the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Nov 2019
| 27 Nov 2019
Wintertime aerosol properties in Beijing
Misti Levy Zamora
Jianfei Peng
Wilmarie Marrero-Ortiz
Dongjie Shang
Jing Zheng
Zhuofei Du
Zhijun Wu
Severe wintertime haze events with exceedingly high levels of aerosols have occurred frequently in China in recent years, impacting human health, weather, and the climate. A better knowledge of the formation mechanism and aerosol properties during haze events is helpful for the development of effective mitigation policies. In this study, we present field measurements of aerosol properties at an urban site in Beijing during January and February 2015. A suite of aerosol instruments were deployed to measure a comprehensive set of aerosol chemical and physical properties. The evolution of haze events in winter, dependent on meteorological conditions, consistently involves new particle formation during the clean period and subsequently continuous growth from the nucleation mode particles to submicron particles over the course of multiple days. Particulate organic matter is primarily responsible for producing the nucleation mode particles, while secondary organic and inorganic components jointly contribute to the high aerosol mass observed during haze events. The average effective density and hygroscopic parameter (κ) of ambient particles are approximately 1.37 g cm−3 and 0.25 during the clean period and increase to 1.42 g cm−3 and 0.4 during the polluted period, indicating the formation of secondary inorganic species from the continuous growth of nucleation mode particles. Our results corroborate that the periodic cycles of severe haze formation in Beijing during winter are attributed to the efficient nucleation and secondary aerosol growth under high gaseous precursor concentrations and the stagnant air conditions, highlighting that reductions in emissions of aerosol precursor gases are critical for remedying secondary aerosol formation and thereby mitigating haze pollution.
- Article
(2876 KB) - Full-text XML
-
Supplement
(156 KB) - BibTeX
- EndNote





Three decades of rapid industrialization have made China the second largest economy in the world. This rapid economic development has resulted in a deterioration of the quality of air, water, land, and ecosystems (Chang et al., 2009; Su et al., 2012; Wang et al., 2008; WHO et al., 2006). One of the major environmental issues is the severe haze events caused by the elevated mass concentration of fine particulate matter (PM2.5) that frequently occurs in many regions of China (Hu et al., 2015; Guo et al., 2013; Hallquist et al., 2016; Zhang et al., 2015; An et al., 2019). Regional haze not only adversely influences human health, but it also impacts weather and climate (Wang et al., 2011, 2014).
PM2.5 is mainly composed of sulfate, nitrate, ammonium, organics, and elemental carbon in China (Guo et al., 2010; Zheng et al., 2016). To elucidate the mechanism of severe PM2.5 pollution, considerable efforts have been made in the past few years. Huang et al. (2014) demonstrated that severe PM2.5 pollution in four megacities in China (i.e., Beijing, Shanghai, Guangzhou, and Xi'an) was driven to a large extent by the secondary aerosol formation. Guo et al. (2014) elucidated that the formation of severe urban haze in Beijing during autumn could be characterized by two distinct aerosol formation processes: (1) the nucleation of aerosols and (2) the subsequent growth of these aerosols driven by secondary formation. The transition from a clean period to a polluted period can be remarkably fast. Wang et al. (2016) further proposed that the aqueous-phase reaction could be of great importance for secondary aerosol formation during the severe haze stage. Moreover, some other studies also reveal key interactions between haze formation and meteorological conditions, such as humidity and the planetary boundary layer (PBL) (Wang et al., 2013; Ding et al., 2016; Li et al., 2017; Tie et al., 2017; Miao et al., 2018).
Aerosol properties can provide essential information on primary emission sources and the atmospheric evolution of atmospheric aerosols (Guo et al., 2014; Peng et al., 2014). For example, particle hygroscopicity is a key factor in identifying the species of ambient aerosols and the changes aerosols undergo during the aging process (Chang et al., 2010; Guo et al., 2014; Peng et al., 2017). The hygroscopicity of ambient aerosols can be quantified by using the parameter kappa or the hygroscopic growth factor (HGF) (Petters and Kreidenweis, 2007). Particle effective density is also a key parameter to probe primary combustion emission and the aging of primary soot aerosols (Peng et al., 2016). Several measurements have previously investigated the particle density in Beijing via several filter-based methods (Hu et al., 2012; Yue et al., 2010), with a time resolution of about half a day. Few measurements have provided high time-resolution and size-resolved density information (Guo et al., 2014; Qiao et al., 2016), hindering the in-depth investigation of atmospheric aging processes. In addition, most of the previous observational studies focused on aerosol properties and haze formation in Beijing were carried out in summer and autumn (Guo et al., 2012, 2014; Huang et al., 2010; Sun et al., 2014). As the most severe haze episodes often occur during wintertime, more observation of aerosol properties in winter will be helpful for investigating the severe haze formation mechanism (Hu et al., 2016, 2017; Sun et al., 2013).
In this study, a field campaign was conducted to measure ambient particulate matter and gaseous concentration in Beijing from January to February 2015 to better understand the haze formation mechanism during wintertime in Beijing. The physical and chemical properties of ambient aerosols during different pollution stages, including particle size distributions, size-resolved effective density measurements, and chemical composition, were simultaneously measured during the campaign. The contribution of secondary aerosol formation to the PM2.5 mass concentration in Beijing was evaluated according to ambient gas concentrations and aerosol properties. Furthermore, the pollution features observed during winter and autumn were further compared to better understand the haze formation mechanisms in different seasons.
All measurements were conducted at the PeKing University urban atmosphere Environment monitoRing Station (PKUERS; 39∘59′21′′ N, 116∘18′25′′ E) located in the northwestern Beijing urban area. The site is located outside the 4th Ring Road with no significant stationary sources or mobile sources within 200 m and is likely representative of the Beijing urban area (Wu et al., 2008). The instruments were located in an air-conditioned room on the roof of a building about 15 m above ground level. A suite of state-of-the-art instruments was deployed to simultaneously measure gaseous species and aerosol properties, including particle mass concentration, size distribution, chemical composition, and size-resolved effective density and hygroscopicity.
A tapered element oscillating microbalance (TEOM, 1400a, Thermo, USA) with a PM2.5 cyclone inlet was used to measure the ambient PM2.5 mass concentration. The sampling flow was 16.7 L min−1, of which 1 L min−1 was introduced to the instrument. The TEOM measures the mass collected on a filter by monitoring the corresponding frequency changes of a tapered element. As the mass concentration increased on the replaceable filter, the tube's natural frequency of oscillation decreased. The mass concentration was determined from the change in the oscillation frequency.
An Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) was employed to measure the size-resolved chemical compositions of submicron particles (Hu et al., 2016). The HR-ToF-AMS operated in 5 min cycles, including a V mode to obtain the mass concentrations of non-refractory species such as ammonium, sulfate, nitrate, organics, and chloride; a W mode to obtain high-resolution mass spectral data; and a particle time-of-flight mode to determine size distributions of species measured with the V mode. The HR-ToF-AMS was calibrated for inlet flow, ionization efficiency, and particle size at the beginning, middle, and end of the measurements (Hu et al., 2016). The calibration of ionization efficiency was conducted with size-selected pure ammonium nitrate particles. The detection limit was defined as 3 times the standard deviations of the observed signals. The detection limits of sulfate, nitrate, ammonium, chloride, and organics were determined to be 0.008, 0.004, 0.026, 0.004, and 0.033 µg m−3, respectively.
A scanning mobility particle sizer (SMPS) and a nano-SMPS were used to concurrently measure the number size distribution of particles between 3 and 600 nm in the ambient air (Wang et al., 2013). Ambient aerosols were sampled at a rate of 16.7 L min−1 through a 3 m long, thermally insulated 0.5 in. stainless steel tube. Before being measured by the SMPS and nano-SMPS, an airflow of 1.8 L min−1 passed through a series of Nafion driers (Perma Pure, Inc.) to reduce the relative humidity of the aerosols to less than 30 %. In the SMPS and nano-SMPS system, polydisperse aerosols were brought to charge equilibrium and passed through the differential mobility analyzer (DMA) and condensation particle counter (CPC) to determine particle size and concentration. The sheath flow rate was maintained at 3 L min−1 for the DMA and 15 L min−1 for nano-DMA. The particle size was calibrated with monodisperse polystyrene latex spheres (PSLs, Duke Scientific, Palo Alto, California, USA) with nominal diameters of 100–500 nm.
A combination DMA–APM (aerosol particle mass analyzer, model 3600, Kanomax Inc., Japan) system was employed to measure the particle size-resolved density (Qiu et al., 2012; Khalizov et al., 2009, 2013). Ambient airflow first passed through a DMA, where the voltage was fixed to provide continuous monodisperse particles. Then, the monodisperse aerosol flow passed through an APM, where the particle mass distribution could be obtained. Effective density is defined as the measured mass over the volume where the particle is assumed to be a sphere based on the measured particle size; therefore, compounds with the same mass but different shapes may have extremely different effective densities. The effective densities for five particle sizes were obtained hourly. Since the mobility diameter corresponds to a unique voltage, if the applied voltage and effective density of PSLs are known, the effective density of the sample aerosols can be calculated by the following equation:
where VAPM and VAPM, PSL are the fitted peak voltages in APM corresponding to the masses of sample and PSL particles, respectively. The material density of the PSL particles is 1.054 g cm−3. Each effective density distribution scans from 0.1 to 2.2 g cm−3. Effective density peaks at 1.76, 1.78, and 1.73 g cm−3 correspond to ammonium sulfate, ammonium bisulfate, and ammonium nitrate, respectively. The effective density of organic aerosols can vary significantly due to the numerous compounds in this category, the different emission sources, and the distinct structures of the compounds. Turpin and Lim (2001) reported that the density of secondary organic aerosols from the oxidation of aromatics and alkanes was between 1.20 and 1.40 g cm−3. The effective densities of fresh black carbon (BC), mineral dust, and sea salt were found to be 0.1–0.6, 2.65, and 2.2 g cm−3, respectively (Geller et al., 2006; Khalizov et al., 2009). This methodology has been utilized and discussed previously (Levy et al., 2014).
The hygroscopicity of ambient aerosols was parameterized by kappa (κ), which is derived using the following formula (Petters and Kreidenweis, 2007):
where M, ρ, ν, and ε, are the molecular weight, molecular densities, van 't Hoff factor, and volume fraction of the component in the aerosols mass, respectively. The subscripts “s” and “w” represent the parameters for solute and water, respectively. Using the volume-weighted average fraction determined by the AMS measurement and the εi values of 0.0, 0.09, 0.48, 0.58, 0.55, and 0.246 for BC, organic aerosols, , , , and Cl−, respectively, the chemical composition-derived kappa was calculated.
The PBL was determined by utilizing the National Oceanic and Atmospheric Administration's (NOAA) Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model. HYSPLIT identifies the mixing layer height, which we assumed to be the PBL, as the height at which the potential temperature is at least 2 ∘C greater than the minimum potential temperature.
3.1 Observed haze cycles
Four PM pollution cycles were documented during the measurement in the winter of 2015 (Fig. S1 in the Supplement). Frequently, the large-scale meteorology governed the length and severity of haze events due to the periodical occurrence of exceedingly stagnant air masses and strong, cleansing winds. During the observation period, a low PM2.5 concentration was accomplished with strong northerly winds. The episodes with the highest PM2.5 concentration (i.e., 22 and 26 January) exhibited calm winds and low PBL heights (Fig. S1), which trapped primary emission near their sources and secondary pollutants within the formation area. Temporal evolutions of the particle mass and number concentration, chemical composition, and size distribution and the average diameter of the first and second pollution cycles (between 21 and 27 January 2015) are illustrated in Fig. 1. The first haze event began with a new particle formation (NPF) event (Zhang et al., 2004), which was followed by the continuous particle growth over 3 d (Fig. 1a). On the clean days (i.e., 21 and 27 January) the strong wind and the high PBL diluted the pollutants both vertically and horizontally (Fig. S1). The total number concentration exceeded 300 000 cm−3 during the NPF event, but this value generally decreased after the NPF event and remained around 100 000 cm−3 (Fig. 1c). The average diameter of all ambient particles was only about 10 nm during the NPF event and steadily increased until their average diameter was near 150 nm (Fig. 1d). During nighttime of 22 January, the particle mass concentration increased but the diameter decreased, which is possibly due to the combination of the primary emission of smaller particles and the compressed PBL during nighttime. Minor mass growth was observed on 21 January (Fig. 1b), with an increase from 8 to 30 µg m−3. The most rapid mass concentration increase occurred on 22 January, with an increase from 30 to 300 µg m−3.
