the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 Jun 2023
| 28 Jun 2023
Photoaging of phenolic secondary organic aerosol in the aqueous phase: evolution of chemical and optical properties and effects of oxidants
Wenqing Jiang
Christopher Niedek
Cort Anastasio
While gas-phase reactions are well established to have significant impacts on the mass concentration, chemical composition, and optical properties of secondary organic aerosol (SOA), the aqueous-phase aging of SOA remains poorly understood. In this study, we performed a series of long-duration photochemical aging experiments to investigate the evolution of the composition and light absorption of the aqueous SOA (aqSOA) from guaiacyl acetone (GA), a semivolatile phenolic carbonyl that is common in biomass burning smoke. The aqSOA was produced from reactions of GA with hydroxyl radical (•OH-aqSOA) or a triplet excited state of organic carbon (3C∗-aqSOA) and was then photoaged in water under conditions that simulate sunlight exposure in northern California for up to 48 h. The effects of increasing aqueous-phase •OH or 3C∗ concentration on the photoaging of the aqSOA were also studied. High-resolution aerosol mass spectrometry (HR-AMS) and UV–Vis spectroscopy were utilized to characterize the composition and the light absorptivity of the aqSOA and to track their changes during aging.
Compared to •OH-aqSOA, the 3C∗-aqSOA is produced more rapidly and shows less oxidation, a greater abundance of oligomers, and higher light absorption. Prolonged photoaging promotes fragmentation and the formation of more volatile and less light-absorbing products. More than half of the initial aqSOA mass is lost, and substantial photobleaching occurs after 10.5 h of prolonged aging under simulated sunlight illumination for 3C∗-aqSOA and 48 h for •OH-aqSOA. By performing positive matrix factorization (PMF) analysis of the combined HR-AMS and UV–Vis spectral data, we resolved three generations of aqSOA with distinctly different chemical and optical properties. The first-generation aqSOA shows significant oligomer formation and enhanced light absorption at 340–400 nm. The second-generation aqSOA is enriched in functionalized GA species and has the highest mass absorption coefficients in 300–500 nm, while the third-generation aqSOA contains more fragmented products and is the least light absorbing. These results suggest that intermediately aged phenolic aqSOA is more light absorbing than other generations, and that the light absorptivity of phenolic aqSOA results from a competition between brown carbon (BrC) formation and photobleaching, which is dependent on aging time. Although photoaging generally increases the oxidation of aqSOA, a slightly decreased of the •OH-aqSOA is observed after 48 h of prolonged photoaging with additional •OH exposure. This is likely due to greater fragmentation and evaporation of highly oxidized compounds. Increased oxidant concentration accelerates the transformation of aqSOA and promotes the decay of BrC chromophores, leading to faster mass reduction and photobleaching. In addition, compared with •OH, photoaging by 3C∗ produces more low-volatility functionalized products, which counterbalances part of the aqSOA mass loss due to fragmentation and evaporation.
- Article
(2851 KB) - Full-text XML
-
Supplement
(1777 KB) - BibTeX
- EndNote




Phenols, which are emitted from biomass burning (BB) through lignin pyrolysis (Schauer et al., 2001) and formed from the oxidation of aromatic hydrocarbons (Berndt and Böge, 2006), are important precursors for atmospheric secondary organic aerosol (SOA) and brown carbon (BrC) (Bruns et al., 2016; Mabato et al., 2022; Misovich et al., 2021; Smith et al., 2016; Sun et al., 2011; Yee et al., 2013). These compounds can form aqueous-phase SOA (aqSOA) at fast rates in atmospheric waters, through photoreactions with oxidants such as hydroxyl radical (•OH), excited triplet states of organic carbon (3C∗), and reactive nitrogen species (e.g., •NO, •NO2, NO+, and NO (Jiang et al., 2021; Li et al., 2022b; Mabato et al., 2022, 2023; Pang et al., 2019; Yang et al., 2021; Yu et al., 2014). The mass yields of aqSOA from the phenolic precursors in atmospheric waters range from 50 % to 140 %, and the proposed formation pathways include oligomerization, functionalization (e.g., hydroxylation), and fragmentation (Arciva et al., 2022; Huang et al., 2018; Jiang et al., 2021; Ma et al., 2021; Smith et al., 2014, 2015, 2016; Sun et al., 2010; Yu et al., 2014, 2016). Reactions involving reactive nitrogen species can also lead to nitration and nitrophenol formation (Heal et al., 2007; Mabato et al., 2022; Pang et al., 2019; Yang et al., 2021). The resulting phenolic oligomers, multifunctional derivatives, and nitrophenols can absorb near-UV and visible light and contribute significantly to BrC formation in biomass burning emissions (Gilardoni et al., 2016; Li et al., 2022a; Misovich et al., 2021; Palm et al., 2020; Pang et al., 2019). In addition, humic-like substances (HULIS), which can induce oxidative stress and cause adverse health effects (Deng et al., 2022), are observed in phenolic aqSOA as well (Chang and Thompson, 2010).
Despite extensive research on the formation of aqSOA from phenols, the aging and degradation of phenolic SOA in water remain poorly characterized. Atmospheric lifetimes of SOA range from hours to weeks (Wagstrom and Pandis, 2009), during which chemical reactions can occur, leading to continuous aging and evolution of SOA. Functionalization (i.e., the addition of functional groups to the molecules) and fragmentation (i.e., the breaking of bonds within the molecules to form smaller species) are critical mechanisms in the aging of SOA that can greatly change the chemical composition and loading of aerosols (Kroll et al., 2009; Leresche et al., 2021; Shrivastava et al., 2017). Chemical aging can also influence the optical properties of SOA, as some reactions increase the light absorptivity, while others cause photobleaching by destroying chromophores (Lee et al., 2014). Furthermore, fragmentation can result in the formation of volatile and semivolatile products, causing a loss of SOA mass and photobleaching (Kroll et al., 2015). Yu et al. (2016) studied the aqueous-phase photooxidation of phenol and methoxyphenols and observed that, as aging progresses, fragmentation reactions become increasingly dominant in comparison to oligomerization and functionalization reactions. However, a portion of the aqSOA appears to be resistant to fragmentation and remains chemically unchanged even after prolonged exposure to simulated sunlight in the aqueous phase (Yu et al., 2016). Similarly, in an environmental chamber study, O'Brien and Kroll (2019) reported that 70 %–90 % of the α-pinene SOA mass remained in particles after an initial decay during photochemical aging.
