the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Jun 2019
| 03 Jun 2019
Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions
Sophia M. Charan
Kelvin H. Bates
Yuanlong Huang
Tran B. Nguyen
Huajun Mai
Weimeng Kong
Richard C. Flagan
John H. Seinfeld
Recent advances in our knowledge of the gas-phase oxidation of isoprene, the impact of chamber walls on secondary organic aerosol (SOA) mass yields, and aerosol measurement analysis techniques warrant reevaluating SOA yields from isoprene. In particular, SOA from isoprene oxidation under high-NOx conditions forms via two major pathways: (1) low-volatility nitrates and dinitrates (LV pathway) and (2) hydroxymethyl-methyl-α-lactone (HMML) reaction on a surface or the condensed phase of particles to form 2-methyl glyceric acid and its oligomers (2MGA pathway). These SOA production pathways respond differently to reaction conditions. Past chamber experiments generated SOA with varying contributions from these two unique pathways, leading to results that are difficult to interpret. This study examines the SOA yields from these two pathways independently, which improves the interpretation of previous results and provides further understanding of the relevance of chamber SOA yields to the atmosphere and regional or global modeling. Results suggest that low-volatility nitrates and dinitrates produce significantly more aerosol than previously thought; the experimentally measured SOA mass yield from the LV pathway is ∼0.15. Sufficient seed surface area at the start of the reaction is needed to limit the effects of vapor wall losses of low-volatility compounds and accurately measure the complete SOA mass yield. Under dry conditions, substantial amounts of SOA are formed from HMML ring-opening reactions with inorganic ions and HMML organic oligomerization processes. However, the lactone organic oligomerization reactions are suppressed under more atmospherically relevant humidity levels, where hydration of the lactone is more competitive. This limits the SOA formation potential from the 2MGA pathway to HMML ring-opening reactions with water or inorganic ions under typical atmospheric conditions. The isoprene SOA mass yield from the LV pathway measured in this work is significantly higher than previous studies have reported, suggesting that low-volatility compounds such as organic nitrates and dinitrates may contribute to isoprene SOA under high-NOx conditions significantly more than previously thought and thus deserve continued study.
- Article
(2503 KB) - Full-text XML
-
Supplement
(2550 KB) - BibTeX
- EndNote





In the atmosphere, submicrometer particulate matter is composed of a significant fraction of organic aerosol (Zhang et al., 2007). There are two forms of organic aerosol: primary, which is directly emitted into the atmosphere, and secondary, which is formed when gas-phase compounds partition to the particle phase. Processes governing secondary organic aerosol (SOA) formation are particularly complex (Kroll and Seinfeld, 2008; Hallquist et al., 2009). SOA yields, the ratio of the mass of SOA formed to the mass of the parent volatile organic compound (VOC) reacted, are measured in environmental chambers and are used in models to reduce the complexity of SOA formation.
Isoprene is the dominant non-methane biogenic VOC emitted into the atmosphere. Because of the large flux of isoprene (∼535 Tg yr−1) into the atmosphere (Guenther et al., 2012), oxidation of isoprene is a significant source of SOA even though SOA yields measured in chambers are relatively low (Carlton et al., 2009). Despite numerous experimental studies of isoprene SOA formation under varying conditions (Pandis et al., 1991; Edney et al., 2005; Kroll et al., 2005, 2006; Dommen et al., 2006; Kleindienst et al., 2006; Ng et al., 2006; Paulot et al., 2009; Chan et al., 2010; Chhabra et al., 2010; Surratt et al., 2010; Nguyen et al., 2011, 2014b, 2015; Zhang et al., 2011, 2012; Lin et al., 2013; Xu et al., 2014; Krechmer et al., 2015; Lambe et al., 2015; Brégonzio-Rozier et al., 2015; Clark et al., 2016, etc.), a consensus on the magnitude of SOA formed from isoprene oxidation by the hydroxyl radical (OH) is still lacking (Carlton et al., 2009; Brégonzio-Rozier et al., 2015; Clark et al., 2016). This lack of consensus in the experimental data leads recent global modeling studies (Marais et al., 2016; Stadtler et al., 2018) to implement SOA schemes that produce significantly different overall isoprene SOA yields. Isoprene SOA yields have been shown to depend on a variety of factors including RO2 fate, NO2∕NO ratio, relative humidity, degree of oxidation, temperature, seed surface area, particle acidity, and chamber irradiation source (Carlton et al., 2009). These experimental conditions have not always been controlled or reported, which is likely a major reason for the variability seen in past isoprene SOA yields. By measuring isoprene SOA yields while controlling for seed surface area, RO2 fate, NO2∕NO ratio, relative humidity, and temperature, we seek to resolve uncertainties in SOA formation in past yields.
Recent advances have improved our understanding of how chamber SOA yields should be measured and analyzed. This includes accounting carefully for particle wall deposition (Loza et al., 2012), vapor wall deposition (Zhang et al., 2014; Ehn et al., 2014), and particle coagulation (Nah et al., 2017). Advances have also taken place in the data processing of aerosol size distribution measurements by the differential mobility analyzer coupled to a condensation particle counter (DMA-CPC), the main instrument used to measure SOA yields (Mai and Flagan, 2018; Mai et al., 2018). Because isoprene SOA yields tend to be relatively small, the DMA data inversion technique and correction for CPC response time are quite important.

Figure 1Simplified chemical mechanism of isoprene OH-initiated oxidation under high-NO conditions, largely based on schemes in Wennberg et al. (2018), emphasizing SOA generated from the LV pathway, which includes low-volatility organic nitrates and dinitrates in red. Compounds detected in the gas phase by the chemical ionization mass spectrometer (CIMS) are highlighted with a blue square.
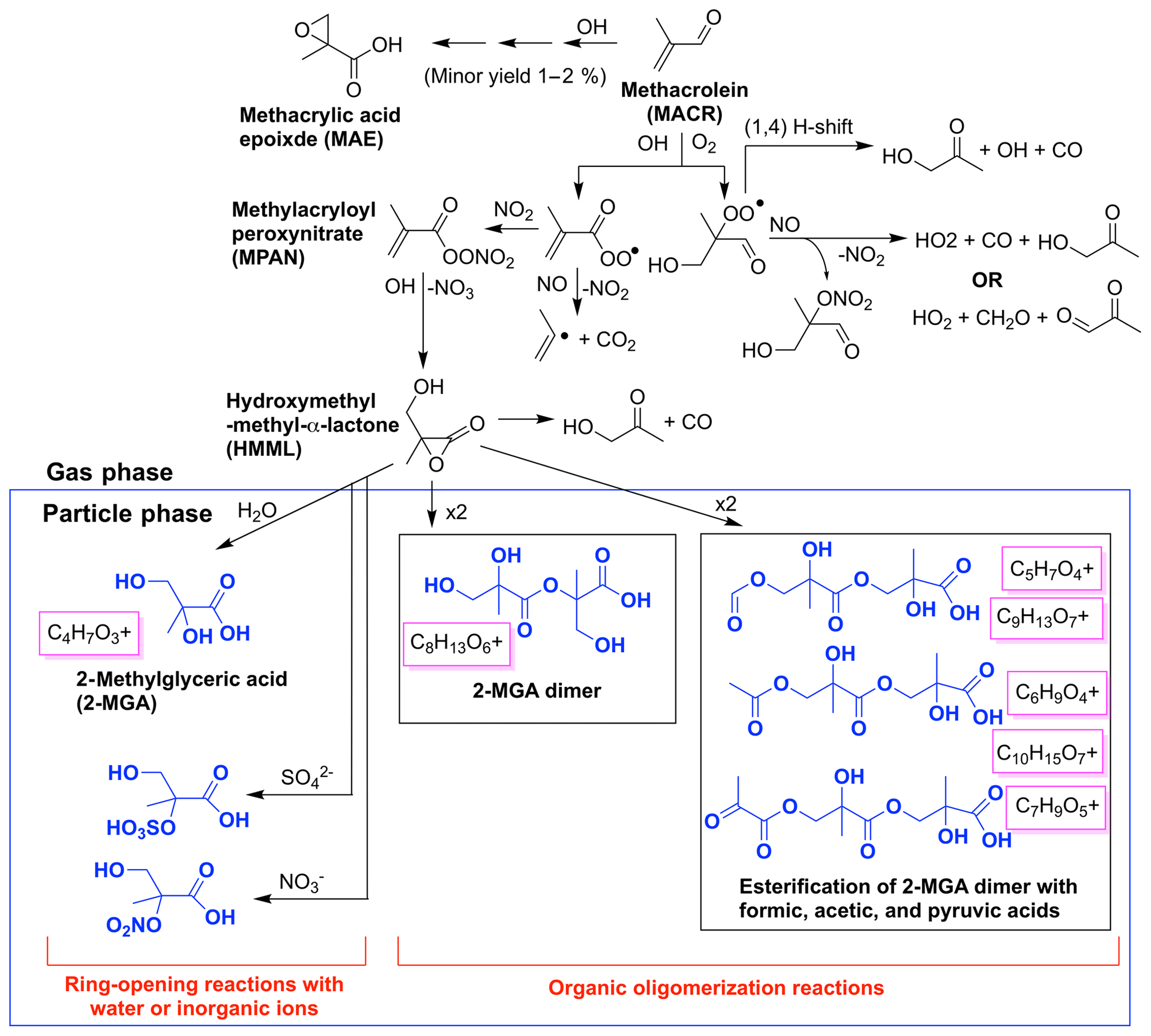
Figure 2Simplified chemical mechanism of methacrolein OH-initiated oxidation under high-NO2 conditions, largely based on schemes in Wennberg et al. (2018), emphasizing SOA generated from the 2MGA pathway including 2-methyl glyceric acid (2-MGA) and its oligomers in blue. Aerosol mass spectrometer (AMS) fragments likely corresponding to each compound are boxed in magenta.
Additionally, there have been major recent advances in our understanding of isoprene gas-phase oxidation (Wennberg et al., 2018, and references therein) including theoretical (e.g., Peeters et al., 2009, 2014; Kjaergaard et al., 2012) and experimental (e.g., Teng et al., 2017; Nguyen et al., 2015; Lee et al., 2014; Jacobs et al., 2014) studies. This improved understanding of isoprene gas-phase chemistry influences the processes governing isoprene SOA formation and informs the experimental design of the present work. This work focuses on the production of SOA from OH-initiated isoprene oxidation under high-NOx conditions, which occurs via two major chemical pathways (Figs. 1 and 2). The first we define throughout as the low-volatility (LV) pathway representing all aerosol formed from the equilibrium gas–particle partitioning of compounds with sufficiently low volatility, which mostly include functionalized nitrates and dinitrates (e.g., red compounds in Fig. 1). The second we define as the 2-methyl glyceric acid (2MGA) pathway representing aerosol formed from 2-MGA, its oligomers, its organosulfates, and its organonitrates (blue compounds in Fig. 2). There are many definitions for high-NOx conditions (Wennberg, 2013). Here we test two different high-NOx chemical regimes. Experiments targeting the LV pathway are designed such that all peroxy radicals including acyl peroxy radicals dominantly react with NO and experiments targeting the 2-MGA pathway are designed such that all acyl peroxy radicals dominantly react with NO2 and all other peroxy radicals dominantly react with NO.
Aerosol from the LV pathway is believed to be composed largely of isoprene dihydroxy dinitrates, which are produced from the first-generation hydroxy nitrate reacting with OH to form a peroxy radical that then reacts with NO. The gas-phase yield of isoprene dihydroxy dinitrates is quite uncertain (Lee et al., 2014). In general, the nitrate yields from highly functionalized RO2 radicals have not been well studied (Wennberg et al., 2018) due mostly to difficulties in measuring such low-volatility compounds. The formation of some organic nitrate SOA precursors are summarized in Fig. 1, which is largely adapted from schemes presented in Wennberg et al. (2018) with the exception of the isoprene dihydroxy nitrooxy alkoxy radical 1,5 H shift. Wennberg et al. (2018) suggests the importance of a similar peroxy radical 1,5 H shift, which will not form in the present experiments due to the high levels of NO. However, based on past studies largely on alkane oxidation (Orlando et al., 2003; Atkinson, 2007), the equivalent alkoxy radical 1,5 H shift is expected to occur and has the potential to form low-volatility nitrates as further described in Sect. 5.1.
Throughout the text we use low volatility as a general term representing gas-phase compounds with a potential to exist partially in the particle phase. In this work, low-volatility compounds include the following volatility classes from Donahue et al. (2012): IVOC (intermediate), SVOC (semi), LVOC (low), and ELVOC (extremely low). When referring to specific volatility classes, the acronyms defined above are used.
Aerosol from the 2MGA pathway forms when methacrolein is oxidized under high-NO2 conditions to form methylacryloyl peroxynitrate (MPAN). MPAN reacts with OH to form hydroxymethyl-methyl-α-lactone (HMML), and HMML either decomposes in the gas phase to form hydroxy acetone or interacts with a wet surface to form 2-methyl glyceric acid (2-MGA) (Kjaergaard et al., 2012; Nguyen et al., 2015). A minor channel to form methacrylic acid epoxide (MAE) also exists from methacrolein oxidation (Lin et al., 2013) but not from pure MPAN oxidation (Nguyen et al., 2015). Nguyen et al. (2015) demonstrated that MAE does not easily undergo ring-opening reactions to form particles. Thus, the yield of MAE from methacrolein (MACR) oxidation reported in Lin et al. (2013) should be adjusted to include only MAE detected in the gas phase, which corresponds to a yield of ∼1 %–2 %.
Because SOA formed from the LV and 2MGA pathways is chemically distinct both in the route of formation and composition, the experiments reported here probed these chemical pathways separately. This experimental design is aimed to resolve inconsistencies associated with previously reported isoprene SOA yields (Carlton et al., 2009) and improve our understanding of isoprene SOA formation. Additionally, we seek to report updated isoprene SOA yields and trace the SOA yields to known gas-phase SOA precursors.
Chamber experiments were performed to study SOA formation from isoprene oxidation under high-NOx conditions from two distinct pathways: (1) low-volatility nitrates and dinitrates (LV pathway) and (2) 2-MGA and its oligomers (2MGA pathway). Experiments targeting the LV pathway were performed using isoprene as the precursor, and an NO2∕NO ratio <1.5 was maintained throughout the entire experiment (as verified by the kinetic mechanism) in order to favor the formation of nitrates and dinitrates and limit the formation of MPAN (Fig. 1). Experiments targeting the 2MGA pathway were performed using methacrolein as the precursor, and an NO2∕NO ratio >11 was maintained throughout the entire experiment (as verified by the kinetic mechanism) with the exception of experiment M9, which maintained an NO2∕NO ratio >8. This high NO2∕NO ratio accentuated the formation of MPAN, and thereby 2-MGA (Fig. 2), and was important for reducing variability between the experiments. If a lower NO2∕NO ratio was used, small fluctuations in the initial NO2 or NO would result in large differences in the NO2∕NO ratio. In order to completely separate the LV and 2MGA pathways, methacrolein had to be used as the VOC precursor for the 2MGA pathway experiments. If isoprene was used, even at the high NO2∕NO ratios used in the 2MGA pathway experiments, the SOA precursors from the LV pathway would form resulting in a mixed regime (i.e., chemistry in Fig. 1 is not dependent on NO2 concentration). In each case, the effect of seed surface area, temperature, and humidity on the SOA yield was independently determined.
