the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Apr 2026
| 27 Apr 2026
Rapid secondary organic aerosol formation at the air–water interface from methoxyphenols in wildfire emissions: UVA-driven S(IV) photooxidation to organosulfates
Baohua Cai
Yuanlong Huang
Wenqing Jiang
Yanchen Li
Yali Li
Jinghao Zhai
Yaling Zeng
Jianhuai Ye
Huizhong Shen
Chen Wang
Tzung-May Fu
Wildfire emissions release large amounts of methoxyphenols, which serve as key precursors of aqueous-phase secondary organic aerosols (SOA). Their transformation is closely coupled with aqueous S(IV) oxidation, jointly driving the formation of sulfate and organosulfates; however, the underlying mechanisms remain poorly understood. Here, we identify a metal-free, UVA-driven mechanism for sulfate radicals () generation at 370 nm, supported by laboratory experiments and quantum chemical calculations. Photolysis of the [] complex yields a [] pair; the radical subsequently reacts with O2 to form peroxomonosulfate (), which then oxidizes S(IV) to produce . These sulfate radicals rapidly oxidize guaiacol, a representative biomass burning phenol, in bulk solution, producing SOA enriched in organosulfates. Microdroplet experiments show ∼100-fold rate enhancement due to interfacial effects. Box modeling indicates that this aqueous UVA pathway represents a potentially important and previously underappreciated source of sulfate. This work establishes a photochemical link between S(IV) oxidation and SOA formation, with implications for aerosol composition, oxidative capacity, and climate-relevant processes.
- Article
(3473 KB) - Full-text XML
-
Supplement
(2235 KB) - BibTeX
- EndNote




Wildfires are occurring with increasing frequency, intensifying climate perturbation and exacerbating human health risks (Zhao et al., 2025; Teymoor Seydi et al., 2025). Their emissions, rich in methoxyphenols and other semi-volatile organic compounds, readily partition into cloud and aerosol water, where they undergo rapid transformations that produce substantial amounts of SOA (He et al., 2024; Li et al., 2023a; Liu et al., 2022). These aqueous-phase reactions are not isolated; rather, they are intricately coupled with other atmospheric chemical processes, resulting in complex multiphase chemistry that remains poorly understood.
Sulfate is a major component of fine particulate matter (PM), with significant impacts on air quality and public health (Wang et al., 2016; Abbatt et al., 2006). In the atmosphere, sulfate forms primarily through gas-phase SO2 oxidation by hydroxyl radicals (•OH) and aqueous-phase oxidation of S(IV) species, such as dissolved SO2, , and , in cloud, fog, or aerosol water. The aqueous-phase pathway includes direct oxidation by H2O2 (Liu et al., 2020), O3 (Hoffmann, 1986; Lan et al., 2011), and NO2 (Zhang and Chan, 2023; Gao et al., 2022; Liu and Abbatt, 2021); catalytic oxidation mediated by transition metal ions (e.g., Fe and Mn) (Zuo et al., 2005; Wang et al., 2021; Harris et al., 2013; Brandt and van Eldik, 1995); and photocatalytic processes involving humic-like substances (HULIS) in the presence of O2 (Wang et al., 2024; Pan et al., 2024). Despite extensive research, substantial discrepancies remain between observed sulfate levels and model predictions, indicating missing or poorly characterized pathways (Zheng et al., 2015).
Beyond sulfate formation, aqueous S(IV) oxidation can also form organosulfates (OSs) in the presence of volatile organic compounds (VOCs) (Passananti et al., 2016; Duporté et al., 2020; Surratt et al., 2008; Iinuma et al., 2007; Darer et al., 2011; Riva et al., 2015). OSs constitute a substantial fraction (e.g., 5 %–30 %) of the organic mass in atmospheric PM (Shakya and Peltier, 2015; Tolocka, 2012; Hughes et al., 2021; Romero and Oehme, 2005) and provide an important chemical link between sulfur and organic aerosols. As amphiphilic molecules, OSs affect aerosol surface activity and hygroscopicity (Riva et al., 2019), thereby enhancing their potential to act as cloud condensation nuclei (CCN) (Peng et al., 2021). Some OSs are also linked to adverse health outcomes, including oxidative stress and proinflammatory responses in human lung cells (Khan et al., 2023).
The sulfate radical () is a highly reactive intermediate in aqueous-phase S(IV) oxidation (Rudzinski et al., 2009), capable of rapidly oxidizing a wide variety of VOCs, including aldehydes (Coddens et al., 2018; Tran et al., 2022), olefins (Schindelka et al., 2013; Ren et al., 2021), phenols (Cope et al., 2022), and polycyclic aromatic hydrocarbons (Wang et al., 2008). These reactions produce oxidized organics that can subsequently form OSs through acid-catalyzed esterification or radical termination reactions. Solar radiation is a key driver of such radical chemistry, including the generation of (George et al., 2015; Herrmann et al., 2015). For example, in high-ionic-strength aerosol solutions (e.g., 3.7 M ammonium sulfate), forms under UVB (∼310 nm) irradiation, reaching steady-state concentrations near 10−12 M (Cope et al., 2022). While direct photolysis of S(IV) species by UVC radiation can also yield (Cao et al., 2021), UVC is largely absorbed by the stratosphere and thus negligible in the troposphere.
Although UVA radiation is the dominant ultraviolet solar band at the Earth's surface, its role in aqueous S(IV) oxidation remains poorly understood. A recent study suggests that UVA light can promote SO2 oxidation at the air-water interface (Gong et al., 2022), but the mechanisms and broader implications are unclear. To address this gap, we combined laboratory experiments with quantum chemical calculations to investigate a novel, metal-free UVA-induced pathway for generation. Using guaiacol (GUA), a representative biomass burning phenol, as a molecular probe, we tracked radical activity and OSs formation. We also explored how droplet microphysics and interfacial effects enhance this chemistry. Our findings reveal a previously overlooked UVA-driven mechanism for sulfate and OSs formation, with important implications for atmospheric chemistry, air quality, and climate.
2.1 Materials
Guaiacol (99 %), sodium sulfite (Na2SO3, >99 %), 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO, 98 %), ethanol (99 %), and tert-butanol (99 %) were purchased from Macklin. Zero air is made up of 21 % O2 and 79 % N2. All water used in the experiments was ultrapure Milli-Q water (18.2 MΩ cm−1).
2.2 Experimental methods
Bulk aqueous experiment. All experiments were performed in a 25 mL airtight Pyrex tube equipped with a magnetic stir bar and a gas inlet tube for feeding high-purity zero air or nitrogen (∼0.4 L min−1) under 370 nm light or Xenon lamp (300w) irradiation. Based on preliminary experiments, we identified the critical role of UVA irradiation in driving the reaction; therefore, a high-power UVA lamp was employed to ensure sufficient photon flux and to obtain reliable kinetic data (see Supplement, Figs. S1–S4). A 20 mL reaction solution containing guaiacol, Na2SO3, and other reactants was prepared. The pH of the reaction solution was adjusted using H2SO4 and NaOH and measured with a pH meter REDOX potentiometer Conductivity meter (AZ-86555) that was calibrated with commercial pH standards. In experiments requiring the measurement of total inorganic sulfur, the pH is adjusted with either phosphoric acid or phosphate. Aliquots (3 mL) were sampled every 20 min for 1 h, with 0.30 mL MeOH added immediately to quench the reaction. Each experiment was repeated at least twice.
HPLC analysis. The concentrations of the guaiacol and phenol were determined using an HPLC (Thermo Scientific™ UltiMate™ 3000) equipped with a diode array detector (DAD) and an Agilent 5 TC-C18 column (150×4.60 mm, 5 µm). The column temperature was maintained at 25 °C, and the flow rate was set to 1 mL min−1. Detection was performed at 274 nm. The mobile phase consisted of () acetonitrile/water acidified with 0.1 % trifluoroacetic acid (TFA).
Direct infusion HRMS. Reaction solutions were filtered through a membrane and then directly introduced into an Agilent 6546 quadrupole time-of-flight mass spectrometer (QTOF-MS, Santa Clara, CA) with electrospray ionization (ESI) source in negative mode. The MS parameters were as follows: nebulizer, 25 psi; gas flow, 10 L min−1; sheath gas temperature, 330 °C; capillary voltage, 3500 V; sheath gas flow, 12 L min−1. MS data were collected in an range of 90–500. Agilent MassHunter Qualitative Analysis software (version 10.0) was used for data analysis.
UV-vis spectroscopy. An ultraviolet-visible Spectrophotometer (Youke, T2602, Shanghai, China) was used to monitor guaiacol absorbance during reaction with Na2SO3 and to record sample spectra from 200 to 500 nm. The reaction solution was directly loaded without dilution or modification. Spectra of the guaiacol – Na2SO3 reaction were collected every 20 min for 1 h.
IC measurements. Sulfite () and sulfate () concentrations were analyzed using a Metrohm 883 Basic IC system quipped with a Metrosep A supply 5-250/4.0 analytical column and a conductivity detector (Liu et al., 2025). Prior to analysis, 2 % isopropanol and 1.0 mM NaOH was added into the reaction solution. The eluent used was 3.2 mM Na2CO3/1.0 mM NaHCO3, with a flow rate of 0.8 mL min−1. For total inorganic sulfur analysis, samples were pre-oxidized to using hydrogen peroxide (H2O2) before IC detection.
