the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Nov 2022
| 16 Nov 2022
Oligomer formation from the gas-phase reactions of Criegee intermediates with hydroperoxide esters: mechanism and kinetics
Long Chen
Yu Huang
Yonggang Xue
Zhihui Jia
Wenliang Wang
Hydroperoxide esters, formed in the reactions of carbonyl oxides (also called Criegee intermediates, CIs) with formic acid, play a crucial role in the formation of secondary organic aerosol (SOA) in the atmosphere. However, the transformation mechanism of hydroperoxide esters in the presence of stabilized Criegee intermediates (SCIs) is not well understood. Herein, the oligomerization reaction mechanisms and kinetics of distinct SCI (CH2OO, syn-CH3CHOO, anti-CH3CHOO, and (CH3)2COO) reactions, with their respective hydroperoxide esters and with hydroperoxymethyl formate (HPMF), are investigated in the gas phase using quantum chemical and kinetics modeling methods. The calculations show that the addition reactions of SCIs with hydroperoxide esters proceed through successive insertion of SCIs into hydroperoxide ester to form oligomers that involve SCIs as the repeated chain unit. The saturated vapor pressure and saturated concentration of the formed oligomers decrease monotonically as the number of SCIs is increased. The exothermicity of oligomerization reactions decreases significantly when the number of methyl substituents increases, and the exothermicity of anti-methyl substituted carbonyl oxides is obviously higher than that of syn-methyl substituted carbonyl oxides. The −OOH insertion reaction is energetically more feasible than the −CH insertion pathway in the SCI oligomerization reactions, and the barrier heights increase with increasing the number of SCIs added to the oligomer, except for syn-CH3CHOO. For the reactions of distinct SCIs with HPMF, the barrier of the −OOH insertion pathway shows a dramatic decrease when a methyl substituent occurs at the anti-position, while it reveals a significant increase when a methyl group is introduced at the syn-position and dimethyl substituent. Compared with the rate coefficients of the CH2OO + HPMF reaction, the rate coefficients increase by about 1 order of magnitude when a methyl substituent occurs at the anti-position, whereas the rate coefficients decrease by 1–2 orders of magnitude when a methyl group is introduced at the syn-position. These new findings advance our current understanding of the influence of Criegee chemistry on the formation and growth processes and the chemical compositions of SOA.
- Article
(1342 KB) - Full-text XML
-
Supplement
(2292 KB) - BibTeX
- EndNote





Alkenes are an important class of volatile organic compounds (VOCs) that are emitted into the atmosphere from large quantities of biogenic and anthropogenic sources (Lester and Klippenstein, 2018). The reaction with ozone is one of the dominant degradation pathways for alkenes in the atmosphere (Johnson and Marston, 2008; Atkinson and Arey, 2003). Ozonolysis of alkene proceeds through the electrophilic 1,3-cycloaddition of ozone to a C=C bond of alkenes to form a primary ozonide (POZ), and then it rapidly decomposes into a carbonyl compound and a carbonyl oxide (also called Criegee intermediates, CIs; Criegee, 1975; Osborn and Taatjes, 2015; Giorio et al., 2017). Part of the initially energized CIs (∼ 37 %–50 %) may promptly dissociate to OH radicals, which are thought to be an important nonphotolytic source of OH radicals in the atmosphere (Novelli et al., 2014; Liu et al., 2014). The remaining CIs (∼ 63 %–50 %) are collisionally stabilized prior to the thermal unimolecular decay (Lester and Klippenstein, 2018; Novelli et al., 2014; Anglada and Solé, 2016). The stabilized Criegee intermediates (SCIs) can proceed with bimolecular reactions with various trace species such as H2O, NO2, SO2, and HCOOH to generate secondary organic aerosol (SOA), thus profoundly influencing the air quality, global climate, and human health (Osborn and Taatjes, 2015; Khan et al., 2018; Lin and Chao, 2017; Liu et al., 2019; Chhantyal-Pun et al., 2018; Gong and Chen, 2021; Taatjes, 2017).
Formic acid (HCOOH), one of the most abundant carboxylic acids, has a significant influence on rainwater acidity in remote areas, where pH reduces by 0.25–0.5 in the presence of HCOOH (Stavrakou et al., 2012; Wang et al., 2020; Chaliyakunnel et al., 2016). It also plays an important role in the formation of cloud condensation nuclei (CCN), indirectly influencing radiative forcing and climate change (Yu, 2000). The primary sources of HCOOH include biomass burning, human activities, tropical and boreal forests, as well as the secondary sources involving the photochemical oxidation of non-methane hydrocarbons, such as ketene enols, vinyl alcohol, isoprene, and terpenoids (Stavrakou et al., 2012; Wang et al., 2020; Chaliyakunnel et al., 2016; So et al., 2014; Paulot et al., 2011). According to satellite measurements, the production of HCOOH is up to 100–120 Tg yr−1, and the value is expected to increase due to the acceleration of industrialization and urbanization (Stavrakou et al., 2012). Recent kinetics measurements have revealed that the reaction with HCOOH is a more important loss process for SCI than is presently assumed, especially in terrestrial equatorial areas and in high SCI concentration areas (Welz et al., 2014; Chung et al., 2019). The formed hydroperoxide esters have been identified as the low-volatility and high-oxygenated compounds, contributing to the formation and growth of SOA (Welz et al., 2014; Vansco et al., 2021; Sakamoto et al., 2017; Riva et al., 2017).
Welz et al. (2014) directly determined the rate coefficients for the reactions of CH2OO and CH3CHOO with formic and acetic acid by employing multiplexed photoionization mass spectrometry and cavity-enhanced broadband ultraviolet absorption spectroscopy. They found that the measured rate coefficients are in the excess of cm3 molec.−1 s−1, which are several orders of magnitude greater than those derived from previous experimental studies (Johnson et al., 2001; Tobias and Ziemann, 2001). Sipilä et al. (2014) conducted a competitive reaction kinetics experiment to investigate the reactions of acetone oxide ((CH3)2OO) with SO2, HCOOH, and CH3COOH, and they concluded that the rate coefficients of the (CH3)2OO + reactions are faster than that of the (CH3)2OO + SO2 system by about 3 times (Sipilä et al., 2014). These high rate coefficients could make the reaction with carboxylic acids a substantial dominant chemical sink for carbonyl oxides in the atmosphere (Welz et al., 2014; Taatjes et al., 2019; Chhantyal-Pun et al., 2017). Quantum chemical calculations show that the reaction of CH2OO with HCOOH proceeds through a facile transfer of the hydrogen atom from the acidic OH group to the terminal oxygen of CH2OO to form hydroperoxymethyl formate (HPMF; Long et al., 2009; Vereecken, 2017; Porterfield et al., 2019). Chen et al. (2018) concluded the same by investigating the reactions of various carbonyl oxides with HCOOH and finding that the barrierless 1,4 insertion reaction is the most favorable pathway, and that the primary products are hydroperoxide esters. Caravan et al. (2020) employed high-level ab initio CCSD(T)-F12 methods to study the reaction of methyl vinyl ketone oxide (MVK oxide) with HCOOH, and they found that the barrierless net insertion of MVK oxide into HCOOH, leading to the formation of a functionalized hydroperoxide, is dominant over fragmentation to produce an alkoxy radical and OH radicals. Moreover, oligomerization reactions with hydroperoxides and peroxy radicals are identified as being one of the dominant loss processes for carbonyl oxides under atmospheric conditions (Sakamoto et al., 2013; Sadezky et al., 2008; Zhao et al., 2015; Chen et al., 2017, 2019). All the above milestone investigations provide important information for understanding the chemistry of Criegee intermediate in the presence of carboxylic acids. However, to the best of our knowledge, there are few studies on the oligomerization reactions of SCIs with hydroperoxide esters, which are important with regard to organic new particle and cloud condensation nuclei formations. Moreover, the relationship between the reactivity of SCIs and the nature of substituents remains uncertain in the SCIs oligomerization reactions.
In the present study, we mainly focus on the oligomerization reaction mechanisms and kinetics of four carbonyl oxides reactions with their respective hydroperoxide esters and with HPMF by employing quantum chemical calculations and kinetics modeling methods. For the initiation reactions of carbonyl oxides with formic acid, four kinds of pathways, including 1,4 O-H insertion, 1,2 O-H insertion, C-H insertion, and C=O cycloaddition, are considered. For the oligomerization reactions of the successive insertion of carbonyl oxides into hydroperoxide esters, two types of reactions involving −OOH and −CH insertions are taken into account. The selected carbonyl oxides, including CH2OO, syn- and anti-CH3CHOO, and (CH3)2CHOO, are anticipated upon the ozonolysis of ethylene, propylene, and 2,3-dimethyl-2-butene, whereas the hydroperoxide esters are assumed to arise from the bimolecular reactions of carbonyl oxides with formic acid in the atmosphere.
2.1 Electronic structure and energy calculations
The geometries of all stationary points, including reactants (Rs), intermediates (IMs), transition states (TSs), and products (Ps), are optimized at the M06-2X/6-311+G(2df,2p) level of theory, since the M06-2X functional has a reliable performance for predicting thermochemistry, kinetics, and hydrogen bonding interactions (Zhao and Truhlar, 2008). Harmonic vibrational frequencies are performed at the same level to verify the nature of transition state (NIMAG = 1) and minimum (NIMAG = 0), and to provide zero-point vibrational energy (ZPVE) and Gibbs free energy corrections (Gcorr), which are scaled by a factor of 0.98 (Alecu et al., 2010). Intrinsic reaction coordinate (IRC) calculations are carried out to verify that each transition state is connected to the desired reactant and product (Fukui, 1981). The single point energy (SPE) calculations are performed at the M06-2X/ma-TZVP level of theory based on the M06-2X/6-311+G(2df,2p) optimized geometries. Moreover, the basis set superposition error (BSSE) is performed by using the counterpoise method proposed by Boys and Bernardi (1970) to evaluate the stability of the pre-reactive complex (RC). Herein, the Gibbs free energy (G) is defined as the sum of the SPE and Gibbs correction (). Electronic energy (ΔE≠) and Gibbs free energy (ΔG≠) barriers are defined as the difference in energy between a TS and a RC ( and . Reaction Gibbs free energy (ΔG) is defined as the difference in energy between a P and a R (.
To further assess the reliability of the selected M06-2X/ma-TZVP method for SPE calculations, the single point energies of all stationary points involved in the initiation reactions of distinct SCIs with HCOOH are recalculated at the high-accuracy CCSD(T)/6-311+G(2df,2p) and QCISD(T)/6-311+G(2df,2p) levels of theory. The calculated results are summarized in Table S1 in the Supplement. This table shows that the ΔE≠ and ΔG≠ obtained using the QCISD(T) method are in excellent agreement with those obtained using the CCSD(T) approach. It is therefore the reason why the energies obtained using the CCSD(T) method are used as the benchmark for comparison. The mean absolute deviations (MADs) of ΔE≠ and ΔG≠ between the CCSD(T) and M06-2X methods are 0.43 and 0.41 kcal mol−1, respectively; the largest deviations of ΔE≠ and ΔG≠ are 1.0 and 1.1 kcal mol−1, respectively. These results reveal that the energies obtained using the M06-2X method are close to those obtained using the CCSD(T) approach. Therefore, the M06-2X/ma-TZVP method is suitable for investigating the SCI oligomerization reactions. In the following discussion, the energies are applied in terms of Gibbs free energy to describe the reaction mechanism, unless otherwise stated. All electronic structure calculations are carried out by using the Gaussian09 program (Frisch et al., 2009). The Multiwfn program and Visual Molecular Dynamics (VMD) are utilized to analyze and visualize the molecular orbitals of the relevant species (Lu and Chen, 2012; Humphrey et al., 1996).