Table 1Initial conditions and SOA yield for all experiments.
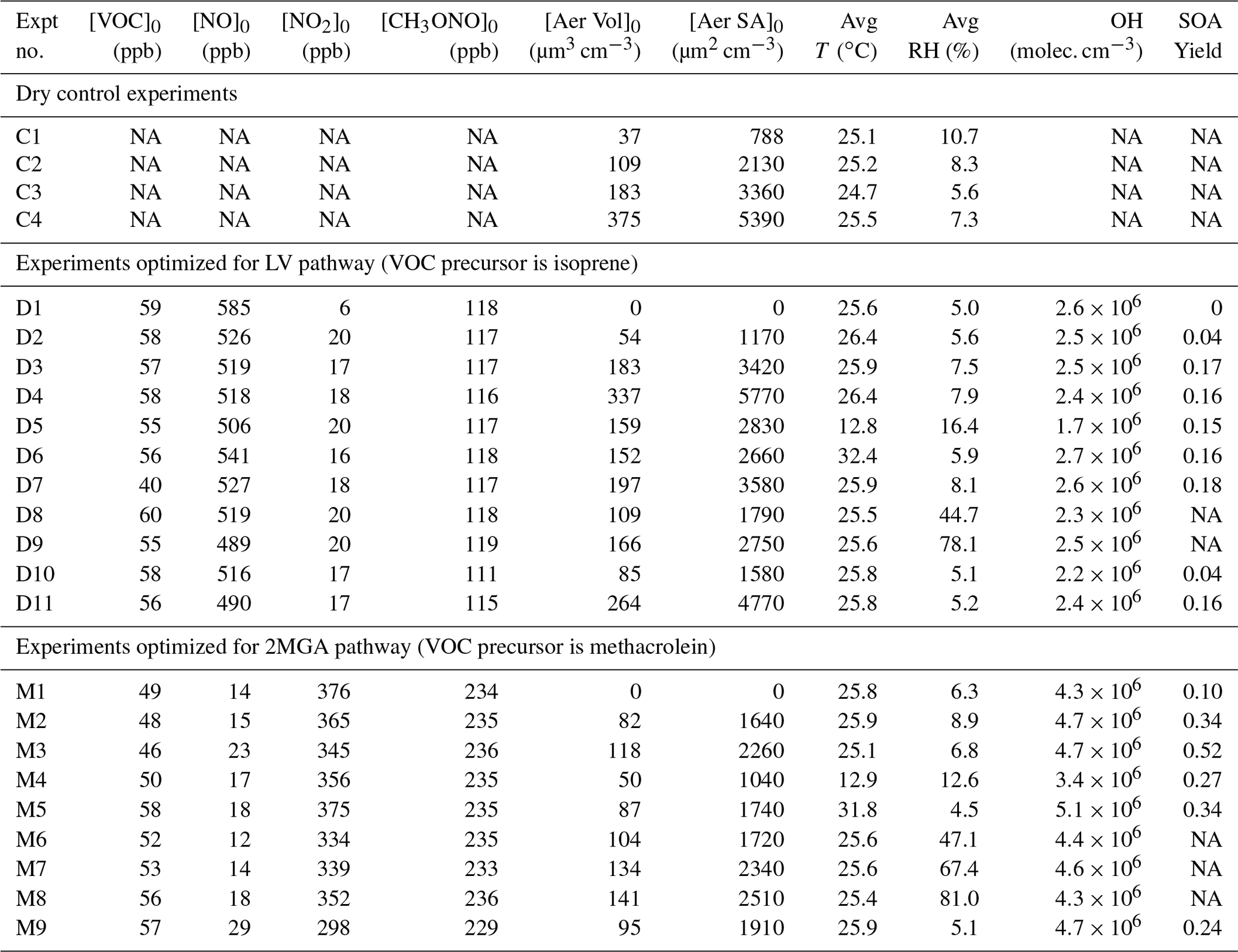
Acronyms are defined as follows: VOC – volatile organic compound; NO – nitric oxide; NO2 – nitrogen dioxide; CH3ONO – methyl nitrite; T – temperature; RH – relative humidity. OH (hydroxyl radical) is estimated from the VOC decay over the first 3 h of each experiment. [Aer Vol]0 is the particle wall-loss-corrected seed volume at the start of photooxidation, which is used to determine the uncertainty in the particle wall loss correction as explained in Sect. 4.1. [Aer SA]0 is the surface area of the seed aerosol at the start of photooxidation not corrected for particle wall loss and is used to understand how the SOA yield changes depending on the surface area of the suspended particles (e.g., Fig. 5). The SOA yield is the mass fraction after 10 h of photooxidation.
2.1 Experimental conditions
Experiments (see Table 1) were conducted in the Caltech dual chamber facility using a 21 m3 Teflon chamber. Prior to each experiment, the chamber was flushed with dry, purified air for 24 h. For humid experiments, the chamber was humidified prior to all injections. Ultrapure water (18 MΩ, Millipore Milli-Q) at 25 ∘C was recirculated through a Nafion membrane humidifier (FC200, Permapure LLC) while purified air flowed through the humidifier and into the chamber. First, isoprene (99 % purity) or methacrolein (95 % purity) was injected into a glass bulb using a gas-tight syringe and was carried by a flow of dry nitrogen into the chamber.
Second, methyl nitrite (CH3ONO) was injected into the chamber. CH3ONO was synthesized using the technique described in Taylor et al. (1980) and Chan et al. (2010) and was stored in liquid nitrogen. Prior to each experiment, an evacuated glass bulb was filled with CH3ONO to the desired pressure, as measured by a capacitance manometer (MKS Baratron™). This bulb was then backfilled with nitrogen and flushed into the chamber. The bulb pressure was used to calculate the CH3ONO mixing ratio in the chamber (see Table 1). After CH3ONO was injected, pulses of purified air were added to the chamber to enhance mixing. Once the chamber was adequately mixed, NO (501 ppm in N2, Scott Specialty Gases) or NO2 (488 ppm in N2, Scott Specialty Gases) was injected into the chamber through a calibrated mass flow controller. Again the chamber was mixed by pulses of purified air.
Seed particles were generated from an atomizer using 0.06 M (NH4)2SO4 seed solution. The seed aerosol was directed through a soft X-ray neutralizer (TSI Model 3088) prior to injection into the chamber to ensure a consistent initial particle charge distribution for all experiments. For humid experiments, the seed aerosol was directed through a wet-wall denuder after exiting the neutralizer in order to ensure the particles were deliquesced. After seed injection, mixing air was turned on for 1 min to enhance mixing. The seed aerosol particle number concentration had an approximately lognormal diameter distribution centered on average ∼100 nm.
After injecting all gas-phase precursors and seed aerosol, photooxidation was delayed by 1 h for experiments with no initial seed aerosol and 4 h for experiments with initial seed aerosol. The rate of particle wall deposition was measured for each experiment during this 4 h delay. Although NO2 was not intentionally added for the LV pathway experiments, a modest NO2 signal was observed to form during the 4 h delay and is reported in Table 1. This “NO2” signal may be NO2 itself or an interference in the NOx monitor from an NOy compound (e.g., known interferences include organic nitrates, nitrous acid, and CH3ONO). The small signal of NO2, or a different NOy compound, is not expected to influence the results given the significantly larger initial NO levels (Table 1). When NO2 or CH3ONO were injected into the chamber, an NO signal was observed on the NOx monitor. As the NOx monitor has few interferences for NO, a small fraction of NO was likely formed from NO2 or CH3ONO photolysis in the Teflon injection lines. Thus, the slight increase in NO with the NO2 and CH3ONO injection was assumed and reported to be initial NO (Table 1).
The Caltech chamber uses ultraviolet (UV) broadband lights with the main emission peak centered at ∼350 nm. Only 10 % of full light capacity ( s−1) was used for these experiments because CH3ONO photolyzes rapidly, and the lower light intensity minimizes chamber temperature increases (∼0.4 ∘C on average) caused by the UV lights. In all experiments, OH was produced by the photolysis of CH3ONO as shown in the following reactions:
Relative to other OH precursors, CH3ONO has a low Henry's law constant (15 M atm−1, calculated by theory) (Sander, 2015). During experiments with high relative humidity (RH), unlike other OH precursors, CH3ONO is not expected to enhance OH production in the particle phase beyond atmospherically relevant levels.
2.2 Instrumentation
Temperature and RH were measured using a Vaisala HMM211 probe. NO and NO2 were monitored using a Teledyne NOx analyzer (T200). Because the Teledyne NOx monitor detects CH3ONO, organic nitrates, and other NOy compounds as NO2, only initial NO2 measurements can be constrained with this instrument. For some experiments, NO2 was also monitored using a luminol NO2 and acyl peroxynitrate analyzer developed by Fitz Aerometric Technologies. This instrument separates NO2 via chromatography at room temperature using a deactivated DB-5 column. NO2 then reacts with luminol to produce a chemiluminescence response (Gaffney et al., 1998). The NO2 measured by the luminol NO2 and acyl peroxynitrate analyzer compares reasonably well with the simulated NO2 from the kinetic model (Fig. S3 in the Supplement).
A gas chromatograph with a flame ionization detector (GC-FID; HP 6890N, column HP-Plot-Q) was used to measure the decay of isoprene and methacrolein. The GC-FID was calibrated with ∼50–60 ppm of isoprene or methacrolein generated from analytical standards (Aldrich 95 %–99 % purity) and cross-calibrated by Fourier transform infrared absorption (FT-IR) spectroscopy (pathlength 19 cm) using the absorption cross sections measured by Pacific Northwest National Laboratory (PNNL) for isoprene or methacrolein (Sharpe et al., 2004). Linearity in the GC-FID calibration was determined to an error of ∼1 % across a factor of 150 in dilution.
Aerosol organic and inorganic composition was recorded in situ using a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS; Aerodyne Research, Inc.). The HR-AMS switched every 1 min between the high-resolution W mode and the lower-resolution, higher-sensitivity V mode. The data were analyzed with Igor Pro (Wave Metrics, Inc.), utilizing the Squirrel 1.56D and PIKA 1.15D analysis toolkits (from http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/index.html, last access: 14 April 2017). In-line filter runs conducted prior to each experiment were used to correct for air interferences (Aiken et al., 2008). Bulk SOA elemental composition was calculated following the methods and recommendations of Aiken et al. (2008) and Canagaratna et al. (2015).
Aerosol volume and number concentration were monitored using a differential mobility analyzer (DMA; TSI 3081 column) coupled with a condensation particle counter (CPC; TSI 3010), which measures all particles with a diameter between 20 and 800 nm. The voltage scan used by the DMA was 1 min hold at 15 V, 4 min increase to 9850 V, 1 min hold at 9850 V, and 0.5 min decrease back to 15 V. Only the up-scan data were used for the analysis. The longer up-scan and hold times used here, compared to previous studies (e.g., Loza et al., 2012 and Zhang et al., 2014), reduced biases caused by mixing in the CPC. Such CPC mixing biases particularly impact the measurement of large particles, which are important in SOA yield experiments as they contribute significantly to the total SOA volume.
The DMA data analysis includes an improved data inversion and a correction for particle mixing in the condensation particle counter, which influences the CPC response time (Mai and Flagan, 2018; Mai et al., 2018). The inversion technique is applicable only to particles ≤600 nm. Particle concentration between 600 and 800 nm was calculated assuming a nonlinear least squares lognormal fit applied to particles from 400 to 600 nm. For experiments with no initial seed aerosol, the inversion inconsistently determined the presence of particles beyond 400 nm. Such large particles are unlikely to be the result of nucleation and more likely to represent an artifact of the inversion; thus, only particles <400 nm were used in the analysis of these experiments. Corrections to the DMA data for coagulation and particle wall loss are addressed in Sect. 4.1.
Isoprene oxidation products were measured using a CF3O− chemical ionization mass spectrometer (CIMS), which utilizes a custom-modified triple quadrupole mass analyzer (Varian 1200) (St. Clair et al., 2010). CF3O− interacts with a gas-phase compound (A) to form a complex that is detected at the molecular weight of A+85 or, in some cases, to fragment. Various fragmentation products can form as explained in previous work (e.g., Paulot et al., 2009; Praske et al., 2015; Schwantes et al., 2017). In this work, the CIMS results are only used to identify the presence of highly functionalized organic nitrates and not for quantification, so only signals from the complex (i.e., ) and not from fragmentation are reported.
All relevant reactions included in the Master Chemical Mechanism (MCM) v3.3.1 (http://mcm.leeds.ac.uk/MCM, last access: 7 September 2018) were used in the current kinetic model (Jenkin et al., 1997; Saunders et al., 2003). Isoprene oxidation chemistry was recently updated in MCM v3.3.1 by Jenkin et al. (2015). Additional reactions included in the kinetic model but not in MCM v3.3.1 are listed in Table S1. Updates include inorganic reactions needed for chamber studies with large NOx levels (e.g., CH3ONO photolysis) and small changes to the isoprene chemistry based largely on Wennberg et al. (2018) and consistent with Figs. 1 and 2. As shown in Table S1, these updates include the first-generation isoprene hydroxy nitrate yields, the rates and branching ratios for the oxidation of the first-generation isoprene hydroxy nitrates, and the HMML yield from the MPAN + OH reaction. In some cases, δ-isoprene hydroxy alkoxy radicals in MCM v3.3.1 decompose through peroxy radical H shifts directly to products that would not form under the high-NO conditions in this work. For simplicity, we change these reactions, so that the δ-isoprene hydroxy alkoxy radicals form unity yields of hydroxy aldehydes. BOXMOX, a box-model software package using the Kinetic PreProcessor (Knote et al., 2015), was used to simulate the chamber experiments. As listed in Table 1, the kinetic model was initialized for each experiment with the measured initial concentration of VOC, NO, NO2, and CH3ONO as well as the measured average temperature and relative humidity.
Saturation mass concentration (C∗) and the fraction of each compound in the particle phase (FP) at 13, 26, and 32 ∘C are estimated for relevant organic nitrates and dinitrates produced in MCM v3.3.1 and listed in Table S2. C∗ was calculated with the vapor pressure estimated from Nannoolal et al. (2004, 2008) using the online calculator located at http://www.aim.env.uea.ac.uk/aim/ddbst/pcalc_main.php (last access: 1 March 2019). FP was calculated from the C∗ values and gas-particle equilibrium theory as further explained in Sect. S1 (Seinfeld and Pandis, 2016).
