals could be formed from BPRs (CxHy+1O5); thus the lower rate constant for may again be explained by the steric influence of double rings.
3.3 OOM contribution to SOA
Figure 6 shows the average elemental ratios of SOA and detected gaseous OOMs as well as the estimated contributions of the detected OOMs to the SOA mass for the experiments having AMS and SMPS particle measurements. For benzene oxidation, greater O:C ratios and lower H:C ratios are observed for SOA produced at higher OH exposures, which can be explained by typical functionalization process (Kroll et al., 2011). For toluene oxidation, the O:C ratios of SOA increase and then decrease as OH exposures increase, while the H:C ratios show the opposite, indicating significant fragmentation at high OH exposures. The lowest OH exposure in the OFR (> 0.9 d atmospheric equivalent photochemical age) is greater than chamber conditions. Correspondingly, the O:C ratios at lowest OH exposure for both types of SOA are greater, and the H:C ratios are lower compared to the reported values in previous chamber studies (Nakao et al., 2013; Chhabra et al., 2010) (Fig. 6a). Unlike benzene and toluene SOA, the H:C ratios for naphthalene SOA increase as OH exposures increase, and the O:C ratios at lowest OH exposure are lower than the reported ratios in chamber study (Chhabra et al., 2010), suggesting more difference in the oxidation conditions between the OFR and chamber studies for naphthalene. Our results agree with the reported O:C ratios in another OFR study (i.e., 0.86–1.40 for toluene SOA and 0.48–1.59 for naphthalene SOA) (Lambe et al., 2011). The detected OOMs produced from naphthalene have lower O:C and lower H:C ratios than those from benzene and toluene (Fig. 6b). When the OH exposure increases, the average O:C ratios of the detected OOMs increase, and the H:C ratios decrease (Fig. S6). The increase in O:C ratios of OOMs is more significant for the oxidation of naphthalene, whereas the decrease in H:C ratios of OOMs is more evident for the oxidation of benzene and toluene. The much broader ranges of O:C and H:C for each type of SOA than those for detected OOMs highlight the important contributions of undetected products to SOA. Notably, the O:C ratios of naphthalene-derived OOMs are much lower than those of benzene-derived OOMs, but the naphthalene-derived OOMs contain more low-volatility fractions than those of benzene-derived OOMs as shown in the volatility distribution (Fig. S7).
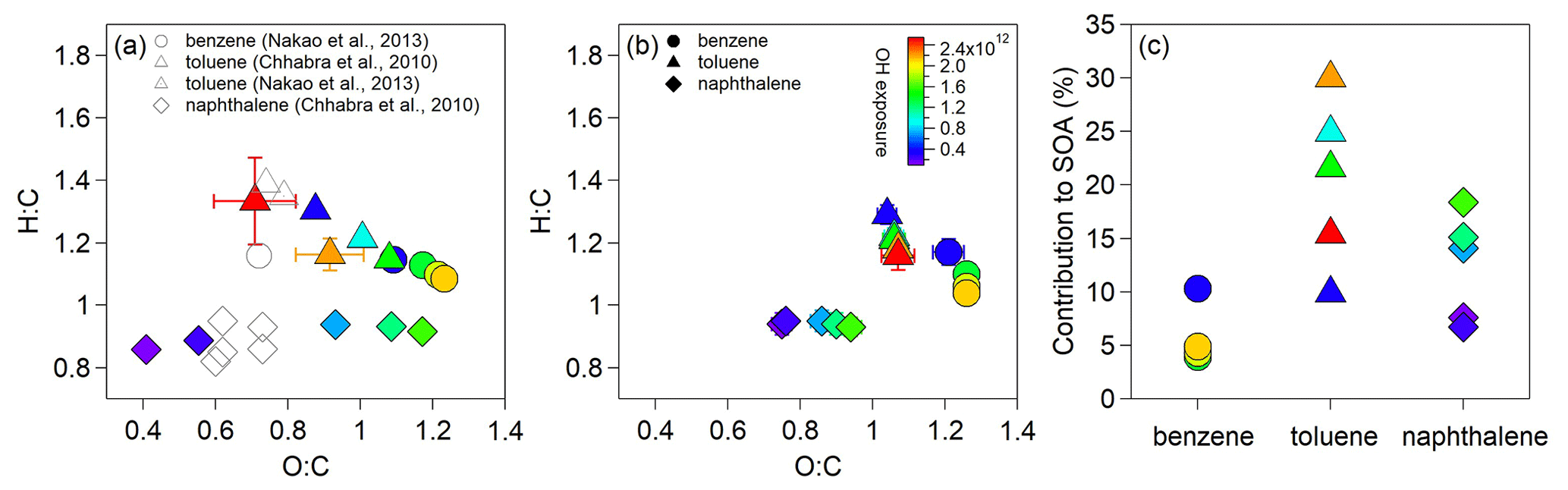
Figure 6(a) The averaged H:C and O:C ratios of the resulted SOA for the oxidation of benzene, toluene and naphthalene. (b) The averaged H:C and O:C ratios of gaseous OOMs detected in these experiments. (c) Estimated contributions of the detected OOMs to the SOA mass. The volatilities of OOMs were calculated using the method described in M. Wang et al. (2020).