The impacts of aging on the concentrations and properties of SOA in the atmosphere have been widely observed in biomass burning emissions (Brege et al., 2018; Chen et al., 2021; Garofalo et al., 2019; Kleinman et al., 2020; Zhou et al., 2017). For instance, aged wildfire plumes subjected to aqueous processing experience substantial losses in organic aerosol (OA) mass, increases in SOA oxidation, and changes in optical properties (Che et al., 2022; Farley et al., 2022; Sedlacek et al., 2022). Aqueous-phase oxidation of organic molecules, including phenols, and the formation of SOA have been observed in residential wood burning smoke in both urban and rural environments as well (Brege et al., 2018; Kim et al., 2019; Stefania et al., 2016; Sun et al., 2010). In addition, in remote regions where aerosols are generally highly aged and have been subjected to more extensive aqueous-phase and heterogeneous processing, SOA is significantly more oxidized, less volatile, and more hygroscopic compared to those in urban areas (Jimenez et al., 2009; Morgan et al., 2010; Ng et al., 2011; Zhang et al., 2011; Zhou et al., 2019).
Understanding the chemical aging process of SOA in the aqueous phase is important for better predicting the concentration of SOA in ambient air and assessing its potential impacts on climate and human health. Sunlight-triggered aqueous-phase reactions, such as direct photolysis of organics, nitrate, nitrite, and hydrogen peroxide, as well as energy and charge-transfer reactions driven by 3C∗, significantly impact the chemical aging of SOA, leading to changes in particle composition and properties (Corral Arroyo et al., 2018; Ervens et al., 2011; Herrmann et al., 2015; Mabato et al., 2022). The extent of exposure of aqSOA to oxidants in atmospheric waters can vary widely, influenced by the concentration and residence time of oxidants. For example, the steady-state concentration of •OH can vary from 10−16 to 10−12 M (Herrmann et al., 2010), while that of 3C∗ can vary from 10−14 to 10−11 M (Kaur et al., 2019), depending on the solute concentration, which ranges from dilute fog/cloud droplets to highly concentrated solutions in particle water. Exposure to elevated levels of oxidants can promote the formation of highly oxygenated SOA (Daumit et al., 2016; Kang et al., 2011; Lambe et al., 2015; Ng et al., 2010) but can also decrease SOA mass and facilitate a shift from the functionalization-dominant regime to the fragmentation-dominant regime (Lambe et al., 2012).
This study investigates the long-timescale aqueous aging of the aqSOA formed from the photooxidation of guaiacyl acetone (GA). GA is a common component of biomass burning emissions and has been widely used as a model compound to study SOA formation in BB emissions. In previous work (Arciva et al., 2022; Jiang et al., 2021; Ma et al., 2021; Misovich et al., 2021; Smith et al., 2016), we examined the kinetics and mechanisms of aqSOA formation from the photoreactions of GA. Here, we extend this research to investigate the aqueous-phase photoreactions of GA with •OH and 3C∗ and propose reaction pathways of GA with •OH and 3C∗. The focus of our investigation is to study the impact of prolonged aqueous aging on the chemical composition and optical properties of the aqSOA. Specifically, we examine the effects of •OH and 3C∗ reactions induced by simulated sunlight for up to 72 and 14 h, respectively, which correspond to approximately 21 and 4 d of winter solstice sunlight exposure in northern California (George et al., 2015). Furthermore, we examine the effects of light and additional oxidant exposure on the aging of the aqSOA.
2.1 Formation and aging of phenolic aqSOA
The initial reaction solution was prepared with 100 µM of guaiacyl acetone and either 100 µM of hydrogen peroxide (H2O2; as a source of •OH) or 5 µM of 3,4-dimethoxybenzaldehyde (3,4-DMB; as a source of 3C∗) in Milli-Q water. The pH of the solution was adjusted to 4.6 using sulfuric acid. These conditions were set to mimic wood-burning-influenced cloud and fog waters (Jiang et al., 2021). The reaction solution was placed in a 400 mL Pyrex tube, continuously stirred, and illuminated inside a RPR-200 photoreactor system equipped with three different types of bulbs to roughly mimic sunlight (George et al., 2015). The steady-state concentration of •OH ([•OH]) is M in the •OH-mediated reaction, similar to the values observed in fog water (Kaur and Anastasio, 2017), and the [3C∗] is M in the 3C∗-mediated reaction, about 2 times higher than in fog water (Kaur and Anastasio, 2018) (see Sect. 3.1 for more details). When ∼95 % of the initial GA has reacted (i.e., after 24 h of irradiation for the •OH reaction and 3.5 h for the 3C∗ reaction), the solution was separated into four aliquots and moved into separate 110 mL Pyrex tubes for further aging. This aging occurred under four different conditions: (1) aging in the dark (tube wrapped with aluminum foil), (2) continued illumination without the addition of extra oxidant, (3) photoaging with the addition of 100 µM of H2O2, and (4) photoaging with the addition of 5 µM of 3,4-DMB. Small aliquots of the solutions were then periodically taken from each tube to measure the chemical composition and optical properties.
During the photoreaction, the solutions were continuously stirred. The Pyrex tubes were capped but not hermetically sealed, and the caps were briefly removed during sample collection. Due to the presence of oxygen in the reaction system, secondary reactive oxygen species (ROS) such as singlet oxygen (1O2*), superoxide/hydroperoxyl radicals (O2•−/HO2•), and •OH can be generated in the solution via energy transfer from 3C∗ to dissolved O2 (Vione et al., 2014; Zepp et al., 1977), electron transfer from the intramolecular charge-transfer complex of DMB (DMB••−) to O2 (Dalrymple et al., 2010; Li et al., 2022b), and the reactions between DMB ketyl radical and O2 (Anastasio et al., 1997). However, according to our previous studies (Smith et al., 2014), singlet oxygen is expected to contribute only minimally to the oxidation of GA in this reaction system. In addition, the negligible loss of GA and DMB in the dark controls suggests there was negligible evaporation of the precursor or the photosensitizer during the experiments.
2.2 Chemical and optical analyses
The concentrations of GA and 3,4-DMB were determined by a high-performance liquid chromatograph equipped with a diode array detector (HPLC-DAD, Agilent Technologies Inc.). A ZORBAX Eclipse XDB-C18 column (150×4.6 mm, 5 µm) was used with a mobile phase consisting of a mixture of acetonitrile and water (20 : 80), and the flow rate was 0.7 mL min−1. Both GA and DMB were detected at 280 nm, and their retention times were 8.349 and 15.396 min, respectively. The mass concentration and chemical composition of the aqSOA products were characterized using a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS; Aerodyne Res. Inc). The liquid samples were atomized in argon (Ar, industrial grade, 99.997 %), followed by diffusion drying (Jiang et al., 2021). This process allowed volatile and semivolatile products to evaporate, leaving only the low-volatility products in the particle phase, which were characterized by AMS.
