the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Jul 2022
| 13 Jul 2022
Observations of gas-phase products from the nitrate-radical-initiated oxidation of four monoterpenes
Michelia Dam
Danielle C. Draper
Andrey Marsavin
Juliane L. Fry
Chemical ionization mass spectrometry with the nitrate reagent ion ( CIMS) was used to investigate the products of the nitrate radical (NO3) initiated oxidation of four monoterpenes in laboratory chamber experiments. α-Pinene, β-pinene, Δ-3-carene, and α-thujene were studied. The major gas-phase species produced in each system were distinctly different, showing the effect of monoterpene structure on the oxidation mechanism and further elucidating the contributions of these species to particle formation and growth. By comparing groupings of products based on the ratios of elements in the general formula CwHxNyOz, the relative importance of specific mechanistic pathways (fragmentation, termination, and radical rearrangement) can be assessed for each system. Additionally, the measured time series of the highly oxidized reaction products provide insights into the ratio of relative production and loss rates of the high-molecular-weight products of the Δ-3-carene system. The measured effective O:C ratios of reaction products were anticorrelated with new particle formation intensity and number concentration for each system; however, the monomer : dimer ratios of products had a small positive trend. Gas-phase yields of oxidation products measured by CIMS correlated with particle number concentrations for each monoterpene system, with the exception of α-thujene, which produced a considerable amount of low-volatility products but no particles. Species-resolved wall loss was measured with CIMS and found to be highly variable among oxidized reaction products in our stainless steel chamber.
- Article
(2533 KB) - Full-text XML
-
Supplement
(3061 KB) - BibTeX
- EndNote





The largest uncertainty in modern climate models is attributed to the radiative effect of aerosols (Stocker et al., 2013). Their chemical complexity makes it challenging to predict their formation as well as properties that determine their direct and indirect impacts on climate. A significant fraction of total global aerosol is secondary organic aerosol (SOA), which originates from the oxidation of gas-phase volatile organic compounds (VOCs) to form highly oxidized species that may partition into particles or small clusters (Kroll and Seinfeld, 2008; Ehn et al., 2014; Bianchi et al., 2019). Many SOA formation pathways have been widely studied, such as the ozone (O3) and hydroxyl radical (OH) initiated oxidation of biogenic volatile organic compounds (BVOCs) (Berndt et al., 2016; Lee et al., 2006; Atkinson and Arey, 2003). One such system that has been shown to contribute significantly to SOA formation but has not been as comprehensively studied is nitrate radical (NO3) initiated oxidation of BVOCs (Ng et al., 2017). The NO3 radical, produced by the oxidation of nitrogen dioxide (NO2) with O3, is mainly anthropogenic in origin and is most abundant at night when photolysis does not occur (Brown and Stutz, 2012). BVOCs are emitted naturally by plants and comprise a large fraction of global VOCs, but BVOC concentrations are highest in forested regions (Acosta Navarro et al., 2014). Therefore, nitrate-radical-initiated oxidation of BVOCs is an SOA-generating system that couples anthropogenic oxidants with biogenic precursors. This chemistry has also been shown to be important in areas like the southeastern United States (Ayres et al., 2015) and the Colorado Rocky Mountains (Fry et al., 2013).
Monoterpenes (MTs), unsaturated C10H16 compounds, comprise a large fraction of global BVOCs and have been shown to have a high SOA production potential from nitrate-radical-initiated oxidation (Ng et al., 2017; Sindelarova et al., 2014; Ayres et al., 2015). However, the large range in SOA yield in laboratory studies of the most abundant MTs, α-pinene (0 %–16 %), β-pinene (27 %–104 %), and Δ-3-carene (68 %–77 %) (Ng et al., 2017; Fry et al., 2014; Boyd et al., 2015; Hallquist et al., 1999), indicates that the oxidation mechanisms of these MTs have key differences. As α-pinene is often used as the representative MT in regional and global models, these oxidation mechanisms need further investigation to improve model predictions of SOA yield from MT + NO3 systems and the concomitant impacts on climate.
Recently, computational and experimental studies have shed light on the initial steps of nitrate-radical-initiated oxidation of α-pinene, β-pinene, and Δ-3-carene – all of which are bicyclic monoterpenes with a single double bond (Kurtén et al., 2017; Draper et al., 2019). These studies concluded that, following NO3 addition onto the carbon–carbon double bond and rapid O2 addition to the alkyl radical, first-generation peroxy radical isomerization reactions are too slow to contribute to overall oxidation product distributions and instead rapidly reduce to alkoxy groups through bimolecular reactions with NO3, HO2, or RO2 in the nighttime atmosphere. In the oxygen-rich atmosphere (O2 concentration > 1015 cm−3), O2 addition to nitroxy-alkyl radical compounds is expected to be much faster than radical decomposition (Berndt and Böge, 1995). First-generation alkoxy scission plays an important role in determining the potential for further radical propagation for these monoterpenes and may help explain why the α-pinene + NO3 system produces much lower SOA yields than β-pinene + NO3.
First-generation alkoxy scission is affected by the position of the endocyclic or exocyclic double bond with respect to the secondary ring in these bicyclic monoterpenes (Vereecken and Peeters, 2009). The most favorable, lowest-energy, first-generation alkoxy scission pathway for the α-pinene + NO3 system leads to the formation of pinonaldehyde – a closed-shell species that is not very highly oxidized and, thus, is not expected to contribute to the formation of new SOA. This contrasts with the Δ-3-carene and β-pinene systems, which form alkyl radicals that allow for further radical propagation, oxidation, and internal isomerization. These processes can lead to the formation of highly oxidized gas-phase products that can readily partition into small particles. Additionally, other unimolecular processes have been shown to be competitive on the timescale of these reactions, including internal hydrogen-shift isomerization and radical rearrangement by opening the secondary ring (Vereecken and Peeters, 2010). The size of the secondary ring strongly influences the energy barrier for ring opening: four-membered rings (α-pinene, β-pinene) are unlikely to open, but strained three-membered rings (Δ-3-carene) are much more susceptible to ring opening (Kurtén et al., 2017). Understanding the prevalence of these early unimolecular processes is important in determining the potential for further radical propagation and oxidation.

Figure 1Scheme of the proposed α-thujene + NO3 oxidation mechanism. Orange arrows indicate the formation of thujenaldehyde – a volatile product that is not expected to contribute to new particle formation. The downward arrow indicates a potential alkyl radical rearrangement that leads to a product that can undergo additional oxidation to form highly oxygenated molecules (HOMs).
For this experimental study, we investigate nitrate-radical-initiated oxidation of four monoterpenes, α-pinene, β-pinene, Δ-3-carene, and α-thujene, in a reaction chamber. The first three monoterpenes are abundant in the atmosphere (Sindelarova et al., 2014), and their oxidation mechanisms have been previously studied in laboratory experiments and theoretical computational studies. α-Thujene, a key component of frankincense oil, is less naturally abundant, but studying the α-thujene system presents a unique opportunity to assess early unimolecular processes because of its structure. α-Thujene has a three-membered secondary ring, similar to Δ-3-carene, with an adjacent double bond position, similar to α-pinene. Following NO3 addition to the double bond, oxidation of the alkyl radical to a peroxy radical and subsequent reduction to an alkoxy radical through a bimolecular reaction is expected to occur. The first-generation alkoxy scission pathways available for α-thujene mirror those of α-pinene (Fig. 1). The figure shows that cleaving the C1–C2 bond, losing the top methyl group, or the left-side C2–C7 bond, leading to an unstable alkyl radical on a three-membered ring, would be unfavorable. Cleaving the right-side C2–C3 bond generates an alkyl radical on the nitrate-substituted carbon, which would lead to rapid radical termination with loss of NO2, forming thujenaldehyde. If, instead, NO3 addition onto the double bond was followed by radical rearrangement by opening the three-membered ring (C5–C7 cleavage) and subsequent tertiary alkyl radical formation, radical propagation pathways become available and can potentially lead to further oxidation and condensable species (Vereecken and Peeters, 2009). By studying the oxidation of α-thujene in addition to the previously studied MT systems, we can assess the prevalence of this ring-opening reaction in the early mechanism.