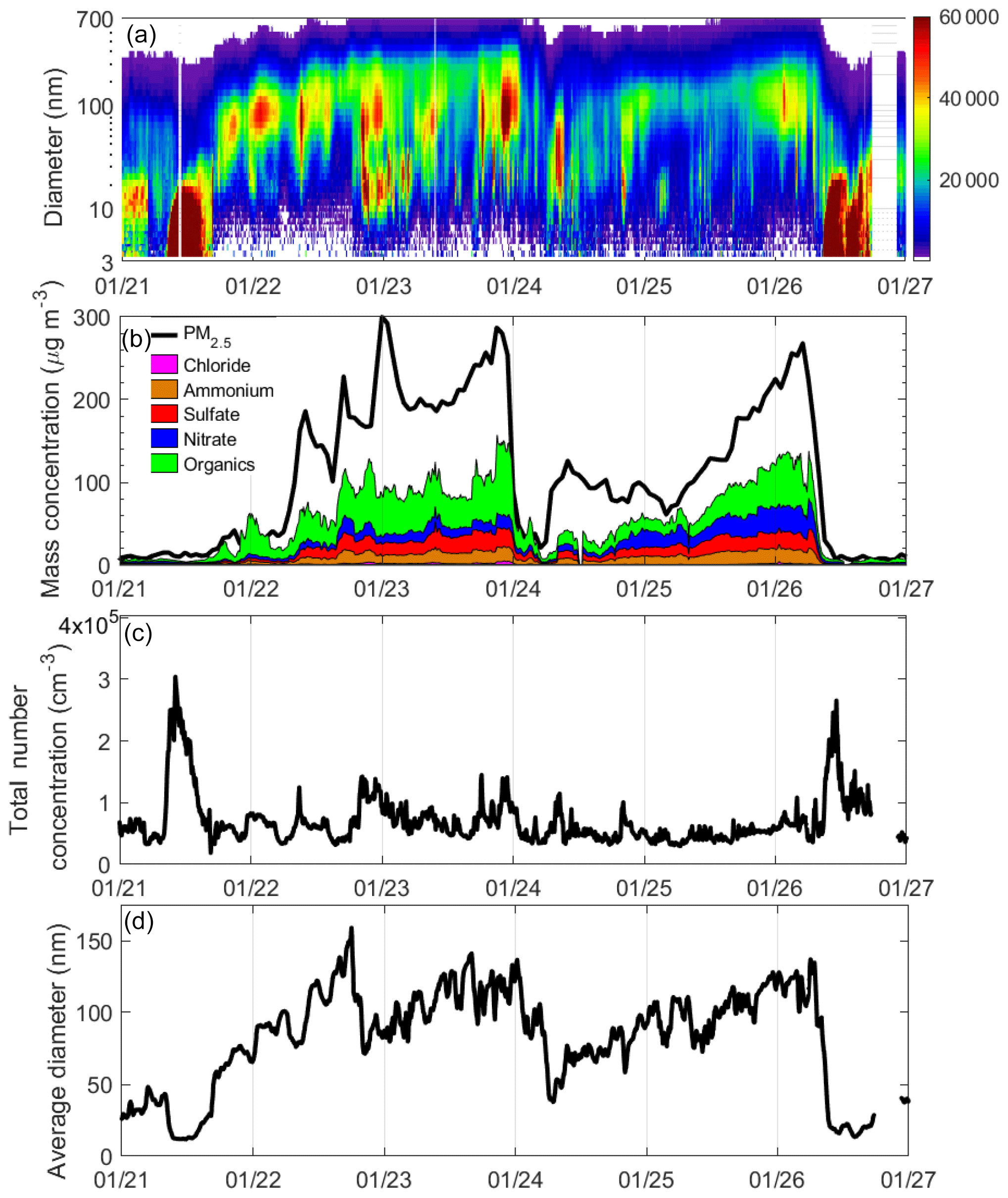
Figure 1Temporal evolutions of (a) the number size distribution, (b) PM2.5 mass concentration and chemical composition, (c) the total number concentration, and (d) the mean diameter between 21 and 27 January 2015. Dates in this figure are given in the month/day format.
During the early morning of January 24, a weak front passed through the sampling site, which led to weak winds from the north and an increased PBL to around 700 m (Fig. S1), diluting the ambient PM2.5 concentration (to about 30 µg m−3 on average during the daytime on 24 January). Meanwhile, a large amount of primary emission in the morning rush hour lowered the average particle diameter to less than 50 nm. After the frontal passage, the winds shifted to the south, which recirculated the polluted air mass back to the sampling site. Though the mass concentration exhibited two distinct pollution cycles, the air mass of this event as a whole was likely the same. On 25 January, weak southerly winds continued, and the boundary layer height decreased to about 300 m. The stagnant conditions led to a rapid accumulation of pollutants. The total number and mass concentrations of ambient PM2.5 gradually grew to 130 nm and 250 µg m−3 during the following 2 d, respectively (Fig. 1b, d). Meanwhile, the aerosol constituents generally remained similar during this episode (Fig. 1b).
3.2 Aerosol chemical composition, hygroscopicity, and density
Organic aerosols accounted for the largest percentage of the aerosol mass during both the clean (58 %, 21 January) and the polluted periods (52 %, 23 January). Nitrate, ammonium, and sulfate accounted for 12 %, 12 %, and 16 %, respectively, during the clean periods, and remained relatively constant at 20 %, 14 %, and 12 %, respectively, throughout the polluted period of the first and the second cycles. The contribution of chloride to PM remained constant at about 2 %.
The average kappa values of ambient aerosols between 21 January and 29 January 2015 are shown in Fig. 3a. Both the diurnal cycles and the overall trend governed by the pollution cycle are evident in the aerosol hygroscopicity. Diurnally, the least hygroscopic aerosols occurred overnight, while the most hygroscopic aerosols were found near sunset. The reduced nighttime hygroscopicity was concurrently observed with the transiently elevated organic aerosol mass concentration (Fig. 1) and reduced effective density of the larger particles (Fig. 3b), which may be due to the increase in vehicle emissions of primary organic aerosols and black carbon in the city overnight. The increased hygroscopicity in the mid-morning and early afternoon hours (09:00 LT to 13:00 LT) was due to the rapid increase in the mass concentrations of nitrate, ammonium, and sulfate aerosols (Fig. 1b), consistent with previous studies in Mexico City which showed that ammonium nitrate rapidly formed in the morning (Hennigan et al., 2008). From the onset of the first pollution cycle on 21 January to the morning of 25 January, the kappa value increased from 0.15 to 0.42.
