the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Sep 2019
| 05 Sep 2019
Chamber-based insights into the factors controlling epoxydiol (IEPOX) secondary organic aerosol (SOA) yield, composition, and volatility
Emma L. D'Ambro
Siegfried Schobesberger
Cassandra J. Gaston
Felipe D. Lopez-Hilfiker
Ben H. Lee
Jiumeng Liu
Alla Zelenyuk
David Bell
Christopher D. Cappa
Taylor Helgestad
Alex Guenther
Jian Wang
Matthew Wise
Ryan Caylor
Jason D. Surratt
Theran Riedel
Noora Hyttinen
Vili-Taneli Salo
Galib Hasan
Theo Kurtén
John E. Shilling
Joel A. Thornton
We present measurements utilizing the Filter Inlet for Gases and Aerosols (FIGAERO) applied to chamber measurements of isoprene-derived epoxydiol (IEPOX) reactive uptake to aqueous acidic particles and associated secondary organic aerosol (SOA) formation. Similar to recent field observations with the same instrument, we detect two molecular components desorbing from the IEPOX SOA in high abundance: C5H12O4 and C5H10O3. The thermal desorption signal of the former, presumably 2-methyltetrols, exhibits two distinct maxima, suggesting it arises from at least two different SOA components with significantly different effective volatilities. Isothermal evaporation experiments illustrate that the most abundant component giving rise to C5H12O4 is semi-volatile, undergoing nearly complete evaporation within 1 h while the second, less volatile component remains unperturbed and even increases in abundance. We thus confirm, using controlled laboratory studies, recent analyses of ambient SOA measurements showing that IEPOX SOA is of very low volatility and commonly measured IEPOX SOA tracers such as C5H12O4 and C5H10O3, presumably 2-methyltetrols and C5-alkene triols or 3-MeTHF-3,4-diols, result predominantly from thermal decomposition in the FIGAERO-CIMS. We infer that other measurement techniques using thermal desorption or prolonged heating for analysis of SOA components may also lead to reported 2-methyltetrols and C5-alkene triols or 3-MeTHF-3,4-diol structures. We further show that IEPOX SOA volatility continues to evolve via acidity-enhanced accretion chemistry on the timescale of hours, potentially involving both 2-methyltetrols and organosulfates.
- Article
(1919 KB) - Full-text XML
- BibTeX
- EndNote





Aerosols less than 1 µm in diameter play particularly important roles in the Earth's radiative balance and air quality, a large fraction of which is organic carbonaceous material of biogenic origin (∼70 %) (Hallquist et al., 2009). Isoprene, with a global emission rate of 500 TgC yr−1 (Guenther et al., 2012), has the potential to form significant quantities of secondary organic aerosol (SOA). The ability of a volatile organic compound (VOC) to form SOA depends on either of two factors: the efficiency of its oxidative conversion to lower-volatility products that can partition to the condensed phase or the reaction of gas-phase oxidation products in the condensed phase to form products that remain in the condensed phase. Each of these two SOA formation mechanisms has been heavily studied in the case of isoprene.
The atmospheric oxidation of isoprene under low NO conditions typically proceeds through OH radicals, leading to the formation of first-generation isoprene hydroxy hydroperoxides (ISOPOOH) in high yield (70 %) (Paulot et al., 2009). The remaining double bond of isoprene can then undergo another OH addition, leaving a carbon-centered radical adjacent to a hydroperoxide moiety. This radical can internally rearrange to form an epoxydiol (IEPOX) at a yield of ∼70 %–80 % (St Clair et al., 2016) or undergo the addition of O2 to form a peroxy radical. The peroxy radical undergoes bimolecular reactions to form closed-shell hydroperoxide or nitrate products, or it undergoes unimolecular H shifts to form carbonyl- and epoxide-containing products (Paulot et al., 2009; D'Ambro et al., 2017b). The bimolecular peroxy radical reaction products have been shown to be of sufficiently low volatility to partition to the aerosol phase (D'Ambro et al., 2017a; Liu et al., 2016), and IEPOX has been shown to react in aqueous acidic particles, forming SOA. Commonly measured species from IEPOX reactive uptake include 2-methyltetrols (Lin et al., 2012; Surratt et al., 2006, 2010; Wang et al., 2005), C5-alkene triols (Lin et al., 2012; Surratt et al., 2010, 2006; Wang et al., 2005), organosulfates (Lin et al., 2012; Surratt et al., 2010, 2007a, b; Riva et al., 2019), 3-methyltetrahydrofuran-3,4-diols (3-MeTHF-3,4-diols) (Lin et al., 2012), and oligomers (Lin et al., 2012, 2014; Surratt et al., 2010).
Recently, studies have called into question whether these commonly measured monomeric products of IEPOX multiphase chemistry exist in the particle phase as measured (Isaacman-VanWertz et al., 2016; Hu et al., 2016). We showed previously that components of organic aerosol in the southeast US with compositions of C5H10O3 and C5H12O4 desorbed at much higher temperatures, and therefore much lower volatilities, than would be expected based on their composition (Lopez-Hilfiker et al., 2016). We concluded that the measured compositions arise from lower-volatility material in the SOA that thermally decomposes rather than evaporating as the native species and that, based on the relative abundance of these lower-volatility components, IEPOX SOA as a whole is typically comprised of very low-volatility material. When IEPOX was reacted in bulk solutions and analyzed via nuclear magnetic resonance, no evidence was found for the production of C5-alkene triols or 3-MeTHF-3,4-diols, and although a second isomer of the MeTHF diol was observed (3-MeTHF-2,4-diols), the formation rate from IEPOX was calculated to be so slow relative to nucleophilic addition that its formation would be limited to situations in which the aerosol had low water content (Watanabe et al., 2018). These findings were further corroborated by comparing a novel chromatography technique that does not involve heating to traditional GC/EI-MS analysis of IEPOX SOA compositions, finding that alkene triols and 3-MeTHF-3,4-diols were in fact formed via thermal decomposition of 2-methyltetrol sulfates and 3-methyltetrol sulfates, respectively (Cui et al., 2018), and that organosulfates contribute significantly to the particle phase (Riva et al., 2019).
An additional challenge to understanding IEPOX SOA is that there remains a large gap in carbon closure resulting from IEPOX reactive uptake to aqueous acidic aerosol particles. Although the reactivity of IEPOX in acidic particles is high (Gaston et al., 2014), the SOA yield per reactive loss of IEPOX to particles is relatively low and varies greatly depending on aerosol composition, from approximately 3 % to 21 % (Riedel et al., 2015). This disconnect may present an inconsistency in models that simulate both IEPOX and IEPOX-derived SOA. If such low yields are indeed realistic, models that adjust the rate of IEPOX reactive uptake to match the IEPOX SOA tracer concentrations without accounting for the lower yield may not correctly simulate IEPOX distributions for which reactive uptake is a dominant sink for IEPOX (Gaston et al., 2014).
In this work we seek to understand the nature of products formed via the reactive uptake of IEPOX in aqueous acidic particles and the flow of carbon between the gas and particle phases. We present measurements from the Pacific Northwest National Laboratory (PNNL; Richland, WA) environmental chamber during the 2015 SOA Formation from Forest Emissions Experiment (SOAFFEE) campaign. Batch- and continuous-flow-mode experiments were performed with authentic trans-β-IEPOX, which is the dominant isomer (Bates et al., 2014), and wet acidic seed to study the products of IEPOX uptake and the resulting aerosol properties. The properties of commonly measured IEPOX uptake products, C5H12O4 (presumably 2-methyltetrols) and C5H10O3 (presumably C5-alkene triols or 3-MeTHF-3,4-diols), such as volatility and solubility, are examined in the context of experiments utilizing isothermal evaporation of the formed SOA. The effect of acidity versus liquid water content on the products formed is also discussed, along with implications for modeling atmospheric IEPOX and its conversion into SOA.
The data presented herein were taken at the Pacific Northwest National Laboratory (PNNL) as part of the Secondary Organic Aerosol Formation from Forest Emissions Experiments (SOAFFEE) campaign held during the summer of 2015. PNNL's 10.6 m3 fluorinated ethylene propylene (FEP) environmental chamber has been described in detail previously (Liu et al., 2012). The chamber was run in both batch mode and continuous-flow mode. In batch mode the experiments lasted ∼10 h, while in continuous-flow mode the total flow rate was 48.2 L min−1, resulting in a residence time of ∼3.7 h.
Aliquots of an authentic trans-β-IEPOX standard, which was synthesized according to Zhang et al. (2012) and dissolved in ethyl acetate, were injected into a glass bulb connected to the chamber via ∼10 cm of OD polytetrafluoroethylene (PTFE) tubing. Enough IEPOX was added to the empty chamber before seed addition to achieve 2 ppb in steady state and at the beginning of batch modes. The bulb and transfer line were heated to 30–40 ∘C and a 100–300 sccm flow of zero air passed over the IEPOX to vaporize and carry it into the chamber. Typically, we measured ∼10 µg m−3 of IEPOX prior to aqueous seed addition. Given calibration uncertainties of % from repetitive injections and analytical errors, as well as potential losses on chamber and sampling surfaces, we estimate an uncertainty of approximately ±50 % for gas-phase IEPOX concentrations and other tracers. Wet, polydisperse acidic ammonium sulfate seed was generated by atomizing an ammonium bisulfate solution acidified with additional H2SO4. The solution was made by mixing ammonium sulfate (0.1308 g) with sulfuric acid (8.02 mL of 0.2465 M) and diluting to a total volume of 1 L with ultrapure water. The average molar ratio measured by the AMS was approximately 0.93 for all experiments, though due to the experimental procedure there may be some interference from organic sulfate formation. The measured ratio is significantly higher than was present in the atomized solution, implying that excess gas-phase ammonia present in the chamber partially neutralized the injected seed. The seed surface area concentrations were approximately 37 600 and 24 000–27 000 cm−3 and the volume-weighted-mode diameters were 106 and 244–254 nm in continuous and batch modes, respectively. Continuous-flow experiments were conducted at 50 % RH, while the RH of batch-mode experiments was either 30 % or 50 %.
