the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Apr 2019
| 02 Apr 2019
Mechanistic and kinetics investigations of oligomer formation from Criegee intermediate reactions with hydroxyalkyl hydroperoxides
Long Chen
Yu Huang
Yonggang Xue
Zhenxing Shen
Junji Cao
Wenliang Wang
Although secondary organic aerosol (SOA) is a major component of PM2.5 and organic aerosol (OA) particles and therefore profoundly influences air quality, climate forcing, and human health, the mechanism of SOA formation via Criegee chemistry is poorly understood. Herein, we perform high-level theoretical calculations to study the gas-phase reaction mechanism and kinetics of four Criegee intermediate (CI) reactions with four hydroxyalkyl hydroperoxides (HHPs) for the first time. The calculated results show that the consecutive reactions of CIs with HHPs are both thermochemically and kinetically favored, and the oligomers contain CIs as chain units. The addition of an −OOH group in HHPs to the central carbon atom of CIs is identified as the most energetically favorable channel, with a barrier height strongly dependent on both CI substituent number (one or two) and position (syn- or anti-). In particular, the introduction of a methyl group into the anti-position significantly increases the rate coefficient, and a dramatic decrease is observed when the methyl group is introduced into the syn-position. These findings are expected to broaden the reactivity profile and deepen our understanding of atmospheric SOA formation processes.
- Article
(3612 KB) - Full-text XML
-
Supplement
(5136 KB) - BibTeX
- EndNote





Alkenes are the most abundant volatile organic compounds (VOCs) in the atmosphere after methane and primarily originate from anthropogenic and biogenic sources (Lester and Klippenstein, 2018; Guenther et al., 2000). Alkene ozonolysis produces a carbonyl oxide (also called Criegee intermediates; CIs) and a carbonyl moiety (Donahue et al., 2011a; Aplincourt and Ruiz-López, 2000; Johnson and Marston, 2008; Welz et al., 2012; Criegee, 1975; Taatjes et al., 2013), which is thought to be an important source of radicals, whose subsequent reactions lead to the formation of hydroperoxides, organic peroxides, and secondary organic aerosol (SOA) (Donahue et al., 2011a; Becker et al., 1990; Kroll and Seinfeld, 2008; Hallquist et al., 2009; Tobias and Ziemann, 2001). It thus influences air quality, climate forcing, and human health (Rissanen et al., 2014; Donahue et al., 2011b, 2012). Criegee intermediates were first proposed by Rudolph Criegee as early as 1975 (Criegee, 1975), and their direct synthesis in a laboratory experiment was performed by the photolysis of organic iodides in the presence of O2 (Welz et al., 2012; Taatjes et al., 2008).
In view of the highly exothermic nature of alkene ozonolysis, nascent CIs often possess a considerable amount of internal energy and are thus highly reactive (Y. Li et al., 2018; Li et al., 2014). The thermal unimolecular decay of vibrationally excited CIs is thought to be an important nonphotolytic source of atmospheric hydroxyl (OH) radicals, particularly in low light conditions, urban environments, and heavily forested areas (Lester and Klippenstein, 2018; Foreman et al., 2016; Kidwell et al., 2016; Green et al., 2017; Zhang and Zhang, 2002; Cremer et al., 2001; Anglada et al., 2002). The OH radical is one of the most powerful oxidants that participates in the atmospheric photochemical oxidation of VOCs (Gligorovski et al., 2015); it thus contributes to tropospheric ozone formation by being involved in the production of organic peroxy radicals (RO2), which, in turn, facilitate the cycling of NO to NO2 (Zhang and Zhan, 2002; Gligorovski et al., 2015). The remaining CIs become collisionally stabilized Criegee intermediates (SCIs) that can undergo further bimolecular reactions with a number of atmospheric trace gases, such as H2O, NO2, and SO2 (Chen et al., 2016a, b, 2018; Mauldin et al., 2012; Berndt et al., 2014; Kuwata et al., 2015; Lin et al., 2016; Lin and Chao, 2017; Ouyang et al., 2013; Stone et al., 2014; Chao et al., 2015; Taatjes, 2017; Long et al., 2018), and contribute to the nucleation and growth of secondary aerosol (e.g., nitrate, sulfate, SOA) by partitioning between gas and particle phases (Foreman et al., 2016; Vereecken, 2017; Huang et al., 2014, 2015; Berndt et al., 2012; Zhang et al., 2015; Li et al., 2018; Ji et al., 2017; Xu et al., 2014). The bimolecular processes of SCIs at the air–water interface have been extensively studied both experimentally and theoretically (Zhu et al., 2016; Kumar et al., 2017, 2018; Zhong et al., 2017, 2018; Enami and Colussi, 2017; Heine et al., 2017), and the reaction with atmosphere-abundant water vapor in the gas phase or at the air–water interface has been identified as one of the dominant degradation pathways of SCI removal from the atmosphere (Taatjes et al., 2013; Chen et al., 2016a, b; Lin et al., 2016; Chao et al., 2015; Huang et al., 2015; Zhu et al., 2016; Zhong et al., 2017; Anglada and Solé, 2016).
Experimentally, Taatjes et al. (2013) studied the kinetics of CH3CHOO reactions with H2O, SO2, and NO2 and found that the reactivity of anti-CH3CHOO toward water and SO2 is substantially higher than syn-CH3CHOO with an upper limit rate coefficient of ( cm3 molecule−1 s−1. Also, Smith et al. (2015) obtained a similar conclusions for the UV absorption of the CH2OO + H2O reaction system with the rate coefficient determined as ( cm3 molecule−1 s−1 at 298 K, and it exhibits a large negative T dependence at temperatures from 283 to 324 K. Winterhalter et al. (2001) studied the mechanisms and products of the gas-phase ozonolysis of β-pinene and found that the main products are the excited C9-CI plus HCHO. Moreover, oligomerization reactions of CIs with typical atmospheric species are identified as one of the dominate pathways leading to the formation of highly oxygenated and high-molecular-weight oligomers that have remarkably low vapor pressure, contributing to SOA formation and growth (Bonn et al., 2008; Heaton et al., 2007; Wang et al., 2016; Inomata et al., 2014). For example, Sakamoto et al. (2013) performed laboratory-scale ethylene ozonolysis in a Teflon bag reactor and revealed that the sequential addition of CH2OO to hydroperoxides leads to oligomeric hydroperoxides and finally affords SOA. Sadezky et al. (2008) proposed that SOA formation is initiated by the reaction of SCI with an RO2 radical, followed by the sequential addition of SCIs, and chain termination by reaction with the HO2 radical.
Zhao et al. (2015) studied the ozonolysis of trans-3-hexene in a flow reactor and static Teflon chambers in the absence and presence of an OH or SCI scavenger at 295±1 K, arriving at the same conclusion as above. In particular, oligomers having SCIs as chain units were identified as one of the dominant components of atmospheric SOA and were produced by the sequential addition of C2H5CHOO to the RO2 radical. More recently, Wang et al. (2016) investigated the heterogeneous ozonolysis of oleic acid (OL) using an aerosol flow tube and found that reactions of particulate SCIs generate high-molecular-weight oligomers with low volatility that are preferentially partitioned into the particle phase to promote SOA formation. They confirmed that the SCI-based mechanism is the dominant pathway in the formation of high-molecular-weight oligomers.
On the other hand, Ehn et al. (2014) reported a large source of low-volatility SOA generated from the ozonolysis of α-pinene and other endocyclic monoterpenes under atmospheric conditions and proposed that the mechanism of extremely low-volatility organic compound (ELVOC) formation is driven by RO2 autoxidation. The RO2 autoxidation pathway mainly includes intramolecular hydrogen shift and the sequential O2 addition steps (Rissanen et al., 2014). Also, several groups obtained similar conclusions that highly oxygenated molecules (HOMs) are produced via RO2 autoxidation in cyclohexene and terpene ozonolysis systems (Kirkby et al., 2016; Berndt et al., 2018). Moreover, HOMs are major contributors to aerosol particle formation and growth on a global scale (Tröstl et al., 2016; Stolzenburg et al., 2018). Compared with the RO2 autoxidation pathways, oligomerization reactions involving CIs as the repeat units preserve carbon oxidation state and increase the number of carbon backbone moieties, therefore leading to a large reduction in volatility (Wang et al., 2016). Moreover, oligomerization reaction proceeds over a shorter period of time during the early stage of biogenic SOA formation and growth (Heaton et al., 2007). Therefore, we think that it is essential to investigate the gas-phase Criegee-chemistry-based mechanism of SOA formation and growth.
Aplincourt and Ruiz-López (2000) investigated the mechanism of CH2OO reactions with CH2O, H2O, SO2, and CO2 at the CCSD(T) level of theory and found that the reactions with H2O, CH2O, and SO2 are preferable, whereas that with CO2 is unlikely to occur. Because of the high concentration of H2O ([H2O] ≈ 7.0×1017 molecules cm−3) in the atmosphere (Zhang et al., 2014; T. Zhang et al., 2015), reaction with water vapor is the dominant chemical sink (Aplincourt and Ruiz-López, 2000). Also, Ryzhkov and Ariya (2003, 2004, 2006) obtained a similar conclusion that the most energetically favorable pathway of carbonyl oxide reaction with water vapor is the formation of hydroxyalkyl hydroperoxides (HHPs). Kumar et al. (2014) proposed that the gas-phase decomposition of HHPs has two competitive pathways: (i) → aldehyde or ketone + H2O2 and (ii) → carboxylic acid + H2O. They found that the barrier of an acid-catalyzed former reaction is dramatically lower than that of water-catalyzed and uncatalyzed systems. Aplincourt and Anglada (2003) investigated the unimolecular decay and water-catalyzed decomposition of HHPs generated from isoprene ozonolysis and found that the main products are methyl vinyl ketone (MVK) or methacrolein (MAC) plus H2O2. Unfortunately, little is known of the reactivity of SCIs toward HHPs generated from reaction with water vapor. Moreover, the effect of substituents on the reactivity of carbonyl oxides is still poorly understood.
Recently, Zhao et al. (2017a) investigated the mechanisms and kinetics of four SCIs reactions with four RO2 radicals and found that the addition of terminal oxygen in RO2 to central carbon in SCI is the most kinetically favorable channel. An analogous conclusion was obtained by investigating the reactions of anti-CH3CHOO with HO2 and H2O2 molecules; i.e., the sequential addition of SCIs is a favorable reaction mode for SOA formation (Chen et al., 2017). Vereecken (2017) also investigated the reactions of CH2OO with acids and enols using the CCSD(T)//M06-2X/aug-cc-pVTZ method and found that the 1,4-insertion mechanism is preferable. The above milestone investigations provide fundamental insights and lay solid foundations for further studies of the Criegee-chemistry-based mechanism of SOA formation.