2.2 Kinetics calculations
The rate coefficients for the barrierless 1,4 O-H insertion reactions are computed by employing the variable–reaction–coordinate variational transition state theory (VRC-VTST; Bao and Truhlar, 2017), in which the potential energies are calculated by direct dynamics using the M06-2X/6-311+G(2df,2p) method. Rate coefficients for the SCIs + HCOOH reactions are calculated using the E,J-resolved microcanonical variational theory (E,J-μVT) using a single-faceted dividing surface. In the VRC-VTST calculations, the reaction coordinate s is defined by pivot points, which are used to orient the reactants 1 and 2. s is defined as the minimal value of rij, where rij is the distance between pivot points i and j, i is a pivot point on reactant 1, and j is a pivot point on reactant 2. Two of the pivot points are located at a distance ±d from the center of mass (COM) of SCIs, and the other two pivot points are located at a distance ±d from the COM of HCOOH, with a fixed length of 0.05, 0.10, 0.15, 0.2, and 0.25 Å. Then, for a given choice of pivot points, the variationally lowest rate coefficients are minimized with respect to s at each of the temperatures. We observed that d=0.05 produces the best variation results, and only its value is reported in the present study.
The rate coefficients for the bimolecular reactions with the tight transition states are calculated by using the canonical transition state theory (CTST) along with one-dimensional asymmetric Eckart tunneling correction (Truhlar et al., 1996; Eckart, 1930). As shown in Fig. 1, the entrance pathway Entry 2 of the R1R2COO reaction with HCOOH consists of two steps, where (i) an intermediate IMent2 is formed via a barrierless process, and (ii) then it rearranges to the product Pent2 through a tight transition state TSent2. The whole reaction process can be described as Reaction (R1), as follows:
assuming that the rapid equilibrium is established between the IMent2 and reactants. According to the steady-state approximation (SSA), the total rate coefficient is approximately expressed as Eq. (1) (Zhang et al., 2012):
The equilibrium constant Keq is written as Eq. (2):
where σ refers to reaction symmetry number, QIM(T), , and denote the partition functions of intermediate, reactants R1 and R2 are equal to the multiplication of translational, rotational, vibrational, and electronic partition functions (Q=QrotQvibQtransQelec) (Mendes et al., 2014), T is the temperature in Kelvin, R is the ideal gas constant, and GR and GIM are the total Gibbs free energies of reactant and intermediate, respectively. The rate coefficient calculations are performed with the Polyrate 2017-C and KiSThelP 2019 programs (Canneaux et al., 2013; Zheng et al., 2018).
3.1 Initiation reactions of distinct SCIs with HCOOH
The reaction with HCOOH is one of the dominant loss processes for SCIs and is expected to trigger the formation of SOA in the atmosphere (Chhantyal-Pun et al., 2018; Cabezas and Endo, 2020; Zhao et al., 2018; Zhou et al., 2019). The potential energy surface (PES) of distinct SCI (CH2OO, syn- and anti-CH3CHOO, and (CH3)2COO) reactions with HCOOH is drawn in Fig. 1. The geometries of all stationary points are displayed in Fig. S1 in the Supplement. The relative free energy of each stationary point and free energy barrier (ΔG≠) of each elementary reaction are summarized in Table 1. As shown in Fig. 1, the bimolecular reaction of distinct SCIs with HCOOH proceeds via four possible pathways, namely (1) 1,4 O-H insertion (Entry 1), (2) 1,2 O-H insertion (Entry 2), (3) C-H insertion (Entry 3), and (4) C=O cycloaddition (Entry 4). For Entry 1, the addition reaction of CH2OO with HCOOH proceeds through the 1,4 O-H insertion of CH2OO into HCOOH to form a hydroperoxide ester HC(O)O-CH2OO-H with an exoergicity of 37.6 kcal molec.−1. The formation of HC(O)O-CH2OO-H is obtained through a concerted process of O2-H2 bond breaking in the HCOOH and O4-H2 and C2-O1 bonds forming. Despite an attempt by various methods, the corresponding transition state is still not located through the efforts of optimization. To further validate the barrierless process of 1,4 O-H insertion reaction, a relaxed scan over the O-H and C-O bonds is performed at the M06-2X/6-311+G(2df,2p) level of theory. The scans start from the optimized structure of the adduct products, and the O-H and C-O bond lengths are then increased in steps of 0.10 Å, while other geometric parameters are fully optimized. The zero of energy is set to be the energy of the adduct products. The electronic potential energy profiles along the O-H and C-O dissociation coordinate are presented in Fig. S2. As seen in Fig. S2a, the electronic potential energy of the minimum potential energy path (MEP) decreases monotonically when the bond length of the O-H and C-O bonds decreases, suggesting that the 1,4 O-H insertion reaction of CH2OO with HCOOH is indeed barrierless. A similar conclusion is also obtained from the electronic potential energy profiles for the anti-CH3CHOO + HCOOH, syn-CH3CHOO + HCOOH, and (CH3)2COO + HCOOH (Fig. S2b–d) reactions that 1,4 O-H insertion reactions are barrierless. This conclusion is further supported by the analogous reaction systems that the 1,4 O-H insertion reactions of carbonyl oxides with carboxylic acids are a barrierless process, including the concerted hydrogen atom transfer and new C-O bond formation (Chhantyal-Pun et al., 2017; Long et al., 2009; Vereecken, 2017; Cabezas and Endo, 2019; Lin et al., 2019).
The exothermicities of 1,4 O-H insertion reactions of distinct SCIs with HCOOH are assessed by the reaction enthalpies (), which are defined as the difference between the enthalpies of formation () of the products and reactants (). To the best of our knowledge, there are no literature values available on the enthalpies of formation of carbonyl oxides and hydroperoxide esters, except the simplest carbonyl oxide CH2OO. Therefore, the isodesmic reaction method is adopted to obtain the enthalpies of formation, and the results are listed in Table S2. An isodesmic reaction is a hypothetical reaction in which the type of chemical bonds in the reactants is the similar to those of chemical bonds in the products. The following isodesmic reaction is constructed because the experimental values of H2, CH4, and H2O are available ((H2) = 0.00 kcal mol−1; (CH4) = −17.82 kcal mol−1; (H2O) = −57.79 kcal mol−1).
As seen in Table S2, the enthalpy of formation of CH2OO is calculated to be 23.23 kcal mol−1, which is in good agreement with the available literature values (Chen et al., 2016; Karton et al., 2013). This result implies that the theoretical method employed herein is reasonable for the prediction of the thermochemical parameters. The enthalpies of formation of carbonyl oxides and hydroperoxide esters significantly decrease when increasing the number of methyl groups. Notably, the decreased values in the enthalpies of formation of carbonyl oxides are greater than those of hydroperoxide esters under the condition of the same number of methyl groups. For example, the enthalpy of formation of anti-CH3CHOO decreases by 12.95 kcal mol−1 compared to the enthalpy of formation of CH2OO (23.23 kcal mol−1), and the enthalpy of formation of Pent1b decreases by 12.12 kcal mol−1 compared to the enthalpy of formation of Pent1a (−112.08 kcal mol−1). The reaction enthalpies of the reactions of distinct SCIs with HCOOH have decreases of the order of −44.69 (CH2OO + HCOOH → Pent1a) < −43.86 (anti-CH3CHOO + HCOOH → Pent1b) < −38.13 (syn-CH3CHOO + HCOOH → Pent1c) < −37.12 kcal mol−1 ((CH3)2COO + HCOOH → Pent1d), indicating that the reaction enthalpies are highly dependent on the number and location of methyl groups. The trend in reaction enthalpies is consistent with the trend in the enthalpies of formation of carbonyl oxides.
For Entry 2, each addition reaction starts with the formation of a pre-reactive hydrogen bonded complex, IMent2, in the entrance channel. Then it immediately converts into product Pent2 through the 1,2 O-H insertion transition state. The formation of Pent2 is obtained via a concerted process of O2-H2 bond rupture in the HCOOH and O4-H2 and C2-O2 bonds forming. The reaction barrier ΔG≠ has increases of the order of 10.0 (CH2OO) < 13.0 (anti-CH3CHOO) < 14.6 (syn-CH3CHOO) ≈ 14.4 ((CH3)2COO) kcal mol−1, suggesting that the parent CH2OO + HCOOH reaction is favored kinetically. Compared with the barrier of the parent system, the barrier increases by 3.0 kcal mol−1 when a methyl substitution occurs at the R1 position, and the barrier increases by ∼ 5 kcal mol−1 when a methyl group is introduced at the R2 position and dimethyl substituent. The aforementioned result implies that the methyl-substituted CH2OO hinders the 1,2 O-H insertion of carbonyl oxides into formic acid. Notably, the exothermicity decreases significantly as the number of the methyl group is increased. The products Pent1 and Pent2 formed from Entries 1 and 2 are two conformations that differ in the orientation of the −C(O)H moiety over the −OOH group. The calculated result shows that Pent1 is more stable than Pent2 in energy due to the existence of intramolecular hydrogen bond between the hydrogen atom of −OOH group and carbonyl oxygen atom.
For Entry 3, the addition reaction begins with the formation of a pre-reactive complex, IMent3, in the entrance channel, and then it surmounts a barrier to reaction. However, the barriers of C-H insertion reactions are significantly high (21.8–27.6 kcal mol−1), such that they are of less importance in the atmosphere. The high reaction barriers might be attributed to the large bond dissociation energy (BDE) of C-H bond in the formic acid. For Entry 4, the addition reaction proceeds through a cyclization process of a C2-O1 and O4-C1 bond forming to produce a five-membered ring compound Pent4. The barrier of the C=O cycloaddition reaction in the CH2OO + HCOOH reaction is 5.8 kcal mol−1, which is lower than that of the corresponding channels in Entry 2 and Entry 3 by 4.2 and 16.0 kcal mol−1, respectively. The result reveals that the C=O cycloaddition reaction is feasible kinetically. A similar conclusion is also obtained from the reactions of HCOOH with syn- and anti-CH3CHOO and (CH3)2COO in that the C=O cycloaddition reactions are favored over 1,2 O-H and C-H insertion reactions.
The rate coefficients of each elementary pathway included in the initiation reactions of distinct SCIs with HCOOH are tabulated in Tables S3–S6. The total rate coefficient is equal to the sum of the rate coefficient of each elementary pathway. As shown in Table S3, the total rate coefficients of CH2OO reaction with HCOOH are in excess of cm3 molec.−1 s−1, and they exhibit a slightly negative temperature dependence in the temperature range of 273–400 K. At room temperature, is estimated to be cm3 molec.−1 s−1, which is in good agreement with the experimental values reported by Welz et al. (2014; [1.1±0.1] × 10−10), Chung et al. (2019; [1.4±0.3] × 10−10), and Peltola et al. (2020; [1.0±0.03] × 10−10). k(TSent1) is approximately equal to in the whole temperature range, and it decreases in the range from (273 K) to (400 K) cm3 molec.−1 s−1 with increasing temperature. k(TSent1) is several orders of magnitude greater than k(TSent2), k(TSent3), and k(TSent4) over the temperature range from 273 to 400 K. The result again shows that the barrierless 1,4 O-H insertion reaction is predominant. It should be noted that, although the barrier of Entry 2 is 4.2 kcal mol−1 higher than that of Entry 4, k(TSent2) is merely about 1–2 fold smaller than k(TSent4). The reason is ascribed to the fact that the C=O cycloaddition reaction is entropically unfavorable (Vereecken, 2017).
