the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Apr 2026
| 13 Apr 2026
Mechanistic investigations of the formation of multifunctional products from the multi-generation ●OH oxidation of styrene
Long Chen
Yu Huang
Yonggang Xue
Long Cui
Zhihui Jia
Styrene is a highly reactive aromatic hydrocarbon that has been identified as a key secondary organic aerosol (SOA) precursor. Recent laboratory chamber experiments have identified C7 and C8 series compounds as the main components of SOA in the photooxidation of styrene. However, their molecular structures and formation pathways remain largely uncharacterized. Herein, the formation mechanisms of multifunctional products from the multi-generation •OH oxidation of styrene are studied using the quantum chemistry methods. The calculations show that the first generation RO2 radicals can either proceed unimolecular decomposition to yield benzaldehyde (C7H6O), or undergo bimolecular reactions with HO2•/NO to form the first generation closed-shell C7- and C8-products, hydroperoxide 1st-ROOH (C8H10O3), benzaldehyde, and organic nitrate 1st-RONO2 (C8H9NO3). For the second generation •OH oxidation, OH-addition reaction occurring at the ortho-site of 1st-ROOH and 1st-RONO2 has a significant dominance. The ortho-OH-addition products can proceed through two O2-addition steps and a cyclization process to produce the peroxide bicyclic peroxy radicals (BPR). BPR can further react with HO2•/NO to form the second generation closed-shell C8-products, hydroperoxide 2nd-ROOH (C8H12O8), organic nitrate 2nd-RONO2 (C8H10N2O10), and other multifunctional products, in which the first two products have fractional yields of 41.4 % and 4.8 %, respectively. For the third generation •OH oxidation, OH-addition occurring at the C=C double bond of 2nd-ROOH and 2nd-RONO2 has the lowest barrier. The major third generation closed-shell C8-products are the multifunctional hydroperoxides and organic nitrates. These findings carry important implications for advancing our understanding of the chemical composition and formation mechanisms of aromatic SOA.
- Article
(2659 KB) - Full-text XML
-
Supplement
(2987 KB) - BibTeX
- EndNote




Aromatic compounds are recognized as the significant secondary organic aerosol (SOA) precursors, accounting for 20 %–30 % of the total volatile organic compounds (VOCs) and up to ∼60 % of the urban atmosphere (Xu et al., 2020; Yan et al., 2019; Yu et al., 2022a, b; Cabrera-Perez et al., 2016; Iyer et al., 2023; Wang et al., 2017; Bloss et al., 2005; Forstner et al., 1997). The primary sources include the incomplete combustion, solvent evaporation, and industrial emission, and the secondary sources involve the biofuel and biomass burning (Xu et al., 2020; Cabrera-Perez et al., 2016; Li et al., 2019). The most abundant aromatic compounds, including benzene, toluene, ethylbenzene, xylenes, styrene and trimethylbenzenes, are highly present in urban environments (Cabrera-Perez et al., 2016; Koppmann, 2008). The degradation of aromatic compounds initiated by the atmospheric oxidants (e.g., OH radicals, NO3 radicals, O3, and Cl atom) leads to the production of multifunctional molecules (e.g., nitroaromatics, dicarbonyls, cresols, epoxides) (Ji et al., 2017; Wu et al., 2014; Fu et al., 2023; Wang and Li, 2021; Wang et al., 2013, 2020b; Zaytsev et al., 2019), significantly contributing to new particle formation (NPF) and SOA formation (up to 50 % in eastern China) in the atmosphere (Wang et al., 2017, 2020a; Garmash et al., 2020; Molteni et al., 2018; Nie et al., 2022).
The secondary organic aerosol formation potential (SOAP) of aromatics is significantly greater than that of alkanes and alkenes during haze episodes in Beijing (Sun et al., 2016). Among these precursors, toluene is the predominant SOA-forming species, contributing more than 16 % of the total SOA, followed by styrene (15 %) and ethylbenzene (9.5 %) (Sun et al., 2016). Styrene is primarily emitted from the anthropogenic activities such as solvent usage and vehicle exhaust (Cho et al., 2014; Wu et al., 2021), which is detected at the ppb levels in urban environments, with the mixing ratios of 0.06–4.50 ppb (Cho et al., 2014; Huang et al., 2019). Styrene has been classified as a hazardous air pollutant in the 1990 Clean Air Act due to the potential mutagen and carcinogen (Environmental Protection Agency (EPA), 1990). Therefore, it is very necessary to investigate the degradation mechanisms of styrene under atmospheric conditions. In general, the atmospheric oxidation of styrene initiated by OH radicals is anticipated to be the dominant daytime sink, and the lifetime is estimated to be ∼8 h under the conditions of typical OH radicals concentrations ( ) (Wu et al., 2021; Shen et al., 2022). Due to the existence of highly reactive vinyl and aromatic groups, OH-initiated oxidation of styrene mainly comprise two kinds of pathways: H-abstraction and OH-addition, in which Cβ-site OH-addition reaction is expected to be the predominant pathway (Wu et al., 2021; Wang et al., 2015; Zhang et al., 2024). The formed products can combine with an O2 molecule leading to the first generation peroxyl radicals, which can further react with NO resulting in the formation of benzaldehyde and formaldehyde. The barrier of the rate-limiting step is predicted to be 28.4 kcal mol−1 (Wang et al., 2015), implying that benzaldehyde is unlikely to be the sole primary product in the oxidation of styrene due to their higher barriers. Additionally, carbonyl oxides, formed in the ozonolysis of styrene, serve as the chain units participating in the formation of oligomers (Yu et al., 2022a). The volatility of oligomers decreases dramatically as the successive addition of carbonyl oxides increases, eventually transforming into extremely low volatility organic compounds (ELVOC) and directly participating in NPF.
Experimentally, Cho et al., investigated the kinetics of the reaction styrene +•OH at 240–340 K and 1–3 Torr using the mass spectrometry technique (Cho et al., 2014). They found that the addition of OH radicals to the vinyl carbons is dominant, and the determined rate coefficient is at room temperature. In the smog chamber experiments, Tajuelo et al. (2019a, b, c) found that the SOA yields from the photolysis and photooxidation of styrene and its homologous species increase with the concentration of initial reactants increasing, and benzaldehyde, benzoyl chloride, acetophenone and formaldehyde are expected to be the primary gas phase products. Yu et al. (2022b) investigated the formation of SOA from styrene in an indoor chamber under different NOx and RH conditions, and found the SOA yields decrease with increasing RH in both the H2O2 and NOx systems. The C7 and C8 species are the main products in the H2O2 system, while organic nitrates are the major components in the NOx system. Although the possible molecular formula and chemical composition of the oxidation products from the reaction styrene +•OH are given in the aforementioned studies, the specific molecular structures and formation pathways remain ambiguous. Additionally, to the best of our knowledge, the majority of studies mainly focus on the first generation •OH oxidation products to date, while the formation mechanisms of multifunctional products from the multi-generation •OH oxidation of styrene are still limited.
In the present study, the multi-generation •OH oxidation mechanisms of styrene in the presence of HO2•/NO are investigated using the quantum chemistry methods. The calculated results arising from the first generation •OH oxidation reactions are presented herein for comparison with the available literatures to ascertain the reliability of the employed theoretical method. For the multi-generation •OH oxidation reactions of styrene, all the possible pathways, including H-abstraction, OH-addition, O2-addition, cyclization, ring-opening, intramolecular H-shifts, C−C bond and O−O bond scission, and HO2-elimination, are taken into account. Additionally, the saturated concentrations of the formed multifunctional products are estimated to identify the volatility classes.
2.1 Electronic structures and energy calculations
The electronic structures and energy calculations of all stationary points, including reactants (R), intermediates (IM), transition states (TS) and products (P), are performed using the Gaussian 16 program (Frisch et al., 2016). Geometric optimizations of all stationary points on the potential energy surfaces (PESs) are carried out at the M06-2X/6-31+g(d,p) level of theory, since it has reliable performance for describing the noncovalent interactions, thermochemical, and kinetics (Zhao and Truhlar, 2008). Harmonic vibrational frequencies are determined at the M06-2X/6-31+g(d,p) theoretical level to confirm the characteristics of all stationary points (a local minimum or a saddle point). The zero-point vibrational energy (ZPVE) is scaled by a factor of 0.967 (Alecu et al., 2010). Intrinsic reaction coordinate (IRC) calculations are carried out to ascertain the connection of the given TS between the designated local minima R and P (Fukui, 1981). Single point energy calculations are performed at the M06-2X/6-311++G(3df,3pd) level based on the M06-2X/6-31+g(d,p) optimized geometries.
In order to further evaluate the reliability of the computational method employed herein, the single point energies of all the stationary points involved in the initial addition of OH radicals to styrene and intramolecular H-shift reactions of the first generation peroxyl radicals S2-1-x are recalculated using the DLPNO-CCSD(T)/aug-cc-pVTZ method performed using the Orca 6.1 program (Neese, 2025). As shown in Table S1 in the Supplement, the ΔEa values obtained using the M06-2X/6-311++G(3df,3pd) method are consistent with those derived from the DLPNO-CCSD(T)/aug-cc-pVTZ method. The largest deviation and the average absolute deviation are 1.2 and 0.6 kcal mol−1, respectively, indicating that the computational method employed in this study is reliable. Considering the computational cost, the M06-2X/6-311++G(3df,3pd) method is employed to investigate the formation mechanism of multifunctional products from the multi-generation •OH oxidation of styrene. The energy barrier (ΔEa) and reaction energy (ΔEr) are defined as the difference in energy between TS and IM, as well as between P and R.
2.2 Conformer research
RO2 radicals formed from the addition of O2 to the carbon-centered site of alkyl radicals R have multiple possible conformers due to the different orientations of O2 attack (Chen et al., 2021; Fu et al., 2020; Møller et al., 2016, 2020). An initial structure of RO2 radicals is optimized at the B3LYP/6-31+G(d) level and subsequently used as the starting geometry to perform the conformer search conducted using the Molclus program (Lu, 2024). The resulting structures are initially optimized at the B3LYP/6-31+G(d) level, as this method accurately predicts the relative energy ordering of different conformers (Møller et al., 2016, 2020). For the intramolecular H-shift reactions of RO2 radicals, the lengths of the O−O, C−H and O−H bonds in the conformational sampling of TSs are constrained to retain the cyclic TS structure. All unique conformers of R, TS and P within 5.0 kcal mol−1 with respect to the lowest energy conformer are further optimized at the M06-2X/6-31+g(d,p) level of theory. Then, the single point energy calculations are performed at the M06-2X/6-311++G(3df,3pd) level of theory. RO radicals formed by the bimolecular reactions of RO2 radicals with HO2 radicals and NO also have multiple conformers. In order to obtain the lowest energy conformer, a similar methodology is employed in the present study.
2.3 Kinetics calculations
The rate coefficients of unimolecular reactions, including intramolecular H-shifts, cyclization, HO2-elimination, and C−C bond and C−O bond scissions, are calculated using the RRKM theory along with energy-grained master equation (RRKM-ME) (Holbrook et al., 1996). The rate coefficients of bimolecular reactions, involving H-abstraction and OH-addition, are determined using the traditional transition state theory (TST) (Fernández-Ramos et al., 2007). An asymmetric one-dimensional Eckart model (Eckart, 1930) is employed to consider the tunneling correction factors in the rate coefficient calculations based on RRKM-ME and TST. A single exponential down model in the RRKM-ME calculations is utilized to approximate the collision transfer (〈ΔE〉down=200 cm−1). The Lennard–Jones parameters of all intermediate species are estimated using the empirical formula as proposed by Gilbert and Smith (1990).
For the intramolecular H-shifts of RO2 and RO radicals, the rate coefficients are computed using the multiconformer transition state theory (MC-TST) (Møller et al., 2016), which is expressed as Eq. (1) (Møller et al., 2016, 2020; Pasik et al., 2024):
where κ is the Eckart tunneling coefficient, h is Planck's constant, kB is Boltzmann's constant, and T is the absolute temperature (298.15 K). QTS,i and QR,j refer to the partition functions of the corresponding transition state i and reactant j conformers, respectively. ΔEi and ΔEj represent the relative electronic energies between the corresponding transition state i and reactant j conformers and the lowest energy conformers, respectively. ETS and ER stand for the electronic energies of the lowest energy transition state and reactant conformers, respectively. All kinetics calculations are carried out using the KiSThelP 2021 and MESMER 6.0 programs (Glowacki et al., 2012; Canneaux et al., 2013).
3.1 First generation •OH oxidation mechanisms of styrene
Styrene is composed of a benzene ring and a vinyl group, and its oxidation initiated by OH radicals may proceed either on the vinyl group or on the benzene ring. Previous literature has demonstrated that the addition of OH radicals to terminal carbon (Cβ-site) of a vinyl group in styrene is the dominant pathway, with the branching ratio of 88.2 % (Wu et al., 2021). Therefore, the Cβ-site OH-addition reaction is mainly considered in the present study. Figure 1 depicts that this reaction starts with the formation of a pre-reactive complex IM1, and then transforms into an alkyl radical S1-1 via transition state TS1 with a ΔEa of 0.8 kcal mol−1. The rate coefficient of Cβ-site OH-addition reaction is estimated to be at ambient temperature, which is approximately consistent with the experimental (1.2– ) and theoretical values (1.7– ) for the total rate coefficient of the reaction styrene +•OH (Wu et al., 2021; Zhang et al., 2024).
