the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Feb 2022
| 01 Feb 2022
Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing
Jesse H. Kroll
Siyuan Wang
Duseong S. Jo
Andrew Gettelman
Zheng Lu
Xiaohong Liu
Rahul A. Zaveri
Eric C. Apel
Donald R. Blake
Jose-Luis Jimenez
Pedro Campuzano-Jost
Patrick R. Veres
Timothy S. Bates
John E. Shilling
Maria Zawadowicz
Aerosol indirect radiative forcing (IRF), which characterizes how aerosols alter cloud formation and properties, is very sensitive to the preindustrial (PI) aerosol burden. Dimethyl sulfide (DMS), emitted from the ocean, is a dominant natural precursor of non-sea-salt sulfate in the PI and pristine present-day (PD) atmospheres. Here we revisit the atmospheric oxidation chemistry of DMS, particularly under pristine conditions, and its impact on aerosol IRF. Based on previous laboratory studies, we expand the simplified DMS oxidation scheme used in the Community Atmospheric Model version 6 with chemistry (CAM6-chem) to capture the OH-addition pathway and the H-abstraction pathway and the associated isomerization branch. These additional oxidation channels of DMS produce several stable intermediate compounds, e.g., methanesulfonic acid (MSA) and hydroperoxymethyl thioformate (HPMTF), delay the formation of sulfate, and, hence, alter the spatial distribution of sulfate aerosol and radiative impacts. The expanded scheme improves the agreement between modeled and observed concentrations of DMS, MSA, HPMTF, and sulfate over most marine regions, based on the NASA Atmospheric Tomography (ATom), the Aerosol and Cloud Experiments in the Eastern North Atlantic (ACE-ENA), and the Variability of the American Monsoon Systems (VAMOS) Ocean-Cloud-Atmosphere-Land Study Regional Experiment (VOCALS-REx) measurements. We find that the global HPMTF burden and the burden of sulfate produced from DMS oxidation are relatively insensitive to the assumed isomerization rate, but the burden of HPMTF is very sensitive to a potential additional cloud loss. We find that global sulfate burden under PI and PD emissions increase to 412 Gg S (+29 %) and 582 Gg S (+8.8 %), respectively, compared to the standard simplified DMS oxidation scheme. The resulting annual mean global PD direct radiative effect of DMS-derived sulfate alone is −0.11 W m−2. The enhanced PI sulfate produced via the gas-phase chemistry updates alone dampens the aerosol IRF as anticipated (−2.2 W m−2 in standard versus −1.7 W m−2, with updated gas-phase chemistry). However, high clouds in the tropics and low clouds in the Southern Ocean appear particularly sensitive to the additional aqueous-phase pathways, counteracting this change (−2.3 W m−2). This study confirms the sensitivity of aerosol IRF to the PI aerosol loading and the need to better understand the processes controlling aerosol formation in the PI atmosphere and the cloud response to these changes.
- Article
(6769 KB) - Full-text XML
-
Supplement
(4207 KB) - BibTeX
- EndNote




The IPCC AR5 (Fifth Assessment Report of the United Nations Intergovernmental Panel on Climate Change; Myhre et al., 2013) and the recent preliminary release of AR6 (Sixth Assessment Report; https://www.ipcc.ch/report/sixth-assessment-report-cycle/, last access: 30 November 2021) indicate that atmospheric aerosol particles are a dominant source of uncertainty in global climate forcing. Aerosols interact with incoming and outgoing radiation directly (via scattering and absorption) and indirectly (via changing cloud properties and lifetime). In particular, the aerosol indirect radiative forcing (IRF) via interactions with clouds is driven by the fractional enhancement of aerosol burden from a preindustrial (PI; 1850) atmosphere to a present-day (PD; 2000) one, with a cleaner PI atmosphere producing a larger IRF (Menon et al., 2002). Carslaw et al. (2013) confirm that the estimated uncertainty in aerosol IRF is dominated by uncertainty in natural aerosols. It is, therefore, critically important to accurately determine the formation of natural aerosols and their radiative impacts in both PD and PI atmosphere.
Marine dimethyl sulfide (DMS; CH3SCH3) accounts for > 50 % of natural gas-phase sulfur emissions (Chin et al., 1996; Andreae, 1990; Kilgour et al., 2021). Once emitted into the troposphere, oxidation of DMS takes place within 1–2 d, forming other sulfur-containing products, such as sulfuric acid (H2SO4) and methane sulfonic acid (MSA; CH3SO3H; Boucher et al., 2003; Breider et al., 2010). These gaseous products can facilitate the formation of new particles and cloud condensation nuclei (CCN), especially in the marine boundary layer (MBL; Charlson et al., 1987; von Glasow and Crutzen, 2004; Kulmala et al., 2000). Sulfate and MSA formed in the particle phase can directly impact the size distribution of aerosols and alter cloud microphysics (Kaufman and Tanré, 1994). DMS is estimated to be responsible for up to 11 %–18 % of global sulfate burden in PD (Yang et al., 2017; Gondwe et al., 2003) and > 48 % of atmospheric sulfur burden in PI (Tilmes et al., 2019). Though the crucial role of DMS oxidation as a source of natural aerosols has been acknowledged for decades, its oxidation mechanisms are still not well understood (Barnes et al., 2006; Hoffmann et al., 2016).
Global/regional models often simplify the DMS oxidation processes for the sake of computational costs. For example, the Community Atmosphere Model with chemistry (CAM-chem) includes only the oxidation of DMS by OH and NO3 radicals, directly producing SO2, which further oxidizes to produce sulfate (Emmons et al., 2020; Lamarque et al., 2012). This simplification ignores some potentially important reaction intermediates and pathways. For instance, previous studies suggest that BrO contributes up to 30 % of the DMS sink in the remote MBL (Boucher et al., 2003; Breider et al., 2010; von Glasow and Crutzen, 2004; Khan et al., 2016). MSA has been found to form efficiently via the multiphase OH-addition DMS oxidation pathway followed by a reaction with OH(aq) to form sulfate aerosol in the MBL (von Glasow and Crutzen, 2004; Milne et al., 1989; Zhu et al., 2006). Recently, both theoretical and laboratory studies have proposed that a pristine environment favors the H-abstraction reaction when DMS is oxidized by OH, generating methylthiomethylperoxy radicals (MSP; CH3SCH2OO), which further undergo a series of rapid intramolecular H-shift isomerization reactions, yielding a stable intermediate hydroperoxymethyl thioformate (HPMTF; HOOCH2SCHO; Wu et al., 2015; Berndt et al., 2019). Recent in situ measurements report HPMTF concentrations that are, on average, 50 % of DMS concentrations in the MBL during the day, but can exceed DMS concentrations at times, confirming the importance of the isomerization branch for capturing the fate of oxidized DMS (Veres et al., 2020; Vermeuel et al., 2020).
The lifetimes of stable intermediates from DMS oxidation can be up to days. As a result, these intermediates can delay the formation of DMS-derived sulfate, affecting not only the spatial distribution of sulfate aerosols but also the effective sulfate yield from DMS as unreacted sulfate precursors may be subject to physical removal through wet or dry deposition. Thus, neglecting these intermediates could lead to misrepresentation of the spatial distribution of sulfate aerosol loading and limit our ability to accurately determine aerosol radiative forcing.
Here, we implement a more detailed multigenerational and multiphase chemical mechanism to describe DMS oxidation within the Community Atmosphere Model version 6 with chemistry (CAM6-chem; Emmons et al., 2020) – the atmosphere component of the Community Earth System Model version 2.1 (CESM2.1; Danabasoglu et al., 2020). The expanded chemistry captures the formation of stable intermediates such as MSA and HPMTF alongside SO2. We perform multiple sensitivity tests to investigate how the uncertainty in modeling the newly confirmed HPMTF could influence the DMS chemistry and the resulting atmospheric sulfate burden. The model results are compared against an array of in situ observations. Finally, we examine how the natural aerosol background from DMS oxidation simulated with the modified model impacts estimates of aerosol radiative forcing.
CESM2.1 consists of model components that quantitatively describe the atmosphere, land, sea ice, land ice, rivers, and ocean (Danabasoglu et al., 2020). Fluxes and state variables are exchanged through a coupler to describe the co-evolution of these Earth system components. Here, we run with a coupled atmosphere (CAM6-chem) and land (Community Land Model – CLM5) model and use prescribed data for the remaining Earth system components. In particular, sea surface temperature (SST) and sea ice conditions (Hurrell et al., 2008), as well as the mixing ratios of greenhouse gases (Meinshausen et al., 2017), are all fixed to present-day conditions. Following similar practices in previous studies (Gettelman, 2015; Gettelman et al., 2019), this configuration aims to constrain the potential environmental feedbacks, such that the aerosol effects (on atmospheric composition, cloud, and radiation) are due to the change in emissions and chemistry only.
2.1 Model configuration
In this work, CAM6-chem is run in an online configuration with free dynamics at 1.9∘ × 2.5∘ (latitude by longitude) horizontal resolution and 32 vertical layers (surface to 3 hPa or ∼ 45 km), with a model time step of 30 min. The default chemistry scheme is the Model for Ozone and Related Chemical Tracers with representations of both tropospheric and stratospheric chemistry (MOZART-TS1; Emmons et al., 2020), with a volatility basis set (VBS) scheme specifically for the gas-phase intermediate semi-volatile organic precursors of secondary organic aerosols (SOAs; Tilmes et al., 2019). The DMS chemistry is described in further detail in Sect. 2.3. Aerosols are simulated using the Modal Aerosol Model with four modes (MAM4; Liu et al., 2016) coupled with the Model for Simulating Aerosol Interactions and Chemistry (MOSAIC; Zaveri et al., 2008, 2021; Lu et al., 2021), for sulfate (SO), ammonium (NH), nitrate (NO), primary organic matter (POM), SOA, sea salt, and mineral dust. MAM4 classifies aerosols into three size-dependent modes (Aitken, accumulation, and coarse), with an additional primary carbon mode for handling the aging of fine POM and black carbon (BC). Size distributions of aerosols in each mode are assumed to be lognormal, with fixed geometric standard deviations and a varying mode dry or wet radius, depending on the particle number and changes in total dry or wet volume (Liu et al., 2012). MAM4 defines the cut-off size ranges of 0.015–0.053 µm for aerosol in the Aitken mode, 0.058–0.27 µm for the accumulation mode, and 0.80–3.65 µm for the coarse mode. Dynamic partitioning of H2SO4, HNO3, HCl, and NH3 to each mode, related particle-phase thermodynamics, and water content and pH of interstitial aerosols are computed using the MOSAIC module (Zaveri et al., 2008, 2021; Lu et al., 2021). Further details describing the dry and wet deposition, aerosol optical properties, radiative transfer, and aerosol–cloud microphysics are described in the Supplement; these processes are all based on the standard CAM6-chem.

We perform four sets of simulations for the PD and PI atmospheric conditions with the standard and our modified chemical schemes. Details of the runs are tabulated in Table 1. Each run is performed for 10 years, with the first year as spin-up, and the averages over the latter 9 years are presented in our results.
2.2 Emissions
DMS emissions from the ocean (EDMS) are simulated via the Online Air–Sea Interface for Soluble Species (OASISS) model developed for CAM6-chem, which has been validated against observations for acetaldehyde (S. Wang et al., 2019a), acetone (Wang et al., 2020), and organohalogens (e.g., CHBr3 and CH2Br2; S. Wang et al., 2019b). OASISS employs a two-layer framework that considers transfer velocities both through air and through water (kair and kwater) as follows (Johnson, 2010):
where k (meters per second; hereafter m s−1) is the overall transfer velocity. The surface seawater concentration, [DMS]water (nanomolar; hereafter nM) is prescribed to follow the Lana et al. (2011) sea surface DMS climatology in both our PI and PD simulations. DMS mixing ratio in the air, [DMS]air, and its Henry's law constant are from CAM6-chem. kair is based on the NOAA COARE algorithm (Jeffery et al., 2010), which is a function of surface wind speed, with an additional adjustment for the diffusivity of still air (Mackay and Yeun, 1983). kwater is based on Nightingale et al. (2000), which considers sea surface temperature and salinity. Last, fcice is the fraction of sea ice coverage in each grid cell, such that DMS emission is suppressed from sea-ice-covered surfaces.
On average, the global annual total marine EDMS is 21.5 Tg S yr−1 in [STD_2000]. Meteorological variability has little impact on the interannual variability of emissions (4 %). Our EDMS is higher than the 18 Tg S yr−1 from the model default inventory (Kettle and Andreae, 2000) but lower than the 28 Tg S yr−1 reported in the original model study by Lana et al. (2011) and within the range of 11–28 Tg S yr−1 simulated by Goddard Earth Observing System (GEOS)-Chem, TOMCAT-GLOMAP software, and other models (Lennartz et al., 2015; Spracklen et al., 2005; Hezel et al., 2011). The estimation of EDMS is sensitive to the choice of sea surface DMS climatology; Chen et al. (2018) show that emissions vary from 18 Tg S yr−1, with the Kettle et al. (1999) DMS climatology, to 22 Tg S yr−1, with the Lana et al. (2011) DMS climatology.
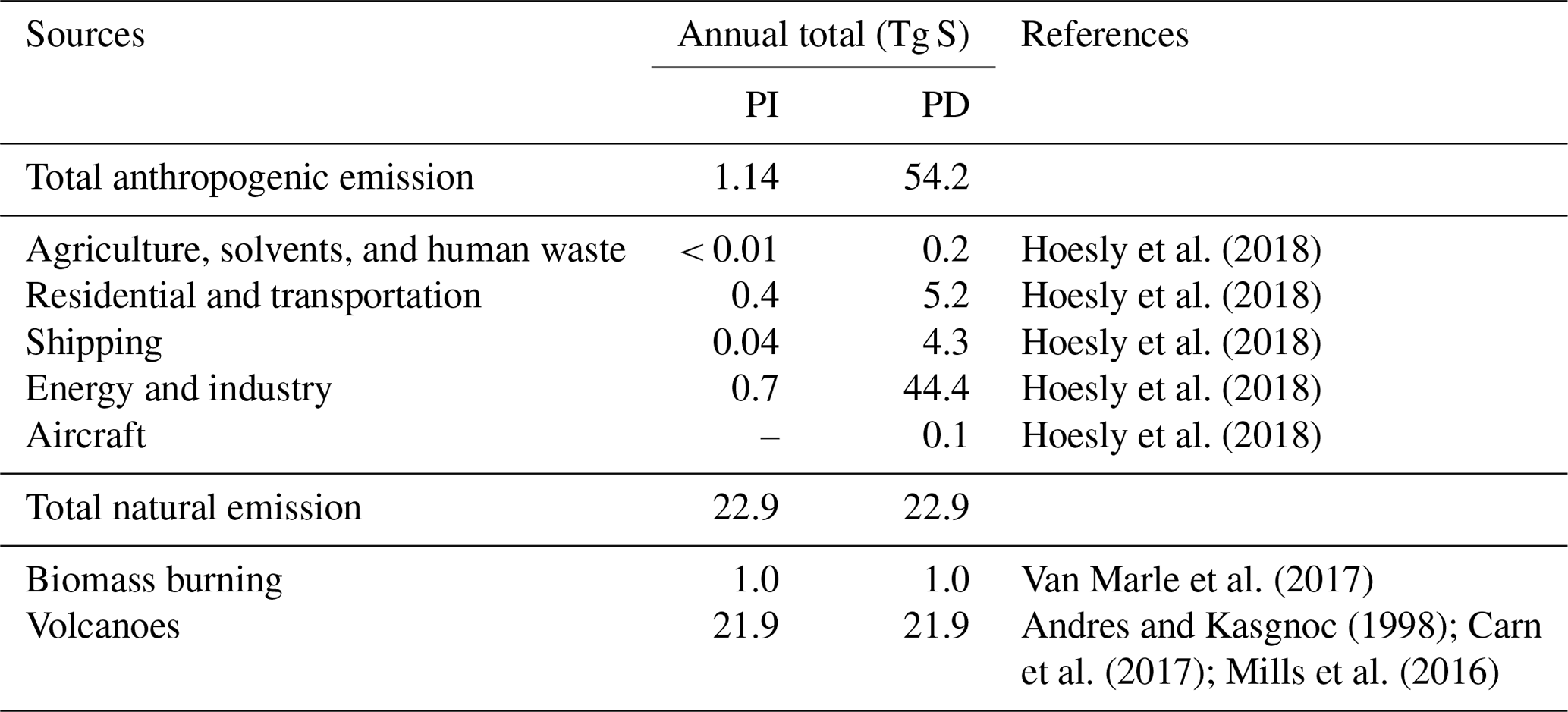
In all simulations, anthropogenic emissions are from the Community Emissions Data System (CEDS; Hoesly et al., 2018), and biomass burning emissions are from the CMIP6 inventory (van Marle et al., 2017). Biogenic emissions are estimated online from CLM5, using the Model of Emissions of Gases and Aerosols from Nature (MEGAN) version 2.1 (Guenther et al., 2012). CAM6-chem assumes that 2.5 % by molar of sulfur emitted from the energy and industry sector is already in the form of primary sulfate aerosols (in accumulation mode). Volcanic emissions are fixed at the same level in both PI and PD simulations. Emissions from continuously outgassing volcanoes are constant (97.5 % as SO2 and 2.5 % emitted as primary sulfate aerosols) based on the GEIA (Global Emissions InitiAtive) inventory (Andres and Kasgnoc, 1998). We use time-averaged (1995–2005) eruptive volcanic emissions of SO2 to impose an average forcing from volcanic eruptions reaching the stratosphere, derived from the database of Volcanic Emissions for Earth System Models (VolcanEESM), version 3.10 (Neely and Schmidt, 2016). SO2 emissions from aircraft (up to ∼ 15 km) and SO2 and primary sulfate emissions from volcanoes (up to ∼ 30 km) are considered as elevated emissions, while other sources of SO2 emissions and oceanic DMS emissions are at the surface. A breakdown of SO2 emissions in this study is summarized in Table 2. We use the same emissions for other species as those in the standard CMIP6 simulations (Emmons et al., 2020).
2.3 Expanded DMS oxidation scheme
The standard CAM6-chem contains three gas-phase DMS oxidation reactions (Table 3; Barth et al., 2000; Emmons et al., 2010). These reactions simplify the DMS oxidation chemistry by treating only gas-phase reactions and producing SO2 directly, neglecting the role of multiphase chemistry and other key chemical products and intermediates found in chamber and field studies (e.g., Hoffmann et al., 2016; Wu et al., 2015). We note that the second reaction does not conserve sulfur.
Table 3The three DMS oxidation reactions in the standard CAM6-chem.

∗ (cm3 molec.−1 s−1).