Equivalent to the case of the reaction of CH2OO with HCOOH, the total rate coefficient ktot-anti of the anti-CH3CHOO reaction with HCOOH also decreases slightly with the temperature increase (Table S4). This table in the Supplement shows that Entry 1 is kinetically favored over Entries 2, 3, and 4, and Entry 2 is competitive with Entry 4 in the temperature range of 273–400 K. A similar conclusion is also obtained from the results of the rate coefficients for the reactions of syn-CH3CHOO and (CH3)2COO with HCOOH in that Entry 1 is the dominant pathway (Tables S5–S6). It is worth mentioning that the competition of Entry 2 is significantly greater than that of Entry 4 in the syn-CH3CHOO + HCOOH and (CH3)2COO + HCOOH systems. Based on the above discussions, it can be concluded that the relative importance of different pathways is highly dependent on the number and location of methyl substituents in the carbonyl oxides. Notably, the rate coefficient of each elementary pathway included in the anti-CH3CHOO + HCOOH reaction is several orders of magnitude greater than that of the corresponding channel involved in the other SCIs + HCOOH systems. It is because anti-CH3CHOO is substantially more reactive toward HCOOH than other SCIs. A similar phenomenon has also observed from the reactivity of anti-CH3CHOO toward water and SO2 (Taatjes et al., 2013; Long et al., 2016; Huang et al., 2015; Cabezas and Endo, 2018). At ambient temperature, the total rate coefficients of HCOOH reactions with anti-CH3CHOO, syn-CH3CHOO, and (CH3)2COO are estimated to be 5.22, 2.18, and cm3 molec.−1 s−1, respectively, which are consistent with the prior experimental measurements of 5±3, 2.5±0.3, and cm3 molec.−1 s−1 (Welz et al., 2014; Sipilä et al., 2014).
In summary, the barrierless 1,4 O-H insertion reaction is the dominant pathway in the initiation reactions of distinct SCIs with HCOOH. This conclusion is consistent with the recent experimental results derived from the reactions of formic acid with methacrolein oxide (MACR-OO) and methyl vinyl ketone oxide (MVK-OO) that the 1,4 addition mechanism is energetically favorable (Vansco et al., 2021; Caravan et al., 2020). Therefore, in the present study, the adduct products Pent1 forming the barrierless 1,4 O-H insertion of carbonyl oxides into HCOOH are selected as the model compounds to investigate the oligomerization reaction mechanisms of carbonyl oxides reactions with hydroperoxide esters.

Figure 1Schematic PES for the possible entrance pathways of the initiation reactions of HCOOH with various SCIs (black, pink, blue, and red lines represent the 1,4 O-H insertion, 1,2 O-H insertion, C-H insertion, and C=O cycloaddition reactions, respectively).
Table 1Relative free energies of stationary points and free energy barriers (ΔG≠) at 298 K (in kcal mol−1) for the various SCI (R1R2COO, R1, and R2 = H, CH3) reactions with HCOOH calculated at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2 The reactions of distinct SCIs with their respective hydroperoxide esters
The formed hydroperoxide ester has two possible unimolecular decay pathways. The first is the direct O-O bond rupture, resulting in the formation of oxylmethylformate and OH radicals (Vereecken, 2017). The second is the −OH fragment binding to adjacent hydrogen atom, leading to the formation of anhydride and H2O (Aplincourt and Ruiz-López, 2000; Neeb et al., 1998). However, the barriers of these two unimolecular reactions are extremely high, such that they are of less importance in the atmosphere. The formed hydroperoxide ester possess −OOH and −OC(O)H groups, and both of them can serve as the reactive moieties to react with carbonyl oxides, giving rise to the formation of oligomers. In the present study, we mainly consider two types of pathways, i.e., (a) −OOH insertion and (b) −CH insertion, while the C=O cycloaddition reaction is not taken into account because it is entropically unfavorable (Vereecken, 2017; Lin et al., 2019). The aforementioned reactions are discussed in detail in the following subsections.
3.2.1 The reactions of 2CH2OO with Pent1a
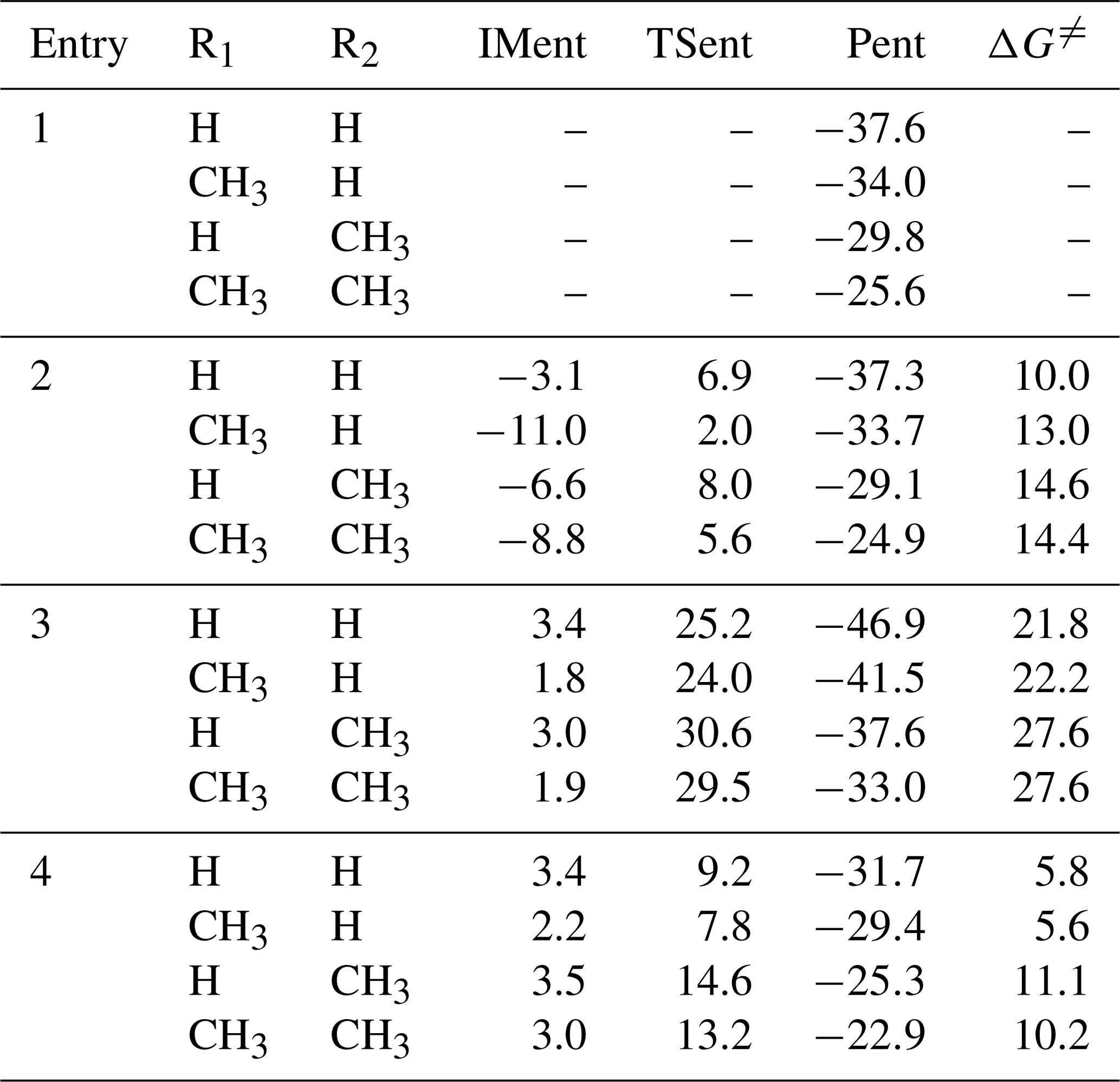
The simplest carbonyl oxide, CH2OO, originates from the reaction of all terminal alkenes with ozone (ozonolysis) in the atmosphere (Lin and Chao, 2017). The reaction with HCOOH is expected to be one of the dominant loss processes for CH2OO, and the main product is Pent1a (also called HPMF; Welz et al., 2014; Cabezas and Endo, 2019). A schematic PES for the addition reaction 2CH2OO + Pent1a is shown in Fig. 2, and the optimized geometries of all stationary points are displayed in Fig. S3. As seen in Fig. 2, the successive insertion of CH2OO into Pent1a eventually leads to the formation of oligomers P2a and P2b composed of CH2OO as the repeat unit. These oligomerization reactions are strongly exothermic and spontaneous (>83 kcal mol−1), implying that they are feasible thermodynamically.
The addition reaction 2CH2OO + Pent1a initially proceeds through two possible pathways, namely (1) −OOH insertion reaction R1a and (2) −CH insertion reaction R1b. For the −OOH insertion reaction R1a, the pre-reactive intermediate IM1a with a seven-membered ring structure is formed in the entrance channel, which is stabilized by the hydrogen bond interactions between the H4 atom of Pent1a and the O6 atom of CH2OO ( Å) and between the H6 atom of CH2OO and the O3 atom of Pent1a ( Å). Then IM1a converts into P1a (C3H6O6, HC(O)O–(CH2OO)2–H) via a concerted process of the O4-H4 bond breaking in the Pent1a and O4-C3 and H4-O6 bonds forming with a barrier of 8.1 kcal mol−1. For the −CH insertion reaction R1b, the pre-reactive intermediate IM1b with a seven-membered ring structure is formed in the entrance channel, which is stabilized by the van der Waals (vdW) interactions between the O3 atom of Pent1a and the C3 atom of CH2OO ( Å) and between the O6 atom of CH2OO and the C1 atom of Pent1a ( Å). Due to the absence of the hydrogen bond in IM1b, the energy of IM1b is lower than that of IM1a by 3.0 kcal mol−1. IM1b transforms into P1b (C3H6O6, HO2CH2OC(O)CH2OOH) via a concerted process of a C1-H1 bond breaking in the Pent1a and C1-C3 and H1-O6 bonds forming with a barrier of 21.5 kcal mol−1. By comparing the barriers of reactions R1a and R1b, it can be concluded that the −OOH insertion reaction is favored over the −CH insertion reaction. The high reaction barrier of reaction R1b is attributed to the large bond dissociation energy (BDE) of the C-H bond in the Pent1a. To gain further insight into the reaction mechanism of reaction R1a, the natural bond orbital (NBO) analysis of the donor–accepter orbitals involved in the TS1a is performed using the M06-2X wave function. The possible donor–accepter interactions are estimated by using the second-order perturbation theory. As illustrated in Fig. S4, the strong interactions are identified as the interaction of the lone pair orbital of O6 atom and the antibonding orbital of O4-H4 bond and the interaction of the lone pair orbital of O4 atom and the antibonding orbital of C3-O5 bond.
Similarly, the addition reaction CH2OO + P1a proceeds through the formation of the pre-reactive intermediates IM2a and IM2b in the entrance channel, which are stabilized by a hydrogen bond between the terminal oxygen atom of CH2OO and the reacting hydrogen atom of P1a and a van der Waals (vdW) interaction between the central carbon atom of CH2OO and the carbonyl oxygen atom of P1a. The relative energies of IM2a and IM2b, with respect to the separate reactants P1a and CH2OO, are −1.2 and 3.2 kcal mol−1, respectively, below the energies of the initial reactants 2CH2OO, and Pent1a are 41.6 and 37.2 kcal mol−1, respectively. Then they immediately transform into the respective products P2a and P2b through the −OOH and −CH insertion transition states TS2a and TS2b, with the barriers of 10.1 and 21.6 kcal mol−1. This result again shows that the −OOH insertion reaction is favored kinetically. It deserves mentioning that the barrier of −OOH insertion reaction increases as the number of CH2OO is increased. From the viewpoint of the geometrical parameters of TS2a and TS2b, the breaking O-H and C-H bonds are elongated by 14.8 % and 20.6 %, respectively, with respect to the equilibrium structures of IM2a and IM2b, while the forming C-O and C-C bond lengths are 2.013 and 2.264 Å, respectively. The result reveals that TS2a and TS2b are structurally reactant-like, which are consistent with the Hammond's (1955) hypothesis that the earlier transition states are generally exothermic.
