the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Jul 2026
| 02 Jul 2026
Gas-phase degradation of the aroma compound ethyl butyrate and its methylated derivatives: UV-C photolysis and reactions with the hydroxyl radical
Mark A. Blitz
Paul W. Seakins
Nicola Carslaw
The aroma compound ethyl butyrate (EB) and its methylated derivatives ethyl 2-methylbutyrate (EM), ethyl isovalerate (EI), and isopropyl butyrate (IB) are present in many consumer products. To evaluate the environmental and health impacts of these volatile organic compounds, a detailed understanding of their gas-phase photochemical reactivity is required. Here, we performed pulsed laser photolysis/laser-induced fluorescence (PLP-LIF) experiments to investigate the kinetics of their reactions with the hydroxyl radical (OH). Room temperature rate coefficients in units of with 2σ statistical errors were determined as: (5.5±0.2) for EB + OH, (7.0±0.3) for EM + OH, (11.2±0.4) for EI + OH, and (7.5±0.4) for IB + OH. All four reactions exhibited complex kinetics with distinct non-Arrhenius behaviour for temperatures up to about 400 K. This behaviour was attributed to pre-reaction complexes and is consistent with site-specific reactivities as predicted by an established structure-activity-relationship (SAR). In a second series of experiments, quasi-gas-phase UV-vis. spectroscopy and time-dependent density functional theory predictions were used to obtain absorption cross-sections. All four esters displayed an absorption band at around 213 nm (spin-forbidden transition), but did not absorb appreciably in the visible or UV-A part of the spectrum where light is abundant at ground level. Therefore, the reaction with OH was considered the main loss process, with lifetimes for tropospheric removal ranging from 22–45 h. Photochemical ozone creation potentials were estimated to be in a moderate range between 28 and 34.
- Article
(2386 KB) - Full-text XML
-
Supplement
(1335 KB) - BibTeX
- EndNote




Ethyl butyrate (ethyl butanoate; henceforth EB) and its methylated derivatives ethyl 2-methylbutyrate (ethyl 2-methylbutanoate; EM), ethyl isovalerate (ethyl 3-methylbutanoate; EI), and isopropyl butyrate (propan-2-yl butanoate; IB) are a group of volatile compounds with widespread occurrence and versatile applications but largely unknown gas-phase behaviour. Their structures are shown in Fig. 1. In industrial contexts, EB and its derivatives have been explored as promising second-generation biofuels that could support the transition away from petroleum-based fuels (Badawy et al., 2016; El-Nahas et al., 2010; Zhang et al., 2021). Furthermore, fatty acid esters with C4–C5 acid chains are potent solvents (Knothe and Steidley, 2011) that can be synthesised from bio-derived alcohols and acids and may therefore be sourced sustainably at large scales (Xu et al., 2020).
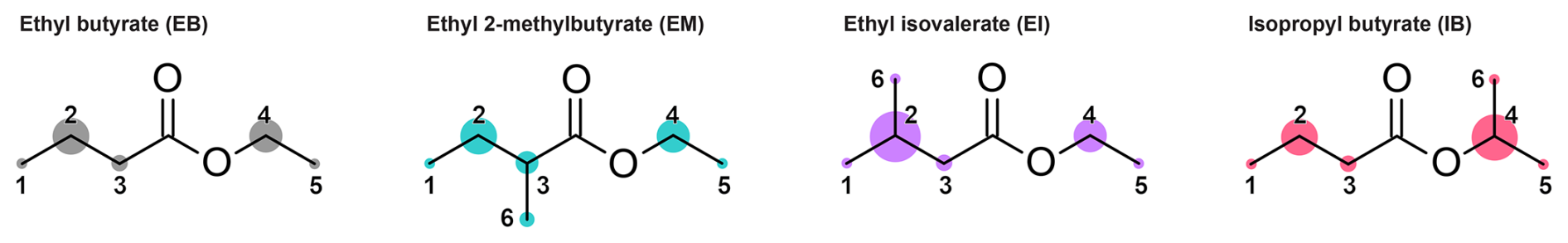
Figure 1Molecular structures of EB and its derivatives with numbered H abstraction sites. The areas of the circles are proportional to the room temperature partial rate coefficients for the reaction with OH at each site, as predicted by the structure-activity relationship (SAR) by Jenkin et al. (2018). The absolute and relative values of these rate coefficients are listed in Table S5 in the Supplement.
Importantly, EB and its derivatives are also present in many consumer products. They occur naturally in many plants and food products (Bicas et al., 2011; Kim et al., 2013; Sosa-Moguel et al., 2018), and can enhance sweetness perception and are thus used as sugar substitutes in food and as active ingredients in e-cigarettes (Barba et al., 2018; Labbe et al., 2007; Rao et al., 2018; Xiao et al., 2023, 2024). Moreover, EB and its derivatives exhibit the distinct fruity odour typical of many esters, and they fall into the range of molecules with a particular strong aroma, i.e. ≤C8 aliphatic compounds with a C3–C5 acyl moiety and a C1–C5 alkoxy group (Boelens et al., 1983; Kraft et al., 2000). With a smell reminiscent of pineapple, apple, or pear, they are a popular additive for a range of household items and consumer products. For instance, this includes their use in room fragrance products or scented personal care products (Nematollahi et al., 2018; Uhde and Schulz, 2015), but also in cleaning products such as washing-up liquids (Fig. S1 with Table S1 for methodological details, both in the Supplement). They are also described in perfumery reference libraries and supplier catalogues, as well as in fragrances databases where they are listed as active ingredients in cosmetics and perfume (e.g. The Good Scents Company Information System, 2026).
As aroma compounds need to reach the human olfactory system to induce scent, they are volatile to some degree and can be classified as volatile organic compounds (VOCs). This is also true for EB and its derivatives, which exhibit vapour pressures in the range of approx. 11–19 mbar at room temperature (US Environmental Protection Agency, 2026). Therefore, they exist mainly in the gas phase after emission, where they are exposed to different sources of light and atmospheric oxidants. Critically, consumer products are mostly used indoors, meaning that the components of the formulations are emitted into confined spaces. The presence of these VOCs in indoor environments can have numerous effects on ambient air quality, including direct toxicity as well as the formation of harmful secondary pollutants after photooxidation (Weschler and Carslaw, 2018; Wolkoff et al., 1997). Inadequate ventilation can slow the dilution of emitted VOCs and their photochemical products, leading to accumulation of these compounds indoors. Since most people spend the vast majority of their time indoors (Klepeis et al., 2001), the cumulative exposure to critical pollutants can represent a substantial health concern. Equally, indoor-outdoor exchange can lead to the occurrence of VOCs emitted from consumer products outdoors (Yeoman et al., 2020), where photooxidation conditions can be different. For both indoor and outdoor air quality, it is therefore important to characterize the photochemical reactivity of emitted compounds.
Since EB and its derivatives do not possess C=C double bonds, rapid ozonolysis or reactions that proceed via the addition of a radical to the molecular structure can be disregarded. Loss processes that can be relevant, on the other hand, include photolysis or radical-initiated H abstraction. For photolytic decay to occur, a chromophore must be present, which in case of esters is the carbonyl group (Lary and Shallcross, 2000). The typical carbonyl transitions tend to be blue-shifted for esters due to the additional alkoxy oxygen, but the exact position and magnitude of the absorption bands depend very much on the specific substituents and molecular structure (Caswell et al., 1976; Closson and Haug, 1964). For EB and its derivatives in particular, the UV-vis. absorption spectra or photolysis quantum yields have not yet been determined to the best of our knowledge, meaning that the relevance of photolytic decay is still poorly constrained.
Oxidation via abstraction of an H-atom can be initiated by reactive free radicals such as OH or Cl (Calvert et al., 2011; Mellouki et al., 2003). Cl atoms are scarce in the troposphere (Wang et al., 2019), yet their reactions are often significantly faster than the equivalent OH radical reactions (Liang et al., 2010; Notario et al., 1998). The room temperature rate coefficients are known for most of the compounds studied here and have been reported for EB + Cl (Cometto et al., 2009; Ifang et al., 2015; Ramya and Rajakumar, 2019), EM + Cl (Ifang et al., 2015), and EI + Cl (Ifang et al., 2015). In contrast, OH radicals are more abundant in the troposphere and are considered the dominant oxidant outdoors (Lelieveld et al., 2004) while also playing an important role indoors (Weschler and Carslaw, 2018). OH is generally known to react with esters (e.g. Ferrari et al., 1996; Wallington et al., 1988; Williams et al., 1993), which makes it a likely reaction partner for EB and its derivatives as shown in Reactions (R1)–(R4).
However, rate coefficients have only been reported for EB + OH (Cometto et al., 2009; Ferrari et al., 1996; Gour et al., 2014; Wallington et al., 1988) and remain unknown for the methylated EB derivatives. While structure-activity-relationships (SARs) are available to predict VOC+OH rate coefficients (Jenkin et al., 2018; Kwok and Atkinson, 1995), these are often limited in their capability to accurately describe oxygenated compounds (e.g. Mapelli et al., 2022, 2023).
Given the lack of available kinetic data, this study aims to assess the gas-phase atmospheric fate and potential air quality impacts of EB and its derivatives by evaluating their photochemical reactivities and atmospheric lifetimes. To this end, we performed experiments to determine UV–vis. absorption spectra as well as temperature-dependent rate coefficients for reactions with OH radicals (R1–R4).
The photochemical properties of EB and its derivatives were explored by experimental and computational approaches for the determination of absorption cross-sections (Sect. 2.1), as well as by pulsed laser photolysis (PLP)/laser induced fluorescence (LIF) experiments conducted at the University of York and the University of Leeds to investigate radical kinetics (Sect. 2.2). In a preliminary study to this work, EB and its derivatives were identified from headspace samples of washing up liquid (see Sect. S1 in the Supplement for details). The organics were purchased as neat compounds from Fisher Scientific (EB, EM, EI), Scientific Laboratory Supplies (IB), and Sigma Aldrich (ethyl acetate), all with a stated purity of at least ≥99 %.
2.1 UV-vis. absorption spectra
2.1.1 Quasi-gas-phase spectroscopy
As direct gas-phase measurements were not feasible with the available instrumentation, the UV-vis. absorption properties of EB and its derivatives were probed experimentally by recording solution-phase spectra, dilute in cyclohexane (D'Souza Metcalf et al., 2025). As demonstrated by Nakashima et al. (1982), the difference in absorption intensity between solution-phase and gas-phase spectra is smallest when the selected solvent has low molecular polarizability and the dispersion forces between the target compound and solvent are minimized. In such cases, solution-phase spectra can be considered “quasi-gas-phase”. Cyclohexane (C6H12) is a non-polar solvent with low polarizability. As such, it has been used in previous studies to record UV-vis. spectra of oxygenated compounds and has been shown to be a suitable choice when a small correction factor for the absorption intensity is applied (D'Souza Metcalf et al., 2025; Mapelli et al., 2023). Furthermore, cyclohexane absorbs mainly below 200 nm and in particular below 180 nm (Doner et al., 2019), and therefore does not interfere with the expected absorption maxima of carbonyl-containing VOCs. To validate the method and to determine a suitable correction factor for this class of compounds, a quasi-gas-phase spectrum of ethyl acetate (ethyl ethanoate; EA) was recorded along with the spectra of EB and its derivatives. EA is structurally and chemically similar to EB and its derivatives, and its UV absorption in the gas phase is well-established (McMillan, 1966; Śmialek et al., 2016).
Each compound was dissolved in cyclohexane to obtain dilute solutions in the range of 0.004–0.016 M. Working with these low concentrations, interactions of the target compound molecules amongst each other were minimized. The solutions were prepared using micropipettes and volumetric flasks to an estimated accuracy of ±10 %. All spectra were recorded with a resolution of 0.2 nm in the range between 200–600 nm (EB) or 200–350 nm (EA, EM, EI, IB) using a pair of quartz cuvettes (pathlength 1 cm) in a UV-vis. spectrometer (UV-2600, Shimadzu). Solvent-vs-solvent baselines were recorded to account for differences in the optical properties of the cuvettes, and were subtracted from all spectra.
To obtain the gas-phase absorption cross-section, we first derived the extinction coefficient according to the Beer Lambert law shown in Eq. (1):
where A(λ) is the absorbance recorded by the spectrometer (unitless), I0(λ) is the reference beam intensity (a.u.), I(λ) is the sample beam intensity (a.u.), c is the concentration in M, l is the pathlength in cm, and ε(λ) is the molar extinction coefficient in . A linear regression of the absorbance against concentration × pathlength was performed for each wavelength interval, resulting in a robust estimate of the extinction coefficient (see also Fig. S2 in the Supplement). In the next step, the extinction coefficient was converted to the gas-phase absorption cross-section using Eq. (2), based on the relationship between Eq. (1) and the Beer Lambert law for gas-phase conditions as shown in Eq. (3):
where σ(λ) is the gas-phase absorption cross-section in , NA is Avogadro's constant , Ag(λ) is the gas-phase absorption (unitless), and n is the concentration in .