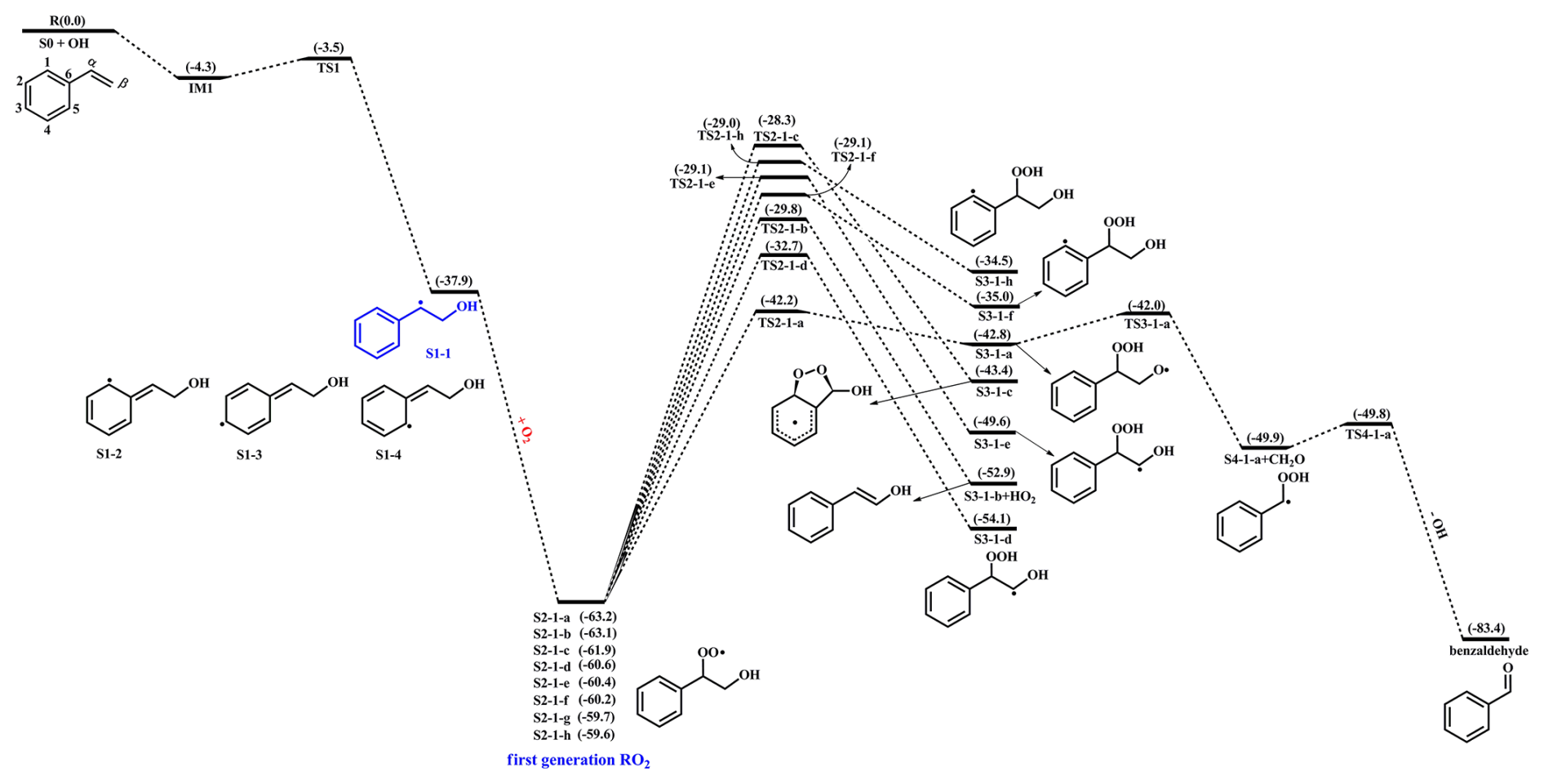
Figure 1PES for the first-stage oxidation of styrene initiated by OH radicals and the isomerization reactions of S2-1-x at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
Due to the existence of resonance structures with radical character on the aromatic ring, the resulting S1-1 can readily isomerize into three other species, namely, S1-2, S1-3 and S1-4. The attack of an O2 molecule on the C-center site of S1-1 leads to the formation of the first generation peroxy radicals S2-1-x ( kcal mol−1). The formed S2-1-x includes eight energetically similar conformers due to the different orientations of O2 attack. In order to distinguish the different conformers, the subscript letter x is used in the present study. The energy ordering of different conformers follows an alphabetical sequence, in which letter a denotes the lowest energy conformer. The Boltzmann population of different conformers in S2-1-x is listed in Table S2 in the Supplement.
For the unimolecular decomposition reactions of S2-1-x, there are three kinds of pathways. One is the intramolecular H-shift reactions, where the hydrogen atom migrates from the −CH2, −CH and −OH groups to the terminal oxygen atom of the −OO group leading to various alkyl and alkoxyl radicals. Among these competing H-shift reactions, the hydrogen atom at the −OH group can be transferred via a six-membered ring transition state (1,5-H shift) to yield an alkoxyl radical S3-1-a, which exhibits the lowest barrier (ΔEa=21.0 kcal mol−1). The resulting S3-1-a can undergo the Cα−Cβ bond cleavage to produce a formaldehyde and an alkyl radical S4-1-a (ΔEa=0.8 kcal mol−1), followed by an OH radical release to form benzaldehyde (ΔEa=0.1 kcal mol−1). The rate coefficients for the aforementioned three pathways are calculated to be , 4.6×1010 and 7.2×1010 s−1, respectively. Based on the values of ΔEa and the corresponding rate coefficients, it can be concluded that the 1,5-H shift reaction is the rate-determining step in the formation of benzaldehyde. The other is the cyclization, where the −OO group attacks the C=C double bond in the benzene ring forming a cyclic peroxide alkyl radical S3-1-c (ΔEa=33.6 kcal mol−1). The last is the HO2-elimination, where a concerted process of Cα−O and Cβ−H bonds scission forms a closed-shell species S3-1-b and a HO2 radical byproduct (ΔEa=33.3 kcal mol−1). The aforementioned results show that the cyclization and HO2-eliminaiton reactions are less importance due to their higher barriers.
As depicted in Fig. S1 in the Supplement, the formations of the first generation peroxy radicals S2-2-x from the association reaction S1-2 + O2 are strongly endothermic ( kcal mol−1), suggesting that they have a significant potential to redissociate back to reactants S1-2 and O2. The resulting S2-2-x can undergo through various intramolecular H-shifts to yield distinct C-centered and O-centered radicals. Among these competing H-shift pathways, hydrogen transfer from the −OH group to the terminal oxygen of −OO group has the lowest barrier (ΔEa= 17.4 kcal mol−1). A similar conclusion is also obtained from the association reactions S1-3 + O2 ( kcal mol−1) and S1-4 + O2 ( kcal mol−1) that the formations of the first generation peroxyl radicals S2-3-x and S2-4-x are thermochemically unfavorable, and their subsequent intramolecular H-shift barriers are considerably high (Figs. S2 and S3 in the Supplement). Therefore, in the present study, we mainly focus on the subsequent reaction mechanisms of S2-1-x under both low and high NOx conditions.
In the low-NOx conditions, the bimolecular reaction with HO2 radicals is expected to be the dominant sink for RO2 radicals (Orlando and Tyndall, 2012; Vereecken et al., 2015). Previous studies have reported that the rate coefficient for the reactions of alkyl peroxyl radicals with HO2 radicals is (Atkinson and Arey, 2003; Boyd et al., 2003). The typical atmospheric concentration of HO2 radicals is 20–40 pptv (Wang et al., 2017; Bianchi et al., 2019), resulting in the pseudo-first-order rate constant [HO2] of 0.01–0.02 s−1. The isomerization reaction of RO2 radicals is competitive with the bimolecular reactions with HO2 radicals only when the rate coefficient of intramolecular H-shifts exceeds 0.01–0.02 s−1. In the high-NOx conditions, the bimolecular reaction of RO2 radicals with NO is considered to be a dominant sink (Orlando and Tyndall, 2012; Vereecken et al., 2015). The rate coefficient for the reaction of alkyl peroxyl radicals with NO is determined to be (Atkinson and Arey, 2003; Bianchi et al., 2019). The typical atmospheric concentration of NO is 0.4–40 ppbv (Wang et al., 2017; Bianchi et al., 2019), leading to the pseudo-first-order rate constant [NO] of 0.1–10 s−1. The intramolecular H-shift reaction of RO2 radicals can compete with the bimolecular reaction with NO only when the rate coefficient of the former case exceeds 10 s−1. Therefore, we use the (0.01–0.02 s−1) and (0.1–10 s−1) values as thresholds to evaluate the relative importance of the isomerization reactions of RO2 radicals under both low- and high-NOx conditions. Previous studies have also employed the same methodology to evaluate the relative importance of isomerization and bimolecular reactions of RO2 radicals during the OH-initiated oxidation of organophosphate esters and alkylbenzenes (Wang et al., 2017; Fu et al., 2024). For the intramolecular H-shift reactions of S2-1-x, the rate coefficient kMC-TST is estimated to be s−1, which is 2–4 orders of magnitude lower than and , indicating that the isomerization reaction of S2-1-x is less competitive than the bimolecular reactions with HO2 radicals and NO.
In the presence of NO, the bimolecular reactions of with NO initially proceed via oxygen-to-oxygen coupling to yield organic nitrites ROONO, which subsequently decompose into benzaldehyde and CH2OH radical or isomerize to organic nitrates RONO2. The energy barrier of the rate-limiting step predicted in Wang's study for the formation of benzaldehyde is 28.4 kcal mol−1, which is approximately 4.0 kcal mol−1 greater than that for the formation of RONO2 (Wang et al., 2015). In the absence of NO, the hydroperoxides ROOH formed from the bimolecular reaction of S2-1-x with HO2 radicals are anticipated to be the dominate products. The aforementioned results are further confirmed by the recent smog chamber experiment study that C7 and C8 series products, as well as organic nitrates are the main components of SOA in the OH-initiated oxidation of styrene under different NOx conditions (Yu et al., 2022b). Considering that the extensive studies on the OH-initiated oxidation of benzaldehyde have done (Sebbar et al., 2011; Zhao et al., 2022; Iuga et al., 2008), this study primarily focuses on the multi-generation •OH oxidation mechanisms of ROOH and RONO2 under both low- and high-NOx conditions.
3.2 Second generation •OH oxidation mechanisms of 1st-ROOH and 1st-RONO2
The first generation products, including hydroperoxides 1st-ROOH and organic nitrates 1st-RONO2, include multiple conformers. To obtain the global minimum of 1st-ROOH and 1st-RONO2, the conformer search is performed by using the Molclus program. The resulting structures are initially optimized at the M06-2X/6-31+g(d,p) level, then the single point energies are calculated at the M06-2X/6-311++G(3df,3pd) level. The global minimum structures of 1st-ROOH (S4) and 1st-RONO2 (S5) are presented in Fig. S4 in the Supplement.
3.2.1 The oxidation mechanism of 1st-ROOH initiated by OH radicals
The reaction 1st-ROOH (S4) +•OH proceeds through the addition of OH radicals to either side of the benzene ring to yield various alkyl radicals, as depicted in Fig. 2. In the present study, syn-OH-addition is defined as the scenario in which the addition of OH radicals occurs at the same side as the −OOH group, while anti-OH-addition is referred to the scenario in which the addition of OH radicals occurs at the opposite side as the −OOH group. For the syn-OH-addition reactions, the addition of OH radicals to the C1-site of 1st-ROOH (S4) exhibits the lowest barrier (ΔEa=3.6 kcal mol−1) due to the stability of the formed product, P. A similar conclusion is also obtained from the anti-OH-addition reactions that the OH-addition pathway occurring at the C1-site is favorable (ΔEa=0.8 kcal mol−1). Notably, the preferred OH-addition pathway in the anti-OH-addition reactions exhibits greater competitiveness compared to that in the syn-OH-addition reactions. It can be explained by the greater steric hindrance present in the latter reaction. In order to further evaluate the reliability of our results, ΔEa of all the syn-OH-addition and anti-OH-addition reactions are recalculated using the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X/6-311+G(d,p) method. As shown in Table S3 in the Supplement, the ΔEa values obtained using the M06-2X/6-311++G(3df,3pd) method are in good agreement with those derived from the DLPNO-CCSD(T)/aug-cc-pVTZ method. The largest deviation and the average absolute deviation are 1.2 and 0.9 kcal mol−1, respectively, indicating that the M06-2X/6-311++G(3df,3pd) method employed in this study is reliable. Based on the values of ΔEa obtained using the DLPNO-CCSD(T)/aug-cc-pVTZ method, it can also be concluded that the addition of OH radicals to C1-site, occurring at the opposite direction relative to the −OOH group, is energetically favorable. The rate coefficients of the addition of OH radicals to the different sites of 1st-ROOH are calculated to be (C1-site), (C2-site), (C3-site), (C4-site), (C5-site) and (C6-site) , respectively. The branching ratios for •OH addition to the C1, C5 and C6 sites are predicted to be 72.4 %, 23.8 % and 3.6 %, respectively, while the sum of branching ratios for •OH addition to other carbon sites is less than 1 %.