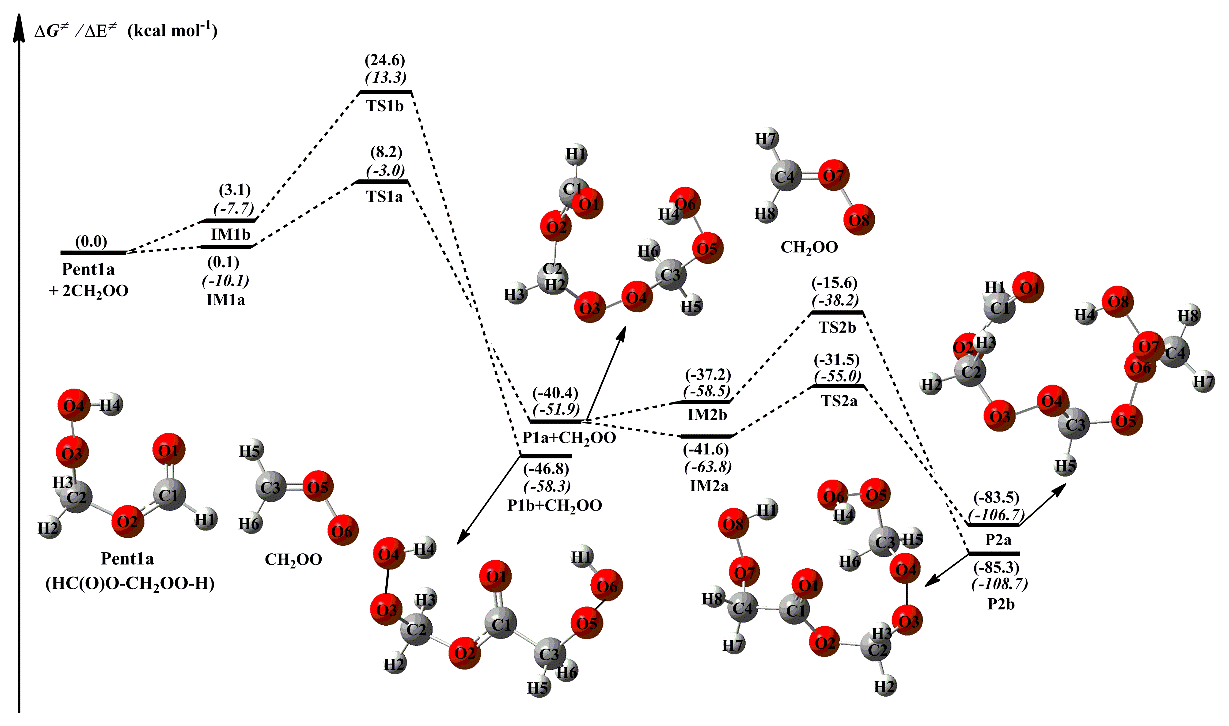
Figure 2PES (ΔG and ΔE) for the 2CH2OO + Pent1a reaction at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2.2 The reactions of anti-CH3CHOO with Pent1b
The methyl-substituted CH2OO has two conformers, syn- and anti-CH3CHOO, that distinguish by the orientation of methyl group relative to the terminal oxygen (Taatjes et al., 2013). syn-CH3CHOO is more stable than anti-CH3CHOO in energy due to the existence of intramolecular hydrogen bond (Long et al., 2016). The activation enthalpy of the interconversion between syn-CH3CHOO and anti-CH3CHOO is up to 38.5 kcal mol−1, implying that they can be treated as independent species in the atmosphere (Long et al., 2016; Yin and Takahashi, 2017). A schematic PES for the addition reaction 2anti-CH3CHOO + Pent1b is presented in Fig. 3, and the optimized geometries of all stationary points are shown in Fig. S5. As shown in Fig. 3, the addition reaction 2anti-CH3CHOO + Pent1b proceeds through successive insertion of anti-CH3CHOO into Pent1b, leading to the formation of oligomers P4a and P4b that contain anti-CH3CHOO as chain unit. The first anti-CH3CHOO addition reaction begins with the formation of IM3a and IM3b in the entrance channel, which lie at −2.2 and 2.4 kcal mol−1, respectively, with respect to the separate reactants. Then the IM3a and IM3b transform into P3a and P3b via −OOH and −CH insertion transition states TS3a and TS3b, with the barriers of 5.6 and 20.3 kcal mol−1, respectively. This result shows that the −OOH insertion reaction is more favorable than the −CH insertion pathway. Compared with the barriers of R1a and R1b in the 2CH2OO + Pent1a reaction, the barriers of R3a and R3b decrease by 2.5 and 1.2 kcal mol−1 when a methyl group is introduced at the anti-position. The result reveals that the reactivity of anti-CH3CHOO is substantially higher than that of CH2OO. This conclusion is further supported by the findings of other studies, which have reported that anti-CH3CHOO is more reactive toward H2O, SO2, and H2O2 than CH2OO (Chen et al., 2017; Taatjes et al., 2013; Huang et al., 2015). Similarly, the secondary anti-CH3CHOO addition reaction starts with the formation of IM4a and IM4b in the entrance channel with the 0.1 and 3.7 kcal mol−1 stability, followed by conversion to the final products P4a and P4b through the −OOH and −CH insertion reactions R4a and R4b. The transition states TS4a and TS4b lie at 7.0 and 21.0 kcal mol−1, respectively, above the energies of the respective intermediates of IM4a and IM4b. This result again shows that the −OOH insertion reaction is the most favorable channel, and the barrier increases as the number of anti-CH3CHOO is increased.
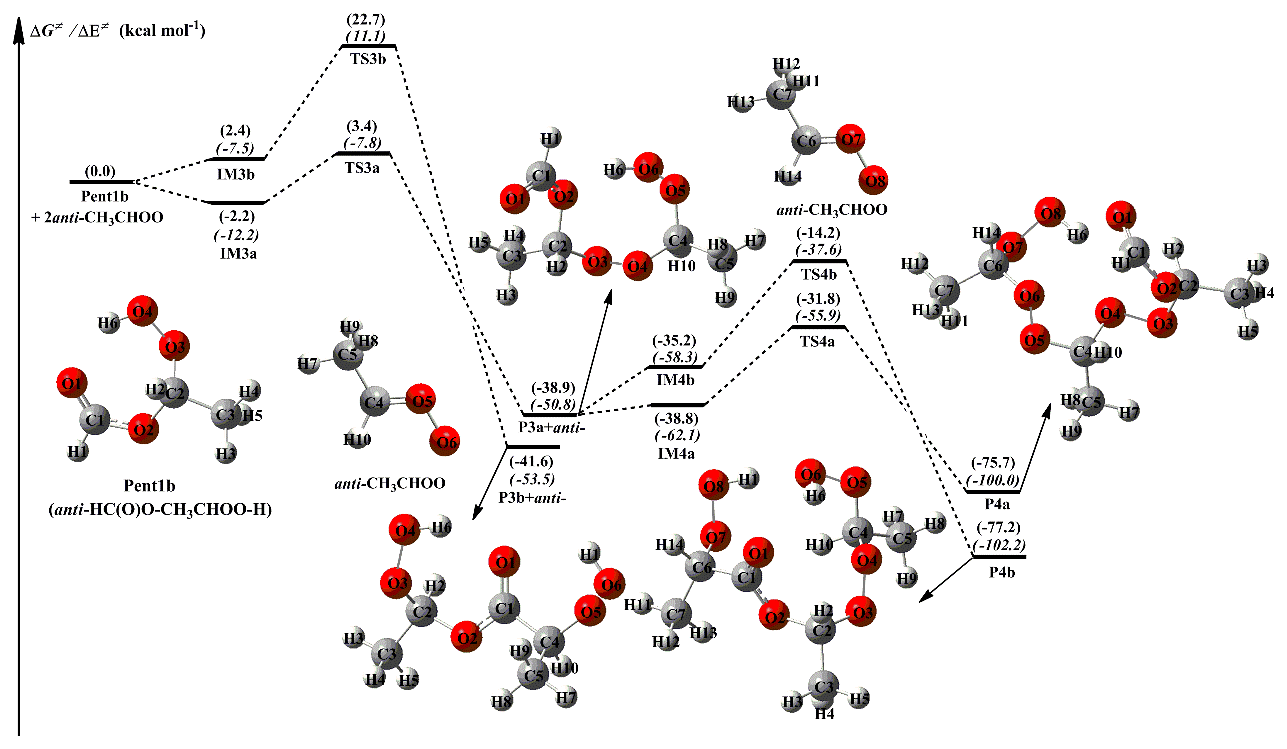
Figure 3PES (ΔG and ΔE) for the 2anti-CH3CHOO + Pent1b reaction at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2.3 The reactions of syn-CH3CHOO with Pent1c
Equivalent to the 2anti-CH3CHOO + Pent1b reaction, the addition reaction 2syn-CH3CHOO + Pent1c has similar transformation pathways and is thus briefly discussed in the present study. From Fig. 4, it can be seen that the addition reaction 2syn-CH3CHOO + Pent1c undergoes change via the successive insertion of syn-CH3CHOO into Pent1c to form P6a and P6b that involves syn-CH3CHOO as the repeating unit. The most favorable pathway is that the breakage of the O4-H6 bond in the −OOH group of Pent1c occurs simultaneously with the insertion of the first syn-CH3CHOO into Pent1c to form P5a, followed by the insertion of the secondary syn-CH3CHOO into P5a to produce P6a. The barriers of these two −OOH insertion reactions R5a and R6a are 13.8 and 11.8 kcal mol−1, respectively, which are higher than those of R3a and R4a in the 2anti-CH3CHOO + Pent1b system by 8.2 and 4.8 kcal mol−1, respectively. The result reveals that the reactivity of syn-CH3CHOO is substantially lower than that of anti-CH3CHOO. Notably, the barrier of the favorable −OOH insertion pathway decreases with increasing the number of syn-CH3CHOO in the 2syn-CH3CHOO + Pent1c reaction, which is contrary to the case of the 2CH2OO + Pent1a and 2anti-CH3CHOO + Pent1b reactions.
Figure 4PES (ΔG and ΔE, in italics) for the 2syn-CH3CHOO + Pent1c reaction at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2.4 The reactions of 2(CH3)2COO with Pent1d
The dimethyl-substituted Criegee intermediate, (CH3)2COO, is generated from the ozonolysis of 2,3-dimethyl-2-butene in the atmosphere (Lester and Klippenstein, 2018; Drozd et al., 2017; Long et al., 2018). The bimolecular reaction of (CH3)2COO with water is not fast enough ( cm3 molec.−1 s−1), while the reaction of (CH3)2COO with HCOOH has a near-gas-kinetic-limit rate ( cm3 molec.−1 s−1; Huang et al., 2015). The result implies that a fraction of (CH3)2COO may survive under high humidity environments and react with HCOOH, leading to the formation of hydroperoxide ester Pent1d. A schematic PES for the addition reaction 2 (CH3)2COO + Pent1d is plotted in Fig. 5, and the optimized geometries of all stationary points are shown in Fig. S7.
As seen in Fig. 5, the addition reaction 2 (CH3)2COO + Pent1d starts with the formation of complexes IM7a and IM7b, which lie at 1.9 and 2.4 kcal mol−1, respectively, above the energies of the separate reactants. Then they subsequently transform into products P7a and P7b through the −OOH and −CH insertion transition states TS7a and TS7b, with the barriers of 12.2 and 26.4 kcal mol−1. This result again shows that the −OOH insertion reaction is favored over the −CH insertion pathway. A similar conclusion is also obtained from the secondary (CH3)2COO addition reaction that the −OOH insertion reaction is the dominant pathway. It is of interest to compare the barriers of −OOH insertion reactions in the (CH3)2COO + Pent1d system with those of the analogous reactions in other SCIs + Pent1 reactions. It can be found that the barriers have decreases of the order of syn-CH3CHOO > (CH3)2COO > CH2OO > anti-CH3CHOO in the first-step SCI addition reaction, while they become (CH3)2COO > syn-CH3CHOO > CH2OO > anti-CH3CHOO in the second-step SCI addition pathway. The result shows that the reactivity of SCIs is significantly affected by the number and location of methyl substituents. A similar conclusion is also obtained from the thermodynamic parameters that the exothermicity of −OOH insertion reactions significantly decreases with increasing the number of methyl substituents, and the exothermicity of anti-methyl substituted carbonyl oxide is obviously higher than that of syn-methyl substituted carbonyl oxide.
