the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 24 Jun 2026
| 24 Jun 2026
Influence of tropospheric temperature on the formation and aging of secondary organic aerosol from biogenic vapor mixtures
Stella E. I. Manavi
Claudia Mohr
Junwei Song
Thomas Leisner
Spyros N. Pandis
Atmospheric temperature and composition variations significantly influence secondary organic aerosol (SOA) formation and aging, and thus fine particulate matter levels and properties relevant for climate, air quality, and human health. However, the temperature dependence of SOA formation and aging from mixed volatile organic compounds (VOCs) remains insufficiently understood. Therefore, we investigated SOA formation from the oxidation of isoprene and α-pinene mixtures covering the range of tropospheric temperatures (213–313 K). We further examine the aging of the resulting SOA by gradually warming to mimic their atmospheric transport and diurnal aging processes. Notably, at 213 K, isoprene most strongly suppresses α-pinene dimer (C18–20) formation, with isoprene-α-pinene cross dimers appearing 3.5 times more frequently than at 273 K, while the suppression is not temperature-sensitive above 273 K. Upon subsequent warming, particles formed at different temperature ranges undergo distinct aging processes including aerosol evaporation and water uptake. Surprisingly, particles formed at higher temperatures are more oxidized yet more volatile than those formed at lower temperatures and subsequently warmed. Chemical transport modeling accounting for temperature-depended simultaneous oxidation of isoprene and α-pinene predicts higher SOA levels across Europe, aligning more closely with observations. These findings highlight the need to consider both temperature and the interaction of biogenic VOCs to accurately describe SOA formation, aging, and global burden.
- Article
(4773 KB) - Full-text XML
-
Supplement
(2676 KB) - BibTeX
- EndNote




Aerosol particles are ubiquitous in the atmosphere, significantly impacting climate and having adverse effects on air quality and human health (Paasonen et al., 2013; Mahowald, 2011; Aubry et al., 2021). Organic aerosol (OA) makes up 20 %–90 % of the total fine particulate mass in the troposphere (Jimenez et al., 2009). An important contributor to the global OA burden is secondary organic aerosol (SOA), which emerges from the condensation of organic compounds formed by the oxidation of volatile organic compounds (VOCs) (Kroll and Seinfeld, 2008). Generally, the key precursors for global SOA are biogenic VOCs, of which isoprene (C5H8) and monoterpenes (C10H16) are the most abundant (Kanakidou et al., 2005). Consequently, large efforts (Kanakidou et al., 2005; Carlton et al., 2009; Kroll and Seinfeld, 2008; Zhang et al., 2015; Hallquist et al., 2009; Lopez-Hilfiker et al., 2014; McFiggans et al., 2019; Takeuchi et al., 2022) have been put into investigating their formation chemistry and particle physicochemical properties of biogenic SOA.
Most of these studies were done at or near room temperature (Zhang et al., 2015; Lopez-Hilfiker et al., 2015; Kourtchev et al., 2015; Takeuchi et al., 2022; McFiggans et al., 2019). The troposphere however covers a wide temperature range between 310 to 200 K. In the near-surface atmosphere, VOCs can be oxidized at varying ambient temperatures throughout the day, depending on the season and region. By convective systems, VOCs could reach to higher altitudes where they can be oxidized at lower temperatures (Schulz et al., 2018; Liu et al., 2023). This is important for the prediction of SOA levels especially in the free troposphere. Temperature affects the reaction rates and pathways (Bilde et al., 2015; Bianchi et al., 2019) of VOCs oxidation as well as the gas-to-particle partitioning of oxidation products (Sheehan and Bowman, 2001; Donahue et al., 2006; Jonsson et al., 2008; Simon et al., 2020), thereby altering the formation, chemical composition, and physicochemical properties of aerosol particles. Thus, developing a comprehensive study covering tropospheric conditions is essential for understanding SOA formation and aging processes in the real atmosphere.
Isoprene makes up the largest portion of the global biogenic VOC emissions (Owen et al., 2003; Sindelarova et al., 2014), making it an important SOA precursor despite its relatively low individual mass yield of < 5 % (Xu et al., 2014; Lamkaddam et al., 2021; Carlton et al., 2009). Previously, Kiendler-Scharr et al. (2009) and McFiggans et al. (2019) found that at room temperature, the presence of isoprene reduces SOA formation from the oxidation of α-pinene. This is due to the competition of isoprene and α-pinene for reacting with hydroxyl radicals (OH) (McFiggans et al., 2019) and the formation of more volatile C15 dimers from the reaction of C10 peroxy radicals (RO2) of α-pinene and C5 RO2 of isoprene, instead of less volatile C20 dimers from self-reactions of C10 RO2 from α-pinene alone. However, the temperature dependence of RO2 cross reactions in the isoprene and α-pinene systems as well as the effects of temperature changes on SOA aging during atmospheric processes such as transport and diurnal aging remains to be fully understood. This knowledge gap is critical given the varying atmospheric abundances of these compounds across different ecosystems. For instance, in the Amazonian rainforest, summertime isoprene mixing ratios range from 0.1 to 20 ppb (Yáñez-Serrano et al., 2020; Yáñez-Serrano et al., 2018), while monoterpenes are typically below 1 ppb but can reach up to 5.5 ppb. In contrast, European forests exhibit lower isoprene levels, typically below 1 ppb but reaching up to ∼5 ppb during warm daytime periods (Li et al., 2021; Petersen et al., 2023). Monoterpene concentrations in these forests are also generally below 1 ppb but can reach several tenths of ppb during summer across a typical temperature range of 10–35 °C (Li et al., 2021). Therefore, investigating the impact of temperature on the oxidation of isoprene and α-pinene mixtures at atmospherically relevant concentrations is essential to accurately predict SOA formation in diverse environmental conditions (Tripathi et al., 2025; Curtius et al., 2024).
We thoroughly investigated the temperature-dependent formation and the properties of SOA from the oxidation of the mixture of isoprene and α-pinene at 213 K (SOA213 K), 243 K (SOA243 K), 273 K (SOA273 K), 298 K (SOA298 K), and 313 K (SOA313 K). The SOA formed at each temperature was subsequently warmed with increments of 15–30 K over 10 h to investigate the aging processes (e.g., diurnal cycle) of SOA over a wider tropospheric temperature range (i.e., SOA213→243 K, SOA243→273 K, SOA273→298 K, and SOA298→313 K). A series of cross dimers from the two precursor VOCs were identified by making use of carbon isotope (13C) labelling experiments, as well as by comparison with the sole α-pinene oxidation experiment. We demonstrated the effect of temperature on the suppression of α-pinene dimers by isoprene and the formation of two-precursor cross dimers. By studying the effect of warming on aged particles, we distinguished the impact of temperature on both the chemistry and phase partitioning of organic molecules and provided evidence that particles at different temperature ranges undergo distinct aging processes (i.e., evaporation and water uptake) during warming.
2.1 Simulation Chamber Experiments
The data presented here was measured in two campaigns in 2019 (SOA19b) and 2021 (SOA21a) covering 213–313 K in the Aerosol Interaction and Dynamics in the Atmosphere (AIDA) aerosol and cloud simulation chamber at the Karlsruhe Institute of Technology (KIT). The chamber is an 84.5 m3 aluminium vessel equipped with a LED solar radiation simulator and with precisely controlled temperature, humidity, and gas mixtures. A fan allows all components to be mixed well within 90 s (Saathoff et al., 2009). Details about the AIDA chamber are given by previous studies (Möhler et al., 2003; Vallon et al., 2022; Wagner et al., 2006).
Two types of SOA were generated in batch mode from dark oxidation of: (1) sole α-pinene at 273 K (SOAap-273), (2) isoprene mixed with α-pinene at 213, 243, 273, 298, and 313 K (SOA213 K, SOA243 K, SOA273 K, SOA298 K, SOA313 K), respectively. The experimental conditions are summarized in Table 1. Well defined amounts of isoprene and α-pinene were added to the AIDA chamber with a flow of 10 L min−1 of synthetic air. Ozone was injected subsequently after the biogenic VOC were mixed well inside the chamber, followed by the continuous addition of tetramethyl ethylene (TME) generating OH radicals by its reaction with ozone. The OH concentrations were (0.8–1.5) × 107 molecules cm−3 in all experiments. The initial concentration ratios of isoprene to α-pinene were kept at 1.0±0.1 for all two-precursor experiments, while the ratios of O3 to α-pinene were 14±3 among all experiments with the exception of Exp 1 at 213 K (O3: α-pinene = 38). At 213 K, the initial concentrations of isoprene and α-pinene of 6.7 ppb led to a relatively small amount of SOA mass. To generate sufficient SOA mass for the longer warming experiment we generated more SOA mass in a second oxidation step with about twice the VOC concentrations of 13.5 ppb. We note that the two times of injections of precursors may have impact on the chemical regimes during the SOA formation at 213 K compared to other experiments. Seed particles and OH scavengers were not used in this work. Additionally, to investigate the cross-dimers formed from the oxidation of α-pinene and isoprene, we used 13C-labelled isoprene (> 98 %, Merck) in Experiments 6 and 7 (Table 1). This isotopic labelling enabled us to identify products with a shift of one nominal mass-to-charge unit (i.e., ), which unambiguously marks those dimers containing one skeleton from the labelled isoprene.
Table 1Experimental conditions for SOA from sole α-pinene and mixtures of isoprene and α-pinene.
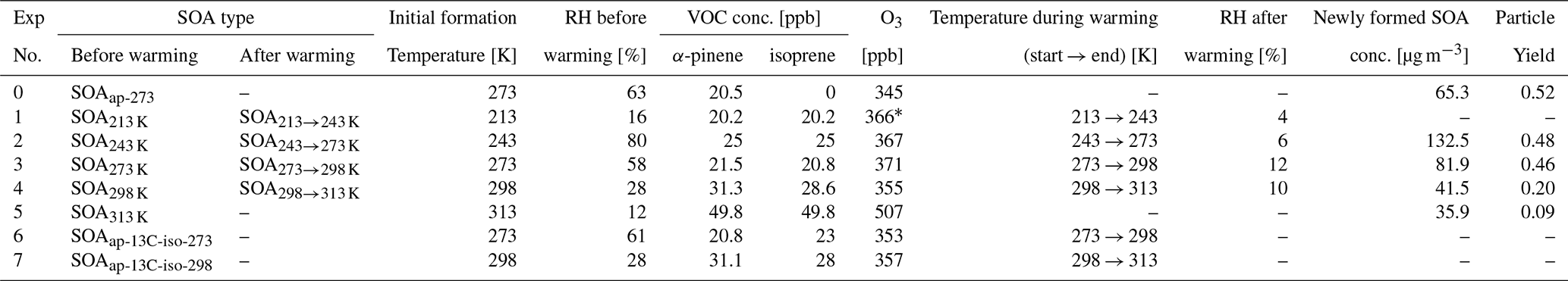
* Total amount of O3 of 366 ppb is summed from two rounds of injections: 253 ppb at the first injection, and 113 ppb at the second injection.
The initial reaction lasted 90 min, then the VOC precursors were depleted. The subsequent course of the experiment consisted of one hour of photochemical aging by illumination and then 14 h of warming the entire chamber at a constant rate. The increment of temperature before and after warming is shown in Table 1. To evaluate the effect of dilution, we injected CO2 which is a chemistry bystander before warming. The loss of CO2 was less than 4 % for all experiments after 14 h of warming. Therefore, the dilution effect is neglectable.
