the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Oct 2018
| 11 Oct 2018
Heterogeneous OH oxidation of secondary brown carbon aerosol
Elijah G. Schnitzler
Jonathan P. D. Abbatt
Light-absorbing organic aerosol, or brown carbon (BrC), has significant but poorly constrained effects on climate; for example, oxidation in the atmosphere may alter its optical properties, leading to absorption enhancement or bleaching. Here, we investigate for the first time the effects of heterogeneous OH oxidation on the optical properties of a laboratory surrogate of aqueous, secondary BrC in a series of photo-oxidation chamber experiments. The BrC surrogate was generated from aqueous resorcinol, or 1,3-dihydroxybenzene, and H2O2 exposed to >300 nm radiation that is atomized, passed through trace gas denuders, and injected into the chamber, which was conditioned to either 15 % or 60 % relative humidity (RH). Aerosol absorption and scattering coefficients and single scattering albedo (SSA) at 405 nm were measured using a photoacoustic spectrometer. At 60 % RH, upon OH exposure, absorption first increased, and the SSA decreased sharply. Subsequently, absorption decreased faster than scattering, and SSA increased gradually. Comparisons to the modelled trend in SSA, based on Mie theory calculations, confirm that the observed trend is due to chemical evolution, rather than slight changes in particle size. The initial absorption enhancement is likely due to molecular functionalization and/or oligomerization and the bleaching to fragmentation. By contrast, at 15 % RH, slow absorption enhancement was observed without appreciable bleaching. A multi-layer kinetics model, consisting of two surface reactions in series, was constructed to provide further insights regarding the RH dependence of the optical evolution. Candidate parameters suggest that the oxidation is efficient, with uptake coefficients on the order of unity. The parameters also suggest that, as RH decreases, reactivity decreases and aerosol viscosity increases, such that particles are well-mixed at 60 % RH but not at 15 % RH. These results further the current understanding of the complex processing of BrC that may occur in the atmosphere.
- Article
(549 KB) - Full-text XML
-
Supplement
(1125 KB) - BibTeX
- EndNote





Among all atmospheric constituents, aerosols have the most uncertain radiative forcing, partly due to an incomplete understanding of carbonaceous aerosols (Chung et al., 2012). In particular, the climate effects of light-absorbing organic aerosol, or brown carbon (BrC) (Bond, 2001; Kirchstetter et al., 2004), are poorly constrained, compared to those of elemental black carbon (BC) (Ramanathan and Carmichael, 2008).
One source of this uncertainty is the wide range of sources of BrC (Laskin et al., 2015). Low-temperature biomass burning results in the formation of primary BrC (Bahadur et al., 2012; Chen and Bond, 2010; Lewis et al., 2008; Radney et al., 2017). Many classes of compounds, including nitroaromatics, polyphenols, and substituted polycyclic aromatic hydrocarbons (Lin et al., 2016), have been identified in primary BrC, but their concentrations and absorptivities vary significantly. At a site strongly influenced by biomass burning in Germany, the contribution of nitroaromatics to the absorption of 370 nm light by BrC was roughly 1 % (Teich et al., 2017); in contrast, in a field campaign in Israel during a bonfire festival, the contribution of nitroaromatics to the total absorption of >400 nm light was greater than 50 % (Lin et al., 2017), in good agreement with results from controlled burns during the fourth Fire Lab at Missoula Experiment (Lin et al., 2016). In recent applications of size-exclusion chromatography, BrC constituents with molecular masses across the range of 102–104 Da have been observed (Di Lorenzo et al., 2017; Di Lorenzo and Young, 2016; Wong et al., 2017). Larger, oligomeric compounds (>1000 Da) have been observed to contribute most of the absorbance (Di Lorenzo and Young, 2016). If the primary BrC particles subsequently pass through a high-temperature region of biomass burning (Tóth et al., 2014), they may form tar balls (Chakrabarty et al., 2010; Hoffer et al., 2016), which are significantly more absorptive (Alexander et al., 2008).
BrC may also form from secondary processes. For example, the ozonolysis of catechol and guaiacol, abundant emissions from biomass burning (Schauer et al., 2001), in the gas phase leads to the formation of lower-volatility products that partition into the condensed phase to form secondary organic aerosol (SOA) that is light-absorbing (Ofner et al., 2011). Light-absorbing SOA has also been observed to form from the gas-phase photo-oxidation of other precursors, e.g. naphthalene (Lambe et al., 2013; Lee et al., 2014). In a recent field campaign, reactions in the condensed phase of cloud droplets or aqueous aerosols have also been shown to result in the formation of light-absorbing SOA (Gilardoni et al., 2016). In the laboratory, the heterogeneous oxidation of catechol has been studied at air–solid (Pillar et al., 2015) and air–water (Pillar et al., 2014; Pillar and Guzman, 2017) interfaces; at the air–water interface, functionalization, which leads to polyphenols and hydroxylated quinones that are expected to be highly absorptive, followed by fragmentation was observed (Pillar and Guzman, 2017). Heterogeneous reactions are sensitive to particle diameter, so they may in part be responsible for the observation of higher concentrations of BrC in particles in the accumulation mode compared to those in the coarse mode (Liu et al., 2013). Reactions of nitrogen-containing species (e.g. ammonium sulfate and methylamine) with aldehydes (e.g. glyoxal and methylglyoxal) have also been shown to result in BrC (De Haan et al., 2009, 2011; Lee et al., 2013; Yu et al., 2011). Furthermore, the formation of intra- or inter-molecular charge transfer complexes, similar to what has been proposed to occur in natural waters (Del Vecchio and Blough, 2004), may enhance the absorption of BrC (Phillips and Smith, 2014, 2015; Rincón et al., 2009).
In addition to the wide range of classes of BrC, the evolution of BrC upon atmospheric aging contributes to the uncertainty in its climate forcing. During the lifetime of the particles or cloud droplets, BrC constituents are photolyzed or react with oxidants; the resulting chemical evolution leads to the evolution of the optical properties of the aerosol. Field measurements have demonstrated that the absorption by BrC may decay drastically during transport, although a small fraction may be recalcitrant (Forrister et al., 2015). On the other hand, in a recent field campaign, absorption at 365 nm by BrC was the same in a fresh convective storm outflow and in its one-day-aged plume, either suggesting that photobleaching was minimal or that secondary chemistry produced new chromophores that compensated for any photobleaching (Zhang et al., 2017).
In the laboratory, the evolution of absorption induced by photolysis and oxidation has been observed for a variety of BrC surrogates in the solution phase, including extracts of biomass burning BrC (Lin et al., 2016; Wong et al., 2017), extracts of SOA derived from naphthalene under high-NOx conditions (Lee et al., 2014), products of (methyl)glyoxal and ammonium sulfate (Wong et al., 2017; Zhao et al., 2015), products of pyruvic acid polymerization (Rincón et al., 2009), and nitrophenols (Zhao et al., 2015). Fewer studies have investigated the evolution of BrC aerosol upon photolysis and heterogeneous oxidation. The aging of particle and gas emissions from biomass burning results in the formation of BrC or non-absorbing SOA coatings on BC particles, leading to enhanced absorption (Saleh et al., 2013; Tasoglou et al., 2017). In flow-tube experiments, BrC aerosol from atomized methylglyoxal and ammonium sulfate has been shown to increase in absorptivity upon exposure to ozone, due to carbonyl products (Sareen et al., 2013). BrC from biomass burning exposed to natural sunlight has been shown to photobleach; additionally, the rate of photobleaching decreased in the presence of NOx, possibly due to formation of secondary BrC constituents that slightly compensate for photobleaching (Zhong and Jang, 2014). Most recently, in potential aerosol mass reactor experiments, primary BrC from biomass burning exposed to irradiation, ozone, and hydroxyl radicals (OH) has been shown to lose almost 50 % of its absorption at 375 and 405 nm after the equivalent of 4.5 days of residence time in the atmosphere (Sumlin et al., 2017).
Here, we consider the fate of secondary BrC constituents from cloud processing upon droplet evaporation and subsequent exposure to OH radicals. The first question to raise is whether the optical properties of secondary BrC aerosol evolve at atmospherically relevant OH exposures; another broad question is whether this evolution is dependent on relative humidity (RH). To address these questions, we investigate for the first time the heterogeneous OH oxidation of a secondary BrC surrogate aerosol, generated from the product mixture of aqueous OH oxidation of resorcinol, or 1,3-dihydroxybenzene. Experiments were conducted in a 1 m3 photo-oxidation chamber at 15 % and 60 % RH, and aerosol absorption, scattering, and single scattering albedo (SSA) at 405 nm were measured in situ using a photoacoustic spectrometer with an integrated reciprocal nephelometer. Changes in the optical properties were observed, raising another question of whether these changes were due to the chemical evolution of the particles or simply slight changes in the size distributions. Mie theory calculations were used to demonstrate that these changes were due to the evolution of the chemical composition and, in turn, the complex refractive index of the particles. A final question is whether the features observed for the evolution of optical properties at each RH can be reproduced by a simple kinetics model. Consequently, a multi-layer kinetics model was constructed, and potential parameters are proposed.

Figure 1Experimental setup during aqueous and heterogeneous OH oxidation. CPC represents the condensational particle counter, DMA the differential mobility analyzer, and PASS the photoacoustic spectrometer.
2.1 Experimental methods
2.1.1 Preparation of BrC surrogate by aqueous OH oxidation
The BrC surrogate was prepared by aqueous OH oxidation of resorcinol. An aqueous solution of 10 mM resorcinol and 10 mM H2O2 was prepared and placed in a cylindrical photochemical reactor (Rayonet, RPR-200), where it was irradiated with UV-B black lights (Ushio, G8T5E) with peak emission at 306 nm. The reactor is equipped with a cooling fan and magnetic stirrer. The solution was housed in a glass bottle, so it was not exposed to light with wavelengths <300 nm. The solution was irradiated for 4 h, resulting in an orange product mixture. When H2O2 was not added to solution, no colouration was observed in the same period of irradiation. UV–vis spectra were measured by passing light from a broadband source, with coupled deuterium and tungsten halogen lamps (Ocean Optics, DT-Mini-2), through a 1 cm quartz cuvette to a grating-based UV–vis spectrometer (Ocean Optics, USB2000+ UV–VIS-ES). Each solution was prepared with deionized water (18 MΩ cm), and the pH was not adjusted. After irradiation, the solution was atomized immediately to inject BrC aerosol into the chamber. A fresh solution was prepared for each chamber experiment.
2.1.2 Preparation of yellow dye aerosol
The heterogeneous OH oxidation of a yellow azo dye, Cibacron Brilliant Yellow 3G-P (CBY; Sigma), was also investigated. The dye has a high molecular mass, 831.02 g mol−1, so it is non-volatile. It contains sulfonate groups, so it is hygroscopic, similar to Solvent Black 5, which is commonly used in calibrations of aerosol optical instruments (Bluvshtein et al., 2017; Lack et al., 2006; Wiegand et al., 2014). CBY aerosol was prepared by atomizing 1 mM CBY solutions. The absorption spectrum of a diluted solution is shown in Fig. S1 in the Supplement. Peak absorbance occurs at 404 nm.