2.1.2 Computational experiments
In addition to the spectroscopic experiments, UV-vis. spectra were predicted computationally using Gaussian 16, revision C.02 (Frisch et al., 2016). In a first step, the molecular geometries were optimised to find the most stable structures, using the B3LYP functional with a 6-311++G(d,p) basis set and gd3bj dispersion correction. Tight convergence criteria were employed for higher accuracy. The fully converged stationary points were verified as true energy minima by vibrational frequency analysis. Imaginary frequencies, which would reveal saddle points on the potential energy surface, were absent in all cases. Using the optimised structures, vertical excitation energies for the lowest 30 singlet states were obtained by time-dependent density functional theory (TD-DFT) calculations at the at the CAM-B3LYP/6-311++G(d,p) level of theory. For evaluating UV-vis. absorption in solution, both the geometry optimisation and the energy calculation were performed with inclusion of cyclohexane as the solvent (CPCM method). For all computational steps, our choice of functionals and basis sets was aligned with the configurations used in similar applications (e.g. Anhaia-Machado et al., 2023; Pandey et al., 2021; Tang et al., 2024).
2.2 PLP-LIF experiments
2.2.1 Experimental setup at University of York
Most PLP-LIF experiments were performed at the University of York, using the apparatus described in detail by Mapelli et al. (2022). Briefly, the flows of the target VOC, the OH precursor, and the bath gas N2 (>99.9999 %, obtained directly as boil-off from liquid N2) were each regulated by mass flow controllers (MFCs), and mixed in a glass manifold before entering the 400 cm3 spherical Pyrex reaction cell. The laser beams entered the cell through quartz Brewster windows, and additional arms were available for the introduction of the gas mix and the vacuum pump. The temperature inside the reaction cell was regulated with electric heating tape, maintained with glass fibre insulation, and monitored with a K-type thermocouple that could be moved along a vertical gradient and was placed in the centre of the cell before and after each experiment. Two capacitance manometers (MKS, 10 and 1000 Torr) were available to monitor the pressure in the reaction cell. The experiments were performed at pressures between 20 and 37 Torr, with a total flow of a few hundred sccm, resulting in approx. 10 sccm Torr−1.
OH radicals were generated through the photolysis of gaseous H2O2 in Reaction (R5), which can be regarded as a clean source of OH radicals.
As a source of H2O2, a 1:1 mixture of H2O2-urea (97 %, Sigma Aldrich) and sand (pure, 40-100 mesh, Fisher Scientific) was placed in a glass container that was heated to approx. 45 °C in a water bath. At this temperature, H2O2-urea decomposes and provides water-free H2O2 vapour. The sand was added to prevent agglomeration. This method has been used in numerous previous studies (e.g. Chen et al., 2024; Hong et al., 2010; Ludwig et al., 2006). To photolyze H2O2, the fourth harmonic output from a Nd:YAG laser (Quantel, Q-Smart) was used to provide a 266 nm laser beam with 10 Hz frequency and a fluence of .
OH radicals were detected via off-resonant LIF, with excitation at 282 nm via the transition. The 282 nm laser beam was supplied from a frequency-doubled dye laser (Radiant Dyes, NarrowScan) that was pumped at 532 nm from the second harmonic output of a Nd:YAG laser (Continuum, Surelite II) and tuned for a Rhodamine-6G emission at 564 nm. The 309 nm fluorescence of OH radicals was collected perpendicular to the laser beams by biconvex lenses and directed through an interference filter onto a photomultiplier tube (PMT, Hamamatsu).
The organics were degassed by repeated freeze-pump-thaw cycles and room temperature degassing. To enable a steady supply of the gaseous compounds in the PLP-LIF experiments, they were diluted and stored in 12 dm3 Pyrex bulbs (mixing ratios of <1 % in N2, with the pressure of the organic in the bulb remaining below the room temperature vapour pressure). Except for the experiments without added ester, all experiments were carried out under [ester]≫[OH] conditions to enable the analysis of the OH decay as a pseudo-first-order reaction. The removal rate of OH radicals in the absence of any organic (see intercept in Fig. 3) suggested a concentration of of H2O2 in the reaction cell. To obtain a decay profile of OH, the timing between the PLP laser and the LIF laser was successively increased. The entire trace was recorded several times and averaged to reduce noise. A LabVIEW programme was used to control the lasers via a delay generator (BNC, 575) and to collect and digitalise the LIF data via an oscilloscope (Picoscope, 5000).
2.2.2 Experimental setup at University of Leeds
Additional PLP-LIF experiments for EB were carried out at the University of Leeds in order to evaluate rate coefficients at higher temperatures, to compare OH and OD radical kinetics for further mechanistic insights, and to probe for OH recycling. The experimental set-up is generally similar to the one described above, with comparable pressures and total flows. Significant differences are noted as follows: the reaction cell was constructed from stainless steel, argon was used as the bath gas, and on-resonant LIF was employed (see below). For further technical specifications, the reader is referred to previous publications (e.g. Blitz et al., 2025; Onel et al., 2012) where the apparatus has been described in detail.
As for the York PLP-LIF experiments, OH radicals were generated from the photolysis of H2O2 at 266 nm, using the same H2O2-urea precursor to introduce H2O2. The OH probe beam at approx. 308 nm was created by doubling a 616 nm tuneable dye laser output (dye: DCM special with pyridine 1). For experiments including OD radicals, D2O was either added to a wet peroxide source (50 % H2O2 in H2O), or flown over the H2O2-urea precursor in an enriched carrier gas flow, forming D2O2 almost instantly in both cases. The amount of D2O was adjusted so that a sufficient amount of H2O2 was converted to D2O2. The photolysis of the gaseous H2O2D2O2 mixture yielded both OH and OD radicals. The kinetics of these radicals with EB were probed in the same reaction cell in immediate succession. To alternate between OH and OD radical lines, the dye laser wavelength was adjusted slightly in between runs.
The relevance of photolytic degradation of EB and its derivatives in different settings was evaluated based on the gas-phase absorption cross-sections in Sect. 3.1. In terms of radical reactions, the rate coefficients of each of the esters with OH are detailed as a function of temperature in Sect. 3.2, and these results are put in the context of SAR predictions of site-specific reactivities and probable abstraction mechanisms. Complementary material is provided in Sects. S2 and S3 in the Supplement, respectively.
3.1 UV absorption cross-sections and rates of photolysis
For method validation and refinement, we compared the experimentally determined solution-phase spectrum (200–350 nm) for EA from this work to available gas-phase spectra from the literature and to gas-phase and solution-phase TD-DFT predictions. For details of this proof-of-concept study, the reader is referred to Sect. S2.1 with Fig. S2 and Table S2 in the Supplement. Importantly, the experimental solution-phase spectrum showed excellent agreement with the literature gas-phase spectra in terms of the peak position and peak shape, but the magnitude of the absorption was amplified due to solvent effects. A robust correction factor of 0.86±0.01 which aligns well with previous studies (Mapelli et al., 2023; Nakashima et al., 1982) was derived from the observed differences in peak areas. We conclude that the experimental solution-phase spectra for EB and its derivatives can provide an accurate proxy for gas-phase behaviour when this correction factor is applied.
The UV absorption spectra of EB and its derivatives are displayed in Fig. 2a. The absorption maxima and corresponding wavelengths for the absorption bands of the individual compounds are listed in Table S3 in the Supplement. The absorption band at around 213 nm is attributable to the forbidden transition. A weak absorption band at around 210 nm is a typical feature of many esters and results from the interaction of the lone pair of electrons of the alkoxy oxygen with the carbonyl π orbitals and the associated blue-shift of the carbonyl transition (Closson and Haug, 1964 and references therein). In terms of intensity, the molar extinction coefficients of (experimental data) are comparable to the values reported for other esters at around 210 nm, which are generally small and remain mostly in the range below ε=100 as a result of the low likelihood with which the forbidden transition occurs (Caswell et al., 1976; Closson and Haug, 1964). There was no absorption in the visible range (Fig. S3 in the Supplement). In the gas-phase spectra predicted by TD-DFT, the peaks appear slightly blue-shifted but agree within 4 nm with the experimental data. Additionally, the peak profiles are a bit narrower, which was previously observed for EA as well (Fig. S2 in the Supplement). The predicted maximum absorption intensities are between 1.3 and 2.8 times lower than the experimental values, suggesting that the computational approach used in this work provides a qualitative rather than quantitative description of the studied compounds.
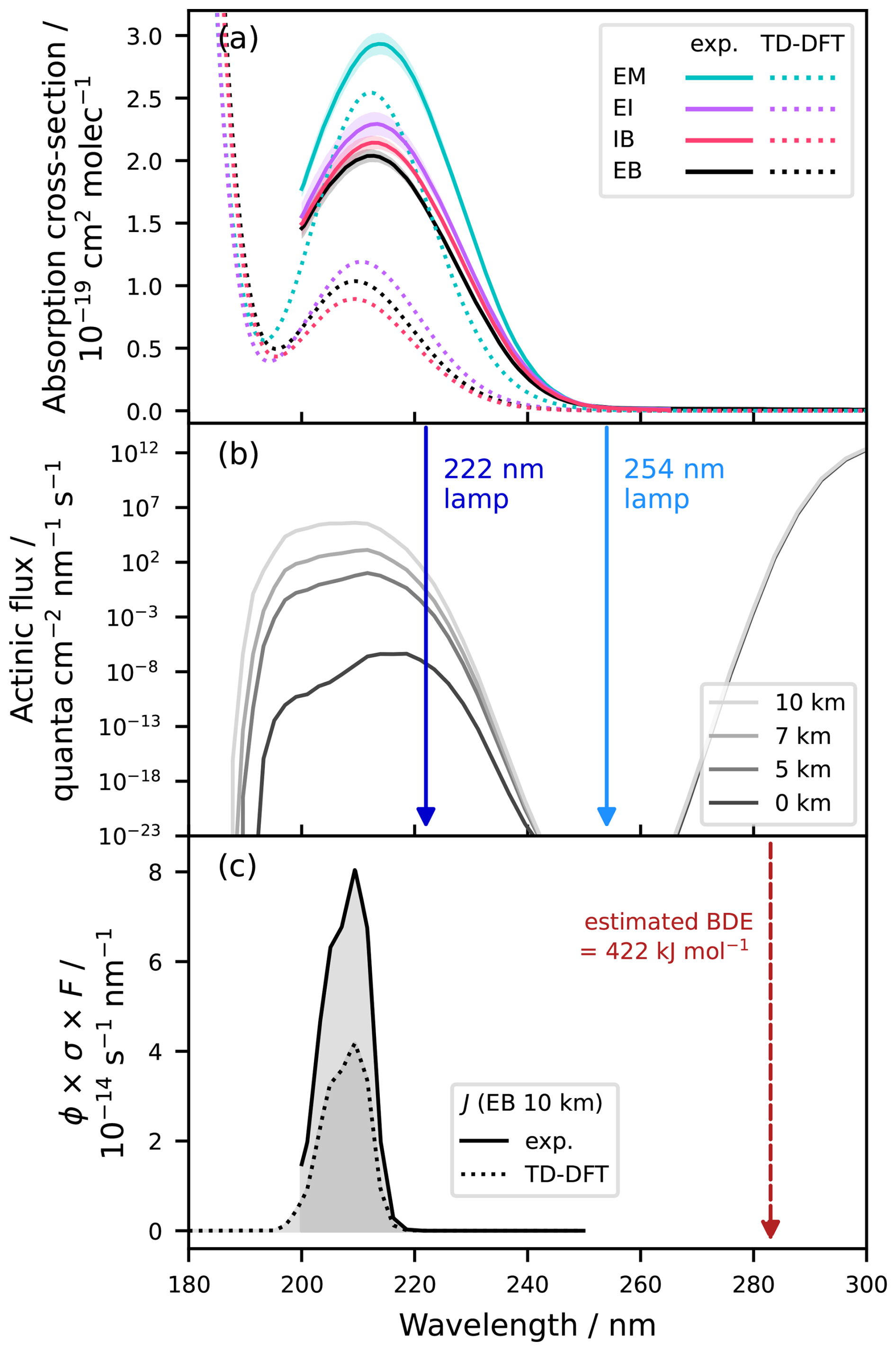
Figure 2UV absorption and potential for photolytic decay for EB and its derivatives. (a) UV-vis. spectra of EB, EM, EI, and IB, recorded as dilute solution-phase spectra (0.004–0.016 M in cyclohexane, n=4 excl. blanks, 2σ statistical errors as shaded areas) and corrected to gas-phase conditions using the scaling factor 0.86. TD-DFT predications are shown as dotted lines (CAM-B3LYP/6-311++G(d,p) level of theory; 30 singlet states). (b) Solar actinic flux at 51° N latitude in peak summer at altitudes of 0, 5, 7, and 10 km (sea level towards upper troposphere) according to the NCAR TUV model v5.3. The wavelengths of different UV-C germicidal lamps are shown as vertical arrows. (c) Product of the EB absorption cross-section, the photon flux at 10 km altitude, and an assumed quantum yield of 1, resulting in the upper limit photolysis rate after integration, shown for the experimental data and the TD-DFT predictions of the absorption cross-section. The wavelength corresponding to the estimated bond dissociation energy (BDE; 422 kJ mol−1 for 283 nm) for the single C–O bond in the ester group is shown as the dashed vertical arrow.