In this study, we mainly focus on the gas-phase oligomerization reaction of carbonyl oxides with HHPs, leading to the formation of high-molecular-weight oligomers under atmospheric conditions. This reaction represents the initial step of oligomer formation and growth during alkene ozonolysis and therefore needs to be extensively characterized to gain deeper insights into the fundamental chemical composition of these oligomers in the atmosphere. Moreover, structure–reactivity relationship plays an important role in determining the rates and outcomes of bimolecular processes. Herein, we employ high-level theoretical calculations in conjunction with kinetics analysis to study the mechanisms and kinetics of the reactions of four carbonyl oxides with four HHPs and describe the effects of carbonyl oxide conformations on reaction rate. The carbonyl oxides considered in this work (CH2OO, syn- and anti-CH3CHOO, and (CH3)2COO) are anticipated upon the ozonolysis of ethylene, propylene, isobutene, and 2,3-dimethyl-2-butene, while the investigated HHPs are assumed to arise from bimolecular reactions with water vapor in the troposphere.
The geometries of all stationary points on a potential energy surface (PES) are optimized and characterized by the M06-2X functional (Zhao and Truhlar, 2006) in combination with the 6-311+G(2df,2p) basis set (Zheng and Truhlar, 2009), since the M06-2X functional allows one to reliably describe the London dispersion energy (Zhao and Truhlar, 2008b) and can produce excellent results in predicting non-covalent interaction energy involving the main-group elements (Zhao et al., 2017a, b; Zhao and Truhlar, 2008a, b). This functional has the best performance for predicting hydrogen bonding, weak interactions, thermochemistry, and kinetics (Zhao and Truhlar, 2008a; Schenker et al., 2011; Ji et al., 2018). Harmonic vibrational frequencies are performed at the same level of theory to verify that the nature of each structure is either a minimum (NIMAG = 0) or a transition state (NIMAG = 1) and to provide the zero-point vibrational energy (ZPVE) corrections. A scale factor of 0.98 is applied to scale all the M06-2X/6-311+G(2df,2p) frequencies to account for the thermodynamic contribution to Gibbs free energy and enthalpy at 298 K and 1 atm (Zheng and Truhlar, 2009). The reactant–product connectivity on either side is established by intrinsic reaction coordinate (IRC) calculations (Fukui, 1981). Then, the single-point energies are calculated at the M06-2X/def2-TZVP level of theory. The predicted free energies are equal to the thermal correction to Gibbs free energies at the M06-2X/6-311+G(2df,2p) level plus the electronic energies obtained at the M06-2X/def2-TZVP level. In order to obtain a better evaluation of the reliability of the M06-2X functional in computing energies, the single-point energies of species included in some selected elementary reactions (R1a-R1d, R3a-R3d and R5a-R5d) are recalculated at the CCSD(T)/6-311+G(2df,2p) level based on the M06-2X/6-311+G(2df,2p) optimized geometries. Furthermore, the basis set superposition error (BSSE) is calculated using the counterpoise method described by Boys and Bernardi (1970) to estimate the stability of the pre-reactive complexes. The electronic-energy ( and free-energy ( barrier comparisons of both approaches for considering and not considering BSSE correction are listed in Table S1 in the Supplement. As shown in Table S1, the contributions of BSSE corrections to the barriers are ∼1.4 (CCSD(T)) and ∼0.4 (M06-2X) kcal mol−1, respectively. The mean absolute deviations (MADs) of both approaches are 0.98 ( and 0.96 ( kcal mol−1 without considering BSSE correction, while they become 0.38 ( and 0.34 ( kcal mol−1 when considering BSSE correction. The result shows that the M06-2X method in combination with the BSSE correction affords energies similar to those determined by the accurate and well-recognized CCSD(T)-level calculation. Considering the computational costs, the M06-2X/def2-TZVP method is selected to perform the single-point energy calculation for the title reaction system. Finally, the rate coefficients are calculated using a combination of canonical transition state theory (CTST) and an asymmetric Eckart tunneling correction at temperatures between 273 and 400 K (Zhao et al., 2017a; Chen et al., 2017).
As shown in Fig. 5, the CH2OO + HO–CH2OO–H (Pa1) reaction proceeds according to a two-step mechanism: (i) a fast thermal equilibrium between the reactants and intermediate IM1a and (ii) the addition of CH2OO, leading to the formation of product P1a. The whole reaction process is expressed as follows.
Applying the steady-state approximation (SSA) for the intermediate IM1a, the overall rate coefficient is extrapolated to Eq. (2) (Zhang et al., 2012; Liu et al., 2015).
If , the overall rate coefficient is written as follows (Chen et al., 2016b; Ryzhkov and Ariya, 2006).
The equilibrium coefficient Keq is expressed as Eq. (4):
where σ is the reaction symmetry number, QIM(T), QA(T), and QB(T) denote the products of electronic, translational, rotational, torsional, and vibrational canonical partition functions for the intermediate and reactants A and B (Mendes et al., 2014), T is the temperature in Kelvin, and GR and GIM are the total free energies of the reactant and complex, respectively. The quantum chemical calculations are executed using the Gaussian 09 program suite (Frisch et al., 2009). Atoms in molecules (AIM) analysis at the bond-critical point (BCP) for the pre-reactive complex and product is performed by using the AIM2000 software (Biegler-König et al., 2000). The rate coefficients are calculated by implementing the KiSThelP program (Canneaux et al., 2014)
3.1 Bimolecular reaction of SCIs with water vapor
Equations (8) and (9) represent the two types of bimolecular reactions between carbonyl oxides and water vapor. Previous investigations have shown that some carbonyl oxides are largely removed by their reactions with water dimer (Taatjes et al., 2013; Chen et al., 2016b; Chao et al., 2015; Anglada and Solé, 2016) to generate HHPs (Chen et al., 2016a, b; Anglada et al., 2011), which are important atmospheric oxidants initiating vegetation damage (Becker et al., 1990; Kumar et al., 2014). Further mechanistic details of the above reaction can be found in our previous works (Chen et al., 2016a, b).
Figure 1 presents a simplified scheme for the reactions of several distinct carbonyl oxides (CH2OO, syn- and anti-CH3CHOO, (CH3)2COO) with water dimer to form HHPs. In all cases, each reaction begins with the formation of a strong pre-reactive complex and then surmounts a small barrier that is still lower in energy than the reactants before product generation. Table 1 lists the relative energies of stationary points and the activation energies of elementary reactions. In Fig. 1 and Table 1, labels A, B, C, and D correspond to the relative energies of the pre-reactive complex (RC), transition state (TS), post-reactive complex (PC), and product (P). R1 and R2 denote the syn- and anti-positions of the substituents, respectively. These four transition states are located by the rotation of two dihedral angles (DO2H4O4H3, DO4H2O3H1). As shown in Table 1, the barrier differences between the computational and literature values that were derived from the CCSD(T)/aug-cc-pVTZ//B3LYP/6-311+G(2df,2p) method (Anglada and Solé, 2016) are 0.5–1.0 kcal mol−1. Such discrepancies may be attributed to the different theoretical method used in computing energies. The barrier of Entry 4 is 3.3 kcal mol−1, which is lower than the corresponding CCSD(T)/CBS result by 0.5 kcal mol−1 (Anglada and Solé, 2016). The results show that the M06-2X method provides energies similar to those determined by the CCSD(T)-level calculation. Based on the energies given in Table 1, product HHPs (Pa and Pb) are near-isoenergetic conformers differing only in the orientation of the H1 atom along the C1–O3 bond. As mentioned above, HHPs are key reactive intermediates that possess −OH and −OOH functional groups and can therefore sequentially react with carbonyl oxide to generate oligomers. Considering the fact that Pa and Pb are structurally and energetically similar, the former is judiciously selected for studying oligomerization reactions, whereas the latter is merely listed in Figs. S1–S3.
Table 1Relative free energies (kcal mol−1) for the stationary points and free-energy ( barriers for the elementary pathways of distinct carbonyl oxide reactions with water dimer calculated at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory. Labels A, B, C, and D are defined in Fig. 1.
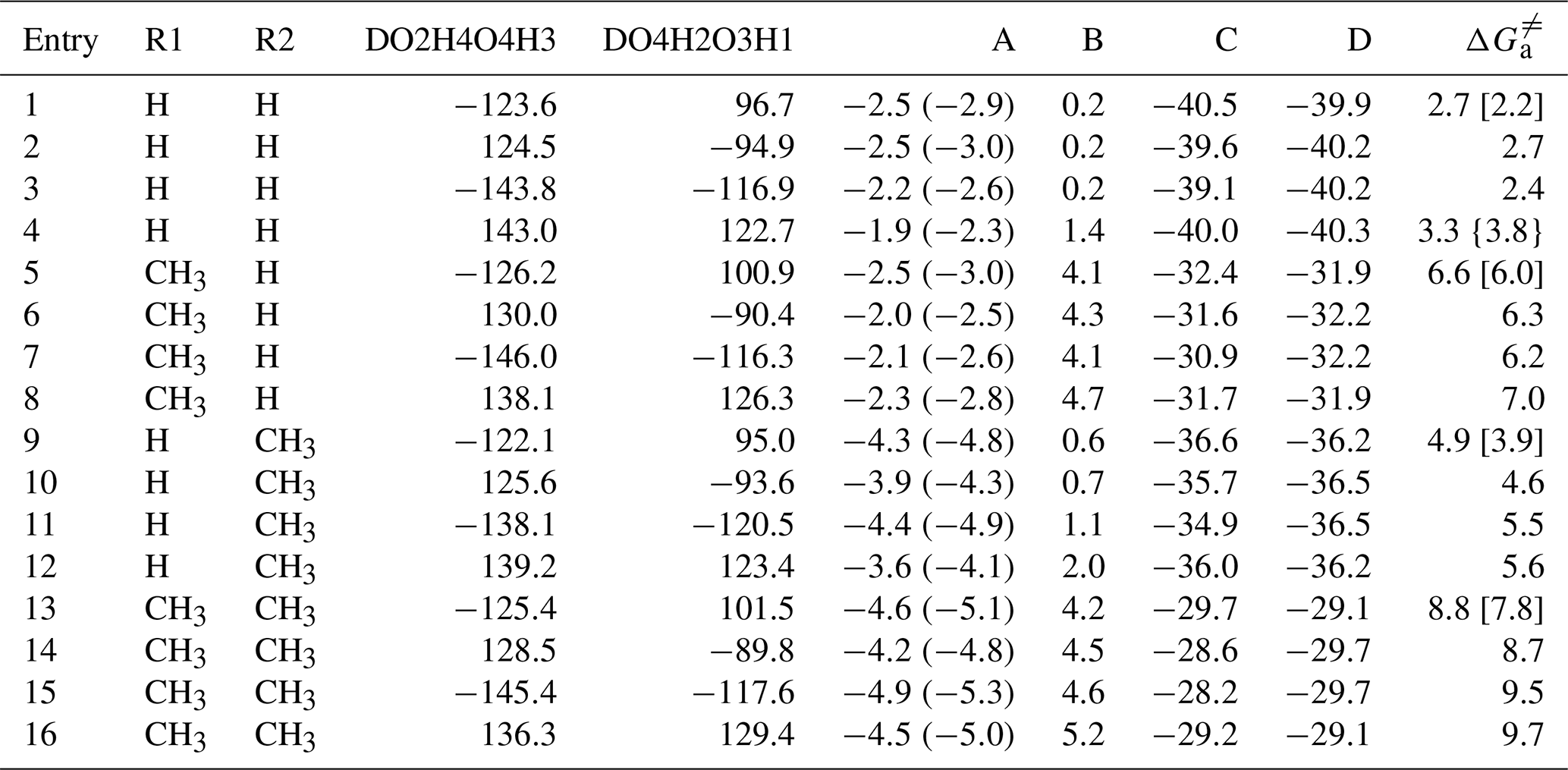
Values in parentheses correspond to no consideration of the BSSE correction, values in brackets correspond to CCSD(T)/aug-cc-pVTZ//B3LYP/6-311+G(2df,2p), and values in braces correspond to CCSD(T)/CBS//B3LYP/6-311+G(2df, 2p).
