the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Aug 2018
| 16 Aug 2018
Production of N2O5 and ClNO2 in summer in urban Beijing, China
Wei Zhou
Jian Zhao
Bin Ouyang
Archit Mehra
Weiqi Xu
Yuying Wang
Thomas J. Bannan
Stephen D. Worrall
Michael Priestley
Asan Bacak
Conghui Xie
Qingqing Wang
Junfeng Wang
Yingjie Zhang
Xinlei Ge
Penglin Ye
James D. Lee
Pingqing Fu
Zifa Wang
Douglas Worsnop
Roderic Jones
Carl J. Percival
The heterogeneous hydrolysis of dinitrogen pentoxide (N2O5) has a significant impact on both nocturnal particulate nitrate formation and photochemistry on the following day through the photolysis of nitryl chloride (ClNO2), yet these processes in highly polluted urban areas remain poorly understood. Here we present measurements of gas-phase N2O5 and ClNO2 by high-resolution time-of-flight chemical ionization mass spectrometer (ToF-CIMS) during summer in urban Beijing, China as part of the Air Pollution and Human Health (APHH) campaign. N2O5 and ClNO2 show large day-to-day variations with average (±1σ) mixing ratios of 79.2±157.1 and 174.3±262.0 pptv, respectively. High reactivity of N2O5, with τ (N2O5)−1 ranging from 0.20 × 10−2 to 1.46 × 10−2 s−1, suggests active nocturnal chemistry and a large nocturnal nitrate formation potential via N2O5 heterogeneous uptake. The lifetime of N2O5, τ (N2O5), decreases rapidly with the increase in aerosol surface area, yet it varies differently as a function of relative humidity with the highest value peaking at ∼ 40 %. The N2O5 uptake coefficients estimated from the product formation rates of ClNO2 and particulate nitrate are in the range of 0.017–0.19, corresponding to direct N2O5 loss rates of 0.00044–0.0034 s−1. Further analysis indicates that the fast N2O5 loss in the nocturnal boundary layer in urban Beijing is mainly attributed to its indirect loss via NO3, for example through the reactions with volatile organic compounds and NO, while the contribution of the heterogeneous uptake of N2O5 is comparably small (7–33 %). High ClNO2 yields ranging from 0.10 to 0.35 were also observed, which might have important implications for air quality by affecting nitrate and ozone formation.
- Article
(909 KB) - Full-text XML
-
Supplement
(444 KB) - BibTeX
- EndNote





Dinitrogen pentoxide (N2O5) is an efficient nocturnal sink for nitrogen oxides (NOx; Dentener and Crutzen, 1993; Brown et al., 2006). N2O5 exists in a rapid temperature-dependent thermal equilibrium with the nitrate radical (NO3) – one of the most important oxidants at nighttime (Wayne et al., 1991). Although NO3 and N2O5 levels can be suppressed by the rapid titration of NO3 against NO and volatile organic compounds (VOCs) in urban areas (Brown et al., 2003b), heterogeneous uptake by aerosol particles, fog, and cloud droplets is often found to be the major pathway for direct N2O5 removal (Bertram and Thornton, 2009; Wagner et al., 2013; Brown et al., 2006; Chang et al., 2011; Thornton et al., 2003). N2O5 can produce nitryl chloride (ClNO2) on chloride-containing aerosols, which serves as an important reservoir of NOx (Finlayson-Pitts et al., 1989; Thornton et al., 2010; Phillips et al., 2012). It has been found that levels of particulate nitrate formed through the hydrolysis of N2O5 at nighttime were comparable to those produced from the reaction of NO2 with the OH radical during daytime (Geyer et al., 2001). Furthermore, ClNO2 can be photolyzed into NO2 and atomic chlorine (Cl) after sunrise, resulting in significant impacts on daytime photochemistry, for example trace gas degradation and ozone formation (Osthoff et al., 2008; Sarwar et al., 2014; Riedel et al., 2012; Mielke et al., 2013). Thus, it is of great importance to understand N2O5 and ClNO2 chemistry in the nocturnal boundary layer of various environments.
The heterogeneous reaction of N2O5 and activation of ClNO2 are parameterized by the N2O5 uptake coefficient ( and ClNO2 product yield (ø), which are defined as the reaction probability of N2O5 upon its collision on an aerosol surface and the number of ClNO2 molecules formed per lost N2O5 molecule upon uptake, respectively (Wagner et al., 2013; Brown, 2006; Roberts et al., 2009). Previous laboratory studies have shown a large variability of (0.0002–0.3) depending on the physical characteristics of the substrates (e.g., aerosol surfaces, water droplets, and ice and crystal surfaces), environmental conditions (e.g., acidity, relative humidity, and temperature), and chemical composition of aerosol particles (e.g., nitrate, sulfate, black carbon, and organic coating; Sander et al., 2006; Chang et al., 2011; Anttila et al., 2006; Cosman et al., 2008; Thornton and Abbatt, 2005; McNeill et al., 2006). To reveal the effects of each factor on N2O5∕ClNO2 chemistry, several parameterizations of and ø have been proposed during the last decade (Riemer et al., 2003; Evans and Jacob, 2005; Anttila et al., 2006; Davis et al., 2008; Riemer et al., 2009; Griffiths et al., 2009). For example, Bertram and Thornton (2009) constructed a parameterization of as a function of aerosol liquid water, nitrate, and chloride content based on the measurements of laboratory-generated internally mixed chloride–nitrate particles. Similarly, ø was parameterized as a function of aerosol liquid water content and aerosol chloride (Roberts et al., 2009). These results have great implications for regional and global chemical transport models that aim to improve the simulations of nitrate and ozone (Evans and Jacob, 2005; Sarwar et al., 2014). However, the field-derived values of and ø often exhibit large inconsistencies with laboratory results, suggesting a more complex nature of heterogeneous N2O5 uptake in the ambient atmosphere (Brown et al., 2006; Chang et al., 2011).
N2O5 and NO3 can be measured by various different techniques, which have been summarized in Chang et al. (2011). For example, N2O5 can be derived from thermal equilibrium with NO2 and NO3 that are simultaneously measured by differential optical absorption spectroscopy (DOAS; Platt and Stutz, 2008; Stutz et al., 2004). Another indirect measurement of N2O5 is subtracting ambient NO3 from the total measured NO3 after converting N2O5 to NO3 in a heated inlet and then detected by cavity ring-down spectroscopy (CRDS), cavity-enhanced absorption spectroscopy (CEAS), or laser-induced fluorescence (LIF; O'Keefe and Deacon, 1988; Brown et al., 2001; Smith et al., 1995; Wood et al., 2003; Stutz et al., 2010). Simultaneous indirect measurements of N2O5 and NO3 can be implemented using thermal dissociation–chemical ionization mass spectrometer (TD–CIMS) with high sensitivity and time resolution (Stutz et al., 2004), although the interference of m∕z 62 (NO3) from the thermal decomposition of peroxy acetyl nitrate (PAN) and other related species needs to be considered (Wang et al., 2014). Recently, the CIMS using iodide reagent ions (I-CIMS) with an unheated inlet configuration allowed for direct measurements of N2O5 (Kercher et al., 2009; Tham et al., 2014, 2016; Wang et al., 2016). The I-CIMS is also widely used to measure ClNO2 in both laboratory and field studies (Thornton and Abbatt, 2005; McNeill et al., 2006; Osthoff et al., 2008; Tham et al., 2014, 2016; Wang et al., 2016). A large amount of ClNO2 was first observed in polluted coastal regions owing to the abundant chloride from sea salt aerosol, for example in the Gulf of Mexico and the Los Angeles basin (Osthoff et al., 2008; Riedel et al., 2012; Kercher et al., 2009). High levels of ClNO2 from anthropogenic chloride sources were also reported in some inland areas (Thornton et al., 2010; Mielke et al., 2011; Phillips et al., 2012, 2016; Bannan et al., 2015). More recently some studies in Hong Kong (Tham et al., 2014; Brown et al., 2016a; Wang et al., 2016) and in the North China Plain (NCP; Tham et al., 2016; X. Wang et al., 2017; Z. Wang et al., 2017; Wang et al., 2018) observed consistently high mixing ratios of N2O5 and ClNO2. In particular, ClNO2 can be rapidly formed in the plumes of coal-fired power plants in the NCP, which serves as an important source of chloride in non-ocean regions. Besides these measurement efforts, recently, some modeling studies have also evaluated the impacts of N2O5 and ClNO2 chemistry on the ozone formation and regional air quality in China (Xue et al., 2015; Wang et al., 2016; Li et al., 2016). Despite this, our understanding of N2O5 and ClNO2 chemistry in highly polluted urban regions with high levels of NOx, O3, and high particulate matter is far from complete.
Beijing has been suffering from severe haze pollution during the last 2 decades (Chan and Yao, 2008). As a result, extensive studies have been conducted to characterize the sources and formation mechanisms of haze episodes (Huang et al., 2014; Guo et al., 2014; Li et al., 2017). The results show that nitrate and its precursors have been playing increasingly important roles in pollution events since 2006 mainly due to the continuous decrease in SO2 (van der A et al., 2017). While the formation mechanisms of nitrate are relatively well known, the relative contributions of different mechanisms can have large variability and uncertainties. Pathak et al. (2009) found that the heterogeneous hydrolysis of N2O5 contributed 50 %–100 % of the nighttime enhancement of nitrate concentration in Beijing. WRF-Chem model simulations showed only 21 % enhancement of nitrate during highly polluted days (Su et al., 2016). A recent study also observed a large nocturnal nitrate formation potential from N2O5 heterogeneous uptake, which is comparable to and even higher than that from the partitioning of HNO3 in rural Beijing in autumn (H. Wang et al., 2017). A large contribution of the heterogeneous hydrolysis of N2O5 to the high PM2.5 nitrate, even in the daytime due to persistently high NO2, was also reported in Hong Kong (Xue et al., 2014a). All these results highlight the fact that N2O5 heterogeneous uptake might be an important pathway of nitrate formation in Beijing. A recent modeling study has evaluated the impacts of heterogeneous ClNO2 formation on next-day ozone formation in Beijing (Xue et al., 2014b). However, the roles of N2O5 in nitrate formation and of N2O5 and ClNO2 in nighttime and daytime chemistry in summer in urban Beijing during field campaigns are not characterized yet, except for one measurement in suburban Beijing in the summer of 2016 (Wang et al., 2018).
In this work, two high-resolution time-of-flight CIMSs using the same iodide ionization system operated by the Institute of Atmospheric Physics (IAP-CIMS) and University of Manchester (UoM-CIMS), respectively, were deployed in urban Beijing for real-time measurements of gas-phase N2O5 and ClNO2. A broadband cavity-enhanced absorption spectrometer (BBCEAS) operated by the University of Cambridge was also deployed synchronously for the intercomparison of N2O5. The temporal variations of N2O5 and ClNO2 in summer and their relationships are characterized. The heterogeneous N2O5 uptake coefficients and ClNO2 production yields are estimated, and their implications in nitrate formation are elucidated.
2.1 Field campaign site and meteorology
The measurements were conducted during the Air Pollution and Human Health (APHH) summer campaign from 11 to 16 June 2017 at the Institute of Atmospheric Physics (IAP), Chinese Academy of Sciences (39∘58′28 N, 116∘22′16 E, 49 m a.s.l.), which is an urban site located between the north 3rd and 4th ring roads in Beijing. The meteorological variables including wind direction (WD), wind speed (WS), relative humidity (RH), and temperature (T) at 15 and 100 m were obtained from the Beijing 325 m meteorological tower (BMT) at the sampling site. The hourly average RH ranged from 12.9 % to 82.8 %, with an average value of 36.8±15.9 %, and the hourly average temperature ranged from 17.9 to 38.7 ∘C averaged at 26.7±4.9 ∘C. All IAP instruments were deployed on the roof of a two-story building (∼ 10 m), while UoM-CIMS and BBCEAS were housed in two containers at ground level (∼ 4 m) approximately 20 m away. More details about the sampling site can be found in previous studies (Sun et al., 2012). All data in this study are reported in Beijing local time.