Peak positions for the methyl-substituted derivatives appear slightly red-shifted, by approx. 1–2 nm when compared to EB. There is a trend of for both the experimental data and the TD-DFT predictions, even though λmax(IB) is slightly smaller than λmax(EB) in the TD-DFT predictions. The observed red-shift of the methylated derivatives is in accordance with the work by Closson and Haug (1964) who have observed that the absorption maximum tends to be shifted to longer wavelengths when a methyl group is attached to either the carbon adjacent to the carbonyl functional group or to the carbon adjacent to the alkoxy oxygen. Here, this is valid for EM and IB, respectively. The red-shift resulting from methyl substitution in the acyl group is likely to result from the electron-donating effect of the methyl group and the resulting increase in energy of the lone pair electrons of the oxygen, thus reducing the energy difference in the transition (Closson and Haug, 1964). The red-shift caused by methyl substitution in the alkoxy group, in turn, is believed to be related to steric effects and the interaction between the methyl group and the carbonyl oxygen, either directly increasing the energy of the lone pair electrons, or lowering π* by reducing coplanarity or increasing the C–O–C bond angle (Closson and Haug, 1964).
For the methylated esters, the absorption intensity tends to increase along with the red-shift, suggesting that similar mechanisms may contribute to both effects. Indeed, Caswell et al. (1976) found that the values of ε in their study were dependent on the availability of electron-donating alkyl group. In both the experimental data and the TD-DFT predictions, EM has a distinctly higher cross-section than the other three compounds which in turn are relatively similar in their absorption intensities. The factor by which σmax(EM) exceeds is 1.4 in the experimental data and 2.5 in the TD-DFT predictions. This suggests that the electron-donating effect of the methyl group is strongest in EM, which can be rationalised by its immediate proximity to the carbonyl carbon.
Photolysis only becomes a competitive atmospheric loss process if the incoming radiation aligns with the absorption bands of EB and its derivatives and if it contains sufficient energy to break intra-molecular bonds. The scission of the ester functional group () has been shown to be the most relevant photolysis pathway for esters exposed to unfiltered light, accounting for at least half of the observed photolysis products of esters such as methyl propionate or methyl butyrate close to room temperature (Ausloos, 1958). The bond dissociation energy (BDE) for the scission of the bond between the carbonyl carbon and the alkoxy oxygen has been calculated to be 420 kJ mol−1 for ethyl propanoate and 424 kJ mol−1 for methyl butanoate (El-Nahas et al., 2007). Assuming an average 422 kJ mol−1 for EB and its derivatives, the wavelength corresponding to this estimated BDE is 283 nm (Fig. 2c). This essentially means that radiation with longer wavelengths will be ineffective in photolyzing the molecules even if they did absorb in the near-UV or visible range, whereas wavelengths shorter than that would be able to break the bond. Within their absorption band, photolysis is hence principally possible. At λmax of EB and its derivatives of around 213 nm, the photon energy is roughly 561 kJ mol−1 and far above the estimated BDE.
However, under standard ground-level conditions outdoors or in conventional indoor environments, there is effectively no radiation in the UV-C range where EB and its derivatives absorb. Light sources used to irradiate living spaces are designed to emit in the visible range, and sunlight reaching the Earth's surfaces contains mainly radiation with wavelengths above 300 nm since most of the UV-B and UV-C radiation is absorbed by optically active gases within the atmosphere. At around 200–220 nm, however, there is less attenuation and UV-C light can penetrate further through the atmosphere. As a result, there is a height-dependent photon flux and a higher availability of UV-C radiation – and hence a higher likelihood for photolysis – in the upper troposphere and stratosphere than at ground level (Fig. 2b).
For relevant spectral ranges, photolysis rates can be calculated as the product of the photon flux, the absorption cross-section of the molecule, and the photolysis quantum yield, integrated over the relevant wavelength range as shown in Eq. (4):
where J is the first-order photolysis rate in s−1, ϕ(λ) is the wavelength-dependent quantum yield, σ(λ) is the wavelength- dependent absorption cross-section in , and F(λ) is the wavelength-dependent photon flux in quanta . As ϕ(λ) is not known for EB (or any of its derivatives), in common with many other VOCs, a value needs to be assumed for the calculation of photolysis rates. Since λmax of the EB absorption band and the spectral range of the atmospheric window in the UV-C range are far below the threshold wavelength corresponding to the estimated BDE and since the incoming energy is hence significantly higher than the energy needed to break the bond (Fig. 2c), a quantum yield of 1 is principally possible. Using an assumed quantum yield of 1 in Eq. (4) results in the photon absorption rate, which is an upper limit to the photolysis rate. Here, we calculated these rates for EB using the recorded UV-vis. absorption spectrum and the actinic fluxes as predicted by the Tropospheric Ultraviolet and Visible (TUV) model v5.3 (NCAR, 2025) for altitudes of 0, 5, 7, and 10 km at 51° N latitude in peak summer. The upper limit photolysis rate of EB increases by numerous orders of magnitude from between sea level (0 km altitude) and the upper troposphere (10 km altitude), but the corresponding (minimum) photolytic lifetime is in the range of millennia for all altitudes (Table 1). Notably, the experimental UV spectra from this work are only available down to 200 nm, but absorption still occurs below that wavelength. However, evaluating the photon absorption rate based on the TD-DFT prediction (which has a smaller absorption cross-section by about a factor of 2, explaining the absolute difference in the photon absorption rate) reveals that the product of the absorption cross-section and the photon flux quickly declines below 200 nm even for a height of 10 km (Fig. 2c). This is mainly due to the sharp decrease of the photon flux (note the logarithmic scale in Fig. 2b).
Table 1Upper limit photolysis rate of EB throughout the troposphere, calculated using the flux predicted by the NCAR TUV model v5.3 for peak summer and 51° N latitude. The lifetime is given by the reciprocal of the photolysis rate.

In spite of the insignificance of photolysis in most settings, the absorption of EB and its derivatives in the UV-C range can still be relevant in specific experimental and environmental contexts. In terms of kinetic work, the absorption of the esters below 260 nm can be associated with additional scatter or bias in PLP-LIF experiments if VUV light sources or – to a lesser extent – 248 nm excimer lasers are used, whereas no interference is expected from 266 nm lasers such as were employed in this work. Beyond such technical considerations, the absorption of EB and its derivatives could be relevant in the context of germicidal UV-C technology. UV-C lamps which emit at 254 or 222 nm have sterilising properties (Naito et al., 2022), and have seen a surge in interest and usage since the onset of the COVID-19 pandemic. For 222 nm lamps in particular, the emitted light overlaps substantially with the absorption band of EB and its derivatives (Fig. 2b). As such, they may induce photolytic degradation of EB and its derivatives in indoor environments where they may be employed next to VOC sources. While the potential impact of UV-C lamps on indoor air quality has been recognised increasingly in recent years, most studies were limited to observations of ozone formation (e.g. Link et al., 2023; Peng et al., 2023) rather than investigations of direct VOC photolysis. Given the high spectral significance not only for EB and its derivatives but for many oxygenated or conjugated VOCs absorbing in the UV-C range, direct photolysis may be an underestimated phenomenon in indoor air chemistry that should be investigated in more detail.
3.2 Kinetics of OH with EB and its derivatives
For each PLP-LIF experiment, the pseudo first order OH decay rate coefficient k′ was determined for a range of different VOC concentrations. All experiments were conducted with the VOC in excess, so that the OH decay could be evaluated according to Eq. (5):
where [OH]t and [OH]0 are the OH signals at time t and time 0 respectively (a.u.), k′ is the pseudo first order rate coefficient in s−1, and t is the time in s. The rate coefficient k in units of could then be derived as the slope of the linear regression of k′ against the VOC concentration [VOC] in , as shown exemplarily in Fig. 3 and as described by Eq. (6). The intercept of the linear regression represents kloss which is the loss term for other losses of OH in s−1, and which was dominated here by the Reaction (R6) between OH radicals and H2O2. Uncertainties of k quoted throughout this work are 2σ statistical errors only, unless specifically stated otherwise. Systematic errors due to unwanted radical chemistry or VOC photolysis were considered negligible, as the experiments were performed at low H2O2 (and hence OH) concentrations and the studied esters do not absorb at 266 nm.
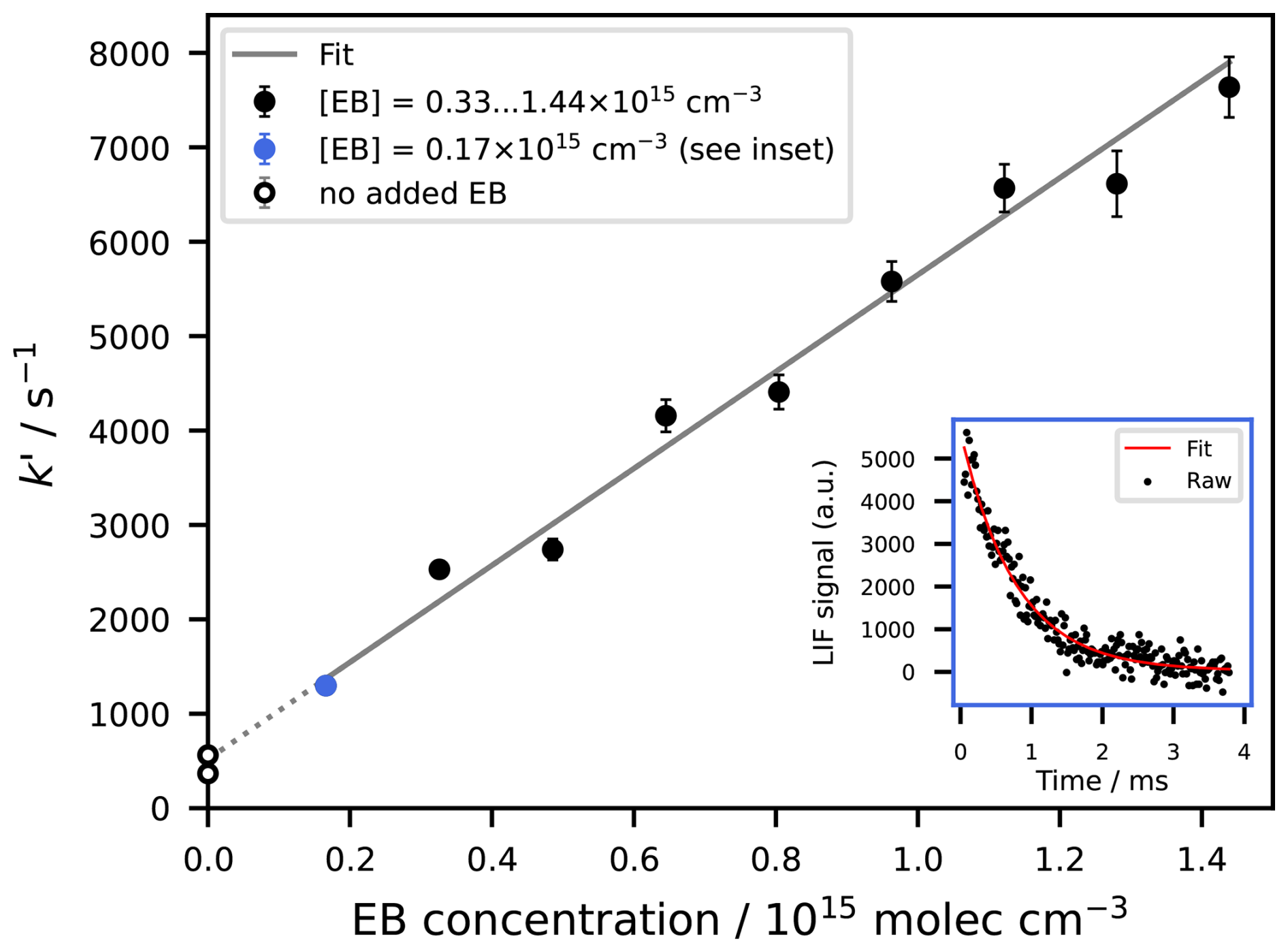
Figure 3Exemplary PLP-LIF dataset, showing the determination of k1 at 340 K. The individual data points are experiments performed at different EB concentrations, for each of which the pseudo first order decay rate of OH radicals has been obtained by exponentially fitting the OH signal (as shown in the inset for the data point ). A linear regression of the data results in the value of k1 (here: with 2σ error, ). The experiments conducted without any added EB were not used in the fit.