2.2 Instrumentation
The concentrations of VOC and semi-volatile organic particles were measured by a Proton-Transfer-Reaction-Time-of-Flight-Mass-Spectrometer coupled with a Chemical Analysis of Aerosol Online (CHARON-PTR-ToF-MS, Ionicon Analytik GmbH) particle inlet.
Bulk SOA was online detected by a high-resolution time-of-flight Aerosol Mass Spectrometer (HR-AMS, Aerodyne Inc.), while the particle-phase chemical composition of SOA at molecular level was detected by a chemical ionization mass spectrometer (CIMS) coupled with a filter inlet for gas and aerosols (FIGAERO) using iodide (I−) as reagent ions with 1 Hz time resolution (Lopez-Hilfiker et al., 2014; Lee et al., 2014). The CIMS data presented in this work stems from offline analysis. The filter samples were analyzed using a FIGAERO-iodide-CIMS. We also note that the sensitivity of FIGAERO-iodide-CIMS is highly dependent on the functionalities of the organic compounds and can vary by orders of magnitudes (Lopez-Hilfiker et al., 2016; Lee et al., 2014; Riva et al., 2019). Therefore, the results shown in this work are based on signal intensities but not mass concentrations. The detailed description of instruments, filter sample collection, and data analysis are described in Sect. S1 and Fig. S1 in the Supplement.
O3 was detected by a gas monitor (O341M, Environment SA). Particle size distributions and number concentrations were measured by a scanning mobility particle sizer (SMPS) utilizing a differential mobility analyzer (DMA, 3071 TSI Inc.) connected to a condensation particle counter (CPC, 3772, TSI Inc.). The total particle number concentrations were monitored by two condensation particle counters (CPC, 3776 and 3022A, TSI Inc.).
Typically, background measurements for both gas and particle phase are done before and after the addition of VOC to identify any contaminations inside the chamber. Gas background confirms that there were no significant gas-phase contaminations for all the experiments. Most of the particle background signals were coming from filter matrix contaminations mainly due to fluorinated constituents of low relevance. Please note that the background in all experiments was measured in the same way as described previously (Gao et al., 2022).
2.3 Determination of SOA particle volatility and glass transition temperature (Tg)
The large number of organic compounds detected in the particle phase are presented in a one-dimensional volatility basis set (1D-VBS) (Donahue et al., 2006), based on the effective saturation concentration (Csat, µg m−3). In this work, 298 K Csat (Csat,298 K, µg m−3) values of individual compounds are determined according to their measured elemental formulas applying a parameterization using molecular corridors (Li et al., 2016). The saturation concentration of species at other temperatures (Csat,T, µg m−3) can be derived from Csat,298 K according to the Clausius-Clapeyron relation:
where T is the experimental temperature in K; ΔHvap is the evaporation enthalpy in kJ mol−1, which can be estimated based on Csat,298 K by Stark et al. (2017)
In the volatility basis set, we use the following volatility classes: ultra-low VOC (ULVOC, log10Csat<−8.5), extremely low VOC (ELVOC, ), low VOC (LVOC, ), semi VOC (SVOC, −0.5<log 10Csat<2.5), intermediate VOC (IVOC, 2.5<log 10Csat<6.5), and VOC (log10Csat>6.5).Based on the same dataset as the volatility prediction, the glass transition temperature (Tg) of CHO compounds is estimated by the parameterization method expressed by the Eq. (3) (DeRieux et al., 2018):
where nC, nH, nO are the number of molecular C, H, O atoms, respectively; is the reference carbon number; bC, bH and bO refers to the contribution of each atom to Tg; and bCH and bCO are coefficients reflecting contributions from carbon–hydrogen and carbon–oxygen bonds, respectively. The values of all parameters used can be found in the published paper (DeRieux et al., 2018).
2.4 Transport Model Simulations
The simulation chamber results are implemented to PMCAMx (Murphy and Pandis, 2009), a chemical transport model (CTM) which utilizes the SOA volatility bases set approach (Lane et al., 2008) to simulate the formation of secondary aerosol from biogenic and anthropogenic VOCs. A brief description of PMCAMx is provided in Sect. S2 of the Supplement. The model-incorporated stoichiometric yields are based on the molecular composition of the particles measured by FIGARO-CIMS. The volatility of the produced aerosol is determined following the approach described in Sect. 2.3, however, to minimize the computational cost the species are re-distributed to four VBS bins (100, 101, 102, 103 µg m−3) rather than using the whole volatility range. The stoichiometric yields of both isoprene and α-pinene are temperature dependent based on the parameterization of Exp 1–5. Specifically: T<243 K, parameters based on Exp 1 (213 K); 243 K ≤ T<273 K, parameters from Exp 2 (243 K); 273 K ≤T<298 K, parameters from Exp 3 (273 K); 298 K ≤ T<313 K, parameters from Exp 4 (298 K); T≥313 K, parameters from Exp 5 (313 K). For the partitioning of the secondary organic species between the gas and the aerosol phase, PMCAMx assumes that there is equilibrium between the two phases and that the organic compounds form a pseudo-ideal solution. Specifically, in the model the partitioning of the organics between the gas and the aerosol phase depends on two parameters, the temperature and the total OA concentration of the simulated cell. Therefore, the model can replicate the concentration changes that occur in the atmosphere due to both warming and cooling. Utilizing the experimental data after warming rather than before warming commenced inside the chamber would result in the same predicted SOA concentrations by PMCAMx. Nevertheless, the warming stage of the experiments is better represented in the model by adopting the mass-stoichiometric yields derived from the initial temperatures. Values for the Base and New cases are provided in Table S1 and S2 in the Supplement. Utilizing the stoichiometric yields of Table S2, Fig. S2 depicts the secondary organic aerosol mass fraction derived as a function of the total organic aerosol mass together with the experimentally measured values.
The period considered in this model application is 5 June–8 July 2012 (PEGASOS campaign) in a European domain (5400×5832 km2, Figs. S3 and S4) with 36×36 km grid resolution, and 14 vertical layers extending up to 7.5 km above ground. The temperature together with other metrological parameters are provided by the Weather Research and Forecasting meteorological model (WRF). Biogenic emissions are calculated by the MEGAN model (Guenther et al., 2006), while anthropogenic and wildfire emissions are based on the GEMS (Visschedijk et al., 2007) and IS4FIRES (Sofiev et al., 2008) inventories, respectively. In our application, the domain average concentrations of isoprene and terpene in the simulated European domain are 0.15 and 0.04 ppb, respectively, with maximum predicted values of 2.8 and 0.5 ppb. The spatial distributions of their average ground-level concentrations over Europe are showed in Fig. S3.
3.1 Influence of temperature on particle-phase chemical composition
In each experiment, the molecular composition of fresh SOA particles was characterized by FIGAERO-iodide-CIMS using iodide as the reagent ion. The evolution of trace gases as well as particle mass and size distribution for the oxidation of isoprene α-pinene mixture at all temperatures are shown in Fig. 1. The beginning of the ozone addition is considered time zero for each experiment. The addition of ozone and the subsequent production of OH radicals, resulted in the complete depletion of the initial α-pinene and isoprene. The SOA particle diameters increased typically to 65–100 nm and the mass concentrations of the newly formed SOA ranged between 33–133 µg m−3 (mass yield: 9 %–52 %) depending on temperature. Subsequently, the fresh SOA was exposed to simulated solar radiation for 1 h. This photochemical aging resulted in no significant chemical change (cf. Fig. S9). This aging age was followed by 10–12 h of warming, which will be discussed in Sect. 3.2.
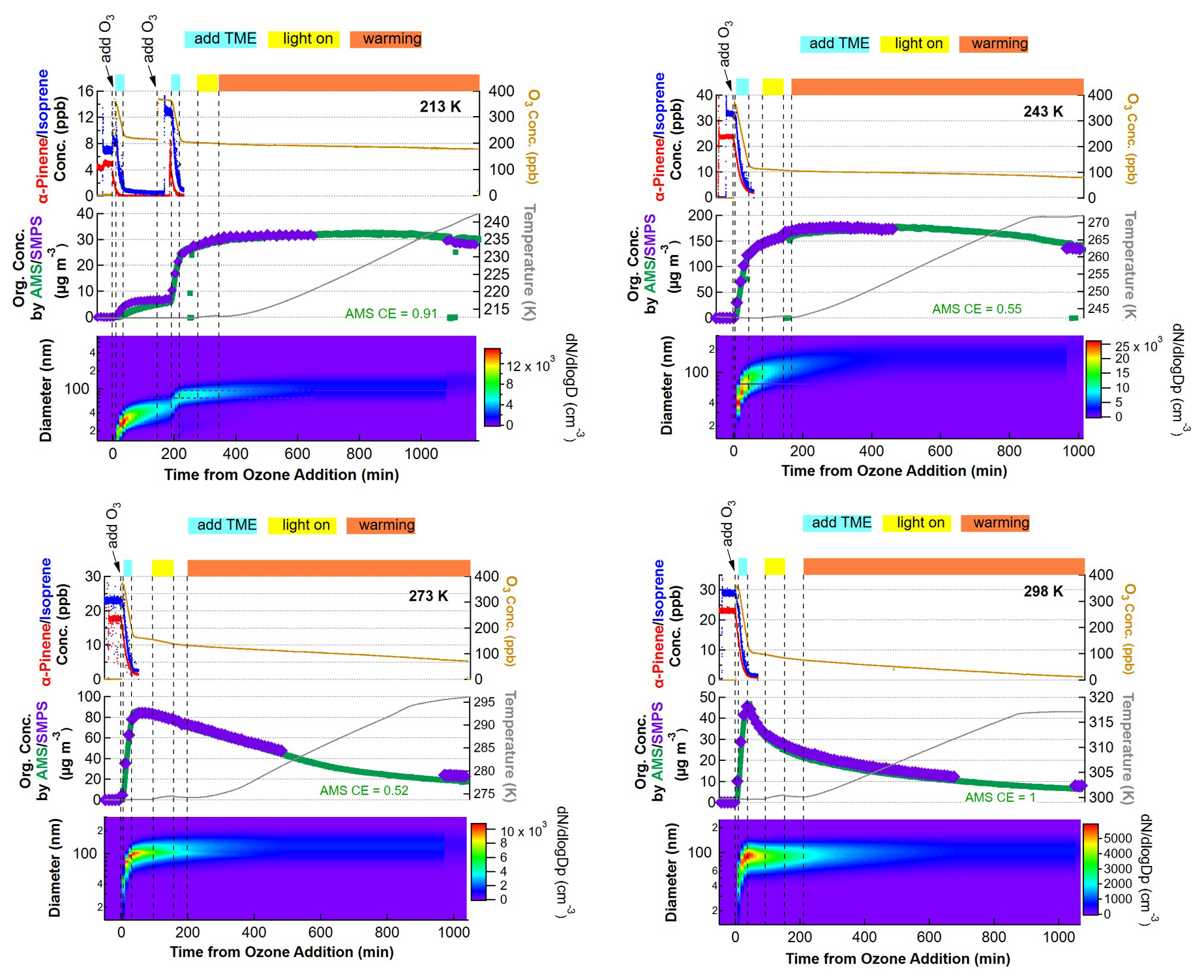
Figure 1Evolution of trace gases as well as particle mass and size for the oxidation of isoprene and α-pinene mixtures at 213, 243, 273, and 298 K. The time axis is relative to the first addition of ozone. The top shaded area of each plot shows the addition of TME to form OH radicals (blue), light on (yellow), as well as warming period (orange).
