the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Sep 2020
| 22 Sep 2020
Tropospheric ozone radiative forcing uncertainty due to pre-industrial fire and biogenic emissions
Matthew J. Rowlinson
Alexandru Rap
Douglas S. Hamilton
Richard J. Pope
Stijn Hantson
Steve R. Arnold
Jed O. Kaplan
Almut Arneth
Martyn P. Chipperfield
Piers M. Forster
Lars Nieradzik
Tropospheric ozone concentrations are sensitive to natural emissions of precursor compounds. In contrast to existing assumptions, recent evidence indicates that terrestrial vegetation emissions in the pre-industrial era were larger than in the present day. We use a chemical transport model and a radiative transfer model to show that revised inventories of pre-industrial fire and biogenic emissions lead to an increase in simulated pre-industrial ozone concentrations, decreasing the estimated pre-industrial to present-day tropospheric ozone radiative forcing by up to 34 % (0.38 to 0.25 W m−2). We find that this change is sensitive to employing biomass burning and biogenic emissions inventories based on matching vegetation patterns, as the co-location of emission sources enhances the effect on ozone formation. Our forcing estimates are at the lower end of existing uncertainty range estimates (0.2–0.6 W m−2), without accounting for other sources of uncertainty. Thus, future work should focus on reassessing the uncertainty range of tropospheric ozone radiative forcing.
- Article
(2299 KB) - Full-text XML
- BibTeX
- EndNote





Tropospheric ozone (O3) is a short-lived greenhouse gas formed in the atmosphere through photochemical oxidation of volatile organic compounds (VOCs) in the presence of nitrogen oxides (NOx). These precursor gases have both natural and anthropogenic sources, and increased anthropogenic emissions are thought to have caused an increase in global tropospheric O3 of 25 %–50 % since 1900 (Gauss et al., 2006; Lamarque et al., 2010; Young et al., 2013). The Intergovernmental Panel on Climate Change (IPCC) current best estimate for tropospheric O3 radiative forcing (RF) over the industrial era is 0.4±0.2 W m−2 with a 5 %–95 % confidence interval, making tropospheric O3 the third most important anthropogenic greenhouse gas after CO2 and CH4 (Myhre et al., 2013). The present-day (PD) radiative effect (RE) of tropospheric O3 is relatively well constrained (Rap et al., 2015). The large uncertainty range (0.2–0.6 W m−2) is caused by a number of factors such as the radiative transfer scheme employed, the model used to simulate tropospheric O3 and tropopause definition; however, it is primarily associated with a poor understanding of pre-industrial (PI) O3 concentrations (Myhre et al., 2013; Stevenson et al., 2013). Although measurements of tropospheric O3 exist as far back as the late 19th century, there are limited reliable quantitative measurements of tropospheric O3 prior to the 1970s (Volz and Kley, 1988; Cooper et al., 2014). Recently, Checa-Garcia et al. (2018) found that differences in PI estimates between Coupled Model Intercomparison Project phase 5 (CMIP5) and CMIP6 cause an 8 %–12 % variation in O3 RF estimates but did not explicitly assess uncertainty in natural PI emissions. Recent analysis of oxygen isotopes in polar ice cores indicates that tropospheric O3 in the Northern Hemisphere increased by less than 40 % between 1850 and 2005, suggesting that O3 RF may be lower than the 0.4 W m−2 estimate (Yeung et al., 2019).
As well as anthropogenic sources, O3 precursor gases such as methane (CH4), carbon monoxide (CO) and NOx have natural emission sources, e.g. wildfires, wetlands, lightning and biogenic emissions. Wildfires, for example, emit large quantities of CO, NOx, CH4 and non-methane volatile organic compounds (NMVOCs) (van der Werf et al., 2010; Voulgarakis and Field, 2015), which influence the chemical production of O3 (Wild, 2007). Changes in the natural environment therefore influence the concentration and distribution of tropospheric O3 (Monks et al., 2015; Hollaway et al., 2017). While human activities such as deforestation, land-use change and fire management are known to affect natural emission sources of O3 precursor gases, their impact on emissions net change remains very uncertain (Mickley et al., 2001; Arneth et al., 2010). An accurate representation of PI natural emissions is therefore very important for quantifying the PI-to-PD tropospheric O3 RF calculations.
Recent studies suggest that the relationship between humans and fire (Bowman et al., 2009) is more complex than previously assumed (Doerr and Santín, 2016). The expansion of agriculture and land-cover fragmentation since PI has decreased the abundance and continuity of fuel, inhibiting fire spread (Marlon et al., 2008; Swetnam et al., 2016) and hence total emissions. Furthermore, at the global scale, increased population density results in declining fire frequency (Knorr et al., 2014; Andela et al., 2017). Increased agricultural land coupled with active fire suppression and management policies mean that human activity has likely caused total fire emissions to decline since the PI (Daniau et al., 2012; Marlon et al., 2016; Hamilton et al., 2018). Paleoenvironmental archives of fire activity also reflect a decline of fire over the industrial era in many regions (Marlon et al., 2016; Rubino et al., 2016; Swetnam et al., 2016). This change in the understanding of PI fire emissions has been shown to have a strong influence on aerosol RF: Hamilton et al. (2018) estimated a 35 %–91 % decrease in global mean cloud albedo forcing over the industrial era when using revised PI fire emission inventories.
Emissions of biogenic VOCs (BVOCs), such as isoprene and monoterpenes, from vegetation also affect tropospheric O3 formation. Isoprene contributes to the formation of peroxyacetylnitrate (PAN), which has a lifetime of several months in the upper troposphere (Singh, 1987), allowing long-range transport of reactive nitrogen and enhancing O3 formation in remote regions. PAN formation is also highly dependent on NOx concentrations, meaning that changes in the distribution of emissions and the magnitude will impact O3 formation. Previous studies of PI tropospheric O3 have often assumed that PI BVOC emissions were equivalent to or lower than those in PD (Stevenson et al., 2013). In Stevenson et al. (2013), only one model of the ensemble included PI isoprene emissions that were larger than in the PD simulation. However, BVOC emissions are sensitive to climate, CO2 concentrations, vegetation type and foliage density, each of which has changed since the PI (Laothawornkitkul et al., 2009; Hantson et al., 2017) and needs to be accounted for when calculating O3 RF.
The aim of this study is to examine the effect of revised PI fire and BVOC emission inventories on PI-to-PD tropospheric O3 RF estimates. We use a global chemical transport model (CTM) and a radiative transfer model to investigate the impact of these revised natural PI emission inventories on tropospheric O3 PI concentrations and its PI–PD RF. The IPCC 5th assessment report moved to the concept of effective radiative forcing (ERF) (Myhre et al., 2013) to more completely capture the expected global energy budget change from a given driver. However, here we employ the more traditional stratospherically adjusted RF as it can be estimated with more certainty than ERF, and previous studies suggest that ERF and RF are likely to be similar for O3 change (Myhre et al., 2013; Shindell et al., 2013). We note that a number of factors not considered here also introduce uncertainty when simulating PI tropospheric O3 concentrations, e.g. changes to lightning and soil NOx emissions, O3 deposition and atmospheric transport. However, the purpose of this study is to address and focus on the uncertainty associated with natural emissions in the pre-industrial era by utilising the revised inventories of fire and biogenic emissions.
2.1 TOMCAT–GLOMAP
We used the TOMCAT global three-dimensional offline chemical transport model (CTM) (Chipperfield, 2006) coupled to the GLOMAP modal aerosol microphysics scheme (Mann et al., 2010) to simulate tropospheric composition and its response to emissions changes. The model used a horizontal resolution with 31 vertical levels from the surface to 10 hPa. All simulations were run with 6-hourly 2008 meteorology from European Centre for Medium-Range Weather Forecasts (ECMWF) ERA-Interim reanalyses with a 1-year spin-up (Dee et al., 2011). The model includes a detailed representation of hydrocarbon chemistry and isoprene oxidation, and it has previously been shown to accurately reproduce observed concentrations and distributions of key tropospheric species such as O3, CO, NOx and VOCs (Monks et al., 2017; Rowlinson et al., 2019). The annual global mean surface CH4 mixing ratio is scaled in TOMCAT–GLOMAP based on observed global mean surface concentrations for the year being simulated; however, the spatial variation in CH4 concentrations is maintained in the model. Biomass burning and biogenic emissions are emitted into the lowest model level, which extends from the surface to 951 hPa.
2.2 Radiative transfer model
Tropospheric O3 RFs were calculated using the SOCRATES radiative transfer model (Edwards and Slingo, 1996) with six bands in the shortwave (SW) and nine in the longwave (LW). This version has been used extensively in conjunction with TOMCAT–GLOMAP for calculating O3 radiative effects (Bekki et al., 2013; Rap et al., 2015; Kapadia et al., 2016; Scott et al., 2018). We used the fixed dynamical heating approximation (Fels et al., 1980) to account for stratospheric temperature adjustments, i.e. changes in the stratospheric heating rate calculated in the model due to the O3 perturbation are applied to the temperature field, with the model run iteratively until stratospheric temperatures reach equilibrium (Forster and Shine, 1997; Rap et al., 2015).
2.3 Simulations
We investigate the effect of natural PI emissions on PI-to-PD changes in tropospheric O3 concentrations by contrasting PI against PD model simulations (Table 1). All simulations are run with PD meteorology and global mean surface CH4 concentrations scaled to be 722 ppb in the PI and 1789 ppb in PD (Etheridge et al., 1998; Dlugokencky et al., 2005; Hartmann et al., 2013; McNorton et al., 2016).
Table 1Details of the TOMCAT–GLOMAP simulations performed in this study.