HR-AMS data were processed using standard toolkits (SQUIRREL v1.56D and PIKA 1.15D). Since Ar was used as the carrier gas, the CO+ signal of aqSOA was quantified directly (Yu et al., 2016). While the organic H2O+ signal (org-H2O+) can also be directly determined for dry aerosols, it tends to be noisy due to high sulfate H2O+ (SO4-H2O+) signal interference. Therefore, org-H2O+ was parameterized as org-H2O × CO, based on the linear regression between the determined org-H2O+ signal (measured H2O SO4-H2O+) and the measured organic CO signal (Jiang et al., 2021). The other org-H2O+ related signals were parameterized as org-OH 0.25 × org-H2O+ and org-O 0.04× org-H2O+ (Aiken et al., 2008). Atomic ratios of oxygen to carbon () and hydrogen to carbon (), and organic mass-to-carbon () ratios, were subsequently determined (Aiken et al., 2008), and the average oxidation state of carbon (OSC) of aqSOA was calculated as OS - (Kroll et al., 2011). The aqSOA concentration in the solution ([Org]solution, µg mL−1) was calculated using sulfate as the internal standard:
where [Org]AMS and [Sulfate]AMS are the AMS-measured concentrations (µg m−3) of aqSOA and sulfate in the aerosolized solution, and [Sulfate]solution is the spiked concentration (µg mL−1) of sulfate in the solution. The aqSOA mass yield (YSOA) after a given time of illumination (t) was calculated as
where [GA]0 is the initial GA concentration (µg mL−1) in the solution, and [Org]t and [GA]t denote the concentrations of the aqSOA and GA, respectively, in the solution after a period of irradiation.
The light absorbance of the reaction solution was measured using a UV–Vis spectrophotometer (UV-2501PC, Shimadzu). The mass absorption coefficient, the absorption Ångström exponent, and the rate of sunlight absorption of the aqSOA were calculated (Sect. S1 in the Supplement).
Positive matrix factorization (PMF) was performed on the combined matrix of the high-resolution mass spectra ( 12–360) and the UV–Vis spectra (280–600 nm) of the •OH-aqSOA and 3C∗-aqSOA separately (Jiang et al., 2023). The PMF results were evaluated using the PMF Evaluation Toolkit (PET v3.08 downloaded from http://cires1.colorado.edu/jimenez-group/wiki/index.php/PMF-AMS_Analysis_Guide, last access: May, 2023). A three-factor solution with fPeak = 0 was chosen based on the evaluation criteria (Ulbrich et al., 2009; Zhang et al., 2011) for •OH- and 3C∗-aqSOA. Figures S1 and S2 summarize the diagnostic plots for the three-factor PMF solutions for •OH- and 3C∗-aqSOA, respectively.
3.1 Formation and characteristics of the aqSOA from photooxidation of GA by •OH and 3C∗
Figure 1a and h demonstrate that the loss of GA follows first-order kinetics in both •OH- and 3C∗-mediated photoreactions. The pseudo-first-order rate constants were determined to be 0.14 and 0.73 h−1, respectively, under our experimental conditions. Based on a previous study (Smith et al., 2016), direct photolysis of GA is expected to be negligible in this study. The fact that the reaction of GA with 3C∗ is much faster than with •OH is consistent with previously reported kinetics for other phenols (Smith et al., 2014; Yu et al., 2016) and can be attributed to the higher oxidant concentration in the 3C∗-mediated reaction. Based on the second-order rate constants for GA reacting with •OH (1.5×1010 M−1 s−1) (Arciva et al., 2022) and with 3C∗ (1.8×109 M−1 s−1) (Ma et al., 2021), we estimate that the steady-state concentrations of oxidants are [•OH] M under the •OH-mediated reaction condition and [3C∗] M under the 3C∗-mediated reaction condition. These oxidant concentrations are comparable to previously observed values in fog waters ([•OH] M and [3C∗] M) (Kaur and Anastasio, 2017, 2018) and in water extracts of ambient particles ([•OH] M and [3C∗] M) (Kaur et al., 2019).
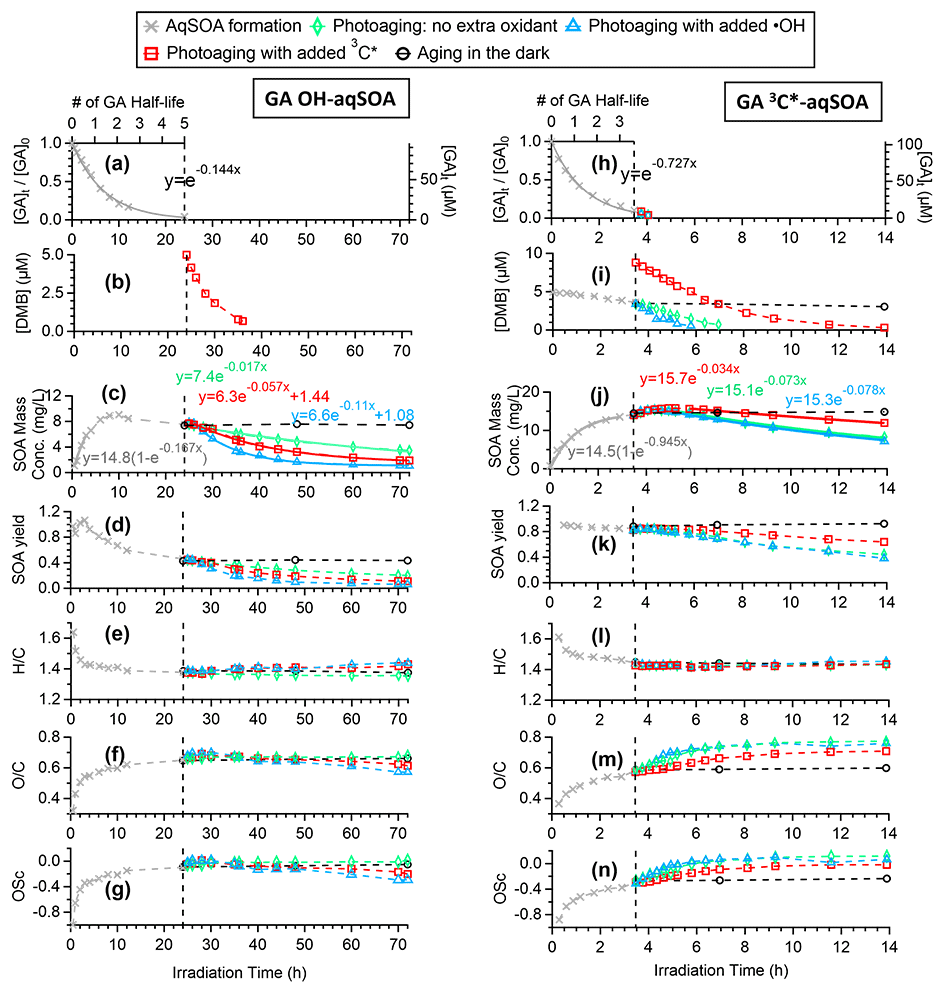
Figure 1Overview of aqSOA formation and aging in •OH- and 3C∗-initiated photoreactions of GA. Decay of (a, h) GA and (b, i) 3,4-DMB in the solution. Trends of aqSOA (c, j) mass concentration and (d, k) mass yield and (e, l) , (f, m) , and (g, n) OSC determined by HR-ToF-AMS. These measured values are also shown in Tables S1 and S2.