As shown in Table 1, the inferred OH concentration was larger in experiments with higher temperatures. Because the temperature dependence of the CH3ONO absorption cross section and quantum yield are not well established, the CH3ONO photolysis rate constant was calculated from the CH3ONO decay curve as measured by the GC-FID. Unfortunately, the GC-FID sensitivity to CH3ONO was low, so only the 2MGA experiments produced a sufficiently large signal for this approach. The average CH3ONO photolysis rate constant from experiments M1–M3, M4, M5, and M6–M8 were used for dry ∼25 ∘C ( s−1), dry ∼13 ∘C ( s−1), dry ∼32 ∘C ( s−1), and humid ∼25 ∘C ( s−1) experiments, respectively. This approach accurately captured the reaction of isoprene and methacrolein with OH in all experiments (Figs. S1 and S2), which implies that the simulated OH in the kinetic model is reasonably accurate even over varying temperature. All other photolysis rate constants are calculated from the absorption cross sections and quantum yields reported in Burkholder et al. (2015) and Jenkin et al. (2015). Additionally, the kinetic model captures NO and NO2 reasonably well for both the LV and 2MGA pathway experiments (Fig. S3).
First, corrections for particle coagulation and particle wall deposition, which are required for accurate calculation of SOA yields, are addressed (Sect. 4.1). Next, SOA produced from the LV (Sect. 4.2) and 2MGA (Sect. 4.3) pathways is discussed.
4.1 Corrections for particle coagulation and particle wall deposition
Past studies reporting particle wall deposition coefficients apply the measured particle number decay rate in each size bin to produce a wall deposition coefficient, (β(Dp,t)), that is a function of particle size (Dp) (Loza et al., 2012). Because larger seed particle number and surface area concentrations were used in these experiments, corrections to β(Dp,t) that account for coagulation are needed (Pierce et al., 2008; Nah et al., 2017). The current work uses an approach similar to that of Nah et al. (2017) and Sunol et al. (2018) with updates to account for electrostatic charges on the chamber walls as described by Charan et al. (2018). To reduce the experimental uncertainty associated with these processes, particle wall deposition was calculated during each experiment. This approach accounted for the day-to-day fluctuations in particle coagulation processes, chamber wall charging, and chamber mixing. We summarize these approaches and describe any changes required for this analysis in Sect. S2 of the Supplement.
Four particle wall loss experiments were performed under dry conditions (RH <10 %) at varying seed surface areas as controls to verify the technique used to correct for particle wall loss, particle coagulation, and electrostatic charges on the chamber walls. These particle wall deposition experiments were performed by injecting ammonium sulfate seed into the chamber, as described in Sect. 2.1. Mixing air was added, and the ammonium sulfate seed aerosol was monitored in the dark chamber for at least 14 h. These controls confirmed that the wall loss correction calculated over the first 3.5 h could be extrapolated for an additional 10 h. Beyond 10 h, the wall loss correction was more uncertain, so only results from the first 10 h of each experiment are reported. The percent change between the aerosol volume over 10 h and the aerosol volume at the start of the control experiment was between +4 % and −6 % for all dry control experiments (Fig. 3).
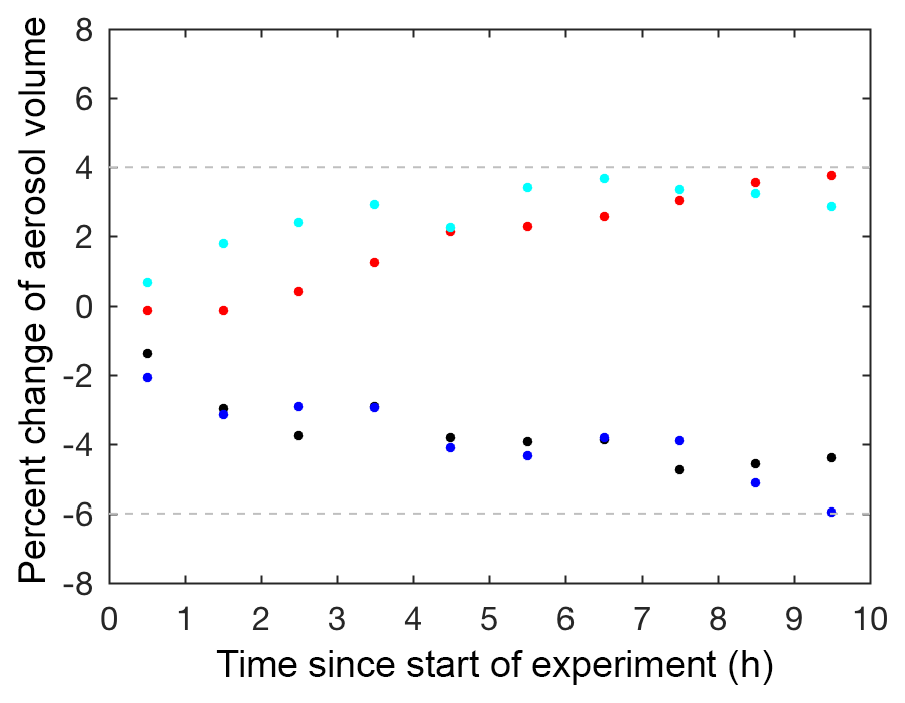
Figure 3Percent change between the corrected aerosol volume over 10 h and the corrected aerosol volume at the start of photooxidation (60 min averages) for the following particle wall deposition control experiments: C1 (V=37 µm3 cm−3; black dot), C2 (V=109 µm3 cm−3; blue dot), C3 (V=183 µm3 cm−3; red dot), and C4 (V=375 µm3 cm−3; cyan dot), where V is the initial corrected particle volume.
The results of these control experiments verified the robustness of the correction technique and provided an estimate for the uncertainty. The reported uncertainty for the particle wall deposition correction is +4 % and −6 % of the corrected aerosol volume at the start of photooxidation. Experiments with larger seed aerosol volumes exhibit larger uncertainty in the reported SOA yield. However, such experiments are necessary despite the extra uncertainty, as larger seed surface areas minimize low biases in SOA yields due to vapor wall deposition of low-volatility compounds (Zhang et al., 2014; Ehn et al., 2014). For experiments with no initial seed aerosol, particle wall loss corrections were applied assuming the particles coagulated and deposited similarly to the lowest aerosol loading control experiment (C1). No uncertainty for the particle wall deposition correction was added to these experiments because the uncertainty derived here is applicable only to experiments with initial seed aerosol.
In experiments C1–C4, D1–D9, and M1–M8, electrostatic charges on the chamber walls were inferred to be present. After these experiments were completed, new Teflon chambers were acquired with negligible electrostatic charges on the chamber walls (Charan et al., 2018) likely due to their smaller volume (18 m3). Three additional new experiments (D10, D11, and M9) were completed using one of these new Teflon chambers to confirm that we had accurately corrected for the chamber wall charging effects. For the LV pathway experiments (Sect. 4.2), results for the new experiments were quite similar and within uncertainties of the old experiments. The new 2MGA pathway experiment produced slightly lower SOA yields than the old experiments but not necessarily because of the chamber wall charging corrections as described in Sect. 4.3.
Five control experiments were also performed under humid conditions. The DMA cannot measure hydrated particles owing to arcing in the DMA column at high RH. Thus, a Nafion dryer was used to dry particles before measurement. For the coagulation correction, the volume of the hydrated seed was calculated based on the dry DMA particle measurement, the RH in the chamber, and the hygroscopic growth curve for ammonium sulfate measured by Sjogren et al. (2007). The percent change for the aerosol volume was higher and less consistent in the humid control experiments than in the dry control experiments. Also, the optimized value of the electric field () was higher in many of the humid experiments than in the dry experiments (Sect. S2, Table S3). Increased humidity is expected to decrease the electrostatic charges on the chamber walls (e.g., Ribeiro et al., 1992), but the inferred suggests the opposite. Possibly, the humidifying process enhanced the electrostatic charges on the chamber walls or nitric acid, which is enhanced in the particle phase in the humid experiments under high-NOx conditions, impacts the coagulation or particle wall loss processes.
The aerosol mass spectrometer (AMS) data confirm that during the humid experiments, nitric acid partitioned to the particle phase and that organic aerosol was produced during photooxidation for all experiments. Nevertheless, the particle wall loss corrected volume measured by the DMA decayed below zero during photooxidation in the humid experiments. Potentially, this DMA volume decay suggests that nitric acid present in the particle phase changes the particle coagulation or wall loss characteristics. Even if we understood the impact of nitric acid on the particle coagulation or wall loss corrections, assessing how much of the particle growth is due to nitric acid versus organics would be difficult with the DMA, which measures only total aerosol volume and not composition. Further chamber characterization is required in order to assess isoprene SOA yields measured by the DMA from humid experiments under high-NOx conditions. Thus, in this work, only the AMS results will be discussed for the humid experiments and SOA yields are only reported for experiments performed under dry conditions (Table 1). None of the dry experiments exhibited the odd behavior observed in the humid experiments, and the AMS results confirm that under dry conditions minimal nitric acid partitioned to the aerosols (Fig. S10). For the dry experiments, the uncertainties are characterized well by the dry control experiments presented in Fig. 3.
4.2 SOA formation from the LV pathway
The SOA mass yields from isoprene for all LV pathway experiments (i.e., experiments targeting low-volatility compounds) are shown in Figs. 4 and 5. To convert aerosol volume measured by the DMA to aerosol mass, a density of 1.4 g cm−3 was assumed, consistent with past work (Dommen et al., 2006; Kroll et al., 2005, 2006; Brégonzio-Rozier et al., 2015). The kinetic mechanism suggests that in all experiments targeting the LV pathway, the formation of HMML was <0.12 ppb even in experiments performed under cold conditions (13 ∘C). The AMS results also confirm that 2-MGA and its oligomers are not present in the LV pathway experiments (Sect. 5.2). Thus, the kinetic mechanism and AMS results verify that the experimental design correctly separates the two chemical regimes and 2-MGA is not substantially adding to the aerosol mass in the LV pathway experiments.
Figure 4SOA mass yield (20 min averages) as measured by the DMA assuming a density of 1.4 g cm−3 for all LV pathway experiments: seed surface area (SA) – D1 (SA = 0 µm2cm−3; blue dot), D2 (SA = 1170 µm2 cm−3; magenta dot), D3 (SA = 3420 µm2 cm−3; green dot), and D4 (SA = 5770 µm2 cm−3; gray dot); temperature – D5 (13 ∘C; inverted cyan triangle) and D6 (32 ∘C; red triangle); isoprene loading – D7 (initial isoprene 110 µg m−3; yellow square); and new chamber with less wall charging – D10 (SA = 1580 µm2 cm−3, orange star) and D11 (SA = 4770 µm2 cm−3; teal star).
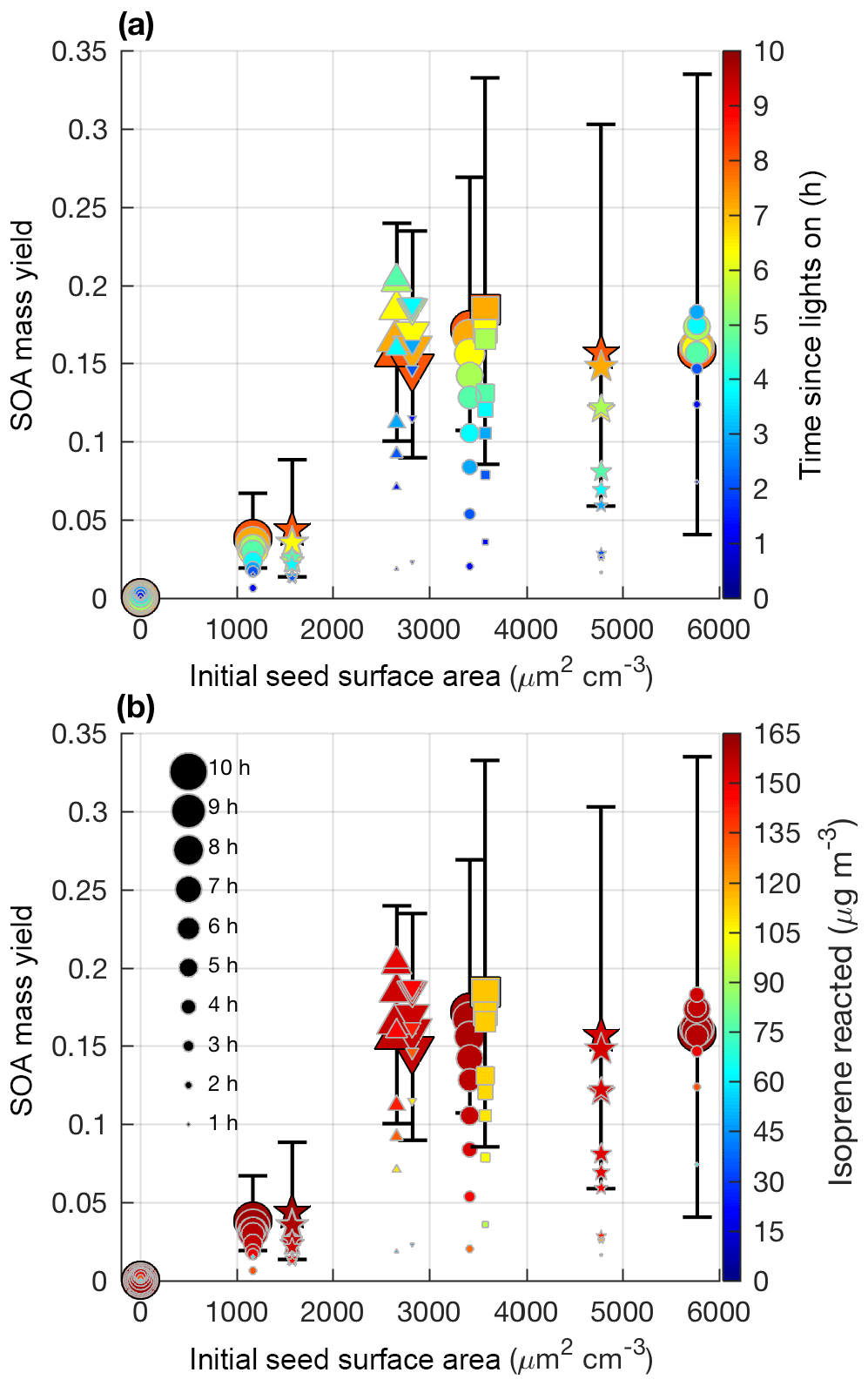
Figure 5SOA mass yield (60 min averages) as a function of initial seed surface area for all LV pathway experiments. Colors represent time since lights on in panel (a) and extent of isoprene reacted in panel (b). Marker size represents time since lights on. Uncertainty is shown in black lines as described in Sect. 4.1. Marker types indicate 25–26 ∘C (dot), 13 ∘C (inverted triangle), 32 ∘C (triangle), lower loadings of isoprene (square), and new chamber with less wall charging (star).
Aerosol growth in the absence of seed aerosols was not observed in the LV pathway experiments (Fig. 4). As expected, SOA formed from gas–particle partitioning in the LV pathway exhibited a large dependence on seed surface area (Fig. 5). Without sufficient seed aerosol, low-volatility nitrates partition primarily to the chamber walls, and the resulting SOA yields are biased low. With the addition of inorganic seed aerosol like ammonium sulfate, vapor species are expected to partition more to particles relative to the chamber wall (Zhang et al., 2014). The gas-particle equilibrium is not expected to be dependent on the concentration of inorganic seed aerosol but instead is dependent on the concentration of organic aerosol. Depending on the saturation mass concentration (C∗), as the concentration of organic aerosol rises, vapors are present more in the particle phase relative to the gas phase (Seinfeld and Pandis, 2016). C∗ and the fraction of a compound expected to be in the particle phase (FP) were estimated for a variety of organic nitrates and dinitrates in MCM v3.3.1 at 13, 26, and 32 ∘C (Table S2).