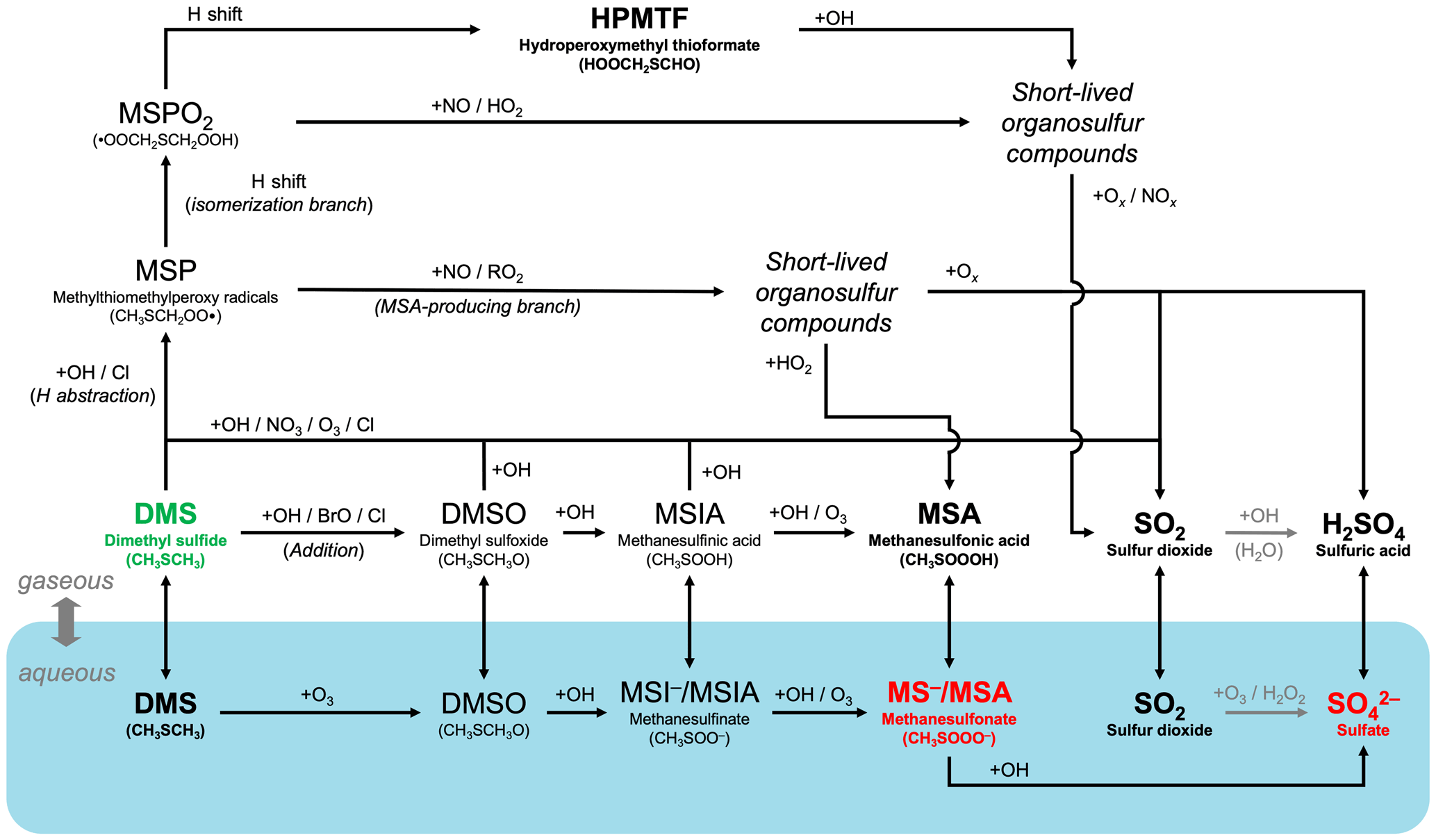
Figure 1A schematic summary of our expanded atmospheric chemistry of DMS oxidation in CAM6-chem (Tables 4, 5, 6, and 7). Key relatively long-lived species (DMS, MSA, HPMTF, SO2, and sulfate), with lifetimes of > 0.5 d, are highlighted in bold. The blue shadings denote species and reactions in the aqueous phase in interstitial aerosols and cloud droplets. DMS (highlighted in green) can undergo O-atom addition (rightward path) or H abstraction (upper paths). The H-abstraction channel further diverts into the isomerization branch (top path) and the MSA-producing branch. SO2 is the dominant product of most gas-phase pathways, while MSA is formed mainly via the aqueous-phase oxidation of DMS. Oxidation of SO2 to sulfate or sulfuric acid is handled by the CAM6-chem standard chemistry. The resultant particulate MSA and sulfate (highlighted in red) are key species with important radiative impacts.
To improve the representation of DMS oxidation in CAM6-chem, we add a suite of new reactions that describe the chemical evolution from DMS to SO2 and, ultimately, sulfate via the H-abstraction and OH-addition pathways. Fig. 1 illustrates the expanded chemistry schematically. Our additions are based on recent laboratory studies and field observations and are discussed in detail in what follows.
2.3.1 The H-abstraction pathway
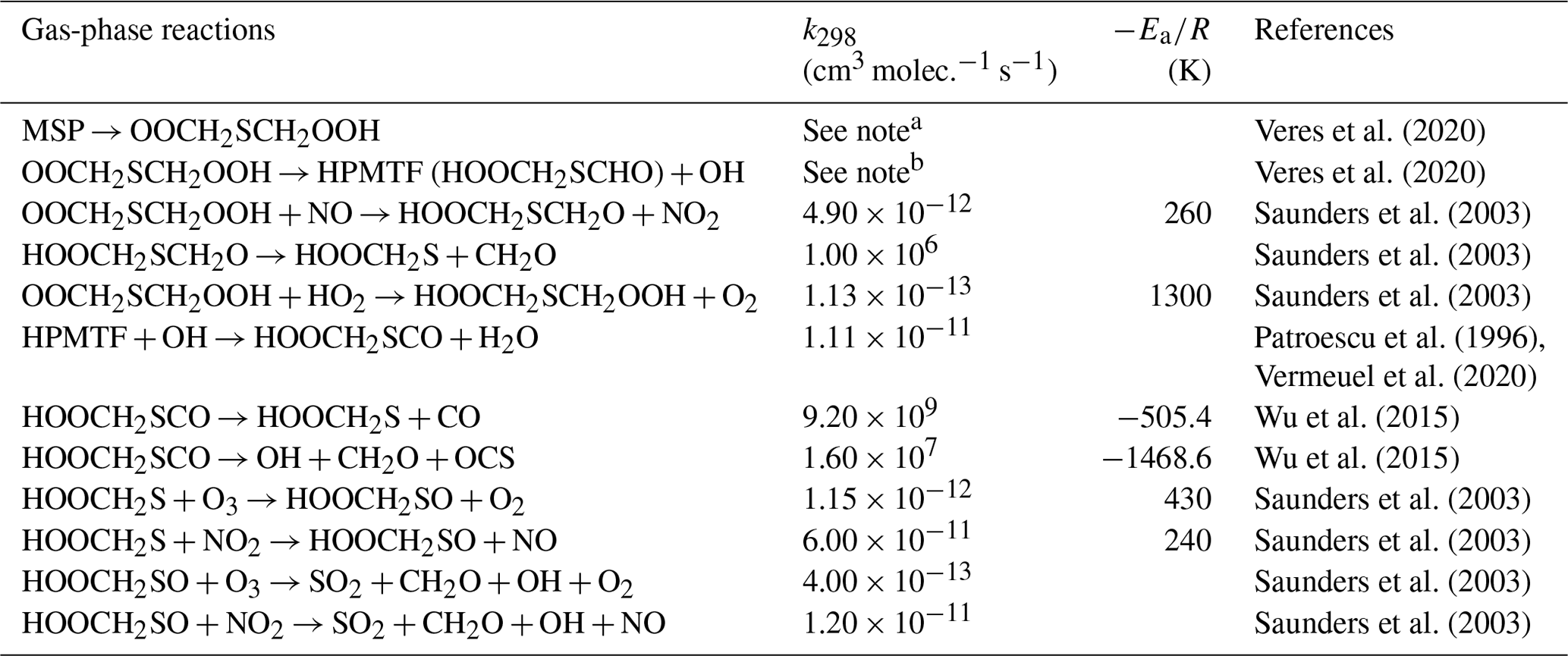
The H-abstraction reactions of DMS with OH or Cl generate MSP, which then either reacts with NO or RO2, forming MSA and H2SO4, or undergoes consecutive intramolecular H-shift reactions (isomerization), yielding HPMTF and SO2. Hence, we group these two serial reactions into two branches, namely the MSA-producing branch and the isomerization branch. The reactions of the MSA-producing branch are tabulated in Table 4. These reactions are largely based on Hoffmann et al. (2016), who combined the chemical mechanism from the Master Chemical Mechanism version 3 (MCM v3; Saunders et al., 2003) and other laboratory and computational studies. The reactions in the isomerization branch are detailed in Table 5. Here, we use the only currently available temperature-dependent isomerization rate of MSP (kiso) of 0.04 s−1 at 293 K, as estimated by Veres et al. (2020). This new kiso is slower than the previously determined values of 0.23 s−1 at 295 K (Berndt et al., 2019) to 2.1 s−1 at 293 K (Wu et al., 2015), delaying the formation of HPMTF. A recent chamber experiment estimates an intermediate kiso value of 0.12 s−1 at 293 K (Ye et al., 2021). We investigate the impact of the uncertainty in kiso in Sect. 3. The only chemical loss process of HPMTF in our model is oxidation by OH at a rate of 1.11 × 10−11 cm3 molec.−1 s−1, as recommended by Vermeuel et al. (2020), which is an experimentally determined OH-oxidation rate of methyl thioformate (MTF; CH3SCHO; a structurally similar proxy to HPMTF) by Patroescu et al. (1996). Oxidizing HPMTF at this rate was shown to match better with recent measurements (Vermeuel et al., 2020) than the 1.4 × 10−12 cm3 molec.−1 s−1 suggested by a computational study (Wu et al., 2015). Recent studies (Veres et al., 2020; Vermeuel et al., 2020) suggest that cloud uptake is another important sink of HPMTF; we include a series of sensitivity tests based on [MOD_2000] to address the uncertainty in the HPMTF budget arising from this potential loss process. Vermeuel et al. (2020) report that using a cloud uptake rate (kHPMTF+cloud) at 5 × 10−3 s−1 results in a better match of the diurnal variability in HPMTF with their local measurements. Due to the lack of detailed measurement, we use this kHPMTF+cloud and a substantially slower hypothetical value at 5 × 10−5 s−1 for our sensitivity tests.
Table 4Summary of the MSA-producing branch of the H-abstraction pathway in the DMS chemistry implemented into CAM6-chem.
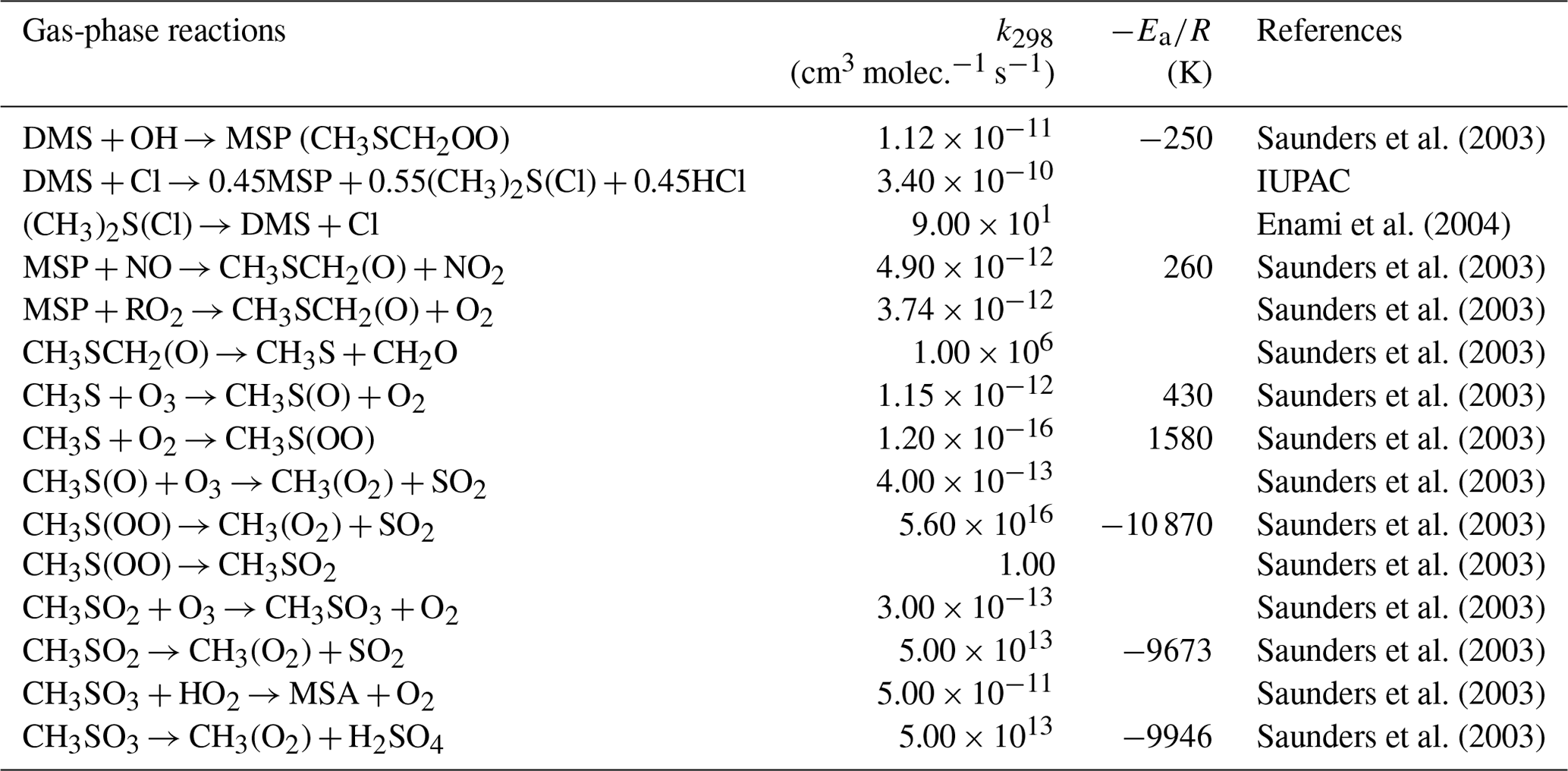
Table 5Summary of the isomerization branch of the H-abstraction pathway in the DMS chemistry implemented into CAM6-chem.
a 2.24 , b 6.09 , where T is air temperature.
2.3.2 Gas-phase reactions of the OH-addition pathway
Table 6 summarizes the new gas-phase reactions in the OH-addition pathway of DMS oxidation. We update the gas-phase reactions in the model to consider the oxidation of DMS by not only OH and NO3 but also BrO, O3, and Cl, as recommended or reported in previous studies (e.g., Barnes et al., 2006; Hoffmann et al., 2016; Chen et al., 2018). The new reactions producing dimethyl sulfoxide (DMSO; CH3SCH3O), methanesulfinic acid (MSIA; CH3SOOH), and MSA intermediates are added to the model as new advected chemical tracers which undergo not only chemical production and loss but also transport and deposition. MSA and SO2 are terminating products of these new gas-phase OH-addition pathway reactions, which is consistent with various modeling studies (e.g., Pham et al., 1995; Spracklen et al., 2005; Chen et al., 2018). All oxidants (OH, O3, H2O2, BrO, and HOBr) are simulated online by the standard gas-phase chemistry scheme of CAM6-chem.
Table 6Gas-phase DMS oxidation (OH-addition pathway) implemented into CAM6-chem in this study.

∗ (cm3 molec.−1 s−1).
2.3.3 Aqueous-phase reactions of the OH-addition pathway
We also introduce new aqueous-phase reactions of the OH-addition pathway, as shown in Table 7.
Table 7Aqueous-phase DMS oxidation (OH-addition pathway) implemented into CAM6-chem in this study.
Following a similar treatment employed by the Community Multiscale Air Quality (CMAQ) model version 5.1 (Fahey et al., 2017), we calculate, for each species involved in the new aqueous-phase reactions, the phase transfer equations for gas-aqueous partitioning, as follows:
where Cg and Ca are the gas-phase and aqueous-phase concentration of a species involving in reactions in Table 7. k(g)→(a) and k(a)→(g) (per second) are its gas-to-aqueous and aqueous-to-gas phase transfer coefficients. kt (per second) is its base phase transfer coefficient. H (molar per standard atmosphere; hereafter M atm−1) is its effective Henry's law constant. r (centimeters) is its mean particle radius, and Dg is the gas-phase diffusion coefficient (assumed at 0.1 cm2 s−1; here following Dentener and Crutzen, 1993). c (centimeters per second) is its thermal speed, and α is its mass accommodation coefficient. Values of H and α for DMS, DMSO, MSIA, and MSA are given in Table 8. LWC (cubic centimeter of water per cubic centimeter of air; hereafter cm3-water (cm3-air)−1) is liquid water content of interstitial aerosol determined by MOSAIC (Zaveri et al., 2021) or cloud liquid water content calculated by CAM6; R=0.082 (liter standard atmosphere per Kelvin per mole; hereafter L atm K−1 mol−1) is the universal gas constant, and T (Kelvin) is air temperature.
Table 8Summary of parameters of DMS and its oxidation intermediates used in this study.
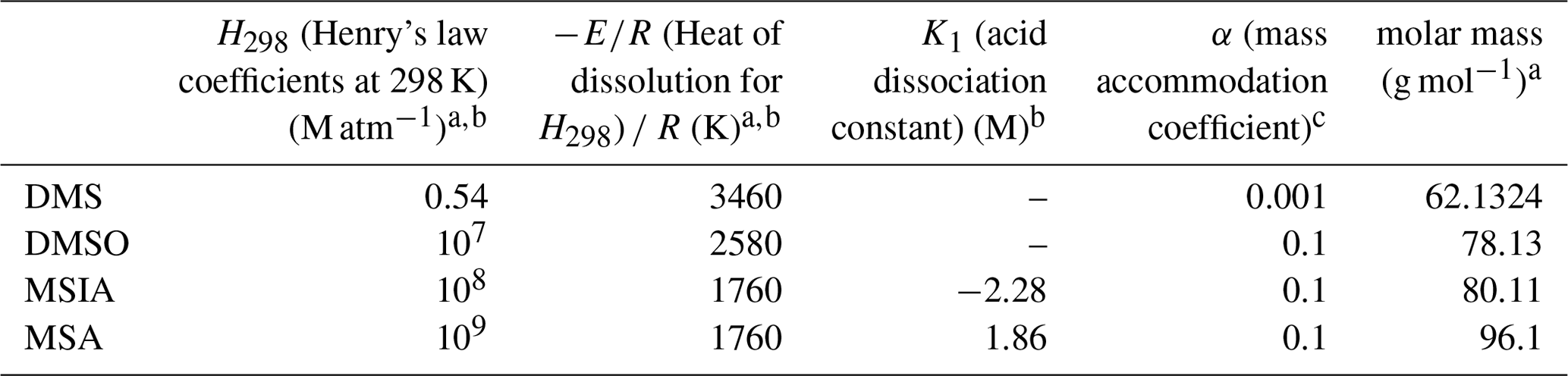
a Lamarque et al. (2012); b Chen et al. (2018); c Zhu et al. (2006).