2.2 Instruments
2.2.1 IAP-CIMS
Ambient air was drawn into the sampling room through ∼ 2 m Teflon perfluoroalkoxy tubing (PFA; 1/4 inch inner diameters) at a flow rate of 10 standard liters per minute (SLM), from which ∼ 2 SLM was subsampled into the CIMS. Methyl iodide gas (CH3I) from a heated CH3I permeation tube cylinder (VICI, 170-015-4600-U50) was ionized by flowing through a soft X-ray ionization source (Tofwerk AG, type P) under an ultrahigh-purity nitrogen (N2, 99.999 %) flow (2.5 SLM). This flow enters an ion molecule reaction (IMR) chamber, which was maintained at a pressure of 200 mbar using an SH-112 pump fitted with a Tofwerk blue pressure control box to account for changes in ambient pressure. A short segmented quadrupole (SSQ) positioned behind the IMR was held at a pressure of 2 mbar using a Tri scroll 600 pump. Note that the voltage settings used for the guidance of ions were carefully tuned to avoid declustering as much as possible (Lopez-Hilfiker et al., 2016). The gas-phase background was determined once during the campaign by passing dry N2 into the inlet for 5 min.
2.2.2 UoM-CIMS
The UoM-CIMS setup has been described elsewhere (Priestley et al., 2018); a Filter Inlet for Gases and AEROsols (FIGAERO; Lopez-Hilfiker et al., 2014) was additionally used in this study. The gas-phase inlet of UoM-CIMS consisted of 5 m 1/4 ID PFA tubing connected to a fast inlet pump with a total flow rate of 13 SLM from which the ToF-CIMS subsampled 2 SLM. CH3I gas mixtures in N2 were made in the field using a custom-made manifold (Bannan et al., 2014). A total of 20 standard cubic centimeters per minute (SCCM) of the CH3I mixture was diluted in 4 SLM N2 and ionized by flowing through a Tofwerk X-ray ionization source. This flow enters into the IMR, which was maintained at a pressure of 400 mbar using an SSH-112 pump also fitted with a Tofwerk blue pressure control box, while the subsequent SSQ was held at a pressure of 2 mbar using a Tri scroll 600 pump. During the campaign, gas-phase backgrounds were established through regularly overflowing the inlet with dry N2 for 5 min continuously every 45 min as has been performed previously.
The ambient target molecules were first ionized by reagent ions in the IMR and then detected as adduction products with iodide, for instance ClNO2 as at m∕z 208 and 210 (), and N2O5 as at m∕z 235 (Slusher et al., 2004; Kercher et al., 2009) at a time resolution of 1 s. Data analysis is performed using the “Tofware” package (version 2.5.11) running in the IGOR Pro (WaveMetrics, OR, USA) environment. The mass axis of UoM-CIMS was calibrated using I−, I, and I, while that of IAP-CIMS was calibrated using , I−, , , , and I covering a wide range from m∕z 62 to 381. Examples of high-resolution peak fittings of m∕z 208, 210, and 235 for IAP-CIMS are presented in Fig. S1 in the Supplement.
2.2.3 Broadband cavity-enhanced absorption spectrometer (BBCEAS)
A detailed description of BBCEAS has been given in Kennedy et al. (2011). Briefly, ambient air is first heated to 140 ∘C to thermally dissociate N2O5 into NO3 and then enters the observational cavity that consists of two high-reflectivity mirrors. The sum of N2O5 and NO3 is determined using the measured optical absorption of NO3 in the wavelength of 640–680 nm. The temperature of the cavity is kept at 85±1 ∘C to prohibit the recombination of NO3 and NO2 and to maintain the stability of the optical transmission signal. A very fast flow rate of 20 L min−1 is adopted to minimize the residence time of gases through PFA tubes. The loss of NO3 through the system was estimated to be approximately 10 %.
Considering that the relatively high aerosol loadings in Beijing can attenuate the intracavity light intensity and thus deteriorate instrument sensitivity, a poly tetrafluoroethylene (PTFE) filter of pore size 1 µm was used to remove aerosol particles from the airstream. This filter also acts a point loss (∼ 10 %) for NO3 but has a negligible impact on N2O5 (Dube et al., 2006). Because the mixing ratio of N2O5 is higher than NO3 by a factor of >10 during the APHH summer campaign, the influence of filter loss on the measurements of N2O5 + NO3 is expected to be small. Aging of aerosol particles on the filter may potentially introduce uncertainties for the transmission efficiencies of NO3 and N2O5, but was found to be insignificant in this study.
2.3 Calibrations and intercomparisons
During the campaign, field calibrations for UoM-CIMS were regularly carried out using known concentration formic acid gas mixtures made in the custom-made manifold. A range of other species were calibrated after the campaign, and relative calibration factors were derived using the measured formic acid sensitivity during these calibrations as has been performed previously (Le Breton et al., 2014, 2017; Bannan et al., 2014, 2015).
The UoM-CIMS was calibrated post-campaign for both N2O5 and ClNO2 relative to formic acid that was calibrated and measured throughout the campaign. This is completed assuming that the ratio between formic acid and ClNO2 sensitivity remains constant. ClNO2 was calibrated using the method described in Kercher et al. (2009). Briefly, a stable source of N2O5 is generated and passed over a salt slurry in which excess chloride reacts to produce gaseous ClNO2. The N2O5 for this process was synthesized based on the methodology described by Le Breton et al. (2014). Excess O3 is generated through flowing 200 SCCM O2 (BOC) through an ozone generator (BMT, 802N) into a 5 L glass volume containing NO2 (Sigma-Aldrich, >99.5 %). The outflow from this reaction vessel is cooled in a cold trap held at −78 ∘C (195 K) by a dry ice–glycerol mixture in which N2O5 is condensed and frozen. The trap is allowed to reach room temperature and the flow is reversed whereby it is then condensed in a second trap held at 220 K. This process is repeated several times to purify the mixture. The system is first purged by flowing O3 for 10 min before usage. To ascertain the N2O5 concentration in the line, the flow is diverted through a heated line to decompose the N2O5 and into to a Thermo Scientific 42i NOx analyzer in which it is detected as NO2. According to the intercomparisons with the BBCEAS, including this study and others (e.g., Le Breton et al., 2014; Bannan et al., 2017), the possible interference of NOy on the NOx analyzer is not deemed important in terms of our reported N2O5 concentrations.
ClNO2 was produced by flowing a known concentration of N2O5 in dry N2 through a wetted NaCl scrubber. Conversion of N2O5 to ClNO2 can be as efficient as 100 % on sea salt, but it can also be lower, for example if ClNO2 is converted to Cl2 (Roberts et al., 2008). In this calibration we have followed the accepted methods of Osthoff et al. (2008) and Kercher et al. (2009) that show a conversion yield of 100 % and have assumed this yield in the calibrations of this study.
The second method used to verify our ClNO2 calibration is cross-calibration with a turbulent flow tube chemical ionization mass spectrometer (TF-CIMS; Leather et al., 2012). A known concentration of 0–20 SCCM Cl2 (99.5 % purity Cl2 cylinder, Aldrich) from a diluted (in N2) gas mix is flowed into an excess constant flow of 20 SCCM NO2 (99.5 % purity NO2 cylinder, Aldrich) from a diluted (in N2) gas mix, to which the TF-CIMS has been calibrated. This flow is carried in 52 SLM N2 that is purified by flowing through two heated molecular sieve traps. This flow is subsampled by the ToF-CIMS in which the adduct is observed. The TF-CIMS is able to quantify the concentration of ClNO2 generated in the flow tube as the equivalent drop in NO signal. This indirect measurement of ClNO2 is similar in its methodology to ClNO2 calibration by quantifying the loss of N2O5 reacted with Cl− (e.g., Kercher et al., 2009). The TF-CIMS method gives a calibration factor 58 % greater than that of the N2O5 synthesis method; therefore this is taken as our measurement uncertainty. This calibration was scaled to those in the field using formic acid calibrations carried out in the laboratory by overflowing the inlet with various known concentrations of gas mixtures (Bannan et al., 2014).
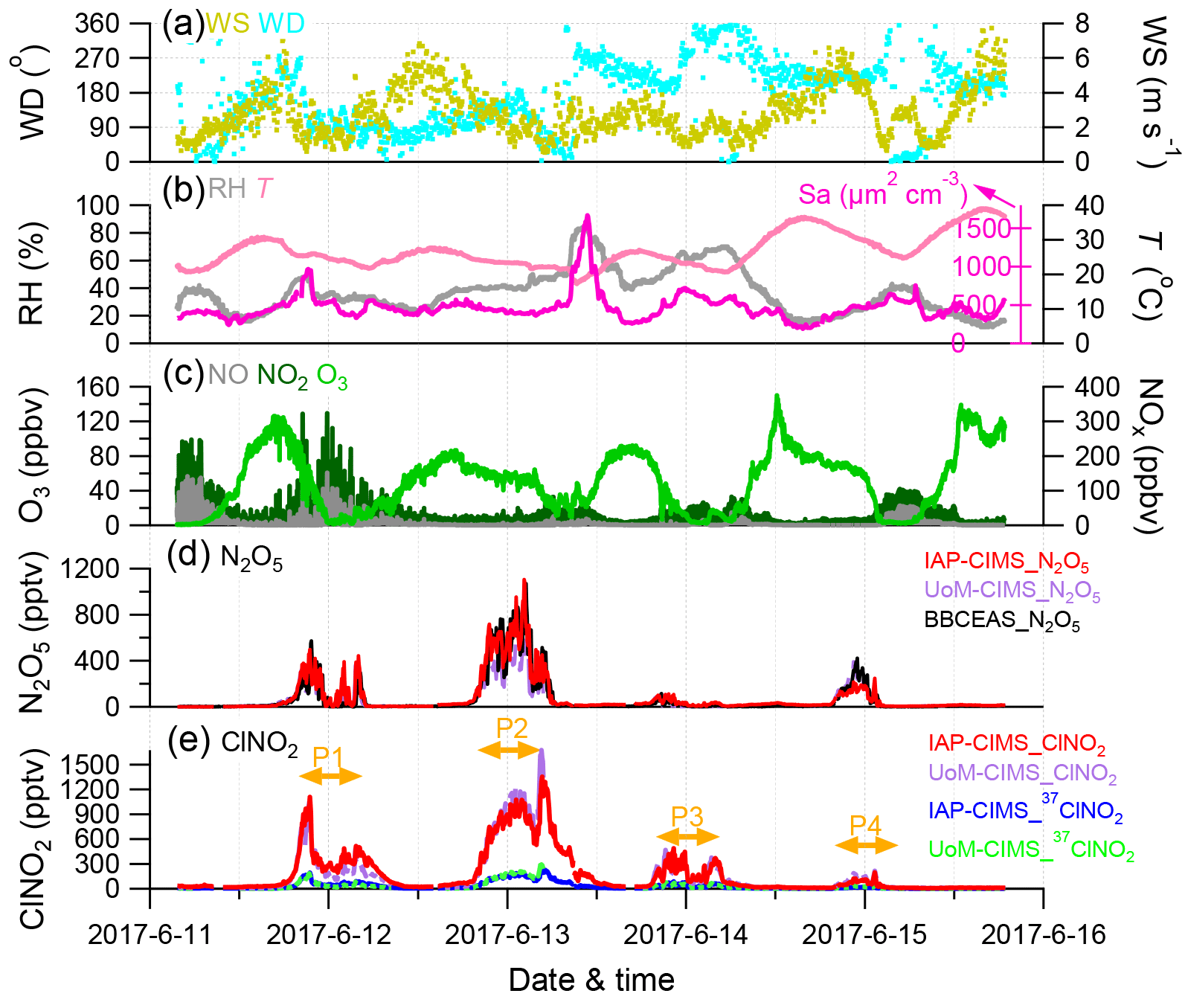
Figure 1Time series of (a–b) meteorological parameters (WS, WD, RH, T) and surface area density (Sa), (c) trace gases (O3, NO, NO2), and (d–e) IAP-CIMS species (N2O5, ClNO2). The UoM-CIMS and BBCEAS measurements are also shown for intercomparison. The four nights (i.e., P1, P2, P3, and P4) are marked for further discussions.