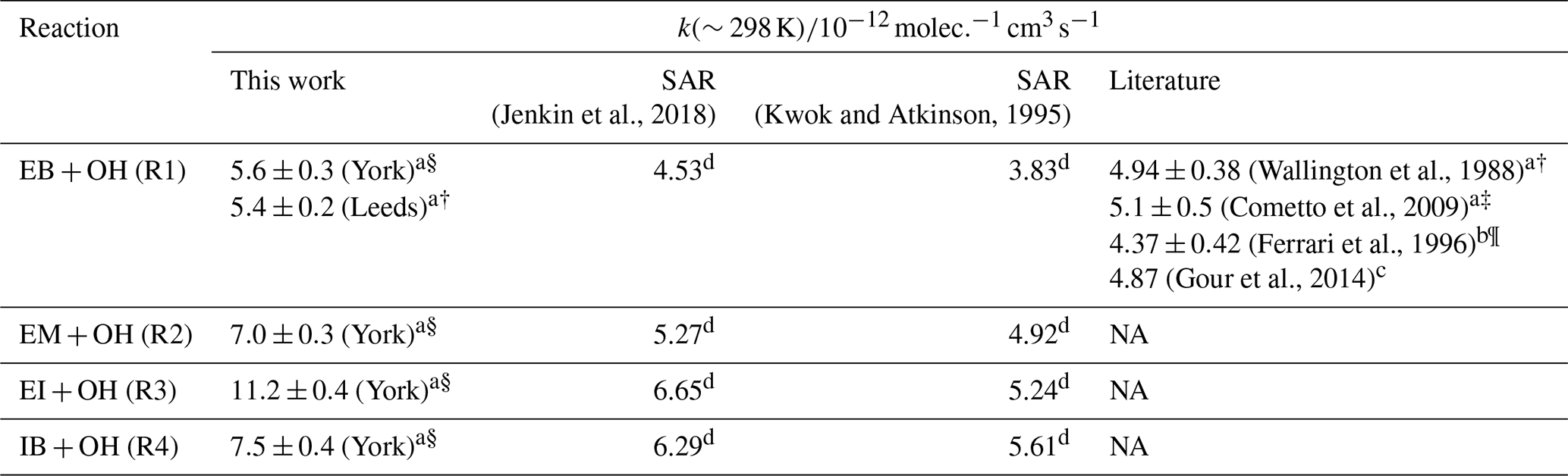
The conditions and resulting rate coefficients for all PLP-LIF experiments conducted in this work are listed in Table S4 in the Supplement. The room temperature rate coefficients of the studied esters are listed along with SAR predictions and literature values (for EB only) in Table 2. Generally, there is very good agreement between the values of k1 determined in York and Leeds, respectively, averaging to (weighted mean with statistical error). There is also good agreement between these experimental values and the body of literature. Notably, the two studies based on absolute rate determinations (Cometto et al., 2009; Wallington et al., 1988) show the greatest consistency with the experimental data from this work. Although a VUV light source (Wallington et al., 1988) and a 248 laser (Cometto et al., 2009) which can potentially trigger the photolysis of the ester (see Sect. 3.1) were used, all absolute values reported in this work or in previous literature agree within their uncertainties. In contrast, the DFT study (Gour et al., 2014) and the study employing a relative rate approach (Ferrari et al., 1996) report lower rate coefficients. The work by Ferrari et al. (1996) was done relative to OH+n-octane, using the absolute rate coefficient of reported by Greiner (1970). Both lower (e.g. Anderson et al., 2003) and higher (e.g. Shaw et al., 2020) rate coefficients have been reported for this reaction based on relative rate techniques since, so that a meaningful re-assessment of k1 based on an updated n-octane reactivity is currently not possible.
Table 2Room temperature rate coefficients for the reactions of the esters with OH radicals, as obtained with the PLP-LIF setup in this work, according to two structure-activity-relationships (SARs), and as reported in the literature. Different methods of obtaining rate coefficients are labelled as absolute rate methods (a), relative rate methods (b), density functional theory calculations (c), or SAR predictions (d). For values based on experimental methods, different bath gases are denoted as nitrogen (§), argon (†), helium (‡), or air (¶). All temperatures are within ±4 K of 298 K. The room temperature coefficients obtained in this work are reported as weighted means from two determinations with statistical errors (see all single results and associated uncertainties in Table S4 in the Supplement). NA: not available.
The room-temperature values of k1 reported in this work were obtained at 26 and 29 Torr (N2 bath gas) using the York set-up and at 37 and 38 Torr (Ar bath gas) using the Leeds set-up. Their agreement with the PLP-LIF results by Cometto et al. (2009) at 100 Torr (He bath gas) and with the flash photolysis resonance fluorescence (FP-RF) results by Wallington et al. (1988) at 25–50 Torr (Ar bath gas) suggests that all of these measurements were obtained in the high-pressure limit. This is in accordance with other reactions of oxygenated VOCs with OH, such as acetone where strong curvature was observed but no pressure dependence was found between 20 and 100 Torr (Wollenhaupt et al., 2000).
Both SARs (Jenkin et al., 2018; Kwok and Atkinson, 1995) underestimate the absolute rate coefficients as obtained by the PLP-LIF experiments for all four compounds, but correctly predict the methylated derivatives to be more reactive than EB. The SAR by Jenkin et al. (2018) furthermore correctly reflects the increase in reactivity in the specific order EB . It should be noted that EB was used in the training set of the Jenkin et al. (2018) SAR with a training value of (taken from Calvert et al., 2011, in turn based on Wallington et al., 1988 and Ferrari et al., 1996) which is very closely matched by the SAR output of . Although the methylated EB derivatives were not included in the SAR training set, the optimisation of EB towards a value which is lower than all absolute rate coefficients reported in the literature may partly explain why the predicted reactivities fall below the experimental values for all compounds studied here. The Kwok and Atkinson (1995) SAR, on the other hand, does not specifically aim to describe esters, and is stated to have limited applicability to compounds outside the training set.
The most reactive sites in EB as predicted by the Jenkin et al. (2018) SAR are the ones β to the carbonyl group (Fig. 1). At these positions, 7-membered hydrogen-bonded pre-reaction complexes involving the OH radical and the carbonyl oxygen can be formed. Such complexes lead to faster reactions by stabilizing the transition state and increasing the probability of quantum mechanical tunnelling (Galano and Raúl Alvarez-Idaboy, 2008; Shannon et al., 2014). The high reactivity of the H atoms attached to these β carbons has previously been referred to in the context of the activating effect of the ester group (Mellouki et al., 2003) or the anomalously high reactivity of butyrates compared to propionates (Wallington et al., 1988). Reflecting these site-specific differences, the relative reactivities observed for the other esters may be explained by the existence and position of their respective methyl substituents. If a methyl group is attached to one of the carbons in β position to the carbonyl group, such as is the case for EI and IB, the overall reactivity of the molecule increases further, not only because of the higher total number of oxidisable bonds in the molecule but also because the remaining H atom attached to the β carbon becomes still more reactive (Table S5 in the Supplement). As the methyl group of IB is located on the alkoxy side of the molecule (Fig. 1), and the value of kOH(298 K) is substantially higher for EI compared to IB in both the experimental dataset and the Jenkin et al. (2018) SAR prediction (Table 2), one might infer that the involvement of the alkoxy oxygen decreases the stability of the pre-reaction complexes. In case of EM, the methyl group is attached to the α carbon and the remaining H atom is oriented away from the carbonyl oxygen. While a pre-reaction complex with the alkoxy oxygen via a 6-membered ring might be possible, the alkoxy oxygen has a smaller partial negative charge than the carbonyl oxygen and is therefore less likely to participate in hydrogen bonding. This description is consistent with the lower value of kOH(298 K) in both the experimental and the SAR data, but in contrast to the DFT study by Gour et al. (2014) where the α carbon is predicted to be the most reactive site.
For evaluating the effect of temperature changes on the reactivity of EB (Fig. 4), we combined and compared the dataset from this work with the available literature data and SAR predictions. The only previous study that evaluated the values of k1for different temperatures (Cometto et al., 2009) covered the temperature range of , whereas the data from this work describe the range of and therefore include higher temperatures that have not been investigated before. In their overlapping range, the two datasets agree well. Together, the cold-temperature and high-temperature data form a consistent pattern that resembles a U-shape with a minimum at 387 K. Using all available absolute data for k1 (this work; Cometto et al., 2009; Wallington et al., 1988), this temperature dependency can be described by the following four-parameter expression (unit ): . The fact that there is a negative temperature dependence for T<387 K at all supports the idea of the prevalence of mechanisms other than direct H abstraction. Similar near-zero or negative temperature dependencies have been found for a range of esters in other studies and have been ascribed to the formation of hydrogen-bonded pre-reaction complexes (Cometto et al., 2009; Liang et al., 2010 and references therein; Mellouki et al., 2003 and references therein; Wallington et al., 1988). Especially at low temperatures, these complexes can dominate the overall reactivity. The seemingly negative activation energy has been related to either well-trapping of the complex or a two-step reaction sequence including the reversible formation of the pre-reaction complex followed by the irreversible decomposition of the complex to the products (Galano and Raúl Alvarez-Idaboy, 2008). For EB specifically, the importance of hydrogen-bonded pre-reaction complexes has been demonstrated theoretically using DFT approaches (Gour et al., 2014).
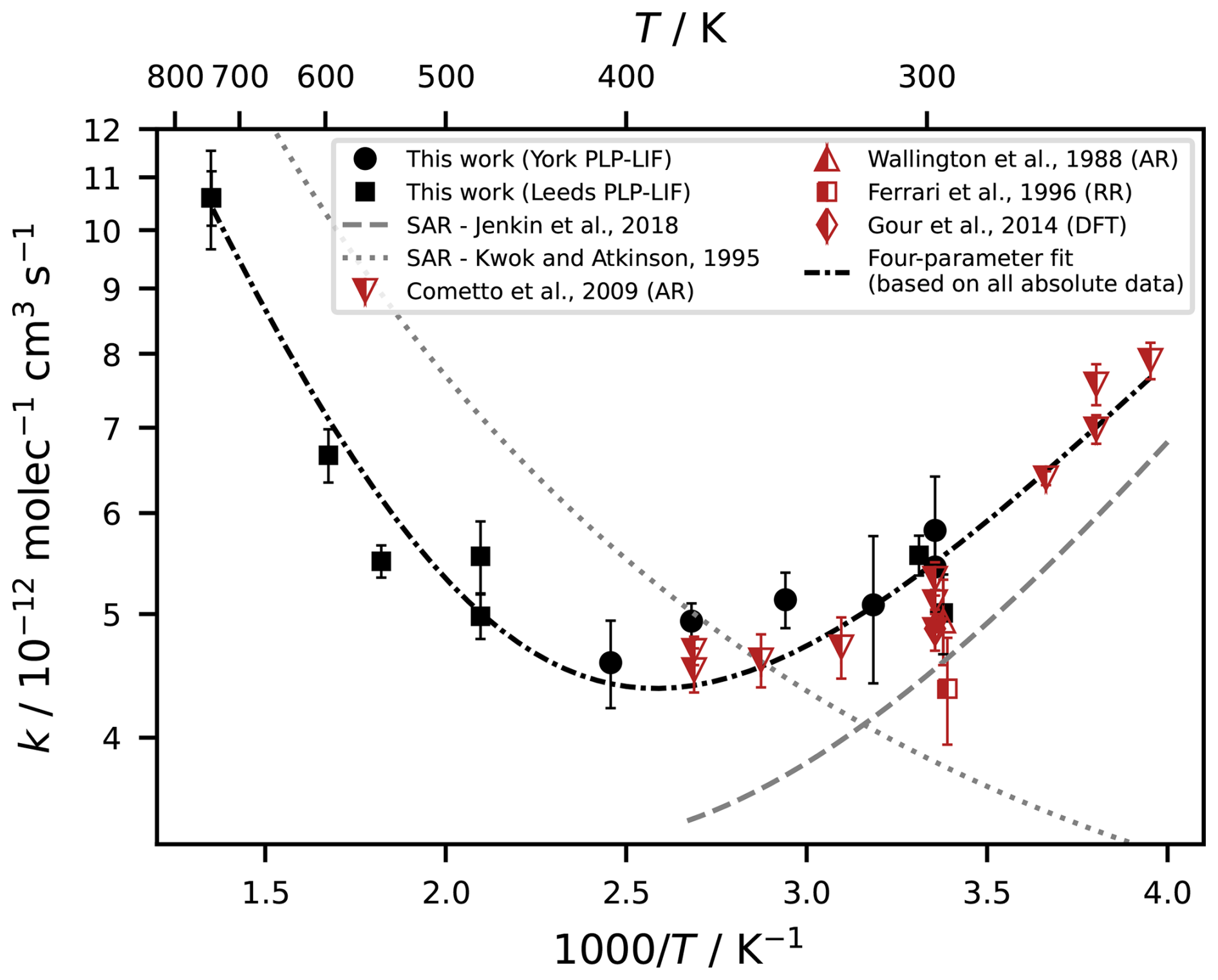
Figure 4Temperature-dependence of the reaction of EB with OH. Comparison of two SAR predictions, experimental data from this work (uncertainties are 2σ statistical errors), and experimental and theoretical values from the literature. The four-parameter fit is based on all kinetic data obtained with absolute methods (this work; Cometto et al., 2009; Wallington et al., 1988).