2.1.3 Heterogeneous OH oxidation of BrC surrogate aerosol
The experimental setup during heterogeneous OH oxidation is illustrated in Fig. 1. The aqueous solutions of BrC surrogate were aerosolized using a constant output atomizer (TSI, 3076). To obtain appreciable aerosol absorption, it was necessary to atomize for roughly 3 h, and during this time the resorcinol product solution became significantly more absorptive. Since particles injected at the end of this period may be more absorptive than those injected at the beginning, the absorption and scattering coefficients must be qualified as time-integrated properties. Two denuders consisting of tubular mesh packed in granular activated carbon were placed in series downstream of the atomizer to remove trace gases. In preliminary experiments without the denuders in place, significant particle growth occurred upon exposure to OH, due to gas phase photo-oxidation of volatile species in the resorcinol product mixture, increasing the scattering of the aerosol. In the yellow dye experiments, the activated carbon denuders were not used, because the large dye did not partition out of the aerosol phase following atomization, and no particle growth occurred.
The aerosol was then injected into the Mobile Oxidative Chamber for Aging (MOCA), which has been described in the past (Wong et al., 2015). Briefly, the chamber consists of a 1 m3 bag composed of fluorinated ethylene propylene (FEP) film surrounded by an array of 24 UV-B black lights (Philips, TL 40W/12 RS) with peak emission at 310 nm. Experiments were conducted at roughly 15 % or 60 % RH. Before each experiment, the chamber was cleaned by bubbling air through H2O2 (Sigma, 30 % w∕w in water) at about 12 L min−1 and irradiating the chamber for at least 12 h. About 6 h before the start of each experiment, the chamber was flushed with either dry or humidified air at 12 L min−1 to achieve 15 % or 60 % RH, respectively. The temperature and RH were measured in the chamber using a capacitance probe (Vaisala, HMP75B). Air was supplied by a clean air generator (Aadco, 737).
During and after particle injection, aerosol size distributions were monitored using a scanning mobility particle sizer (SMPS), consisting of a differential mobility analyzer (DMA; TSI, 3081) and a condensational particle counter (CPC; TSI, 3772). The sample flow rate of the CPC is 1 L min−1, but only 0.3 L min−1 was sampled through the DMA; the difference was sampled through a filter. The DMA sheath flow rate was set to 3 L min−1.
Aerosol absorption and scattering were measured using a Photoacoustic Soot Spectrometer (PASS, Droplet Measurement Technologies), equipped with 405 and 781 nm lasers and a reciprocal integrating nephelometer (Sharma et al., 2013) with a low truncation angle (Abu-Rahmah et al., 2006). Absorption and scattering coefficients were taken as averages of 120 samples, each with an integration time of 2 s. Acoustic and zero calibrations, the latter consisting of 30 samples, were performed before each average, so the interval between averaged coefficients was 5 min. The zero calibrations account for slight changes in the gas phase background – in particular, water vapour. In the following discussion, we characterize the optical properties of the aerosol in terms of the directly measured absorption and scattering coefficients (βabs and βsca) and the single scattering albedo (). Relative SSA is calculated by taking the ratio of the current and initial values, e.g. SSA∕SSA0.
Following aerosol injection, the size distribution and optical properties of the BrC aerosol were monitored for about 1 h before passing air through a glass bubbler containing H2O2 into the chamber at a flow rate of 1.5 L min−1. Following 1 h of bubbling, the black lights were turned on to produce OH from the photolysis of H2O2. Oxidation was monitored for about 3 h.
One potential source of bias in photoacoustic measurements is evaporation of water from particles (Baker, 1976; Raspet et al., 2003; Langridge et al., 2013); through evaporation, some of the energy of the absorbed photons may be lost and may not contribute to the detected pressure wave. On this basis, the higher RH was selected to be lower than the maximum operating RH of the PASS (70 %). To verify that evaporation of water did not influence the measurements of absorption coefficients, we alternately sampled with and without a diffusion dryer downstream of the chamber in one experiment.
2.1.4 Determination of OH concentration
In a separate set of high-RH experiments, o-xylene (Sigma, ≥98 %) was injected into the chamber as a tracer for OH, following the aerosol injection. The concentration of the tracer was measured using a proton-transfer-reaction mass spectrometer (PTR-MS; Ionicon). Following the injection of o-xylene, air was bubbled through H2O2 into the chamber at 1.5 L min−1 for 1 h; subsequently, the black lights were turned on to initiate photo-oxidation. The concentration decayed exponentially, due to dilution and a pseudo-first-order reaction with OH. Dividing the experimental reaction rate constant by the second-order reaction rate constant (Atkinson and Arey, 2003) gave a steady-state OH concentration of molecule cm−3. Three experiments were performed with different initial o-xylene concentrations to verify that the tracer did not contribute significantly to the total OH sink; i.e. the steady-state OH concentration did not systematically increase as the o-xylene concentration decreased. The tracer could not be added to all experiments, because the photo-oxidation products partitioned onto the pre-existing particles to form secondary organic aerosol (SOA), which drastically increased the scattering coefficient. The average OH concentration is used to present results in terms of OH exposure, in addition to reaction time.
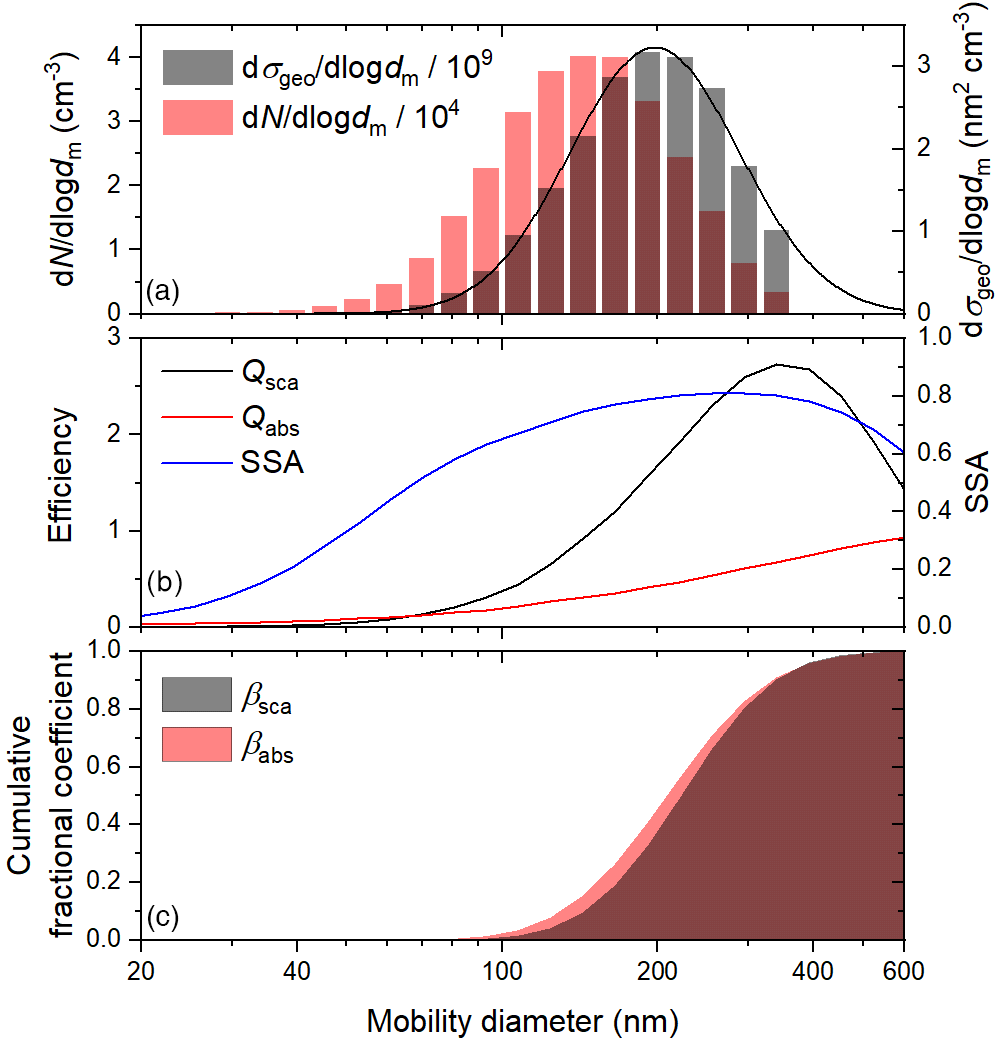
Figure 2Representative (a) number- and geometric-cross-section-weighted size distributions, (b) predicted scattering and absorption efficiencies and SSA, based on an assumed complex refractive index of , and (c) cumulative fractional scattering and absorption coefficients, based on the observed size distribution and predicted efficiencies.
2.2 Computational methods
2.2.1 Mie theory calculations
To verify that changes in SSA during heterogeneous OH oxidation were induced by chemical changes to the aerosol rather than small changes in the size distributions, we compared the observed trends to modelled trends, based on the observed size distributions and an assumed, constant complex refractive index of , where n and k are the real and imaginary parts, respectively. Lognormal curves were fit to the raw geometric-cross-section-weighted size distributions (see Fig. 2a), and the absorption and scattering efficiencies for 405 nm light of individual channels between about 20 and 600 nm were calculated from the complex refractive index of the initial BrC particles, which are assumed to be spherical and homogeneously mixed, using the program Mie3Layer (Charamisinau et al., 2005). The absorption and scattering coefficients were calculated for each channel, and total values across the distribution were used to calculate the SSA of the whole aerosol population.
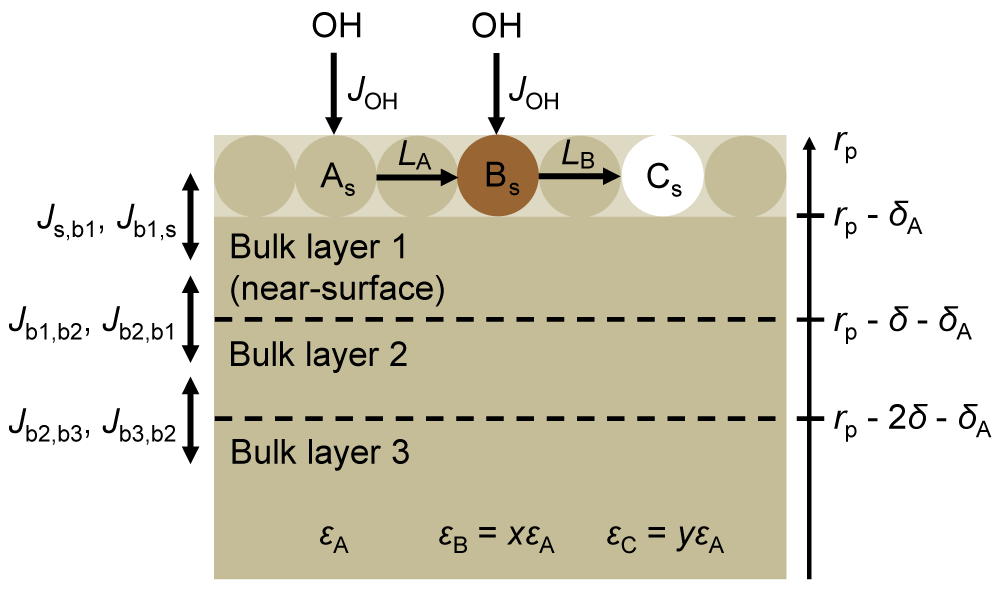
2.2.2 Multi-layer kinetics modelling
To better understand the effects of RH on the evolution of the optical properties of the BrC surrogate, we constructed a multi-layer kinetics model based on the Pöschl–Rudich–Ammann (PRA) framework (Ammann and Pöschl, 2007; Pöschl et al., 2007). The model particles consist of a surface layer, two near-surface bulk layers, and the remaining bulk phase, as illustrated in Fig. 3. The near-surface bulk layers are included to account for concentration gradients in the bulk phase (Shiraiwa et al., 2010); each is 2 nm thick. The diameter and density of the particles are assumed to be 180 nm and 1.3 g cm−3, respectively. The particles are assumed to be initially composed of a single BrC species, A. At the surface, A reacts with OH to form product B, and B reacts to form product C. The loss of species i=A, B due to reaction with OH, Li, is calculated as γOH,iJOHθi, where γOH,i is the probability that a collision between OH and species i leads to a reaction, JOH is the flux of OH to the particles per unit of surface area, and θi is the fractional surface coverage of species i (Shiraiwa et al., 2009).