The IAP-CIMS calibration for N2O5 was performed by comparing with the measurements from the BBCEAS. As shown in Fig. S2, the raw signals of N2O5 from the IAP-CIMS measurements were highly correlated with those from BBCEAS (R2=0.84). Given that the intercomparison between the two instruments was relatively constant throughout the study, the average regression slope of 0.54 was then applied to determine the mixing ratio of N2O5 for the IAP-CIMS. The estimated N2O5 mixing ratios were then compared with those measured by UoM-CIMS. As shown in Fig. 1, the two N2O5 measurements tracked well with each other (R2=0.84, slope = 1.42) although some differences at midnight on 13 June were observed. The raw signals of ClNO2 given by the IAP-CIMS were first converted to mixing ratios by assuming the same sensitivity between ClNO2 and N2O5 (i.e., 0.54 cps pptv−1). The results show that the estimated ClNO2 for the IAP-CIMS agrees well with that measured by UoM-CIMS and calibrated post-campaign (R2=0.93, slope = 0.905, Fig. 1). Overall, the uncertainty is 17 % and 58 %, and the detection limit is 1.7 and 0.7 pptv for N2O5 and ClNO2 of IAP-CIMS, respectively. All the discussions below are based on IAP-CIMS measurements unless otherwise stated.
2.4 Collocated measurements
An Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-AMS hereafter) and an Aethalometer (AE33, Magee Scientific Corp.) were deployed on the roof of the two-story building to measure size-resolved non-refractory submicron aerosol (NR-PM1) species with a time resolution of 5 min, including organics (Org), sulfate (SO, nitrate (NO, ammonium (NH, chloride (Cl−) (DeCarlo et al., 2006; Canagaratna et al., 2007), and black carbon (BC). A more detailed description of the operations and calibrations of this HR-AMS can be found in Xu et al. (2015) and Sun et al. (2016). Other collocated measurements in two containers at ground level included gaseous species of O3 (TEI 49C UV absorption analyzer), NO (TEI 42i TL NO analyzer), and NO2 (CAPS NO2 monitor, Aerodyne Research Inc.) and size-resolved particle number concentrations (11–550 nm) by a scanning mobility particle sizer (SMPS) equipped with a long differential mobility analyzer (DMA, TSI, 3081A) and a condensation particle counter (CPC, TSI, 3772).
2.5 Data analysis
2.5.1 Estimation of
NO3 is formed from the reaction of NO2 with O3 (Reaction R1) with a temperature-dependent reaction rate constant k1. NO3 rapidly photolyzes during daytime, but at night it reacts with NO2 to produce N2O5 (Reaction R2). N2O5 can thermally decompose back to NO3 and NO2, and the equilibrium rate coefficient Keq is a function of ambient temperature. In this study, values of k1 and Keq recommended by Atkinson et al. (2004) and Brown and Stutz (2012) were used. The indirect loss of N2O5 is mainly through reactions of NO3 with either NO or VOCs (Reaction R3), while direct N2O5 loss is predominantly from the heterogeneous hydrolysis on the surface of aerosol particles that contain water (Reaction R4) or chloride (Reaction R5). Note that “het” is an abbreviation of heterogeneous in the equations. The net reaction of Reactions (R4) and (R5) can be described as Reaction (R6) where is the heterogeneous uptake rate of N2O5, and ø is the ClNO2 yield.
When the uptake reaction was not limited by gas-phase diffusion, can be simplified as Eq. (1) (Riemer et al., 2003; Dentener and Crutzen, 1993):
where c is the mean molecular speed of N2O5 (unit, m s−1), and Sa is the aerosol surface area density calculated from the size-resolved particle number concentrations assuming spherical particles (unit, µm cm−3). Note that Sa determined under dry conditions was converted to that under ambient RH levels by using the hygroscopic growth factor in Liu et al. (2013).
The nocturnal mixing ratio of NO3 can be derived from simultaneous measurements of NO2 and N2O5 (Reaction R2) assuming that the equilibrium between NO3 and N2O5 is rapidly established after sunset (Brown et al., 2003a).
The nitrate radical production rate p(NO3) can be calculated from Reaction (R1) assuming that the nitrate radical is solely from Reaction (R1).
With a steady-state assumption for NO3 and N2O5, the inverse N2O5 steady-state lifetime, τ(N2O5)−1, which is defined as the ratio of p(NO3) to the N2O5 mixing ratios, can be expanded to Eq. (4) after the substitution of Eqs. (2) and (3) into the approximate time change rate for N2O5 (Brown et al., 2003a).
represents the contribution to τ(N2O5)−1 from the indirect N2O5 loss pathway, i.e., through NO3 reactions with VOCs and NO, while indicates the direct loss of N2O5 through heterogeneous uptake.
Considering that the production of ClNO2 is predominantly from heterogeneous N2O5 uptake within stable air masses and precursors, the production rate of ClNO2 (pClNO2) can be related to the heterogeneous loss rate of N2O5 by
The production rate of particulate nitrate (pNO was obtained from HR-AMS measurements assuming that the measured p was totally from the production of nitrate by Reaction (R4) (Phillips et al., 2016). Note that the formation of particulate nitrate from regional transport or via the net uptake of HNO3 to aerosol is not taken into consideration.
Only periods with concurrent nighttime formation of ClNO2 and meet the requirement that both are produced only from heterogeneous N2O5 uptake. By combining Eq. (5) with Eq. (6), and ø can be represented as follows.
2.5.2 Parameterization of and ø
Aerosol liquid water content associated with inorganic species was estimated using the ISORROPIA-II thermodynamic equilibrium model (Nenes et al., 1998; Fountoukis and Nenes, 2007), with input data of ambient NR-PM1 species, and RH and T at 15 m. The N2O5 uptake coefficient and ClNO2 yield can also be calculated by the parameterization proposed by Bertram and Thornton (2009).
[H2O], [], and [Cl−] are molar concentrations of liquid water, particle nitrate, and chloride, respectively, and the empirical parameters A and are used.
3.1 Overview of N2O5 and ClNO2 measurements
Figure 1 shows the time series of N2O5 and ClNO2, gaseous species of NO, NO2, and O3, and meteorological parameters during the field campaign. Both N2O5 and ClNO2 exhibited large day-to-day variability with the 5 min average (±1σ) mixing ratios being 79.2±157.1 and 174.3±262.0 pptv, respectively. Such dramatic variations of N2O5 and ClNO2 are consistent with previous observations in various environments, for example at ground sites in Colorado and London (Bannan et al., 2015; Thornton et al., 2010) and the residual layer at Mt. Tai (Z. Wang et al., 2017). Four nights (i.e., P1, P2, P3, and P4 from 20:00 to 04:30) were selected to investigate the nocturnal chemistry of N2O5 and ClNO2 in this study. The first two nights (P1 and P2) showed much higher mixing ratios of N2O5 and ClNO2 than those during P3 and P4, although the NOx and O3 levels during P4 were comparable to those during P2 (Table 1).
Table 1Summary of average (±1σ) meteorological parameters (RH, T, WS), CIMS species (N2O5, ClNO2), the calculated NO3, nitrate radical production rate p(NO3), N2O5 reactivity (τ(N2O5)−1) and NO3 reactivity (τ(NO3)−1), trace gases (O3, NO2, NO), and NR-PM1 species (, Cl−) for the entire study and four nighttime periods (i.e., P1, P2, P3, and P4).
The highest N2O5 mixing ratio (1.10 ppbv, 5 min average) was observed at 02:15 on 13 June (P2), which is comparable to the previous observation in urban Beijing (1.3 ppbv; H. Wang et al., 2017), but much lower than that in the aged air masses in Hong Kong at ∼ 7.8 ppbv (Brown et al., 2016b). A recent measurement at a suburban site in Beijing impacted by the outflow of urban Beijing air masses also reported consistently high N2O5 (1 min maxima 937 pptv; Wang et al., 2018). The mixing ratio of N2O5 was also much higher than that in the nocturnal residual boundary layer at Mt. Tai (167 pptv; Z. Wang et al., 2017), indicating potentially significant nighttime N2O5 chemistry in highly polluted urban areas. One of the reasons for this could be the high mixing ratios of precursors; for instance, the average O3 mixing ratios at nighttime were as high as 18–56 ppbv. The maximal N2O5 that occurred during P2 rather than the rest of the nights was likely due to insignificant titration of NO during P2, e.g., 0.5 vs. 2.3–15.6 ppbv. The lowest nighttime average of N2O5 (∼ 37.8 pptv) was observed during P3 although the NO2 showed a much higher concentration than those during P2 and P4, indicating the joint influences of precursors (NO2 and O3). Fast heterogeneous hydrolysis of N2O5 under high RH (∼ 60.5 %) conditions during P3 could be another reason, which was supported by the higher ClNO2 during P3 than P4.
Similar to N2O5, ClNO2 presented the highest value (1.44 ppbv, 5 min average) before sunrise on 13 June (P2), yet it is lower than the maximum of 2.1 ppbv (1 min average) observed at a rural site located to the southwest of Beijing (Tham et al., 2016) and also the ClNO2 peak of 2.9 ppbv (1 min average) in suburban Beijing (Wang et al., 2018). These results indicated ubiquitously observed ClNO2 in the NCP, although high ClNO2 mixing ratios have also been previously observed in both marine and continental environments in North America, Europe, and Asia (Osthoff et al., 2008; Mielke et al., 2011; Thornton et al., 2010; Phillips et al., 2012; Tham et al., 2014). The average nitrate radical production rate p(NO3) was 2.8 and 3.6 ppbv h−1 during P1 and P2, respectively, which are both higher than those during P3 and P4 (1.7–2.6; Table 1). This result supports a higher production potential for N2O5 during P1 and P2. On average, p(NO3) was 2.6±2.4 ppbv h−1 at nighttime, indicating more active nocturnal chemistry than previous studies in NCP in terms of radical production rates, for example 1.2±0.9 ppbv h−1 in suburban Beijing, 1.7±0.6 ppbv h−1 in Wangdu, and 0.45±0.40 ppb h−1 at Mt. Tai (Tham et al., 2016; Z. Wang et al., 2017; Wang et al., 2018). We also note that the p(NO3) was comparable between P4 and P2 (2.6 vs. 2.8 pptv), yet the N2O5 and ClNO2 mixing ratios during P4 were much lower, likely due to the difference in NO levels, i.e., 0.5 vs. 7.1 ppbv. The favorable dispersing meteorological conditions with higher wind speed and lower relative humidity in P4 than in P2 might also be an explanation (Table 1). Our results illustrate that precursors levels, reaction rates, and meteorological conditions can all affect the variability of N2O5 and ClNO2.