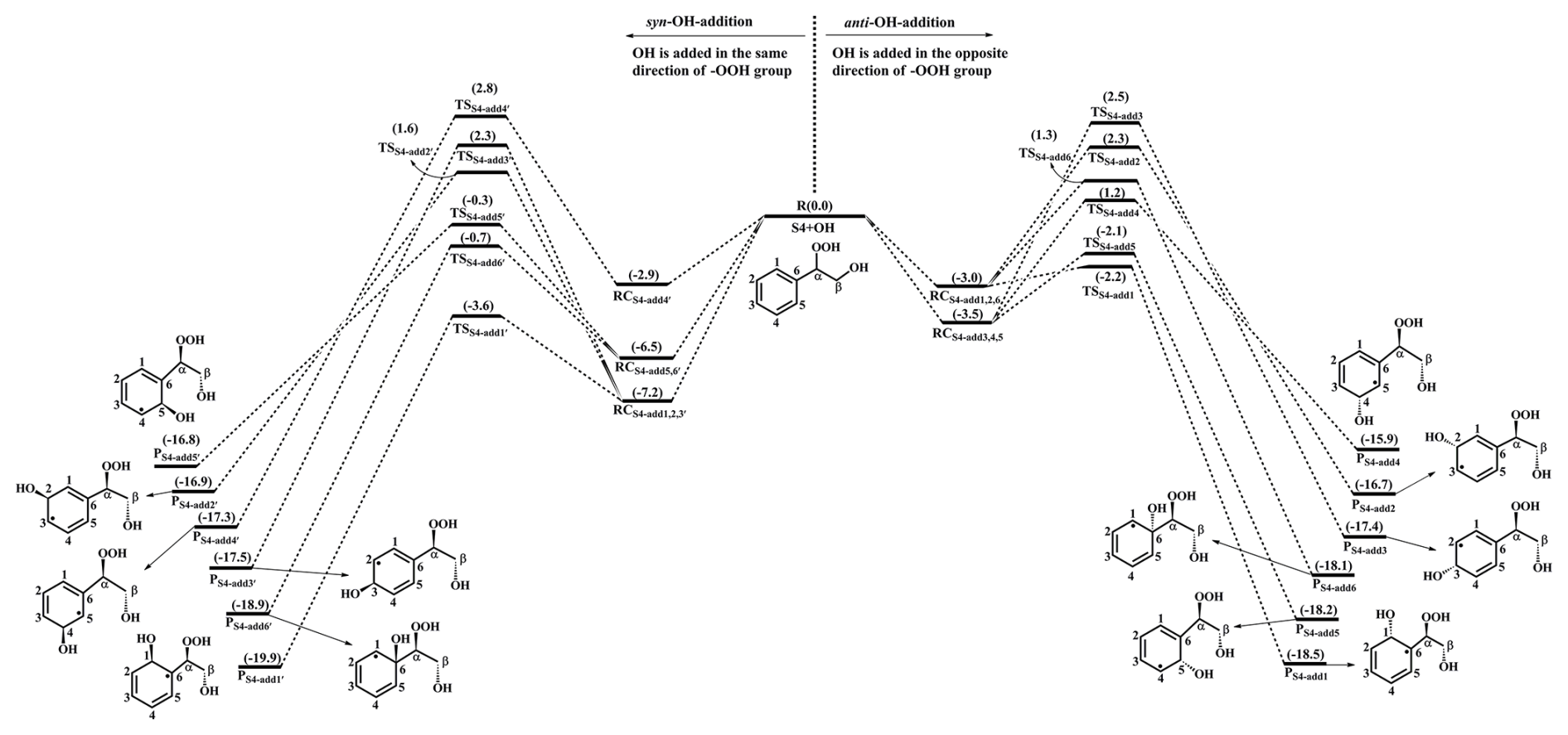
Figure 2PES for the oxidation of 1st-ROOH(S4) initiated by OH radicals at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
Our result is opposite to Zhang's finding that the addition of OH radicals to C6-site would be the most favorable pathway (Zhang et al., 2024). The discrepancy can be explained by the following three factors: (1) The 1st-ROOH conformer selected in the Zhang's study is not the global minimum. In the present study, the global minimum conformer of 1st-ROOH, identified through the conformer search, is found to be 2.2 kcal mol−1 lower than the 1st-ROOH structure selected in the Zhang's study. (2) The pre-reactive complexes are not considered in the Zhang's study. The addition of OH radicals to C1-, C2-, C3- and C6-sites, occurring at the opposite direction relative to the −OOH group, are merely considered in the Zhang's study. They found that the apparent energy barrier of the addition of OH radicals to C6-site is smallest, and is therefore expected to be the favorable pathway. Actually, these OH-addition reactions are modulated by the pre-reactive complexes. It may be inappropriate to determine the favorable pathway based solely on apparent activation energy without considering the pre-reaction complexes. (3) From a geometric perspective, the addition of OH radicals to C6-site is associated with greater steric hindrance compared to other sites, as C6-atom connects with a larger functional group. Base on the aforementioned discussions, we believe that the addition of OH radicals to C6-site is unlikely to be the dominant pathway. Our calculations also confirm that the addition of OH radicals to C6-site is less importance compared to that at the C1-site.
Our conclusion is further supported by the reaction toluene +•OH that the ortho-OH-addition reaction exhibits significant dominance, with the branching ratio of up to 69.8 %–75.8 % (Ji et al., 2017; Zhang, 2019; Wu et al., 2020). Considering the high reactivity of ortho-OH-addition in the reactions toluene +•OH and 1st-ROOH (S4) +•OH, the substitute effects of the −CH3 and −OOH groups are explicitly discussed in the present study. Notably, the −CH3 group in toluene is bonded to the C6 atom, and the −OOH group in 1st-ROOH is bonded to the Cα atom, as depicted in Fig. S5 in the Supplement. The optimized geometries of toluene and 1st-ROOH and the NPA atomic charges of all the carbon atoms in the benzene ring are displayed in Fig. S5. The C−C bond lengths and the bond angles in the benzene ring of toluene are approximately 1.39 Å and 120°, respectively, which are consistent with those in the benzene ring of 1st-ROOH. The aforementioned results show the effect of the −CH3 and −OOH groups on the geometric structure of benzene ring is negligible. From the perspective of NPA atomic charges, the charges on the C1 (−0.246 e) and C5 (−0.246 e) atoms are more greater than those on the other carbon atoms in the benzene ring of toluene. And the OH-adduct formed from the ortho-OH-addition reaction exhibits the greater stability. These results indicate that the −CH3 group is a typical ortho-directing substituent and exerts an activating effect on the ortho-site of the benzene ring, which explains why the ortho-OH-addition reaction is predominant in the reaction toluene +•OH. Compared with the charges on the carbon atoms in the benzene ring of toluene, the charges on C1 and C6 atoms increase by 0.013 e and 0.057 e, respectively, in 1st-ROOH, which can be attributed to the electron-withdrawing effect of the −OOH group. The charge on the C1 atom (−0.259 e) is the highest, and the stability of the resulting OH-adduct is the greatest, implying that the addition of OH radicals to C1-site is dominant in the reaction 1st-ROOH +•OH. Therefore, a direct comparison of the favorable OH-addition pathway in the reactions toluene +•OH and 1st-ROOH (S4) +•OH is performed in this study.
The formed product PS4-add1 includes two conjugate double bonds (C2=C3 and C4=C5), which can readily isomerize to PS4-add2 and PS4-add3, as evident from Fig. S6 in the Supplement. In the present of O2, the attack of an O2 molecule on the C-centered site of PS4-add1, PS4-add2, and PS4-add3 proceed via the barrierless processes to produce the second generation peroxy radicals PS4-add1-a/-s, PS4-add2-a/-s and PS4-add3-a/-s. The O2-addition reaction occurring at the same direction as the −OOH group is defined as syn-O2-addition, while the O2-addition reaction occurring at the opposite direction as the −OOH group is defined as anti-O2-addition. For the reaction , ΔEr of anti-O2-addition is −5.8 kcal mol−1, which is lower than that of syn-O2-addition by 0.4 kcal mol−1, suggesting that anti-O2-addition is preferable over syn-O2-addition in energy. For the reactions and , it can be concluded the same by the ΔEr values that anti-O2-addition reaction is energetically feasible.
The resulting PS4-add1-a/-s can proceed intramolecular cyclization reaction, where the attack of end-site oxygen atom of the −OO group on C2-site of the C2=C3 double bond, leading to the formation of peroxide bicyclic alkyl radicals. ΔEa and ΔEr of the reaction PS4-add1-a→PS4-add1-a-1 are 11.8 and −16.8 kcal mol−1, respectively, which are lower than those of the reaction PS4-add1-s→PS4-add1-s-1 by 3.9 and 2.2 kcal mol−1, respectively. The aforementioned results reveal that the intramolecular cyclization reaction of anti-O2-addition product PS4-add1-a is favorable on both thermochemically and kinetically. A similar conclusion is also derived from the intramolecular cyclization reactions of anti-O2-addition products PS4-add2-a and PS4-add3-a. Notably, the barriers of the intramolecular cyclization reactions PS4-add2-a→PS4-add2-a-1 (ΔEa=31.1 kcal mol−1) and PS4-add2-a→PS4-add2-a-2 (ΔEa=34.6 kcal mol−1) are extremely high, making them insignificant in the atmosphere. The tautomerization between PS4-add1-a-1 and PS4-add3-a-1 readily occurs due to the existence of resonance structures, and it is therefore that the latter conformer is selected as a prototype for the investigating of its subsequent reaction mechanism.
The formed PS4-add3-a-1 can combine with an O2 molecule leading to the third generation peroxy radicals (also called as peroxide bicyclic peroxy radicals, BPR) PS4-add3-a-2, and the lowest energy conformer is presented in Fig. S7 in the Supplement. The isomerization of PS4-add3-a-2 may undergo through a concerted process of the cleavage of bridge bond and C1−C2 bond as well as hydrogen atom transfer from the hydroxyl group to the bridge oxygen atom, yielding a new peroxy radical (ΔEa=28.5 kcal mol−1). The room temperature rate coefficient is calculated to be s−1, which is several orders of magnitude low than the typical pseudo-first-order rate constants (0.01–0.02 s−1) and (0.1–10 s−1), suggesting that the isomerization reaction is less importance in the atmosphere. Therefore, the bimolecular reactions of PS4-add3-a-2 with HO2 radicals with NO are mainly taken into consideration in this study.