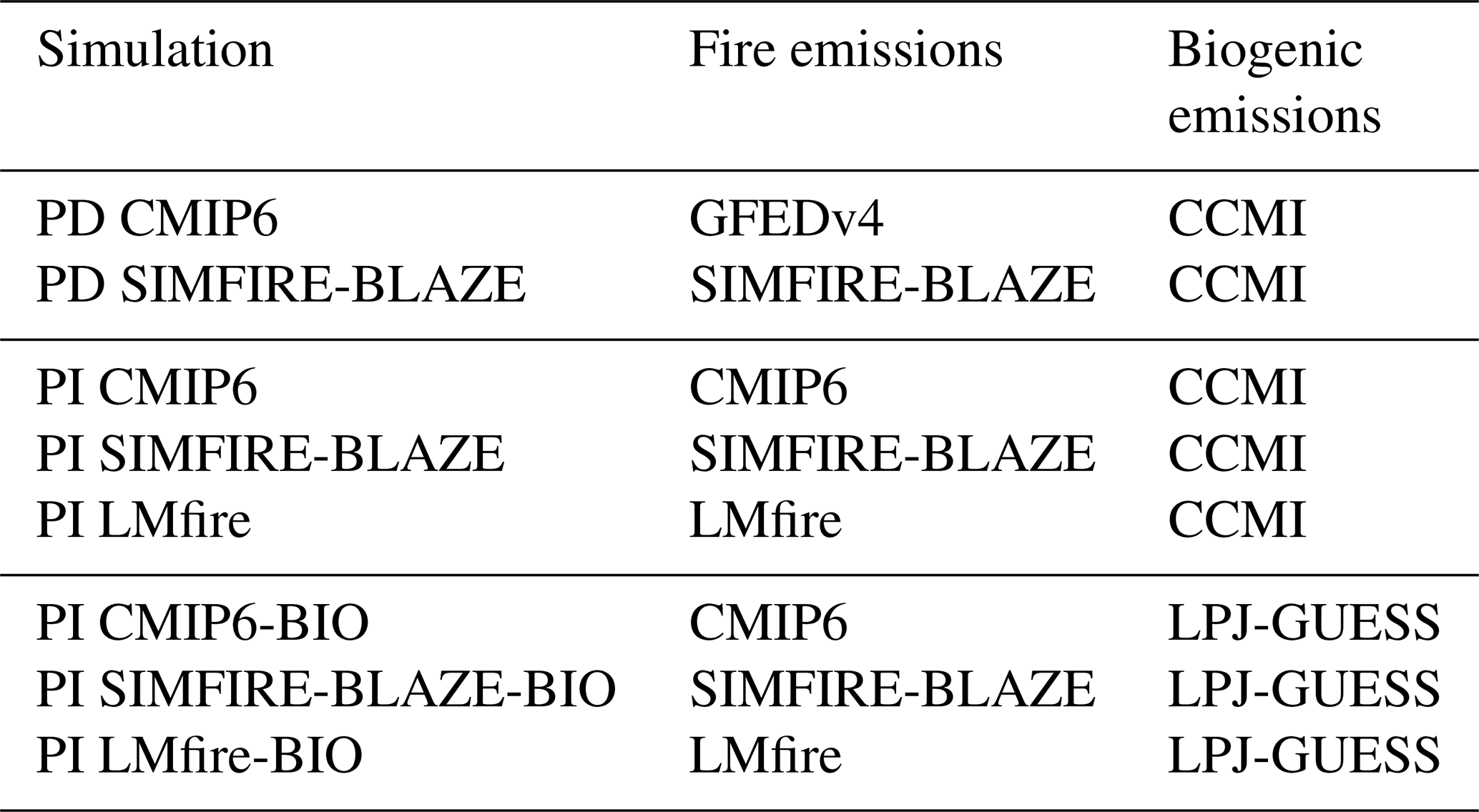
All PI simulations considered anthropogenic emissions set to zero, except for biofuel emissions taken from AeroCom for the year 1750 (Dentener et al., 2006). The first set of three PI simulations (i.e. PI CMIP6, PI SIMFIRE-BLAZE and PI LMfire) investigates the impact of fire emissions only by keeping BVOC emissions (i.e. isoprene and monoterpenes) at their PD values based on the Chemistry–Climate Model Initiative (CCMI) biogenic emissions (Sindelarova et al., 2014). The second set of three PI simulations (i.e. PI CMIP6-BIO, PI SIMFIRE-BLAZE-BIO and PI LMfire-BIO) investigates the additional impact of PI biogenic emissions by combining each PI fire emission inventory with an estimate of PI BVOC emissions from the LPJ-GUESS model (Arneth et al., 2007; Schurgers et al., 2009; Smith et al., 2014).
The PD simulations used anthropogenic emissions from the MACCity emissions dataset (from EU projects MACC/CityZEN; Lamarque et al., 2010) and CCMI biogenic emissions (Sindelarova et al., 2014). Two PD simulations were performed, namely the primary PD simulation (PD CMIP) driven by the Global Fire Emissions Database version 4 with small fires (GFED v4s) inventory as employed in CMIP6 (Randerson et al., 2017; van Marle et al., 2017) and PD SIMFIRE-BLAZE (Knorr et al., 2014). A PD simulation is not available for LMfire, a PI fire model not designed to undertake a PD simulation. To isolate the effect of revised natural PI emissions on PI-to-PD tropospheric ozone RF, we compare the six PI simulations against the main PD CMIP6 simulation. The other PD simulation, i.e. PD SIMFIRE-BLAZE, is also included in our analysis in order to explore the additional uncertainty in RF introduced by PD emission inventories uncertainties. However, as PD tropospheric ozone RE was shown to be well constrained by satellite observations (Rap et al., 2015), this additional uncertainty is known to be small.
2.4 Fire emission inventories
Following Hamilton et al. (2018), we used three PI inventories to investigate the sensitivity of tropospheric O3 RF to PI fire uncertainty. The CMIP6 PI inventory is treated as a control, as this has been widely used in previous studies and was developed from a set of global fire models, with SIMFIRE-BLAZE and LMfire providing PI perturbation scenarios from this baseline.
2.4.1 Pre-industrial and present-day CMIP6
CMIP6 provides monthly mean emissions of CO, NOx, CH4 and VOCs from fires. In the PD, CMIP6 emissions are derived from satellite estimates of global burden area and active fire detections (Randerson et al., 2012; Giglio et al., 2013). In the absence of satellite data, PI CMIP6 fire emissions are generated by merging PD satellite observations with fire proxy records, visibility records and analysis from six fire models (van Marle et al., 2017). The mean of 1750–1770 emissions is used in this study to represent PI emissions. Biomass burning emissions from deforestation and peat fires are assumed to be reduced in the PI, while agricultural fires are kept fairly constant with PD due to a lack of information on the PI environment.
2.4.2 Pre-industrial and present-day SIMFIRE-BLAZE
The SIMFIRE-BLAZE PI fire emission inventory was developed using the LPJ-GUESS-SIMFIRE-BLAZE model. The PI emissions employed here are the mean for the period 1750–1770 (Hamilton et al., 2018). The LPJ-GUESS dynamic vegetation model predicts ecosystem properties for given climate variables (Smith et al., 2014), which, combined with the HYDE 3.1 dataset of human land-use change, allows for the simulation of global PI land cover (Klein Goldewijk et al., 2011). The SIMple fire model (SIMFIRE) calculates total burned area (Knorr et al., 2014), with the total fire carbon flux calculated from BLAZE (BLAZe induced biosphere–atmosphere flux Estimator) (Rabin et al., 2017). Akagi et al. (2011) emissions factors were used with separate treatment of herbaceous and non-herbaceous as well as tropical and extratropical vegetation to produce emission inventories. Agricultural fire emissions are not included. Total PI fire emissions of gas species in the SIMFIRE-BLAZE inventory are 28 % larger than in the PI CMIP6 inventory.
The fire emissions in the PD SIMFIRE-BLAZE model are very similar to the PD CMIP6 inventory, with only slightly increased global NOx emissions (174 Tg yr−1 compared to 171 Tg yr−1 in CMIP6) and CO emissions (1027 Tg yr−1 compared to 970 Tg yr−1). The global distribution of the inventories is also similar (Fig. 1), with slightly larger CO emissions in the SH tropics in PD SIMFIRE-BLAZE but smaller in the NH tropical region. NOx and VOC emissions are similar in both inventories across all latitude bands (Fig. 1b, d). The seasonality of emissions is also consistent across both inventories in terms of NOx and VOC emissions; however, for CO the peak in emissions is slightly later for the SIMFIRE-BLAZE inventory (Fig. 3). The slightly higher emissions in PD SIMFIRE-BLAZE result in a simulated tropospheric O3 burden of 359.9 Tg, an increase of 1 % relative to the PD CMIP6 TOMCAT–GLOMAP simulation (Table 2).
Table 2Annual mean global tropospheric burdens of CO, NOx and O3, mean tropospheric OH concentration, tropospheric column O3 for all model simulations, and 1750–2010 radiative forcing of tropospheric O3 estimated for each PI simulation against the PD CMIP6 simulation.
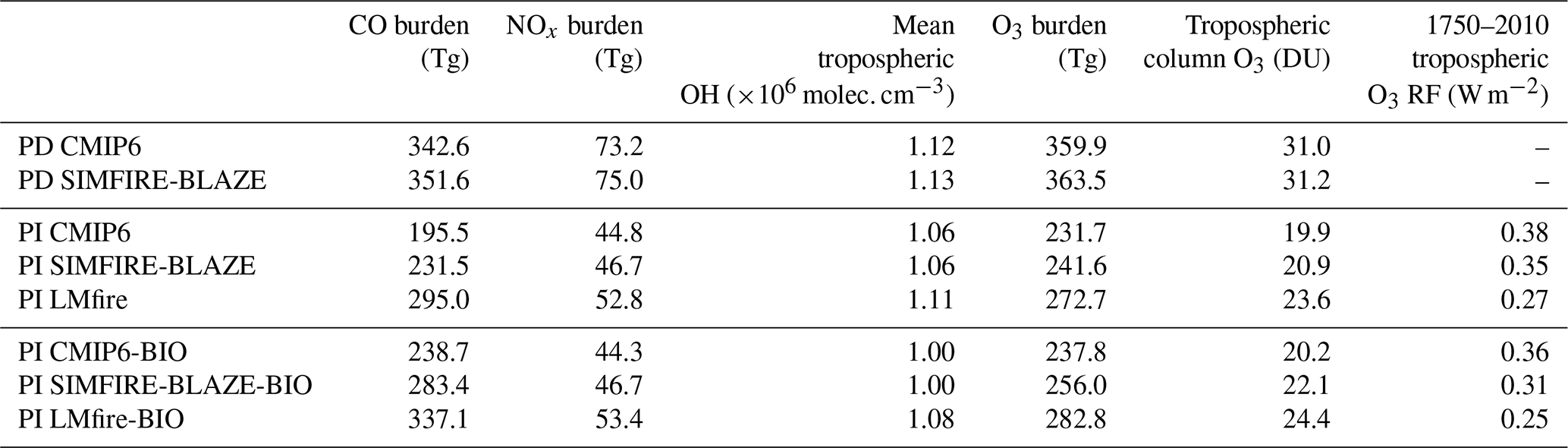
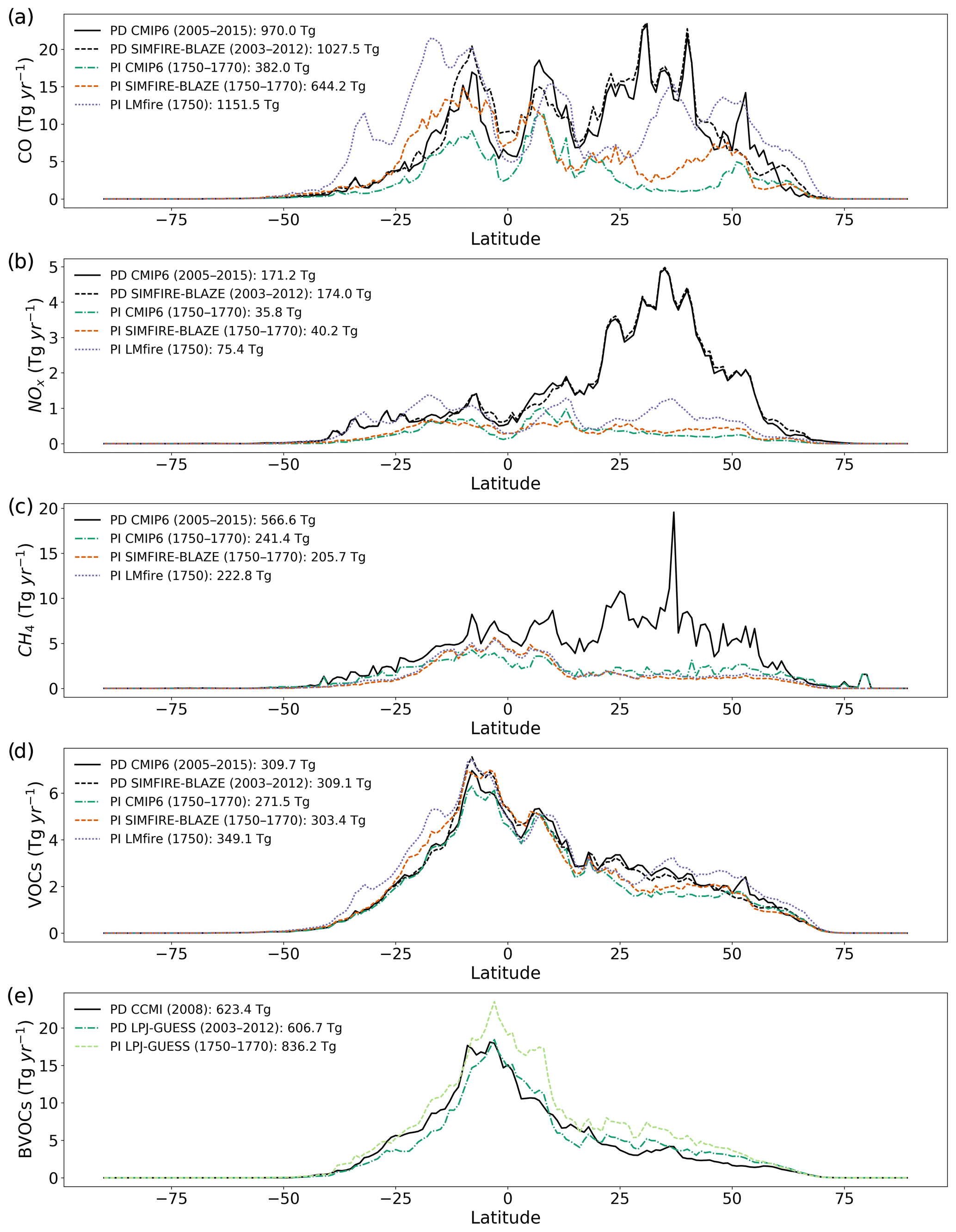
Figure 1Annual latitudinal mean pre-industrial emissions (Tg yr−1) of (a) CO, (b) NOx, (c) CH4 and (d) VOCs in the PD CMIP6 (solid black line), PD SIMFIRE-BLAZE (dashed black), PI CMIP6 (dashed green), PI SIMFIRE-BLAZE (dotted orange) and PI LMfire (dashed purple) inventories. (e) Annual latitudinal mean BVOC emissions (Tg yr−1) in PD CCMI (solid black line), PD LPJ-GUESS (dashed dark green) and PI LPJ-GUESS (dotted light green).