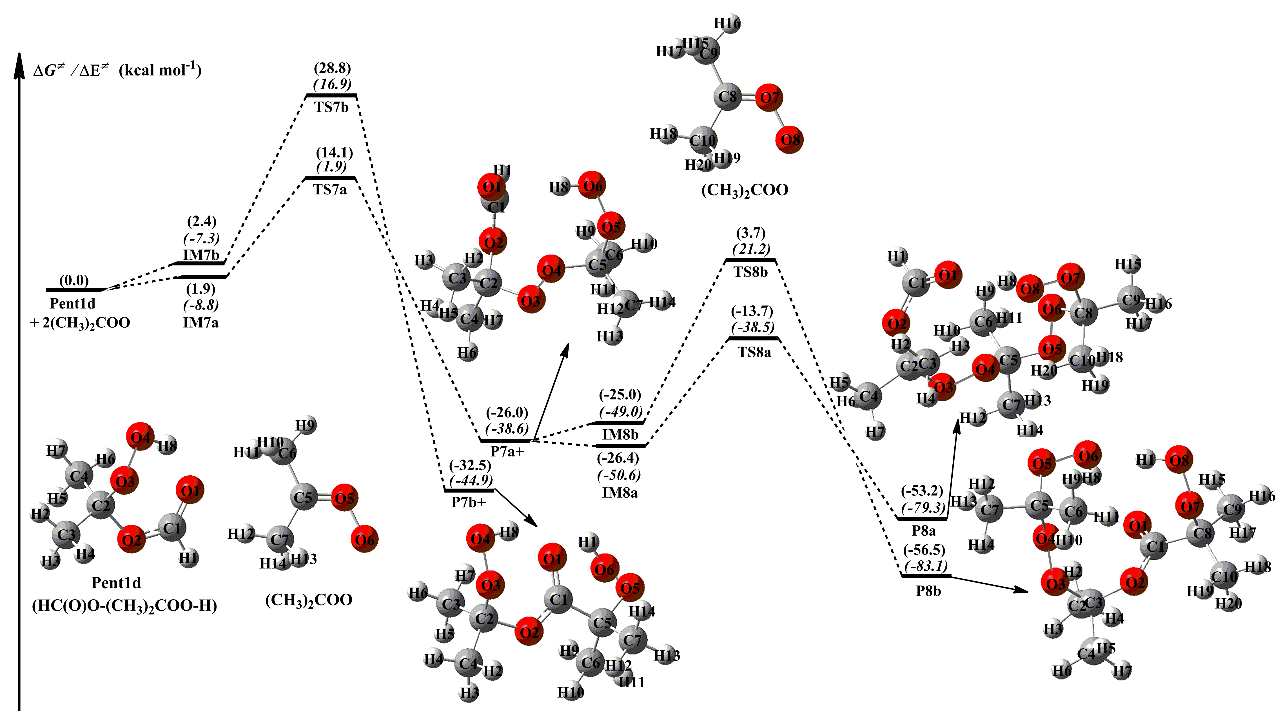
Figure 5PES (ΔG and ΔE) for the 2(CH3)2COO + Pent1d reaction at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.3 The reactions of distinct SCIs with Pent1a and implications in atmospheric chemistry
To further elucidate the effect of the number and location of methyl substituents on the reactivity of carbonyl oxides toward hydroperoxide esters, Pent1a (also called HPMF) is selected as the model compound since it is the simplest hydroperoxide ester formed from the barrierless reaction of the 1,4 O-H insertion of CH2OO into HCOOH. As mentioned above, −OOH insertion reaction in the oligomerization reactions is the most favorable pathway. Therefore, this type of reaction is merely considered in the reactions of distinct SCIs with Pent1a. The corresponding PES and the optimized geometries of all stationary points are displayed in Figs. 6 and S8, respectively. As seen in Fig. 6, each pathway starts with the formation of a pre-reactive intermediate, and then it overcomes a modest barrier to reaction. The barrier of the reaction of CH2OO with Pent1a is calculated to be 8.1 kcal mol−1, which is higher than that of the anti-CH3CHOO + Pent1a reaction by 2.5 kcal mol−1. The reason for the low barrier can be explained by the natural population analysis (NPA) atomic charges, as presented in Fig. S9. As seen in Fig. S9, the charges of the central carbon atom C1 and the terminal oxygen atom O1 of CH2OO are 0.186 e and −0.459 e, respectively, indicating that CH2OO is indeed a zwitterion. The C1 atom charge becomes more positive (0.393 e), while the O1 atom charge becomes more negative (−0.497 e), when a methyl substituent occurs at the anti-position. This result suggests that the anti-methyl substituent enhances the characteristic of the carbonyl oxide zwitterion and reduces the reaction barriers. Compared with the barrier of the CH2OO + Pent1a reaction, the barriers increase by about 3.0 kcal mol−1 when a methyl group is introduced at the syn-position and dimethyl substituent. Although syn-methyl and dimethyl substituent promote the raise of carbonyl oxides zwitterion, the steric hindrance effect and intramolecular hydrogen bond are obviously dominant for syn-CH3CHOO and (CH3)2COO, which is not thus conducive to the nucleophilic attack of hydroperoxide esters. It is worth noting that the exothermicity of distinct SCIs reactions with Pent1a obviously decreases as the number of methyl group is increased, and the exothermicity of anti-methyl substituent is higher than that of syn-methyl substituent.
The rate coefficients of distinct SCIs reactions with Pent1a are calculated in the temperature range of 273–400 K, as summarized in Table S7. This table shows that the rate coefficients kR1a of the CH2OO + Pent1a reaction (R1a) decrease in the range of (273 K) to cm3 molec.−1 s−1 (400 K) with increasing temperature. A similar phenomenon is also observed from the rate coefficients of Pent1a reactions with anti-CH3CHOO (R9), syn-CH3CHOO (R10), and (CH3)2COO (R11) in that they exhibit a slightly negative temperature dependence. kR9 is several orders of magnitude greater than kR1a, kR10 and kR11 in the whole temperature range, suggesting that the bimolecular reaction anti-CH3CHOO + Pent1a (R9) is favored kinetically. Compared with the rate coefficients of R1a, the rate coefficients increase by about 1 order of magnitude when a methyl substituent occurs at the anti-position, whereas the rate coefficients decrease by 1 to 2 orders of magnitude when a methyl group is introduced at the syn-position. It should be noted that, although the barrier of R10 is nearly identical to that of R11, kR10 is 1 to 2 orders of magnitude lower than kR11 in the entire temperature range. This is probably because the rate coefficients are mediated by pre-reactive intermediates so that IM11 is more stable than IM10 in energy.
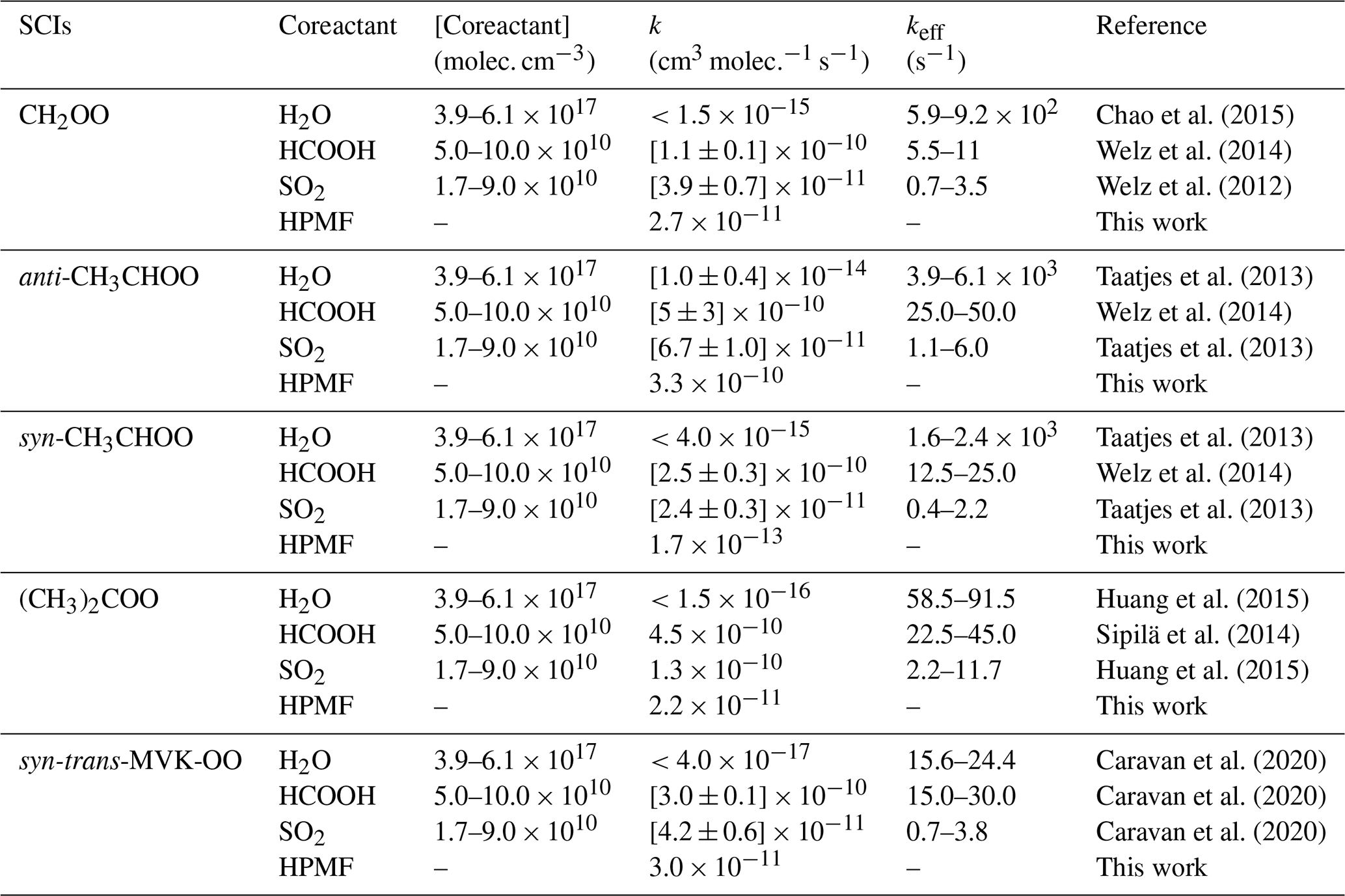
It is of interest to assess whether the reactions of distinct SCIs with HPMF can compete well with the losses to reactions with trace species (e.g., H2O, HCOOH, and SO2) because it is well known that the reactions with trace species are expected to be the dominant chemical sinks for SCIs in the atmosphere (Taatjes et al., 2013; Long et al., 2016). The reported concentrations of coreactant, the rate coefficients k, and the effective pseudo-first-order rate constants (keff=k[coreactant]) for the reactions of distinct SCIs with H2O, HCOOH, SO2, and HPMF are summarized in Table 2. As seen in Table 2, the rate coefficient of a particular SCI reaction with trace species is strongly dependent on its structure. The methyl group substitution may alter the rate coefficient by several to tens of times. The atmospheric concentrations of H2O, HCOOH, and SO2 in tropical forest environments are measured to be 3.9–6.1 ×1017, 5.0–10 × 1010 and 1.7–9.0 × 1010 molec. cm−3, respectively (Vereecken et al., 2012). For the reactions of CH2OO with H2O, HCOOH, and SO2, the experimental rate coefficients are determined to be , [1.1±0.1] × 10−10 and [3.9±0.7] × 10−11 cm3 molec.−1 s−1, respectively (Welz et al., 2012, 2014; Chao et al., 2015), which translate into , , and of 5.9–9.2 × 102, 5.5–11, and 0.7–3.5 s−1, respectively. The result reveals that the reaction of CH2OO with H2O is the most important bimolecular reaction. is greater, by a factor of 3–8, than , indicating that the CH2OO reaction with HCOOH is favored over reaction with SO2. A similar conclusion is also obtained from the results of keff for the reactions of anti-CH3CHOO, syn-CH3CHOO, and (CH3)2COO with H2O, HCOOH, and SO2 in that SCI reactions with H2O are faster than with HCOOH, which, in turn, are faster than with SO2.