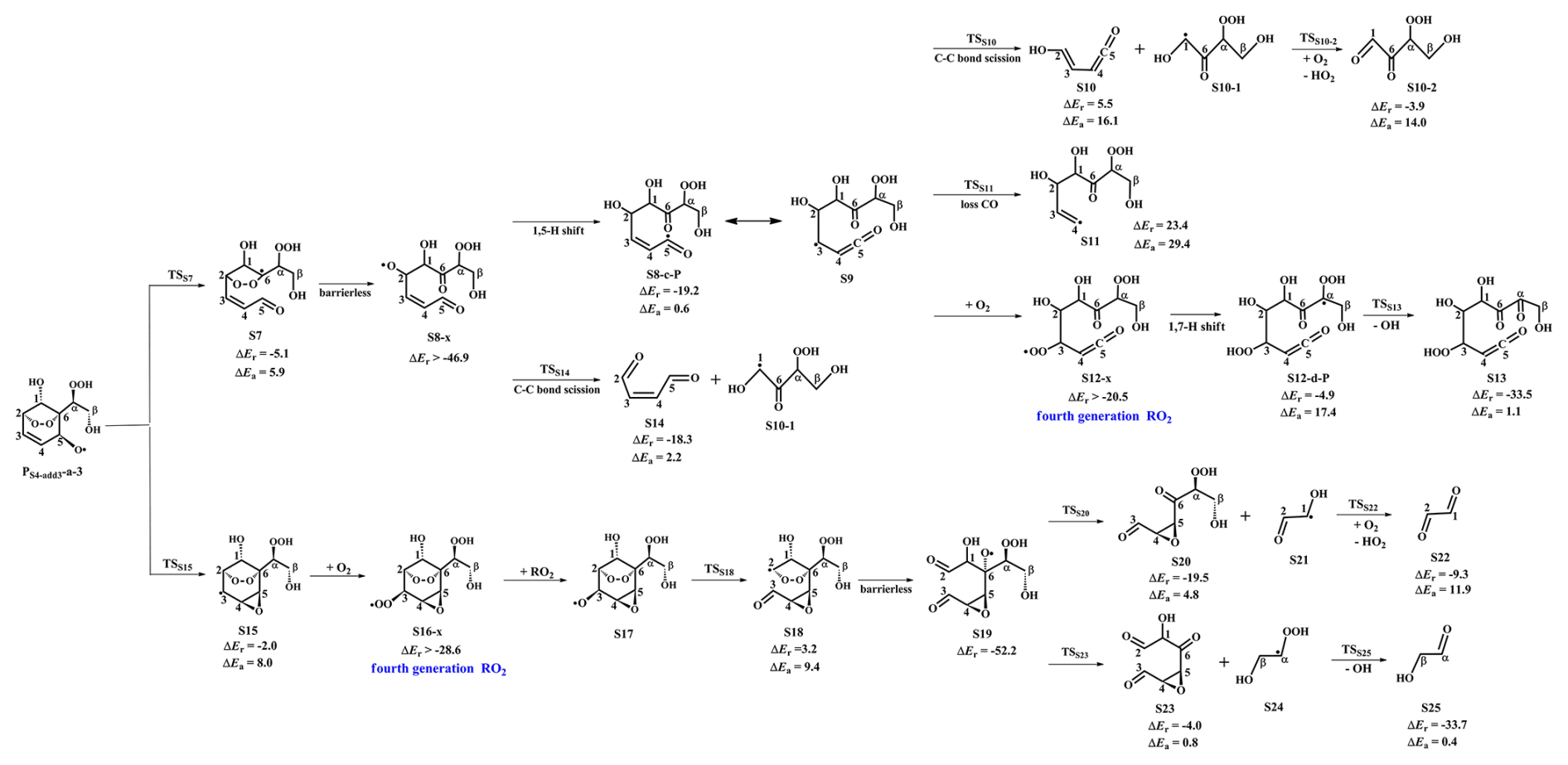
Figure 3PES for the unimolecular decomposition of PS4-add3-a-3 and its subsequent reactions at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
In the pristine environments, PS4-add3-a-2 can react with HO2 radicals resulting in the formation of the second generation products, bicyclic hydroperoxide 2nd-ROOH (S6) and peroxide bicyclic alkoxy radical (BAR) PS4-add3-a-3, as depicted in Fig. S7. For the subsequent reactions of S6 initiated by OH radicals, the detailed mechanisms are discussed in Sect. 3.3.1. From Fig. 3, it can be seen that the unimolecular decomposition of PS4-add3-a-3 involves two kinds of pathways. One is the ring-opening reaction, where the breakage of C5−C6 bond produces an alkyl radical S7 (ΔEa=5.9 kcal mol−1). The other is cyclization reaction, where the attack of oxygen atom of O-centered site on the C4-site of the C3=C4 double bond generates the ring-retaining alkyl radical S15 (Ea=8.0 kcal mol−1). The branching ratios for the formation of S7 and S15 are predicted to be 74.7 % and 25.3 %, respectively.
As shown in Fig. 3, S7 decomposes through the barrierless rupture of bridge bond to form alkoxy radical S8-x, which includes five possible conformers as presented in Fig. S8 in the Supplement. The Boltzmann populations of different conformers are listed in Table S4 in the Supplement. S8-x can undergo various intramolecular H-shifts, in which a hydrogen atom is transferred from different carbon atoms to O-centered site, forming the alkyl radicals. Among the competing H-shift reactions, 1,5 H-shift occurring at the −C5(O)H group exhibits the smallest barrier (ΔEa=0.6 kcal mol−1), and kMC-TST is calculated to be 8.2×109 s−1 at ambient temperature (Table S5 in the Supplement). The formed S8-c-P can readily isomerize to S9 due to its resonance stabilized structure. The unimolecular decomposition of S9 can proceed through the C1−C2 bond scission to produce a ketene-enol S10 and an alkyl radical S10-1 (ΔEa=16.1 kcal mol−1), followed by reaction with O2 leading to a HO2 radical and a 1,2-dicarbonyl compound S10-2 (ΔEa=14.0 kcal mol−1). Alternatively, S9 may undergo via the elimination of CO to generate an alkyl radicals S11 (ΔEa=29.4 kcal mol−1). The aforementioned results show that the formation of S10 and S10-1 is energetically favorable, with the rate coefficient kS10 of 26.1 s−1.
In the presence of O2, the attack of an O2 molecule on the C-centered sites of S9 leads to the fourth generation peroxyl radical S12-x ( kcal mol−1). Adopting the rate coefficient of for the reactions of alkyl radicals with O2, and the atmospheric O2 concentration of 5×1018 (Ma et al., 2021), the pseudo-first-order rate constant [O2] is 3.0×107 s−1. The unimolecular decomposition of alkyl radicals is competitive only when their decay rate exceeds 3.0×107 s−1. is about six orders of magnitude greater than kS10, indicating that the unimolecular decomposition of S9 is less importance. As shown in Fig. S9 in the Supplement, S12-x can proceed various intramolecular H-shift reactions, where hydrogen atom migrates from the different carbon sites or hydroxyl groups to the terminal oxygen atom of the −OO group, resulting in the formation of QOOH radicals and alkoxyl radicals. Among these competing H-shift reactions, the 1,7-H transfer at the Cα-site leading to the formation of S12-d-P exhibits the smallest barrier (ΔEa=17.4 kcal mol−1). Then, it decomposes to yield an OH radical and a closed-shell product S13 containing a hydroperoxide, three hydroxyl and three carbonyl groups (ΔEa=1.1 kcal mol−1).
S8-x can proceed through the C1−C2 bond scission to yield an unsaturated 1,4-dicarbonyl species S14 and an alkyl radical S10-1 (ΔEa=2.2 kcal mol−1), with the rate coefficient of 2.1×1010 s−1. Notably, both the 1,5 aldehyde H-shift and C1−C2 bond scission reactions yield a closed-shell species S10-2 with up to five oxygen atoms, and the branching ratios are predicted to be 28.1 % and 71.9 %, respectively. The result is further supported by the previous study that the proportion of aldehyde H-shift products constitutes about one third of the total products in the reaction benzene +•OH (Wang et al., 2020b).
As shown in Fig. 3, S15 can further react with O2 leading to the fourth generation peroxy radical S16-x, which can proceed either intramolecular H-shifts forming QOOH radicals (Fig. S10 in the Supplement), or reactions with RO2 radicals and NO forming alkoxyl radical S17. Notably, the barriers of intramolecular H-shifts are extremely high (ΔEa>34.6 kcal mol−1), making them less importance in the atmosphere. The transformation of S17 undergoes through the breakage of C2−C3 bond to produce an alkyl radical S18 (ΔEa=9.4 kcal mol−1), followed by fragmentation into an alkoxyl radical S19 via the barrierless rupture of the bridge bond. Then, S19 dissociates to an OH radical, a glycolaldehyde S25 and a C6-epoxide product S23 bearing a hydroxy and three carbonyl groups, being the dominant pathway. The regeneration of OH radicals drives the successive autoxidation of styrene, eventually leading to the production of multifunctional products.
3.2.2 The oxidation mechanism of 1st-RONO2 initiated by OH radicals
The OH-initiated oxidation of 1st-RONO2 (S5) proceeds through the addition of OH radicals to different carbon sites in the benzene ring to form various alkyl radicals PS5-addx, as depicted in Fig. 4. Among the competing OH-addition reactions, the OH-addition reaction at the C1-site, which proceeds on the opposite direction as the −ONO2 group, has the smallest barrier (RS5-add1, ΔEa=0.4 kcal mol−1) due to the stability of the formed product PS5-add1. The result again shows that the ortho-addition reaction is energetically feasible. PS5-add1 may isomerize to two other resonance structures, namely, PS5-add2 and PS5-add3. For the reaction PS5-add1+O2, O2 may add on either the opposite (anti-O2-addition) or the same direction (syn-O2-addition) relative to the −NO3 group, leading to the second generation peroxyl radicals PS5-add1-a and PS5-add1-s (Fig. S11 in the Supplement). The exoergicity of these two reactions are −6.7 and −4.4 kcal mol−1, respectively, suggesting that the anti-O2-addition reaction is thermochemically favorable. Next, they can isomerize via a cyclization process to yield PS5-add1-a-1 and PS5-add1-s-1 with the ΔEa of 13.3 and 18.1 kcal mol−1. This result shows that the cyclization reaction of anti-O2-addition product PS5-add1-a is kinetically feasible. A similar conclusion is also obtained from the reaction PS5-add3+O2 that the formation of anti-O2-addition product PS5-add3-a-1 is dominant. Due to the existence of the conjugate double bond, it facilitates the tautomerization between PS5-add1-a-1 and PS5-add3-a-1. Therefore, we mainly focus on the subsequent chemistry of PS5-add3-a-1 in the present study.
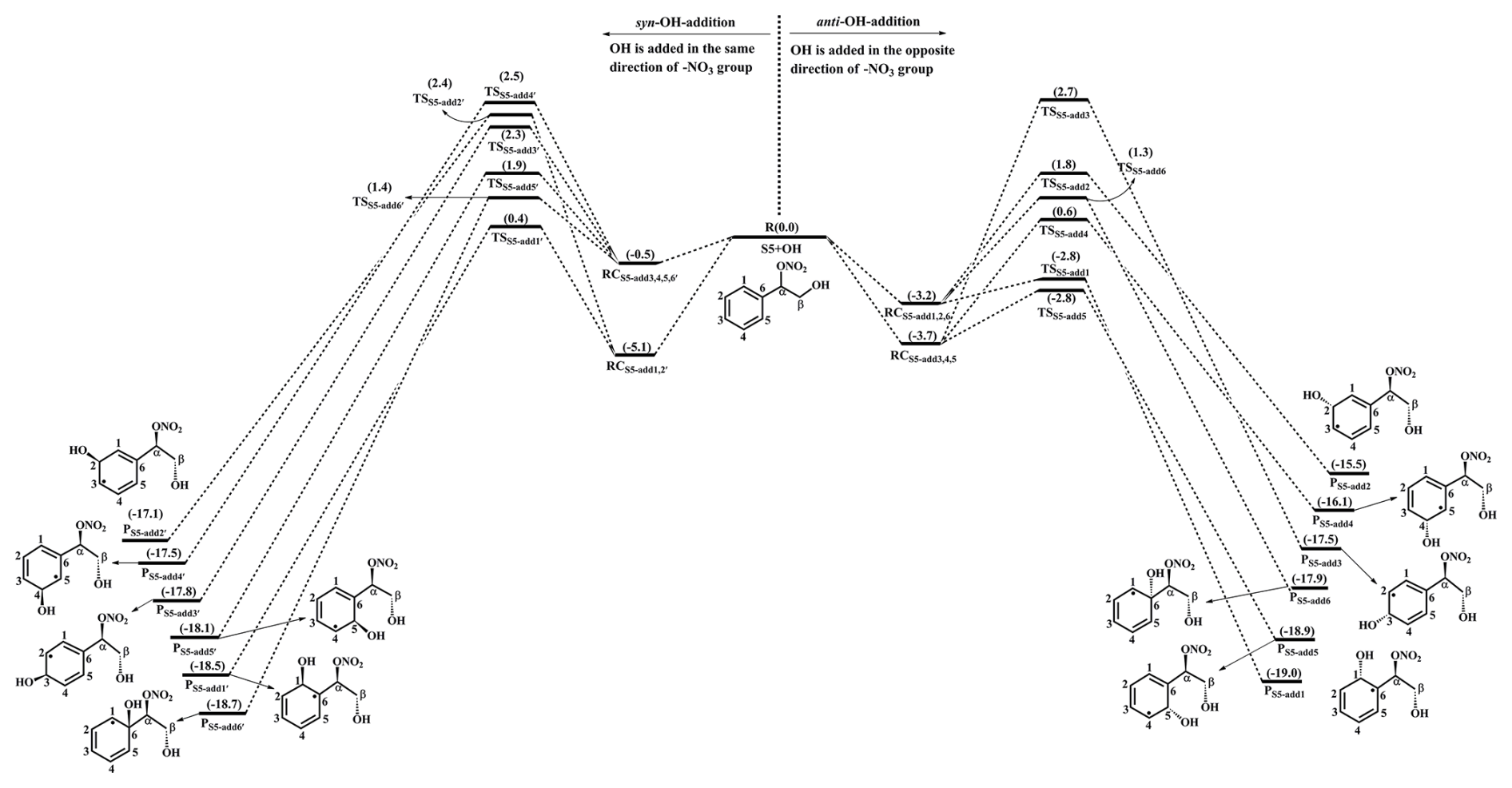
Figure 4PES for the oxidation of 1st-RONO2(S5) initiated by OH radicals at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
PS5-add3-a-1 can further react with an O2 molecule leading to the third generation peroxyl radicals PS5-add3-a-2, which include multiple conformers. The lowest energy conformer resulting from conformer search is presented in Fig. S12 in the Supplement. In urban environments, the bimolecular reaction of PS5-add3-a-2 with NO yields the second generation products, a bicyclic organic nitrate 2nd-RONO2 (S26) and a BAR PS5-add3-a-3, as displayed in Fig. S12. The detailed mechanism of OH-initiated oxidation of S26 is discussed in Sect. 3.3.2. As shown in Fig. 5, PS5-add3-a-3 can either proceed via a ring opening process to form an alkyl radical S27 (ΔEa=7.3 kcal mol−1), or undergo through a cyclization process to generate an epoxide species S35 (ΔEa=8.5 kcal mol−1). The branching ratios of these two reactions are predicted to be 69.2 % and 30.8 %, respectively. Notably, the branching ratio of cyclization reaction of PS5-add3-a-3 increases by 5.5 % compared to that of cyclization reaction of PS4-add3-a-3, suggesting that the −ONO2 substitution is beneficial to cyclization reaction.