The rate constant for diffusion between bulk layers, , is calculated as , where Db,i is the bulk diffusion coefficient of species i, and δ is the layer thickness (Shiraiwa et al., 2010). For the transfer from the near-surface bulk layer 1 to the surface, , δ above is substituted with , where δA is the effective molecular diameter of A (Shiraiwa et al., 2010). Though B may form from oligomerization of compounds grouped into species A, we assume that the effective molecular diameters of all species are the same. We also assume the species have the same bulk diffusion coefficients, ensuring that the total concentration in the surface layer is the same as the number of surface sites. The rate constant for diffusion from the surface to the near-surface bulk layer 1, , is taken as .
The above considerations allow us to calculate concentrations of A, B, and C in the surface and bulk layers. However, to compare the modelled and experimental results, we must calculate the relative absorption of the model particles. We derive the relative absorption of the particles solely from the concentrations and molar absorptivities, as in a bulk solution; i.e. the absorption that would be measured upon particle-into-liquid sampling. This modelled relative absorption is compared to the experimental absorption coefficient normalized to that which would be expected if the initial complex refractive index (m) did not change during the experiment, accounting for size dependence. Experimental time series of this normalized absorption were derived for photolysis and heterogeneous OH oxidation experiments, and the latter were corrected for the effect of photolysis.
Processes other than directly OH-initiated oxidation also occur. For example, radical products of the initial OH reaction likely form RO2 species by a reaction with O2. These RO2 species may react with each other in a termination step or react with NO, for example, to form RO (Richards-Henderson et al., 2015, 2016), which may initiate further oxidation of the brown carbon constituents. They may also facilitate auto-oxidation by abstracting hydrogen from adjacent groups on the same or neighbouring molecules. We do not consider these processes in our simple multi-phase kinetics model, since many other parameters, such as the diffusion coefficient of O2 in the particles and the absorptivities of any additional products, would have to be introduced, but they may play a role in the apparent values of uptake coefficients.
3.1 Initial BrC surrogate
To produce a laboratory surrogate of secondary BrC, we generated a mixture of light-absorbing products by the aqueous photo-oxidation of resorcinol and H2O2 exposed to UV-B radiation. Initially, the solution of resorcinol and H2O2 exhibited no absorption of light at visible wavelengths, as shown in Fig. S2. Following 4 h of photo-oxidation, the solution was highly coloured, exhibiting broad absorption features that were strongly dependent on wavelength, such that absorbance across a 1 cm path length was about 0.6 at 400 nm but was negligible at >600 nm (see Fig. S2). In the past, light-absorbing products of the aqueous photo-oxidation of resorcinol were also observed by Chang and Thompson (2010), who used 1 mM H2O2. Here, the concentration of H2O2 was 1 order of magnitude greater, and the colour developed slightly faster, as illustrated by the time series of absorbance at 450 nm (see Fig. S3).
More generally, light-absorbing products of aqueous photo-oxidation have been observed for a wide range of phenolic species, including those with methyl, methoxy, and carbonyl substituents (Chang and Thompson, 2010; Gelencsér et al., 2003; Smith et al., 2016). In detailed mechanistic studies, products of both hydroxylation and oligomerization have been identified (Hoffer et al., 2004; Li et al., 2014; Sun et al., 2010; Yu et al., 2014, 2016). Oligomers form by C–C or C–O radical coupling (Kobayashi and Higashimura, 2003). C–C coupling might be expected to lead to greater absorption enhancement, since the resulting biphenyls and larger oligomers may have some degree of delocalization across the rings (Zhang et al., 2010). Indeed, based on density functional theory calculations, Magalhães et al. (2017) have shown that the absorptivity of bi- and terphenyls is greater in water than in the gas phase, because the planarity and delocalization increase. Interestingly, 5 of the 10 most abundant products of the aqueous photo-oxidation of 0.1 mM syringol with 0.1 mM H2O2 have been shown to be biphenyls (Yu et al., 2014). The concentration of the phenolic precursor used here was 2 orders of magnitude greater, a condition that may favour oligomerization over hydroxylation. Fragmentation may also occur during aqueous photo-oxidation, but the volatile products are removed in the activated carbon denuders following atomization. Based on these considerations, the secondary BrC aerosol is likely composed primarily of oligomers, and we assume the average molecular mass is 326 g mol−1, representative of a terphenyl product of resorcinol. Before investigating the effects of photolysis and heterogeneous OH oxidation on the optical properties of the BrC surrogate, we performed control experiments without either H2O2 or irradiation to constrain the size dependence of the optical properties. Results from a deposition experiment at 60 % RH, in which BrC was injected into the chamber and diluted with clean humidified air, are shown in Fig. 4. The initial geometric mean number diameter and standard deviation were 125 nm and 1.5, respectively, and the initial SSA was about 0.76. When the particles were unperturbed by radiation or OH, they were simply lost by dilution and deposition. Because smaller particles deposit faster than larger ones, the geometric mean surface diameter gradually increased from about 170 to 190 nm over the course of 3 h. Both the absorption and scattering coefficients steadily decreased as particles were continuously lost, but the SSA increased slightly (see Fig. 2b) due to the increase in the geometric mean surface diameter.
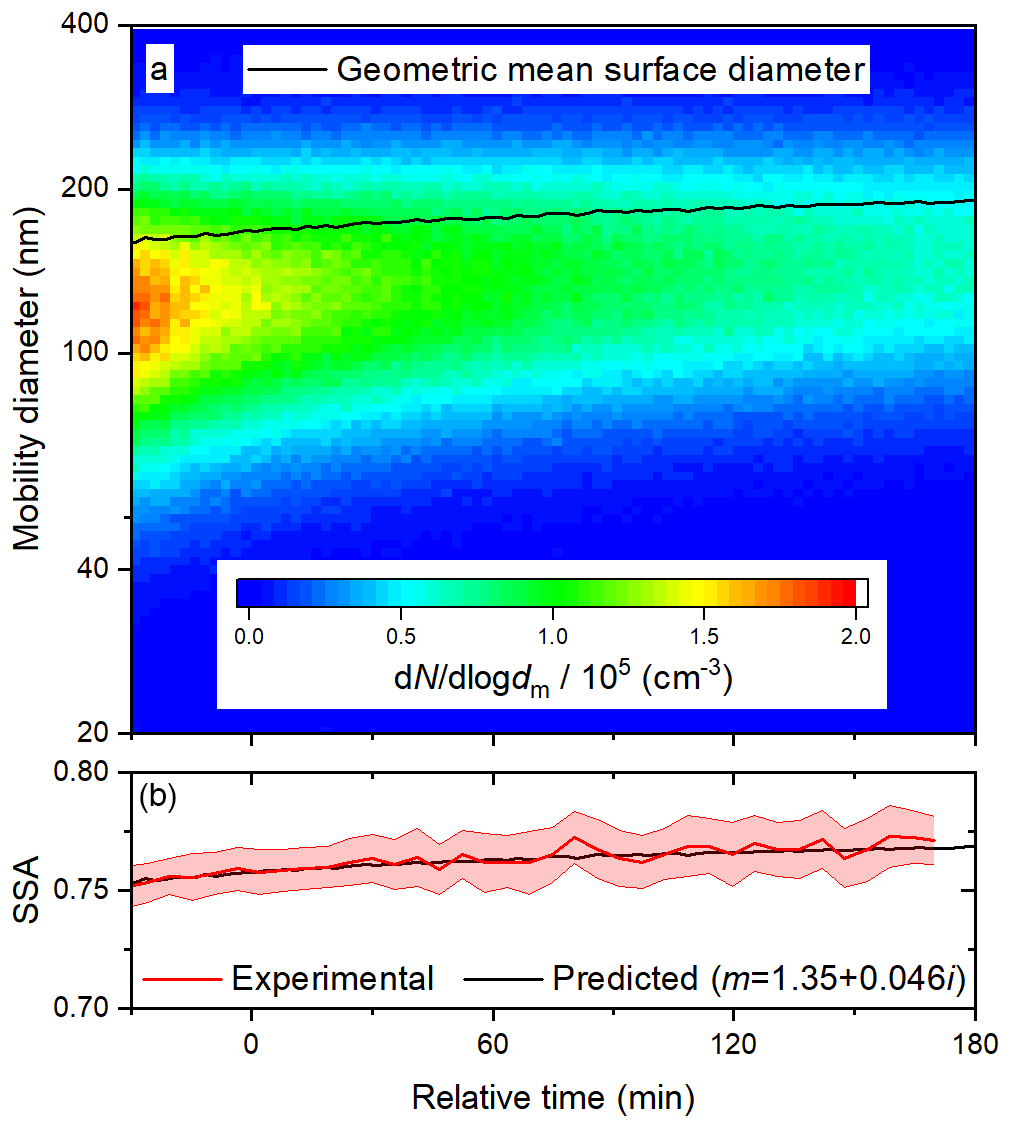
Figure 4Time series of (a) size distribution and geometric surface diameter and (b) predicted (based on size distributions) and observed SSA during a deposition experiment at 60 % RH. In (b), the upper and lower bounds illustrate one standard deviation about the 5 min averages.
This observed size dependence of SSA can be compared to Mie theory calculations, based on the measured size distributions and an assumed complex refractive index. The complex refractive index should not change without photolysis or heterogeneous OH oxidation, so we manually scanned values of n and k to apply for the duration of the experiment. We found that n and k values of 1.35 and 0.046, respectively, are suitable for reproducing the evolution of SSA in this particular experiment (see Fig. 4b). Stepping n from 1.40 to 1.55 and adjusting k accordingly demonstrates that the good agreement for this refractive index is unique, as shown in Fig. S4. We stress that even an approximate value of the refractive index is adequate to distinguish whether changes in the SSA are due to changes in the size distribution or chemical composition. For SOA generated from anthropogenic and biogenic volatile organic compounds, n values ranging from 1.35 to 1.61 have been observed (Kim and Paulson, 2013). The value of k is in the regime of previous observations for BrC at 405 nm, which vary from very low values of 0.004 (Cappa et al., 2012) and 0.007 (Lack et al., 2012) to 0.112 (at 400 nm) (Kirchstetter et al., 2004). In a recent field study in India, a similar value of 0.037 for ambient BrC was observed (Shamjad et al., 2016).
In one experiment, BrC particles were sampled from the chamber, alternately, with and without a diffusion dryer upstream of the DMA. The conditioning was alternated every 15 min, allowing time for the RH of the sheath flow to stabilize. As shown in Fig. S5, the size distribution slowly shifts to larger mobility diameters, because smaller particles are lost faster than larger particles. Besides this trend, the variation between the conditions is negligible, so there is not significant water uptake at 60 % RH.