3.1 Global sulfur budget and distribution in the present day
The global burden of DMS in [MOD_2000] is 50 Gg S. It is 38 % lower than the standard run, [STD_2000], but remains within the range of 9.6–140 Gg S from other studies (Faloona, 2009; Kloster et al., 2006). Figure 2 shows that the reduction is mainly over the Southern Ocean and is attributable to faster chemical losses via DMS + BrO and DMS(aq) + O3(aq) (Fig. 3). The global lifetime of DMS decreases from 1.5 d in [STD_2000] to 0.8 d in [MOD_2000]. These values are comparable to the range of 1.2–2.1 d reported in Chen et al. (2018).
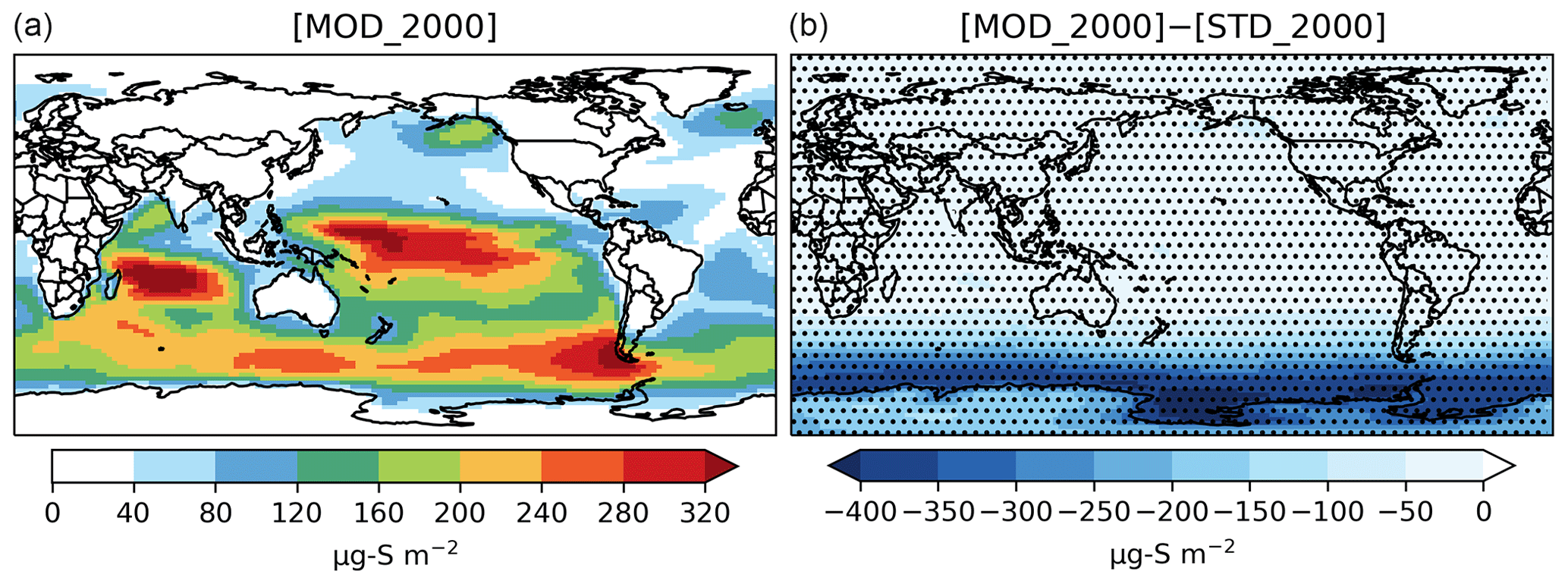
Figure 2Spatial distribution of the annual mean column concentration (micrograms of sulfur per meter squared; hereafter µg S m−2) for DMS simulated by [MOD_2000] (a) and its difference from the baseline run, i.e., [MOD_2000]–[STD_ 2000] (b). Dotted regions (nearly worldwide) indicate where statistically significant differences are identified by grid-by-grid two-sample t tests with p values < 0.05.
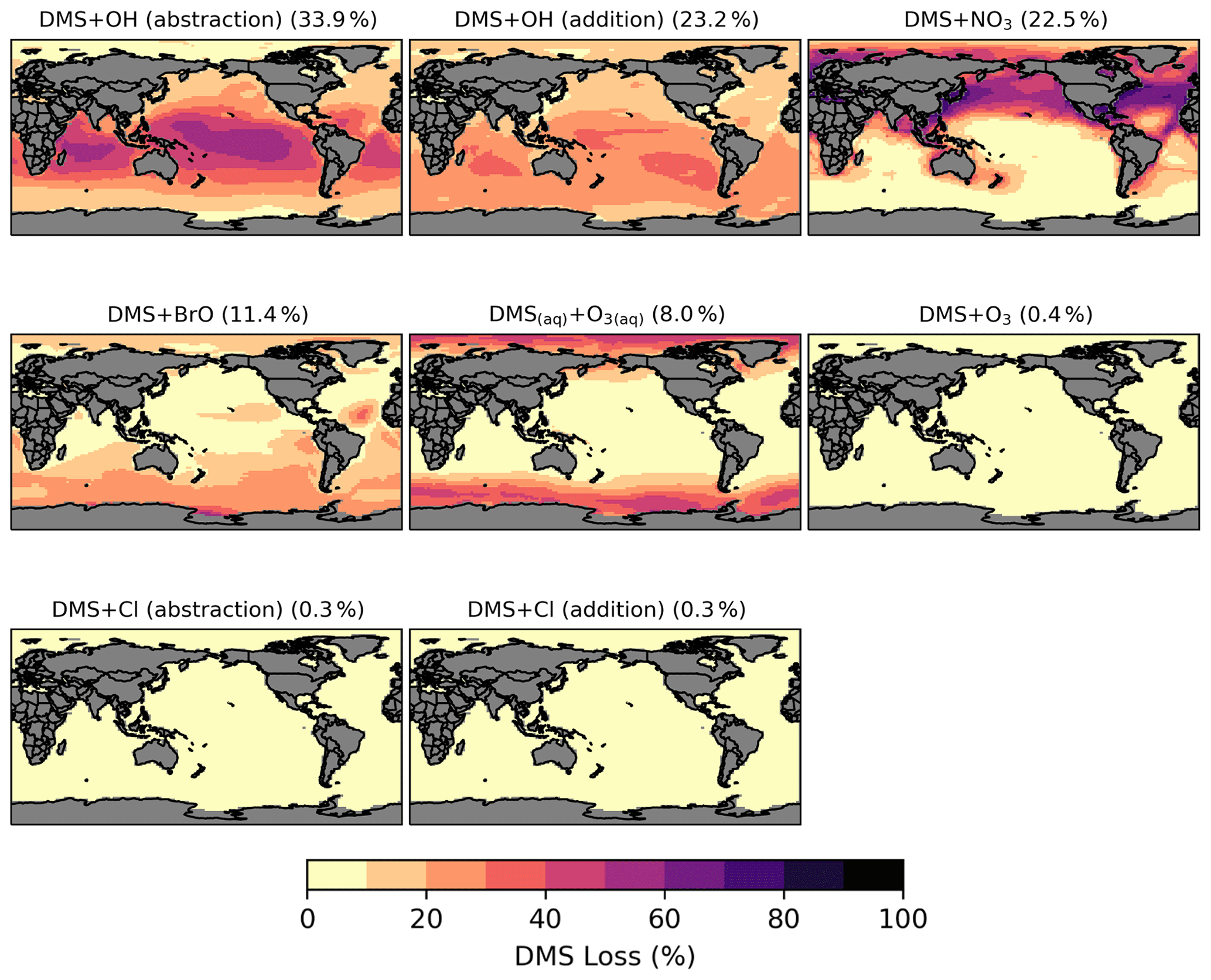
Figure 3Spatial distribution of the fractional DMS oxidation (percent) from [MOD_2000] through DMS + OH (abstraction), DMS + OH (addition), DMS + NO3, DMS + BrO, DMS(aq) + O3(aq), DMS + O3, DMS + Cl (abstraction), and DMS + Cl (addition). Percentages in parentheses denote contribution to global chemical loss. Subplots are arranged in descending order of their annual total oxidation rates.
Globally, chemical loss is the largest sink of DMS (∼ 24 Tg S yr−1) in both PD simulations. The model default chemistry in [STD_2000] predicts that OH oxidation makes up 40 % (H abstraction) and 39 % (OH addition) of DMS chemical removal globally, while the remaining portion is attributed to NO3 oxidation. These three reactions are only responsible for 80 % of global DMS loss in [MOD_2000]. Figure 3 shows that, in [MOD_2000], DMS is mainly oxidized by OH in the gas phase (34 % via the H-abstraction channel and 23 % via the OH-addition pathway), which contributes up to 80 % of local loss over the tropical oceans where surface OH is the highest. The annual mean surface concentrations of all oxidants which react with DMS in our updated scheme are summarized in Fig. S1 in the Supplement. NO3 oxidation of DMS accounts for 23 % of global DMS chemical loss and is dominant in Northern Hemisphere mid-latitudes, where the outflow of nitrogen oxides (NOx) – precursors of atmospheric NO3 – from the land are substantial (Miyazaki et al., 2012). DMS oxidation by NO3 contributes < 10 % over most marine environments in the Southern Hemisphere. Previous studies estimate that the global contribution of OH and NO3 to the DMS oxidation ranges from ∼ 50 %–70 % to 15 %–30 %, respectively (Berglen, 2004; Boucher et al., 2003; Khan et al., 2016; Chen et al., 2018).
Oxidation by BrO is responsible for 11 % of the global DMS removal, which falls midway within the previously estimated range of 8 %–29 % (Boucher et al., 2003; Khan et al., 2016; Chen et al., 2018). Regionally, its importance can be up to 50 %–60 % over the high latitudes in the Southern Hemisphere, which is close to a previous box model experiment (Hoffmann et al., 2016).
DMS + O3 is the only multiphase DMS oxidation reaction in this study, accounting for 8 % (aqueous phase) and 0.4 % (gas phase) of global DMS depletion. The oxidation rates via these reactions, estimated by Boucher et al. (2003), were 6 % and 3 % of the total DMS sink calculated, respectively. Our lower gas-phase DMS + O3 reaction rate could be due to the inclusion of the BrO oxidation, which is missing in their study. Regionally, the fractional contribution of aqueous-phase DMS + O3 to DMS oxidation can be up to 20 %–30 % over high-latitude oceans, which is on the upper end of the range of 5 %–30 % high-latitude DMS losses previously reported (von Glasow and Crutzen, 2004; Chen et al., 2018).
Lastly, the Cl oxidation reactions via either the addition or abstraction channels contribute equally (0.3 % each, globally) to the chemical removal of DMS, which is consistent with the proposal of Atkinson et al. (2004). Our estimated values are much lower than the 4 % found in a global model study (Chen et al., 2018) and the 8 %–18 % from box model studies (von Glasow and Crutzen, 2004; Hoffmann et al., 2016).
We note that recent studies (e.g., X. Wang et al., 2021) have shown that large discrepancies in Cl and BrO are found within the same models and/or sets of measurements. Further investigation of how uncertainties in the representation of the halogen cycle feed back onto DMS chemistry is, hence, warranted.
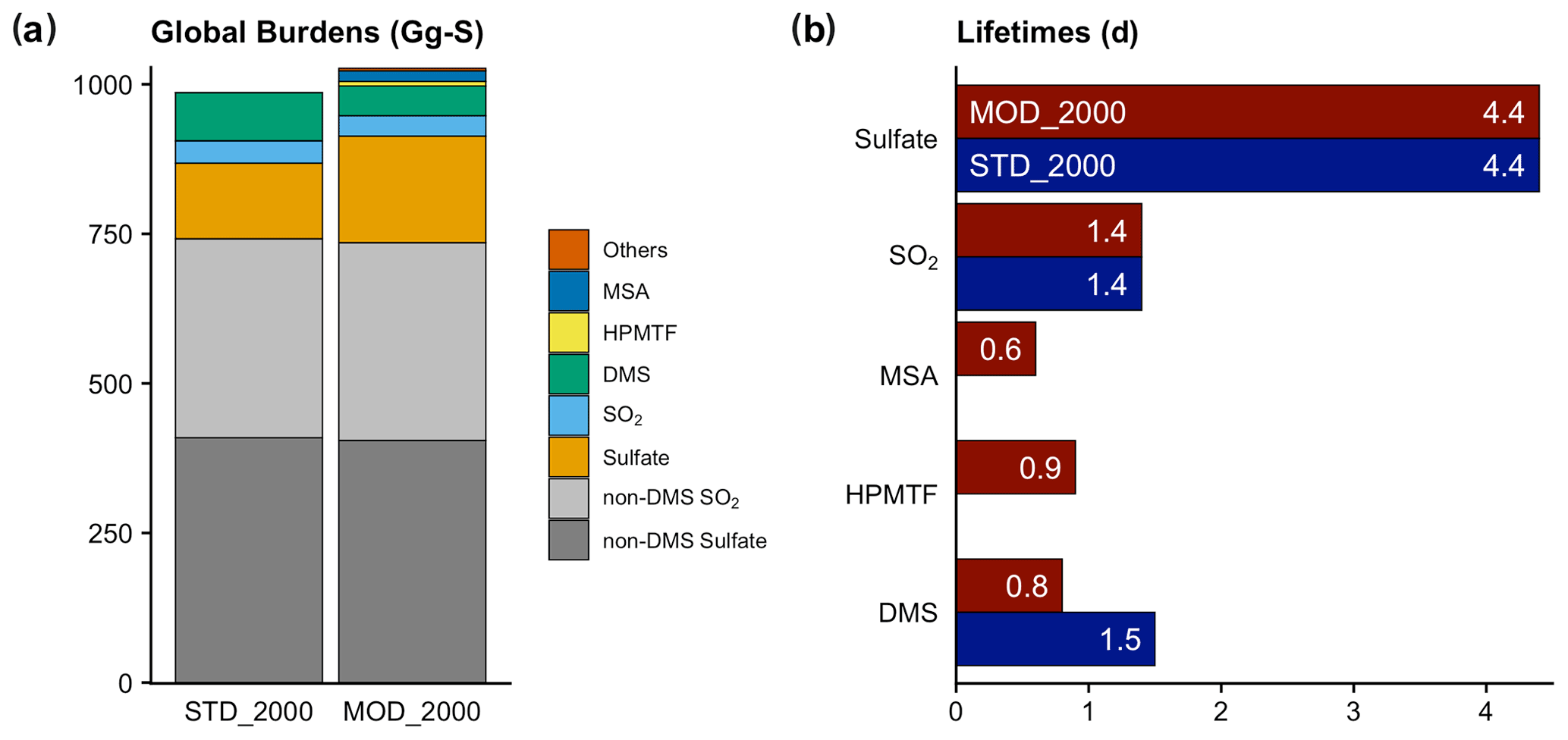
Figure 4(a) Global burdens of various atmospheric sulfur species in our simulations. The category of “Others” includes all other sulfur-containing intermediates in the new chemistry (DMSO, MSIA, etc.). SO2 (blue) and sulfate (orange) refer to the burden of these species that originate from DMS oxidation only; non-DMS contributions are shown in gray. (b) Total lifetimes of the atmospheric sulfur species to both physical and chemical losses.
The global atmospheric sulfur burden is increased by 41 Gg S (or 4.1 %) from [STD_2000] to [MOD_2000] (Fig. 4 and Table S1). Approximately half (23 Gg S) of this increment is associated with the recovery of the missing sulfur associated with the OH-addition reaction in the standard chemistry (the second reaction in Table 3), which does not conserve sulfur. The remaining total sulfur burden increase is attributable to the extended chemistry scheme. As discussed above, the DMS burden in [MOD_2000] is lower than [STD_2000] by 38 % due to faster oxidation. This oxidation produces intermediates with a wide range of lifetimes. The addition of intermediates with relatively long physical lifetimes (to dry and wet deposition only) of HPMTF (1300 d) and MSA (8.5 d) delays the formation of SO2 (2.6 d) and sulfate (4.4 d) compared to the standard reactions in [STD_2000], which increases the export of sulfur-containing intermediates.