3.2 PES for the reaction of CH2OO with
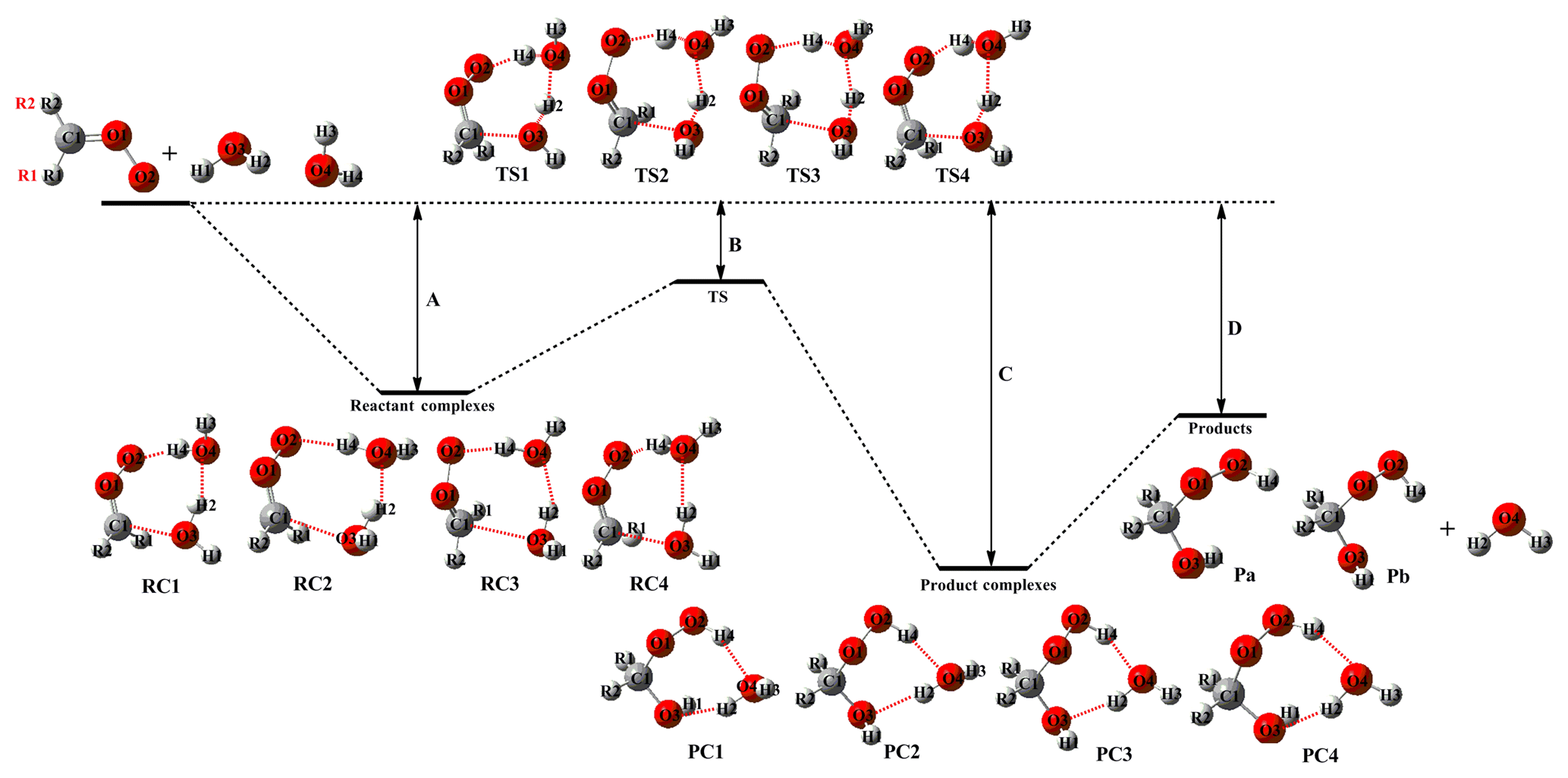
CH2OO, the simplest Criegee intermediate, originates from the ozonolysis of all exocyclic alkenes, e.g., isoprene, monoterpenes, and sesquiterpenes (Nguyen et al., 2016), which makes its chemistry particularly important for forest and urban environments. The largest sink of CH2OO corresponds to its bimolecular reaction with water dimer in the troposphere, which generates HO–CH2OO–H as the dominant product (Kumar et al., 2014; Lewis et al., 2015). Figure 2 shows the schematic PES for the reaction of CH2OO with HO–CH2OO–H, with the optimized geometries of all stationary points on this PES given in Fig. S4.
Figure 2 shows that the differences between the relative free energies and the electronic energies for all stationary points are significant (∼10–24 kcal mol−1), implying that the addition reactions of the parent carbonyl oxide with Pa1 are characterized by obvious contributions of the entropy effect. Similar behaviors are also observed for oligomerization reactions of other carbonyl oxides with HHPs (see Figs. 3–5). The partition function of every vibrational mode involved in the complex IM1a is listed in Table S2. As shown in Table S2, the partition function of the low-frequency vibrational mode (<200 cm−1) is significantly higher than the high-frequency vibrational mode (>200 cm−1), and their contribution is up to 70.5 %. The result implies that the low-frequency vibrational mode contributes to the entropic factor significantly. A similar conclusion is also obtained from the IM2a case (Table S3). Thus, unless otherwise stated, the discussion in the following sections refers to free-energy barriers (.

Figure 2PES (ΔG and ΔE, in italics) for the reaction of CH2OO with (Pa1) computed at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
The formation of oligomers P2a, P2b, P2c, and P2d (containing CH2OO as the repeating unit) is strongly exothermic (>81 kcal mol−1), and the apparent activation energies Eapp observed for all elementary reactions are negative values, signifying that these reactions are both thermochemically and dynamically feasible under atmospheric condition. Product Pa1 () formed in the reaction of CH2OO with water dimer has two functional groups (−OH and −OOH), both of which can be involved in addition reactions. The addition reactions of 2CH2OO + Pa1 begin with the formation of loosely bound pre-reactive complexes IM1a and IM1b, with 3.1 and 2.6 kcal mol−1 stability. They are formed by a hydrogen bond between the terminal CH2OO oxygen atom and the hydrogen atom of the −OOH group in Pa1 and a van der Waals (vdW) bond between the central carbon atom of CH2OO and the oxygen atom of the −OH group in Pa1. The above complexes are immediately converted into products P1a and P1b via transition states TS1a and TS1b with barriers of 8.4 and 11.7 kcal mol−1, respectively, while the corresponding reaction exothermicities are estimated as 43.4 and 40.5 kcal mol−1, respectively. The above result shows that the most favorable channel is the addition of the −OOH group of Pa1 to the parent carbonyl oxide. The detailed mechanism mainly involves the HO−CH2OO moiety released from the breaking O−H bond in Pa1 binding to the central carbon atom of CH2OO, and simultaneously the remnant hydrogen atom transfers to the terminal oxygen, leading to product P1a. The electronic density (ρ), Laplacian (∇2), and the three eigenvalues of the Hessian of the complexes IM1a and IM1b are displayed in Table S4. As shown in Table S4, the ∇2 values of all forming bonds (B1(O5−H4), B2(O3−C4), B3(O5−H3), and B1(O1−C4)) are negative, indicating that they are covalent bonds. The ρ values of B1 and B3 are significantly higher than those of B2 and B4, showing that the bond strength of the former case is higher than the latter case.
The addition pathway opens the door for other subsequent reactions leading to SOA via Criegee chemistry, which may result in aerosol formation and thus impact climate. As pointed out in previous studies (Chen et al., 2016b; Kumar et al., 2014), the thermal unimolecular decay of Pa1 can occur via two competitive pathways, namely (i) → CH2O + H2O2 and (ii) → HCOOH + H2O. The barriers of these two pathways in the absence of water and formic acid molecules are 44.7 and 57.9 kcal mol−1, respectively (Kumar et al., 2014). The result reveals that the formaldehyde-forming channel is preferable. The of the formaldehyde-forming channel in the presence of a single formic acid molecule is 18.5 kcal mol−1, which is 12.7 and 26.2 kcal mol−1 lower than the water-catalyzed and uncatalyzed reactions. However, since the corresponding barriers are higher than that of the bimolecular reaction with CH2OO, the thermal unimolecular decay of HHPs is not taken into consideration in this work. Moreover, the bimolecular reaction of Pa1 with the OH radical is investigated at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory, and the corresponding PES is shown in Fig. S5. As seen from Fig. S5, the hydrogen abstraction reactions between OH and Pa1 are strongly exothermic and spontaneous, indicating that the occurrence of these reactions in the atmosphere is thermochemically feasible. One can notice that the exothermic reaction RHγ releasing energy is significantly higher than that of RHα and RHβ. The pre-reactive complexes (IMαH-a, βH-a, and γH-a) are formed in the entrance channel and are followed by direct hydrogen abstraction processes leading to the products PHα, PHβ, and PHγ plus H2O. The barriers predict TSHα, TSHβ, and TSHγ to be 6.8, 4.8, and 4.4 kcal mol−1 above the energies of the corresponding pre-reactive complexes IMαH-a, βH-a, and γH-a. The result again shows that the reaction RHγ is the most energetically favorable channel. One can notice that the barrier of RHγ is lower than that of the corresponding addition reaction R1a 4.0 kcal mol−1, indicating that the photochemical oxidation of HHPs is important in the atmosphere. In the present study, we mainly focus on the gas-phase reaction mechanism and kinetics of oligomer formation from carbonyl oxide reactions with HHPs. This is because the type of reaction studied is very important for understanding the first step of new particle formation form alkene ozonolysis, particularly in heavily forested areas.