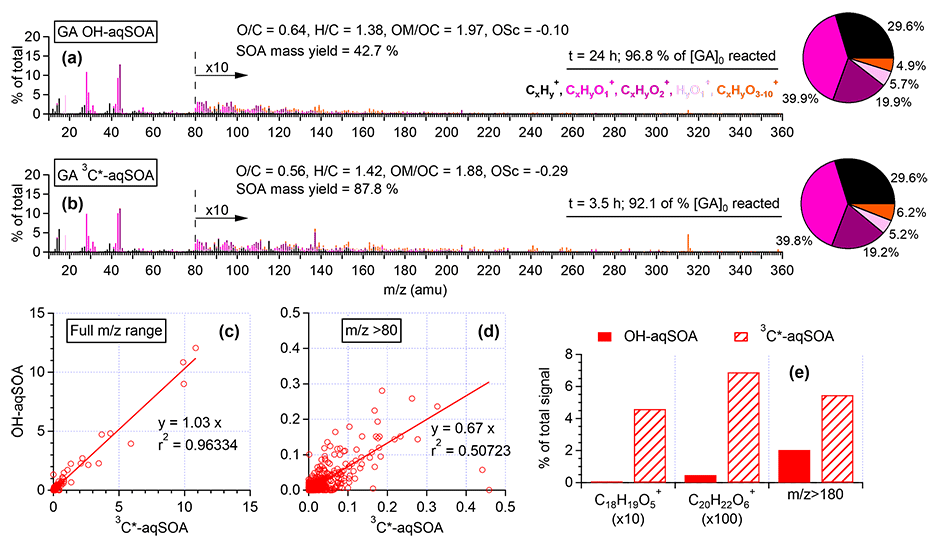
Figure 2HR-AMS mass spectra of (a) •OH-aqSOA and (b) 3C∗-aqSOA after nearly all the initial GA has reacted. Scatter plots that compare the mass spectra of the •OH-aqSOA with 3C∗-aqSOA for (c) all ions and for (d) ions with . (e) Relative abundances of the GA-oligomer tracer ions and high mass ions () in the HR-AMS spectra of the aqSOA.
As GA is transformed, the mass of the aqSOA increases (Fig. 1c and j). For both •OH- and 3C∗-mediated reactions, the aqSOA formation rate relative to the GA decay rate is similar initially, giving a relatively constant mass yield of ∼ 90 % until one GA half-life (, which is 4.8 h for the •OH reaction and 0.95 h for the 3C∗ reaction). However, in the •OH reaction, the formation of aqSOA slows down after t, resulting in a reduction in SOA yield to as low as 46 % when ∼95 % of the initial GA has reacted (Fig. 1d). In contrast, in the 3C∗ reaction, the aqSOA yield stabilizes in the range of 85 %–90 % until GA has been completely consumed (Fig. 1d and k). These results suggest that the aqSOA reacts with •OH to produce volatile products, which leads to mass loss and slower mass growth. In addition, the results suggest that the photodegradation of •OH-aqSOA of GA has a higher tendency than 3C∗-aqSOA to form volatile and semivolatile compounds that evaporate from the condensed phase. This finding is confirmed by prolonged photoaging experiments, which are presented in Sect. 3.3 and 3.4.
The chemical composition of GA aqSOA changes continuously during photoreaction. In both the •OH and 3C∗ reactions, the , , and OSC of the aqSOA increase, while slightly decreases until all the GA has been consumed (Fig. 1e–g and l–n). The HR-AMS spectra of the aqSOA (Fig. 2a and b) show that when ∼95 % of the initial GA has reacted (at 24 h for the •OH reaction and 3.5 h for the 3C∗ reaction), the •OH-aqSOA ( = 0.64 and OS) is more oxidized than the 3C∗-aqSOA ( = 0.56 and OS). In addition, compared with the •OH-aqSOA, the 3C∗-aqSOA spectrum shows a significantly greater abundance of high ions (Fig. 2c–e), including the marker ions of GA oligomers (e.g., C18H19O and C20H22O at 315 and 358, respectively) (Jiang et al., 2021), suggesting a higher production of oligomers with 3C∗. This observation aligns with the trend we observed previously in the aqueous-phase oxidations of phenol and methoxyphenols, where more oligomerization occurred in photoreactions initiated by 3C∗, while •OH reactions promoted the breakdown of aromatic rings and formation of smaller organic acids (Sun et al., 2010; Yu et al., 2014).
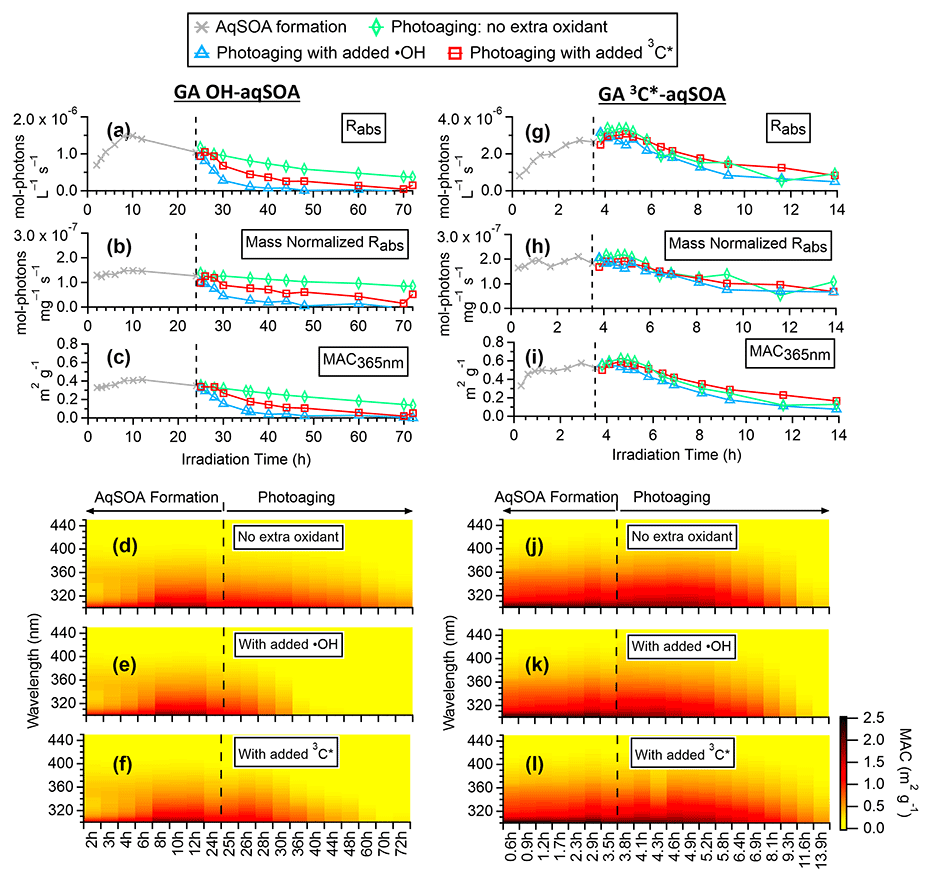
Figure 3Evolution of the optical properties of •OH-aqSOA and 3C∗-aqSOA during the course of the photoreactions: (a, g) rate of sunlight absorption, (b, h) rate of sunlight absorption normalized by aqSOA mass, (c, i) mass absorption coefficient (MAC) at 365 nm, and (d–f, j–l) MAC spectra in the wavelength range of 300–450 nm.