This study is an observation of detailed compositional differences of observed gas-phase oxidation products as they relate to the current understanding of oxidation mechanisms using high-resolution time-of-flight chemical ionization mass spectrometry (HR-TOF-CIMS) with the reagent ion. CIMS has been used to measure the composition of oxidized organics in laboratory studies (Berndt et al., 2015; Rissanen et al., 2014; Hyttinen et al., 2015; Mentel et al., 2015; Riva et al., 2019) and in ambient air (Ehn et al., 2014), but it has also been used specifically to probe nitrogen-containing oxidized monoterpenes (Draper et al., 2019). The reagent ion has been shown to cluster with highly oxidized compounds that contain at least two hydrogen bond donor sites. Therefore, we do not expect to be able to measure highly volatile aldehyde products (thujenaldehyde/pinonaldehyde) with CIMS, but we can probe the formation of highly oxidized products and compare the differences in composition among the four MT systems. Nitrate-radical-initiated oxidation of α-thujene has not been previously studied; thus, the results of this particular system are unique observations. Iodide (I−) has been used extensively in previous studies as a CIMS reagent ion, specifically to measure oxidized organics and inorganic nitrogen species (Brophy and Farmer, 2015; Aljawhary et al., 2013; Lee et al., 2014; Lopez-Hilfiker et al., 2016). The use of both I− and reagent ions could provide additional mechanistic information and is planned for future studies.
2.1 Experimental methods
We ran chamber experiments using a darkened 560 L stainless steel chamber in flow-through mode with a total flow of 17 L min−1. Previously, we confirmed that operating in this manner resulted in conditions in which the chamber was well mixed after ∼ 100 s with a residence time of ∼ 33 min (see Sect. S6 in the Supplement). A schematic of our experimental setup is shown in Fig. 2. All experiments were performed under dry conditions. We generated NO3 radical by oxidizing NO2 with O3 inside the chamber and allowing the oxidants to reach steady state (∼ 2 h) before adding MT. O3 was generated by UV photolysis of air scrubbed of NOx (NO + NO2) and VOCs by a zero-air generator (Aadco Instruments, model 737-13) at 1.5 L min−1. NO2 was introduced directly from a commercially prepared cylinder (Praxair, Inc., 2.5 ppm in purified air) at 1.5 L min−1. Before the addition of MT, the mixing ratios of oxidants and nitrogen compounds in the chamber were as follows: [O3] ≈ 240 ppb, [NO2] ≈ 240 ppb, [NO3] ≈ 0.2 ppb, and [N2O5] ≈ 25 ppb. O3 was measured using an absorption gas analyzer (2B Technologies, model 106-L). NO2 was measured with both an absorption gas analyzer (2B Technologies, model 405nm) and with a homebuilt thermal-dissociation–cavity ring-down spectrometer (TD-CRDS; Keehan et al., 2020). NO3 radical and N2O5 concentrations were modeled using the kinetic box model, KinSim (Peng and Jimenez, 2019), which was run on the Igor Pro computing platform (WaveMetrics, Inc., version 7). N2O5 was also measured by I− CIMS and the TD-CRDS, but those measurements were performed primarily to estimate wall losses and were, thus, not calibrated. We introduced MT into the chamber from gas cylinders that were prepared by injecting liquid MT (Sect. S2 in the Supplement) into the cylinders and then pressurizing with ultrapure nitrogen. The mixing ratio inside the cylinders (11–20 ppm) was estimated using the mass of injected liquid and confirmed using a gas chromatograph with a flame-ionization detector and a homebuilt cryogenic preconcentrator (GC-FID). The small MT flow (25 cm3 min−1) was fed into the center of a larger zero-air flow (1 L min−1) with tee fitting to more effectively carry it into the chamber. Monoterpene concentrations (∼ 41 ppb) inside the chamber were estimated from the flow dilution; the concentrations were confirmed using GC-FID and modeled with KinSim. The remainder of the flow, 12 L min−1, was zero air that was introduced into the chamber with a Teflon “shower head” consisting of a capped tube with holes drilled perpendicularly along the length of the tube to facilitate mixing in the chamber. We ran experiments under continuous flow and measured precursor and product concentrations for 1–2 h until the gas-phase products reached steady state, as detected with CIMS. The sample lines extended ∼ 20 cm into the chamber in order to minimize the possibility of sampling in a gradient caused by loss of low-volatility species to the chamber walls.
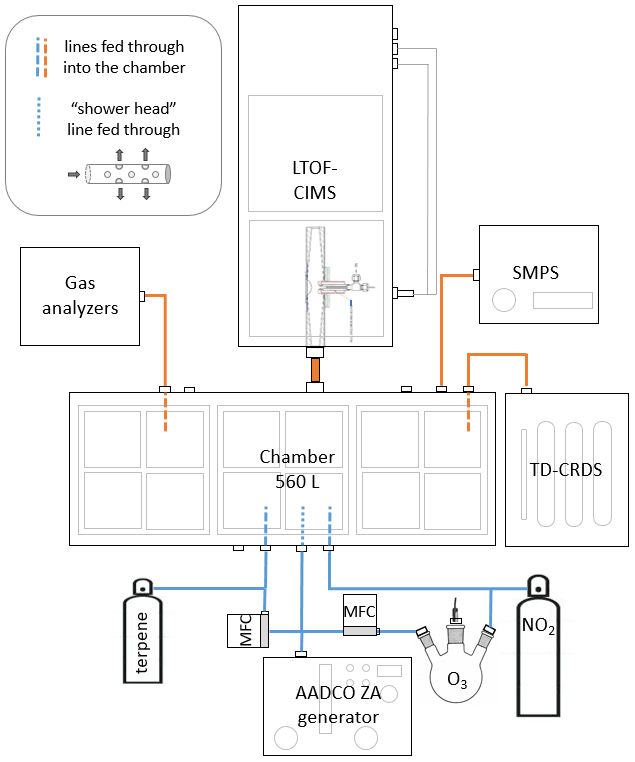
Figure 2Experimental setup for chamber studies. Blue lines indicate flow going into the chamber, and orange lines indicate where flow is being removed.
The TD-CRDS was used to measure nitrogen-containing species (nitric acid, alkyl nitrates – ANs, peroxy nitrates – PNs, and NO2) in both the gas and particle phases (Keehan et al., 2020). The TD-CRDS measured total ANs + PNs during this experiment, and the NO2 channel was used to parameterize the KinSim model. Additionally, a scanning mobility particle sizer (SMPS), which consisted of a differential mobility analyzer (TSI, Inc., model 3081), a condensation particle counter (CPC, TSI, Inc., model 3020), and a homebuilt flow and voltage controller, was used to measure the particle number size distributions. We probed the formation of low-volatility oxidation products using CIMS, consisting of a high-resolution time-of-flight mass spectrometer (Tofwerk AG, model LTOF) operating in V mode. We used a homebuilt transverse ionization CIMS inlet, in which the sample flow is perpendicular to the flow of the reagent ions into the entrance orifice of the mass spectrometer (Li et al., 2019). The inlet minimizes the wall losses of sampled gases as well as clustering with neutral compounds such as water vapor in the ion source, with an average ionization reaction time of ∼ 80 ms. reagent ion was generated by flowing ultrahigh-purity N2 gas over the headspace of a small glass vial filled with nitric acid (HNO3). The reagent ion flow (8 cm3 min−1) was diluted with a larger flow (1 L min−1) of pure N2. Peaks in mass spectra were fitted and assigned using the Tofware software (Aerodyne Research, version 7). The reagent ion monomer (), dimer (), and trimer () were used as calibration peaks for the low mass range. High-mass calibrations were determined from the highest intensity single peaks in the monomer and dimer region clustered with the NO3 ion, and reasonable formulas were predicted from the base MT formula (C10H16). The total gas-phase product concentration was estimated using the sum of the abundance of all species detected by CIMS integrated over the duration of the experiment. The integrated ion signal was converted to concentration using a calibration factor (6 × 1010 ). This calibration factor was determined experimentally using, as a proxy, the reaction of sulfuric acid (H2SO4) and ion, which is estimated to be at the collision limit, as detailed by Kurtén et al. (2016). A calculation is shown in Sect. S4 in the Supplement. We recognize that this calibration factor result is likely an upper limit for the actual concentration of organic nitrate compounds.