A suite of online gas- and particle-phase instrumentation was used to monitor concentrations throughout the experiments. Aerosol number and volume concentrations were measured with a scanning mobility particle sizer (SMPS; TSI model 3936), and O3 and concentrations were monitored with commercial instrumentation (Thermo Environmental Instruments models 49C and 42C, respectively). An Aerodyne high-resolution time-of-flight mass spectrometer (HRToF-AMS) was utilized to measure bulk submicron organic and inorganic aerosol composition.
A high-resolution time-of-flight chemical ionization mass spectrometer (HRToF-CIMS) using iodide adduct ionization was deployed for the detection of both semi- and low-volatility organic compounds (Lee et al., 2014). The HRToF-CIMS was used to monitor the evolving concentrations of both the precursor (IEPOX) and reaction products in both the gas and particle phases when coupled to a Filter Inlet for Gases and Aerosols (FIGAERO), from here on referred to as FIGAERO-CIMS. The coupling, optimization, and operation of this combination has been described in detail previously (Lopez-Hilfiker et al., 2014) and is nearly identical to previous operations (D'Ambro et al., 2017a, b). Briefly, the FIGAERO is an inlet manifold that allows for semi-continuous measurements of both gases and aerosols with approximately hourly resolution. Aerosol was collected on a 24 mm PTFE filter for 43 min at 2.5 L min−1, during which the gases were measured in real time. Following collection, programmatically heated ultrahigh-purity (UHP) N2 was passed over the filter, while the temperature was ramped from ambient to 200 ∘C at a rate of 10 ∘C min−1 in order to thermally desorb compounds from the particle phase to the gas phase to be carried into the CIMS for detection. After the temperature was ramped, it was held at 200 ∘C for 50 min to allow species to desorb and signals to return to background levels. Particle blanks were conducted approximately every fourth desorption during continuous-flow mode or at the beginning and end of each batch-mode experiment by inserting a secondary filter upstream of the primary FIGAERO filter in order to get a measure of the gas adsorption artifact on the primary filter. Gas zeros were conducted every 2 min by overblowing the CIMS pinhole flow with UHP N2. The specific coupling to a HRToF-CIMS with iodide ions allows for a detailed molecular analysis of hundreds of oxygenated organic compounds via a clustering, fragmentation-free ionization process.
The FIGAERO-CIMS was also utilized to perform isothermal evaporation experiments as has been described previously (D'Ambro et al., 2018). In normal operation, the programmatic thermal desorption is begun immediately after moving the FIGAERO filter under the heating tube. During isothermal evaporation experiments, however, the aerosol is instead exposed for 1 h to a stream of ambient-temperature UHP N2 humidified to 50 % RH by using a water bubbler and two mass-flow controllers (Fig. 1). After the hour of exposure, the N2 flow is reverted to its normal dry state and the programmatic heating proceeds as normal. See Fig. 1 for the experimental setup. The RH of 50 % during evaporation periods was chosen to match that of the chamber to keep the phase state of the collected aerosols the same (i.e., so as to not drive efflorescence). Only the isothermal evaporation portion of the experiment was humidified; during the thermal desorption the bubbler was isolated using two solenoid valves. Passing excess humidified N2 over the aerosol and into the instrument resulted in a constant dilution of the vapor phase, which allowed any semi-volatile material to evaporate and also be carried into the CIMS for detection.

3.1 Chamber-generated IEPOX SOA composition and volatility
Our primary goals with this experiment were to assess whether chamber-generated IEPOX SOA had a composition and volatility similar to that inferred from field measurements using the FIGAERO-CIMS of various IEPOX tracers and to test whether IEPOX SOA as a whole or some of its components were semi-volatile as expected from mechanistic and kinetic considerations. Overall, the estimated mass yield of OA from IEPOX exposure to aqueous acidic seed was generally less than unity, of order 0.5 to less than 0.25, somewhat higher than estimates from Riedel et al. (2015). Mean organic aerosol (OA) mass concentrations generated with IEPOX and aqueous acidic seed were 2.5 µg m−3 for steady-state conditions, 5.5 µg m−3 at the beginning of the 30 % RH batch experiment, and 4.5 µg m−3 at the beginning of the 50 % RH batch experiment. The OA-to-sulfate ratio was observed to evolve during a batch experiment (e.g., decreasing by 20 % from the peak), and given the uncertainties associated with potential vapor wall losses of IEPOX and its reaction products, we refrain from quantitatively interpreting the SOA yield behaviors in detail.
Regardless of the conditions, the uptake of an authentic IEPOX standard onto acidic seeds in these experiments results in a rather simple observed particle-phase composition upon thermal desorption. As shown in Fig. 2, a few compositions dominate the average particle-phase mass spectra, most predominantly C5H12O4 and C5H10O3. Species with these compositions have been repeatedly shown to be major components of IEPOX SOA (Lin et al., 2012; Surratt et al., 2010, 2006; Wang et al., 2005), although the relative abundances could change with time or conditions. In ambient aerosol in the southeast US, the same FIGAERO-CIMS instrument detected C5H12O4 and C5H10O3 in SOA, and these tracers correlated with and explained ∼50 % of the IEPOX SOA mass derived from factor analysis of aerosol mass spectrometer (AMS) data (Lopez-Hilfiker et al., 2016). Our laboratory chamber experiments, starting with an authentic IEPOX standard and acidic seed without photochemical oxidants, therefore support the use of these FIGAERO-CIMS compositions as tracers of IEPOX SOA in atmospheric particles. As these two compositions are such a large component of the particle-phase signal (97.5 %) measured by FIGAERO-CIMS in chamber-generated IEPOX SOA, the properties of the corresponding SOA are presumably similar to their properties.
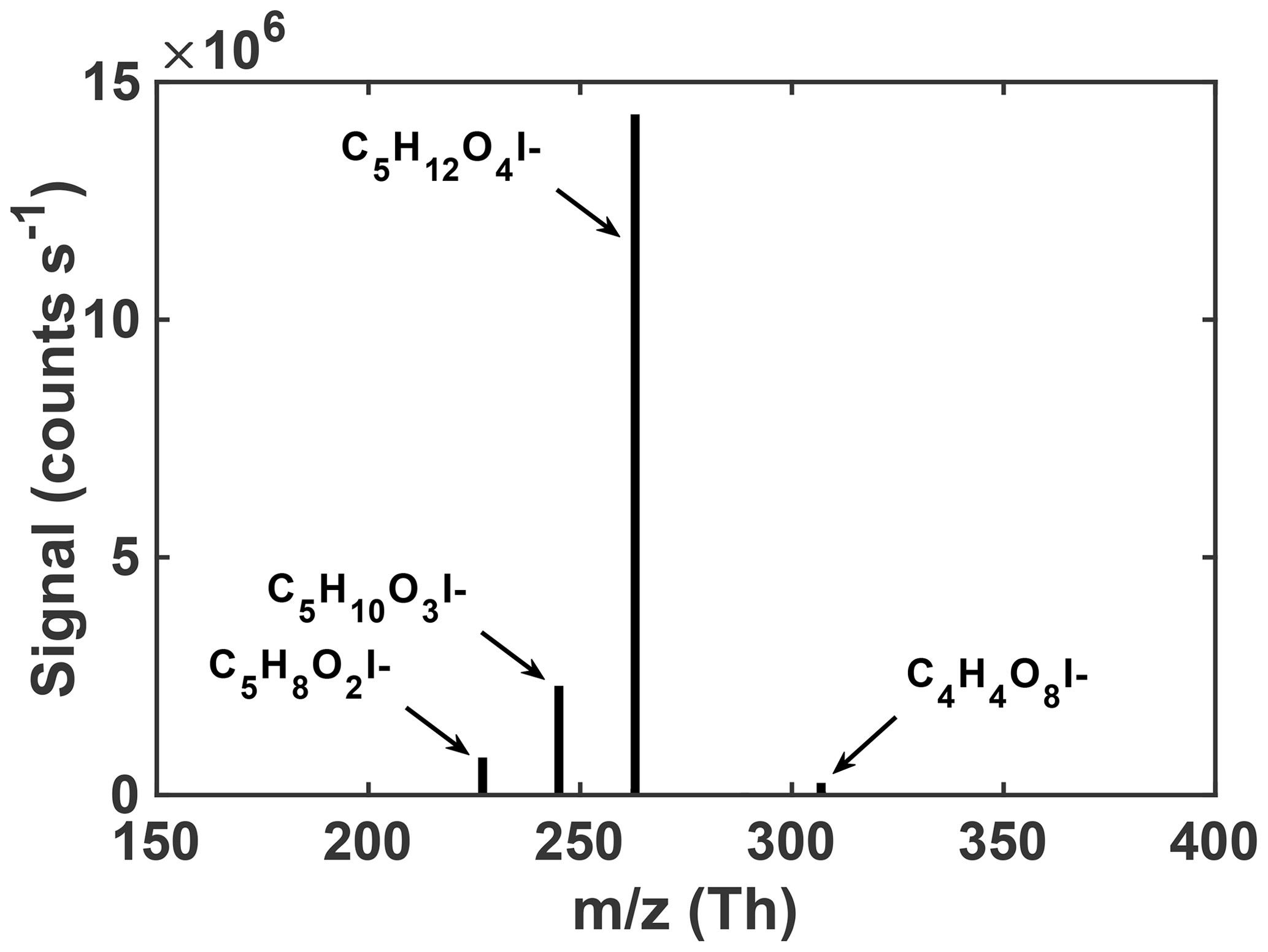
In previous work, we have related the temperature at which the ion signal for a given molecular composition reaches a maximum during a desorption, known as the Tmax, to the enthalpy of vaporization (or sublimation) and thus effective volatility (Lopez-Hilfiker et al., 2014). Effective volatility refers to the fact that the thermal desorption signal is a convolution of evaporation, thermal decomposition, particle viscosity, and mass transfer of desorbed vapors through the apparatus and not solely saturation vapor pressure. Notably, the thermogram of C5H12O4 for chamber-generated IEPOX SOA is bimodal (Fig. 3a); i.e., it exhibits two distinct maxima, one occurring at Tmax=55 ∘C (∼40 % of signal) and a second mode at Tmax=90 ∘C. These two maxima indicate SOA components with orders of magnitude different effective volatilities contribute to the desorption of C5H12O4. For example, using the calibration curve in Lopez-Hilfiker et al. (2014) relating saturation vapor concentration (c*) to Tmax, the two modes correspond to SOA components with an effective c* of 50 and 0.005 µg m−3. The lower-temperature, higher-volatility mode of the C5H12O4 desorption has a Tmax consistent with that of an authentic 2-methyltetrol standard, synthesized according to Bondy et al. (2018), deposited on the filter (gray dashed line, Fig. 3a). For comparison, if the structure of C5H12O4 were assumed to be a 2-methyltetrol, group contribution methods predict the c* to be 34 µg m−3 (Compernolle et al., 2011), remarkably similar to what the FIGAERO relationship predicts for the lower Tmax mode of the C5H12O4 thermogram.