The compositional differences between the •OH-aqSOA and 3C∗-aqSOA can be attributed to their different reaction mechanisms (as discussed in Sect. 3.2). In the •OH experiment, the reaction can start either by •OH addition to the aromatic ring to generate OH adducts or by H-atom abstraction from the hydroxyl group to generate a phenoxy radical. The subsequent coupling of phenoxy radicals leads to the formation of oligomers (Kobayashi and Higashimura, 2003). According to previous studies on phenol oxidation in the gas phase (Atkinson, 1986; Olariu et al., 2002), it has been observed that at room temperature only ∼10 % of the phenol +•OH reaction involves H-atom abstraction that leads to the formation of phenoxy radical, whereas ∼90 % of the •OH reaction proceeds through OH addition. Moreover, modeling studies have indicated that in both gas-phase and aqueous-phase •OH oxidation of phenols, the OH addition pathways exhibit considerably lower activation energy than the H-abstraction pathway (Kılıç et al., 2007). As a result, it is highly likely that the primary products of the •OH reaction with phenols are hydroxyphenols. On the other hand, the 3C∗ reaction primarily proceeds through electron transfer and/or H-atom abstraction, which produces a phenoxy radical (Anastasio et al., 1997; Canonica et al., 2000; Yu et al., 2014). The more pronounced production of phenoxy radicals in the 3C∗ reaction can lead to more prominent oligomerization. Although the reactions of phenols with 3C∗ also produce •OH radical, the amount generated is relatively small (Anastasio et al., 1997; Smith et al., 2014) and the •OH addition pathway in the 3C∗ reaction is expected to be less important than in the •OH reaction.
Figure 3d and j (and Figs. S3a and S4a) show the mass absorption coefficient spectra of the •OH-aqSOA and the 3C∗-aqSOA. Both aqSOAs are more light absorbing than the parent GA (Fig. S5), which is likely due to the formation of GA oligomers and functionalized products containing conjugated structures. Phenolic dimers and higher oligomers formed through the coupling of phenoxyl radicals and monomeric phenol derivatives formed through •OH and carbonyl addition to the aromatic ring are effective light absorbers (Jiang et al., 2021; Misovich et al., 2021; Yu et al., 2014). In addition, the 3C∗-aqSOA exhibits greater light absorption than the •OH-aqSOA for a similar extent of GA decay, reflecting the fact that the 3C∗-aqSOA is generally enriched with more high-molecular-weight conjugated species.
3.2 Aqueous-phase reaction pathways of guaiacyl acetone
Drawing from the results of this study, our previous research (Jiang et al., 2021), and existing literature, we present in Schemes 1 and 2 proposed chemical mechanisms for the aqueous-phase reactions of GA with •OH and 3C∗. As a phenolic carbonyl, GA has two reactive sites, i.e., the phenol functional group and the carbonyl functional group. Scheme 1 outlines the main reaction pathways triggered by the phenol functional group of GA. In •OH-mediated reactions, •OH can either abstract a H atom from the hydroxyl group to form a phenoxyl radical or add to the aromatic ring to form a dihydroxycyclohexadienyl radical (Olariu et al., 2002). The phenoxyl radical can couple to produce dimers and higher oligomers (Sun et al., 2010) or react with HO2• to produce quinonic and hydroxylated products (D'Alessandro et al., 2000). The dihydroxycyclohexadienyl radical reacts with O2 to form a peroxyl radical, which can subsequently eliminate a HO2• to produce hydroxylated products (Barzaghi and Herrmann, 2002). Furthermore, the peroxyl radical can undergo further O2 addition and cyclization to generate a bicyclic peroxyl radical, leading to the cleavage of the aromatic ring via C–C and O–O bond scission and producing fragmented products such as small carboxylic acids, aldehydes, and ketones (Dong et al., 2021; Suh et al., 2003).