3.2 Evolution of BrC due to photolysis
Having constrained the size dependence of the optical properties, based on the closure between measured and calculated SSA values, we now consider the effects of photolysis on the optical properties of the BrC surrogate. The emission of the UV-B black lights in the chamber, with a peak at 310 nm, is not representative of natural sunlight, so we do not draw direct comparisons between the timescale of our experiments and that of photolysis in the atmosphere. Rather, we performed photolysis experiments to account for the effects of photolysis during the heterogeneous OH oxidation experiments. Wong et al. (2015) have shown that for this chamber equipped with UV-B bulbs, the photon flux at wavelengths below 310 nm is much greater than for a clear-sky summer day; for example, at 300 nm, the photon flux from the bulbs is close to its peak value of about 1×1014 , while the photon flux outside is negligible. Light at wavelengths below 310 nm is likely driving most of the photochemistry during the photolysis experiments, so we believe that the relative impact of photolysis would be small in the atmosphere, even considering that the ambient OH concentration is lower than in the chamber.
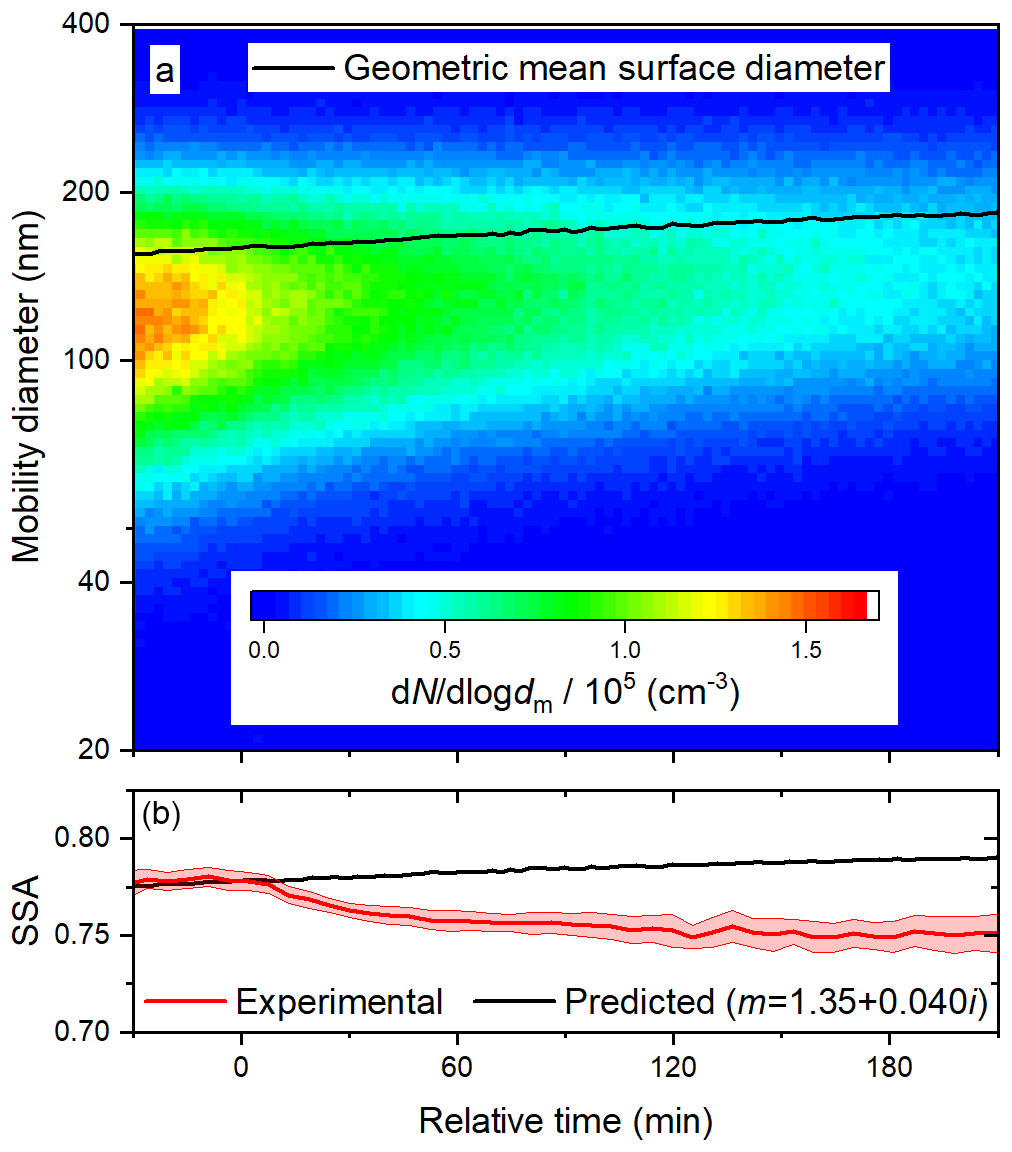
Figure 5Time series of (a) size distribution and geometric surface diameter and (b) predicted (based on size distributions) and observed SSA during a photolysis experiment at 60 % RH. In (b), the upper and lower bounds illustrate one standard deviation about the 5 min averages.
Results from a photolysis experiment at 60 % RH are illustrated in Fig. 5. In each experiment, the value of n was taken as 1.35, as described above; the assumption that n is constant for the duration of the experiment is supported by the previous observation that, for BrC generated from ammonium sulfate and methylglyoxal, n did not change as the reaction proceeded (Tang et al., 2016). Despite closely reproducing the preparation of the BrC surrogate solution and injecting it to approximately the same total particle volume in each experiment, we observed small differences in initial size distributions and SSA values, so it was necessary to vary k slightly from experiment to experiment. For the calculated SSA to follow the trend observed before the lights are turned on, at relative time zero, a k value of 0.040 must be adopted. When strict comparisons are made between experiments, the relative SSA (the current SSA divided by the initial SSA) is presented. In 3 h, the geometric mean surface diameter increased from about 160 to 180 nm, similar to the deposition experiment, so little or no volatilization of the particles can be inferred. Because the diameter increased uniformly, the predicted SSA increases steadily. In contrast, the observed SSA gradually decreased once the lights were turned on, indicating that the particles slowly became more absorbing.
In the past, photolysis has been observed to lead to the absorption enhancement of other BrC surrogates (Saleh et al., 2013; Wong et al., 2017; Zhao et al., 2015; Zhong and Jang, 2014). For example, the absorbance at 400 nm of water-soluble species produced by the combustion of kaoliang stalks has been shown to increase during the first 30 min of irradiation in a solar simulator (Zhao et al., 2015). Recently, the peak absorption enhancement due to the photolysis of biomass burning BrC constituents has been shown to increase with their molecular mass (Wong et al., 2017). Since the trace gas denuders remove volatile species after atomization, the composition of the particles is likely skewed towards an average molecular mass that is larger than that of the species in solution, possibly making the particles more susceptible to photolysis. In general, an absorption enhancement may occur due to direct photolysis of chromophores, followed by radical recombination leading to larger, more absorptive oligomers or reactions with OH from the dissociation of H2O2 generated from the photolysis of carbonyl compounds in the condensed phase (Anastasio et al., 1997; von Sonntag and Schuchmann, 1991). In the past, the absorption enhancement of biomass burning BrC exposed to natural sunlight has been shown to increase with RH (Zhong and Jang, 2014), a trend that was attributed to the increasing production and dissociation of H2O2 with increasing RH. Here, we observe similar trends in relative SSA under both low and high RH conditions (see Fig. 6b). It is unlikely that sufficient OH could be produced in the particle phase at the lower RH, so the indirect photolysis of carbonyl compounds likely does not have a significant role in the observed absorption enhancement.
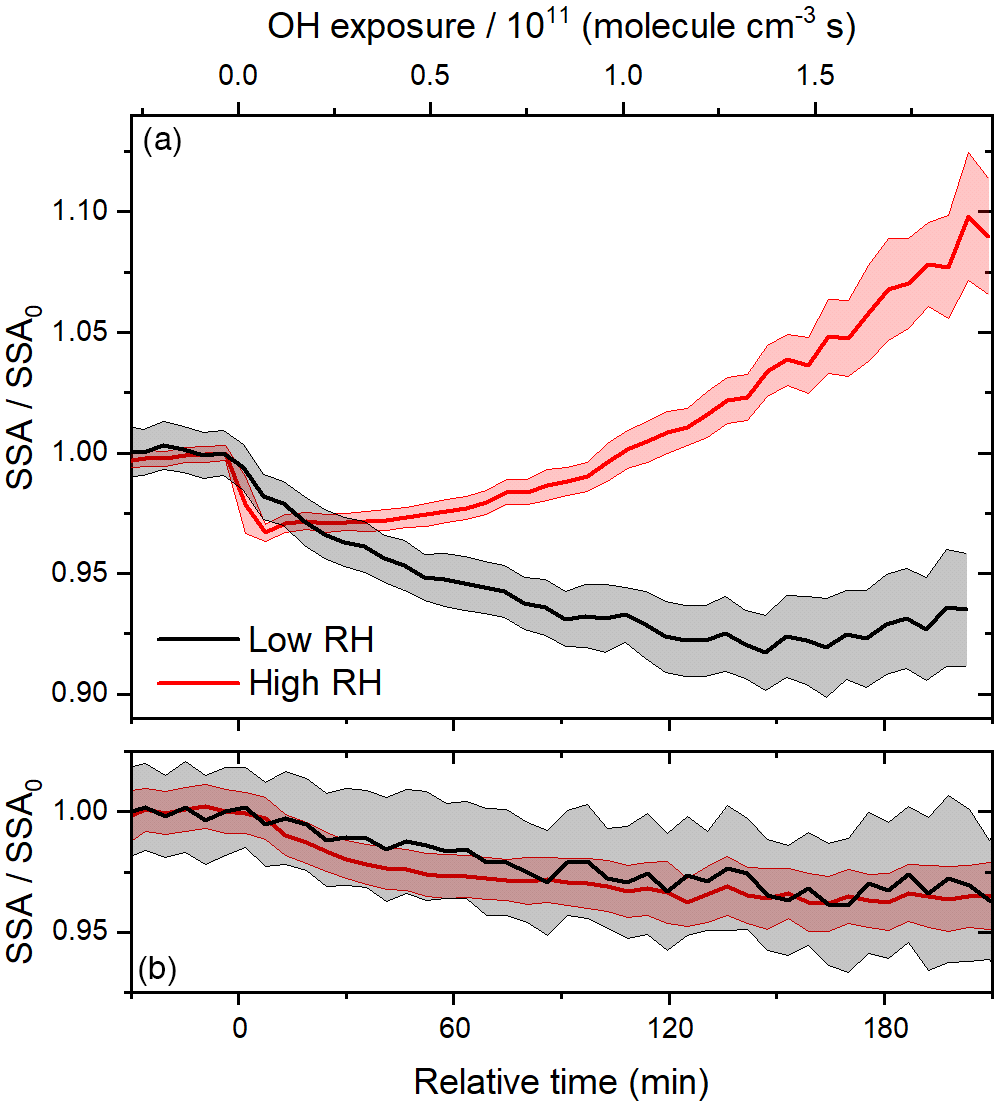
Figure 6Time series of relative SSA during (a) heterogeneous OH oxidation and (b) photolysis experiments at 15 % and 60 % RH.
In past studies, the period of absorption enhancement due to photolysis has been shown to be followed by bleaching (Wong et al., 2017; Zhao et al., 2015; Zhong and Jang, 2014). In the present experiments, the irradiation time is limited by the particle losses; after about 3 h, the scattering and absorption coefficients approach the detection limits of the PASS. Near the end of the photolysis experiments, SSA levelled off, and this period may coincide with the inflection point between absorption enhancement and bleaching.