The observed temperature dependence of EB resembles a superposition of the two SAR predictions. The SAR proposed by Kwok and Atkinson (1995) was originally tested and validated for oxygenated compounds over a very extensive temperature range, and is applied here for temperatures between 250 and 700 K. It follows the observed Arrhenius behaviour at higher temperatures albeit with a large shift of around −150 K compared to the experimental data. However, it fails to describe the strong curvature and the non-Arrhenius behaviour at lower temperatures. This is likely due to the fact that the SAR was not designed or trained for esters, and the activating effect of the ester functional group is only considered for the α carbon which is much less likely to be involved in pre-reaction complexes than the β carbons. In contrast, the SAR by Jenkin et al. (2018), which does contain an explicit description of the neighbouring ester group for the β carbons, provides a very good match with the observed temperature dependence in the range between 250 and around 360 K, except for an offset that may relate to the above-mentioned optimisation of k(298 K) towards . The good qualitative fit of the negative temperature dependence suggests that the more holistic consideration of the ester functional group is appropriate and that the branching ratios and relative site reactivities are reliable. The Jenkin et al. (2018) SAR is targeted towards atmospherically relevant temperatures rather than combustion-regime temperatures, hence the ability of the SAR to describe the shift towards an Arrhenius behaviour at higher temperatures is not evaluated here. Specifically, we restricted our calculations to the range between 250 and 375 K, based on the temperature range covered by the training data set for saturated esters of the SAR. There is a slight disagreement at the higher end of this range, where the SAR appears to underestimate the curvature observed in the experimental data. This behaviour is not unexpected, as SAR predictions tend to become less accurate towards the limits of their designated temperature ranges.
For all three methylated EB derivatives, the investigated temperature range was dominated by a negative temperature dependence (Fig. 5). The non-Arrhenius behaviour generally seems more pronounced for the methylated esters than for EB, especially so for EI and IB. In the predictions of the SAR by Jenkin et al. (2018) as well, the negative temperature dependence is clearly strongest for EI and IB, followed by EM (Fig. S4 in the Supplement). This is consistent with the observation that the room temperature reactivity, which in turn is driven by the feasibility of pre-reaction-complexes that are especially important at lower temperature, is highest for EI and IB. The site-specific reactivities predicted by the Jenkin et al. (2018) SAR support the idea that it is the H atoms attached to the β carbons which are largely responsible for the negative temperature dependence (Fig. S5 in the Supplement). It can be concluded that the order of applies to all of (i) the ability of the compound to form pre-reaction complexes, (ii) the magnitude of the non-Arrhenius behaviour, and (iii) the room temperature rate coefficient for the reaction with OH.
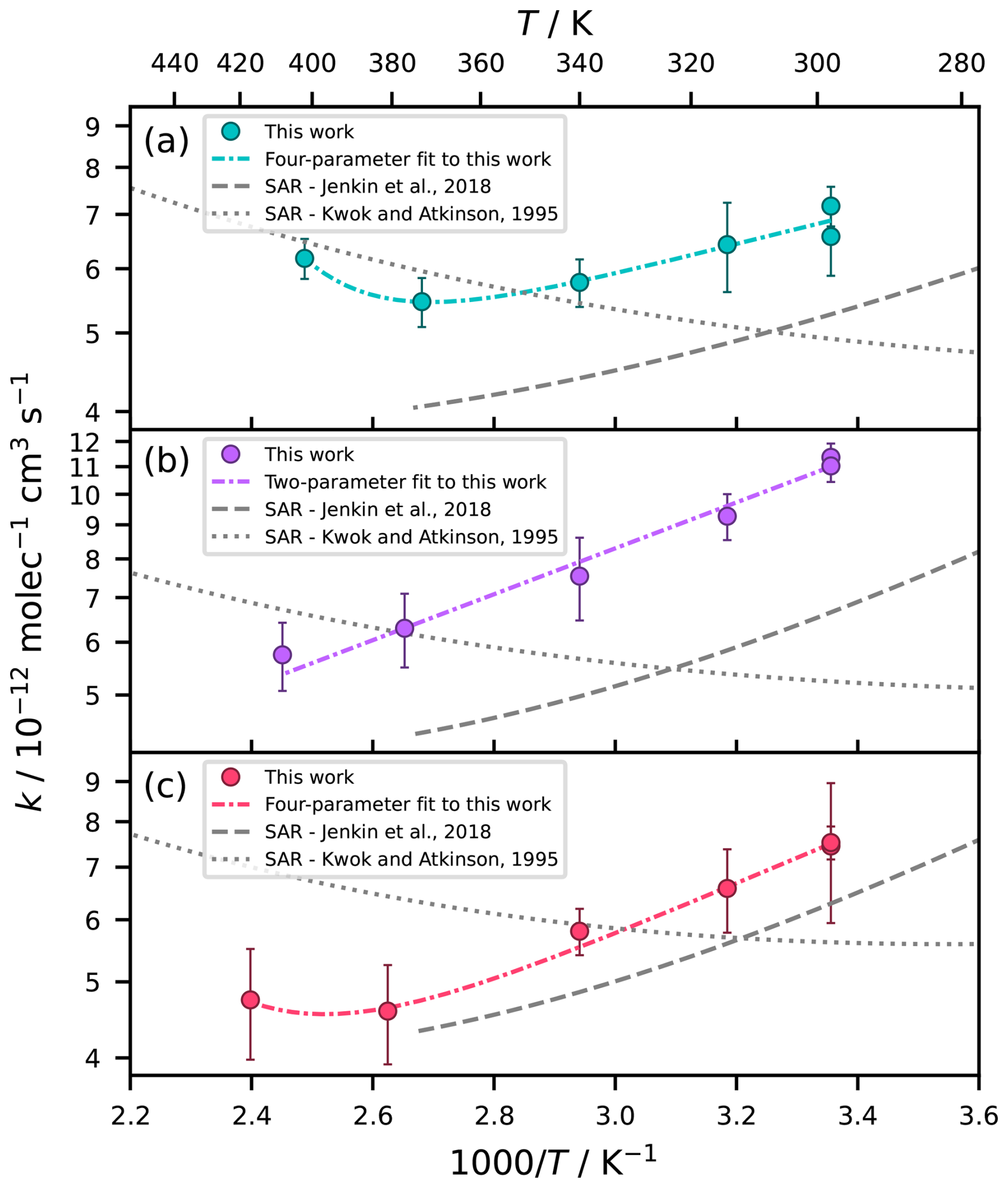
Figure 5Temperature-dependence of the reaction of (a) EM, (b) EI, and (c) IB with OH. Comparison of two SAR predictions and experimental data from this work, fitted with a four-parameter expression in case of EM and IB and a two-parameter expression in case of EI. Uncertainties are 2σ statistical errors.
Similar to EB, EM and IB show a U-shaped temperature dependence, with minima at 371 and 397 K respectively, and were therefore fitted with four-parameter expressions (units ), resulting in and . For EI, there is no clear minimum, and fitting a four-parameter expression resulted in two terms that each described a negative temperature dependence. Hence, we used a two-parameter expression for EI instead (unit ): . Considering that EI is the compound with the highest tendency to form pre-reaction complexes, it seems plausible that these complexes could be relevant over an extended temperature range, shifting the transition between the non-Arrhenius and Arrhenius regimes towards a higher temperature. Given the limited scope of the datasets for the methylated esters, we would like to emphasize that the reported temperature dependencies are only valid within their specified ranges and that any extrapolations may be subject to large uncertainty. This is particularly true at the high-temperature end, where more data would be needed to accurately describe the curvature.
Secondary isotope effects were investigated in a further series of PLP-LIF experiments including the Reaction (R7) between EB and deuterated hydroxyl radicals OD.
In particular, we evaluated the ratio of the rate coefficients for the reactions of EB with OD radicals and OH radicals respectively. This ratio, k7:k1, does not show a distinct temperature dependence over the range between 298 and 673 K, averaging to 1.09 (Fig. S6 in the Supplement). For the structurally related compound methyl formate, marginally faster reactions with OD radicals compared to OH radicals have been explained by the slightly lower activation barrier height, which in turn results from differences in the vibrational frequency modes (Robertson et al., 2024). While all k7:k1 datapoints in this work agree within their uncertainties, the data still hint at a possible increase of k7:k1 at decreasing temperature. This observation is consistent with previous studies on methyl formate (Robertson et al., 2024), methylglyoxal (Baeza-Romero et al., 2007), and limonene (Braure et al., 2014), and points at a kinetic isotope effect and a greater influence of the H or D atom of the radical than would occur in direct H abstraction. In turn, this would support the idea that H-bonded pre-reaction complexes involving the OH or OD radical (and/or addition reactions in case of unsaturated compounds) become increasingly relevant at lower temperatures. For T>387 K, i.e. over most of the temperature range for which we determined k7:k1, the kinetics of EB show Arrhenius behaviour and are hence dominated by direct H abstraction (Fig. 4). It can be hypothesized that at temperatures below room temperature where the contribution of pre-reaction complexes to the overall abstraction mechanism is higher, the ratio would increase further and reveal a stronger secondary kinetic isotope effect.
Finally, we investigated whether OH recycling occurs in the oxidation of EB. OH recycling following the initial photooxidation reaction is not only relevant for assessing the overall gas-phase chemistry mechanism and impact of a given compound (Lelieveld et al., 2008), but can also falsify PLP-LIF experiments that rely on observing the decay rate of OH. Substantial OH recycling causes the OH signal to become biexponential, which may however be difficult to resolve and instead appear as a slower monoexponential decay, such that fitting the signal without accounting for recycling would lead to an underestimation of the VOC + OH rate coefficients (Baeza-Romero et al., 2007; Potter et al., 2018, 2019). Generally, OH recycling proceeds via intermediate peroxy radicals and therefore requires the presence of oxygen (O2). To probe for OH recycling, we therefore evaluated the OH signal as a function of added O2 while the EB concentration remained constant. Since OH recycling is generally more likely to occur at high temperatures (Potter et al., 2018), we performed these experiments at 477 K and at 741 K (Fig. S7 in the Supplement). In both cases, the OH decay rate was not affected by the added O2 and proceeded monoexponentially with the same decay rate as in the absence of O2. Hence, OH recycling did not affect the experiments performed in this work and is negligible under all standard tropospheric or indoor conditions.
In this work we investigated the principal gas-phase removal processes for EB and its derivatives, including radical reactions and photolysis. In the visible and near-UV range, there was essentially no photon absorption and insufficient energy to break the ester functional group. Photolysis can therefore be disregarded at standard ground-level conditions or in typical indoor environments. Notably, EB and its derivatives show an absorption band from the forbidden transition which peaks at around 213 nm. Even at higher altitudes and under conditions of increased photon fluxes in the UV-C range, however, the calculated upper limit photolysis rates remain too low to be of significance. As such, while the photolysis quantum yields of EB and its derivatives are not known, they are of minor real-world relevance.
Reactions with OH, on the other hand, were rapid in the range of at room temperature. Under standard oxidative conditions of the troposphere, they are therefore likely the main sink. The room temperature rate coefficients decreased in the order . This hierarchy of reactivity was attributed to the enhancement of the reactivity of specific sites following methylation, notably when the sites are in β position to the carbonyl group and can hence facilitate the formation of pre-reaction complexes between the molecule and the radical. These pre-reaction complexes stabilize the transition state and therefore speed up the reaction. As they are especially prevalent at cold temperatures, they may explain the complex non-Arrhenius temperature dependencies observed for EB and its derivatives. Together with more conventional Arrhenius like behaviour at high temperatures, the result was a U-shaped temperature dependence, which was especially evident over the extended temperature range studied for EB.
The tropospheric lifetimes of EB and its derivatives were calculated solely against the reaction with OH radicals as their main loss process, using Eq. (7).
where τtotal is the total lifetime in s, τOH is the lifetime with regard to OH radical reactions, kOH is the rate coefficient of the reaction of the ester with OH in units of , and [OH] is the OH radical concentration in . We adopted an average tropospheric OH concentration of (Lelieveld et al., 2016), resulting in lifetimes in the range of 22–45 h (Table 3). These lifetimes are a rough estimate and may differ substantially depending on the conditions and depending on where and when the emission occurs. As OH radicals in the troposphere are mainly created photolytically and hence much more abundant during daytime, the actual lifetime can be much longer and dispersion much further if EB and its derivatives are released at nighttime. Furthermore, the lifetimes listed here are valid at room temperatures, but higher rate coefficients were found for all esters when the temperature decreases. Hence, the lifetimes could be shorter in cold environments, although this effect could be compensated by lower OH concentrations.