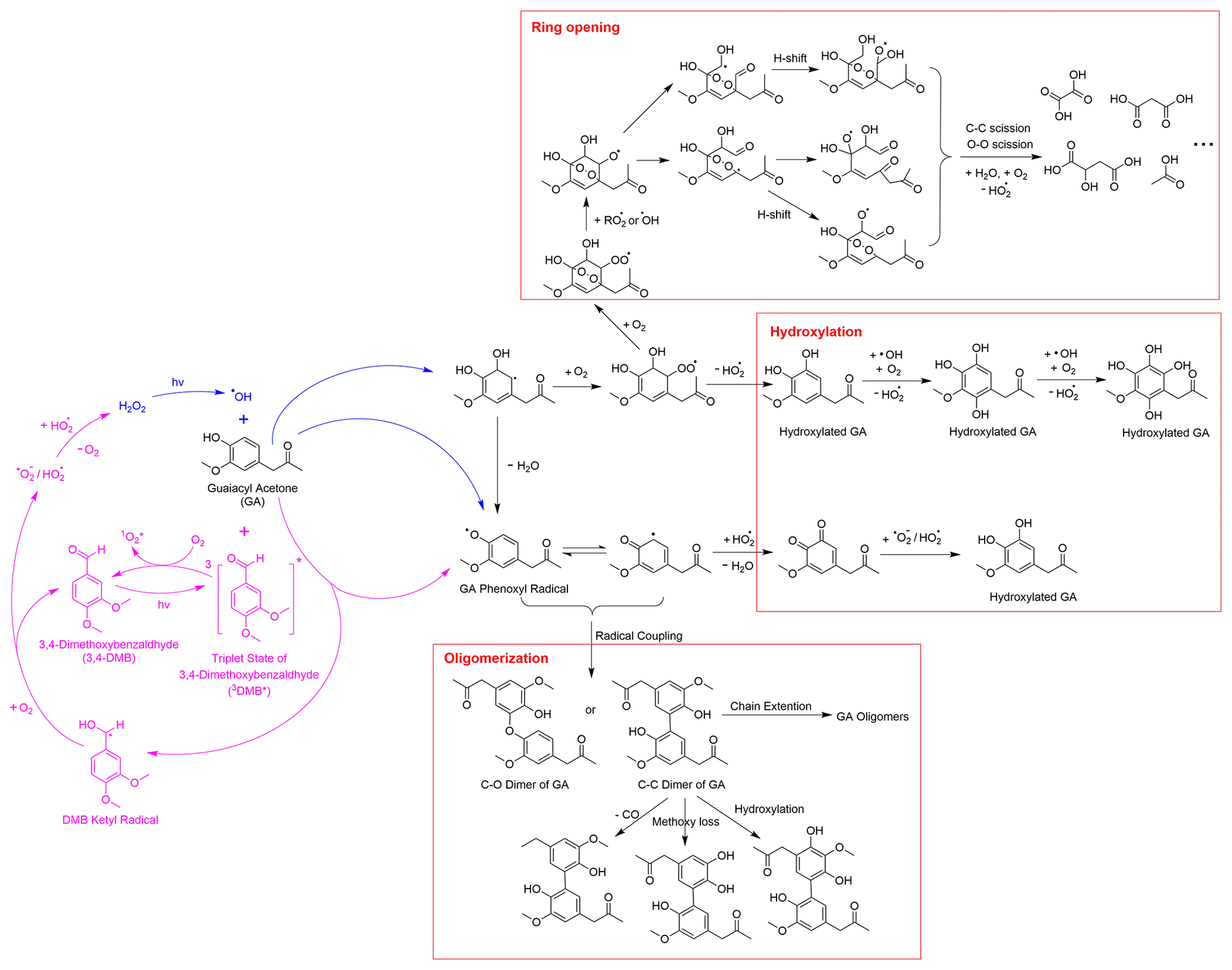
Scheme 1Postulated aqueous-phase reaction pathways triggered by the phenol functional group of GA.
In 3C∗-mediated reactions, 3C∗ can oxidize GA via H-atom abstraction and/or electron transfer to form a phenoxyl radial and a ketyl radical (Anastasio et al., 1997; Smith et al., 2014; Yu et al., 2014). The ketyl radical can react with O2 to produce superoxide/hydroperoxyl radicals (O2• −/HO2•), which subsequently react to produce H2O2 (Anastasio et al., 1997). The photolysis of H2O2 can serve as a source of •OH in the aqueous phase. Additionally, energy transfer from 3C∗ to ground state O2 can lead to the formation of 1O2* (Anastasio and McGregor, 2001; McNeill and Canonica, 2016; Zepp et al., 1977). However, according to our previous studies (Smith et al., 2014; Yu et al., 2014), the amount of •OH and 1O2* generated in the reaction of phenols with 3C∗ is small, and they are expected to be minor oxidants compared to 3C∗.
Scheme 2 outlines the reaction pathways that can be triggered by the carbonyl functional group of GA. The α position of the ketone group of GA can undergo H-atom abstraction by •OH or 3C∗, which generates alkyl radicals (Talukdar et al., 2003; Wagner and Park, 2017). The alkyl radicals can react with O2 to produce peroxyl radicals, which can further react to form dicarbonyls (Kamath et al., 2018). These dicarbonyls can then undergo photo-dissociation or hydration in the aqueous phase, forming diols and tetrols, which can further react to produce oligomers and functionalized products (Lim et al., 2013; Parandaman et al., 2018; Tan et al., 2010; Zhang et al., 2022). In our previous study (Jiang et al., 2021), we observed that the majority of the GA aqSOA products were formed through reactions triggered by the phenol functional group. The importance of the reactions initiated by the carbonyl functional group may need to be evaluated in future work.
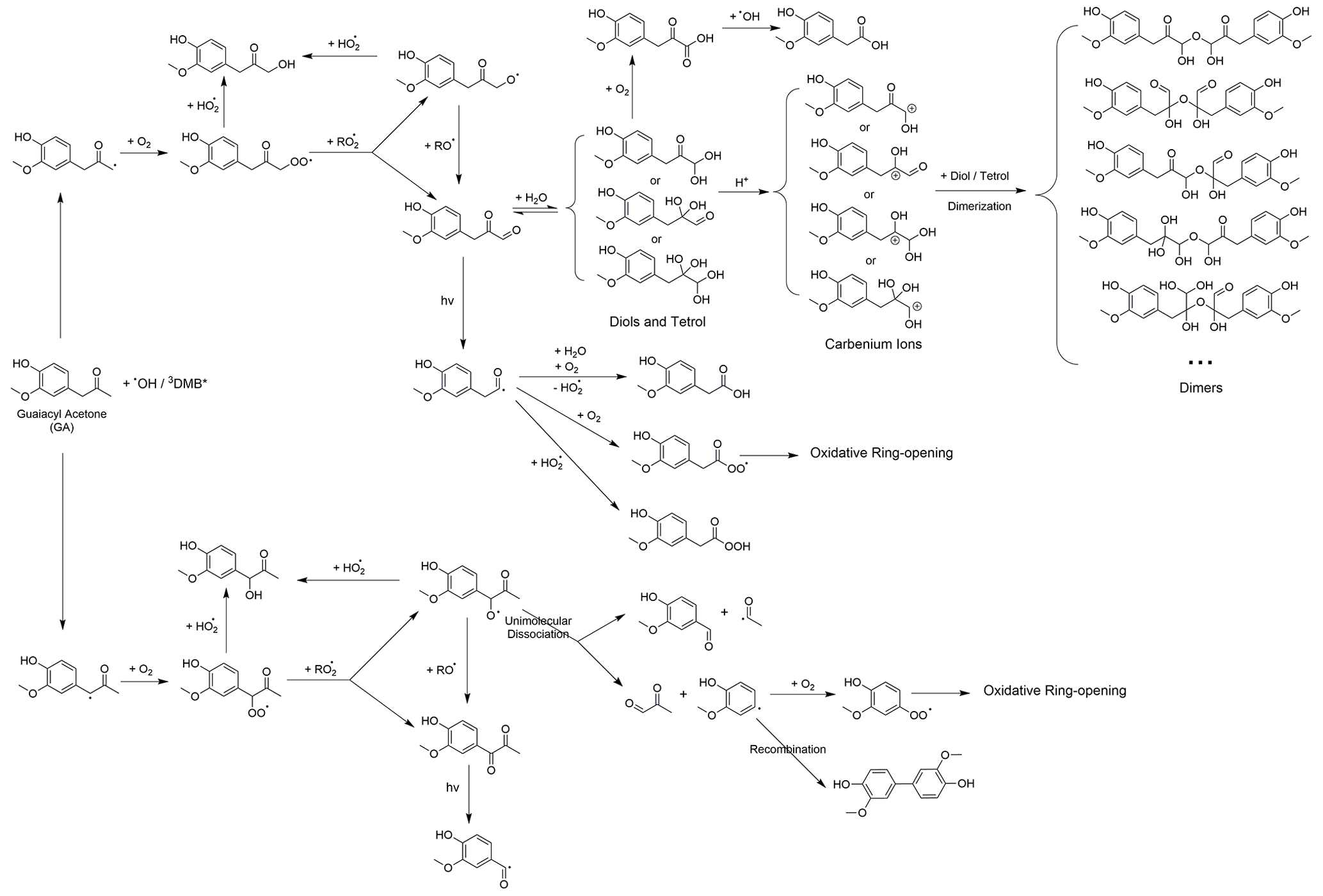
Scheme 2Postulated aqueous-phase reaction pathways triggered by the ketone functional group of GA.