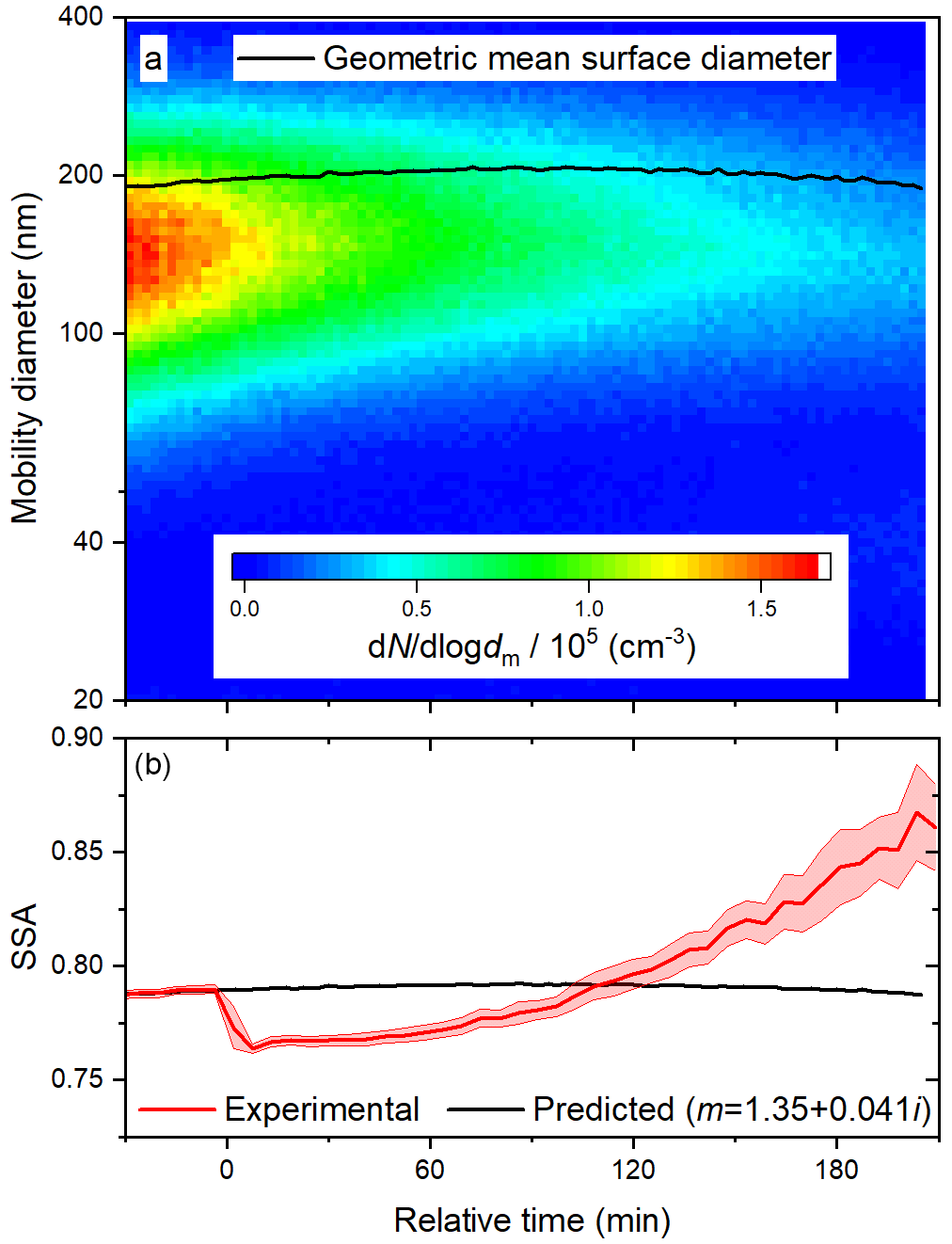
Figure 7Time series of (a) size distribution and geometric surface diameter and (b) predicted (based on size distributions) and observed SSA during a photo-oxidation experiment at 60 % RH. In (b), the upper and lower bounds illustrate one standard deviation about the 5 min averages.
3.3 Evolution of BrC due to heterogeneous OH oxidation
The evolution of the optical properties of the BrC surrogate during a photo-oxidation experiment is affected by deposition, photolysis, and heterogeneous OH oxidation. Results of a representative photo-oxidation experiment at 60 % RH are shown in Fig. 7; replicate experiments were performed at each RH. The initial geometric mean surface diameter was slightly higher than in the photolysis experiment described above (about 196 nm compared to 160 nm), so although the initial aerosol was slightly more scattering (higher SSA), the value of k required to reproduce the SSA before irradiation is very similar (0.041 compared to 0.040). During irradiation, the geometric mean surface diameter first increased slightly due to deposition and then began to decrease due to volatilization. As a result, the predicted SSA first increases slightly and then, begins to decrease at about 90 min. The observed SSA exhibited a sharp decrease within only 10 min of reaction time and an induction period before a significant increase. Both the absorption enhancement and bleaching contrast with the predicted trend of SSA, so they are certainly due to the evolution of particle composition, rather than size. The absorption enhancement was rapid enough to lead to a peak in the absorption coefficient, despite continuous particle losses (see Fig. S6).
As described above, the evaporation of water from particles can result in a negative bias in photoacoustic measurements (Langridge et al., 2013). If heterogeneous OH oxidation significantly increased the hygroscopicity of the particles, the water content of the aerosol would increase during the experiment. The resulting increase in the magnitude of the bias could contribute to apparent bleaching. In addition to this instrumental artifact, progressively more water uptake could lead to genuinely greater scattering coefficients, which would also result in apparent bleaching. We investigated these potential effects by alternately sampling with and without a diffusion dryer downstream of the chamber at 60 % RH. As shown in Fig. S7, the absorption coefficient does not depend on the conditioning. The scattering coefficient is about 10 % lower for the dried particles (see Fig. S8), but this difference is roughly steady during photo-oxidation. In other words, the particles do not become significantly more hygroscopic, and the changes in absorption and scattering coefficients are indeed due to the chemical evolution of the particles.
To ensure that the sequential absorption enhancement and bleaching described above are distinctive features of the BrC surrogate, we also investigated the heterogeneous OH oxidation of a yellow dye aerosol at 60 % RH. In this case, we observed uniform bleaching, as shown in Fig. S9, consistent with bulk aqueous studies of similar azo dyes (Georgiou et al., 2002). There was no decrease in the geometric mean surface diameter, so there was little or no volatilization. It is likely that OH attacks the azo nitrogen–nitrogen bond (Hisaindee et al., 2013). The product fragments would be large enough to remain in the particle phase; for example, the smaller fragment would still contain one sulfonate group and two aromatic rings.
Unlike photolysis, heterogeneous OH oxidation is strongly dependent on RH, as shown in Fig. 6a. In contrast to the rapid absorption enhancement and bleaching observed at 60 % RH, the period of absorption enhancement is prolonged at 15 % RH, on a timescale similar to that of the photolysis experiments. The peak absorption enhancement at 15 % RH results in a lower relative SSA than at 60 % RH. To better understand the effects of RH on heterogeneous OH oxidation, we compare experimental results with those of the multi-layer kinetics model described above. In all, the model has five adjustable parameters; these are two uptake coefficients, γOH,i, the diffusion coefficient, Db,i, and the molar absorptivities of products B and C, εB, and εC, respectively.
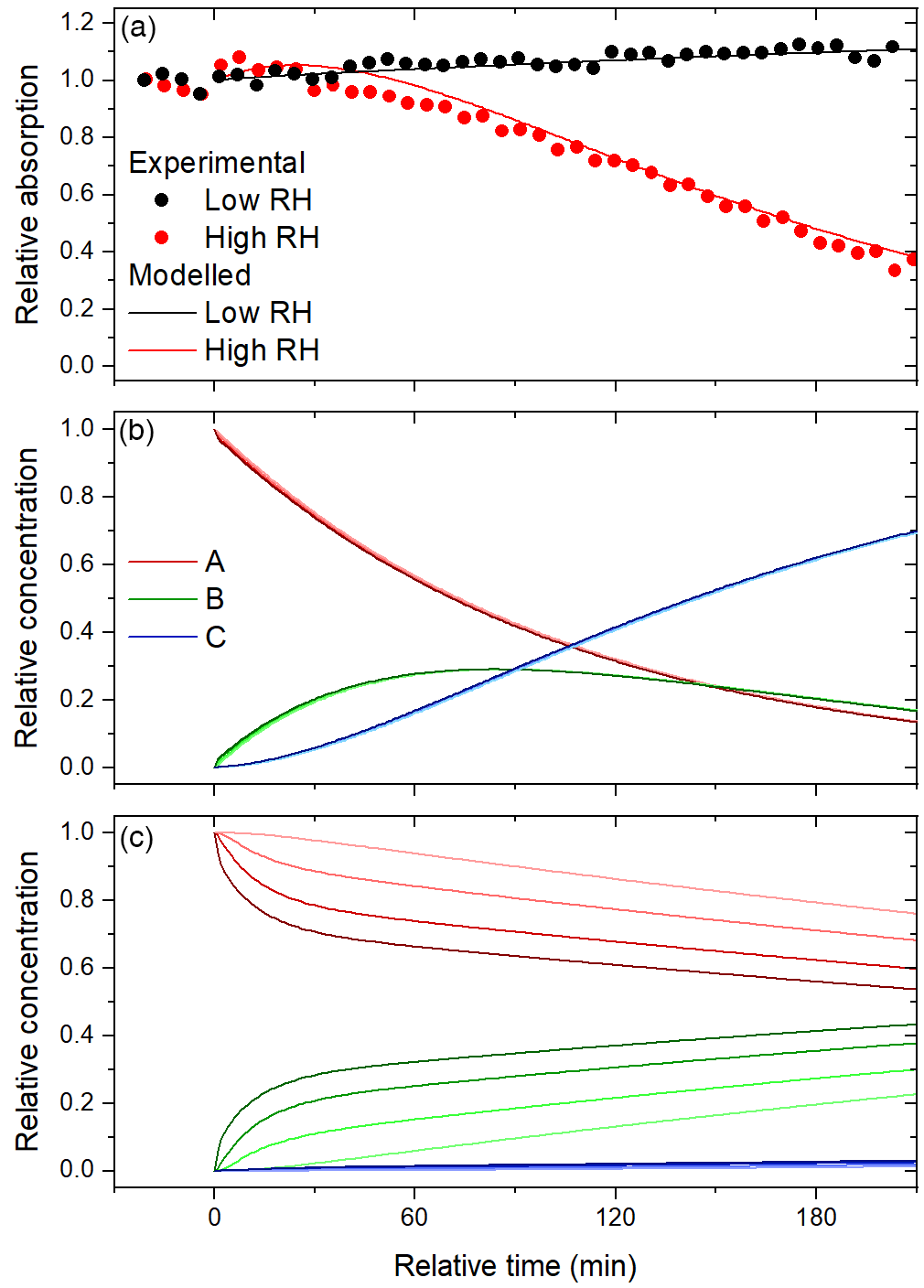
Figure 8Time series of (a) observed and modelled relative absorption at 60 % and 15 % RH and the modelled relative concentrations of A, B, and C at (b) 60 % RH and (c) 15 % RH. The curves are shaded according to layer, with the darkest curves corresponding to the surface layer.