FIDI-MS experiment. The working principle of FIDI-MS (field-induced droplet ionization mass spectrometry) is described as follows (Gong et al., 2022). Droplets approximately 2 mm in diameter (∼4 µL volume) were suspended from the tip of a stainless-steel capillary, which was positioned equidistantly between two parallel plate electrodes separated by 6.3 mm apart. The droplets were formed by injecting the analyte solution through the capillary using a syringe pump. The chemical composition and solute concentrations of the suspended droplets were identical to those of the corresponding bulk stock solutions used for droplet generation. The parallel plates were mounted on a translation stage to align the front electrode's aperture with the atmospheric pressure inlet of a Thermo-Fischer LTQ-XL mass spectrometer (Waltham, MA), which was operated under laboratory ambient air conditions at a relative humidity of approximately 50 %. Once the droplets were formed, a 60 s equilibration period was allowed to enable compound diffusion and achieve equilibrium coverage at the air-water interface.
Sampling of the suspended droplets was accomplished by applying a high-voltage pulse (3–5 kV, 100 ms duration, variable polarity) to the rear electrode and capillary, with half the voltage simultaneously applied to the rear plate, thereby establishing a uniform electric field. This field induced a dipole in the suspended droplets, causing it to elongate and form a double Taylor cone at both ends, which ejected oppositely charged submicron-sized droplets. These negatively charged droplets passed through the aperture of the front plate and entered the mass spectrometer for gas-phase ion detection. Due to the significant disturbance caused by the ionization droplet interface (IDI) sampling, a new droplet was generated for each measurement. In this study, a negative voltage polarity was applied to the rear plate and capillary to facilitate detection of deprotonated guaiacol ions ([GUA]−).
HR-ToF-AMS experiment. During photochemical experiments, reaction solutions were aerosolized with a constant output atomizer (TSI Inc.) using N2 as the carrier gas. The resulting aerosols were dried with a diffusion dryer and then introduced into a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Research, Inc.) for chemical characterization. Drying allowed evaporation of volatile and semi-volatile species; therefore, the AMS primarily measured the mass concentration and bulk composition of the remaining low-volatility products. The operating principles of AMS have been described previously (Decarlo et al., 2006; Canagaratna et al., 2007). Briefly, the AMS analyzes non-refractory aerosols that vaporize at ∼600 °C under high vacuum via 70 eV electron impact ionization. In this study, the AMS was operated in “V” ion optical mode (mass resolution ∼3000) to acquire mass spectra up to 422. The AMS data were processed with the standard AMS toolkits SQUIRREL (v1.67) and PIKA (v1.27), available at http://cires.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/ (last access: 23 April 2026).
2.3 Theoretical calculations
DFT and TDDFT calculations. Geometry optimizations and frequency calculation for all molecular structures (reactants, products, and transition states) were performed using the M06-2X (Zhao and Truhlar, 2007) functional with the ma-TZVP basis set (Zheng et al., 2010), employing the SMD solvation model (Marenich et al., 2009) to simulate aqueous-phase effects in water, as implemented in the Gaussian 16 software package (Frisch et al., 2016). Optimized structures were verified by frequency computations to confirm local minima (zero imaginary frequencies) or transition structures (single imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were performed to ensure that the first-order saddle points found were true transition states (TS) connecting the reactants and the products. Single-point energy calculations and solvation effects were evaluated at the CCSD(T)/aug-cc-pVTZ (Guo et al., 2018; Noga and Bartlett, 1987) level using the SMD solvation model, with geometries optimized at M06-2X/ma-TZVP and zero-point energy (ZPE) correction applied. The calculations were carried out using the ORCA 5.0.3 program package (Neese, 2025). Multiwfn 3.8 (Lu and Chen, 2012) and Shermo 2.4 (Lu and Chen, 2021) were used for further data analysis.
Classical MD calculations. Classical molecular dynamics (MD) calculations were performed using GROMACS 4.5.5 (Hess et al., 2008). In a cubic box with periodic boundary conditions, the system consisted of 1000 SPC/E water molecules and one GUA molecule using the OPLS-AA force field. Electrostatics were treated with the particle-mesh Ewald (PME) method; van der Waals interactions were truncated at 10 Å. A leap-frog integrator was used with a 2 fs timestep, and the trajectories were recorded every 10 steps.
Umbrella Sampling: To determine the average volume for each system, 10 ns simulations were conducted in the NVT ensemble, where the temperature was set to 300 K using the V-rescale method. The potentials of mean force (PMF) were calculated using the Weighted Histogram Analysis Method (WHAM) calculations, which were performed in one additional 10 ns simulation with initial configurations from the preceding simulations. The GUA moved in the z-dimension around their frozen positions under a harmonic restoring force. The force constant was set at 1×103 (), and configurations were recorded every 0.5 ps. Visualization and trajectory analysis were implemented using VMD (Humphrey et al., 1996).
2.4 Model calculation
Box model conditions. Based on the empirically determined apparent photooxidation rate constants of S(IV) under UVA irradiation, the apparent photon efficiency (APE) was calculated (see Supplement). Assuming that the APE remains constant, the apparent rate constants under UVA irradiation corresponding to the AM0 standard solar spectrum (Seinfeld and Pandis, 2016) were then derived (derivation details are provided in the Supplement). Sulfate production rates at 271 K were calculated for different aqueous-phase reaction pathways with O3, H2O2, TMIs, and NO2, following Cheng (Cheng et al., 2016), excluding ionic strength effects.
The Henry's law constants at 271 K for SO2, O3, H2O2, and NO2 are 3.521 M atm−1, 0.025 M atm−1, 1.147×106 M atm−1, and M atm−1, respectively. Equilibrium constants for SO2⋅H2O are KS1=0.025 M and M (Cheng et al., 2016).
Scenario Conditions. “Cloud droplets” scenario: [SO2(g)]=5 ppb, [NO2(g)]=1 ppb, [H2O2(g)]=1 ppb, [O3(g)]=50 ppb, [Fe(III)]=0.3 µM, [Mn(II)]=0.03 µM, liquid water content (LWC)=0.1 g m−3.
“Beijing haze” scenario: [SO2(g)]=40 ppb, [NO2(g)]=66 ppb, [H2O2(g)]=0.01 ppb, [O3(g)]=1 ppb, LWC=300 µg m−3. The concentrations of Fe(III) and Mn(II) were assumed to vary with pH (Cheng et al., 2016).
The sulfate formation rate was calculated using the following equation.
3.1 Photooxidation of Na2SO3 solution under 370 nm irradiation
To investigate the photodegradation of GUA in sodium sulfite (Na2SO3) solutions under UVA irradiation, we first examined the photooxidation behavior of Na2SO3 in the UVA region. Na2SO3 solutions with controlled initial pH were prepared and continuously bubbled with zero air (Fig. S5). At pH 4.0 and 0.5 mM Na2SO3, sulfite loss in the dark was slow, with an observed rate constant of s−1 (Fig. S6) (Brandt and van Eldik, 1995). Under UVA irradiation (370 nm), the sulfite loss rate increased nearly tenfold to s−1, with sulfate () as the primary product (Fig. S7). Increasing Na2SO3 concentration to 2.0 mM had only a moderate effect, with rate constants averaging (2.41 ± 0.79) × 10−4 s−1 (Figs. S8 and S9). In contrast, pH significantly influenced photooxidation kinetics (Fig. S10): the apparent sulfite decay rate increased by nearly 14-fold from pH 4.0 to 7.0, reflecting shifts in dominant S(IV) species ( vs. ) with different photochemical reactivities. These findings demonstrate that UVA light substantially enhances S(IV) oxidation in metal-free systems and that the reaction is strongly pH-dependent.
3.2 Photodegradation of guaiacol in Na2SO3 solution
Guaiacol (GUA), a methoxyphenol emitted primarily from biomass burning (4.7 Tg yr−1 globally) (Liu et al., 2022; Li et al., 2023a), was used as a molecular probe to trace reactive intermediates formed during UVA-driven S(IV) oxidation. Given its Henry's law constant (McFall et al., 2020), up to 40 % of atmospheric GUA can partition into the aqueous phase (Fig. S11), making it a relevant proxy for aqueous-phase organic transformations.
At pH 4.0, GUA (0.1 mM) was added to 2.0 mM Na2SO3 solution under continuous zero-air bubbling. GUA remained stable in the dark, with minor losses attributed to evaporation. Under UVA irradiation (370 nm), however, it degraded rapidly following pseudo-first-order kinetics (k≈0.023 min−1; Fig. S12a) that is approximately 14 times faster than direct photolysis (Fig. S12b), highlighting the critical role of S(IV)-derived reactive intermediates. Suppressing O2 via N2 purging significantly reduced GUA degradation (Fig. 1a), confirming the importance of O2-dependent photochemistry induced by UVA.