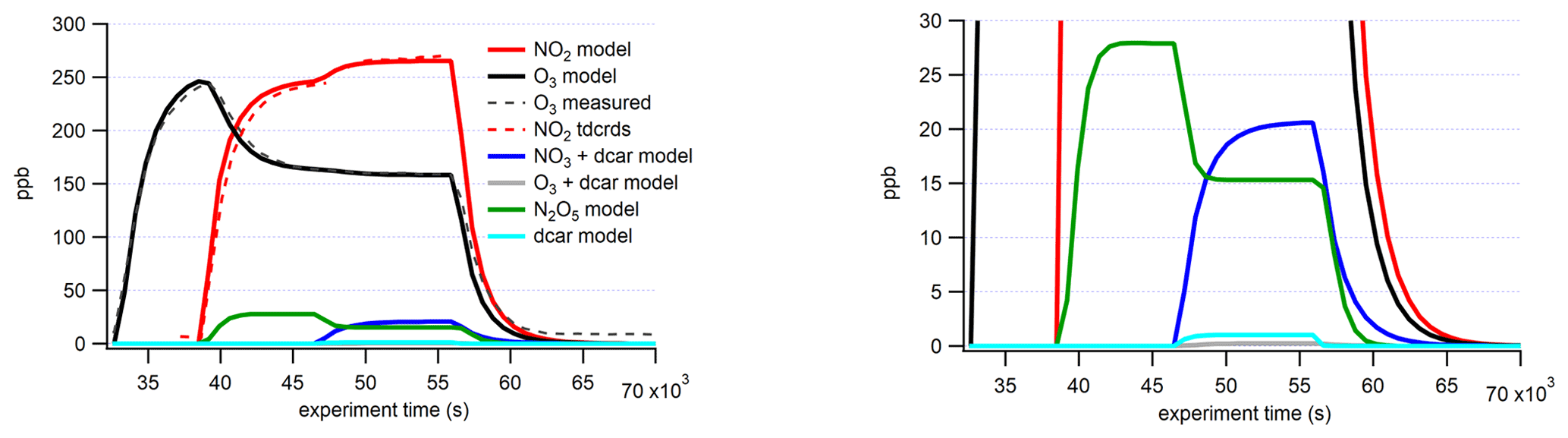
Figure 3KinSim simulation of the Δ-3-carene + NO3 experiment. Measured traces are shown with dashed lines, and modeled results are shown with solid lines. Additional modeling of the other MT systems are shown in Fig. S1 in the Supplement.
2.2 Kinetic modeling
KinSim was used to support our experiments by providing approximate concentrations of unmeasured oxidant species (NO3/N2O5) and helping assess the dominant oxidant chemistry (NO3 vs. O3) in the chamber. The rate constants for all reactions considered in the model are listed in Sect. S1 in the Supplement. The chamber is assumed to be well mixed in the model, and dilution flow is also represented. Experimentally measured time series of oxidants agree well with the modeled concentrations (Fig. 3). Experimentally measured wall loss of N2O5 (1.25 × 10−3 s−1) was also considered in the model. This value was experimentally determined by stopping the flow of oxidants to the chamber and making up the lost flow with additional zero air, thereby “turning off” the chemistry, and using I− CIMS to measure the decay of the N2O5I− cluster (Sect. S6 in the Supplement). The raw N2O5I− decay was exponentially fitted to determine the wall loss rate. DeHaan et al. (1999) found the N2O5 wall loss rate of this chamber to be 9.2 × 10−5 s−1, which is somewhat lower, but the chamber was coated with paraffin wax and was operated under static flow conditions for their measurements. Even though excess O3 remains in the chamber from the generation of the NO3, the model predicts that more than 98 % of oxidation products in all MT systems should be initiated by NO3. This is expected, as the rate constant for the MT + NO3 radical reaction is several orders of magnitude faster than for MT + O3. The rate constants of NO3 and O3 oxidation for α-thujene are unknown and, therefore, cannot be modeled in the same way; hence, we estimated a “worst-case scenario” by taking the slowest NO3 + MT rate constant (β-pinene, 2.5 × 10−12 cm3 molec.−1 s−1, from Ng et al., 2017) and the fastest O3 + MT rate constant (α-pinene, 6.0 × 10−12 cm3 molec.−1 s−1, from Atkinson and Arey, 2003). This resulted in a ratio of O3:NO3 products of 0.1.
3.1 Oxidation product quantification
Mixing ratios of reaction products calculated from integrated concentrations were on the order of 0.1–0.001 ppt. Percent yield is reported for each MT system and shows the following: Δ-3-carene (2.5 × 10−4) > α-thujene (1.0 × 10−4) > β-pinene (5.0 × 10−5) > α-pinene (9.8 × 10−6). Substantial wall losses were found for CIMS-measured species, with high variability observed among individual species and no clear trend with O:C or molecule weight (see Sect. S6 in the Supplement). Therefore, individual wall loss corrections were applied for all species before calculating yield from CIMS measurements. Explicit wall effects were only measured for the Δ-3-carene system; therefore, for the other MTs, average wall loss rates for monomers (5.5 × 10−3 s−1) and dimers (3.4 × 10−3 s−1) were calculated using observations from the Δ-3-carene system experiment and are, thus, subject to greater uncertainty. In contrast, wall losses of the total ANs+PNs measured by TD-CRDS were observed to be negligible relative to the dilution timescale of the chamber, suggesting that the majority of this bulk organic nitrate signal is due to higher-volatility species not measured by the CIMS, not substantially lost to the walls, and likely not contributing substantially to SOA formation. The trend in observed molar yields of total ANs+PNs is consistent with previous measurements (Δ-3-carene > β-pinene > α-pinene), but the magnitudes (20 %, 10 %, and 5 %, respectively) are substantially lower than previous studies. This is puzzling, but it may be due to the TD-CRDS measurement measuring only the subset of high-volatility nitrates in these experiments, while the lower-volatility nitrates have rapid wall losses that prevent the TD-CRDS from measuring them. We note that the CIMS is essentially inlet-less in comparison with a 2 m inlet line for the TD-CRDS in these experiments.
The α-thujene system produced more highly oxidized products than the α-pinene system; especially significant is the amount of highly oxidized dimers that were formed from α-thujene. The Δ-3-carene system generated the most particles, followed by the β-pinene system, and both the α-pinene and α-thujene systems did not generate any particles (Fig. S2 in the Supplement). Observed particle number trends agree with gas-phase product trends except for the α-thujene system, in which highly oxidized products formed but did not nucleate and/or grow effectively to form measurable particles.
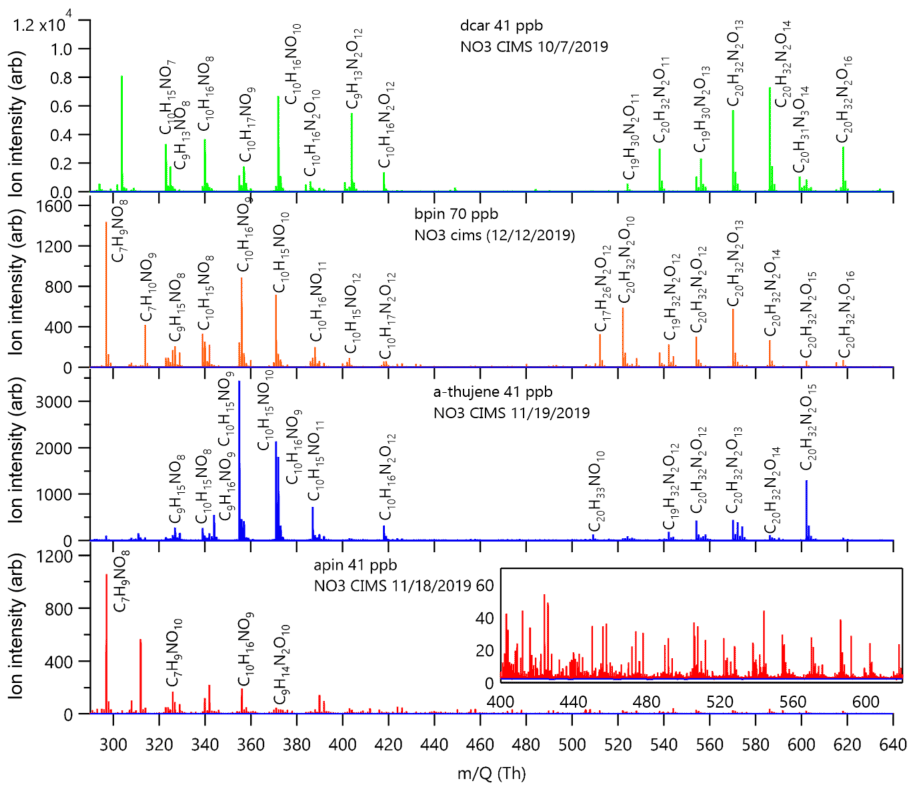
Figure 4Stacked mass spectra from each MT + NO3 system. The reagent ion is not included in assigned formula. The left axes are raw ion counts and are not normalized to the reagent ion.