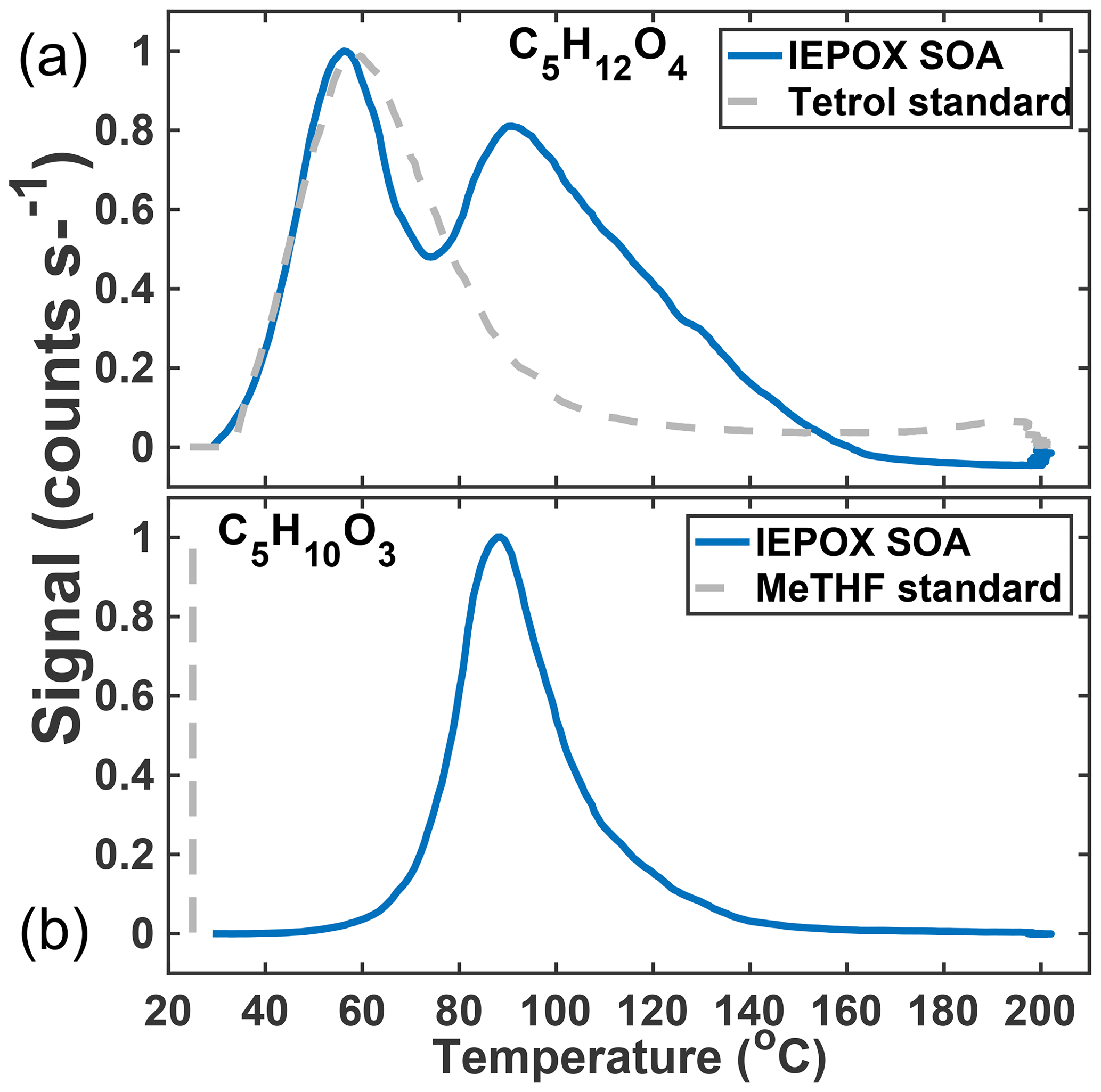
Figure 3Thermal desorption profiles of chamber aerosol (blue) and calibrations with authentic standards (gray, dashed) of the two major particle-phase compounds detected in the chamber: C5H12O4 (a) and C5H10O3 (b).
The C5H10O3 thermogram, in contrast, is monomodal, suggesting a single component giving rise to its desorption. However, the Tmax is much higher than the corresponding authentic cis-3-MeTHF-3,4-diol standard, synthesized according to Zhang et al. (2012) (and also that of an alkane triol standard). This standard desorbs completely from the FIGAERO filter in seconds without heating (gray dashed line, Fig. 3b). The Tmax of C5H10O3 desorbing from IEPOX SOA is 90 ∘C, the same as the higher Tmax mode of the C5H12O4 component, and thus implies an SOA component with an effective c* of at most 0.005 µg m−3, indicative of thermal decomposition during desorption. For comparison, if the structure is assumed to be a C5-alkene triol or 3-MeTHF-3,4-diol, group contribution methods would predict a much larger c* of ∼60 or 1.3×105 µg m−3, respectively (Compernolle et al., 2011). For comparison, the group contribution predicted c* of trans-β-IEPOX is 1.7×104 µg m−3. All of this evidence indicates that C5H10O3, as detected in the chamber-generated SOA, is a result of thermal decomposition of a lower-volatility species, consistent with recent studies that found that the C5-alkene triol was not present as a product of bulk IEPOX reactions (Watanabe et al., 2018) or when the IEPOX SOA was analyzed with novel methods not involving heating (Cui et al., 2018). Taken together, the thermograms of C5H12O4 and C5H10O3 therefore indicate that a large fraction of the chamber-generated IEPOX SOA is composed of very low-volatility material (effective µg m−3) and several orders of magnitude lower volatility than the compounds that are detected upon thermal desorption.
The thermogram behaviors of chamber-generated IEPOX SOA are entirely consistent with those observed for the same tracers measured in ambient aerosol in the southeast US, where C5H12O4 was also detected with two modes in the thermogram, the respective areas of which varied relative to each other over the course of the measurement campaign (Lopez-Hilfiker et al., 2016). Lopez-Hilfiker et al. (2016) demonstrated from these field observations that the abundance and variability of the lower Tmax (semi-volatile) mode was consistent with an organic compound having the measured molecular composition and c* of the 2-methyltetrol undergoing equilibrium gas–particle partitioning, while the higher Tmax mode and that of the C5H10O3 arose from the decomposition of accretion products.
3.2 Insights into volatility via isothermal evaporations
The above considerations of chamber-generated IEPOX SOA suggest that a large fraction (the high Tmax mode) should be relatively stable against evaporation upon dilution of the gas phase, while the lower Tmax mode of the C5H12O4 (likely 2-methyltetrol) component should respond to dilution by evaporating from the particle phase. To test this hypothesis, we conducted isothermal evaporation experiments using IEPOX SOA generated in the chamber.