At 60 % RH, the experimental features can be reproduced (see Fig. 8) by setting γOH,A and γOH,B to 5.0 and 7.5, respectively, suggesting that oxidation is very efficient. An uptake coefficient, γOH,A, greater than unity indicates that each collision between OH and A leads to the formation of more than one molecule of B. In previous heterogeneous OH oxidation experiments, a wide range of OH uptake coefficients have been observed, including values greater than unity (George and Abbatt, 2010). In the particle phase, oxidation initiated by OH may form products directly from reactions with OH or indirectly from organic radicals, RO. The organic radicals may result from free-radical chain reactions that have been shown to lead to very high effective uptake coefficients, in particular, when species like NO and SO2 are present to enhance the conversion of RO2 to RO (Richards-Henderson et al., 2015, 2016). We note that the values of γOH,i are dependent on the assumed average molecular mass, which is set to 326 g mol−1, representative of a terphenyl oligomerization product of resorcinol, as discussed above. A greater molecular mass would result in lower values of γOH,i, so we cannot be sure to what extent free-radical chain reactions are playing a role in the experiments. Scaling the uptake coefficients such that γOH,A decreases from 5.0 to 1.0 requires increasing the molecular weight from 326 to 1600 g mol−1. It is difficult to rationalize such large oligomers forming during the 4 h aqueous photo-oxidation, so secondary radicals are indeed likely contributing to oxidation. Furthermore, we assume that OH undergoes solely gas-surface reactions, following Shiraiwa et al. (2009). Recent measurements indicate that this may be an over-simplification, as the reactive–diffusive length of OH in squalene is similar to that of ozone (Lee and Wilson, 2016), so surface-layer reactions may also occur. Nonetheless, we can state with confidence that the oxidation is very efficient.
We find that the observed trend in relative absorption at 60 % RH – in particular, the significant bleaching – can be reproduced only if the aging particles are taken as well-mixed, so the diffusion of A from the bulk to the surface is not restricted. As shown in Fig. 8b, the concentrations of A, B, and C are the same in all four layers. At 60 % RH, secondary organic material derived from the gas-phase photo-oxidation of toluene has a diffusion coefficient on the order of 10−10 cm2 s−1 (Song et al., 2016). Here, any value greater than cm2 s−1 gives good agreement, so Db,i is set to cm2 s−1 as a lower limit. Though the extent of bleaching is captured by the model, rapid initial colour enhancement is not. We speculate that the other processes, like auto-oxidation, could contribute to this feature. The same diffusion coefficient is assumed for all three species, but in reality Db,i may increase in going from A to B and decrease in going from B to C; if C is a product of fragmentation, it might be expected to have a greater diffusion coefficient in A than the self-diffusion coefficient of A. Indeed, plasticization has been shown to occur during heterogeneous OH oxidation of semi-solid alkane particles at high gas-phase OH concentrations (Wiegel et al., 2017).
Finally, the experimental features are reproduced by setting the molar absorptivities of B and C (εB and εC) to 1.5εA and zero, respectively. We note that this combination of parameters is likely one of several possible solutions. Even for a more tightly constrained set of experimental data regarding consumption of oleic acid by heterogeneous reactions with ozone (Ziemann, 2005), more than one model scenario has been found to adequately reproduce observations of the number of oleic acid molecules in the particle phase (Pfrang et al., 2010).
At 15 % RH, the experimental features are reproduced (see Fig. 8) by setting γOH,A and γOH,B to 1.0 and 0.1, respectively, and Db,i to cm2 s−1. The decreases in the uptake coefficients with RH may be the result of differences in the distribution of initial BrC species in the particle or the orientation of the species at the surface. The decrease in Db,i by 2 orders of magnitude is reasonable, considering the significant RH dependence on viscosity observed for secondary organic material derived from toluene and isoprene (Song et al., 2015, 2016). The diffusion coefficient of secondary organic material derived from toluene was found to be less than 10−17 cm2 s−1 for RH≤17 % (Song et al., 2016). As before, we set εB and εC to 1.5εA and zero. It is possible that fragmentation products volatilize out of the condensed phase to a greater extent than they diffuse to the bulk phase, because the particles are so viscous. In fact, a slight decrease in the mean geometric surface diameter suggests that there is some degree of volatilization. On a related note, because the viscosity is so high, the products B and C are much more concentrated at the surface than in the bulk layers (see Fig. 8c). Consequently, at 15 % RH, the aging particles likely consist of less absorptive cores and highly absorptive shells.
In general, we emphasize that the set of molecular properties presented above, which best fit the experimental data, were selected after multiple trials with different input parameter values and different model scenarios as well. Although the mechanism and final parameter set fit the data remarkably well, we acknowledge that the complex nature of the inherent chemistry, with changes in optical properties occurring alongside concentration, suggests that this parameter set should only be viewed semi-quantitatively; i.e. the solution is most useful for substantiating the mechanism leading to the changes in absorption.
In this study, we have demonstrated for the first time that secondary BrC aerosol derived from a phenolic precursor is susceptible to further photochemical aging after cloud processing and droplet evaporation. Specifically, at 60 % RH, OH exposure induced a rapid absorption enhancement followed by the relatively slow bleaching of the surrogate BrC aerosol; at 15 % RH, OH exposure induced only slow absorption enhancement. Moreover, we have constructed a multi-layer kinetics model that captures the general features of the evolution of the optical properties of the particles. The candidate parameters suggest that the oxidation is very efficient, possibly even involving free-radical chain reactions, and the surrogate BrC aerosol is very viscous at 15 % RH. Free-radical chain reactions may be more important in the atmosphere, where a lower concentration of OH results in a lower concentration of RO2 and a reduced probability of chain termination reactions. Furthermore, the presence of other pollutants like NO may enhance the conversion of RO2 to RO (Richards-Henderson et al., 2015). Since the BrC is very viscous at 15 % RH, it is important that the experimental timescale approximates the atmospheric timescale, so species are allowed sufficient time to diffuse within the particles. In the laboratory, flow-tube experiments with higher OH concentrations and shorter timescales than photo-oxidation chamber experiments may not as accurately account for free-radical chain reactions and diffusion timescales.
Using our measurements and those of others, we can now speculate on the photochemical behaviour of BrC in the atmosphere. Recently, Sumlin et al. (2017) observed bleaching due to the heterogeneous OH oxidation of primary BrC derived from the smoldering of Alaskan peat, which lost almost 50 % of its absorption at 375 and 405 nm after the equivalent of about 4.5 days in the atmosphere. In our experiments at 60 % RH, the SSA of the BrC first increased and then decreased to its initial value after about 2 h of photo-oxidation in the chamber; in the atmosphere, this would correspond to about 30 h, because the average OH concentration in the chamber (1.6×107 molecule cm−3) is considerably greater than typical OH concentrations in the atmosphere (on the order of 106 molecule cm−3). In other words, the initial, rapid absorption enhancement could compensate for more than 1 day of bleaching. In the atmosphere, then, we may expect highly variable evolution of biomass burning plumes. If little or no secondary BrC forms, the bleaching of primary BrC will dominate, and the absorption will decay uniformly. Indeed, Forrister et al. (2015) have observed such a trend in the field. If considerable secondary BrC forms, e.g. by cloud processing, some additional absorption will develop. We have shown that additional absorption may develop after cloud processing as well. These processes will compete with the bleaching of primary BrC, such that the total BrC absorption may persist for longer periods. Indeed, Zhang et al. (2017) observed that the absorption by BrC at 365 nm was largely preserved during convection and 1 day of residence in the upper troposphere. Our results suggest that the persistence of BrC in this field study was the result of absorption enhancement compensating for bleaching, rather than recalcitrance of the BrC. We speculate that, upon further aging of this plume, bleaching would begin to play a greater role than absorption enhancement. In the 15 % RH experiment, an absorption enhancement was only observed after the equivalent of about 40 h in the atmosphere. Though such low RH conditions are less widely applicable in the atmosphere, there may be scenarios in which bleaching, due to heterogeneous OH oxidation, does not occur even days after secondary BrC formation from cloud processing of phenolic species.
The aerosol studied here is a reasonable proxy for the secondary BrC that may form in the atmosphere upon evaporation of cloud droplets. Biomass burning emits BC, primary BrC and non-absorbing organic compounds, in both the gas and particle phases. These organic compounds include phenolic species, derived from the decomposition of lignin during combustion (Simoneit, 2002). Resorcinol is a representative phenolic emission of biomass burning (Simoneit, 2002; Veres et al., 2010; Wang et al., 2009); for example, Schauer et al. (2001) observed about 50 µg of resorcinol from the combustion of 1 kg of wood. Furthermore, about 95 % of the resorcinol was in the particle phase, compared to about 55 and 0 % for the 1,4- and 1,2-dihydroxybenzene isomers, respectively. Whether in the aerosol phase or in cloud droplets, resorcinol and other phenolic species may react with OH to form chromophores, like the bi- and terphenyl products discussed above. Interestingly, biphenyls have recently been identified in ambient cloud water (Cook et al., 2017). The formation of secondary BrC from OH oxidation of phenolic species during the day contrasts with some other routes of secondary BrC formation that occur at night and result in chromophores – e.g. pyruvic acid polymers (Rincón et al., 2009) and imine compounds (Zhao et al., 2015) – that may be bleached comparatively quickly. The photochemical behaviour of these and other classes of secondary BrC chromophores plays an important role in the overall climate effects of BrC, and similar experiments should be performed for other surrogates in the future.
All data presented in the figures in the main text and the supplement are available at the University of Toronto Dataverse (https://doi.org/10.5683/SP2/KGAVEO, Schnitzler and Abbatt, 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-14539-2018-supplement.
EGS and JPDA designed the experiments. EGS performed the experiments and wrote the manuscript with contributions from JPDA.
The authors declare that they have no conflict of interest.
This research was funded by the Natural Sciences and Engineering Research
Council of Canada (NSERC). Elijah G. Schnitzler gratefully acknowledges a
postdoctoral fellowship from NSERC.
Edited by: Rainer Volkamer
Reviewed by: two anonymous referees
Abu-Rahmah, A., Arnott, W. P., and Moosmüller, H.: Integrating nephelometer with a low truncation angle and an extended calibration scheme, Meas. Sci. Technol., 17, 1723, https://doi.org/10.1088/0957-0233/17/7/010, 2006.
Alexander, D. T. L., Crozier, P. A., and Anderson, J. R.: Brown carbon spheres in East Asian outflow and their optical properties, Science, 321, 833–836, https://doi.org/10.1126/science.1155296, 2008.
Ammann, M. and Pöschl, U.: Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions – Part 2: Exemplary practical applications and numerical simulations, Atmos. Chem. Phys., 7, 6025–6045, https://doi.org/10.5194/acp-7-6025-2007, 2007.
Anastasio, C., Faust, B. C., and Rao, C. J.: Aromatic carbonyl compounds as aqueous-phase photochemical sources of hydrogen peroxide in acidic sulfate aerosols, fogs, and clouds. 1. Non-Phenolic methoxybenzaldehydes and methoxyacetophenones with reductants (phenols), Environ. Sci. Technol., 31, 218–232, https://doi.org/10.1021/es960359g, 1997.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bahadur, R., Praveen, P. S., Xu, Y., and Ramanathan, V.: Solar absorption by elemental and brown carbon determined from spectral observations, P. Natl. Acad. Sci. USA, 109, 17366–17371, https://doi.org/10.1073/pnas.1205910109, 2012.
Baker, M. B.: Energy absorption by volatile atmospheric aerosol particles, Atmos. Environ., 10, 241–248, https://doi.org/10.1016/0004-6981(76)90097-4, 1976.
Bluvshtein, N., Flores, J. M., He, Q., Segre, E., Segev, L., Hong, N., Donohue, A., Hilfiker, J. N., and Rudich, Y.: Calibration of a multi-pass photoacoustic spectrometer cell using light-absorbing aerosols, Atmos. Meas. Tech., 10, 1203–1213, https://doi.org/10.5194/amt-10-1203-2017, 2017.