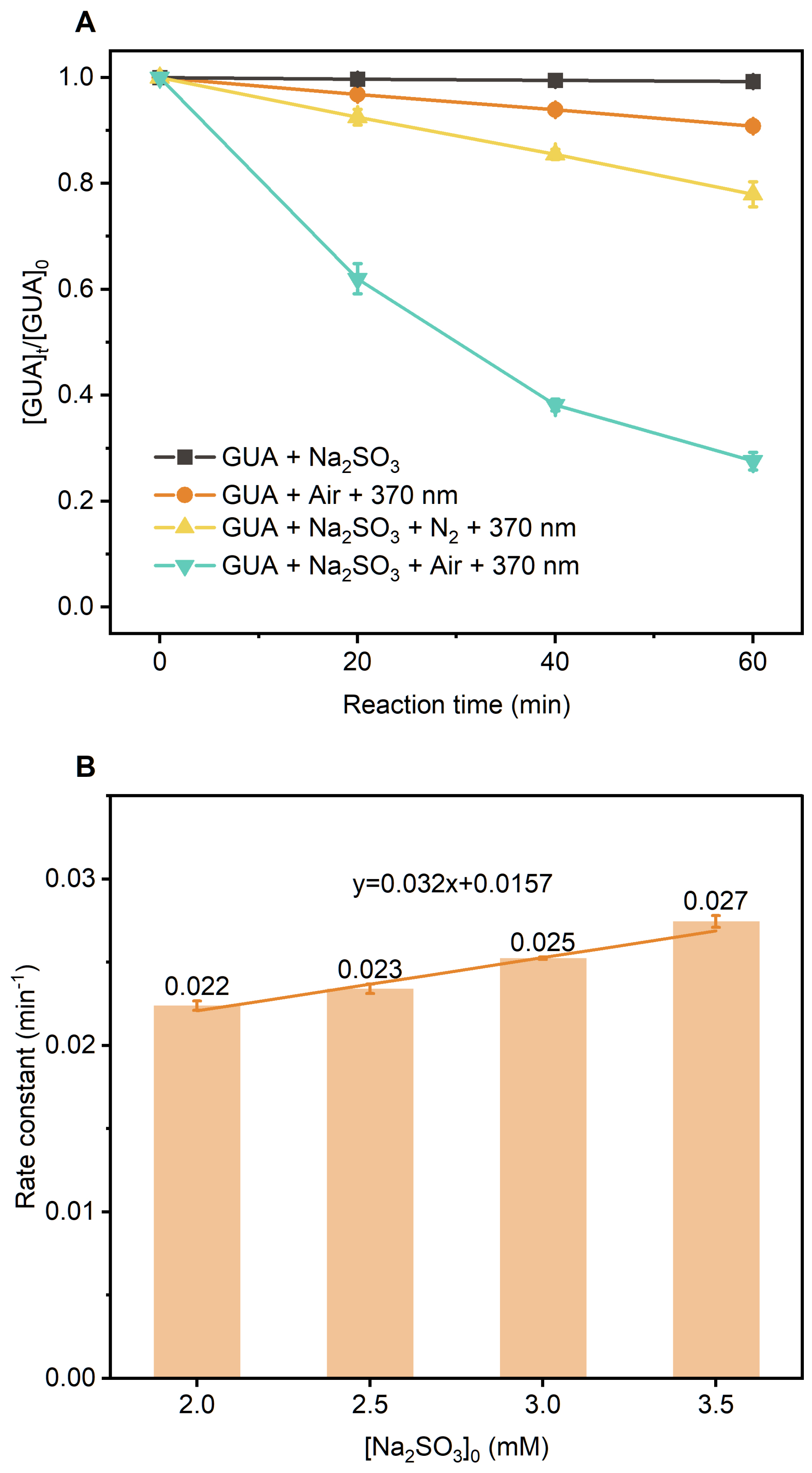
Figure 1(a) Kinetics of the aqueous-phase reaction between guaiacol and Na2SO3 under different conditions. (b) The dependence of the pseudo-first-order rate constant for GUA decay on the concentration of Na2SO3. Error bars represent the standard deviations from independent experiments. Experimental conditions: [guaiacol]=0.1 mM, [Na2SO3]=2.0 mM, pH=4.0 ± 0.1, zero-air bubbling, 370 nm light irradiation, room temperature.
We further investigated how reagent concentrations influence degradation kinetics. At high Na2SO3 : GUA molar ratios (≥20), GUA degradation followed pseudo-first-order kinetics, with rates increasing linearly with Na2SO3 concentration (Fig. 1b). At lower ratios, deviations from first-order behavior were observed (Fig. S13), suggesting a shift in the limiting reagent or changes in radical propagation dynamics.
3.3 Formation of organosulfates and steady-state concentration
Figure S14 presents the kinetics of UVA-irradiated solutions containing 0.1 mM GUA and 0.5 mM Na2SO3. The apparent oxidation rate constant for was s−1 (Fig. S15), about three times higher than that without GUA ( s−1) (Fig. S7b), indicating that GUA significantly promoted S(IV) oxidation. The concurrent decrease in total inorganic sulfur closely tracked GUA degradation, suggesting that GUA reacted with photochemically generated intermediates to form S-containing organic species, such as organosulfates (OSs).
High-resolution mass spectrometry (HRMS; ) was used to identify reaction products. Negative-mode ESI analysis of a solution containing 0.1 mM GUA and 2.0 mM Na2SO3 at pH 4.0 (Fig. 2a) revealed unreacted GUA (, ) along with multiple sulfate ester derivatives: C6H5O5S− (), C7H7O5S− (), C7H7O6S− (), and C7H7O7S− (. These signals indicate OSs formation from GUA reacting with radicals photochemically generated from and O2 under UVA.
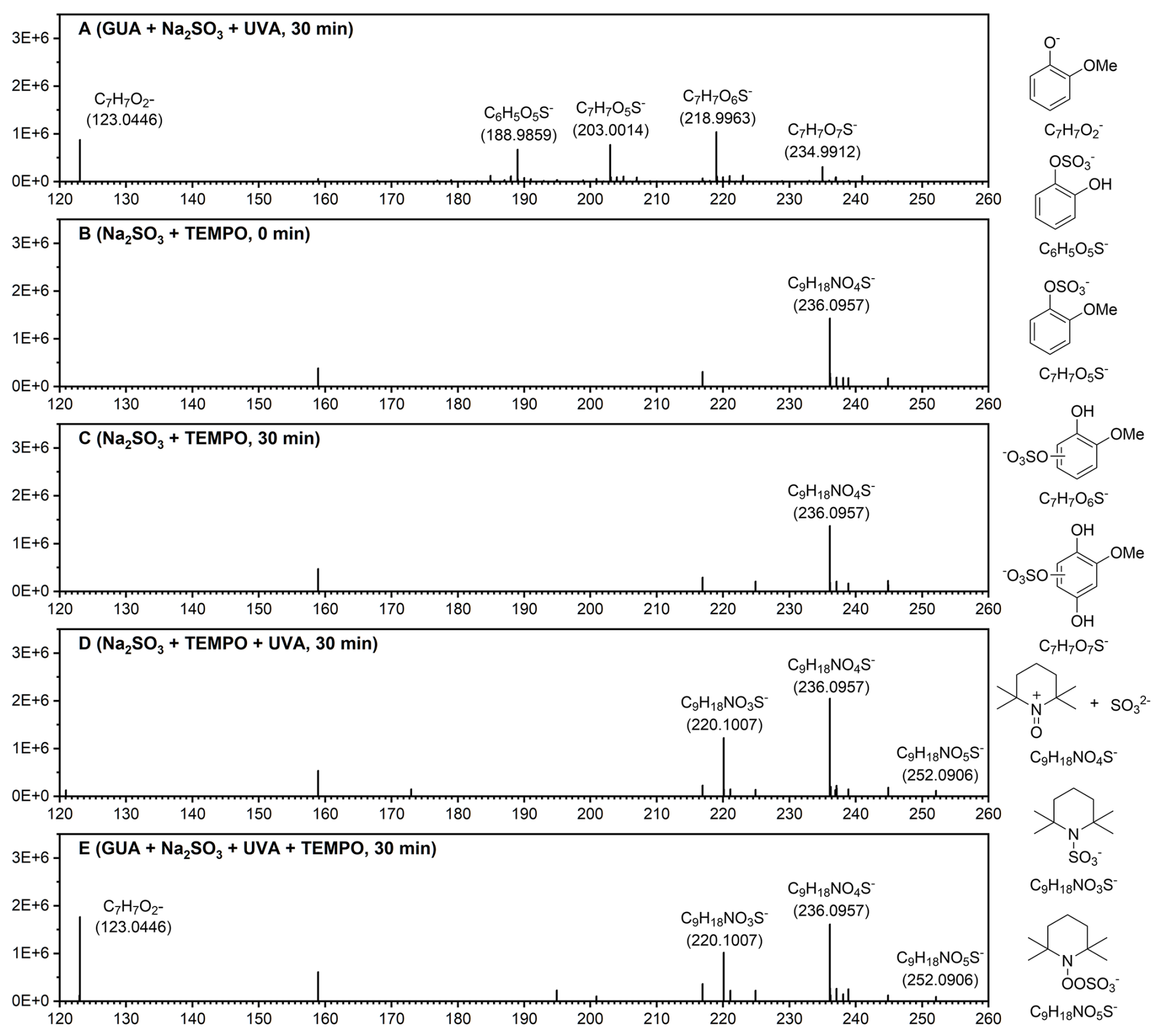
Figure 2High-resolution mass spectra of reaction products from: (a) GUA+Na2SO3 under after 30 min of 370 nm irradiation; (b) Na2SO3+TEMPO at 0 min; (c) Na2SO3+TEMPO after 30 min in the dark; (d) Na2SO3+TEMPO after 30 min of 370 nm irradiation; and (e) after 30 min of 370 nm irradiation. Experimental conditions: [guaiacol]=0.1 mM, [Na2SO3]=2.0 mM, [TEMPO]=4.0 mM, pH=4.0 ± 0.1, zero-air bubbling, and room temperature. Proposed chemical structures corresponding to the key mass spectral peaks are shown to the right of the spectra.
To verify involvement, we introduced 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO; C9H18NO) as a radical scavenger (Bai et al., 2016). In the Na2SO3+TEMPO system without UVA (Fig. 2b) or after 30 min in the dark (Fig. 2c), only the TEMPO+ – adduct (C9H18NO4S−, ) was observed. However, under 370 nm irradiation, new peaks appeared at and 220.1019, corresponding to the TEMPO- adduct (C9H18NO5S−) and its O2-loss fragment (C9H18NO3S−, Fig. 2d), respectively, confirming generation.