2.4.3 Pre-industrial LPJ-LMfire
The LPJ-LMfire model calculates dry matter consumed by fire and simulates natural wildfire ignition from lightning (Pfeiffer et al., 2013; Murray et al., 2014). Land use is prescribed for the year 1770 using the KK10 scenario from Kaplan et al. (2011); climate forcing comes from a 1020-year detrended, interannually variable equilibrium dataset representing late 19th century conditions (see Pfeiffer et al., 2013, Sect. 3.4 for details). Akagi et al. (2011) emissions factors were again used to calculate the gas-phase fire emissions from dry biomass burned in each grid cell. Burned area is calculated based on fuel availability. LMfire includes emissions from managed agricultural burning, with 50 % of the litter on 20 % of croplands burden annually. Also included are emissions from post-harvest agricultural burning, with 10 % of harvested agricultural crop material assumed to be burned each year. Total PI fire emissions in LMfire are approximately double the SIMFIRE-BLAZE inventory and thus 4 times larger than CMIP6 emissions.
2.5 Assessment of PI fire emissions
Although the PI LMfire and PI SIMFIRE-BLAZE emissions are substantially larger than the PI CMIP6 emissions, both inventories fall with the current uncertainty range for fire emissions, deemed to differ by up to a factor of ∼4 (Lee et al., 2013; Hamilton et al., 2018; Pan et al., 2020). In Hamilton et al. (2018), both the SIMFIRE-BLAZE and LMfire PI inventories were shown to compare more favourably than CMIP6 to changes in PI-to-PD ice core BC measurements in the Swiss Alps. Furthermore, the LMfire emissions result in simulated aerosol concentrations that were closer to Northern Hemisphere (NH) ice core records in Greenland and Wyoming than both the CMIP6 and SIMFIRE-BLAZE emissions (Hamilton et al., 2018). In addition to the extensive examination of paleoenvironmental archives with PI fire emissions datasets by Hamilton et al. (2018), here we compared simulated annual mean surface PI CO concentrations in Antarctica for each fire emissions inventory using the Southern Hemisphere (SH) ice core CO record from Wang et al. (2010). Simulated Antarctic CO concentrations using PI CMIP6 emissions are 37 ppb, substantially lower than the Wang et al. (2010) 1750 value of 45±5 ppb. This CMIP6 value is closer to the 650-year minimum that occurred in the mid-17th century (38 ppb). When using SIMFIRE-BLAZE and LMfire emissions, Antarctic CO concentrations for 1750 are estimated at 48 and 61 ppb, respectively. The overestimation when using LMfire suggests that SH CO emissions may be high for 1750; however, they are comparable to the peak CO concentration measured in the late 1800s (55±5 ppb) when fire emissions also peaked (van der Werf et al., 2013). As 1850 is also sometimes used as the PI baseline year when calculating RF, we suggest that LMfire provides a realistic upper bound to possible PI fire emissions.
The combined evaluation of these inventories in Hamilton et al. (2018) and here indicates that although the revised PI fire inventories differ considerably from each other and are substantially larger than CMIP6 in some regions, they result in simulated PI atmospheric concentrations that more closely represent the changes observed in paleoenvironmental archives of changes in industrial era fire activity than CMIP6 estimates do. Therefore, their respective impacts on PI tropospheric O3 concentrations and RF estimates need to be carefully considered.
2.6 Biogenic emission inventories
2.6.1 Present-day CCMI
The PD control biogenic emissions were provided from the CCMI inventory. CCMI mean annual BVOC emissions, comprising isoprene and monoterpenes, are derived using the Model of Emissions of Gases and Aerosols from Nature (MEGAN; Guenther et al., 2012) under the MACC project (Sindelarova et al., 2014). The CCMI inventory estimates global BVOC emissions at 623 Tg yr−1, in reasonable agreement with surface flux measurements and other modelling studies (Arneth et al., 2008; Sindelarova et al., 2014; Rap et al., 2018).
2.6.2 Pre-industrial and present-day LPJ-GUESS
Alternative biogenic emissions were produced using the LPJ-GUESS dynamic vegetation model simulating isoprene and monoterpenes (Arneth et al., 2007; Schurgers et al., 2009). Total PD emissions and distribution in the LPJ-GUESS inventory (i.e. 607 Tg yr−1) are similar to the PD CCMI inventory (Fig. 2). For the PI emissions, the LPJ-GUESS biogenic emissions inventory is based on the mean for the period 1750–1770, estimated to be 836 Tg yr−1. There are large spatial differences between the PI LPJ-GUESS and PD CCMI inventories, with significantly higher emissions in South America and central Africa and lower emissions in South East Asia in the PI LPJ-GUESS inventory (Fig. 2).
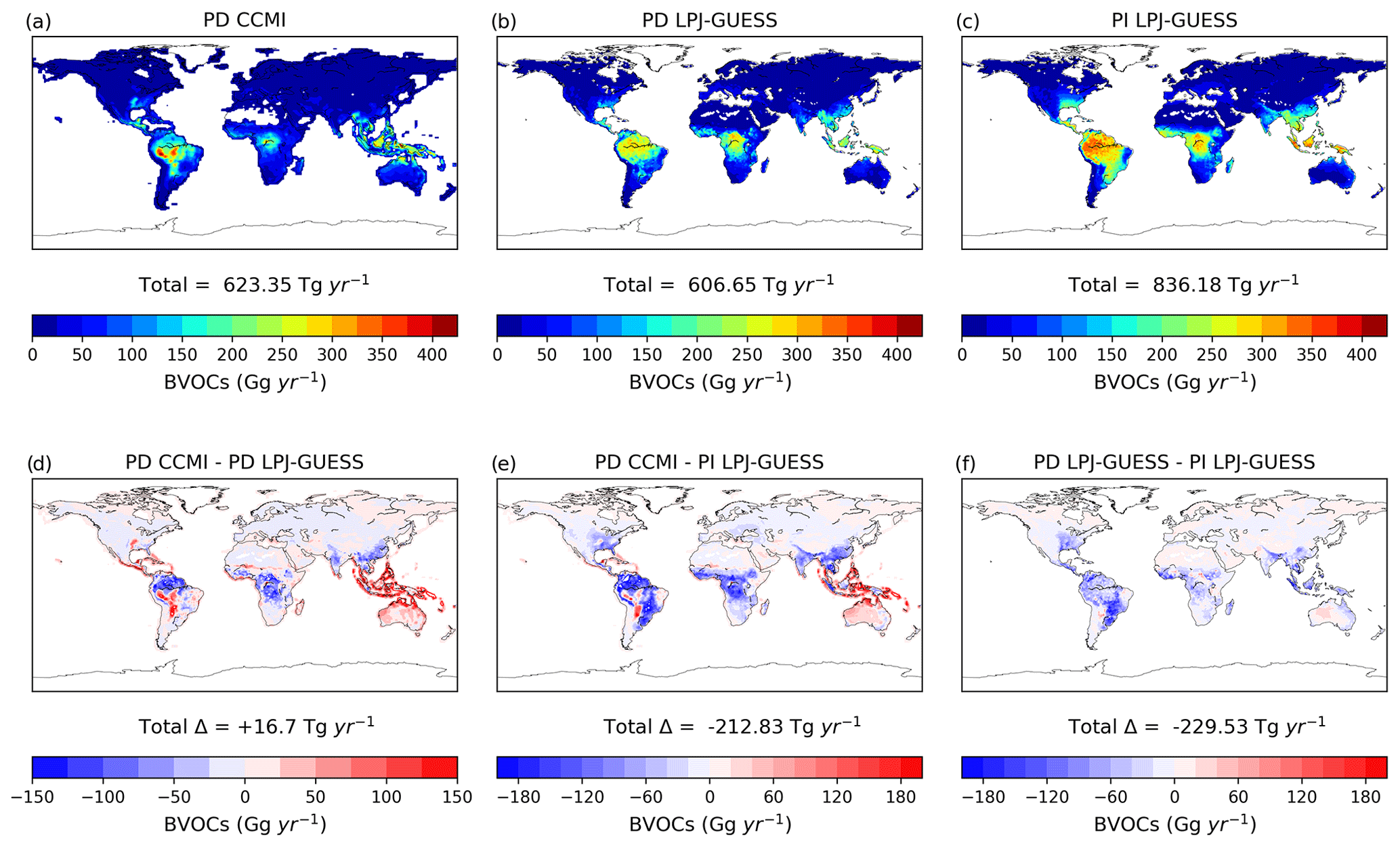
Figure 2Annual BVOC (isoprene + monoterpenes) emissions at resolution in the two present-day biogenic emissions inventories (CCMI and LPJ-GUESS) and the pre-industrial LPJ-GUESS inventory. (a–c) Total emissions per year and (d–f) differences between the three inventories. Total annual emissions and the difference in annual emissions are also shown.