Figure 4 shows an example ion signal time series during an isothermal evaporation experiment for C5H12O4. C5H12O4 is detected in the gas phase during particle collection (panel b, blue shaded area) when chamber air containing IEPOX, acidic aqueous sulfate particles, and IEPOX SOA was being continuously sampled by the FIGAERO-CIMS. The gas-phase sampling included scanning of the dilution ratio, which resulted in varying signals, as well as periodic zeros, resulting in occasional significant short-duration drops in signal. We show the last 15 min portion of the cycle in the gas phase (blue shaded region) when dilution was held constant, but zeros are still visible as the two dips in signal. The chamber was at steady state and the changing gas-phase signal is due to conditioning of the ion–molecule reaction (IMR) region and inlet tubing. During the isothermal evaporation period (yellow shaded area), C5H12O4 is also detected when a continuous flow of humidified UHP N2 (panel a, 100 indicates that desorption flow is on) passes over the particles collected from the chamber on the FIGAERO filter and into the mass spectrometer, consistent with C5H12O4 evaporation from the collected particles at room temperature (i.e., without heating). Finally, during the temperature-programmed thermal desorption, another pulse of C5H12O4 was detected, corresponding to components in the remaining SOA that desorbed at elevated temperature (green shaded area).
The mass concentration of C5H12O4 measured during a normal temperature-programmed thermal desorption is compared to that measured during the isothermal evaporation and subsequent desorption (Fig. 4b). Mass closure is achieved to within the experimental error, driven by variance in normal temperature-programmed thermal desorptions due to chamber conditions and the water vapor effect on CIMS sensitivity (Lee et al., 2014). The observed behaviors, namely detectable gas-phase concentrations of C5H12O4 in the chamber and isothermal evaporation of C5H12O4 from collected particles, indicate that (i) C5H12O4 is produced from IEPOX reactive uptake, as expected given that the 2-methyltetrol is predicted to be a major product (Eddingsaas et al., 2010), and (ii) a portion of the detected C5H12O4 behaves as a semi-volatile organic compound (SVOC), being present in both the gas and particle phases, and evaporating promptly from the particle phase in response to dilution of the surrounding organic vapors at room temperature.
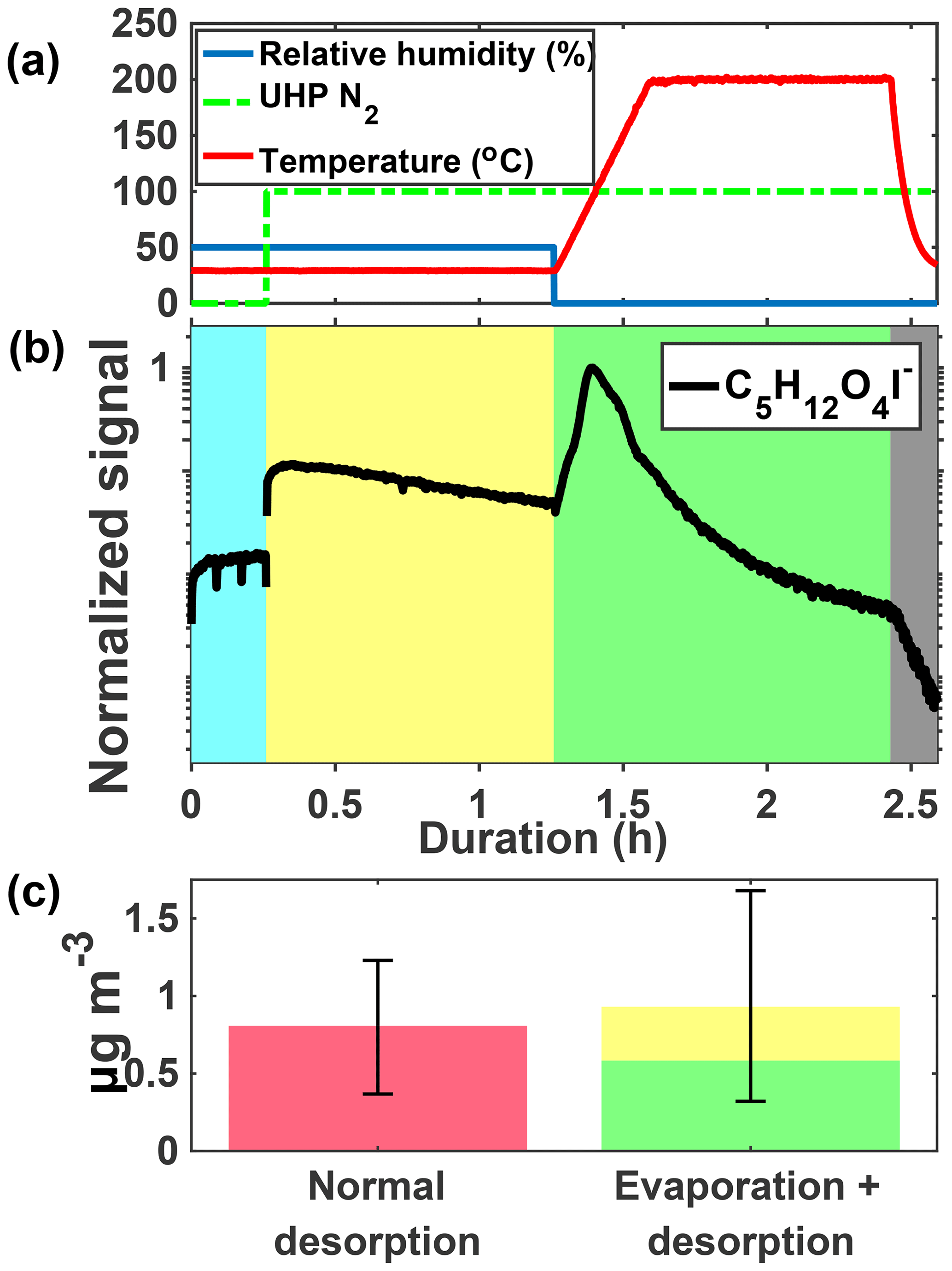
Figure 4Schematic of the isothermal evaporation process. (a) Relative humidity (blue), N2 flow (green, 0: off, 100: on), and temperature (red). (b) C5H12O4I− during a 1 h isothermal evaporation experiment shaded by the phase of the experiments: simultaneous real-time gas-phase sampling and offline aerosol collection (blue), isothermal evaporation whereby compounds are measured as they evaporate off the filter (yellow), temperature-programmed thermal desorption (green), and cooldown of the heating tube (gray). (c) Mass concentration of C5H12O4 measured during a normal desorption (pink, left) versus the isothermal evaporation + desorption (yellow and green, right) for the same chamber conditions.
After 1 h of exposure to UHP N2 at 50 % RH, only 16 % of the lower Tmax mode of the C5H12O4 thermogram remains, suggesting nearly complete evaporation (Fig. 5a). The observed rate of decay (84 % h−1) upon dilution can be related to an effective c*, or, if the structure is 2-methyltetrol, it is likely to partition into the aqueous phase, making an effective Henry's law constant more appropriate. Assuming no particle-phase diffusion limitations but accounting for FIGAERO mass transfer limitations (Schobesberger et al., 2018), we predict a c* of 5–15 µg m−3 for the portion of the C5H12O4 thermogram that evaporates. Utilizing COSMOtherm (2018) with the BP_TZVPD_FINE_18 parameterization as described previously (Kurtén et al., 2018), a Henry's law constant of 4.9×108–1.1×1010 M atm−1 is predicted if all conformers are used (4.9×108 M atm−1) or if the number of internal H bonds is minimized (1.1×1010 M atm−1), which compares well to the value calculated from the observed decay of C5H12O4 during the evaporation (1.8×108 M atm−1). These estimates do not include the likelihood of a salting-out effect expected for 2-methyltetrol (Waxman et al., 2015), which would further lower the Henry's law constant. Whether Raoult's or Henry's law is the appropriate framework for interpretation depends on whether IEPOX reactive uptake results in a phase-separated organic medium, for example an organic coating, or a homogeneous aqueous solution and the competitive partitioning of the C5H12O4 species between two such regimes. Regardless, as we show below, the semi-volatile nature of the low Tmax portion of C5H12O4, assumed to be the 2-methyltetrol, a major product of IEPOX reactive uptake, will cause it to partition strongly to the gas phase under typical atmospheric conditions outside of cloud.