Finally, adding TEMPO to the (Fig. 2e) eliminated all OS peaks, leaving only signals for the TEMPO- adduct and its fragment. This demonstrates that TEMPO scavenged and suppressed GUA-derived OS formation, confirming as the key intermediate driving the observed OSs production.
can also oxidize water or OH− to form hydroxyl radicals (•OH) (Wojnárovits and Takács, 2019), which effectively oxidize GUA in aqueous phase (Yu et al., 2014). To assess the relative contributions of •OH versus , we used ethanol (EtOH) and tert-butyl alcohol (tBuOH) as radical scavengers: EtOH reacts rapidly with both •OH (1.2×109 ) and (1.6×107 ), while tBuOH reacts primarily with •OH (3.8×108 ) and only weakly with (4×105 ) (Liang and Su, 2009). At pH 4.0, adding 0.5 M EtOH significantly suppressed GUA photodegradation, while tBuOH had little effect, supporting as the dominant oxidant (Fig. S16).
The kinetics of the reaction between GUA and were assessed using a relative rate method with phenol as the reference compound (Fig. S17) (Tran et al., 2022; Liang and Su, 2009). After correcting for direct photodegradation, the results indicate that GUA reacts very rapidly with , with an effective rate approaching the diffusion-controlled regime under the experimental conditions. It is important to note that these values represent condition-dependent, relative estimates rather than absolute intrinsic rate constants. Given the uncertainties inherent in the relative rate approach (e.g., reference rate selection, radical distribution, and irradiation heterogeneity), the derived rate should be interpreted as an upper-limit estimate of reactivity. The observed fast kinetics are nevertheless consistent with prior quantum chemical calculations (Li et al., 2023b), supporting the high reactivity of GUA toward sulfate radicals..
3.4 Photochemical pathway of formation from Na2SO3 under UVA irradiation
In aqueous Na2SO3, the primary S(IV) species are SO2⋅H2O, , and , with dominant under the experimental pH range (Fig. S18). At pH 4.0, Na2SO3 showed nearly no UV-vis absorption above 250 nm (Fig. 3a). Adding 0.1 mM GUA introduced a strong 274 nm peak from π–π∗ transitions in GUA's aromatic ring. Although initial UVA absorption was minimal, it increased markedly during irradiation, indicating the formation of new light-absorbing products.
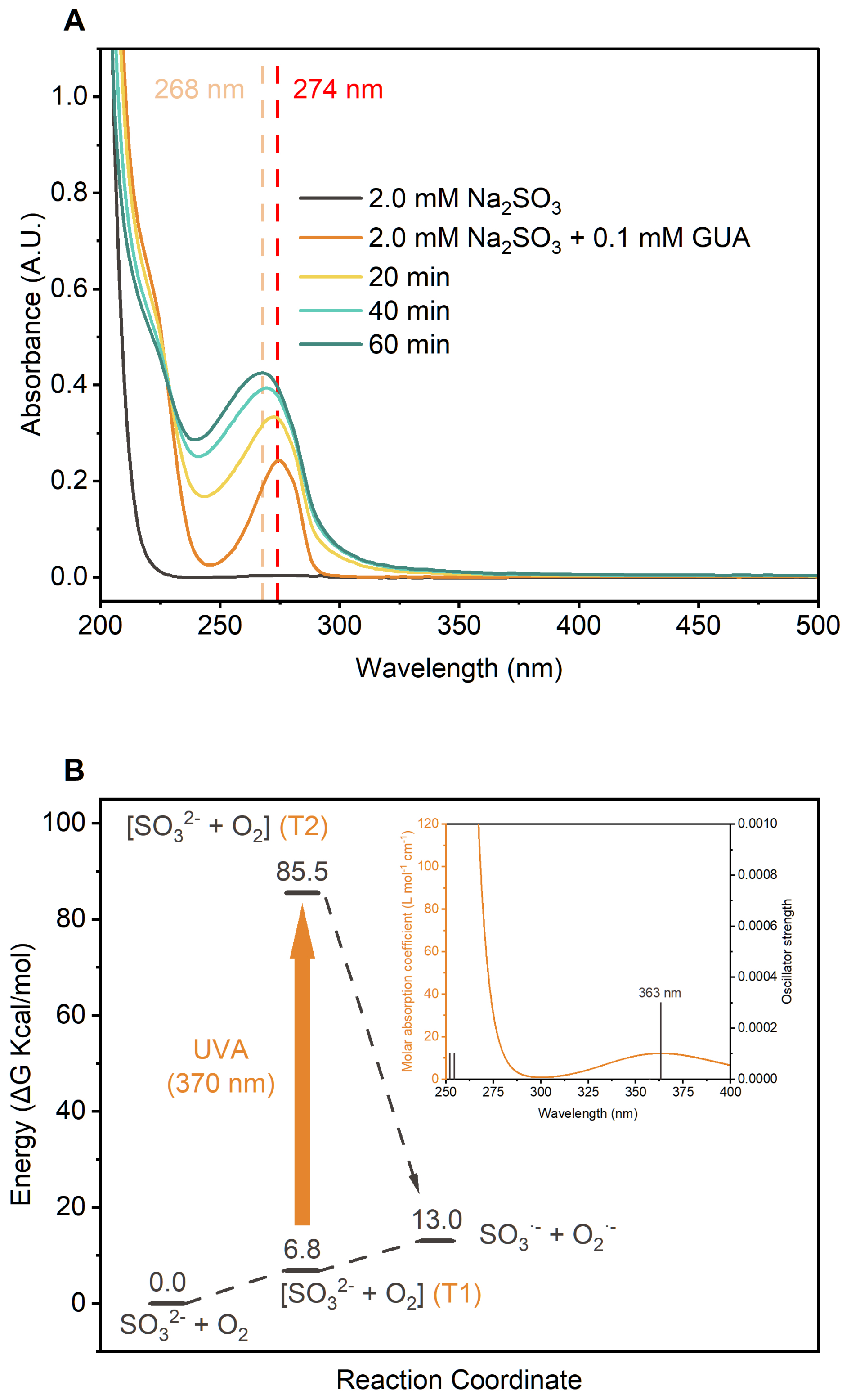
Figure 3(a) UV-vis absorption spectra of the GUA+Na2SO3 reaction solution at different time points. (b) Gibbs free energy profiles (kcal mol−1, 298.15 K) for the reaction, calculated at the CCSD(T)/aug-cc-pVTZ/SMD(water)//M06-2X/ma-TZVP/SMD(water) level with Zero Point Energy (ZPE) correction, with the inset showing the vertical excitation spectra of the complex, calculated using TDDFT at the M06-2X/ma-TZVP/SMD(water) level.
Since 370 nm UVA light (∼3.35 eV) lacks sufficient energy to directly excite either or the -GUA complex, the formation of likely involved photoactivation of intermediate complexes such as [] or []. Time-dependent density functional theory (TDDFT) calculations support this, showing that [] can absorb UVA light (Fig. S19) and subsequently form reactive radicals. This is consistent with previous findings that halide-O2 complexes can be photoexcited by UVA to yield radicals like X• and HO2• (Cao et al., 2024a, b).
Density functional theory (DFT) calculations (Fig. 3b) reveal that electron transfer from the triplet state (T1) of [] to form and is endergonic (∼13 kcal mol−1) and unfavorable without light. TDDFT results indicate that UVA can excite T1 to higher-energy triplet states (T2), enabling this electron transfer. The resulting is oxidized by O2 to , which decomposes to , while further oxidizes S(IV) species.