The effective densities for 81, 97, 151, and 240 nm particles are exhibited in Fig. 3b. During the clean phase (e.g., 22 January), all four particle sizes exhibited effective density values that clustered around 1.37 g cm−3. However, as the haze event developed, the effective densities of particles with the four sizes became increasingly differentiated. When the PM2.5 mass concentration decreased at the end of the haze event (i.e., 27 January), the effective density of the particles again clustered around 1.36 g cm−3. The lower average effective density during the clean period can be attributed to the organic-dominated aerosol composition as well as fresh low-density BC emission. Furthermore, the increasing effective density corresponds to the increasing sulfate and nitrate composition that was observed as the haze events progressed (Fig. 1b). Generally, the effective density distributions were unimodal, indicating that the aerosols were internally mixed. The weighted average density, i.e., the effective density weighted by the total number concentration of particles near each size of the four particles sizes, increased throughout the haze event, but this was primarily due to the changes in the particles smaller than 100 nm. The effective density of smaller particles (i.e., 81 and 97 nm) increased as the ambient atmosphere became more polluted, whereas the densities of larger particles (i.e., 151 and 240 nm) remained near 1.37 g cm−3. Figure 4 shows the diurnal variation of the effective densities of 81, 97, 151, and 240 nm particles during the entire observational campaign as well as the polluted period and the clean period. An evident diurnal variation of effective density was observed, i.e., higher density overnight, lower density during the morning and evening rush hours, and moderately stable density during the daytime. Over the entire campaign, the lowest density was observed after the evening rush hour (i.e., 1.38 g cm−3 for 81 nm particles), and the highest density (∼1.41 g cm−3 for 81 nm particles) was observed in the early morning before the intensification of heavy traffic. During high-traffic periods (i.e., 06:00–09:00 LT and 16:00–20:00 LT), the decreasing effective density was likely due to the increasing concentrations of BC and primary organic aerosol (POA). Overnight, the effective density increased due to the formation of inorganic species on aerosols. The variations in effective density were suppressed during the polluted days. This may be because the newly emitted particles were being mixed into a relatively higher concentration of particles (Levy et al., 2014). Therefore, the average effective density of the entire air mass was not as sensitive to the primary emitted particles during rush hours.
3.3 Comparisons of aerosol properties in autumn and winter
In Fig. 5, a comparison of the mass concentration, total number concentration, and the average diameter of ambient particles from representative pollution cycles from the autumn 2013 and winter 2015 field campaigns are displayed (Guo et al., 2014). The chemical composition during the clean and polluted periods from both campaigns is also exhibited in Fig. 2. Both seasonal haze events began with NPF events, which were typically followed by particle growth over several days. Overall various properties (i.e., particle size, number concentration, chemical composition, and hygroscopicity) appear to be similar in the autumn and winter campaigns, with a few notable caveats.
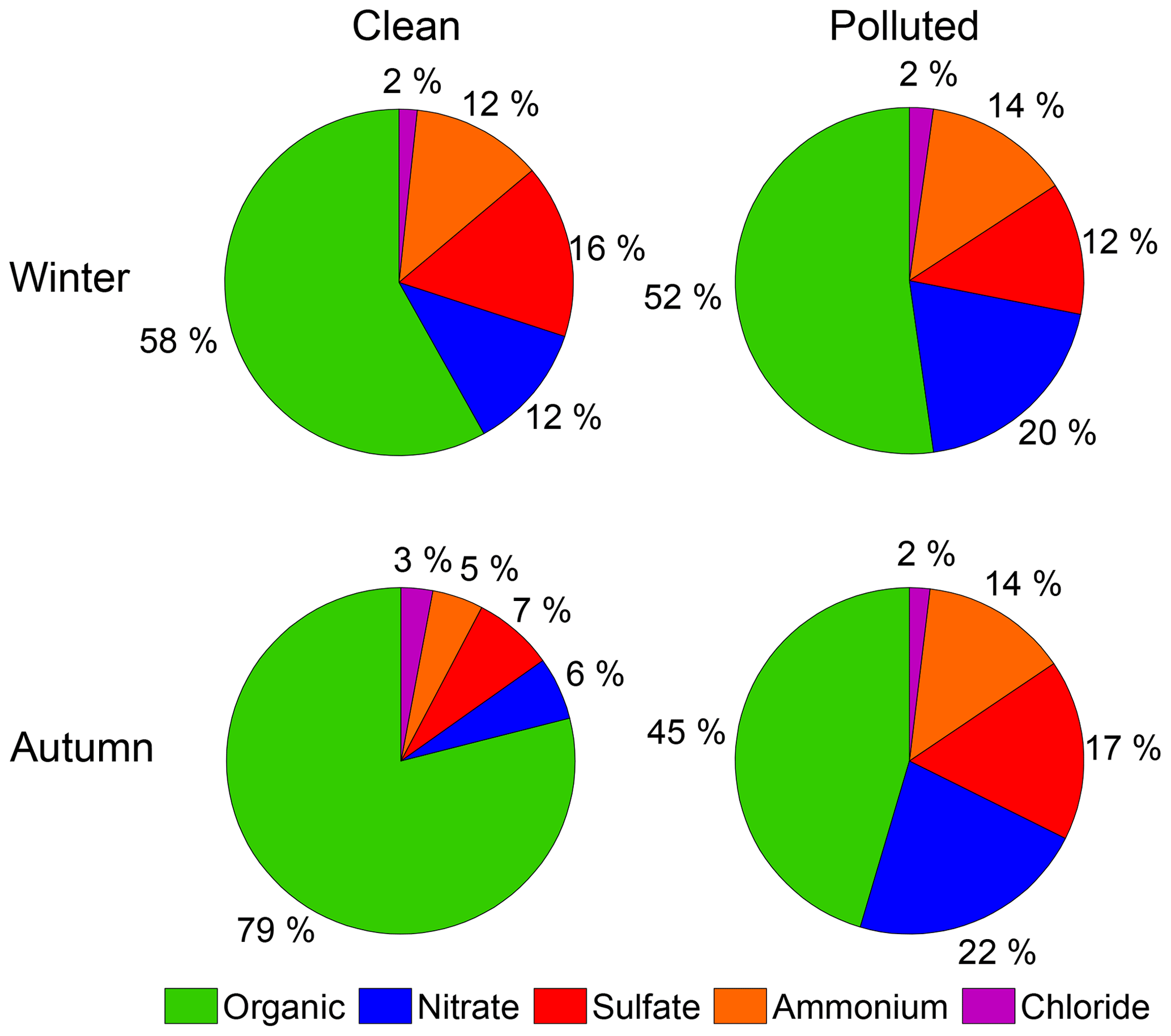
Figure 2A comparison of the chemical composition during the clean and polluted periods from the autumn 2013 and winter 2015 field campaigns.