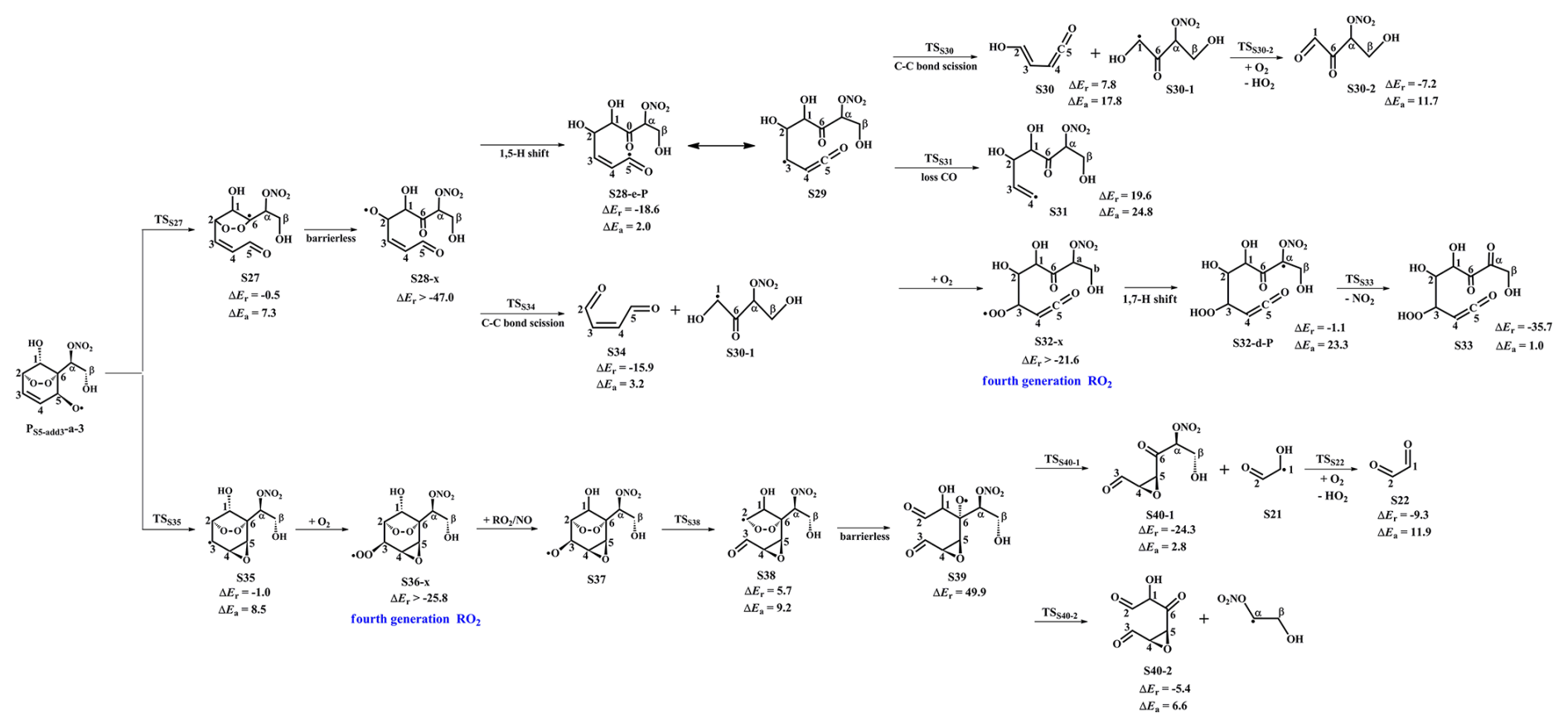
Figure 5PES for the unimolecular decomposition of PS5-add3-a-3 and its subsequent reactions at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
The degradation of S27 proceeds through the barrierless scission of bridge bond to form S28-x, and the Boltzmann populations of different conformers are listed in Table S6 in the Supplement. S28-x can undergo via various intramolecular H-shifts to produce QOOH radicals, in which hydrogen atom transfer from the −C(O)H group to the terminal oxygen atom of the −OO group forming S28-e-P has the smallest barrier (ΔEa=2.0 kcal mol−1) (Fig. S13 in the Supplement). S28-e-P can readily isomerize to S29, which includes two distinct decomposition pathways. One is the C1−C2 bond cleavage, yielding a ketene-enol S30 and an alkyl radical S30-1 (ΔEa=17.8 kcal mol−1), followed by reaction with O2 to form a HO2 radical and a 1,2-dicarbonyl species S30-2 (ΔEa=11.7 kcal mol−1). The other is the elimination of CO to generate an alkyl radical S31 (ΔEa=24.8 kcal mol−1), but the barrier is considerably high, making this pathway less competitive. The rate coefficient for the formation of S30 and S30-1 is calculated to be 14.4 s−1, which is about six orders of magnitude lower than the pseudo-first-order rate constant , indicating that the unimolecular decomposition of S29 is insignificant.
In the presence of O2, the bimolecular reaction of S29 with O2 produces the fourth generation peroxyl radicals S32-x, comprising five energetically similar conformers as shown in Fig. S14 in the Supplement. For the 1,7-H transfer reaction, hydrogen atom at the Cα-site can be transferred through an eight-membered ring transition state to generate an alkyl radical S32-d-P (ΔEa=23.3 kcal mol−1), followed by the elimination of NO2 forming a closed product S33 (ΔEa=1.0 kcal mol−1). S33 and S13 are isomeric species, with the former exhibiting more stability than the latter. S28-x can proceed through the cleavage of C1−C2 bond to generate an unsaturated 1,4-dicarbonyl compound S34 and an alkyl radical S30-1. The rare coefficients of the 1,5 aldehyde H-shift and C1−C2 bond scission reactions are predicted to be 1.7×109 and 5.8×109 s−1 (Table S7 in the Supplement), respectively, with the branching ratios of 23 % and 77 %. S30-1, formed from the above mentioned two pathways, may undergo through H-abstraction by O2 to yield an organic nitrate S30-2 bearing a hydroxyl and two carbonyl groups (ΔEa=11.7 kcal mol−1).
S35 can combine with an O2 molecule forming the fourth generation peroxyl radicals S36-x, which have five possible conformers as shown in Fig. S15 in the Supplement. S36-x can proceed either intramolecular H-shifts forming QOOH radicals, or reaction with RO2 radicals and NO generating alkoxyl radical S37. However, the barriers of intramolecular H-shifts are extremely high (ΔEa>31.3 kcal mol−1), making them less importance in the atmosphere. The degradation of S37 initially proceeds via the breakage of C2−C3 bond to form S38 ( followed by decomposition into an alkoxyl radical S39 via the barrierless scission of bridge bond. The dominant pathway of the unimolecular decomposition of S39 is the formation of a glyoxal and a C6-epoxide species S40-1 bearing a −NO3, a hydroxyl and two carbonyl groups. This process differs from the unimolecular decay of S19, where the favorable pathways is the formation of a tricarbonyl compound S23. The aforementioned results reveal that the preferable pathway is strongly dependent on the breakage of C−C bond associated with the property of substituents in the decomposition of alkoxy radicals.
3.3 Third generation •OH oxidation mechanisms of 2nd-ROOH and 2nd-RONO2
The second generation products, bicyclic hydroperoxide 2nd-ROOH and bicyclic organic nitrate 2nd-RONO2, have multiple possible conformers. The global minimum structures of 2nd-ROOH (S6) and 2nd-RONO2 (S26) resulting from the conformer search are presented in Figs. S7 and S12, respectively.
3.3.1 The oxidation mechanism of 2nd-ROOH initiated by OH radicals
OH-initiated oxidation of 2nd-ROOH (S6) can either undergo through the addition of OH radicals to either side of the C3=C4 double bond to generate the alkyl radicals, or proceed via H-abstraction from the different carbon sites to produce the alkyl radicals and alkoxyl radicals, as shown in Figs. S16 and S17 in the Supplement. For the OH-addition reactions, syn-OH-addition is defined as the addition of OH radicals on the same side as the −OOH group, while anti-OH-addition is referred to the addition of OH radicals on the opposite side as the −OOH group. The addition of OH radicals to the C3-site of the C3=C4 double bond forming the product PS6-abs3 has the smallest barrier (ΔEa=2.4 kcal mol−1) and the exoergicity of −33.5 kcal mol−1. For the H-abstraction reactions, the abstraction of hydrogen atom at the C5-site is the most favorable pathway (ΔEa=3.6 kcal mol−1) and the exoergicity of −20.2 kcal mol−1. It is mainly because that the presence of an allyl group enhances the stability of the resulting product PS6-abs5. Notably, the abstraction of hydrogen atom at the C2-site proceeds through a concerted process of C2−H bond and bridge bond rupture, leading to the formation of an alkoxyl radical PS6-abs2 (ΔEa=7.2 kcal mol−1). This reaction is expected to be less importance due to its higher energy barrier. The rate coefficient of the favorable OH-addition reaction is calculated to be , which is about one order of magnitude greater than that of the preferable H-abstraction reaction ( ). Based on the above discussion, it can be concluded that OH-addition reaction is favorable on both thermochemically and kinetically. This conclusion is further supported by the •OH + alkene reaction systems that OH-addition pathways are predominant (Chen et al., 2021; Yang et al., 2017; Arathala and Musah, 2024).
As depicted in Fig. S18 in the Supplement, the unimolecular decay of the product PS6-add3 resulting from the favorable OH-addition reaction proceeds through a cyclization process to yield an epoxide compound S41 and an OH radical byproduct with the ΔEa of 15.3 kcal mol−1 and the rate coefficient kR41 of 1.8×102 s−1, or undergoes via intramolecular 1,4 H-shift to form a peroxy radical S42 with the ΔEa of 21.8 kcal mol−1 and the rate coefficient kR43 of 1.9 s−1, or proceeds via the elimination of hydrogen atom to produce an alkene S43 with the ΔEa of 37.9 kcal mol−1. Based on the values of ΔEa and the corresponding rate coefficients, the dominant pathway of the unimolecular decomposition of PS6-add3 is the formation of S41. In the presence of O2, the pseudo-first-order rate constant of the reactions of alkyl radicals with O2 is 3.0×107 s−1, which is about five orders of magnitude greater than kR41, suggesting that the unimolecular decomposition of PS6-add3 is insignificant.