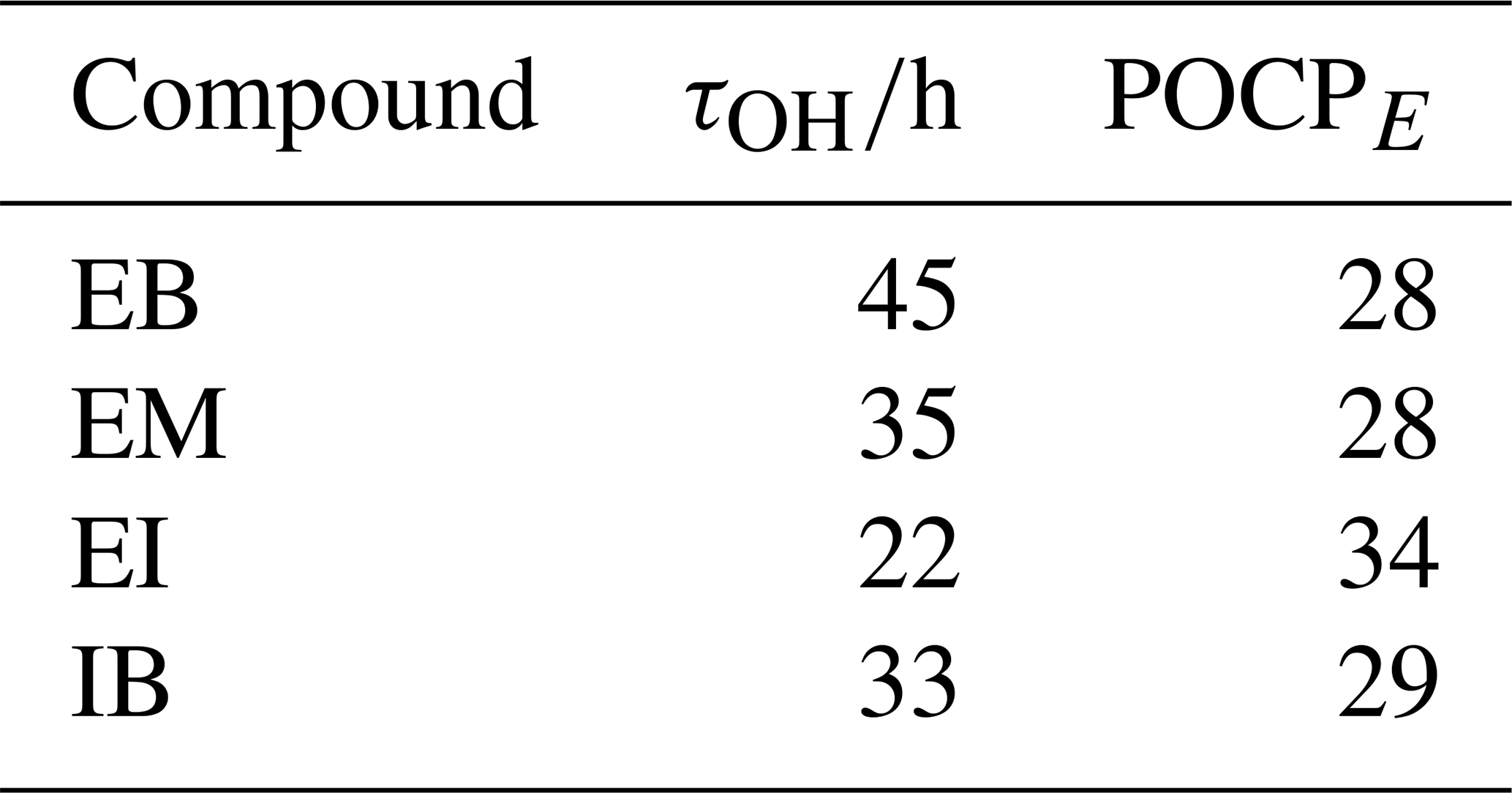
Table 3Lifetimes against the reaction with OH radicals (τOH) and corresponding estimated photochemical ozone creation potentials (POCPE) of EB and its derivatives. POCPE calculations were based on the method by Jenkin et al. (2017). Lifetimes were calculated assuming .
Importantly, the calculated lifetimes are an upper limit as they neglect other losses besides the reaction with OH. Further removal pathways of EB and its derivatives are possible, however, and chlorine atom reactions may be the next most relevant sink. At room temperature, these reactions have been found to be (Cometto et al., 2009), (Ramya and Rajakumar, 2019), (Ifang et al., 2015) for EB; for EM (Ifang et al., 2015); and for EI (Ifang et al., 2015). These rate coefficients are by factors of approx. 10–20 higher than the corresponding rate coefficients for the OH radical reactions determined in this work, which is in accordance with the general trend of VOCs being on average by about one order of magnitude more reactive towards Cl atoms than towards OH radicals (Young et al., 2014). Generally, chlorine atom concentrations in different environments are poorly characterized and highly uncertain. However, on average they tend to be several orders of magnitude lower than tropospheric OH radical concentrations in the troposphere, so that Cl atoms reactions typically have little influence on the overall lifetime under most conditions, even if the rate coefficients are by about one order of magnitude higher (Liang et al., 2010; Notario et al., 1998).
With respect to other potential atmospheric sinks, such as wet removal or reactions with NO3 radicals, it can be assumed that EB and its derivatives behave similarly to other aliphatic esters. The rate coefficients for the reactions of NO3 radicals with a range of different esters have been determined to be in the range of (upper limits) at room temperature, which is not fast enough to make a relevant contribution to the overall degradation rate of the esters but could influence night-time chemistry to some extent (Langer et al., 1993). In terms of wet removal, the high volatility and low water solubility of esters have prompted the conclusion that this mechanism is a negligible sink (Liang et al., 2010). Hence, approximating the overall lifetime by considering only OH radical reactions is reasonable in most settings, although chlorine atom reactions may be of relevance if there is a strong source.
It should be noted that the emission of the aroma compounds EB and its derivatives occurs mainly indoors, where their lifetime and fate depends strongly on the specific activities and settings. For instance, concentrations of OH tend to be about an order of magnitude lower than outdoors, but are also more variable and may peak following household activities such as cleaning or in proximity to windows (Weschler and Carslaw, 2018). Generally, however, the lifetime against the reaction with OH is likely to be significantly longer indoors compared to outdoor conditions. On the other hand, chlorine atom reactions may be highly relevant in cases when the usage of chlorinated products such as bleach releases a substantial amount of chlorinated species (Wong et al., 2017) which may react to form atomic Cl (Faxon and Allen, 2013; Ravishankara, 2009). In that case, Cl atom reactions could influence the overall lifetime, particularly if [Cl] is by less than two orders of magnitude smaller (or even higher) than [OH]. This becomes even more important because combinations of cleaning products co-emit chlorinated species and aroma compounds such as EB and its derivatives, which could result in a highly reactive mixture and fast formation of secondary products. In most other situations, however, air exchange rates are likely to outcompete reactive losses, so that ventilation to the outdoor surroundings would be the dominant fate.
In order to better characterize some of the outdoor air quality impacts that may follow from the reactions of EB and its derivatives, we calculated their estimated photochemical ozone creation potentials (POCPE) according to the methodology proposed by Jenkin et al. (2017). The resulting values of 28–34 (Table 3) are compared to structurally related compounds and other commonly used aroma compounds in Fig. 6. EA has a lower POCPE than EB and its derivatives, due to the smaller molecular size and lower number of oxidisable bonds. Meanwhile, all other compounds shown here for comparison have higher POCPEs than EB and its derivatives, mostly due to the possibility to react with ozone (ethene, dihydromyrcenol, limonene), substantially higher rate coefficients for the reaction with OH radicals (dihydromyrcenol, limonene), a higher number of oxidisable bonds (heptane, 4-heptanone, dihydromyrcenol, limonene), or the possibility for photolysis (4-heptanone). In terms of ozone formation, EB and its derivatives are hence of less concern than some other aroma compounds.
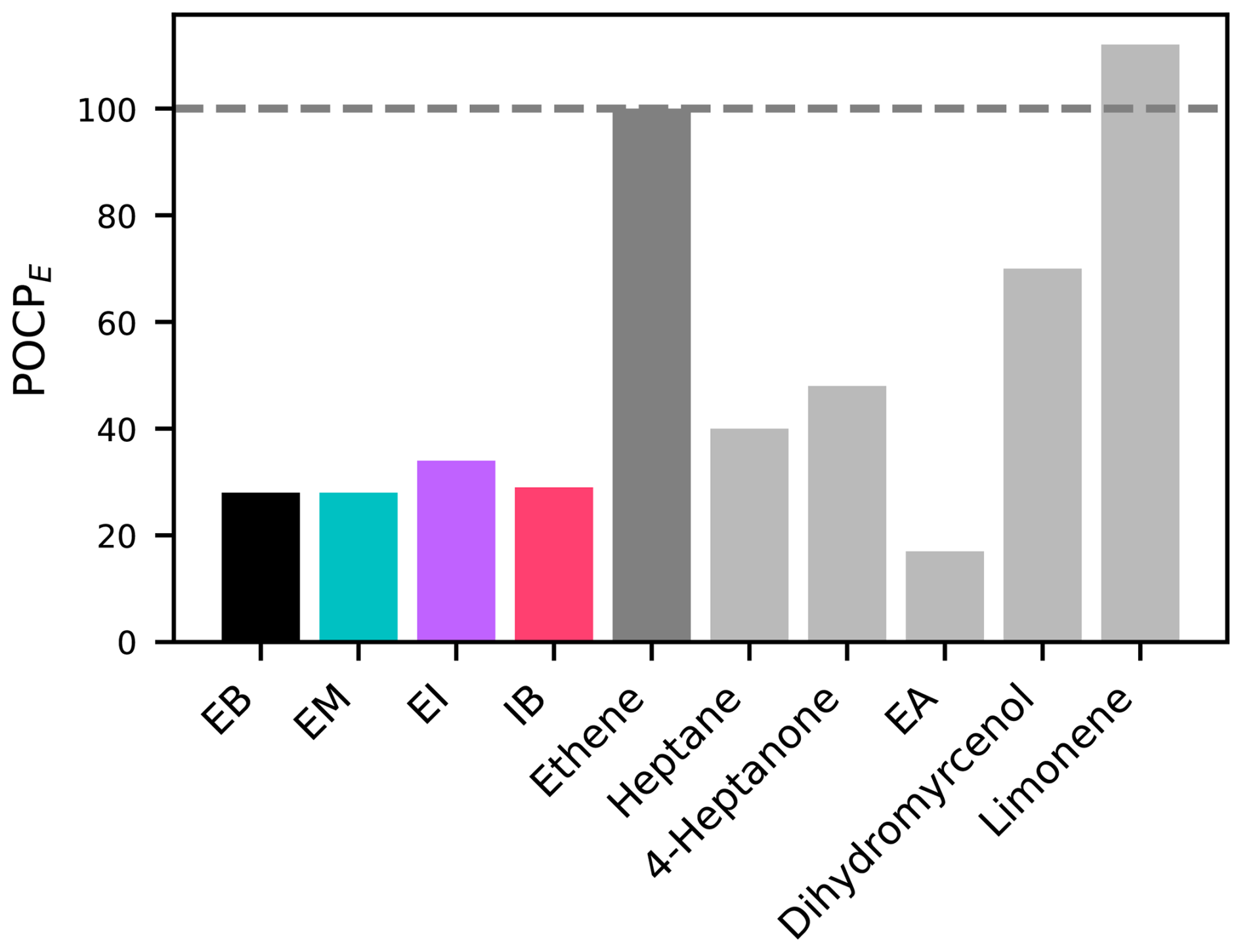
Figure 6Comparison of the POCPE of EB and its derivatives with ethene (POCP reference compound), structurally related compounds, and other commonly used aroma compounds.
However, the production of ground-level ozone represents only one dimension of air pollution. For a range of alkyl esters, Ifang et al. (2015) have pointed out that the formation of acidic products is likely to contribute substantially to the emergence of particulate matter, suggesting that aerosol formation may pose a greater environmental and health concern than ozone production. While detailed product studies – either experimental or computational – were beyond the scope of this work, we evaluated the first steps following the initial reaction of EB with OH at its most reactive sites using the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere, GECKO-A in short (Aumont et al., 2005). As illustrated in Fig. S8 in the Supplement, the products that are predicted to be formed after abstraction of one of the H atoms attached to either of the β carbons are multifunctional. As such, they are likely to have a low volatility and a high potential to form secondary organic aerosol (SOA), which ultimately impacts both indoor and outdoor air quality as well as radiative forcing (Chaturvedi et al., 2023; Hoyle et al., 2009; Mutzel et al., 2015). Additionally, some of the gaseous organic reaction products can induce direct adverse health effects, which has led to a call for a more explicit consideration of these products when assessing air quality impacts. Metrics such as the secondary product creation potential (SPCP) introduced by Carslaw and Shaw (2019) can help to better characterize the potential harm induced by a given VOC, but require knowledge of the formation rates of hazardous products, as well as knowledge of which VOCs are harmful to human health (data exist for relatively few VOCs at present). Hence, future studies focusing on reaction mechanisms and product yields, as well as on the toxicity and particle formation potential of the photooxidation products, would help to refine our understanding of the fate and impact of EB and its derivatives.