The secondary addition reaction CH2OO + P1a is equivalent to the CH2OO + Pa1 reaction and hence features an analogous pathway, i.e., the formation of pre-reactive complexes IM2a and IM2b in entrance channels, followed by the addition of −OH and −OOH groups of P1a to the CH2OO central carbon atom to produce P2a and P2b. The barrier heights predict TS2a and TS2b to be −36.3 and −35.9 kcal mol−1, respectively, below the energies of the separate reactants and 7.1 and 7.0 kcal mol−1 above the energies of the corresponding pre-reactive complexes IM2a and IM2b. The above result shows that these two addition reactions (R2a and R2b) equally contribute to the title reaction system. Compared to the first CH2OO addition reaction, the second one features a lower barrier. Finally, the addition reaction CH2OO + P1b proceeds via a mechanism fairly similar to those described above for the CH2OO + P1a system and is not discussed in detail to avoid redundancy.
3.3 PES for the reaction of CH3CHOO with
The methyl-substituted Criegee intermediate can exist in two conformations, syn- and anti-CH3CHOO, depending on whether the methyl group is located on the same or opposite side of the terminal oxygen (Long et al., 2016). Numerous theoretical studies have proven that the presence of an intramolecular hydrogen bond in the syn-conformer makes it more stable than the anti-conformer (Anglada et al., 2011; Anglada and Solé 2016). The interconversion of these two conformers via rotation around the C−O bond has a very high barrier (∼42 kcal mol−1), which implies that one can treat syn- and anti-CH3CHOO as independent species existing in the atmosphere (Yin and Takahashi, 2017). Long et al. (2016) proposed that the predominant pathway of the unimolecular decay of syn-CH3CHOO is isomerization to vinyl hydroperoxide (VHP) via hydrogen atom migration from the methyl group to the terminal oxygen atom, then the decomposition of VHP produces the OH radical. Also, Donahue et al. (2011a) obtained a similar conclusion that syn-CI isomerization to VHP is preferable due to the low ring strain of the H-atom transfer transition state. Both the prompt decay and thermal unimolecular decay of the energized VHP may dissociate to the OH radical, and their yields are strongly pressure and temperature dependent (Kroll et al., 2001a, b). The preferable route of unimolecular decay of anti-CH3CHOO is ring closure to dioxirane via an oxygen atom transfer (Donahue et al., 2011a; Taatjes et al., 2013; Long et al., 2016). The dioxirane can finally isomerize to acetic acid via the “hot acid” channel (Kroll et al., 2001b). Alternatively, syn- and anti-CH3CHOO may undergo bimolecular reactions with water vapor, leading to the formation of (Anglada et al., 2011). The energy diagram of addition reactions between CH3CHOO and is given in Fig. 3. The optimized geometries of all stationary points are shown in Figs. S6 and S7.
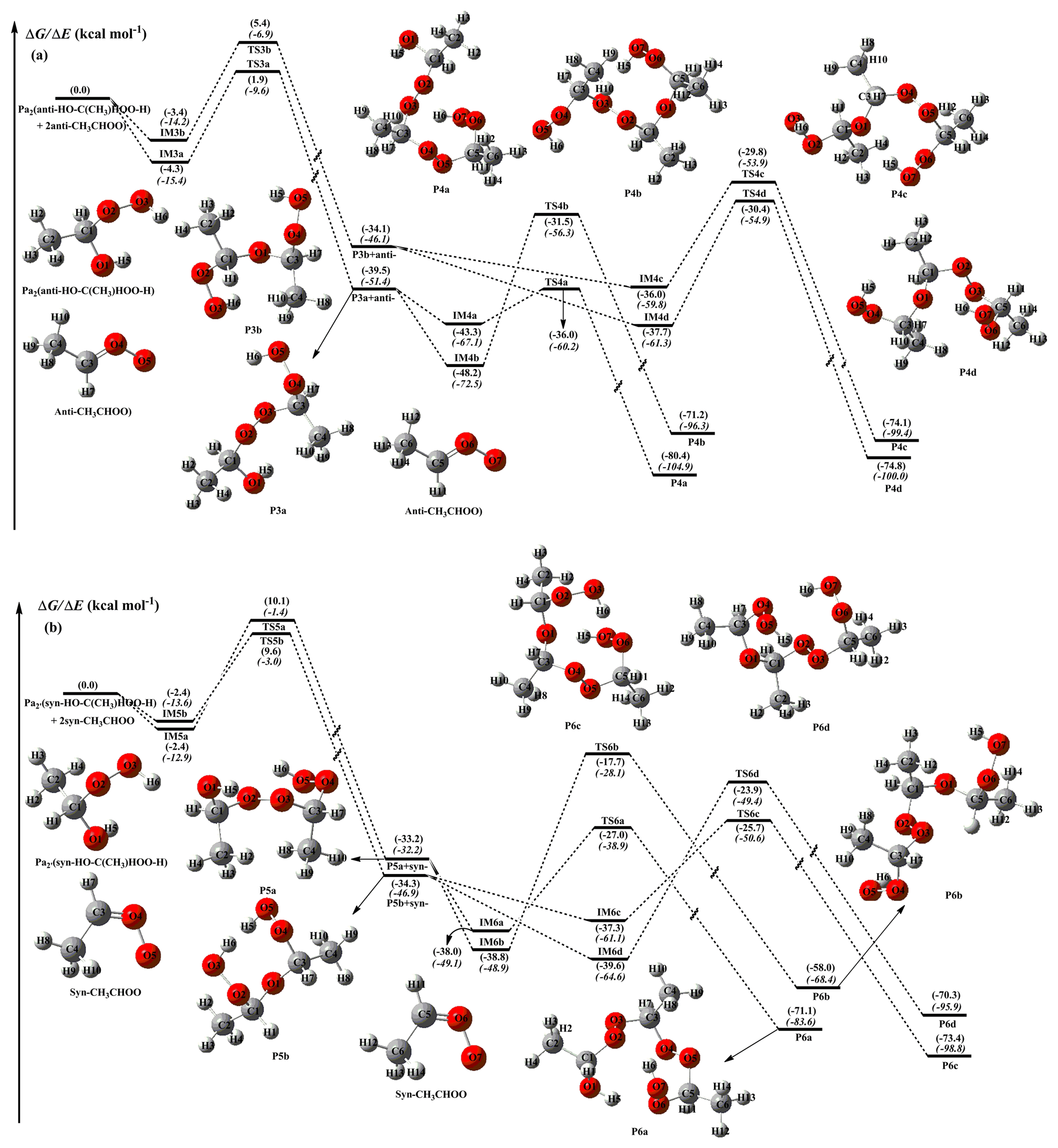
Figure 3PES (ΔG and ΔE, in italics) for the reactions of with anti- (a) and syn-CH3CHOO (b) calculated at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
Figure 3a demonstrates that the sequential additions of anti-CH3CHOO to Pa2 are strongly exothermic and spontaneous, indicating that the occurrence of these consecutive reactions in the atmosphere is thermochemically feasible. The addition reactions of 2-anti-CH3CHOO + Pa2 start with the barrierless formation of pre-reactive complexes IM3a and IM3b held together by weak hydrogen bonds and vdW forces. Subsequently, the −OH and −OOH fragments in Pa2 immediately add to the central carbon atom of anti-CH3CHOO to produce P3a and P3b. The barriers of these two addition reactions are 5.8 and 8.3 kcal mol−1 with the concomitant release of 39.5 and 34.1 kcal mol−1 of energy. This result confirms that the most favorable channel, both thermochemically and dynamically, corresponds to the −OOH group addition pathway. Notably, the above reaction barriers are lower than that of the 2CH2OO + Pa1 system by ∼3.0 kcal mol−1, indicating that anti-CH3CHOO is significantly more reactive than the parent carbonyl oxide. This finding is further corroborated by the results of Anglada and Solé (2016) and Chen et al. (2017), who found that CH2OO is significantly less reactive than anti-CH3CHOO towards H2O, HCOOH, and CH3COOH.
The addition reaction anti-CH3CHOO + Pa2 results in the formation of P3a, which can subsequently react with anti-CH3CHOO via channels R4a and R4b. Both of these pathways start with the formation of pre-reactive complexes IM4a and IM4b in entrance channels, followed by the addition of −OH and −OOH groups of P3a to the central carbon atom of anti-CH3CHOO to produce P4a and P4b. According to the predicted barrier heights, TS4a and TS4b are 6.8 and 16.0 kcal mol−1 above complexes IM4a and IM4b, respectively, which reconfirms that the most favorable reaction channel is the −OOH group addition pathway.
At this point, it is worth noting that the addition reactions in the 2-syn-CH3CHOO + system proceed through a similar mechanism and are thus only briefly discussed in the following section. As revealed by Fig. 3b, both R5a and R5b pathways start with the formation of vdW complexes IM5a and IM5b, which are spontaneously converted into products P5a and P5b. The barriers of these two addition reactions are estimated as 12.1 and 11.5 kcal mol−1, respectively, and are therefore significantly higher than those calculated for the comparable 2-anti-CH3CHOO + Pa2 system. This discrepancy is ascribed to the fact that the steric repulsion between the methyl group and the terminal oxygen in syn-CH3CHOO results in decreased hydrogen transfer ability and hinders the formation of pre-reactive complexes.
The above result is further supported by recent reports, which claim the syn-conformer to be substantially less reactive than the anti-conformer toward key atmospheric species, such as H2O, SO2, and NO2 (Taatjes et al., 2013; Anglada et al., 2011; Sheps et al., 2014). The lowest-energy channel among the P5a–P5b + syn-CH3CHOO reaction pathways, TS6a, involves the addition of the −OOH group with a barrier of only 10.1 kcal mol−1 and a large exothermicity of ∼40 kcal mol−1.
3.4 PES for the reaction of (CH3)2COO with
The dimethyl-substituted Criegee intermediate, (CH3)2COO, is generated in the ozonolysis of 2,3-dimethyl-2-butene (Lester and Klippenstein, 2018; Drozd et al., 2017). Long et al. (2018) proposed that the unimolecular decay of (CH3)2COO is the predominant pathway above 240 K, whereas it can compete with the reaction (CH3)2COO + SO2 below 240 K. Drozd et al. (2017) elucidated the fact that tunneling for both the thermal and prompt unimolecular decay of (CH3)2COO is significant. Also, Lester and Klippenstein (2018) obtained a similar conclusion in the unimolecular decay of (CH3)2COO to the OH radical reaction that the contribution of tunneling to unimolecular decay rates is significant. Alternatively, (CH3)2COO may react with water vapor, leading to the formation of (Anglada and Solé, 2016). The PES of addition reactions (CH3)2COO + is given in Fig. 4, and the optimized geometries of all stationary points are shown in Fig. S8.