The measured SOA mass concentrations are 4.3–22.2 µg m−3 at the exit of OFR. The particle wall loss correction has much greater uncertainties than for chamber seeded experiments because the particle size distributions can only be measured at the exit of the OFR. We therefore did not calculate the SOA yields in this study. The contributions of OOMs with µg m−3 to SOA may be evaluated by assuming instantaneous gas–particle equilibrium. On the basis of partition theory and the vapor pressure of OOMs (Pankow, 1994), the estimated contributions are about 0.3 %–18.3 % for the oxidation of aromatics. For OOMs with C∗ of ≤0.01 µg m−3, their contributions to SOA are usually estimated by calculating the net condensation flux. We can only obtain the net condensation flux at the exit of OFR, which should be greater than the flux inside the OFR considering the particle growth in the OFR. Multiplying this flux with the mean residence time of the OFR, these low-volatility species would explain 1.9 %–14.1 % of the aromatic SOA mass. Together, the identified OOMs by -TOF-CIMS may contribute to 3.9 %–10.3 %, 9.9 %–30.0 % and 6.7 %–18.4 % of the measured SOA mass for the oxidation of benzene, toluene and naphthalene, respectively, under our low-NOx OFR conditions (Fig. 6c). A recent field study suggests ∼ 50 %–70 % of aromatic OOM (also detected by -TOF-CIMS) contributions to SOA in urban environments in China (Nie et al., 2022), highlighting the potential importance of these OOMs to SOA. As shown in Fig. S7, most of the identified OOMs are in the low-volatility range (i.e., ULVOCs, ELVOCs, LVOCs). The volatility distribution of OOMs from naphthalene has a greater ULVOC fraction than that of benzene, which is consistent with the greater estimated contributions of naphthalene-derived OOMs to the SOA mass. Field measurements in urban Shanghai suggest that heavy aromatic VOCs are important SOA precursors (Tian et al., 2022). The results herein provide laboratory evidence that heavy (e.g., double-ring) aromatic VOCs might be more important than light (i.e., monocyclic) aromatic ones in initial particle growth during SOA formation.
The estimation of OOM contributions to the SOA mass has large uncertainties. The main calculation uncertainty is associated with the estimation of OOM volatilities, which could be ±1 bin different (i.e., 1 or 2 orders of magnitude in C∗) or more (Donahue et al., 2011). This may significantly affect the calculation of gas–particle partitioning and net condensation flux with compensation errors among species. On the other hand, the OFR experiments may not mimic the ambient conditions fully. High OH exposures with short residence time may lead to fast RO2+HO2 reactions that limit some RO2 auto-oxidation. Precursors are mixed in the real atmosphere, which may lead to different RO2 chemistry and product distributions compared to single-precursor systems in this study (Chen et al., 2022). Measurement uncertainties are relatively minor and not subject to significant systematic bias. The underestimation of large molecules (450–650 Th) in -TOF-CIMS analysis by a factor of 2 only affects the OOM contribution by ∼ 0.1 %. The quantification uncertainties of VOCs and SOA are usually less than 30 % (Huang et al., 2019; Zheng et al., 2021). In ambient applications, the calculation of OOMs to SOA needs to isolate the observed SOA growth to OOM production, which introduces additional uncertainties related to air mass dilution and transport.