All underlying data are included in the Supplement. Any further information is available upon request from the corresponding authors.
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-9277-2026-supplement.
FL: investigation, formal analysis, visualization, writing (original draft preparation). MAB: investigation, resources, writing (review and editing). PWS: resources, writing (review and editing). NC: supervision, writing (review and editing). TJD: conceptualization, resources, supervision, writing (review and editing).
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
The authors would like to acknowledge technical support by Martyn Ward (GC-MS), Chris Anthony (atmospheric chemistry laboratories), Danny Shaw (physical chemistry laboratories, laser safety), and the University of York Chemistry Teaching Labs (UV-vis spectrometer). The Viking cluster was used during this project, which is a high performance compute facility provided by the University of York. We are grateful for computational support from the University of York, IT Services, and the Research IT team.
This work was supported by the Leeds-York-Hull Natural Environment Research Council (NERC) Doctoral Training Partnership (DTP) Panorama under grant no. NE/S007458/1.
This paper was edited by Eleanor Browne and reviewed by three anonymous referees.
Anderson, R. S., Czuba, E., Ernst, D., Huang, L., Thompson, A. E., and Rudolph, J.: Method for Measuring Carbon Kinetic Isotope Effects of Gas-Phase Reactions of Light Hydrocarbons with the Hydroxyl Radical, J. Phys. Chem. A, 107, 6191–6199, https://doi.org/10.1021/jp034256d, 2003.
Anhaia-Machado, J. O., Soares, A. C. G., de Oliveira Pinto, C. A. S., Barrera, A. I. Á., Baby, A. R., and Trossini, G. H. G.: Molecular Modeling Based on Time-Dependent Density Functional Theory (TD-DFT) Applied to the UV-Vis Spectra of Natural Compounds, Chemistry, 5, 41–53, https://doi.org/10.3390/chemistry5010004, 2023.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Ausloos, P.: The photolysis of alkyl esters, Can. J. Chem., 36, 383–392, https://doi.org/10.1139/v58-054, 1958.
Badawy, T., Williamson, J., and Xu, H.: Laminar burning characteristics of ethyl propionate, ethyl butyrate, ethyl acetate, gasoline and ethanol fuels, Fuel, 183, 627–640, https://doi.org/10.1016/j.fuel.2016.06.087, 2016.
Baeza-Romero, M. T., Glowacki, D. R., Blitz, M. A., Heard, D. E., Pilling, M. J., Rickard, A. R., and Seakins, P. W.: A combined experimental and theoretical study of the reaction between methylglyoxal and OH/OD radical: OH regeneration, Phys. Chem. Chem. Phys., 9, 4114–4128, https://doi.org/10.1039/B702916K, 2007.
Barba, C., Beno, N., Guichard, E., and Thomas-Danguin, T.: Selecting odorant compounds to enhance sweet flavor perception by gas chromatography/olfactometry-associated taste (GC/O-AT), Food Chem., 257, 172–181, https://doi.org/10.1016/j.foodchem.2018.02.152, 2018.
Bicas, J. L., Molina, G., Dionísio, A. P., Barros, F. F. C., Wagner, R., Maróstica, M. R., and Pastore, G. M.: Volatile constituents of exotic fruits from Brazil, Food Res. Int., 44, 1843–1855, https://doi.org/10.1016/j.foodres.2011.01.012, 2011.
Blitz, M. A., Guy, P., Shannon, R., and Seakins, P. W.: Rate Coefficients and Branching Ratios for the Reaction of the OH Radical with Formic Acid under Low-Temperature Combustion Conditions and the Fate of the HOCO Product, J. Phys. Chem. A, 129, 6437–6450, https://doi.org/10.1021/acs.jpca.5c01814, 2025.
Boelens, H., Haring, H. G., and Takken, H. J.: The Influence of Molecular-Structure on Olfactive Quality (a Quantitative Approach), Chem. Ind., 1, 26–30, 1983.
Braure, T., Bedjanian, Y., Romanias, M. N., Morin, J., Riffault, V., Tomas, A., and Coddeville, P.: Experimental Study of the Reactions of Limonene with OH and OD Radicals: Kinetics and Products, J. Phys. Chem. A, 118, 9482–9490, https://doi.org/10.1021/jp507180g, 2014.
Calvert, J., Mellouki, A., Orlando, J., Pilling, M., and Wallington, T.: Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press, Oxford, New York, 1634 pp., ISBN 978-0-19-976707-6, 2011.
Carslaw, N. and Shaw, D.: Secondary product creation potential (SPCP): a metric for assessing the potential impact of indoor air pollution on human health, Environ. Sci. Process. Impacts, 21, 1313–1322, https://doi.org/10.1039/C9EM00140A, 2019.
Caswell, L. R., Howard, M. F., and Onisto, T. M.: Solvent and substituent effects upon the transition of aliphatic carboxylic acids and esters, J. Org. Chem., 41, 3312–3316, https://doi.org/10.1021/jo00882a023, 1976.
Chaturvedi, S., Kumar, A., Singh, V., Chakraborty, B., Kumar, R., and Min, L.: Recent Advancement in Organic Aerosol Understanding: a Review of Their Sources, Formation, and Health Impacts, Water Air Soil Poll., 234, 750, https://doi.org/10.1007/s11270-023-06772-0, 2023.
Chen, I.-Y., Chang, C.-W., Fittschen, C., and Luo, P.-L.: Accurate Kinetic Studies of OH+HO2 Radical–Radical Reaction through Direct Measurement of Precursor and Radical Concentrations with High-Resolution Time-Resolved Dual-Comb Spectroscopy, J. Phys. Chem. Lett., 15, 3733–3739, https://doi.org/10.1021/acs.jpclett.4c00494, 2024.
Closson, W. D. and Haug, Pat.: The Effects of Solvent and Structure on the Low Intensity () Electronic Transition of Carboxylate Esters, J. Am. Chem. Soc., 86, 2384–2389, https://doi.org/10.1021/ja01066a018, 1964.
Cometto, P. M., Daële, V., Idir, M., Lane, S. I., and Mellouki, A.: Reaction Rate Coefficients of OH Radicals and Cl Atoms with Ethyl Propanoate, n-Propyl Propanoate, Methyl 2-Methylpropanoate, and Ethyl n-Butanoate, J. Phys. Chem. A, 113, 10745–10752, https://doi.org/10.1021/jp9061708, 2009.
Doner, A. C., Christianson, M. G., Davis, J. C., Koritzke, A. L., Larsson, A., Frandsen, K., and Rotavera, B.: Vacuum-ultraviolet absorption cross-sections of functionalized cyclic hydrocarbons: Six-membered rings, J. Quant. Spectrosc. Ra., 236, 106603, https://doi.org/10.1016/j.jqsrt.2019.106603, 2019.
D'Souza Metcalf, J., Winkless, R. K., Robinson, A., Smith, S. C., Rickard, A. R., and Dillon, T. J.: Practical atmospheric photochemical kinetics for undergraduate teaching and research, RSC Sustain., 3, 5146–5154, https://doi.org/10.1039/D5SU00681C, 2025.
El-Nahas, A. M., Navarro, M. V., Simmie, J. M., Bozzelli, J. W., Curran, H. J., Dooley, S., and Metcalfe, W.: Enthalpies of Formation, Bond Dissociation Energies and Reaction Paths for the Decomposition of Model Biofuels: Ethyl Propanoate and Methyl Butanoate, J. Phys. Chem. A, 111, 3727–3739, https://doi.org/10.1021/jp067413s, 2007.
El-Nahas, A. M., Heikal, L. A., Mangood, A. H., and El-Shereefy, E.-S. E.: Structures and Energetics of Unimolecular Thermal Degradation of Isopropyl Butanoate as a Model Biofuel: Density Functional Theory and Ab Initio Studies, J. Phys. Chem. A, 114, 7996–8002, https://doi.org/10.1021/jp103397f, 2010.
Faxon, C. B. and Allen, D. T.: Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 10, 221–233, https://doi.org/10.1071/EN13026, 2013.
Ferrari, C., Roche, A., Jacob, V., Foster, P., and Baussand, P.: Kinetics of the reaction of OH radicals with a series of esters under simulated conditions at 295 K, Int. J. Chem. Kinet., 28, 609–614, https://doi.org/10.1002/(SICI)1097-4601(1996)28:8<609::AID-KIN6>3.0.CO;2-Z, 1996.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, J. A., Jr., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 16, Revision C.02, Gaussian Inc., Wallingford CT, https://gaussian.com/gaussian16/ (last access: 30 June 2026), 2016.
Galano, A. and Raúl Alvarez-Idaboy, J.: Atmospheric Reactions of Oxygenated Volatile Organic Compounds + OH Radicals: Role of Hydrogen-Bonded Intermediates and Transition States, in: Advances in Quantum Chemistry, edited by: Goodsite, M. E. and Johnson, M. S., vol. 55, Academic Press, 245–274, https://doi.org/10.1016/S0065-3276(07)00212-2, 2008.
Gour, N. K., Deka, R. C., Singh, H. J., and Mishra, B. K.: Theoretical study on the gas-phase reactions of ethyl butyrate with OH radicals at 298 K, Monatsh. Chem., 145, 1759–1767, https://doi.org/10.1007/s00706-014-1255-0, 2014.
Greiner, N. R.: Hydroxyl Radical Kinetics by Kinetic Spectroscopy, VI. Reactions with Alkanes in the Range 300–500° K, J. Chem. Phys., 53, 1070–1076, https://doi.org/10.1063/1.1674099, 1970.
Hong, Z., Cook, R. D., Davidson, D. F., and Hanson, R. K.: A Shock Tube Study of and using Laser Absorption of H2O and OH, J. Phys. Chem. A, 114, 5718–5727, https://doi.org/10.1021/jp100204z, 2010.
Hoyle, C. R., Myhre, G., Berntsen, T. K., and Isaksen, I. S. A.: Anthropogenic influence on SOA and the resulting radiative forcing, Atmos. Chem. Phys., 9, 2715–2728, https://doi.org/10.5194/acp-9-2715-2009, 2009.
Ifang, S., Benter, T., and Barnes, I.: Reactions of Cl atoms with alkyl esters: kinetic, mechanism and atmospheric implications, Environ. Sci. Pollut. R., 22, 4820–4832, https://doi.org/10.1007/s11356-014-2913-9, 2015.
Jenkin, M. E., Derwent, R. G., and Wallington, T. J.: Photochemical ozone creation potentials for volatile organic compounds: Rationalization and estimation, Atmos. Environ., 163, 128–137, https://doi.org/10.1016/j.atmosenv.2017.05.024, 2017.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018.
Kim, Y.-H., Kim, K.-H., Szulejko, J. E., and Parker, D.: Quantitative Analysis of Fragrance and Odorants Released from Fresh and Decaying Strawberries, Sensors, 13, 7939–7978, https://doi.org/10.3390/s130607939, 2013.
Klepeis, N. E., Nelson, W. C., Ott, W. R., Robinson, J. P., Tsang, A. M., Switzer, P., Behar, J. V., Hern, S. C., and Engelmann, W. H.: The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants, J. Expo. Sci. Env. Epid., 11, 231–252, https://doi.org/10.1038/sj.jea.7500165, 2001.
Knothe, G. and Steidley, K. R.: Fatty Acid Alkyl Esters as Solvents: Evaluation of the Kauri-Butanol Value. Comparison to Hydrocarbons, Dimethyl Diesters, and Other Oxygenates, Ind. Eng. Chem. Res., 50, 4177–4182, https://doi.org/10.1021/ie1023172, 2011.
Kraft, P., Bajgrowicz, J. A., Denis, C., and Fráter, G.: Odds and Trends: Recent Developments in the Chemistry of Odorants, Angew. Chem. Int. Edit., 39, 2980–3010, https://doi.org/10.1002/1521-3773(20000901)39:17%3C2980::AID-ANIE2980%3E3.0.CO;2-%23, 2000.
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995.
Labbe, D., Rytz, A., Morgenegg, C., Ali, S., and Martin, N.: Subthreshold Olfactory Stimulation Can Enhance Sweetness, Chem. Senses, 32, 205–214, https://doi.org/10.1093/chemse/bjl040, 2007.
Langer, S., Ljungström, E., and Wängberg, I.: Rates of reaction between the nitrate radical and some aliphatic esters, J. Chem. Soc. Farad. T., 89, 425–431, https://doi.org/10.1039/FT9938900425, 1993.
Lary, D. J. and Shallcross, D. E.: Central role of carbonyl compounds in atmospheric chemistry, J. Geophys. Res.-Atmos., 105, 19771–19778, https://doi.org/10.1029/1999JD901184, 2000.