Figure 4PES (ΔG and ΔE, in italics) for the reaction of (CH3)2COO with (Pa3) calculated at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
As shown in Fig. 4, the vdW complexes IM7a and IM7b are 4.6 and 4.5 kcal mol−1 lower in energy than the reactants, while the corresponding transition states TS7a and TS7b leading to products P7a and P7b are 9.9 and 13.6 kcal mol−1 higher in energy than the respective complexes. The formations of P7a and P7b are strongly exothermic, with reaction energies of 31.0 and 25.8 kcal mol−1. Again, this result shows that the addition of the −OOH group in Pa3 to the central carbon atom of (CH3)2COO is both thermochemically and dynamically favorable. Compared with the barriers of the 2-anti-CH3CHOO + Pa2 system given in Fig. 3a, one can notice that the dimethyl-substituted parent carbonyl oxide leads to the barrier increasing by ∼4 kcal mol−1. The secondary addition reaction (CH3)2COO + P7a is found to be similar to that described for the (CH3)2COO + Pa3 system. The pre-reactive complexes IM8a and IM8b are formed in entrance channels with over 4.5 kcal mol−1 stabilization energies, followed by the addition of −OH and −OOH groups in P7a to the central carbon atom of (CH3)2COO to generate P8a and P8b. According to the predicted barrier heights, TS8a and TS8b are 11.5 and 12.5 kcal mol−1 above complexes IM8a and IM8b, respectively, which shows that the second addition reactions R8a and R8b are nearly equally accessible.
3.5 PES of distinct SCI reactions with
To gain deeper insights into the substituent-influenced modification of atmospheric oligomer composition, one should elucidate the origin of the substituent influence on the reactivity and kinetics of carbonyl oxides. Therefore, an understanding of structure–reactivity relationships is important for determining bimolecular processes and reaction products. Since the addition of the −OOH group to the central carbon atom of SCIs is shown to be both thermochemically and dynamically preferable, this type of addition reaction is selected to study the effect of substituents on the reactivity of carbonyl oxides. The PES of addition reaction SCIs + is given in Fig. 5, whereas those for bimolecular reactions with other HHPs are displayed in Figs. S9–S11.
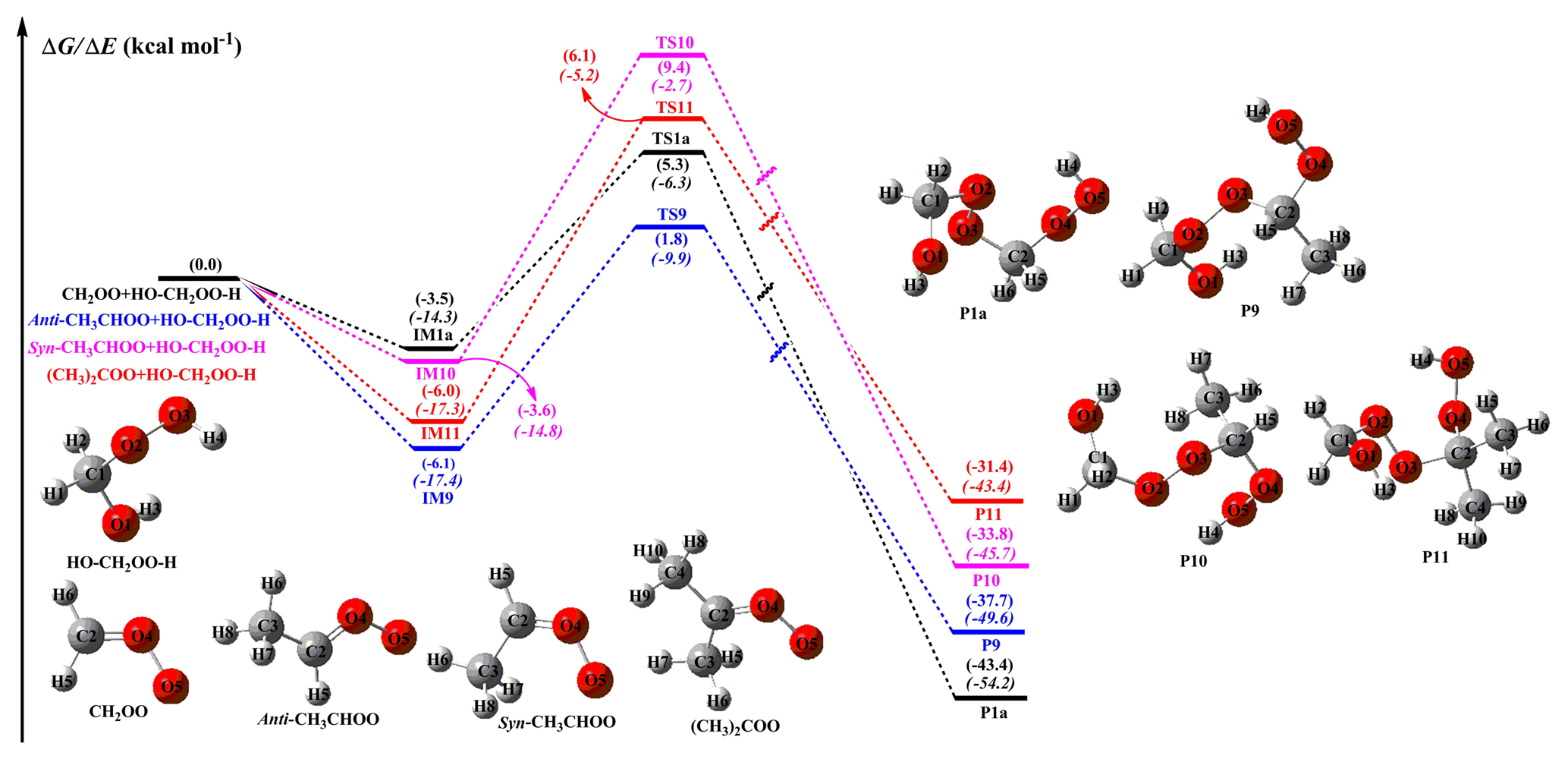
Figure 5PES (ΔG and ΔE, in italics) of distinct SCI reactions with (Pa1) calculated at the M06-2X/def2-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
As shown in Fig. 5, each reaction begins with the formation of a strong pre-reactive complex and then surmounts a medium barrier that is higher in energy relative to the reactants before forming the corresponding products. The bimolecular reaction of CH2OO with to form P1a is characterized by a barrier of 8.4 kcal mol−1 and an exothermicity of 43.4 kcal mol−1. Hence, the small barrier and large stability of the hydroperoxide species imply that its formation is both thermochemically and kinetically favored. Notably, the introduction of a methyl group at the anti-position reduces the barrier by ∼1.0 kcal mol−1 relative to that of the CH2OO + system, whereas the corresponding syn- and dimethyl substitutions increase the above barrier by 4.2 and 3.2 kcal mol−1, respectively. These results indicate that the anti-conformer is substantially more reactive toward than syn-, dimethyl, and parent conformers in the atmosphere. A similar conclusion has been obtained by studying the reactions of syn- and anti-CH3CHOO with water and SO2; i.e., the rate coefficient of the anti-CH3CHOO reaction was calculated to be 1 to 2 orders of magnitude higher than that of the syn-CH3CHOO system (Taatjes et al., 2013; Lin et al., 2016; Huang et al., 2015; Anglada and Solé, 2016). Therefore, it is concluded that the position and number of methyl groups significantly affect barrier heights and reaction rates. On the other hand, the exothermicities of other reaction pathways are lower than that of the parent system, which implies that methyl substitution is thermochemically unfavorable. A similar trend is observed for the bimolecular reactions of SCIs with other HHPs (Figs. S9–S11). In order to avoid redundancy, we do not repeat them here in detail.
3.6 Kinetics and implications in atmospheric chemistry
To better understand the effect of substituents on reaction kinetics, the rate coefficients of distinct SCI reactions with (Pa1) are computed using a combination of canonical transition state theory (CTST) and an asymmetric Eckart tunneling correction based on free energies obtained at the M06-2X level in the temperature range from 273 to 400 K. And the different conformers of stationary points in the kinetics study are taken into account, with the results listed in Tables 2 and S5–S8. In order to assess the reliability of our kinetics study, the rate coefficients of some selected reactions (R1a and R9) are recomputed employing the canonical variational transition state theory (CVTST) with Eckart tunneling correction. The calculated result is listed in Table S9. As shown in Table S9, the difference between kCTST(R1a) and kCVTST(R1a) decreases in the range of 2.3 (273 K) to 1.8 (400 K). Such rate coefficient discrepancies between CTST and CVTST are acceptable. A similar conclusion is also obtained from the rate coefficients between kCTST(R9) and kCVTST(R9). It is concluded that the CTST–Eckart method allows one to reliably describe the kinetics parameters.
Table 2Rate coefficients (cm3 molecule−1 s−1) of distinct SCIs reactions with HO–CH2OO–H(Pa1) computed at different temperatures.
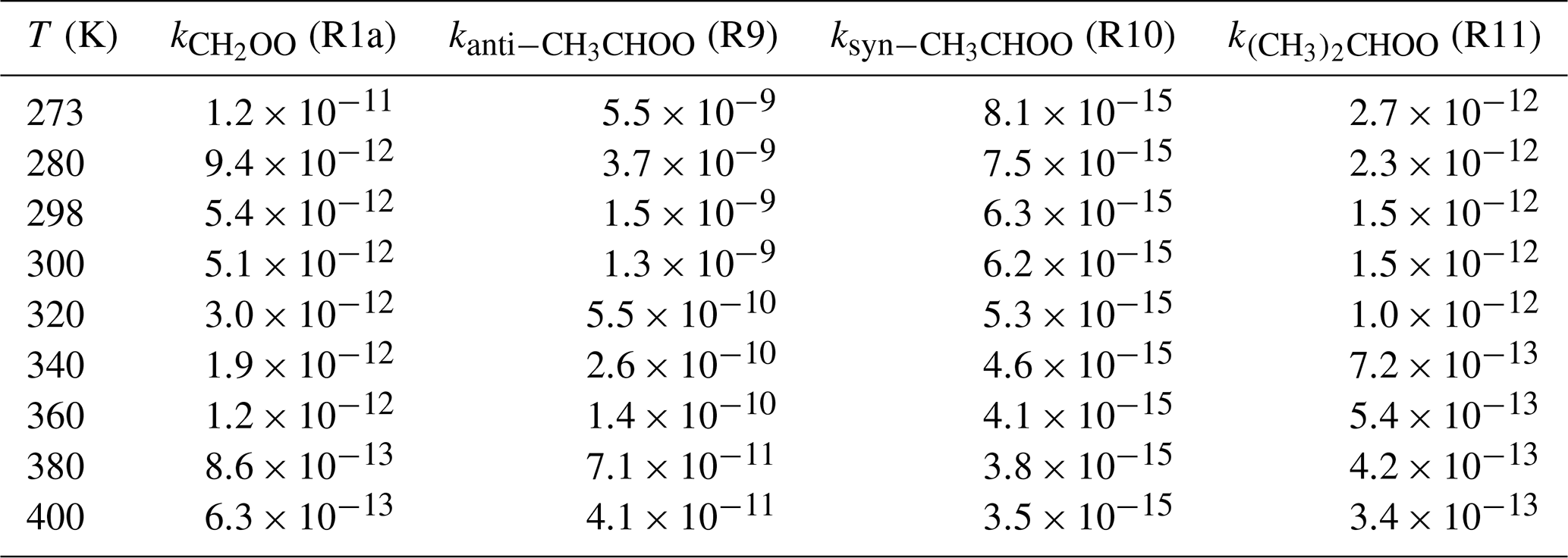
Table 2 shows that the predicted rate coefficients for the reaction of CH2OO with (R1a) decrease with increasing temperature, with a similar trend observed for the syn-CH3CHOO(R9), anti-CH3CHOO(R10), and (CH3)2COO + (R11) systems. The above behavior is ascribed to the fact that the apparent activation barriers Eapp of these four addition reactions are significantly negative, as previously observed for the reaction of CH3O2 with BrO (Shallcross et al., 2015). As shown in Table S5, the rate coefficients of CH2OO + P1a, CH2OO + P1b, CH2OO + P1c, and CH2OO + P1d reactions decrease with increasing temperature, indicating that oligomer formation from CH2OO reactions with HHP is preferable under low temperature conditions. Similar conclusions are also obtained from the anti-CH3CHOO + P3x (Table S6), syn-CH3CHOO + P5x (Table S7), and (CH3)2CHOO + P7x (Table S8) systems. In order to avoid redundancy, we do not repeat them here in detail. Considering the errors of the computed free-energy barriers, the uncertainty in the rate coefficient is estimated within an order of magnitude.