3.2 Comparison of oxidation product composition
3.2.1 Definition of categories for elemental analysis
The experiment-averaged mass spectra for each MT system showed very different peak distributions (Fig. 4). Here, we explore the relative ratios of experiment-averaged ion abundance from categories of products. For comparison, we normalized the integrated area of each peak by the total integrated area of all organic peaks in the mass spectrum. The general formula CwHxNyOz was used to create categories of reaction products that correspond to specific predicted mechanistic pathways that are summarized in Table 1, with the representative mechanisms for each pathway shown in Fig. 5. The carbon number provides an indication of fragmentation caused by C–C bond cleavage for any carbon number that is not equal to 10 (for monomers) or 20 (for dimer formation from peroxy or alkoxy radical additions, e.g., RO2 + RO2). The hydrogen number informs us about the terminal functional group and the associated bimolecular reactions leading to them. The nitrogen number can indicate secondary double bond generation or NO2 + RO2 chemistry from residual NO2 in the chamber. Structure–activity relationships (SARs) and rate constants were found in the literature for relevant pathways (Vereecken and Peeters, 2009, 2010; Novelli et al., 2021; Kurtén et al., 2017; Draper et al., 2019; Jenkin et al., 2019; Crounse et al., 2013), but proposed mechanisms were not explicitly modeled for this study. The oxygen number and O:C ratio provide insights into how much autoxidation chemistry occurred, increasing the O:C ratio of the products. While we cannot completely explain the origin of every observed compound, the observed differences provide valuable information regarding the most important pathways that lead to these low-volatility products. The ratio distributions for all of the MT systems are summarized in Fig. 6. Complete peak lists for each MT system can be found in Sect. S8 in the Supplement. Note that the β-pinene system ratios are the average of an 8 ppb experiment and a 70 ppb experiment (39 ppb average). The full comparison of results from both experiments can be found in Sect. S7 in the Supplement.
Table 1Summary table of reaction product formulas grouped by the mechanistic pathways shown in Fig. 5. Group A details the formation of C10HXN1OZ reaction products, group B details the formation of C9HXN1OZ products, group C details the formation of C7HXN1OZ products, and group D details various pathways for dimer formation.

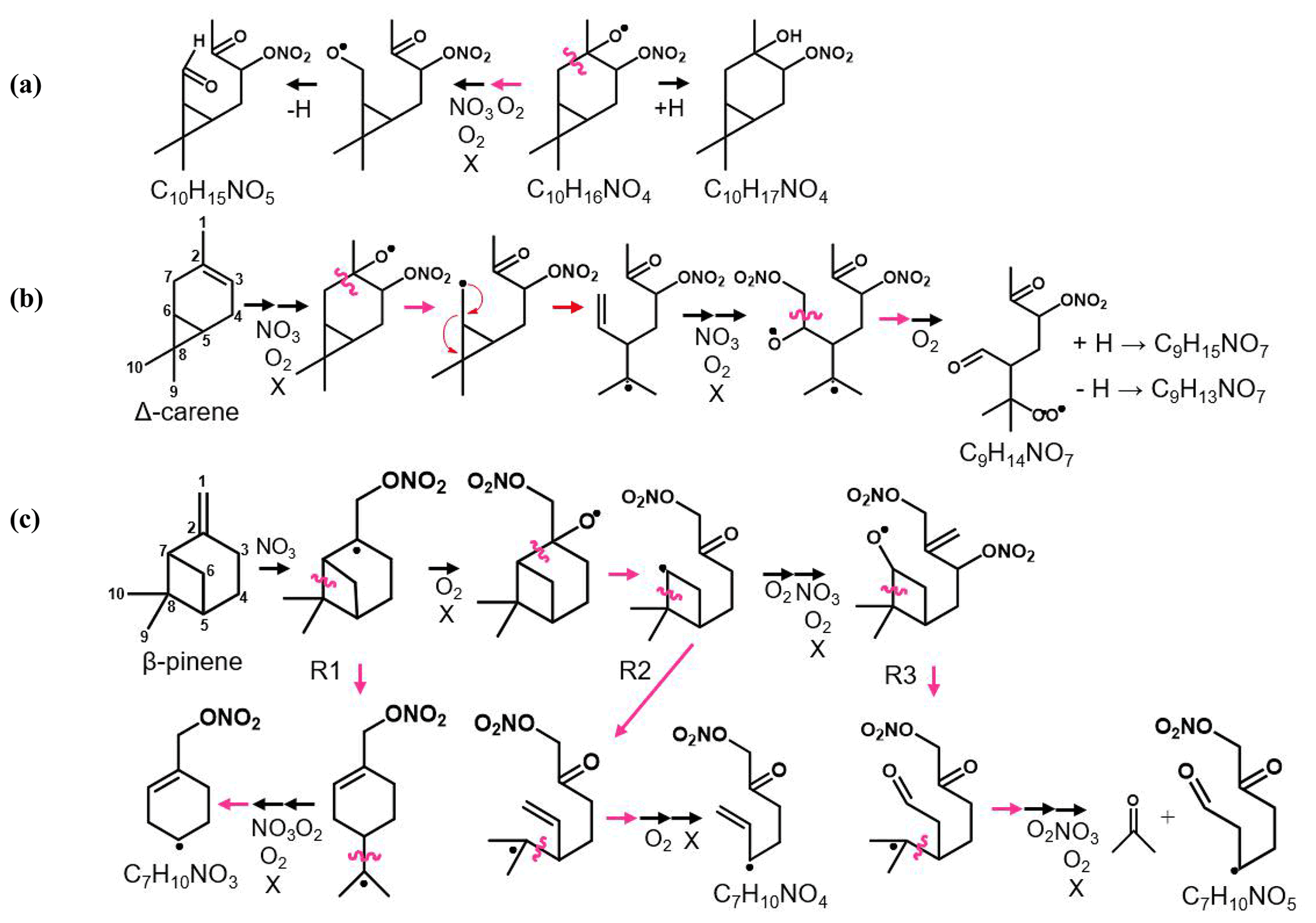
Figure 5Scheme of the mechanistic pathways listed in Table 1. Potential bimolecular reaction partners in a nighttime atmosphere (NO3, RO2, and HO2) are abbreviated as “X”. Panel (a) shows Δ-3-carene oxidation as an example of how the hydrogen number can indicate radical termination. Panel (b) shows a C9 fragment formation pathway available to the Δ-3-carene and α-thujene systems. Panel (c) shows two potential C7 fragment formation pathways available to the β-pinene and α-pinene systems.
3.2.2 Categorized monomer composition and mechanistic implications
The relative abundance of dimers compared with monomers (Fig. 6) correlates with observed particle formation (Sect. S3 in the Supplement) for each MT system, with the exception of α-thujene, for which we observed substantial dimer formation without significant particle formation. We predict that the first alkyl radical formed from NO3 addition onto the α-thujene double bond can potentially rearrange to form a new double bond while opening the strained three-membered ring, forming a six-membered ring and new tertiary alkyl radical (Fig. 1). That structure can then undergo further oxidation to form highly oxygenated, lower-volatility compounds, including dimers. Our observations of highly oxygenated products for α-thujene shows that, under these experimental conditions, there is an additional pathway for the first-generation alkoxy scission (Fig. 1) that is competitive with the formation of thujenaldehyde.
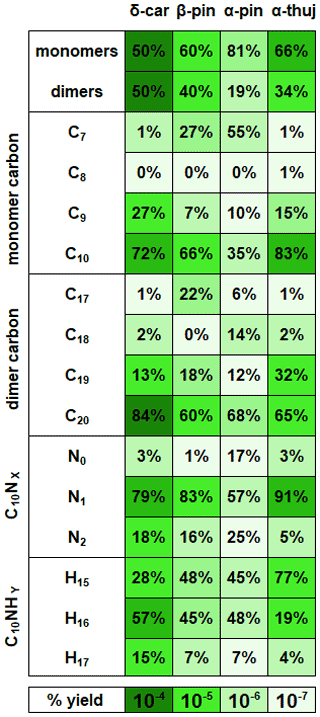
Figure 6Ratios of reaction products separated into categories for each MT system. Each category for each MT system adds up to 100 %. The color axis indicates the magnitude of the total percent yield, which can be compared across the MT systems. β-Pinene percentages are averaged from high-mixing-ratio (70 ppb) and low-mixing-ratio (8 ppb) experiments, but the rest of the MT systems are at the same mixing ratio (41 ppb).
Carbon number
Figure 6 summarizes the monomer carbon number distributions, which provide insights into fragmentation pathways. Detailed schemes for each MT system are shown in Sect. S5 in the Supplement. In general, alkoxy decomposition pathways (C7, C9) are predicted to be competitive with H migration when leaving groups become highly substituted (Novelli et al., 2021; Vereecken and Peeters, 2010). For three of the MT systems studied (Δ-3-carene, β-pinene, and α-thujene), terminal sites that are available in the α position to alkoxy radicals become oxidized and lead to fast alkoxy decomposition (Fig. 5b). For the α-pinene system, however, such a terminal site is not available. Another possible pathway for fragmentation is alkyl radical rearrangement that leads to ring opening. For C7 compounds, radical rearrangement creates new alkyl radicals at tertiary isopropyl sites (Fig. 5c). Oxidation of the alkyl radical site to an RO2 radical and bimolecular reaction to form an RO radical allows alkoxy decomposition to occur, generating a new alkyl radical (Kurtén et al., 2017). For C9 compounds, ring opening and NO3 radical addition creates a new alkyl radical in the α position to a CH3ONO2 group. If the alkyl radical is again oxidized (forming RO2) and decomposed (forming RO), alkoxy scission with an ONO2 group on the α carbon would not have a smaller energy barrier than a site with OH or OOH α substitution, but this barrier would still be smaller than an alkyl-substituted site.