We present first the identified cross dimers formed from concurrent oxidation of isoprene and α-pinene (hereafter “ISO-AP dimers”) at 273 K. By comparison of the particle-phase chemical composition among the experiment for sole α-pinene (Exp 0), α-pinene and isoprene mixture at an equal concentration (Exp 3), and α-pinene and 13C labelled isoprene mixture at an equal concentration (Exp 6) shown in Table 1, we identified ISO-AP dimers such as C15H20O3–7, C15H22O3–9, C15H24O4–9, C15H26O5–9, C15H28O5–9, C14H20O6–8, C14H22O5–9, and C14H24O6–8. The identification of these cross dimers with 3–9 oxygen atoms completes the list of highly oxygenated cross dimers with 9–13 oxygen atoms, which were previously identified by a CIMS using nitrate as the regent ion (McFiggans et al., 2019; Heinritzi et al., 2020). Among all identified ISO-AP C14–15 cross dimers in Exp 3, C15H24O4–9 and C14H22O5–9 contribute most to the total signals (21 %), followed by C15H26O5–9, C15H28O5–9, and C15H22O3–9 with signal fractions of 16 %, 11 %, and 11 %, respectively. The relative abundances of these cross dimers in Exp 0 and Exp 6 are given in Figs. S5–S6. Due to the scavenging of OH and RO2 radicals in the presence of isoprene, the relative contribution of solely α-pinene derived C18−20 dimers from ozonolysis increases, while the contribution of dimers formed via OH radical reactions decrease (Fig. S7). This is qualitatively consistent with previous studies (McFiggans et al., 2019; Heinritzi et al., 2020; Wang et al., 2021) performed at ∼298 K.
As shown in Fig. 2, particle-phase C14–15 ISO-AP cross dimers show higher signal fractions at lower temperatures (in all detected compounds, 16 % and 11 % in total for 213 and 243 K, respectively) compared to those formed at higher temperatures (<4 % for 273, 298, and 313 K). For the dimers formed from α-pinene oxidation alone (C18–20, hereafter “AP-AP dimers”), including those from self- (i.e., both RO2 involved in the dimerization originate either from O3 oxidation or from OH oxidation.) and cross-dimerization (i.e., between the two RO2 radicals involved in the dimerization, one originates from O3 oxidation, while the other originates from OH oxidation) of RO2 derived from α-pinene oxidation initiated by both O3 and OH radicals, lower temperatures exhibit slightly higher fractions with 8 %–9 % at 213–243 K compared to 4 %–6 % at 273–313 K, consistent with previous observations (Zhang et al., 2015). Most interestingly, the ratio of ISO-AP dimers to AP-AP dimers is 3.5 times higher at 213 K (the ratio is 2) than that at 273–313 K (the ratios are 0.6, Fig. 2f). This indicates that the production of ISO-AP dimers plays a progressively more important role in SOA formation at lower temperatures. Shown as Figs. 2g and 1h, the higher ratio of ISO-AP dimers to AP-AP dimers at 213 K than 313 K is mainly contributed by the greater formation of C14–15 compounds. The volatility (expressed by the saturation concentration at 298 K, ) of ISO-AP dimers (C: 10−3.6–102.2 µg m−3) is generally higher than that of AP-AP dimers (C: 10−4.8–100.6 µg m−3), indicating that ISO-AP dimers are more volatile than AP-AP dimers when formed at the same low temperatures. The difference in volatility between both groups of dimers is also shown by their desorption temperature of maximum signal in the FIGAERO thermograms (hereafter “Tmax”) (Lopez-Hilfiker et al., 2014), which is an independent and qualitative indicator of effective volatility compared to the volatility estimated by the parameterization approach (Li et al., 2016) used in this work (Sect. S3, Fig. S8).
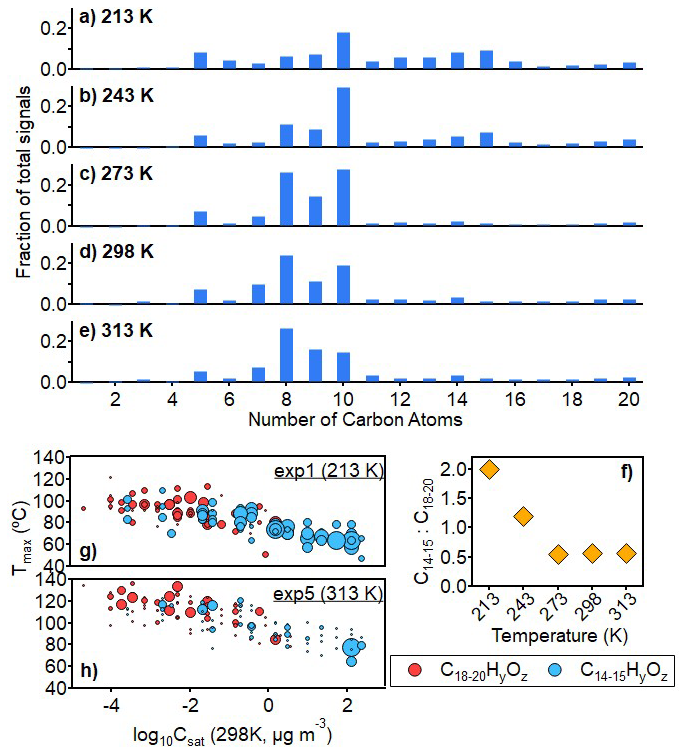
Figure 2Chemical composition of SOA derived from the mixture of isoprene and α-pinene at all temperatures (Exp 1–5): 213 K (a), 243 K (b), 273 K (c), 298 K (d), and 313 K (e); the ratio of C14–15 compound signals to C18–20 compound signals as a function of temperature (f); the distribution of C14−15 compounds and C18–20 compounds with molecular Tmax corresponding to molecular logarithmic 298 K saturation concentration for Exp 1 at 213 K (g) and Exp 5 at 313 K (h). Sizes of symbols in (g) and (h) correspond to the normalised signal abundance of molecules.
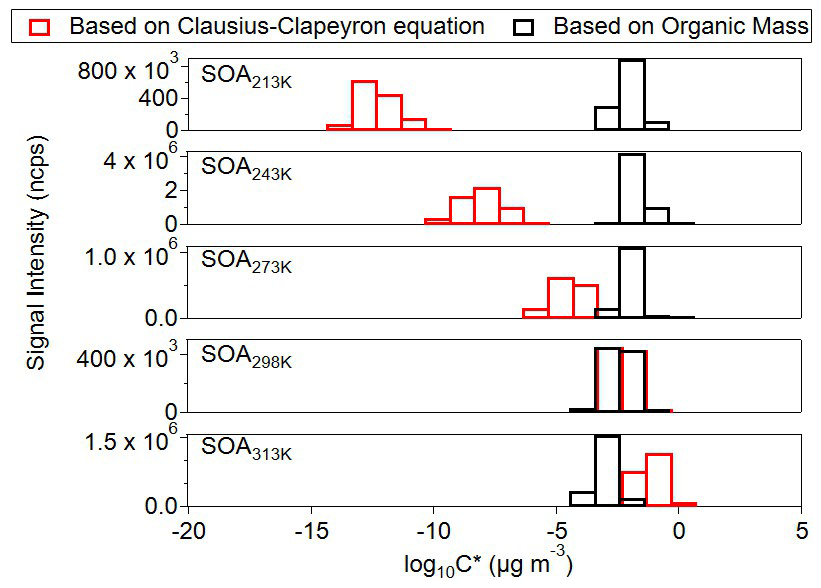
Figure 3Volatility of C14–15 cross dimers formed at all temperatures calculated based on two approaches: (Gkatzelis et al., 2018) and Clausius-Clapeyron equation. The volatility of the bins is at their experiment temperatures.
Figure 3 shows the comparison of the particle volatility distribution at different temperatures. This comparison integrates two approaches: gas- and particle-phase measurements at each temperature (i.e., (Gkatzelis et al., 2018)) and the Clausius–Clapeyron equation. C14–15 dimers span in the less volatile bins from Clausius-Clapeyron equation at temperatures below 273 K, exhibiting a strong temperature dependence, compared with the volatility based on the measured organic mass. This dependence is suggested to be chemistry-driven rather than governed solely by phase partitioning. We interpret this as follows. First, the gas-phase production rates of the two types of dimers may be temperature dependent due to the temperature-affected concentrations of RO2 radicals. The gas-phase dimer formation rate via the bimolecular termination of RO2+ R′O2→ ROOR′ rises strongly with temperature (Quéléver et al., 2019). At lower temperatures, the lower rate coefficient of α-pinene + O3 (Khamaganov and Hites, 2001; Bernard et al., 2012) and higher rate coefficient of isoprene + OH (Campuzano-Jost et al., 2000; Campuzano-Jost et al., 2004; Dillon et al., 2017) lead to higher differential between the concentrations of C10 RO2 from α-pinene and C5 RO2 from isoprene. Therefore, at lower temperatures, higher [C5 RO2][C10 RO2] results in larger production of ISO-AP dimers compared with less formation of C20 AP-AP dimers due to lower [C10 RO2][C10 RO2]. Besides, the other well-established dimer formation pathway for α-pinene derived dimers, condensed-phase combination of acetyl peroxy radicals yielding diacyl peroxides and their subsequent decomposition (Zhang et al., 2015) to produce esters, carboxylic acids, and alcohols, is affected by temperature as well (Leffler and More, 1972; Lamb et al., 1965). Our observation suggests that the formation of ISO-AP dimers via the diacyl peroxides pathway may be faster than that of AP-AP dimers at lower temperatures. Second, previous studies (Trump and Donahue, 2014; Morino et al., 2020) have shown that the decomposition rates of dimers depend on temperature and the type of dimers. Therefore, we cannot exclude that AP-AP dimers decompose faster than ISO-AP dimers at lower temperature, leading to higher condensed-phase ratios of C14−15 C18−20.
It should be noted that the sensitivity of iodide chemical ionization exhibits substantial variability in detecting organic compounds with different functional groups. As a result, signal-based analyses may not accurately represent the actual abundances of these species. Nevertheless, comparing signal fractions across different experiments can provide valuable insights into product distributions and underlying reaction mechanisms.
Overall, the nonmonotonic temperature dependence (Fig. 2f) of the ratio of C14–15 to C18–20 dimers between 213–313 K highlights the importance of the AP-AP dimer suppression by the ISO-AP cross dimers. This is particularly relevant for biogenic particle formation and growth in the real atmosphere especially at lower temperatures (Fu et al., 2009; Andreae et al., 2018).
3.2 Influence of temperature on SOA aging
To study the influence of temperature change on SOA formed at a specific temperature, we warmed the fresh SOA particles up by 1.4–2.4 K h−1 over 10–12 h, which resembles the ambient temperature changing rate (∼0.1–2 K h−1) in the real atmosphere (Hansen et al., 2006). The illumination has no significant effect on the bulk O : C, H : C and OSC (Fig. S9). The molecular chemical composition and volatility of fresh particles before warming are described in the Sect. S3. Most interestingly, by warming, SOA particles formed initially at different temperatures showed distinct increments and/or decrements in bulk O : C and H : C ratios as well as oxidation states (OSC) as measured by HR-AMS (Figs. 4, S10, and Table S3). It indicates these SOA particles underwent distinct aging processes including water uptake and evaporation when being warmed up.