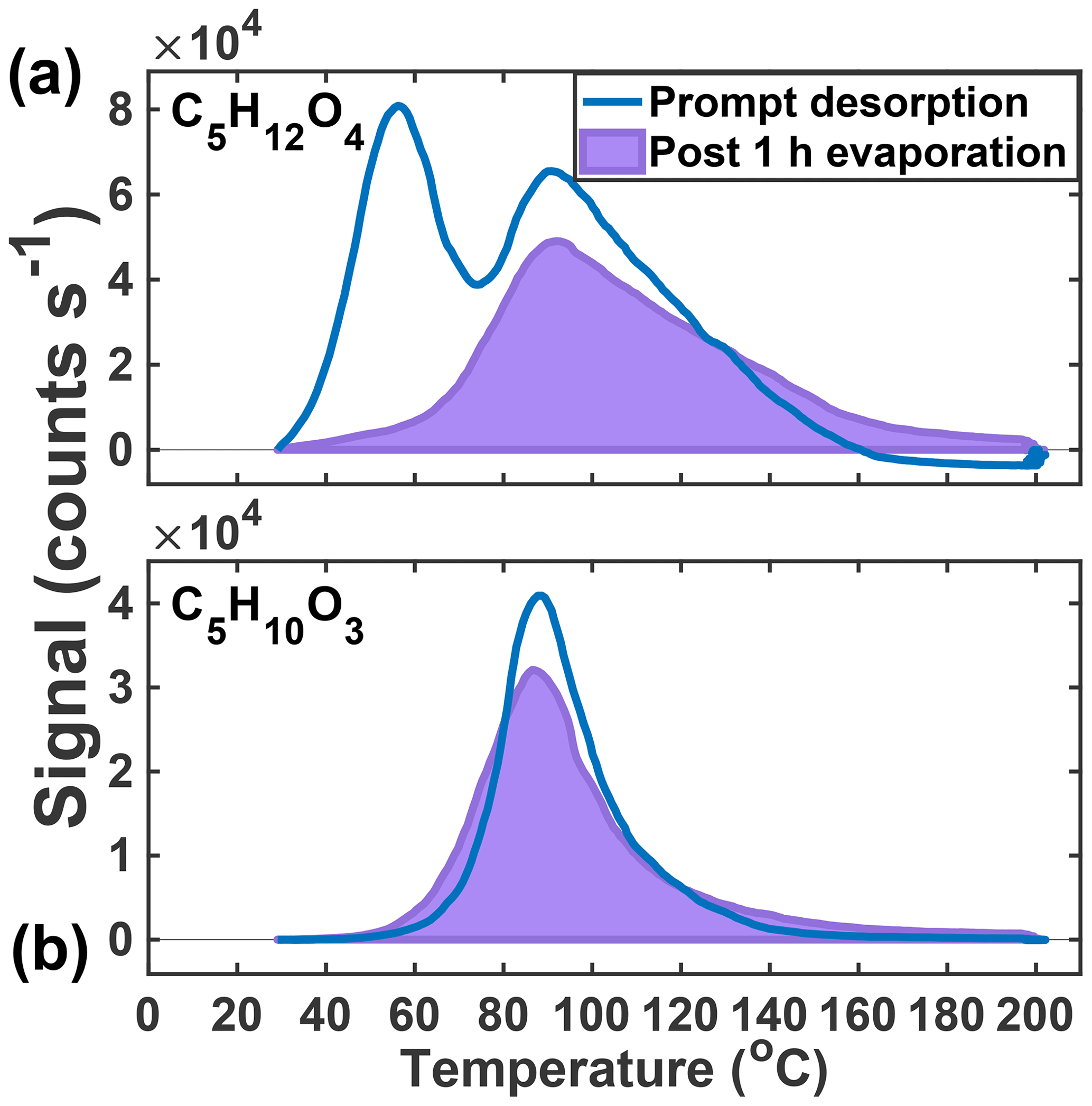
Figure 5Thermograms obtained from prompt desorption of the aerosol (blue) and after 1 h of evaporation (lavender, shaded) of the two major particle-phase compounds detected in the chamber: C5H12O4 (a) and C5H10O3 (b).
The second, higher Tmax mode of the C5H12O4 thermogram and the single Gaussian-like thermogram of C5H10O3 (Fig. 5b) do not change significantly after 1 h of exposure to UHP N2. In the case of the C5H10O3 thermogram, nearly 95 % of the signal remains after the 1 h evaporation period. In both cases, there is a slight broadening of the thermogram and a measurable increase in material desorbing at the highest temperatures (120+ ∘C). Previous isothermal evaporation experiments with the FIGAERO have shown that, for α-pinene ozonolysis SOA, the physical age of the aerosol had a controlling role in the volatility of the bulk SOA and individual desorbing compounds (D'Ambro et al., 2018), consistent with the additional hour of non-oxidative aging in this system resulting in an increase in lower-volatility material. As to the widening of the thermograms, Schobesberger et al. (2018) showed that a shallowing of the low-temperature side of the thermogram and a broadening of the higher-temperature tail correspond to thermal decomposition from a larger suite of bonds with different dissociation energies, consistent with the continued formation of a variety of accretion products during the isothermal evaporation period.
The above evidence supports the previous assertions of Lopez-Hilfiker et al. (2016) that the lower Tmax mode of the C5H12O4 thermogram corresponds to a semi-volatile component, very likely the 2-methyltetrol, and further supports the conclusion that IEPOX SOA in ambient aerosol is very to extremely low volatility (Lopez-Hilfiker et al., 2016; Hu et al., 2016; Riva et al., 2019). The isothermal evaporation experiments presented above provide an explanation as to why the ambient SOA contained such a relatively small fraction of the low Tmax (semi-volatile) 2-methyltetrol component in that it likely had evaporated to maintain gas–particle equilibrium. Furthermore, the high Tmax tracers do not decay in abundance during the evaporation experiments, but rather slightly increase with time at the highest temperatures (>120 ∘C), indicating ongoing accretion chemistry and leading to lower-volatility components.
3.3 Effect of RH and acidity on IEPOX SOA characteristics: mechanistic insights
We performed two time-dependent “batch-mode” chamber experiments using IEPOX and acidic aqueous seed particles, one at 30 % and the other at 50 % RH. By operating in batch mode as opposed to continuous-flow mode, we are able to temporally resolve the formation of SOA. By varying the RH, we varied the liquid water content relative to sulfate and therefore also acidity. Three sequential thermal desorptions of C5H12O4 obtained over the course of experiments (∼10 h total) at 30 % (panels a and c) and 50 % (panels b and d) RH are shown in Fig. 6. At 30 % RH, the lower Tmax (higher-volatility) mode grows in rapidly and is clearly visible in the first desorption (black line). The first desorption occurred after 43 min of particle collection, which began immediately after the seed was injected into the chamber and IEPOX uptake was initiated, resulting in collected aerosols having a variety of ages, and thus the median age of 22 min is assumed. However, this mode does not grow significantly larger as the experiment progresses. During the second and third desorptions at 2 h 22 min and 4 h 22 min, respectively, after the initial exposure, the higher Tmax (lower-volatility) mode is visible and dominates the thermogram.
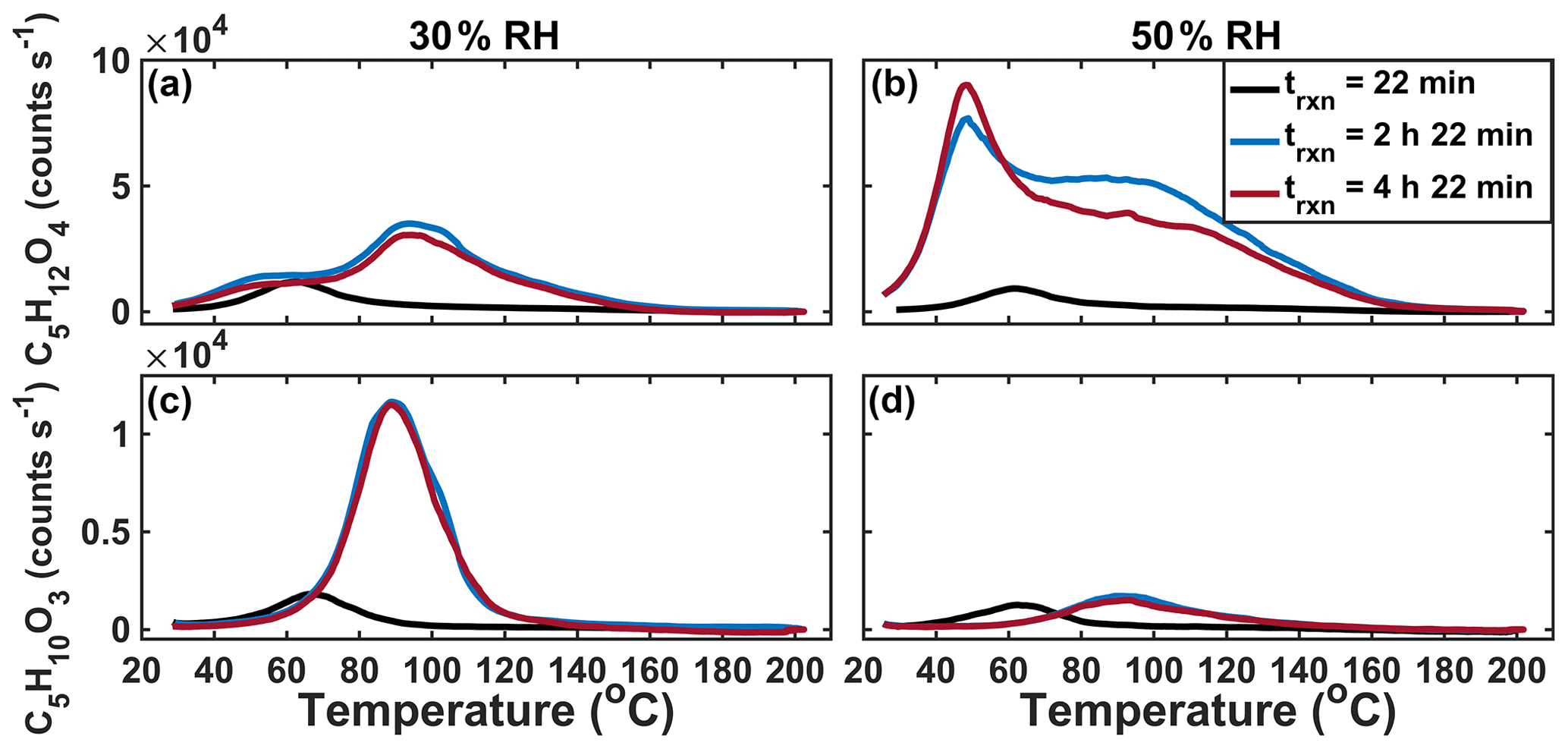
Figure 6Sequential desorptions of C5H12O4 (a, b) and C5H10O3 (c, d) during batch-mode experiments at 30 % RH (a, c) and 50 % RH (b, d).