Although Draper et al. (2019) observed a substantial C7 contribution for a Δ-3-carene + NO3 experiment with the same instrument, we observed a negligible C7 contribution. The conditions of the two experiments were different in that the ratio of MT to oxidant was lower in our experiment (0.004 vs. 0.012) and the residence time for our experiment was longer (33 min vs. 23 min). These differences can lead to different chemistry inside the chamber that could result in different observed reaction product distributions. We plan future studies to further explore the source of these differences. A large C7 contribution was observed for both the α-pinene and β-pinene systems. One possible formation mechanism is shown by the R1 arrow in Fig. 5c, in which the initial alkyl radical formed from NO3 addition rearranges to open the four-membered ring (C7–C8 cleavage), forming a new tertiary alkyl radical. This early alkyl radical rearrangement was proposed by Boyd et al. (2015) for the β-pinene + NO3 system. However, O2 addition to the initial alkyl radical is expected to be fast under our experimental conditions and in the ambient atmosphere (Berndt and Böge, 1995; Kurtén et al., 2017), so we expect this alkyl radical rearrangement not to be significant. R2 in Fig. 5 shows an alkyl radical rearrangement that opens the four-membered ring and generates a new tertiary alkyl radical (C7–C8 cleavage). This specific ring-opening reaction is not currently supported by quantum chemical modeling, so we cannot currently comment on its competitiveness. Another possibility is that this C7 compound comes from the first-generation alkoxy scission of the C2–C7 bond (Fig. 5c). While predicted to be a minor pathway for α-pinene, this C2–C7 scission is expected to be equally, if not more, competitive with the other two first-generation alkoxy scission (C2–C1 and C2–C3) for β-pinene (Claflin and Ziemann, 2018). This branching ratio may be somewhat reflected in the relative yields observed from the CIMS. The C7 product yield from the α-pinene system is roughly half that of the β-pinene system. This early left-side C2–C7 scission makes a secondary radical on the four-membered ring, which is less stable than an acyclic analog because of ring strain (Kurtén et al., 2017). R3 in Fig. 5c shows the proposed mechanism for this newly generated compound. In abundant O2, this alkyl radical will be oxidized to an RO2 radical. Because this new RO2 radical is confined by the rigid structure of the ring, autoxidation could be slow, while in the presence of other radical species (NO3, RO2), bimolecular decomposition from a peroxy radical (RO2) to an alkoxy radical (RO) could be faster. Few cyclic RO2 H migration computational studies exist for relevant systems (Kurtén et al., 2015; Xu et al., 2019; Draper et al., 2019), but bimolecular rate constants with NO3, NO2, and HO2 have recently become better defined for RO2 radicals (Jenkin et al., 2019). Once the RO radical is formed, it is possible that decomposition of a C–C bond (thus opening the strained ring and leading to formation of a ketone and a tertiary alkyl radical) could be fast, with a predicted energy barrier of 0.6 kcal mol−1 from SARs (Vereecken and Peeters, 2009, 2010; Novelli et al., 2021). In this position, the tertiary radical resembles the structure predicted for Δ-3-carene in Kurtén et al. (2017) and could lead to the loss of the isopropyl group from C5–C8 cleavage and formation of a C7 fragment. This pathway is only available for the β-pinene and α-pinene systems, and a significant contribution from C7 compounds is only observed from these two systems.
Instead of abundant C7 compounds, we observe a large contribution from C9 compounds for the Δ-3-carene system and smaller C9 signal for the rest of the MT systems. A possible pathway for C9 formation is through the generation of a secondary double bond and subsequent NO3 addition (Fig. 5b). The CH3ONO2 leaving group could quickly fragment into CH3O and NO2 (Novelli et al., 2021; Vereecken and Peeters, 2010) but is assumed to be more stable than a CH3 radical formed from any other terminal site on the molecule in this case. The most abundant C9 compound for the Δ-3-carene system contains two nitrogen atoms (C9H14N2O12). From our predicted pathway, the resulting product may contain one or two nitrogen atoms because, even if the second ONO2 group is expected to be part of the leaving group, a radical site remains on the molecule, making the RO2 + NO2 pathway possible via reaction with residual NO2 in the chamber. For the α-thujene system, a C9 compound is predicted to be formed in an analogous pathway to the Δ-3-carene system, as terminal double bonds may be generated. For the α-pinene system, C9 formation would be possible if an alkyl radical rearrangement occurred on the four-membered ring, generating a terminal double bond. Finally, for the β-pinene system, a terminal site is available after the initial right-side alkoxy scission (C2–C3), but alkyl radical ring opening could also be possible if a left-side scission (C2–C7) were to occur.
Hydrogen number
As previously mentioned, the hydrogen number is an indication of the bimolecular termination pathway and the presence of the subsequent terminal functional group for RO2 and RO radicals. For C10 compounds with a single nitrogen atom (Fig. 5a), H15 closed-shell compounds indicate aldehyde groups created from hydrogen abstraction on a carbon atom in an α position to an alkoxy radical or by other RO2 + RO2 pathways described by Hasan et al. (2020). C10H16NOz compounds are radicals – most likely peroxy radical compounds, as the lifetimes of alkoxy and alkyl radicals are very short. C10H17NOz compounds are closed-shell compounds, and they contain hydroxy or hydroperoxy terminal groups formed from abstracting a hydrogen atom from a different molecule. C10H16NOz radical compounds were the most abundant for the Δ-3-carene, β-pinene, and α-pinene systems. In contrast, C10H15NOz aldehyde-/ketone-containing compounds were the most abundant for the α-thujene system and were mainly distributed among two compounds (C10H15NO9 and C10H15NO10). If one assumes that the concentration of bimolecular reaction partners is similar for each MT system, it appears that the reaction products generated in the α-thujene system included structures with especially labile hydrogen atoms in an α position to possible alkoxy radical sites. In that case, this bimolecular hydrogen abstraction reaction would be fast and would lead to a high ratio of C10H15NOz compounds relative to the C10H16NOz and C10H17NOz compounds. Additionally, a moderate C10H15NOz contribution was observed for the β-pinene system from a species with the same formula (C10H12NO10).
Analogously, C9 radical compounds with a single nitrogen atom will contain 14 hydrogen atoms if they are formed from the loss of a methyl group, whereas closed-shell C9 compounds will contain 13 or 15 hydrogen atoms (Fig. 5b). For the β-pinene system, hydroperoxy or hydroxy C9H15NOz compounds were most abundant. The dominant C9 species in the α-thujene system was a C9H16NOz species; it is not currently clear how C9H16NOz products can be formed. Surprisingly, the most abundant C9 compounds for the rest of the MT systems do not fall into this C9HxNOz category, although some of these types of compounds make up a fraction of the overall C9 signal. C9 dinitrogen compounds were the most abundant for the Δ-3-carene (C9H14N2O12) and α-pinene (C9H14N2O10) systems, with 14 hydrogen atoms indicative of the formation of peroxy radical intermediates or an RO2NO2 group. Finally, C7 radical compounds with a single nitrogen atom are predicted to have 10 hydrogen atoms if formed by the loss of an isopropyl group, whereas closed-shell C7N1 compounds will have 9 or 11 hydrogen atoms (Fig. 5c). The most abundant C7 compound (C7H9NO8), observed for both the β-pinene and α-pinene system, was a closed-shell compound, and its formula is consistent with the mechanism that we present in Fig. 5c, but a large radical contribution from C7H10NO9 was also measured in the β-pinene system.
Nitrogen number
Monomer compounds with a single nitrogen dominate all MT systems, although with varying abundance. The initial addition of NO3 radical leads to the formation of these single nitrogen-containing compounds. The α-pinene system had much higher contributions from N0 and N2 compounds compared with the other MT systems, but it is expected that the majority of products detected for the α-pinene system come from minor pathways; therefore, unusual pathways that are normally considered to be slow can potentially be competitive and lead to the observed ratios of nitrogen-containing compounds. We are not currently certain what those pathways are. The α-thujene system had a 10 % greater contribution from single nitrogen-containing compounds, compared with the Δ-3-carene and β-pinene systems, and a corresponding 10 % smaller contribution from dinitrogen compounds. α-Thujene and Δ-3-carene both contain strained three-membered rings that can make a ring-opening alkyl radical rearrangement reaction faster than the MTs with less strained four-membered rings (β-pinene, α-pinene) (Kurtén et al., 2017). Therefore, it is not possible to know for certain how many nitrogen atoms have been lost by a molecule in the process of oxidation. It is also not possible to attribute a product containing a single nitrogen to the initial NO3 radical addition to the parent α-thujene molecule. This pathway would be available early in the oxidation mechanism for α-thujene, but Δ-3-carene requires one generation of oxidation to pass before the secondary double bond can be generated (Sect. 1). Additionally, the Δ-3-carene system has other pathways that can lead to highly oxidized species whereas the α-thujene system is currently predicted to only have one pathway that leads to secondary double bond generation. Dinitrogen compounds can be formed via NO2 + RO2 (from excess NO2 in the chamber), and this can also form a product with two nitrogen atoms. The rate constant for this NO2 + RO2 reaction is highly uncertain, making explicit kinetic modeling of the RO2 fate challenging.