3.5 Mechanism of guaiacol photodegradation in Na2SO3 solutions under UVA irradiation
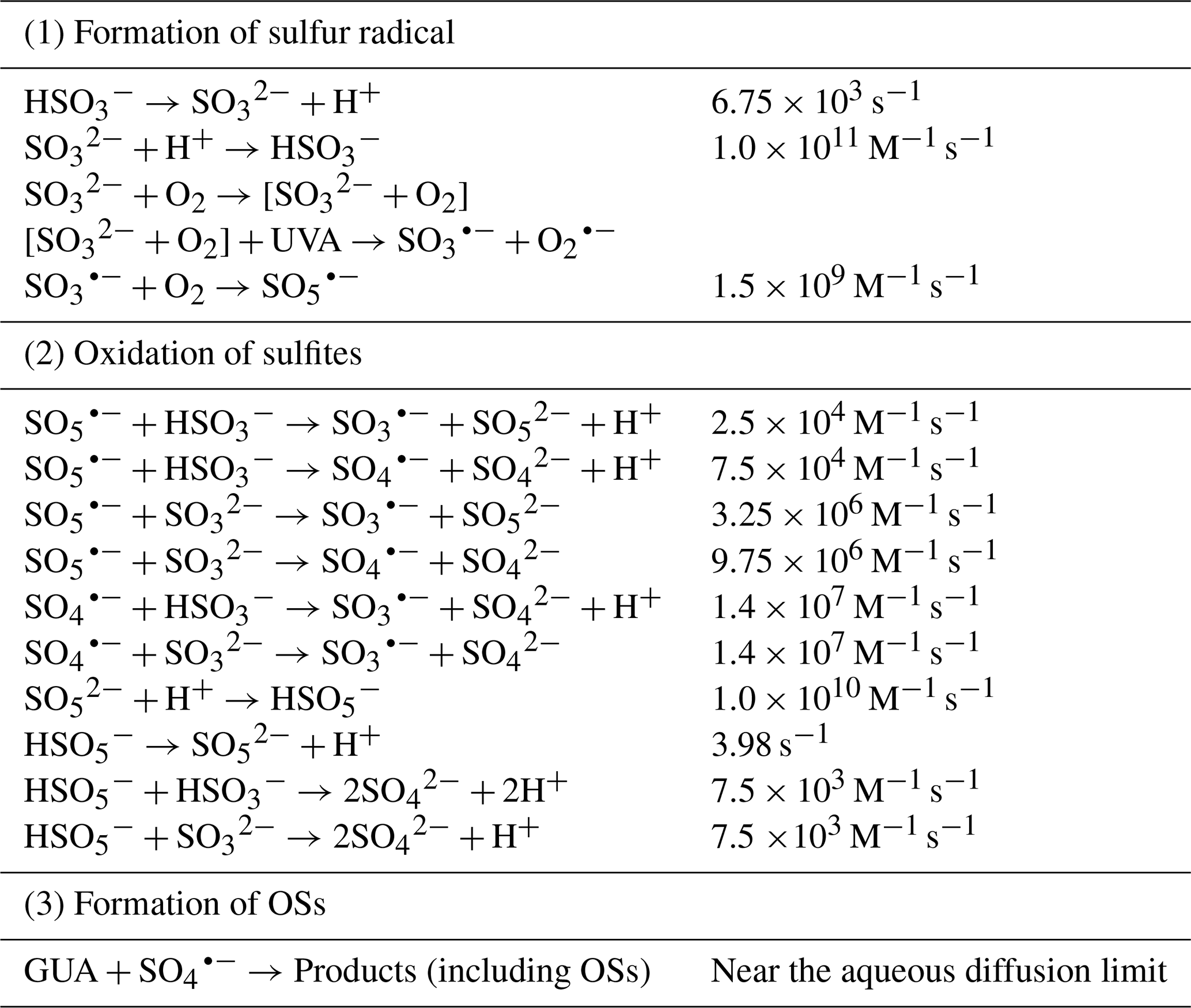
The photodegradation of GUA in aqueous Na2SO3 solution under UVA irradiation proceeds through three major mechanisms, summarized in Table 1:
(1) Formation of sulfur-centered radicals: Under UVA irradiation, the [] complex is photoexcited from the triplet state (T1) to a higher triplet (T2), enabling electron transfer to produce and . Although is the predominant S(IV) species at pH 4.0, it exists in rapid dynamic equilibrium with , which is generated via fast dissociation of ( s−1), ensuring a sufficient concentration of for complex formation. is rapidly oxidized by molecular O2 to form at a high rate ( ).
(2) Oxidation of sulfites to sulfate: reacts with or to produce , and . re-enters the cycle by reacting with O2 to regenerate . protonates to , which continues oxidizing S(IV) species to . also oxidizes S(IV), but more slowly than or (Table S1). Importantly, reacts approximately 100 times faster with than with , leading to a significant acceleration of sulfite photooxidation at pH>4.0 where dominates (Fig. S7).
(3) Formation of organosulfates: reacts with GUA extremely rapidly, much faster than with S(IV) species. This rapid reaction leads to substantial formation of low-volatility organics compounds, including OSs and GUA dimers and derivatives, with a SOA yield of ∼80 % (Fig. S20). GUA also increases the overall rate of sulfite oxidation by nearly threefold, probably via additional reactive radicals generated during its reaction with . Proposed mechanisms for the GUA- reaction are shown in Figs. S21 and S22.
Table 1Reactions and rate constants of GUA photodegradation in Na2SO3 solutions (Seinfeld and Pandis, 2016; Rudzinski et al., 2009).
3.6 Photodegradation of GUA at aqueous interfaces
In atmospheric environments, cloud and fog droplets typically range from a few to tens of micrometers in diameter. Within these microdroplets, surface-active solutes often concentrate at the air-water interface, where reactions are accelerated due to surface enrichment and reduced activation energies (Ruiz-Lopez et al., 2020). Classical molecular dynamics (MD) simulations revealed that GUA is energetically favored at the interface, with an interfacial free energy 2.8 kcal mol−1 lower than in bulk water (Figs. 4a and S23). also shows an interfacial preference, although much smaller (0.17 kcal mol−1 difference) (Xie et al., 2024), suggesting that both species are enriched at the interface.
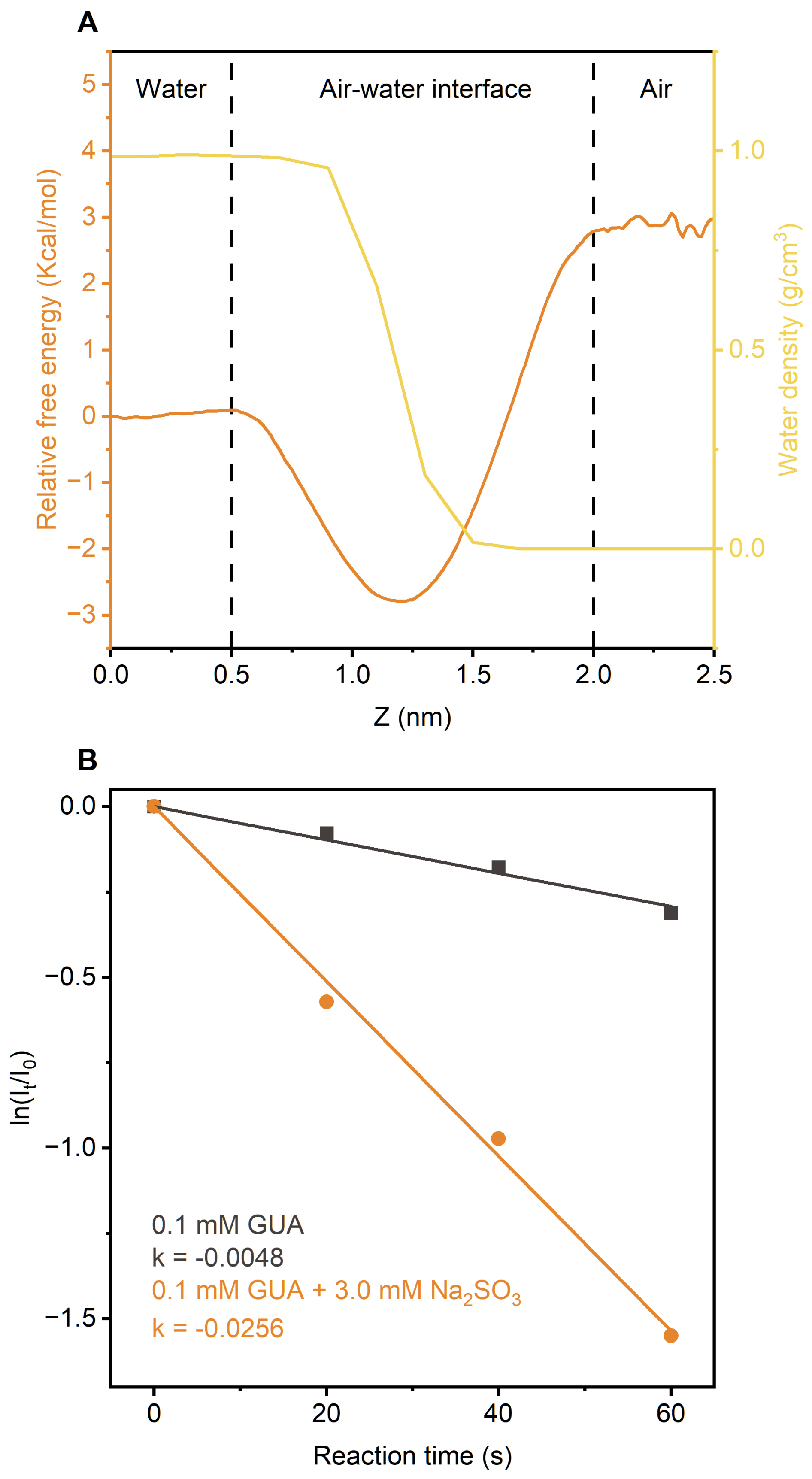
Figure 4(a) Free energy profiles for GUA transfer from the gas phase to bulk water, overlaid with water density distribution at air-water interface. (b) Kinetics of direct photodegradation of GUA in microdroplets, with and without Na2SO3, under UVA irradiation.
Microdroplets also facilitate gas exchange, boosting [] complex formation and production under UVA. Thus, GUA photodegradation is expected to be far greater in microdroplets than in bulk water – potentially by several orders of magnitude.
To test this, we used field-induced droplet ionization mass spectrometry (FIDI-MS) (Huang et al., 2018; Gong et al., 2022; Zhang et al., 2023) to monitor UVA-induced photodegradation of 0.1 mM GUA in microdroplets, with and without 3.0 mM Na2SO3 (see Methods). Figure 4b shows averaged FIDI-MS signals from five droplets, fitted to pseudo-first-order kinetics (Figs. S24 and S25). GUA degraded nearly 200 times faster at the interface than in bulk ( s−1 vs. s−1). With Na2SO3, the rate similarly increased ∼60-fold, indicating interfacial concentrations of M, about two orders magnitude higher than in bulk.
Overall, these findings demonstrate that phenolic compounds like GUA are enriched and highly reactive at air-water interfaces, where UVA-driven formation greatly accelerates photodegradation and OS production.
3.7 Atmospheric implications
The experimental system used in this study represents a simplified aqueous-phase environment designed to isolate key photochemical processes. Consequently, the derived kinetic parameters should be interpreted as condition-dependent estimates rather than direct quantitative representations of atmospheric reaction rates.