In the 50 % RH batch experiment, the lower Tmax (higher-volatility) mode of the C5H12O4 thermogram also dominates in the first desorption, but in contrast to the experiment at 30 % RH, this lower Tmax mode continues to grow and is the dominant portion of the thermogram for all desorptions. While the higher Tmax mode is observed after the first desorption, it is much broader and has an ambiguous peak, unlike at 30 % RH. In the 30 % and 50 % RH experiments, both modes of the thermogram are observed by the second desorption 2.5 h after IEPOX uptake starts, but the relative abundance and shape of the two modes differ with RH. The thermogram shape also changes as a function of time since IEPOX injection in the steady-state experiments, although due to the range in aerosol ages present within the chamber at a given time during steady-state experiments, it is more straightforward to define this feature as a function of time in batch-mode measurements. The corresponding thermograms of C5H10O3 in each of the two batch-mode experiments are shown in Fig. 6c and d. Most obvious is that the amount of C5H10O3 desorbing, relative to the C5H12O4, is highest in the 30 % RH experiment, when sulfate and hydronium ion concentrations are highest. Further work could be done to understand the evolution of IEPOX SOA components as a function of time, but a fairly stable set of products and volatility are reached within a few hours.
Along with the shape, the Tmax of the C5H10O3 and each mode of the C5H12O4 vary slightly in time, and the two are likely related. It has been shown previously that when the IEPOX-derived organosulfate (C5H12SO7) is deposited on and desorbed from the FIGAERO filter, it decomposes into both C5H12O4 and C5H10O3. The corresponding Tmax values of both are colocated and highly dependent on acidity, with higher acidity leading to lower Tmax (Lopez-Hilfiker et al., 2016). This dependence on the inorganic aerosol components, present in much larger excess in these experiments than our previous FIGAERO experiments, could be the cause of the shifts of the lower Tmax modes. Alternatively, the shift could be due to the increasing complexity of the SOA as it evolves in time, leading to different interactions between particle components, which affects volatility.
From these observations we can draw two conclusions regarding the mechanisms that give rise to the dominant components of IEPOX SOA, illustrated in Fig. 7, which shows hypothetical reaction pathways and oligomers that could explain the observed time evolution of detected products. First, the low Tmax, semi-volatile C5H12O4 exists in the aerosol from the first desorption and is thus likely formed promptly from IEPOX uptake, consistent with the formation of 2-methyltetrol via nucleophilic attack by water of the protonated epoxide ring (see Fig. 7). That this semi-volatile mode is more prominent in the higher RH experiment, i.e., higher liquid water content and therefore higher H2O nucleophile content relative to sulfate, further supports this interpretation. Additionally, the higher liquid water content supports a greater amount of 2-methyltetrol remaining partitioned in the aerosol via Henry's law, consistent with offline filter analysis (Riva et al., 2016). Second, the higher Tmax (lower-volatility) modes are mostly produced more slowly over time, indicating a second- or higher-generation product of IEPOX uptake, as these modes are mainly observed 2.5 h after IEPOX uptake has largely ended. Thus, if the higher Tmax modes are from the thermal decomposition of an organosulfate product, as suggested by Lopez-Hilfiker et al. (2016) and as Cui et al. (2018) demonstrate, our experiments suggest it is unlikely to form solely from the nucleophilic addition of (bi-)sulfate to protonated IEPOX, as that reaction should occur concurrently with 2-methyltetrol formation.

Figure 7Hypothetical schematic of IEPOX reactive uptake and particle-phase processes. White region denotes semi-volatile species that actively partition between the gas and particle phases, light green denotes species that are of lower volatility, and dark green outline denotes a coating.
Another possible mechanism of organosulfate formation, as well as sulfate ester oligomers, is via SN2 reactions in which one of the 2-methyltetrol -OH groups is protonated to make H2O the leaving group, while bisulfate, sulfate, or an organosulfate is the substituting group (Fig. 7). At 30 % RH, the particle acidity is higher due to less dilution of the sulfate, which would result in higher organosulfate concentrations and acid-catalyzed accretion chemistry (Jang et al., 2002), consistent with the observation of a more prominent higher Tmax (lower-volatility) mode compared to the 50 % RH experiment. The broader higher Tmax mode at 50 % RH indicates that there is likely an array of compounds breaking apart to give rise to this specific composition, consistent with a greater variety of oligomerization reactions occurring due to the dilution of sulfate and higher 2-methyltetrol concentrations. Previous work has identified several non-sulfur-containing polyol species, both monomers and oligomers, in IEPOX SOA (Surratt et al., 2010; Lin et al., 2012, 2014).
To place our findings into context, we present results from a simple conceptual model simulating IEPOX (initially ∼2 ppb/10 µg m−3) reactive uptake to form the corresponding 2-methyltetrol and organosulfate at yields of 90 % and 10 %, respectively, with an uptake coefficient of 0.05 based on Gaston et al. (2014) and an atmospherically relevant total surface area ( cm2 cm−3) and volume ( cm3 cm−3). We also include a loss of gas-phase species (2-methyltetrol and IEPOX) due to reaction with OH at cm3 molec−1 s−1 (Atkinson, 1987), consistent with previous studies for the IEPOX + OH rate constant (Bates et al., 2014; Jacobs et al., 2013). The processes in the model are simplified from the reaction scheme discussed above; i.e., it does not include particle-phase processes, but its purpose is to capture the salient factors that control the reactive uptake and partitioning. The chosen branching between 2-methyltetrol and organosulfate yields from IEPOX reactive uptake to aerosol is somewhat arbitrary and only to illustrate the behavior of the system. The Henry's law constant found via COSMOtherm for the 2-methyltetrol is used to simulate gas–particle partitioning of the 2-methyltetrol, while the organosulfate is a proxy for all low-volatility products, including the promptly formed organosulfates, sulfate esters from further accretion, and polyol oligomers. Vapor wall loss is not considered in the model, which might be resulting in more tetrol evaporating from the particles in our measurements than would occur in the atmosphere. However, operating in continuous-flow mode helps to mitigate these issues, and in batch mode we do not observe a significant loss of the low Tmax, semi-volatile mode. In the atmosphere, photochemical losses of the gas-phase tetrol and the smaller aqueous volume of the aerosol would lead to partitioning of the tetrol out of the aerosol, as illustrated by the model in Fig. 8, which does include these atmospheric processes. Thus, while vapor wall loss in the chamber potentially leads to lower particle-phase tetrol, the chamber experiments neglect oxidation of gas-phase tetrol, which would have a similar effect in the atmosphere.
The simulated loss of IEPOX and formation of IEPOX SOA are shown in Fig. 8. IEPOX is almost completely consumed after 1 h of reaction, corresponding to rapid formation of SOA. The composition of the aerosol changes significantly as a function of time. Initially, the SOA is composed primarily of 2-methyltetrol. However, despite the relatively high Henry's law constant, much of the 2-methyltetrol evaporates into the gas phase to maintain equilibrium with the gas-phase 2-methyltetrol, which is subjected to loss by gas-phase bimolecular reactions with the hydroxyl radical (OH). This behavior supports our findings herein that on a typical aerosol lifetime, the dominant IEPOX reactive uptake product, 2-methyltetrol, will be a small component of IEPOX SOA, and organosulfates and other low-volatility material, including oligomers of 2-methyltetrol, will dominate, albeit at a smaller overall SOA yield (Fig. 8) per IEPOX reacting on aerosol.