Similar to previous studies (e.g., Zhang et al., 2014), at a certain point increased seed surface area no longer substantially impacts the SOA yield (i.e., Fig. 5 after 2500 µm2 cm−3). This point will heavily depend on the system and the saturation mass concentration (C∗) of the SOA precursors. As shown in Table S2, the isoprene SOA precursors are mostly classified as IVOCs and SVOCs (Donahue et al., 2012). Reaching a point where most of the vapors are in particles relative to the chamber wall is expected for IVOCs and SVOCs, which have moderate vapor wall losses in Teflon chambers especially under dry conditions (Zhang et al., 2014; Huang et al., 2018).
As expected the isoprene dihydroxy dinitrates had the lowest C∗ values and high FP at all temperatures. Based on the kinetic model, even assuming all of the isoprene dihydroxy dinitrates exist in the particle phase, the SOA formed would be much less than that detected in this study (Fig. S4). This is likely caused by too low of a production of the isoprene dihydroxy dinitrates and/or the importance of other SOA precursors. There are many additional compounds largely produced from hydroxy aldehyde oxidation or alkoxy [1,5]-H shifts (Fig. 1) with FP at 26 ∘C between 0.05 and 0.4 (Table S2). The vapor pressures may be overpredicted for these specific compounds, but past studies suggest that in general, vapor pressure estimation methods like Nannoolal et al. (2004, 2008) underpredict rather than overpredict vapor pressure (Kurten et al., 2016). From the C∗ calculations (Table S2), none of the multifunctional organic nitrates are expected to be appreciably in the particle phase. However, in a recent study, Lee et al. (2016) detected many multifunctional organic nitrates in aerosols in the ambient atmosphere, which has lower organic aerosol concentrations than chamber studies. Possibly, MCM underpredicts the formation of these IVOC and SVOC products (Fig. S4), such that even if only a fraction exists in the particle phase relative to the gas phase, an appreciable mass of aerosol still forms and/or these results suggest that volatility is not the only driver for aerosol formation from the LV pathway. All of the multifunctional nitrates here with estimated FP at 26 ∘C between 0.05 and 0.4 have at least one hydroxy or aldehyde group (Table S2). Alcohols and aldehydes are well known to combine in particles to produce hemiacetals, whose vapor pressure is significantly lower than that of the initial reactants (Kroll and Seinfeld, 2008). Several past studies have confirmed that NOx in general, but not necessarily linearly, decreases the volatility of isoprene SOA (Kleindienst et al., 2009; Xu et al., 2014; D'Ambro et al., 2017). This decrease in volatility is likely due to accretion reactions. Whether the accretion reactions from hemiacetal formation versus those from 2MGA oligomerization are responsible for the decrease in volatility is yet unknown.
Differences in the SOA yield at 10 h of photooxidation by varying temperatures (13–32 ∘C) lie within the experimental uncertainty. SOA forms earlier (i.e. with less isoprene reacted) at 13 ∘C than at 26 or 32 ∘C at comparable seed surface areas. This is consistent with the C∗ values estimated in Table S2 and the above explanation demonstrating the likelihood of accretion reactions. Vapors that are only moderately in the particle phase at 26 ∘C (e.g., FP=0.05–0.4) will exist more appreciably in the particle phase at 13 ∘C (e.g., FP=0.2–0.8). From the above discussion, we expect that many of these compounds are SOA precursors not based only on their volatility but also on their potential to react in the particle phase to form lower-volatility products such as hemiacetals. Thus, if accretion reactions are the main factor, reducing temperature is expected to increase the rate of SOA production but not necessarily to impact the overall SOA yield.
Clark et al. (2016) have also measured isoprene SOA formation under high-NOx conditions at varying temperatures. Under the high-NOx conditions of their study, SOA is produced from both the 2MGA and LV pathways combined. Similarly to our study, Clark et al. (2016) do not find appreciable differences for temperatures from 27 to 40 ∘C. Contrary, to our work, Clark et al. (2016) found that reducing the temperature to 5 ∘C increases the SOA yield by a factor of 4. Unfortunately, there are no experiments between 5 ∘C and 27 ∘C to determine whether this shift is exponential or linear, so direct comparison of our results at 13 ∘C is difficult. Under the high-NOx conditions used by Clark et al. (2016), at colder temperatures MPAN will be more stable and so more HMML will form, which produces more SOA. Under this mixed regime, determining how much of the SOA increase is due to the LV versus the 2MGA pathway for direct comparison to this study is difficult. Additionally, Clark et al. (2016) start with significantly more isoprene than in our experiment, which enhances the concentration of organic aerosol, which will increase the fraction of a compound in the particle phase relative to the gas phase.
While vapor wall losses of LV compounds are expected to increase at colder temperatures (Zhang et al., 2015; Schwantes et al., 2017), the organic nitrate yields are also expected to be enhanced under colder temperatures (Orlando and Tyndall, 2012). Thus, the effects of these two temperature-dependent processes might cancel each other out. The increase in organic nitrate yield is expected to be moderate. For example, a ∼30 % increase from 32 to 13 ∘C is estimated for the yield of isoprene hydroxy nitrates (Wennberg et al., 2018). The loss of vapors to the walls could be much higher at colder temperatures, but this is hard to constrain as vapor wall deposition is dependent on the compound itself and the chamber used. The chamber used by Clark et al. (2016) (90 m3) is larger than our chamber (21 m3). Vapor wall losses are expected to be lower in larger chambers, which have a lower chamber surface-area-to-volume ratio (Zhang et al., 2015). Significant seed aerosol is added into our chamber to reduce the influence of vapor wall deposition, but vapor wall deposition could certainly explain some of the differences at cold temperatures between our results and those from Clark et al. (2016).
Consistent with past work (e.g., Kroll et al., 2005; Ng et al., 2006), aerosol from the LV pathway is produced only after most of the isoprene is consumed, implying that aerosol from the LV pathway largely forms from later-generation chemistry (Fig. 4). As shown in Fig. 4, generally, SOA formation begins earlier (i.e., with less isoprene reacted) in experiments with larger seed aerosol. This is consistent with vapors partitioning more to particles relative to the chamber wall when seed aerosol is enhanced. The extent to which later-generation products are oxidized (i.e., the degree of oxidation) impacts the SOA yield as demonstrated by the varying slope (i.e., SOA yield) during each experiment in Fig. 4. We tested the OH∕isoprene ratio on the SOA yield. Experiment D7 was performed with 40 ppb of isoprene compared to 55–60 ppb used in the other experiments, while the OH precursor concentration was kept constant. The kinetic model predicts that the production of important gas-phase SOA precursors from the LV pathway (e.g., isoprene dihydroxy dinitrates), when corrected for total isoprene reacted, is similar in experiment D7 to the other experiments (Fig. S4). The empirical results are consistent with these predictions. Although a lower isoprene loading decreases the competition of isoprene with OH, other compounds also react with OH quickly (e.g., NO). Under the conditions used in this study, differences in isoprene loading are not expected to greatly influence the isoprene SOA mass yield. However, detailed kinetic modeling of past experimental conditions would be necessary to understand how the degree of oxidation of later-generation products in this study compares to other studies.
In summary, the results (Fig. 5) suggest that one of the most important metrics for understanding the variability in SOA production from the LV pathway in various chamber experiments may be the initial seed surface area, instead of temperature or OH∕isoprene ratio. Other parameters such as humidity and seed composition may also be important for SOA yields but were not tested in this study. Future experiments examining SOA yields should report the initial seed surface area and use a sufficient seed loading to reduce the impact of vapor wall deposition.
4.3 SOA formation from the 2MGA pathway
The SOA mass yields from methacrolein for all 2MGA pathway experiments (i.e., experiments targeting 2MGA and its oligomers) are shown in Figs. 6 and 7. Results from past experiments (Chan et al., 2010) have already demonstrated that fluctuations in the NO2∕NO ratio impact SOA formation through the production of MPAN. In this work, the NO2∕NO ratio is kept as consistent as possible to isolate other influences on SOA production. The kinetic model suggests that the conditions for each experiment produce a consistent level of HMML (Fig. S4). Interestingly, because the experimental conditions heavily favored MPAN formation, the level of OH available to react with MPAN became the limiting reactant for aerosol formation in each experiment.
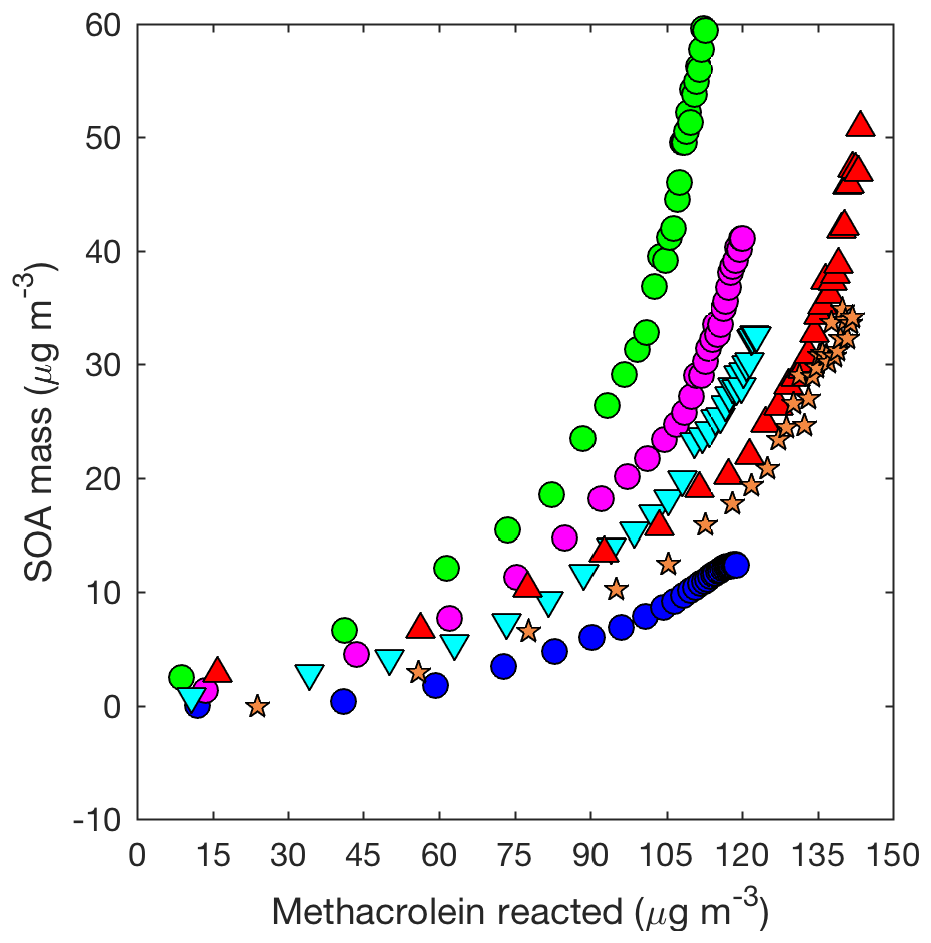
Figure 6SOA mass yield (20 min averages) as measured by the DMA assuming a density of 1.4 µg cm−3 for 2MGA pathway experiments: seed surface area (SA) – M1 (SA = 0 µm2 cm−3; blue dot), M2 (SA = 1640 µm2 cm−3; magenta dot), and M3 (SA = 2260 µm2 cm−3; green dot), temperature – M5 (13 ∘C; inverted cyan triangle) and M6 (32 ∘C; red triangle), and new chamber with less wall charging – M9 (SA = 1910 µm2 cm−3; orange star).
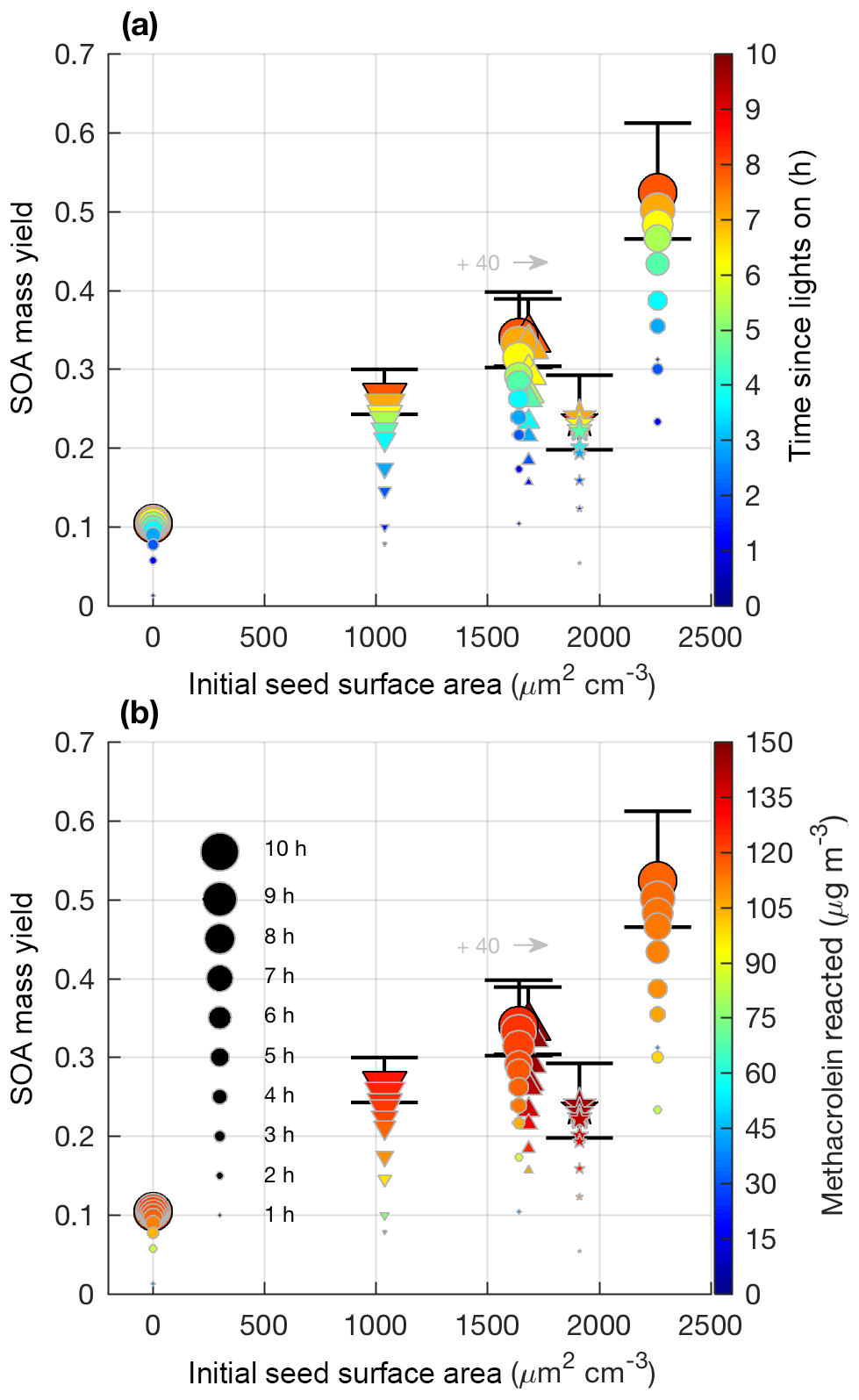
Figure 7SOA mass yield (60 min averages) versus initial seed surface area for all 2MGA pathway experiments. Colors represent time since lights on (a) and extent of methacrolein reacted (b). Marker size represents time since lights on. Uncertainty is shown in black lines and described in Sect. 4.1. Markers represent 25–26 ∘C (dot), 13 ∘C (inverted triangle), 32 ∘C (triangle), and new chamber with less wall charging (star). Two experiments were performed at nearly the same seed surface area. To enhance viewing, experiment M6 (32 ∘C; triangle) is shifted to the right by 40 µm2 cm−3.