Figure 4Van-Krevelen diagram for SOA particles during the warming periods of Exp 1 (213 to 243 K), Exp 2 (243 to 273 K), Exp 3 (273 to 298 K), Exp 4 (298 to 313 K), and Exp 5 (313 K) from HR-AMS measurements (left) and FIGAERO-iodide-CIMS measurements mean values (right, symbols of diamonds and circles for warming start and end, respectively). Arrows are for guiding from the start to end of the warming periods. Symbols are coloured by temperatures. The carbon oxidation state (OSC= 2 O : C – H : C) is shown with a grey dashed line. The pink lines with different slopes represent various reaction pathways: slope = 2 (hydration); slope = 0 (formation of hydroxy/peroxy groups); slope 1 (formation of carboxylic acids, or addition of both hydroxy and carbonyl groups).
We observed a clear increase of O : C ratios (from 0.36 to 0.54 from HR-AMS measurements, from 0.5 to 0.6 from FIGAERO-iodide-CIMS measurements) of fresh SOA particles formed between 243 and 313 K (Fig. 4). One exception is the particles formed at 213 K. The O : C ratio of the fresh SOA213 K (0.45 from HR-AMS measurement, 0.55 from FIGAERO-iodide-CIMS measurement) is higher than that of SOA243 K, contrary to the lower O : C ratios of particles formed at lower temperatures. This may result from the higher ratios of initial O3 to VOCs concentrations (∼19) in Exp 1 at 213 K compared to other experiments (∼7) (details in Method).
According to the HR-AMS measurements, the bulk O : C and H : C ratios of SOA particles formed at 243 K (SOA243 K) increases from 0.36 to 0.4 and from 1.69 to 1.82, respectively, during gradual warming to 273 K (SOA243→273 K). Although the incremental O : C change is small, the online HR-AMS measurements showed a significant trend during the warming process (Fig. 4). The ratio (= 3.25) of the H : C increment and O : C increment in the Van-Krevelen diagram indicates hydration reactions during warming (Schilling Fahnestock et al., 2015; Heald et al., 2010). Correspondingly, the oxidation state (OSC) of bulk SOA243 K decreases from −0.97 to −1.02 for SOA243→273 K (Fig. S10), while the organic particle mass decayed 17 % as measured by HR-AMS. According to the FIGAERO-iodide-CIMS measurements, 18 % fraction of the particle-phase CxHyOz signals are lost during warming of SOA from 243 K (SOA243 K) to 273 K (SOA243→273 K) (Fig. 5d). The loss of oxygenated organic compounds mainly involves C5, C8–10, and C14–15 compounds (Fig. 5h), which are identified as monomeric products from sole isoprene and sole α-pinene, and their ISO-AP dimers, respectively. However, the loss of these oxygenated organic compounds leads to no significant change in the H : C ratio (from 1.63 to 1.62) and O : C ratio (from 0.50 to 0.50) of the particle-phase CxHyOz measured by FIGAERO-iodide-CIMS. Therefore, the increase in bulk O : C and H : C ratios of SOA measured by HR-AMS indicates not only hydration reactions (Schilling Fahnestock et al., 2015; Heald et al., 2010) but also potential losses of more oxidized compounds with low H : C ratios. The bulk SOA aging towards higher H : C and O : C ratios during warming is likely due not only to sample evaporation but also to the change in the particle phase state.
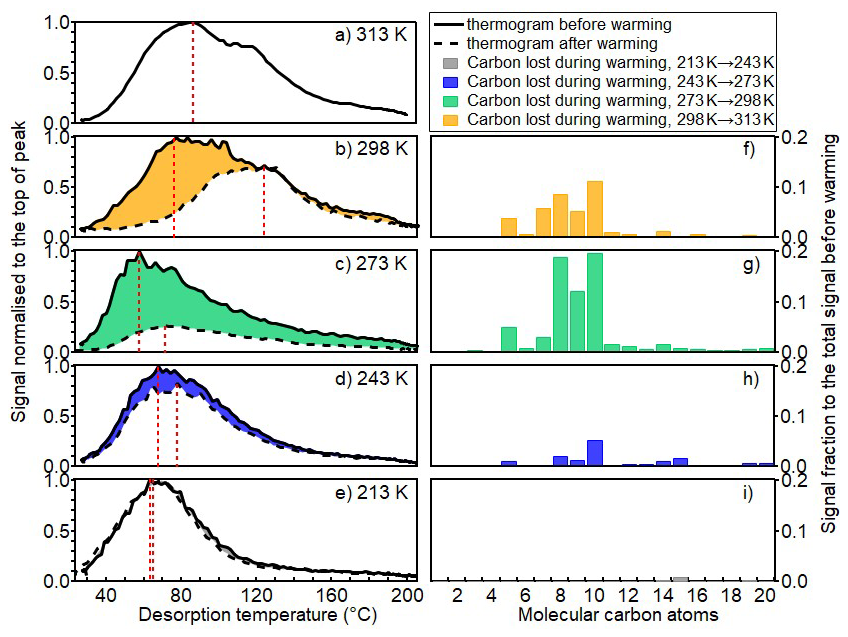
Figure 5FIGAERO-iodide-CIMS thermograms of particles before and after warming process for all experiments ((a) for 313 K, (b) for 298 K, (c) for 273 K, (d) for 243 K, and (e) for 213 K), and the corresponding carbon distributions of molecules lost during warming (f–i). Black solid and dashed lines show the thermograms of particles sampled before and after warming, respectively. The thermograms are normalized to the peak signals of each thermogram before warming. Red vertical dashed lines indicate the Tmax.
Similar changes of O : C and H : C ratios were observed for SOA213 K warmed to SOA213→243 K. Therefore, the SOA213 K warmed to SOA213→243 K seems to undergo similar aging processes like SOA243 K being warmed to SOA243→273 K.
We characterized the viscosity of SOA particles by using the glass transition temperature, Tg which is defined as the temperature at which an amorphous material transitions from a liquid-like or semi-solid state to a glassy solid state. As the ambient temperature approaches or drops below the Tg of a particle, its viscosity increases dramatically, often by several orders of magnitude. Therefore, the phase state and viscosity of SOA can be inferred by characterizing the Tg values. Tg was calculated for all detected organic compounds by FIGAERO-iodide-CIMS using a parameterization approach (DeRieux et al., 2018). In this study, SOA243 K is estimated to be in a glassy solid state with a Tg of 289 K, comparable to the Tg values for sole isoprene- or α-pinene-derived SOA reported in previous studies (DeRieux et al., 2018; Ladino et al., 2014). We note that the Tg values may be underestimated, as the water content in the particles was not taken into account due to a lack of measurements. The high viscosity at low temperature kinetically inhibits the diffusion of water and large organic molecules within the particle. Upon warming, the particle transitions from a glassy to a semi-solid or liquid state, which facilitates the uptake and internal mixing of water. This process can promote aqueous-phase reactions (e.g., hydrolysis, oxidation) that alter the organic composition, increasing the H : C and O : C ratios. Therefore, water uptake and the potential change of particle hygroscopicity (Shiraiwa et al., 2017; Pajunoja et al., 2015; Shiraiwa et al., 2011) may contribute to increasing H : C and O : C ratios of bulk SOA243 K during warming from 243 to 273 K.
In contrast, from the HR-AMS measurement, the bulk SOA273 K loses 75 % of mass, and show a significant increase of OSC (from −0.87 to −0.75) and O : C ratios (from 0.44 to 0.48) but a decrease of H : C ratios (from 1.76 to 1.73) during warming to 298 K (Fig. 4). This tendency is consistent with the changes of O : C ratios and H : C ratios for the oxygenated constitutes measured by FIGAERO-iodide-CIMS. During warming of SOA273 K to 298 K, 72.5 % of all particle-phase CxHyOz compounds are lost, with OSC increasing from −0.52 to −0.41, O : C from 0.54 to 0.57, and H : C ratios decreasing from 1.60 to 1.55. This indicates that evaporation is the main loss process with higher losses of less oxidized compounds. This is consistent with the analysis of HR-AMS spectra before and after warming (Fig. S11) The Tg we estimated for SOA273 K is 278 K, which is in between the temperatures of warming at the start (273 K) and at the end (298 K). Therefore, the diffusion and evaporation of organic molecules are gradually less hindered when particle phase state transits from solid/semi-solid to liquid.
For the bulk SOA298 K, its estimated Tg is 283 K, which is evidently lower than the temperatures during warming from 298 to 313 K. This indicates that the SOA298 K remain in the liquid phase during the whole warming process. As illustrated in Fig. 4, warming of all SOA compounds formed at 298 K leads to lower H : C (from 1.63 to 1.57) and O : C ratios (weekly from 0.53 to 0.52), resulting in slightly higher OSC values (from −0.57 to −0.53). During warming from 298 to 313 K, 40.2 % of all particle-phase CxHyOz compounds and 71 % of organic particle mass were lost, by substantial evaporation. However, the evaporation induces only small changes of O : C ratio from 0.60 (SOA298 K) to 0.59 (SOA298→313 K), H : C ratio from 1.51 to 1.49, and OSC from −0.31 to −0.32 for oxygenated organics, even though the trend is clear as shown in Fig. 4. As the reduction in H : C ratio is around 2 times higher than the reduction in O : C ratio for bulk SOA298 K particles during the warming to 313 K, we infer that there might be water evaporation due to the potential particle-phase dehydration reactions involving elimination of H2O.
As illustrated in Fig. 5, before warming, volatility indicated by the Tmax of the fresh particles showed a non-monotonic trend. This is similar with previous findings in the β-caryophyllene system (Gao et al., 2023), which is due to favoured condensation or oligomerization reactions at lower temperature and less production of low volatile HOMs which show larger fractions at warmer temperatures. During warming, the volatility of the particles is influenced by changes in their chemical composition as the gas-particle equilibrium is re-established through phase partitioning. As the temperature increases, more volatile organic compounds evaporate from the particle phase. Consequently, the particle composition becomes enriched in the remaining lower-volatile organic species. This is corroborated by the higher Tmax values observed in the thermograms: SOA243→273 K (78 °C) compared to SOA243 K (68 °C), SOA273→298 K (71 °C) compared to SOA273 K (57 °C), and SOA298→313 K (124 °C) compared to SOA298 K (77 °C), as illustrated in Fig. 5b–d. The results indicate that the overall effect of warming on the SOA particle volatility is governed by the initial temperature-dependent chemical composition and the corresponding glass transition temperature.
Further support comes from the distribution of volatility groups estimated by the parameterization approach (Li et al., 2016) based on measured numbers of molecular carbon, hydrogen, and oxygen atoms. During warming, evaporation leads to compositional changes that enrich the relatively lower-volatility compounds. Concurrently, rising temperatures shift the entire VBS toward higher apparent volatility, following the Clausius-Clapeyron relation. For instance, despite the evaporation of some volatile components during warming from 243 to 273 K, the resulting SOA243→273 K particles exhibit higher overall apparent volatility, containing only 35 % of LVOC/ELVOC/ULVOC (Figs. S12 and 6). Similarly, in the warming experiment from 273 to 298 K, the fraction of LVOC/ELVOC/ULVOC decreased slightly from 16 % to 15 %, and from 18 % to 14 % in the case of SOA298→313 K compared to SOA298 K. These results underscore the significant role of ambient temperature on the apparent volatility of SOA particles.