As shown in the proposed reaction pathways, dissolved O2 plays an important role in the aqueous-phase reactions of GA and can influence the reactions in several ways. Firstly, the presence of O2 is essential for the formation of peroxyl radical, which serves as a crucial intermediate in hydroxylation and ring-opening pathways. Therefore, high O2 concentration in the aqueous phase can lead to enhanced hydroxylation and fragmentation, while suppressing oligomer formation from phenoxyl radical (Dong et al., 2021). Additionally, in 3C∗-mediated reactions, the involvement of O2 can generate secondary ROS (e.g., 1O2*, O2•−/HO2•, and •OH) via energy transfer from 3C∗ to ground-state O2 (Zepp et al., 1977), electron transfer from DMB••− to O2 (Dalrymple et al., 2010), and reactions between DMB ketyl radical and O2 (Anastasio et al., 1997). These secondary ROS can act as potential oxidants for GA. For instance, 1O2* reacts with phenols mainly through the 1,4-cycloaddition route to produce quinonic products (Al-Nu'airat et al., 2019; García, 1994), whereas •OH and O2•−/HO2• are important contributors to the hydroxylation and ring cleavage of phenols. Therefore, the presence of O2 is expected to facilitate functionalization and ring-opening pathways while inhibiting oligomerization in 3C∗-initiated reactions.
3.3 Photo-transformation of aqSOA and influence of prolonged photoaging on SOA yield and composition
After ∼95 % of the initial GA has reacted, the aqSOA was subjected to additional aging under different conditions: (1) aging in the dark, (2) continued illumination without the addition of extra oxidant, and (3) continued illumination with the addition of an oxidant (•OH or 3C∗). As shown in Figs. 1c–g, 1j–n, and S6, the mass concentration, elemental ratios, and HR-AMS spectra of the aqSOA remain unchanged during dark aging, indicating negligible dark chemical reactions. In contrast, continued exposure to simulated sunlight results in a 46 % reduction in the mass of 3C∗-aqSOA over about 10.5 h of prolonged aging (i.e., 14 h of irradiation in total). More than 60 % of the •OH-aqSOA mass is degraded after 48 h of extended photoaging (i.e., 72 h of irradiation in total). These observations indicate that phenolic aqSOA is susceptible to photodegradation and that fragmentation reactions and evaporation of volatile products likely play important roles in the photoaging process.
The fitted pseudo-first-order decay rate constant (k) is 0.073 h−1 for the 3C∗-aqSOA and 0.017 h−1 for the •OH-aqSOA (Figs. 1c, 1j, and 4). The faster decay of the 3C∗-aqSOA is likely due to the higher oxidant concentration in the 3C∗ reaction during aqSOA aging. Here, we assume that the steady-state concentrations of •OH and 3C∗ at the onset of the prolonged photoaging are approximately the same as in the initial solutions, and thus the [3C∗ ] in the 3C∗ reaction is about 40 times higher than the [•OH] in the •OH reaction during aqSOA aging. This assumption is proved by the first-order decay behavior of GA and the relatively stable 3,4-DMB concentration during the aqSOA formation period. Additionally, •OH production from 3C∗ becomes increasingly important during the prolonged photoaging (Anastasio et al., 1997), which may lead to an increased oxidant concentration in the 3C∗ solution. Another possible reason for the faster decay of the 3C∗-aqSOA compared to the •OH-aqSOA may be related to its higher light absorptivity, which can contribute to faster direct photodegradation. However, it is important to note that the rate of photodegradation is also dependent on the quantum yield of photodegradation (i.e., the ratio of the number of compounds destroyed to the number of photons absorbed) (Smith et al., 2016).
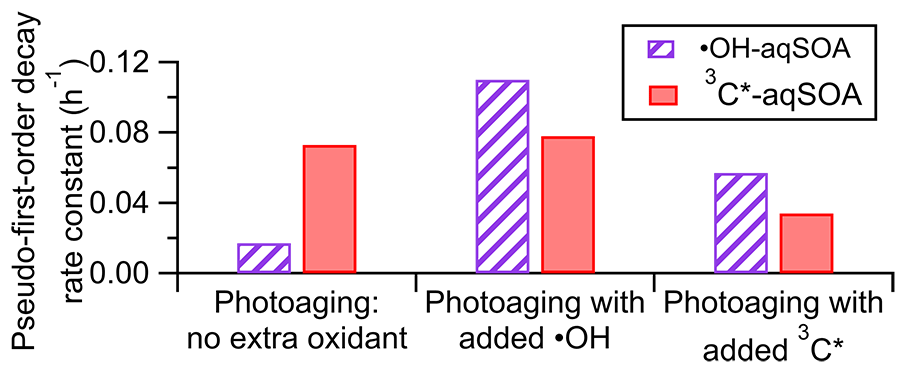
Figure 4Pseudo-first-order decay rate constants for loss of mass of •OH-aqSOA and 3C∗-aqSOA under different photoaging conditions.