3.1 Pre-industrial emission inventories
Figure 1a–d show annual latitudinal fire emissions of CO, NOx, CH4 and VOCs from all sources for the different fire inventories considered, while Fig. 1e compares BVOC emissions (i.e. isoprene and all monoterpenes) from the biogenic inventories. There is large variation in simulated CO emissions between the three PI fire inventories: 644 Tg yr−1 in SIMFIRE-BLAZE (69 % larger than CMIP6) and 1152 Tg yr−1 in LMfire (200 % larger). Estimates of CO emissions using LMfire result in total global emissions which are larger than the PD estimate, which also includes anthropogenic sources. The larger PI biomass burning emissions in LMfire are a result of a number of factors not present in the other PI inventories such as the inclusion of high-latitude fire occurrence, agricultural fire emissions and differing emission factors (Hamilton et al., 2018). The largest increase occurs due to increased SH burning in the LMfire inventory, substantially increasing CO emissions from Australia and South America (particularly eastern Amazonia and Argentina). In the CMIP6 simulations, global CO emissions are increased by a factor of 2.5 between PI and PD from 382 to 970 Tg yr−1. The main driver of this increase is industrial emissions, particularly in the NH mid-latitudes.
Global NOx emissions also vary considerably between PI inventories, with values in the SIMFIRE-BLAZE inventory increasing 13 % compared to the CMIP6 inventory (36 compared to 32 Tg yr−1). This difference is largely due to increased emissions in NH mid-latitudes within SIMFIRE-BLAZE. NOx emissions in LMfire are 112 % larger than the CMIP6 total (68 Tg yr−1), with the most significant increases in the extratropics.
As CH4 emissions from fires are significantly smaller than CO emissions (Voulgarakis and Field, 2015), increased PI fire estimates do not substantially alter total CH4 emissions. CH4 emissions in SIMFIRE-BLAZE and LMfire are similar in amount and distribution: 15 % and 9 % lower than CMIP6, respectively. There is an increase in SH CH4 emissions in both SIMFIRE-BLAZE and LMfire compared to CMIP6 but a decrease in the NH and SH mid-latitudes. Total PI CH4 emissions are greatest in CMIP6 at 241 Tg yr−1, approximately 43 % of PD emissions. PD SIMFIRE-BLAZE emissions of CH4 from biomass burning were not available; therefore, PD CMIP6 CH4 was applied in the PD SIMFIRE-BLAZE simulation. Due to the scaling of global mean surface CH4 concentrations in TOMCAT–GLOMAP, the effect of changes in the amount of CH4 emitted is likely small; however, the change in distribution may impact the formation and loss rates of tropospheric O3.
In terms of fire-emitted VOC species, their magnitude and distribution of emissions are fairly consistent between PD and PI inventories. PI CMIP6 is 87 % of PD CMIP6 values, with PI SIMFIRE-BLAZE at 97 % (303 Tg yr−1). Total global VOC emissions are largest in LMfire at 349 Tg yr−1, 29 % larger than PI CMIP6 (271 Tg yr−1) and 13 % larger than PD CMIP6 (310 Tg yr−1). The distribution of total global VOC emissions is relatively uniform across all inventories; however, individual species do have larger variability between inventories. Formaldehyde and acetylene, for example, have substantially increased SH emissions in SIMFIRE-BLAZE and LMfire due to differences in emission factors, vegetation type and burned area between the fire models.
The BVOC emissions in the two PD inventories (CCMI and LPJ-GUESS) are similar (Fig. 1e), although a small positive NH gradient exists in PD LPG-GUESS compared to PD CCMI. Total BVOC emissions are 16.7 Tg larger in the PD CCMI inventory than PD LPJ-GUESS (Fig. 2). However, the PI LPJ-GUESS BVOC estimate (836 Tg yr−1) is 37 % larger than its PD equivalent and 34 % larger than PD CCMI, although with a similar spatial distribution (Fig. 2). The largest difference is in South American emissions, for which PI LPJ-GUESS emissions are up to 120 Tg larger than PD. The reduction of BVOC emissions between PI and PD is due to a combination of crop expansion, land-cover changes and CO2 inhibition (Hantson et al., 2017). Our results are consistent with previous studies reporting between ∼25 % (Lathière et al., 2010; Pacifico et al., 2012; Hollaway et al., 2017) and ∼35 % (Unger, 2014) larger PI values than PD.
The seasonality of fire emissions in the PD and PI inventories used here is demonstrated in Fig. 3. CMIP6 PI and PD emissions have an extremely similar seasonal cycle for all species, with monthly values offset by larger emissions in PD. This is expected as the PI CMIP6 emissions are based on GFED4s climatology and monthly patterns were assumed not to have changed over time (van Marle et al., 2017). The seasonal cycle of CO emissions (Fig. 3a) varies substantially across the three PI inventories, with LMfire estimating peak emissions in May–June as opposed to July–August in CMIP6 and SIMFIRE-BLAZE. This may be a result of increased emissions from SH Africa and Central America, where large fire events are common in late spring. The inclusion of high-latitude fire occurrence and agricultural burning in LMfire may also play a role, as these contribute to fire emissions in the boreal spring season (Hamilton et al., 2018). The SIMFIRE-BLAZE CO emissions exhibit a similar but more pronounced seasonal cycle than that in CMIP6, with peak emissions in August. Similarly, NOx and VOC emissions peak earlier in the year in the LMfire inventory relative to SIMFIRE-BLAZE and CMIP6, again with a larger peak in August in SIMFIRE-BLAZE. Monthly CH4 emissions are broadly consistent across all inventories, with peak emissions in July or August and lower emissions over the NH winter. The seasonality of BVOC emissions is also consistent across all PI inventories and PD CMIP6, with a peak in July–August. Isoprene emissions are heavily dependent on temperature and photosynthetic active radiation (Malik et al., 2018), therefore reaching a maximum in NH summer when these parameters are optimum for vegetation emissions.
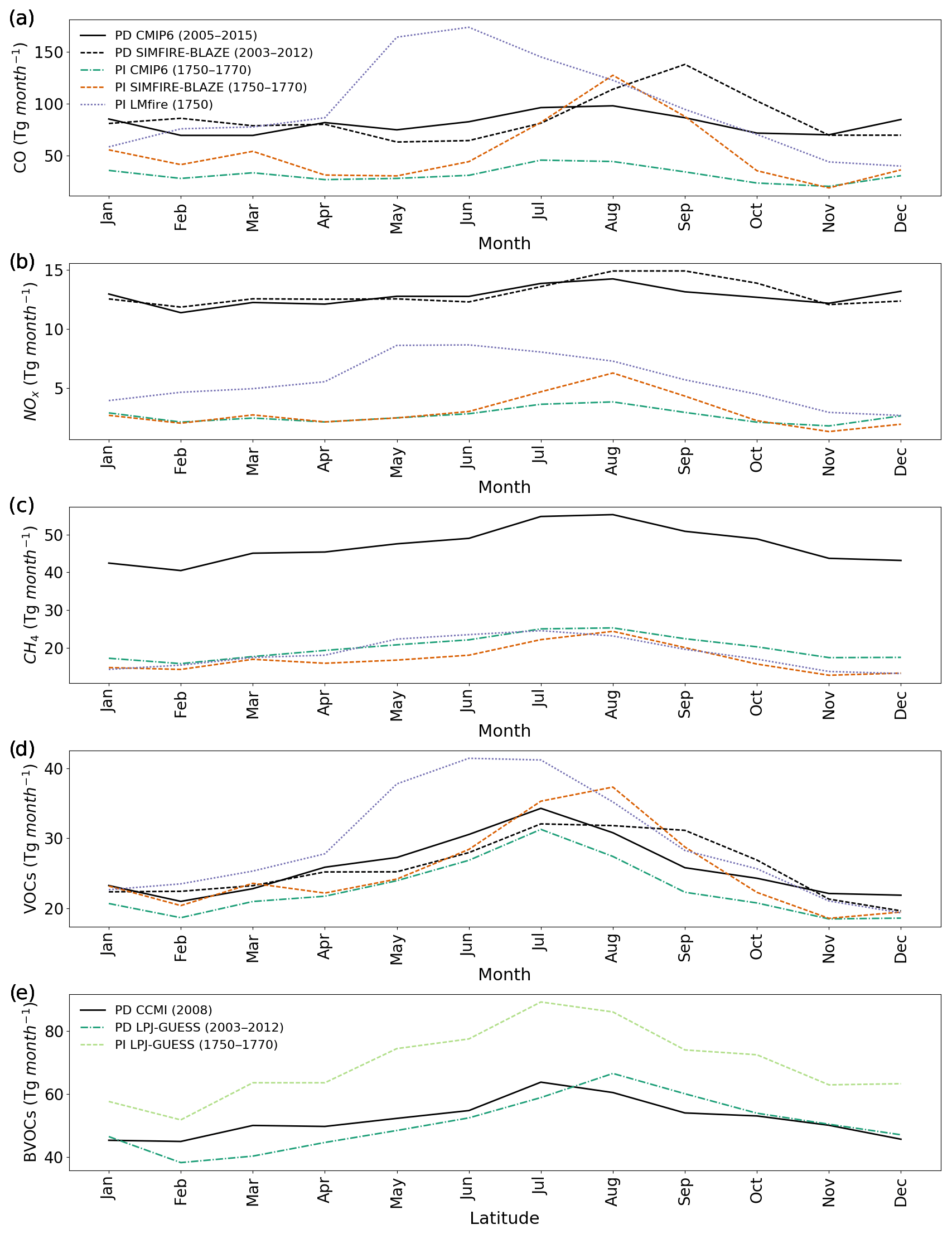
Figure 3Total monthly emissions (Tg per month) of (a) CO, (b) NOx, (c) CH4, (d) VOCs and BVOCs (e) for PD CMIP6 (solid black line), PD SIMFIRE-BLAZE (dashed black), PI CMIP6 (dashed green), PI SIMFIRE-BLAZE (dotted orange), PI LMfire (dashed purple), PD LPJ-GUESS (dashed dark green) and PI LPJ-GUESS (dotted light green). The legend in panel (a) also applies to panels (b), (c) and (d).
Figure 3 indicates similar controls over the modelled seasonality of PI fire occurrence in both PI CMIP6 and PI SIMFIRE-BLAZE, with an increase in estimates of fire extent in SIMFIRE-BLAZE resulting in a more pronounced seasonal cycle. LMfire, on the other hand, estimates a shift in the seasonality of global fire emissions, with larger fire emissions earlier than other inventories, as well as a broader peak period of emissions. The change in the seasonality of precursors will undoubtedly affect the formation and transport of tropospheric O3, as atmospheric chemistry and circulation also strong have seasonal cycles. However, the broadly similar pattern of maximum emissions in the NH summer and a minimum in winter, coinciding with similar climatic conditions, mean that the substantial difference in the volume of precursor emissions across the PI inventories is likely to be more significant than seasonal changes.
3.2 Pre-industrial fire emission effect on O3
Annual emissions of O3 precursors and their contribution to the formation of tropospheric O3 are shown in Fig. 4. The largest difference between simulations is estimates of the global tropospheric CO burden, which varies by up to 100 Tg depending on the PI fire emission inventory employed: 195 Tg in the PI CMIP6 simulation, 232 Tg in PI SIMFIRE-BLAZE (18 % higher than CMIP6) and 295 Tg in PI LMfire (50 % higher) (Table 2).