3.2.3 Categorized dimer composition and mechanistic implications
Carbon number
C20 compounds were the most abundant dimers across all MT systems. However, the β-pinene system had a large (22 %) C17 contribution, whereas the other MT systems produced negligible (< 6 %) C17 dimers. Correspondingly, a large C7 monomer contribution was observed for the β-pinene system but not for the Δ-3-carene or α-thujene systems. C19 dimers were produced in all MT systems, but the α-thujene system had an approximately 20 % greater fractional C19 contribution than β-pinene (10 %) and Δ-3-carene (18 %), even though the Δ-3-carene system produced the highest percentage of C9 monomers. Dimers with carbon numbers other than 20 imply that, after alkoxy decomposition and fragmentation of a C10 monomer, the newly generated monomer fragment must contain an active radical site. This can occur through the generation of a secondary double bond within one of the monomer units through alkyl radical rearrangement. Another possibility is that the smaller leaving group is a closed-shell species, leaving the larger fragment with an alkyl radical. A possible example of this rearrangement is shown in Fig. 5c, in which a C7 radical is generated with acetone as the leaving group in the β-pinene system. That C7 alkyl radical can be oxidized and can possibly form an adduct with a C10 radical, making a C17 dimer that is uniquely observed for the β-pinene system. C19 dimers can potentially be formed through a similar radical rearrangement pathway, as discussed above (Sect. 3.2.2). It is possible to create a C19 dimer from that C9 fragment, as another active radical site could exist on the molecule after NO3 radical addition to the newly generated double bond. An analogous C9 and C19 dimer could be formed from the α-thujene and β-pinene systems.
Hydrogen number
Closed-shell C20 compounds formed from two C10H16 compounds (RO2 + RO2) have 32 hydrogen atoms and were the most abundant type of dimers in all MT systems. If C20 compounds have an alternate hydrogen number, it is assumed that a second double bond was generated at some point during the oxidation process, forming a monomer species with both an active radical site and a terminated site (CHO, OOH, or OH). C19N2H30 compounds make up ∼ 80 % of the total observed C19 signal from the Δ-3-carene system and are predicted to form via the addition of a C10H16NOz radical and a C9H14NOz radical. In contrast, the β-pinene system produced C19H32N2Oz compounds with the highest abundance among C19 species, and the α-thujene system produced both C19H30N2Oz and C19H32N2Oz compounds with equal intensity. As with the C9 monomer units, it is uncertain where the extra two hydrogen atoms are gained. For the β-pinene system, C19H26N2Oz molecules were the most abundant C19 species, which is consistent with a C7H10NOz-RO2 monomer fragment combining with a C10H16NOz-RO2 monomer.
Nitrogen number
For C20 dimers, a nitrogen number of two can correspond to the addition of RO or RO2 radicals with one substituent ONO2 group each. It is expected that products without nitrogen atoms lose them in termination steps. Thus, dimers with two nitrogen atoms cannot be formed by the addition of a dinitrogen monomer and a monomer with no nitrogen atoms unless two radical sites are available on the same molecule and one participates in RO2 + RO2 adduct formation while the other leads to the loss of NO2. This would create a monomer with no nitrogen atoms that is also bonded to another monomer unit. C20 dimers containing two nitrogen atoms are the most abundant across all MT systems. Very small contributions are observed from N3 or N4 C20 species. The α-thujene system has a considerable contribution (13 %) from single nitrogen-containing dimer species. The single nitrogen-containing compounds can possibly be formed by the same combination of monomer units mentioned above for C20N2 compounds, except one of the monomers contains only a single nitrogen atom instead of two. Additionally, it is possible that ozonolysis-RO2 and nitrate-RO2 intermediates engaging in cross-reactions play a role, as the rate constant for α-thujene with ozone or NO3 radical has not been measured.
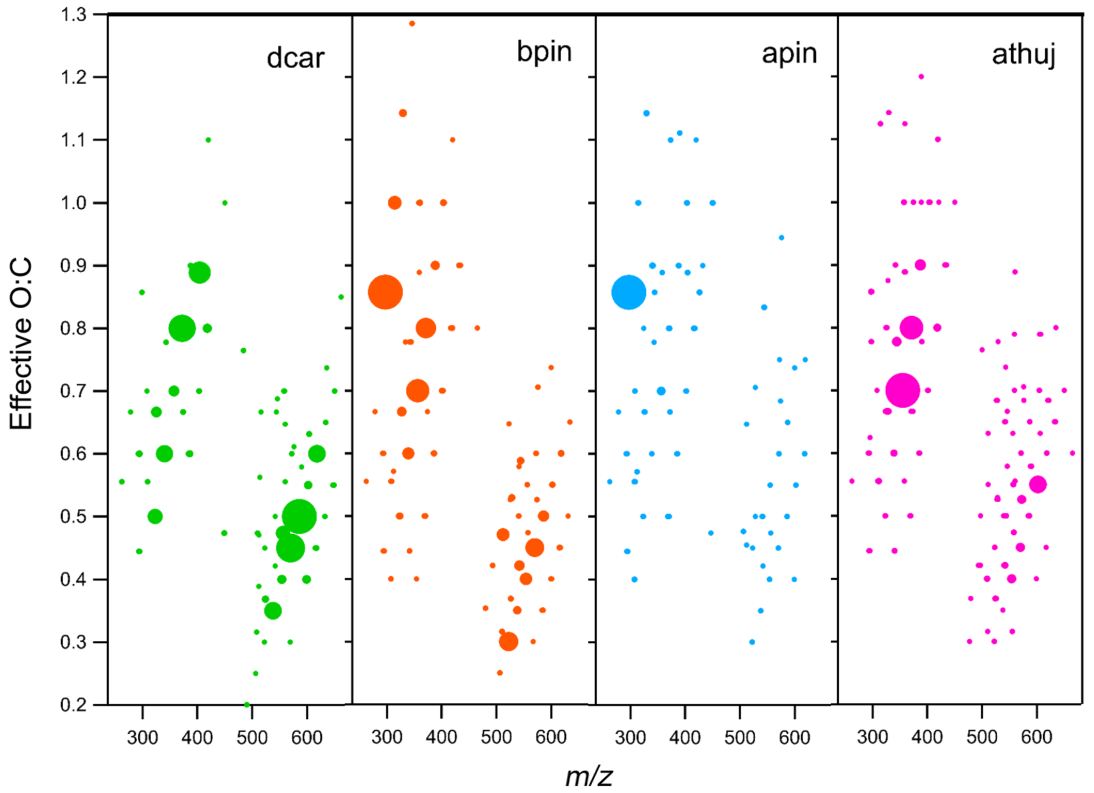
Figure 7Effective O:C ratio plotted vs. for all measured oxidation products. Plots include oxygen atoms only on the carbon backbone. Each marker represents one compound, and the marker area is proportional to the signal intensity. Note that each MT system is scaled to its own maximum intensity; thus, marker sizes cannot be compared across MT systems. The β-pinene plot shown is for the high-mixing-ratio (70 ppb) experiment, and all others are 41 ppb
3.3 Effective O:C ratio of oxidation products
The average, weighted O:C ratios for total detected organic reaction products are as follows: Δ-3-carene, 0.71; β-pinene, 0.62; α-thujene, 0.45; and α-pinene, 0.73. The reported effective O:C ratio for all molecules does not include the oxygen atoms from the nitrate group (two oxygen atoms subtracted for every NO3 group). Even though the average O:C ratio correlates with observed particle formation for every system except α-pinene, the effective O:C ratio distribution is very different for all systems (Fig. 7). Additionally, if products are grouped into monomer and dimer species, the effective O:C ratios do not necessarily correlate with observed particle formation. For monomer species, the average effective O:C ratios were found to have an opposite trend with the intensity of new particle formation events, with the Δ-3-carene reaction products having the lowest effective O:C (0.700), followed by β-pinene (0.724), α-thujene (0.771), and α-pinene (0.780). The dimer products do not show a trend with effective O:C ratios: Δ-3-carene, 0.37; β-pinene, 0.43; α-thujene, 0.37; and α-pinene, 0.50. In general, we observe lower O:C ratios for dimers compared with monomers. If the average O:C for the dimer compounds is around 0.4, they were possibly formed from monomers with an average O:C of 0.6. The average monomer O:C observed from all experiments was 0.7. For the α-thujene system, the majority of the monomer species and the dimer species are centered in a narrow effective O:C range. This is in contrast to the observations from the other MT systems. If the three-membered ring is opened early on in the oxidation mechanism, and the subsequently generated double bond is attacked by an NO3 radical, the molecule will be symmetrical in one plane (Fig. 1). It is possible that this symmetry leads to a lack of diversity of products, reducing the possibility of structurally unique products by half. It is important to note that CIMS is selective towards highly oxidized species, so the oxygen distribution reported here is within the limits of the sensitivity of the reagent ion. Every O:C bin is assumed to be ionized with the same efficiency.