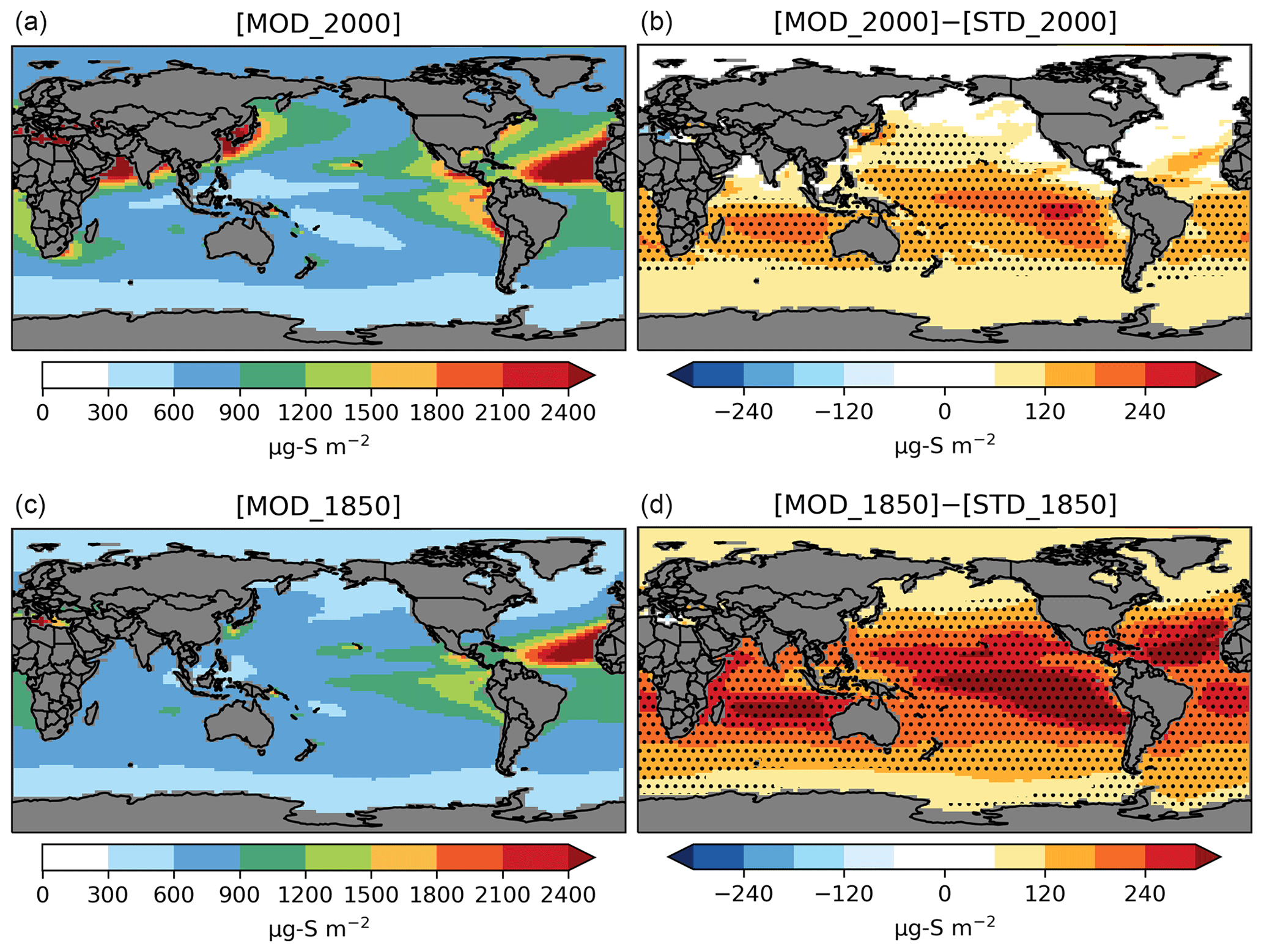
Figure 5Spatial distribution of annual mean column concentrations (µg S m−2) for sulfate aerosol simulated by [MOD] (a, c) and their difference from the baseline run (b, d). Only values over the ocean are shown. Dotted regions indicate where statistically significant differences are identified by grid-by-grid two-sample t tests with p values < 0.05.
The PD global annual mean burden for sulfate aerosol is 582 Gg S in [MOD_2000], with an interannual variability of 46 Gg S (standard deviation of annual means). It is comparable to the 580 Gg S in a previous CAM6-chem study (Tilmes et al., 2019) and is within the estimates (420–660 Gg S) from studies using other models (e.g., Heald et al., 2014; Chen et al., 2018). The new DMS chemistry has increased the global sulfate burden by 47 Gg S (or 8.8 %) from the baseline value of 535 Gg S in [STD_2000]. The statistically significant increases in sulfate resulting from the expanded chemistry are mostly found over the tropical and subtropical oceans in the Southern Hemisphere (Fig. 5). There is no strong seasonality in the additional sulfate produced from our expanded chemistry. We estimate that the sulfate burden attributable to DMS increases by 41 % from 126 Gg S in [STD_2000] to 178 Gg S in [MOD_2000]. Most of this increase in sulfate burden (72 %) comes from the expansion of the gas-phase chemistry with a minor additional contribution from the aqueous-phase chemistry. In a sensitivity test where the isomerization branch reaction (Table 5) is removed from [MOD_2000], the global DMS-derived sulfate burden is reduced by 2.0 % (relative to [MOD_2000]).
The spatial distribution of the product branching ratios of DMS oxidation is shown in Fig. 6. In addition to depositional removal, HPMTF converts into SO2, while SO2 and MSA are then oxidized to form sulfate. We estimate that 33 % of the annual total DMS oxidation will yield HPMTF. This is comparable to the observationally constrained estimates from NASA Atmospheric Tomography (ATom)-3 and ATom-4 flight campaigns, where ∼ 30 %–40 % DMS was oxidized to HPMTF along their flight tracks (Veres et al., 2020). High HPMTF production is typically seen in the summer MBL, coinciding with the HPMTF hotspots over tropical oceans, as shown in Fig. S2. To address the uncertainty in the production and loss of HPMTF as discussed in Sect. 2.3, we run several sensitivity tests using five combinations of kiso and kHPMTF+cloud values based on [MOD_2000]. Table S2 and Fig. S5 summarize the key results of these sensitivity tests. Compared to [MOD_2000], we find that using a faster kiso of 0.12 s−1 at 293 K (Ye et al., 2021) increases the global annual total isomerization rate of MSP by 5.6 %, while the global burden of HPMTF increases by 4.1 %. Increasing the isomerization rate has little impact on the burden of sulfate from DMS (increase of only 4.0 %). We also evaluate the importance of the cloud uptake of HPMTF with two hypothetical values of kHPMTF+cloud at 5 × 10−3 s−1 (Vermeuel et al., 2020) and 5 × 10−5 s−1. At these rates, the cloud uptake becomes an important sink of HPMTF, which is responsible for 68 %–69 % and 28 % of the total HPMTF losses respectively. The corresponding global burdens of HPMTF are substantially lowered by 85 %–86 % and 52 %. For simulation with cloud uptake loss, the burdens of HPMTF and sulfate are much less sensitive to our choice of kiso due to the rapid loss of HPMTF to cloud uptake. In these sensitivity simulations, the sulfur contained in HPMTF is assumed to be removed from the system once taken up by cloud, thus reducing the sequential formation of SO2 and sulfate (by up to 8 %).
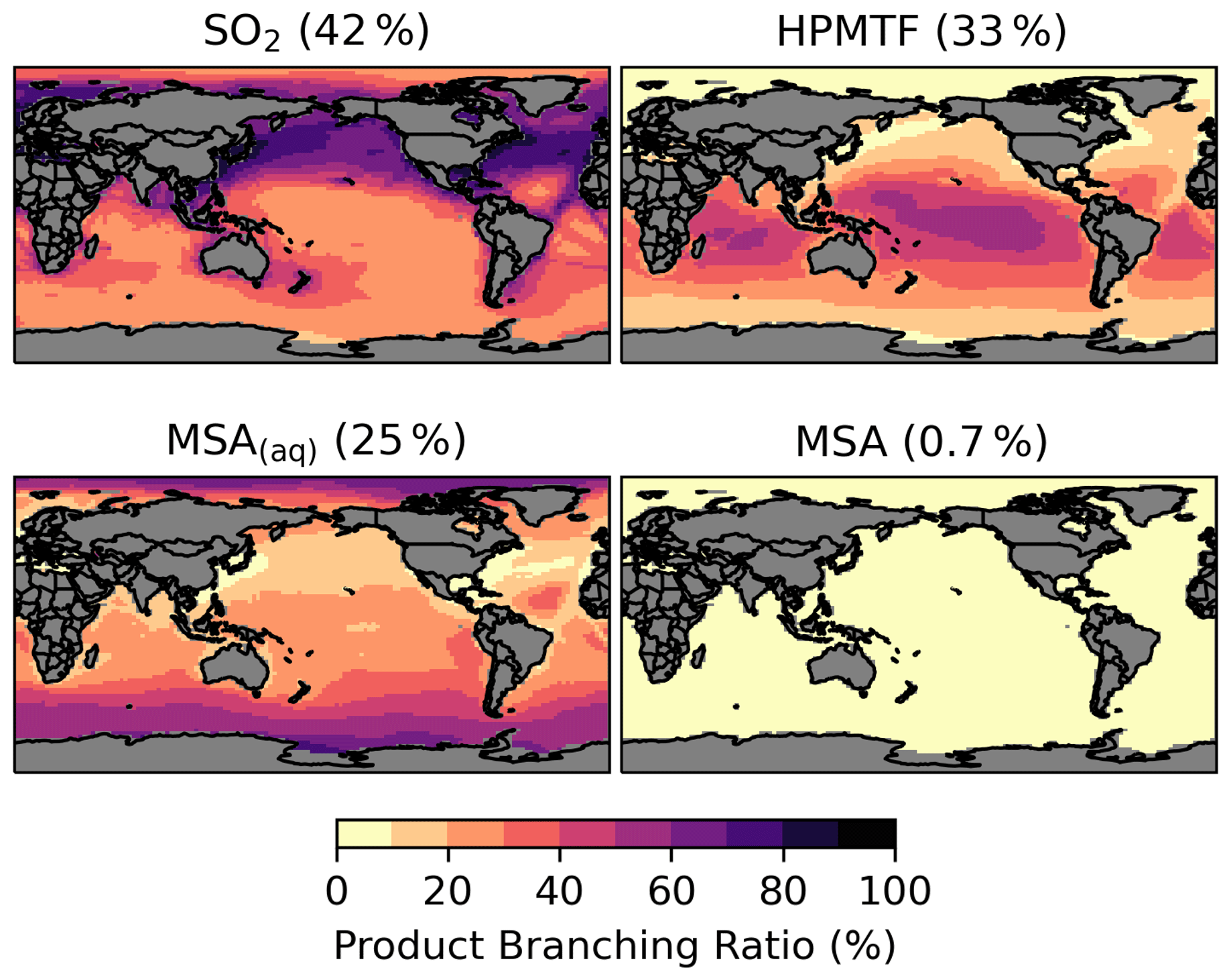
Figure 6Branching ratio (percent) of the multiphase DMS oxidation pathways in [MOD_2000], considering HPMTF, SO2, and MSA as terminating products estimated from their annual total production rates.
MSA is a key intermediate generated from the OH-addition channel of the multiphase DMS oxidation, especially over the remote marine atmosphere. Our result shows that aqueous-phase MSA formation accounts for most of the MSA production as commonly reported (von Glasow and Crutzen, 2004; Barnes et al., 2006; Zhu et al., 2006; Hoffmann et al., 2016; Chen et al., 2018). In [MOD_2000], the global MSA burden is 7.5 Gg S, which is smaller than the range of 13–40 Gg S from previous model studies (Pham et al., 1995; Chin et al., 1996, 2000; Cosme et al., 2002; Hezel et al., 2011; Chen et al., 2018). In [MOD_2000], most MSA is formed over the Southern Ocean (Fig. S3). The lifetime of MSA is 0.6 d globally, shorter than the 5–7 d previously proposed (Chin et al., 1996, 2000; Cosme et al., 2002; Hezel et al., 2011; Pham et al., 1995), likely because we include the aqueous-phase OH oxidation to sulfate, which is a significant loss process for MSA. This oxidation accounts for ∼ 76 % of removal in [MOD_2000], followed by cloud uptake (18 %).
3.2 Comparison with observations
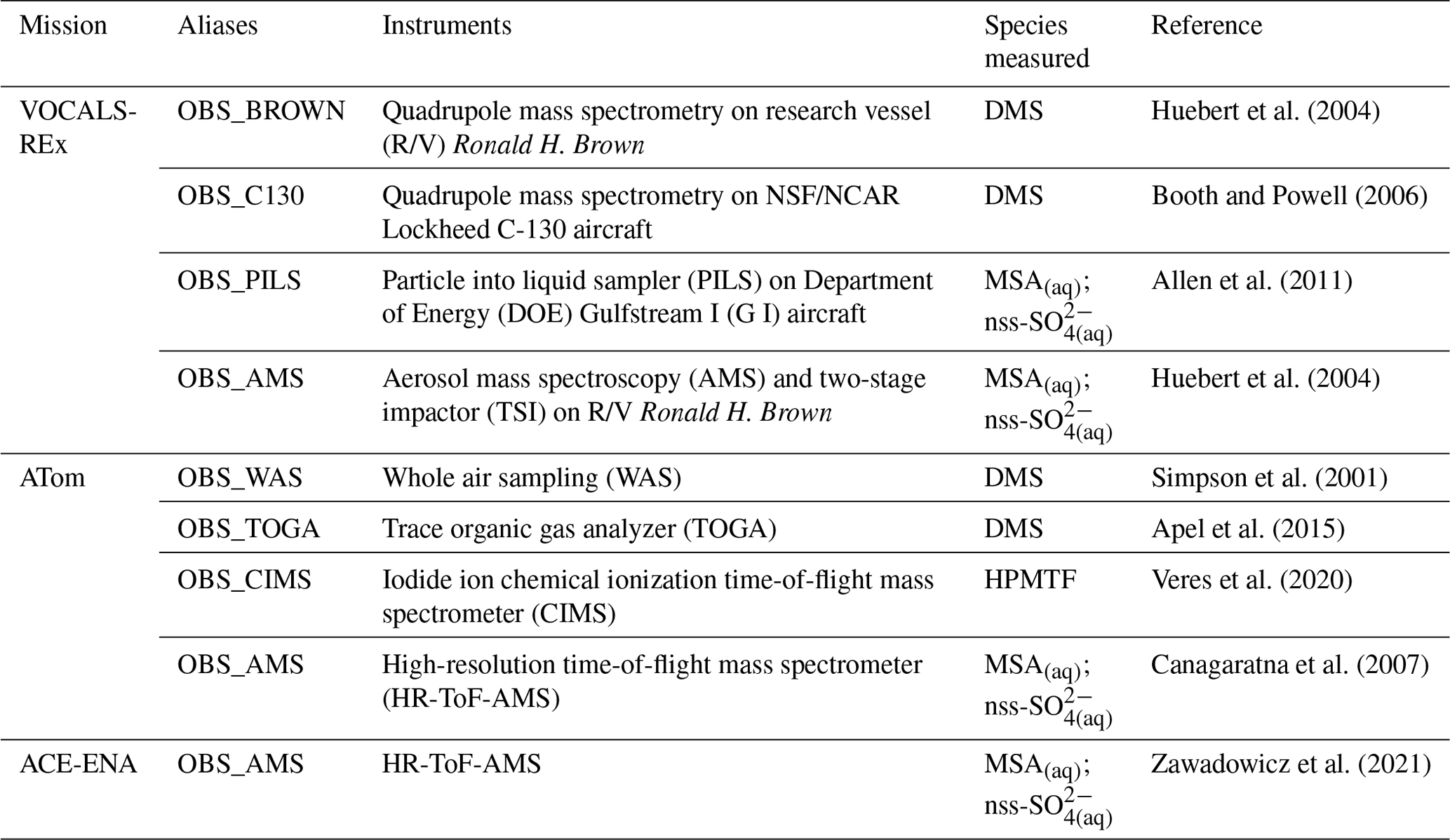
Table 9 summarizes the key observational datasets used here to compare with our PD model simulations for their wide coverage of the remote marine atmosphere. The Variability of the American Monsoon Systems (VAMOS) Ocean-Cloud-Atmosphere-Land Study Regional Experiment (VOCALS-REx) is an international field project that took place during October and November in 2008 over the southeastern Pacific off northern Chile and southern Peru (Wood et al., 2011). VOCAL-REx consists of both ship-based and airborne measurements for lower-atmospheric DMS (MSA(aq)) and non-sea-salt sulfate aerosol (nss-SO). The ATom mission of NASA is a flight campaign spanning from the Arctic to the Antarctic over the remote Pacific and Atlantic oceans between 2016 and 2018 (Wofsy et al., 2018). During ATom, an array of instruments was used to collect and analyze daytime air samples from the remote marine environments, providing measurements of DMS, HPMTF, MSA(aq), and nss-SO. The Aerosol and Cloud Experiments in the Eastern North Atlantic (ACE-ENA) probed the atmosphere surrounding the ENA observatory on Graciosa Island during summer in 2017 and winter in 2018 (J. Wang et al., 2019, 2021). ACE-ENA provides high time resolution in situ measurements of MSA and sulfate aerosol in the lower troposphere (Zawadowicz et al., 2021). We note that the model–measurement comparisons are not exact, given that our simulations are performed using free-running dynamics and, thus, are not paired to the meteorological year of measurements. We, therefore, sample monthly mean values from the model at the location of the observations to provide qualitative comparisons.
Most DMS resides in the lower troposphere (Fig. 7). Annual mean surface DMS from [MOD_2000] ranges from 40–300 ppt (parts per trillion) over much of the ocean but can exceed 320 ppt over the Southern Ocean and northeastern Pacific, which are regions with high DMS emissions. DMS concentrations of ∼ 25–125 ppt were observed at Cape Grim, Tasmania, Australia, in 1990–1993 (Ayers et al., 1995). Sciare et al. (2000) report an annual mean DMS of 181 ppt at Amsterdam Island in the Indian Ocean during the 1990s. Both values are in line with the surface DMS at the corresponding locations modeled by [MOD_2000].
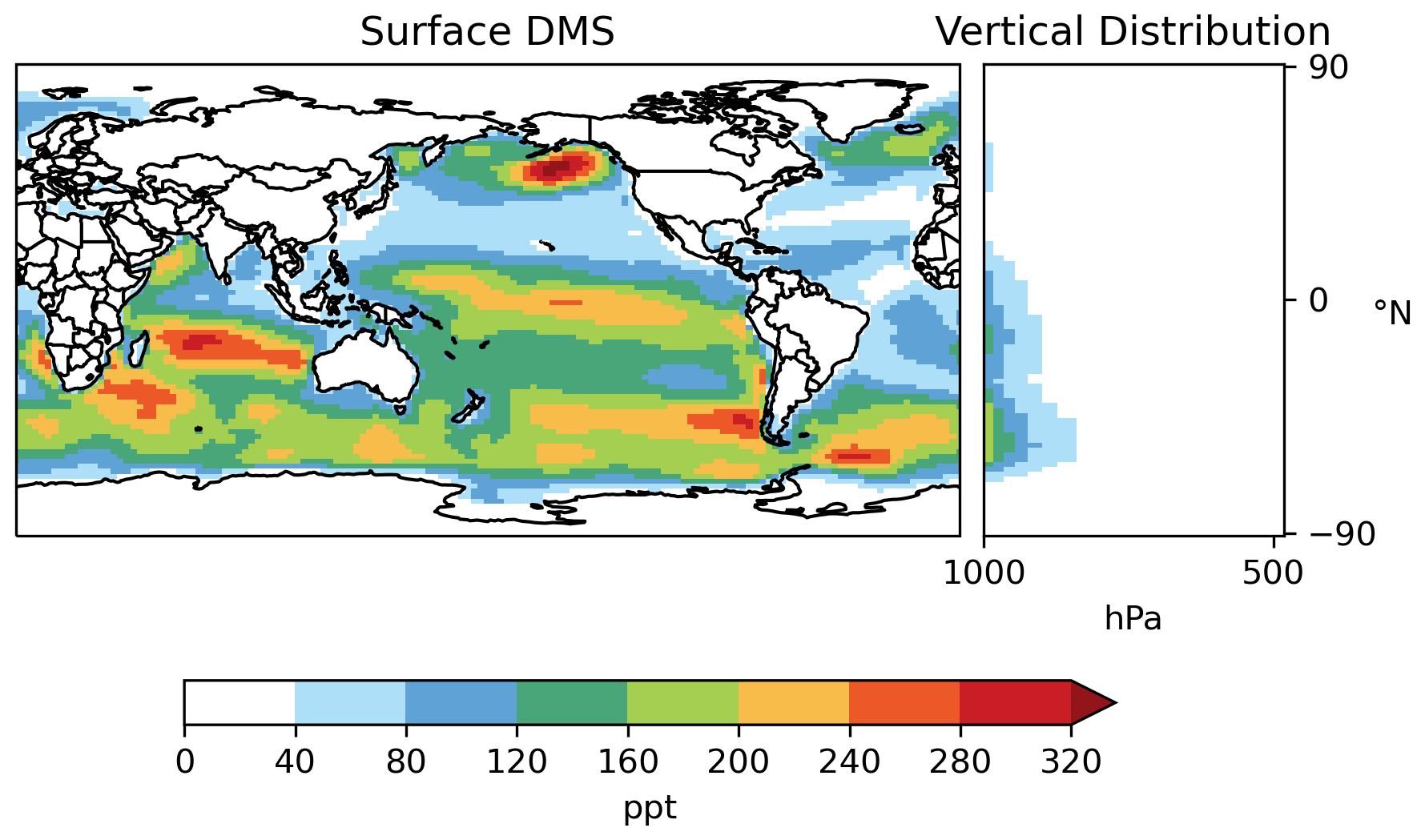
Figure 7Horizontal distribution of annual mean surface mixing ratio and zonal mean vertical distribution of DMS (both in parts per trillion – ppt) modeled by [MOD_2000].