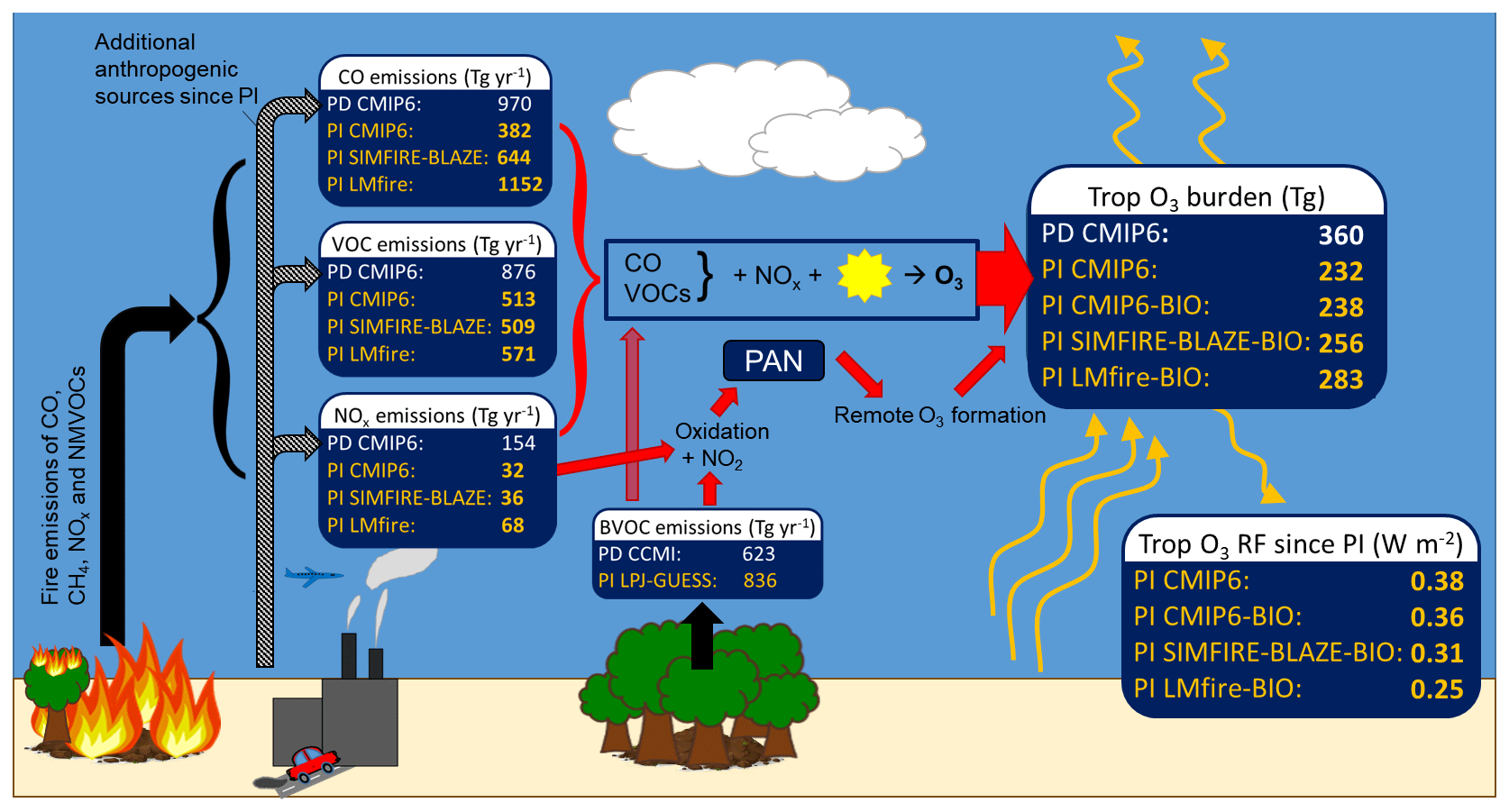
Figure 4Summary schematic showing tropospheric O3 precursor emissions from fire, biogenic and anthropogenic sources, the processes of photochemical O3 formation, the tropospheric O3 burden, and the PI-to-PD RF. The magnitude of CO, NOx, VOC and BVOC precursor emissions used in this study is shown for the PD (white text) and each PI inventory (yellow text). The resulting calculated tropospheric O3 burden and RF when using each emission inventory are also shown.
The difference in global NOx burden between PI simulations is less pronounced, with increases of 4 % and 18 % in PI SIMFIRE-BLAZE and PI LMfire, respectively, relative to PI CMIP6. The annual mean NH ∕ SH ratio of tropospheric NOx burden in PI simulations is 1.09, 1.12 and 1.18 for CMIP6, SIMFIRE-BLAZE and LMfire, respectively. The hydroxyl radical (OH), which plays a key role in regulating tropospheric O3 concentrations, had lower PI concentrations than in PD due to the higher concentrations of the OH precursors NOx and O3 in PD outcompeting the effect of increased CH4 and CO concentrations, which deplete OH (Naik et al., 2013). This is consistent in the TOMCAT PI simulations, with air-mass-weighted global mean concentrations of tropospheric OH at 1.06, 1.06 and 1.11×106 molec. cm−3 in CMIP6, SIMFIRE-BLAZE and LMfire, respectively, compared to 1.12×106 molec. cm−3 in PD CMIP6. Each of these values falls within 1 standard deviation of the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP) multi-model mean of 1.13±0.17 (Naik et al., 2013).
Changes to the atmospheric concentration and distribution of O3 precursor species lead to changes in the tropospheric O3 burden. The PI CMIP6 simulation produced the lowest tropospheric O3 burden at 232 Tg, slightly below the ACCMIP multi-model mean of 239 Tg for 1850 (Young et al., 2013). In PI SIMFIRE-BLAZE the burden is 242 Tg (4 % higher than CMIP6), while in LMfire it is 273 Tg (18 % higher), slightly outside the range of estimates of 1850 tropospheric O3 burden in ACCMIP models (192 to 272 Tg) (Young et al., 2013). The burdens simulated here represent a PI-to-PD tropospheric O3 burden change of 55 %, 49 % and 32 % for CMIP6, SIMFIRE-BLAZE and LMfire, respectively. We note that PI LMfire is the only inventory leading to a simulated PI-to-PD global burden change of less than 40 %, a value consistent with that recently indicated by isotope measurements in ice cores (Yeung et al., 2019). The differences between CMIP6 and SIMFIRE-BLAZE are primarily related to increases in tropospheric O3 within the Amazon region (Fig. 5a). The change in the tropospheric O3 vertical profile in the PI SIMFIRE-BLAZE simulation compared to PI CMIP6 (Fig. 5c) shows increased annual mean concentrations throughout the troposphere, driven by changes at 30∘ S and 50∘ N. Changes between LMfire and CMIP6 simulated tropospheric O3 profiles are larger, with increased O3 at all latitudes. Compared to PI CMIP6, there is a mean global increase in the O3 column of 3.7 DU when using LMfire and 1.0 DU when using SIMFIRE-BLAZE. The largest changes occur over central Asia, Australia and South America where tropospheric column O3 can be as much as 9.0 DU higher in the PI LMfire simulation than the PI CMIP6 simulation (Fig. 5b). This is reflected in the changes to the vertical O3 profile, with the largest increases in the subtropics. The difference between LMfire and CMIP6 simulations is greatest between 600 and 800 hPa in the SH and is roughly constant with respect to changes in altitude over the northern subtropics. The only regions where tropospheric O3 is higher in the CMIP6 simulation are central Africa and Indonesia, likely due to the PI CMIP6 emissions being anchored to PD fire observations and thus transferring these patterns to the PI (van Marle et al., 2017).

Figure 5Difference in simulated PI O3 between the CMIP6 control and the revised inventories SIMFIRE-BLAZE and LMfire. (a, b) A comparison of differences in tropospheric column O3 in DU; (c, d) differences in zonal mean vertical O3 (ppbv).
The effect of different fire emission inventories on the O3 burden is significantly smaller than the impact on CO concentrations (Table 2), as fire emissions are one of several sources of O3 variability (Lelieveld and Dentener, 2000). O3 production is reliant on a number of precursors which do not respond uniformly to the different estimates of fire occurrence in the inventories used here. The relatively minor response of NOx concentrations across the three PI emission estimates (Table 2), and the prevailing NOx-limited state across rural environments in PD (Duncan et al., 2010), suggests that increases in CO and VOCs have only a small impact on O3 production because of NOx availability limitations. Moreover, Stevenson et al. (2013) attributed the majority of the PI-to-PD shift in tropospheric O3 to NOx and CH4 changes, with a relatively small contribution from CO and NMVOCs despite increasing emissions of both. However, the simulated changes still represent significant shifts in the abundance and distribution of tropospheric O3 in the PI atmosphere.
3.3 Pre-industrial BVOC emission effect on O3
We repeated the three PI simulations, replacing the PD biogenic emissions with the PI LPJ-GUESS inventory. In general, the inclusion of PI BVOC emissions increases PI O3 concentrations due to an increased VOC source and hence PAN formation (Fig. 4). For CMIP6 fire emissions, the inclusion of PI BVOCs increases the CO burden by 22 % and the tropospheric O3 burden by 3 %, while the mean tropospheric OH concentration decreases by 6 %. The decrease in OH is likely responsible for the simulated increase in CO, as OH is consumed by VOC oxidation. The increase in global tropospheric O3 indicates that the simulated increases in VOC and CO concentrations are co-located with high NOx concentrations, as in low NOx BVOCs may decrease local O3 concentrations. The inclusion of PI BVOCs in the LMfire fire emission simulation causes a 3 % decrease in tropospheric OH and increases in tropospheric CO and O3 of 14 % and 4 %, respectively.
For SIMFIRE-BLAZE, the inclusion of PI BVOCs decreases OH by 6 % and increases CO and O3 by 22 % and 6 %, respectively. In all simulations the inclusion of PI BVOCs has only a small effect on the NOx burden (∼1 %). The effect on tropospheric O3 of including PI BVOCs is notably larger in the simulation using SIMFIRE-BLAZE fire emissions compared to CMIP6 or LMfire. The SIMFIRE-BLAZE simulation combines fire and biogenic emissions produced using the same land-use model, with consistent vegetation distributions. The co-location of isoprene and NOx emissions promotes PAN formation, enabling long-range transport of NOx and enhancing O3 production (Hollaway et al., 2017). This synergistic effect has been found to amplify the effect of biogenic emissions on tropospheric O3 production (Bossioli et al., 2012). Therefore, if PI biogenic emissions inventories were specifically produced for each fire inventory, the corresponding impact on O3 would likely be larger than presented here. With the inclusion of PI BVOC emissions, both the SIMFIRE-BLAZE and LMfire simulations result in a PI-to-PD tropospheric O3 burden change of 40 % or less, in line with estimates from oxygen isotope measurements from ice cores (Yeung et al., 2019).
3.4 Effect on ozone radiative forcing
The estimated tropospheric O3 RF, based on the CMIP6 PI and PD control simulations, is 0.38 W m−2 (Fig. 4 and Table 2), comparing well with the IPCC AR5 estimate of 0.4±0.2 W m−2 (Myhre et al., 2013; Stevenson et al., 2013). We obtain the same 0.38 W m−2 RF value when contrasting the PI CMIP6 simulation against the other the other PD simulation (PD SIMFIRE-BLAZE). This is consistent with the fact that PD tropospheric O3 is well constrained by satellite observations (Rap et al., 2015). Given the similarity of the PD simulations, the main PD CMIP6 simulation is used here as the PD for RF calculations in this section. When PI SIMFIRE-BLAZE and PI LMfire emissions are used instead of PI CMIP6 fire emissions, larger PI tropospheric O3 concentrations lead to 8 % (to 0.35 W m−2) and 29 % (to 0.27 W m−2) decreases in O3 RF, respectively. When the PI BVOC emission inventory is used in conjunction with each PI fire emission inventory, O3 RF is further reduced compared to the control by 5 % (to 0.36 W m−2), 18 % (to 0.31 W m−2) and 34 % (to 0.25 W m−2) for CMIP6, SIMFIRE-BLAZE and LMfire, respectively (Fig. 4). While these reductions in O3 RF are still within the IPCC uncertainty range, they are caused entirely by uncertainty in PI precursor emissions from wildfires and vegetation. Other key sources of uncertainty (e.g. inter-model spread, use of different radiative transfer schemes) are not accounted for here and would therefore alter estimates further, potentially outside the current 5 %–95 % confidence range. The most important region for changes to the RF of O3 is the upper troposphere at subtropical latitudes (Fig. 5d), where there are substantially higher O3 concentrations in the LMfire simulation. O3 changes in this region are up to 10 times more efficient at altering the radiative flux than in other regions (Rap et al., 2015). However, the lack of a vertical distribution to fire emissions in TOMCAT affects the simulated changes to the O3 vertical profile. Previous studies, which introduced an injection height scheme, found small increases in O3 production downwind of emission sources (Jian and Fu, 2014), although the change to total O3 and precursors is relatively small (Bossioli et al., 2012; Zhu et al., 2018).