The formation of OOMs from the photooxidation of benzene, toluene, m-xylene, 1,3,5-trimethylbenzene, naphthalene and 1-methylnaphthalene was investigated under low-NOx conditions with a wide range of OH exposure. The detected OOM formulae show diverse oxygen and hydrogen numbers that need both multi-step auto-oxidation and multi-generation OH oxidation to explain, highlighting similar reaction rates of the two mechanisms under atmospheric relevant VOC concentrations. Multi-generation OH oxidation appears to be more prominent at higher OH exposure for the oxidation of double-ring precursors than for the oxidation of single-ring aromatic precursors. The product distributions demonstrate significant steric effects of the methyl substitution and the double-ring structure on the OOM formation. We show that (1) the formation of fragmented products, especially unsaturated small oxygenates, is less efficient for precursors with more methyl groups, and (2) the steric effects of double rings affect the formation of both monomeric and dimeric products. More studies are needed for developing quantitative understanding to improve the CTM representation on the OOM formation. Finally, we found more OOMs produced by the oxidation of double-ring precursors in the lower volatility range than those formed from the oxidation of traditional monocyclic precursors, highlighting the importance of heavy aromatics to SOA formation. The detectable OOMs by the -TOF-CIMS are usually highly oxygenated products and explain at maximum 30.0 % of the aromatic SOA mass herein. More of the SOA mass is perhaps contributed by less oxygenated products that require other types of chemical ionization technique to explore.
Data presented in this paper are available at https://doi.org/10.5281/zenodo.10047716 (Cheng, 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-2099-2024-supplement.
QC and YJL designed the study. XC, YZ and KL conducted the experiments. XC performed the data analysis with the help of all authors. XC, QC and YJL wrote the manuscript, with inputs from all other authors.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors gratefully acknowledge Andrew Lambe and Penglin Ye for instrument support and helpful discussion.
This work is supported by the National Natural Science Foundation of China (grant nos. 41875165, 42250410322, 41961134034), the special fund of the State Key Joint Laboratory of Environmental Simulation and Pollution Control (grant no. 23Y05ESPCP), and the 111 Center of Urban Air Pollution and Health Effects (grant no. B20009). Yong Jie Li has been supported from the Science and Technology Development Fund, Macao SAR (file no. 0023/2021/A1), and a multiyear research grant (grant no. MYRG2022-00027-FST) from the University of Macau.
This paper was edited by Thomas Berkemeier and reviewed by two anonymous referees.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from ozonolysis and OH radical reaction of α-Pinene: mechanistic insight and the influence of isoprene and ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: a key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Birdsall, A. W., Andreoni, J. F., and Elrod, M. J.: Investigation of the role of bicyclic peroxy radicals in the oxidation mechanism of toluene, J. Phys. Chem. A, 114, 10655–10663, https://doi.org/10.1021/jp105467e, 2010.
Chan, A. W. H., Kautzman, K. E., Chhabra, P. S., Surratt, J. D., Chan, M. N., Crounse, J. D., Kürten, A., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 9, 3049–3060, https://doi.org/10.5194/acp-9-3049-2009, 2009.
Chen, Q., Liu, Y., Donahue, N. M., Shilling, J. E., and Martin, S. T.: Particle-phase chemistry of secondary organic material: modeled compared to measured O:C and H:C elemental ratios provide constraints, Environ. Sci. Technol., 45, 4763–4770, https://doi.org/10.1021/es104398s, 2011.
Chen, T., Zhang, P., Chu, B., Ma, Q., Ge, Y., Liu, J., and He, H.: Secondary organic aerosol formation from mixed volatile organic compounds: Effect of RO2 chemistry and precursor concentration, npj Climate and Atmospheric Science, 5, 95, https://doi.org/10.1038/s41612-022-00321-y, 2022.
Cheng, X.: Xi_OFR_LowNOx_AromaticPaper, Version v1, Zenodo [data set], https://doi.org/10.5281/zenodo.10047716, 2023.