Contrary to the LV pathway, SOA in the 2MGA pathway experiments does not require seed particles to form. The process of SOA formation from these two pathways is very different. Lactone SOA precursors may polymerize in the presence of organics and water, which possibly explains why SOA from the 2MGA pathway readily forms particles without significant seed surface area, whereas in the LV pathway experiments volatility-based SOA formation results in aerosol yields that are particularly impacted by vapor partitioning. For the 2MGA pathway experiments, even though SOA formation occurred without initial seed aerosol, larger initial seed loadings still enhanced the SOA yield (Fig. 7). Possibly, similar to the LV pathway, larger seed surface areas limit vapor wall loss of HMML or its oligomerization partners. Alternatively, the presence of higher ammonium sulfate seed aerosol may also increase organosulfate formation, which could impact SOA composition and yield.
Temperature was varied between 13 and 32 ∘C. The NO2∕NO ratio used in this work was sufficiently high such that this temperature change did not greatly influence MPAN or HMML formation (Fig. S4). Thus, these experiments only test whether aerosol properties and SOA yields are affected by temperature, as MPAN thermal decomposition is minimized. At the high NO2∕NO ratios used in this work, temperature does not impact SOA mass yield beyond given uncertainties (Fig. 7). Based on known gas-phase chemistry, past studies (e.g., Clark et al., 2016) with more moderate NO2∕NO ratios than that used in this work are expected to measure an enhanced SOA yield under colder temperatures due to a reduction in MPAN thermal decomposition and thereby an increase in HMML formation.
HMML, based on volatility alone, would exist mostly in the gas phase, but because HMML is very reactive (e.g., oligomerization or reaction with inorganic ions in the particle phase), HMML quickly produces aerosol (Kjaergaard et al., 2012; Nguyen et al., 2015). Based on HMML production simulated by the kinetic mechanism under the conditions used in these experiments, ∼0.21 SOA mass yield from methacrolein is expected purely from the mass contained in HMML (molecular weight = 102 g mol−1, Fig. S4). At first, the molecular weight of HMML itself is used because this is the mass of the majority of the oligomer monomers. This represents about half of the SOA mass yield (∼0.5) measured from the experiment performed with the highest seed surface area. The rest of the aerosol is likely comprised of inorganic or organic compounds that react with HMML in the particle phase. For example, inorganic compounds such as water, nitrate, and sulfate can react with HMML through ring-opening reactions to produce total methacrolein SOA mass yields of ∼0.25, ∼0.34, and ∼0.41, respectively (Fig. 2). Additionally, HMML can react with 2-MGA and other organic compounds through oligomerization processes (e.g., Chan et al., 2010; Nguyen et al., 2015; Zhang et al., 2011, 2012). Some of these organic oligomerization reactions bring into the particle phase additional organic compounds (e.g., organic acids) that ordinarily would exist primarily in the gas phase (Fig. 2). The details of these particle-phase reactions are further discussed in Sect. 5.2.
In general, there is much greater variability in the SOA mass yields measured from the 2MGA pathway than the LV pathway. The additional variability is only partially explained by the initial seed surface area (Fig. 7). Because the SOA yield is larger for experiments in which less methacrolein is oxidized (Figs. 6 and 7b), potentially, the extent of methacrolein oxidization contributes to this variability. The kinetic model suggests that the formation of gas-phase HMML is similar for all of the experiments (Fig. S4), but potentially slight variations in the NO2∕NO ratio and/or OH particularly near the end of each experiment are not well captured by the model. The kinetic model used here only simulates gas-phase oxidation. Chemistry occurring on surfaces such as the chamber walls or in the particle phase may be especially important for capturing the variability in the 2MGA pathway experiments. Considering that the 2MGA pathway experiments are very susceptible to small differences in chamber conditions, regional and global models should parameterize SOA formation from the 2MGA pathway through gas-phase formation of HMML and subsequent particle-phase reactions.
The gas-phase compounds measured by the CIMS (Sect. 5.1) and aerosol composition measured by the AMS (Sect. 5.2) provide important insight into isoprene SOA chemical composition formed from both the LV and 2MGA pathways. Additionally, a comparison of the AMS and DMA results lends insight into possible biases in the AMS measurements of organic aerosol in Sect. 5.3. The SOA yields measured in this study are compared with past measurements in Sect. 5.4, and the atmospheric contribution of the LV versus 2MGA pathways toward SOA formation from isoprene OH-initiated oxidation under high-NOx conditions is estimated in Sect. 5.5.
5.1 Specific low-volatility nitrates and dinitrates detected in the gas phase
Numerous nitrates and dinitrates are detected in the gas phase by the CF3O− CIMS (i.e., compounds highlighted in blue boxes in Fig. 1). Many of these nitrates have been identified in previous studies (e.g., Lee et al., 2014). Yields for the low-volatility later-generation nitrates are either highly uncertain or unknown. Quantification is difficult for these low-volatility compounds due to high losses to sampling lines or chamber walls and lack of available standards. One study, Lee et al. (2014), was able to quantify the yield of dinitrates from the first-generation isoprene hydroxy nitrate standards. Assuming a sensitivity similar to the isoprene hydroxy nitrate standards, Lee et al. (2014) measured a dinitrate yield of 0.03–0.04 from OH-initiated oxidation of the δ-1-hydroxy,4-nitrate isomer.
Although most past studies have focused on dihydroxy dinitrates as the main contributor to isoprene high-NO SOA, other low-volatility nitrates are likely also important. In Fig. 8, the CIMS signals for the other low-volatility nitrates are comparable or larger than the dihydroxy dinitrate signal. The relative sensitivities for these compounds are unknown, but these results suggest that detection and quantification of all low-volatility dinitrates and nitrates is important. The peroxy radical formed from OH-initiated oxidation of an isoprene hydroxy nitrate can undergo a 1,5 or 1,6 α-hydroxy H shift to form a number of low-volatility nitrates that would occur in the ambient atmosphere (Wennberg et al., 2018). The NO concentrations are too high in these experiments for such shifts to occur. However, similarly, certain isomers of the alkoxy radical, formed from OH-initiated oxidation of a isoprene hydroxy nitrate, can undergo a 1,5 α-hydroxy H shift to form a dihydroxy carbonyl nitrate detected by the CIMS at m∕z (-) 264 (Fig. 1 and 8). Additionally, various low-volatility nitrates in the gas phase are detected, which are potentially oxidation products from the δ-isoprene hydroxy alkoxy radical as depicted in Fig. 1.
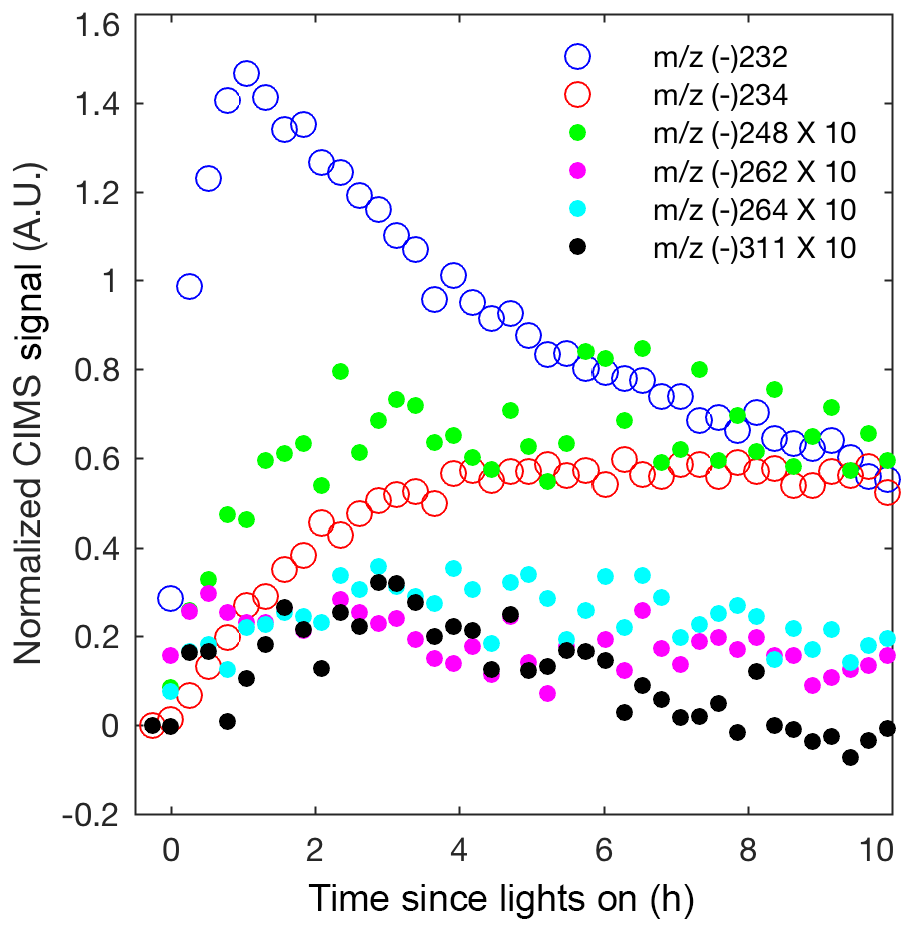
Figure 8Normalized CIMS signal for known nitrates: C5 hydroxy nitrate (m∕z (–) 232; blue circle), methyl vinyl ketone or methacrolein nitrate (m∕z (–) 234; red circle), and C5 dihydroxy dinitrate (m∕z (–) 311; black filled circle) and unknown nitrates, which are postulated in Fig. 1 as C5 dihydroxy nitrate (m∕z (–) 248; green filled circle), unknown (m∕z (–) 262; magenta filled circle), and C5 dihydroxy carbonyl nitrate (m∕z (–) 264; cyan filled circle). As indicated in the legend, signals represented by filled circles are multiplied by 10.
Many multifunctional isoprene-derived organic nitrates have been detected in ambient aerosol (Lee et al., 2016). Although these low-volatility nitrates and dinitrates have low molar yields from isoprene OH-initiated oxidation, their mass is substantially larger than isoprene and so their contribution to the isoprene SOA mass yield is significant. The nitrate yield from straight-chain hydrocarbons is reasonably well understood, but few experimental measurements of the nitrate yield from highly oxidized compounds exist (Orlando and Tyndall, 2012; Wennberg et al., 2018). Further measurements of the yield of these low-volatility nitrates and dinitrates in the gas phase will be crucial for a better understanding of isoprene SOA formation under high-NO conditions.
5.2 Aerosol composition of high-NO isoprene SOA
Pieber et al. (2016) determined that inorganic aerosol such as ammonium nitrate or ammonium sulfate causes an interference on the AMS for the ion signal. Although this interference is small for ammonium sulfate aerosol (<1 %, Pieber et al., 2016), a correction may be needed for experiments with high initial seed aerosol loadings. Here organic signals from the AMS rise when ammonium sulfate seed is injected into the chamber. We expect that this is due to the same interferences described in Pieber et al. (2016) and not due to contamination in ammonium sulfate solution or atomization technique. The background organic signal caused by the ammonium sulfate is subtracted from the overall results to produce Figs. 9, 10, and 11.

Figure 9High-resolution AMS organic mass spectra (averaged over 10 h of photooxidation – the sulfate background) for experiment D3 (RH = 8 %, panel a) and D9 (RH = 78 %, panel b) in gray. Fragments are labeled as 2-MGA monomer or dimer (cyan), esterification of 2-MGA with acids (red), isoprene epoxydiol (IEPOX) tracers (dark green), and examples of organonitrate fragments – CxHyNOz (purple).

Figure 10High-resolution AMS organic mass spectra (averaged over 10 h of photooxidation – the sulfate background) for experiment M2 (RH = 9 %, panel a) and M8 (RH = 81 %, panel b) in gray. Fragments are labeled as 2-MGA monomer or dimer (cyan), esterification of 2-MGA with acids (red), examples of organosulfate fragments (dark green), and examples of organonitrate fragments – CxHyNOz (purple).