Revised inventories of PI fire and biogenic emissions substantially decrease estimates of PI-to-PD tropospheric O3 RF. When using PI LMfire fire emissions, which represent a plausible upper emissions limit, O3 RF is reduced to 0.27 W m−2, 29 % smaller than the CMIP6 simulation. Large increases in estimated PI fire occurrence drive increases in PI O3 concentrations (3.7 DU global mean tropospheric column O3 increase for LMfire inventory) through larger emissions of CO, NOx and VOCs. PI CO increases by up to 51 % depending on the PI inventory, but the effect on O3 production is limited by the relatively small increase in NOx (∼4 %). Using PI biogenic emissions, rather than assuming PD values, further increases simulated PI tropospheric O3, though the magnitude of this depends on the fire inventory. When accounting for revised emissions from fire and biogenic sources, both the LMfire and SIMFIRE-BLAZE inventories simulated a PI-to-PD change in the tropospheric O3 burden of approximately 40 % or less, in good agreement with estimates from Yeung et al. (2019). Consequently, we find that the estimate of O3 RF since PI decreases by up to 34 % (to 0.25 W m−2) when considering the uncertainty in PI emissions of both fires and BVOCs.
The impact on tropospheric O3 from uncertainty in PI natural emissions suggests that previous estimates of O3 RF over the industrial era are likely too large. Our revised tropospheric O3 RF estimates are at the lower end of the existing uncertainty range, without yet taking into account other sources of uncertainty. We therefore argue that the impact of uncertainty on PI natural emissions should be further investigated using more models in order to reassess the current best estimate and uncertainty range of O3 RF.
The LMfire aerosol inventory is available at https://doi.org/10.1594/PANGAEA.896425 (Kaplan and Hamilton, 2018). Other datasets are available via the Open Science Framework (https://doi.org/10.17605/OSF.IO/98C2N, Rowlinson, 2019) or by request from the authors.
MJR, AR, DSH and RJP conceptualised the study and planned the model experiments. Emission inventories were produced by DH, SH, JOK, AA and LN, and they were processed for use in TOMCAT–GLOMAP by RJP and DSH. All model runs and analysis were performed by MJR with guidance from AR, RJP and SRA. The paper was written by MJR with comments and advice from all co-authors.
The authors declare that they have no conflict of interest.
Matthew J. Rowlinson is funded by a NERC SPHERES DTP (NE/L002574/1) studentship. This work used the UK ARCHER (http://www.archer.ac.uk, last access: 19 June 2020) and Leeds ARC3 high-performance computing facilities. Richard J. Pope is funded by the UK National Centre for Earth Observation (NCEO). Stijn Hantson and Almut Arneth acknowledge support from EU FP7 projects BACCHUS (grant agreement no. 603445) and LUC4C (grant agreement no. 603542). Douglas S. Hamilton is funded by the Atkinson Center for a Sustainable Future at Cornell University. Piers M. Forster is supported by NERC grant NE/N006038/1 (SMURPHS) and EU Horizon 2020 programme grant agreement number 820829 (CONSTRAIN).
This research has been supported by the Leeds York NERC DTP (grant no. NE/L002574/1), the UK National Centre for Earth Observation (NCEO), the European Commission Seventh Framework Programme (BACCHUS (grant no. 603445) and LUC4C (grant no. 603542)), H2020 Research Infrastructures (CONSTRAIN (grant no. 820829)), the David R. Atkinson Center for a Sustainable Future, Cornell University and NERC (grant no. NE/N006038/1 (SMURPHS)).
This paper was edited by Kostas Tsigaridis and reviewed by two anonymous referees.
Akagi, S. K., Yokelson, R. J., Wiedinmyer, C., Alvarado, M. J., Reid, J. S., Karl, T., Crounse, J. D., and Wennberg, P. O.: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011, 2011.
Andela, N., Morton, D. C., Giglio, L., Chen, Y., van der Werf, G. R., Kasibhatla, P. S., DeFries, R. S., Collatz, G. J., Hantson, S., Kloster, S., Bachelet, D., Forrest, M., Lasslop, G., Li, F., Mangeon, S., Melton, J. R., Yue, C., and Randerson, J. T.: A human-driven decline in global burned area, Science, 356, 1356–1362, https://doi.org/10.1126/science.aal4108, 2017.
Arneth, A., Niinemets, Ü., Pressley, S., Bäck, J., Hari, P., Karl, T., Noe, S., Prentice, I. C., Serça, D., Hickler, T., Wolf, A., and Smith, B.: Process-based estimates of terrestrial ecosystem isoprene emissions: incorporating the effects of a direct CO2-isoprene interaction, Atmos. Chem. Phys., 7, 31–53, https://doi.org/10.5194/acp-7-31-2007, 2007.
Arneth, A., Monson, R. K., Schurgers, G., Niinemets, Ü., and Palmer, P. I.: Why are estimates of global terrestrial isoprene emissions so similar (and why is this not so for monoterpenes)?, Atmos. Chem. Phys., 8, 4605–4620, https://doi.org/10.5194/acp-8-4605-2008, 2008.
Arneth, A., Sitch, S., Bondeau, A., Butterbach-Bahl, K., Foster, P., Gedney, N., de Noblet-Ducoudré, N., Prentice, I. C., Sanderson, M., Thonicke, K., Wania, R., and Zaehle, S.: From biota to chemistry and climate: towards a comprehensive description of trace gas exchange between the biosphere and atmosphere, Biogeosciences, 7, 121–149, https://doi.org/10.5194/bg-7-121-2010, 2010.
Bekki, S., Rap, A., Poulain, V., Dhomse, S., Marchand, M., Lefevre, F., Forster, P. M., Szopa, S., and Chipperfield, M. P.: Climate impact of stratospheric ozone recovery, Geophys. Res. Lett., 40, 2796–2800, https://doi.org/10.1002/grl.50358, 2013.
Bossioli, E., Tombrou, M., Karali, A., Dandou, A., Paronis, D., and Sofiev, M.: Ozone production from the interaction of wildfire and biogenic emissions: a case study in Russia during spring 2006, Atmos. Chem. Phys., 12, 7931–7953, https://doi.org/10.5194/acp-12-7931-2012, 2012.
Bowman, D. M. J. S., Balch, J. K., Artaxo, P., Bond, W. J., Carlson, J. M., Cochrane, M. A., D'Antonio, C. M., DeFries, R. S., Doyle, J. C., Harrison, S. P., Johnston, F. H., Keeley, J. E., Krawchuk, M. A., Kull, C. A., Marston, J. B., Moritz, M. A., Prentice, I. C., Roos, C. I., Scott, A. C., Swetnam, T. W., van der Werf, G. R., and Pyne, S. J.: Fire in the Earth System, Science, 324, 481–484, https://doi.org/10.1126/science.1163886, 2009.
Checa-Garcia, R., Hegglin, M. I., Kinnison, D., Plummer, D. A., and Shine, K. P.: Historical Tropospheric and Stratospheric Ozone Radiative Forcing Using the CMIP6 Database, Geophys. Res. Lett., 45, 3264–3273, https://doi.org/10.1002/2017GL076770, 2018.
Chipperfield, M. P.: New version of the TOMCAT/SLIMCAT off-line chemical transport model: Intercomparison of stratospheric tracer experiments, Q. J. Roy. Meteor. Soc., 132, 1179–1203, https://doi.org/10.1256/qj.05.51, 2006.
Cooper, O. R., Parrish, D. D., Ziemke, J., Balashov, N. V., Cupeiro, M., Galbally, I. E., Gilge, S., Horowitz, L., Jensen, N. R., Lamarque, J.-F., Naik, V., Oltmans, S., Schwab, J., Shindell, D. T., Thompson, A. M., Thouret, V., Wang, Y., and Zbinden, R. M.: Global distribution and trends of tropospheric ozone: An observation-based review, Elementa: Science of the Anthropocene, 2, 000029, https://doi.org/10.12952/journal.elementa.000029, 2014.
Daniau, A. L., Bartlein, P. J., Harrison, S. P., Prentice, I. C., Brewer, S., Friedlingstein, P., Harrison-Prentice, T. I., Inoue, J., Izumi, K., Marlon, J. R., Mooney, S., Power, M. J., Stevenson, J., Tinner, W., Andrič, M., Atanassova, J., Behling, H., Black, M., Blarquez, O., Brown, K. J., Carcaillet, C., Colhoun, E. A., Colombaroli, D., Davis, B. A. S., D'Costa, D., Dodson, J., Dupont, L., Eshetu, Z., Gavin, D. G., Genries, A., Haberle, S., Hallett, D. J., Hope, G., Horn, S. P., Kassa, T. G., Katamura, F., Kennedy, L. M., Kershaw, P., Krivonogov, S., Long, C., Magri, D., Marinova, E., McKenzie, G. M., Moreno, P. I., Moss, P., Neumann, F. H., Norström, E., Paitre, C., Rius, D., Roberts, N., Robinson, G. S., Sasaki, N., Scott, L., Takahara, H., Terwilliger, V., Thevenon, F., Turner, R., Valsecchi, V. G., Vannière, B., Walsh, M., Williams, N., and Zhang, Y.: Predictability of biomass burning in response to climate changes, Global Biogeochem. Cy., 26, GB4007, https://doi.org/10.1029/2011GB004249, 2012.
Dee, D. P., Uppala, S. M., Simmons, A. J., Berrisford, P., Poli, P., Kobayashi, S., Andrae, U., Balmaseda, M. A., Balsamo, G., Bauer, P., Bechtold, P., Beljaars, A. C. M., van de Berg, L., Bidlot, J., Bormann, N., Delsol, C., Dragani, R., Fuentes, M., Geer, A. J., Haimberger, L., Healy, S. B., Hersbach, H., Hólm, E. V., Isaksen, L., Kållberg, P., Köhler, M., Matricardi, M., McNally, A. P., Monge-Sanz, B. M., Morcrette, J.-J., Park, B.-K., Peubey, C., de Rosnay, P., Tavolato, C., Thépaut, J.-N., and Vitart, F.: The ERA-Interim reanalysis: configuration and performance of the data assimilation system, Q. J. Roy. Meteor. Soc., 137, 553–597, https://doi.org/10.1002/qj.828, 2011.
Dentener, F., Kinne, S., Bond, T., Boucher, O., Cofala, J., Generoso, S., Ginoux, P., Gong, S., Hoelzemann, J. J., Ito, A., Marelli, L., Penner, J. E., Putaud, J.-P., Textor, C., Schulz, M., van der Werf, G. R., and Wilson, J.: Emissions of primary aerosol and precursor gases in the years 2000 and 1750 prescribed data-sets for AeroCom, Atmos. Chem. Phys., 6, 4321–4344, https://doi.org/10.5194/acp-6-4321-2006, 2006.