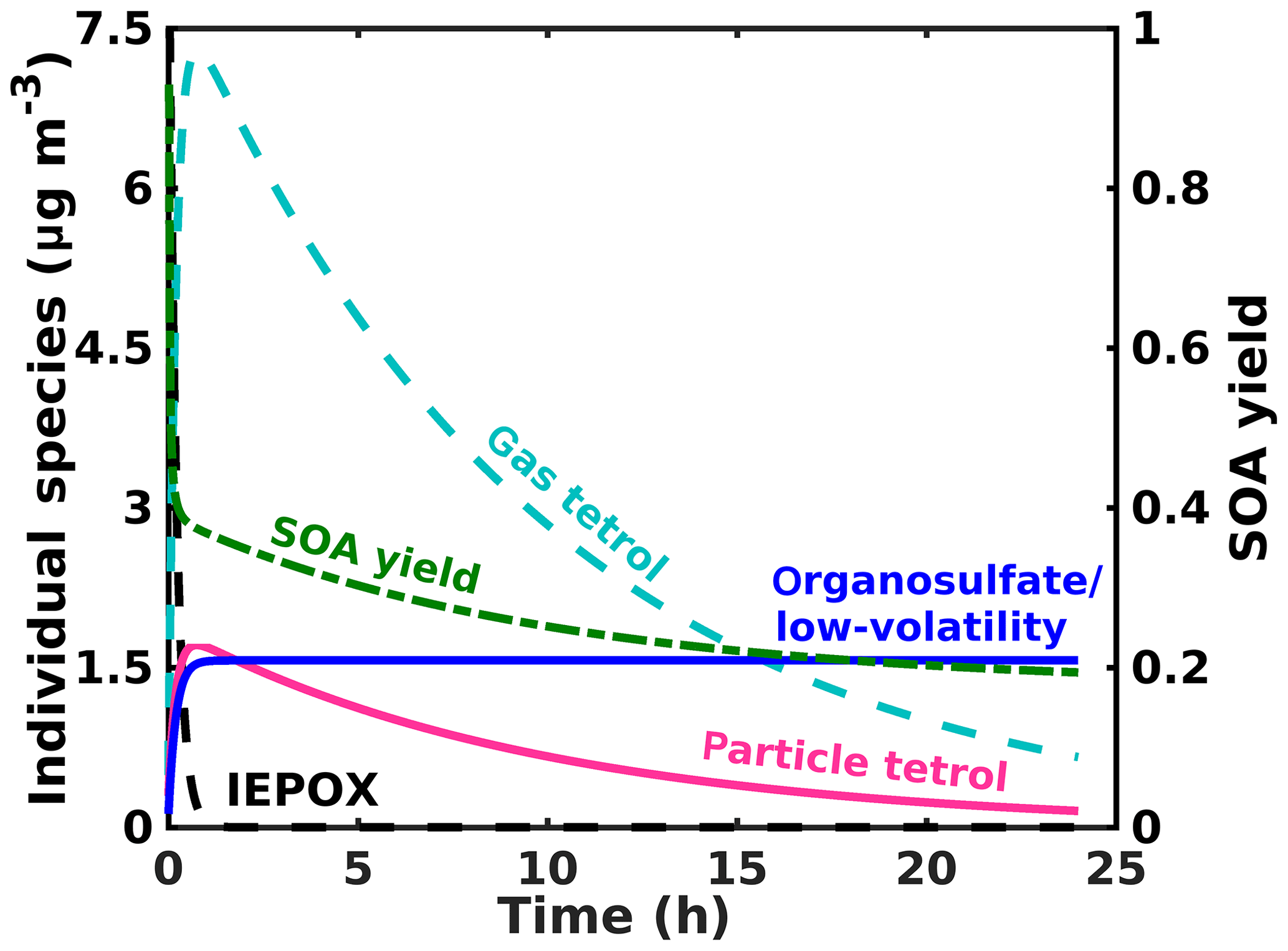
Figure 8Model results for the major gas- and particle-phase species of IEPOX reactive uptake for typical atmospheric aerosol and typical IEPOX mixing ratios.
Fundamental chamber studies of IEPOX reactive uptake to aqueous acidic seed were performed, and we find that the resulting molecular composition and volatility of the formed SOA suggests that the vast majority of IEPOX SOA in the atmosphere is of very low volatility in the form of organosulfates and polyol oligomers, which is supported by the recent findings of Riva et al. (2019). We show that the major product expected from IEPOX reactive uptake in acidic aqueous solutions, C5H12O4 (most probably 2-methyltetrol), is semi-volatile and will likely partition out of the aerosol and thus contribute relatively little to IEPOX SOA mass under atmospheric conditions. We further confirm that the observed properties of C5H10O3 are not consistent with the structure of C5-alkene triols and/or 3-MeTHF-3,4-diols, and thus these structures cannot be components of IEPOX SOA but are likely artifacts of thermal decomposition during analytical workup. While our results are specific to the FIGAERO, we predict that the issue is more general and affects other methods, as indicated, for example, in Cui et al. (2018). A direct intercomparison is required to definitively determine whether all instruments (those reporting C5H12O4, C5H10O3, 2-methyltetrols, C5-alkene triols, and/or 3-MeTHF-3,4-diols) are in fact measuring the same species and that prior estimates of IEPOX SOA have not been overestimated due to “double counting” carbon in these tracers, which might be derived from organosulfates and oligomers measured separately.
The evidence presented herein, as well as in independent experiments (Cui et al., 2018), indicates that the C5H10O3, regardless of structure, and a significant portion of the C5H12O4 are not actual components of the SOA but rather derived from other related components during the analysis. Therefore, we do not recommend that these species be included as products in mechanistic models of IEPOX SOA formation and evolution. Finally, we provide evidence that a portion of the low-volatility IEPOX SOA is composed of oligomers formed in part from slower particle-phase accretion chemistry, likely involving the first-generation organosulfates and possibly also the 2-methyltetrol, though we cannot distinguish between these possible combinations. The distribution of products and formation timescales depend upon aerosol water, sulfate, and hydronium ion activities and thus ultimately on ambient RH and particle alkalinity sources. However, the low volatility of the majority of IEPOX SOA will make it less susceptible to subsequent changes in RH or dilution.
These findings help to explain properties of IEPOX SOA observed in the field and to resolve inconsistencies in the descriptions of IEPOX SOA formation from gas-phase IEPOX reactive uptake. The AMS-IEPOX-related positive matrix factorization (PMF) factor has been shown to correlate with sulfate (Xu et al., 2015; Hu et al., 2015). If the majority of IEPOX SOA is in the form of low-volatility components, such as organosulfates and oligomers thereof as our observations indicate, then a correlation of IEPOX SOA with sulfate, and not water content or acidity, is expected given the direct coupling to inorganic sulfate and similar lifetimes against removal. Moreover, the low SOA yield per reactive uptake of IEPOX on aqueous acidic seed can be explained as C5H12O4, the dominant product and likely 2-methyltetrol, being semi-volatile and largely partitioning out of the particle phase, especially in a system with gas-phase oxidation or in chambers with vapor wall loss. Measured reactive uptake probabilities of IEPOX (γIEPOX) on aqueous acidic seed are an order of magnitude or more higher than often used in models (Gaston et al., 2014; Eddingsaas et al., 2010; Pye et al., 2013; Marais et al., 2016). This discrepancy partly reflects a lower SOA yield per IEPOX lost than accounted for in models, for which we provide an explanation, and also a role for organic coatings that likely exist in the atmosphere, which slow reactive uptake relative to pure aqueous acidic seed (Gaston et al., 2014; Zhang et al., 2018).
These aspects likely have some cancelation of errors but could lead to errors in models of IEPOX abundance, IEPOX SOA production rate, and thus their corresponding spatial variability. For example, in regions with relatively fresh, uncoated aqueous acidic particles, models using a low reaction probability would underestimate the IEPOX loss rate from the gas phase, sustaining IEPOX SOA formation over a larger area than in reality. A model that ignored the role of organic coatings and 2-methyltetrol evaporation would potentially overestimate IEPOX SOA formation on regional scales. Models that treat products of IEPOX reactive uptake in a volatility basis set would need to utilize the volatility inferred from the thermograms herein or thermal denuder measurements, rather than that inferred from commonly reported IEPOX SOA tracers that utilize high-temperature analytical methods. Finally, without accounting for slower accretion chemistry involving organosulfates and polyols, IEPOX SOA predicted from a treatment of first-generation products alone could be underestimated.
ELD analyzed FIGAERO data; ELD, JAT, and SS conducted modeling; and ELD and JAT wrote the paper. JES and JAT designed the chamber experiments. NH, V-TS, GH, and TK made COSMOtherm predictions. All other coauthors participated in data collection, experiment operations, and paper discussions.
The authors declare that they have no conflict of interest.
We thank Avram Gold and Zhenfa Zhang for synthesizing the trans-β-IEPOX, 2-methyltetrol, and 3-MeTHG-3,4-diol standards used in this work.
This work was supported by the U.S. Department of Energy ASR grants DE-SC0011791 and DE-SC0018221. Emma L. D'Ambro was supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1256082. PNNL authors were supported by the U.S. Department of Energy, Office of Biological and Environmental Research, as part of the ASR program. Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under contract DE-AC05-76RL01830. UC Davis authors were supported by NSF grant ATM-1151062.
This paper was edited by Jacqui Hamilton and reviewed by three anonymous referees.
Atkinson, R.: A Structure-Activity Relationship for the Estimation of Rate Constants for the Gas-Phase Reactions of OH Radicals with Organic-Compounds, Int. J. Chem. Kinet., 19, 799–828, https://doi.org/10.1002/kin.550190903, 1987.
Bates, K. H., Crounse, J. D., St Clair, J. M., Bennett, N. B., Nguyen, T. B., Seinfeld, J. H., Stoltz, B. M., and Wennberg, P. O.: Gas phase production and loss of isoprene epoxydiols, J. Phys. Chem. A, 118, 1237–1246, https://doi.org/10.1021/jp4107958, 2014.