Cheng, X., Chen, Q., Li, Y., Huang, G., Liu, Y., Lu, S., Zheng, Y., Qiu, W., Lu, K., Qiu, X., Bianchi, F., Yan, C., Yuan, B., Shao, M., Wang, Z., Canagaratna, M. R., Zhu, T., Wu, Y., and Zeng, L.: Secondary production of gaseous nitrated phenols in polluted urban environments, Environ. Sci. Technol., 55, 4410–4419, https://doi.org/10.1021/acs.est.0c07988, 2021a.
Cheng, X., Chen, Q., Jie Li, Y., Zheng, Y., Liao, K., and Huang, G.: Highly oxygenated organic molecules produced by the oxidation of benzene and toluene in a wide range of OH exposure and NOx conditions, Atmos. Chem. Phys., 21, 12005–12019, https://doi.org/10.5194/acp-21-12005-2021, 2021b.
Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Elemental analysis of chamber organic aerosol using an aerodyne high-resolution aerosol mass spectrometer, Atmos. Chem. Phys., 10, 4111–4131, https://doi.org/10.5194/acp-10-4111-2010, 2010.
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A semiempirical correlation between enthalpy of vaporization and saturation concentration for organic aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008.
Huang, G., Liu, Y., Shao, M., Li, Y., Chen, Q., Zheng, Y., Wu, Z., Liu, Y., Wu, Y., Hu, M., Li, X., Lu, S., Wang, C., Liu, J., Zheng, M., and Zhu, T.: Potentially important contribution of gas-phase oxidation of naphthalene and methylnaphthalene to secondary organic aerosol during haze events in Beijing, Environ. Sci. Technol., 53, 1235–1244, https://doi.org/10.1021/acs.est.8b04523, 2019.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Kautzman, K. E., Surratt, J. D., Chan, M. N., Chan, A. W. H., Hersey, S. P., Chhabra, P. S., Dalleska, N. F., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of gas- and aerosol-phase products from the photooxidation of naphthalene, J. Phys. Chem. A, 114, 913–934, https://doi.org/10.1021/jp908530s, 2010.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T., Onasch, T. B., Massoli, P., Croasdale, D. R., Wright, J. P., Ahern, A. T., Williams, L. R., Worsnop, D. R., Brune, W. H., and Davidovits, P.: Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA), Atmos. Chem. Phys., 11, 8913–8928, https://doi.org/10.5194/acp-11-8913-2011, 2011.
Li, L., Tang, P., Nakao, S., Chen, C.-L., and Cocker III, D. R.: Role of methyl group number on SOA formation from monocyclic aromatic hydrocarbons photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 2255–2272, https://doi.org/10.5194/acp-16-2255-2016, 2016a.
Li, L., Tang, P., Nakao, S., and Cocker III, D. R.: Impact of molecular structure on secondary organic aerosol formation from aromatic hydrocarbon photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 10793–10808, https://doi.org/10.5194/acp-16-10793-2016, 2016b.
Li, Y. and Wang, L.: The atmospheric oxidation mechanism of 1,2,4-trimethylbenzene initiated by OH radicals, Phys. Chem. Chem. Phys., 16, 17908–17917, https://doi.org/10.1039/c4cp02027h, 2014.