Lelieveld, J., Dentener, F. J., Peters, W., and Krol, M. C.: On the role of hydroxyl radicals in the self-cleansing capacity of the troposphere, Atmos. Chem. Phys., 4, 2337–2344, https://doi.org/10.5194/acp-4-2337-2004, 2004.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Liang, P., Mu, Y., Daële, V., and Mellouki, A.: Rate Coefficients for Reactions of OH and Cl with Esters, ChemPhysChem, 11, 4097–4102, https://doi.org/10.1002/cphc.201000262, 2010.
Link, M. F., Shore, A., Hamadani, B. H., and Poppendieck, D.: Ozone Generation from a Germicidal Ultraviolet Lamp with Peak Emission at 222 nm, Environ. Sci. Tech. Let., 10, 675–679, https://doi.org/10.1021/acs.estlett.3c00318, 2023.
Ludwig, W., Brandt, B., Friedrichs, G., and Temps, F.: Kinetics of the Reaction C2H5+HO2 by Time-Resolved Mass Spectrometry, J. Phys. Chem. A, 110, 3330–3337, https://doi.org/10.1021/jp0557464, 2006.
Mapelli, C., Schleicher, J. V., Hawtin, A., Rankine, C. D., Whiting, F. C., Byrne, F., McElroy, C. R., Roman, C., Arsene, C., Olariu, R. I., Bejan, I. G., and Dillon, T. J.: Atmospheric breakdown chemistry of the new “green” solvent 2,2,5,5-tetramethyloxolane via gas-phase reactions with OH and Cl radicals, Atmos. Chem. Phys., 22, 14589–14602, https://doi.org/10.5194/acp-22-14589-2022, 2022.
Mapelli, C., Donnelly, J. K., Hogan, Ú. E., Rickard, A. R., Robinson, A. T., Byrne, F., McElroy, C. R., Curchod, B. F. E., Hollas, D., and Dillon, T. J.: Atmospheric oxidation of new “green” solvents – Part 2: methyl pivalate and pinacolone, Atmos. Chem. Phys., 23, 7767–7779, https://doi.org/10.5194/acp-23-7767-2023, 2023.
McMillan, V.: Photochemistry, personal communication to J. G. Calvert, J. N. Pitts Jr., John Wiley & Sons, New York, p. 429, 1966.
Mellouki, A., Le Bras, G., and Sidebottom, H.: Kinetics and Mechanisms of the Oxidation of Oxygenated Organic Compounds in the Gas Phase, Chem. Rev., 103, 5077–5096, https://doi.org/10.1021/cr020526x, 2003.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M., Böge, O., Richters, S., Spindler, G., Sipilä, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Naito, K., Sawadaishi, K., and Kawasaki, M.: Photobiochemical mechanisms of biomolecules relevant to germicidal ultraviolet irradiation at 222 and 254 nm, Sci. Rep., 12, 18217, https://doi.org/10.1038/s41598-022-22969-5, 2022.
Nakashima, K., Uchida-Kai, K., Koyanagi, M., and Kanda, Y.: Solvent Effects on the Intensities of Forbidden Bands of Molecules. Absorption Spectra of Acetone and Cyclopentanone, B. Chem. Soc. Jpn., 55, 415–419, https://doi.org/10.1246/bcsj.55.415, 1982.
NCAR: Tropospheric Ultraviolet and Visible (TUV) Radiation Model by the National Center for Atmospheric Research (NCAR), v5.3, web version of model, https://www.acom.ucar.edu/Models/TUV/Interactive_TUV/ (last access: 28 Nov 2025), 2025.
Nematollahi, N., Doronila, A., Mornane, P. J., Duan, A., Kolev, S. D., and Steinemann, A.: Volatile chemical emissions from fragranced baby products, Air Qual. Atmos. Hlth., 11, 785–790, https://doi.org/10.1007/s11869-018-0593-1, 2018.
Notario, A., Le Bras, G., and Mellouki, A.: Absolute Rate Constants for the Reactions of Cl Atoms with a Series of Esters, J. Phys. Chem. A, 102, 3112–3117, https://doi.org/10.1021/jp980416n, 1998.
Onel, L., Blitz, M. A., and Seakins, P. W.: Direct Determination of the Rate Coefficient for the Reaction of OH Radicals with Monoethanol Amine (MEA) from 296–510 K, J. Phys. Chem. Lett., 3, 853–856, https://doi.org/10.1021/jz300200c, 2012.
Pandey, N., Mehata, M. S., Pant, S., and Tewari, N.: Structural, Electronic and NLO Properties of 6-aminoquinoline: A DFT/TD-DFT Study, J. Fluoresc., 31, 1719–1729, https://doi.org/10.1007/s10895-021-02788-z, 2021.
Peng, Z., Day, D. A., Symonds, G. A., Jenks, O. J., Stark, H., Handschy, A. V., de Gouw, J. A., and Jimenez, J. L.: Significant Production of Ozone from Germicidal UV Lights at 222 nm, Environ. Sci. Tech. Let., 10, 668–674, https://doi.org/10.1021/acs.estlett.3c00314, 2023.
Potter, D. G., Wiseman, S., Blitz, M. A., and Seakins, P. W.: Laser Photolysis Kinetic Study of OH Radical Reactions with Methyl tert-Butyl Ether and Trimethyl Orthoformate under Conditions Relevant to Low Temperature Combustion: Measurements of Rate Coefficients and OH Recycling, J. Phys. Chem. A, 122, 9701–9711, https://doi.org/10.1021/acs.jpca.8b09122, 2018.
Potter, D. G., Blitz, M. A., and Seakins, P. W.: A generic method for determining R+O2 rate parameters via OH regeneration, Chem. Phys. Lett., 730, 213–219, https://doi.org/10.1016/j.cplett.2019.06.014, 2019.
Ramya, C. B. and Rajakumar, B.: Cl atom initiated tropospheric chemistry of ethyl butyrate, Chem. Phys. Lett., 731, 136594, https://doi.org/10.1016/j.cplett.2019.136594, 2019.
Rao, P. D., Husile, N., Strasser, A. A., and Wise, P. M.: Pilot Experiment: the Effect of Added Flavorants on the Taste and Pleasantness of Mixtures of Glycerol and Propylene Glycol, Chemosens. Percept., 11, 1–9, https://doi.org/10.1007/s12078-017-9231-9, 2018.
Ravishankara, A. R.: Are chlorine atoms significant tropospheric free radicals?, P. Natl. Acad. Sci. USA, 106, 13639–13640, https://doi.org/10.1073/pnas.0907089106, 2009.
Robertson, N. C. K., Onel, L., Blitz, M. A., Shannon, R., Stone, D., Seakins, P. W., Robertson, S. H., Kühn, C., Pazdera, T. M., and Olzmann, M.: Temperature-Dependent, Site-Specific Rate Coefficients for the Reaction of OH (OD) with Methyl Formate Isotopologues via Experimental and Theoretical Studies, J. Phys. Chem. A, 128, 5028–5040, https://doi.org/10.1021/acs.jpca.4c02524, 2024.
Shannon, R. J., Caravan, R. L., Blitz, M. A., and Heard, D. E.: A combined experimental and theoretical study of reactions between the hydroxyl radical and oxygenated hydrocarbons relevant to astrochemical environments, Phys. Chem. Chem. Phys., 16, 3466–3478, https://doi.org/10.1039/C3CP54664K, 2014.
Shaw, J. T., Rickard, A. R., Newland, M. J., and Dillon, T. J.: Rate coefficients for reactions of OH with aromatic and aliphatic volatile organic compounds determined by the multivariate relative rate technique, Atmos. Chem. Phys., 20, 9725–9736, https://doi.org/10.5194/acp-20-9725-2020, 2020.
Śmialek, M. A., Łabuda, M., Guthmuller, J., Hubin-Franskin, M.-J., Delwiche, J., Hoffmann, S. V., Jones, N. C., Mason, N. J., and Limão-Vieira, P.: Electronic state spectroscopy by high-resolution vacuum ultraviolet photoabsorption, He(I) photoelectron spectroscopy and ab initio calculations of ethyl acetate, Eur. Phys. J. D, 70, 138, https://doi.org/10.1140/epjd/e2016-70239-9, 2016.
Sosa-Moguel, O., Pino, J. A., Sauri-Duch, E., and Cuevas-Glory, L.: Characterization of odor-active compounds in three varieties of ciruela (Spondias purpurea L.) fruit, Int. J. Food Prop., 21, 1008–1016, https://doi.org/10.1080/10942912.2018.1479858, 2018.
Tang, X., Zhang, Y., Wang, L., Sun, C., Wang, L., and Huang, Z.: Excited-state antioxidant capacity of Flavonoids based on solvent effect: A TD-DFT study, J. Mol. Liq., 404, 124973, https://doi.org/10.1016/j.molliq.2024.124973, 2024.
The Good Scents Company Information System: https://www.thegoodscentscompany.com/, last access: 20 January 2026.
Uhde, E. and Schulz, N.: Impact of room fragrance products on indoor air quality, Atmos. Environ., 106, 492–502, https://doi.org/10.1016/j.atmosenv.2014.11.020, 2015.
US Environmental Protection Agency: EPI Suite™ Version 1.0.1-beta, Module MPBPVP™, https://episuite.dev/#/ (last access: 25 January 2026), 2026.
Wallington, T. J., Dagaut, P., Liu, R., and Kurylo, M. J.: The gas phase reactions of hydroxyl radicals with a series of esters over the temperature range 240–440 K, Int. J. Chem. Kinet., 20, 177–186, https://doi.org/10.1002/kin.550200210, 1988.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Weschler, C. J. and Carslaw, N.: Indoor Chemistry, Environ. Sci. Technol., 52, 2419–2428, https://doi.org/10.1021/acs.est.7b06387, 2018.
Williams, D. C., O'Rji, L. N., and Stone, D. A.: Kinetics of the reactions of OH radicals with selected acetates and other esters under simulated atmospheric conditions, Int. J. Chem. Kinet., 25, 539–548, https://doi.org/10.1002/kin.550250704, 1993.
Wolkoff, P., Clausen, P. A., Jensen, B., Nielsen, G. D., and Wilkins, C. K.: Are We Measuring the Relevant Indoor Pollutants?, Indoor Air, 7, 92–106, https://doi.org/10.1111/j.1600-0668.1997.t01-2-00003.x, 1997.
Wollenhaupt, M., Carl, S. A., Horowitz, A., and Crowley, J. N.: Rate Coefficients for Reaction of OH with Acetone between 202 and 395 K, J. Phys. Chem. A, 104, 2695–2705, https://doi.org/10.1021/jp993738f, 2000.
Wong, J. P. S., Carslaw, N., Zhao, R., Zhou, S., and Abbatt, J. P. D.: Observations and impacts of bleach washing on indoor chlorine chemistry, Indoor Air, 27, 1082–1090, https://doi.org/10.1111/ina.12402, 2017.
Xiao, Z., Zhang, S., Zhu, J., Niu, Y., Xiong, W., and Chen, F.: Identification of key aromas of grapefruit juice and study of their contributions to the enhancement of sweetness perception, Eur. Food Res. Technol., 249, 537–551, https://doi.org/10.1007/s00217-022-04151-3, 2023.
Xiao, Z., Hu, Y., Niu, Y., Zhang, J., and Yang, B.: Five representative esters and aldehydes from fruits can enhance sweet perception, LWT, 194, 115804, https://doi.org/10.1016/j.lwt.2024.115804, 2024.
Xu, Y., Minhazul, K. A. H. M., and Li, X.: The occurrence, enzymatic production, and application of ethyl butanoate, an important flavor constituent, Flavour Frag. J., 35, 601–615, https://doi.org/10.1002/ffj.3613, 2020.
Yeoman, A. M., Shaw, M., Carslaw, N., Murrells, T., Passant, N., and Lewis, A. C.: Simplified speciation and atmospheric volatile organic compound emission rates from non-aerosol personal care products, Indoor Air, 30, 459–472, https://doi.org/10.1111/ina.12652, 2020.
Young, C. J., Washenfelder, R. A., Edwards, P. M., Parrish, D. D., Gilman, J. B., Kuster, W. C., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., Roberts, J. M., Griffith, S., Dusanter, S., Stevens, P. S., Flynn, J., Grossberg, N., Lefer, B., Holloway, J. S., Peischl, J., Ryerson, T. B., Atlas, E. L., Blake, D. R., and Brown, S. S.: Chlorine as a primary radical: evaluation of methods to understand its role in initiation of oxidative cycles, Atmos. Chem. Phys., 14, 3427–3440, https://doi.org/10.5194/acp-14-3427-2014, 2014.
Zhang, H., Kang, S., Li, J., Li, Z., Chang, J., Xu, Y., Max Lu, G., and Sun, C.: Near zero-waste biofuel production from bioderived polyhydroxybutyrate, Fuel, 286, 119405, https://doi.org/10.1016/j.fuel.2020.119405, 2021.