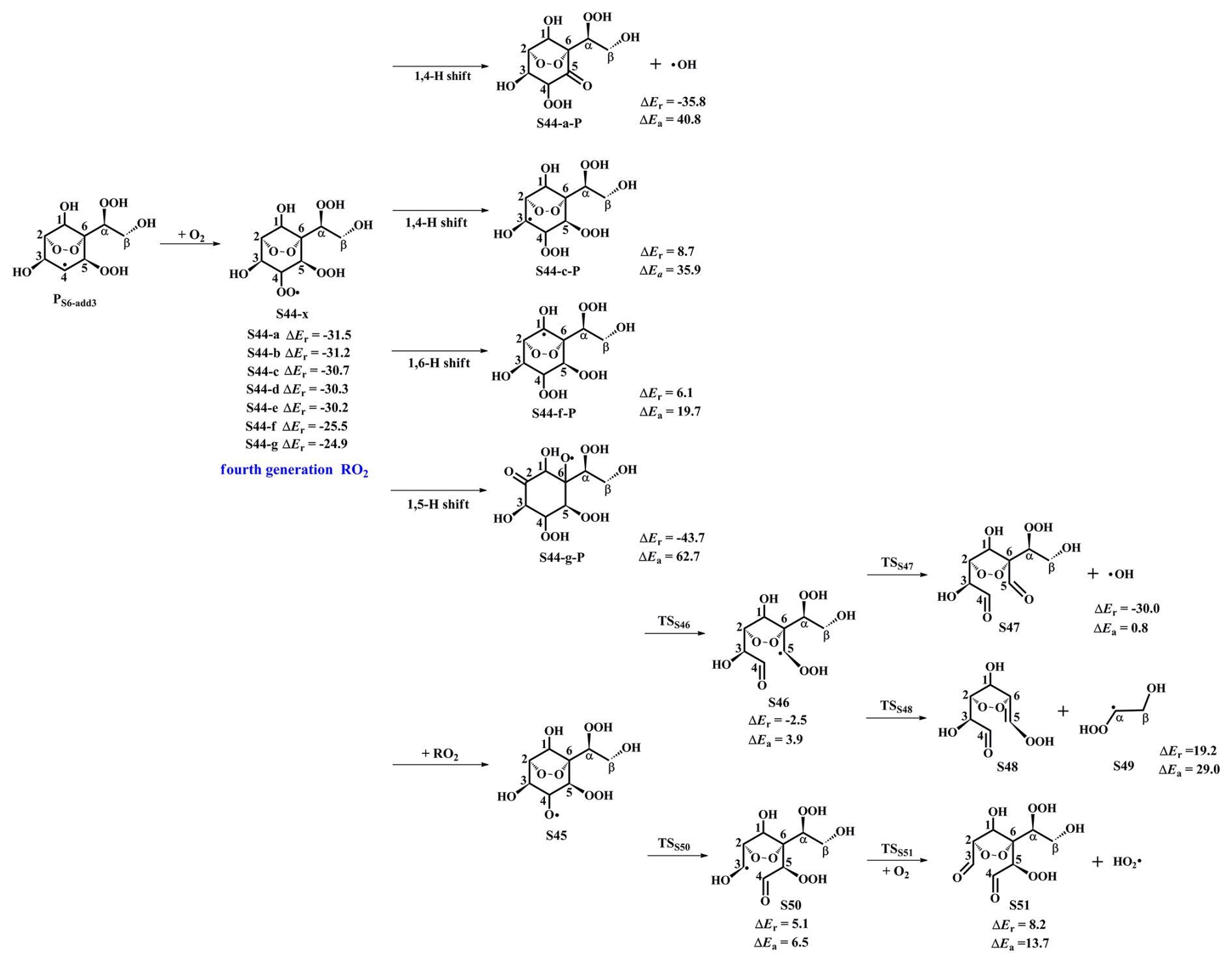
Figure 6PES for the subsequent reactions of PS6-add3 in the presence of O2 at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
As shown in Fig. 6, the fourth generation peroxy radicals S44-x formed in the addition reaction PS6-add3+O2 can either proceed via intramolecular H-shits to form QOOH, or undergo through self- or cross-reactions to yield an alkoxy radical S45. Due to the considerably high barriers of intramolecular H-shifts, they are deemed to be negligible under atmospheric conditions. S45 can convert into an alkyl radical S46 through the cleavage of C4−C5 bond, or dissociate to an alkyl radical S50 via the rupture of C3−C4 bond. The barrier of the former reaction is 3.9 kcal mol−1, which is lower than that of the latter pathway by 2.6 kcal mol−1, indicating that the formation of S46 is kinetically preferable. Then, S46 decomposes into an OH radical byproduct and a C8-product S47 bearing a −OOH, a peroxide bridge, two carbonyls, and three hydroxy groups, which is expected to be the dominant pathway owing to its lower barrier. The rate coefficient kRS47 is estimated to be 1.8×109 s−1, which is about two orders of magnitude greater than the pseudo-first-order rate constant (3.0×107 s−1). The result reveals that the unimolecular decomposition of S46 is more competitive than the bimolecular reaction with O2. The formed OH radicals can once again participate in the oxidations of styrene and its multifunctional products, continuing these processes until they are completely consumed.
3.3.2 The oxidation mechanism of 2nd-RONO2 initiated by OH radicals
OH-initiated oxidation of 2nd-RONO2 (S26) includes four different OH-addition pathways and five different H-abstraction pathways, as displayed in Figs. S19 and S20 in the Supplement. For the OH-addition reactions, the attack of OH radicals on the C3-site of the C3=C4 double bond forming the product PS26-add3, occurring on the same direction relative to the −ONO2 group, is found to be the favorable pathway (ΔEa=2.4 kcal mol−1, kcal mol−1). For the H-abstraction reactions, the abstraction of hydrogen atom at the C5-site is identified as the preferable pathway (ΔEa=5.7 kcal mol−1, kcal mol−1) due to the enhanced stability of the resulting product PS26-add5 by the presence of an allyl group. By comparing the values of ΔEa and ΔEr of the favorable OH-addition and H-abstraction pathways, it can concluded that the former case is dominant on both thermochemically and kinetically. This conclusion is consistent with the result from the reaction 2nd-ROOH (S6) +•OH that OH-addition is more competitive than H-abstraction.
The product PS26-add3 arising from the favorable OH-addition pathway has three potential unimolecular decay pathways, as depicted in Fig. S21 in the Supplement: (1) PS26-add3 dissociates to an epoxide S52 and a NO2 molecule through a cyclization process with the ΔEa of 18.5 kcal mol−1 and the rate coefficient kR52 of 0.4 s−1; (2) PS26-add3 isomerizes to an alkyl radical S53 via the intramolecular 1,2 H-shift (ΔEa=40.0 kcal mol−1); (3) PS26-add3 converts into an alkene S54 via the elimination of hydrogen atom (ΔEa=39.1 kcal mol−1). Based on the value of ΔEa and the corresponding rate coefficient, the dominant pathway of the unimolecular decomposition of PS26-add3 is the formation of S52. kR52 is about seven orders of magnitude lower than the pseudo-first-order rate constant , indicating that the unimolecular decomposition of PS26-add3 is less importance.
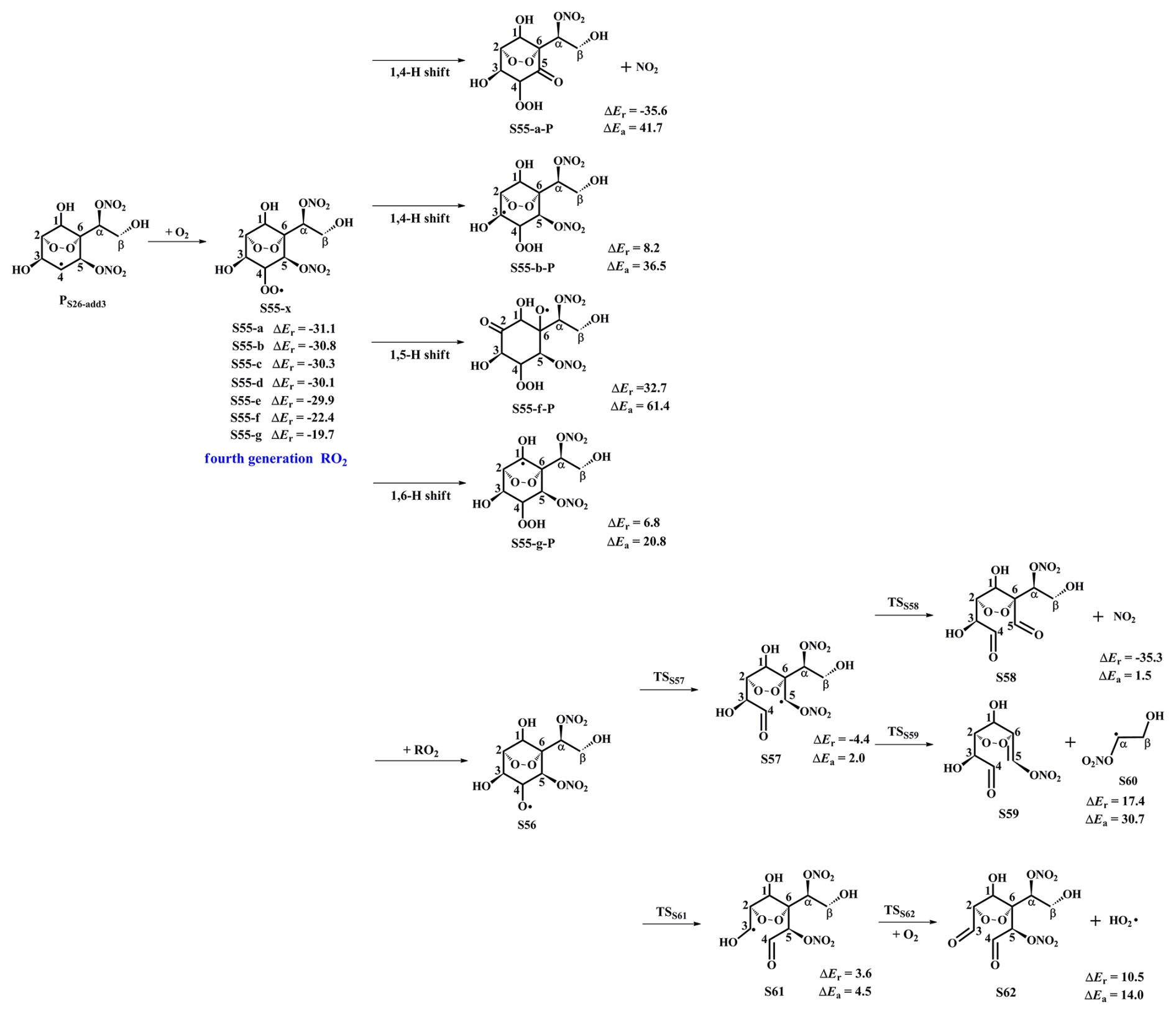
Figure 7PES for the subsequent reactions of PS26-add3 in the presence of O2 at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+g(d,p) level.
In the presence of O2, PS26-add3 can react with an O2 molecule leading to the formation of the fourth generation peroxy radicals S55-x, comprising seven possible conformers as shown in Fig. 7. For the intramolecular H-shifts of S55-x, not all of reactants (S55-c, S55-d and S55-e) have the suitable conformers that allow for the pathways across the reaction barriers. The barriers of intramolecular H-shifts are considerably high (ΔEa=20.8 kcal mol−1), making them uncompetitive in the atmosphere. Alternatively, S55-x can react with other RO2 radicals forming an alkoxyl radical S56, followed by decomposition into an alkyl radical S57 via the breakage of C4−C5 bond (ΔEa=2.0 kcal mol−1), or fragmentation into an alkyl radical S61 through the cleavage of C3−C4 bond (ΔEa=4.5 kcal mol−1). The aforementioned results reveal that the formation of S57 is energetically favorable, which is consistent with the conclusion derived from the unimolecular decomposition of S45 that the breakage of C4−C5 bond is feasible. Next, S57 dissociates to a NO2 coproduct and a C8-product S58 that possessed a −NO3, a peroxide bridge, two carbonyls, and three hydroxy groups. This pathway is expected to be the dominant one (ΔEa=1.5 kcal mol−1), with the rate coefficient kRS58 of 1.2×109 s−1. The resulting NO2 can further participate in the cycling of NOx, ultimately generating tropospheric ozone and SOA.
The overall reaction mechanism and the fractional yields of the major products in the multi-generation •OH oxidation of styrene under different NOx conditions are presented in Figs. S22 and S23 in the Supplement. In the low-NOx conditions, the fractional yield of the first generation closed-shell product 1st-ROOH (S4) formed from the reaction S2-1-x + HO2• is predicted to be 71.6 %. For the second generation •OH oxidation, the reaction of the peroxyl radical PS4-add3-a-2 with HO2 radicals produces the second generation closed-shell product 2nd-ROOH (S6) and an alkoxyl radical PS4-add3-a-3, with the fractional yields of 41.4 % and 10.4 %, respectively. The formed PS4-add3-a-2 can either proceed through the C5−C6 bond scission to produce an alkyl radical S7 with the fractional yield of 7.8 %, or undergo via a cyclization process to generate an alkyl radical S15 with the fractional yield of 2.6 %. S7 and S15 can be transformed via a series of reactions, ultimately leading to the formation of second generation closed-shell product S10-2, S13 and S23, with the fractional yields of 5.6 %, 2.2 % and 1.3 %, respectively. For the third generation •OH oxidation, the degradation of 2nd-ROOH (S6) ultimately yields the third generation closed-shell products S47 and S51, with the fractional yields of 26.3 % and 0.3 %, respectively. As a result, the major closed-shell products are 1st-ROOH (S4), 2nd-ROOH (S6), S10-2, S13 and S47 in the multi-generation •OH oxidation of styrene in the low-NOx conditions.
In the high-NOx conditions, the fractional yield of the first generation closed-shell product 1st-RONO2 (S5) formed from the reaction S2-1-x + NO is predicted to be 26.5 %, as shown in Fig. S23. As the •OH oxidation reactions proceed, 1st-RONO2 (S5) can be initially transformed into the peroxyl radical PS5-add3-a-2, followed by reaction with NO to form the second generation closed-shell product 2nd-RONO2 (S26) and an alkoxyl radical PS5-add3-a-3, with the fractional yields of 4.8 % and 11.2 %, respectively. The decomposition of PS5-add3-a-3 undergoes via two distinct pathways. One is the C5−C6 bond cleavage, leading to an alkyl radical S27 with the fractional yield of 7.8 %. The other is the cyclization, resulting in an alkyl radical S35 with the fractional yield of 3.4 %. The resulting S27 and S35 undergo multiple oxidation steps, finally leading to the formation of the second generation closed-shell products S30-2, S33 and S40-1, with the fractional yields of 6.0 %, 1.8 %, and 1.7 %, respectively. 2nd-RONO2 (S26) can be further oxidized to yield the third generation closed-shell products S58 and S62, with the fractional yields of 2.6 % and 0.03 %, respectively. In summary, the major closed-shell products are 1st-RONO2 (S5), 2nd-RONO2 (S26), S30-2 and S58 in the multi-generation •OH oxidation of styrene in the high-NOx conditions.