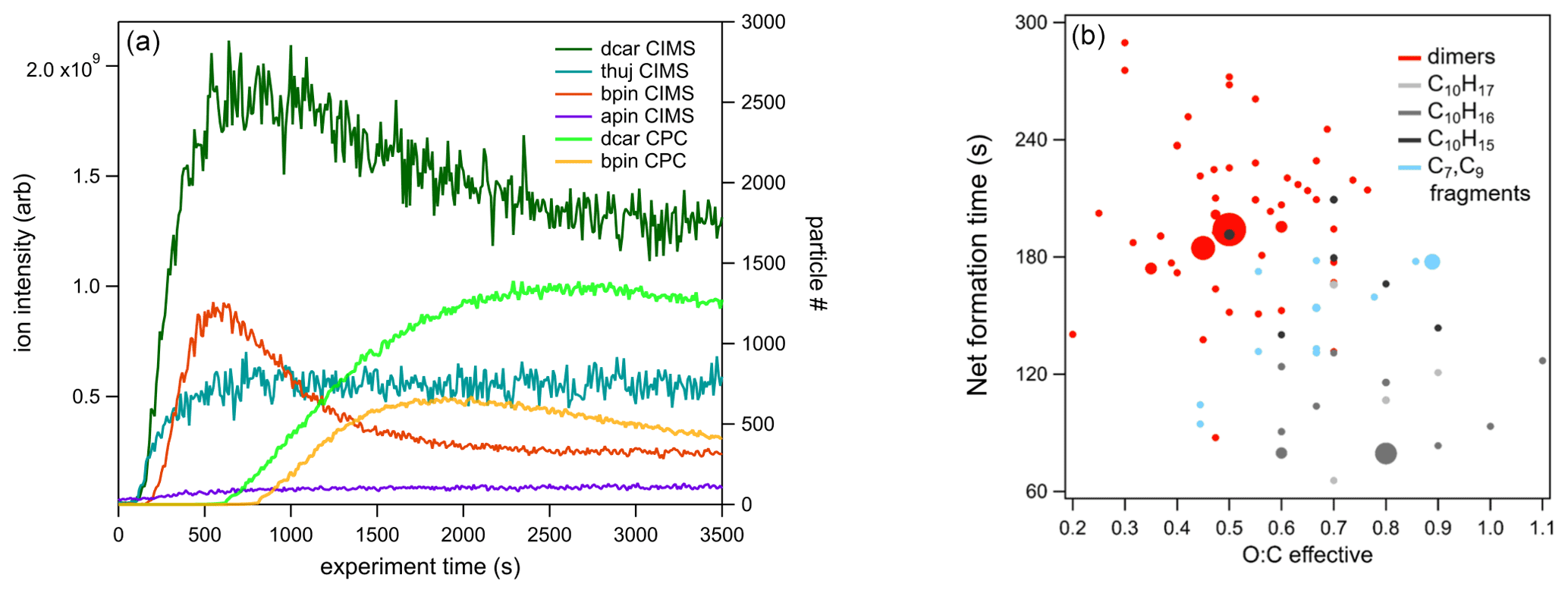
Figure 8(a) Time series of total organic compounds for each MT system measured with CIMS and plotted with the associated particle number concentration. The β-pinene traces shown are from the high-concentration (70 ppb) experiment. (b) The formation : sink ratio of reaction products of the Δ-3-carene system plotted against the effective O:C ratio.
3.3.1 Temporal analysis of oxidation products
When comparing the uncorrected (not corrected for wall losses) time series traces for each MT system, a decrease in product signal after the initial increase was observed for the two systems (Δ-3-carene, β-pinene) that exhibited new particle formation (Fig. 8). These decreases roughly correspond to the increasing particle number. More quantitative analysis of particle growth rates is beyond the scope of the current study but is planned for future studies. The decrease in gas-phase products over the course of the experiment was not observed for the MT systems that did not produce particles (α-pinene and α-thujene).
Different reaction products have different time series because of different formation rates and sinks, as observed in the Δ-3-carene system. At this time, this analysis is only available for the Δ-3-carene system; however, we expect similar behavior for the products of the other MT systems. By grouping the individual species into the categories detailed in Sect. 3.1, insights can be gained into the net formation time (), which considers wall loss but not loss due to the condensation sink on particles. The time series curves were fitted to sigmoidal curves to determine the time it took for the signal to reach one-half the maximum (). In general, monomers were found to have faster net formation time than dimers (Fig. 8). C10H16NOz compounds have the fastest and are the most abundant monomer species. Most of these species are also single nitrogen-containing compounds. Within the N1 category, C10H16NOz compounds and C10H17NOz compounds have similar times, whereas the C10H15NOz species have a slower overall, falling into the same regime as the dimer compounds. If one assumes that these C10 compounds all have a similar condensation sink rate, it appears that H16 radicals and H17 hydroxy and hydroperoxy compounds form faster than the H15 carbonyl compounds. C9 compounds have more variable times but are, in general, slower than the fastest C10 monomers. Additionally, the most abundant C9 species (C9H14N2O12) has an value in the dimer region (note that this compound contains two nitrogen atoms). Additionally, for C10 compounds with one nitrogen atom, there is an almost imperceptible increasing trend relating and the effective O:C ratio (Fig. 8b). A similar slight trend can be observed for C20 compounds. It is possible that, because the formation and sink times cannot be isolated, more highly oxidized molecules take longer to form but also condense more rapidly.
CIMS has been used to probe the composition of NO3-MT oxidation products in laboratory chamber experiments in order to gain mechanistic insights. The major detected species formed in each system were distinctly different, showing the effect of MT structure on the oxidation mechanism. We initially hypothesized that the structural similarities between α-thujene and α-pinene would lead to the dominance of the relatively high-volatility oxidation products thujenaldehyde and pinonaldehyde, respectively. Our results, however, suggest that an alkyl radical rearrangement can lead to an intermediate that can undergo additional oxidation and form highly oxygenated molecule (HOM) monomers and dimers (Fig. 1) in α-thujene oxidation. The lack of measurable new particle formation in spite of the presence of these dimers indicates a more complex relationship between HOMs and new particle formation. This should be studied in greater detail to provide insights into the ability of HOMs to participate in nanoparticle formation and growth.
For all systems, the HOM carbon number, an indicator of fragmentation pathways, supports the notion that decomposition is more likely when leaving groups become highly substituted. The presence of substantial amounts of C7 fragments for the β-pinene and α-pinene systems is consistent with the loss of an isopropyl group from those species, and we have hypothesized the mechanism by which this occurs (Fig. 5c). The hydrogen numbers for C10N1 compounds, an indicator of termination pathways and the presence of closed-shell or radical intermediates, show the dominance of peroxy radical H16 compounds for all but the α-thujene system – the latter of which was dominated by closed-shell H15 aldehydes or ketones. For C9N products, closed-shell H15 hydroperoxy or hydroxy compounds dominated the β-pinene system, and H16 species dominate the α-thujene system by a mechanism that is unclear to us. For the α-pinene and Δ-3-carene systems, the dominant C9 compounds detected were species containing 2 nitrogen atoms and 14 hydrogen atoms. The dominant C7 fragment observed for α-pinene and β-pinene was a closed-shell C7H9NO8 compound with possible isomers, again consistent with the mechanism we propose in Fig. 5c. The nitrogen number for all monomers was dominated by those containing a single nitrogen atom, which arises from the initial NO3 radical addition. Some later-generation monomeric dinitrogen compounds were detected in all systems with the exception of α-thujene – the latter of which has fewer pathways for the formation of a second double bond. This limitation may be partly responsible for the lack of observed new particle formation for α-thujene despite the abundance of HOMs in that system.