Bond, T. C.: Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion, Geophys. Res. Lett., 28, 4075–4078, https://doi.org/10.1029/2001GL013652, 2001.
Cappa, C. D., Onasch, T. B., Massoli, P., Worsnop, D. R., Bates, T. S., Cross, E. S., Davidovits, P., Hakala, J., Hayden, K. L., Jobson, B. T., Kolesar, K. R., Lack, D. A., Lerner, B. M., Li, S.-M., Mellon, D., Nuaaman, I., Olfert, J. S., Petäjä, T., Quinn, P. K., Song, C., Subramanian, R., Williams, E. J., and Zaveri, R. A.: Radiative absorption enhancements due to the mixing state of atmospheric black carbon, Science, 337, 1078–1081, https://doi.org/10.1126/science.1223447, 2012.
Chakrabarty, R. K., Moosmüller, H., Chen, L.-W. A., Lewis, K., Arnott, W. P., Mazzoleni, C., Dubey, M. K., Wold, C. E., Hao, W. M., and Kreidenweis, S. M.: Brown carbon in tar balls from smoldering biomass combustion, Atmos. Chem. Phys., 10, 6363–6370, https://doi.org/10.5194/acp-10-6363-2010, 2010.
Chang, J. L. and Thompson, J. E.: Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds, Atmos. Environ., 44, 541–551, https://doi.org/10.1016/j.atmosenv.2009.10.042, 2010.
Charamisinau, I., Happawana, G., Evans, G., Rosen, A., His, R. A., and Bour, D.: Semiconductor laser insert with uniform illumination for use in photodynamic therapy, Appl. Optics, 44, 5055–5068, https://doi.org/10.1364/AO.44.005055, 2005.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Chung, C. E., Ramanathan, V., and Decremer, D.: Observationally constrained estimates of carbonaceous aerosol radiative forcing, P. Natl. Acad. Sci. USA, 109, 11624–11629, https://doi.org/10.1073/pnas.1203707109, 2012.
Cook, R. D., Lin, Y.-H., Peng, Z., Boone, E., Chu, R. K., Dukett, J. E., Gunsch, M. J., Zhang, W., Tolic, N., Laskin, A., and Pratt, K. A.: Biogenic, urban, and wildfire influences on the molecular composition of dissolved organic compounds in cloud water, Atmos. Chem. Phys., 17, 15167–15180, https://doi.org/10.5194/acp-17-15167-2017, 2017.
De Haan, D. O., Tolbert, M. A., and Jimenez, J. L.: Atmospheric condensed-phase reactions of glyoxal with methylamine, Geophys. Res. Lett., 36, L11819, https://doi.org/10.1029/2009GL037441, 2009.
De Haan, D. O., Hawkins, L. N., Kononenko, J. A., Turley, J. J., Corrigan, A. L., Tolbert, M. A., and Jimenez, J. L.: Formation of nitrogen-containing oligomers by methylglyoxal and amines in simulated evaporating cloud droplets, Environ. Sci. Technol., 45, 984–991, https://doi.org/10.1021/es102933x, 2011.
Del Vecchio, R. and Blough, N. V.: On the origin of the optical properties of humic substances, Environ. Sci. Technol., 38, 3885–3891, https://doi.org/10.1021/es049912h, 2004.
Di Lorenzo, R. A. and Young, C. J.: Size separation method for absorption characterization in brown carbon: Application to an aged biomass burning sample, Geophys. Res. Lett., 43, 2015GL066954, https://doi.org/10.1002/2015GL066954, 2016.
Di Lorenzo, R. A., Washenfelder, R. A., Attwood, A. R., Guo, H., Xu, L., Ng, N. L., Weber, R. J., Baumann, K., Edgerton, E., and Young, C. J.: Molecular-size-separated brown carbon absorption for biomass-burning aerosol at multiple field sites, Environ. Sci. Technol., 51, 3128–3137, https://doi.org/10.1021/acs.est.6b06160, 2017.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 2015GL063897, https://doi.org/10.1002/2015GL063897, 2015.
Gelencsér, A., Hoffer, A., Kiss, G., Tombácz, E., Kurdi, R., and Bencze, L.: In-situ formation of light-absorbing organic matter in cloud water, J. Atmos. Chem., 45, 25–33, https://doi.org/10.1023/A:1024060428172, 2003.
George, I. J. and Abbatt, J. P. D.: Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2, 713–722, https://doi.org/10.1038/nchem.806, 2010.
Georgiou, D., Melidis, P., Aivasidis, A., and Gimouhopoulos, K.: Degradation of azo-reactive dyes by ultraviolet radiation in the presence of hydrogen peroxide, Dyes Pigments, 52, 69–78, https://doi.org/10.1016/S0143-7208(01)00078-X, 2002.
Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Hisaindee, S., Meetani, M. A., and Rauf, M. A.: Application of LC-MS to the analysis of advanced oxidation process (AOP) degradation of dye products and reaction mechanisms, TrAC Trends Anal. Chem., 49, 31–44, https://doi.org/10.1016/j.trac.2013.03.011, 2013.
Hoffer, A., Kiss, G., Blazsó, M., and Gelencsér, A.: Chemical characterization of humic-like substances (HULIS) formed from a lignin-type precursor in model cloud water, Geophys. Res. Lett., 31, L06115, https://doi.org/10.1029/2003GL018962, 2004.
Hoffer, A., Tóth, A., Nyiro-Kósa, I., Pósfai, M., and Gelencsér, A.: Light absorption properties of laboratory-generated tar ball particles, Atmos. Chem. Phys., 16, 239–246, https://doi.org/10.5194/acp-16-239-2016, 2016.
Kim, H. and Paulson, S. E.: Real refractive indices and volatility of secondary organic aerosol generated from photooxidation and ozonolysis of limonene, a-pinene and toluene, Atmos. Chem. Phys., 13, 7711–7723, https://doi.org/10.5194/acp-13-7711-2013, 2013.
Kirchstetter, T. W., Novakov, T., and Hobbs, P. V.: Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon, J. Geophys. Res.-Atmos., 109, D21208, https://doi.org/10.1029/2004JD004999, 2004.
Kobayashi, S. and Higashimura, H.: Oxidative polymerization of phenols revisited, Prog. Polym. Sci., 28, 1015–1048, https://doi.org/10.1016/S0079-6700(03)00014-5, 2003.
Lack, D. A., Lovejoy, E. R., Baynard, T., Pettersson, A., and Ravishankara, A. R.: Aerosol absorption measurement using photoacoustic spectroscopy: Sensitivity, calibration, and uncertainty developments, Aerosol Sci. Technol., 40, 697–708, https://doi.org/10.1080/02786820600803917, 2006.
Lack, D. A., Langridge, J. M., Bahreini, R., Cappa, C. D., Middlebrook, A. M., and Schwarz, J. P.: Brown carbon and internal mixing in biomass burning particles, P. Natl. Acad. Sci. USA, 109, 14802–14807, https://doi.org/10.1073/pnas.1206575109, 2012.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between oxidation level and optical properties of secondary organic aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Langridge, J. M., Richardson, M. S., Lack, D. A., Brock, C. A., and Murphy, D. M.: Limitations of the photoacoustic technique for aerosol absorption measurement at high relative humidity, Aerosol Sci. Technol., 47, 1163–1173, https://doi.org/10.1080/02786826.2013.827324, 2013.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of atmospheric brown carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lee, A. K. Y., Zhao, R., Li, R., Liggio, J., Li, S.-M., and Abbatt, J. P. D.: Formation of light absorbing organo-nitrogen species from evaporation of droplets containing glyoxal and ammonium sulfate, Environ. Sci. Technol., 47, 12819–12826, https://doi.org/10.1021/es402687w, 2013.
Lee, H. J., Aiona, P. K., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Effect of solar radiation on the optical properties and molecular composition of laboratory proxies of atmospheric brown carbon, Environ. Sci. Technol., 48, 10217–10226, https://doi.org/10.1021/es502515r, 2014.
Lee, L. and Wilson, K.: The reactive–diffusive length of OH and ozone in model organic aerosols, J. Phys. Chem. A, 120, 6800–6812, https://doi.org/10.1021/acs.jpca.6b05285, 2016.
Lewis, K., Arnott, W. P., Moosmüller, H., and Wold, C. E.: Strong spectral variation of biomass smoke light absorption and single scattering albedo observed with a novel dual-wavelength photoacoustic instrument, J. Geophys. Res.-Atmos., 113, D16203, https://doi.org/10.1029/2007JD009699, 2008.
Li, Y. J., Huang, D. D., Cheung, H. Y., Lee, A. K. Y., and Chan, C. K.: Aqueous-phase photochemical oxidation and direct photolysis of vanillin – a model compound of methoxy phenols from biomass burning, Atmos. Chem. Phys., 14, 2871–2885, https://doi.org/10.5194/acp-14-2871-2014, 2014.
Lin, P., Aiona, P. K., Li, Y., Shiraiwa, M., Laskin, J., Nizkorodov, S. A., and Laskin, A.: Molecular characterization of brown carbon in biomass burning aerosol particles, Environ. Sci. Technol., 50, 11815–11824, https://doi.org/10.1021/acs.est.6b03024, 2016.
Lin, P., Bluvshtein, N., Rudich, Y., Nizkorodov, S. A., Laskin, J., and Laskin, A.: Molecular chemistry of atmospheric brown carbon inferred from a nationwide biomass burning event, Environ. Sci. Technol., 51, 11561–11570, https://doi.org/10.1021/acs.est.7b02276, 2017.
Liu, J., Bergin, M., Guo, H., King, L., Kotra, N., Edgerton, E., and Weber, R. J.: Size-resolved measurements of brown carbon in water and methanol extracts and estimates of their contribution to ambient fine-particle light absorption, Atmos. Chem. Phys., 13, 12389–12404, https://doi.org/10.5194/acp-13-12389-2013, 2013.
Magalhães, A. C. O., Esteves da Silva, J. C. G., and da Silva, L. P.: Density functional theory calculation of the absorption properties of brown carbon chromophores generated by catechol heterogeneous ozonolysis, ACS Earth Space Chem., 1, 353–360, https://doi.org/10.1021/acsearthspacechem.7b00061, 2017.
Ofner, J., Krúger, H.-U., Grothe, H., Schmitt-Kopplin, P., Whitmore, K., and Zetzsch, C.: Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS, Atmos. Chem. Phys., 11, 1–15, https://doi.org/10.5194/acp-11-1-2011, 2011.
Pfrang, C., Shiraiwa, M., and Pöschl, U.: Coupling aerosol surface and bulk chemistry with a kinetic double layer model (K2-SUB): oxidation of oleic acid by ozone, Atmos. Chem. Phys., 10, 4537–4557, https://doi.org/10.5194/acp-10-4537-2010, 2010.
Phillips, S. M. and Smith, G. D.: Light absorption by charge transfer complexes in brown carbon aerosols, Environ. Sci. Technol. Lett., 1, 382–386, https://doi.org/10.1021/ez500263j, 2014.
Phillips, S. M. and Smith, G. D.: Further evidence for charge transfer complexes in brown carbon aerosols from excitation–emission matrix fluorescence spectroscopy, J. Phys. Chem. A, 119, 4545–4551, https://doi.org/10.1021/jp510709e, 2015.