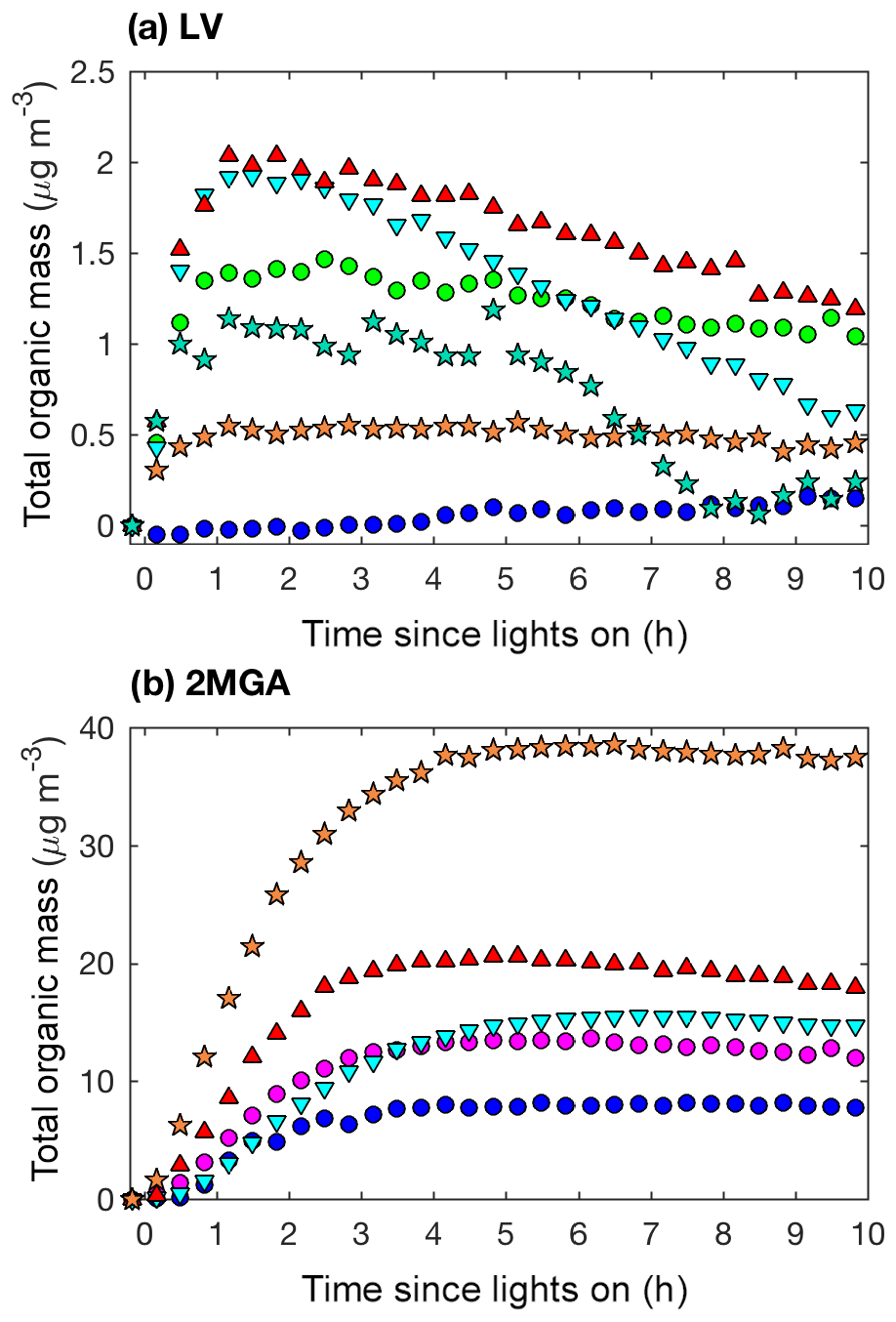
Figure 11Total organic mass (20 min averages) as measured by the AMS for LV pathway experiments (a): seed surface area (SA) – D1 (SA = 0 µm2 cm−3; blue dot) and D3 (SA = 3420 µm2 cm−3; green dot); temperature – D5 (13 ∘C; inverted cyan triangle) and D6 (32 ∘C; red triangle); and new chamber with less wall charging – D10 (SA = 1580 µm2 cm−3; orange star) and D11 (SA = 4770 µm2 cm−3; teal star). Total organic mass as measured by the AMS for 2MGA pathway experiments (b): seed surface area – M1 (SA = 0 µm2 cm−3; blue dot) and M2 (SA = 1640 µm2 cm−3; magenta dot); temperature – M4 (13 ∘C; inverted cyan triangle) and M5 (32 ∘C; red triangle); and new chamber with less wall charging – M9 (SA = 1910 µm2 cm−3; orange star).
The AMS spectra from the LV pathway confirm that SOA formed from the LV pathway is not dominated by 2-MGA and its oligomers (cyan and red bars in Figs. 9 and S11). This is an important confirmation that isoprene SOA formed from the 2MGA and LV pathways are distinct. A small yield of isoprene epoxydiol (IEPOX) is produced from OH-initiated oxidation of isoprene hydroxy nitrates (Jacobs et al., 2014), and IEPOX SOA can be formed when particle liquid water is present (Nguyen et al., 2014a). NO levels remained high (>100 ppb) throughout all LV pathway experiments (Fig. S3), ensuring that the RO2 fate in these experiments was always RO2+NO. AMS fragments associated with IEPOX, which were identified by Lin et al. (2012), are slightly enhanced under humid conditions in the LV pathway experiments (Fig. 9). Some examples of organonitrate fragments () are highlighted in Fig. 9. Some of these organonitrate fragments are enhanced under humid conditions (e.g., CH3NO+). In general, the AMS spectra are similar between all LV pathway conditions (i.e. varied humidity – Fig. 9; varied temperature – Fig. S11).
Prominent peaks in the AMS spectra from the 2MGA pathway clearly indicate that under dry conditions aerosol is comprised of various oligomerization products as mechanistically summarized in Fig. 2. These oligomerization processes include 2-MGA oligomerization with HMML (cyan bars in Fig. 10a) and, possibly, esterification of 2-MGA with carboxylic acids including formic, acetic, and pyruvic acids (red bars in Fig. 10a), which yield products that have been detected in numerous studies (Chan et al., 2010; Zhang et al., 2011, 2012). Based on the AMS spectra, 2-MGA oligomerization appears to be more dominant without the presence of ammonium sulfate seed aerosol (Fig. S13). Varying temperature from 13 to 32 ∘C does not appear to substantially change the extent of 2-MGA oligomerization (Fig. S14).
Past studies have determined that HMML reaction with 2-MGA to form oligomers decreases under humid conditions, while HMML ring-opening reactions with water and inorganic ions to form organic nitrates and organic sulfates increase (Zhang et al., 2011, 2012; Nguyen et al., 2015). Consistent with these past studies, the 2-MGA oligomer fragments on the AMS (cyan and red) are no longer prominent signals for all humid experiments (Figs. 10 and S12). 2-MGA oligomer fragments are not substantially different at 47 %, 67 %, or 81 % RH, suggesting that the HMML oligomerization processes are impeded as soon as aerosol particles become deliquesced. Because isoprene is mostly emitted in regions with relatively high humidity, in the ambient atmosphere, HMML will more likely react with water and inorganic ions than undergo the various organic oligomerization reactions summarized in Fig. 2.
5.3 Comparison of AMS and DMA results
Based on the DMA measurements when assuming the same density, the SOA mass produced from the 2MGA pathway experiments is ∼2 times higher in magnitude than that from the LV pathway experiments (Figs. 4 and 6). However, the AMS results (Fig. 11) suggest that the SOA mass produced from the 2MGA pathway experiments is ≥8 times larger than that from the LV pathway experiments. This implies that the collection efficiency (CE) and/or the ionization efficiency of the AMS is quite different between these two regimes. Because the AMS is significantly more sensitive to aerosol formed from the 2MGA pathway, and not to SOA formed from the LV pathway, even ambient organic aerosol measurements have the potential to be impacted. Understanding whether the AMS is systematically underestimating organic aerosol from organic nitrates and dinitrates in general, or if this is only relevant to the isoprene system is crucial as the AMS is used throughout the world to quantify organic aerosol. Moreover, ambient measurements over the isoprene-rich southeastern United States of particulate organic nitrates measured by the AMS are a factor of ∼5 lower than those measured by the thermal dissociation laser-induced fluorescence instrument (TD-LIF) (Lee et al., 2016). The relative CE differences between the LV and 2MGA pathways in this study and these field campaign results suggest that further AMS calibration of organic nitrates is necessary.
In this work, a CE of 0.5 is assumed for both regimes consistent with past work (Nguyen et al., 2014b). The exact CE is not relevant as no mass yields are reported here from the AMS. Docherty et al. (2013) determined that the CE could be estimated based on the f44∕f57 ratio. The f44∕f57 ratio for all experiments (2MGA and LV) is ≥6, which is where the CE vs. f44∕f57 curve plateaus at 0.2. Thus, the CE vs. f44∕f57 relationship developed by Docherty et al. (2013) is not able to explain the large difference in AMS sensitivity between aerosol formed from the LV and 2MGA pathways.
Edney et al. (2005)Kroll et al. (2005)Dommen et al. (2006)Kleindienst et al.2006Chan et al. (2010)Chhabra et al. (2010)Zhang et al. (2011)Nguyen et al. (2011)Xu et al. (2014)Brégonzio-Rozier et al.2015Clark et al. (2016)Chan et al. (2010)Brégonzio-Rozier et al.2015Table 2Reported SOA mass yields and chamber conditions for isoprene and methacrolein OH-initiated oxidation under high-NOx conditions.

CV: chamber volume. Acronyms are defined as follows:
NR – not reported; UV – ultraviolet lights; N – natural; X – xenon arc lamps; AS – ammonium sulfate seed aerosol
volume.
a Chamber was operated in dynamic mode (residence
time = 6 h). b Ammonium sulfate was injected throughout the
experiment to generate the lower limit of initial seed
aerosol.
SO2 was added in some experiments to generate the upper limit of
initial seed aerosol. c Secondary organic carbon is converted to SOA using factor
(2.47) reported in
Kleindienst et al. (2007). d VOC reacted was
reported and tabulated instead of VOC initial.
5.4 Comparison to previously reported SOA yields
SOA mass yields reported from past environmental chamber studies of OH-initiated oxidation of isoprene under high-NOx conditions vary over the range of 0.001–0.41 (Brégonzio-Rozier et al., 2015; Clark et al., 2016), suggesting isoprene SOA yields are highly dependent on chamber conditions (Carlton et al., 2009). In Table 2, past reported SOA mass yields are summarized along with the chamber conditions for both isoprene and methacrolein OH-initiated oxidation under high-NOx conditions. Only experiments that explicitly measure an SOA mass yield are listed in Table 2. Overall, our results suggest that the initial seed surface area has the greatest impact on SOA yield. Unfortunately, the initial seed surface area was not commonly reported in past studies. The closest metric is aerosol volume, which can roughly be used to understand differences.
As shown in Table 2, the range for isoprene SOA yields under high-NOx conditions even from the two most recent studies at comparable temperatures spans over an order of magnitude (0.004 at ∼21 ∘C for Brégonzio-Rozier et al., 2015, and 0.1 at 27 ∘C for Clark et al., 2016). Our results are most consistent with those of Clark et al. (2016). As shown in Table 2, a variety of NOx regimes (i.e., non-consistent NO2∕NO ratios) are all labeled as high-NOx in these past studies. Each study likely produces SOA in varying degrees from the LV and 2MGA pathways, which greatly complicates direct comparison between these past studies. By varying a large number of conditions and completely separating SOA production between the 2MGA and LV pathways, our results lend insight into the variation in these past experiments.
Many of the past SOA yield measurements were performed with no seed aerosol. Consistent with past results, when no seed aerosol was injected into the chamber (experiments D1 and M1), the SOA mass yield for the LV pathway (0 from isoprene) and 2MGA pathway (0.1 from methacrolein) were quite low. Past experiments performed with no seed aerosol only measured SOA from the 2MGA pathway, which is highly dependent on the NO2∕NO ratio (Chan et al., 2010), which varied greatly between these past studies. Clark et al. (2016), who measured high SOA yields (0.1 at 27 ∘C) in unseeded experiments, are the exception. Possibly, the larger chamber volume (90 m3) used by Clark et al. (2016) compared to most studies listed in Table 2 reduced vapor wall losses and contributed to the enhanced SOA yield. However, other chamber characteristics might also be important because Zhang et al. (2011) measured quite low isoprene SOA yields (0.007–0.03) using a chamber larger than the one used in the Clark et al. (2016) study.
While the zero or low seed aerosol loading experiments in this study generally compare well with the past, SOA yields measured here using higher initial seed surface areas are substantially greater than most studies, especially for the LV pathway. The SOA yield from the LV pathway is ∼0.15 in this study, while past isoprene SOA yields are largely ≤0.07 with the exception of studies optimizing for high RO2+NO2 reactions (Chan et al., 2010) or mixed regimes – (Xu et al., 2014). The SOA yield from the LV pathway in this work is even larger than the SOA yield from Clark et al. (2016) (0.1 at 27 ∘C), which includes SOA from both the LV and 2MGA pathways. Possibly the larger chamber volume used by Clark et al. (2016) reduces vapor wall losses but not to the extent that enhanced seed surface area does in this work. The higher yields measured in this study are not unexpected given that recent publications have recognized the importance of using high initial seed surface areas when measuring SOA yields to reduce the impact of vapor wall deposition (e.g., Zhang et al., 2014; Ehn et al., 2014). The methacrolein SOA yields measured in this study from the 2MGA pathway are comparable to those measured by Chan et al. (2010) but larger than those measured by Brégonzio-Rozier et al. (2015).
Brégonzio-Rozier et al. (2015) measured low isoprene (0.001–0.01) and methacrolein (0.005–0.042) SOA mass yields and proposed that these lower yields were due to using xenon arc lamps as a light source, which are more representative of natural sunlight than the UV lamps used here and in most other studies. Dommen et al. (2006) also used xenon arc lamps and reported low yields. However, both of these studies used chambers with moderate to low chamber volumes (27–4.2 m3) unlike the chamber used by Clark et al. (2016) and low levels of initial seed aerosol (0–16 µm3 cm−3) unlike this work, which could also cause this low bias. Additionally, the stainless steel chamber used by Brégonzio-Rozier et al. (2015) may have higher vapor wall losses than the Teflon chambers used in other studies. Further work is necessary to understand how vapor wall losses compare across different types of environmental chambers.
As discussed by Carlton et al. (2009), isoprene SOA forms mostly from oxidation of second and later-generation products (e.g., Ng et al., 2006). Towards the end of the experiment, SOA continues to grows even when isoprene is no longer reacting (e.g., the characteristic hook in Fig. 4). Differences in the level of oxidation of second and later-generation products could also explain some of the discrepancies between our results and past results. The isoprene SOA mass yields from the LV pathway are particularly sensitive to the extent of oxidation. More studies measuring the gas-phase yields and formation processes of low-volatility nitrates and dinitrates will be critical for further understanding isoprene SOA.
Many of the previous studies listed in Table 2 report the VOC∕NO ratio when comparing experiments. A more useful metric is understanding the RO2 fate and RO2 lifetime. Simply injecting NO and/or NO2 and reporting the initial concentrations are not sufficient to confirm that SOA was dominantly produced from the RO2+NO channel or in the case of HMML formation from the RO2+NO2 channel. For example, if NO decreases to zero before the end of the experiment, SOA has formed in a mixed regime; RO2+NO reactions dominate in the beginning and RO2+HO2 reactions dominate at the end. If large initial VOC loadings are used in the beginning of the experiment without comparable increases in NO, RO2+RO2 reactions may become dominant.
Experiments here are specifically designed to test two different RO2 fates, and the kinetic mechanism is used to confirm the fate of the RO2. In the LV pathway experiments, high NO levels are maintained such that NO2∕NO ratio remains <1.5 throughout the entire experiment, and RO2 dominantly and consistently across the experiments reacts with NO. In the 2MGA pathway experiments, high NO2 levels are used such that the acyl radical derived from methacrolein dominantly and consistently across experiments reacts with NO2. By controlling for the RO2 fate, the effects of temperature, seed surface area, and relative humidity on SOA formation become easier to resolve. The design of future experiments should optimize and report the RO2 fate for which the experiment was designed, in addition to key reaction parameters such as seed surface area, rather than simply reporting an initial VOC∕NO ratio.
5.5 Estimating the atmospheric contribution of the LV versus 2MGA pathways
This work was not only designed to independently study SOA formation from the two high-NOx regimes (the 2MGA and LV pathways) but also to suggest alternative methods for parameterizing isoprene SOA under high-NOx conditions in regional and global models. Because obtaining constant NO2∕NO ratios similar to the ambient atmosphere is near impossible for a chamber study (e.g., temporal variation in Fig. S3), creating isoprene SOA parameterizations based on NO2∕NO ratio that realistically extrapolate to the ambient atmosphere is not realistic. Instead, this work highlights a potential alternative. Aerosol from the 2MGA pathway could be incorporated directly from gas-phase HMML formation, and aerosol from the LV pathway could be included either from the formation of surrogate compounds such as isoprene dihydroxy dinitrates or with a volatility basis set scheme. By treating the SOA from these two independent regimes separately, this study sets up the experimental basis for such an approach.