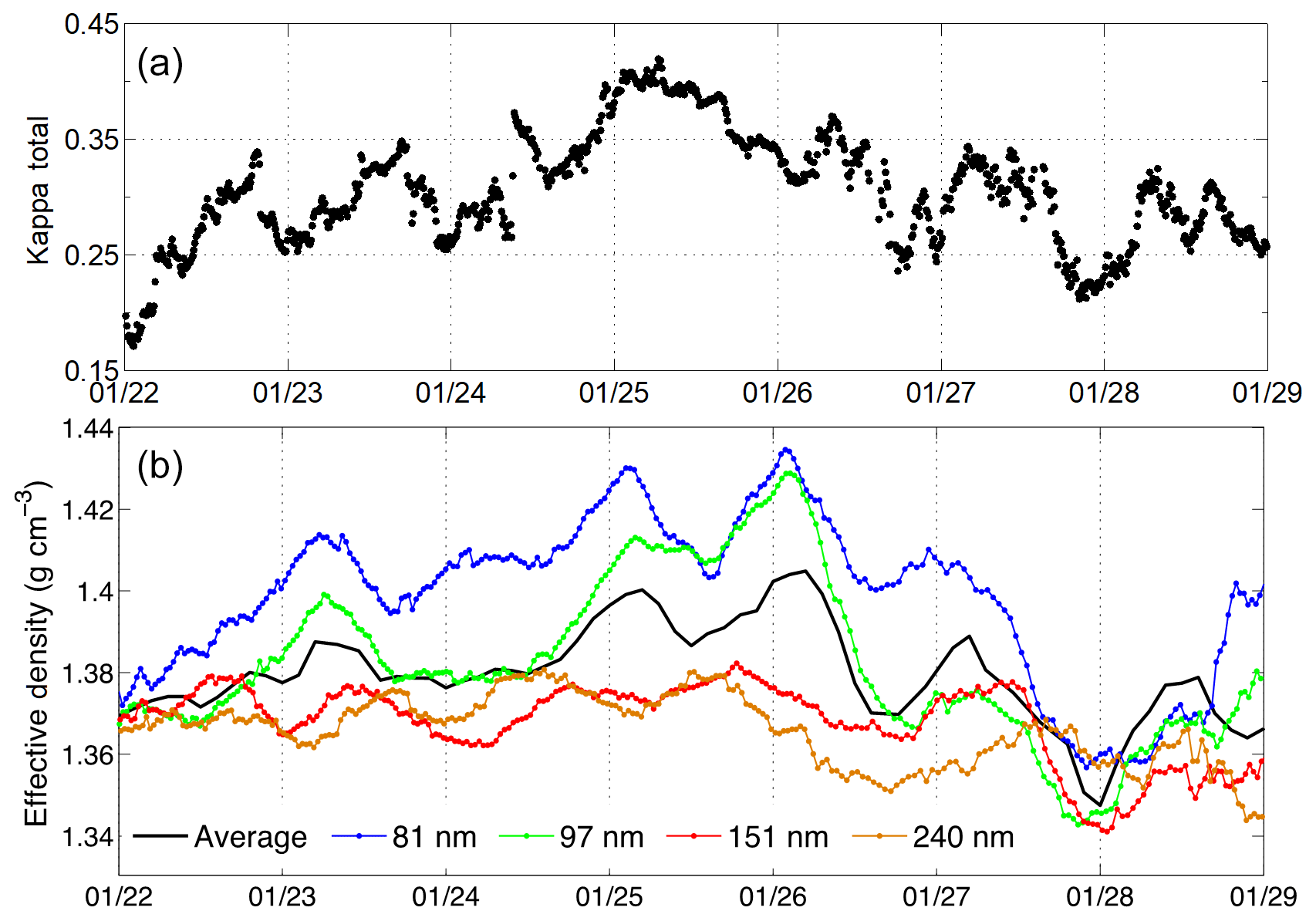
Figure 3(a) The determined hygroscopicity based on chemical composition (i.e., kappa) of the aerosols. (b) Temporal evolutions of the effective density of particles with a diameter of 81 (blue), 97 (green), 151 (red) and 240 nm (orange) and the weighted average of the four particle sizes (black) between 21 and 29 January 2015. Dates in this figure are given in the month/day format.
The particle growth and formation were more efficient in the autumn campaign: i.e., the NPF events resulted in a higher total number concentration in the autumn, and the final average particle diameter was near 180 nm compared to 150 nm in the winter, which may be attributable to the stronger solar irradiation in autumn than in winter. Studies have demonstrated that large particle mass growth always occurs concurrently with elevated daily ozone concentrations and solar irradiation (Guo et al., 2014; Peng et al., 2016), particularly during clean periods, suggesting the importance of photochemical activity in the growth of particles. Furthermore, such faster particle growth in autumn was observed when the PBL in autumn was nearly twice as high compared with that in winter (Fig. S1). If the PBL was the same, the particle growth and the secondary aerosol formation in autumn would likely be even more efficient. Organics were the dominant species during clean episodes in both autumn and winter at 79 % and 58 %, respectively (Fig. 2), while inorganic species, i.e., sulfate, nitrate, and ammonium, were more abundant in the polluted episodes in both campaigns. More organic aerosols were observed in the winter polluted episode, likely due to the increased POA emissions from residential heating during winter. The trends in the hygroscopicity of particles are similar during the two seasons, with the only significant differences occurring on the clean days (i.e., 21 and 27 January, 25 September, and 1 October). The hygroscopicity of the autumn aerosols (25 September) during the clean phase was much lower (0.2) than the hygroscopicity of the wintertime aerosols (0.35). This is likely due to the higher proportion of sulfate in PM during the winter (7 % in the autumn, 16 % in the winter) caused by the higher SO2 emission from residential heating in the winter and the lower PBL (1100 m compared to 2200 m; Fig. S1), which suppressed the horizontal dilution of pollutants and thereby accelerated sulfate formation (Wang et al., 2016).