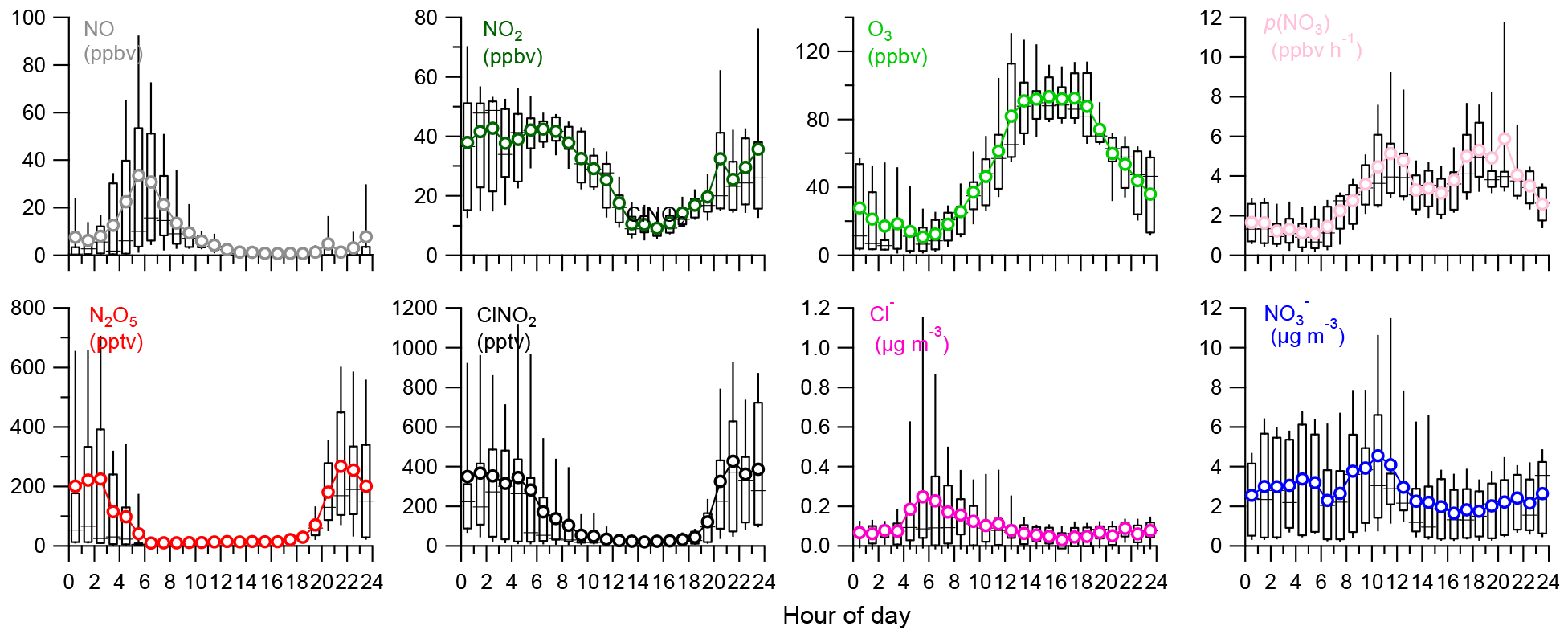
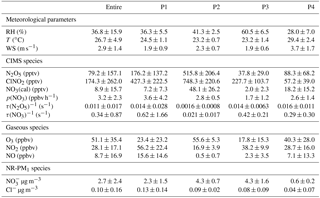
Figure 2Diurnal variations of trace gases (NO, NO2, O3), IAP-CIMS species (N2O5, ClNO2), nitrate radical production rate p(NO3), and NR-PM1 species (Cl−, NO.
The average diurnal variations of trace gases, N2O5, ClNO2, and submicron nitrate and chloride are depicted in Fig. 2. O3 showed a pronounced peak of 93.3 ppbv between 14:00 and 16:00 corresponding to a minimum mixing ratio of NO2 (9.1 ppbv). As a consequence, p(NO3) showed relatively high values around noon with a decrease in the middle of the afternoon owing to the depletion of NO2 and then reached a maximum of 5.9 ppbv h−1 before sunset. A similar diurnal pattern of p(NO3) was also observed at a rural site in the autumn in Beijing (H. Wang et al., 2017). Both NO and NO2 showed pronounced diurnal cycles with lowest concentrations in the afternoon. In addition to the rising boundary layer, the formation of NOz is another important reason for the low levels of NOx during this time period in urban Beijing (Sun et al., 2011). Nitrate and chloride also showed lowest concentrations in the late afternoon, mainly due to the evaporative loss under high temperature conditions (Sun et al., 2012).
Figure 3(a–b) Average reactivity of N2O5 (τ(N2O5)−1) and NO3 (τ(NO3)−1) for different nights (i.e., P1, P2, P3, and P4). The error bar represents the standard deviation. (c) Variations of the nocturnal τ(N2O5) as a function of aerosol surface area density (Sa) and (d) variations of the nocturnal τ(N2O5) as a function of relative humidity (RH). The data were binned according to Sa (50 µm2 cm−3 increment) or RH (5 % increment). Mean (triangle), median (horizontal line), 25 and 75th percentiles (lower and upper box), and 10 and 90th percentiles (lower and upper whiskers) are shown for each bin.
N2O5 was rapidly formed after sunset. The mixing ratio of N2O5 peaked approximately at 22:00 and then remained at a consistently high level (∼ 200–300 pptv) until 03:00. After that, N2O5 showed a rapid decrease due to significant titration by NO. Similar loss of N2O5 due to the injection of NO-containing air was also reported at sites near urban areas (Brown et al., 2003b). Because NO is predominantly from local emissions as supported by the tight correlation (R2=0.64–0.73, Fig. S3) with black carbon, a tracer for combustion emissions, our results demonstrated that local NO emissions serve as an important scavenger of N2O5 before sunrise in urban Beijing. In comparison, the decrease in N2O5 due to NO titration only occurred during the second half of the night with low O3 in suburban Beijing (Wang et al., 2018). This study also found high N2O5 after midnight due to the incomplete titration of O3, for instance ∼ 52.9 ppbv after midnight on 13 June, which is different from previous findings that high N2O5 mixing ratios were typically observed before midnight due to the rapid depletion of O3 (H. Wang et al., 2017; Z. Wang et al., 2017). The high nocturnal mixing ratios of O3 and NO2 (Fig. 2) highlight much higher oxidative capacity at night in summer in urban Beijing compared to the other seasons and/or rural locations.
ClNO2 showed clear nocturnal formation from heterogeneous processing and decreased rapidly after sunrise, mainly due to photolysis (Fig. 2). Note that ClNO2 peaked at a similar time (21:00–22:00) as that of N2O5 without showing a time lag of 1–3 h as previously observed in Jinan (X. Wang et al., 2017), indicating that either particulate Cl− was sufficient for the heterogeneous reactions or other chlorine sources (e.g., HCl) contributed to the formation of ClNO2 in urban Beijing. According to previous studies, the partitioning of HCl to particulate Cl− could substantially contribute to ClNO2 formation at urban sites (Thornton et al., 2010; Riedel et al., 2012). In addition, Wang et al. (2018) also speculated that large particulate chloride during the campaign was possibly replenished by gas-phase HCl due to high emissions from human activities. We also found that ClNO2 was well correlated with chlorine (Cl2) derived from IAP-CIMS (R2=0.90–0.99) rather than particulate chloride (Cl−) (R2=0.01–0.44) at nighttime, indicating that ClNO2 might act as an intermediate during the formation of Cl2 under sufficient chloride conditions (Roberts et al., 2008). Indeed, the much lower particulate Cl− than ClNO2 also indicated other chlorine sources. Therefore, we need simultaneous measurements to further support such a conclusion in this study, e.g., HCl.
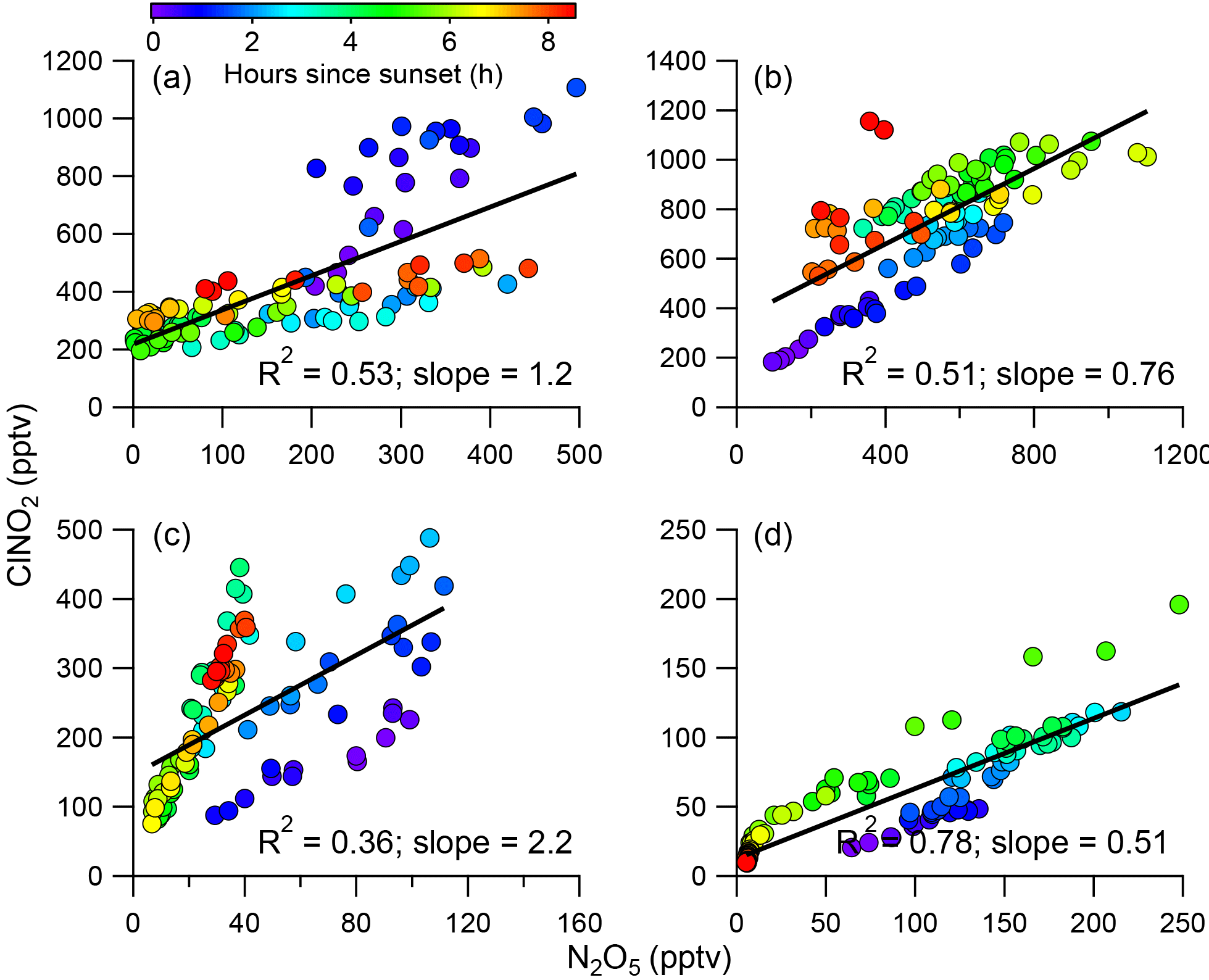
Figure 4Correlations between ClNO2 and N2O5 for four different nights, i.e., P1, P2, P3, and P4. The data are color coded by the hours since sunset. Also shown are the correlation coefficients and slopes.