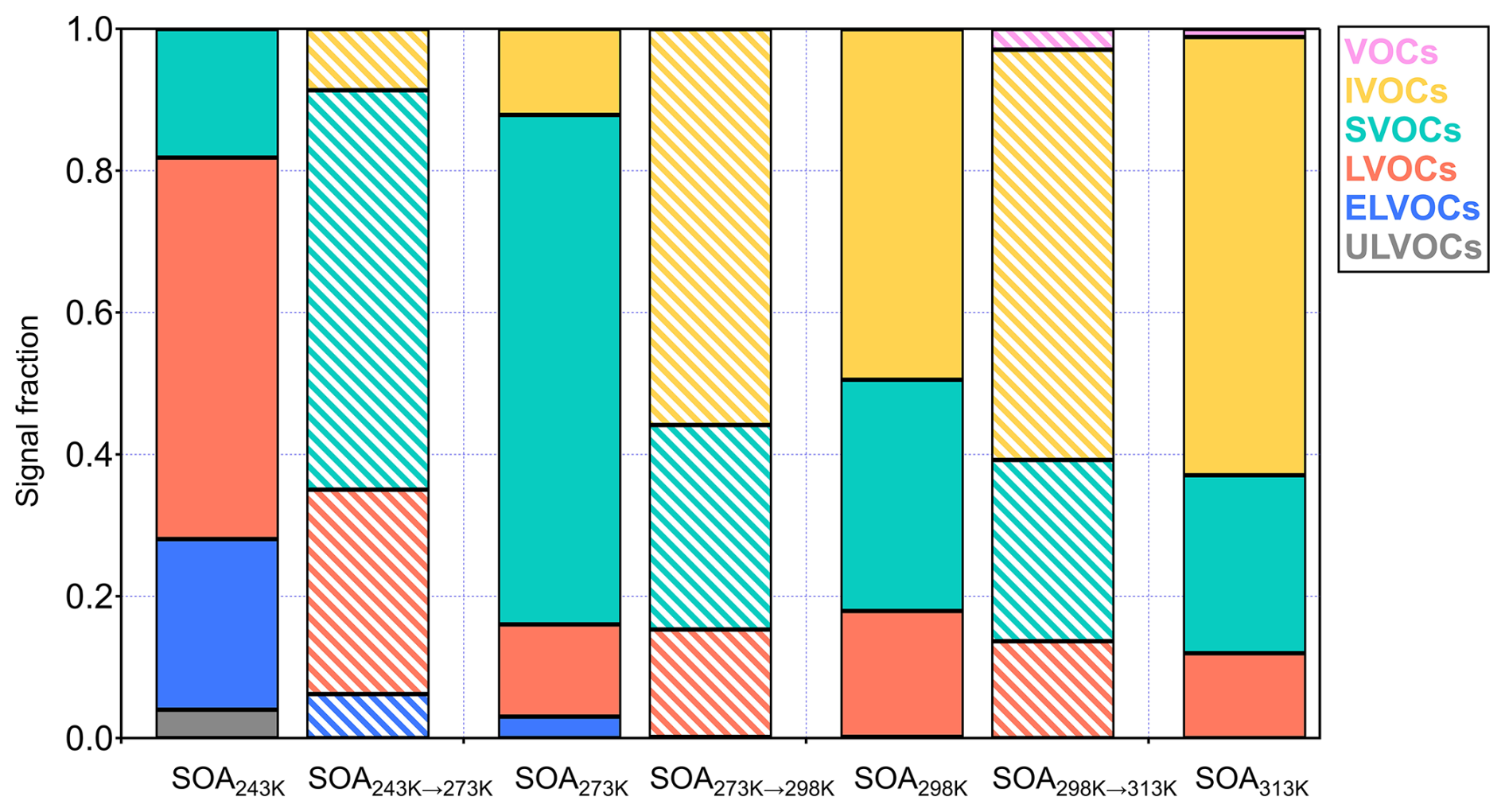
Figure 6Signal fraction of volatility groups (ULVOC, ELVOC, LVOC, SLVOC, IVOC, and VOC) in the SOA particles before (solid bars) and after (striped bars) warming process, respectively. Colors refer to the volatility groups. Volatility groups are identified based on the saturation concentrations as described in the Method. Please note that the volatility groups in this Figure are related to their chamber temperatures but not 298 K.
It should be noted that the FIGAERO-iodide-CIMS exhibits higher sensitivity toward moderate oxygenated compounds (e.g., 2–9 oxygen atoms) (Riva et al., 2019), which may introduce bias in the VBS. Nevertheless, comparisons of signal-weighted VBS distributions across different experimental conditions remain indicative of the effects of temperature and warming on particle volatility and the related chemical processes. Here, we note that the precursor concentrations used in this study are substantially higher than typical atmospheric levels. While such conditions are typical in chamber experiments to generate sufficient particle mass for instrument detection and > 10 h aging by warming, they may influence the underlying chemical processes. Specifically, elevated VOC and O3 concentrations can enhance the rates of bimolecular reactions, potentially favoring radical–radical recombination, accelerating oligomer formation (Zhao et al., 2023), and increasing SOA yields relative to ambient conditions. These conditions may also shift the partitioning of semi-volatile species toward the particle phase. Consequently, the volatility distributions reported here likely represent an upper bound of reactivity and should be interpreted with caution when extrapolating to atmospheric conditions.
Please note, that other condensed-phase chemical reactions may play a role during warming process as well, e.g., dimers may decompose due to their chemical instability (Pospisilova et al., 2020; Surdu et al., 2024), and the formation and condensation of molecules such as peroxyhemiacetal and aldol has been found to be reversible and temperature-dependent. However, it requires further studies to address this question systematically.
Furthermore, the oxidation states (OSC) of SOA particles after warming were lower than that of the particles formed directly at these temperatures. As mentioned above, increasing temperatures during warming facilitate the evaporation of more volatile compounds. This results in the organic components remaining in the particles being generally less volatile and higher oxidized corresponding to a higher oxidation state of bulk SOA as measured by the HR-AMS.
However, it cannot compensate for the composition difference (e.g., due to autoxidation (Bianchi et al., 2019)) between SOA243 K and SOA273 K caused by different reaction pathways and product distributions at the different formation temperatures. For example, the saturation concentrations of the same compounds in SOA243→273 K and SOA273 K systems are the same because they are both at 273 K. We compared the molecular chemical composition of SOA243→273 K and SOA273 K (Fig. S13c and g). The C11–20 products make up a higher signal fraction in SOA243→273 K (66 %) compared to the corresponding compound groups in the SOA273 K (43 %), and vice versa for C4–10 products. Higher mean OSC values are found for dimeric groups of C13–15 as well as C20, and other products of C6 and C16 in SOA273 K, leading to higher OSC for bulk SOA particles. Thus, we emphasize that, besides promoting the condensation of condensable components, lower temperatures chemically enhance the formation of ISO-AP cross dimers, while higher temperatures may favor the formation of higher oxidized products, e.g., via the autoxidation mechanism (Bianchi et al., 2019).
For instance, SOA298→313 K has OSC values of −0.53 (HR-AMS) and −0.31 (FIGAERO-iodide-CIMS), much lower than those of SOA313 K (−0.49 by HR-AMS and −0.27 by FIGAERO-iodide-CIMS). By comparing the molecular chemical composition of both particles at 313 K (Fig. S13a and e), the higher mean OSC values for carbon groups of C4–10 in SOA formed directly at 313 K cause the increase of overall OSC of oxygenated products. This confirms again that higher temperatures favour the formation of higher oxidized products.
In addition, C8−10 compounds (mean formula C8.4H12.4O5.0) in SOA298→313 K have a Tmax of 100 °C. However, C8–10 compounds have a similar mean formula (C8.5H12.5O5.3) in SOA313 K but have a lower Tmax of 82 °C. This implies that C8–10 compounds consist of varying monomeric isomers with significantly different volatilities, being less volatile in SOA298→313 K and more volatile in SOA313 K.
Besides, although SOA273→298 K has the largest OSC increment (−0.75 for bulk, −0.41 for oxygenated constitutes) from its initial particles before warming among all particles discussed, its oxidation state is still significantly lower than SOA298 K (−0.56 for bulk, −0.31 for oxygenated constituents). This is consistent with the somewhat higher volatility of SOA273→298 K (Tmax of 71 °C) than SOA298 K (Tmax of 77 °C, Fig. 5b and c) observed. All compound groups remaining after warming have higher mean OSC except for C4 (Fig. S13). This means that the compounds in most compound groups are more oxidized when formed at 298 K. Thus, we conclude that besides promoting the evaporation of particle-phase compounds, higher temperatures also enhance the formation of higher oxidized products in SOA from oxidation of α-pinene and isoprene mixtures.
3.3 Modelling the impact of cross dimers for real world scenarios
The relevance of these findings is corroborated by model simulations that incorporates the new VBS parameterization derived below, including mixed dimers. The results discussed above are based on experiments with equal initial amounts of α-pinene and isoprene. However, in the natural atmosphere, the α-pinene to isoprene ratios can vary substantially for different temperatures (seasons, day night) and in different regions, e.g., boreal forest, tropical forest, and temperate regions. The averaged ratio of isoprene to terpenes (including other monoterpene compounds) over Europe during the simulation period is predicted to be 3.75 (Fig. S3). With α-pinene representing terpenes, simulations using the PMCAMx chemical transport model (Tsimpidi et al., 2010; Fountoukis et al., 2011; Murphy and Pandis, 2009) show that when the C14–15 ISO-AP dimers are considered under different temperatures, the predicted mass concentration of organics over Europe is significantly enhanced. The mean ground-level PM1 biogenic SOA mass concentrations over Europe for the simulated period are found to increase by 47 % from 0.23 µg m−3 with the original setup (Base case, Table S1) to 0.41 µg m−3 by utilizing the new VBS parameterization including the temperature dependent C14–15 ISO-AP cross dimers (New case, Table S2) (Fig. 7a, b, c). The contribution of biogenic SOA to the total OA mass increased from 9 % to 14 % (Fig. 7d, e). Specifically, in the New case, the predicted ground-level OA mass concentrations are on average higher by 0.6 µg m−3 for four measuring stations located in Italy (Fig. S4), as shown in Fig. S14.
During the simulated period, the isoprene emissions are greater over Croatia than other European areas. As a result, the difference in monthly averaged ground-level biogenic SOA concentrations between the two simulations is larger in Croatia (0.9 µg m−3) than the domain average (0.4 µg m−3). Figure S15 shows the correlation between isoprene concentrations and the enhancement of biogenic SOA mass predicted in the New case compared to the Base case over Croatia. In addition, due to the synergetic effect of relatively high concentrations of biogenic precursors and higher temperatures (Fig. 7g–h), the simulated biogenic SOA concentrations at the site of Monte Cimone (2165 m a.s.l.) show a better fit with the observed values compared to the Base case (Fig. 7f).