In this study, direct comparison of the results from the 2MGA and LV pathways is difficult due to the difference in the extent of oxidation between the two regimes caused by the use of different VOC precursors and the variation in OH levels (Table 1). Thus, the kinetic model is used here to estimate the contribution of each pathway to the total under consistent oxidant levels. A detailed global modeling study is needed to precisely capture the contribution of the LV versus the 2MGA pathways toward SOA formation from isoprene OH-initiated oxidation under high-NOx conditions. However, in order to demonstrate the significance of the new isoprene SOA yield from the LV pathway measured in this work, we roughly approximate the contribution of each pathway under typical atmospheric conditions. We use the same kinetic mechanism described in Sect. 3 but hold the following constant: RH = 70 %; T = 298 K; NO2 = 0.3 ppb; NO = 0.05 ppb; isoprene = 5 ppb; OH molec. cm−3; CO = 135 ppb; O3 = 37 ppb; and HO2 = 25 ppt (Sanchez et al., 2018; Feiner et al., 2016; Pajunoja et al., 2016). Then gas-phase HMML and the gas-phase dinitrate SOA precursors are simulated as done for the experimental results in Fig. S4.
To estimate the aerosol contribution from the LV pathway, we assume that SOA production from the LV pathway scales with the production of isoprene dihydroxy dinitrates. Organic aerosol concentrations are higher in chamber experiments than the ambient atmosphere. By using low levels of VOC precursors compared to previous studies, this study attempts to reduce the organic aerosol concentrations to produce results more relevant to the ambient atmosphere. However, due to limitations in the DMA sensitivity, reducing the organic aerosol concentrations further to ambient levels is not possible. The ratio of the measured SOA yield (Fig. 5) versus the simulated gas-phase dihydroxy dinitrate SOA precursor yield (Fig. S4) is about 5. FP is decreased by a factor of 2 for the dihydroxy dinitrates when COA is reduced from ∼ 25 µg cm−3 in the chamber to ∼4 µg cm−3 measured in the southeast US (Zhang et al., 2018). Thus, we multiply the dihydroxy dinitrate SOA precursors by 2.5 and we convert to mass by multiplying by the molecular weight of dihydroxy dinitrate. MCM v3.3.1 assumes a nitrate yield of 0.087–0.104 from NO reacting with the peroxy radical derived from OH+isoprene hydroxy nitrate. Low-volatility nitrates such as dihydroxy hydroperoxy nitrates form when HO2 reacts with the peroxy radical derived from OH+isoprene hydroxy nitrate. Such products would not form in the chamber conditions used in this work, where NO levels remained above 100 ppb, but would form in the ambient atmosphere. Considering these low-volatility species from mixed chemical regimes would further increase the SOA mass generated from the LV pathway.
For the 2MGA pathway, we convert to mass by multiplying gas-phase HMML by the molecular weight of 2-MGA (120 g mol−1), 2-MGA nitrate (165 g mol−1), and 2-MGA sulfate (200 g mol−1), which are the expected condensed-phase products under the high humidity levels in the atmosphere. Laboratory studies confirm that 2-MGA forms under humid conditions and some of the 2-MGA partitions to the gas phase as expected based on its volatility (Nguyen et al., 2015). For simplicity, we assume most of the HMML forms 2-MGA nitrate and 2-MGA sulfate but acknowledge that further experimental and modeling studies are needed to fully understand HMML and 2-MGA aqueous phase chemistry.
Then based on the gas-phase SOA precursor distribution from the kinetic model and assumptions above, under typical atmospheric conditions the fraction of the total SOA mass from isoprene OH-initiated oxidation under high-NOx conditions is ∼0.7 from the LV pathway and ∼0.3 from the 2MGA pathway. This assumes that the dihydroxy dinitrates are valid surrogates for the isoprene SOA. Considering that many multifunctional isoprene-derived organic nitrates have been detected in ambient aerosol (Lee et al., 2016), all SOA precursors in Table S2 with FP>0.05 at 26 ∘C are combined and converted to mass. Extrapolating these to ambient organic aerosol concentrations is more difficult because these compounds are more likely to exist in the particle phase because of accretion reactions and not volatility. When these products are assumed to exist entirely in the particle phase and no factor is applied to correct for differences in organic aerosol concentration or for these products only representing about one-third of the isoprene SOA yield measured in this study (Fig. S4), the LV pathway is estimated to contribute to ∼0.6 of the SOA formed under high-NOx conditions.
Thus, based on the simple calculations summarized above, the LV pathway may produce moderately more SOA mass than the 2MGA pathway in the atmosphere and consequently deserves equal attention. The conditions chosen here represent average atmospheric conditions around noon as measured during the Southern Oxidant and Aerosol Study (SOAS) field campaign, which occurred in the isoprene-rich southeastern United States (Sanchez et al., 2018; Feiner et al., 2016; Pajunoja et al., 2016). A more complete assessment using global and regional modeling is needed to more definitively determine the fraction of SOA formed via the LV versus 2MGA pathways as location, time of day, season, ambient aerosol concentration, and composition, etc. will all impact the amount of SOA formed from each pathway. Additional studies addressing organic nitrate hydrolysis and aerosol acidity are also necessary to fully understand the relative impact of the two pathways on SOA formation. Additionally, the kinetic model used in this work only estimates gas-phase potential SOA precursors. Future analysis using a more complex model that explicitly simulates both the gas and particle phases would be useful for extrapolating the SOA yields measured here to the ambient atmosphere, which typically has lower organic aerosol concentrations than chamber experiments. This would need to be combined with additional analysis of the chemical constituents in the particle phase. From past work (Kleindienst et al., 2009; Xu et al., 2014; D'Ambro et al., 2017) demonstrating that isoprene-derived SOA under high-NOx conditions is lower in volatility than that derived under low-NOx conditions and the C∗ values estimated in this work (Table S2), accretion reactions appear to be important even in the LV pathway experiments. The degree to which accretion reactions occur in the LV pathway experiments to form even lower-volatility products is quite uncertain and will greatly impact future analysis on how best to extrapolate isoprene SOA yields measured in chambers to the ambient atmosphere.
SOA from OH-initiated isoprene oxidation under high-NOx conditions forms from two major pathways: (1) low-volatility nitrates and dinitrates (LV pathway) and (2) 2-methyl glyceric acid and its oligomers (2MGA pathway). These SOA production pathways respond differently to experimental conditions, so this work examines the SOA yields from these two pathways independently. Results suggest that low-volatility nitrates and dinitrates produce significantly more aerosol than previously thought, with the isoprene SOA mass yield from the LV pathway being ∼0.15. Sufficient initial seed aerosol is necessary to reduce the impact of vapor wall losses of low-volatility compounds and accurately measure the entire SOA mass yield. Even though previous studies have assumed that isoprene high-NOx SOA largely forms from 2-MGA and its oligomers (Chan et al., 2010; Zhang et al., 2011, 2012), results from this study confirm that low-volatility compounds are also important for isoprene SOA formed under high-NOx conditions. The fate of isoprene's RO2 radicals and the environmental conditions will determine which pathways are active in the atmosphere at a certain time and location.
Under dry conditions, substantial amounts of SOA form from HMML reaction with 2-MGA to produce oligomers. The AMS results confirm that under humid conditions, these low-volatility oligomers are diminished in favor of higher-volatility monomer formation (and potentially subsequent volatilization of 2-MGA) to reduce the SOA mass. Thus, under atmospherically relevant humid conditions, aerosol formed from the 2MGA pathway is limited to HMML reaction with water or inorganic ions such as nitrate and sulfate. The importance of SOA from the 2MGA pathway will also depend on the NO2∕NO ratio, while SOA formed from the LV pathway will be important under all NO2∕NO ratios. Under typical atmospheric conditions (RH = 70 %, T = 298 K, NO2∕NO = 6, NO = 0.05 ppb, isoprene = 5 ppb, and OH molec. cm−3), based on the simple assumptions discussed in Sect. 5.5 we now estimate that the LV pathway produces moderately more SOA mass than the 2MGA pathway due to the high isoprene SOA yield from the LV pathway measured in this work.
Given the high isoprene SOA mass yield from the LV pathway (∼0.15) measured here, low-volatility compounds are as important as 2MGA-based compounds for isoprene SOA formed under high-NOx conditions. Thus, further studies investigating the formation rates and yields of these low-volatility compounds are needed. Consistent with past work (e.g., Lee et al., 2014), a number of low-volatility nitrates and dinitrates, which are likely important precursors for SOA formed from the LV pathway, were detected in the gas phase by the CF3O− CIMS (Sect. 5.1). These low-volatility compounds are likely derived from OH-initiated oxidation of the first-generation isoprene hydroxy nitrates. Synthetic pathways toward standards of many of the isoprene hydroxy nitrates exist (Lee et al., 2014; Teng et al., 2017). Now that this study has confirmed that low-volatility products contribute significantly to isoprene SOA, measuring SOA mass yields under varying RO2 fates using these isoprene hydroxy nitrate standards as the initial precursor instead of isoprene could be particularly valuable for decreasing the uncertainty in isoprene SOA yields. Additionally, an improved mechanistic understanding of isoprene SOA is needed. This would include an improved understanding of gas-phase reactions including measurements of highly functionalized peroxy radical isomerization rate constants, quantification of nitrate and hydroperoxide yields from highly functionalized RO2 radicals reacting with NO or HO2, respectively, and additional constraints on possible particle-phase accretion reactions leading to lower-volatility products (e.g., hemiacetal formation).
There are some limitations for how results from this study should be interpreted. In the atmosphere, the RO2 lifetime is longer than that in chamber experiments from this study and most past studies measuring SOA yields. Due to limitations in the sensitivity of the DMA and high NO levels needed to control the RO2 fate, performing SOA yield chamber experiments at conditions that favor a long RO2 lifetime is difficult. At longer RO2 lifetimes, the hydroxy nitrate isomer distribution shifts toward a higher percentage of β-isomers over δ-isomers (Peeters et al., 2014; Teng et al., 2017). Additionally, NOx emissions are decreasing across many regions of the world due to improvements in emissions controls creating mixed regimes in the ambient atmosphere where a later-generation gas-phase product could form from RO2+NO reaction during the first generation and RO2+HO2 during the second generation. Field measurements confirm the presence of such products. For example, Xiong et al. (2015) discuss the presence of dihydroxy hydroperoxy nitrates detected in the particle phase by Lee et al. (2016) during SOAS, a field campaign that took place during the summer in the southeastern United States. Dihydroxy hydroperoxy nitrates likely form when hydroxy nitrates, produced from the RO2+NO pathway, react with OH and O2 to form a peroxy radical that then reacts with HO2. Additionally, because isoprene SOA from the LV pathway only forms once later-generation products become oxidized, the extent of oxidation is important but also difficult to compare across different studies.
Comparisons of the DMA and AMS results imply that the collection and/or ionization efficiency on the AMS for SOA formed from the LV pathway is significantly lower than that formed from the 2MGA pathway. This could have important consequences for the interpretation of ambient organic aerosol measured by the AMS. Further work calibrating organic hydroxy nitrates on the AMS is needed to better understand why the organic fraction analysis varied so significantly between the two pathways.
Results from this work combined with past work provide further insight into how isoprene SOA should be parameterized in global and regional atmospheric chemistry models. Under humid conditions, SOA formation from the 2MGA pathway is produced mostly from HMML ring-opening reactions to form monomer compounds 2-MGA, 2-MGA nitrate, and 2-MGA sulfate, which simplifies the parameterization of SOA from the 2MGA pathway as the organic oligomerization reactions can be ignored. The particle's liquid water and pH will be important to consider, as these metrics shift the equilibrium of 2MGA and its carboxylate and change the hydrolysis rates for the 2-MGA nitrate and 2-MGA sulfate. The gas-phase kinetics for MPAN formation and reaction with OH to form HMML have been reasonably well studied (e.g., Orlando et al., 1999, 2002; Nguyen et al., 2015). HMML formation and generation of SOA in the atmosphere would be best incorporated into models by directly forming SOA through the MPAN + OH reaction. This would best parameterize the effects of temperature and NO2∕NO ratio on MPAN formation and also the influence of OH on HMML formation. This study confirms the need to perform experiments with adequate seed aerosol to limit vapor wall deposition processes when measuring SOA yields from the LV pathway. When regional chemical transport models use SOA yields that account for vapor wall deposition, there are differences in the contribution of isoprene to the total SOA budget and improvements in the agreement between simulated and observed total SOA and diurnal variability (Cappa et al., 2016). Incorporating the isoprene SOA yields from the LV pathway measured in this work into models will further improve the accuracy of simulated isoprene SOA. Moreover, the results from this study along with future experiments studying the formation of low-volatility nitrates and dinitrates on a mechanistic basis will be important for incorporating more explicit SOA formation into global models as has recently been done (e.g., Marais et al., 2016; Stadtler et al., 2018), thus replacing previous parameterizations that were typically based on a single chamber condition (e.g., Henze and Seinfeld, 2006; Henze et al., 2008; Heald et al., 2008).
We welcome future collaboration with those who wish to use this data set for additional modeling purposes (e.g., creating volatility basis set parameters for global or regional models or for evaluating the results with a more complex box model that includes aerosol chemistry). Please contact Rebecca H. Schwantes (rschwant@ucar.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7255-2019-supplement.
RHS designed the experiments. RHS and SMC performed the experiments. RHS analyzed the data with help from SMC, KHB, TBN, JHS, and YH. RHS did the kinetic modeling. YH, HM, WK, and RCF assisted RHS with DMA operation and data analysis. RHS wrote the paper with assistance from KHB, TBN, JHS, SMC, and YH.
The authors declare that they have no conflict of interest.
We thank Dennis Fitz for assistance with maintenance and data analysis of the luminol NO2/acyl peroxynitrate analyzer.
This research has been supported by the National Science Foundation, Division of Atmospheric and Geospace Sciences (grant no. AGS-1523500) and the National Science Foundation (grant no. 1745301). This material is based upon work supported by the National Center for Atmospheric Research, which is a major facility sponsored by the National Science Foundation under Cooperative Agreement no. 1852977.