Dlugokencky, E. J., Myers, R. C., Lang, P. M., Masarie, K. A., Crotwell, A. M., Thoning, K. W., Hall, B. D., Elkins, J. W., and Steele, L. P.: Conversion of NOAA atmospheric dry air CH4 mole fractions to a gravimetrically prepared standard scale, J. Geophys. Res.-Atmos., 110, D18306, https://doi.org/10.1029/2005JD006035, 2005.
Doerr, S. H. and Santín, C.: Global trends in wildfire and its impacts: perceptions versus realities in a changing world, Philos. T. R. Soc. B, 371, 20150345, https://doi.org/10.1098/rstb.2015.0345, 2016.
Duncan, B. N., Yoshida, Y., Olson, J. R., Sillman, S., Martin, R. V., Lamsal, L., Hu, Y., Pickering, K. E., Retscher, C., and Allen, D. J.: Application of OMI observations to a space-based indicator of NOx and VOC controls on surface ozone formation, Atmos. Environ., 44, 2213–2223, https://doi.org/10.1016/j.atmosenv.2010.03.010, 2010.
Edwards, J. M. and Slingo, A.: Studies with a flexible new radiation code. I: Choosing a configuration for a large-scale model, Q. J. Roy. Meteor. Soc., 122, 689–719, https://doi.org/10.1002/qj.49712253107, 1996.
Etheridge, D. M., Steele, L. P., Francey, R. J., and Langenfelds, R. L.: Atmospheric methane between 1000 A.D. and present: Evidence of anthropogenic emissions and climatic variability, J. Geophys. Res.-Atmos., 103, 15979–15993, https://doi.org/10.1029/98JD00923, 1998.
Fels, S. B., Mahlman, J. D., Schwarzkopf, M. D., and Sinclair, R. W.: Stratospheric Sensitivity to Perturbations in Ozone and Carbon Dioxide: Radiative and Dynamical Response, J. Atmos. Sci., 37, 2265–2297, https://doi.org/10.1175/1520-0469(1980)037<2265:Sstpio>2.0.Co;2, 1980.
Forster, P. and Shine, K. P.: Radiative forcing and temperature trends from stratospheric ozone changes, Geophys. Res. Lett., 102, 10841–10855, https://doi.org/10.1029/96jd03510, 1997.
Gauss, M., Myhre, G., Isaksen, I. S. A., Grewe, V., Pitari, G., Wild, O., Collins, W. J., Dentener, F. J., Ellingsen, K., Gohar, L. K., Hauglustaine, D. A., Iachetti, D., Lamarque, F., Mancini, E., Mickley, L. J., Prather, M. J., Pyle, J. A., Sanderson, M. G., Shine, K. P., Stevenson, D. S., Sudo, K., Szopa, S., and Zeng, G.: Radiative forcing since preindustrial times due to ozone change in the troposphere and the lower stratosphere, Atmos. Chem. Phys., 6, 575–599, https://doi.org/10.5194/acp-6-575-2006, 2006.
Giglio, L., Randerson, J. T., and van der Werf, G. R.: Analysis of daily, monthly, and annual burned area using the fourth-generation global fire emissions database (GFED4), J. Geophys. Res.-Biogeo., 118, 317–328, https://doi.org/10.1002/jgrg.20042, 2013.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hamilton, D. S., Hantson, S., Scott, C. E., Kaplan, J. O., Pringle, K. J., Nieradzik, L. P., Rap, A., Folberth, G. A., Spracklen, D. V., and Carslaw, K. S.: Reassessment of pre-industrial fire emissions strongly affects anthropogenic aerosol forcing, Nat. Commun., 9, 3182, https://doi.org/10.1038/s41467-018-05592-9, 2018.
Hantson, S., Knorr, W., Schurgers, G., Pugh, T. A. M., and Arneth, A.: Global isoprene and monoterpene emissions under changing climate, vegetation, CO2 and land use, Atmos. Environ., 155, 35–45, https://doi.org/10.1016/j.atmosenv.2017.02.010, 2017.
Hartmann, D. L., Klein Tank, A. M. G., Rusticucci, M., Alexander, L. V., Brönnimann, S., Charabi, Y., Dentener, F. J., Dlugokencky, E. J., Easterling, D. R., Kaplan, A., Soden, B. J., Thorne, P. W., Wild, M., and Zhai, P. M.: Observations: Atmosphere and Surface, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 159–254, 2013.
Hollaway, M. J., Arnold, S. R., Collins, W. J., Folberth, G., and Rap, A.: Sensitivity of midnineteenth century tropospheric ozone to atmospheric chemistry-vegetation interactions, J. Geophys. Res., 122, 2452–2473, https://doi.org/10.1002/2016jd025462, 2017.
Jian, Y. and Fu, T.-M.: Injection heights of springtime biomass-burning plumes over peninsular Southeast Asia and their impacts on long-range pollutant transport, Atmos. Chem. Phys., 14, 3977–3989, https://doi.org/10.5194/acp-14-3977-2014, 2014.
Kapadia, Z. Z., Spracklen, D. V., Arnold, S. R., Borman, D. J., Mann, G. W., Pringle, K. J., Monks, S. A., Reddington, C. L., Benduhn, F., Rap, A., Scott, C. E., Butt, E. W., and Yoshioka, M.: Impacts of aviation fuel sulfur content on climate and human health, Atmos. Chem. Phys., 16, 10521–10541, https://doi.org/10.5194/acp-16-10521-2016, 2016.
Kaplan, J. O. and Hamilton, D. S.: Global aerosol emissions from biomass burning for the late preindustrial Holocene, link to data in NetCDF format, PANGAEA, https://doi.org/10.1594/PANGAEA.896425, 2018.
Kaplan, J. O., Krumhardt, K. M., Ellis, E. C., Ruddiman, W. F., Lemmen, C., and Goldewijk, K. K.: Holocene carbon emissions as a result of anthropogenic land cover change, The Holocene, 21, 775–791, https://doi.org/10.1177/0959683610386983, 2011.
Klein Goldewijk, K., Beusen, A., van Drecht, G., and de Vos, M.: The HYDE 3.1 spatially explicit database of human-induced global land-use change over the past 12,000 years, Global Ecol. Biogeogr., 20, 73–86, https://doi.org/10.1111/j.1466-8238.2010.00587.x, 2011.
Knorr, W., Kaminski, T., Arneth, A., and Weber, U.: Impact of human population density on fire frequency at the global scale, Biogeosciences, 11, 1085–1102, https://doi.org/10.5194/bg-11-1085-2014, 2014.
Lamarque, J.-F., Bond, T. C., Eyring, V., Granier, C., Heil, A., Klimont, Z., Lee, D., Liousse, C., Mieville, A., Owen, B., Schultz, M. G., Shindell, D., Smith, S. J., Stehfest, E., Van Aardenne, J., Cooper, O. R., Kainuma, M., Mahowald, N., McConnell, J. R., Naik, V., Riahi, K., and van Vuuren, D. P.: Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: methodology and application, Atmos. Chem. Phys., 10, 7017–7039, https://doi.org/10.5194/acp-10-7017-2010, 2010.
Laothawornkitkul, J., Taylor, J. E., Paul, N. D., and Hewitt, C. N.: Biogenic volatile organic compounds in the Earth system, New Phytol.,183, 27–51, https://doi.org/10.1111/j.1469-8137.2009.02859.x, 2009.
Lathière, J., Hewitt, C. N., and Beerling, D. J.: Sensitivity of isoprene emissions from the terrestrial biosphere to 20th century changes in atmospheric CO2 concentration, climate, and land use, Global Biogeochem. Cy., 24, GB1004, https://doi.org/10.1029/2009gb003548, 2010.
Lee, L. A., Pringle, K. J., Reddington, C. L., Mann, G. W., Stier, P., Spracklen, D. V., Pierce, J. R., and Carslaw, K. S.: The magnitude and causes of uncertainty in global model simulations of cloud condensation nuclei, Atmos. Chem. Phys., 13, 8879–8914, https://doi.org/10.5194/acp-13-8879-2013, 2013.
Lelieveld, J. and Dentener, F. J.: What controls tropospheric ozone?, J. Geophys. Res., 105, 3531–3551, https://doi.org/10.1029/1999jd901011, 2000.
Malik, T., Gajbhiye, T., and Pandey, S.: Seasonality in emission patterns of isoprene from two dominant tree species of Central India: Implications on terrestrial carbon emission and climate change, Proceedings of the International Academy of Ecology and Environmental Science, 8, 204–212, 2018.
Mann, G. W., Carslaw, K. S., Spracklen, D. V., Ridley, D. A., Manktelow, P. T., Chipperfield, M. P., Pickering, S. J., and Johnson, C. E.: Description and evaluation of GLOMAP-mode: a modal global aerosol microphysics model for the UKCA composition-climate model, Geosci. Model Dev., 3, 519–551, https://doi.org/10.5194/gmd-3-519-2010, 2010.
Marlon, J. R., Bartlein, P. J., Carcaillet, C., Gavin, D. G., Harrison, S. P., Higuera, P. E., Joos, F., Power, M. J., and Prentice, I. C.: Climate and human influences on global biomass burning over the past two millennia, Nat. Geosci., 1, 697, https://doi.org/10.1038/ngeo313, 2008.
Marlon, J. R., Kelly, R., Daniau, A.-L., Vannière, B., Power, M. J., Bartlein, P., Higuera, P., Blarquez, O., Brewer, S., Brücher, T., Feurdean, A., Romera, G. G., Iglesias, V., Maezumi, S. Y., Magi, B., Courtney Mustaphi, C. J., and Zhihai, T.: Reconstructions of biomass burning from sediment-charcoal records to improve data–model comparisons, Biogeosciences, 13, 3225–3244, https://doi.org/10.5194/bg-13-3225-2016, 2016.
McNorton, J., Chipperfield, M. P., Gloor, M., Wilson, C., Wuhu, F., Hayman, G. D., Rigby, M., Krummel, P. B., O'Doherty, S., Prinn, R. G., Weiss, R. F., Young, D., Dlugokencky, E., and Montzka, S. A.: Role of OH variability in the stalling of the global atmospheric CH4 growth rate from 1999 to 2006, Geophys. Res. Lett., 16, 1–24, 2016.
Mickley, L. J., Jacob, D. J., and Rind, D.: Uncertainty in preindustrial abundance of tropospheric ozone: Implications for radiative forcing calculations, J. Geophys. Res., 106, 3389–3399, https://doi.org/10.1029/2000jd900594, 2001.
Monks, P. S., Archibald, A. T., Colette, A., Cooper, O., Coyle, M., Derwent, R., Fowler, D., Granier, C., Law, K. S., Mills, G. E., Stevenson, D. S., Tarasova, O., Thouret, V., von Schneidemesser, E., Sommariva, R., Wild, O., and Williams, M. L.: Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer, Atmos. Chem. Phys., 15, 8889–8973, https://doi.org/10.5194/acp-15-8889-2015, 2015.
Monks, S. A., Arnold, S. R., Hollaway, M. J., Pope, R. J., Wilson, C., Feng, W., Emmerson, K. M., Kerridge, B. J., Latter, B. L., Miles, G. M., Siddans, R., and Chipperfield, M. P.: The TOMCAT global chemical transport model v1.6: description of chemical mechanism and model evaluation, Geosci. Model Dev., 10, 3025–3057, https://doi.org/10.5194/gmd-10-3025-2017, 2017.