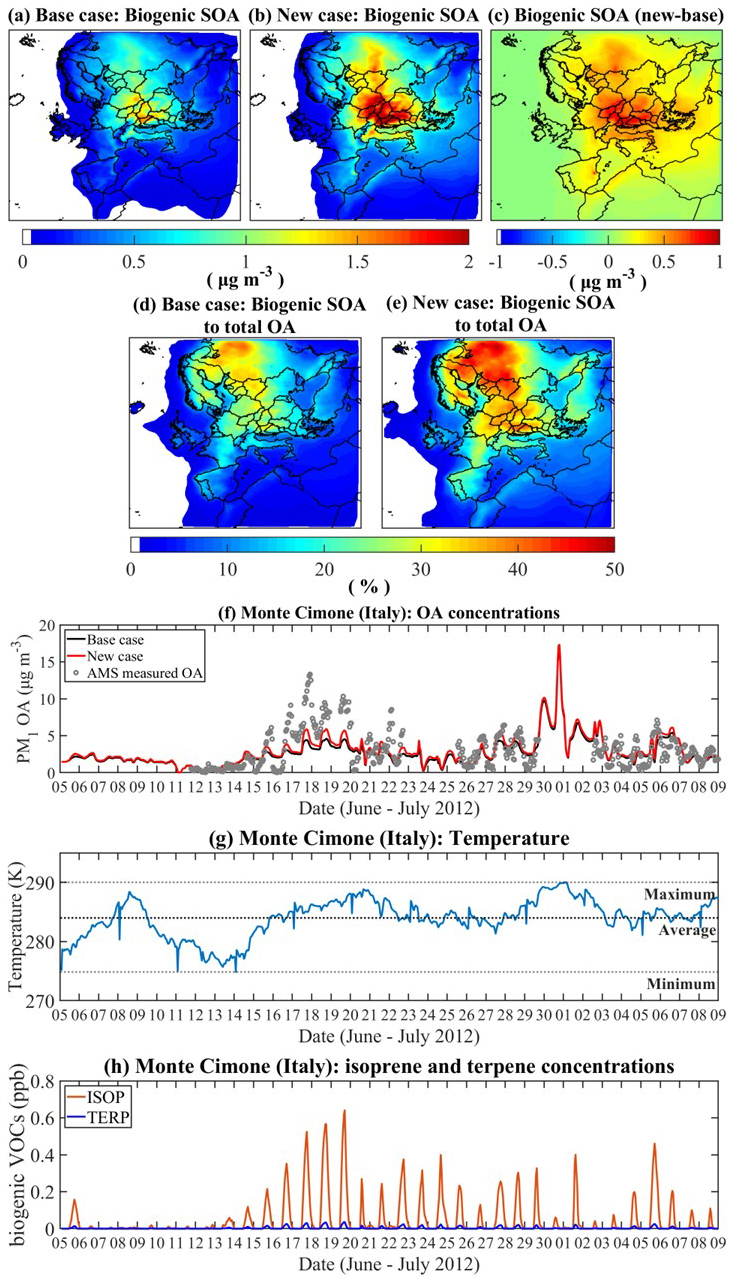
Figure 7The effect of the newly developed VBS parametrization on the predicted ground-level concentrations of biogenic SOA and total OA in PM1 over Europe. Spatial distribution of the domain average biogenic SOA concentrations predicted utilizing (a) the original parametrization (Base case); (b) the new parameters derived considering the temperature dependent ISO-AP cross dimers (New case) and (c) the difference between the two simulations (New case – Base case); the contribution of the predicted biogenic SOA to the total OA concentrations over Europe in (d) the Base case and (e) the New case; (f) hourly ground-level OA concentrations predicted by the two simulations and measured by HR-AMS in the measuring site of Monte Cimone (Italy) during the PEGASUS campaign (June–July 2012); (g) the simulated hourly temperature profile (1st simulation layer) and (h) the predicted isoprene (ISOP) and terpene (TERP) ground-level concentrations over the measuring site for the simulated period.
This study utilized 13C-labeled isoprene to identify cross-dimeric products from parallel oxidation of isoprene and α-pinene while examining SOA composition and volatility covering most tropospheric conditions with temperatures between 213 and 313 K. The identified C14–15 ISO-AP cross dimers suppress the formation of the α-pinene self-dimers (AP-AP). This is achieved by competing with α-pinene-derived peroxy radicals (which are generated via OH-oxidation), thereby inhibiting their reactions. This suppression effect is more pronounced at lower temperatures between 213–273 K, while it is not temperature-sensitive above 273 K. SOA components observed are more oxidized at higher temperatures which is consistent with previous studies, potentially driven by a larger contribution from autoxidation pathways (Gao et al., 2022; Bianchi et al., 2019; Ye et al., 2019). Warming experiments reveal significant volatility changes: SOA formed at 243 K is and remains solid/semi-solid, losing only 18 % of particle-phase compounds upon warming to 273 K. In contrast, SOA formed at 273 K undergoes substantial transition from a semi-solid to a liquid phase when being warmed to 298 K, losing 72.5 % of all the particle-phase CxHyOz compounds detected by FIGAERO-CIMS. At 298 K, SOA shows lower O : C ratios and a 42 % loss of oxygenated organics when warmed to 313 K. Except for particles formed at 273 K and subsequently warmed to 298 K, in all other temperature regimes, SOA formed directly at higher temperatures is both more oxidized and more volatile than SOA formed at lower temperatures and then warmed. This observation has implications for SOA evolution over diurnal cycles. For example, in mid-latitude or tropical environments, SOA formed during warmer daytime hours may be more oxidized yet more volatile to reversible evaporation. In contrast, SOA generated during cooler nighttime periods would be less oxidized but more persistent, forming a low-volatility reservoir. This daily alternation could significantly influence the overall lifetime, chemical aging, and mass yield of SOA particles on a regional scale.
Implementing this new mechanistic and volatility information in the PMCAMx model resulted in higher predicted SOA over Europe. Future studies should extend this modelling to a broader ground-level temperature range and evaluate it with extensive field data. Furthermore, although this study focuses on isoprene and α-pinene mixtures, other biogenic and anthropogenic VOC mixtures may exhibit similar temperature-sensitive behaviour. Therefore, further studies are warranted to elucidate the impact of temperature on SOA formation from diverse VOC mixtures, which will extend our understanding and improve predictions in the context of a warming climate.
Dataset used in this manuscript are accessible from the KITopen data repository (https://doi.org/10.35097/9zvc7fvj505n9d93, Gao et al., 2026).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-8875-2026-supplement.
L.G. and H.S. designed the study. Chamber experiments were carried out by L.G., H.S., and J.S. Data analysis and interpretation were performed by L.G., J.S., H.S., C.M., and C.W. Model simulations and interpretation were done by S.E.I.M., S.N.P., L.G., and H.S. The manuscript was written by L.G, with input from S.E.I.M., C.M., J.S., C.W., T.L., S.N.P., and H.S. All co-authors commented on the manuscript.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
L.G. and J.S. acknowledge the China Scholarship Council (CSC) for financial support and the Graduate School for Climate and Environment (GRACE). The authors thank the IMK-AAF technicians at the KIT for their support of this work.
This research has been supported by the EU H2020 European Research Council (CHAPAs (grant no. 850614)).
The article processing charges for this open-access publication were covered by the Karlsruhe Institute of Technology (KIT).
This paper was edited by Manabu Shiraiwa and reviewed by two anonymous referees.
Andreae, M. O., Afchine, A., Albrecht, R., Holanda, B. A., Artaxo, P., Barbosa, H. M. J., Borrmann, S., Cecchini, M. A., Costa, A., Dollner, M., Fütterer, D., Järvinen, E., Jurkat, T., Klimach, T., Konemann, T., Knote, C., Krämer, M., Krisna, T., Machado, L. A. T., Mertes, S., Minikin, A., Pöhlker, C., Pöhlker, M. L., Pöschl, U., Rosenfeld, D., Sauer, D., Schlager, H., Schnaiter, M., Schneider, J., Schulz, C., Spanu, A., Sperling, V. B., Voigt, C., Walser, A., Wang, J., Weinzierl, B., Wendisch, M., and Ziereis, H.: Aerosol characteristics and particle production in the upper troposphere over the Amazon Basin, Atmos. Chem. Phys., 18, 921–961, https://doi.org/10.5194/acp-18-921-2018, 2018.
Aubry, T. J., Staunton-Sykes, J., Marshall, L. R., Haywood, J., Abraham, N. L., and Schmidt, A.: Climate change modulates the stratospheric volcanic sulfate aerosol lifecycle and radiative forcing from tropical eruptions, Nat. Commun., 12, 4708, https://doi.org/10.1038/s41467-021-24943-7, 2021.
Bernard, F., Fedioun, I., Peyroux, F., Quilgars, A., Daële, V., and Mellouki, A.: Thresholds of secondary organic aerosol formation by ozonolysis of monoterpenes measured in a laminar flow aerosol reactor, J. Aerosol Sci., 43, 14–30, https://doi.org/10.1016/j.jaerosci.2011.08.005, 2012.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bilde, M., Barsanti, K., Booth, M., Cappa, C. D., Donahue, N. M., Emanuelsson, E. U., McFiggans, G., Krieger, U. K., Marcolli, C., Topping, D., Ziemann, P., Barley, M., Clegg, S., Dennis-Smither, B., Hallquist, M., Hallquist, Å. M., Khlystov, A., Kulmala, M., Mogensen, D., Percival, C. J., Pope, F., Reid, J. P., Ribeiro da Silva, M. A. V., Rosenoern, T., Salo, K., Soonsin, V. P., Yli-Juuti, T., Prisle, N. L., Pagels, J., Rarey, J., Zardini, A. A., and Riipinen, I.: Saturation Vapor Pressures and Transition Enthalpies of Low-Volatility Organic Molecules of Atmospheric Relevance: From Dicarboxylic Acids to Complex Mixtures, Chem. Rev., 115, 4115–4156, https://doi.org/10.1021/cr5005502, 2015.
Campuzano-Jost, P., Williams, M. B., O'Ottone, L., and Hynes, A. J.: Kinetics of the OH-initiated oxidation of isoprene, Geophys. Res. Lett., 27, 693–696, https://doi.org/10.1029/1999GL010995, 2000.
Campuzano-Jost, P., Williams, M. B., D'Otton, L., and Hynes, A. J.: Kinetics and Mechanism of the Reaction of the Hydroxyl Radical with h8-Isoprene and d8-Isoprene: Isoprene Absorption Cross Sections, Rate Coefficients, and the Mechanism of Hydroperoxyl Radical Production, J. Phys. Chem. A, 108, 1537–1551, https://doi.org/10.1021/jp0363601, 2004.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Curtius, J., Heinritzi, M., Beck, L. J., Pöhlker, M. L., Tripathi, N., Krumm, B. E., Holzbeck, P., Nussbaumer, C. M., Hernández Pardo, L., Klimach, T., Barmpounis, K., Andersen, S. T., Bardakov, R., Bohn, B., Cecchini, M. A., Chaboureau, J.-P., Dauhut, T., Dienhart, D., Dörich, R., Edtbauer, A., Giez, A., Hartmann, A., Holanda, B. A., Joppe, P., Kaiser, K., Keber, T., Klebach, H., Krüger, O. O., Kürten, A., Mallaun, C., Marno, D., Martinez, M., Monteiro, C., Nelson, C., Ort, L., Raj, S. S., Richter, S., Ringsdorf, A., Rocha, F., Simon, M., Sreekumar, S., Tsokankunku, A., Unfer, G. R., Valenti, I. D., Wang, N., Zahn, A., Zauner-Wieczorek, M., Albrecht, R. I., Andreae, M. O., Artaxo, P., Crowley, J. N., Fischer, H., Harder, H., Herdies, D. L., Machado, L. A. T., Pöhlker, C., Pöschl, U., Possner, A., Pozzer, A., Schneider, J., Williams, J., and Lelieveld, J.: Isoprene nitrates drive new particle formation in Amazon's upper troposphere, Nature, 636, 124–130, https://doi.org/10.1038/s41586-024-08192-4, 2024.
DeRieux, W.-S. W., Li, Y., Lin, P., Laskin, J., Laskin, A., Bertram, A. K., Nizkorodov, S. A., and Shiraiwa, M.: Predicting the glass transition temperature and viscosity of secondary organic material using molecular composition, Atmos. Chem. Phys., 18, 6331–6351, https://doi.org/10.5194/acp-18-6331-2018, 2018.