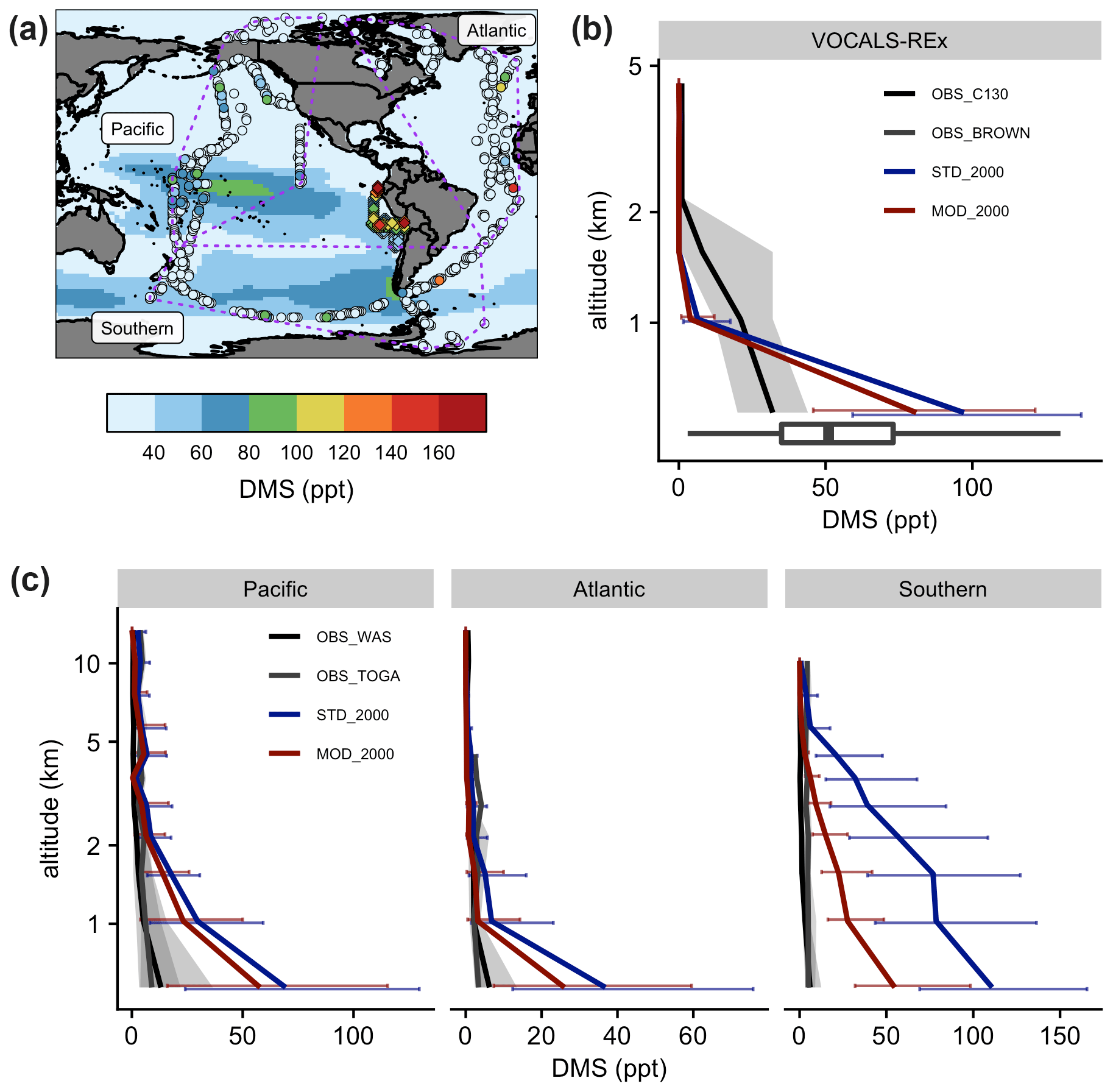
Figure 8(a) Measurements of DMS during VOCALS-REx (diamonds in the southeastern Pacific near the coastline of Peru) and ATom (dots) missions. Measured values are showing local 90th percentiles above oceans. ATom data are grouped into three regions, as shown in the purple dashed polygons. Marine-only annual mean near-surface (> 500 hPa) DMS mixing ratios from [MOD_2000] are shown in the background as a reference. Vertically binned modeled and observed medians of DMS, during (b) VOCALS-REx and (c) ATom, are shown. Error bars and gray shadings indicate that the data ranged between corresponding upper and lower quantiles.
Figure 8 summarizes the spatial difference between the observed DMS from the VOCALS-REx and ATom missions and the simulated DMS. The model captures the peaks over the tropical Pacific and the Southern oceans off the coast of South America, but aircraft measurements detect hotspots that are not simulated by the model (Fig. 8a). During VOCALS-REx the ship-based measurements (BROWN) recorded a range of near-surface DMS from 18 to 111 ppt, while the airborne measurements (C130) reveal a vertically decreasing trend of DMS mixing ratios, from 33 ppt at ∼ 500 m to 2.0 ppt at ∼ 2 km (Fig. 8b). Modeled surface DMS falls in the range of the ship measurements. Compared to the aircraft observations, simulated DMS is biased high at the surface and declines more abruptly, which may indicate biases in vertical mixing or cloud processing. DMS concentrations are slightly lower in the simulation with updated chemistry [MOD_2000] but follow the same vertical profile. We disaggregate the ATom observations into three regional groups, namely Pacific, Atlantic, and Southern oceans as in Fig. 8a. DMS concentrations were measured by two instruments during ATom (WAS and TOGA; the former generally reported higher values), and both are compared with model values in Fig. 8c. Observed DMS concentrations during ATom are substantially lower than measured during VOCALS, and lower than any region simulated by the model. Modeled DMS is biased high in all three regions, especially over the Southern Ocean region where the discrepancy extends up to 5 km. The new chemistry increases DMS losses and shortens the DMS lifetime, reducing the model bias in [MOD_2000]. The decrease in simulated DMS is largest over the Southern Ocean (−49 % at the surface), where oxidation by BrO and O3 in the aqueous phase are important and the model–observation bias is substantially reduced. The remaining model biases during ATom exceed the uncertainty of the kinetics for the current DMS oxidation scheme and are likely attributable, at least in part, to the uncertainty in DMS emission. A sensitivity test, where we reduce the sea surface DMS concentration by 50 % in regions south of 30∘ S in [MOD_2000] produces, as expected, a comparable decrease in DMS mixing ratios in the lower atmosphere (< 5 km), and the model–observation deviations are further narrowed (see Fig. S4 and the Supplement for details). Constraining DMS emissions is beyond the scope of this work but is clearly a major source of uncertainty that may impact the sulfur budget discussed in Sect. 3.1 and the climate response discussed below.

Figure 9Measured (ATom) and modeled values of HPMTF are vertically binned. The thick lines show the medians. Error bars and gray shadings indicate that the data ranged between corresponding upper and lower quantiles. The thin red line indicates the results from a sensitivity test with kiso at 0.04 s−1 and kHPMTF+cloud at 5 × 10−5 s−1.
Figure 9 compares the mean vertical profile of HPMTF mixing ratios observed during ATom against the model [MOD_2000]. Over the Pacific and Atlantic regions, HPMTF mixing ratios are largest at lower altitudes and decrease to < 1 ppt in the middle and upper troposphere. The model generally reproduces the observed magnitude and vertical profile. The model [MOD_2000] is biased high over the Southern Ocean region, particularly in the lower troposphere. Such high biases are consistent with the aforementioned overestimation of DMS over this region. In the lower atmosphere over tropical and mid-latitude oceans, the modeled DMS : HPMTF ratios range from 5 : 1 to 2 : 1, which is generally larger than the average 2 : 1 ratio observed during ATom (Veres et al., 2020), suggesting that the model may underestimate the DMS-to-HPMTF conversion rate or overestimate the HPMTF loss. Our model predicts that OH oxidation to SO2 is dominant in the removal of HPMTF while dry and wet deposition are negligible. The addition of cloud uptake (discussed in Sect. 2.3.2) can dramatically decrease HPMTF concentration (by up to > 73 % when assuming moderate cloud uptake rate of 5 × 10−5 s−1), resulting in a better model–observation agreement in the lower troposphere over the Southern Ocean but low biases over the Pacific and Atlantic (Fig. 9). In light of the DMS biases in Fig. 8, this irreversible cloud uptake may overcorrect the HPMTF concentrations.
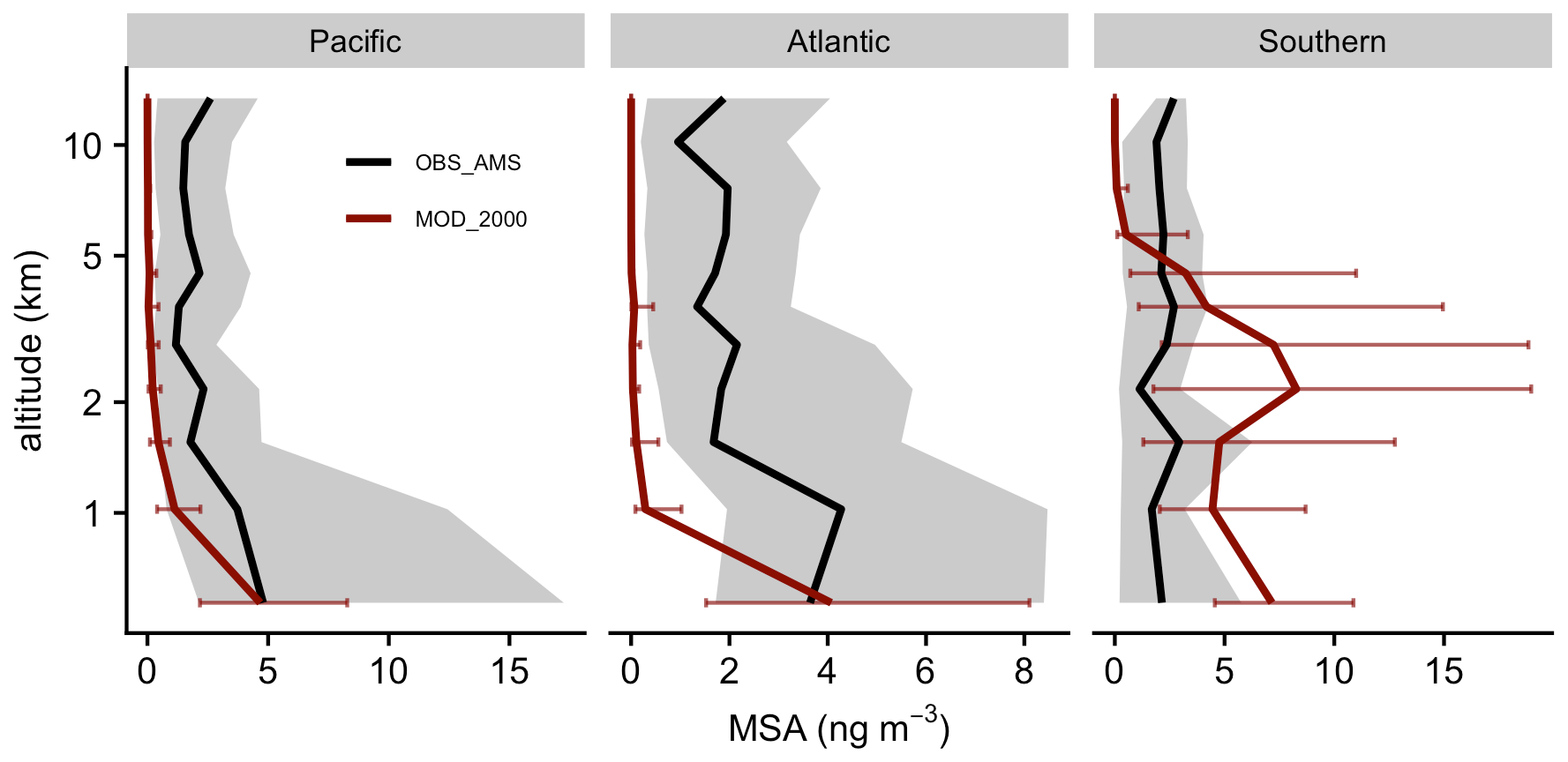
Figure 10Medians of observed (ATom) and modeled concentration of MSA aerosol are vertically binned. The thick lines show the medians. Error bars and gray shadings indicate that the data ranged between the corresponding upper and lower quantiles.
Our simulation shows that the gas-phase MSA formation is small compared to aqueous-phase formation, in line with previous work (Barnes et al., 2006; von Glasow and Crutzen, 2004; Zhu et al., 2006; Hoffmann et al., 2016; Chen et al., 2018; Hoffmann et al., 2021). Near the sea surface, simulated gas-phase MSA is < 0.03 ppt, even in Southern Hemisphere summer, while recent ship-based measurements reported concentrations ranging from 1.4 to 25 ppt (Yan et al., 2019). The model also substantially underestimates gas-phase MSA (< 0.001 ppt) when compared to a wintertime site measurements in Germany (0.5–10 ppt; Stieger et al., 2021). Figure 10 shows the concentration of submicron particulate MSA measured during ATom and the co-located concentration of MSA aerosol in Aitken and accumulated modes modeled by [MOD_2000]. The model overestimates the mid-tropospheric MSA concentrations in the Southern Ocean during ATom. Conversely, over the Pacific and Atlantic, the model underestimates MSA at mid- and low altitudes. Over the southeastern Pacific, the measured submicron MSA from VOCALS-REx ranged from 50–80 ng m−3 at lower altitudes (< 1 km; Wood et al., 2011), while the location-matched simulated MSA was considerably lower (6–15 ng m−3). Above Graciosa Island in the North Atlantic, ACE-ENA-observed MSA ranged from 10 to 20 ng m−3 at the lower troposphere (< 1 km) and gradually reduced to ∼ 5 ng m−3 in the mid-troposphere (2–5 km; Zawadowicz et al., 2021), whereas the model estimates a negligible amount of MSA (< 0.5 ng m−3) near the Azores as a result of limited DMS emissions in that region (as reflected by low DMS concentrations in Fig. 2). Outside of the Southern Ocean, the mean simulated concentration of MSA is underestimated compared to all observations, which suggests that the MSA-forming branches of DMS oxidation (H abstraction, where MSP reacts with NO or multiphase OH-addition reactions of DMS) may be underrepresented in our simulations or that the loss of MSA (by reaction with OH, the reaction rate for which is still highly uncertain; Milne et al., 1989; Zhu et al., 2006; Chen et al., 2018) may be overestimated.
Concentrations of the sulfate aerosol simulated with both [STD_2000] and [MOD_2000] generally agree well with measurements from ATom (Fig. S6). Our model also performs well at the surface when compared against VOCAL-REx and ACE-ENA but is biased high above 1 km, likely reflecting biases in anthropogenic sulfate exported from continental regions.
3.3 Global sulfur budget and distribution in the preindustrial era
As seen under PD conditions, the formation of intermediates expands the overall lifetime of sulfur-containing species in the PI atmosphere, thereby increasing the natural sulfate aerosol background. A summary of the burdens and lifetimes of the sulfur-containing species from the PI simulations is given in Table S1. The DMS burden in the PI from [MOD_1850] is 84 % larger than its PD counterpart, due to slower oxidation which prolongs the atmospheric lifetime. Oxidation by OH via the H abstraction (38 % of total DMS oxidation in [MOD_1850]) and the OH-addition channels (27 %) are still the primary loss pathways of DMS (Fig. S7). DMS + NO3 becomes less important (23 % in PD vs. 6.0 % in PI), given the reduced sources of NOx, resulting in a lowered DMS-to-SO2 conversion rate at 27 %, compared to 47 % in [MOD_2000] (Fig. S8). Reduced NOx also limits the reaction rates of MSP + NO, thereby favoring the isomerization pathway (92 % of total loss of MSP in PD vs. 96 % in PI). Hence, the conversion of DMS to HPMTF becomes more important (39 % of total DMS oxidation in PI), leading to a doubling of the HPMTF burden in the PI compared to PD. The addition pathway producing MSA becomes more dominant over the tropical ocean via oxidation by OH and the high latitudes by DMS + BrO, raising the MSA burden by 59 % compared to PD. Lastly, the expanded DMS oxidation chemistry increases the PI global annual mean sulfate burden by 29 % from 319 Gg S [STD_1850] to 412 Gg S ([MOD_1850]; Fig. 5), of which 57 % is derived from DMS, significantly larger than the 31 % in PD, confirming that DMS is a relatively more important source of sulfate in PI. Similar to PD, the majority (66 %) of this additional sulfate in the PI is produced via the expanded gas-phase oxidation pathways, and this addition is largely aseasonal. The absolute burden of sulfate produced from DMS oxidation is higher in the PI (236 Gg S) compared to the PD (178 Gg S).
Changes to particle-phase sulfate and MSA due to the expanded DMS chemistry, as described above, may alter both aerosol–radiation and aerosol–cloud interactions. Given that particulate MSA is not included in the current CAM6-chem aerosol scheme, to account for its radiative impacts, we assume MSA interacts with radiation like sulfate optically by implementing an artificial rapid conversion of MSA to sulfate. These two adjusted cases are aliased as [MOD_RE_1850] and [MOD_RE_2000], respectively. Details of this implementation are described in the Supplement.
