the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 Nov 2019
| 28 Nov 2019
Impact of anthropogenic emissions on biogenic secondary organic aerosol: observation in the Pearl River Delta, southern China
Yu-Qing Zhang
Duo-Hong Chen
Tao Zhang
Jun-Qi Wang
Qian Cheng
Hao Jiang
Wei Song
Yu-Bo Ou
Peng-Lin Ye
Gan Zhang
Xin-Ming Wang
Secondary organic aerosol (SOA) formation from biogenic precursors is affected by anthropogenic emissions, which are not well understood in polluted areas. In this study, we accomplished a year-round campaign at nine sites in polluted areas located in the Pearl River Delta (PRD) region during 2015. We measured typical biogenic SOA (BSOA) tracers from isoprene, monoterpenes, and β-caryophyllene, as well as major gaseous and particulate pollutants and investigated the impact of anthropogenic pollutants on BSOA formation. The concentrations of BSOA tracers were in the range of 45.4 to 109 ng m−3 with the majority composed of products from monoterpenes (SOAM, 47.2±9.29 ng m−3), isoprene (SOAI, 23.1±10.8 ng m−3), and β-caryophyllene (SOAC, 3.85±1.75 ng m−3). We found that atmospheric oxidants, Ox (O3 plus NO2), and sulfate correlated well with later-generation SOAM tracers, but this was not the case for first-generation SOAM products. This suggested that high Ox and sulfate levels could promote the formation of later-generation SOAM products, which probably led to the relatively aged SOAM that we observed in the PRD. For the SOAI tracers, both 2-methylglyceric acid (NO/NO2-channel product) and the ratio of 2-methylglyceric acid to 2-methyltetrols (HO2-channel products) exhibit NOx dependence, indicating the significant impact of NOx on SOAI formation pathways. The SOAC tracer was elevated in winter at all sites and was positively correlated with levoglucosan, Ox, and sulfate. Thus, the unexpected increase in SOAC in wintertime might be highly associated with the enhancement of biomass burning, O3 chemistry, and the sulfate component in the PRD. The BSOAs that were estimated using the SOA tracer approach showed the highest concentration in fall and the lowest concentration in spring with an annual average concentration of 1.68±0.40 µg m−3. SOAM dominated the BSOA mass all year round. We also found that BSOA correlated well with sulfate and Ox. This implied a significant effect from anthropogenic pollutants on BSOA formation and highlighted that we could reduce BSOA by controlling the anthropogenic emissions of sulfate and Ox precursors in polluted regions.
- Article
(7424 KB) - Full-text XML
-
Supplement
(607 KB) - BibTeX
- EndNote





Secondary organic aerosols (SOAs) that are produced through homogenous and heterogeneous processes of volatile organic compounds (VOCs) have significant effects on global climate change and regional air quality (Von Schneidemesser et al., 2015). Globally, the emissions of biogenic VOCs (BVOCs) are dominant over anthropogenic VOCs (AVOCs). Thus, biogenic SOA (BSOA) is predominant with respect to anthropogenic SOA. In the past decade, laboratorial, field, and modeling studies have demonstrated that BSOA formation is highly affected by anthropogenic emissions (Zhang et al., 2015; Hoyle et al., 2011; Carlton et al., 2010). Increasing NOx shifts isoprene oxidation from low-NOx conditions to high-NOx conditions (Surratt et al., 2010) and enhances nighttime SOA formation via nitrate radical oxidation of monoterpenes (Xu et al., 2015). High SO2 emission leads to abundant sulfate and acidic particles, which accelerate BSOA production due to the “salting-in” effect and acid-catalyzed reactions (Offenberg et al., 2009; Xu et al., 2016). In polluted regions, the increase in O3 levels due to high emissions of NOx and VOCs likely results in significant SOA formation via the ozonolysis of BVOCs (Sipilä et al., 2014; Riva et al., 2017). In addition, the large emission and formation of anthropogenic organic matter (OM) in urban areas enhance the incorporation of BVOC oxidation products into the condensed phase (Donahue et al., 2006). Recently, Carlton et al. (2018) found that the removal of anthropogenic emissions of NOx, SO2, and primary OA in the Community Multiscale Air Quality Model (CMAQ) simulations could reduce BSOA by 23 %, 14 %, and 8 % in summertime, respectively.
The Pearl River Delta (PRD) region (Fig. 1a) is the most developed area in China. Rapid economic growth in this region during the past 3 decades has resulted in high anthropogenic emissions (Lu et al., 2013). Our observation during the fall–winter season in 2008 at a regional site in the PRD showed that daily PM2.5 was as high as 150 µg m−3 (Ding et al., 2012). Fortunately, due to increasingly strict and effective pollution controls in the PRD, PM2.5 concentrations have significantly decreased over the last decade and have met the national ambient air quality standard (NAAQS) for annual mean PM2.5 (35 µg m−3) since the year 2015 (Figure 1b). However, O3 and oxidant species (Ox, Ox = O3 + NO2) are still at high levels and have shown no apparent decrease (Fig. 1b). Hofzumahaus et al. (2009) observed extremely high OH concentrations in the PRD and proposed a recycling mechanism that increases the stability of OH in the air of polluted regions. All of the abovementioned results indicate a high atmospheric oxidative capacity in the PRD, as O3, NOx, and OH are intimately linked in atmospheric chemistry. Moreover, BVOC emissions in the PRD are expected to be high year-round due to the area's subtropical climate (Zheng et al., 2010). During the process of such dramatic changes in air pollution characteristics (e.g., PM2.5 and O3), BSOA origins and formation mechanisms in the PRD should have been profoundly affected over the last decade.
In this study, year-round PM2.5 samples were collected at nine sites in the PRD during 2015. We investigated SOA tracers from typical BVOCs (isoprene, monoterpenes, and β-caryophyllene) across the PRD for the first time. We checked seasonal variations in the concentrations and compositions of these BSOA tracers and evaluated the impact of anthropogenic pollutants on BSOA formation in the PRD. We also accessed the SOA origins and discussed the possibility of further reducing BSOA by controlling anthropogenic emissions.
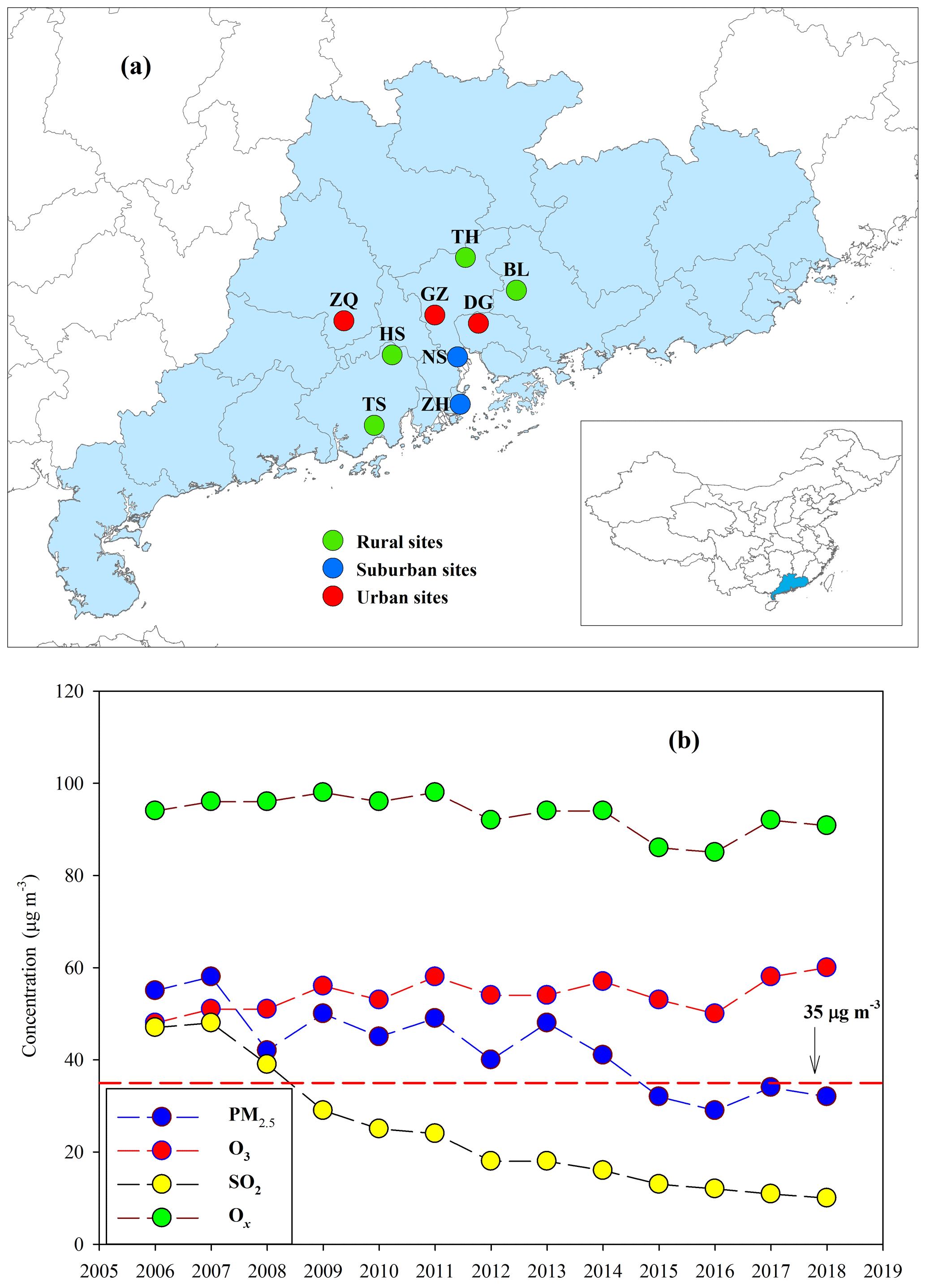
Figure 1(a) Sampling sites in the PRD, including Zhaoqing (ZQ), Guangzhou (GZ), Dongguan (DG), Nansha (NS), Zhuhai (ZH), Tianhu (TH), Boluo (BL), Heshan (HS), and Taishan (TS), and (b) long-term trends of annual mean PM2.5, O3, SO2, and Ox recorded by the Guangdong–Hong Kong–Macao regional air quality monitoring network (http://gdee.gd.gov.cn/kqjc/, last access: 26 November 2019) . The red dashed line indicates the NAAQS for annual mean PM2.5 concentrations (35 µg m−3).
2.1 Field sampling
Concurrent sampling was performed at 9 of the 23 Guangdong–Hong Kong–Macao regional air quality monitoring network sites (http://gdee.gd.gov.cn/kqjc/, last access: 26 November 2019, Fig. 1a), including 3 urban sites in Zhaoqing (ZQ), Guangzhou (GZ), and Dongguan (DG); 2 suburban sites in Nansha (NS) and Zhuhai (ZH); and 4 rural sites in Tianhu (TH), Boluo (BL), Heshan (HS), and Taishan (TS).
At each site, 24 h sampling was conducted every 6 d from January to December in 2015 using a PM2.5 sampler equipped with quartz filters (8 in. × 10 in.) at a flow rate of 1.1 m3 min−1. Additionally, field blanks were collected at all sites at a monthly interval. Blank filters were covered with aluminum foil, baked at 500 ∘C for 12 h, and stored in a container with silica gel. After sampling, the filter samples were stored at −20 ∘C.
In this study, the filters collected in January, April, July, and October 2015 were selected to represent winter, spring, summer, and fall, respectively. A total of 170 field samples (4–5 samples for each season at each site) were analyzed in the current study.
2.2 Chemical analysis
For each filter, organic carbon (OC) and elemental carbon (EC) were measured using an OC–EC aerosol analyzer (Sunset Laboratory Inc.). Water-soluble ions were analyzed by ion chromatography (Metrohm). All of these species are major components in PM2.5 (see Fig. 2). Meteorological parameters (temperature and relative humidity) and gaseous pollutants (SO2, CO, NO2, NO, and O3) at each site were recorded hourly. We also calculated the daily averages to probe the potential influence of air pollutants on BSOA formation.
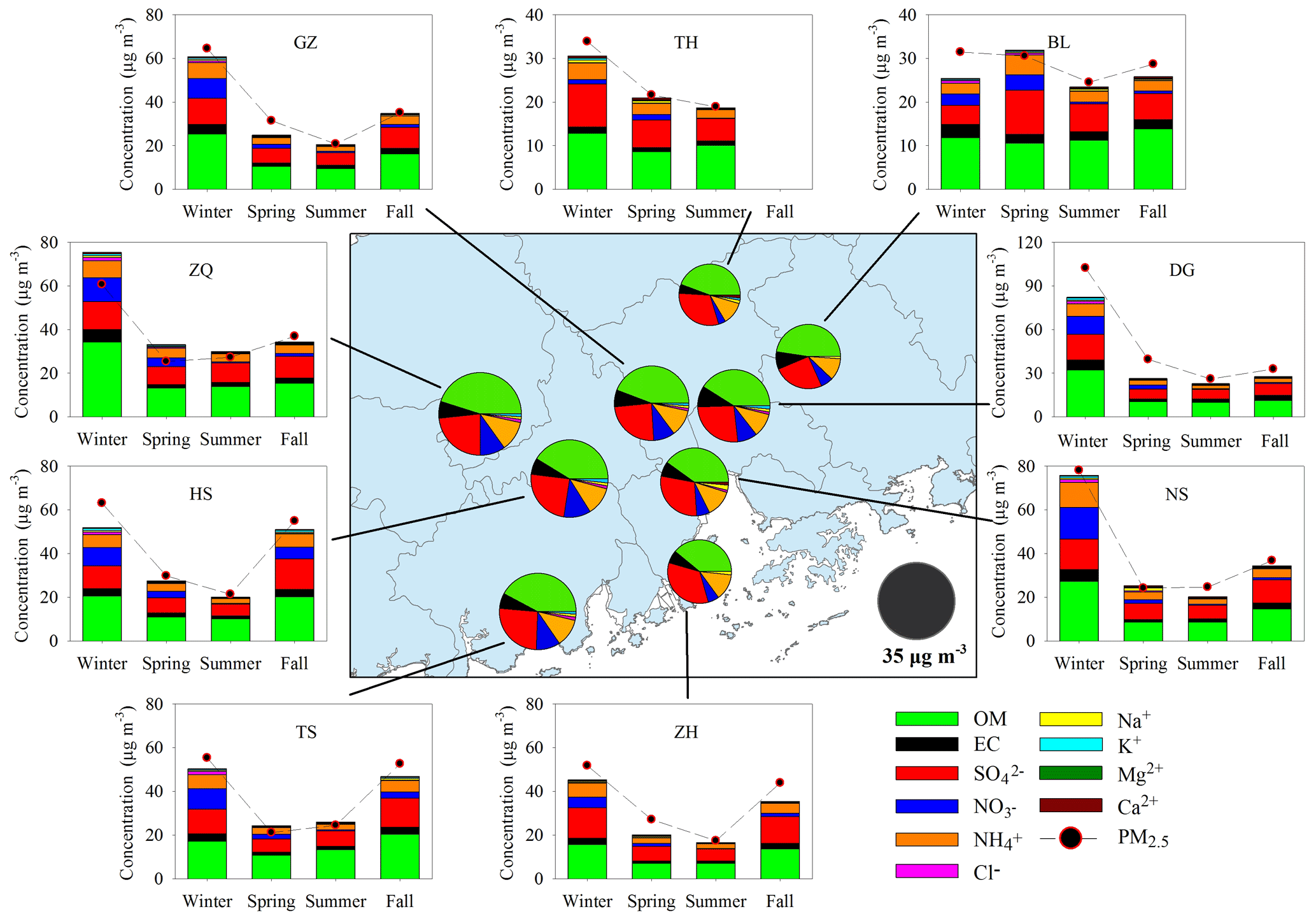
Figure 2Major components in PM2.5 and their seasonal variation at nine sites. The pie charts in the central panel represent the annual average of the major components. High levels of PM2.5 and major components were observed in wintertime.
For BSOA tracer analysis, detailed information regarding the processes used is given in the existing literature (Shen et al., 2015; Ding et al., 2012). Isotope-labeled standard mixtures, including dodecanoic acid-d23, hexadecanoic acid-d31, docosanoic acid-d43, and levoglucosan-13C6 were added into each sample as internal standards. Samples were then extracted by sonication with the mixed solvents of dichloride methane (DCM)∕hexane (1:1, v∕v) and DCM∕methanol (1:1, v∕v), sequentially. The extraction solutions of each sample were combined, filtered, and concentrated to ∼2 mL. Each concentrated sample was split into two parts for silylation and methylation, respectively.
We analyzed 14 BSOA tracers in the derivatized samples using GC-MSD (Agilent 7890/5975C). The isoprene-derived SOA (SOAI) tracers were composed of 2-methyltetrols (2-MTLs, 2-methylthreitol, and 2-methylerythritol) (Claeys et al., 2004a), 2-methylglyceric acid (2-MGA) (Claeys et al., 2004b), 3-MeTHF-3,4-diols (cis-3-methyltetrahydrofuran-3,4-diol and trans-3-methyltetrahydrofuran-3,4-diol) (Lin et al., 2012), and C5-alkene triols (cis-2-methyl-1,3,4-trihydroxy-1-butene, trans-2-methyl-1,3,4-trihydroxy-1-butene, and 3-methyl-2,3,4-trihydroxy-1-butene) (Wang et al., 2005). The monoterpene-derived SOA (SOAM) tracers included 3-hydroxy-4,4-dimethylglutaric acid (HDMGA), 3-hydroxyglutaric acid (HGA) (Claeys et al., 2007), pinic acid (PA), cis-pinonic acid (PNA) (Christoffersen et al., 1998), and 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) (Szmigielski et al., 2007). The β-caryophyllene-derived SOA (SOAC) tracer was β-caryophyllenic acid (CA) (Jaoui et al., 2007). Due to the lack of authentic standards, surrogate standards were used to quantify BSOA tracers except PNA. Specifically, erythritol, PNA, and octadecanoic acid were used for the quantification of SOAI tracers (Ding et al., 2008), other SOAM tracers (Ding et al., 2014), and CA (Ding et al., 2011), respectively. The method detection limits (MDLs) for erythritol, PNA, and octadecanoic acid were 0.01, 0.02, and 0.02 ng m−3, respectively. Table S1 in the Supplement summarizes BSOA data from each site in the PRD.
2.3 Quality assurance/quality control
The target BSOA tracers were not detected or were lower than the MDLs in the field blanks. The results of spiked samples (erythritol-, PNA-, and octadecanoic-acid-spiked in prebaked quartz filters) indicated that the recoveries were 65 % ±14 % for erythritol, 101 % ±3 % for PNA, and 83 % ±7 % for octadecanoic acid. The results of paired duplicate samples indicated that all of the relative differences for target BSOA tracers were lower than 15 %.
It should be noted that the application of surrogate quantification introduces additional errors to the results. Based on the empirical approach of calculating uncertainties from surrogate quantification (Stone et al., 2012), we estimated the errors in analyte measurement that were propagated from the uncertainties in field blanks, spike recoveries, repeatability, and surrogate quantification. As Table S2 shows, the estimated uncertainties in the tracers' measurement ranged from 15 % (PNA) to 157 % (CA).
3.1 PM2.5 and gaseous pollutants
Figure 2 presents the spatial and seasonal variations of PM2.5 and its major components. Although annual mean PM2.5 (34.8±6.1 µg m−3) in the PRD met the NAAQS value of 35 µg m−3, PM2.5 concentrations at the urban sites (ZQ, GZ, and DG) all exceeded the NAAQS value. The rural TH site in the northern part of the PRD witnessed the lowest PM2.5 concentration (25.0 µg m−3) among the nine sites. PM2.5 levels were highest in winter (60.1±21.6 µg m−3 on average) and lowest in summer (22.8±3.3 µg m−3 on average). Carbonaceous aerosols and water-soluble ions together explained 98 % ±11 % of the PM2.5 mass. OM (OC×1.6) was the most abundant component in PM2.5, followed by sulfate, ammonium, nitrate, and EC. Similar to PM2.5, the five major components all increased in winter and fall (Fig. S1), suggesting severe PM2.5 pollution during the fall–winter season in the PRD.
In the gas phase, SO2, CO, NO2, and NOx presented similar seasonal trends to PM2.5; i.e., higher levels occurred during fall and winter and lower concentrations during spring and summer (Fig. 3a, b, c, d). Annual mean SO2 and NO2 in the PRD both met the NAAQS values of 60 and 40 µg m−3, respectively (Fig. 3a, c). As a typical secondary pollutant, O3 was highest in summer (Fig. 3e), probably due to the strong photochemistry. Due to the compromise of opposite seasonal trends of O3 and NO2, Ox showed less seasonal variation (Fig. 3f) compared with other gaseous pollutants, and the annual mean for Ox reached 96.1±14.9 µg m−3. This indicated significant year-round O3 pollution in the PRD.
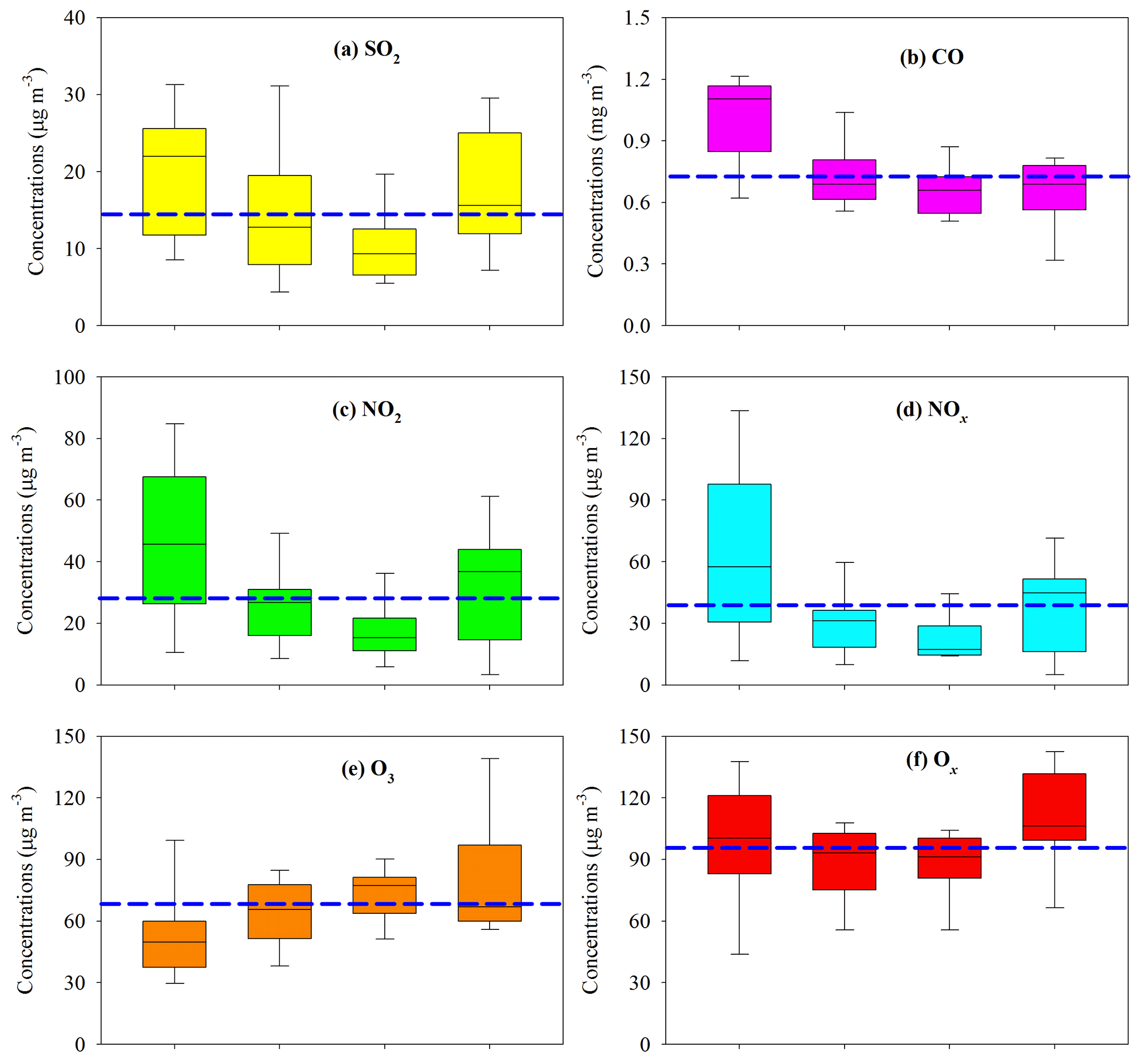
Figure 3Seasonal variation of gaseous pollutants in the PRD. The boxes with error bars represent the 10th, 25th, 75th, and 90th percentiles for each pollutant. The line in each box represents the median value. The blue dashed lines indicate the annual average concentrations of SO2 (14.9 µg m−3), CO (0.74 mg m−3), NO2 (28.5 µg m−3), NOx (39.0 µg m−3), O3 (67.7 µg m−3), and Ox (96.1 µg m−3).
3.2 Spatial distribution and seasonal variation of SOA tracers
The total concentrations of BSOA tracers ranged from 45.4 to 109 ng m−3 among the nine sites. SOAM tracers (47.2±9.29 ng m−3) were predominant, followed by SOAI tracers (23.1±10.8 ng m−3) and SOAC tracers (3.85±1.75 ng m−3).
3.2.1 Monoterpene-derived SOA tracers
Annual averages of total SOAM tracers at the nine sites were in the range of 26.5 to 57.4 ng m−3 (Table S1). Figures 4 and S2a show the spatial distribution of SOAM tracers and monoterpene emissions in the PRD (Zheng et al., 2010). The highest concentration of SOAM tracers was observed at the rural TH site, where monoterpene emissions were high. Figure 4 also presents seasonal variations of SOAM tracers. At most sites, high levels occurred in summer and fall. Monoterpene emission rates are influenced by temperature and solar radiation (Guenther et al., 2012). Thus, high temperature and intensive solar radiation during summer and fall in the PRD (Zheng et al., 2010) could stimulate monoterpene emissions and subsequent SOAM formation.

Figure 4Spatial and seasonal variations of SOAM tracers at nine sites in the PRD. The bars in the central panel represent the annual average concentrations of the SOAM tracers.
Among the five SOAM tracers, HGA (20.1±4.28 ng m−3) showed the highest concentration, followed by HDMGA (14.7±2.93 ng m−3), MBTCA (7.63±1.49 ng m−3), PNA (3.75±2.72 ng m−3), and PA (1.01±0.48 ng m−3). SOAM formation undergoes multigenerational reactions. The first-generation SOAM (SOAM_F) products, PNA and PA, can be further oxidized and form the later-generation (SOAM_L) products, e.g., MBTCA (Müller et al., 2012). Thus, the (PNA + PA) / MBTCA ratio has been used to probe SOAM aging (Haque et al., 2016; Ding et al., 2014). The (PNA + PA) / MBTCA ratios in chamber-generated α-pinene SOA samples were reported to be in the range of 1.51 to 5.91, depending on different oxidation conditions (Offenberg et al., 2007; Eddingsaas et al., 2012). In this study, the median values of (PNA + PA) / MBTCA varied from 0.27 at ZH to 1.67 at TH. The ratios observed in this study were consistent with our previous observations at the regional site, Wanqingsha (WQS) in the PRD (Ding et al., 2012), but lower than those in fresh α-pinene SOA samples from chamber experiments (Fig. S3), indicating relatively aged SOAM in the air of the PRD.
Moreover, the levels of SOAM_L tracers (HGA + HDMGA + MBTCA) were much higher than those of SOAM_F tracers (PNA + PA), with mean mass fractions of SOAM_L tracers reaching 86 % (Fig. 4). Mass fractions of SOAM_F tracers decreased in the summer samples (Fig. 4), probably resulting from strong photochemistry and more intensive subsequent oxidation during summer. High abundances of SOAM_L tracers in the PRD were different from our year-round observations at 12 sites across China (Ding et al., 2016b). Ding et al. (2016b) found that the (PNA + PA) / MBTCA ratio suggested generally fresh SOAM (Fig. S3) and that SOAM_F tracers were predominant. Thus, we see more aged SOAM in the PRD.
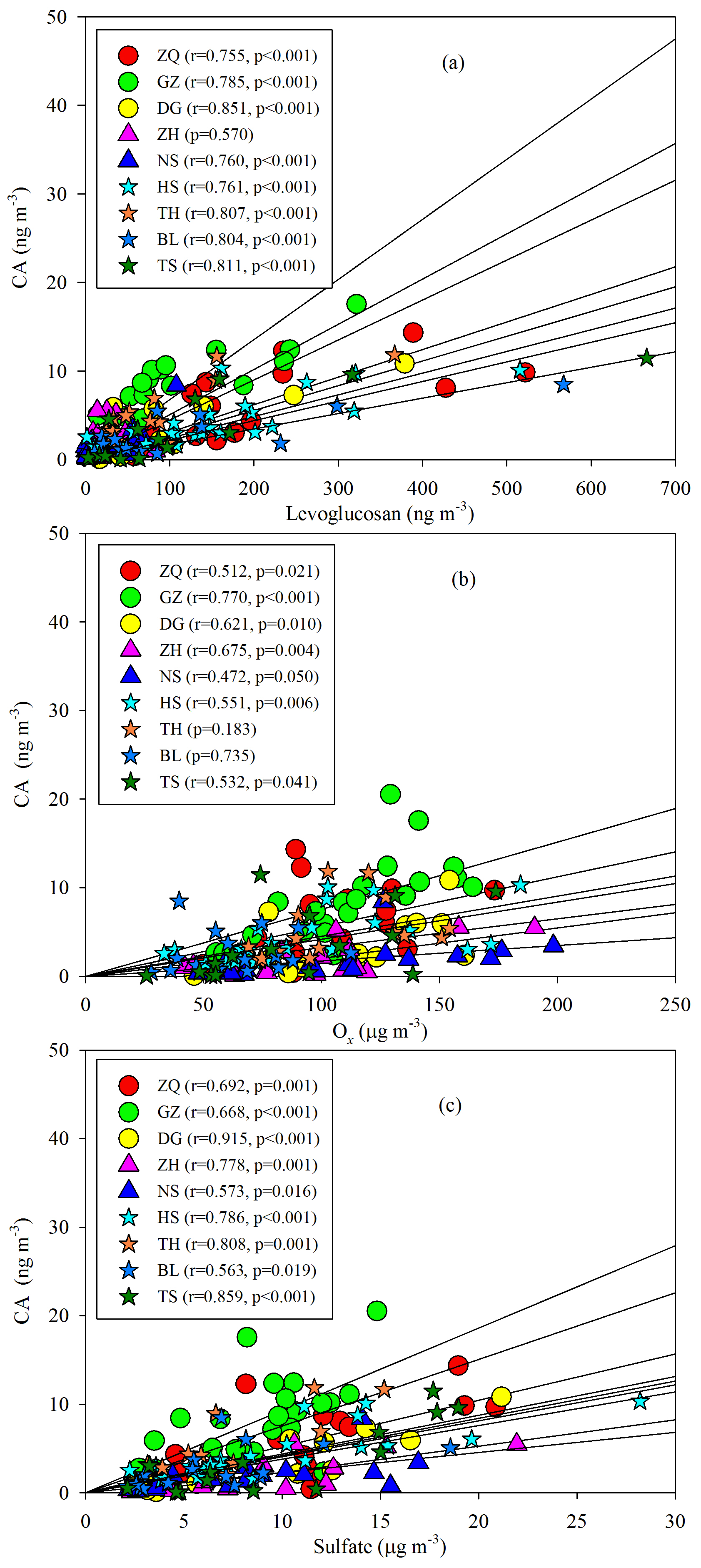
As Figs. 5a–b, S4, and S5 show, the SOAM_F tracers did not show good correlations with Ox at most sites, whereas the SOAM_L tracers exhibited significant Ox dependence. When Ox is high, strong photooxidation of PNA and PA could reduce their concentrations and promote the formation of SOAM_L tracers (Müller et al., 2012). Thus, the levels of SOAM_L tracers would increase with increasing Ox, whereas this is not the case for SOAM_F tracers. Moreover, sulfate could influence SOA formation. Sulfate is a key species in particles that determines the aerosol liquid water amount, aerosol acidity, and particle surface area (Xu et al., 2015, 2016). Thus, an increase in sulfate could promote aqueous and heterogeneous reactions. In this study, the SOAM_F tracers poorly correlated with sulfate (Fig. 5c), whereas the SOAM_L tracers positively correlated with sulfate at all nine sites (Fig. 5d). At each site, the SOAM_L tracers exhibited more sulfate dependence than the SOAM_F tracers (Fig. S5). This suggested that sulfate also played a critical role in forming SOAM_L tracers through particle-phase reactions. In addition to gas-phase OH oxidation (Müller et al., 2012), the heterogeneous OH oxidation of PNA could also produce SOAM_L tracers (Lai et al., 2015). Aljawhary et al. (2016) reported the kinetics and mechanism of PNA oxidation in acidic solutions and found that the molar yields of MBTCA via aqueous-phase reactions were similar to those in gas-phase oxidation. Here, we conclude that high concentrations of Ox and sulfate could stimulate SOAM_L tracer production and thereby lead to aged SOAM in the PRD.

3.2.2 Isoprene-derived SOA tracers
Annual averages of total SOAI tracers at the nine sites were in the range of 10.8 to 49.3 ng m−3 (Table S1). Figures 6 and S2b show the spatial distribution of SOAI tracers and isoprene emissions in the PRD (Zheng et al., 2010), respectively. The highest concentration occurred at ZQ, where emissions were high. Figure 6 also presents seasonal variations of SOAI tracers at the nine sites. High levels occurred in summer and fall. Similar to monoterpenes, the emission rate of isoprene is influenced by temperature and solar radiation (Guenther et al., 2012), which are both expected to be higher in summer and fall in the PRD (Zheng et al., 2010). Among these SOAI tracers, 2-MTLs (14.2±5.61 ng m−3) were the most abundant products, followed by C5-alkene triols (6.81±5.05 ng m−3), 2-MGA (1.99±0.72 ng m−3), and 3-MeTHF-3,4-diols (0.19±0.08 ng m−3).
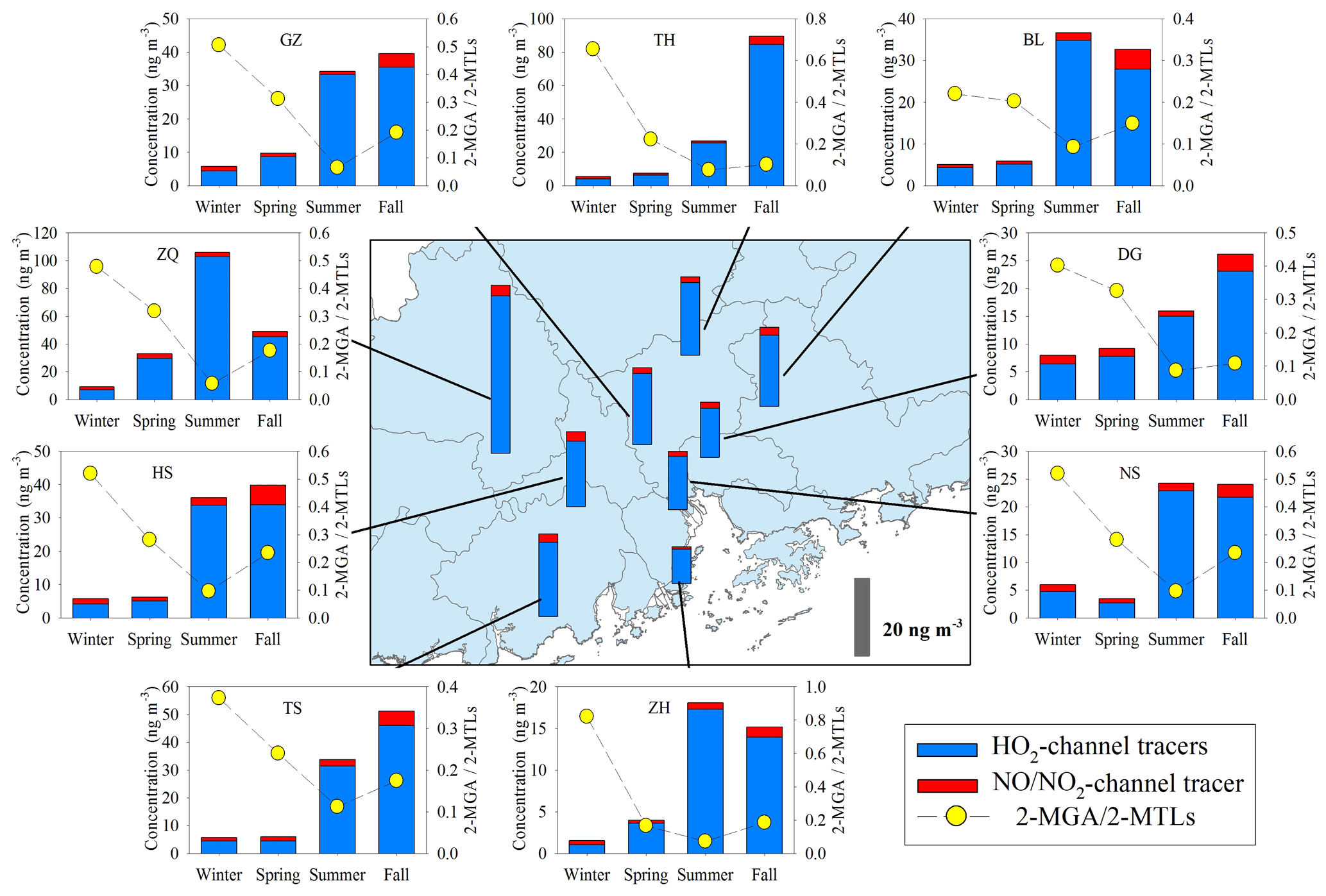
Figure 6Spatial and seasonal variations of SOAI tracers at nine sites in the PRD. The bars in the central panel represent the annual average concentrations of the SOAI tracers.
SOAI formation is highly affected by NOx (Surratt et al., 2010). Under low-NOx or NOx-free conditions, isoprene is oxidized by the OH and HO2 radicals via the HO2 channel which generates a hydroxy hydroperoxide (ISOPOOH) and then forms epoxydiols (IEPOX) (Paulot et al., 2009). Reactive uptake of IEPOX on acidic particles eventually produces 2-MTLs, C5-alkene triols, 3-MeTHF-3,4-diols, 2-MTLs-organosulfates, and oligomers (Lin et al., 2012). Under high-NOx conditions, isoprene undergoes oxidation by NOx via the NO/NO2 channel, generates methacrolein (MACR), and then forms peroxymethylacrylic nitric anhydride (MPAN). Further oxidation of MPAN by the OH radical produces hydroxymethyl-methyl-α-lactone (HMML) and/or methacrylic acid epoxide (MAE). HMML and MAE are the direct precursors of 2-MGA, 2-MGA-organosulfate, and its corresponding oligomers (Nguyen et al., 2015). As Fig. 6 shows, the concentrations of HO2-channel tracers (2-MTLs + C5-alkene triols + 3-MeTHF-3,4-diols) were much higher than those of the NO/NO2-channel product (2-MGA) at all the nine sites. The dominance of HO2-channel products was also observed at another regional site (WQS) in the PRD (He et al., 2018).
Figure 6 also shows seasonal trends of the 2-MGA to 2-MTLs ratio (2-MGA/2-MTLs) which is often applied to probe the influence of NOx on the formation of SOAI (Ding et al., 2013, 2016a; Pye et al., 2013). The ratios were highest in wintertime and lowest in summertime; these results are consistent with the seasonal trend of NOx during our campaign (Fig. 3d). As Table 1 shows, 2-MGA was positively correlated with NO2, probably due to the enhanced formation of MPAN from the peroxymethacryoyl (PMA) radical which reacted with NO2 (Worton et al., 2013; Chan et al., 2010). Previous laboratory studies have shown that increasing the NO2∕NO ratio could promote the formation of 2-MGA and its corresponding oligoesters (Chan et al., 2010; Surratt et al., 2010). However, we did not see a significant correlation between 2-MGA and the NO2∕NO ratio in the PRD. Instead, the 2-MGA/2-MTLs ratio correlated well with NO, NO2, and the NO2∕NO ratio (Table 1). Increasing NO limits the formation of ISOPOOH but prefers the production of MACR, and increasing NO2 enhances MPAN formation. Thus, it is expected that the 2-MGA/2-MTLs ratio will show stronger NOx dependence than 2-MGA. These findings demonstrate the significant impact of NOx on SOAI formation pathways in the atmosphere. We also checked the correlations of SOAI tracers with Ox and sulfate (Fig. S6). The NO∕NO2-channel product exhibited more Ox and sulfate dependance than the HO2-channel products.

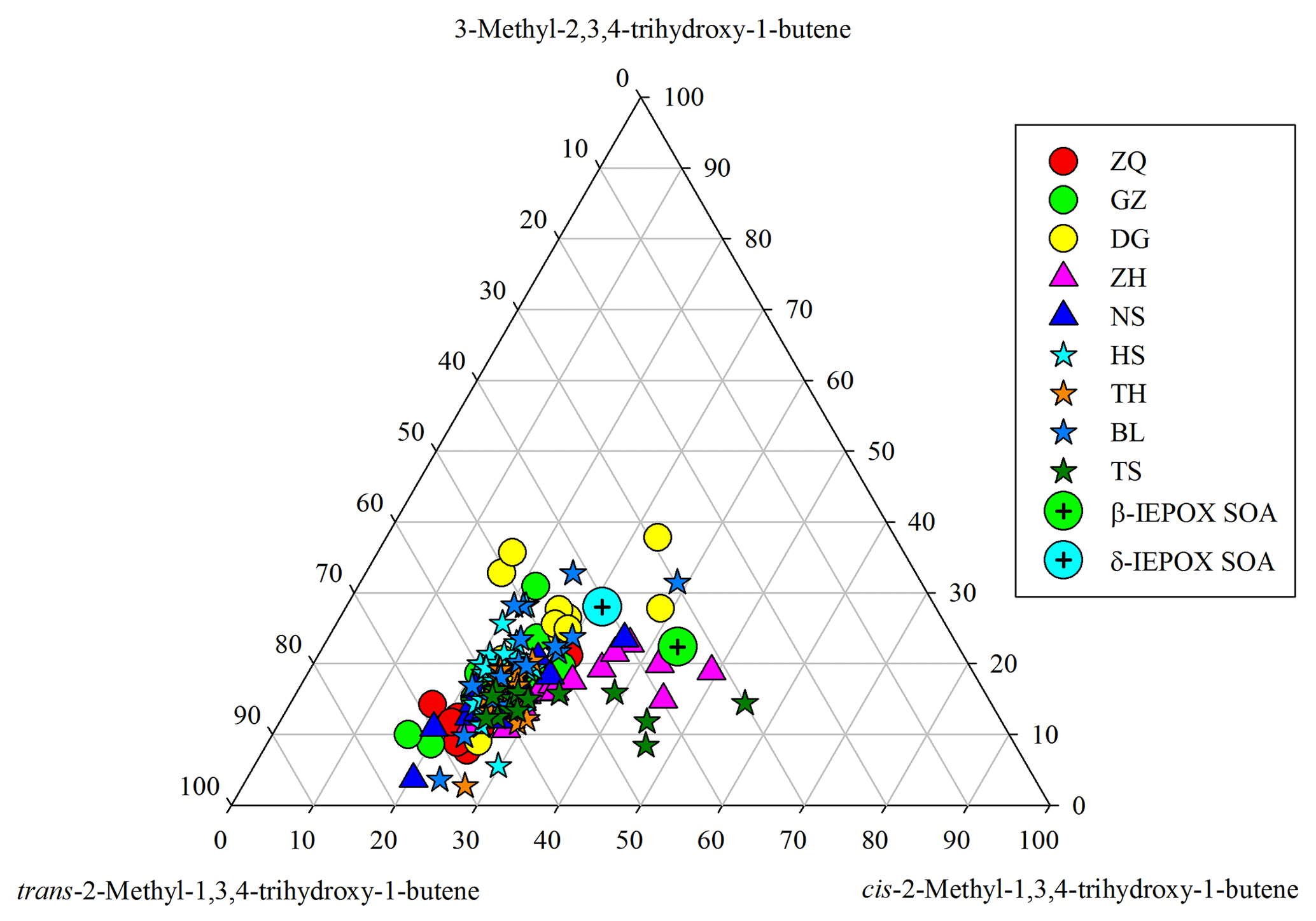
Figure 7Ternary plot of C5-alkene triol isomers in the PRD samples and in the β-IEPOX- and δ-IEPOX-derived SOA (Lin et al., 2012).
Recent studies have indicated that isoprene ozonolysis might play a role in SOAI formation in the ambient air. Riva et al. (2016) found that isoprene ozonolysis with acidic particles could produce substantial 2-MTLs, but this was not the case for C5-alkene triols and 3-MeTHF-3,4-diols. Li et al. (2018) observed a positive correlation between 2-MTLs and O3 on the North China Plain. In the PRD, we also saw weak but significant correlations of 2-MTLs with O3 (Table S3). However, 3-MeTHF-3,4-diols and C5-alkene triols were detected in all samples, and 2-MTLs, C5-alkene triols, and 3-MeTHF-3,4-diols correlated well with each other (Table S4), which was apparently different from the results reported by Riva et al. (2016). Moreover, the ratios of 2-MTLs isomers in the PRD samples (2.00–2.85) were much lower than those (10–22; Fig. S7) reported in the SOA from isoprene ozonolysis (Riva et al., 2016). Furthermore, isoprene oxidation by the OH radical is much faster than oxidation by O3 under polluted PRD conditions (Table S5), and IEPOX yields via ISOPOOH oxidation by the OH radical are more than 75% in the atmosphere (St. Clair et al., 2016). Thus, isoprene ozonolysis might be not the major formation pathway of SOAI, even though the annual mean O3 level reached 67.7 µg m−3 in the PRD (Table S1).
Previous studies have found that thermal decomposition of low-volatility organics in IEPOX-derived SOA could produce SOAI tracers, e.g., 2-MTLs, C5-alkene triols, and 3-MeTHF-3,4-diols (Lopez-Hilfiker et al., 2016; Watanabe et al., 2018). This means that these tracers detected by GC-MSD might be generated from the thermal decomposition of IEPOX-derived SOA. As estimated by Cui et al. (2018), 14.7 %–42.8 % of C5-alkene triols, 11.1 % of 2-MTLs, and approximately all 3-MeTHF-3,4-diols measured by GC-MSD could be attributed to the thermal degradation of 2-MTL-derived organosulfates (MTL-OSs). We also measured MTL-OSs in two samples at the HS and TS sites (Table S6), respectively, utilizing the widely used LC-MS approach (He et al., 2014, 2018). Assuming that all MTL-OSs decomposed to these tracers, the thermal decomposition of MTL-OSs would account for 15.1 %–31.6 % of C5-alkene triols, 6.0 %–10.0 % of 2-MTLs, and all 3-MeTHF-3,4-diols measured by GC/MSD. Thus, C5-alkene triols and 2-MTLs are major products of isoprene oxidation rather than products of the thermal decomposition of MTL-OSs, whereas 3-MeTHF-3,4-diols are only present in trace amount in the air and might be produced largely from thermal degradation.
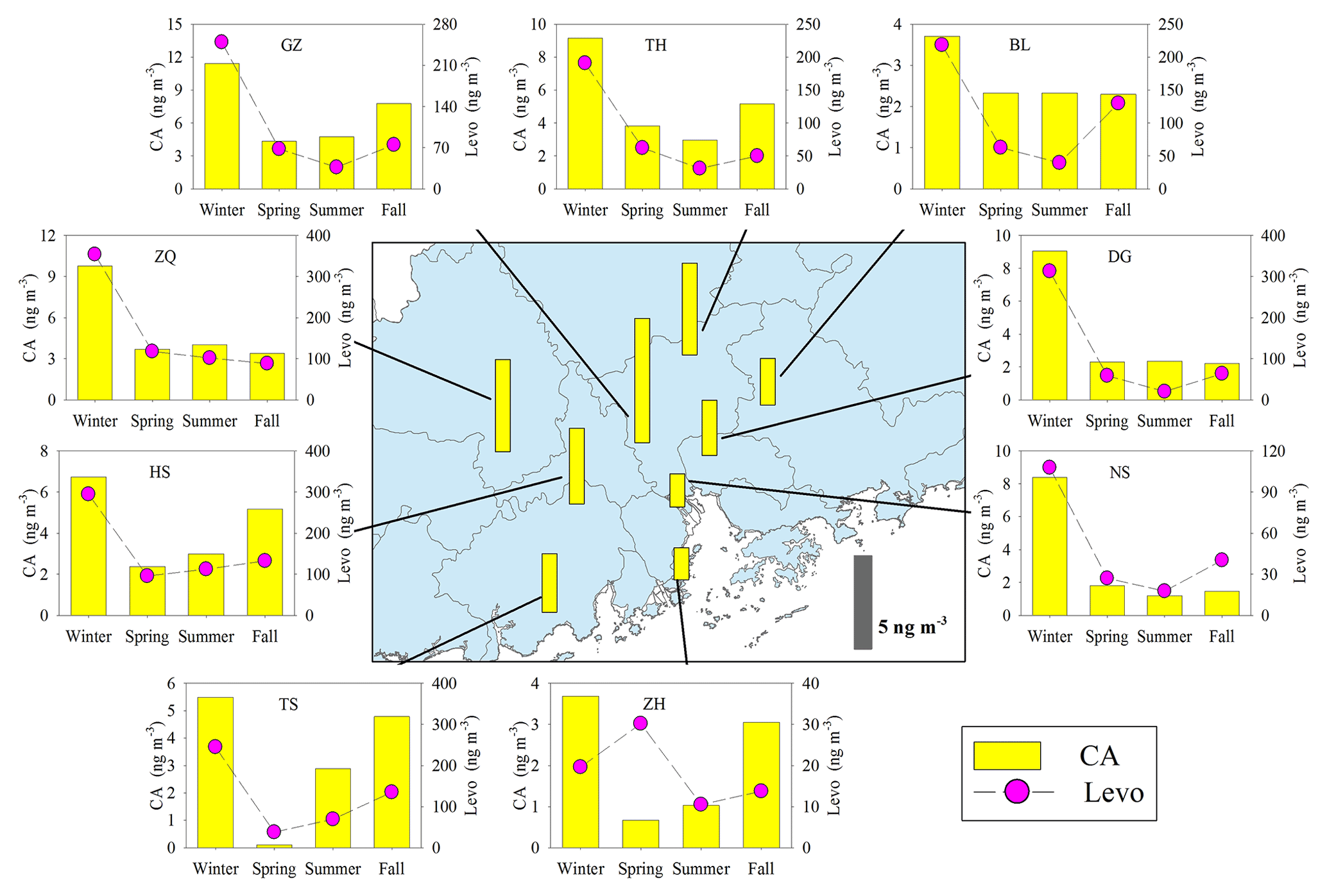
Figure 8Spatial and seasonal variations of SOAc tracer (CA) at nine sites in the PRD. The bars in the central panel represent the annual average concentration of the SOAc tracers. The pink circle indicates the BB tracer, levoglucosan (Levo).
Moreover, we see significant variations in SOAI tracer compositions in the PRD. For instance, C5-alkene triols have three isomers. If these tracers were mainly generated from a thermal process, their compositions should be similar in different samples. In fact, the relative abundances of the three C5-alkene triol isomers significantly changed from site to site (Fig. 7) and from season to season (Fig. S8), and their compositions in the PRD were different from those measured in the chamber samples (Lin et al., 2012). In addition, the slopes of linear correlations among these IEPOX-derived SOA tracers also varied from site to site (Fig. S9). Coupled with the seasonal trend of 2-MGA/2-MTLs ratios, the apparent variations in SOAI tracer compositions demonstrate that these SOAI tracers are mainly formed via different pathways in the ambient atmosphere; however, they might partly arise from the thermal decomposition of different dimers/OSs, and the parent dimers/OSs vary with site and seasons.
3.2.3 Sesquiterpene-derived SOA tracer
Annual averages of CA at the nine sites ranged from 1.82 to 7.07 ng m−3. The levels of CA at the inland sites (e.g., GZ, ZQ, and TH) were higher than those at the coastal sites (ZH and NS; Fig. 8). As sesquiterpenes are typical BVOCs, it is unexpected that the concentrations of CA were highest during winter in the PRD (Fig. 8). Interestingly, the seasonal trend of CA was consistent with that of the biomass burning (BB) tracer, levoglucosan (Fig. 8), and CA correlated well with levoglucosan at eight sites in the PRD (Fig. 9a). Sesquiterpenes are stored in plant tissues, partly to protect the plants from insects and pathogens (Keeling and Bohlmann, 2006). BB can not only stimulate sesquiterpene emissions (Ciccioli et al., 2014) but can also substantially alter SOA formation and yields (Mentel et al., 2013). Emissions inventories in the PRD showed that BB emissions were enhanced during winter (He et al., 2011). This suggested that the unexpected increase in SOAC in wintertime could be highly associated with BB emissions in the PRD.
Besides the impact of BB, we also found positive correlations of CA with Ox (Fig. 9b) and sulfate (Fig. 9c). The oxidation of β-caryophyllene by the OH radical and O3 is very rapid. Under typical oxidation conditions in the air of the PRD, the lifetimes of β-caryophyllene are only several minutes (Table S5). Once emitted from vegetation or biomass burning, β-caryophyllene will react rapidly and form CA immediately. This partly explains the positive correlations between CA and levoglucosan in this region. The unexpected high levels of CA in the winter indicated that biomass burning could be an important source of SOAC in the PRD, especially in wintertime. In addition, the increase in sulfate could raise aerosol acidity and thereby promote aqueous and heterogeneous reactions to form SOAC. In the PRD, both Ox (Fig. 3f) and sulfate (Fig. S1) increased during winter, which could promote SOAC formation. Here, we conclude that the enhancement of BB emissions as well as the increase in Ox and sulfate in wintertime led to high SOAC production during this season in the PRD.
3.3 Source apportionment and atmospheric implications
We further attributed BSOA using the SOA tracer approach, which was first developed by Kleindienst et al. (2007). This method has been applied to SOA apportionment at multiple sites across the United States (Lewandowski et al., 2013) and China (Ding et al., 2016b), and over global oceans from the Arctic to the Antarctic (Hu et al., 2013). Details of the SOA tracer method and its application in this study as well as the uncertainty of the estimation procedure are described in Sect. S1. Table S1 lists the results of estimated SOAs from different BVOCs.
Figure 10a exhibits the spatial distribution of BSOA components (SOAM + SOAI + SOAC). The annual average at the nine sites ranged from 0.97 µg m−3 (NS) to 2.19 µg m−3 (ZQ), accounting for 9 %–15 % of OM. SOAM was the largest BSOA contributor with an average contribution of 64 % ±7 %, followed by SOAC (21 % ±6 %), and SOAI (14 % ±4 %). Figure 10b presents the seasonal variation of BSOA components. The levels were highest in fall (2.35±0.95 µg m−3) and lowest in spring (1.06±0.42 µg m−3). SOAM contributions ranged from 57 % in winter to 68 % in spring. The contribution of SOAI was only 5 % in winter and reached up to 22 % in summer. The contribution of SOAC increased to 40 % in wintertime.
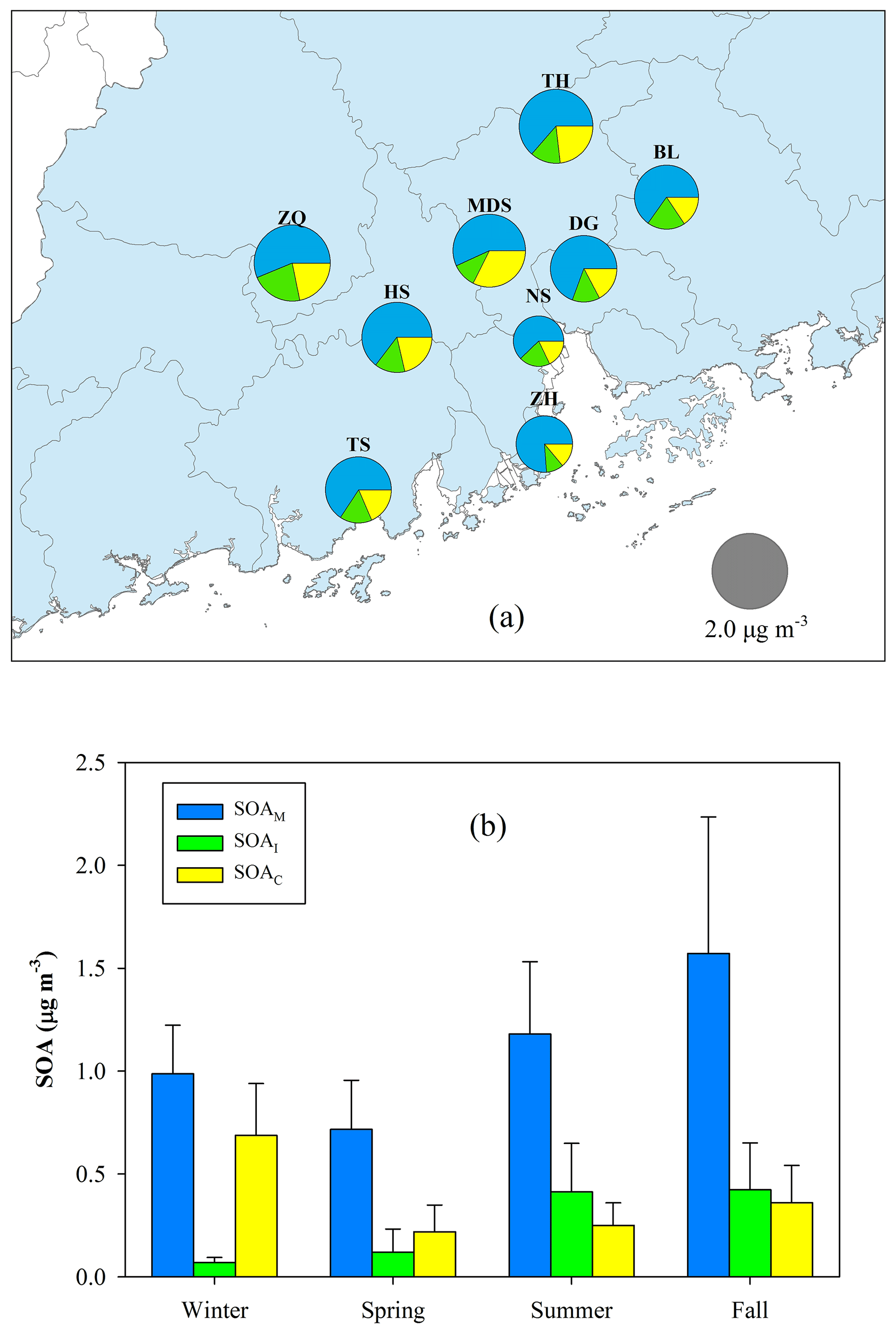
Figure 10Spatial (a) and seasonal (b) variations in BSOA components.
It is interesting to note that SOAM, SOAI, and SOAC all positively correlated with sulfate and Ox in the PRD (Table 2). As anthropogenic emissions can enhance BSOA formation (Hoyle et al., 2011), the reduction of anthropogenic emissions indeed lowers BSOA production (Carlton et al., 2018). As the oxidation product of SO2, sulfate is a key species in particles and determines aerosol acidity and surface area (Xu et al., 2015, 2016); thus, sulfate could promote BSOA formation via acid-catalyzed heterogeneous reactions. A recent study found that SO2 could directly react with organic peroxides from monoterpene ozonolysis and form substantial organosulfates (Ye et al., 2018). Thus, a decrease in SO2 emissions indeed reduces SO2 and sulfate in the ambient air, which leads to less acidic particles and a reduction in BSOA production. For Ox, the increase in O3 likely results in significant SOA formation via BVOC ozonolysis (Sipilä et al., 2014; Riva et al., 2017). Hence, the decrease in Ox resulting from the control of VOC and NOx emissions could reduce BSOA formation through O3 chemistry. Based on the observed sulfate and Ox dependence of BSOA in this study, a reduction of 1 µg m−3 in sulfate and Ox in the air of the PRD could lower BSOA levels by 0.17 and 0.02 µg m−3, respectively. If both concentrations decline by 50 %, the reduction of Ox is more efficient than the reduction of sulfate with respect to reducing BSOA in the PRD (Table 2).
Table 2Correlations of BSOA with sulfate and Ox.
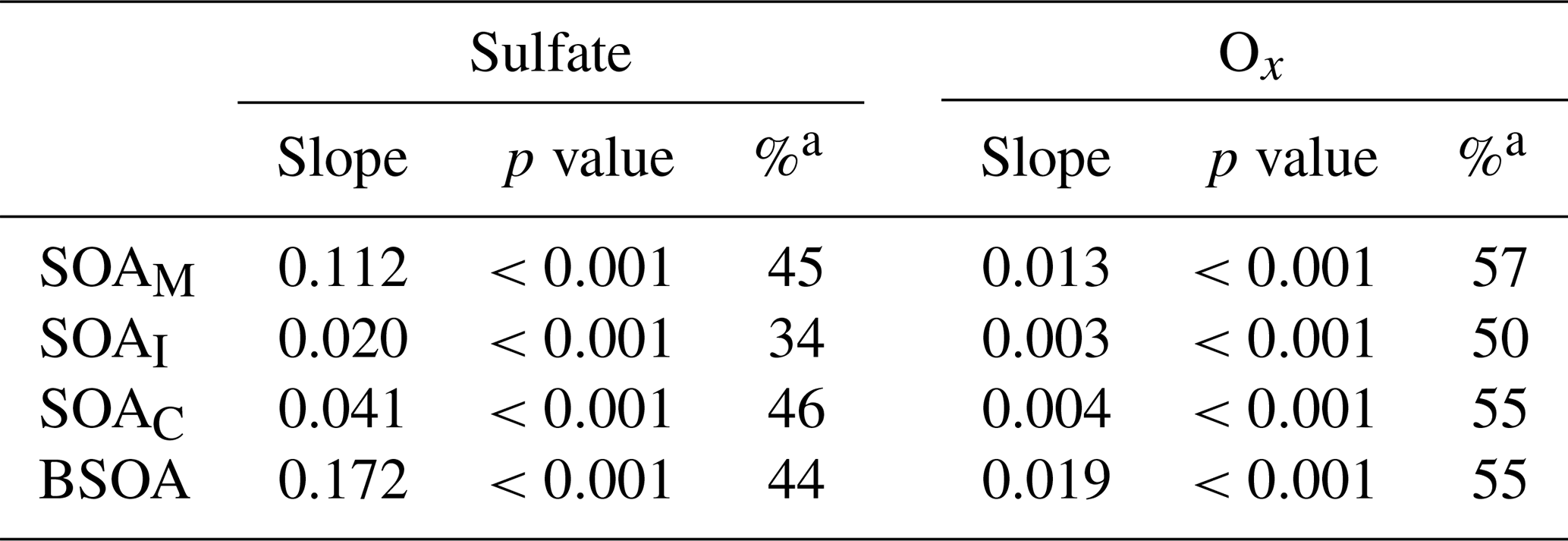
a Percentage reduction of SOA due to a 50 % decline in sulfate or Ox.
We further compared the results from 2015 with those from the fall–winter season of 2008 at WQS (Ding et al., 2012). We found that all BSOA species positively correlated with sulfate but exhibited no Ox dependence (Table S7). Thus, BSOA formation was largely influenced by sulfate in 2008, probably due to high sulfate levels during this period (as high as 46.8 µg m−3). Owing to subsequent strict control of SO2 emissions (Wang et al., 2013), ambient SO2 has significantly decreased over the PRD (Fig. 1b). Our long-term observation during the fall–winter season at WQS also witnessed a decreasing trend in sulfate from 2007 to 2016 (Fig. S10). However, Ox levels have not decreased during the past decade (Fig. 1b), and Ox concentrations were much higher than sulfate in 2015 in the PRD (96.1±14.9 µg m−3 vs. 8.44±1.09 µg m−3 on average). These results emphasize the current importance of Ox in BSOA formation in the PRD. At present, short-term despiking and long-term attainment of O3 concentrations are challenges for air pollution control in the PRD (Ou et al., 2016). Thus, lowering Ox is critical to improve the air quality in the PRD. Our results highlight the importance of future reductions of anthropogenic pollutant emissions (e.g., SO2 and Ox precursors) in order to considerably reduce the BSOA burden in polluted regions.
The experimental data in this study are available upon request to the corresponding author by email.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-14403-2019-supplement.
XD, D-HC, and JL conceived the project and designed the study. Y-QZ and D-HC performed the data analysis and wrote the paper. D-HC, TZ, and Y-BO arranged the sample collection and assisted with the data analysis. J-QW, QC, and HJ analyzed the samples. XD, P-LY, WS, GZ, and X-MW performed the data interpretation and edited the paper. All authors contributed to the development of the final paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Multiphase chemistry of secondary aerosol formation under severe haze”. It is not associated with a conference.
This study was supported by the National Key Research and Development Program (grant no. 2018YFC0213902), the National Natural Science Foundation of China (grant nos. 41722305, 41603070, and 41473099), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant no. 2017BT01Z134). We would like to thank Sasho Gligorovski for his helpful suggestion on the discussion of atmospheric oxidation process. The gaseous pollutants, major components in PM2.5, and BSOA tracers data can be found in the Supplement.
This research has been supported by the National Key Research and Development Program (grant no. 2018YFC0213902), the National Natural Science Foundation of China (grant nos. 41722305, 41603070, 41473099), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant no. 2017BT01Z134).
This paper was edited by Jingkun Jiang and reviewed by two anonymous referees.
Aljawhary, D., Zhao, R., Lee, A. K. Y., Wang, C., and Abbatt, J. P. D.: Kinetics, mechanism, and secondary organic aerosol yield of aqueous phase photo-oxidation of α-pinene oxidation products, J. Phys. Chem. A., 120, 1395–1407, https://doi.org/10.1021/acs.jpca.5b06237, 2016.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To what extent can biogenic SOA be controlled?, Environ. Sci. Technol., 44, 3376–3380, https://doi.org/10.1021/es903506b, 2010.
Carlton, A. G., Pye, H. O. T., Baker, K. R., and Hennigan, C. J.: Additional benefits of federal air-quality rules: Model estimates of controllable biogenic secondary organic aerosol, Environ. Sci. Technol., 52, 9254–9265, https://doi.org/10.1021/acs.est.8b01869, 2018.
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010.
Christoffersen, T. S., Hjorth, J., Horie, O., Jensen, N. R., Kotzias, D., Molander, L. L., Neeb, P., Ruppert, L., Winterhalter, R., Virkkula, A., Wirtz, K., and Larsen, B. R.: Cis-pinic acid, a possible precursor for organic aerosol formation from ozonolysis of α-pinene, Atmos. Environ., 32, 1657–1661, https://doi.org/10.1016/S1352-2310(97)00448-2, 1998.
Ciccioli, P., Centritto, M., and Loreto, F.: Biogenic volatile organic compound emissions from vegetation fires, Plant Cell Environ., 37, 1810–1825, https://doi.org/10.1111/pce.12336, 2014.
Claeys, M., Graham, B., Vas, G., Wang, W., Vermeylen, R., Pashynska, V., Cafmeyer, J., Guyon, P., Andreae, M. O., Artaxo, P., and Maenhaut, W.: Formation of secondary organic aerosols through photooxidation of isoprene, Science, 303, 1173–1176, https://doi.org/10.1126/science.1092805, 2004a.
Claeys, M., Wang, W., Ion, A. C., Kourtchev, I., Gelencsér, A., and Maenhaut, W.: Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide, Atmos. Environ., 38, 4093–4098, https://doi.org/10.1016/j.atmosenv.2004.06.001, 2004b.
Claeys, M., Szmigielski, R., Kourtchev, I., Van der Veken, P., Vermeylen, R., Maenhaut, W., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., and Edney, E. O.: Hydroxydicarboxylic acids:? Markers for secondary organic aerosol from the photooxidation of α-pinene, Environ. Sci. Technol., 41, 1628–1634, https://doi.org/10.1021/es0620181, 2007.
Cui, T., Zeng, Z., dos Santos, E. O., Zhang, Z., Chen, Y., Zhang, Y., Rose, C. A., Budisulistiorini, S. H., Collins, L. B., Bodnar, W. M., de Souza, R. A. F., Martin, S. T., Machado, C. M. D., Turpin, B. J., Gold, A., Ault, A. P., and Surratt, J. D.: Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci.: Process. Imp., 20, 1524–1536, https://doi.org/10.1039/C8EM00308D, 2018.
Ding, X., Zheng, M., Yu, L., Zhang, X., Weber, R. J., Yan, B., Russell, A. G., Edgerton, E. S., and Wang, X.: Spatial and seasonal trends in biogenic secondary organic aerosol tracers and water-soluble organic carbon in the southeastern United States, Environ. Sci. Technol., 42, 5171–5176, https://doi.org/10.1021/es7032636, 2008.
Ding, X., Wang, X., and Zheng, M.: The influence of temperature and aerosol acidity on biogenic secondary organic aerosol tracers: Observations at a rural site in the central Pearl River Delta region, South China, Atmos. Environ., 45, 1303–1311, https://doi.org/10.1016/j.atmosenv.2010.11.057, 2011.
Ding, X., Wang, X., Gao, B., Fu, X., He, Q., Zhao, X., Yu, J., and Zheng, M.: Tracer based estimation of secondary organic carbon in the Pearl River Delta, South China, J. Geophys. Res.-Atmos., 117, D05313, https://doi.org/10.1029/2011JD016596, 2012.
Ding, X., Wang, X., Xie, Z., Zhang, Z., and Sun, L.: Impacts of Siberian biomass burning on organic aerosols over the North Pacific Ocean and the Arctic: Primary and secondary organic tracers, Environ. Sci. Technol., 47, 3149–3157, https://doi.org/10.1021/es3037093, 2013.
Ding, X., He, Q. F., Shen, R. Q., Yu, Q. Q., and Wang, X. M.: Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer, J. Geophys. Res.-Atmos., 119, 11877–11891, https://doi.org/10.1002/2014JD021748, 2014.
Ding, X., He, Q. F., Shen, R. Q., Yu, Q. Q., Zhang, Y. Q., Xin, J. Y., Wen, T. X., and Wang, X. M.: Spatial and seasonal variations of isoprene secondary organic aerosol in China: Significant impact of biomass burning during winter, Sci. Rep., 6, 20411, https://doi.org/10.1038/srep20411, 2016a.
Ding, X., Zhang, Y. Q., He, Q. F., Yu, Q. Q., Shen, R. Q., Zhang, Y., Zhang, Z., Lyu, S. J., Hu, Q. H., Wang, Y. S., Li, L. F., Song, W., and Wang, X. M.: Spatial and seasonal variations of secondary organic aerosol from terpenoids over China, J. Geophys. Res.-Atmos., 121, 14661–14678, https://doi.org/10.1002/2016JD025467, 2016b.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Haque, M. M., Kawamura, K., and Kim, Y.: Seasonal variations of biogenic secondary organic aerosol tracers in ambient aerosols from Alaska, Atmos. Environ., 130, 95–104, https://doi.org/10.1016/j.atmosenv.2015.09.075, 2016.
He, M., Zheng, J., Yin, S., and Zhang, Y.: Trends, temporal and spatial characteristics, and uncertainties in biomass burning emissions in the Pearl River Delta, China, Atmos. Environ., 45, 4051–4059, https://doi.org/10.1016/j.atmosenv.2011.04.016, 2011.
He, Q. F., Ding, X., Wang, X. M., Yu, J. Z., Fu, X. X., Liu, T. Y., Zhang, Z., Xue, J., Chen, D. H., Zhong, L. J., and Donahue, N. M.: Organosulfates from pinene and isoprene over the Pearl River Delta, South China: Seasonal variation and implication in formation mechanisms, Environ. Sci. Technol., 48, 9236–9245, https://doi.org/10.1021/es501299v, 2014.
He, Q. F., Ding, X., Fu, X. X., Zhang, Y. Q., Wang, J. Q., Liu, Y. X., Tang, M. J., Wang, X. M., and Rudich, Y.: Secondary organic aerosol formation from isoprene epoxides in the Pearl River Delta, South China: IEPOX- and HMML- derived tracers, J. Geophys. Res.-Atmos., 123, 6999–7012, https://doi.org/10.1029/2017JD028242, 2018.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., Petäjä, T., Rosenoern, T., and Sullivan, A. P.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Hu, Q. H., Xie, Z. Q., Wang, X. M., Kang, H., He, Q. F., and Zhang, P.: Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic, Sci. Rep., 3, 2280, https://doi.org/10.1038/srep02280, 2013.
Jaoui, M., Lewandowski, M., Kleindienst, T. E., Offenberg, J. H., and Edney, E. O.: β-caryophyllinic acid: An atmospheric tracer for β-caryophyllene secondary organic aerosol, Geophys. Res. Let., 34, L05816, https://doi.org/10.1029/2006gl028827, 2007.
Keeling, C. I. and Bohlmann, J.: Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens, New Phytol., 170, 657–675, https://doi.org/10.1111/j.1469-8137.2006.01716.x, 2006.
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007.
Lai, C., Liu, Y., Ma, J., Ma, Q., Chu, B., and He, H.: Heterogeneous kinetics of cis-pinonic acid with hydroxyl radical under different environmental conditions, J. Phys. Chem. A., 119, 6583–6593, https://doi.org/10.1021/acs.jpca.5b01321, 2015.
Lewandowski, M., Piletic, I. R., Kleindienst, T. E., Offenberg, J. H., Beaver, M. R., Jaoui, M., Docherty, K. S., and Edney, E. O.: Secondary organic aerosol characterisation at field sites across the United States during the spring–summer period, Int. J. Environ. An. Ch., 93, 1084–1103, https://doi.org/10.1080/03067319.2013.803545, 2013.
Li, J., Wang, G., Wu, C., Cao, C., Ren, Y., Wang, J., Li, J., Cao, J., Zeng, L., and Zhu, T.: Characterization of isoprene-derived secondary organic aerosols at a rural site in North China Plain with implications for anthropogenic pollution effects, Sci. Rep., 8, 535, https://doi.org/10.1038/s41598-017-18983-7, 2018.
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S., Knipping, E., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic standards, Environ. Sci. Technol., 46, 189–195, https://doi.org/10.1021/es202554c, 2012.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular composition and volatility of organic aerosol in the southeastern U.S.: Implications for IEPOX derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Lu, Q., Zheng, J., Ye, S., Shen, X., Yuan, Z., and Yin, S.: Emission trends and source characteristics of SO2, NOx, PM10 and VOCs in the Pearl River Delta region from 2000 to 2009, Atmos. Environ., 76, 11–20, https://doi.org/10.1016/j.atmosenv.2012.10.062, 2013.
Mentel, Th. F., Kleist, E., Andres, S., Dal Maso, M., Hohaus, T., Kiendler-Scharr, A., Rudich, Y., Springer, M., Tillmann, R., Uerlings, R., Wahner, A., and Wildt, J.: Secondary aerosol formation from stress-induced biogenic emissions and possible climate feedbacks, Atmos. Chem. Phys., 13, 8755–8770, https://doi.org/10.5194/acp-13-8755-2013, 2013.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/C5CP02001H, 2015.
Offenberg, J. H., Lewis, C. W., Lewandowski, M., Jaoui, M., Kleindienst, T. E., and Edney, E. O.: Contributions of toluene and α-pinene to SOA formed in an irradiated toluene/α-pinene/NOx/air mixture: Comparison of results using 14C content and SOA organic tracer methods, Environ. Sci. Technol., 41, 3972–3976, https://doi.org/10.1021/es070089+, 2007.
Offenberg, J. H., Lewandowski, M., Edney, E. O., Kleindienst, T. E., and Jaoui, M.: Influence of aerosol acidity on the formation of secondary organic aerosol from biogenic precursor hydrocarbons, Environ. Sci. Technol., 43, 7742–7747, https://doi.org/10.1021/es901538e, 2009.
Ou, J., Yuan, Z., Zheng, J., Huang, Z., Shao, M., Li, Z., Huang, X., Guo, H., and Louie, P. K. K.: Ambient ozone control in a photochemically active region: Short-term despiking or long-term attainment?, Environ. Sci. Technol., 50, 5720–5728, https://doi.org/10.1021/acs.est.6b00345, 2016.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Pye, H. O. T., Pinder, R. W., Piletic, I. R., Xie, Y., Capps, S. L., Lin, Y.-H., Surratt, J. D., Zhang, Z., Gold, A., Luecken, D. J., Hutzell, W. T., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Epoxide pathways improve model predictions of isoprene markers and reveal key role of acidity in aerosol formation, Environ. Sci. Technol., 47, 11056–11064, https://doi.org/10.1021/es402106h, 2013.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., and Surratt, J. D.: Chemical characterization of secondary organic aerosol constituents from isoprene ozonolysis in the presence of acidic aerosol, Atmos. Environ., 130, 5–13, https://doi.org/10.1016/j.atmosenv.2015.06.027, 2016.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., Thornton, J. A., Turpin, B. J., and Surratt, J. D.: Multiphase reactivity of gaseous hydroperoxide oligomers produced from isoprene ozonolysis in the presence of acidified aerosols, Atmos. Environ., 152, 314–322, https://doi.org/10.1016/j.atmosenv.2016.12.040, 2017.
Shen, R.-Q., Ding, X., He, Q.-F., Cong, Z.-Y., Yu, Q.-Q., and Wang, X.-M.: Seasonal variation of secondary organic aerosol tracers in Central Tibetan Plateau, Atmos. Chem. Phys., 15, 8781–8793, https://doi.org/10.5194/acp-15-8781-2015, 2015.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jørgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and products of the reaction of the first-generation isoprene hydroxy hydroperoxide (ISOPOOH) with OH, J. Phys. Chem. A., 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2016.
Stone, E. A., Nguyen, T. T., Pradhan, B. B., and Man Dangol, P.: Assessment of biogenic secondary organic aerosol in the Himalayas, Environ. Chem., 9, 263–272, https://doi.org/10.1071/EN12002, 2012.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Szmigielski, R., Surratt, J. D., Gómez-González, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Let., 34, L24811, https://doi.org/10.1029/2007gl031338, 2007.
Von Schneidemesser, E., Monks, P. S., Allan, J. D., Bruhwiler, L., Forster, P., Fowler, D., Lauer, A., Morgan, W. T., Paasonen, P., Righi, M., Sindelarova, K., and Sutton, M. A.: Chemistry and the linkages between air quality and climate change, Chem. Rev., 115, 3856–3897, https://doi.org/10.1021/acs.chemrev.5b00089, 2015.
Wang, W., Kourtchev, I., Graham, B., Cafmeyer, J., Maenhaut, I., and Claeys, M.: Characterization of oxygenated derivatives of isoprene related to 2-methyltetrols in Amazonian aerosols using trimethylsilylation and gas chromatography/ion trap mass spectrometry, Rapid Commun. Mass Spectrom., 19, 1343–1351, https://doi.org/10.1002/rcm.1940, 2005.
Wang, X., Liu, H., Pang, J., Carmichael, G., He, K., Fan, Q., Zhong, L., Wu, Z., and Zhang, J.: Reductions in sulfur pollution in the Pearl River Delta region, China: Assessing the effectiveness of emission controls, Atmos. Environ., 76, 113–124, https://doi.org/10.1016/j.atmosenv.2013.04.074, 2013.
Watanabe, A. C., Stropoli, S. J., and Elrod, M. J.: Assessing the potential mechanisms of isomerization reactions of isoprene epoxydiols on secondary organic aerosol, Environ. Sci. Technol., 52, 8346–8354, https://doi.org/10.1021/acs.est.8b01780, 2018.
Worton, D. R., Surratt, J. D., LaFranchi, B. W., Chan, A. W. H., Zhao, Y., Weber, R. J., Park, J.-H., Gilman, J. B., de Gouw, J., Park, C., Schade, G., Beaver, M., Clair, J. M. S., Crounse, J., Wennberg, P., Wolfe, G. M., Harrold, S., Thornton, J. A., Farmer, D. K., Docherty, K. S., Cubison, M. J., Jimenez, J.-L., Frossard, A. A., Russell, L. M., Kristensen, K., Glasius, M., Mao, J., Ren, X., Brune, W., Browne, E. C., Pusede, S. E., Cohen, R. C., Seinfeld, J. H., and Goldstein, A. H.: Observational insights into aerosol formation from isoprene, Environ. Sci. Technol., 47, 11396–11402, https://doi.org/10.1021/es4011064, 2013.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Xu, L., Middlebrook, A. M., Liao, J., de Gouw, J. A., Guo, H., Weber, R. J., Nenes, A., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., Brock, C. A., Neuman, J. A., Nowak, J. B., Pollack, I. B., Welti, A., Graus, M., Warneke, C., and Ng, N. L.: Enhanced formation of isoprene-derived organic aerosol in sulfur-rich power plant plumes during Southeast Nexus, J. Geophys. Res.-Atmos., 121, 11137–11153, https://doi.org/10.1002/2016JD025156, 2016.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Zhang, R. Y., Wang, G. H., Guo, S., Zarnora, M. L., Ying, Q., Lin, Y., Wang, W. G., Hu, M., and Wang, Y.: Formation of urban fine particulate matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015.
Zheng, J., Zheng, Z., Yu, Y., and Zhong, L.: Temporal, spatial characteristics and uncertainty of biogenic VOC emissions in the Pearl River Delta region, China, Atmos. Environ., 44, 1960–1969, https://doi.org/10.1016/j.atmosenv.2010.03.001, 2010.