According to the results shown in the Table 2, the room temperature rate coefficient for the reaction of CH2OO with HPMF is calculated to be cm3 molec.−1 s−1. However, to the best of our knowledge, the atmospheric concentration of HPMF has not been reported up to now. We assume that the concentration of HPMF is approximately equal to the atmospheric concentration of SCIs, since the SCIs are the deficient reactant in the bimolecular reaction of SCIs with HCOOH. Previous model measurement studies have estimated the surface-level SCI concentrations to be in the range of 1.0×104 to 1.0×105 molec. cm−3 (Khan et al., 2018; Novelli et al., 2017). is calculated to be 2.7–27 × 10−7 s−1, which is several orders of magnitude lower than , , and . A similar conclusion is also obtained from the reactions of anti-CH3CHOO, syn-CH3CHOO, and (CH3)2COO with HPMF.
To further evaluate the relative importance of the complex SCI reactions with the coreactant, the bimolecular reactions of methyl vinyl ketone oxide (MVK-OO) with H2O, HCOOH, SO2, and HPMF are taken into account. MVK-OO, formed with 21 % to 23 % yield from the ozonolysis of isoprene, is a four-carbon, asymmetric, resonance-stabilized Criegee intermediate (Barber et al., 2018). MVK-OO has four conformers, syn-trans-, syn-cis-, anti-trans-, and anti-cis-, as shown in Fig. S10. Herein, syn- and anti- refer to the orientation of the −CH3 group relative to the terminal oxygen of MVK-OO, whereas cis- and trans- refer to the orientation of the C8 = C9 bond relative to the C1 = O2 bond. According to the results shown in the Fig. S10, the lowest-energy conformer is syn-trans-MVK-OO, which is lower than syn-cis-, anti-trans-, and anti-cis-MVK-OO by 1.42, 2.43, and 2.69 kcal mol−1, respectively. Therefore, the lowest-energy conformer syn-trans-MVK-OO is selected as the model compound to study its bimolecular reactions. As shown in Table 2, the rate coefficient of the H2O reaction with syn-trans-MVK-OO is lower than with other SCIs by 2 to 3 orders of magnitude. The reason is likely to be that the existence of methyl and vinyl groups hinders the occurrence of bimolecular reaction with water vapor. Consequently, a fraction of syn-trans-MVK-OO may survive in the presence of water vapor and react with other species. is nearly identical to keff(MVK-OO+HCOOH), which is greater than , which, in turn, is greater than keff(MVK-OO+HPMF) when the concentration of HPMF is equal to the atmospheric concentration of SCIs. Based on the above discussions, it can be concluded that the reactions of SCIs with HPMF are of minor importance in the atmosphere. These reactions may play a certain role in the formation and growth of organic new particle in some regions where low concentration of water vapor and high concentration of hydroperoxide esters occur.
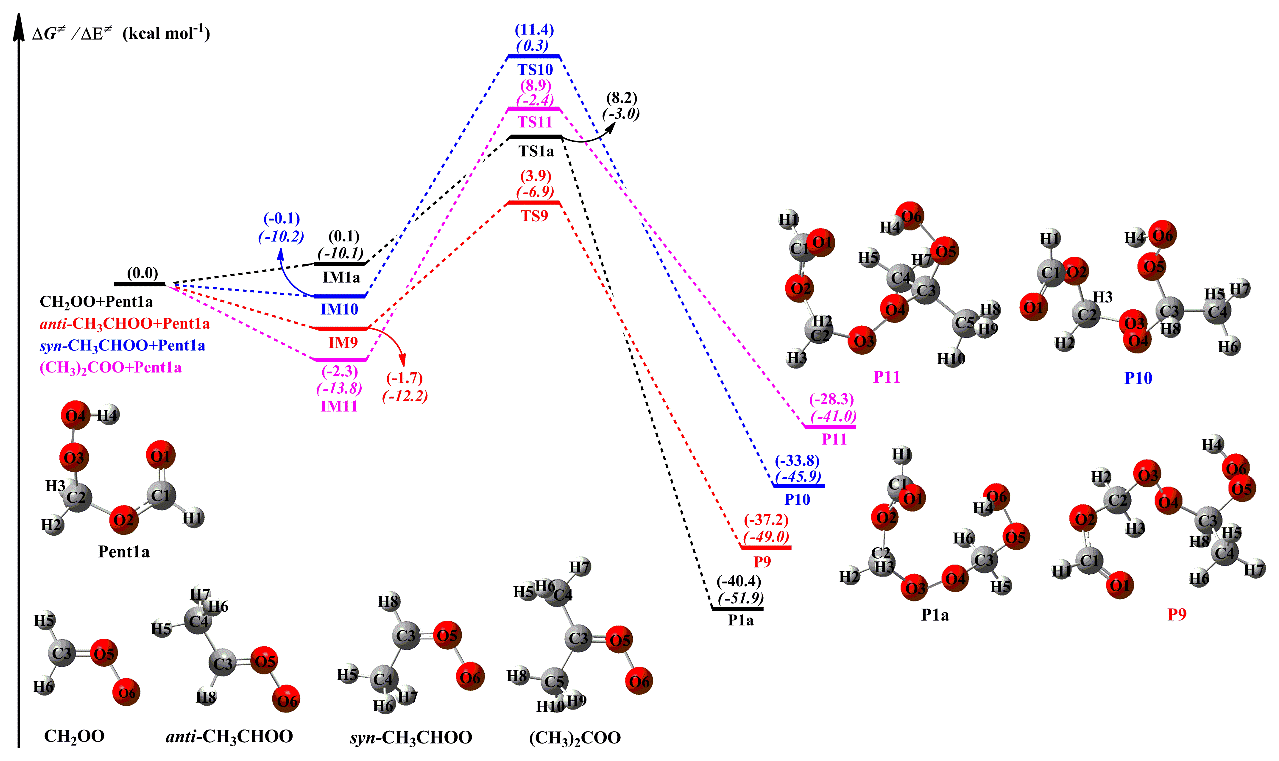
Figure 6PES (ΔG and ΔE) for the distinct SCI + Pent1a reactions at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
Table 2The reported concentrations of the coreactant, the rate coefficients k, and the effective pseudo-first-order rate constants (keff = k[coreactant]) for distinct SCI reactions with HPMF, H2O, HCOOH, and SO2 in tropical forest environments.
3.4 Vapor pressure and volatility of the adduct products
The saturated vapor pressure (P0) of the adduct products formed from the successive reactions of SCIs with HCOOH is estimated by using the EVAPORATION (Estimation of VApour Pressure of ORganics, Accounting for Temperature, Intramolecular, and Non-additivity effects) method proposed by Compernolle et al. (2011), and the room temperature results are summarized in Table S8. This table shows that the P0 of the adduct products decreases significantly as the number of SCIs is increased. Notably, the P0 of the adduct products decreases when the size of SCIs increases. For example, the P0 of the adduct product HC(O)O(CH2OO)3H in the nCH2OO + HCOOH reaction is estimated to be atm, which is greater than those of the corresponding adduct products in the nanti-CH3CHOO + HCOOH ( atm), nsyn-CH3CHOO + HCOOH ( atm), and n(CH3)2COO + HCOOH ( atm) reactions by 7.21, 7.21, and 33.11 times, respectively.
A classify scheme of various organic compounds is based on their volatility, as presented by Donahue et al. (2012). The volatility of organic compounds is described by their effective saturation concentrations. The saturated concentrations (c0) of the adduct products formed from the successive reactions of SCIs with HCOOH are listed in Table S8. As shown in Table S8, the c0 of the adduct products decreases significantly as the number of SCIs is increased. It deserves mentioning that the c0 of the adduct products decreases with the increasing the size of the SCIs. For the nCH2OO + HCOOH reaction, the c0 of the adduct products is estimated to be 1.03×108 (n=1), 5.42×106 (n=2), 2.53×105 (n=3), 1.11×104 (n=4), and 4.67×102 (n=5) µg m−3, respectively. According to the volatility basis set (VBS) of organic compounds (Donahue et al., 2012), the adduct products belong to volatile organic compounds (VOCs; µg m−3) when the number of SCIs is less than or equal to two, while they belong to intermediate volatility organic compounds (IVOCs; 300 < c0 < 3 × 106 µg m−3) when the number of SCIs is greater than or equal to three. Similarly, the adduct products in the nanti-CH3CHOO + HCOOH, nsyn-CH3CHOO + HCOOH, and n(CH3)2COO + HCOOH reactions belong to IVOCs when the number of SCIs ranges from 2 to 4, whereas they belong to semivolatile organic compounds (SVOCs; 0.3 < c0 < 300 µg m−3) when the number of SCIs is equal to 5. Based on the above discussion, it can be concluded that the volatility of the adduct products is significantly affected by the number and size of SCIs in the successive reaction of SCIs with HCOOH. The formed adduct products may participate in the formation and growth processes of organic new particle in the atmosphere.
The oligomerization reaction mechanism and kinetics of Criegee intermediate reactions with their respective hydroperoxide esters and HPMF are investigated using quantum chemical calculations and kinetics modeling methods. The main conclusion is summarized as follows.
- a.
For the initiation reactions of distinct SCIs with HCOOH, the barrierless 1,4 O-H insertion reaction, leading to the formation of hydroperoxide esters, is the most favorable pathway. The exothermicity of distinct SCIs reactions with HCOOH decreases when the number of methyl groups increases, and the exothermicity of the anti-CH3CHOO + HCOOH reaction is higher than that of the syn-CH3CHOO + HCOOH system.
- b.
The addition reactions of SCIs with hydroperoxide esters proceed through successive insertion of SCIs into hydroperoxide ester to form oligomers that involve SCIs as the repeating unit. These oligomerization reactions are strongly exothermic and spontaneous. The exothermicity of oligomerization reactions significantly decreases when the number of methyl substituents increases, and the exothermicity of anti-methyl substituted carbonyl oxides is higher than that of syn-methyl substituted carbonyl oxides.
- c.
The −OOH insertion reaction is favored over the −CH insertion pathway in the SCIs oligomerization reactions, and the barrier heights increase with increasing the number of SCIs added to the oligomer, except for syn-CH3CHOO. The barrier of −OOH insertion pathway shows a dramatic decrease when a methyl substituent occurs at the anti-position, while it reveals a significant increase when a methyl group is introduced at the syn-position and dimethyl substituent.
- d.
Compared with the barrier of CH2OO reaction with HPMF (8.1 kcal mol−1), the barrier decreases by 2.5 kcal mol−1 when a methyl substituent occurs at the anti-position, while the barrier increases by about 3.0 kcal mol−1 when a methyl group is introduced at the syn-position and dimethyl substituent. The rate coefficients increase by about 1 order of magnitude when a methyl substituent occurs at the anti-position, whereas the rate coefficients decrease by 1 to 2 orders of magnitude when a methyl group is introduced at the syn-position compared to the rate coefficients of the CH2OO + HPMF reaction.
- e.