The obtained data show that the rate coefficient depends on the relative position and number of methyl groups in the parent carbonyl oxide; e.g., the rate coefficient increases by 2 orders of magnitude when methyl substitution occurs at the anti-position, whereas a reduction by 4–6 orders of magnitude is observed for methyl substitution at the syn-position. Thus, the relative position of the methyl group plays an important role in determining SCI reactivity; in particular, anti-substitution promotes reaction with HHPs and accelerates the formation of oligomers in the atmosphere. Anglada et al. (2011) and Anglada and Solé (2016) arrived at the same conclusion by studying the reactions of SCIs with water vapor, showing that the anti-conformer is significantly more reactive than the parent carbonyl oxide and the syn-conformer. On the other hand, the introduction of two methyl groups does not result in a marked rate coefficient change compared to the parent system, since the addition reaction R11 is mediated by the pre-reactive hydrogen-bonded complex.
As discussed above, the anti-CH3CHOO + (Pa1) reaction (R9) is preferred over the other three pathways (R1a, R10, and R11). Therefore, it would be interesting to investigate whether the anti-CH3CHOO + Pa1 reaction can compete well with the anti-CH3CHOO + (H2O)2 (R12) system because the latter reaction is the dominant chemical sink (Taatjes et al., 2013; Anglada and Solé, 2016). The ratio of the reaction rates of R9 and R12 is expressed as follows.
The room temperature rate coefficient kR9 is cm3 molecule−1 s−1, assuming that the concentration of Pa1 is approximately equal to that of SCIs ( molecules cm−3, within an order of magnitude uncertainty) in the boreal forest and rural environments of Finland and Germany (Novelli et al., 2017). The atmospheric lifetime of anti-CH3CHOO reactivity toward Pa1 can be estimated as 1.3–13×103 s. The experimental rate coefficient of reaction R12 approximately equals cm3 molecule−1 s−1 at 298 K (Lin et al., 2016). The concentration of water dimer is 5.5×1013 molecules cm−3 at 3 km of altitude (Long et al., 2016). The atmospheric lifetime of anti-CH3CHOO reactivity toward water dimer is s. The result implies that the reactions studied are minor loss processes in the atmosphere. However, [(H2O)2] decreases significantly with increasing altitude (Long et al., 2016). For example, at 15 km of altitude, [(H2O)2] is 2.7×106 molecules cm−3, the atmospheric lifetime of anti-CH3CHOO is 3.4×104 s. As discussed above, the anti-CH3CHOO + Pa1 reaction can compete with the anti-CH3CHOO + (H2O)2 system in some regions where the altitude is above 15 km.
The νR9∕νR12 ratio is less than 1.4 % when [(H2O)2] is ∼1013 molecules cm−3, meaning that the anti-CH3CHOO + Pa1 reaction is a minor loss process in the atmosphere. However, [(H2O)2] is very low at the altitude above 15 km, and the anti-CH3CHOO + Pa1 reaction can compete well with the anti-CH3CHOO + (H2O)2 reaction, thus contributing to the formation and growth of SOA. Kumar et al. (2014) proposed that the gas-phase decomposition of Pa1 has two competitive pathways: (i) → CH2O+H2O2 and (ii) → HCOOH + H2O. The values in the presence of a single water molecular are 31.2 and 47.8 kcal mol−1, respectively, which are 13.5 and 10.2 kcal mol−1 lower than the uncatalyzed reactions. The result reveals that the formaldehyde-forming channel is preferable in the absence and presence of water molecules, and the role of water catalysis on the gas-phase decomposition of Pa1 is significant. The of the bimolecular reaction of anti-CH3CHOO with Pa1 is 7.3 kcal mol−1, which is 23.9 kcal mol−1 lower than the formaldehyde-forming channel. It is concluded that the Pa1 + H2O reaction is less competitive compared to the anti-CH3CHOO + Pa1 system.
The gas-phase reaction mechanism and kinetics of Criegee intermediate reactions with HHPs are studied using quantum chemical methodologies in conjunction with statistical theory calculations. The main conclusions are summarized as follows.
-
Oligomerization reactions of SCIs with HHPs are strongly exothermic and spontaneous, signifying that consecutive reactions are thermochemically feasible in the atmosphere.
-
The addition of the −OOH group in HHPs to the central carbon atom of SCIs is both thermochemically and dynamically preferable compared with the −OH group addition pathway.
-
The reaction barrier and kinetics strongly depend on both the number of substituents in the Criegee intermediate and its position (syn- or anti-).
-
The rate coefficients show a significant increase when adding a methyl group on the anti-position, whereas they display a dramatic decrease on the syn-position. On the other hand, the addition of the dimethyl group does not cause much variation in the rate coefficients.
All of the research data have been included in the supplement, which are available on the EGU Publications website at https://doi.org/10.5194/acp-19-1-2019 (Fu et al., 2019).
The following information is provided in the Supplement: the barriers for the addition reactions of carbonyl oxides with HHPs, partition function of vibrational mode, AIM properties at the bond-critical points, rate coefficients of CH2OO + P1x, anti-CH3CHOO + P3x, syn-CH3CHOO + P5x, and (CH3)2CHOO + P7x reactions, PESs for addition reactions of CH2OO + (Pb1), syn- and anti-CH3CHOO + , (CH3)2COO + (Pb3), (Pa1)+ OH, SCIs + (Pa2), SCIs + (Pa, and SCIs + (Pa3), optimized geometries, z matrices, and vibrational frequencies of all stationary points. The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-4075-2019-supplement.
LC designed the study. LC and YH wrote the paper. LC performed theoretical calculations. YX, ZS, JC, and WW analyzed the data. All authors reviewed and commented on the paper.
The authors declare that they have no conflict of interest.
This work was supported by the National Key Research and Development Program of China (2016YFA0203000) and the National Natural Science Foundation of China (nos. 41805107, 41573138, and 21473108). It was also partially supported by the Key Project of International Cooperation of the Chinese Academy of Sciences (GJHZ1543), the Research Grants Council of Hong Kong (PolyU 152083/14E), the Open Foundation of State Key Laboratory of Loess and Quaternary Geology (SKLLQG1627), and the Shaanxi Province Postdoctoral Science Foundation (no. 2017BSHEDZZ62). Yu Huang is also supported by the “Hundred Talents Program” of the Chinese Academy of Sciences.
This paper was edited by Neil M. Donahue and reviewed by two anonymous referees.
Anglada, J. M. and Solé, A.: Impact of water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 18, 17698–17712, https://doi.org/10.1039/c6cp02531e, 2016.
Anglada, J. M., Aplincourt, P., Bofill, J. M., and Cremer, D.: Atmospheric formation of OH radicals and H2O2 from alkene ozonolysis under humid conditions, Chem. Phys. Chem., 2, 215–221, https://doi.org/10.1002/1439-7641(20020215)3:2<215::AID-CPHC215>3.0.CO;2-3, 2002.
Anglada, J. M., González, J., and Torrent-Sucarrat, M.: Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 13, 13034–13045, https://doi.org/10.1039/c1cp20872a, 2011.
Aplincourt, P. and Anglada, J. M.: Theoretical studies of the isoprene ozonolysis under tropospheric conditions. 2. unimolecular and water-assisted decomposition of the α-hydroxy hydroperoxides, J. Phys. Chem. A, 107, 5812–5820, https://doi.org/10.1021/jp034203w, 2003.
Aplincourt, P. and Ruiz-López, M. F.: Theoretical investigation of reaction mechanisms for carboxylic acid formation in the atmosphere, J. Am. Chem. Soc., 122, 8990–8997, https://doi.org/10.1021/ja000731z, 2000.
Becker, K. H., Brockmann, K. J., and Bechara, J.: Production of hydrogen peroxide in forest air by reaction of ozone with terpenes, Nature, 346, 256–258, https://doi.org/10.1038/346256a0, 1990.
Berndt, T., Jokinen, T., Mauldin, R. L., Petäjä, T., Herrmann, H., Junninen, H., Paasonen, P., Worsnop, D. R., and Sipilä, M.: Gas-phase ozonolysis of selected olefins: the yield of stabilized Criegee intermediate and the reactivity toward SO2, J. Phys. Chem. Lett., 3, 2892–2896, https://doi.org/10.1021/jz301158u, 2012.
Berndt, T., Voigtländer, J., Stratmann, F., Junninen, H., Mauldin, I. R. L., Sipilä, M., Kulmala, M., and Herrmann, H.: Competing atmospheric reactions of CH2OO with SO2 and water vapour, Phys. Chem. Chem. Phys., 16, 19130–19136, https://doi.org/10.1039/c4cp02345e, 2014.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion productformation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Ed., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Biegler-König, F., Schönbohm, J., Derdau, R., Bayles, D., and Bader, R. F. W.: AIM2000, Version 1.0, McMaster University, 2000.
Bonn, B., Kulmala, M., Riipinen, I., Sihto, S. L., and Ruuskanen, T. M.: How biogenic terpenes govern the correlation between sulfuric acid concentrations and new particle formation, J. Geophys. Res.-Atmos., 113, D12209, https://doi.org/10.1029/2007JD009327, 2008.
Boys, S. F. and Bernardi, F.: The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 19, 553–566, https://doi.org/10.1080/00268977000101561, 1970.
Canneaux, S., Bohr, F., and Henon, E.: KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 35, 82–93, https://doi.org/10.1002/jcc.23470, 2014.
Chao, W., Hsieh, J. T., Chang, C. H., and Lin, J. J. M.: Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 347, 751–754, https://doi.org/10.1126/science.1261549, 2015.