3.2 Reactivity of N2O5 and NO3
Considering the time needed to meet the steady-state assumption, only the data 2 h after sunset were used to calculate N2O5 steady-state lifetime via Eq. (4) (Wagner et al., 2013). High N2O5 reactivity was observed and the average τ (N2O5)−1 was 0.16–1.58 s−1 during these four nights corresponding to a short nighttime N2O5 lifetime between 1.1 and 10.7 min (Fig. 3), with τ(N2O5)−1 ranging from 0.20 × 10−2 to 1.46 × 10−2 s−1 throughout the campaign. Such values are overall consistent with those measured at surface sites and in the nocturnal residual layer in NCP, for example 1.30 × 10−2 s−1 in Wangdu (Tham et al., 2016) and 1.30–1.40 × 10−2 s−1 at Mt. Tai (Z. Wang et al., 2017). In comparison, the N2O5 loss is much more rapid than that previously reported in southern China (1–5 h; Brown et al., 2016b) and the USA (a few hours; Wagner et al., 2013), mainly due to the high aerosol loading in NCP leading to an enhanced N2O5 sink through both indirect and direct pathways. Correspondingly, the average τ(NO3)−1 values calculated from the inferred NO3 were 0.02–0.62 s−1 during the four nights, indicating active NO3 nighttime chemistry through reactions with NO and VOCs in the polluted nocturnal boundary. Note that P2 and P4 showed comparable p(NO3) (2.8 vs. 2.6 ppbv h−1; Table 1), yet the N2O5 reactivity during P4 (1.58 × 10−2 s−1) was significantly higher than that during P2 (0.16 × 10−2 s−1), likely due to the higher NO level, and the enhanced N2O5 heterogeneous loss might also be an explanation. Consistently, τ(NO3)−1 showed similar patterns to τ(N2O5)−1. Indeed, the N2O5 reactivity presented a nonlinear dependence on aerosol surface area (Sa) and relative humidity (Fig. 3c and d). Although P3 showed much higher RH than P4 (60.5 % vs. 28.0 %), the N2O5 reactivity was comparable between P3 and P4 (0.014 vs. 0.016 s−1), illustrating the complex heterogeneous process of N2O5.
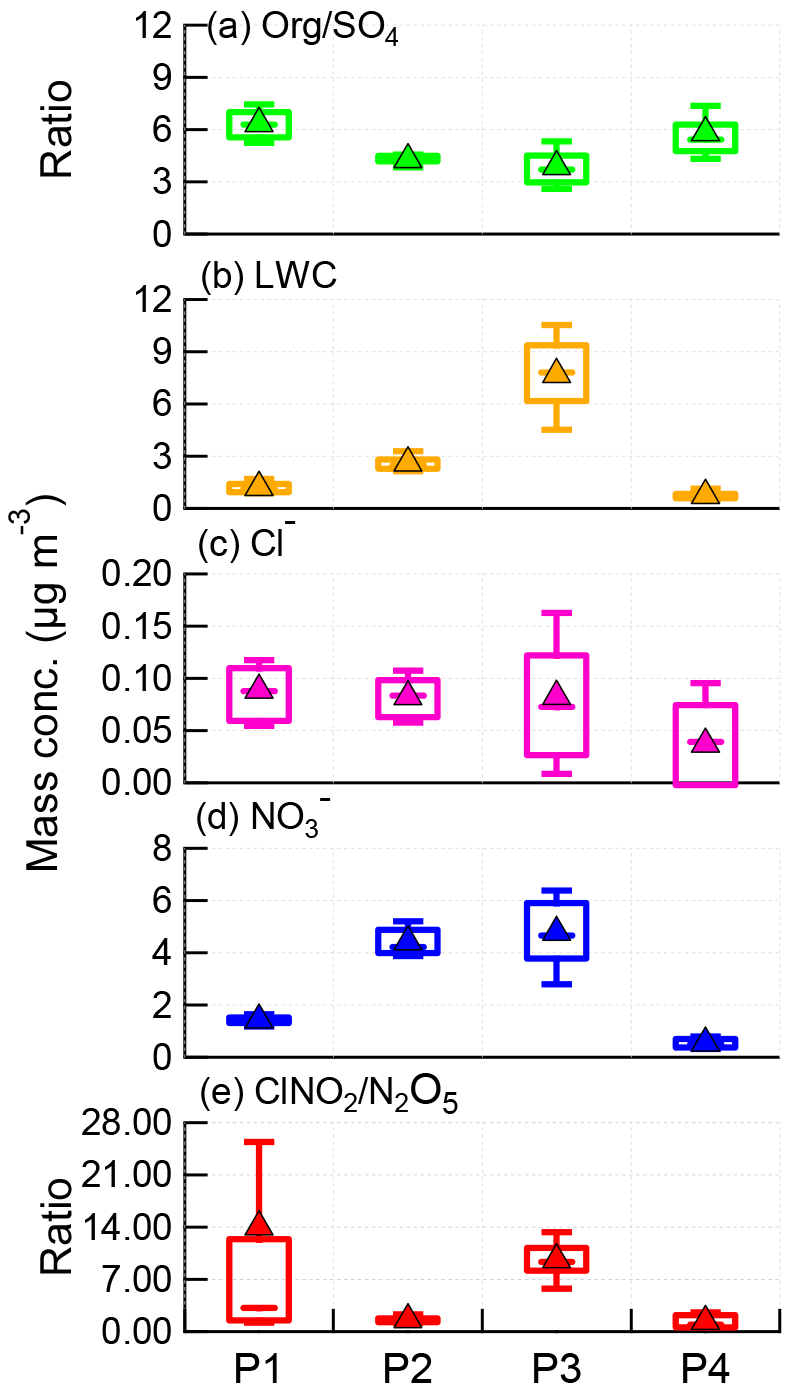
Figure 5Box plots of (a) Org ∕ SO4 ratio, (b) LWC, (c) particulate chloride, (d) particulate nitrate, and (e) ClNO2∕N2O5 ratio for each night, i.e., P1, P2, P3, and P4. The mean (triangle), median (horizontal line), 25 and 75th percentiles (lower and upper box), and 10 and 90th percentiles (lower and upper whiskers) are shown.
Figure 3c shows the N2O5 lifetime as a function of surface area density (Sa) with the data being binned according to the 50 µm2 cm−3Sa increment. τ(N2O5) decreased rapidly from 11.8 to 2.2 min as Sa increased up to 500 µm2 cm−3 and then remained at relatively constant levels at Sa>500 µm2 cm−3. Such an Sa dependence of τ(N2O5) is consistent with previous observations in Hong Kong (Brown et al., 2016b). Large variations in τ(N2O5) as a function of RH were also observed. As shown in Fig. 3d, the N2O5 lifetime decreased by nearly a factor of 5 from 11.3 to 2.2 min as RH increased from 40 % to 50 %. We noticed that the aerosol surface area exhibits an increase as a function of RH at RH > 40 % (Fig. S4). These results suggested that the decrease in τ(N2O5) at high RH levels (RH > 40 %) might be caused by increased N2O5 uptake rates due to the higher Sa. In addition, the increasing aerosol liquid water content at high RH might be another reason (Fig. S4). Comparatively, the N2O5 lifetime showed an increase as a function of RH at RH < 40 %, while the variations in Sa were small, suggesting additional contributions from other factors, for example aerosol loading and composition (Morgan et al., 2015). Considering that the period of this study is relatively short, long-term measurements are needed in future studies to better characterize the parameterizations of τ(N2O5) as a function of Sa and RH.
3.3 Relationship between N2O5 and ClNO2
Previous studies have found that N2O5 and ClNO2 were generally positively correlated in predominantly continental air masses, whereas they were negatively correlated in marine air masses with high chloride content (Bannan et al., 2015). Phillips et al. (2012) also reported large variability in N2O5 and ClNO2 correlations and ClNO2∕N2O5 ratios in air masses from continental or marine origins due to the changes in particle Cl−. In this study, ClNO2 was well and positively correlated with N2O5 during all four nights (Fig. 4, R2=0.36–0.78), and only slight changes in ClNO2∕N2O5 ratios were observed after sunset. These results are different from previous observations showing large variability in the correlations (Osthoff et al., 2008), which indicates that there are sufficient chloride sources for ClNO2 formation during this study period. The differences in regression coefficients among the four nights can be explained by different air masses originating from different regions, which were calculated using the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT; NOAA) model (Draxler and Hess, 1997; Fig. S5). For example, ClNO2 tracked much better with N2O5 after midnight (R2=0.69) than before midnight (R2=0.16) during P2 (Fig. S6), suggesting the influence of air masses from different regions (Fig. S5). Comparatively, P4 and P1 showed similar tight correlations between ClNO2 and N2O5 before and after midnight, consistent with their similar back trajectories during the two different periods.
The ClNO2∕N2O5 ratios varied significantly throughout the study ranging from 0.3 to 95.5 (30 min average). The average (±1σ) ratio of ClNO2∕N2O5 was 6.9±7.4, consistent with previous studies in NCP, for example 0.4–131.3 in Jinan and Wangdu (X. Wang et al., 2017; Tham et al., 2016). However, the ratios are substantially higher than those measured in other megacities, e.g., Hong Kong (0.1–2.0; Wang et al., 2016), London (0.02–2.4; Bannan et al., 2015), and Los Angeles, California (0.2–10.0; Mielke et al., 2013). These results indicate ubiquitously high ClNO2∕N2O5 ratios in the NCP, consistent with another measurement in suburban Beijing (Wang et al., 2018), which might result from the high ClNO2 production rate due to high aerosol loadings. We also note that the relatively low N2O5 associated with high N2O5 reactivity might be another possible explanation. Furthermore, we compared the ClNO2∕N2O5 ratios with particulate concentrations and compositions during the four nights (Fig. 5). P3 showed the highest median ratio of 9.4, which is much higher than during the other three nights (1.0–3.2). This can be explained by the correspondingly high liquid water content that facilitated N2O5 heterogeneous uptake (Morgan et al., 2015). In comparison, the particle chloride concentrations were relatively close during the four nights, with slightly lower concentrations during P4, further supporting the fact that the ClNO2∕N2O5 ratios were independent of particle chloride in this study due to the sufficient chloride source for ClNO2 production, e.g., HCl gas–particle partitioning. The lower ClNO2∕N2O5 ratios during P2 compared with P1 can be explained by the “nitrate effect”, which suppressed N2O5 uptake (Mentel and Wahner, 1999) as P2 showed much higher nitrate concentrations than P1 (4.2 vs. 1.4 µg m−3). Note that the ClNO2∕N2O5 ratios were also characterized by a dependence on Org ∕ SO4 ratios in our campaign, similar to other studies (Evans and Jacob, 2005; Riemer et al., 2009).