This paper was edited by Jacqui Hamilton and reviewed by two anonymous referees.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, R. M., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperer, U., and Jimenez, J. L.: O∕C and OM∕OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008. a, b
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007. a
Brégonzio-Rozier, L., Siekmann, F., Giorio, C., Pangui, E., Morales, S. B., Temime-Roussel, B., Gratien, A., Michoud, V., Ravier, S., Cazaunau, M., Tapparo, A., Monod, A., and Doussin, J.-F.: Gaseous products and secondary organic aerosol formation during long term oxidation of isoprene and methacrolein, Atmos. Chem. Phys., 15, 2953–2968, https://doi.org/10.5194/acp-15-2953-2015, 2015. a, b, c, d, e, f, g, h, i, j, k, l
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, Tech. Rep. JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, CA, http://jpldataeval.jpl.nasa.gov (last access: 24 March 2017), 2015. a
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015. a
Cappa, C. D., Jathar, S. H., Kleeman, M. J., Docherty, K. S., Jimenez, J. L., Seinfeld, J. H., and Wexler, A. S.: Simulating secondary organic aerosol in a regional air quality model using the statistical oxidation model – Part 2: Assessing the influence of vapor wall losses, Atmos. Chem. Phys., 16, 3041–3059, https://doi.org/10.5194/acp-16-3041-2016, 2016. a
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009. a, b, c, d, e, f
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010. a, b, c, d, e, f, g, h, i, j, k
Charan, S. M., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Effect of particle charge on aerosol dynamics in Teflon environmental chambers, Aerosol Sci. Tech., 52, 854–871, https://doi.org/10.1080/02786826.2018.1474167, 2018. a, b
Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Elemental analysis of chamber organic aerosol using an aerodyne high-resolution aerosol mass spectrometer, Atmos. Chem. Phys., 10, 4111–4131, https://doi.org/10.5194/acp-10-4111-2010, 2010. a, b
Clark, C. H., Kacarab, M., Nakao, S., Asa-Awuku, A., Sato, K., and Cocker, D. R.: Temperature effects on secondary organic aerosol (SOA) from the dark ozonolysis and photo-oxidation of isoprene, Environ. Sci. Technol., 50, 5564–5571, https://doi.org/10.1021/acs.est.5b05524, 2016. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t
D'Ambro, E. L., Lee, B. H., Liu, J., Shilling, J. E., Gaston, C. J., Lopez-Hilfiker, F. D., Schobesberger, S., Zaveri, R. A., Mohr, C., Lutz, A., Zhang, Z., Gold, A., Surratt, J. D., Rivera-Rios, J. C., Keutsch, F. N., and Thornton, J. A.: Molecular composition and volatility of isoprene photochemical oxidation secondary organic aerosol under low- and high-NOx conditions, Atmos. Chem. Phys., 17, 159–174, https://doi.org/10.5194/acp-17-159-2017, 2017. a, b
Docherty, K. S., Jaoui, M., Corse, E., Jimenez, J. L., Offenberg, J. H., Lewandowski, M., and Kleindienst, T. E.: Collection efficiency of the aerosol mass spectrometer for chamber-generated secondary organic aerosols, Aerosol Sci. Tech., 47, 294–309, https://doi.org/10.1080/02786826.2012.752572, 2013. a, b
Dommen, J., Metzger, A., Duplissy, J., Kalberer, M., Alfarra, M. R., Gascho, A., Weingartner, E., Prevot, A. S. H., Verheggen, B., and Baltensperer, U.: Laboratory observation of oligomers in the aerosol from isoprene/NOx photooxidation, Geophys. Res. Lett., 33, L13805, https://doi.org/10.1029/2006GL026523, 2006. a, b, c, d
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012. a, b
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005. a, b
Ehn, M., Thornton, J. A., E., K., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014. a, b, c
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., De Gouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing atmospheric oxidation in an Alabama forest, J. Atmos. Sci., 73, 4699–4710, https://doi.org/10.1175/JAS-D-16-0044.1, 2016. a, b
Gaffney, J. S., Bornick, R. M., Chen, Y.-H., and Marley, N. A.: Capillary gas chromatographic analysis of nitrogen dioxide and PANs with luminol chemiluminescent detection, Atmos. Environ., 32, 1445–1454, https://doi.org/10.1016/S1352-2310(97)00098-8, 1998. a
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009. a
Heald, C. L., Henze, D. K., Horowitz, L. W., Feddema, J., Lamarque, J.-F., Guenther, A., Hess, P. G., Vitt, F., Seinfeld, J. H., Goldstein, A. H., and Fung, I.: Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res. Atmos., 113, D05211, https://doi.org/10.1029/2007JD009092, 2008. a
Henze, D. K. and Seinfeld, J. H.: Global secondary organic aerosol from isoprene oxidation, Geophys. Res. Lett., 33, L09812, https://doi.org/10.1029/2006GL025976, 2006. a
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008. a
Huang, Y., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified theory of vapor-wall mass transport in Teflon-walled environmental chambers, Environ. Sci. Technol., 52, 2134–2142, https://doi.org/10.1021/acs.est.7b05575, 2018. a
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014. a, b
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a, b
Kjaergaard, H. G., Knap, H. C., Ornso, K. B., Jorgensen, S., Crounse, J. D., Paulot, F., and Wennberg, P. O.: Atmospheric fate of methacrolein. 2. Formation of lactone and implications for organic aerosol production, J. Phys. Chem. A., 116, 5763–5768, https://doi.org/10.1021/jp210853h, 2012. a, b, c
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006. a, b, c
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007. a
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Jaoui, M., and Edney, E. O.: The formation of secondary organic aerosol from the isoprene + OH reaction in the absence of NOx, Atmos. Chem. Phys., 9, 6541–6558, https://doi.org/10.5194/acp-9-6541-2009, 2009. a, b
Knote, C., Tuccella, P., Curci, G., Emmons, L., Orlando, J. J., Madronich, S., Baro, R., Jimenez-Guerrero, P., Luecken, D., Hogrefe, C., Forkel, R., Werhahn, J., Hirtl, M., Perez, J. L., San Jose, R., Giordano, L., Brunner, D., Yahya, K., and Zhang, Y.: Influence of the choice of gas-phase mechanism on predictions of key gaseous pollutants during the AQMEII phase-2 intercomparison, Atmos. Environ., 115, 553–568, https://doi.org/10.1016/j.atmosenv.2014.11.066, 2015. a
Krechmer, J. E., Coggon, M. E., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Janye, J. T., Sipila, M., Junninen, H., St. Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015. a
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008. a, b
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation under high-NOx conditions, Geophys. Res. Lett., 32, L18808, https://doi.org/10.1029/2005GL023637, 2005. a, b, c, d
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006. a, b
Kurten, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene autoxidation products may not have extremely low saturation vapor pressures despite high O:C ratios, J. Phys. Chem. A., 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016. a
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015. a
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016. a, b, c, d, e
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On rates and mechanisms of OH and O3 reactions with isoprene-derived hydroxy nitrates, J. Phys. Chem. A, 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014. a, b, c, d, e, f, g
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012. a
Lin, Y.-H., Zhang, H., Pye, H. O. T., Zhang, Z., Marth, W. J., Park, S., Arashiro, M., Cui, T., Budisulistiorini, S. H., Sexton, K. G., Vizuete, W., Xie, Y., Luecken, D. J., Piletic, I. R., Edney, E. O., Bartolotti, L. J., Gold, A., and Surratt, J. D.: Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, P. Natl. Acad. Sci. USA, 110, 6718–6723, https://doi.org/10.1073/pnas.1221150110, 2013. a, b, c
Loza, C. L., Chhabra, P. S., Yee, L. D., Craven, J. S., Flagan, R. C., and Seinfeld, J. H.: Chemical aging of m-xylene secondary organic aerosol: laboratory chamber study, Atmos. Chem. Phys., 12, 151–167, https://doi.org/10.5194/acp-12-151-2012, 2012. a, b, c
Mai, H. and Flagan, R. C.: Scanning DMA Data Analysis I. Classification Transfer Function, Aerosol Sci. Tech., 52, 1382–1399, https://doi.org/10.1080/02786826.2018.1528005, 2018. a, b
Mai, H., Kong, W., Seinfeld, J. H., and Flagan, R. C.: Scanning DMA Data Analysis II. Integrated DMA-CPC Instrument Response and Data Inversion, Aerosol Sci. Tech., 52, 1400–1414, https://doi.org/10.1080/02786826.2018.1528006, 2018. a, b
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016. a, b
Nah, T., McVay, R. C., Pierce, J. R., Seinfeld, J. H., and Ng, N. L.: Constraining uncertainties in particle-wall deposition correction during SOA formation in chamber experiments, Atmos. Chem. Phys., 17, 2297–2310, https://doi.org/10.5194/acp-17-2297-2017, 2017. a, b, c
Nannoolal, Y., Rarey, J., Ramjugernath, D., and Cordes, W.: Estimation of pure component properties Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilibr., 226, 45–63, https://doi.org/10.1016/j.fluid.2004.09.001, 2004. a, b
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilibr., 269, 117–133, https://doi.org/10.1016/j.fluid.2008.04.020, 2008. a, b
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Contribution of first-versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006. a, b, c
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011. a, b
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014a. a
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014b. a, b
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/C5CP02001H, 2015. a, b, c, d, e, f, g, h, i, j
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/C2CS35166H, 2012. a, b
Orlando, J. J., Tyndall, G. S., and Paulson, S. E.: Mechanism of the OH-initiated oxidation of methacrolein, Geophys. Res. Lett., 26, 2191–2194, https://doi.org/10.1029/1999GL900453, 1999. a
Orlando, J. J., Tyndall, G. S., Bertman, S. B., Chen, W., and Burkholder, J. B.: Rate coefficient for the reaction of OH with CH2=C(CH3)C(O)OONO2 (MPAN), Atmos. Environ., 36, 1895–1900, https://doi.org/10.1016/S1352-2310(02)00090-0, 2002. a
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The atmospheric chemistry of alkoxy radicals, Chem. Rev., 103, 4657–4689, https://doi.org/10.1021/cr020527p, 2003. a
Pajunoja, A., Hu, W., Leong, Y. J., Taylor, N. F., Miettinen, P., Palm, B. B., Mikkonen, S., Collins, D. R., Jimenez, J. L., and Virtanen, A.: Phase state of ambient aerosol linked with water uptake and chemical aging in the southeastern US, Atmos. Chem. Phys., 16, 11163–11176, https://doi.org/10.5194/acp-16-11163-2016, 2016. a, b
Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C.: Aerosol formation in the photooxidation of isoprene and β-pinene, Atmos. Environ., 25A, 997–1008, https://doi.org/10.1016/0960-1686(91)90141-S, 1991. a
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kurten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009. a, b
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009. a
Peeters, J., Muller, J.-F., Stavrakou, T., and Nguyen, V.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A., 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014. a, b
Pieber, S. M., Haddad, I. E., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. S., Frohlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jimenez, J. L., Worsnop, D. R., Baltensperger, U., and Prevot, A. S. H.: Inorganic salt interference on in Aerodyne AMS and ACSM organic aerosol composition studies, Environ. Sci. Technol., 50, 10494–10503, https://doi.org/10.1021/acs.est.6b01035, 2016. a, b, c
Pierce, J. R., Engelhart, G. J., Hildebrandt, L., Weitkamp, E. A., Pathak, R. K., Donahue, N. M., Robinson, A. L., Adams, P. J., and Pandis, S. N.: Constraining particle evolution from wall losses, coagulation, and condensation-evaporation in smog-chamber experiments: Optimal estimation based on size distribution measurements, Aerosol Sci. Tech., 42, 1001–1015, https://doi.org/10.1080/02786820802389251, 2008. a
Praske, E., Crounse, J. D., Bates, K. H., Kurten, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric fate of methyl vinyl ketone: Peroxy radical reactions with NO and HO2, J. Phys. Chem. A., 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015. a
Ribeiro, P. A., Raposo, M., Marat-Mendes, J. N., and Giacometti, J. A.: Constant-current corona charging of biaxially stretched PVDF films in humidity-controlled atmospheres, IEEE T. Electr. Insul., 27, 744–750, https://doi.org/10.1109/14.155791, 1992. a
Sanchez, D., Jeong, D., Seco, R., Wrangham, I., Park, J.-H., Brune, W., Koss, A., Gilman, J., de Gouw, J., Misztal, P., Goldstein, A., Baumann, K., Wennberg, P. O., Keutsch, F. N., Guenther, A., and Kim, S.: Intercomparison of OH and OH reactivity measurements in a high isoprene and low NO environment during the Southern Oxidant and Aerosol Study (SOAS), Atmos. Environ., 174, 227–236, https://doi.org/10.1016/j.atmosenv.2017.10.056, 2018. a, b
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017. a, b
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3 edn., John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2016. a, b
Sharpe, S. W., Johnson, T. J., Sams, R. L., Chu, P. M., Rhoderick, G. C., and Johnson, P. A.: Gas-phase databases for quantitative infrared spectroscopy, composite spectrum for ISOPREN_25T (Ver 1.0, Oct, 01) and composite spectrum for ISOBUTENAL_25T (Ver 1.0, Dec, 04), Appl. Spectrosc., 58, 1452–1461, https://doi.org/10.1366/0003702042641281, 2004. a
Sjogren, S., Gysel, M., Weingartner, E., Baltensperger, U., Cubison, M., Coe, H., Zardini, A., Marcolli, C., Krieger, U., and Peter, T.: Hygroscopic growth and water uptake kinetics of two-phase aerosol particles consisting of ammonium sulfate, adipic and humic acid mixtures, J. Aerosol Sci., 38, 157–171, https://doi.org/10.1016/j.jaerosci.2006.11.005, 2007. a
Stadtler, S., Kühn, T., Schröder, S., Taraborrelli, D., Schultz, M. G., and Kokkola, H.: Isoprene-derived secondary organic aerosol in the global aerosol–chemistry–climate model ECHAM6.3.0–HAM2.3–MOZ1.0, Geosci. Model Dev., 11, 3235–3260, https://doi.org/10.5194/gmd-11-3235-2018, 2018. a, b
St. Clair, J. M., McCabe, D. C., Crounse, J. D., Steiner, U., and Wennberg, P. O.: Chemical ionization tandem mass spectrometer for the in situ measurement of methyl hydrogen peroxide, Rev. Sci. Instrum., 81, 094102, https://doi.org/10.1063/1.3480552, 2010. a
Sunol, A. M., Charan, S. M., and Seinfeld, J. H.: Computational simulation of the dynamics of secondary organic aerosol formation in an environmental chamber, Aerosol Sci. Tech., 52, 470–482, https://doi.org/10.1080/02786826.2018.1427209, 2018. a
Surratt, J. D., Chan, a. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, a. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010. a
Taylor, W. D., Allston, T. D., Moscato, M. J., Fazekas, G. B., Kozlowski, R., and Takacs, G. A.: Atmospheric photo-dissociation lifetimes for nitromethane, methyl nitrite, and methyl nitrate, Int. J. Chem. Kinet, 12, 231–240, https://doi.org/10.1002/kin.550120404, 1980. a
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017. a, b, c
Wennberg, P. O.: Let's abandon the high NOx and low NOx terminology, IGACnews, 50, 3–4, 2013. a
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018. a, b, c, d, e, f, g, h, i, j
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015. a
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the volatility of secondary organic aerosol from isoprene photooxidation, Environ. Sci. Tech., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014. a, b, c, d, e
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J., and Kamens, R. M.: Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions, Atmos. Chem. Phys., 11, 6411–6424, https://doi.org/10.5194/acp-11-6411-2011, 2011. a, b, c, d, e, f, g
Zhang, H., Lin, Y. H., Zhang, Z., Zhang, X., Shaw, S. L., Knipping, E. M., Weber, R. J., Gold, A., Kamens, R. M., and Surratt, J. D.: Secondary organic aerosol formation from methacrolein photooxidation: roles of NOx level, relative humidity and aerosol acidity, Environ. Chem., 9, 247–262, https://doi.org/10.1071/EN12004, 2012. a, b, c, d, e
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018. a
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007. a
Zhang, X., Cappa, C. D., Jather, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014. a, b, c, d, e, f, g
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015. a, b