In tropical forest environments, the effective pseudo-first-order rate constants for the reactions of distinct SCIs with HPMF (keff(SCIs+HPMF)) are several orders of magnitude lower than those for the reactions of distinct SCIs with H2O (), HCOOH, (keff(SCIs+HCOOH)) and SO2 (). is greater than keff(SCIs+HCOOH), which, in turn, is greater than .
- f.
The saturated vapor pressure and saturated concentration of the adduct products formed from the successive reactions of SCIs with HCOOH decrease significantly as the number of SCIs is increased. The adduct products in the nCH2OO + HCOOH reactions belong to IVOCs when the number of SCIs is greater than or equal to 3. The adduct products in the nanti-CH3CHOO + HCOOH, nsyn-CH3CHOO + HCOOH, and n(CH3)2COO + HCOOH reactions belong to IVOCs when the number of SCIs ranges from 2 to 4, whereas they belong to SVOCs when the number of SCIs is equal to 5.
The data are accessible by contacting the corresponding author (huangyu@ieecas.cn).
The following information is provided in the Supplement:
-
The electronic energy (ΔE≠) and Gibbs free energy (ΔG≠) barriers for the initiation reactions of distinct SCIs with HCOOH predicted at different levels.
-
Enthalpies of formation for the various carbonyl oxides and hydroperoxide esters.
-
Rate coefficients of initiation reactions of distinct SCIs with HCOOH.
-
Rate coefficients of distinct SCIs reactions with HPMF.
-
Predicted saturated vapor pressure (P0) and saturated concentrations (c0) for the adduct products.
-
Electronic potential energy along the O-H and C-O distance calculated by the M06-2X/6-311+G(2df,2p) method for the barrierless 1,4 insertion reactions.
-
Natural bond orbital (NBO) analysis of the donor–acceptor orbitals involved in the TS1a.
-
NPA charges of different atoms in the distinct SCIs.
-
Optimized geometries of all the stationary points.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-14529-2022-supplement.
LC designed the study. LC and YH wrote the paper. LC performed theoretical calculation. YX, ZJ, and WW analyzed the data. All authors reviewed and commented on the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Makroni Lily, for valuable discussions on the barrierless 1,4 O-H insertion reactions.
This research has been supported by the National Natural Science Foundation of China (grant nos. 42175134, 41805107, and 22002080), the Strategic Priority Research Program of the Chinese Academy of Sciences, China (grant nos. XDA23010300 and XDA23010000), and CAS “Light of West China” Program (grant no. XAB2019B01).
This paper was edited by Ivan Kourtchev and reviewed by Keith Kuwata and two anonymous referees.
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G.: Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries, J. Chem. Theory Comput., 6, 2872–2887, https://doi.org/10.1021/ct100326h, 2010.
Anglada, J. M. and Solé, A.: Impact of the water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 18, 17698–17712, https://doi.org/10.1039/C6CP02531E, 2016.
Aplincourt, P. and Ruiz-López, M. F.: Theoretical study of formic acid anhydride formation from carbonyl oxide in the atmosphere, J. Phys. Chem. A, 104, 380–388, https://doi.org/10.1021/jp9928208, 2000.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bao, J. L. and Truhlar, D. G.: Variational transition state theory: theoretical framework and recent developments, Chem. Soc. Rev., 46, 7548–7596, https://doi.org/10.1039/c7cs00602k, 2017.
Barber, V. P., Pandit, S., Green, A. M., Trongsiriwat, N., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Four-carbon Criegee intermediate from isoprene ozonolysis: methyl vinyl ketone oxide synthesis, infrared spectrum, and OH production, J. Am. Chem. Soc., 140, 10866–10880, https://doi.org/10.1021/jacs.8b06010, 2018.
Boys, S. F. and Bernardi, F.: The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 19, 553–566, https://doi.org/10.1080/00268977000101561, 1970.
Cabezas, C. and Endo, Y.: The reactivity of the Criegee intermediate CH3CHOO with water probed by FTMW spectroscopy, J. Chem. Phys., 148, 014308–014315, https://doi.org/10.1063/1.5009033, 2018.
Cabezas, C. and Endo, Y.: The Criegee intermediate-formic acid reaction explored by rotational spectroscopy, Phys. Chem. Chem. Phys., 21, 18059–18064, https://doi.org/10.1039/c9cp03001h, 2019.
Cabezas, C. and Endo, Y.: Observation of hydroperoxyethyl formate from the reaction between the methyl Criegee intermediate and formic acid, Phys. Chem. Chem. Phys., 22, 446–454, https://doi.org/10.1039/C9CP05030B, 2020.
Canneaux, S., Bohr, F., and Henon, E.: KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 35, 82–93, https://doi.org/10.1002/jcc.23470, 2013.
Caravan, R. L., Vansco, M. F., Au, K., Khan, M. A. H., Li, Y. L., Winiberg, F. A. F., Zuraski, K., Lin, Y. H., Chao, W., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Lin, J. J. M., Shallcross, D. E., Sheps, L., Klippenstein, S. J., Taatjes, C. A., and Lester, M. I.: Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate, P. Natl. Acad. Sci. USA, 117, 9733–9740, https://doi.org/10.1073/pnas.1916711117, 2020.
Chaliyakunnel, S., Millet, D. B., Wells, K. C., Cady-Pereira, K. E., and Shephard, M. W.: A large underestimate of formic acid from tropical fires: constraints from space-borne measurements, Environ. Sci. Technol., 50, 5631–5640, https://doi.org/10.1021/acs.est.5b06385, 2016.
Chao, W., Hsieh, J. T., Chang, C. H., and Lin, J. J. M.: Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 347, 751–754, https://doi.org/10.1126/science.1261549, 2015.
Chen, L., Wang, W., Wang, W., Liu, Y., Liu, F., Liu, N., and Wang, B: Water-catalyzed decomposition of the simplest Criegee intermediate CH2OO, Theor. Chem. Acc., 135, 131–143, https://doi.org/10.1007/s00214-016-1894-9, 2016.
Chen, L., Huang, Y., Xue, Y., Cao, J., and Wang, W.: Competition between HO2 and H2O2 reactions with CH2OO/anti-CH3CHOO in the oligomer formation: a theoretical perspective, J. Phys. Chem. A, 121, 6981–6991, https://doi.org/10.1021/acs.jpca.7b05951, 2017.
Chen, L., Huang, Y., Xue, Y., Cao, J., and Wang, W.: Effect of oligomerization reactions of Criegee intermediate with organic acid/peroxy radical on secondary organic aerosol formation from isoprene ozonolysis, Atmos. Environ., 187, 218–229, https://doi.org/10.1016/j.atmosenv.2018.06.001, 2018.
Chen, L., Huang, Y., Xue, Y., Shen, Z., Cao, J., and Wang, W.: Mechanistic and kinetics investigations of oligomer formation from Criegee intermediate reactions with hydroxyalkyl hydroperoxides, Atmos. Chem. Phys., 19, 4075–4091, https://doi.org/10.5194/acp-19-4075-2019, 2019.
Chhantyal-Pun, R., McGillen, M. R., Beames, J. M., Khan, M. A. H., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Temperature Dependence of the Rates of Reaction of Trifluoracetic Acid with Criegee Intermediates, Angew. Chem. Int. Edit., 129, 9172–9175, https://doi.org/10.1002/anie.201703700, 2017.
Chhantyal-Pun, R., Rotavera, B., Mcgillen, M. R., Khan, M. A. H., Eskola, A. J., Caravan, R. L., Blacker, L., Tew, D. P., Osborn, D. L., Percival, C. J., Taatjes, C. A., Shallcross D. E., and Orr-Ewing, A. J.: Criegee intermediate reactions with carboxylic acids: a potential source of secondary organic aerosol in the atmosphere, ACS Earth Space Chem., 2, 833–842, https://doi.org/10.1021/acsearthspacechem.8b00069, 2018.
Chung, C. A., Su, J. W., and Lee, Y. P.: Detailed mechanism and kinetics of the reaction of Criegee intermediate CH2OO with HCOOH investigated via infrared identification of conformers of hydroperoxymethyl formate and formic acid anhydride, Phys. Chem. Chem. Phys., 21, 21445–21455, https://doi.org/10.1039/c9cp04168k, 2019.
Compernolle, S., Ceulemans, K., and Müller, J.-F.: EVAPORATION: a new vapour pressure estimation methodfor organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Criegee, R.: Mechanism of ozonolysis, Angew. Chem. Int. Edit., 14, 745–752, https://doi.org/10.1002/anie.197507451, 1975.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Drozd, G. T., Kurtén, T., Donahue, N. M., and Lester, M. I.: Unimolecular decay of the dimethyl-substituted Criegee intermediate in alkene ozonolysis: decay time scales and the importance of tunneling, J. Phys. Chem. A, 121, 6036–6045, https://doi.org/10.1021/acs.jpca.7b05495, 2017.
Eckart, C.: The penetration of a potential barrier by electrons, Phys. Rev., 35, 1303–1309, https://doi.org/10.1103/PhysRev.35.1303, 1930.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A. Jr., Vreven, T., Kudin, K. N., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci. B., Cossi, M., Scalmani, G., Rega, N., Petersson, G. A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J. E., Hratchian, H. P., Cross, J. B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Ayala, P. Y., Morokuma, K., Voth, G. A., Salvador, P., Dannenberg, J. J., Zakrzewski, V. G., Dapprich, S., Daniels, A. D., Strain, M. C., Farkas, O., Malick, D. K., Rabuck, A. D., Raghavachari, K., Foresman, J. B., Ortiz, J. V., Cui, Q., Baboul, A. G., Clifford, S., Cioslowski, J., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R. L., Fox, D. J., Keith, T., Al-Laham, M. A., Peng, C. Y., Nanayakkara, A., Challacombe, M., Gill, P. M. W., Johnson, B., Chen, W., Wong, M. W., Gonzalez, C., and Pople, J. A.: Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, https://www.gaussian.com (last access: 9 November 2022), 2009.
Fukui, K.: The path of chemical reactions – the IRC approach, Accounts Chem. Res., 14, 363–368, https://doi.org/10.1021/ar00072a001, 1981.
Giorio, C., Campbell, S. J., Bruschi, M., Tampieri, F., Barbon, A., Toffoletti, A., Tapparo, A., Paijens, C., Wedlake, A. J., Grice, P., Howe, D. J., and Kalbere, M.: Online quantification of Criegee intermediates of α-pinene ozonolysis by stabilization with spin traps and proton-transfer reaction mass spectrometry detection, J. Am. Chem. Soc., 139, 3999–4008, https://doi.org/10.1021/jacs.6b10981, 2017.
Gong, Y. and Chen, Z.: Quantification of the role of stabilized Criegee intermediates in the formation of aerosols in limonene ozonolysis, Atmos. Chem. Phys., 21, 813–829, https://doi.org/10.5194/acp-21-813-2021, 2021.
Hammond, G. S.: A correlation of reaction rates, J. Am. Chem. Soc., 77, 334–338, https://doi.org/10.1021/ja01607a027, 1955.
Huang, H. L., Chao, W., and Lin, J. J. M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
Humphrey, W., Dalke, A., and Schulten, K.: VMD: Visual molecular dynamics, J. Mol. Graphics, 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5, 1996.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/B704260B, 2008.
Johnson, D., Lewin, A. G., and Marston, G.: The effect of Criegee-intermediate scavengers on the OH yield from the reaction of ozone with 2-methylbut-2-ene, J. Phys. Chem. A, 105, 2933–2935, https://doi.org/10.1021/jp003975e, 2001.
Karton, A., Kettner, M., and Wild, D. A.: Sneaking up on the Criegee intermediate from below: Predicted photoelectron spectrum of the CH2OO− anion and W3-F12 electron affinity of CH2OO, Chem. Phys. Lett., 585, 15–20, https://doi.org/10.1016/j.cplett.2013.08.075, 2013.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci.-Proc. Imp., 20, 437–453, https://doi.org/10.1039/C7EM00585G, 2018.