When gas-phase SO2 dissolves into cloud and fog droplets, it hydrates to form S(IV) species such as . In the presence of O2 and UVA irradiation, can be oxidized to through radical pathways. Based on our experimental measurements, the apparent photon efficiency (APE) of S(IV) oxidation under UVA irradiation was estimated. Using this experimentally derived APE as a constant parameter, we simulated sulfate formation induced by UVA under the AM0 standard solar spectrum, representing an upper-limit estimate of the sulfate production efficiency via this pathway. This efficiency was then compared with sulfate formation driven by conventional atmospheric oxidants, including NO2, O3, and transition metal ions (TMIs) (Cheng et al., 2016) (Fig. 5, see Methods). Under “Cloud droplets” conditions (Seinfeld and Pandis, 2016; Herrmann et al., 2015) (Fig. 5a), sulfate formation induced by UVA in the bulk solution was comparable in magnitude to that driven by NO2. Under “Beijing haze” conditions (Cheng et al., 2016), where the photonic flux in the UVA range is reduced to 34 %, UVA-induced sulfate formation remained comparable to the O3 oxidation pathways (Fig. 5b).
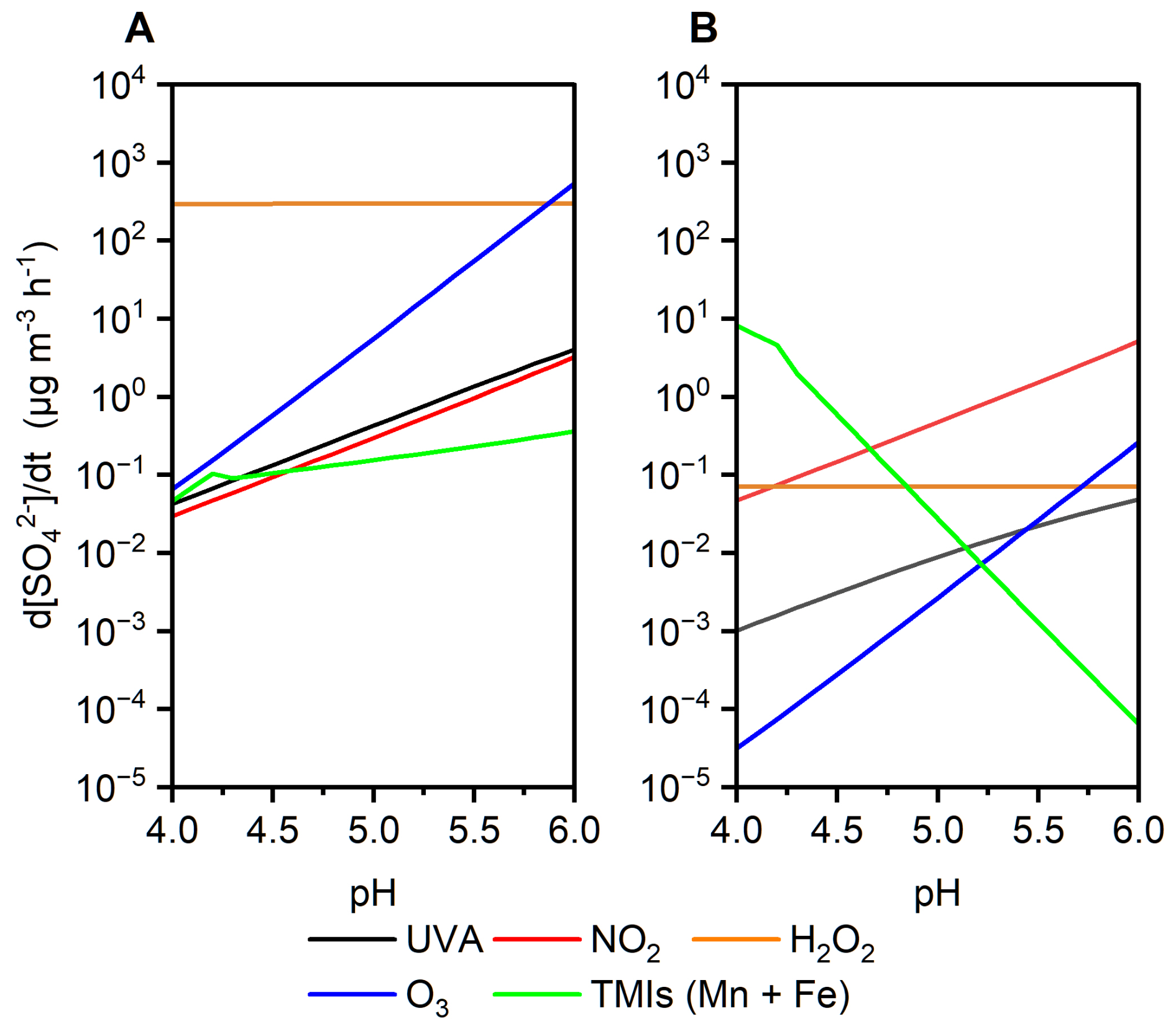
Figure 5Simulated aqueous-phase sulfate production rates from SO2 oxidation as a function of pH under two atmospheric scenarios: (a) “Cloud droplets” scenario with full UVA intensity (AM0 standard). (b) “Beijing haze” scenario with 34 % reduced UVA intensity (AM0 standard). Colored lines represent contributions from individual oxidants.
At the same time, it should be noted that the UVA-driven pathway identified here is not intended to replace or dominate established sulfate formation mechanisms, such as transition metal ion (TMI)-catalyzed oxidation. Rather, it represents a complementary pathway that may contribute to sulfate formation under specific conditions, particularly in aqueous environments where phenolic compounds and UVA irradiation coexist. Therefore, this pathway should be viewed as a complementary and condition-dependent source of sulfate, rather than a dominant process in complex atmospheric systems.
In summary, our results reveal a metal-free pathway for SO2 oxidation to sulfate in atmospheric aqueous phases under UVA irradiation. Unlike traditional mechanisms that rely on metal catalysts or high-energy UVB/UVC lights, we show that the [] complex can initiate sulfate radical production under UVA – wavelengths far more prevalent in the solar spectrum.
In the presence of guaiacol – a common phenolic compound from biomass burning, these sulfate radicals drive rapid GUA oxidation, producing low-volatility organic compounds, including organosulfates. Moreover, microdroplet experiments show that GUA photodegradation is dramatically accelerated in small droplets under UVA light due to intensified interfacial chemistry. The high surface-area-to-volume ratio of microdroplets promotes efficient generation of reactive oxidants, particularly sulfate radicals, which accelerate both S(IV) oxidation and organics transformations. Together, these findings uncover a sunlight-accessible, metal-free pathway for sulfate and SOA formation, especially relevant to slightly acidic, sunlit, and water-rich atmospheric environments.
The results presented in this study should be interpreted within the context of the specific experimental and modeling framework employed. The apparent photon efficiency (APE) reported here is defined based on incident photon flux and is not equivalent to a true photochemical quantum yield, which would require quantification of absorbed photons. In addition, the normalization of light intensity is inherently geometry-dependent, reflecting the dual-lamp configuration and spherical reactor used in this work, and may not be directly transferable to other experimental or atmospheric systems. Furthermore, the kinetic parameters and modeled sulfate formation rates are derived under controlled laboratory conditions and should be regarded as condition-dependent estimates. The modeling results presented here are intended to provide sensitivity-based or upper-limit estimates of UVA-driven S(IV) oxidation, rather than definitive quantitative predictions of atmospheric sulfate production. As such, caution should be exercised when extrapolating these findings to complex atmospheric environments.
The data that support the findings of this study are available in the Supplement of this article.
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-5713-2026-supplement.
BC, XY, and QZ designed research; BC, YH, WJ, XY, and QZ performed research; BC, YH, WJ, YanL, YaL, JZ, YZ, JY, HS, CW, LZ, TMF, QZ, and XY analyzed data; BC, YH, QZ, and XY wrote the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
Supported by Center for Computational Science and Engineering at Southern University of Science and Technology. Qi Zhang acknowledges support from the Donald G. Crosby Endowed Chair at the University of California at Davis.
This work was supported by Shenzhen Key Laboratory of Precision Measurement and Early Warning Technology for Urban Environmental Health Risks (grant no. ZDSYS20220606100604008), Guangdong Provincial Observation and Research Station for Coastal Atmosphere and Climate of the Greater Bay Area (grant no. 2021B1212050024), Shenzhen Science and Technology Program (grant nos. KQTD20210811090048025 and KCXFZ20230731093601003).
This paper was edited by Jason Surratt and reviewed by two anonymous referees.
Abbatt, J. P. D., Benz, S., Cziczo, D. J., Kanji, Z., Lohmann, U., and Möhler, O.: Solid Ammonium Sulfate Aerosols as Ice Nuclei: A Pathway for Cirrus Cloud Formation, Science, 313, 1770–1773, https://doi.org/10.1126/science.1129726, 2006.
Bai, C.-B., Wang, N.-X., Lan, X.-W., Wang, Y.-J., Xing, Y., Wen, J.-L., Gao, X.-W., and Zhang, W.: An Unexpected Controlled New Oxidant: , Sci. Rep.-UK, 6, 20163, https://doi.org/10.1038/srep20163, 2016.
Brandt, C. and van Eldik, R.: Transition Metal-Catalyzed Oxidation of Sulfur(IV) Oxides. Atmospheric-Relevant Processes and Mechanisms, Chem. Rev., 95, 119–190, https://doi.org/10.1021/cr00033a006, 1995.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Cao, Y., Qiu, W., Li, J., Jiang, J., and Pang, S.: Review on UV/sulfite process for water and wastewater treatments in the presence or absence of O2, Sci. Total Environ., 765, 142762, https://doi.org/10.1016/j.scitotenv.2020.142762, 2021.
Cao, Y., Liu, J., Ma, Q., Zhang, C., Zhang, P., Chen, T., Wang, Y., Chu, B., Zhang, X., Francisco, J. S., and He, H.: Photoactivation of Chlorine and Its Catalytic Role in the Formation of Sulfate Aerosols, J. Am. Chem. Soc., 146, 1467–1475, https://doi.org/10.1021/jacs.3c10840, 2024a.
Cao, Y., Wang, Z., Liu, J., Ma, Q., Li, S., Liu, J., Li, H., Zhang, P., Chen, T., Wang, Y., Chu, B., Zhang, X., Saiz-Lopez, A., Francisco, J. S., and He, H.: Spontaneous molecular bromine production in sea salt aerosols, Angew. Chem. Int. Edit., 63, e202409779, https://doi.org/10.1002/anie.202409779, 2024b.
Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Gao, M., Zhang, Q., He, K., Carmichael, G., Pöschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Coddens, E. M., Huang, L., Wong, C., and Grassian, V. H.: Influence of Glyoxal on the Catalytic Oxidation of S(IV) in Acidic Aqueous Media, ACS Earth Space Chem., 3, 142–149, https://doi.org/10.1021/acsearthspacechem.8b00168, 2018.
Cope, J. D., Bates, K. H., Tran, L. N., Abellar, K. A., and Nguyen, T. B.: Sulfur radical formation from the tropospheric irradiation of aqueous sulfate aerosols, P. Natl. Acad. Sci. USA, 119, e2202857119, https://doi.org/10.1073/pnas.2202857119, 2022.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1029/2001jd001213, 2006.
Duporté, G., Flaud, P. M., Kammer, J., Geneste, E., Augagneur, S., Pangui, E., Lamkaddam, H., Gratien, A., Doussin, J. F., Budzinski, H., Villenave, E., and Perraudin, E.: Experimental Study of the Formation of Organosulfates from α-Pinene Oxidation. 2. Time Evolution and Effect of Particle Acidity, J. Phys. Chem. A, 124, 409–421, https://doi.org/10.1021/acs.jpca.9b07156, 2020.
Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Robb, M., Cheeseman, J., Scalmani, G., Barone, V., Petersson, G., and Nakatsuji, H.: Gaussian 16, Revision A.03, Gaussian [code], https://gaussian.com/gaussian16/ (last access: 23 April 2026), 2016.
Gao, Y., Zhang, M., Guo, J., and Xu, L.: Impact of the oxidation of SO2 by NO2 on regional sulfate concentrations over the North China Plain, Atmos. Pollut. Res., 13, 101337, https://doi.org/10.1016/j.apr.2022.101337, 2022.
George, C., Ammann, M., D'Anna, B., Donaldson, D. J., and Nizkorodov, S. A.: Heterogeneous photochemistry in the atmosphere, Chem. Rev., 115, 4218–4258, https://doi.org/10.1021/cr500648z, 2015.
Gong, C., Yuan, X., Xing, D., Zhang, D., Martins-Costa, M. T. C., Anglada, J. M., Ruiz-Lopez, M. F., Francisco, J. S., and Zhang, X.: Fast Sulfate Formation Initiated by the Spin-Forbidden Excitation of SO2 at the Air-Water Interface, J. Am. Chem. Soc., 144, 22302–22308, https://doi.org/10.1021/jacs.2c10830, 2022.
Guo, Y., Riplinger, C., Becker, U., Liakos, D. G., Minenkov, Y., Cavallo, L., and Neese, F.: Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)], J. Chem. Phys., 148, 011101, https://doi.org/10.1063/1.5011798, 2018.
Harris, E., Sinha, B., van Pinxteren, D., Tilgner, A., Fomba, K. W., Schneider, J., Roth, A., Gnauk, T., Fahlbusch, B., Mertes, S., Lee, T., Collett, J., Foley, S., Borrmann, S., Hoppe, P., and Herrmann, H.: Enhanced Role of Transition Metal Ion Catalysis During In-Cloud Oxidation of SO2, Science, 340, 727–730, https://doi.org/10.1126/science.1230911, 2013.
He, Y., Zhao, B., Wang, S., Valorso, R., Chang, X., Yin, D., Feng, B., Camredon, M., Aumont, B., Dearden, A., Jathar, S. H., Shrivastava, M., Jiang, Z., Cappa, C. D., Yee, L. D., Seinfeld, J. H., Hao, J., and Donahue, N. M.: Formation of secondary organic aerosol from wildfire emissions enhanced by long-time ageing, Nat. Geosci., 17, 124–129, https://doi.org/10.1038/s41561-023-01355-4, 2024.
Herrmann, H., Schaefer, T., Tilgner, A., Styler, S. A., Weller, C., Teich, M., and Otto, T.: Tropospheric aqueous-phase chemistry: kinetics, mechanisms, and its coupling to a changing gas phase, Chem. Rev., 115, 4259–4334, https://doi.org/10.1021/cr500447k, 2015.
Hess, B., Kutzner, C., van der Spoel, D., and Lindahl, E.: GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation, J. Chem. Theory Comput., 4, 435–447, https://doi.org/10.1021/ct700301q, 2008.
Hoffmann, M. R.: On the kinetics and mechanism of oxidation of aquated sulfur dioxide by ozone, Atmos. Environ., 20, 1145–1154, https://doi.org/10.1016/0004-6981(86)90147-2, 1986.
Huang, Y., Barraza, K. M., Kenseth, C. M., Zhao, R., Wang, C., Beauchamp, J. L., and Seinfeld, J. H.: Probing the OH Oxidation of Pinonic Acid at the Air–Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry (FIDI-MS), J. Phys. Chem. A, 122, 6445–6456, https://doi.org/10.1021/acs.jpca.8b05353, 2018.
Hughes, D. D., Christiansen, M. B., Milani, A., Vermeuel, M. P., Novak, G. A., Alwe, H. D., Dickens, A. F., Pierce, R. B., Millet, D. B., Bertram, T. H., Stanier, C. O., and Stone, E. A.: PM2.5 chemistry, organosulfates, and secondary organic aerosol during the 2017 Lake Michigan Ozone Study, Atmos. Environ., 244, 117939, https://doi.org/10.1016/j.atmosenv.2020.117939, 2021.
Humphrey, W., Dalke, A., and Schulten, K.: VMD: Visual molecular dynamics, J. Mol. Graphics, 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5, 1996.
Iinuma, Y., Müller, C., Berndt, T., Böge, O., Claeys, M., and Herrmann, H.: Evidence for the Existence of Organosulfates from β-Pinene Ozonolysis in Ambient Secondary Organic Aerosol, Environ. Sci. Technol., 41, 6678–6683, https://doi.org/10.1021/es070938t, 2007.
Khan, F., Chen, Y., Hartwell, H. J., Yan, J., Lin, Y.-H., Freedman, A., Zhang, Z., Zhang, Y., Lambe, A. T., Turpin, B. J., Gold, A., Ault, A. P., Szmigielski, R., Fry, R. C., and Surratt, J. D.: Heterogeneous Oxidation Products of Fine Particulate Isoprene Epoxydiol-Derived Methyltetrol Sulfates Increase Oxidative Stress and Inflammatory Gene Responses in Human Lung Cells, Chem. Res. Toxicol., 36, 1814–1825, https://doi.org/10.1021/acs.chemrestox.3c00278, 2023.
Lan, Y., Wheeler, S. E., and Houk, K. N.: Extraordinary Difference in Reactivity of Ozone (OOO) and Sulfur Dioxide (OSO): A Theoretical Study, J. Chem. Theory Comput., 7, 2104–2111, https://doi.org/10.1021/ct200293w, 2011.
Li, F., Zhou, S., Du, L., Zhao, J., Hang, J., and Wang, X.: Aqueous-phase chemistry of atmospheric phenolic compounds: A critical review of laboratory studies, Sci. Total Environ., 856, 158895, https://doi.org/10.1016/j.scitotenv.2022.158895, 2023a.
Li, M., Duan, P., Huo, Y., Jiang, J., Zhou, Y., Ma, Y., Jin, Z., Mei, Q., Xie, J., and He, M.: The multiple roles of phenols in the degradation of aniline contaminants by sulfate radicals: A combined study of DFT calculations and experiments, J. Hazard. Mater., 443, 130216, https://doi.org/10.1016/j.jhazmat.2022.130216, 2023b.
Liang, C. and Su, H.-W.: Identification of Sulfate and Hydroxyl Radicals in Thermally Activated Persulfate, Ind. Eng. Chem. Res., 48, 5558–5562, https://doi.org/10.1021/ie9002848, 2009.
Liu, C., Chen, D., and Chen, X.: Atmospheric Reactivity of Methoxyphenols: A Review, Environ. Sci. Technol., 56, 2897–2916, https://doi.org/10.1021/acs.est.1c06535, 2022.
Liu, T. and Abbatt, J. P. D.: Oxidation of sulfur dioxide by nitrogen dioxide accelerated at the interface of deliquesced aerosol particles, Nat. Chem., 13, 1173–1177, https://doi.org/10.1038/s41557-021-00777-0, 2021.
Liu, T., Clegg, S. L., and Abbatt, J. P. D.: Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles, P. Natl. Acad. Sci. USA, 117, 1354–1359, https://doi.org/10.1073/pnas.1916401117, 2020.
Liu, Y., Li, X., Ge, Q., Fang, X., Wang, T., You, W., Wang, W., Xie, L., Li, K., Gong, K., Yang, L., Wang, R., Wang, J., Wang, L., Ma, M., Huang, T., Fu, H., Chen, J., Dong, X., and Zhang, L.: Carbonate radical ion as a key driver of rapid atmospheric sulfate formation, npj Clim. Atmos. Sci., 8, 45, https://doi.org/10.1038/s41612-025-00905-4, 2025.
Lu, T. and Chen, F.: Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 33, 580–592, https://doi.org/10.1002/jcc.22885, 2012.
Lu, T. and Chen, Q.: Shermo: A general code for calculating molecular thermochemistry properties, Comput. Theor. Chem., 1200, 113249, https://doi.org/10.1016/j.comptc.2021.113249, 2021.
Marenich, A. V., Cramer, C. J., and Truhlar, D. G.: Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies, J. Phys. Chem. B, 113, 4538–4543, https://doi.org/10.1021/jp809094y, 2009.
McFall, A. S., Johnson, A. W., and Anastasio, C.: Air–Water Partitioning of Biomass-Burning Phenols and the Effects of Temperature and Salinity, Environ. Sci. Technol., 54, 3823–3830, https://doi.org/10.1021/acs.est.9b06443, 2020.
Neese, F.: Software update: the ORCA program system – Version 6.0, WIRes Comput. Mol. Sci., 15, e70019, https://doi.org/10.1002/wcms.70019, 2025.
Noga, J. and Bartlett, R. J.: The full CCSDT model for molecular electronic structure, J. Chem. Phys., 86, 7041–7050, https://doi.org/10.1063/1.452353, 1987.
Pan, Y., Zhang, F., Tan, W., and Feng, X.: New insight into wastewater treatment by activation of sulfite with humic acid under visible light irradiation, Water Res., 258, 121773, https://doi.org/10.1016/j.watres.2024.121773, 2024.
Passananti, M., Kong, L., Shang, J., Dupart, Y., Perrier, S., Chen, J., Donaldson, D. J., and George, C.: Organosulfate Formationthrough the Heterogeneous Reactionof Sulfur Dioxide with Unsaturated Fatty Acids and Long-Chain Alkenes, Angew. Chem. Int. Edit., 55, 10336–10339, https://doi.org/10.1002/anie.201605266, 2016.
Peng, C., Razafindrambinina, P. N., Malek, K. A., Chen, L., Wang, W., Huang, R.-J., Zhang, Y., Ding, X., Ge, M., Wang, X., Asa-Awuku, A. A., and Tang, M.: Interactions of organosulfates with water vapor under sub- and supersaturated conditions, Atmos. Chem. Phys., 21, 7135–7148, https://doi.org/10.5194/acp-21-7135-2021, 2021.
Ren, H., Sedlak, J. A., and Elrod, M. J.: General Mechanism for Sulfate Radical Addition to Olefinic Volatile Organic Compounds in Secondary Organic Aerosol, Environ. Sci. Technol., 55, 1456–1465, https://doi.org/10.1021/acs.est.0c05256, 2021.
Riva, M., Tomaz, S., Cui, T., Lin, Y. H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015.
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Ribeiro, I. O., Oliveira, R. L., Dos Santos, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sa, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin Junior, S., de Souza, R. A. F., Palm, B. B., Jimenez, J. L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing Isoprene Epoxydiol-to-Inorganic Sulfate Aerosol Ratio Results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019.
Romero, F. and Oehme, M.: Organosulfates – A New Component of Humic-Like Substances in Atmospheric Aerosols?, J. Atmos. Chem., 52, 283–294, https://doi.org/10.1007/s10874-005-0594-y, 2005.
Rudziński, K. J., Gmachowski, L., and Kuznietsova, I.: Reactions of isoprene and sulphoxy radical-anions – a possible source of atmospheric organosulphites and organosulphates, Atmos. Chem. Phys., 9, 2129–2140, https://doi.org/10.5194/acp-9-2129-2009, 2009.
Ruiz-Lopez, M. F., Francisco, J. S., Martins-Costa, M. T. C., and Anglada, J. M.: Molecular reactions at aqueous interfaces, Nat. Rev. Chem., 4, 459–475, https://doi.org/10.1038/s41570-020-0203-2, 2020.
Schindelka, J., Iinuma, Y., Hoffmann, D., and Herrmann, H.: Sulfate radical-initiated formation of isoprene-derived organosulfates in atmospheric aerosols, Faraday Discuss., 165, 237–259, https://doi.org/10.1039/c3fd00042g, 2013.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, in: 3rd Edn., Wiley, ISBN 9781119221166, 2016.
Shakya, K. M. and Peltier, R. E.: Non-sulfate sulfur in fine aerosols across the United States: Insight for organosulfate prevalence, Atmos. Environ., 100, 159–166, https://doi.org/10.1016/j.atmosenv.2014.10.058, 2015.
Surratt, J. D., Gómez-González, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate Formation in Biogenic Secondary Organic Aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Teymoor Seydi, S., Abatzoglou, J. T., Jones, M. W., Kolden, C. A., Filippelli, G., Hurteau, M. D., AghaKouchak, A., Luce, C. H., Miao, C., and Sadegh, M.: Increasing global human exposure to wildland fires despite declining burned area, Science, 389, 826–829, https://doi.org/10.1126/science.adu6408, 2025.
Tolocka, M. P.: Contribution of Organosulfur Compounds to Organic Aerosol Mass, Environ. Sci. Technol., 46, 7978–7983, https://doi.org/10.1021/es300651v, 2012.
Tran, L. N., Abellar, K. A., Cope, J. D., and Nguyen, T. B.: Second-Order Kinetic Rate Coefficients for the Aqueous-Phase Sulfate Radical () Oxidation of Some Atmospherically Relevant Organic Compounds, J. Phys. Chem. A, 126, 6517–6525, https://doi.org/10.1021/acs.jpca.2c04964, 2022.
Wang, D., Li, Y., Yang, M., and Han, M.: Decomposition of polycyclic aromatic hydrocarbons in atmospheric aqueous droplets through sulfate anion radicals: An experimental and theoretical study, Sci. Total Environ., 393, 64–71, https://doi.org/10.1016/j.scitotenv.2007.11.036, 2008.
Wang, G., Zhang, R., Gomez, M. E., Yang, L., Levy Zamora, M., Hu, M., Lin, Y., Peng, J., Guo, S., Meng, J., Li, J., Cheng, C., Hu, T., Ren, Y., Wang, Y., Gao, J., Cao, J., An, Z., Zhou, W., Li, G., Wang, J., Tian, P., Marrero-Ortiz, W., Secrest, J., Du, Z., Zheng, J., Shang, D., Zeng, L., Shao, M., Wang, W., Huang, Y., Wang, Y., Zhu, Y., Li, Y., Hu, J., Pan, B., Cai, L., Cheng, Y., Ji, Y., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, https://doi.org/10.1073/pnas.1616540113, 2016.
Wang, W., Liu, M., Wang, T., Song, Y., Zhou, L., Cao, J., Hu, J., Tang, G., Chen, Z., Li, Z., Xu, Z., Peng, C., Lian, C., Chen, Y., Pan, Y., Zhang, Y., Sun, Y., Li, W., Zhu, T., Tian, H., and Ge, M.: Sulfate formation is dominated by manganese-catalyzed oxidation of SO2 on aerosol surfaces during haze events, Nat. Commun., 12, 1993, https://doi.org/10.1038/s41467-021-22091-6, 2021.
Wang, W., Liu, Y., Wang, T., Ge, Q., Li, K., Liu, J., You, W., Wang, L., Xie, L., Fu, H., Chen, J., and Zhang, L.: Significantly Accelerated Photosensitized Formation of Atmospheric Sulfate at the Air–Water Interface of Microdroplets, J. Am. Chem. Soc., 146, 6580–6590, https://doi.org/10.1021/jacs.3c11892, 2024.
Wojnárovits, L. and Takács, E.: Rate constants of sulfate radical anion reactions with organic molecules: A review, Chemosphere, 220, 1014–1032, https://doi.org/10.1016/j.chemosphere.2018.12.156, 2019.
Xie, R., Guo, K., Li, Y., Zhang, Y., Zhong, H., Leung, D. Y. C., and Huang, H.: Harnessing air-water interface to generate interfacial ROS for ultrafast environmental remediation, Nat. Commun., 15, 8860, https://doi.org/10.1038/s41467-024-53289-z, 2024.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Zhang, D., Wang, J., Chen, H., Gong, C., Xing, D., Liu, Z., Gladich, I., Francisco, J. S., and Zhang, X.: Fast Hydroxyl Radical Generation at the Air-Water Interface of Aerosols Mediated by Water-Soluble PM2.5 under Ultraviolet A Radiation, J. Am. Chem. Soc., 145, 6462–6470, https://doi.org/10.1021/jacs.3c00300, 2023.
Zhang, R. and Chan, C. K.: Simultaneous formation of sulfate and nitrate via co-uptake of SO2 and NO2 by aqueous NaCl droplets: combined effect of nitrate photolysis and chlorine chemistry, Atmos. Chem. Phys., 23, 6113–6126, https://doi.org/10.5194/acp-23-6113-2023, 2023.
Zhao, J., Zheng, B., Ciais, P., Chen, Y., Gasser, T., Canadell, J. G., Zhang, L., and Zhang, Q.: Global warming amplifies wildfire health burden and reshapes inequality, Nature, https://doi.org/10.1038/s41586-025-09612-9, 2025.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2007.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015.
Zheng, J., Xu, X., and Truhlar, D. G.: Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 128, 295–305, https://doi.org/10.1007/s00214-010-0846-z, 2010.
Zuo, Y., Zhan, J., and Wu, T.: Effects of Monochromatic UV-Visible Light and Sunlight on Fe(III)-Catalyzed Oxidation of Dissolved Sulfur Dioxide, J. Atmos. Chem., 50, 195–210, https://doi.org/10.1007/s10874-005-2813-y, 2005.