The observed dimers included major peaks containing 20, 19, and 17 carbon atoms, which is consistent with the observed monomers containing 10, 9, and 7 carbon atoms. The hydrogen numbers for all systems indicate that C20 dimers form predominantly closed-shell compounds with 32 hydrogen atoms. In general, our observations of the hydrogen and nitrogen number in detected dimers are consistent with the composition of detected monomers, which suggests dimer formation by cross-reactions between nitrate-containing RO2 species.
Detected O:C ratios of gas-phase products provide some insights into new particle formation mechanisms. In general, monomer O:C ratios share a very small trend with new particle formation intensity. It is possible that monomers with higher O:C ratios are preferentially partitioning into growing nanoparticles, and, indeed, we observe a decrease in HOMs coincident with an increase in the concentration of newly formed particles, as discussed in Sect. 3.3.1.
Finally, our temporal analysis of oxidation products from the Δ-3-carene system shows unique, species-dependent formation rates and provide insights into wall loss rates. In general, dimers formed more slowly than monomers. As dimers had lower O:C ratios, there was a weak anticorrelation between O:C ratio and the net formation time. This correlation is not apparent for monomers, but C10 monomers did display some trends such as H16 radicals and H17 hydroxy and hydroperoxy compounds forming faster than H15 carbonyl compounds. Additional applications of this temporal analysis approach for the other MT systems would be an interesting extension of this work.
The information gained from this detailed comparison of gas-phase composition with currently established mechanisms provides new information on these oxidation processes and further elucidates the effect of these species on particle formation and growth. A wider range of oxidation products (semi-volatile organic compounds) need to be measured to observe the compounds not detected by CIMS in order to more comprehensively draw conclusions about particle formation potential. Further analysis of the particle formation rate, particle composition, and modeling of energy barriers for some of the proposed mechanistic pathways is needed. Additional spectroscopy can also be useful for confirming the presence of functional groups.
KinSim software (version 4) runs on the Igor Pro programming environment and is freely available from the following web portal: http://tinyurl.com/kinsim-release (last access: 27 June 2022; Peng and Jimenez, 2022).
Data are publicly available and are archived at https://doi.org/10.7280/D1XX2R (Smith, 2022).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-9017-2022-supplement.
MD and JNS conducted chamber experiments and curated, analyzed, investigated, and visualized data from all measurements. DCD, AM, and JLF helped design chamber studies and conducted the TD-CRDS analysis and kinetic modeling. MD prepared the manuscript with contributions from DCD, AM, JLF, and JNS. JNS and JLF acquired funding and supervised the project.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research was supported by funding from the US National Science Foundation (NSF; grant no. AGS-1762098) and from the US Department of Energy (DOE; grant no. DE-SC0019000). The authors would like to acknowledge Emily McLaughlin Santa Maria, Mike Lawler, Sabrina Chee, Hayley Glicker, Deanna Myers, Adam Thomas, Jeremy Wakeen, and Paulus Bauer for their contributions to discussions regarding this project.
This research has been supported by the National Science Foundation (grant no. AGS-1762098) and the US Department of Energy (grant no. DE-SC0019000).
This paper was edited by Arthur Chan and reviewed by two anonymous referees.
Acosta Navarro, J. C., Smolander, S., Struthers, H., Zorita, E., Ekman, A. M. L., Kaplan, J. O., Guenther, A., Arneth, A., and Riipinen, I.: Global emissions of terpenoid VOCs from terrestrial vegetation in the last millennium, J. Geophys. Res.-Atmos., 119, 6867–6885, https://doi.org/10.1002/2013JD021238, 2014. a
Aljawhary, D., Lee, A. K. Y., and Abbatt, J. P. D.: High-resolution chemical ionization mass spectrometry (ToF-CIMS): application to study SOA composition and processing, Atmos. Meas. Tech., 6, 3211–3224, https://doi.org/10.5194/amt-6-3211-2013, 2013. a
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, Special Issue: The 1997 Southern California Ozone Study (SCOS97-NARSTO). Dedicated to the Memory of Dr. Glen Cass (1947–2001), 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003. a, b
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015. a, b
Berndt, T. and Böge, O.: Products and mechanism of the reaction of NO3 with selected acyclic monoalkenes, J. Atmos. Chem., 21, 275–291, https://doi.org/10.1007/BF00696759, 1995. a, b
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015. a
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kumala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015. a, b
Brophy, P. and Farmer, D. K.: A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: characterization from the 2013 southern oxidant and aerosol study, Atmos. Meas. Tech., 8, 2945–2959, https://doi.org/10.5194/amt-8-2945-2015, 2015. a
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012. a
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018. a
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a
DeHaan, D. O., Brauers, T., Oum, K., Stutz, J., Nordmeyer, T., and Finlayson-Pitts, B. J.: Heterogeneous chemistry in the troposphere: Experimental approaches and applications to the chemistry of sea salt particles, Int. Rev. Phys. Chem., 18, 343–385, https://doi.org/10.1080/014423599229910, 1999. a
Draper, D. C., Myllys, N., Hyttinen, N., Møller, K. H., Kjaergaard, H. G., Fry, J. L., Smith, J. N., and Kurtén, T.: Formation of Highly Oxidized Molecules from NO3 Radical Initiated Oxidation of δ-3-Carene: A Mechanistic Study, ACS Earth and Space Chemistry, 3, 1460–1470, https://doi.org/10.1021/acsearthspacechem.9b00143, 2019. a, b, c, d, e
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014. a, b
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013. a
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014. a
Hallquist, M., Wangberg, I., Ljungstrom, E., Barnes, I., and Becker, K.: Aerosol and Product Yields from NO3 radical-Initiated Oxidation of Selected Monoterpenes, Environ. Sci. Technol., 33, 553–559, https://doi.org/10.1021/es980292s, 1999. a
Hasan, G., Salo, V.-T., Valiev, R. R., Kubečka, J., and Kurtén, T.: Comparing Reaction Routes for 3(RO…OR′) Intermediates Formed in Peroxy Radical Self- and Cross-Reactions, J. Phys. Chem. A, 124, 8305–8320, https://doi.org/10.1021/acs.jpca.0c05960, 2020. a
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015. a
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019. a, b
Keehan, N. I., Brownwood, B., Marsavin, A., Day, D. A., and Fry, J. L.: A thermal-dissociation–cavity ring-down spectrometer (TD-CRDS) for the detection of organic nitrates in gas and particle phases, Atmos. Meas. Tech., 13, 6255–6269, https://doi.org/10.5194/amt-13-6255-2020, 2020. a, b
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008. a
Kurtén, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jørgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in α-Pinene Ozonolysis Products, J. Phys. Chem. A, 119, 11366–11375, https://doi.org/10.1021/acs.jpca.5b08948, 2015. a
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene Autoxidation Products May Not Have Extremely Low Saturation Vapor Pressures Despite High O:C Ratios, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016. a
Kurtén, T., Møller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields from α-Pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, https://doi.org/10.1021/acs.jpclett.7b01038, 2017. a, b, c, d, e, f, g, h
Lee, A., Goldstein, A. H., Keywood, M. D., Varutbangkul, V., Bahreini, R., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res.-Atmos., 111, D7, https://doi.org/10.1029/2005JD006437, 2006. a
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014. a
Li, X., Chee, S., Hao, J., Abbatt, J. P. D., Jiang, J., and Smith, J. N.: Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 19, 1555–1570, https://doi.org/10.5194/acp-19-1555-2019, 2019. a
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016. a
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015. a
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017. a, b, c, d
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/D0CP05555G, 2021. a, b, c, d
Peng, Z. and Jimenez, J. L.: KinSim: A Research-Grade, User-Friendly, Visual Kinetics Simulator for Chemical-Kinetics and Environmental-Chemistry Teaching, J. Chem. Educ., 96, 806–811, https://doi.org/10.1021/acs.jchemed.9b00033, 2019. a
Peng, Z. and Jimenez, J. L.: KinSim, http://tinyurl.com/kinsim-release, last access: 27 June 2022. a
Rissanen, M., Kurtén, T., Sipilä, M., Thornton, J., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M., Taipale, R., Springer, M., Mentel, T., Ruuskanen, T., Petäjä, T., Worsnop, D., Kjaergaard, H., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014. a
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019. a
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014. a, b
Smith, J.: Supplemental Data for “Observations of gas-phase products from the nitrate radical-initiated oxidation of four monoterpenes”, Dryad [data set], https://doi.org/10.7280/D1XX2R, 2022. a
Stocker, T., Qin, D., Plattner, M., Tignor, S., Allen, J., Boschung, A., Nauels, Y., Xia, V., Midgley, B., and P. M.: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Geneva, Switzerland, p. 1535, 2013. a
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – Part I: A generalized structure-activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/b909712k, 2009. a, b, c, d
Vereecken, L. and Peeters, J.: A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/c0cp00387e, 2010. a, b, c, d, e
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of α-Pinene and β-Pinene by Hydroxyl Radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019. a