Lester, M. I. and Klippenstein, S. J.: Unimolecular decay of Criegee intermediates to OH radical products: prompt and thermal decay processes, Accounts Chem. Res., 51, 978–985, https://doi.org/10.1021/acs.accounts.8b00077, 2018.
Lin, J. J. M. and Chao, W.: Structure-dependent reactivity of Criegee intermediates studied with spectroscopic methods, Chem. Soc. Rev., 46, 7483–7497, https://doi.org/10.1039/c7cs00336f, 2017.
Lin, X., Meng, Q., Feng, B., Zhai, Y., Li, Y., Yu, Y., Li, Z., Shan, X., Liu, F., Zhang, L., and Sheng, L.: Theoretical study on Criegee intermediate's role in ozonolysis of acrylic acid, J. Phys. Chem. A, 123, 1929–1936, https://doi.org/10.1021/acs.jpca.8b11671, 2019.
Liu, F., Beames, J. M., Petit, A. S., McCoy, A. B., and Lester, M. I.: Infrared-driven unimolecular reaction of CH3CHOO Criegee intermediates to OH radical products, Science, 345, 1596–1598, https://doi.org/10.1126/science.1257158, 2014.
Liu, L., Bei, N., Wu, J., Liu, S., Zhou, J., Li, X., Yang, Q., Feng, T., Cao, J., Tie, X., and Li, G.: Effects of stabilized Criegee intermediates (sCIs) on sulfate formation: a sensitivity analysis during summertime in Beijing–Tianjin–Hebei (BTH), China, Atmos. Chem. Phys., 19, 13341–13354, https://doi.org/10.5194/acp-19-13341-2019, 2019.
Long, B., Cheng, J. R., Tan, X. F., and Zhang, W. J.: Theoretical study on the detailed reaction mechanisms of carbonyl oxide with formic acid, J. Mol. Struc.-Theochem., 916, 159–167, https://doi.org/10.1016/j.theochem.2009.09.028, 2009.
Long, B., Bao, J. L., and Truhlar, D. G.: Atmospheric chemistry of Criegee intermediates: unimolecular reactions and reactions with water, J. Am. Chem. Soc., 138, 14409–14422, https://doi.org/10.1021/jacs.6b08655, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere, P. Natl. Acad. Sci. USA, 115, 6135–6140, https://doi.org/10.1073/pnas.1804453115, 2018.
Lu, T. and Chen, F.: Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 33, 580–592, https://doi.org/10.1002/jcc.22885, 2012.
Mendes, J., Zhou, C. W., and Curran, H. J.: Theoretical chemical kinetic study of the H-atom abstraction reactions from aldehydes and acids by H atoms and OH, HO2, and CH3 radicals, J. Phys. Chem. A, 118, 12089–12104, https://doi.org/10.1021/jp5072814, 2014.
Neeb, P., Horie, O., and Moortgat, G. K.: The ethene-ozone reaction in the gas phase, J. Phys. Chem. A, 102, 6778–6785, https://doi.org/10.1021/jp981264z, 1998.
Novelli, A., Vereecken, L., Lelieveld, J., and Harder, H.: Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 16, 19941–19951, https://doi.org/10.1039/c4cp02719a, 2014.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Osborn, D. L. and Taatjes, C. A.: The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 34, 309–360, https://doi.org/10.1080/0144235X.2015.1055676, 2015.
Paulot, F., Wunch, D., Crounse, J. D., Toon, G. C., Millet, D. B., DeCarlo, P. F., Vigouroux, C., Deutscher, N. M., González Abad, G., Notholt, J., Warneke, T., Hannigan, J. W., Warneke, C., de Gouw, J. A., Dunlea, E. J., De Mazière, M., Griffith, D. W. T., Bernath, P., Jimenez, J. L., and Wennberg, P. O.: Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 11, 1989–2013, https://doi.org/10.5194/acp-11-1989-2011, 2011.
Peltola, J., Seal, P., Inkilä, A., and Eskola, A.: Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 22, 11797–11808, https://doi.org/10.1039/d0cp00302f, 2020.
Porterfield, J. P., Lee, K. L. K., Dell'Isola, V., Carroll, P. B., and McCarthy, M. C.: Characterization of the simplest hydroperoxide ester, hydroperoxymethyl formate, a precursor of atmospheric aerosols, Phys. Chem. Chem. Phys., 21, 18065–18070, https://doi.org/10.1039/c9cp03466h, 2019.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., Thornton, J. A., Turpin, B. J., and Surratt, J. D.: Multiphase reactivity of gaseous hydroperoxide oligomers produced from isoprene ozonolysis in the presence of acidified aerosols, Atmos. Environ., 152, 314–322, https://doi.org/10.1016/j.atmosenv.2016.12.040, 2017.
Sadezky, A., Winterhalter, R., Kanawati, B., Römpp, A., Spengler, B., Mellouki, A., Le Bras, G., Chaimbault, P., and Moortgat, G. K.: Oligomer formation during gas-phase ozonolysis of small alkenes and enol ethers: new evidence for the central role of the Criegee Intermediate as oligomer chain unit, Atmos. Chem. Phys., 8, 2667–2699, https://doi.org/10.5194/acp-8-2667-2008, 2008.
Sakamoto, Y., Inomata, S., and Hirokawa, J.: Oligomerization reaction of the Criegee intermediate leads to secondary organic aerosol formation in ethylene ozonolysis, J. Phys. Chem. A, 117, 12912–12921, https://doi.org/10.1021/jp408672m, 2013.
Sakamoto, Y., Yajima, R., Inomatad, S., and Hirokawa, J.: Water vapour effects on secondary organic aerosol formation in isoprene ozonolysis, Phys. Chem. Chem. Phys., 19, 3165–3175, https://doi.org/10.1039/c6cp04521a, 2017.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
So, S., Wille, U., and Silva, G. D.: Atmospheric chemistry of enols: a theoretical study of the vinyl alcohol + OH + O2 reaction mechanism, Environ. Sci. Technol., 48, 6694–6701, https://doi.org/10.1021/es500319q, 2014.
Stavrakou, T., Müller, J. F., Peeters, J., Razavi, A., Clarisse, L., Clerbaux, C., Coheur, P. F., Hurtmans, D., Mazière, M. D., Vigouroux, C., Deutscher, N. M., Griffith, D. W. T., Jones, N., and Paton-Walsh, C.: Satellite evidence for a large source of formic acid from boreal and tropical forests, Nat. Geosci., 5, 26–30, https://doi.org/10.1038/ngeo1354, 2012.
Taatjes, C. A.: Criegee intermediates: what direct production and detection can teach us about reactions of carbonyl oxides, Annu. Rev. Phys. Chem., 68, 183–207, https://doi.org/10.1146/annurev-physchem-052516-050739, 2017.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO, Science, 340, 177–180, https://doi.org/10.1126/science.1234689, 2013.
Taatjes, C. A., Khan, M. A. H., Eskola, A. J., Percival, C. J., Osborn, D. L., Wallington, T. J., and Shallcross, D. E.: Reaction of perfluorooctanoic acid with Criegee intermediates and implications for the atmospheric fate of perfluorocarboxylic acids, Environ. Sci. Technol., 53, 1245–1251, https://doi.org/10.1021/acs.est.8b05073, 2019.
Tobias, H. J. and Ziemann, P. J.: Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids, and water with the C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene, J. Phys. Chem. A, 105, 6129–6135, https://doi.org/10.1021/jp004631r, 2001.
Truhlar, D. G., Hase, W. L., and Hynes, J. T.: Current status of transition-state theory, J. Phys. Chem., 87, 2664–2682, https://doi.org/10.1021/jp953748q, 1996.
Vansco, M. F., Zuraski, K., Winiberg, F. A. F., Au, K., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Klippenstein, S. J., Taatjes, C. A., Lester, M. I., and Caravan, R. L.: Functionalized hydroperoxide formation from the reaction of methacrolein-oxide, an isoprene-derived Criegee intermediate, with formic acid: experiment and theory, Molecules, 26, 3058–3072, https://doi.org/10.3390/molecules26103058, 2021.
Vereecken, L.: The reaction of Criegee intermediates with acids and enols, Phys. Chem. Chem. Phys., 19, 28630–28640, https://doi.org/10.1039/c7cp05132h, 2017.
Vereecken, L., Harder, H., and Novelli, A.: The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 14, 14682–14695, https://doi.org/10.1039/c2cp42300f, 2012.
Wang, S., Newland, M. J., Deng, W., Rickard, A. R., Hamilton, J. F., Muñoz, A., Ródenas, M., Vázquez, M. M., Wang, L., and Wang, X.: Aromatic photo-oxidation, a new source of atmospheric acidity, Environ. Sci. Technol., 54, 7798–7806, https://doi.org/10.1021/acs.est.0c00526, 2020.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, https://doi.org/10.1126/science.1213229, 2012.
Welz, O., Eskola, A. J., Sheps, L., Rotavera, B., Savee, J. D., Scheer, A. M., Osborn, D. L., Lowe, D., Booth, A. M., Xiao, P., Khan, M. A. H., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Rate coefficients of C(1) and C(2) Criegee intermediate reactions with formic and acetic Acid near the collision limit: direct kinetics measurements and atmospheric implications, Angew. Chem. Int. Edit., 53, 4547–4550, https://doi.org/10.1002/anie.201400964, 2014.
Yin, C. and Takahashi, K.: How does substitution affect the unimolecular reaction rates of Criegee intermediates? Phys. Chem. Chem. Phys., 19, 12075–12084, https://doi.org/10.1039/c7cp01091e, 2017.
Yu, S.: Role of organic acids (formic, acetic, pyruvic and oxalic) in the formation of cloud condensation nuclei (CCN): a review, Atmos. Res., 53, 185–217, https://doi.org/10.1016/S0169-8095(00)00037-5, 2000.
Zhang, P., Wang, W., Zhang, T., Chen, L., Du, Y., Li, C., and Lv, J.: Theoretical study on the mechanism and kinetics for the self-reaction of C2H5O2 radicals, J. Phys. Chem. A, 116, 4610–4620, https://doi.org/10.1021/jp301308u, 2012.
Zhao, R., Kenseth, C. M., Huang, Y., Dalleska, N. F., Kuang, X. M., Chen, J., Paulson, S. E., and Seinfeld, J. H.: Rapid aqueous-phase hydrolysis of ester hydroperoxides arising from Criegee intermediates and organic acids, J. Phys. Chem. A, 122, 5190–5201, https://doi.org/10.1021/acs.jpca.8b02195, 2018.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.
Zhao, Y., Wingen, L. M., Perraud, V., Greaves, J., and Finlayson-Pitts, B. J.: Role of the reaction of stabilized Criegee intermediates with peroxy radicals in particle formation and growth in air, Phys. Chem. Chem. Phys., 17, 12500–12514, https://doi.org/10.1039/c5cp01171j, 2015.
Zheng, J., Bao, L. J., Meana-Paneda, R., Zhang, S., Lynch, B. J., Corchado, J. C., Chuang, Y. Y., Fast, P. L., Hu, W. P., Liu, Y. P., Lynch, G. C., Nguyen, K. A., Jackels, C. F., Fernandez-Ramos, A., Ellingson, B. A., Melissas, V. S., Villa, J., Rossi, I., Coitino, L., Pu, J., Albu, T. V., Steckler, R., Garrett, B. C., Issacson, A. D., and Truhlar, D. G.: Polyrate, version 2017-C, University of Minnesota, Minneapolis, MN, https://comp.chem.umn.edu/polyrate/ (last access: 9 November 2022), 2018.
Zhou, S., Joudan, S., Forbes, M. W., Zhou, Z., and Abbatt, J. P. D.: Reaction of condensed-phase Criegee intermediates with carboxylic acids and perfluoroalkyl carboxylic acids, Environ. Sci. Tech. Let., 6, 243–250, https://doi.org/10.1021/acs.estlett.9b00165, 2019.