Dillon, T. J., Dulitz, K., Groß, C. B. M., and Crowley, J. N.: Temperature-dependent rate coefficients for the reactions of the hydroxyl radical with the atmospheric biogenics isoprene, alpha-pinene and delta-3-carene, Atmos. Chem. Phys., 17, 15137–15150, https://doi.org/10.5194/acp-17-15137-2017, 2017.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Fountoukis, C., Racherla, P. N., Denier van der Gon, H. A. C., Polymeneas, P., Charalampidis, P. E., Pilinis, C., Wiedensohler, A., Dall'Osto, M., O'Dowd, C., and Pandis, S. N.: Evaluation of a three-dimensional chemical transport model (PMCAMx) in the European domain during the EUCAARI May 2008 campaign, Atmos. Chem. Phys., 11, 10331–10347, https://doi.org/10.5194/acp-11-10331-2011, 2011.
Fu, P., Kawamura, K., Chen, J., and Barrie, L. A.: Isoprene, Monoterpene, and Sesquiterpene Oxidation Products in the High Arctic Aerosols during Late Winter to Early Summer, Environ. Sci. Technol., 43, 4022–4028, https://doi.org/10.1021/es803669a, 2009.
Gao, L., Song, J., Mohr, C., Huang, W., Vallon, M., Jiang, F., Leisner, T., and Saathoff, H.: Kinetics, SOA yields, and chemical composition of secondary organic aerosol from β-caryophyllene ozonolysis with and without nitrogen oxides between 213 and 313 K, Atmos. Chem. Phys., 22, 6001–6020, https://doi.org/10.5194/acp-22-6001-2022, 2022.
Gao, L., Buchholz, A., Li, Z., Song, J., Vallon, M., Jiang, F., Möhler, O., Leisner, T., and Saathoff, H.: Volatility of Secondary Organic Aerosol from β-Caryophyllene Ozonolysis over a Wide Tropospheric Temperature Range, Environ. Sci. Technol., 57, 8965–8974, https://doi.org/10.1021/acs.est.3c01151, 2023.
Gao, L., Manavi, S. E. I., Mohr, C., Song, J., Wu, C., Leisner, T., Pandis, S. N., and Saathoff, H.: Dataset on the manuscript: Influence of Tropospheric Temperature on the Formation and Aging of Secondary Organic Aerosol from Biogenic Vapor Mixtures, Karlsruhe Institute of Technology [data set], https://doi.org/10.35097/9zvc7fvj505n9d93, 2026.
Gkatzelis, G. I., Hohaus, T., Tillmann, R., Gensch, I., Müller, M., Eichler, P., Xu, K.-M., Schlag, P., Schmitt, S. H., Yu, Z., Wegener, R., Kaminski, M., Holzinger, R., Wisthaler, A., and Kiendler-Scharr, A.: Gas-to-particle partitioning of major biogenic oxidation products: a study on freshly formed and aged biogenic SOA, Atmos. Chem. Phys., 18, 12969–12989, https://doi.org/10.5194/acp-18-12969-2018, 2018.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansen, J., Sato, M., Ruedy, R., Lo, K., Lea, D. W., and Medina-Elizade, M.: Global temperature change, P. Natl. Acad. Sci. USA, 103, 14288–14293, https://doi.org/10.1073/pnas.0606291103, 2006.
Heald, C. L., Kroll, J. H., Jimenez, J. L., Docherty, K. S., DeCarlo, P. F., Aiken, A. C., Chen, Q., Martin, S. T., Farmer, D. K., and Artaxo, P.: A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 37, https://doi.org/10.1029/2010GL042737, 2010.
Heinritzi, M., Dada, L., Simon, M., Stolzenburg, D., Wagner, A. C., Fischer, L., Ahonen, L. R., Amanatidis, S., Baalbaki, R., Baccarini, A., Bauer, P. S., Baumgartner, B., Bianchi, F., Brilke, S., Chen, D., Chiu, R., Dias, A., Dommen, J., Duplissy, J., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Granzin, M., El Haddad, I., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kangasluoma, J., Keber, T., Kim, C., Kürten, A., Lamkaddam, H., Laurila, T. M., Lampilahti, J., Lee, C. P., Lehtipalo, K., Leiminger, M., Mai, H., Makhmutov, V., Manninen, H. E., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Molteni, U., Müller, T., Nie, W., Nieminen, T., Onnela, A., Partoll, E., Passananti, M., Petäjä, T., Pfeifer, J., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Schobesberger, S., Scholz, W., Scholze, K., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Vazquez-Pufleau, M., Virtanen, A., Vogel, A. L., Volkamer, R., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Zhou, X., Amorim, A., Baltensperger, U., Hansel, A., Kulmala, M., Tomé, A., Winkler, P. M., Worsnop, D. R., Donahue, N. M., Kirkby, J., and Curtius, J.: Molecular understanding of the suppression of new-particle formation by isoprene, Atmos. Chem. Phys., 20, 11809–11821, https://doi.org/10.5194/acp-20-11809-2020, 2020.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: The effect of temperature and water on secondary organic aerosol formation from ozonolysis of limonene, Δ3-carene and α-pinene, Atmos. Chem. Phys., 8, 6541–6549, https://doi.org/10.5194/acp-8-6541-2008, 2008.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Khamaganov, V. G. and Hites, R. A.: Rate Constants for the Gas-Phase Reactions of Ozone with Isoprene, α- and β-Pinene, and Limonene as a Function of Temperature, J. Phys. Chem. A, 105, 815–822, https://doi.org/10.1021/jp002730z, 2001.
Kiendler-Scharr, A., Wildt, J., Maso, M. D., Hohaus, T., Kleist, E., Mentel, T. F., Tillmann, R., Uerlings, R., Schurr, U., and Wahner, A.: New particle formation in forests inhibited by isoprene emissions, Nature, 461, 381–384, https://doi.org/10.1038/nature08292, 2009.
Kourtchev, I., Doussin, J.-F., Giorio, C., Mahon, B., Wilson, E. M., Maurin, N., Pangui, E., Venables, D. S., Wenger, J. C., and Kalberer, M.: Molecular composition of fresh and aged secondary organic aerosol from a mixture of biogenic volatile compounds: a high-resolution mass spectrometry study, Atmos. Chem. Phys., 15, 5683–5695, https://doi.org/10.5194/acp-15-5683-2015, 2015.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Ladino, L. A., Zhou, S., Yakobi-Hancock, J. D., Aljawhary, D., and Abbatt, J. P. D.: Factors controlling the ice nucleating abilities of α-pinene SOA particles, J. Geophys. Res., 119, 9041–9051, https://doi.org/10.1002/2014JD021578, 2014.
Lamb, R. C., Pacifici, J. G., and Ayers, P. W.: Organic Peroxides. IV. Kinetics and Products of Decompositions of Cyclohexaneformyl and Isobutyryl Peroxides. BDPA as a Free-Radical Scavenger1, J. Am. Chem. Soc., 87, 3928–3935, https://doi.org/10.1021/ja01095a024, 1965.
Lamkaddam, H., Dommen, J., Ranjithkumar, A., Gordon, H., Wehrle, G., Krechmer, J., Majluf, F., Salionov, D., Schmale, J., Bjelić, S., Carslaw, K. S., El Haddad, I., and Baltensperger, U.: Large contribution to secondary organic aerosol from isoprene cloud chemistry, Sci. Adv., 7, eabe2952, https://doi.org/10.1126/sciadv.abe2952, 2021.
Lane, T. E., Donahue, N. M., and Pandis, S. N.: Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model, Atmos. Environ., 42, 7439–7451, https://doi.org/10.1016/j.atmosenv.2008.06.026, 2008.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Leffler, J. E. and More, A. A.: Decomposition of bicyclo[2.2.2]-1-formyl and pivaloyl peroxides, J. Am. Chem. Soc., 94, 2483–2487, https://doi.org/10.1021/ja00762a048, 1972.
Li, H., Canagaratna, M. R., Riva, M., Rantala, P., Zhang, Y., Thomas, S., Heikkinen, L., Flaud, P.-M., Villenave, E., Perraudin, E., Worsnop, D., Kulmala, M., Ehn, M., and Bianchi, F.: Atmospheric organic vapors in two European pine forests measured by a Vocus PTR-TOF: insights into monoterpene and sesquiterpene oxidation processes, Atmos. Chem. Phys., 21, 4123–4147, https://doi.org/10.5194/acp-21-4123-2021, 2021.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Liu, Y., Su, H., Wang, S., Wei, C., Tao, W., Pöhlker, M. L., Pöhlker, C., Holanda, B. A., Krüger, O. O., Hoffmann, T., Wendisch, M., Artaxo, P., Pöschl, U., Andreae, M. O., and Cheng, Y.: Strong particle production and condensational growth in the upper troposphere sustained by biogenic VOCs from the canopy of the Amazon Basin, Atmos. Chem. Phys., 23, 251–272, https://doi.org/10.5194/acp-23-251-2023, 2023.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Carrasquillo, A. J., Daumit, K. E., Hunter, J. F., Kroll, J. H., Worsnop, D. R., and Thornton, J. A.: Phase partitioning and volatility of secondary organic aerosol components formed from α-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds, Atmos. Chem. Phys., 15, 7765–7776, https://doi.org/10.5194/acp-15-7765-2015, 2015.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
Mahowald, N.: Aerosol Indirect Effect on Biogeochemical Cycles and Climate, Science, 334, 794–796, https://doi.org/10.1126/science.1207374, 2011.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Möhler, O., Stetzer, O., Schaefers, S., Linke, C., Schnaiter, M., Tiede, R., Saathoff, H., Krämer, M., Mangold, A., Budz, P., Zink, P., Schreiner, J., Mauersberger, K., Haag, W., Kärcher, B., and Schurath, U.: Experimental investigation of homogeneous freezing of sulphuric acid particles in the aerosol chamber AIDA, Atmos. Chem. Phys., 3, 211–223, https://doi.org/10.5194/acp-3-211-2003, 2003.
Morino, Y., Sato, K., Jathar, S. H., Tanabe, K., Inomata, S., Fujitani, Y., Ramasamy, S., and Cappa, C. D.: Modeling the Effects of Dimerization and Bulk Diffusion on the Evaporative Behavior of Secondary Organic Aerosol Formed from α-Pinene and 1,3,5-Trimethylbenzene, ACS Earth Space Chem., 4, 1931–1946, https://doi.org/10.1021/acsearthspacechem.0c00106, 2020.
Murphy, B. N. and Pandis, S. N.: Simulating the Formation of Semivolatile Primary and Secondary Organic Aerosol in a Regional Chemical Transport Model, Environ. Sci. Technol., 43, 4722–4728, https://doi.org/10.1021/es803168a, 2009.
Owen, S. M., MacKenzie, A. R., Stewart, H., Donovan, R., and Hewitt, C. N.: BIOGENIC VOLATILE ORGANIC COMPOUND (VOC) EMISSION ESTIMATES FROM AN URBAN TREE CANOPY, Ecol. Appl., 13, 927–938, https://doi.org/10.1890/01-5177, 2003.
Paasonen, P., Asmi, A., Petäjä, T., Kajos, M. K., Äijälä, M., Junninen, H., Holst, T., Abbatt, J. P. D., Arneth, A., Birmili, W., van der Gon, H. D., Hamed, A., Hoffer, A., Laakso, L., Laaksonen, A., Richard Leaitch, W., Plass-Dülmer, C., Pryor, S. C., Räisänen, P., Swietlicki, E., Wiedensohler, A., Worsnop, D. R., Kerminen, V.-M., and Kulmala, M.: Warming-induced increase in aerosol number concentration likely to moderate climate change, Nat. Geosci., 6, 438–442, https://doi.org/10.1038/ngeo1800, 2013.