Pillar, E. A. and Guzman, M. I.: Oxidation of substituted catechols at the air–water interface: Production of carboxylic acids, quinones, and polyphenols, Environ. Sci. Technol., 51, 4951–4959, https://doi.org/10.1021/acs.est.7b00232, 2017.
Pillar, E. A., Camm, R. C., and Guzman, M. I.: Catechol oxidation by ozone and hydroxyl radicals at the air–water interface, Environ. Sci. Technol., 48, 14352–14360, https://doi.org/10.1021/es504094x, 2014.
Pillar, E. A., Zhou, R., and Guzman, M. I.: Heterogeneous oxidation of catechol, J. Phys. Chem. A, 119, 10349–10359, https://doi.org/10.1021/acs.jpca.5b07914, 2015.
Pöschl, U., Rudich, Y., and Ammann, M.: Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions – Part 1: General equations, parameters, and terminology, Atmos. Chem. Phys., 7, 5989–6023, https://doi.org/10.5194/acp-7-5989-2007, 2007.
Radney, J. G., You, R., Zachariah, M. R., and Zangmeister, C. D.: Direct in situ mass specific absorption spectra of biomass burning particles generated from smoldering hard and softwoods, Environ. Sci. Technol., 51, 5622–5629, https://doi.org/10.1021/acs.est.7b00810, 2017.
Ramanathan, V. and Carmichael, G.: Global and regional climate changes due to black carbon, Nat. Geosci., 1, 221–227, https://doi.org/10.1038/ngeo156, 2008.
Raspet, R., Slaton, W. V., Arnott, W. P., and Moosmüller, H.: Evaporation–condensation effects on resonant photoacoustics of volatile aerosols, J. Atmos. Ocean. Technol., 20, 685–695, https://doi.org/10.1175/1520-0426(2003)20<685:ECEORP>2.0.CO;2, 2003.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Large enhancement in the heterogeneous oxidation rate of organic aerosols by hydroxyl radicals in the presence of nitric oxide, J. Phys. Chem. Lett., 6, 4451–4455, https://doi.org/10.1021/acs.jpclett.5b02121, 2015.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Sulfur dioxide accelerates the heterogeneous oxidation rate of organic aerosol by hydroxyl radicals, Environ. Sci. Technol., 50, 3554–3561, https://doi.org/10.1021/acs.est.5b05369, 2016.
Rincón, A. G., Guzmán, M. I., Hoffmann, M. R., and Colussi, A. J.: Optical absorptivity versus molecular composition of model organic aerosol matter, J. Phys. Chem. A, 113, 10512–10520, https://doi.org/10.1021/jp904644n, 2009.
Saleh, R., Hennigan, C. J., McMeeking, G. R., Chuang, W. K., Robinson, E. S., Coe, H., Donahue, N. M., and Robinson, A. L.: Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions, Atmos. Chem. Phys., 13, 7683–7693, https://doi.org/10.5194/acp-13-7683-2013, 2013.
Sareen, N., Moussa, S. G., and McNeill, V. F.: Photochemical aging of light-absorbing secondary organic aerosol material, J. Phys. Chem. A, 117, 2987–2996, https://doi.org/10.1021/jp309413j, 2013.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 3. C1–C29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Schnitzler, E. G. and Abbatt, J. P. D.: Supplementary Data for Heterogeneous OH Oxidation of Secondary Brown Carbon Aerosol, available at: https://doi.org/10.5683/SP2/KGAVEO, last access: 5 October 2018.
Shamjad, P. M., Tripathi, S. N., Thamban, N. M., and Vreeland, H.: Refractive index and absorption attribution of highly absorbing brown carbon aerosols from an urban Indian city-Kanpur, Sci. Rep., 6, 37735, https://doi.org/10.1038/srep37735, 2016.
Sharma, N., Arnold, I. J., Moosmüller, H., Arnott, W. P., and Mazzoleni, C.: Photoacoustic and nephelometric spectroscopy of aerosol optical properties with a supercontinuum light source, Atmos. Meas. Tech., 6, 3501–3513, https://doi.org/10.5194/amt-6-3501-2013, 2013.
Shiraiwa, M., Garland, R. M., and Pöschl, U.: Kinetic double-layer model of aerosol surface chemistry and gas-particle interactions (K2-SURF): Degradation of polycyclic aromatic hydrocarbons exposed to O3, NO2, H2O, OH and NO3, Atmos. Chem. Phys., 9, 9571–9586, https://doi.org/10.5194/acp-9-9571-2009, 2009.
Shiraiwa, M., Pfrang, C., and Pöschl, U.: Kinetic multi-layer model of aerosol surface and bulk chemistry (KM-SUB): the influence of interfacial transport and bulk diffusion on the oxidation of oleic acid by ozone, Atmos. Chem. Phys., 10, 3673–3691, https://doi.org/10.5194/acp-10-3673-2010, 2010.
Simoneit, B. R. T.: Biomass burning – A review of organic tracers for smoke from incomplete combustion, Appl. Geochem., 17, 129–162, https://doi.org/10.1016/S0883-2927(01)00061-0, 2002.
Smith, J. D., Kinney, H., and Anastasio, C.: Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products, Atmos. Environ., 126, 36–44, https://doi.org/10.1016/j.atmosenv.2015.11.035, 2016.
Song, M., Liu, P. F., Hanna, S. J., Li, Y. J., Martin, S. T., and Bertram, A. K.: Relative humidity-dependent viscosities of isoprene-derived secondary organic material and atmospheric implications for isoprene-dominant forests, Atmos. Chem. Phys., 15, 5145–5159, https://doi.org/10.5194/acp-15-5145-2015, 2015.
Song, M., Liu, P. F., Hanna, S. J., Zaveri, R. A., Potter, K., You, Y., Martin, S. T., and Bertram, A. K.: Relative humidity-dependent viscosity of secondary organic material from toluene photo-oxidation and possible implications for organic particulate matter over megacities, Atmos. Chem. Phys., 16, 8817–8830, https://doi.org/10.5194/acp-16-8817-2016, 2016.
Sumlin, B. J., Pandey, A., Walker, M. J., Pattison, R. S., Williams, B. J., and Chakrabarty, R. K.: Atmospheric photooxidation diminishes light absorption by primary brown carbon aerosol from biomass burning, Environ. Sci. Technol. Lett., 4, 540–545, https://doi.org/10.1021/acs.estlett.7b00393, 2017.
Sun, Y. L., Zhang, Q., Anastasio, C., and Sun, J.: Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 10, 4809–4822, https://doi.org/10.5194/acp-10-4809-2010, 2010.
Tang, M., Alexander, J. M., Kwon, D., Estillore, A. D., Laskina, O., Young, M. A., Kleiber, P. D., and Grassian, V. H.: Optical and physicochemical properties of brown carbon aerosol: Light scattering, FTIR extinction spectroscopy, and hygroscopic growth, J. Phys. Chem. A, 120, 4155–4166, https://doi.org/10.1021/acs.jpca.6b03425, 2016.
Tasoglou, A., Saliba, G., Subramanian, R., and Pandis, S. N.: Absorption of chemically aged biomass burning carbonaceous aerosol, J. Aerosol Sci., 113, 141–152, https://doi.org/10.1016/j.jaerosci.2017.07.011, 2017.
Teich, M., van Pinxteren, D., Wang, M., Kecorius, S., Wang, Z., Müller, T., Mocnik, G., and Herrmann, H.: Contributions of nitrated aromatic compounds to the light absorption of water-soluble and particulate brown carbon in different atmospheric environments in Germany and China, Atmos. Chem. Phys., 17, 1653–1672, https://doi.org/10.5194/acp-17-1653-2017, 2017.
Tóth, A., Hoffer, A., Nyiro-Kósa, I., Pósfai, M., and Gelencsér, A.: Atmospheric tar balls: aged primary droplets from biomass burning?, Atmos. Chem. Phys., 14, 6669–6675, https://doi.org/10.5194/acp-14-6669-2014, 2014.
Veres, P., Roberts, J. M., Burling, I. R., Warneke, C., de Gouw, J., and Yokelson, R. J.: Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry, J. Geophys. Res.-Atmos., 115, D23302, https://doi.org/10.1029/2010JD014033, 2010.
von Sonntag, C. and Schuchmann, H.-P.: The elucidation of peroxyl radical reactions in aqueous solution with the help of radiation-chemical methods, Angew. Chem. Int. Ed. Engl., 30, 1229–1253, https://doi.org/10.1002/anie.199112291, 1991.
Wang, Z., Bi, X., Sheng, G., and Fu, J.: Characterization of organic compounds and molecular tracers from biomass burning smoke in South China I: Broad-leaf trees and shrubs, Atmos. Environ., 43, 3096–3102, https://doi.org/10.1016/j.atmosenv.2009.03.012, 2009.
Wiegand, J. R., Mathews, L. D., and Smith, G. D.: A UV–Vis photoacoustic spectrophotometer, Anal. Chem., 86, 6049–6056, https://doi.org/10.1021/ac501196u, 2014.
Wiegel, A. A., Liu, M. J., Hinsberg, W. D., Wilson, K. R., and Houle, F. A.: Diffusive confinement of free radical intermediates in the OH radical oxidation of semisolid aerosols, Phys. Chem. Chem. Phys., 19, 6814–6830, https://doi.org/10.1039/C7CP00696A, 2017.
Wong, J. P. S., Zhou, S., and Abbatt, J. P. D.: Changes in secondary organic aerosol composition and mass due to photolysis: Relative humidity dependence, J. Phys. Chem. A, 119, 4309–4316, https://doi.org/10.1021/jp506898c, 2015.
Wong, J. P. S., Nenes, A., and Weber, R. J.: Changes in light absorptivity of molecular weight separated brown carbon due to photolytic aging, Environ. Sci. Technol., 51, 8414–8421, https://doi.org/10.1021/acs.est.7b01739, 2017.
Yu, G., Bayer, A. R., Galloway, M. M., Korshavn, K. J., Fry, C. G., and Keutsch, F. N.: Glyoxal in aqueous ammonium sulfate solutions: Products, kinetics and hydration effects, Environ. Sci. Technol., 45, 6336–6342, https://doi.org/10.1021/es200989n, 2011.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Yu, L., Smith, J., Laskin, A., George, K. M., Anastasio, C., Laskin, J., Dillner, A. M., and Zhang, Q.: Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: competition among oligomerization, functionalization, and fragmentation, Atmos. Chem. Phys., 16, 4511–4527, https://doi.org/10.5194/acp-16-4511-2016, 2016.
Zhang, L., Peslherbe, G. H., and Muchall, H. M.: A general measure of conjugation in biphenyls and their radical cations, Can. J. Chem., 88, 1175–1185, https://doi.org/10.1139/V10-126, 2010.
Zhang, Y., Forrister, H., Liu, J., Dibb, J., Anderson, B., Schwarz, J. P., Perring, A. E., Jimenez, J. L., Campuzano-Jost, P., Wang, Y., Nenes, A., and Weber, R. J.: Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere, Nat. Geosci., 10, 486–489, https://doi.org/10.1038/ngeo2960, 2017.
Zhao, R., Lee, A. K. Y., Huang, L., Li, X., Yang, F., and Abbatt, J. P. D.: Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 15, 6087–6100, https://doi.org/10.5194/acp-15-6087-2015, 2015.
Zhong, M. and Jang, M.: Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 14, 1517–1525, https://doi.org/10.5194/acp-14-1517-2014, 2014.
Ziemann, P. J.: Aerosol products, mechanisms, and kinetics of heterogeneous reactions of ozone with oleic acid in pure and mixed particles, Faraday Discuss., 130, 469–490, https://doi.org/10.1039/B417502F, 2005.