4.1 Direct radiative effect (DRE)
Following the recommendation in Ghan (2013), we focus our analyses on the shortwave (SW) DRE. The PD global annual mean sulfate DRE modeled with [MOD_RE_2000] and [STD_2000] are −0.32 and −0.31 W m−2, respectively, which are slightly less negative than previous estimations of −0.36 to −0.42 W m−2 (Heald et al., 2014; Myhre et al., 2013; Yang et al., 2017). The global annual mean cloud fraction in the model is 77 %, while the all-sky DRE is 59 % of the estimate of clear-sky DRE. The more frequent cloudiness in CAM6-chem may explain the lower DRE compared to the AeroCom II models, which report a typical all-sky to clear-sky ratio of 1 : 2 (Myhre et al., 2013). The DMS-associated sulfate DRE in PD is −0.11 W m−2 in [MOD_RE_2000], which is slightly stronger than the value of −0.074 W m−2 in a CAM5-chem study using the standard DMS oxidation chemistry (Yang et al., 2017) but substantially weaker than the −0.23 W m−2 reported by Rap et al. (2013) using a different model. The DRE contribution of MSA alone is small (−0.8 mW m−2; estimated by the DRE difference of [MOD_RE_2000] minus [MOD_2000]). The sulfate DRE is not sensitive to HPMTF cloud loss, given that this loss has a modest impact on the sulfate burden in our simulations. Our new DMS chemistry has strengthened the PD sulfate direct cooling by 0.01 W m−2 or 4 % of the contribution attributed to DMS relative to [STD_2000].

Figure 11Contrasting the zonal means of (a) sulfate column concentration, (b) sulfate aerosol optical depth (AOD), and (c) all-sky SW sulfate DRE modeled with [MOD_RE] and [STD] chemistry at the PI and PD emission levels. Note that particulate MSA is included as sulfate in the MOD_RE simulations.
The rise in the sulfate burden from PI to PD, driven by anthropogenic emissions, occurs mainly over the land in the Northern Hemisphere; this impact is much larger than the increase in sulfate produced by the expanded DMS chemistry (Fig. 11). The zonal extrema of the PD sulfate burden, aerosol optical depth (AOD), and DRE are co-located around 30∘ N (Fig. 11). The larger difference in sulfate load in the southern tropics (30∘ S to 0∘ N) due to the new DMS oxidation reactions, also translates to a larger DRE difference in those latitudes (Fig. S9).
The direct radiative forcing (DRF) is estimated by differencing the DRE estimated with anthropogenic emissions at 1850 and 2000 levels. The DRF of [MOD_RE] and [STD] attributed to sulfate and MSA aerosols are calculated as −0.11 and −0.13 W m−2, respectively. This difference indicates a relatively linear relationship between sulfate loading and DRF.
4.2 Impacts on aerosol–cloud interactions and indirect radiative forcing (IRF)
While the central estimate of the IRF of aerosols from the AR5, which reflects constraints from selected satellite and general circulation model (GCM) analyses, is −0.45 W m−2, the IRF estimated by the majority of models reported in AR5 (see Figs. 7–19 of Boucher et al., 2013) ranges from −1.0 to −2.5 W m−2. This suggests that aerosol–cloud interactions may be substantially overestimated by the majority of models; an overly pristine preindustrial era may contribute to this (Menon et al., 2002; Carslaw etal., 2013). We anticipate that the increase in the PI sulfate, following the expansion of DMS chemistry (Fig. 11a), may dampen the IRF in our simulations. We evaluate IRF by calculating changes in the SW cloud radiative effects (ΔCRE) from PI to PD conditions, following previous studies (Gettelman et al., 2019; Ghan, 2013). Our estimates of IRF encapsulate not only the Twomey and the Albrecht effects (Twomey, 1977; Albrecht, 1989) but also cloud feedbacks in response to meteorological changes driven by different sulfate aerosol loadings. For instance, our simulations do not fix air temperature and wind fields. Deeper mixing and stronger turbulence may affect cloud microphysics and limit/promote the precipitation efficiency of clouds and, hence, alter the cloud lifetime (Gettelman and Sherwood, 2016). These cloud feedback mechanisms are still poorly constrained and contribute to the uncertainty in cloud radiative effect (CRE) estimations in both the PI and PD eras.
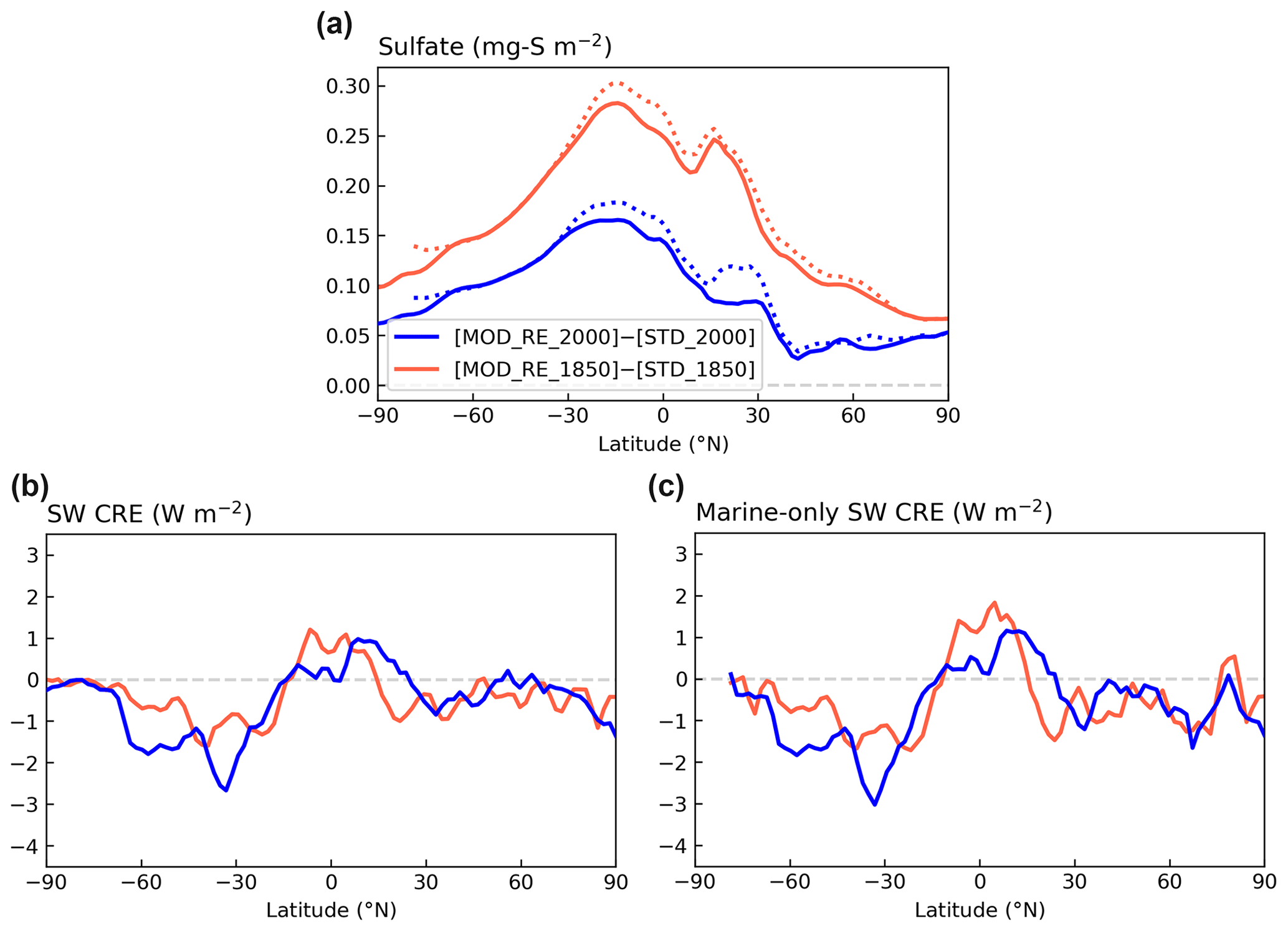
Figure 12Contrasting the zonal means of changes in (a) sulfate column concentration, (b) SW CRE, and (c) marine-only SW CRE modeled with [MOD_RE] and [STD] chemistry for PI and PD simulations. Dotted lines in panel (a) show the zonal mean marine grid cells only.
The global mean ΔCRE estimated by differencing [STD_2000] and [STD_1850] is −2.2 W m−2, comparable to a previous study, using CAM6 (Gettelman et al., 2019), which reported SW ΔCRE of −2.1 W m−2 with CMIP6 emissions. They also showed that, under CMIP5 emissions, the magnitude of the SW ΔCRE drops to −1.4 W m−2, which is closer to the AR5 range. Our expanded DMS chemistry leads to a modest (∼ 5 %) strengthening in the simulated IRF (−2.3 W m−2). However, due to the high variability in the cloud effects, these differences, both in the global values and the zonal means shown in Fig. 12c, are not statistically significant.
The strengthening of the IRF is opposite in sign to the expected response to the increase in PI sulfate. Carslaw et al. (2013) describe how cloud albedo is much more strongly sensitive to CCN in the PI, suggesting that higher PI aerosol burden may decrease the cloud response to anthropogenic increases. Figure 13 illustrates the change in SW CRE and sulfate burden from multiple simulations from the PI to the PD eras. Counter to expectations, we find that the SW CRE is more sensitive to each unit increment of sulfate burden (steeper slope) when the PI aerosol burden is higher (in [MOD_RE]; shown in dark red) compared to our baseline simulations ([STD]; shown in blue). Figure S10 shows that the PI-to-PD changes in CCN and cloud properties are also more sensitive to the change in sulfate burden in [MOD_RE] than [STD]. Hence, each unit PI-to-PD increase in sulfate burden in [MOD_RE] appears to induce more numerous and smaller cloud droplets and enrich cloud water content, thus enhancing cloud albedo. This could contribute to the enlarged change in SW CRE (or the IRF) in [MOD_RE], even though its PI-to-PD sulfate burden increment is smaller than [STD].

Figure 13PI-to-PD changes in the SW CRE and sulfate burden of simulations in this study. [STD] (blue) refers to the simulation with model default chemistry. [MOD_RE] (dark red) denote the cases with our expanded DMS chemistry implemented with all gas-phase, aerosol-phase, and in-cloud reactions. [GAS_RE] (yellow), which only includes the expanded gas-phase reactions, is also shown. Arrows indicate changes from PI (tails) to PD (heads). Horizontal and vertical error bars span the 1σ variabilities in the burden and SW CRE in the simulations, respectively.
This sensitivity may also be related to the spatial distribution in the change in DMS-derived sulfate burden. Figure 12 shows that the increase in sulfate burden from PI to PD is stronger in the marine atmosphere, as expected. In a sensitivity test, [GAS_RE], where only the gas-phase reactions of the new DMS oxidation scheme are enabled but not the aqueous-phase reactions (with the exception of aqueous-phase oxidation of SO2), produces a less negative IRF of 1.7 W m−2. Figure S11 illustrates that this sensitivity simulation follows the anticipated response, with an increase in the PI sulfate decreasing the change in the liquid water path (LWP) from the PI and the PD (particularly in low clouds) and, thus, dampening the IRF, indicating the important climate implications of marine stratocumulus clouds (Wood, 2012). Contrasting [MOD_RE] and [GAS_RE] reveals that the introduction of the aqueous-phase pathway contributes to large changes in the PD–PI sulfate co-located with high clouds over the tropics and low clouds over the Southern Ocean. Though the change in sulfate in both regions produces stronger regional cooling IRF, this appears to be the result of two different processes. In the tropics, decreases in sulfate in the presence of high clouds modify ice nucleation (Gettelman et al., 2010), leading to increased ice water path (IWP) and strengthened cloud cooling over the tropical oceans. Over the Southern Ocean, decreases in sulfate attributable to the aqueous-phase chemistry are associated with even higher LWP in low clouds, further exaggerating the local cooling IRF compared to the gas-phase-only simulation. Even though the amount of sulfate produced by the aqueous-phase pathway is relatively small (∼ 8 % of DMS-derived sulfate), it appears to have a disproportionate impact on clouds and the estimate of aerosol IRF, suggesting a strong sensitivity of cloud properties to the spatial distribution of natural marine sulfate. More work is needed to better understand this response.
We expand the chemical mechanism in CAM6-chem to better describe DMS oxidation in the atmosphere, determine the formation of the intermediate sulfur products, and estimate the aerosol radiative implications under the PI and PD periods.
Uncertainty in our estimate of sulfate response to the new DMS chemistry is largely associated with estimated reaction rates. Some rate constants for the multiphase reactions are obtained from a limited set of box model and laboratory studies which have not been validated with field measurements. For example, our rate constant for MS + OH(aq) from Zhu et al. (2003) is 4.7 times smaller than another lab study (Milne et al., 1989), potentially leading to a higher global tropospheric MSA burden by ∼ 30 % (Chen et al., 2018). As discussed in Sect. 3.2, it is likely that our model overestimates the concentration of particulate MSA and underestimate gaseous MSA when compared with in situ measurements (e.g., ATom; Yan et al., 2019) in the Southern Ocean. This could result in an overestimate of sulfate, given that gas-phase MSA is expected to have a longer lifetime than particulate MSA and H2SO4 vapor (Berresheim, 2002). Our comparisons with observations also suggest that emissions of DMS, in particular a likely overestimate over the Southern Ocean, play an important role in dictating the regional loading of secondary oxidation products; the climate response to these changes should be further investigated.
This study included a relatively new chemical mechanism for the formation and loss of HPMTF. The rate of isomerization of MSP (kiso) controls the production of HPMTF. The analyses reported here are based on a theoretically calculated value of kiso (0.04 s−1 at 293 K; Veres et al., 2020), which is slower than previous experiment- and model-based estimates of 0.23–2.1 s−1 (Wu et al., 2015; Berndt et al., 2019). We find that a faster kiso (0.12 s−1 at 293 K, based on Ye et al., 2021) has a negligible impact on the HPMTF burden (+4.1 %) and resulting sulfate formation. However, the HPMTF burden is quite sensitive to the loss of HPMTF due to the cloud uptake (kHPMTF+cloud), which was recently suggested as being a particularly important sink of HPMTF in the MBL (Veres et al., 2020; Vermeuel et al., 2020). These large changes suggest that further field measurements are needed to better understand the cloud uptake process of HPMTF and the resulting formation of in-cloud sulfur products.
In this study, we dramatically expand the DMS oxidation mechanism within an Earth system model. Doing so increases the global sulfate burden by 8.8 % in PD and 29 % in PI. While we anticipated that a larger PI burden of sulfate would dampen the aerosol IRF, our simulations instead suggested that the role of aqueous-phase chemistry, though modest in terms of the sulfate burden, confounds this effect. In a simulation with only updated gas-phase chemistry, the higher PI burden decreases the magnitude of the IRF, as anticipated (−2.2 W m−2 in standard chemistry vs. −1.7 W m−2 with updated gas-phase chemistry). However, high clouds in the tropics and low clouds in the Southern Ocean appear to be particularly sensitive to the sulfate produced via the aqueous-phase pathway, counteracting the effect of the additional sulfate formed via the gas-phase pathways (net −2.3 W m−2). These large differences confirm the high sensitivity of aerosol indirect effects to the natural aerosol background, while revealing complex cloud responses to aerosol produced in different geographical regions via different pathways. More work is needed to understand these responses (e.g., better understanding of cloud responses to aerosols formed in the aqueous phase and observational constraints on the cloud-uptake process of HPMTF via both laboratory and field measurements). While our new chemistry increases the computational costs by ∼ 15 % (in core hours per simulation year), this study suggests that a detailed description of the chemical oxidation of DMS and its products, and particularly the chemistry relevant to pristine conditions, is needed to accurately represent the abundance of natural sulfur species in the marine atmosphere and changes in natural aerosol burden over time.
The modified codes of CESM2 developed in this study will be made available upon request to the corresponding authors (Ka Ming Fung at kamingfung@mit.edu and Colette L. Heald at heald@mit.edu).
Our simulation results are reproducible using the setup described. ACE-ENA data were obtained from the Atmospheric Radiation Measurement (ARM) User Facility, a U.S. DOE Office of Science user facility managed by the Biological and Environmental Research Program (https://www.arm.gov/research/campaigns/aaf2017ace-ena, last access: 30 November 2021; J. Wang et al., 2021).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-1549-2022-supplement.
KMF, CLH, and JHK formulated the overarching research goals and aims. KMF and CLH designed the methodology. KMF implemented the new code into CAM6-chem, based on the standard model developed by SW, DSJ, AG, ZL, XL, and RAZ. KMF validated the model results against observational data provided by ECA, DRB, JLJ, PCJ, PRV, TSB, JES, and MZ. KMF analyzed the data and created the figures. KMF and CLH wrote the initial draft of this paper. All authors reviewed this paper.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Rebecca Schwantes, Simone Tilmes, and Louisa Emmons, for their contribution to the modeling of this study. This material is based upon work supported by the National Center for Atmospheric Research (NCAR), which is a major facility sponsored by the National Science Foundation (NSF; grant no. 1852977). The high-performance computing was conducted on Cheyenne (https://doi.org/10.5065/D6RX99HX, Computational and Information Systems Laboratory, 2019), provided by NCAR's Computational and Information Systems Laboratory, sponsored by NSF. We also thank Andy Neuman, Alan Bandy, Barry J. Huebert, and Stephen R. Springston, for their contribution to the measurements used in this study.
This research has been supported by the U.S. Department of Energy (DOE; grant no. DE-SC0018934); the U.S. DOE Office of Science, Office of Biological and Environmental Research (BER), and Earth and Environmental System Modeling (EESM) program as part of its Earth System Model Development (ESMD) activity; NASA (grant nos. 80NSSC18K0630, 80NSSC19K0124, and 80NSSC21K1451); Atmospheric Radiation Measurement (ARM) and the DOE's Atmospheric System Research, an Office of Science Biological and Environmental Research program.
This paper was edited by Anja Schmidt and reviewed by two anonymous referees.
Albrecht, B. A.: Aerosols, Cloud Microphysics, and Fractional Cloudiness, Science, 245, 1227–1230, https://doi.org/10.1126/science.245.4923.1227, 1989.
Allen, G., Coe, H., Clarke, A., Bretherton, C., Wood, R., Abel, S. J., Barrett, P., Brown, P., George, R., Freitag, S., McNaughton, C., Howell, S., Shank, L., Kapustin, V., Brekhovskikh, V., Kleinman, L., Lee, Y.-N., Springston, S., Toniazzo, T., Krejci, R., Fochesatto, J., Shaw, G., Krecl, P., Brooks, B., McMeeking, G., Bower, K. N., Williams, P. I., Crosier, J., Crawford, I., Connolly, P., Allan, J. D., Covert, D., Bandy, A. R., Russell, L. M., Trembath, J., Bart, M., McQuaid, J. B., Wang, J., and Chand, D.: South East Pacific atmospheric composition and variability sampled along 20∘ S during VOCALS-REx, Atmos. Chem. Phys., 11, 5237–5262, https://doi.org/10.5194/acp-11-5237-2011, 2011.