Murray, L. T., Mickley, L. J., Kaplan, J. O., Sofen, E. D., Pfeiffer, M., and Alexander, B.: Factors controlling variability in the oxidative capacity of the troposphere since the Last Glacial Maximum, Atmos. Chem. Phys., 14, 3589–3622, https://doi.org/10.5194/acp-14-3589-2014, 2014.
Myhre, G., Shindell, D., Bréon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Takemura, T., and Zhang, H.: Anthropogenic and Natural Radiative Forcing, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 659–740, 2013.
Naik, V., Voulgarakis, A., Fiore, A. M., Horowitz, L. W., Lamarque, J.-F., Lin, M., Prather, M. J., Young, P. J., Bergmann, D., Cameron-Smith, P. J., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R., Eyring, V., Faluvegi, G., Folberth, G. A., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., van Noije, T. P. C., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R., Shindell, D. T., Stevenson, D. S., Strode, S., Sudo, K., Szopa, S., and Zeng, G.: Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 5277–5298, https://doi.org/10.5194/acp-13-5277-2013, 2013.
Pacifico, F., Folberth, G. A., Jones, C. D., Harrison, S. P., and Collins, W. J.: Sensitivity of biogenic isoprene emissions to past, present, and future environmental conditions and implications for atmospheric chemistry, J. Geophys. Res., 117, D22302, https://doi.org/10.1029/2012jd018276, 2012.
Pan, X., Ichoku, C., Chin, M., Bian, H., Darmenov, A., Colarco, P., Ellison, L., Kucsera, T., da Silva, A., Wang, J., Oda, T., and Cui, G.: Six global biomass burning emission datasets: intercomparison and application in one global aerosol model, Atmos. Chem. Phys., 20, 969–994, https://doi.org/10.5194/acp-20-969-2020, 2020.
Pfeiffer, M., Spessa, A., and Kaplan, J. O.: A model for global biomass burning in preindustrial time: LPJ-LMfire (v1.0), Geosci. Model Dev., 6, 643–685, https://doi.org/10.5194/gmd-6-643-2013, 2013.
Rabin, S. S., Melton, J. R., Lasslop, G., Bachelet, D., Forrest, M., Hantson, S., Kaplan, J. O., Li, F., Mangeon, S., Ward, D. S., Yue, C., Arora, V. K., Hickler, T., Kloster, S., Knorr, W., Nieradzik, L., Spessa, A., Folberth, G. A., Sheehan, T., Voulgarakis, A., Kelley, D. I., Prentice, I. C., Sitch, S., Harrison, S., and Arneth, A.: The Fire Modeling Intercomparison Project (FireMIP), phase 1: experimental and analytical protocols with detailed model descriptions, Geosci. Model Dev., 10, 1175–1197, https://doi.org/10.5194/gmd-10-1175-2017, 2017.
Randerson, J. T., Chen, Y., van der Werf, G. R., Rogers, B. M., and Morton, D. C.: Global burned area and biomass burning emissions from small fires, J. Geophys. Res., 117, G04012, https://doi.org/10.1029/2012jg002128, 2012.
Randerson, J. T., Van Der Werf, G. R., Giglio, L., Collatz, G. J., and Kasibhatla, P. S.: Global Fire Emissions Database, Version 4.1 (GFEDv4), ORNL Distributed Active Archive Center, 2017.
Rap, A., Richards, N. A. D., Forster, P. M., Monks, S. A., Arnold, S. R., and Chipperfield, M. P.: Satellite constraint on the tropospheric ozone radiative effect, Geophys. Res. Lett., 42, 5074–5081, https://doi.org/10.1002/2015GL064037, 2015.
Rap, A., Scott, C. E., Reddington, C. L., Mercado, L., Ellis, R. J., Garraway, S., Evans, M. J., Beerling, D. J., MacKenzie, A. R., Hewitt, C. N., and Spracklen, D. V.: Enhanced global primary production by biogenic aerosol via diffuse radiation fertilization, Nat. Geosci., 11, 640–644, https://doi.org/10.1038/s41561-018-0208-3, 2018.
Rowlinson, M.: Tropospheric ozone radiative forcing uncertainty due to pre-industrial fire and biogenic emissions (emissions in text files), OSFHOME, https://doi.org/10.17605/OSF.IO/98C2N, 2019.
Rowlinson, M. J., Rap, A., Arnold, S. R., Pope, R. J., Chipperfield, M. P., McNorton, J., Forster, P., Gordon, H., Pringle, K. J., Feng, W., Kerridge, B. J., Latter, B. L., and Siddans, R.: Impact of El Niño–Southern Oscillation on the interannual variability of methane and tropospheric ozone, Atmos. Chem. Phys., 19, 8669–8686, https://doi.org/10.5194/acp-19-8669-2019, 2019.
Rubino, M., D'Onofrio, A., Seki, O., and Bendle, J. A.: Ice-core records of biomass burning, The Anthropocene Review, 3, 140–162, https://doi.org/10.1177/2053019615605117, 2016.
Schurgers, G., Arneth, A., Holzinger, R., and Goldstein, A. H.: Process-based modelling of biogenic monoterpene emissions combining production and release from storage, Atmos. Chem. Phys., 9, 3409–3423, https://doi.org/10.5194/acp-9-3409-2009, 2009.
Scott, C. E., Monks, S. A., Spracklen, D. V., Arnold, S. R., Forster, P. M., Rap, A., Äijälä, M., Artaxo, P., Carslaw, K. S., Chipperfield, M. P., Ehn, M., Gilardoni, S., Heikkinen, L., Kulmala, M., Petäjä, T., Reddington, C. L. S., Rizzo, L. V., Swietlicki, E., Vignati, E., and Wilson, C.: Impact on short-lived climate forcers increases projected warming due to deforestation, Nat. Commun., 9, 157, https://doi.org/10.1038/s41467-017-02412-4, 2018.
Shindell, D. T., Pechony, O., Voulgarakis, A., Faluvegi, G., Nazarenko, L., Lamarque, J.-F., Bowman, K., Milly, G., Kovari, B., Ruedy, R., and Schmidt, G. A.: Interactive ozone and methane chemistry in GISS-E2 historical and future climate simulations, Atmos. Chem. Phys., 13, 2653–2689, https://doi.org/10.5194/acp-13-2653-2013, 2013.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Singh, H. B.: Reactive nitrogen in the troposphere, Environ. Sci. Technol., 21, 320–327, https://doi.org/10.1021/es00158a001, 1987.
Smith, B., Wårlind, D., Arneth, A., Hickler, T., Leadley, P., Siltberg, J., and Zaehle, S.: Implications of incorporating N cycling and N limitations on primary production in an individual-based dynamic vegetation model, Biogeosciences, 11, 2027–2054, https://doi.org/10.5194/bg-11-2027-2014, 2014.
Stevenson, D. S., Young, P. J., Naik, V., Lamarque, J.-F., Shindell, D. T., Voulgarakis, A., Skeie, R. B., Dalsoren, S. B., Myhre, G., Berntsen, T. K., Folberth, G. A., Rumbold, S. T., Collins, W. J., MacKenzie, I. A., Doherty, R. M., Zeng, G., van Noije, T. P. C., Strunk, A., Bergmann, D., Cameron-Smith, P., Plummer, D. A., Strode, S. A., Horowitz, L., Lee, Y. H., Szopa, S., Sudo, K., Nagashima, T., Josse, B., Cionni, I., Righi, M., Eyring, V., Conley, A., Bowman, K. W., Wild, O., and Archibald, A.: Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 3063–3085, https://doi.org/10.5194/acp-13-3063-2013, 2013.
Swetnam, T. W., Farella, J., Roos, C. I., Liebmann, M. J., Falk, D. A., and Allen, C. D.: Multiscale perspectives of fire, climate and humans in western North America and the Jemez Mountains, USA, Philos. T. Roy. Soc. B, 371, 20150168, https://doi.org/10.1098/rstb.2015.0168, 2016.
Unger, N.: Human land-use-driven reduction of forest volatiles cools global climate, Nat. Clim. Change, 4, 907, https://doi.org/10.1038/nclimate2347, 2014.
van der Werf, G. R., Randerson, J. T., Giglio, L., Collatz, G. J., Mu, M., Kasibhatla, P. S., Morton, D. C., DeFries, R. S., Jin, Y., and van Leeuwen, T. T.: Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009), Atmos. Chem. Phys., 10, 11707–11735, https://doi.org/10.5194/acp-10-11707-2010, 2010.
van der Werf, G. R., Peters, W., van Leeuwen, T. T., and Giglio, L.: What could have caused pre-industrial biomass burning emissions to exceed current rates?, Clim. Past, 9, 289–306, https://doi.org/10.5194/cp-9-289-2013, 2013.
van Marle, M. J. E., Kloster, S., Magi, B. I., Marlon, J. R., Daniau, A.-L., Field, R. D., Arneth, A., Forrest, M., Hantson, S., Kehrwald, N. M., Knorr, W., Lasslop, G., Li, F., Mangeon, S., Yue, C., Kaiser, J. W., and van der Werf, G. R.: Historic global biomass burning emissions for CMIP6 (BB4CMIP) based on merging satellite observations with proxies and fire models (1750–2015), Geosci. Model Dev., 10, 3329–3357, https://doi.org/10.5194/gmd-10-3329-2017, 2017.
Volz, A. and Kley, D.: Evaluation of the Montsouris series of ozone measurements made in the nineteenth century, Nature, 332, 240–242, https://doi.org/10.1038/332240a0, 1988.
Voulgarakis, A. and Field, R. D.: Fire Influences on Atmospheric Composition, Air Quality and Climate, Current Pollution Reports, 1, 70–81, https://doi.org/10.1007/s40726-015-0007-z, 2015.
Wang, Z., Chappellaz, J., Park, K., and Mak, J. E.: Large Variations in Southern Hemisphere Biomass Burning During the Last 650 Years, Science, 330, 1663–1666, https://doi.org/10.1126/science.1197257, 2010.
Wild, O.: Modelling the global tropospheric ozone budget: exploring the variability in current models, Atmos. Chem. Phys., 7, 2643–2660, https://doi.org/10.5194/acp-7-2643-2007, 2007.
Yeung, L. Y., Murray, L. T., Martinerie, P., Witrant, E., Hu, H., Banerjee, A., Orsi, A., and Chappellaz, J.: Isotopic constraint on the twentieth-century increase in tropospheric ozone, Nature, 570, 224–227, https://doi.org/10.1038/s41586-019-1277-1, 2019.
Young, P. J., Archibald, A. T., Bowman, K. W., Lamarque, J.-F., Naik, V., Stevenson, D. S., Tilmes, S., Voulgarakis, A., Wild, O., Bergmann, D., Cameron-Smith, P., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R. M., Eyring, V., Faluvegi, G., Horowitz, L. W., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R. B., Shindell, D. T., Strode, S. A., Sudo, K., Szopa, S., and Zeng, G.: Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 2063–2090, https://doi.org/10.5194/acp-13-2063-2013, 2013.
Zhu, L., Val Martin, M., Gatti, L. V., Kahn, R., Hecobian, A., and Fischer, E. V.: Development and implementation of a new biomass burning emissions injection height scheme (BBEIH v1.0) for the GEOS-Chem model (v9-01-01), Geosci. Model Dev., 11, 4103–4116, https://doi.org/10.5194/gmd-11-4103-2018, 2018.