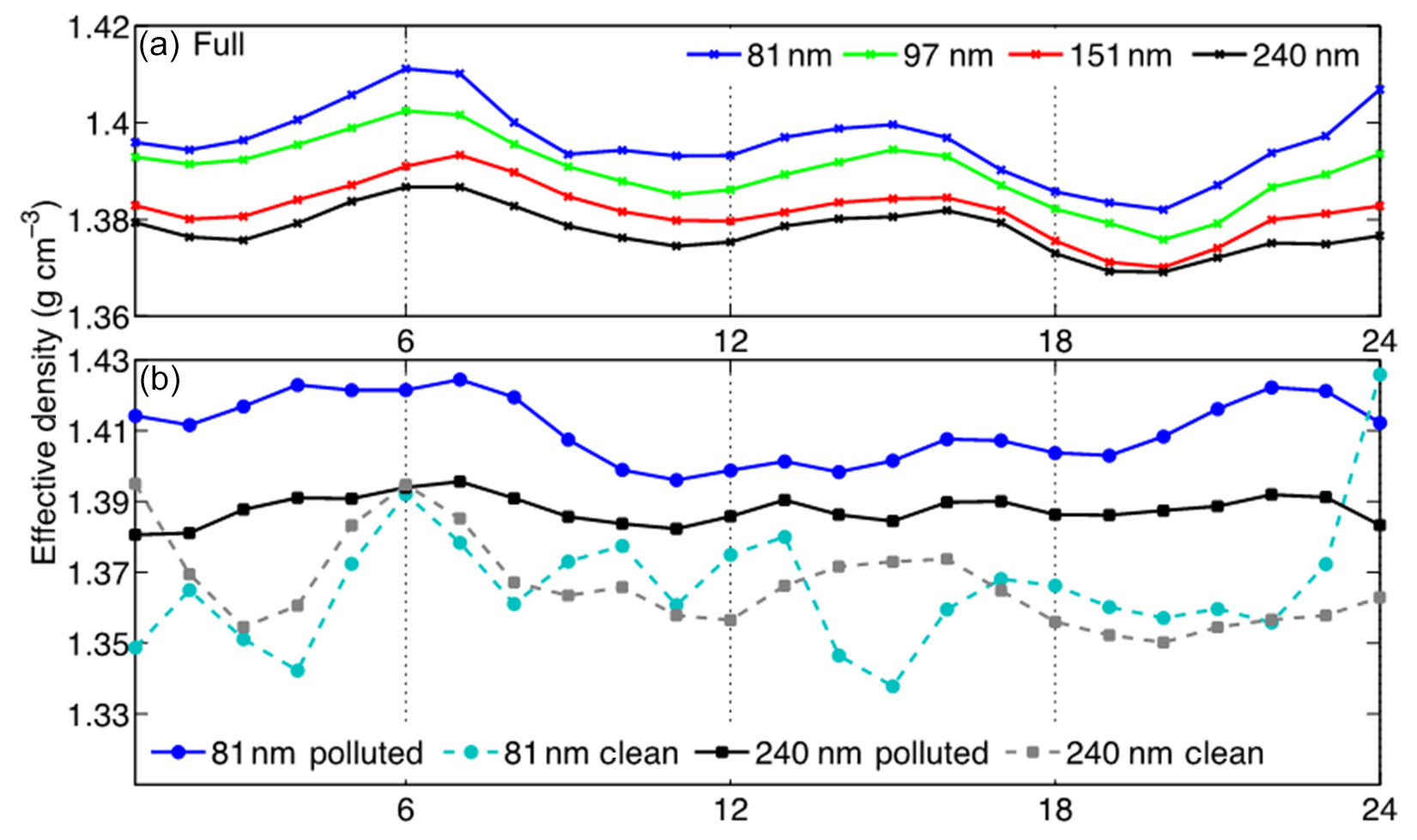
Figure 4(a) The diurnal variation of the effective density (g cm−3) of the four particle sizes from the full observational period. (b) The effective density (g cm−3) diurnal cycle of the 81 nm (blue circles) and 240 nm (black squares) particles during the clean (dashed, lighter colors) and polluted (solid, darker colors) periods.

Figure 5A comparison of the (a) mass concentration, (b) total number concentration, (c) average diameter, and (d) hygroscopicity between autumn 2013 (blue, bottom axis) and winter 2015 (black, top axis). Dates in this figure are given in the month/day format.
We have evaluated the influence of meteorology, local emissions, and aerosol processes on severe haze events in Beijing during winter by conducting comprehensive aerosol properties measurements. We show that the periodic cycles of haze episodes during the winter in Beijing are regulated by meteorological conditions. Formation of severe haze is comprised of two distinct processes of secondary aerosol formation, i.e., the nucleation that initially produces high concentrations of nanoparticles and the subsequent continuous growth from the nucleation mode particles to submicron particles. Our analysis of the aerosol chemical compositions suggests that organic aerosols are primarily responsible for producing the nucleation mode particles, while secondary organic aerosols and inorganic salts contribute jointly to particle growth. The combination of high aerosol nucleation potential and efficient subsequent growth over several days uniquely differentiates the severe PM2.5 episodes in Beijing from those typically observed in other regions worldwide. The average effective density and kappa value of ambient particles are approximately 1.37 g cm−3 and 0.25 during the clean days and 1.42 g cm−3 and 0.4 during the severe haze episodes, respectively. The higher effective density and kappa value during hazy days indicate the formation of secondary inorganic species during the continuous growth of nucleation mode particles.
From the perspective of pollution control, it may be feasible to suppress the aerosol growth processes to reduce the PM2.5 levels in Beijing. Our results imply that the reductions in the emissions of the aerosol precursor gases, i.e., volatile organic compounds, NOx, and SO2, are critical for suppressing aerosol growth and thereby the remediation of haze pollution in Beijing. Such a viewpoint of severe haze formation is critical for improving the formulation of effective regulatory policies by decision makers at the central and local government levels.
The data set is available upon request by contacting Renyi Zhang (renyi-zhang@tamu.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-14329-2019-supplement.
MH, SG, ZW, and RZ organized the field campaign. MLZ, JP, WM, DS, JZ, and ZD conducted the online measurement and analyzed the data. MLZ and JP wrote the paper with input from RZ. All authors contributed to the discussion of the results.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Regional transport and transformation of air pollution in eastern China”. It is not associated with a conference.
Wilmarie Marrero-Ortiz acknowledges support by the Louis Stokes Alliance for Minority Participation – Bridge to the Doctorate and a National Science Foundation Graduate Research Fellowship Program (GRFP) fellowship.
This research has been supported by the National Natural Science Foundation of China (grant nos. 91544214 and 91844301), the National Research Program for Key Issues in Air Pollution Control (grant no. DQGG0103), the National Key Research and Development Program of China (grant no. 2016YFC0202000: Task 3), the Ministry of Science and Technology of China (grant no. 2013CB955800), and the Robert A. Welch Foundation (grant no. A-1417).
This paper was edited by Leiming Zhang and reviewed by two anonymous referees.
An, Z., Huang, R.-J., Zhang, R., Tie, X., Li, G., Cao, J., Zhou, W., Shi, Z., Han, Y., Gu, Z., and Ji, Y.: Severe haze in Northern China: A synergy of anthropogenic emissions and atmospheric processes, P. Natl. Acad. Sci. USA, 116, 8657–8666, https://doi.org/10.1073/pnas.1900125116, 2019.
Chang, D., Song, Y., and Liu, B.: Visibility trends in six megacities in China 1973–2007, Atmos. Res., 94, 161–167, 2009.
Chang, R. Y.-W., Slowik, J. G., Shantz, N. C., Vlasenko, A., Liggio, J., Sjostedt, S. J., Leaitch, W. R., and Abbatt, J. P. D.: The hygroscopicity parameter (κ) of ambient organic aerosol at a field site subject to biogenic and anthropogenic influences: relationship to degree of aerosol oxidation, Atmos. Chem. Phys., 10, 5047–5064, https://doi.org/10.5194/acp-10-5047-2010, 2010.
Ding, A. J., Huang, X., Nie, W., Sun, J. N., Kerminen, V. M., Petaja, T., Su, H., Cheng, Y. F., Yang, X. Q., Wang, M. H., Chi, X. G., Wang, J. P., Virkkula, A., Guo, W. D., Yuan, J., Wang, S. Y., Zhang, R. J., Wu, Y. F., Song, Y., Zhu, T., Zilitinkevich, S., Kulmala, M., and Fu, C. B.: Enhanced haze pollution by black carbon in megacities in China, Geophys. Res. Lett., 43, 2873–2879, 2016.
Geller, M., Biswas, S., and Sioutas, C.: Determination of particle effective density in urban environments with a differential mobility analyzer and aerosol particle mass analyzer, Aerosol Sci. Tech., 40, 709–723, https://doi.org/10.1080/02786820600803925, 2006.
Guo, S., Hu, M., Wang, Z. B., Slanina, J., and Zhao, Y. L.: Size-resolved aerosol water-soluble ionic compositions in the summer of Beijing: implication of regional secondary formation, Atmos. Chem. Phys., 10, 947–959, https://doi.org/10.5194/acp-10-947-2010, 2010.
Guo, S., Hu, M., Guo, Q. F., Zhang, X., Zheng, M., Zheng, J., Chang, C. C., Schauer, J. J., and Zhang, R. Y.: Primary Sources and Secondary Formation of Organic Aerosols in Beijing, China, Environ. Sci. Technol., 46, 9846–9853, 2012.
Guo, S., Hu, M., Guo, Q., Zhang, X., Schauer, J. J., and Zhang, R.: Quantitative evaluation of emission controls on primary and secondary organic aerosol sources during Beijing 2008 Olympics, Atmos. Chem. Phys., 13, 8303–8314, https://doi.org/10.5194/acp-13-8303-2013, 2013.
Guo, S., Hu, M., Zamora, M. L., Peng, J. F., Shang, D. J., Zheng, J., Du, Z. F., Wu, Z., Shao, M., Zeng, L. M., Molina, M. J., and Zhang, R. Y.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Hallquist, M., Munthe, J., Hu, M., Wang, T., Chan, C. K., Gao, J., Boman, J., Guo, S., Hallquist, A. M., Mellqvist, J., Moldanova, J., Pathak, R. K., Pettersson, J. B. C., Pleijel, H., Simpson, D., and Thynell, M.: Photochemical smog in China: scientific challenges and implications for air-quality policies, Nat. Sci. Rev., 3, 401–403, https://doi.org/10.1093/nsr/nww080, 2016.
Hennigan, C. J., Sullivan, A. P., Fountoukis, C. I., Nenes, A., Hecobian, A., Vargas, O., Peltier, R. E., Case Hanks, A. T., Huey, L. G., Lefer, B. L., Russell, A. G., and Weber, R. J.: On the volatility and production mechanisms of newly formed nitrate and water soluble organic aerosol in Mexico City, Atmos. Chem. Phys., 8, 3761–3768, https://doi.org/10.5194/acp-8-3761-2008, 2008.
Hu, M., Peng, J., Sun, K., Yue, D., Guo, S., Wiedensohler, A., and Wu, Z.: Estimation of Size-Resolved Ambient Particle Density Based on the Measurement of Aerosol Number, Mass, and Chemical Size Distributions in the Winter in Beijing, Environ. Sci. Technol., 46, 9941–9947, https://doi.org/10.1021/es204073t, 2012.
Hu, M., Guo, S., Peng, J., and Wu, Z.: Insight into characteristics and sources of PM2.5 in the Beijing-Tianjin-Hebei region, China, Nat. Sci. Rev., 2, 257–258, 2015.
Hu, W., Hu, M., Hu, W., Jimenez, J. L., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Zeng, L., and Shao, M.: Chemical composition, sources and aging process of sub-micron aerosols in Beijing: contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, https://doi.org/10.1002/2015JD024020, 2016.
Hu, W., Hu, M., Hu, W.-W., Zheng, J., Chen, C., Wu, Y., and Guo, S.: Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 17, 9979–10000, https://doi.org/10.5194/acp-17-9979-2017, 2017.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Huang, X.-F., He, L.-Y., Hu, M., Canagaratna, M. R., Sun, Y., Zhang, Q., Zhu, T., Xue, L., Zeng, L.-W., Liu, X.-G., Zhang, Y.-H., Jayne, J. T., Ng, N. L., and Worsnop, D. R.: Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer, Atmos. Chem. Phys., 10, 8933–8945, https://doi.org/10.5194/acp-10-8933-2010, 2010.
Khalizov, A. F., Zhang, R., Zhang, D., Xue, H., Pagels, J., and McMurry, P. H.: Formation of highly hygroscopic soot aerosols upon internal mixing with sulfuric acid vapor, J. Geophys. Res.-Atmos., 114, D05208, https://doi.org/10.1029/2008JD010595, 2009.
Khalizov, A. F., Lin, Y., Qiu, C., Guo, S., Collins, D., and Zhang, R. Y.: Role of OH-Initiated Oxidation of Isoprene in Aging of Combustion Soot, Environ. Sci. Technol., 47, 2254–2263, https://doi.org/10.1021/Es3045339, 2013.
Levy, M. E., Zhang, R. Y., Zheng, J., Tan, H. B., Wang, Y., Molina, L. T., Takahama, S., Russell, L. M., and Li, G. H.: Measurements of submicron aerosols at the California-Mexico border during the Cal-Mex 2010 field campaign, Atmos. Environ., 88, 308–319, 2014.
Li, Z. Q., Guo, J. P., Ding, A. J., Liao, H., Liu, J. J., Sun, Y. L., Wang, T. J., Xue, H. W., Zhang, H. S., and Zhu, B.: Aerosol and boundary-layer interactions and impact on air quality, Natl. Sci. Rev., 4, 810–833, 2017.
Miao, Y. C., Liu, S. H., Guo, J. P., Huang, S. X., Yan, Y., and Lou, M. Y.: Unraveling the relationships between boundary layer height and PM2.5 pollution in China based on four-year radiosonde measurements, Environ. Pollut., 243, 1186–1195, 2018.
Peng, J., Hu, M., Guo, S., Du, Z., Shang, D., Zheng, J., Zheng, J., Zeng, L., Shao, M., Wu, Y., Collins, D., and Zhang, R.: Ageing and hygroscopicity variation of black carbon particles in Beijing measured by a quasi-atmospheric aerosol evolution study (QUALITY) chamber, Atmos. Chem. Phys., 17, 10333–10348, https://doi.org/10.5194/acp-17-10333-2017, 2017.
Peng, J. F., Hu, M., Wang, Z. B., Huang, X. F., Kumar, P., Wu, Z. J., Guo, S., Yue, D. L., Shang, D. J., Zheng, Z., and He, L. Y.: Submicron aerosols at thirteen diversified sites in China: size distribution, new particle formation and corresponding contribution to cloud condensation nuclei production, Atmos. Chem. Phys., 14, 10249–10265, https://doi.org/10.5194/acp-14-10249-2014, 2014.
Peng, J. F., Hu, M., Guo, S., Du, Z. F., Zheng, J., Shang, D. J., Zamora, M. L., Zeng, L. M., Shao, M., Wu, Y. S., Zheng, J., Wang, Y., Glen, C. R., Collins, D. R., Molina, M. J., and Zhang, R. Y.: Markedly enhanced absorption and direct radiative forcing of black carbon under polluted urban environments, P. Natl. Acad. Sci. USA, 113, 4266–4271, https://doi.org/10.1073/pnas.1602310113, 2016.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Qiao, K., Wu, Z. J., Pei, X. Y., Liu, Q. Y., Shang, D. J., Zheng, J., Du, Z. F., Zhu, W. F., Wu, Y. S., Lou, S. R., Guo, S., Chan, C. K., Pathak, R. K., Hallquist, M., and Hu, M.: Size-resolved effective density of submicron particles during summertime in the rural atmosphere of Beijing, China, J. Environ. Sci., 73, 69–77, 2018.
Qiu, C., Khalizov, A. F., and Zhang, R. Y.: Soot Aging from OH-Initiated Oxidation of Toluene, Environ. Sci. Technol., 46, 9464–9472, https://doi.org/10.1021/Es301883y, 2012.
Su, S., Xiao, R., Jiang, Z., and Zhang, Y.: Characterizing landscape pattern and ecosystem service value changes for urbanization impacts at an eco-regional scale, Appl. Geogr., 34, 295–305, https://doi.org/10.1016/j.apgeog.2011.12.001, 2012.
Sun, Y., Jiang, Q., Wang, Z., Fu, P., Li, J., Yang, T., and Yin, Y.: Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013, J. Geophys. Res.-Atmos., 119, 4380–4398, 2014.
Sun, Y. L., Wang, Z. F., Fu, P. Q., Yang, T., Jiang, Q., Dong, H. B., Li, J., and Jia, J. J.: Aerosol composition, sources and processes during wintertime in Beijing, China, Atmos. Chem. Phys., 13, 4577–4592, https://doi.org/10.5194/acp-13-4577-2013, 2013.
Tie, X. X., Huang, R. J., Cao, J. J., Zhang, Q., Cheng, Y. F., Su, H., Chang, D., Poschl, U., Hoffmann, T., Dusek, U., Li, G. H., Worsnop, D. R., and O'Dowd, C. D.: Severe Pollution in China Amplified by Atmospheric Moisture, Sci. Rep., 7, 15760, https://doi.org/10.1038/s41598-017-15909-1, 2017.
Turpin, B. J. and Lim, H. J.: Species contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass, Aerosol Sci. Tech., 35, 602–610, https://doi.org/10.1080/02786820119445, 2001.
Wang, G. H., Zhang, R. Y., Gomez, M. E., Yang, L. X., Zamora, M. L., Hu, M., Lin, Y., Peng, J. F., Guo, S., Meng, J. J., Li, J. J., Cheng, C. L., Hu, T. F., Ren, Y. Q., Wang, Y. S., Gao, J., Cao, J. J., An, Z. S., Zhou, W. J., Li, G. H., Wang, J. Y., Tian, P. F., Marrero-Ortiz, W., Secrest, J., Du, Z. F., Zheng, J., Shang, D. J., Zeng, L. M., Shao, M., Wang, W. G., Huang, Y., Wang, Y., Zhu, Y. J., Li, Y. X., Hu, J. X., Pan, B., Cai, L., Cheng, Y. T., Ji, Y. M., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, 2016.
Wang, J. Y., Da, L. J., Song, K., and Li, B. L.: Temporal variations of surface water quality in urban, suburban and rural areas during rapid urbanization in Shanghai, China, Environ. Pollut., 152, 387–393, 2008.
Wang, Y., Wan, Q., Meng, W., Liao, F., Tan, H., and Zhang, R.: Long-term impacts of aerosols on precipitation and lightning over the Pearl River Delta megacity area in China, Atmos. Chem. Phys., 11, 12421–12436, https://doi.org/10.5194/acp-11-12421-2011, 2011.
Wang, Y., Khalizov, A., Levy, M., and Zhang, R.: Light absorbing aerosols and their atmospheric impacts, Atmos. Environ., 81, 713–715, https://doi.org/10.1016/j.atmosenv.2013.09.034, 2013.
Wang, Y., Zhang, R., and Saravanan, R.: Asian pollution climatically modulates mid-latitude cyclones following hierarchical modeling and observational analysis, Nat. Commun., 4, 3098, https://doi.org/10.1038/ncomms4098, 2014.
Wang, Z. B., Hu, M., Wu, Z. J., Yue, D. L., He, L. Y., Huang, X. F., Liu, X. G., and Wiedensohler, A.: Long-term measurements of particle number size distributions and the relationships with air mass history and source apportionment in the summer of Beijing, Atmos. Chem. Phys., 13, 10159–10170, https://doi.org/10.5194/acp-13-10159-2013, 2013.
WHO (World Health Organization): Regional Office for Europe, & World Health Organization, Air quality guidelines: global update 2005: particulate matter, ozone, nitrogen dioxide, and sulfur dioxide, 3–11, 2006.
Wu, Z., Hu, M., Lin, P., Liu, S., Wehner, B., and Wiedensohler, A.: Particle number size distribution in the urban atmosphere of Beijing, China, Atmos. Environ., 42, 7967–7980, https://doi.org/10.1016/j.atmosenv.2008.06.022, 2008.
Yue, D., Hu, M., Wu, Z., Wang, Z., Guo, S., Wehner, B., Nowak, A., Achtert, P., Wiedensohler, A., and Jung, J.: Characteristics of aerosol size distributions and new particle formation in the summer in Beijing, J. Geophys. Res.-Atmos., 114, 1159–1171, 2010. Zhang, R., Suh, I., Zhao, J., Zhang, D., Fortner, E.C., Tie, X., Molina, L.T. and Molina, M.J.: Atmospheric new particle formation enhanced by organic acids. Science, 304, 1487-1490, doi:10.1126/science.1095139, 2004.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of urban fine particulate matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015.
Zheng, J., Hu, M., Peng, J. F., Wu, Z. J., Kumar, P., Li, M. R., Wang, Y. J., and Guo, S.: Spatial distributions and chemical properties of PM2.5 based on 21 field campaigns at 17 sites in China, Chemosphere, 159, 480–487, 2016.