Mehra, A., Wang, Y., Krechmer, J. E., Lambe, A., Majluf, F., Morris, M. A., Priestley, M., Bannan, T. J., Bryant, D. J., Pereira, K. L., Hamilton, J. F., Rickard, A. R., Newland, M. J., Stark, H., Croteau, P., Jayne, J. T., Worsnop, D. R., Canagaratna, M. R., Wang, L., and Coe, H.: Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation, Atmos. Chem. Phys., 20, 9783–9803, https://doi.org/10.5194/acp-20-9783-2020, 2020.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kuerten, A., Lehtipalo, K., Makhmutov, V., Petaja, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tome, A., Trostl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of highly oxygenated organic molecules from α-pinene ozonolysis: chemical characteristics, mechanism, and kinetic model development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Nakao, S., Tang, P., Tang, X., Clark, C. H., Qi, L., Seo, E., Asa-Awuku, A., and Cocker III, D.: Density and elemental ratios of secondary organic aerosol: application of a density prediction method, Atmos. Environ., 68, 273–277, https://doi.org/10.1016/j.atmosenv.2012.11.006, 2013.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Nie, W., Yan, C., Huang, D. D., Wang, Z., Liu, Y. L., Qiao, X. H., Guo, Y. S., Tian, L. H., Zheng, P. G., Xu, Z. N., Li, Y. Y., Xu, Z., Qi, X. M., Sun, P., Wang, J. P., Zheng, F. X., Li, X. X., Yin, R. J., Dallenbach, K. R., Bianchi, F., Petaja, T., Zhang, Y. J., Wang, M. Y., Schervish, M., Wang, S. N., Qiao, L. P., Wang, Q., Zhou, M., Wang, H. L., Yu, C. A., Yao, D. W., Guo, H., Ye, P. L., Lee, S. C., Li, Y. J., Liu, Y. C., Chi, X. G., Kerminen, V. M., Ehn, M., Donahue, N. M., Wang, T., Huang, C., Kulmala, M., Worsnop, D., Jiang, J. K., and Ding, A. J.: Secondary organic aerosol formed by condensing anthropogenic vapours over China's megacities, Nat. Geosci., 15, 255–261, https://doi.org/10.1038/s41561-022-00922-5, 2022.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The atmospheric chemistry of alkoxy radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Pankow, J. F.: An absorption model of gas/particle partitioning of organic compounds in the atmosphere, Atmos. Environ., 28, 185-188, https://doi.org/10.1016/1352-2310(94)90093-0, 1994.
Peng, Z. and Jimenez, J. L.: Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 49, 2570–2616, https://doi.org/10.1039/c9cs00766k, 2020.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z., Palm, B. B., Day, D. A., Talukdar, R. K., Hu, W. W., Lambe, A. T., Brune, W. H., and Jimenez, J. L.: Model evaluation of new techniques for maintaining high-NO conditions in oxidation flow reactors for the study of OH-initiated atmospheric chemistry, ACS Earth Space Chem., 2, 72–86, https://doi.org/10.1021/acsearthspacechem.7b00070, 2018.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Pye, H. O. T. and Pouliot, G. A.: Modeling the role of alkanes, polycyclic aromatic hydrocarbons, and their oligomers in secondary organic aerosol formation, Environ. Sci. Technol., 46, 6041–6047, https://doi.org/10.1021/es300409w, 2012.
Schervish, M. and Donahue, N. M.: Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 20, 1183–1199, https://doi.org/10.5194/acp-20-1183-2020, 2020.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 8, eabp8702, https://doi.org/10.1126/sciadv.abp8702, 2022.
Tian, L., Huang, D. D., Wang, Q., Zhu, S., Wang, Q., Yan, C., Nie, W., Wang, Z., Qiao, L., Liu, Y., Qiao, X., Guo, Y., Zheng, P., Jing, S. a., Lou, S., Wang, H., Yu, J. Z., Huang, C., and Li, Y. J.: Underestimated contribution of heavy aromatics to secondary organic aerosol revealed by comparative assessments using new and traditional methods, ACS Earth Space Chem., 7, 110–119, https://doi.org/10.1021/acsearthspacechem.2c00252, 2022.
Tian, L., Huang, D. D., Li, Y. J., Yan, C., Nie, W., Wang, Z., Wang, Q., Qiao, L., Zhou, M., Zhu, S., Liu, Y., Guo, Y., Qiao, X., Zheng, P., Jing, S. a., Lou, S., Wang, H., and Huang, C.: Enigma of urban gaseous oxygenated organic molecules: precursor type, role of NOx, and degree of oxygenation, Environ. Sci. Technol., 57, 64–75, https://doi.org/10.1021/acs.est.2c05047, 2023.