Bondy, A. L., Craig, R. L., Zhang, Z., Gold, A., Surratt, J. D., and Ault, A. P.: Isoprene-Derived Organosulfates: Vibrational Mode Analysis by Raman Spectroscopy, Acidity-Dependent Spectral Modes, and Observation in Individual Atmospheric Particles, J. Phys. Chem. A, 122, 303–315, https://doi.org/10.1021/acs.jpca.7b10587, 2018.
Compernolle, S., Ceulemans, K., and Muller, J. F.: EVAPORATION: a new vapour pressure estimation method for organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Cui, T., Zeng, Z., dos Santos, E. O., Zhang, Z., Chen, Y., Zhang, Y., Rose, C. A., Budisulistiorini, S. H., Collins, L. B., Bodnar, W. M., de Souza, R. A. F., Martin, S. T., Machado, C. M. D., Turpin, B. J., Gold, A., Ault, A. P., and Surratt, J. D.: Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci. Process. Imp., 20, 1524–2536, https://doi.org/10.1039/c8em00308d, 2018.
D'Ambro, E. L., Lee, B. H., Liu, J., Shilling, J. E., Gaston, C. J., Lopez-Hilfiker, F. D., Schobesberger, S., Zaveri, R. A., Mohr, C., Lutz, A., Zhang, Z., Gold, A., Surratt, J. D., Rivera-Rios, J. C., Keutsch, F. N., and Thornton, J. A.: Molecular composition and volatility of isoprene photochemical oxidation secondary organic aerosol under low- and high-NOx conditions, Atmos. Chem. Phys., 17, 159–174, https://doi.org/10.5194/acp-17-159-2017, 2017a.
D'Ambro, E. L., Moller, K. H., Lopez-Hilfiker, F. D., Schobesberger, S., Liu, J. M., Shilling, J. E., Lee, B., Kjaergaard, H. G., and Thornton, J. A.: Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields, Environ. Sci. Technol., 51, 4978–4987, https://doi.org/10.1021/acs.est.7b00460, 2017b.
D'Ambro, E. L., Schobesberger, S., Zaveri, R. A., Shilling, J. E., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: Isothermal Evaporation of α-Pinene Ozonolysis SOA: Volatility, Phase State, and Oligomeric Composition, ACS Earth Space Chem., 2, 1058–1067, https://doi.org/10.1021/acsearthspacechem.8b00084, 2018.
Eddingsaas, N. C., VanderVelde, D. G., and Wennberg, P. O.: Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols, J. Phys. Chem. A, 114, 8106–8113, https://doi.org/10.1021/jp103907c, 2010.
Gaston, C. J., Riedel, T. P., Zhang, Z. F., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sa, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Hu, W. W., Palm, B. B., Day, D. A., Campuzano-Jost, P., Krechmer, J. E., Peng, Z., de Sa, S. S., Martin, S. T., Alexander, M. L., Baumann, K., Hacker, L., Kiendler-Scharr, A., Koss, A. R., de Gouw, J. A., Goldstein, A. H., Seco, R., Sjostedt, S. J., Park, J. H., Guenther, A. B., Kim, S., Canonaco, F., Prevot, A. S. H., Brune, W. H., and Jimenez, J. L.: Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 16, 11563–11580, https://doi.org/10.5194/acp-16-11563-2016, 2016.
Isaacman-VanWertz, G., Yee, L. D., Kreisberg, N. M., Wernis, R., Moss, J. A., Hering, S. V., de Sa, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W. W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Riva, M., Surratte, J. D., Viegas, J., Manzi, A., Edgerton, E., Baumann, K., Souza, R., Artaxo, P., and Goldstein, A. H.: Ambient Gas-Particle Partitioning of Tracers for Biogenic Oxidation, Environ. Sci. Technol., 50, 9952–9962, https://doi.org/10.1021/acs.est.6b01674, 2016.
Jacobs, M. I., Darer, A. I., and Elrod, M. J.: Rate Constants and Products of the OH Reaction with Isoprene-Derived Epoxides, Environ. Sci. Technol., 47, 12868–12876, https://doi.org/10.1021/es403340g, 2013.
Jang, M. S., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Kurtén, T., Hyttinen, N., D'Ambro, E. L., Thornton, J., and Prisle, N. L.: Estimating the saturation vapor pressures of isoprene oxidation products C5H12O6 and C5H10O6 using COSMO-RS, Atmos. Chem. Phys., 18, 17589–17600, https://doi.org/10.5194/acp-18-17589-2018, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurten, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lin, Y. H., Zhang, Z. F., Docherty, K. S., Zhang, H. F., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012.
Lin, Y. H., Budisulistiorini, H., Chu, K., Siejack, R. A., Zhang, H. F., Riva, M., Zhang, Z. F., Gold, A., Kautzman, K. E., and Surratt, J. D.: Light-absorbing oligomer formation in secondary organic aerosol from reactive uptake of isoprene epoxydiols, Environ. Sci. Technol., 48, 12012–12021, https://doi.org/10.1021/es503142b, 2014.
Liu, J. M., D'Ambro, E. L., Lee, B. H., Lopez-Hilfiker, F. D., Zaveri, R. A., Rivera-Rios, J. C., Keutsch, F. N., Iyer, S., Kurten, T., Zhang, Z. F., Gold, A., Surratt, J. D., Shilling, J. E., and Thornton, J. A.: Efficient isoprene secondary organic aerosol formation from a non-IEPOX pathway, Environ. Sci. Technol., 50, 9872–9880, https://doi.org/10.1021/acs.est.6b01872, 2016.
Liu, S., Shilling, J. E., Song, C., Hiranuma, N., Zaveri, R. A., and Russell, L. M.: Hydrolysis of organonitrate functional groups in aerosol particles, Aerosol Sci. Technol., 46, 1359–1369, https://doi.org/10.1080/02786826.2012.716175, 2012.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, T. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular composition and volatility of organic aerosol in the southeastern US: Implications for IEPOX derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kurten, A., St Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Pye, H. O. T., Pinder, R. W., Piletic, I. R., Xie, Y., Capps, S. L., Lin, Y. H., Surratt, J. D., Zhang, Z. F., Gold, A., Luecken, D. J., Hutzell, W. T., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Epoxide pathways improve model predictions of isoprene markers and reveal key role of acidity in aerosol formation, Environ. Sci. Technol., 47, 11056–11064, https://doi.org/10.1021/es402106h, 2013.
Riedel, T. P., Lin, Y. H., Budisulistiorini, H., Gaston, C. J., Thornton, J. A., Zhang, Z. F., Vizuete, W., Gold, A., and Surrattt, J. D.: Heterogeneous reactions of isoprene-derived epoxides: reaction probabilities and molar secondary organic aerosol yield estimates, Environ. Sci. Technol. Lett., 2, 38–42, https://doi.org/10.1021/ez500406f, 2015.
Riva, M., Budisulistiorini, S. H., Chen, Y. Z., Zhang, Z. F., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., Canagaratna, M. R., and Surratt, J. D.: Chemical characterization of secondary organic aerosol from oxidation of isoprene hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016.
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Ribeiro, I. O., e Oliveira, R. L., dos Santos, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sá, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin Junior, S., de Souza, R. A. F., Palm, B. B., Jimenez, J.-L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing Isoprene Epoxydiol-to-Inorganic Sulfate Aerosol Ratio Results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019.
Schobesberger, S., D'Ambro, E. L., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: A model framework to retrieve thermodynamic and kinetic properties of organic aerosol from composition-resolved thermal desorption measurements, Atmos. Chem. Phys., 18, 14757–14785, https://doi.org/10.5194/acp-18-14757-2018, 2018.
St Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jørgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and products of the reaction of the first-generation isoprene hydroxy hydroperoxide (ISOPOOH) with OH, J. Phys. Chem. A, 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2016.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007a.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007b.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Wang, W., Kourtchev, I., Graham, B., Cafmeyer, J., Maenhaut, W., and Claeys, M.: Characterization of oxygenated derivatives of isoprene related to 2-methyltetrols in Amazonian aerosols using trimethylsilylation and gas chromatography/ion trap mass spectrometry, Rapid Commun. Mass Spectrom., 19, 1343–1351, https://doi.org/10.1002/rcm.1940, 2005.
Watanabe, A. C., Stropoli, S. J., and Elrod, M. J.: Assessing the Potential Mechanisms of Isomerization Reactions of Isoprene Epoxydiols on Secondary Organic Aerosol, Environ. Sci. Technol., 52, 8346–8354, https://doi.org/10.1021/acs.est.8b01780, 2018.
Waxman, E. M., Elm, J., Kurtén, T., Mikkelsen, K. V., Ziemann, P. J., and Volkamer, R.: Glyoxal and Methylglyoxal Setschenow Salting Constants in Sulfate, Nitrate, and Chloride Solutions: Measurements and Gibbs Energies, Environ. Sci. Technol., 49, 11500–11508, https://doi.org/10.1021/acs.est.5b02782, 2015.
Xu, L., Guo, H. Y., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Technol. Lett., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018.
Zhang, Z., Lin, Y. H., Zhang, H., Surratt, J. D., Ball, L. M., and Gold, A.: Technical Note: Synthesis of isoprene atmospheric oxidation products: isomeric epoxydiols and the rearrangement products cis- and trans-3-methyl-3,4-dihydroxytetrahydrofuran, Atmos. Chem. Phys., 12, 8529–8535, https://doi.org/10.5194/acp-12-8529-2012, 2012.