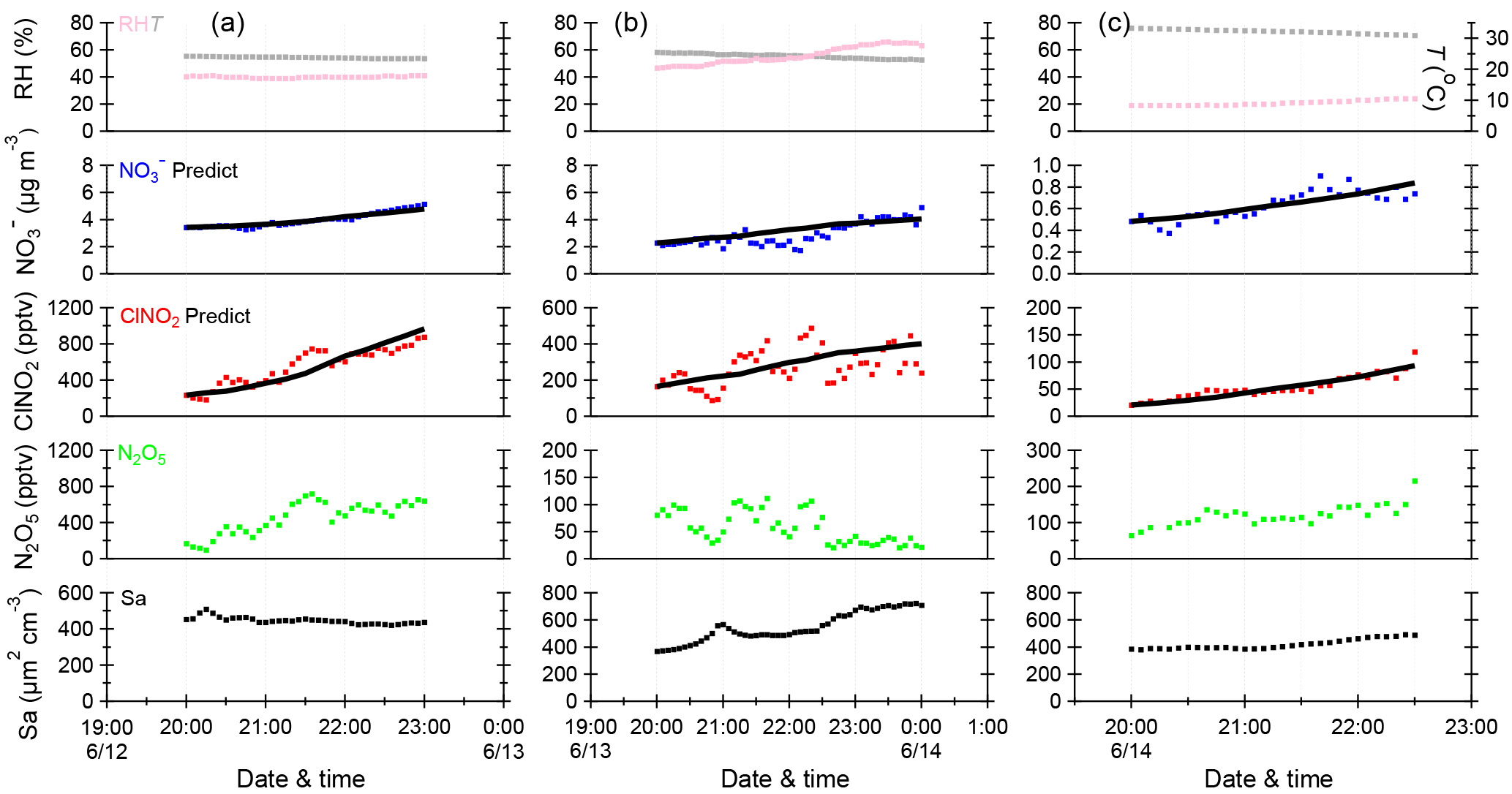
Figure 6Time series of meteorological parameters (RH, T), particulate nitrate (NO, mixing ratios of N2O5 and ClNO2, and aerosol surface area density (Sa) for the selected periods on three nights. The black solid lines are the predicted integration concentrations of NO and ClNO2 calculated using the estimated method.
3.4 N2O5 uptake coefficient and ClNO2 production yield
To quantity the relative contributions of different pathways to N2O5 loss, three periods with relatively stable air masses and concurrent increases in ClNO2 and (Fig. 6; 20:00–23:00 on 12 June, 20:00–00:00 on 13 June, and 20:00–22:30 on 14 June) were selected for the calculations of and ø. A rigorous method as suggested by Phillips et al. (2016) was used in this study. Briefly, the predicted concentrations of ClNO2 and were derived by integrating pClNO2 and p with average Sa and N2O5 over each time step (∼ 15 min) and initial estimations for and ø. The integration was repeated by changing and ø until good agreements between observed and predicted values of ClNO2 and were reached. The derived heterogeneous uptake coefficient, ClNO2 yield, and N2O5 loss rate kd following this method are listed in Table 2.
Table 2Estimated uptake coefficient of N2O5, ClNO2 production yield, and related parameters for the selected periods on three nights.
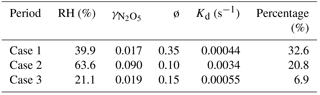
The estimated values for the three selected periods were 0.017–0.09, which was generally comparable to previous values (0.014–0.092) derived from the steady-state assumption method in the NCP (H. Wang et al., 2017; X. Wang 2017; Z. Wang et al., 2017; Tham et al., 2016) and also consistent with recent measurements (0.012–0.055) using the same method in suburban Beijing (Wang et al., 2018). However, the determined in our campaign was 1–2 orders of magnitude higher than obtained in the laboratory (Thornton et al., 2003) and also much higher than those in Hong Kong and Germany (Brown et al., 2016b; Phillips et al., 2016). We also found that the parameterized values (0.0014–0.012) determined from Eq. (9) (the Bertram–Thornton parameterization) were significantly lower than the observed values, suggesting that more field measurements are needed to improve the parameterization schemes. Note that values appeared to increase with rising relative humidity, which was also observed at other sites (X. Wang et al., 2017; Thornton et al., 2003). For example, values increased from 0.019 to 0.090 when RH increased from 21.1 % to 63.6 %. However, the values were comparable at low RH levels (<40 %; 0.019 vs. 0.017 in Table 2) although RH differed by a factor of 2 (21 % vs. 40 %). These results further supported the fact that the influences of hygroscopic growth on were mainly caused by increasing aerosol liquid water content. The direct N2O5 loss rates estimated from the uptake coefficient were in the range of 0.00044–0.0034 s−1, which contributed 7–33 % to the total N2O5 loss with the rest being indirect loss. The uncertainty of the direct N2O5 loss rate contributions is estimated to be ∼ 40 %, associated with Sa (∼ 30 %), O3 and NO2 (∼ 5 %), and N2O5 (∼ 17 %). Our results indicated that the fast N2O5 loss in the nocturnal boundary in urban Beijing was predominantly from the indirect loss of NO3 rather than the heterogeneous uptake of N2O5, mainly due to active NO3 reaction in summer. Such a conclusion was different from previous results in autumn in Beijing that found N2O5 loss to be dominated by N2O5 heterogeneous hydrolysis (69.1 %–98.8 %; H. Wang et al., 2017). Several studies also revealed the importance of heterogeneous N2O5 uptake in N2O5 loss in the NCP by using steady-state-derived (Tham et al., 2016; X. Wang et al., 2017; Z. Wang et al., 2017). While the uncertainties in different analysis methods, e.g., due to product formation rates or the steady-state assumption, the high NO concentration could be an important reason for the dominant N2O5 loss pathway. The higher VOC emissions, particularly biogenic emissions (e.g., isoprene and terpene), in summer than in other seasons might be another reason for the differences in the dominant N2O5 loss pathway. Indeed, indirect N2O5 loss via NO3 + VOCs was also found to dominate the total loss of N2O5 (67 %) in summer in suburban Beijing (Wang et al., 2018). Our results highlight significant nighttime NOx loss through reactions of NO3 with VOCs in summer in urban Beijing.
The ClNO2 yields ø derived for the three cases were 0.35, 0.10, and 0.15, respectively. The production yields in this study are substantially larger than those in urban Jinan (0.014–0.082; X. Wang et al., 2017), yet comparable to those reported at Mt. Tai (0.02–0.90; Z. Wang et al., 2017) and continental Colorado (0.07–0.36; Thornton et al., 2010). However, the significantly lower ø than that in suburban Beijing (0.50–1.0; X. Wang et al., 2017) indicated more effective ClNO2 production in suburban regions than urban regions to some extent. Indeed, the product of and ø ( in this study ranged from 0.006–0.009 and was much lower than in X. Wang et al. (2017; 0.008–0.035). We noticed that ø values were much lower than those parameterized from Eq. (10) (0.55–0.97), indicating that the Bertram–Thornton parameterization scheme might overestimate the ClNO2 yield substantially. Note that might be overestimated, associated with an underestimation of ø if assuming particulate nitrate is completely from N2O5 heterogeneous uptake. Possible contribution from gas-phase HNO3 repartitioning to the particulate phase was not considered, mainly due to the lack of observational data for HNO3 and NH3. Indeed, a recent study found that the nocturnal nitrate formation potential by N2O5 heterogeneous uptake was comparable to that formed by gas-phase HNO3 repartitioning in Beijing (H. Wang et al., 2017). In addition, was higher on 13 June than the other two days (e.g., 0.009 vs. 0.003–0.006), which might explain the correspondingly higher ClNO2∕N2O5 ratio on this day (on average 8.2 vs. 1.2–1.4). Our results overall suggest fast heterogeneous N2O5 uptake and a high ClNO2 production rate in summer in urban Beijing, which might have great implications for models to improve simulations for nocturnal nitrate and daytime ozone.
We present the simultaneous measurement of gas-phase N2O5 and ClNO2 by I-CIMS during the APHH summer campaign to investigate the nocturnal chemistry in urban Beijing. The average (±1σ) mixing ratios of N2O5 and ClNO2 were 79.2±157.1 and 174.3±262.0 pptv, with maximum values of 1.17 and 1.44 ppbv, respectively. Differing from previous studies with negligible N2O5 after midnight at surface level, our measurements showed high nocturnal levels of N2O5 across the entire night, suggesting a high oxidative capacity in summer in urban Beijing. N2O5 and ClNO2 exhibited clear diurnal variations with significant nocturnal formation due to heterogeneous uptake. The average nighttime nitrate radical production rate p(NO3) was 2.6±2.4 ppbv h−1, and the τ(N2O5)−1 was in the range of 0.20– s−1, corresponding to a nighttime N2O5 lifetime of 1.1–10.7 min. We also observed a decrease in τ(N2O5) under high relative humidity (RH > 40 %) conditions due to higher N2O5 uptake rates with higher available surface area and liquid water content. N2O5 and ClNO2 were positively correlated, although the ClNO2∕N2O5 ratios changed significantly from 0.3 to 95.5. The high ClNO2∕N2O5 ratios in this study might result from a high ClNO2 production rate and fast N2O5 loss due to the sufficient chloride source supply.
The N2O5 uptake coefficients estimated on the basis of the product formation rates of ClNO2 and were 0.017–0.09 in this study. Correspondingly, the direct N2O5 loss rates via heterogeneous uptake were in the range of 0.00044–0.0034 s−1, contributing 7 %–33 % to the total N2O5 loss. Our results indicated that fast N2O5 loss in the nocturnal boundary in urban Beijing was mainly due to indirect pathways through NO3 reactions with NO and VOCs rather than the heterogeneous uptake of N2O5. We also noticed that the derived ClNO2 production yields (0.10–0.35) were substantially lower than those from the Bertram–Thornton parameterization, indicating that future studies are needed to address these discrepancies.
The data in this study are available from the authors upon request (sunyele@mail.iap.ac.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-11581-2018-supplement.
YS and HC designed research; WZ and JZ, BO, AM, WX, YW, TW, MP, AB, QC, CX, QW, JW, WD, YZ, XG, PY, JL, PF, DW, RJ, and CP performed research; WZ, JZ, BO, AM, WX, YW, TW, MP, WD, and JL analyzed data; and WZ, JZ, BO, AM, YS, and HC wrote the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
This work was supported by the National Key Project of Basic Research (2014CB447900) and
the National Natural Science Foundation of China
(41571130034, 91744207). The University of Manchester work was supported
through NERC grants for AIRPOLL and AIRPRO (NE/N007123/1, NE/N00695X/1).