Pajunoja, A., Lambe, A. T., Hakala, J., Rastak, N., Cummings, M. J., Brogan, J. F., Hao, L., Paramonov, M., Hong, J., Prisle, N. L., Malila, J., Romakkaniemi, S., Lehtinen, K. E. J., Laaksonen, A., Kulmala, M., Massoli, P., Onasch, T. B., Donahue, N. M., Riipinen, I., Davidovits, P., Worsnop, D. R., Petäjä, T., and Virtanen, A.: Adsorptive uptake of water by semisolid secondary organic aerosols, Geophys. Res. Lett., 42, 3063–3068, https://doi.org/10.1002/2015GL063142, 2015.
Petersen, R., Holst, T., Mölder, M., Kljun, N., and Rinne, J.: Vertical distribution of sources and sinks of volatile organic compounds within a boreal forest canopy, Atmos. Chem. Phys., 23, 7839–7858, https://doi.org/10.5194/acp-23-7839-2023, 2023.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Science Advances, 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922, 2020.
Quéléver, L. L. J., Kristensen, K., Normann Jensen, L., Rosati, B., Teiwes, R., Daellenbach, K. R., Peräkylä, O., Roldin, P., Bossi, R., Pedersen, H. B., Glasius, M., Bilde, M., and Ehn, M.: Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis, Atmos. Chem. Phys., 19, 7609–7625, https://doi.org/10.5194/acp-19-7609-2019, 2019.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Saathoff, H., Naumann, K.-H., Möhler, O., Jonsson, Å. M., Hallquist, M., Kiendler-Scharr, A., Mentel, Th. F., Tillmann, R., and Schurath, U.: Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 9, 1551–1577, https://doi.org/10.5194/acp-9-1551-2009, 2009.
Schilling Fahnestock, K. A., Yee, L. D., Loza, C. L., Coggon, M. M., Schwantes, R., Zhang, X., Dalleska, N. F., and Seinfeld, J. H.: Secondary Organic Aerosol Composition from C12 Alkanes, J. Phys. Chem. A, 119, 4281–4297, https://doi.org/10.1021/jp501779w, 2015.
Schulz, C., Schneider, J., Amorim Holanda, B., Appel, O., Costa, A., de Sá, S. S., Dreiling, V., Fütterer, D., Jurkat-Witschas, T., Klimach, T., Knote, C., Krämer, M., Martin, S. T., Mertes, S., Pöhlker, M. L., Sauer, D., Voigt, C., Walser, A., Weinzierl, B., Ziereis, H., Zöger, M., Andreae, M. O., Artaxo, P., Machado, L. A. T., Pöschl, U., Wendisch, M., and Borrmann, S.: Aircraft-based observations of isoprene-epoxydiol-derived secondary organic aerosol (IEPOX-SOA) in the tropical upper troposphere over the Amazon region, Atmos. Chem. Phys., 18, 14979–15001, https://doi.org/10.5194/acp-18-14979-2018, 2018.
Sheehan, P. E. and Bowman, F. M.: Estimated Effects of Temperature on Secondary Organic Aerosol Concentrations, Environ. Sci. Technol., 35, 2129–2135, https://doi.org/10.1021/es001547g, 2001.
Shiraiwa, M., Ammann, M., Koop, T., and Pöschl, U.: Gas uptake and chemical aging of semisolid organic aerosol particles, P. Natl. Acad. Sci. USA, 108, 11003–11008, https://doi.org/10.1073/pnas.1103045108, 2011.
Shiraiwa, M., Li, Y., Tsimpidi, A. P., Karydis, V. A., Berkemeier, T., Pandis, S. N., Lelieveld, J., Koop, T., and Pöschl, U.: Global distribution of particle phase state in atmospheric secondary organic aerosols, Nat. Commun., 8, 15002, https://doi.org/10.1038/ncomms15002, 2017.
Simon, M., Dada, L., Heinritzi, M., Scholz, W., Stolzenburg, D., Fischer, L., Wagner, A. C., Kürten, A., Rörup, B., He, X.-C., Almeida, J., Baalbaki, R., Baccarini, A., Bauer, P. S., Beck, L., Bergen, A., Bianchi, F., Bräkling, S., Brilke, S., Caudillo, L., Chen, D., Chu, B., Dias, A., Draper, D. C., Duplissy, J., El-Haddad, I., Finkenzeller, H., Frege, C., Gonzalez-Carracedo, L., Gordon, H., Granzin, M., Hakala, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kong, W., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Mai, H., Manninen, H. E., Marie, G., Marten, R., Mentler, B., Molteni, U., Nichman, L., Nie, W., Ojdanic, A., Onnela, A., Partoll, E., Petäjä, T., Pfeifer, J., Philippov, M., Quéléver, L. L. J., Ranjithkumar, A., Rissanen, M. P., Schallhart, S., Schobesberger, S., Schuchmann, S., Shen, J., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Tomé, A. R., Vazquez-Pufleau, M., Vogel, A. L., Wagner, R., Wang, M., Wang, D. S., Wang, Y., Weber, S. K., Wu, Y., Xiao, M., Yan, C., Ye, P., Ye, Q., Zauner-Wieczorek, M., Zhou, X., Baltensperger, U., Dommen, J., Flagan, R. C., Hansel, A., Kulmala, M., Volkamer, R., Winkler, P. M., Worsnop, D. R., Donahue, N. M., Kirkby, J., and Curtius, J.: Molecular understanding of new-particle formation from α-pinene between −50 and +25 °C, Atmos. Chem. Phys., 20, 9183–9207, https://doi.org/10.5194/acp-20-9183-2020, 2020.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Sofiev, M., Vankevich, R., Lanne, M., Koskinen, J., and Kukkonen, J.: On Integration Of A Fire Assimilation System And A Chemical Transport Model For Near-realtime Monitoring Of The Impact Of Wild-land Fires On Atmospheric Composition And Air Quality, WIT Trans. Ecol. Envir., 119, 343–351, 2008.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Surdu, M., Top, J., Yang, B., Zhang, J., Slowik, J. G., Prévôt, A. S. H., Wang, D. S., el Haddad, I., and Bell, D. M.: Real-Time Identification of Aerosol-Phase Carboxylic Acid Production Using Extractive Electrospray Ionization Mass Spectrometry, Environ. Sci. Technol., 58, 8857–8866, https://doi.org/10.1021/acs.est.4c01605, 2024.
Takeuchi, M., Berkemeier, T., Eris, G., and Ng, N. L.: Non-linear effects of secondary organic aerosol formation and properties in multi-precursor systems, Nat. Commun., 13, 7883, https://doi.org/10.1038/s41467-022-35546-1, 2022.
Tripathi, N., Krumm, B. E., Edtbauer, A., Ringsdorf, A., Wang, N., Kohl, M., Vella, R., Machado, L. A. T., Pozzer, A., Lelieveld, J., and Williams, J.: Impacts of convection, chemistry, and forest clearing on biogenic volatile organic compounds over the Amazon, Nat. Commun., 16, 4692, https://doi.org/10.1038/s41467-025-59953-2, 2025.
Trump, E. R. and Donahue, N. M.: Oligomer formation within secondary organic aerosols: equilibrium and dynamic considerations, Atmos. Chem. Phys., 14, 3691–3701, https://doi.org/10.5194/acp-14-3691-2014, 2014.
Tsimpidi, A. P., Karydis, V. A., Zavala, M., Lei, W., Molina, L., Ulbrich, I. M., Jimenez, J. L., and Pandis, S. N.: Evaluation of the volatility basis-set approach for the simulation of organic aerosol formation in the Mexico City metropolitan area, Atmos. Chem. Phys., 10, 525–546, https://doi.org/10.5194/acp-10-525-2010, 2010.
Vallon, M., Gao, L., Jiang, F., Krumm, B., Nadolny, J., Song, J., Leisner, T., and Saathoff, H.: LED-based solar simulator to study photochemistry over a wide temperature range in the large simulation chamber AIDA, Atmos. Meas. Tech., 15, 1795–1810, https://doi.org/10.5194/amt-15-1795-2022, 2022.
Visschedijk, A., Zandveld, P., and Denier Van Der Gon, H.: A High Resolution Gridded European Emission Database for the EU Integrated Project GEMS, TNO-Report, Bedford, A771, 2007.
Wagner, R., Bunz, H., Linke, C., Möhler, O., Naumann, K.-H., Saathoff, H., Schnaiter, M., and Schurath, U.: Chamber Simulations of Cloud Chemistry: The AIDA Chamber, in: Environmental Simulation Chambers: Application to Atmospheric Chemical Processes. Nato Science Series: IV: Earth and Environmental Science, edited by: Barnes, I. and Rudzinski, K. J., vol. 62, Springer, Dordrecht, https://doi.org/10.1007/1-4020-4232-9_5, 2006.
Wang, Y., Zhao, Y., Li, Z., Li, C., Yan, N., and Xiao, H.: Importance of Hydroxyl Radical Chemistry in Isoprene Suppression of Particle Formation from α-Pinene Ozonolysis, ACS Earth Space Chem., 5, 487–499, https://doi.org/10.1021/acsearthspacechem.0c00294, 2021.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the Volatility of Secondary Organic Aerosol from Isoprene Photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Yáñez-Serrano, A. M., Nölscher, A. C., Bourtsoukidis, E., Gomes Alves, E., Ganzeveld, L., Bonn, B., Wolff, S., Sa, M., Yamasoe, M., Williams, J., Andreae, M. O., and Kesselmeier, J.: Monoterpene chemical speciation in a tropical rainforest:variation with season, height, and time of dayat the Amazon Tall Tower Observatory (ATTO), Atmos. Chem. Phys., 18, 3403–3418, https://doi.org/10.5194/acp-18-3403-2018, 2018.
Yáñez-Serrano, A. M., Bourtsoukidis, E., Alves, E. G., Bauwens, M., Stavrakou, T., Llusià, J., Filella, I., Guenther, A., Williams, J., Artaxo, P., Sindelarova, K., Doubalova, J., Kesselmeier, J., and Peñuelas, J.: Amazonian biogenic volatile organic compounds under global change, Glob. Change Biol., 26, 4722–4751, https://doi.org/10.1111/gcb.15185, 2020.
Ye, Q., Wang, M., Hofbauer, V., Stolzenburg, D., Chen, D., Schervish, M., Vogel, A., Mauldin, R. L., Baalbaki, R., Brilke, S., Dada, L., Dias, A., Duplissy, J., El Haddad, I., Finkenzeller, H., Fischer, L., He, X., Kim, C., Kürten, A., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Manninen, H. E., Marten, R., Mentler, B., Partoll, E., Petäjä, T., Rissanen, M., Schobesberger, S., Schuchmann, S., Simon, M., Tham, Y. J., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Wu, Y., Xiao, M., Baltensperger, U., Curtius, J., Flagan, R., Kirkby, J., Kulmala, M., Volkamer, R., Winkler, P. M., Worsnop, D., and Donahue, N. M.: Molecular Composition and Volatility of Nucleated Particles from α-Pinene Oxidation between −50 °C and +25 °C, Environ. Sci. Technol., 53, 12357–12365, https://doi.org/10.1021/acs.est.9b03265, 2019.
Zhang, X., McVay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 112, 14168–14173, https://doi.org/10.1073/pnas.1517742112, 2015.
Zhao, J., Häkkinen, E., Graeffe, F., Krechmer, J. E., Canagaratna, M. R., Worsnop, D. R., Kangasluoma, J., and Ehn, M.: A combined gas- and particle-phase analysis of highly oxygenated organic molecules (HOMs) from α-pinene ozonolysis, Atmos. Chem. Phys., 23, 3707–3730, https://doi.org/10.5194/acp-23-3707-2023, 2023.