Tomaz, S., Wang, D., Zabalegui, N., Li, D., Lamkaddam, H., Bachmeier, F., Vogel, A., Monge, M. E., Perrier, S., Baltensperger, U., George, C., Rissanen, M., Ehn, M., El Haddad, I., and Riva, M.: Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry, Nat. Commun., 12, 300, https://doi.org/10.1038/s41467-020-20532-2, 2021.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Tsigaridis, K., Daskalakis, N., Kanakidou, M., Adams, P. J., Artaxo, P., Bahadur, R., Balkanski, Y., Bauer, S. E., Bellouin, N., Benedetti, A., Bergman, T., Berntsen, T. K., Beukes, J. P., Bian, H., Carslaw, K. S., Chin, M., Curci, G., Diehl, T., Easter, R. C., Ghan, S. J., Gong, S. L., Hodzic, A., Hoyle, C. R., Iversen, T., Jathar, S., Jimenez, J. L., Kaiser, J. W., Kirkevåg, A., Koch, D., Kokkola, H., Lee, Y. H., Lin, G., Liu, X., Luo, G., Ma, X., Mann, G. W., Mihalopoulos, N., Morcrette, J.-J., Müller, J.-F., Myhre, G., Myriokefalitakis, S., Ng, N. L., O'Donnell, D., Penner, J. E., Pozzoli, L., Pringle, K. J., Russell, L. M., Schulz, M., Sciare, J., Seland, Ø., Shindell, D. T., Sillman, S., Skeie, R. B., Spracklen, D., Stavrakou, T., Steenrod, S. D., Takemura, T., Tiitta, P., Tilmes, S., Tost, H., van Noije, T., van Zyl, P. G., von Salzen, K., Yu, F., Wang, Z., Wang, Z., Zaveri, R. A., Zhang, H., Zhang, K., Zhang, Q., and Zhang, X.: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 14, 10845–10895, https://doi.org/10.5194/acp-14-10845-2014, 2014.
Wang, M., Chen, D., Xiao, M., Ye, Q., Stolzenburg, D., Hofbauer, V., Ye, P., Vogel, A. L., Mauldin, R. L., Amorim, A., Baccarini, A., Baumgartner, B., Brilke, S., Dada, L., Dias, A., Duplissy, J., Finkenzeller, H., Garmash, O., He, X.-C., Hoyle, C. R., Kim, C., Kvashnin, A., Lehtipalo, K., Fischer, L., Molteni, U., Petäjä, T., Pospisilova, V., Quéléver, L. L. J., Rissanen, M., Simon, M., Tauber, C., Tomé, A., Wagner, A. C., Weitz, L., Volkamer, R., Winkler, P. M., Kirkby, J., Worsnop, D. R., Kulmala, M., Baltensperger, U., Dommen, J., El-Haddad, I., and Donahue, N. M.: Photo-oxidation of aromatic hydrocarbons produces low-volatility organic compounds, Environ. Sci. Technol., 54, 7911–7921, https://doi.org/10.1021/acs.est.0c02100, 2020.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.
Zheng, P., Chen, Y., Wang, Z., Liu, Y., Pu, W., Yu, C., Xia, M., Xu, Y., Guo, J., Guo, Y., Tian, L., Qiao, X., Huang, D. D., Yan, C., Nie, W., Worsnop, D. R., Lee, S., and Wang, T.: Molecular characterization of oxygenated organic molecules and their dominating roles in particle growth in Hong Kong, Environ. Sci. Technol., 57, 7764–7776, https://doi.org/10.1021/acs.est.2c09252, 2023.
Zheng, Y., Cheng, X., Liao, K., Li, Y., Li, Y. J., Huang, R.-J., Hu, W., Liu, Y., Zhu, T., Chen, S., Zeng, L., Worsnop, D. R., and Chen, Q.: Characterization of anthropogenic organic aerosols by TOF-ACSM with the new capture vaporizer, Atmos. Meas. Tech., 13, 2457–2472, https://doi.org/10.5194/amt-13-2457-2020, 2020.
Zheng, Y., Chen, Q., Cheng, X., Mohr, C., Cai, J., Huang, W., Shrivastava, M., Ye, P., Fu, P., Shi, X., Ge, Y., Liao, K., Miao, R., Qiu, X., Koenig, T. K., and Chen, S.: Precursors and Pathways Leading to Enhanced Secondary Organic Aerosol Formation during Severe Haze Episodes, Environ. Sci. Technol., 55, 15680–15693, https://doi.org/10.1021/acs.est.1c04255, 2021.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/C2CS35122F, 2012.