Andreae, M. O.: Ocean-atmosphere interactions in the global biogeochemical sulfur cycle, Mar. Chem., 30, 1–29, https://doi.org/10.1016/0304-4203(90)90059-L, 1990.
Andres, R. J. and Kasgnoc, A. D.: A time-averaged inventory of subaerial volcanic sulfur emissions, J. Geophys. Res., 103, 25251–25261, https://doi.org/10.1029/98JD02091, 1998.
Apel, E. C., Hornbrook, R. S., Hills, A. J., Blake, N. J., Barth, M. C., Weinheimer, A., Cantrell, C., Rutledge, S. A., Basarab, B., Crawford, J., Diskin, G., Homeyer, C. R., Campos, T., Flocke, F., Fried, A., Blake, D. R., Brune, W., Pollack, I., Peischl, J., Ryerson, T., Wennberg, P. O., Crounse, J. D., Wisthaler, A., Mikoviny, T., Huey, G., Heikes, B., O'Sullivan, D., and Riemer, D. D.: Upper tropospheric ozone production from lightning NOx-impacted convection: Smoke ingestion case study from the DC3 campaign, J. Geophys. Res.-Atmos., 120, 2505–2523, https://doi.org/10.1002/2014JD022121, 2015.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Ayers, G. P., Bentley, S. T., Ivey, J. P., and Forgan, B. W.: Dimethylsulfide in marine air at Cape Grim, 41∘ S, J. Geophys. Res.-Atmos., 100, 21013–21021, https://doi.org/10.1029/95JD02144, 1995.
Bardouki, H., Berresheim, H., Vrekoussis, M., Sciare, J., Kouvarakis, G., Oikonomou, K., Schneider, J., and Mihalopoulos, N.: Gaseous (DMS, MSA, SO2, H2SO4 and DMSO) and particulate (sulfate and methanesulfonate) sulfur species over the northeastern coast of Crete, Atmos. Chem. Phys., 3, 1871–1886, https://doi.org/10.5194/acp-3-1871-2003, 2003.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere, Chem. Rev., 36, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Barth, M. C., Rasch, P. J., Kiehl, J. T., Benkovitz, C. M., and Schwartz, S. E.: Sulfur chemistry in the National Center for Atmospheric Research Community Climate Model: Description, evaluation, features, and sensitivity to aqueous chemistry, J. Geophys. Res., 105, 1387–1415, https://doi.org/10.1029/1999JD900773, 2000.
Berglen, T. F.: A global model of the coupled sulfur/oxidant chemistry in the troposphere: The sulfur cycle, J. Geophys. Res., 109, D19310, https://doi.org/10.1029/2003JD003948, 2004.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019.
Berresheim, H.: Gas-aerosol relationships of H2 SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res., 107, 8100, https://doi.org/10.1029/2000JD000229, 2002.
Booth, M. M. and Powell, D. H.: Trace Organic Analysis by Gas Chromatography with Quadrupole Mass Spectrometry, in: Encyclopedia of Analytical Chemistry, American Cancer Society, https://doi.org/10.1002/9780470027318.a0876, 2006.
Boucher, O., Moulin, C., Belviso, S., Aumont, O., Bopp, L., Cosme, E., von Kuhlmann, R., Lawrence, M. G., Pham, M., Reddy, M. S., Sciare, J., and Venkataraman, C.: DMS atmospheric concentrations and sulphate aerosol indirect radiative forcing: a sensitivity study to the DMS source representation and oxidation, Atmos. Chem. Phys., 3, 49–65, https://doi.org/10.5194/acp-3-49-2003, 2003.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., and Lohmann, U.: Clouds and aerosols, in: Climate change 2013: the physical science basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 571–657, https://doi.org/10.1017/CBO9781107415324.016, 2013.
Breider, T. J., Chipperfield, M. P., Richards, N. A. D., Carslaw, K. S., Mann, G. W., and Spracklen, D. V.: Impact of BrO on dimethylsulfide in the remote marine boundary layer: IMPACT OF BRO + DMS IN RMBL, Geophys. Res. Lett., 37, L02807, https://doi.org/10.1029/2009GL040868, 2010.
Burkholder, J. B., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies: evaluation number 18, Jet Propulsion Laboratory, National Aeronautics and Space Administration, Pasadena, CA, USA, http://hdl.handle.net/2014/45510 (last access: 30 November 2021), 2015.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Carn, S. A., Fioletov, V. E., McLinden, C. A., Li, C., and Krotkov, N. A.: A decade of global volcanic SO2 emissions measured from space, Sci. Rep., 7, 44095, https://doi.org/10.1038/srep44095, 2017.
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., and Warren, S. G.: Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, Nature, 326, 655–661, https://doi.org/10.1038/326655a0, 1987.
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018.
Chin, M., Jacob, D. J., Gardner, G. M., Foreman-Fowler, M. S., Spiro, P. A., and Savoie, D. L.: A global three-dimensional model of tropospheric sulfate, J Geophys. Res.-Atmos., 101, 18667–18690, https://doi.org/10.1029/96JD01221, 1996.
Chin, M., Rood, R. B., Lin, S.-J., Müller, J.-F., and Thompson, A. M.: Atmospheric sulfur cycle simulated in the global model GOCART: Model description and global properties, J Geophys. Res.-Atmos., 105, 24671–24687, https://doi.org/10.1029/2000JD900384, 2000.
Computational and Information Systems Laboratory: HPE/SGI ICE XA System (University Community Computing), National Center for Atmospheric Research, Cheyenne, https://doi.org/10.5065/D6RX99HX, 2019.
Cosme, E., Genthon, C., Martinerie, P., Boucher, O., and Pham, M.: The sulfur cycle at high-southern latitudes in the LMD-ZT General Circulation Model, J. Geophys. Res.-Atmos., 107, ACH 7-1–ACH 7-19, https://doi.org/10.1029/2002JD002149, 2002.
Danabasoglu, G., Lamarque, J. -F., Bacmeister, J., Bailey, D. A., DuVivier, A. K., Edwards, J., Emmons, L. K., Fasullo, J., Garcia, R., Gettelman, A., Hannay, C., Holland, M. M., Large, W. G., Lauritzen, P. H., Lawrence, D. M., Lenaerts, J. T. M., Lindsay, K., Lipscomb, W. H., Mills, M. J., Neale, R., Oleson, K. W., Otto-Bliesner, B., Phillips, A. S., Sacks, W., Tilmes, S., Kampenhout, L., Vertenstein, M., Bertini, A., Dennis, J., Deser, C., Fischer, C., Fox-Kemper, B., Kay, J. E., Kinnison, D., Kushner, P. J., Larson, V. E., Long, M. C., Mickelson, S., Moore, J. K., Nienhouse, E., Polvani, L., Rasch, P. J., and Strand, W. G.: The Community Earth System Model Version 2 (CESM2), J. Adv. Model. Earth Syst., 12, e2019MS001916, https://doi.org/10.1029/2019MS001916, 2020.
Dentener, F. J. and Crutzen, P. J.: Reaction of N2O5 on tropospheric aerosols: Impact on the global distributions of NOx, O3, and OH, J. Geophys. Res.-Atmos., 98, 7149–7163, https://doi.org/10.1029/92JD02979, 1993.
Emmons, L. K., Walters, S., Hess, P. G., Lamarque, J.-F., Pfister, G. G., Fillmore, D., Granier, C., Guenther, A., Kinnison, D., Laepple, T., Orlando, J., Tie, X., Tyndall, G., Wiedinmyer, C., Baughcum, S. L., and Kloster, S.: Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4), Geosci. Model Dev., 3, 43–67, https://doi.org/10.5194/gmd-3-43-2010, 2010.
Emmons, L. K., Schwantes, R. H., Orlando, J. J., Tyndall, G., Kinnison, D., Lamarque, J., Marsh, D., Mills, M. J., Tilmes, S., Bardeen, C., Buchholz, R. R., Conley, A., Gettelman, A., Garcia, R., Simpson, I., Blake, D. R., Meinardi, S., and Pétron, G.: The Chemistry Mechanism in the Community Earth System Model Version 2 (CESM2), J. Adv. Model. Earth Syst., 12, e2019MS001882, https://doi.org/10.1029/2019MS001882, 2020.
Enami, S., Nakano, Y., Hashimoto, S., Kawasaki, M., Aloisio, S., and Francisco, J. S.: Reactions of Cl atoms with dimethyl sulfide: A theoretical calculation and an experimental study with cavity ring-down spectroscopy, J. Phys. Chem. A, 108, 7785–7789, 2004.
Fahey, K. M., Carlton, A. G., Pye, H. O. T., Baek, J., Hutzell, W. T., Stanier, C. O., Baker, K. R., Appel, K. W., Jaoui, M., and Offenberg, J. H.: A framework for expanding aqueous chemistry in the Community Multiscale Air Quality (CMAQ) model version 5.1, Geosci. Model Dev., 10, 1587–1605, https://doi.org/10.5194/gmd-10-1587-2017, 2017.
Faloona, I.: Sulfur processing in the marine atmospheric boundary layer: A review and critical assessment of modeling uncertainties, Atmos. Environ., 43, 2841–2854, https://doi.org/10.1016/j.atmosenv.2009.02.043, 2009.
Flyunt, R., Makogon, O., Schuchmann, M. N., Asmus, K.-D., and von Sonntag, C.: OH-Radical-induced oxidation of methanesulfinic acid. The reactions of the methanesulfonyl radical in the absence and presence of dioxygen, J. Chem. Soc., Perkin Trans. 2, 787–792, https://doi.org/10.1039/b009631h, 2001.
Gershenzon, M., Davidovits, P., Jayne, J. T., Kolb, C. E., and Worsnop, D. R.: Simultaneous Uptake of DMS and Ozone on Water, J. Phys. Chem. A, 105, 7031–7036, https://doi.org/10.1021/jp010696y, 2001.
Gettelman, A.: Putting the clouds back in aerosol–cloud interactions, Atmos. Chem. Phys., 15, 12397–12411, https://doi.org/10.5194/acp-15-12397-2015, 2015.
Gettelman, A. and Sherwood, S. C.: Processes Responsible for Cloud Feedback, Curr. Clim. Change Rep., 2, 179–189, https://doi.org/10.1007/s40641-016-0052-8, 2016.
Gettelman, A., Liu, X., Ghan, S. J., Morrison, H., Park, S., Conley, A. J., Klein, S. A., Boyle, J., Mitchell, D. L., and Li, J.-L. F.: Global simulations of ice nucleation and ice supersaturation with an improved cloud scheme in the Community Atmosphere Model, J. Geophys. Res., 115, D18216, https://doi.org/10.1029/2009JD013797, 2010.
Gettelman, A., Hannay, C., Bacmeister, J. T., Neale, R. B., Pendergrass, A. G., Danabasoglu, G., Lamarque, J. -F., Fasullo, J. T., Bailey, D. A., Lawrence, D. M., and Mills, M. J.: High Climate Sensitivity in the Community Earth System Model Version 2 (CESM2), Geophys. Res. Lett., 46, 8329–8337, https://doi.org/10.1029/2019GL083978, 2019.
Ghan, S. J.: Technical Note: Estimating aerosol effects on cloud radiative forcing, Atmos. Chem. Phys., 13, 9971–9974, https://doi.org/10.5194/acp-13-9971-2013, 2013.
Gondwe, M., Krol, M., Gieskes, W., Klaassen, W., and de Baar, H.: The contribution of ocean-leaving DMS to the global atmospheric burdens of DMS, MSA, SO2, and NSS SO, Global Biogeochem. Cy., 17, 1056, https://doi.org/10.1029/2002GB001937, 2003.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Heald, C. L., Ridley, D. A., Kroll, J. H., Barrett, S. R. H., Cady-Pereira, K. E., Alvarado, M. J., and Holmes, C. D.: Contrasting the direct radiative effect and direct radiative forcing of aerosols, Atmos. Chem. Phys., 14, 5513–5527, https://doi.org/10.5194/acp-14-5513-2014, 2014.
Hezel, P. J., Alexander, B., Bitz, C. M., Steig, E. J., Holmes, C. D., Yang, X., and Sciare, J.: Modeled methanesulfonic acid (MSA) deposition in Antarctica and its relationship to sea ice, J. Geophys. Res., 116, D23214, https://doi.org/10.1029/2011JD016383, 2011.
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, Proc. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hoffmann, E. H., Heinold, B., Kubin, A., Tegen, I., and Herrmann, H.: The Importance of the Representation of DMS Oxidation in Global Chemistry-Climate Simulations, Geophys. Res. Lett., 48, e2021GL094068, https://doi.org/10.1029/2021GL094068, 2021.
Huebert, B. J., Blomquist, B. W., Hare, J. E., Fairall, C. W., Johnson, J. E., and Bates, T. S.: Measurement of the sea-air DMS flux and transfer velocity using eddy correlation, Geophys. Res. Lett., 31, L23113, https://doi.org/10.1029/2004GL021567, 2004.
Hurrell, J. W., Hack, J. J., Shea, D., Caron, J. M., and Rosinski, J.: A new sea surface temperature and sea ice boundary dataset for the community atmosphere model, J. Climate, 21, 5145–5153, https://doi.org/10.1175/2008JCLI2292.1, 2008.
Jeffery, C. D., Robinson, I. S., and Woolf, D. K.: Tuning a physically-based model of the air–sea gas transfer velocity, Ocean Model., 31, 28–35, https://doi.org/10.1016/j.ocemod.2009.09.001, 2010.
Johnson, M. T.: A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas, Ocean Sci., 6, 913–932, https://doi.org/10.5194/os-6-913-2010, 2010.
Kaufman, Y. J. and Tanré, D.: Effect of variations in super-saturation on the formation of cloud condensation nuclei, Nature, 369, 45–48, https://doi.org/10.1038/369045a0, 1994.
Kettle, A. J. and Andreae, M. O.: Flux of dimethylsulfide from the oceans: A comparison of updated data sets and flux models, J. Geophys. Res.-Atmos., 105, 26793–26808, 2000.
Kettle, A. J., Andreae, M. O., Amouroux, D., Andreae, T. W., Bates, T. S., Berresheim, H., Bingemer, H., Boniforti, R., Curran, M. A. J., DiTullio, G. R., Helas, G., Jones, G. B., Keller, M. D., Kiene, R. P., Leck, C., Levasseur, M., Malin, G., Maspero, M., Matrai, P., McTaggart, A. R., Mihalopoulos, N., Nguyen, B. C., Novo, A., Putaud, J. P., Rapsomanikis, S., Roberts, G., Schebeske, G., Sharma, S., Simó, R., Staubes, R., Turner, S., and Uher, G.: A global database of sea surface dimethylsulfide (DMS) measurements and a procedure to predict sea surface DMS as a function of latitude, longitude, and month, Global Biogeochem. Cy., 13, 399–444, https://doi.org/10.1029/1999GB900004, 1999.
Khan, M. A. H., Gillespie, S. M. P., Razis, B., Xiao, P., Davies-Coleman, M. T., Percival, C. J., Derwent, R. G., Dyke, J. M., Ghosh, M. V., Lee, E. P. F., and Shallcross, D. E.: A modelling study of the atmospheric chemistry of DMS using the global model, STOCHEM-CRI, Atmos. Environ., 127, 69–79, https://doi.org/10.1016/j.atmosenv.2015.12.028, 2016.
Kilgour, D. B., Novak, G. A., Sauer, J. S., Moore, A. N., Dinasquet, J., Amiri, S., Franklin, E. B., Mayer, K., Winter, M., Morris, C. K., Price, T., Malfatti, F., Crocker, D. R., Lee, C., Cappa, C. D., Goldstein, A. H., Prather, K. A., and Bertram, T. H.: Marine gas-phase sulfur emissions during an induced phytoplankton bloom, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-615, in review, 2021.
Kloster, S., Feichter, J., Maier-Reimer, E., Six, K. D., Stier, P., and Wetzel, P.: DMS cycle in the marine ocean-atmosphere system – a global model study, Biogeosciences, 3, 29–51, https://doi.org/10.5194/bg-3-29-2006, 2006.
Kulmala, M., Pirjola, L., and Mäkelä, J.: Stable sulfate clusters as a source of new atmospheric particles, Nature, 404, 66–69, https://doi.org/10.1038/35003550, 2000.
Lamarque, J.-F., Emmons, L. K., Hess, P. G., Kinnison, D. E., Tilmes, S., Vitt, F., Heald, C. L., Holland, E. A., Lauritzen, P. H., Neu, J., Orlando, J. J., Rasch, P. J., and Tyndall, G. K.: CAM-chem: description and evaluation of interactive atmospheric chemistry in the Community Earth System Model, Geosci. Model Dev., 5, 369–411, https://doi.org/10.5194/gmd-5-369-2012, 2012.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004 https://doi.org/10.1029/2010GB003850, 2011.
Lennartz, S. T., Krysztofiak, G., Marandino, C. A., Sinnhuber, B.-M., Tegtmeier, S., Ziska, F., Hossaini, R., Krüger, K., Montzka, S. A., Atlas, E., Oram, D. E., Keber, T., Bönisch, H., and Quack, B.: Modelling marine emissions and atmospheric distributions of halocarbons and dimethyl sulfide: the influence of prescribed water concentration vs. prescribed emissions, Atmos. Chem. Phys., 15, 11753–11772, https://doi.org/10.5194/acp-15-11753-2015, 2015.
Liu, X., Easter, R. C., Ghan, S. J., Zaveri, R., Rasch, P., Shi, X., Lamarque, J.-F., Gettelman, A., Morrison, H., Vitt, F., Conley, A., Park, S., Neale, R., Hannay, C., Ekman, A. M. L., Hess, P., Mahowald, N., Collins, W., Iacono, M. J., Bretherton, C. S., Flanner, M. G., and Mitchell, D.: Toward a minimal representation of aerosols in climate models: description and evaluation in the Community Atmosphere Model CAM5, Geosci. Model Dev., 5, 709–739, https://doi.org/10.5194/gmd-5-709-2012, 2012.
Liu, X., Ma, P.-L., Wang, H., Tilmes, S., Singh, B., Easter, R. C., Ghan, S. J., and Rasch, P. J.: Description and evaluation of a new four-mode version of the Modal Aerosol Module (MAM4) within version 5.3 of the Community Atmosphere Model, Geosci. Model Dev., 9, 505–522, https://doi.org/10.5194/gmd-9-505-2016, 2016.