3.4 Volatility classes
The volatility classes for various organic compounds are based on their saturation concentration, as proposed by Donahue et al. (2012). The saturated vapour pressure (P0) and saturated concentration (c0) of styrene and its multi-generation •OH oxidation products are predicted by using the SIMPOL.1 method (Pankow et al., 2008). As show in Table S8 in the Supplement, the P0 and c0 of the first generation closed-shell product benzaldehyde (C7H6O) are atm and 2.89×106 ug m−3, respectively, which are 3–4 orders of magnitude greater than those of S4 (C8H10O3, atm and ug m−3) and S5 (C8H9NO3, atm and ug m−3). Based on the values of c0, benzaldehyde is classified as the volatile organic compounds (VOCs), whereas S4 and S5 are classified as the intermediate volatility organic compounds (IVOCs). These first generation closed-shell products exist exclusively in the gas phase under atmospheric conditions (Bianchi et al., 2019).
For the second generation closed-shell products, S6 (C8H12O8, ug m−3) and S26 (C8H10N2O10, c0=0.18 ug m−3) formed from the bimolecular reactions with HO2 radicals and NO are classified as the low volatility organic compounds (LVOCs). Similarly, S13 (C8H10O8, ug m−3) and S33 (C8H10O8, ug m−3), formed through the ring-opening and subsequent intramolecular H-shift reactions of PS4-add3-a-3 and PS5-add3-a-3, respectively, are also classified as LVOCs, which can condense onto the existing large particles (Bianchi et al., 2019). The c0 values of the remaining closed-shell products are significantly greater than those of the aforementioned four products, for example, the c0 values of S20 (C6H8O6) and S40-1 (C6H7NO7), formed by the cyclization and decomposition reactions of PS4-add3-a-3 and PS5-add3-a-3, are 42.21 and 75.86 ug m−3, respectively, classifying them as the semivolatile organic compounds (SVOC).
For the third generation closed-shell products, the c0 values of S47 (C8H12O9, ug m−3) and S51 (C8H10O10, ug m−3), formed through the O2-addition and subsequent decomposition reactions of PS6-add3, are about two orders of magnitude lower than those of the second generation closed-shell products S6 and S13, despite being classified as LVOCs. Similarly, S58 (C8H11NO10, ug m−3) and S62 (C8H10N2O12, ug m−3), formed via the O2-addition and subsequent decomposition reactions of PS26-add3, exhibit lower c0 values compared to the second generation closed-shell products S26 and S33. The aforementioned results reveal that the volatility of the multi-generation •OH oxidation products significantly decreases with increasing the number of •OH oxidation steps. As the oxidation reactions of the third generation closed-shell products proceed further, the formed products may possess sufficiently low volatility to participate in the formation and growth of new aerosol particle.
The results reveal that the first generation RO2 radicals, formed from the addition of OH radicals to the Cβ-site of a vinyl group in styrene followed by O2-addition, can proceed intramolecular H-shifts to generate various alkyl and alkoxyl radicals. The rate coefficient kMC-TST is calculated to be s−1. Among the competing H-shift pathways, the hydrogen atom transfer from the −OH group to the terminal oxygen atom of the −OO group has the lowest barrier. The resulting alkoxy radical can further decompose into benzaldehyde through the successive elimination of HCHO and an OH radical. The 1,5-H shift reaction occurring at the −OH group is the rate-determining step in the formation of benzaldehyde. Alternatively, the first generation RO2 radicals can proceed bimolecular reactions with HO2 radicals and NO, leading to the formation of the first generation closed-shell C7- and C8-products 1st-ROOH (C8H10O3), benzaldehyde (C7H6O), and 1st-RONO2 (C8H9NO3).
For the second generation •OH oxidation, OH-addition reaction occurring at the ortho-site of 1st-ROOH and 1st-RONO2 has a significant dominance. This is consistent with the analogous reaction systems, toluene +•OH and phenol +•OH, in which ortho-OH-addition reaction is energetically favorable (Wu et al., 2020; Xu and Wang, 2013). The resulting alkyl radicals may undergo two O2-addition steps and a cyclization process to form BPR, which can react with HO2 radicals and NO to yield the corresponding BAR, and the second generation closed-shell C8-product 2nd-ROOH (C8H12O8) and 2nd-RONO2 (C8H10N2O10), with the fractional yields of 41.4 % and 4.8 %. The unimolecular decomposition of BAR formed in the reaction 1st-ROOH +•OH includes two distinct pathways: (1) ring-opening and followed by decomposition, yielding the multifunctional products S10-2 (C4H6O5) and S13 (C8H10O8) with the fractional yields of 5.6 % and 2.2 %, respectively; or (2) cyclization and followed by dissociation, generating the closed-shell C6-product S23 (C6H6O5) with the fractional yield of 1.3 %. The major products formed from the unimolecular decomposition of BAR in the reaction 1st-RONO2+•OH are the multifunctional products S30-2 (C4H5NO6), S33 (C8H10O8) and S40-1 (C6H7NO7), with the fractional yields of 6.0 %, 1.8 % and 1.7 %, respectively.
For the third generation •OH oxidation, the addition of OH radicals to the C=C bond in 2nd-ROOH and 2nd-RONO2 is the dominant pathway. The resulting alkyl radicals can proceed a series of reactions to produce the alkoxyl radicals, which subsequently decompose into an OH radical byproduct and a closed-shell C8-product S47 (C8H12O9), identified as the favorable pathway in the reaction 2nd-ROOH +•OH. S47 contains a −OOH, a peroxide bridge, two carbonyls, and three hydroxy groups. The major product formed in the reaction 2nd-RONO2+•OH is a closed-shell C8-product S58 (C8H11NO10), which contain a −NO3, a peroxide bridge, two carbonyls, and three hydroxy groups. The fractional yields of S47 and S58 are 26.3 % and 2.6 %, respectively. The volatility of the oxidation products significantly decreases with increasing the number of •OH oxidation steps in the multi-generation •OH oxidation of styrene.
In the laboratory chamber experiments, the structures of some specific oxidation products remain uncharacterized but are merely inferred from the exact mass and fragmentation data. Using high-level quantum chemistry methods, we identify the molecular structures of multifunctional products and elucidate their formation pathways in the multi-generation •OH oxidation of styrene. The mechanistic insights derived from this work are broadly applicable to the photooxidation of structurally analogous aromatics. Furthermore, we quantify the yields of multifunctional products and demonstrate that their volatility decreases significantly with increasing the number of •OH oxidation steps. The resulting multifunctional products may undergo a series of oxidation reactions to form low volatility compounds, thereby contributing to the formation and growth of new aerosol particle. In the future, more detailed experimental and theoretical studies need to be conducted to identify the molecular structures and formation pathways of multifunctional products formed through the photooxidation of other aromatics under both low and high-NOx conditions. These studies will facilitate a more accurate characterization of the chemical composition and formation yields of aromatic SOA, and thereby help narrow the gap between the measured and modeled SOA concentrations in urban environments.
Datasets are accessible by contacting the corresponding author, Yu Huang, (huangyu@ieecas.cn).
Tables S1 and S3 list the energy barriers of all the elementary reactions involved in the addition of OH radicals to styrene and 1st-ROOH (S4) predicted at different levels. Tables S2, S4 and S6 list the relative electronic energy, free energy and Boltzmann population of different conformers involved in S2-1-x, S8-x and S28-x. Tables S5 and S7 list the MC-TST rate coefficients for the intramolecular H-shift reactions of S8-x and S28-x. Table S8 summaries the saturated vapour pressure and saturated concentrations of styrene and its multiple generation •OH oxidation closed-shell products. Figures S1–S3 display the PESs for the unimolecular reactions of S2-2-x, S2-3-x and S2-4-x. Figure S4 shows the global minimum structures of 1st-ROOH(S4) and 1st-RONO2(S5). Figure S5 depicts the geometric parameters of toluene and 1st-ROOH (S4) and the NPA atomic charges of all the carbon atoms. Figures S6 and S11 show the PESs for the addition reactions PS4-add1+O2 and PS5-add1+O2. Figures S7 and S12 present the lowest energy conformers of third generation peroxy radicals PS4-add3-a-2 and PS5-add3-a-2. Figures S8–S10 depict the PESs for the intramolecular hydrogen transfer reactions of S8-x, S12-x and S16-x. Figures S13–S15 depict the PESs for the intramolecular hydrogen transfer reactions of S28-x, S32-x and S36-x. Figures S16–18 show the PESs for the OH-initiated oxidation of 2nd-ROOH (S6) and unimolecular decomposition of PS6-add3. Figures S19–S21 show the PESs for the OH-initiated oxidation of 2nd-RONO2 (S26) and unimolecular decomposition of PS26-add3. Figures S22 and S23 show the overall reaction mechanism of the multi-generation •OH oxidation of styrene in the low- and high-NOx conditions. The supplement related to this article is available online at https://doi.org/10.5194/acp-26-4823-2026-supplement.
LC and YH conceptualized the study. LC conducted quantum chemical calculation. YX and ZJ analyzed the data. LC conducted the volatility estimation. All authors discussed the results and commented on the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This study was supported by the National Natural Science Foundation of China (grant no. 42175134) and the Youth Innovation Promotion Association of the Chinese Academy of Sciences (grant no. 2022415).
This paper was edited by Arthur Chan and reviewed by two anonymous referees.
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G.: Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries, J. Chem. Theory Comput., 6, 2872–2887, https://doi.org/10.1021/ct100326h, 2010.
Arathala, P. and Musah, R. A.: Atmospheric chemistry of chloroprene initiated by OH radicals: combined Ab initio/DFT calculations and kinetics analysis, J. Phys. Chem. A, 128, 8983–8995, https://doi.org/10.1021/acs.jpca.4c05428, 2024.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: a key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Boyd, A. A., Flaud, P. M., Daugey, N., and Lesclaux, R.: Rate constants for RO2 + HO2 reactions measured under a large excess of HO2, J. Phys. Chem. A, 107, 818–821, https://doi.org/10.1021/jp026581r, 2003.
Cabrera-Perez, D., Taraborrelli, D., Sander, R., and Pozzer, A.: Global atmospheric budget of simple monocyclic aromatic compounds, Atmos. Chem. Phys., 16, 6931–6947, https://doi.org/10.5194/acp-16-6931-2016, 2016.
Canneaux, S., Bohr, F., and Henon, E.: KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 35, 82–93, https://doi.org/10.1002/jcc.23470, 2013.
Chen, L., Huang, Y., Xue, Y., Jia, Z., and Wang, W.: Atmospheric oxidation of 1-butene initiated by OH radical: Implications for ozone and nitrous acid formations, Atmos. Environ., 244, 118010–118021, https://doi.org/10.1016/j.atmosenv.2020.118010, 2021.
Cho, J., Roueintan, M., and Li, Z.: Kinetic and dynamic investigations of OH reaction with styrene, J. Phys. Chem. A, 118, 9460–9470, https://doi.org/10.1021/jp501380j, 2014.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Eckart, C.: The penetration of a potential barrier by electrons, Phys. Rev., 35, 1303–1309, https://doi.org/10.1103/PhysRev.35.1303, 1930.
Environmental Protection Agency (EPA): Clean Air Act: Title I-Air Pollution Prevention and Control. U.S., ISBN: 978-0314835024, 1990.
Fernández-Ramos, A., Ellingson, B. A., Meana-Pañeda, R., Marques, J. M. C., and Truhlar, D. G.: Symmetry numbers and chemical reaction rates, Theor. Chem. Acc., 118, 813–826, https://doi.org/10.1007/s00214-007-0328-0, 2007.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: molecular composition, Environ. Sci. Technol., 31, 1345–1358, https://doi.org/10.1021/es9605376, 1997.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, J. A., Peralta, J. J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, https://www.gaussian.com (last access: 20 June 2025), 2016.
Fu, Z., Xie, H. B., Elm, J., Guo, X., Fu, Z., and Chen, J.: Formation of low-volatile products and unexpected high formaldehyde yield from the atmospheric oxidation of methylsiloxanes, Environ. Sci. Technol., 54, 7136–7145, https://doi.org/10.1021/acs.est.0c01090, 2020.
Fu, Z., Ma, F., Liu, Y., Yan, C., Huang, D., Chen, J., Elm, J., Li, Y., Ding, A., Pichelstorfer, L., Xie, H. B., Nie, W., Francisco, J. S., and Zhou, P.: An overlooked oxidation mechanism of toluene: computational predictions and experimental validations, Chem. Sci., 14, 13050–13059, https://doi.org/10.1039/D3SC03638C, 2023.
Fu, Z., Guo, S., Xie, H. B., Zhou, P., Boy, M., Yao, M., and Hu, M.: A near-explicit reaction mechanism of chlorine-initiated limonene: implications for health risks associated with the concurrent use of cleaning agents and disinfectants, Environ. Sci. Technol., 58, 19762–19773, https://doi.org/10.1021/acs.est.4c04388, 2024.
Fukui, K.: The path of chemical reactions – the IRC approach, Accounts Chem. Res., 14, 363–368, https://doi.org/10.1021/ar00072a001, 1981.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Gilbert, R. G. and Smith, S. C.: Theory of unimolecular and recombination reactions, Blackwell Scientific, Carlton, Australia, ISBN: 978-0632027491, 1990.
Glowacki, D. R., Liang, C. H., Morley, C., Pilling, M. J., and Robertson, S. H.: MESMER: an open-source master equation solver for multi-energy well reactions, J. Phys. Chem. A, 116, 9545–9560, https://doi.org/10.1021/jp3051033, 2012.
Holbrook, K. A., Pilling, M. J., Robertson, S. H., and Robinson, P. J.: Unimolecular reactions, 2nd edn., Wiley, New York, ISBN: 978-0471922681, 1996.
Huang, Y., Su, T., Wang, L., Wang, N., Xue, Y., Dai, W., Lee, S. C., Cao, J., and Ho, S. S. H.: Evaluation and characterization of volatile air toxics indoors in a heavy polluted city of northwestern China in wintertime, Sci. Total Environ., 662, 470–480, https://doi.org/10.1016/j.scitotenv.2019.01.250, 2019.
Iuga, C., Galano, A., and Vivier-Bunge, A.: Theoretical investigation of the OH-initiated oxidation of benzaldehyde in the troposphere, Chem. Phys. Chem., 9, 1453–1459, https://doi.org/10.1002/cphc.200800144, 2008.
Iyer, S., Kumar, A., Savolainen, A., Barua, S., Daub, C., Pichelstorfer, L., Roldin, P., Garmash, O., Seal, P., Kurtén, T., and Rissanen, M.: Molecular rearrangement of bicyclic peroxy radicals is a key route to aerosol from aromatics, Nat. Commun., 14, 4984–4991, https://doi.org/10.1038/s41467-023-40675-2, 2023.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Koppmann, R.: Volatile organic compounds in the atmosphere, John Wiley & Sons, ISBN: 978-1405131155, 2008.
Li, M., Zhang, Q., Zheng, B., Tong, D., Lei, Y., Liu, F., Hong, C., Kang, S., Yan, L., Zhang, Y., Bo, Y., Su, H., Cheng, Y., and He, K.: Persistent growth of anthropogenic non-methane volatile organic compound (NMVOC) emissions in China during 1990–2017: drivers, speciation and ozone formation potential, Atmos. Chem. Phys., 19, 8897–8913, https://doi.org/10.5194/acp-19-8897-2019, 2019.
Lu, T.: Molclus program, Version 1.9.3., http://www.keinsci.com/research/molclus.html (last access: 21 May 2024), 2019.
Ma, F., Guo, X., Xia, D., Xie, H. B., Wang, Y., Elm, J., Chen, J., and Niu, J.: Atmospheric chemistry of allylic radicals from isoprene: a successive cyclization-driven autoxidation mechanism, Environ. Sci. Technol., 55, 4399–4409, https://doi.org/10.1021/acs.est.0c07925, 2021.
Møller, K. H., Otkjær, R. V., Hyttinen, N., Kurtén, T., and Kjaergaard, H. G.: Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions, J. Phys. Chem. A, 120, 10072–10087, https://doi.org/10.1021/acs.jpca.6b09370, 2016.
Møller, K. H., Berndt, T., and Kjaergaard, H. G.: Atmospheric autoxidation of amines, Environ. Sci. Technol., 54, 11087–11099, https://doi.org/10.1021/acs.est.0c03937, 2020.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Neese, F.: Software update: the ORCA program system – version 6.0, Wires Comput. Mol. Sci., 15, e70019, https://doi.org/10.1002/wcms.70019, 2025.
Nie, W., Yan, C., Huang, D. D., Wang, Z., Liu, Y., Qiao, X., Guo, Y., Tian, L., Zheng, P., Xu, Z., Li, Y., Xu, Z., Qi, X., Sun, P., Wang, J., Zheng, F., Li, X., Yin, R., Dallenbach, K. R., Bianchi, F., Petäjä, T., Zhang, Y., Wang, M., Schervish, M., Wang, S., Qiao, L., Wang, Q., Zhou, M., Wang, H., Yu, C., Yao, D., Guo, H., Ye, P., Lee, S., Li, Y. J., Liu, Y., Chi, X., Kerminen, V. M., Ehn, M., Donahue, N. M., Wang, T., Huang, C., Kulmala, M., Worsnop, D, Jiang, J., and Ding, A.: Secondary organic aerosol formed by condensing anthropogenic vapours over China's megacities, Nat. Geosci., 15, 255–261, https://doi.org/10.1038/s41561-022-00922-5, 2022.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/C2CS35166H, 2012.
Pankow, J. F. and Asher, W. E.: SIMPO L.1: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Pasik, D., Frandsen, B. N., Meder, M., Iyer, S., Kurtén, T., and Myllys, N.: Gas-phase oxidation of atmospherically relevant unsaturated hydrocarbons by acyl peroxy radicals, J. Am. Chem. Soc., 146, 13427–13437, https://doi.org/10.1021/jacs.4c02523, 2024.
Sebbar, N., Bozzelli, J. W., and Bockhorn, H.: Thermochemistry and reaction paths in the oxidation reaction of benzoyl radical: C6H5C•(=O), J. Phys. Chem. A, 115, 11897–11914, https://doi.org/10.1021/jp2078067, 2011.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 8, eabp8702, https://doi.org/10.1126/sciadv.abp8702, 2022.
Sun, J., Wu, F., Hu, B., Tang, G., Zhang, J., and Wang, Y.: VOC characteristics, emissions and contributions to SOA formation during hazy episodes, Atmos. Environ., 141, 560–570, https://doi.org/10.1016/j.atmosenv.2016.06.060, 2016.
Tajuelo, M., Rodríguez, D., Baeza-Romero, M. T., Díaz-de-Mera, Y., Aranda, A., and Rodríguez, A.: Secondary organic aerosol formation from styrene photolysis and photooxidation with hydroxyl radicals, Chemosphere, 231, 276–286, https://doi.org/10.1016/j.chemosphere.2019.05.136, 2019a.
Tajuelo, M., Rodríguez, A., Baeza-Romero, M. T., Aranda, A., Díaz-de-Mera, Y., and Rodríguez, D.: Secondary organic aerosol formation from α-methylstyrene atmospheric degradation: Role of NOx level, relative humidity and inorganic seed aerosol, Atmos. Res., 230, 104631–104640, https://doi.org/10.1016/j.atmosres.2019.104631, 2019b.
Tajuelo, M., Bravo, I., Rodríguez, A., Aranda, A., Díaz-de-Mera, Y., and Rodríguez, D.: Atmospheric sink of styrene, α-methylstyrene, trans-β-methylstyrene and indene: Rate constants and mechanisms of Cl atom-initiated degradation, Atmos. Environ., 200, 78–89, https://doi.org/10.1016/j.atmosenv.2018.11.059, 2019c.
Vereecken, L., Glowacki, D. R., and Pilling, M. J.: Theoretical chemical kinetics in tropospheric chemistry: methodologies and applications, Chem. Rev., 115, 4063–4114, https://doi.org/10.1021/cr500488p, 2015.
Wang, H., Ji, Y., Gao, Y., Li, G., and An, T.: Theoretical model on the formation possibility of secondary organic aerosol from OH initialed oxidation reaction of styrene in the presence of , Atmos. Environ., 101, 1–9, https://doi.org/10.1016/j.atmosenv.2014.10.042, 2015.
Wang, L., Wu, R., and Xu, C.: Atmospheric oxidation mechanism of benzene. Fates of alkoxy radical intermediates and revised mechanism, J. Phys. Chem. A, 117, 14163–14168, https://doi.org/10.1021/jp4101762, 2013.
Wang, M., Chen, D., Xiao, M., Ye, Q., Stolzenburg, D., Hofbauer, V., Ye, P., Vogel, A. L., Mauldin, R. L., Amorim, A., Baccarini, A., Baumgartner, B., Brilke, S., Dada, L., Dias, A., Duplissy, J., Finkenzeller, H., Garmash, O., He, X. C., Hoyle, C. R., Kim, C., Kvashnin, A., Lehtipalo, K., Fischer, L., Molteni, U., Petäjä, T., Pospisilova, V., Quéléver, L. L. J., Rissanen, M., Simon, M., Tauber, C., Tomé, A., Wagner, A. C., Weitz, L., Volkamer, R., Winkler, P. M., Kirkby, J., Worsnop, D. R., Kulmala, M., Baltensperger, U., Dommen, J., El-Haddad, I., and Donahue, N. M.: Photo-oxidation of aromatic hydrocarbons produces low-volatility organic compounds, Environ. Sci. Technol., 54, 7911–7921, https://doi.org/10.1021/acs.est.0c02100, 2020a.
Wang, S. and Li, H.: NO3-initiated gas-phase formation of nitrated phenolic compounds in polluted atmosphere, Environ. Sci. Technol., 55, 2899–2907, https://doi.org/10.1021/acs.est.0c08041, 2021.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzene, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, S., Newland, M. J., Deng, W., Rickard, A. R., Hamilton, J. F., Muñoz, A., Ródenas, M., Vázquez, M. M., Wang, L., and Wang, X.: Aromatic photo-oxidation, a new source of atmospheric acidity, Environ. Sci. Technol., 54, 7798–7806, https://doi.org/10.1021/acs.est.0c00526, 2020b.
Wu, R., Pan, S., Li, Y., and Wang, L.: Atmospheric oxidation mechanism of toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/jp500077f, 2014.
Wu, X., Huang, C., Niu, S., and Zhang, F.: New theoretical insights into the reaction kinetics of toluene and hydroxyl radicals, Phys. Chem. Chem. Phys., 22, 22279–22288, https://doi.org/10.1039/D0CP02984J, 2020.
Wu, X., Hou, Q., Huang, J., Chai, J., and Zhang, F.: Exploring the OH-initiated reactions of styrene in the atmosphere and the role of van der Waals complex, Chemosphere, 282, 131004–131012, https://doi.org/10.1016/j.chemosphere.2021.131004, 2021.
Xu, C. and Wang, L.: Atmospheric oxidation mechanism of phenol initiated by OH radical, J. Phys. Chem. A, 117, 2358–2364, https://doi.org/10.1021/jp308856b, 2013.
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Yan, Y., Cabrera-Perez, D., Lin, J., Pozzer, A., Hu, L., Millet, D. B., Porter, W. C., and Lelieveld, J.: Global tropospheric effects of aromatic chemistry with the SAPRC-11 mechanism implemented in GEOS-Chem version 9-02, Geosci. Model Dev., 12, 111–130, https://doi.org/10.5194/gmd-12-111-2019, 2019.
Yang, F., Deng, F., Pan, Y., Zhang, Y., Tang, C., and Huang, Z.: Kinetics of hydrogen abstraction and addition reactions of 3-hexene by OH radicals, J. Phys. Chem. A, 121, 1877–1889, https://doi.org/10.1021/acs.jpca.6b11499, 2017.
Yu, S., Jia, L., Xu, Y., and Pan, Y.: Formation of extremely low-volatility organic compounds from styrene ozonolysis: Implication for nucleation, Chemosphere, 305, 135459–135467, https://doi.org/10.1016/j.chemosphere.2022.135459, 2022a.
Yu, S., Jia, L., Xu, Y., and Pan, Y.: Molecular composition of secondary organic aerosol from styrene under different NOx and humidity conditions, Atmos. Res., 266, 105950–10604, https://doi.org/10.1016/j.atmosres.2021.105950, 2022b.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhang, H., Wang, J., Dong, B., Xu, F., Liu, H., Zhang, Q., Zong, W., and Shi, X.: New mechanism for the participation of aromatic oxidation products in atmospheric nucleation, Sci. Total Environ., 917, 170487–170494, https://doi.org/10.1016/j.scitotenv.2024.170487, 2024.
Zhang, R. M., Truhlar, D. G., and Xu, X.: Kinetics of the toluene reaction with OH radical, Research, 2019, 5373785, https://doi.org/10.34133/2019/5373785, 2019.
Zhao, H., Zhang, Y., Zhao, Q., Li, Y., and Huang, Z.: A theoretical study of H-abstractions of benzaldehyde by H, O3(P), 3O2, OH, HO2, and CH3 radicals: Ab initio rate coefficients and their uncertainty quantification, J. Phys. Chem. A, 126, 7523–7533, https://doi.org/10.1021/acs.jpca.2c02384, 2022.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.