Edited by: Yongjie Li
Reviewed by: two anonymous referees
Anttila, T., Kiendler-Scharr, A., Tillmann, R., and Mentel, T. F.: On the reactive uptake of gaseous compounds by organic-coated aqueous aerosols: Theoretical analysis and application to the heterogeneous hydrolysis of N2O5, J. Phys. Chem. A., 110, 10435–10443, https://doi.org/10.1021/jp062403c, 2006.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Bannan, T. J., Bacak, A., Muller, J. B. A., Booth, A. M., Jones, B., Le Breton, M., Leather, K. E., Ghalaieny, M., Xiao, P., Shallcross, D. E., and Percival, C. J.: Importance of direct anthropogenic emissions of formic acid measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012, Atmos. Environ., 83, 301–310, https://doi.org/10.1016/j.atmosenv.2013.10.029, 2014.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B. A., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prévôt, A. S. H., Lee, J., Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp, T., Stone, D., Heard, D. E., Fleming, Z. L., Leigh, R., Shallcross, D. E., and Percival, A. C. J.: The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.-Atmos., 120, 5638–5657, https://doi.org/10.1002/2014JD022629, 2015.
Bannan, T. J., Bacak, A., Le Breton, M., Flynn, M., Ouyang, B., McLeod, M., Jones, R., Malkin, T. L., Whalley, L. K., Heard, D. E., Bandy, B., Khan, M. A. H., Shallcross, D. E., and Percival, C. J.: Ground and Airborne U.K. Measurements of Nitryl Chloride: An Investigation of the Role of Cl Atom Oxidation at Weybourne Atmospheric Observatory, J. Geophys. Res.-Atmos., 122, 11154–11165, https://doi.org/10.1002/2017JD026624, 2017.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Brown, S. S., Stark, H., Ciciora, S. J., and Ravishankara, A. R.: In-situ measurement of atmospheric NO3 and N2O5 via cavity ring-down spectroscopy, Geophys. Res. Lett., 28, 3227–3230, https://doi.org/10.1029/2001gl013303, 2001.
Brown, S. S., Stark, H., and Ravishankara, A. R.: Applicability of the steady state approximation to the interpretation of atmospheric observations of NO3 and N2O5, J. Geophys. Res., 108, 4539, https://doi.org/10.1029/2003jd003407, 2013a.
Brown, S. S., Stark, H., Ryerson, T. B., Williams, E. J., Nicks, D. K., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Nitrogen oxides in the nocturnal boundary layer: Simultaneous in situ measurements of NO3, N2O5, NO2, NO, and O3, J. Geophys. Res.-Atmos., 108, 4099, https://doi.org/10.1029/2002jd002917, 2013b.
Brown, S. S., Ryerson, T. B., Wollny, A. G., Brock, C. A., Peltier, R., Sullivan, A. P., Weber, R. J., Dubé, W. P., Trainer, M., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. R.: Variability in Nocturnal Nitrogen Oxide Processing and Its Role in Regional Air Quality, Science, 311, 67–70, https://doi.org/10.1126/science.1120120, 2006.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Brown, S. S., Dube, W. P., Tham, Y. J., Zha, Q. Z., Xue, L. K., Poon, S., Wang, Z., Blake, D. R., Tsui, W., Parrish, D. D., and Wang, T.: Nighttime chemistry at a high altitude site above Hong Kong, J. Geophys. Res.-Atmos., 121, 2457–2475, https://doi.org/10.1002/2015jd024566, 2016.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Chan, C. K. and Yao, X.: Air pollution in mega cities in China, Atmos. Environ., 42, 1–42, https://doi.org/10.1016/j.atmosenv.2007.09.003, 2008.
Chang, W. L., Bhave, P. V., Brown, S. S., Riemer, N., Stutz, J., and Dabdub, D.: Heterogeneous Atmospheric Chemistry, Ambient Measurements, and Model Calculations of N2O5: A Review, Aerosol Sci. Technol., 45, 665–695, https://doi.org/10.1080/02786826.2010.551672, 2011.
Cosman, L. M., Knopf, D. A., and Bertram, A. K.: N2O5 Reactive Uptake on Aqueous Sulfuric Acid Solutions Coated with Branched and Straight-Chain Insoluble Organic Surfactants, J. Phys. Chem. A., 112, 2386–2396, https://doi.org/10.1021/jp710685r, 2008.
Davis, J. M., Bhave, P. V., and Foley, K. M.: Parameterization of N2O5 reaction probabilities on the surface of particles containing ammonium, sulfate, and nitrate, Atmos. Chem. Phys., 8, 5295–5311, https://doi.org/10.5194/acp-8-5295-2008, 2008.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Dentener, F. J. and Crutzen, P. J.: Reaction of N2O5 on tropospheric aerosols: Impact on the global distributions of NOx, O3, and OH, J. Geophys. Res., 98, 7149–7163, https://doi.org/10.1029/92JD02979, 1993.
Draxler, R. R. and Hess, G.: Description of the HYSPLIT4 modeling system, Air Resources Laboratory, Silver Spring, Maryland, 1997.
Dube, W. P., Brown, S. S., Osthoff, H. D., Nunley, M. R., Ciciora, S. J., Paris, M. W., McLaughlin, R. J., and Ravishankara, A. R.: Aircraft instrument for simultaneous, in situ measurement of NO3 and N2O5 via pulsed cavity ring-down spectroscopy, Rev. Sci. Instrum., 77, 034101, https://doi.org/10.1063/1.2176058, 2006.
Evans, M. J. and Jacob, D. J.: Impact of new laboratory studies of N2O5 hydrolysis on global model budgets of tropospheric nitrogen oxides, ozone, and OH, Geophys. Res. Lett., 32, L09813, https://doi.org/10.1029/2005gl022469, 2005.
Finlayson-Pitts, B. J., Ezell, M. J., and Pitts, J. N.: Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2, Nature, 337, 241–244, https://doi.org/10.1038/337241a0, 1989.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+–NH4+–Na+–SO––Cl−–H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Geyer, A., Alicke, B., Konrad, S., Schmitz, T., Stutz, J., and Platt, U.: Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.-Atmos., 106, 8013–8025, https://doi.org/10.1029/2000jd900681, 2001.
Griffiths, P. T., Badger, C. L., Cox, R. A., Folkers, M., Henk, H. H., and Mentel, T. F.: Reactive Uptake of N2O5 by Aerosols Containing Dicarboxylic Acids. Effect of Particle Phase, Composition, and Nitrate Content, J. Phys. Chem. A., 113, 5082–5090, https://doi.org/10.1021/jp8096814, 2009.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kennedy, O. J., Ouyang, B., Langridge, J. M., Daniels, M. J. S., Bauguitte, S., Freshwater, R., McLeod, M. W., Ironmonger, C., Sendall, J., Norris, O., Nightingale, R., Ball, S. M., and Jones, R. L.: An aircraft based three channel broadband cavity enhanced absorption spectrometer for simultaneous measurements of NO3, N2O5 and NO2, Atmos. Meas. Tech., 4, 1759–1776, https://doi.org/10.5194/amt-4-1759-2011, 2011.
Kercher, J. P., Riedel, T. P., and Thornton, J. A.: Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry, Atmos. Meas. Tech., 2, 193–204, https://doi.org/10.5194/amt-2-193-2009, 2009.
Le Breton, M., Bacak, A., Muller, J. B. A., Xiao, P., Shallcross, B. M. A., Batt, R., and Percival, C. J.: Simultaneous airborne nitric acid and formic acid measurements using a chemical ionization mass spectrometer around the UK: Analysis of primary and secondary production pathways, Atmos. Environ., 83, 166–175, https://doi.org/10.1016/j.atmosenv.2013.10.008, 2014.
Le Breton, M., Wang, Y., Hallquist, Å. M., Pathak, R. K., Zheng, J., Yang, Y., Shang, D., Glasius, M., Bannan, T. J., Liu, Q., Chan, C. K., Percival, C. J., Zhu, W., Lou, S., Topping, D., Wang, Y., Yu, J., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 18, 10355–10371, https://doi.org/10.5194/acp-18-10355-2018, 2018.
Leather, K. E., Bacak, A., Wamsley, R., Archibald, A. T., Husk, A., Shallcross, E., and Percival, C. J.: Temperature and pressure dependence of the rate coefficient for the reaction between ClO and CH3O2 in the gas-phase, Phys. Chem. Chem. Phys., 14, 3425–3434, https://doi.org/10.1039/c2cp22834c, 2012.
Li, Q., Zhang, L., Wang, T., Tham, Y. J., Ahmadov, R., Xue, L., Zhang, Q., and Zheng, J.: Impacts of heterogeneous uptake of dinitrogen pentoxide and chlorine activation on ozone and reactive nitrogen partitioning: improvement and application of the WRF-Chem model in southern China, Atmos. Chem. Phys., 16, 14875–14890, https://doi.org/10.5194/acp-16-14875-2016, 2016.
Li, Y. J., Sun, Y., Zhang, Q., Li, X., Li, M., Zhou, Z., and Chan, C. K.: Real-time chemical characterization of atmospheric particulate matter in China: A review, Atmos. Environ., 158, 270–304, https://doi.org/10.1016/j.atmosenv.2017.02.027, 2017.
Liu, X., Gu, J., Li, Y., Cheng, Y., Qu, Y., Han, T., Wang, J., Tian, H., Chen, J., and Zhang, Y.: Increase of aerosol scattering by hygroscopic growth: Observation, modeling, and implications on visibility, Atmos. Res., 132–133, 91–101, https://doi.org/10.1016/j.atmosres.2013.04.007, 2013.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
McNeill, V. F., Patterson, J., Wolfe, G. M., and Thornton, J. A.: The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol, Atmos. Chem. Phys., 6, 1635–1644, https://doi.org/10.5194/acp-6-1635-2006, 2006.
Mentel, T. F. and Wahner, M. S. A.: Nitrate effect in the heterogeneous hydrolysis of dinitrogen pentoxide on aqueous aerosols, Phys. Chem. Chem. Phys., 1, 5451–5457, https://doi.org/10.1039/A905338G, 1999.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of ClNO2 in a mid-continental urban environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Mielke, L. H., Stutz, J., Tsai, C., Hurlock, S. C., Roberts, J. M., Veres, P. R., Froyd, K. D., Hayes, P. L., Cubison, M. J., Jimenez, J. L., Washenfelder, R. A., Young, C. J., Gilman, J. B., de Gouw, J. A., Flynn, J. H., Grossberg, N., Lefer, B. L., Liu, J., Weber, R. J., and Osthoff, H. D.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res., 118, 610638–610652, https://doi.org/10.1002/jgrd.50783, 2013.
Morgan, W. T., Ouyang, B., Allan, J. D., Aruffo, E., Di Carlo, P., Kennedy, O. J., Lowe, D., Flynn, M. J., Rosenberg, P. D., Williams, P. I., Jones, R., McFiggans, G. B., and Coe, H.: Influence of aerosol chemical composition on N2O5 uptake: airborne regional measurements in northwestern Europe, Atmos. Chem. Phys., 15, 973–990, https://doi.org/10.5194/acp-15-973-2015, 2015.
Nenes, A., Pandis, S. N., and Pilinis, C.: ISORROPIA: A New Thermodynamic Equilibrium Model for Multiphase Multicomponent Inorganic Aerosols, Aquat. Geochem., 4, 123–152, https://doi.org/10.1023/A:1009604003981, 1998.
O'Keefe, A. and Deacon, D. A. G.: Cavity ring-down optical spectrometer for absorption measurements using pulsed laser sources, Rev. Sci. Instrum., 59, 2544, https://doi.org/10.1063/1.1139895, 1988.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Pathak, R. K., Wu, W. S., and Wang, T.: Summertime PM2.5 ionic species in four major cities of China: nitrate formation in an ammonia-deficient atmosphere, Atmos. Chem. Phys., 9, 1711–1722, https://doi.org/10.5194/acp-9-1711-2009, 2009.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012gl051912, 2012.
Phillips, G. J., Thieser, J., Tang, M., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Platt, U. and Stutz, J.: Differential Optical Absorption Spectroscopy Principles and Applications, Springer Berlin Heidelberg, New York, 2008.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., De Vocht, F., Shallcross, B. M. A., Brazier, T., Anwar Khan, M., Allan, J., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of isocyanate, amide, nitrate and nitro compounds from an anthropogenic biomass burning event using a ToF-CIMS, J. Geophy. Res.-Atmos., https://doi.org/10.1002/2017jd027316, 2018.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl chloride and molecular chlorine in the coastal marine boundary layer, Environ. Sci. Technol., 46, 10463–10470, https://doi.org/10.5194/acp-12-2959-2012, 2012.
Riemer, N., Vogel, H., Vogel, B., Schell, B., Ackermann, I., Kessler, C., and Hass, H.: Impact of the heterogeneous hydrolysis of N2O5 on chemistry and nitrate aerosol formation in the lower troposphere under photosmog conditions., J. Geophys. Res., 108, 4144, https://doi.org/10.1029/2002jd002436, 2003.
Riemer, N., Vogel, H., Vogel, B., Anttila, T., Kiendler-Scharr, A., and Mentel, T. F.: Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe, J. Geophys. Res., 114, D17307, https://doi.org/10.1029/2008jd011369, 2009.
Roberts, J. M., Osthoff, H. D., Brown, S. S., and Ravishankara, A. R.: N2O5 Oxidizes Chloride to Cl2 in Acidic Atmospheric Aerosol, Science, 321, 1059, https://doi.org/10.1126/science.1158777, 2008.
Roberts, J. M., Osthoff, H. D., Brown, S. S., Ravishankara, A. R., Coffman, D., Quinn, P., and Bates, T.: Laboratory studies of products of N2O5 uptake on Cl-containing substrates, Geophys. Res. Lett., 36, L20808, https://doi.org/10.1029/2009gl040448, 2009.
Sander, S. P., Friedl, R. R., Ravishankara, A. R., Golden, D. M., Kolb, C. E., Kurylo, M. J., Molina, M. J., Moortgat, G. K., Keller-Rudek, H., FinlaysonPitts, B. J., Wine, P. H., Huie, R. E., and Orkin, V. L.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation Number 15, NASA/JPL Publication 06-2, Pasadena, CA, 2006.
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 40, 4050–4058, https://doi.org/10.1002/2014GL059962, 2014.
Slusher, D. L., Huey, L. G., Tanner, D. J., Flocke, F. M., and Roberts, J. M.: A thermal dissociation–chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res., 109, D19315, https://doi.org/10.1029/2004jd004670, 2004.
Smith, N., Plane, J. M. C., Nien, C. F., and Solomon, P. A.: Nighttime Radical Chemistry in the San-Joaquin Valley, Atmos. Environ., 29, 2887–2897, https://doi.org/10.1016/1352-2310(95)00032-T, 1995.
Stutz, J., Alicke, B., Ackermann, R., Geyer, A., White, A., and Williams, E.: Vertical profiles of NO3, N2O5, O3, and NOx in the nocturnal boundary layer: 1. Observations during the Texas Air Quality Study 2000, J. Geophys. Res., 109, D12306, https://doi.org/10.1029/2003jd004209, 2004.
Stutz, J., Wong, K. W., Lawrence, L., Ziemba, L., Flynn, J. H., Rappenglück, B., and Lefer, B.: Nocturnal NO3 radical chemistry in Houston, TX, Atmos. Environ., 44, 4099–4106, https://doi.org/10.1016/j.atmosenv.2009.03.004, 2010.
Su, X., Tie, X., Li, G., Cao, J., Huang, R., Feng, T., Long, X., and Xu, R.: Effect of hydrolysis of N2O5 on nitrate and ammonium formation in Beijing China: WRF-Chem model simulation, Sci. Total Environ, 579, 221–229, https://doi.org/10.1016/j.scitotenv.2016.11.125, 2016.
Sun, Y., Wang, L., Wang, Y., Quan, L., and Zirui, L.: In situ measurements of SO2, NOx, NOy, and O3 in Beijing, China during August 2008, Sci. Total Environ., 409, 933–940, https://doi.org/10.1016/j.scitotenv.2010.11.007, 2011.
Sun, Y., Wang, Z., Dong, H., Yang, T., Li, J., Pan, X., Chen, P., and Jayne, J. T.: Characterization of summer organic and inorganic aerosols in Beijing, China with an Aerosol Chemical Speciation Monitor, Atmos. Environ., 51, 250–259, https://doi.org/10.1016/j.atmosenv.2012.01.013, 2012.
Sun, Y., Du, W., Fu, P., Wang, Q., Li, J., Ge, X., Zhang, Q., Zhu, C., Ren, L., Xu, W., Zhao, J., Han, T., Worsnop, D. R., and Wang, Z.: Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 16, 8309–8329, https://doi.org/10.5194/acp-16-8309-2016, 2016.
Tan, H., Xu, H., Wan, Q., Li, F., Deng, X., Chan, P. W., Xia, D., and Yin, Y.: Design and Application of an Unattended Multifunctional H-TDMA System, J. Atmos. Ocean. Technol., 30, 1136–1148, https://doi.org/10.1175/jtech-d-12-00129.1, 2013.
Tham, Y. J., C. Yan, L. Xue, Q. Zha, X. W., and Wang, A. T.: Presence of high nitryl chloride in Asian coastal environment and its impact on atmospheric photochemistry, Chin. Sci. Bull., 59, 356–359, https://doi.org/10.1007/s11434-013-0063-y, 2014.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Thornton, J. A., Braban, C. F., and Abbatt, J. P. D.: N2O5 hydrolysis on sub-micron organic aerosols: the effect of relative humidity, particle phase, and particle size, Phys. Chem. Chem. Phys., 5, 4593–4603, https://doi.org/10.1039/b307498f, 2003.
Thornton, J. A. and Abbatt, J. P. D.: N2O5 Reaction on Submicron Sea Salt Aerosol: Kinetics, Products, and the Effect of Surface Active Organics, J. Phys. Chem. A., 109, 10004–10012, https://doi.org/10.1021/jp054183t, 2005.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
van der A, R. J., Mijling, B., Ding, J., Koukouli, M. E., Liu, F., Li, Q., Mao, H., and Theys, N.: Cleaning up the air: effectiveness of air quality policy for SO2 and NOx emissions in China, Atmos. Chem. Phys., 17, 1775–1789, https://doi.org/10.5194/acp-17-1775-2017, 2017.
Wagner, N. L., Riedel, T. P., Young, C. J., Bahreini, R., Brock, C. A., Dubé, W. P., Kim, S., Middlebrook, A. M., Öztürk, F., Roberts, J. M., Russo, R., Sive, B., Swarthout, R., Thornton, J. A., VandenBoer, T. C., Zhou, Y., and Brown, S. S.: N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements, J. Geophys. Res.-Atmos., 118, 9331–9350, https://doi.org/10.1002/jgrd.50653, 2013.
Wang, X., Wang, T., Yan, C., Tham, Y. J., Xue, L., Xu, Z., and Zha, Q.: Large daytime signals of N2O5 and NO3 inferred at 62 amu in a TD-CIMS: chemical interference or a real atmospheric phenomenon?, Atmos. Meas. Tech., 7, 1–12, https://doi.org/10.5194/amt-7-1-2014, 2014.
Wang, T., Tham, Y. J., Xue, L., Li, Q., Zha, Q., Wang, Z., Poon, S. C. N., Dubé, W. P., Blake, D. R., Louie, P. K. K., Luk, C. W. Y., Tsui, W., and Brown, A. S. S.: Observations of nitryl chloride and modeling its source and effect on ozone in the planetary boundary layer of southern China, J. Geophys. Res., 121, 2457–2475, https://doi.org/10.1002/2015jd024556, 2016.
Wang, H., Lu, K., Chen, X., Zhu, Q., Chen, Q., Guo, S., Jiang, M., Li, X., Shang, D., Tan, Z., Wu, Y., Wu, Z., Zou, Q., Zheng, Y., Zeng, L., Zhu, T., Hu, M., and Zhang, Y.: High N2O5 Concentrations Observed in Urban Beijing: Implications of a Large Nitrate Formation Pathway, Environ. Sci. Tech. Lett., 4, 416–420, https://doi.org/10.1021/acs.estlett.7b00341, 2017.
Wang, X., Wang, H., Xue, L., Wang, T., Wang, L., Gu, R., Wang, W., Tham, Y. J., Wang, Z., Yang, L., Chen, J., and Wang, W.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Environ., 156, 125–134, https://doi.org/10.1016/j.atmosenv.2017.02.035, 2017.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Wang, H., Lu, K., Guo, S., Wu, Z., Shang, D., Tan, Z., Wang, Y., Le Breton, M., Lou, S., Tang, M., Wu, Y., Zhu, W., Zheng, J., Zeng, L., Hallquist, M., Hu, M., and Zhang, Y.: Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing, Atmos. Chem. Phys., 18, 9705–9721, https://doi.org/10.5194/acp-18-9705-2018, 2018.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosa-Mas, C. E., Hjorth, J., Le Bras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The nitrate radical: Physics, chemistry, and the atmosphere, Atmos. Environ., 25, 1–203, https://doi.org/10.1016/0960-1686(91)90192-A, 1991.
Wood, E. C., Wooldridge, P. J., Freese, J. H., Albrecht, T., and Cohen, R. C.: Prototype for In Situ Detection of Atmospheric NO3 and N2O5 via Laser-Induced Fluorescence, Environ. Sci. Technol., 37, 5732–5738, https://doi.org/10.1021/es034507w, 2003.
Xu, W. Q., Sun, Y. L., Chen, C., Du, W., Han, T. T., Wang, Q. Q., Fu, P. Q., Wang, Z. F., Zhao, X. J., Zhou, L. B., Ji, D. S., Wang, P. C., and Worsnop, D. R.: Aerosol composition, oxidation properties, and sources in Beijing: results from the 2014 Asia-Pacific Economic Cooperation summit study, Atmos. Chem. Phys., 15, 13681–13698, https://doi.org/10.5194/acp-15-13681-2015, 2015.
Xue, J., Z. Yuan, A. K. H. Lau, and J. Z. Yu: Insights into factors affecting nitrate in PM2.5 in a polluted high NOx environment through hourly observations and size distribution measurements, J. Geophys. Res.-Atmos., 119, 4888–4902, https://doi.org/10.1002/2013JD021108, 2014a.
Xue, L. K., Wang, T., Gao, J., Ding, A. J., Zhou, X. H., Blake, D. R., Wang, X. F., Saunders, S. M., Fan, S. J., Zuo, H. C., Zhang, Q. Z., and Wang, W. X.: Ground-level ozone in four Chinese cities: precursors, regional transport and heterogeneous processes, Atmos. Chem. Phys., 14, 13175–13188, https://doi.org/10.5194/acp-14-13175-2014, 2014b.
Xue, L. K., Saunders, S. M., Wang, T., Gao, R., Wang, X. F., Zhang, Q. Z., and Wang, W. X.: Development of a chlorine chemistry module for the Master Chemical Mechanism, Geosci. Model Dev., 8, 3151–3162, https://doi.org/10.5194/gmd-8-3151-2015, 2015.