Lu, Z., Liu, X., Zaveri, R. A., Easter, R. C., Tilmes, S., Emmons, L. K., Vitt, F., Singh, B., Wang, H., Zhang, R., and Rasch, P. J.: Radiative Forcing of Nitrate Aerosols From 1975 to 2010 as Simulated by MOSAIC Module in CESM2-MAM4, J. Geophys. Res.-Atmos., 126, e2021JD034809, https://doi.org/10.1029/2021JD034809, 2021.
Lucas, D. D.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res., 107, 4201, https://doi.org/10.1029/2001JD000843, 2002.
Mackay, D. and Yeun, A. T. K.: Mass transfer coefficient correlations for volatilization of organic solutes from water, Environ. Sci. Technol., 17, 211–217, https://doi.org/10.1021/es00110a006, 1983.
Meinshausen, M., Vogel, E., Nauels, A., Lorbacher, K., Meinshausen, N., Etheridge, D. M., Fraser, P. J., Montzka, S. A., Rayner, P. J., Trudinger, C. M., Krummel, P. B., Beyerle, U., Canadell, J. G., Daniel, J. S., Enting, I. G., Law, R. M., Lunder, C. R., O'Doherty, S., Prinn, R. G., Reimann, S., Rubino, M., Velders, G. J. M., Vollmer, M. K., Wang, R. H. J., and Weiss, R.: Historical greenhouse gas concentrations for climate modelling (CMIP6), Geosci. Model Dev., 10, 2057–2116, https://doi.org/10.5194/gmd-10-2057-2017, 2017.
Menon, S., Genio, A. D. D., Koch, D., and Tselioudis, G.: GCM simulations of the aerosol indirect effect: Sensitivity to cloud parameterization and aerosol burden, J. Atmos. Chem., 59, 692–713, 2002.
Mills, M. J., Schmidt, A., Easter, R., Solomon, S., Kinnison, D. E., Ghan, S. J., Neely, R. R., Marsh, D. R., Conley, A., Bardeen, C. G., and Gettelman, A.: Global volcanic aerosol properties derived from emissions, 1990-2014, using CESM1(WACCM), J. Geophys. Res.-Atmos., 121, 2332–2348, https://doi.org/10.1002/2015JD024290, 2016.
Milne, P. J., Zika, R. G., and Saltzman, E. S.: Rate of Reaction of Methanesulfonic Acid, Dimethyl Sulfoxide, and Dimethyl Sulfone with Hydroxyl Radical in Aqueous Solution, in: Biogenic Sulfur in the Environment, American Chemical Society, vol. 393, 518–528, https://doi.org/10.1021/bk-1989-0393.ch033, 1989.
Miyazaki, K., Eskes, H. J., and Sudo, K.: Global NOx emission estimates derived from an assimilation of OMI tropospheric NO2 columns, Atmos. Chem. Phys., 12, 2263–2288, https://doi.org/10.5194/acp-12-2263-2012, 2012.
Myhre, G., Shindell, D., Bréon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Zhang, H.: Anthropogenic and Natural Radiative Forcing, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, https://doi.org/10.1017/CBO9781107415324.018, 2013.
Neely, R. R. and Schmidt, A.: VolcanEESM: Global volcanic sulphur dioxide (SO2) emissions database from 1850 to present – Version 1.0 (1.0), Centre for Environmental Data Analysis [data set], https://doi.org/10.5285/76EBDC0B-0EED-4F70-B89E-55E606BCD568, 2016.
Nightingale, P. D., Malin, G., Law, C. S., Watson, A. J., Liss, P. S., Liddicoat, M. I., Boutin, J., and Upstill-Goddard, R. C.: In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers, Global Biogeochem. Cy., 14, 373–387, https://doi.org/10.1029/1999GB900091, 2000.
Patroescu, I. V., Barnes, I., and Becker, K. H.: FTIR Kinetic and Mechanistic Study of the Atmospheric Chemistry of Methyl Thiolformate, J. Phys. Chem., 100, 17207–17217, https://doi.org/10.1021/jp961452u, 1996.
Pham, M., Müller, J.-F., Brasseur, G. P., Granier, C., and Mégie, G.: A three-dimensional study of the tropospheric sulfur cycle, J. Geophys. Res., 100, 26061, https://doi.org/10.1029/95JD02095, 1995.
Rap, A., Scott, C. E., Spracklen, D. V., Bellouin, N., Forster, P. M., Carslaw, K. S., Schmidt, A., and Mann, G.: Natural aerosol direct and indirect radiative effects, Geophys. Res. Lett., 40, 3297–3301, https://doi.org/10.1002/grl.50441, 2013.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Sciare, J., Mihalopoulos, N., and Dentener, F. J.: Interannual variability of atmospheric dimethylsulfide in the southern Indian Ocean, J. Geophys. Res., 105, 26369–26377, https://doi.org/10.1029/2000JD900236, 2000.
Sehested, K. and Holcman, J.: A pulse radiolysis study of the OH radical induced autoxidation of methanesulfinic acid, Radiat. Phys. Chem., 47, 357–360, https://doi.org/10.1016/0969-806X(95)00115-E, 1996.
Simpson, I. J., Colman, J. J., Swanson, A. L., Bandy, A. R., Thornton, D. C., Blake, D. R., and Rowland, F. S.: Aircraft measurements of dimethyl sulfide (DMS) using a whole air sampling technique, J. Atmos. Chem., 39, 191–213, https://doi.org/10.1023/A:1010608529779, 2001.
Spracklen, D. V., Pringle, K. J., Carslaw, K. S., Chipperfield, M. P., and Mann, G. W.: A global off-line model of size-resolved aerosol microphysics: I. Model development and prediction of aerosol properties, Atmos. Chem. Phys., 5, 2227–2252, https://doi.org/10.5194/acp-5-2227-2005, 2005.
Stieger, B., van Pinxteren, D., Tilgner, A., Spindler, G., Poulain, L., Grüner, A., Wallasch, M., and Herrmann, H.: Strong Deviations from Thermodynamically Expected Phase Partitioning of Low-Molecular-Weight Organic Acids during One Year of Rural Measurements, ACS Earth Space Chem., 5, 500–515, https://doi.org/10.1021/acsearthspacechem.0c00297, 2021.
Tilmes, S., Hodzic, A., Emmons, L. K., Mills, M. J., Gettelman, A., Kinnison, D. E., Park, M., Lamarque, J. -F., Vitt, F., Shrivastava, M., Campuzano-Jost, P., Jimenez, J. L., and Liu, X.: Climate Forcing and Trends of Organic Aerosols in the Community Earth System Model (CESM2), J. Adv. Model. Earth Syst., 11, 4323–4351, https://doi.org/10.1029/2019MS001827, 2019.
Twomey, S.: The Influence of Pollution on the Shortwave Albedo of Clouds, J. Atmos. Sci., 34, 1149–1152, 1977.
van Marle, M. J. E., Kloster, S., Magi, B. I., Marlon, J. R., Daniau, A.-L., Field, R. D., Arneth, A., Forrest, M., Hantson, S., Kehrwald, N. M., Knorr, W., Lasslop, G., Li, F., Mangeon, S., Yue, C., Kaiser, J. W., and van der Werf, G. R.: Historic global biomass burning emissions for CMIP6 (BB4CMIP) based on merging satellite observations with proxies and fire models (1750–2015), Geosci. Model Dev., 10, 3329–3357, https://doi.org/10.5194/gmd-10-3329-2017, 2017.
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, Proc. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020.
Vermeuel, M. P., Novak, G. A., Jernigan, C. M., and Bertram, T. H.: Diel Profile of Hydroperoxymethyl Thioformate: Evidence for Surface Deposition and Multiphase Chemistry, Environ. Sci. Technol., 54, 12521–12529, https://doi.org/10.1021/acs.est.0c04323, 2020.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wang, J., Wood, R., Jensen, M., Azevedo, E., Bretherton, C., and Chand, D.: Aerosol and Cloud Experiments in Eastern North Atlantic (ACE-ENA) Field Campaign Report, 18,https://www.osti.gov/servlets/purl/1526025 (last access: 30 November 2021), 2019.
Wang, J., Wood, R., Jensen, M. P., Chiu, J. C., Liu, Y., Lamer, K., Desai, N., Giangrande, S. E., Knopf, D. A., Kollias, P., Laskin, A., Liu, X., Lu, C., Mechem, D., Mei, F., Starzec, M., Tomlinson, J., Wang, Y., Yum, S. S., Zheng, G., Aiken, A. C., Azevedo, E. B., Blanchard, Y., China, S., Dong, X., Gallo, F., Gao, S., Ghate, V. P., Glienke, S., Goldberger, L., Hardin, J. C., Kuang, C., Luke, E. P., Matthews, A. A., Miller, M. A., Moffet, R., Pekour, M., Schmid, B., Sedlacek, A. J., Shaw, R. A., Shilling, J. E., Sullivan, A., Suski, K., Veghte, D. P., Weber, R., Wyant, M., Yeom, J., Zawadowicz, M., and Zhang, Z.: Aerosol and Cloud Experiments in the Eastern North Atlantic (ACE-ENA), B. Am. Meteorol. Soc., 1, 1–51, https://doi.org/10.1175/BAMS-D-19-0220.1, 2021 (data available at: https://www.arm.gov/research/campaigns/aaf2017ace-ena, last access: 30 November 2021).
Wang, S., Hornbrook, R. S., Hills, A., Emmons, L. K., Tilmes, S., Lamarque, J., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Crounse, J. D., Wennberg, P. O., Kim, M., Allen, H., Ryerson, T. B., Thompson, C. R., Peischl, J., Moore, F., Nance, D., Hall, B., Elkins, J., Tanner, D., Huey, L. G., Hall, S. R., Ullmann, K., Orlando, J. J., Tyndall, G. S., Flocke, F. M., Ray, E., Hanisco, T. F., Wolfe, G. M., St. Clair, J., Commane, R., Daube, B., Barletta, B., Blake, D. R., Weinzierl, B., Dollner, M., Conley, A., Vitt, F., Wofsy, S. C., Riemer, D. D., and Apel, E. C.: Atmospheric Acetaldehyde: Importance of Air-Sea Exchange and a Missing Source in the Remote Troposphere, Geophys. Res. Lett., 46, 5601–5613, https://doi.org/10.1029/2019GL082034, 2019a.
Wang, S., Kinnison, D., Montzka, S. A., Apel, E. C., Hornbrook, R. S., Hills, A. J., Blake, D. R., Barletta, B., Meinardi, S., Sweeney, C., Moore, F., Long, M., Saiz-Lopez, A., Fernandez, R. P., Tilmes, S., Emmons, L. K., and Lamarque, J.: Ocean Biogeochemistry Control on the Marine Emissions of Brominated Very Short-Lived Ozone-Depleting Substances: A Machine-Learning Approach, J. Geophys. Res.-Atmos., 124, 12319–12339, https://doi.org/10.1029/2019JD031288, 2019b.
Wang, S., Apel, E. C., Schwantes, R. H., Bates, K. H., Jacob, D. J., Fischer, E. V., Hornbrook, R. S., Hills, A. J., Emmons, L. K., Pan, L. L., Honomichl, S., Tilmes, S., Lamarque, J., Yang, M., Marandino, C. A., Saltzman, E. S., Bruyn, W., Kameyama, S., Tanimoto, H., Omori, Y., Hall, S. R., Ullmann, K., Ryerson, T. B., Thompson, C. R., Peischl, J., Daube, B. C., Commane, R., McKain, K., Sweeney, C., Thames, A. B., Miller, D. O., Brune, W. H., Diskin, G. S., DiGangi, J. P., and Wofsy, S. C.: Global Atmospheric Budget of Acetone: Air-Sea Exchange and the Contribution to Hydroxyl Radicals, J. Geophys. Res.-Atmos., 125, e2020JD032553, https://doi.org/10.1029/2020JD032553, 2020.
Wang, X., Jacob, D. J., Downs, W., Zhai, S., Zhu, L., Shah, V., Holmes, C. D., Sherwen, T., Alexander, B., Evans, M. J., Eastham, S. D., Neuman, J. A., Veres, P. R., Koenig, T. K., Volkamer, R., Huey, L. G., Bannan, T. J., Percival, C. J., Lee, B. H., and Thornton, J. A.: Global tropospheric halogen (Cl, Br, I) chemistry and its impact on oxidants, Atmos. Chem. Phys., 21, 13973–13996, https://doi.org/10.5194/acp-21-13973-2021, 2021.
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E. C., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgeois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., Chin, M., Commane, R., Correa, G., Crounse, J .D., Cullis, P. D., Daube, B. C., Day, D. A., Dean-Day, J. M., Dibb, J. E., DiGangi, J. P., Diskin, G. S., Dollner, M., Elkins, J. W., Erdesz, F., Fiore, A. M., Flynn, C. M., Froyd, K. D., Gesler, D. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hills, A. J., Hintsa, E. J., Hoffman, A., Hornbrook, R. S., Huey, L. G., Hughes, S., Jimenez, J. L., Johnson, B. J., Katich, J. M., Keeling, R. F., Kim, M. J., Kupc, A., Lait, L. R., Lamarque, J.-F., Liu, J., McKain, K., Mclaughlin, R. J., Meinardi, S., Miller, D. O., Montzka, S. A., Moore, F. L., Morgan, E. J., Murphy, D. M., Murray, L. T., Nault, B. A., Neuman, J. A., Newman, P. A., Nicely, J. M., Pan, X., Paplawsky, W., Peischl, J., Prather, M. J., Price, D. J., Ray, E. A., Reeves, J. M., Richardson, M., Rollins, A. W., Rosenlof, K. H., Ryerson, T. B., Scheuer, E., Schill, G. P., Schroder, J. C., Schwarz, J. P., St. Clair, J. M., Steenrod, S. D., Stephens, B. B., Strode, S. A., Sweeney, C., Tanner, D., Teng, A. P., Thames, A. B., Thompson, C. R., Ullmann, K., Veres, P. R., Vieznor, N., Wagner, N. L., Watt, A., Weber, R., Weinzierl, B., Wennberg, P. O., Williamson, C. J., Wilson, J. C., Wolfe, G. M., Woods, C. T., and Zeng, L. H.: Atmospheric Tomography Mission (ATom)ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, ORNL DAAC, Oak Ridge, Tennessee, USA [data set], https://doi.org/10.3334/ORNLDAAC/1581, 2018.
Wood, R.: Stratocumulus Clouds, Mon. Weather Rev., 140, 2373–2423, https://doi.org/10.1175/MWR-D-11-00121.1, 2012.
Wood, R., Mechoso, C. R., Bretherton, C. S., Weller, R. A., Huebert, B., Straneo, F., Albrecht, B. A., Coe, H., Allen, G., Vaughan, G., Daum, P., Fairall, C., Chand, D., Gallardo Klenner, L., Garreaud, R., Grados, C., Covert, D. S., Bates, T. S., Krejci, R., Russell, L. M., de Szoeke, S., Brewer, A., Yuter, S. E., Springston, S. R., Chaigneau, A., Toniazzo, T., Minnis, P., Palikonda, R., Abel, S. J., Brown, W. O. J., Williams, S., Fochesatto, J., Brioude, J., and Bower, K. N.: The VAMOS Ocean-Cloud-Atmosphere-Land Study Regional Experiment (VOCALS-REx): goals, platforms, and field operations, Atmos. Chem. Phys., 11, 627–654, https://doi.org/10.5194/acp-11-627-2011, 2011.
Wu, R., Wang, S., and Wang, L.: New Mechanism for the Atmospheric Oxidation of Dimethyl Sulfide. The Importance of Intramolecular Hydrogen Shift in a CH3 SCH2 OO Radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015.
Yan, J., Jung, J., Zhang, M., Xu, S., Lin, Q., Zhao, S., and Chen, L.: Significant Underestimation of Gaseous Methanesulfonic Acid (MSA) over Southern Ocean, Environ. Sci. Technol., 53, 13064–13070, https://doi.org/10.1021/acs.est.9b05362, 2019.
Yang, Y., Wang, H., Smith, S. J., Easter, R., Ma, P.-L., Qian, Y., Yu, H., Li, C., and Rasch, P. J.: Global source attribution of sulfate concentration and direct and indirect radiative forcing, Atmos. Chem. Phys., 17, 8903–8922, https://doi.org/10.5194/acp-17-8903-2017, 2017.
Ye, Q., Goss, M. B., Isaacman-VanWertz, G., Zaytsev, A., Massoli, P., Lim, C., Croteau, P., Canagaratna, M., Knopf, D. A., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Organic Sulfur Products and Peroxy Radical Isomerization in the OH Oxidation of Dimethyl Sulfide, ACS Earth Space Chem., 5, 2013–2020, https://doi.org/10.1021/acsearthspacechem.1c00108, 2021.
Zaveri, R. A., Easter, R. C., Fast, J. D., and Peters, L. K.: Model for Simulating Aerosol Interactions and Chemistry (MOSAIC), J. Geophys. Res., 113, D13204, https://doi.org/10.1029/2007JD008782, 2008.
Zaveri, R. A., Easter, R. C., Singh, B., Wang, H., Lu, Z., Tilmes, S., Emmons, L. K., Vitt, F., Zhang, R., Liu, X., Ghan, S. J., and Rasch, P. J.: Development and Evaluation of Chemistry-Aerosol-Climate Model CAM5-Chem-MAM7-MOSAIC: Global Atmospheric Distribution and Radiative Effects of Nitrate Aerosol, J. Adv. Model Earth Syst., 13, e2020MS002346, https://doi.org/10.1029/2020MS002346, 2021.
Zawadowicz, M. A., Suski, K., Liu, J., Pekour, M., Fast, J., Mei, F., Sedlacek, A. J., Springston, S., Wang, Y., Zaveri, R. A., Wood, R., Wang, J., and Shilling, J. E.: Aircraft measurements of aerosol and trace gas chemistry in the eastern North Atlantic, Atmos. Chem. Phys., 21, 7983–8002, https://doi.org/10.5194/acp-21-7983-2021, 2021.
Zhu, L., Nicovich, J. M., and Wine, P. H.: Temperature-dependent kinetics studies of aqueous phase reactions of hydroxyl radicals with dimethylsulfoxide, dimethylsulfone, and methanesulfonate, Aquat. Sci. – Research Across Boundaries, 65, 425–435, https://doi.org/10.1007/s00027-003-0673-6, 2003.
Zhu, L., Nenes, A., Wine, P. H., and Nicovich, J. M.: Effects of aqueous organosulfur chemistry on particulate methanesulfonate to non–sea salt sulfate ratios in the marine atmosphere, J. Geophys. Res., 111, D05316, https://doi.org/10.1029/2005JD006326, 2006.
- Abstract
- Introduction
- Model description
- Implications of the extended DMS oxidation mechanism
- Global radiative impacts of updated DMS chemistry
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Model description
- Implications of the extended DMS oxidation mechanism
- Global radiative impacts of updated DMS chemistry
- Conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement