the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Dec 2024
| 02 Dec 2024
Opinion: Challenges and needs of tropospheric chemical mechanism development
Bernard Aumont
William P. L. Carter
Max McGillen
Abdelwahid Mellouki
John Orlando
Bénédicte Picquet-Varrault
Paul Seakins
William R. Stockwell
Luc Vereecken
Timothy J. Wallington
Chemical mechanisms form the core of atmospheric models to describe degradation pathways of pollutants and ultimately inform air quality and climate policymakers and other stakeholders. The accuracy of chemical mechanisms relies on the quality of their input data, which originate from experimental (laboratory, field, chamber) and theoretical (quantum chemistry, theoretical kinetics, machine learning) studies. The development of robust mechanisms requires rigorous and transparent procedures for data collection, mechanism construction and evaluation and the creation of reduced or operationally defined mechanisms. Developments in analytical techniques have led to a large number of identified chemical species in the atmospheric multiphase system that have proved invaluable for our understanding of atmospheric chemistry. At the same time, advances in software and machine learning tools have enabled automated mechanism generation. We discuss strategies for mechanism development, applying empirical or mechanistic approaches. We show the general workflows, how either approach can lead to robust mechanisms and that the two approaches complement each other, resulting in reliable predictions. Current challenges are discussed related to global change, including shifts in emission scenarios that result in new chemical regimes (e.g., low-NO scenarios, wildfires, mega- and gigacities) and that require the development of new or expanded gas- and aqueous-phase mechanisms. In addition, new mechanisms should be developed to also target oxidation capacity and aerosol chemistry impacting climate, human and ecosystem health.




The troposphere is a highly complex dynamic chemical system composed of multiple phases (gaseous, aqueous, organic matrices) containing millions of chemical compounds that constantly mix and interact. The atmospheric degradation of these compounds is complex and nonlinear and results in the formation of a wide range of potentially harmful secondary pollutants including ozone and secondary organic aerosol (SOA). Understanding this chemistry requires numerical models with chemical mechanisms that include kinetic and mechanistic information describing the formation, cycling and degradation of chemical compounds. Thus, chemical mechanisms merge information from a variety of fundamental research activities including laboratory, theoretical and field studies. The reactions that make up these mechanisms are incorporated in models in the form of ordinary differential equations, allowing for the prediction of temporal changes in species concentrations due to chemical processing, which can be computationally challenging. Chemical mechanisms need to be applicable over a wide range of spatial and temporal scales (local, regional and global) and conditions, including indoor and outdoor environments. Chemistry models can either be applied separately (e.g., box models) or form modules in larger model frameworks that also consider additional processes, possibly in multiple dimensions, such as transport, dynamics, emissions and deposition, and convection.
The development of gas-phase chemical mechanisms describing the oxidation of volatile organic compounds (VOCs) can be traced back to the 1950s, arising from interest in photochemical reactions resulting in high ozone levels in Los Angeles (Haagen-Smit, 1952). In the 1970s, acid rain was recognized as a major threat to ecosystems (Likens and Bormann, 1974), where the acidification was ascribed to the production of sulfate, nitrate and organic acids (Calvert and Stockwell, 1983). Research showed that gas-phase oxidation reactions alone were not sufficient to explain the measured acidification levels in rain and fog water, suggesting that aqueous-phase conversion processes could not be ignored (Hoffmann and Jacob, 1984). These conclusions initiated the expansion of atmospheric mechanism development to include chemical processes in cloud droplets. The resulting coupled gas–aqueous (multiphase) mechanisms in regional atmospheric models led to a comprehensive understanding of the relevant factors involved in precipitation acidity and ultimately to policy measures for its mitigation (Grennfelt et al., 2020). The role of multiphase chemistry for chemical budgets in the atmosphere has since been further explored and expanded towards the inclusion of oxidants (e.g., reactive oxygen species, epoxides, 1O2) and organic compounds (Abbatt and Ravishankara, 2023).
Recognition that a major fraction of atmospheric aerosol mass is secondary, i.e., formed by in situ chemical processes, combined with evidence of the effects of aerosols on climate, ecosystems and human health has stimulated research during the last decades (Pye et al., 2023). As a consequence, atmospheric chemical models have become increasingly more complex, in particular in terms of the consideration of multiple phases. At the same time, huge advances in theoretical, analytical and numerical techniques provided a wealth of detailed information on the variety, abundance and properties of organic species. The simultaneous development in computational power and capabilities have allowed the implementation of increasingly complex chemical mechanisms into models, up to a point where it may become unfeasible to include all available information. Instead, the computational requirements in chemical transport models and global climate models make the use of parameterizations necessary that take into account the phenomenological descriptions of the underlying chemical processes. Therefore, targeted and sophisticated strategies are needed to collect, filter, evaluate and use the relevant information to answer urgent and emerging research questions on the chemical composition of the atmosphere and its evolution in a changing climate and society. Such strategies will be only successful and efficient if reliable data are available and accessible and if their use and implementation in chemical mechanisms is transparent.
In the current opinion article, we discuss the different strategies used for chemical mechanism development and the individual steps and activities they comprise. We do not aim to provide a detailed review of the investigation or description of individual chemical systems; we refer the reader to the numerous (review) articles on these topics cited throughout the text. Instead, we frame our discussion based on the complementary differences and applicability of the main mechanism development strategies (Sect. 2). In Sect. 3, we systematically outline the various steps needed to develop chemical mechanisms while ensuring their reliability and robustness, together with an overview of the progress and status for each individual step. The spectrum of research activities, in terms of the theoretical and experimental studies that are required to inform mechanism developers of reliable fundamental data, is described in Sect. 4, briefly summarizing recent advancements, the current status and perspectives. We discuss insufficiently characterized chemical systems and regimes, their related challenges and the emerging needs for future chemical mechanism development in Sect. 5. We conclude with an outlook on future needs and directions of chemical mechanism developments (Sect. 6).
The choice of the approach for mechanism development depends on (1) the intended purpose of the mechanism and (2) how well we understand the chemical system being represented. Figure 1 shows the approach if the mechanism is to be used primarily to compile what we know about the system from a scientific perspective. This has primarily been used in the initial development of explicit “master mechanisms” such as the Master Chemical Mechanism (MCM, https://mcm.york.ac.uk/MCM/, last access: 25 November 2024), which has proven valuable in summarizing current knowledge, and for the analysis of detailed chemical systems and mechanism development research. Master mechanisms have been initially used to predict specific ozone-relevant metrics such as maximum incremental reactivity (MIR) or photochemical ozone creation potentials (POCP), where they are particularly useful for simulating multiday photochemical ozone formation (Derwent and Jenkin, 1991; Carter, 1994). The choice of such metrics depends on the specific application of the chemical mechanism and on the research questions to be addressed.
Explicit mechanisms can be derived manually, as is the case for the National Center for Atmospheric Research (NCAR) Master Mechanism (Madronich and Calvert, 1989), Master Chemical Mechanism (MCM) (Jenkin et al., 1997, 2003, 2015), Chemical Aqueous Phase Radical Mechanism (CAPRAM) (e.g., Ervens et al., 2003; Hoffmann et al., 2020) and Cloud Explicit Physico-chemical Scheme (CLEPS) (Mouchel-Vallon et al., 2017) or automatically, as is the case for the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) (e.g., Lannuque et al., 2018) and SAPRC-Mechgen (Carter, 2024a, b). Both methods are based on existing fundamental chemical data and theories and rely on the expert judgments and estimates of mechanism developers. The difference is that automated methods require developing pre-defined estimation protocols for all reactions that may occur, while manual methods allow for separate examination and evaluation of the specifics of each chemical system. Regardless of how they are derived, these explicit mechanisms tend to be very large owing to the numbers of possible reactions, in particular for organic compounds. Manually derived mechanisms necessarily have to either ignore or approximate relatively minor processes, which may not be negligible when taken as an aggregate or may be important in the formation of minor but key species. Even computer-generated mechanisms require use of a “minimum yield” parameter to eliminate processes considered to be negligible and also must use approximate methods, e.g., to represent cross-reactions of the many peroxy radicals predicted to be formed. Therefore, most scientifically based mechanisms are necessarily “semi-explicit” and reduced, at least to some extent.
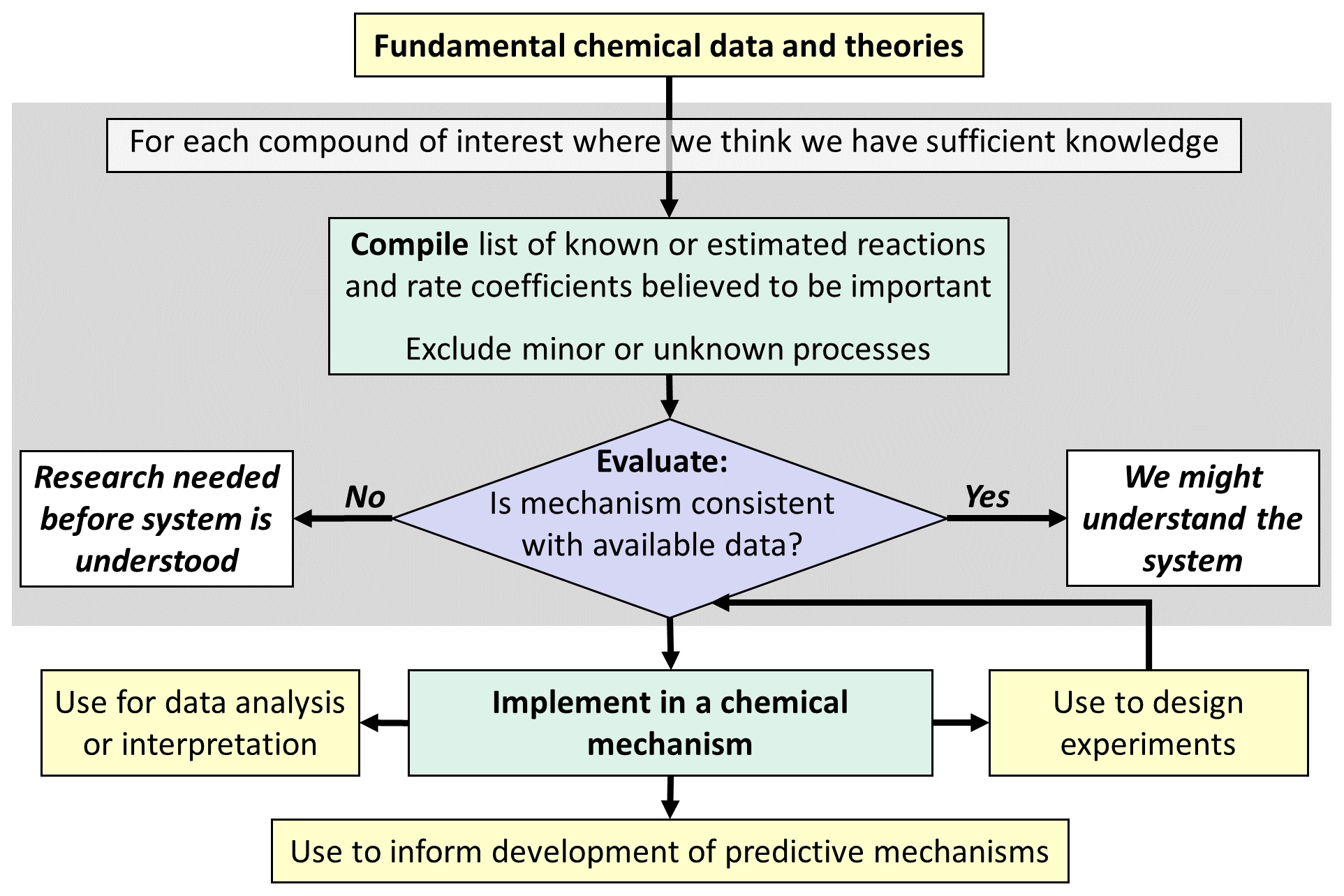
Figure 1Outline of approach for development of (semi-)explicit mechanisms to be used primarily to compile scientific knowledge or interpret scientific data.
There is a valid concern that neglecting or over-simplifying chemical details in a model may introduce errors or biases into its predictions. However, Stockwell et al. (2020) expressed the opposite concern that the creation of very large, explicit mechanisms may involve an extensive over-extrapolation of the underlying structure–activity relationships (SARs). This may occur when SARs are used to estimate unknown reaction parameters outside of an acceptable range of the chemical databases used to derive them. These mechanistic uncertainties may ultimately propagate into the predictions based on the atmospheric models. In fact, Puy et al. (2024) showed that increasing model detail greatly beyond current knowledge does not improve model estimates and in the worst case makes a model unverifiable. Therefore, a reasonable limit to the level of detail in a chemical mechanism due to deficiencies in the available empirical and theoretical chemical databases should be acknowledged, to appropriately inform public policymakers and other stakeholders. However, highly detailed mechanisms may provide strong guidance for prioritizing experiments and quantum chemistry calculations for the further development of databases.
Explicit mechanisms, derived as indicated in Fig. 1, generally do not attempt to represent chemical systems where we lack sufficient understanding to justify estimating explicit reactions. Instead, chemical systems with large uncertainties are judged as areas where more research and evaluation are needed before they can be represented in scientifically based chemical mechanisms. Although valuable for scientific research, this is not sufficient for predictive modeling, which is necessary for assessing how air quality is affected by emissions or what pollutants might be formed and how they evolve.
The need for predictive modeling capability has been a major justification for continued funding of mechanism development research. An outline of an approach for developing predictive mechanisms is shown in Fig. 2. Ideally, we understand the system sufficiently to be able to use the approach shown on the right side of Fig. 2 for all portions of the mechanism, as advocated by Kaduwela et al. (2015). This would result in the most scientifically justifiable mechanisms that are least likely to have compensating errors, even if some uncertain rate coefficients have to be modified to improve fits to available data. However, we are not able to derive predictive explicit mechanisms for many types of compounds, so representing them requires use of (often highly) simplified or parameterized mechanisms with uncertain parameters adjusted to fit atmospheric simulation chamber data. In addition, the chemistry and physics of SOA formation and processing in both the gas and aqueous phases are sufficiently uncertain and complex that the use of largely parameterized models is necessary for any hope of predictive capability for specific SOA properties. In these cases, direct comparisons with experimental data and making adjustments to improve fits are a necessary part of mechanism development, but such adjustments are subject to uncertainties due to possible compensating errors or due to the conditions of the experiment not representing the full range of conditions in the environment.
An additional consideration is that many more types of compounds are emitted into or formed within the atmosphere than can be represented explicitly in practical modeling applications, which makes some degree of aggregation or lumping necessary. This requires (1) developing mechanisms for lumped model species to represent a range of chemically similar compounds and (2) developing rules for assigning these model species to specific compounds. The mechanisms for lumped model species could be based on those derived for selected representative compounds, or they can be constructed manually based on general considerations. The former approach has the advantage that mechanisms for individual compounds can be evaluated using experimental data since experiments cannot be carried out with lumped model species. In any case, the optimum set of lumped model species and the rules for assigning compounds to them will depend on the model application. Lumping approaches for most current mechanisms used in predictive models are generally based on considerations of O3 predictions, which are not optimal for predicting SOA and other secondary pollutants.
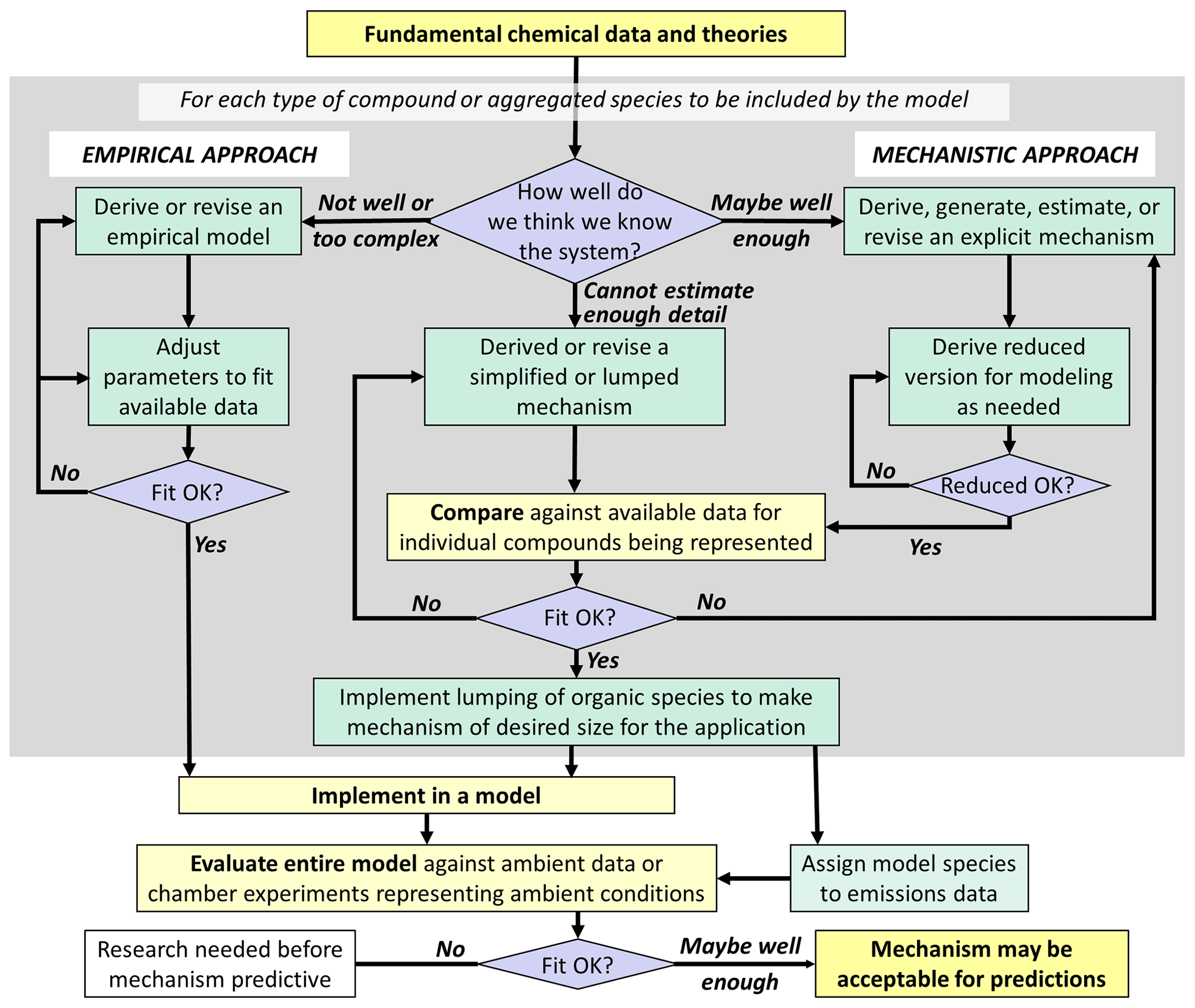
Figure 2Outline of approaches for development of predictive mechanisms to be used for analyses of environmental impacts.
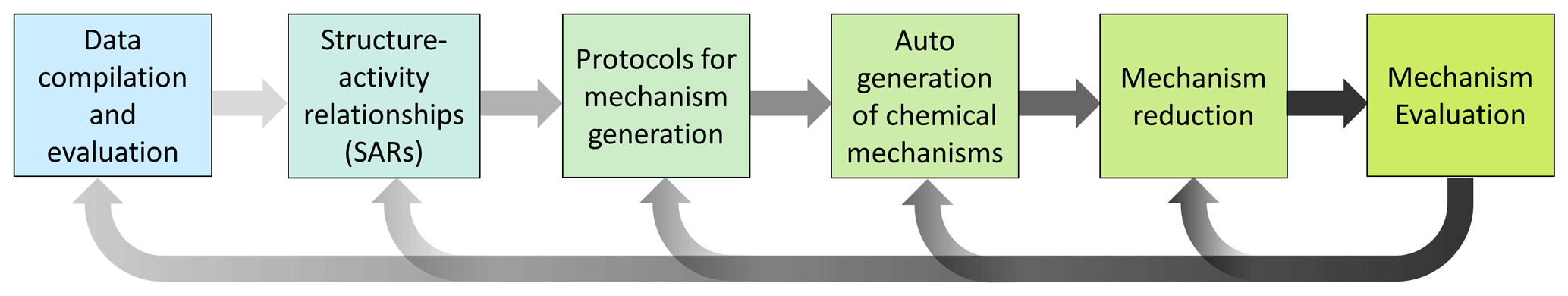
Both strategies generally imply a sequence of similar steps to build high-quality chemical mechanisms that can be applied to a range of chemical systems and research questions (Sect. 3). The underlying research questions determine the focus and its potential importance. For example, a high-molecular-weight VOC present in low concentration may have a limited effect on controlling OH reactivity and secondary ozone formation, but it might be key for new particle formation.
These strategies, as highlighted in Figs. 1 and 2, are not exclusive but rather complementary. They have some fundamental principle elements in common, namely (i) the need for fundamental chemical databases; (ii) mechanism generation, which may include the development and use of SARs and/or parameterizations; (iii) reduction in the mechanism to a manageable size and/or a specific focus; and (iv) evaluation to ensure robustness and accuracy (Fig. 3). Assessments of predicted differences from models using mechanisms based on the two approaches in Fig. 1 may eventually lead to the identification of uncertainty and knowledge gaps that warrant further research. However, this requires that each individual step (or even intermediate steps) be carefully performed, taking advantage of and combining insights from various research activities (Sect. 4). Thus, a delicate balance between completeness and practicability should be the target so that models maintain their utility for users and stakeholders, including policymakers, while enabling easy and transparent incorporation of the most up-to-date information on the underlying chemical processes.
Developments during the last decade in terms of targeted cheminformatics methods based on data mining, machine learning, data assimilation and/or similar mathematical approaches may provide new opportunities for developing reduced mechanisms or optimizing lumping approaches. These will ultimately and efficiently lead to reliable modeling tools integrating the data from a range of concerted research activities.
In general, mechanism development should follow five iterative steps, as outlined in Fig. 3 and the subsections below, in order to build robust, reliable state-of-the-art chemical schemes.
3.1 Data compilations and evaluations
Over the past ∼ 50 years, a rich suite of experimental and theoretical techniques has been developed and employed to provide kinetic and mechanistic data for key gas-phase, heterogeneous and aqueous-phase processes. This has resulted in extensive atmospheric chemical kinetic databases that contain literature data for several thousand processes and continue to grow, with approximately 100 new papers each year. The gas- and solution-phase NIST Chemical Kinetics Databases (Manion et al., 2015), MPI-Mainz UV/VIS Spectral Atlas (Keller-Rudek et al., 2013) and Henry’s Law Constants database (Sander, 2023) are regularly updated online compendia of reaction kinetics that are extremely valuable. However, they do not provide expert evaluation of the measured data and thus do not provide recommendations.
In contrast, expert panels have been convened to critically evaluate and provide recommended data for use in atmospheric science and policy models. Examples include the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation (Mellouki et al., 2021), the NASA Panel for Data Evaluation (Burkholder et al., 2020), and groups that reviewed and evaluated literature data on (i) atmospherically relevant gas-phase reactions of organic compounds available as of 2015 (Calvert et al., 2015); (ii) aqueous-phase kinetic reactions (Ervens et al., 2003; Herrmann, 2003; Herrmann et al., 2015; Herrmann et al., 2010); and (iii) atmospherically relevant gas-phase reactions of organic compounds with OH and NO3 radicals, Cl atoms, and ozone (McGillen et al., 2020). Compiling and evaluating data are complementary efforts. Evaluated fundamental chemical data are the foundation of all air quality and climate models and need to be updated on an ongoing basis to ensure that models are based on the latest science. Data evaluation is critically important for continued progress in the field of atmospheric chemistry and for the development of effective policy and regulations to address air pollution and climate change. As such, data compilation and evaluation need to be funded accordingly.
Aside from the non-trivial task of maintaining and updating these databases, the future challenges for data compilation and evaluation efforts are to harness the growing power of information technology to gather and assess input data and to provide recommendations in machine-readable formats to facilitate their use in atmospheric models. As an example of this effort, the IUPAC Task Group is developing machine-readable data sheets to enable automatic updates of chemical mechanisms with the latest IUPAC recommended data. Such efforts in data evaluation and recommendations are also essential to inform machine learning applications (Sect. 4.5) to make their use in future mechanism creation as reliable and valuable as possible.
3.2 Structure–activity relationships (SARs)
The development of structure–activity relationships (SARs) to make estimates needed for developing chemical mechanisms for atmospheric models relies on accurate data for the rate coefficients and mechanisms of relevant atmospheric processes. If the underlying chemical mechanisms are understood, it is possible to create SARs to predict them, as it is neither necessary nor feasible to measure kinetic rate coefficients and/or branching ratios for all individual reactions.
SAR approaches range from the purely empirical methods that rely largely on chemical intuition to those that possess a more theoretical basis. The role of SARs in larger kinetic models, together with emerging needs and current limitations of gas-phase SARs, has been discussed extensively, e.g., Vereecken et al. (2018). The expected increased importance of auto-generation or software-assisted building of more explicit mechanisms will put greater emphasis on reliable and machine-readable SARs to provide kinetic information, including reaction rate coefficients, product distribution and branching ratios as a function of the reaction conditions. This may lead to future challenges in providing SARs for all reaction classes, with a wider scope of applicability that covers the full range of molecular structures in large explicit models. This will also test our ability to validate these SARs to avoid incorrect extrapolation of reactivity trends and to quantify the uncertainty in the SAR predictions to help in model validation.
In the early 1980s, Atkinson et al. (1982) developed an empirical SAR for estimating rate coefficients that relies on base rate coefficients modified for substituents in the immediate environment of the reactive site. This approach has been widely adopted, together with several variants, owing to its many advantages including its ease of use, its site-specificity and the fidelity with which it often reproduces the rate coefficients. This approach is, however, not without its challenges. Multiple functionalities can interact to influence the reactivity non-additively (steric effects, H-bonding, site-specific reactivity, etc.), and training data to derive higher-order parameters describing these cooperative effects are typically not abundantly available. As a result, large uncertainties still remain in all SARs. Even when a VOC is expected to be oxidized by “conventional chemistry”, automated oxidation mechanisms can have significant cumulative uncertainties in the product spectrum, and SARs often do not work well for even the initial OH abstraction or addition reactions (Kwok and Atkinson, 1995; Jenkin et al., 2018). Novel concepts or strategies may help to tackle this problem to provide workable, although not necessarily simple, SAR models. One such strategy is to incorporate procedurally generated a priori descriptions of substitutions, which may lead to a more robust estimation method, as discussed in McGillen et al. (2024).
Another area of research is the use of more and/or different molecular descriptors, where pure molecular descriptors (e.g., number of atoms, bonding and positioning of substituents, hydration effects) are supplemented with computational descriptors (e.g., fingerprints, Wiener or Randić indices, topological indices, Fukui indices, wave function hardness/softness, or electro-topological state). The latter are derived from the molecular wave function, typically calculated using an inexpensive methodology (semi-empirical or molecular modeling tools). Another approach that is receiving increased attention is abandoning the traditional SAR setup as a parameterized set of simple equations in favor of the more versatile mathematical models used in machine learning, such as neural networks. Both these approaches make the resulting SAR harder to interpret by a human and may even appear to be black-box approaches. Therefore, the improved predictive power of such methods needs to be balanced against the potential for inadvertently generating incorrect predictions by extrapolating reactivity trends beyond their scope of applicability.
Contrary to a decade ago, when SARs were primarily developed based on experimental data, current SAR development aims to combine experimental and theoretical data. A particular strength of theoretical studies is that they are well-suited to probe systematic molecular variations; even if absolute accuracy is not achieved, reactivity trends may still be apparent. The use of theoretical studies has opened up new possibilities for the development of SARs for radicals and other unstable intermediates, such as alkoxy and peroxy radicals or Criegee intermediates. In addition, the use of high-level quantum chemical calculations has recently been extended to derive photolysis data (i.e., absorption cross sections and quantum yields) of atmospherically photolabile species, which have previously been difficult to measure (Prlj et al., 2022; Janoš et al., 2023).
The abundance, variety and role of multifunctional compounds in both the gas and aqueous phases pose another problem for SARs since there are insufficient training data available to cover the combinatorially high number of possible compounds. The combinatorics also makes systematic studies to derive sufficient training data challenging, for both experimental and theoretical reasons. Novel approaches such as machine learning may help to focus explicit studies on those compounds that have the highest benefit for SAR development.
Reaction rules in the gas phase are often transferable to the aqueous phase, as shown, for example, in correlations of the OH rate coefficients of aqueous- vs. gas-phase reactions (Monod et al., 2005). However, hydration effects and acid dissociation cause deviations from these relationships (Doussin and Monod, 2013). SARs for oxidation reactions of organics are available for other aquatic environments, including those for aromatic compounds by OH, 1O2 and O3 based on the theories by Marcus-Hush and Rehm-Weller (Canonica and Tratnyek, 2003) and for OH reactions with organics derived by machine learning algorithms (Zhong et al., 2021). It seems promising to extend such SARs towards more atmospherically relevant organic species. Accounting for non-ideal (high ionic strength) conditions present in atmospheric aqueous particles poses a specific challenge because most kinetic data are experimentally derived for dilute aqueous solutions. Herrmann (2003) proposed an ion-pairing model accounting for the ionic-strength dependence of rate coefficients. To date, such physicochemically based concepts remain hard to generalize to atmospherically relevant solute mixtures due to the lack of comprehensive data. They should become the focus of future studies to explore the validity of simplifications in multiphase chemistry models that often apply the same kinetic data to both dilute (cloud) and high-ionic-strength (aqueous aerosol) solutions. In addition, the mechanisms of organic reactions in aerosol particles, including non-radical and accretion reactions and the role of photosensitizers (Ervens et al., 2011; McNeill et al., 2012) resulting in different products than in (more) dilute cloud droplets, are far from being understood sufficiently to develop rule-based SARs.
3.3 Protocols for mechanism generation
The design of any chemical mechanism relies on a well-defined protocol comprising a set of rules and datasets that govern the construction process. The reliability of these mechanisms rests upon the accuracy and relevance of the prescribed protocol. Conceptually, the protocol should include sufficient information so that its meticulous and independent application to the same distribution of primary species results in identical mechanisms. It is often the protocol itself that requires regular updates and maintenance to align with the latest understanding of atmospheric chemistry. The protocol must ideally be transparent and accessible so that the scientific community can actively keep track of and contribute to the maintenance and revision of the mechanisms. The concept of the protocol is not limited to the construction of chemical mechanisms but also extends to their use. In particular, the mechanisms employed in air quality and chemical transport models often involve the use of model (or surrogate) species that are partially or completely anonymized and lack a direct equivalent in emission inventories (Sect. 2). As a result, it is imperative to associate these mechanisms with a transparent protocol that precisely outlines how the diversity of emitted species should be adequately distributed among the variables within the mechanism.
Protocols have played a pivotal role in the development of detailed mechanisms, particularly in the context of atmospheric oxidation of organic compounds in both the gas and aqueous phases. These mechanisms can be represented by a limited number of reaction types, iterated multiple times until the complete oxidation of the original parent compound is achieved. The inherent redundancy in these reaction sequences enables the design of generic protocols, which facilitate the systematic construction of chemical mechanisms. This approach relies heavily on the utilization of SARs to estimate unknown kinetic, mechanistic or thermodynamic parameters (Sect. 3.2).
Rigorous application of protocol rules can lead to an unmanageably large number of reactions (Aumont et al., 2005), and practical considerations demand substantial reductions in complexity. These reductions are, for example, achieved through the use of isomer or surrogate species (lumping) and by selectively excluding certain chemical reactions. Consequently, simplifications and reduction rules have become an integral component of the overall protocol (Sect. 3.5). The development and application of these protocols have led to various families of mechanisms for gas-phase oxidation of organic compounds, such as the MCM (Jenkin et al., 1997), the SAPRC mechanisms (Carter, 2000, 2010, 2023; Carter and Heo, 2013; Carter et al., 2023) and GECKO-A (Aumont et al., 2005). The same approach has also been proposed and has started to be applied for the development of oxidation mechanisms in the atmospheric aqueous phase (Mouchel-Vallon et al., 2013, 2017; Bräuer et al., 2019).
3.4 Automated generation of chemical mechanisms
The application of protocols for mechanism development has often relied on manual construction, even for detailed mechanisms like the MCM (Jenkin et al., 1997), the NCAR master mechanism (Madronich and Calvert, 1989, 1990; Aumont et al., 2000) and CAPRAM (Herrmann et al., 2000; Ervens et al., 2003). This manual approach not only is time-consuming and subject to human errors but also severely limits modifications and updates or sensitivity tests. However, more recently automated mechanism generation tools have been developed that, guided by the protocol rules, systematically and comprehensively describe the reactivity of various compounds. Conceptually, auto-generated mechanisms do not differ from those created manually. The significant advantages of such automated mechanism generation lies in its ability to easily and efficiently create chemical mechanisms to test novel hypotheses or ideas. Moreover, updating a mechanism can be a laborious task, particularly when the entire mechanism must be revisited due to changes in underlying SARs. In contrast, updating or correcting the coded SAR rules is relatively straightforward, enabling easy (re-)generation of new mechanisms.
Two generators are commonly used for describing the gaseous oxidation of organic compounds: Mechgen for SAPRC mechanism generation (Carter, 2023, 2024b) and GECKO-A for near-explicit mechanism generation (Aumont et al., 2005). These generators can be considered computer tools that emulate the process of chemical mechanism development as it is carried out by chemists. For a given precursor, generators analyze the chemical structure of the compounds to determine the appropriate reaction pathways. For each identified reaction, rate coefficients and/or products are searched for in an experimental database. In the absence of such data, estimates are made based on defined SAR rules. Alternatively, reaction products may be substituted or lumped using predefined surrogate species (as in SAPRC) or using existing species within the mechanism (as in GECKO-A). This lumping process also follows a well-defined protocol.
The auto-generation approach enables the development of highly detailed (semi-)explicit oxidation mechanisms, even for parent compounds with complex molecular structures such as monoterpenes, e.g., Valorso et al. (2011), or long-chain branched aliphatic compounds, e.g., Aumont et al. (2013) and La et al. (2016). For instance, in the case of C7 alkanes, this may result in 105 species and 106 reactions (Aumont et al., 2005). These mechanisms encompass hundreds of thousands of species and track the identity and reactivity of organic carbon throughout various stages, leading to final oxidation products. Consequently, these mechanisms may serve as valuable tools for exploring the state and behavior of organic matter during oxidation, and examining its impact on pollution episodes, climate and tropospheric oxidizing capacity. Moreover, the resulting near-explicit mechanisms are rooted in first principles, aiming to reflect the current fundamental understanding of atmospheric transformations. Thus, they provide objective means to quantify our knowledge of atmospheric processes by assessing the disparities between model results and observations. This approach offers great potential to investigate the underlying causes of model discrepancies and may inspire the design of new or modified experiments for further testing. However, automatically generated mechanisms are, at least in part, based on relatively crude approximations (e.g., SARs), most of which have significant uncertainties. Furthermore, automated mechanism generators must continue to be updated with new processes as they are discovered and proven to be significant. For example, within the realm of RO2 chemistry alone, autoxidation (Nozière and Vereecken, 2024) and the formation of the organic peroxides ROOR′ and other accretion products in even relatively simple RO2 R′O2 reaction systems (Murphy et al., 2023) and bimolecular reactions of acyl peroxy radicals (Nozière et al., 2023) are the subject of current laboratory investigation, but inclusion of these processes leads to explosions in the size of the explicit mechanisms and complicates reduction schemes. It is important to undertake sensitivity analyses in order to identify key steps in the reaction pathways, including those where there is competition between reaction routes.
3.5 Mechanism reduction
As mentioned in Sect. 2, near-explicit mechanisms serve as a benchmark for developing reduced mechanisms, offering a systematic route to design simpler mechanisms for specific applications. Such an approach enables easy traceability of the simpler mechanism (or parameterizations) back to the reference explicit mechanism, which is, in turn, linked to the current understanding of atmospheric chemistry (Fig. 3). Manual application of various reduction protocols has previously been employed to assess their relevance in the gas phase (Aumont et al., 1996; Kirchner, 2005; Utembe et al., 2009) and aqueous phase (Ervens et al., 2003). The reduction process can also be automated in mechanism generation tools, ensuring easy maintenance and traceability of the reduced mechanisms (Szopa et al., 2005; Carter, 2023, 2024b; Carter et al., 2023).
Model simulation results obtained with near-explicit mechanisms over a range of conditions can also be used as a foundation for the development of simplified mechanisms. The general approach is based on box model simulation results used as a database to train parameterizations. Highly simplified mechanisms can then be developed for organic compounds, in which the products and the stoichiometric coefficients of a set of pseudo-reactions are optimized to best reproduce a particular target, e.g., the evolution of the SOA mass concentration (Lannuque et al., 2018).
Recently, model results obtained with a near-explicit mechanism were successfully used to train a random forest that emulates SOA formation and aging (Mouchel-Vallon and Hodzic, 2023). In addition, a reduction tool was designed that iteratively and progressively tests various reduction strategies for organic aerosol mechanisms on an initial detailed benchmark mechanism based on the elimination of species or reactions and the substitution or lumping of species (Wang et al., 2022). Such approaches allow more accurate representation of the complexity of organic multiphase processes in air quality and climate models (Woo and McNeill, 2015). However, from a practical point of view, maintaining such mechanisms can present significant challenges due to the necessity of regenerating the training database with each update of the reference mechanism and initiating the development process afresh using the updated database.
3.6 Mechanism evaluation
It is essential to continually evaluate chemical mechanisms against theoretical, laboratory, chamber and field data to ensure accuracy and identify knowledge gaps. Mechanism development has to occur synergistically with developments of instruments and techniques, allowing more stringent testing of mechanisms for a continuously increasing number of species and processes that occur in the respective phase (gas, aqueous) or multiphase system. Evaluations must be done over the relevant ranges of conditions and chemical regimes and for target criteria, e.g., concerning air quality, human or ecosystem health, and/or climate.
The availability of datasets from unexplored or understudied areas led to the identification of new chemical regimes (Sect. 5.2) for which the applicability of existing chemical mechanisms needs to be carefully evaluated and potentially updated. In certain circumstances, evaluation can be performed based on field studies, but the controlled environments of simulation chambers generally provide better (and more accessible) test beds (Sect. 4.3). Improving the representativeness of experimental conditions in lab and chamber studies involves, e.g., lowering the reactant concentrations but also increasing the complexity of the mixtures. For example, SOA experiments have pointed to non-additive effects resulting in lower yields when mixtures of VOCs are used compared to single-precursor experiments (McFiggans et al., 2019). Newer measurement techniques, such as proton-transfer-reaction and chemical ionization mass spectrometry (PTR-MS, CI-MS), have led to the identification of multi-generation products and to the elucidation of chemical mechanisms for the formation and processing of aerosol particles. The direct detection of stabilized Criegee intermediates has allowed for the quantitative studying of their kinetics and reaction mechanisms; similarly, speciated RO2 measurements have helped establish the importance of specific precursor VOCs and their oxidation products. These oxidation products may include autoxidation and/or highly oxygenated organic molecules (HOMs) that have been identified as forming new particles (Bianchi et al., 2019), e.g., in RO2 unimolecular reactions, dimer and accretion product formation and/or bimolecular reactions of acyl peroxy radicals (Sect. 3.4).
The evaluation of multiphase chemical mechanisms is relatively more difficult and complex. Kinetic data for aqueous-phase processes are usually measured in bulk aqueous phases. However, the atmospheric multiphase system contains dispersed particles or droplets. While the kinetics in the homogeneous gas and aqueous phases are not affected by this physical state, the phase transfer processes and phase states pose major challenges. The sparsity of suitable lab setups or chambers for cloud and aqueous aerosol chemistry studies makes it difficult to test multiphase chemical mechanisms under (near-) atmospheric conditions.
Major advances in measurement techniques and data analysis over the last decades have led to the detection of a multitude of atmospheric chemical compounds, in particular organics, in the gas and condensed phases. At the same time, developments in theoretical, laboratory, field and chamber studies allowed experiments to derive detailed kinetic and mechanistic data of individual species/products under controlled conditions. Figure 4 illustrates the way that chemical mechanism developments result from a wide range of insights coming from various research activities, including theoretical studies (ab initio, quantum mechanics) but also field observations, laboratory/chamber studies and machine learning. It is evident that monitoring is essential to test the resulting models. Selected aspects and examples of these research activities are discussed in the following subsections; they have triggered the development of new or the expansion of existing chemical mechanisms or have been used to identify gaps in our knowledge that require more research efforts.
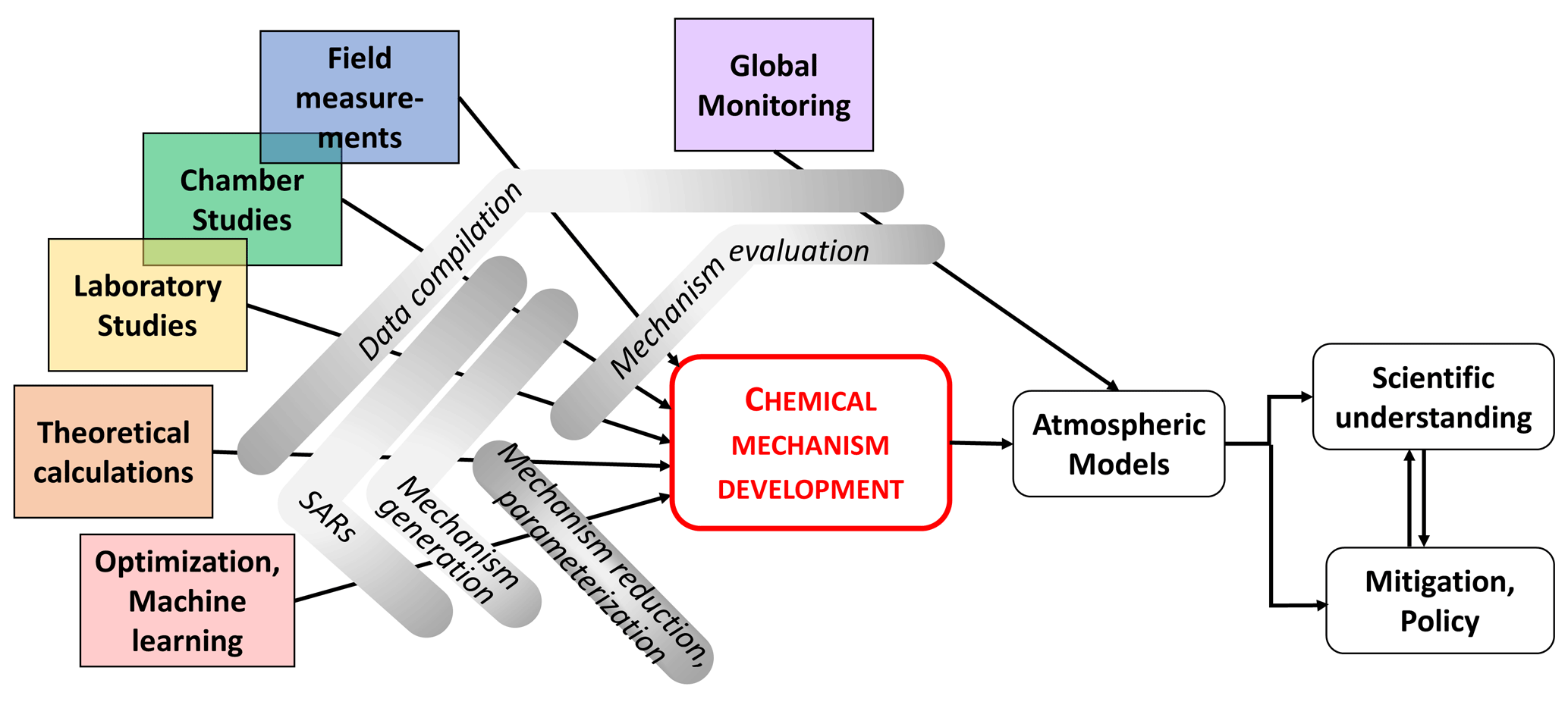
Figure 4Research activities connected to chemical mechanism development for implementation in atmospheric models, leading to an enhanced understanding of atmospheric implications and eventually resulting in adequate air quality and climate policies.
4.1 Theoretical calculations
Theoretical calculations can provide significant insights into the mechanisms of specific reactions and can predict the kinetic parameters needed in models. Calculations are highly complementary to experimental studies and provide information for species and/or reaction conditions that are difficult to probe experimentally. Recent developments have led to an increase in the accuracy and range of theoretical calculations for gas-phase reactions, e.g., Vereecken and Francisco (2012) and Vereecken et al. (2015). From being semi-quantitative at best 3 decades ago, current high-level ab initio calculations are now able to calculate activation barriers predicted at chemical accuracy (to ∼ 4 kJ mol−1 or better) for most systems, and agreement between theory and experiment for rate coefficients is often well within a factor of 2. Some systems, for which our understanding has been driven by theoretical studies, include the complex isoprene OH regeneration chemistry, RO2 autoxidation and the chemistry of Criegee intermediates. Many of these reactions are described well by a density functional theory (DFT) characterization of the potential energy system, followed by a higher-level wave function refinement of the energies, e.g., by Coupled Cluster, CCSD(T) or an equivalent. However, not all systems are amenable to precise calculations. Exceptions include calculations involving decomposition reactions of QOOH radicals (i.e., carbon-centered radicals containing an -OOH group) that are relevant to autoxidation and highly oxygenated organic molecule (HOM) formation, as they are subject to significant spin contamination. For such systems, post-CCSD(T) and/or multi-reference calculations are necessary, often at a prohibitive cost, but the agreement between experiment and theory in terms of barrier heights is currently still poor.
Calculations for aqueous-phase or multiphase processes are likewise complex, as these must describe not only the surrounding matrix (e.g., hydration effect) but also the impact of non-ideality of the aqueous or organic phases and of the interface with the gas phase. As a result, such calculations are much sparser than for gas-phase processes and are harder to generalize to the range of atmospheric conditions.
Theoretically calculated data are particularly useful in filling in the data gaps needed for the development of the SARs that are required for explicit mechanism development. This would be greatly aided if there were a readily available database of evaluated theoretical results, analogous to what is currently available for experimental results, as discussed in Sect. 3.1. However, such an effort is not straightforward because of the many levels of theory that can be employed and the resulting variable reliability of the results. The rapid advances in accuracy made by theory over a comparatively short period of time require expertise to assess the reliability of the results. Any database for theoretical data must, therefore, provide ample metadata and evaluation to avoid interpretation or use of kinetic data based on incorrect assumptions of their uncertainty interval for the reaction conditions examined.
4.2 Laboratory studies of elementary reactions
Elementary reactions include thermal chemical (uni-, bi- or termolecular) and photolysis reactions. Notable recent developments in laboratory-based measurement techniques include time- and energy-resolved photoionization (Osborn et al., 2008), IR and NIR absorption techniques (Thiébaud and Fittschen, 2006), CI-MS methodologies, multiplexed UV absorption spectroscopy (Stone et al., 2018), and frequency comb techniques (Bjork et al., 2016; Roberts et al., 2020). However, conventional laser-induced fluorescence coupled to laser flash photolysis, discharge flow and relative rate methods still account for most kinetic studies of gas- and aqueous-phase reactions. Their application has been extended beyond the traditional measurement of overall rate coefficients of “simple” radical reactions with stable molecules (e.g., the OH reaction with VOCs) to site-specific chemistry (McGillen et al., 2016; Onel et al., 2015) for more refined SAR and mechanism developments (Sect. 3.2). Specific pathways and mechanisms related to Criegee intermediates and their roles in SOA formation have been elucidated (Chhantyal-Pun et al., 2020; Lin and Chao, 2017; Taatjes, 2017). Despite these recent advances, our understanding of site-specific chemistry remains limited, and further experimental studies should be a priority. Increased efforts to combine theoretical and experimental studies have contributed immensely to our understanding of isoprene (Berndt et al., 2019a; Fuchs et al., 2013; Medeiros et al., 2022) and dimethyl sulfide oxidation (Berndt et al., 2019b; Jacob et al., 2024) and need to be applied more widely to improve our fundamental understanding of gas- and aqueous-phase reactions.
Photolysis rate coefficients for the eventual building of SARs and incorporation into chemical mechanisms remain a challenge to determine in the laboratory. Given the key role of these reactions in atmospheric radical cycles, more research is needed in this area. Determining photolysis rate coefficients for atmospherically relevant species requires quantification of absorption cross sections and quantum yields, both of which vary with wavelength, temperature, pressure and phase (gas, aqueous, heterogeneous). General features of absorption spectra (cross sections) for organic species are relatively well-known, depend on the nature of the chromophore and are often additive for multifunctional species. However, cross sections in the actinic region of the spectrum are often weak (i.e., in the long-wavelength “tail”), are strong functions of temperature, are difficult to measure, can depend on substituents to an uncertain extent and are very difficult to predict. Despite this, promising new developments and strategies in computational photochemistry have recently been used to build new tools to predict photolysis cross sections for atmospherically important VOCs (Hollas and Curchod, 2024). Some quantum yield data are relatively simple to measure or predict, for example, values of unity for organic hydroperoxides and nitrates due to the nature of the electronic state being accessed and the presence of sufficiently weak bonds. In contrast, photolysis of the atmospherically abundant carbonyl-containing compounds can be far more complex. Multiple photolysis pathways, collisional quenching, inter-system crossing and internal conversion make determination of photodissociation quantum yields over the full parameter space challenging and make predictions difficult or untenable.
Similar to kinetic rate coefficients in the condensed phases, photolysis rate coefficients also often show similar trends to the better-constrained data for gas-phase processes. In addition to such direct photolysis processes, the photosensitizers in and on aqueous particles represent an additional challenge (Bernard et al., 2016; Malecha and Nizkorodov, 2017; Mekic et al., 2019; Felber et al., 2020). More comprehensive studies are warranted to systematically implement these processes into multiphase chemistry models to assess the importance of photosensitizers under different conditions.
4.3 Atmospheric simulation chambers
Atmospheric simulation chambers provide a compromise between the tightly controlled conditions in laboratory experiments and the real atmosphere, and they are also used as excellent test beds for the development, intercomparison and calibration of instrumentation. The European PHOtoREactor (EUPHORE) in Valencia can be considered a pioneering outdoor chamber facility, which, along with more recent large outdoor chambers (e.g., SAPHIR (Simulation of Atmospheric PHotochemistry In a large Reaction Chamber) at Forschungszentrum Jülich), allows for the decrease of the surface-to-volume ratios to minimize chamber wall effects in conjunction with sensitive detection methods for long-duration studies under close-to-ambient concentrations (Rohrer et al., 2005; Jeffries et al., 2013; Li et al., 2021). The huge improvement in the range and sensitivity of chamber instrumentation has led to the ability to comprehensively monitor the chemical evolution of precursor and product species. This includes the use of CI-MS instrumentation noted above for stable organic species and, although not yet commonplace due to cost and complexity, instrumentation for the measurement of radical intermediates such as NO3, HO2 and RO2 (Fuchs et al., 2008; Onel et al., 2017) and measurement of OH and NO3 reactivity (Fuchs et al., 2017; Dewald et al., 2020). Intercomparisons of radical measurements have led to valuable insights into their validity and robustness (e.g., Schlosser et al., 2009; Fuchs et al., 2010; Dorn et al., 2013).
During the past 20 years, the development of a broad range of indoor and outdoor atmospheric simulation chambers has facilitated many fruitful collaborative efforts that also include making the chambers accessible to the community. The European EUROCHAMP network (https://data.eurochamp.org/, last access: 25 November 2024) has done an excellent job promoting and applying best practices in terms of development of new techniques and approaches; creation and curation of publicly available databases; and making European chamber infrastructure more integrated, sustainable and accessible (Doussin et al., 2023). These efforts are sustained under the Aerosol, Clouds and Trace Gases Research Infrastructure (ACTRIS) umbrella (https://www.actris.eu/facilities/national-facilities/exploratory-platforms, last access: 25 November 2024).
The availability and development of chambers for multiphase systems is not as advanced as those for gas-phase chambers. While numerous cloud chambers have been available for some time (e.g., CESAM, https://cesam.cnrs.fr/, last access: 25 November 2024, or AIDA (Aerosol Interaction and Dynamics in the Atmosphere), https://www.eurochamp.org/simulation-chambers/AIDA, last access: 25 November 2024), these are mostly equipped to explore (micro-)physical processes and/or aerosol–cloud interactions or are dedicated to aerosol chemistry experiments under non-cloud conditions. Cloud chemistry chambers would likely yield important new insights into the complex multiphase chemistry system (Sect. 5.4). Data centers, including the EUROCHAMP/ACTRIS ASC (https://www.actris.eu/topical-centre/data-centre, last access: 25 November 2024), give easy and sustainable access to experimental and modeled data obtained from simulation chamber experiments, as well as resources, protocols, mature products and tools that support activities related to measurements and modeling. More recently, the Integrated Chamber Atmospheric data Repository for Unified Science (ICARUS) data center has been developed to give access to data from simulation chambers in the USA (https://icarus.ucdavis.edu/, last access: 25 November 2024).
4.4 Field studies and global monitoring
Data from field studies are the ultimate test to confirm the accuracy and completeness of chemical mechanisms. As with chamber studies, field studies in a broad range of locations, from highly polluted megacities to the remote, pristine rainforest, have advanced enormously due to instrument developments, evaluation and intercomparisons. They have led to immense datasets from long- and short-term measurements and include sensitive radical intermediate and reactivity observations, similar to the wealth of data from chamber experiments. Such valuable datasets require consistent data processing, curating and archiving and need transparent and open provision to the community. Field measurements on the ground and/or by aircraft may be complemented by remote sensing data. Satellites and (low-cost) sensor networks can provide concentration data with wide spatial (global) coverage. Given their limitations in terms of species, resolution and specificity, their immediate role in informing mechanism development efforts seem currently limited. However, new and emerging satellite products and spaceborne platforms are coming online, and the sensitivity and retrieval coverage are improving all the time (Wells et al., 2020).
In addition to complementing existing datasets on previously investigated locations, a large focus of field studies in recent years has been to study locations with particular chemical characteristics (Sect. 5). The growth of urban agglomerations into megacities or even a gigacity (Kulmala et al., 2021) has significant consequences for air quality, human health and climate. The immediate impact of atmospheric processes on air quality and human well-being in such areas motivated numerous field campaigns in densely populated regions, e.g., MILAGRO in 2006 (Molina et al., 2010) and UK APHH Programs in Beijing (Shi et al., 2019) and Delhi (Nelson et al., 2021). Extreme chemical conditions leading to a suppression of NO due to very high O3 levels and hence a change in RO2 chemistry (Zou et al., 2015; Newland et al., 2021) have led to the definition of new chemical regimes or even the questioning of their concept in general (Wennberg, 2024), which requires the adjustment of existing or development of new chemical mechanisms (Sect. 5.2). Anthropogenic climate change and the implementation of net-zero policies are driving significant changes in emissions and atmospheric composition. Sustained and expanded long-term ambient observations from a range of platforms and locations are needed to track the evolution and atmospheric composition and to develop and test our chemical understanding.
4.5 Machine learning
Chemical mechanism development, like many fields in chemistry and physics, has greatly benefited from advances in machine learning; at the same time, the rapid developments in machine learning pose a risk since the assumptions on which the underlying algorithms are based and their evolution are not fully transparent. Machine learning has been successfully applied to build chemical mechanisms (Sanches-Neto et al., 2022) and to predict partitioning coefficients between the gas and the (organic) particle phases (Lumiaro et al., 2021). A combination of quantum chemical mechanisms and machine learning is a promising approach to quantitatively predict the chemical pollutant degradation by homogeneous or heterogeneous processes (Xia et al., 2022). In addition to the creation of chemical mechanisms, machine learning has been applied to reduce chemical schemes and develop surrogate models to limit the huge computational burden of explicit chemical mechanisms for the gas and aqueous phases (Kelp et al., 2022; Berkemeier et al., 2023). Shen et al. (2022) significantly reduced the computational costs of a global model with an adaptive algorithm, splitting the full chemical scheme into sub-mechanisms.
More recently, graph theory has been shown to be a powerful tool for the investigation and interpretation of complex chemical systems (Silva et al., 2021; Wiser et al., 2023; Sander, 2024). These tools visualize chemical mechanisms and illustrate the relative importance of individual pathways depending on conditions, which ultimately allows the removal of unimportant pathways, resulting in smaller mechanisms.
Overall, such tools are certainly a step towards the efficient development of explicit and reduced or empirical chemical mechanisms. However, major developments in their application to SARs or theoretical chemistry are still limited due to the lack of sufficiently complete training datasets. While, generally, their use seems very tempting, their application as a magic black box should be cautioned against: as for any chemical mechanism, the predictions are most robust and certain for parameter ranges that are covered by the training data. Analogous to data reporting for experimental studies, algorithms from machine learning and published results thereof should be documented together with the conditions and limitations from which they were derived. Algorithms that do not allow reconciling or tracing back individual chemical processes or species might be a useful tool to quickly predict chemical concentrations in well-defined chemical regimes; however, they clearly bear the risk of uninformed usage and thus biased predictions beyond their validity limits. Their application should therefore always be accompanied by more explicit models for comparison and testing. Generally, all training datasets should be transparently, continuously and interactively updated based on findings from the complementary research activities summarized in Sect. 4.
5.1 New insights into VOC oxidation
Central and well-studied components of chemical mechanisms such as peroxy radical (RO2) reactions as intermediate steps in VOC oxidation and SOA formation continue to trigger new insights and subsequent research efforts. Until recently, the fate of RO2 radicals in the gas phase was assumed to be dominated by bimolecular reactions with NOx, HO2, NO3 (at night) and other RO2 radicals. However, recent studies have demonstrated the importance of hydrogen-shift reactions, which have been implicated in HOM formation (Bianchi et al., 2019). The rates of these unimolecular reactions and their competitiveness with bimolecular reactions are highly dependent upon molecular structure (Nozière and Vereecken, 2024). The significance of such unimolecular (autoxidation) pathways, for a subset of RO2, was discovered from measurements of large OH concentrations above tropical forests (Lelieveld et al., 2008), which initiated successful collaboration and interactions between laboratory, theory, field and mechanism development activities to further elucidate isoprene oxidation chemistry.
Studies in remote regions, urban areas where high ozone levels efficiently titrate NO to NO2 or during the night have led to an increasing need to understand the rate coefficients and mechanisms for other bimolecular reactions of RO2. These additional RO2 losses under low-NO conditions include the unexpectedly high reactivity of certain combinations of RO2 and alkene reagents under ambient conditions (Nozière et al., 2023), the formation of stable ester and ether accretion products from RO2 self- and cross-reactions (Peräkylä et al., 2023), and the formation of dimers from RO2 + RO2 recombinations (Bates et al., 2022). For some biogenic VOCs, SOA yields were observed to vary from 0 % to >80 %, depending on the dominant RO2 reaction pathway. This strongly highlights the need for experiments on various and more representative RO2 fates and also the need to implement the loss pathways into chemical mechanisms to more accurately interpret data obtained across chemical regimes. Mechanism development can also provide data to other fields, e.g., by identifying the best chemical conditions to experimentally probe specific processes (e.g., Kenagy et al., 2024). Criegee intermediates, formed in the ozonolysis of unsaturated VOCs, represent a new class of oxidants with many branching pathways that have been identified only recently. As with RO2, unimolecular hydrogen shifts are important, as are ring-closure reactions and decomposition, each of which depends strongly upon conformer and substitution patterns (Vereecken et al., 2017). Furthermore, there are a wide and varied range of bimolecular reactions in which Criegee intermediates may also participate. From a mechanistic perspective, the insertion reactions that occur with a broad variety of labile hydrogen-containing compounds (McGillen et al., 2017; Chhantyal-Pun et al., 2019) are of significance since these may lead to a complex matrix of functionalized peroxidic products whose chemistry remains highly uncertain. Updated theory-based SARs for the chemical reactions of Criegee intermediates may be used to identify the dominant channels and eliminate unnecessary reactions, limiting the size of the mechanism. The intricacies of gas-phase ozonolysis chemistry can be attributed to the exothermicity of the initiation step, coupled with a complex landscape of weakly bound intermediates and local potential energy minima. Ensuing competition between unimolecular processes, collisional energy transfer and bimolecular reactions results in a complex product distribution. This was recently demonstrated in the unexpected formation of extremely long-lived products from the ozonolysis of hydrofluoroolefins (McGillen et al., 2023). Consequently, we anticipate that ozonolysis chemistry will be a continuing topic of study into the foreseeable future.
While it seems reasonable to assume that the similar pathways also occur in the aqueous phase, much fewer data and mechanistic insight into such pathways is available there. RO2 recombinations and other (organic) radical reactions are likely to initiate the formation of high-molecular-weight compounds (oligomers, accretion products) that have been identified in field and lab studies as SOA compounds. While oligomer formation in laboratory experiments was shown to be consistent with data on oligomerization for isoprene oxidation products in general (Ervens et al., 2015), these pathways cannot be generalized yet for implementation into detailed chemical mechanisms.
5.2 New chemical regimes
Over the last decades, atmospheric chemists have increasingly recognized the need to investigate chemical regimes with more extreme conditions, such as high temperatures in wildfire plumes and low pressures in the free troposphere but also over large changes in levels and ratios of key pollutants (e.g., NOx, mobile combustion sources). However, mechanisms that were developed and/or optimized for a specific chemical regime may not be applicable – and possibly even misleading – in such regimes. Specifically, the applicability of traditional chemical mechanisms depends on whether the oxidation processes are occurring via established VOC chemistry or whether new chemistry needs to be described because either the VOC has novel functional groups or the conditions are novel.
The emergence of new chemical regimes often arises from changing emission profiles. As vehicular emission standards become more stringent and NOx concentrations continue to decline, urban areas exhibit lower NOx levels. The resulting new mixtures of VOC, NOx and particles may even represent a new “aerosol-inhibited regime”, in addition to the more conventional NOx- and VOC-limited regimes (Ivatt et al., 2022). In this new regime, the HO2 loss is largely controlled by reactive uptake onto aerosol particles, which highlights the need for careful studies of its uptake coefficient for wide ranges of particle compositions. As a further consequence of changes in emission patterns, the acidity of cloud water may be increasingly controlled by organic acids (Lawrence et al., 2023, 2024), which are often neglected in thermodynamic pH calculations.
Megacities are growing in size and number and are environments where established mechanisms for oxidation of known VOCs may not be appropriate. Very high O3 concentrations lead to a suppression of NOx, and the reaction pathways for RO2 species may be very different from those of the more traditional high- or low-NOx environments, including more prevalent unimolecular reactions and bimolecular reactions with non-traditional collision partners such as particles, unsaturated VOCs and various radical species. Chemical models incorporating such chemistry can indicate when and where such novel chemistry would affect the atmosphere (e.g., Färber et al., 2024). As global change is resulting in a warmer, dryer climate in some regions of the planet, increasing occurrences of wildfires in populated areas represent new challenges in terms of air pollution, as they affect local and regional air quality through direct emission of a large set of pollutants. These plumes contain products of incomplete combustion processes of biogenic compounds that are rapidly processed under extreme conditions in terms of temperature and concentration levels, leading to the formation of a wide range of secondary pollutants both in the gas and condensed phases. The highly dynamic nature of such plumes makes measurements at sufficiently high temporal and spatial resolution difficult, which then translates into difficulties in the development and testing of suitable chemical mechanisms. To better constrain the chemical processes occurring in such scenarios, controlled experiments are needed to systematically generate and iterate suitable mechanisms.
5.3 New chemical compounds
“New” atmospheric chemical compounds can be defined in multiple ways: chemical compounds may have been in the atmosphere for a long time, but their presence is new to atmospheric science, and they have only been detected recently thanks to advanced measurement techniques and/or based on evidence from lab or theoretical studies or model–measurement discrepancies. The latter demonstrated that a significant fraction of ambient OH reactivity is unaccounted for, which makes predictions of the overall reactivity and oxidation capacity, and ultimately self-cleansing efficiency, of the atmosphere uncertain. Some of this missing reactivity may be due to heterogeneous processes or inorganic reactions, but it is likely that a significant fraction is due to reactions with VOC compounds, either primary emissions or secondary products, that have not been measured or identified yet and thus are missing in current chemical mechanisms. They include increasing relative contributions of non-vehicular industrial and consumer volatile chemical products (VCPs) (McDonald et al., 2018) and a multitude of higher-carbon-number VOCs that are becoming detectable by new and innovative methods such as multidimensional chromatography (Lewis et al., 2000; Dunmore et al., 2015) and various forms of chemical ionization mass spectrometry. Once they are identified, the protocols for mechanism generation can be applied (Sect. 3.3).
A second group of new species requiring mechanism development also arises from their introduction into the environment via anthropogenic activities. Such compounds include replacements for other chemicals, such as industrially important fluorochemicals (hydrofluoroolefins, hydrofluoroethers, fluorotelomers, etc.) (Michelat et al., 2022; Tokuhashi et al., 2018). The environmental fate and persistence of their oxidation is a potential concern, especially the formation of per- and polyfluoroalkyl substances (PFAS) (Kwiatkowski et al., 2020). Detailed atmospheric chemical models are usually not configured for halogenated species, which limits attempts to understand PFAS chemical interactions and degradation in the environment. Thus, for such compounds the full suite of steps as outlined in Fig. 3 needs to be followed to generate robust and operable mechanisms to be integrated into atmospheric models.
Finally, climate change mitigation may lead to chemical interactions in the atmosphere that are not captured in current mechanisms and, thus, require further study. Examples include amine oxidation chemistry from large-scale introduction of amine-based carbon capture and storage (CCS) schemes and heterogeneous chemistry associated with possible geo-engineering schemes to reduce the solar radiation flux. Chemical processing of amine emissions occurs both in the gas and aqueous phases (Nielsen et al., 2012; Karl et al., 2014), and considerations for multiphase systems (Sect. 5.4) need to be applied to assess impacts of increasing amine concentrations in the atmosphere.
5.4 New considerations for multiphase systems
Given the high computational costs of chemical transport or global models, chemical reactions are often heavily parameterized, e.g., by reactive uptake parameters that obscure the details of the underlying chemical systems. Such parameterizations do not (necessarily) allow extrapolation to all conditions covered by the models, and thus, the role and feedbacks of individual chemical processes may be not correctly accounted for. More details and fundamental descriptions of the chemical reactions involved in multiphase systems represents one of the challenges for future model implementations.
Even in models with explicit representation of individual reactions, intercomparisons of multiphase chemistry models revealed large discrepancies in predicted cloud droplet number concentrations and radiative forcing due to in-cloud sulfate formation (Kreidenweis et al., 2003) and in predicted oxidant concentrations in both phases (Barth et al., 2021). The models participating in these exercises only differed in the chemical mechanisms, i.e., the sets of kinetic and mechanistic data for gas, aqueous and phase-transfer processes. Thus, the metrics for mechanism comparison in these studies targeted key species for aqueous-phase chemistry. Detailed multiphase chemistry models have pointed to the importance of considering drop-size-resolved chemical composition, in particular acidity, that affects sulfate (Barth, 2006) and the phase partitioning of chemical compounds that affects their atmospheric residence time (Tilgner et al., 2021). Furthermore, cloud droplet size may affect the rates of uptake and chemical conversion within the aqueous phase (McVay and Ervens, 2017; Ervens, 2022). Such sensitivity studies demonstrate the challenges associated with model simulations of the atmospheric multiphase system. While robust and evaluated chemical mechanisms are a basic criterion for such models, the dependence of the reaction rates as a function of the chemical composition and microphysical properties of cloud droplets adds another layer of complexity and uncertainty in prediction of the atmospheric oxidative capacity and aerosol radiative forcing. These considerations suggest that parameterizations based on bulk properties (e.g., total cloud liquid water content or monodisperse drop populations) as often applied in large-scale models may not be sufficient for all chemical systems. Machine learning approaches seem well-suited to further map wide parameter spaces of the multiphase systems and their feedback on chemical reaction rates. This applies to not only (relatively) dilute cloud droplets but also less dilute, possibly viscous and/or semi-solid aerosol particles with limited penetration depth (Shiraiwa and Pöschl, 2021; Hua et al., 2022).
The formation and processing of the organic particle fraction that absorbs light (brown carbon, BrC) poses another particular challenge since it comprises a small percentage (∼ 1 %) of the organic particle mass. The light absorption is attributed to only a very small number of individual molecules. As a result, developing explicit chemical mechanisms for their description is challenging. Instead, an empirical approach parameterizing the mass absorption efficiency of ambient BrC may be a more feasible starting point that could be successively expanded to include more information once it becomes available from complementary studies.
In addition to their effects on climate, the role of aerosol particles affecting human health has been demonstrated clearly. The mass concentration of PM2.5 is the most commonly used metric to correlate aerosol particles and health effects. However, enhanced occurrence of mortality due to cardiorespiratory diseases in the presence of biogenic SOA may point to specific precursors or reaction pathways (Pye et al., 2021) that trigger adverse health effects due to reactive oxygen species (ROS) in aerosol particles (Shiraiwa et al., 2012). Combining such empirical trends in adverse health effects with knowledge of explicit ROS reactions in multiphase systems may be a promising avenue to connect atmospheric chemical processes to (physiological, health-related) target parameters.
5.5 FAIR models and tool chains
To perform chemical process modeling, the chemical mechanism needs to be converted into differential equations and run through a suitable integrator. This is done by software able to import a specific notation for the chemical reactions (e.g., Facsimile, KPP or GECKO-A) and the equations for the rate coefficients (e.g., Fortran, C, MATLAB or parameters for a fixed set of equations, together with support for variables, functions and other implementation aides). These numerical integrators, in turn, are typically embedded in a much larger software tool chain that also provides support for the intended use of the kinetic modeling, such as importing experimental data, providing fitting routines, handling time-specific photolysis rates, exporting and transforming modeling results, visualization, statistical analysis, integration in an Earth system model, etc., where each of these is implemented differently depending on the task at hand. Furthermore, the various models use different chemical notation for their mechanisms; e.g., isoprene is “C5H8” in the MCM, “CH3Cd(=CdH2)CdH=CdH2” in GECKO-A, “CH2=CH-C(=CH2)-CH3” in MechGen, “C=C(C)C=C” in SMILES notation and possibly “ISOP” in a locally developed model.
Transferring a chemical mechanism from one tool chain to another is less than obvious and even more so if one would like to combine (parts or) models, e.g., starting with the MCM, add chemical reactions based on theoretical calculations and complete with GECKO-A-generated chemical mechanisms for new reaction products. To date, information exchange between models relies all too often on laborious and error-prone manual effort. This opinion paper lists many aspects for which we need even more elements in the tool chain, such as model reduction; implementation, application and adaptation of SARs; model generation; visualization; evaluation; and optimization. It is unrealistic to hope that all tool chains will implement these aspects independently and even less so that all applications will converge on a single toolchain. Indeed, many tool chains exist because of different needs in various applications; e.g., the spreadsheet-based input for Kintecus is excellent to teach students, benefiting from a familiar interface, but would be a very poor choice in global Earth system modeling where the chemical module is only a small part of the performance-optimized computations.
The scientific community is moving towards free, open and FAIR data (findable, accessible/annotated, interoperable, reusable; https://www.go-fair.org/fair-principles/, last access: 25 November 2024), and many journals and funding agencies now demand that data, models and software be FAIR. Atmospheric mechanisms are usually free, open and findable, as they are described in the literature. They are, however, less accessible, often relying on cryptic notations for molecules without a user-friendly browsing tool. The MCM website (https://www.mcm.york.ac.uk/MCM/, last access: 25 November 2024) is an example of a more human-oriented interface, but all models would benefit from having human-oriented molecular representations as well as unambiguous, traceable and machine-readable notations for the molecules that are all available in a public vocabulary and consistent for gas and aqueous phases. The literature and protocol papers provide annotation and provenance for the individual models, but once models are merged, modified or reduced, this information is ill-matched. Provenance should be traced at the level of the individual reactions, but this is rarely implemented (e.g., Chemistry As A Boxmodel Application–Module Efficiently Calculating the Chemistry of the Atmosphere (CAABA/MECCA) supports adding literature references to specific reactions).
The various tool chains and thus the models that are geared towards a single chain (subset) are essentially non-interoperable, and the models are therefore also not generally interoperable or reusable without significant effort. Without interoperability, the ability to rigorously validate modeling runs is lost. One of the challenges of implementing the many aspects discussed in the current article thus also lies in providing tools to make any solutions available and transparent to the community without excessive duplicate effort across the tool chains, while maintaining transparency for the underlying data and the model build.
This opinion article provides a summary of some of the developments and gaps that define current and future efforts in chemical mechanism development. It is no coincidence that a large number of references cited here have been published in Atmospheric Chemistry and Physics, a journal that covers atmospheric chemical studies of a broad range of research activities, including fundamental, lab, field, chamber and machine learning studies that form the cornerstones for development of chemical mechanisms. While historically gas-phase chemical mechanism developments usually followed one of two strategies, i.e., either explicit mechanisms based on fundamental parameters and processes or empirical, operationally defined mechanisms, we propose that these approaches are not mutually exclusive: different levels of chemical complexity are required to answer the breadth of scientific questions over a range of scales. The wealth of chemical information provided by new analytical instrumentation and by cheminformatics methods, including machine learning, data mining and assimilation, offers opportunities for bridging the approaches, i.e., supplementing empirical approaches with (nearly) explicit information.
The last 2 decades have seen huge developments in analytical techniques and instrumentation that have allowed for the detection of more detailed speciated compounds in lab, field and chamber studies. New state-of-the-art measurement techniques have led to the identification of multigenerational products and elucidation of chemical mechanisms in not only the gas phase but also the condensed phases (cloud droplets, aerosol particles). These new experimental techniques have helped immensely in evaluating and pinning down key processes and knowledge gaps in our understanding of the underlying processes. Continued development of sophisticated instrumentation, in particular to measure the vast suite of partially oxidized, multifunctional organic species, has been and will remain critical. Thus, concerted development and application of instrumentation and chemical mechanisms need to occur to tackle current challenges that include, among others, the branching ratios or site-specific rate coefficients that are particularly challenging to obtain. Developments in the general area of mass spectrometry, including arrays of chemical ionization schemes (H3O+, NH, I−, NO, CF3O−, etc.), time-of-flight and Orbitrap techniques, have been extremely beneficial in this regard. However, calibration of these techniques for the enormous range of multifunctional species is a daunting task that must be dealt with. This requires strong reliance on synthetic chemists to provide authentic standards of (at least) representative species.
Any research efforts in these directions should also extend to the characterization of processes in and on cloud droplets and particles. Elucidating the chemical mechanisms in the atmospheric aqueous (and possibly organic) phase in modifying the chemical composition of aerosol particles is essential, as these processes affect aerosol properties, which in turn affect radiative forcing and human and ecosystem health. As such, the (physico-)chemical feedbacks and interactions between multiple phases must be thoroughly accounted for in mechanism development. To date, the mechanistic understanding and developments for chemical mechanisms in the condensed phases significantly lag behind those in the gas phase. Specifically, such developments are suffering from the higher complexity of the multiphase chemistry system that may also require the consideration of additional physicochemical (e.g., ionic strength) and microphysical (e.g., size of dispersed droplets or particles) parameters to comprehensively and reliably predict chemical formation, processing and degradation.
Rapid increases in computational power have opened new avenues to develop tools and approaches to complement or partially replace time-consuming, tedious, error-prone manual mechanism generation. Such automated techniques are doubtlessly extremely useful tools for mechanism development and also for the creation of reduced mechanisms to address a variety of research questions and to inform policymakers. Despite rapid progress in theoretical calculations, experimental studies are required more than ever to complement theory and to inform mechanism development efforts, by either traditional manual mechanism generation or automated tools. However, parameterizations and/or empirical (surrogate) models derived by theoretical and machine learning approaches bear the risk that they are used as black boxes since the underlying algorithms are not fully transparent. Therefore, we call for similar thoroughness and guidelines in reporting the conditions for which algorithms were trained and their limitations. Clear communication of the validity ranges of chemical mechanisms and specifically the SARs used to build them is even more important since the need to extend current mechanisms to conditions of new chemical regimes (e.g., mega- and gigacities, wildfires) emerged due to societal developments during the last decades and continuing into the future, as the climate continues to change.
In addition to focusing on such new scenarios, it is also particularly important that research funding agencies look sympathetically on and are supportive of applications of new techniques that look to re-visit established chemical systems and parameters (e.g., rate coefficients, photolysis cross sections and quantum yields, vapor pressures, SAR development) that are fundamentally important to atmospheric chemistry. New insights from experimental and theoretical work have revealed significant uncertainties in important inorganic reactions (e.g., OH + NO2) that are relevant to oxidant levels and, in turn, for atmospheric oxidation chemistry in general. The advent of new experimental techniques, such as frequency combs, facilitates the study of these foundation reactions since they can identify potential systematic errors in previous studies and/or improve the precision of rate coefficients.
The vast number of organic species and their pathways leading to highly oxidized and multifunctional products represents a particular challenge for atmospheric chemistry. The reactivity of these products cannot be accurately described by traditional SARs, which calls for dedicated studies to unravel the complexities to ultimately develop suitable SARs and chemical mechanisms. Otherwise, applying insufficiently constrained SARs will propagate into large errors in the prediction of the reactivity and concentration levels of organics and their potential to form SOA.
The most fundamental requirement to create and further develop and update chemical mechanisms is the availability and access to reliable data, along with the associated metadata, from experiments and observations, as enabled by data centers. Careful data curation and evaluation by experts should be supported accordingly. Ensuring free and open access to such data, following FAIR (findable, accessible, interoperable, reusable) principles must become the standard in our scientific community. Furthermore, publishing open code for modeling studies, as is already mandatory in the open-access EGU/Copernicus journal Geoscientific Model Development and strongly encouraged in all EGU journals, should become the standard requirement to make mechanism creation and application transparent and accessible.
Research efforts combining lab, field, chamber, theory and model studies, in which the chemical mechanism is at the core, have been shown to successfully lead to the mitigation of significant environmental issues (e.g., acid rain, stratospheric ozone depletion), demonstrating the success of synergistic open research efforts of the atmospheric chemistry community. In addition, we recommend that chemical mechanism developers should think outside of their atmospheric box and learn from research efforts being carried out in other related fields. One example is work in the combustion community, where they have combined information from multiple sources (e.g., direct data from lab measurements and theory, indirect data from the field and from different chamber experiments) to produce a mechanism and to assess and target uncertainties (e.g., Olm et al., 2017). Overall, it remains critical that we maintain such research capacity, in terms of both experimental and theoretical studies, and maintain blue-sky funding programs to pre-empt potential global problems and to identify and respond rapidly to emerging issues.
No data sets were used in the current article.
BE and AR conceptualized the idea of this article. BE, AR, BA, WPLC, MM, JO, BPV, PS, WRS, LV and TJW contributed to the writing of the paper and its revision.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This article is part of the special issue “20 years of Atmospheric Chemistry and Physics”. It is not associated with a conference.
We gratefully acknowledge the Coordinating Research Council’s Atmospheric Impacts Committee, who have provided valued support to the authors' work on the development and evaluation of databases and estimation methods for predicting air quality impacts of emitted organic compounds, and we appreciate our fruitful discussions with Sasha Madronich (National Center for Atmospheric Research). We would also like to acknowledge the further input and useful discussions from John Wenger and Mike Jenkin, as well as two anonymous referees in the discussion phase of this work.
This paper was edited by Rolf Müller and Gabriele Stiller and reviewed by John Wenger and two anonymous referees.
Abbatt, J. P. D. and Ravishankara, A. R.: Opinion: Atmospheric multiphase chemistry – past, present, and future, Atmos. Chem. Phys., 23, 9765–9785, https://doi.org/10.5194/acp-23-9765-2023, 2023. a
Atkinson, R., Aschmann, S. A., Carter, W. P. L., Winer, A. M., and J. N. Pitts, Jr.: Alkyl Nitrate Formation from the NOx-Air Photooxidation of C2–C8 n-Alkanes, The J. Phys. Chem. A, 86, 4563–4589, https://doi.org/10.1021/j100220a022, 1982. a
Aumont, B., Jaecker-Voirol, A., Martin, B., and Toupance, G.: Tests of some reduction hypotheses made in photochemical mechanisms, Atmos. Environ., 30, 2061–2077, https://doi.org/10.1016/1352-2310(95)00279-0, 1996. a
Aumont, B., Madronich, S., Bey, I., and Tyndall, G.: Contribution of Secondary VOC to the Composition of Aqueous Atmospheric Particles: A Modeling Approach, J. Atmos. Chem., 35, 59–75, https://doi.org/10.1023/a:1006243509840, 2000. a
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005. a, b, c, d
Aumont, B., Camredon, M., Mouchel-Vallon, C., La, S., Ouzebidour, F., Valorso, R., Lee-Taylor, J., and Madronich, S.: Modeling the influence of alkane molecular structure on secondary organic aerosol formation, Faraday Discuss., 165, 105–122, https://doi.org/10.1039/C3FD00029J, 2013. a
Barth, M. C.: The importance of cloud drop representation on cloud photochemistry, Atmos. Res., 82, 294–309, https://doi.org/10.1016/j.atmosres.2005.10.008, 2006. a
Barth, M. C., Ervens, B., Herrmann, H., Tilgner, A., McNeill, V. F., Tsui, W. G., Deguillaume, L., Chaumerliac, N., Carlton, A., and Lance, S. M.: Box Model Intercomparison of Cloud Chemistry, J. Geophys. Res.-Atmos., 126, e2021JD035486, https://doi.org/10.1029/2021JD035486, 2021. a
Bates, K. H., Burke, G. J. P., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022. a
Berkemeier, T., Krüger, M., Feinberg, A., Müller, M., Pöschl, U., and Krieger, U. K.: Accelerating models for multiphase chemical kinetics through machine learning with polynomial chaos expansion and neural networks, Geosci. Model Dev., 16, 2037–2054, https://doi.org/10.5194/gmd-16-2037-2023, 2023. a
Bernard, F., Ciuraru, R., Boréave, A., and George, C.: Photosensitized Formation of Secondary Organic Aerosols above the Air/Water Interface, Environ. Sci. Technol., 50, 8678–8686, https://doi.org/10.1021/acs.est.6b03520, 2016. a
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2, 21, https://doi.org/10.1038/s42004-019-0120-9, 2019a. a
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, The J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019b. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a, b
Bjork, B. J., Bui, T. Q., Heckl, O. H., Changala, P. B., Spaun, B., Heu, P., Follman, D., Deutsch, C., Cole, G. D., Aspelmeyer, M., Okumura, M., and Ye, J.: Direct frequency comb measurement of OD+ CO→DOCO kinetics., Science (New York, N.Y.), 354, 444–448, https://doi.org/10.1126/science.aag1862, 2016. a
Bräuer, P., Mouchel-Vallon, C., Tilgner, A., Mutzel, A., Böge, O., Rodigast, M., Poulain, L., van Pinxteren, D., Wolke, R., Aumont, B., and Herrmann, H.: Development of a protocol for the auto-generation of explicit aqueous-phase oxidation schemes of organic compounds, Atmos. Chem. Phys., 19, 9209–9239, https://doi.org/10.5194/acp-19-9209-2019, 2019. a
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation Number 19, https://jpldataeval.jpl.nasa.gov/pdf/NASA-JPL Evaluation 19-5.pdf (last access: 25 November 2024), 2020. a
Calvert, J. and Stockwell, W. R.: Acid generation in the troposphere by gas phase chemistry, Environ. Sci. Technol., 17, 428A–443A, 1983. a
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, https://doi.org/10.1093/oso/9780190233020.001.0001, 2015. a
Canonica, S. and Tratnyek, P. G.: Quantitative structure-activity relationships for oxidation reactions of organic chemicals in water, Environ. Toxicol. Chem., 22, 1743–1754, https://doi.org/10.1897/01-237, 2003. a
Carter, W. P. L.: Development of Ozone Reactivity Scales for Volatile Organic Compounds, Air Waste, 44, 881–899, https://doi.org/10.1080/1073161X.1994.10467290, 1994. a
Carter, W. P. L.: Documentation of the SAPRC-99 Chemical Mechanism for VOC Reactivity Assessment,” Report to the California Air Resources Board, Tech. rep., Report to the California Air Resources Board, http://www.cert.ucr.edu/~carter/absts.htm#saprc99 (last access: 25 November 2024), 2000. a
Carter, W. P. L.: Development of the SAPRC-07 Chemical Mechanism, Atmos. Environ., 44, 5324–5335, https://doi.org/10.1016/j.atmosenv.2010.01.026, 2010. a
Carter, W. P. L.: Documentation of the SAPRC-22 Mechanism, Tech. rep., Report to California Air Resources Board Contract No. 21AQP011, Sep 9, https://intra.cert.ucr.edu/~carter/SAPRC/22/ (last access: 25 November 2024), 2023. a, b, c
Carter, W. P. L.: Gateway to the SAPRC Mechanism Generation System, for the Atmospheric Reactions of Volatile Organic Compounds in the Presence of NOx, Tech. rep., https://intra.engr.ucr.edu/~carter/MechGen/ (last access: 25 November 2024), 2024a. a
Carter, W. P. L.: Users manual for the SAPRC atmospheric chemical mechanism generation system, Tech. rep., https://intra.engr.ucr.edu/~carter/SAPRC/22/MechGen.pdf (last access: 25 November 2024), 2024b. a, b, c
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013. a
Carter, W. P. L., Jiang, J., Orlando, J. J., and Barsanti, K. C.: Derivation of Atmospheric Reaction Mechanisms for Volatile Organic Compounds by the SAPRC Mechanism Generation System (MechGen), EGUsphere [preprint], https://doi.org/10.5194/egusphere-2023-2343, 2023. a, b
Chhantyal-Pun, R., Khan, M. A. H., Martin, R., Zachhuber, N., Buras, Z. J., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Direct Kinetic and Atmospheric Modeling Studies of Criegee Intermediate Reactions with Acetone, ACS Earth Space Chem., 3, 2363–2371, https://doi.org/10.1021/acsearthspacechem.9b00213, 2019. a
Chhantyal-Pun, R., Khan, M. A. H., Taatjes, C. A., Percival, C. J., Orr-Ewing, A. J., and Shallcross, D. E.: Criegee intermediates: production, detection and reactivity, Int. Rev. Phys. Chem., 39, 385–424, https://doi.org/10.1080/0144235X.2020.1792104, 2020. a
Derwent, R. G. and Jenkin, M. E.: Hydrocarbons and the long-range transport of ozone and pan across Europe, Atmos. Environ. A, 25, 1661–1678, https://doi.org/10.1016/0960-1686(91)90025-3, 1991. a
Dewald, P., Liebmann, J. M., Friedrich, N., Shenolikar, J., Schuladen, J., Rohrer, F., Reimer, D., Tillmann, R., Novelli, A., Cho, C., Xu, K., Holzinger, R., Bernard, F., Zhou, L., Mellouki, W., Brown, S. S., Fuchs, H., Lelieveld, J., and Crowley, J. N.: Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 20, 10459–10475, https://doi.org/10.5194/acp-20-10459-2020, 2020. a
Dorn, H.-P., Apodaca, R. L., Ball, S. M., Brauers, T., Brown, S. S., Crowley, J. N., Dubé, W. P., Fuchs, H., Häseler, R., Heitmann, U., Jones, R. L., Kiendler-Scharr, A., Labazan, I., Langridge, J. M., Meinen, J., Mentel, T. F., Platt, U., Pöhler, D., Rohrer, F., Ruth, A. A., Schlosser, E., Schuster, G., Shillings, A. J. L., Simpson, W. R., Thieser, J., Tillmann, R., Varma, R., Venables, D. S., and Wahner, A.: Intercomparison of NO3 radical detection instruments in the atmosphere simulation chamber SAPHIR, Atmos. Meas. Tech., 6, 1111–1140, https://doi.org/10.5194/amt-6-1111-2013, 2013. a
Doussin, J.-F. and Monod, A.: Structure–activity relationship for the estimation of OH-oxidation rate constants of carbonyl compounds in the aqueous phase, Atmos. Chem. Phys., 13, 11625–11641, https://doi.org/10.5194/acp-13-11625-2013, 2013. a
Doussin, J.-F., Fuchs, H., Kiendler-Scharr, A., Seakins, P., and Wenger, J.: A Practical Guide to Atmospheric Simulation Chambers, Springer International Publishing, Cham, Switzerland, ISBN 978-3-031-22276-4, https://doi.org/10.1007/978-3-031-22277-1, 2023. a
Dunmore, R. E., Hopkins, J. R., Lidster, R. T., Lee, J. D., Evans, M. J., Rickard, A. R., Lewis, A. C., and Hamilton, J. F.: Diesel-related hydrocarbons can dominate gas phase reactive carbon in megacities, Atmos. Chem. Phys., 15, 9983–9996, https://doi.org/10.5194/acp-15-9983-2015, 2015. a
Ervens, B.: Average cloud droplet size and composition: Good assumptions for predicting oxidants in the atmospheric aqueous phase?, J. Phys. Chem. A, 126, 8295–8304, https://doi.org/10.1021/acs.jpca.2c05527, 2022. a
Ervens, B., George, C., Williams, J. E., Buxton, G. V., Salmon, G. A., Bydder, M., Wilkinson, F., Dentener, F., Mirabel, P., Wolke, R., and Herrmann, H.: CAPRAM2.4 (MODAC mechanism): An extended and condensed tropospheric aqueous phase mechanism and its application, J. Geophys. Res., 108, 4426, https://doi.org/10.1029/2002JD002202, 2003. a, b, c, d
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011. a
Ervens, B., Renard, P., Tlili, S., Ravier, S., Clément, J.-L., and Monod, A.: Aqueous-phase oligomerization of methyl vinyl ketone through photooxidation – Part 2: Development of the chemical mechanism and atmospheric implications, Atmos. Chem. Phys., 15, 9109–9127, https://doi.org/10.5194/acp-15-9109-2015, 2015. a
Färber, M., Vereecken, L., Fuchs, H., Gkatzelis, G. I., Rohrer, F., Wedel, S., Wahner, A., and Novelli, A.: Impact of temperature-dependent non-PAN peroxynitrate formation, RO2NO2, on nighttime atmospheric chemistry, Phys. Chem. Chem. Phys., 26, 5183–5194, https://doi.org/10.1039/D3CP04163H, 2024. a
Felber, T., Schaefer, T., and Herrmann, H.: Five-Membered Heterocycles as Potential Photosensitizers in the Tropospheric Aqueous Phase: Photophysical Properties of Imidazole-2-carboxaldehyde, 2-Furaldehyde, and 2-Acetylfuran, The J. Phys. Chem. A, 124, 10029–10039, https://doi.org/10.1021/acs.jpca.0c07028, 2020. a
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, The Rev. Sci. Instrum., 79, 84104, https://doi.org/10.1063/1.2968712, 2008. a
Fuchs, H., Brauers, T., Dorn, H.-P., Harder, H., Häseler, R., Hofzumahaus, A., Holland, F., Kanaya, Y., Kajii, Y., Kubistin, D., Lou, S., Martinez, M., Miyamoto, K., Nishida, S., Rudolf, M., Schlosser, E., Wahner, A., Yoshino, A., and Schurath, U.: Technical Note: Formal blind intercomparison of HO2 measurements in the atmosphere simulation chamber SAPHIR during the HOxComp campaign, Atmos. Chem. Phys., 10, 12233–12250, https://doi.org/10.5194/acp-10-12233-2010, 2010. a
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013. a
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017. a
Grennfelt, P., Engleryd, A., Forsius, M., Hov, Ø., Rodhe, H., and Cowling, E.: Acid rain and air pollution: 50 years of progress in environmental science and policy., Ambio, 49, 849–864, https://doi.org/10.1007/s13280-019-01244-4, 2020. a
Haagen-Smit, A. J.: Chemistry and Physiology of Los Angeles Smog, Indust. Eng. Chem., 44, 1342–1346, https://doi.org/10.1021/ie50510a045, 1952. a
Herrmann, H.: Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 103, 4691–4716, https://doi.org/10.1021/cr020658q, 2003. a, b
Herrmann, H., Schaefer, T., Tilgner, A., Styler, S. A., Weller, C., Teich, M., and Otto, T.: Tropospheric aqueous-phase chemistry: kinetics, mechanisms, and its coupling to a changing gas phase, Chem. Rev., 115, 4259–4334, https://doi.org/10.1021/cr500447k, 2015. a
Herrmann, H., Ervens, B., Jacobi, H.-W., Wolke, R., Nowacki, P., and Zellner, R.: CAPRAM2.3: A Chemical Aqueous Phase Radical Mechanism for Tropospheric Chemistry, J. Atmos. Chem., 36, 231–284, https://doi.org/10.1023/A:1006318622743, 2000. a
Herrmann, H., Hoffmann, D., Schaefer, T., Bräuer, P., and Tilgner, A.: Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, Chem. Phys. Chem., 11, 3796–3822, https://doi.org/10.1002/cphc.201000533, 2010. a
Hoffmann, E. H., Schrödner, R., Tilgner, A., Wolke, R., and Herrmann, H.: CAPRAM reduction towards an operational multiphase halogen and dimethyl sulfide chemistry treatment in the chemistry transport model COSMO-MUSCAT(5.04e), Geosci. Model Dev., 13, 2587–2609, https://doi.org/10.5194/gmd-13-2587-2020, 2020. a
Hoffmann, M. R. and Jacob, D. J.: Kinetics and Mechanisms of the Catalytic Oxidation of Dissolved Sulfur Dioxide in Aqueous Solution: An Application to Nighttime Fog Water Chemistry, Butterworth Publishers, 101–172 pp., https://authors.library.caltech.edu/records/kqb4a-9dd20 (last access: 25 November 2024), 1984. a
Hollas, D. and Curchod, B. F. E.: AtmoSpec – A Tool to Calculate Photoabsorption Cross-Sections for Atmospheric Volatile Organic Compounds, The J. Phys. Chem. A, 128, 8580–8590, https://doi.org/10.1021/acs.jpca.4c05174, 2024. a
Hua, A. K., Lakey, P. S. J., and Shiraiwa, M.: Multiphase Kinetic Multilayer Model Interfaces for Simulating Surface and Bulk Chemistry for Environmental and Atmospheric Chemistry Teaching, J. Chem. Educ., 99, 1246–1254, https://doi.org/10.1021/acs.jchemed.1c00931, 2022. a
Ivatt, P. D., Evans, M. J., and Lewis, A. C.: Suppression of surface ozone by an aerosol-inhibited photochemical ozone regime, Nat. Geosci., 15, 536–540, https://doi.org/10.1038/s41561-022-00972-9, 2022. a
Jacob, L. S. D., Giorio, C., and Archibald, A. T.: Extension, development, and evaluation of the representation of the OH-initiated dimethyl sulfide (DMS) oxidation mechanism in the Master Chemical Mechanism (MCM) v3.3.1 framework, Atmos. Chem. Phys., 24, 3329–3347, https://doi.org/10.5194/acp-24-3329-2024, 2024. a
Janoš, J., Vinklárek, I. S., Rakovský, J., Mukhopadhyay, D. P., Curchod, B. F. E., Fárník, M., and Slavíček, P.: On the Wavelength-Dependent Photochemistry of the Atmospheric Molecule CF3COCl, ACS Earth Space Chem., 7, 2275–2286, https://doi.org/10.1021/acsearthspacechem.3c00196, 2023. a
Jeffries, H. E., Kamens, R. M., and Sexton, K.: Early history and rationale for outdoor chamber work at the University of North Carolina, Environ. Chem., 10, 349–364, https://doi.org/10.1071/EN13901, 2013. a
Jenkin, M. E., M, S. S., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a, b, c
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018. a
Kaduwela, A., Luecken, D., Carter, W., and Derwent, R.: New directions: Atmospheric chemical mechanisms for the future, Atmos. Environ., 122, 609–610, https://doi.org/10.1016/j.atmosenv.2015.10.031, 2015. a
Karl, M., Castell, N., Simpson, D., Solberg, S., Starrfelt, J., Svendby, T., Walker, S.-E., and Wright, R. F.: Uncertainties in assessing the environmental impact of amine emissions from a CO2 capture plant, Atmos. Chem. Phys., 14, 8533–8557, https://doi.org/10.5194/acp-14-8533-2014, 2014. a
Keller-Rudek, H., Moortgat, G. K., Sander, R., and Sörensen, R.: The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest, Earth Syst. Sci. Data, 5, 365–373, https://doi.org/10.5194/essd-5-365-2013, 2013. a
Kelp, M. M., Jacob, D. J., Lin, H., and Sulprizio, M. P.: An Online-Learned Neural Network Chemical Solver for Stable Long-Term Global Simulations of Atmospheric Chemistry, J. Adv. Model. Earth Syst., 14, e2021MS002926, https://doi.org/10.1029/2021MS002926, 2022. a
Kenagy, H. S., Heald, C. L., Tahsini, N., Goss, M. B., and Kroll, J. H.: Can we achieve atmospheric chemical environments in the laboratory? An integrated model-measurement approach to chamber SOA studies, Sci. Adv., 10, eado1482, https://doi.org/10.1126/sciadv.ado1482, 2024. a
Kirchner, F.: The chemical mechanism generation programme CHEMATA – Part 1: The programme and first applications, Atmos. Environ., 39, 1143–1159, https://doi.org/10.1016/j.atmosenv.2004.09.086, 2005. a
Kreidenweis, S. M., Walcek, C. J., Feingold, G., Gong, W., Jacobson, M. J., Kim, C., Liu, X., Penner, J. E., Nenes, A., and Seinfeld, J. H.: Modification of aerosol mass and size distribution due to aqueous phase SO2 oxidation in clouds: Comparisons of several models, J. Geophys. Res., 108, 4213, https://doi.org/10.1029/2002JD002697, 2003. a
Kulmala, M., Kokkonen, T. V., Pekkanen, J., Paatero, S., Petäjä, T., Kerminen, V.-M., and Ding, A.: Opinion: Gigacity – a source of problems or the new way to sustainable development, Atmos. Chem. Phys., 21, 8313–8322, https://doi.org/10.5194/acp-21-8313-2021, 2021. a
Kwiatkowski, C. F., Andrews, D. Q., Birnbaum, L. S., Bruton, T. A., DeWitt, J. C., Knappe, D. R. U., Maffini, M. V., Miller, M. F., Pelch, K. E., Reade, A., Soehl, A., Trier, X., Venier, M., Wagner, C. C., Wang, Z., and Blum, A.: Scientific Basis for Managing PFAS as a Chemical Class, Environ. Sci. Technol. Lett., 7, 532–543, https://doi.org/10.1021/acs.estlett.0c00255, 2020. a
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995. a
La, Y. S., Camredon, M., Ziemann, P. J., Valorso, R., Matsunaga, A., Lannuque, V., Lee-Taylor, J., Hodzic, A., Madronich, S., and Aumont, B.: Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation, Atmos. Chem. Phys., 16, 1417–1431, https://doi.org/10.5194/acp-16-1417-2016, 2016. a
Lannuque, V., Camredon, M., Couvidat, F., Hodzic, A., Valorso, R., Madronich, S., Bessagnet, B., and Aumont, B.: Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: development of the VBS-GECKO parameterization, Atmos. Chem. Phys., 18, 13411–13428, https://doi.org/10.5194/acp-18-13411-2018, 2018. a, b
Lawrence, C., Barth, M., Orlando, J., Casson, P., Brandt, R., Kelting, D., Yerger, E., and Lance, S.: Process Analysis of Elevated Concentrations of Organic Acids at Whiteface Mountain, New York, EGUsphere [preprint], https://doi.org/10.5194/egusphere-2024-715, 2024. a
Lawrence, C. E., Casson, P., Brandt, R., Schwab, J. J., Dukett, J. E., Snyder, P., Yerger, E., Kelting, D., VandenBoer, T. C., and Lance, S.: Long-term monitoring of cloud water chemistry at Whiteface Mountain: the emergence of a new chemical regime, Atmos. Chem. Phys., 23, 1619–1639, https://doi.org/10.5194/acp-23-1619-2023, 2023. a
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, 2008. a
Lewis, A. C., Carslaw, N., Marriott, P. J., Kinghorn, R. M., Morrison, P., Lee, A. L., Bartle, K. D., and Pilling, M. J.: A larger pool of ozone-forming carbon compounds in urban atmospheres, Nature, 405, 778–781, https://doi.org/10.1038/35015540, 2000. a
Li, J., Li, H., Wang, X., Wang, W., Ge, M., Zhang, H., Zhang, X., Li, K., Chen, Y., Wu, Z., Chai, F., Meng, F., Mu, Y., Mellouki, A., Bi, F., Zhang, Y., Wu, L., and Liu, Y.: A large-scale outdoor atmospheric simulation smog chamber for studying atmospheric photochemical processes: Characterization and preliminary application, J. Environ. Sci., 102, 185–197, https://doi.org/10.1016/j.jes.2020.09.015, 2021. a
Likens, G. E. and Bormann, F. H.: Acid rain: a serious regional environmental problem, Science (New York, N.Y.), 184, 1176–1179, https://doi.org/10.1126/science.184.4142.1176, 1974. a
Lin, J. J.-M. and Chao, W.: Structure-dependent reactivity of Criegee intermediates studied with spectroscopic methods, Chem. Soc. Rev., 46, 7483–7497, https://doi.org/10.1039/C7CS00336F, 2017. a
Lumiaro, E., Todorović, M., Kurten, T., Vehkamäki, H., and Rinke, P.: Predicting gas–particle partitioning coefficients of atmospheric molecules with machine learning, Atmos. Chem. Phys., 21, 13227–13246, https://doi.org/10.5194/acp-21-13227-2021, 2021. a
Madronich, S. and Calvert, J. G.: The NCAR Master Mechanism of the Gas Phase Chemistry – Version 2.0, Tech. rep., No. NCAR/TN- 333+STR, University Corporation for Atmospheric Research, https://doi.org/10.5065/D6HD7SKH, 1989. a, b
Madronich, S. and Calvert, J. G.: Permutation reactions of organic peroxy radicals in the troposphere, J. Geophys. Res., 95, 5697–5715, https://doi.org/10.1029/JD095iD05p05697, 1990. a
Malecha, K. T. and Nizkorodov, S. A.: Feasibility of Photosensitized Reactions with Secondary Organic Aerosol Particles in the Presence of Volatile Organic Compounds, The J. Phys. Chem. A, 121, 4961–4967, https://doi.org/10.1021/acs.jpca.7b04066, 2017. a
Manion, J. A., Sheen, D. A., and Awan, I. A.: Evaluated Kinetics of the Reactions of H and CH3 with n-Alkanes: Experiments with n-Butane and a Combustion Model Reaction Network Analysis, The J. Phys. Chem. A, 119, 7637–7658, https://doi.org/10.1021/acs.jpca.5b01004, 2015. a
McDonald, B. C., de Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S.-W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018. a
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019. a
McGillen, M. R., Tyndall, G. S., Orlando, J. J., Pimentel, A. S., Medeiros, D. J., and Burkholder, J. B.: Experimentally Determined Site-Specific Reactivity of the Gas-Phase OH and Cl + i-Butanol Reactions Between 251 and 340 K, The J. Phys. Chem. A, 120, 9968–9981, https://doi.org/10.1021/acs.jpca.6b09266, 2016. a
McGillen, M. R., Curchod, B. F. E., Chhantyal-Pun, R., Beames, J. M., Watson, N., Khan, M. A. H., McMahon, L., Shallcross, D. E., and Orr-Ewing, A. J.: Criegee Intermediate–Alcohol Reactions, A Potential Source of Functionalized Hydroperoxides in the Atmosphere, ACS Earth Space Chem., 1, 664–672, https://doi.org/10.1021/acsearthspacechem.7b00108, 2017. a
McGillen, M. R., Carter, W. P. L., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., and Wallington, T. J.: Database for the kinetics of the gas-phase atmospheric reactions of organic compounds, Earth Syst. Sci. Data, 12, 1203–1216, https://doi.org/10.5194/essd-12-1203-2020, 2020. a
McGillen, M. R., Fried, Z. T. P., Khan, M. A. H., Kuwata, K. T., Martin, C. M., O'Doherty, S., Pecere, F., Shallcross, D. E., Stanley, K. M., and Zhang, K.: Ozonolysis can produce long-lived greenhouse gases from commercial refrigerants, P. Natl. Acad. Sci. USa, 120, e2312714120, https://doi.org/10.1073/pnas.2312714120, 2023. a
McGillen, M. R., Michelat, L., Orlando, J. J., and Carter, W. P. L.: The use of the electrotopological state as a basis for predicting hydrogen abstraction rate coefficients: a proof of principle for the reactions of alkanes and haloalkanes with OH, Environ. Sci.-Atmos., 4, 18–34, https://doi.org/10.1039/D3EA00147D, 2024. a
McNeill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-Phase Secondary Organic Aerosol and Organosulfate Formation in Atmospheric Aerosols: A Modeling Study, Environ. Sci. Technol., 46, 8075–8081, https://doi.org/10.1021/es3002986, 2012. a
McVay, R. and Ervens, B.: A microphysical parameterization of aqSOA and sulfate formation in clouds, Geophys. Res. Lett., 44, 7500–7509, https://doi.org/10.1002/2017GL074233, 2017. a
Medeiros, J. D., Blitz, M. A., Seakins, P. W., and Whalley, L. K.: Direct Measurements of Isoprene Autoxidation: Pinpointing Atmospheric Oxidation in Tropical Forests, JACS Au, 2, 809–818, https://doi.org/10.1021/jacsau.1c00525, 2022. a
Mekic, M., Liu, J., Zhou, W., Loisel, G., Cai, J., He, T., Jiang, B., Yu, Z., Lazarou, Y. G., Li, X., Brigante, M., Vione, D., and Gligorovski, S.: Formation of highly oxygenated multifunctional compounds from cross-reactions of carbonyl compounds in the atmospheric aqueous phase, Atmos. Environ., 219, 117046, https://doi.org/10.1016/j.atmosenv.2019.117046, 2019. a
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥ C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021. a
Michelat, L., Mellouki, A., Ravishankara, A. R., El Othmani, H., Papadimitriou, V. C., Daële, V., and McGillen, M. R.: Temperature-Dependent Structure–Activity Relationship of OH + Haloalkene Rate Coefficients under Atmospheric Conditions and Supporting Measurements, ACS Earth Space Chem., 6, 3101–3114, https://doi.org/10.1021/acsearthspacechem.2c00296, 2022. a
Molina, L. T., Madronich, S., Gaffney, J. S., Apel, E., de Foy, B., Fast, J., Ferrare, R., Herndon, S., Jimenez, J. L., Lamb, B., Osornio-Vargas, A. R., Russell, P., Schauer, J. J., Stevens, P. S., Volkamer, R., and Zavala, M.: An overview of the MILAGRO 2006 Campaign: Mexico City emissions and their transport and transformation, Atmos. Chem. Phys., 10, 8697–8760, https://doi.org/10.5194/acp-10-8697-2010, 2010. a
Monod, A., Poulain, L., Grubert, S., Voisin, D., and Wortham, H.: Kinetics of OH-initiated oxidation of oxygenated organic compounds in the aqueous phase: new rate constants, structure-activity relationships and atmospheric implications, Atmos. Environ., 39, 7667–7688, https://doi.org/10.1016/j.atmosenv.2005.03.019, 2005. a
Mouchel-Vallon, C. and Hodzic, A.: Toward Emulating an Explicit Organic Chemistry Mechanism With Random Forest Models, J. Geophys. Res.-Atmos., 128, e2022JD038227, https://doi.org/10.1029/2022JD038227, 2023. a
Mouchel-Vallon, C., Bräuer, P., Camredon, M., Valorso, R., Madronich, S., Herrmann, H., and Aumont, B.: Explicit modeling of volatile organic compounds partitioning in the atmospheric aqueous phase, Atmos. Chem. Phys., 13, 1023–1037, https://doi.org/10.5194/acp-13-1023-2013, 2013. a
Mouchel-Vallon, C., Deguillaume, L., Monod, A., Perroux, H., Rose, C., Ghigo, G., Long, Y., Leriche, M., Aumont, B., Patryl, L., Armand, P., and Chaumerliac, N.: CLEPS 1.0: A new protocol for cloud aqueous phase oxidation of VOC mechanisms, Geosci. Model Dev., 10, 1339–1362, https://doi.org/10.5194/gmd-10-1339-2017, 2017. a, b
Murphy, S. E., Crounse, J. D., Møller, K. H., Rezgui, S. P., Hafeman, N. J., Park, J., Kjaergaard, H. G., Stoltz, B. M., and Wennberg, P. O.: Accretion product formation in the self-reaction of ethene-derived hydroxy peroxy radicals, Environ. Sci.-Atmos., 3, 882–893, https://doi.org/10.1039/D3EA00020F, 2023. a
Nelson, B. S., Stewart, G. J., Drysdale, W. S., Newland, M. J., Vaughan, A. R., Dunmore, R. E., Edwards, P. M., Lewis, A. C., Hamilton, J. F., Acton, W. J., Hewitt, C. N., Crilley, L. R., Alam, M. S., Şahin, Ü. A., Beddows, D. C. S., Bloss, W. J., Slater, E., Whalley, L. K., Heard, D. E., Cash, J. M., Langford, B., Nemitz, E., Sommariva, R., Cox, S., Shivani, Gadi, R., Gurjar, B. R., Hopkins, J. R., Rickard, A. R., and Lee, J. D.: In situ ozone production is highly sensitive to volatile organic compounds in Delhi, India, Atmos. Chem. Phys., 21, 13609–13630, https://doi.org/10.5194/acp-21-13609-2021, 2021. a
Newland, M. J., Bryant, D. J., Dunmore, R. E., Bannan, T. J., Acton, W. J. F., Langford, B., Hopkins, J. R., Squires, F. A., Dixon, W., Drysdale, W. S., Ivatt, P. D., Evans, M. J., Edwards, P. M., Whalley, L. K., Heard, D. E., Slater, E. J., Woodward-Massey, R., Ye, C., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Hewitt, C. N., Lee, J. D., Cui, T., Surratt, J. D., Wang, X., Lewis, A. C., Rickard, A. R., and Hamilton, J. F.: Low-NO atmospheric oxidation pathways in a polluted megacity, Atmos. Chem. Phys., 21, 1613–1625, https://doi.org/10.5194/acp-21-1613-2021, 2021. a
Nielsen, C. J., Herrmann, H., and Weller, C.: Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS), Chem. Soc. Rev., 41, 6684–6704, https://doi.org/10.1039/C2CS35059A, 2012. a
Nozière, B. and Vereecken, L.: H-shift and cyclization reactions in unsaturated alkylperoxy radicals near room temperature: propagating or terminating autoxidation?, Phys. Chem. Chem. Phys., 26, 25373–25384, https://doi.org/10.1039/D4CP02718C, 2024. a, b
Nozière, B., Durif, O., Dubus, E., Kylington, S., Emmer, Å., Fache, F., Piel, F., and Wisthaler, A.: The reaction of organic peroxy radicals with unsaturated compounds controlled by a non-epoxide pathway under atmospheric conditions, Phys. Chem. Chem. Phys., 25, 7772–7782, https://doi.org/10.1039/D2CP05166D, 2023. a, b
Olm, C., Varga, T., Valkó, É., Curran, H. J., and Turányi, T.: Uncertainty quantification of a newly optimized methanol and https://doi.org/10.1016/j.combustflame.2017.07.029, 2017. a
Onel, L., Blitz, M. A., Breen, J., Rickard, A. R., and Seakins, P. W.: Branching ratios for the reactions of OH with ethanol amines used in carbon capture and the potential impact on carcinogen formation in the emission plume from a carbon capture plant, Phys. Chem. Chem. Phys., 17, 25342–25353, https://doi.org/10.1039/C5CP04083C, 2015. a
Onel, L., Brennan, A., Seakins, P. W., Whalley, L., and Heard, D. E.: A new method for atmospheric detection of the CH3O2 radical, Atmos. Meas. Tech., 10, 3985–4000, https://doi.org/10.5194/amt-10-3985-2017, 2017. a
Osborn, D. L., Zou, P., Johnsen, H., Hayden, C. C., Taatjes, C. A., Knyazev, V. D., North, S. W., Peterka, D. S., Ahmed, M., and Leone, S. R.: The multiplexed chemical kinetic photoionization mass spectrometer: A new approach to isomer-resolved chemical kinetics, Rev. Sci. Instrum., 79, 104103, https://doi.org/10.1063/1.3000004, 2008. a
Peräkylä, O., Berndt, T., Franzon, L., Hasan, G., Meder, M., Valiev, R. R., Daub, C. D., Varelas, J. G., Geiger, F. M., Thomson, R. J., Rissanen, M., Kurtén, T., and Ehn, M.: Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 145, 7780–7790, https://doi.org/10.1021/jacs.2c10398, 2023. a
Prlj, A., Marsili, E., Hutton, L., Hollas, D., Shchepanovska, D., Glowacki, D. R., Slavíček, P., and Curchod, B. F. E.: Calculating Photoabsorption Cross-Sections for Atmospheric Volatile Organic Compounds, ACS Earth Space Chem., 6, 207–217, https://doi.org/10.1021/acsearthspacechem.1c00355, 2022. a
Puy, A., Beneventano, P., Levin, S. A., Lo Piano, S., Portaluri, T., and Saltelli, A.: Models with higher effective dimensions tend to produce more uncertain estimates, Sci. Adv., 8, eabn9450, https://doi.org/10.1126/sciadv.abn9450, 2024. a
Pye, H. O. T., Ward-Caviness, C. K., Murphy, B. N., Appel, K. W., and Seltzer, K. M.: Secondary organic aerosol association with cardiorespiratory disease mortality in the United States, Nat. Commun., 12, 7215, https://doi.org/10.1038/s41467-021-27484-1, 2021. a
Pye, H. O. T., Place, B. K., Murphy, B. N., Seltzer, K. M., D'Ambro, E. L., Allen, C., Piletic, I. R., Farrell, S., Schwantes, R. H., Coggon, M. M., Saunders, E., Xu, L., Sarwar, G., Hutzell, W. T., Foley, K. M., Pouliot, G., Bash, J., and Stockwell, W. R.: Linking gas, particulate, and toxic endpoints to air emissions in the Community Regional Atmospheric Chemistry Multiphase Mechanism (CRACMM), Atmos. Chem. Phys., 23, 5043–5099, https://doi.org/10.5194/acp-23-5043-2023, 2023. a
Roberts, F. C., Lewandowski, H. J., Hobson, B. F., and Lehman, J. H.: A rapid, spatially dispersive frequency comb spectrograph aimed at gas phase chemical reaction kinetics, Mol. Phys., 118, e1733116, https://doi.org/10.1080/00268976.2020.1733116, 2020. a
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005. a
Sanches-Neto, F. O., Dias-Silva, J. R., de Oliveira, V. M., Aquilanti, V., and Carvalho-Silva, V. H.: Evaluating and elucidating the reactivity of OH radicals with atmospheric organic pollutants: Reaction kinetics and mechanisms by machine learning, Atmos. Environ., 275, 119019, https://doi.org/10.1016/j.atmosenv.2022.119019, 2022. a
Sander, R.: Compilation of Henry's law constants (version 5.0.0) for water as solvent, Atmos. Chem. Phys., 23, 10901–12440, https://doi.org/10.5194/acp-23-10901-2023, 2023. a
Sander, R.: MEXPLORER 1.0.0 – a mechanism explorer for analysis and visualization of chemical reaction pathways based on graph theory, Geosci. Model Dev., 17, 2419–2425, https://doi.org/10.5194/gmd-17-2419-2024, 2024. a
Schlosser, E., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Wahner, A., Kanaya, Y., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., and Schurath, U.: Technical Note: Formal blind intercomparison of OH measurements: results from the international campaign HOxComp, Atmos. Chem. Phys., 9, 7923–7948, https://doi.org/10.5194/acp-9-7923-2009, 2009. a
Shen, L., Jacob, D. J., Santillana, M., Bates, K., Zhuang, J., and Chen, W.: A machine-learning-guided adaptive algorithm to reduce the computational cost of integrating kinetics in global atmospheric chemistry models: application to GEOS-Chem versions 12.0.0 and 12.9.1, Geosci. Model Dev., 15, 1677–1687, https://doi.org/10.5194/gmd-15-1677-2022, 2022. a
Shi, Z., Vu, T., Kotthaus, S., Harrison, R. M., Grimmond, S., Yue, S., Zhu, T., Lee, J., Han, Y., Demuzere, M., Dunmore, R. E., Ren, L., Liu, D., Wang, Y., Wild, O., Allan, J., Acton, W. J., Barlow, J., Barratt, B., Beddows, D., Bloss, W. J., Calzolai, G., Carruthers, D., Carslaw, D. C., Chan, Q., Chatzidiakou, L., Chen, Y., Crilley, L., Coe, H., Dai, T., Doherty, R., Duan, F., Fu, P., Ge, B., Ge, M., Guan, D., Hamilton, J. F., He, K., Heal, M., Heard, D., Hewitt, C. N., Hollaway, M., Hu, M., Ji, D., Jiang, X., Jones, R., Kalberer, M., Kelly, F. J., Kramer, L., Langford, B., Lin, C., Lewis, A. C., Li, J., Li, W., Liu, H., Liu, J., Loh, M., Lu, K., Lucarelli, F., Mann, G., McFiggans, G., Miller, M. R., Mills, G., Monk, P., Nemitz, E., O'Connor, F., Ouyang, B., Palmer, P. I., Percival, C., Popoola, O., Reeves, C., Rickard, A. R., Shao, L., Shi, G., Spracklen, D., Stevenson, D., Sun, Y., Sun, Z., Tao, S., Tong, S., Wang, Q., Wang, W., Wang, X., Wang, X., Wang, Z., Wei, L., Whalley, L., Wu, X., Wu, Z., Xie, P., Yang, F., Zhang, Q., Zhang, Y., Zhang, Y., and Zheng, M.: Introduction to the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing)”, Atmos. Chem. Phys., 19, 7519–7546, https://doi.org/10.5194/acp-19-7519-2019, 2019. a
Shiraiwa, M. and Pöschl, U.: Mass accommodation and gas–particle partitioning in secondary organic aerosols: dependence on diffusivity, volatility, particle-phase reactions, and penetration depth, Atmos. Chem. Phys., 21, 1565–1580, https://doi.org/10.5194/acp-21-1565-2021, 2021. a
Shiraiwa, M., Selzle, K., and Pöschl, U.: Hazardous components and health effects of atmospheric aerosol particles: reactive oxygen species, soot, polycyclic aromatic compounds and allergenic proteins, Free Rad. Res., 46, 927–939, https://doi.org/10.3109/10715762.2012.663084, 2012. a
Silva, S. J., Burrows, S. M., Evans, M. J., and Halappanavar, M.: A Graph Theoretical Intercomparison of Atmospheric Chemical Mechanisms, Geophys. Res. Lett., 48, e2020GL090481, https://doi.org/10.1029/2020GL090481, 2021. a
Stockwell, W. R., Saunders, E., Goliff, W. S., and Fitzgerald, R. M.: A perspective on the development of gas-phase chemical mechanisms for Eulerian air quality models, J. Air Waste Manage. Assoc., 70, 44–70, https://doi.org/10.1080/10962247.2019.1694605, 2020. a
Stone, D., Au, K., Sime, S., Medeiros, D. J., Blitz, M., Seakins, P. W., Decker, Z., and Sheps, L.: Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO, Phys. Chem. Chem. Phys., 20, 24940–24954, https://doi.org/10.1039/C8CP05332D, 2018. a
Szopa, S., Aumont, B., and Madronich, S.: Assessment of the reduction methods used to develop chemical schemes: building of a new chemical scheme for VOC oxidation suited to three-dimensional multiscale HOx-NOx-VOC chemistry simulations, Atmos. Chem. Phys., 5, 2519–2538, https://doi.org/10.5194/acp-5-2519-2005, 2005. a
Taatjes, C. A.: Criegee Intermediates: What Direct Production and Detection Can Teach Us About Reactions of Carbonyl Oxides, Annu. Rev. Phys. Chem., 68, 183–207, https://doi.org/10.1146/annurev-physchem-052516-050739, 2017. a
Thiébaud, J. and Fittschen, C.: Near infrared cw-CRDS coupled to laser photolysis: Spectroscopy and kinetics of the HO2 radical, Appl. Phys. B, 85, 383–389, https://doi.org/10.1007/s00340-006-2304-0, 2006. a
Tilgner, A., Schaefer, T., Alexander, B., Barth, M., Collett Jr., J. L., Fahey, K. M., Nenes, A., Pye, H. O. T., Herrmann, H., and McNeill, V. F.: Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 21, 13483–13536, https://doi.org/10.5194/acp-21-13483-2021, 2021. a
Tokuhashi, K., Takizawa, K., and Kondo, S.: Rate constants for the reactions of OH radicals with CF3CX=CY2 (X=H, F, CF3, Y=H, F, Cl), Environ. Sci. Pollut. Res., 25, 15204–15215, https://doi.org/10.1007/s11356-018-1700-4, 2018. a
Utembe, S. R., Watson, L. A., Shallcross, D. E., and Jenkin, M. E.: A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 3: Development of a secondary organic aerosol module, Atmos. Environ., 43, 1982–1990, https://doi.org/10.1016/j.atmosenv.2009.01.008, 2009. a
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011. a
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere., Chem. Soc. Rev., 4119, 6259–6293, https://api.semanticscholar.org/CorpusID:205815849 (last access: 25 November 2024), 2012. a
Vereecken, L., Glowacki, D. R., and Pilling, M. J.: Theoretical Chemical Kinetics in Tropospheric Chemistry: Methodologies and Applications, Chem. Rev., 115, 4063–4114, https://doi.org/10.1021/cr500488p, 2015. a
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, https://doi.org/10.1039/C7CP05541B, 2017. a
Vereecken, L., Aumont, B., Barnes, I., Bozzelli, J. W., Goldman, M. J., Green, W. H., Madronich, S., Mcgillen, M. R., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., Rickard, A. R., Stockwell, W. R., Wallington, T. J., and Carter, W. P. L.: Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kin., 50, 435–469, https://doi.org/10.1002/kin.21172, 2018. a
Wang, Z., Couvidat, F., and Sartelet, K.: GENerator of reduced Organic Aerosol mechanism (GENOA v1.0): an automatic generation tool of semi-explicit mechanisms, Geosci. Model Dev., 15, 8957–8982, https://doi.org/10.5194/gmd-15-8957-2022, 2022. a
Wells, K. C., Millet, D. B., Payne, V. H., Deventer, M. J., Bates, K. H., de Gouw, J. A., Graus, M., Warneke, C., Wisthaler, A., and Fuentes, J. D.: Satellite isoprene retrievals constrain emissions and atmospheric oxidation, Nature, 585, 225–233, https://doi.org/10.1038/s41586-020-2664-3, 2020. a
Wennberg, P. O.: Let's Abandon the “High NOx” and “Low NOx” Terminology, ACS ES&T Air, 1, 3–4, https://doi.org/10.1021/acsestair.3c00055, 2024. a
Wiser, F., Place, B. K., Sen, S., Pye, H. O. T., Yang, B., Westervelt, D. M., Henze, D. K., Fiore, A. M., and McNeill, V. F.: AMORE-Isoprene v1.0: a new reduced mechanism for gas-phase isoprene oxidation, Geosci. Model Dev., 16, 1801–1821, https://doi.org/10.5194/gmd-16-1801-2023, 2023. a
Woo, J. L. and McNeill, V. F.: simpleGAMMA v1.0 – a reduced model of secondary organic aerosol formation in the aqueous aerosol phase (aaSOA), Geosci. Model Dev., 8, 1821–1829, https://doi.org/10.5194/gmd-8-1821-2015, 2015. a
Xia, D., Chen, J., Fu, Z., Xu, T., Wang, Z., Liu, W., Xie, H.-b., and Peijnenburg, W. J. G. M.: Potential Application of Machine-Learning-Based Quantum Chemical Methods in Environmental Chemistry, Environ. Sci. Technol., 56, 2115–2123, https://doi.org/10.1021/acs.est.1c05970, 2022. a
Zhong, S., Zhang, K., Wang, D., and Zhang, H.: Shedding light on “Black Box” machine learning models for predicting the reactivity of HO radicals toward organic compounds, Chem. Eng. J., 405, 126627, https://doi.org/10.1016/j.cej.2020.126627, 2021. a
Zou, Y., Deng, X. J., Zhu, D., Gong, D. C., Wang, H., Li, F., Tan, H. B., Deng, T., Mai, B. R., Liu, X. T., and Wang, B. G.: Characteristics of 1 year of observational data of VOCs, NOx and O3 at a suburban site in Guangzhou, China, Atmos. Chem. Phys., 15, 6625–6636, https://doi.org/10.5194/acp-15-6625-2015, 2015. a
- Abstract
- Introduction
- Strategies for chemical mechanism development
- Steps involved in chemical mechanism development
- Research activities related to chemical mechanism development
- Current and future challenges
- Conclusions and future research needs
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Review statement
- References
- Abstract
- Introduction
- Strategies for chemical mechanism development
- Steps involved in chemical mechanism development
- Research activities related to chemical mechanism development
- Current and future challenges
- Conclusions and future research needs
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Review statement
- References