Chen, L., Wang, W. L., Wang, W. N., Liu, Y. L., Liu, F. Y., Liu, N., and Wang, B. Z.: Water-catalyzed decomposition of the simplest Criegee intermediate CH2OO, Theor. Chem. Acc., 135, 131–143, https://doi.org/10.1007/s00214-016-1894-9, 2016a.
Chen, L., Wang. W. L., Zhou, L. T., Wang, W. N., Liu, F. Y., Li, C. Y., and Lü, J.: Role of water clusters in the reaction of the simplest Criegee intermediate CH2OO with water vapour, Theor. Chem. Acc., 135, 252–263, https://doi.org/10.1007/s00214-016-1998-2, 2016b.
Chen, L., Huang, Y., Xue, Y., Cao, J. J., and Wang, W.: Competition between HO2 and H2O2 reactions with CH2OO/anti-CH3CHOO in the oligomer formation: a theoretical perspective, J. Phys. Chem. A, 121, 6981–6991, https://doi.org/10.1021/acs.jpca.7b05951, 2017.
Chen, L., Huang, Y., Xue, Y., Cao, J. J., and Wang, W.: Effect of oligomerization reactions of Criegee intermediate with organic acid/peroxy radical on secondary organic aerosol formation from isoprene ozonolysis, Atmos. Environ., 187, 218–229, https://doi.org/10.1016/j.atmosenv.2018.06.001, 2018.
Cremer, D., Kraka, E., and Sosa, C.: First evidence for the production of OH radicals by carbonyl oxides in solution phase-A DFT investigation, Chem. Phys. Lett., 337, 199–208, https://doi.org/10.1016/S0009-2614(01)00177-4, 2001.
Criegee, R.: Mechanism of ozonolysis, Angew. Chem. Int. Ed. Engl., 14, 745–752, https://doi.org/10.1002/anie.197507451, 1975.
Donahue, N. M., Drozd, G. T., Epstein, S. A., Presto, A. A., and Kroll, J. H.: Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 13, 10848–10857, https://doi.org/10.1039/c0cp02564j, 2011a.
Donahue, N. M., Trump, E. R., Pierce, J. R., and Riipinen, I.: Theoretical constraints on pure vapor-pressure driven condensation of organics to ultrafine particles, Geophys. Res. Lett., 38, L16801, https://doi.org/10.1029/2011GL048115, 2011b.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Drozd, G. T., Kurtén, T., Donahue, N. M., and Lester, M. I.: Unimolecular decay of the dimethyl-substituted Criegee intermediate in alkene ozonolysis: decay time scales and the importance of tunneling, J. Phys. Chem. A, 121, 6036–6045, https://doi.org/10.1021/acs.jpca.7b05495, 2017.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Enami, S. and Colussi, A. J.: Reactions of Criegee intermediates with alcohols at air-aqueous interfaces, J. Phys. Chem. A, 121, 5175–5182, https://doi.org/10.1021/acs.jpca.7b04272, 2017.
Foreman, E. S., Kapnas, K. M., and Murray, C.: Reactions between Criegee intermediates and the inorganic acids HCl and HNO3: kinetics and atmospheric implications, Angew. Chem. Int. Ed., 55, 10419–10422, https://doi.org/10.1002/anie.201604662, 2016.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery Jr., J. A., Vreven, T., Kudin, K. N., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci. B., Cossi, M., Scalmani, G., Rega, N., Petersson, G. A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J. E., Hratchian, H. P., Cross, J. B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Ayala, P. Y., Morokuma, K., Voth, G. A., Salvador, P., Dannenberg, J. J., Zakrzewski, V. G., Dapprich, S., Daniels, A. D., Strain, M. C., Farkas, O., Malick, D. K., Rabuck, A. D., Raghavachari, K., Foresman, J. B., Ortiz, J. V., Cui, Q., Baboul, A. G., Clifford, S., Cioslowski, J., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R. L., Fox, D. J., Keith, T., Al-Laham, M. A., Peng, C. Y., Nanayakkara, A., Challacombe, M., Gill, P. M. W., Johnson, B., Chen, W., Wong, M. W., Gonzalez, C., and Pople, J. A.: Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009.
Fu, X., Wang, T., Zhang, L., Li, Q., Wang, Z., Xia, M., Yun, H., Wang, W., Yu, C., Yue, D., Zhou, Y., Zheng, J., and Han, R.: The significant contribution of HONO to secondary pollutants during a severe winter pollution event in southern China, Atmos. Chem. Phys., 19, 1–14, https://doi.org/10.5194/acp-19-1-2019, 2019.
Fukui, K.: The path of chemical reactions – the IRC approach, Acc. Chem. Res., 14, 363–368, https://doi.org/10.1021/ar00072a001, 1981.
Gligorovski, S., Strekowski, R., Barbati, S., and Vione, D.: Environmental implications of hydroxyl radicals (⋅ OH), Chem. Rev., 115, 13051–13092, https://doi.org/10.1021/cr500310b, 2015.
Green, A. M., Barber, V. P., Fang, Y., Klippenstein, S. J., and Lester, M. I.: Selective deuteration illuminates the importance of tunneling in the unimolecular decay of Criegee intermediates to hydroxyl radical products, Proc. Natl. Acad. Sci. USA, 114, 12372–12377, https://doi.org/10.1073/pnas.1715014114, 2017.
Guenther, A., Geron, C., Pierce, T., Lamb, B., Harley, P., and Fall, R.: Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America, Atmos. Environ., 34, 2205–2230, https://doi.org/10.1016/S1352-2310(99)00465-3, 2000.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heaton, K. J., Dreyfus, M. A., Wang, S., and Johnston, M. V.: Oligomers in the early stage of biogenic secondary organic aerosol formation and growth, Environ. Sci. Technol., 41, 6129–6136, https://doi.org/10.1021/es070314n, 2007.
Heine, N., Houle, F. A., and Wilson, K. R.: Connecting the elementary reaction pathways of Criegee intermediates to the chemical erosion of squalene interfaces during ozonolysis, Environ. Sci. Technol., 51, 13740–13748, https://doi.org/10.1021/acs.est.7b04197, 2017.
Huang, H. L., Chao, W., and Lin, J. J. M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, Proc. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
Huang, R. J., Zhang, Y., Bozzetti, C., Ho, K. F., Cao, J. J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Inomata, S., Sato, K., Hirokawa, J., Sakamoto, Y., Tanimoto, H., Okumura, M., Tohno, S., and Imamura, T.: Analysis of secondary organic aerosols from ozonolysis of isoprene by proton transfer reaction mass spectrometry, Atmos. Environ., 97, 397–405, https://doi.org/10.1016/j.atmosenv.2014.03.045, 2014.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, Proc. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Ji, Y., Zheng, J., Qin, D., Li, Y., Gao, Y., Yao, M., Chen, X., Li, G., An, T., and Zhang, R.: OH-initiated oxidation of acetylacetone: implications for ozone and secondary organic aerosol formation, Environ. Sci. Technol., 52, 11169–11177, https://doi.org/10.1021/acs.est.8b03972, 2018.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/b704260b, 2008.
Kidwell, N. M., Li, H., Wang, X., Bowman, J. M., and Lester, M. I.: Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products, Nat. Chem., 8, 509–514, https://doi.org/10.1038/NCHEM.2488, 2016.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, Martin., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Clarke, J. S., Donahue, N. M., and Anderson, J. G.: Mechanism of HOx formation in the gas-phase ozone-alkene reaction. 1. direct, pressure-dependent measurements of prompt OH yields, J. Phys. Chem. A, 105, 1554–1560, https://doi.org/10.1021/jp002121r, 2001a.
Kroll, J. H., Hanisco, T. F., Donahue, N. M., Demerjian, K. L., and Anderson, J. G.: Accurate, direct measurements of OH yields from gas-phase ozone-alkene reactions using an in situ LIF instrument, Geophys. Res. Lett., 28, 3863–3866, https://doi.org/10.1029/2001GL013406, 2001b.
Kumar, M., Busch, D. H., Subramaniam, B., and Thompson, W. H.: Role of tunable acid catalysis in decomposition of α-hydroxyalkyl hydroperoxides and mechanistic implications for tropospheric chemistry, J. Phys. Chem. A, 118, 9701–9711, https://doi.org/10.1021/jp505100x, 2014.
Kumar, M., Zhong, J., Francisco, J. S., and Zeng, X. C.: Criegee intermediate-hydrogen sulfide chemistry at the air/water interface, Chem. Sci., 8, 5385–5391, https://doi.org/10.1039/c7sc01797a, 2017.
Kumar, M., Zhong, J., Zeng, X. C., and Francisco, J. S.: Reaction of Criegee intermediate with nitric acid at the air-water interface, J. Am. Chem. Soc., 140, 4913–4921, https://doi.org/10.1021/jacs.8b01191, 2018.
Kuwata, K. T., Guinn, E. J., Hermes, M. R., Fernandez, J. A., Mathison, J. M., and Huang, K.: A computational re-examination of the Criegee intermediate-sulfur dioxide reaction, J. Phys. Chem. A, 119, 10316–10335, https://doi.org/10.1021/acs.jpca.5b06565, 2015.
Lester, M. I. and Klippenstein, S. J.: Unimolecular decay of Criegee intermediates to OH radical products: prompt and thermal decay processes, Acc. Chem. Res., 51, 978–985, https://doi.org/10.1021/acs.accounts.8b00077, 2018.
Lewis, T. R., Blitz, M. A., Heard, D. E., and Seakins, P. W.: Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 17, 4859–4863, https://doi.org/10.1039/c4cp04750h, 2015.
Li, J., Carter, S., Bowman, J. M., Dawes, R., Xie, D., and Guo, H.: High-level, first-principles, full-dimensional quantum calculation of the ro-vibrational spectrum of the simplest criegee intermediate (CH2OO), J. Phys. Chem. Lett., 5, 2364–2369, https://doi.org/10.1021/jz501059m, 2014.
Li, L., Hoffmann, M. R., and Colussi, A. J.: Role of nitrogen dioxide in the production of sulfate during Chinese haze-aerosol episodes, Environ. Sci. Technol., 52, 2686–2693, https://doi.org/10.1021/acs.est.7b05222, 2018.
Li, Y., Gong, Q., Yue, L., Wang, W., and Liu, F.: Photochemistry of the simplest Criegee intermediate, CH2OO: photoisomerization channel toward dioxirane revealed by CASPT2 calculations and trajectory surface-hopping dynamics, J. Phys. Chem. Lett., 9, 978–981, https://doi.org/10.1021/acs.jpclett.8b00023, 2018.
Lin, J. and Chao, W.: Structure-dependent reactivity of Criegee intermediates studied with spectroscopic methods, Chem. Soc. Rev., 46, 7483–7497, https://doi.org/10.1039/C7CS00336F, 2017.
Lin, L. C., Chang, H. T., Chang, C. H., Chao, W., Smith, M. C., Chang, C. H., Lin, J. J., and Takahashi, K.: Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 18, 4557–4568, https://doi.org/10.1039/C5CP06446E, 2016.
Liu, J., Fang, S., Wang, Z., Yi, W., Tao, F. M., and Liu, J. Y.: Hydrolysis of sulfur dioxide in small clusters of sulfuric acid: mechanistic and kinetic study, Environ. Sci. Technol., 49, 13112–13120, https://doi.org/10.1021/acs.est.5b02977, 2015.
Long, B., Bao, J. L., and Truhlar, D. G.: Atmospheric chemistry of Criegee intermediates: unimolecular reactions and reactions with water, J. Am. Chem. Soc., 138, 14409–14422, https://doi.org/10.1021/jacs.6b08655, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere, Proc. Natl. Acad. Sci. USA, 115, 6135–6140, https://doi.org/10.1073/pnas.1804453115, 2018.
Mauldin, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V. M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
Mendes, J., Zhou, C. W., and Curran, H. J.: Theoretical chemical kinetic study of the H-atom abstraction reactions from aldehydes and acids by H atoms and OH, HO2, and CH3 radicals, J. Phys. Chem. A, 118, 12089–12104, https://doi.org/10.1021/jp5072814, 2014.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/C6CP00053C, 2016.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Ouyang, B., McLeod, M. W., Jones, R. L., and Bloss, W. J.: NO3 radical production from the reaction between the Criegee intermediate CH2OO and NO2, Phys. Chem. Chem. Phys., 15, 17070–17075, https://doi.org/10.1039/c3cp53024h, 2013.
Rissanen, M. P., Kurtén,T., Sipila, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Ryzhkov, A. B. and Ariya, P. A.: A theoretical study of the reactions of carbonyl oxide with water in atmosphere: the role of water dimer, Chem. Phys. Lett., 367, 423–429, https://doi.org/10.1016/S0009-2614(02)01685-8, 2003.
Ryzhkov, A. B. and Ariya, P. A.: A theoretical study of the reactions of parent and substituted Criegee intermediates with water and the water dimer, Phys. Chem. Chem. Phys., 6, 5042–5050, https://doi.org/10.1039/B408414D, 2004.
Ryzhkov, A. B. and Ariya, P. A.: The importance of water clusters (H2O)n (n=2, ..., 4) in the reaction of Criegee intermediate with water in the atmosphere, Chem. Phys. Lett., 419, 479–485, https://doi.org/10.1016/j.cplett.2005.12.016, 2006.
Sadezky, A., Winterhalter, R., Kanawati, B., Römpp, A., Spengler, B., Mellouki, A., Le Bras, G., Chaimbault, P., and Moortgat, G. K.: Oligomer formation during gas-phase ozonolysis of small alkenes and enol ethers: new evidence for the central role of the Criegee Intermediate as oligomer chain unit, Atmos. Chem. Phys., 8, 2667–2699, https://doi.org/10.5194/acp-8-2667-2008, 2008.
Sakamoto, Y., Inomata, S., and Hirokawa, J.: Oligomerization reaction of the Criegee intermediate leads to secondary organic aerosol formation in ethylene ozonolysis, J. Phys. Chem. A, 117, 12912–12921, https://doi.org/10.1021/jp408672m, 2013.
Schenker, S., Schneider, C., Tsogoeva, S. B., and Clark, T.: Assessment of popular DFT and semiempirical molecular orbital techniques for calculating relative transition state energies and kinetic product distributions in enantioselective organocatalytic reactions, J. Chem. Theory Comput., 7, 3586–3595, https://doi.org/10.1021/ct2002013, 2011.
Shallcross, D. E., Leather, K. E., Bacak, A., Xiao, P., Lee, E. P. F., Ng, M., Mok, D. K. W., Dyke, J. M., Hossaini, R., Chipperfield, M. P., Khan, M. A. H., and Percival, C. J.: Reaction between CH3O2 and BrO radicals: A new source of upper troposphere lower stratosphere hydroxyl radicals, J. Phys. Chem. A, 119, 4618–4632, https://doi.org/10.1021/jp5108203, 2015.
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 16, 26701–26706, https://doi.org/10.1039/C4CP04408H, 2014.
Smith, M. C., Chang, C. H., Chao, W., Lin, L. C., Takahashi, K., Boering, K. A., and Lin, J. J. M.: Strong negative temperature dependence of the simplest Criegee intermediate CH2OO reaction with water dimer, J. Phys. Chem. Lett., 6, 2708–2713, https://doi.org/10.1021/acs.jpclett.5b01109, 2015.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simonc, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Mazon, S. B., Chen, D., Dias, A., Draper, D. C., Duplissy, J., Haddad, I. E., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, Proc. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Stone, D., Blitz, M., Daubney, L., Howes, N. U. M., and Seakins, P.: Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 16, 1139–1149, https://doi.org/10.1039/c3cp54391a, 2014.
Taatjes, C. A.: Criegee intermediates: what direct production and detection can teach us about reactions of carbonyl oxides, Annu. Rev. Phys. Chem., 68, 183–207, https://doi.org/10.1146/annurev-physchem-052516-050739, 2017.
Taatjes, C. A., Meloni, G., Selby, T. M., Trevitt, A. J., Osborn, D. L., Percival, C. J., and Shallcross, D. E.: Direct observation of the gas-phase criegee intermediate (CH2OO), J. Am. Chem. Soc., 130, 11883–11885, https://doi.org/10.1021/ja804165q, 2008.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO, Science, 340, 177–180, https://doi.org/10.1126/science.1234689, 2013.
Tobias, H. J. and Ziemann, P. J.: Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids, and water with the C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene, J. Phys. Chem. A, 105, 6129–6135, https://doi.org/10.1021/jp004631r, 2001.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R. Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vereecken, L.: The reaction of Criegee intermediates with acids and enols, Phys. Chem. Chem. Phys., 19, 28630–28640, https://doi.org/10.1039/c7cp05132h, 2017.
Wang, M., Yao, L., Zheng, J., Wang, X., Chen, J., Yang, X., Worsnop, D. R., Donahue, N. M., and Wang, L.: Reactions of atmospheric particulate stabilized Criegee intermediates lead to high-molecular-weight aerosol components, Environ. Sci. Technol., 50, 5702–5710, https://doi.org/10.1021/acs.est.6b02114, 2016.
Welz, O., Savee, J. D., Osborn, D. L., Subith, S. V., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, https://doi.org/10.1126/science.1213229, 2012.
Winterhalter, R., Neeb, P., Grossmann, D., Kolloff, A., Horie, O., and Moortgat, G.: Products and mechanism of the gas phase reaction of ozone with ?-pinene, J. Atmos. Chem., 35, 165–197, https://doi.org/10.1023/A:1006257800929, 2000.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the volatility of secondary organic aerosol from isoprene photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Yin, C. and Takahashi, K.: How does substitution affect the unimolecular reaction rates of Criegee intermediates?, Phys. Chem. Chem. Phys., 19, 12075–12084, https://doi.org/10.1039/C7CP01091E, 2017.
Zhang, D. and Zhang, R.: Mechanism of OH formation from ozonolysis of isoprene:? a quantum-chemical study, J. Am. Chem. Soc., 124, 2692–2703, https://doi.org/10.1021/ja011518l, 2002.
Zhang, P., Wang, W. L., Zhang, T. L., Chen, L., Du, Y. M., Li, C. Y., and Lü, J.: Theoretical study on the mechanism and kinetics for the self-reaction of C2H5O2 radicals, J. Phys. Chem. A, 116, 4610–4620, https://doi.org/10.1021/jp301308u, 2012.
Zhang, T., Wang, R., Chen, H., Min, S., Wang, Z., Zhao, C., Xu, Q., Jin, L., Wang, W., and Wang, Z.: Can a single water molecule really affect the hydrogen abstraction reaction of HO2+NO2 under tropospheric conditions? Phys. Chem. Chem. Phys., 17, 15046–15055, https://doi.org/10.1039/C5CP00968E, 2015.
Zhang, W., Du, B., and Qin, Z.: Catalytic effect of water, formic acid, or sulfuric acid on the reaction of formaldehyde with OH radicals, J. Phys. Chem. A, 118, 4797–4807, https://doi.org/10.1021/jp502886p, 2014.
Zhang, X., McVay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, Proc. Natl. Acad. Sci. USA, 117, 14168–14173, https://doi.org/10.1073/pnas.1517742112, 2015.
Zhao, Q., Liu, F., Wang, W., Li, C., Lu, J., and Wang, W.: Reactions between hydroxyl-substituted alkylperoxy radicals and Criegee intermediates: correlations of the electronic characteristics of methyl substituents and the reactivity, Phys. Chem. Chem. Phys., 19, 15073–15083, https://doi.org/10.1039/C7CP00869D, 2017a.
Zhao, Q., Wang, W., Liu, F., Lu, J., and Wang, W.: Oligomerization reactions for precursors to secondary organic aerosol: Comparison between two formation mechanisms for the oligomeric hydroxyalkyl hydroperoxides, Atmos. Environ., 166, 1–8, https://doi.org/10.1016/j.atmosenv.2017.07.008, 2017b.
Zhao, Y. and Truhlar, D. G.: A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 125, 194101–194118, https://doi.org/10.1063/1.2370993, 2006.
Zhao, Y. and Truhlar, D. G.: Density functionals with broad applicability in chemistry, Acc. Chem. Res., 41, 157–167, https://doi.org/10.1021/ar700111a, 2008a.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008b.
Zhao, Y., Wingen, L. M., Perraud, V., Greaves, J., and Finlayson-Pitts, B. J.: Role of the reaction of stabilized Criegee intermediates with peroxy radicals in particle formation and growth in air, Phys. Chem. Chem. Phys., 17, 12500–12514, https://doi.org/10.1039/c5cp01171j, 2015.
Zheng, J. and Truhlar, D. G.: Direct dynamics study of hydrogen-transfer isomerization of 1-pentyl and 1-hexyl radicals, J. Phys. Chem. A, 113, 11919–11925, https://doi.org/10.1021/jp903345x, 2009.
Zhong, J., Kumar, M., Zhu, C. Q., Francisco, J. S., and Zeng, X. C.: Surprising stability of larger Criegee intermediates on aqueous interfaces, Angew. Chem. Int. Ed., 56, 7740–7744, https://doi.org/10.1002/anie.201702722, 2017.
Zhong, J., Kumar, M., Francisco, J. S., and Zeng, X. C.: Insight into chemistry on cloud/aerosol water surfaces, Acc. Chem. Res., 51, 1229–1237, https://doi.org/10.1021/acs.accounts.8b00051, 2018.
Zhu, C., Kumar, M., Zhong, J., Li, L., Francisco, J. S., and Zeng, X. C.: New mechanistic pathways for Criegee-water chemistry at the air/water interface, J. Am. Chem. Soc., 138, 11164–11169, https://doi.org/10.1021/jacs.6b04338, 2016.