As depicted in Figs. 1l–n and S7, the chemical composition of 3C∗-aqSOA evolves continuously during photoaging, with the ratio increasing from 0.59 to 0.77, consistent with previous research demonstrating that SOA becomes more oxidized during chemical aging (Kroll et al., 2015; Yu et al., 2016). In contrast, the and ratios of •OH-aqSOA exhibit negligible changes ( and = 1.36±0.008) during prolonged photoaging (Figs. 1e–g and S7), even though the mass of aqSOA decreases significantly. This can be explained by the simultaneous evaporation of highly oxidized volatile compounds and the transformation of less oxidized species into more oxidized, low-volatility products, thereby maintaining relatively constant bulk elemental ratios. Additionally, as shown in Figs. 5, S8, and S9, both •OH-aqSOA and 3C∗-aqSOA show increasing (mass fraction of CHO in the total organic signal, a tracer of carboxylic acids) and decreasing (mass fraction of C2H3O+, a tracer of non-acid carbonyls) during prolonged photoaging, indicating the importance of acid formation in the aqSOA. Furthermore, the continuous increase of indicates a more pronounced production of carboxylic acids from the prolonged aging of 3C∗-aqSOA compared to the •OH-aqSOA. However, acid formation is comparably important during •OH-aqSOA and 3C∗-aqSOA formation initially.
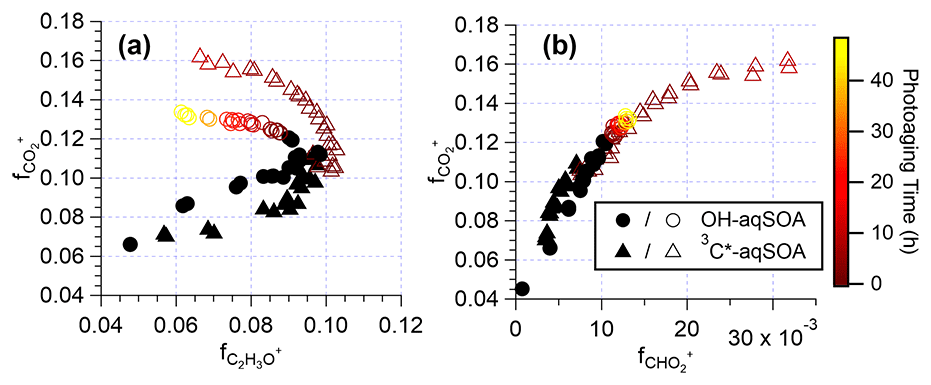
Figure 5The plots of vs. and vs. that illustrate the evolution of the •OH-aqSOA and 3C∗-aqSOA during the formation and the prolonged photoaging periods under the condition of without extra oxidant addition. The solid black markers represent the period of aqSOA formation, while the colored markers represent prolonged aqSOA aging (i.e., after ∼ 95 % of the initial GA is consumed).
To further elucidate the chemical evolution of the aqSOA, we performed PMF analysis on the combined AMS and UV–Vis absorption spectral data and successfully resolved three distinct factors for both •OH-aqSOA and 3C∗-aqSOA, each with different temporal profiles, mass spectra, and absorption spectra that represent different generations of aqSOA products (Figs. 6 and 7). The formation and decay rate constants of different generations of the aqSOA products were determined by performing exponential fits ( +c and , respectively) to the time trends of the aqSOA factors (Figs. 6d, f, h and 7d, f, h). The fitted parameter b (in the unit of h−1) represents the first-order rate constant for the aqSOA factor formation or decay in the photoreactor.
The first-generation 3C∗-aqSOA, which is the least oxidized ( = 0.49 and = 1.48), shows enhanced ion signals corresponding to GA oligomers, such as C18H19O and C20H22O (Figs. 7a, j, k and S11e–f). These products grow rapidly and peak within the first hour of 3C∗-aqSOA formation, but they subsequently decrease and disappear completely when GA is consumed. The second-generation factor ( = 0.59 and = 1.42), in which the oligomer tracer ions are substantially reduced, shows enhanced ion signals corresponding to functionalized GA monomers or ring-opening dimers such as C9H7O and C15H11O (Figs. 7b, j, k, and S11d). The second-generation products build up gradually, peak after GA is consumed, and degrade more slowly than the first-generation 3C∗-aqSOA (k=0.36 h−1 vs 1.8 h−1) during prolonged aging (Fig. 7d).
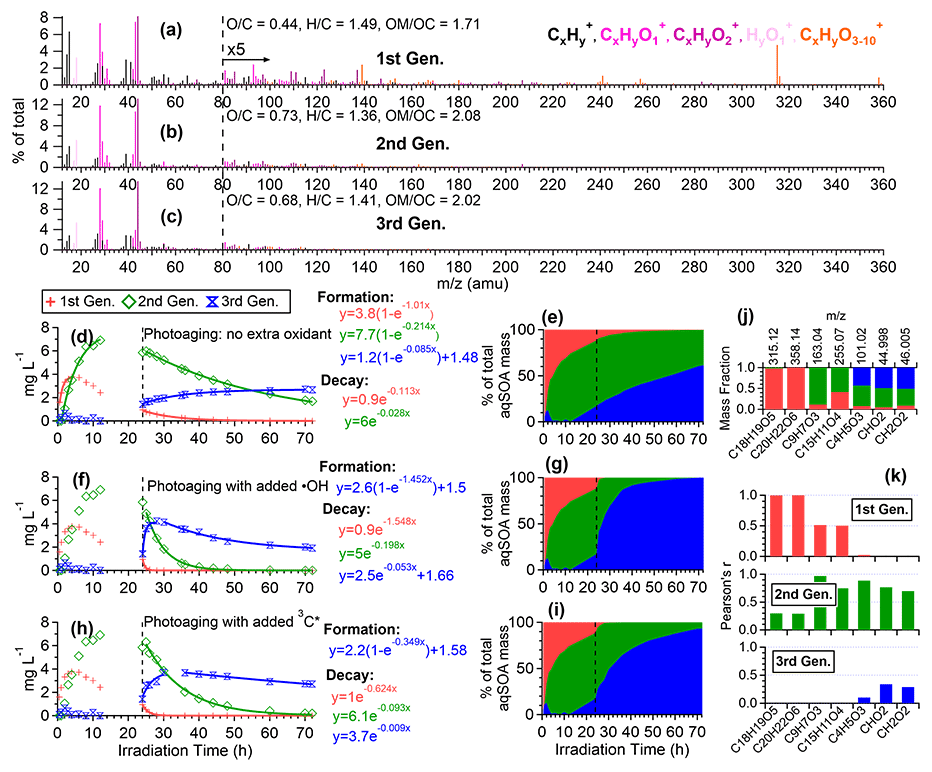
Figure 6Characteristics of the three generations of the •OH-aqSOA products resolved by PMF: (a–c) MS profiles, (d, f, and h) mass concentration time series, and (e, g, and i) fractional contribution time series of the PMF factors. (j) Mass fraction of selected AMS tracer ions attributed to each PMF factor. (k) Correlation between PMF factors and selected AMS tracer ions.
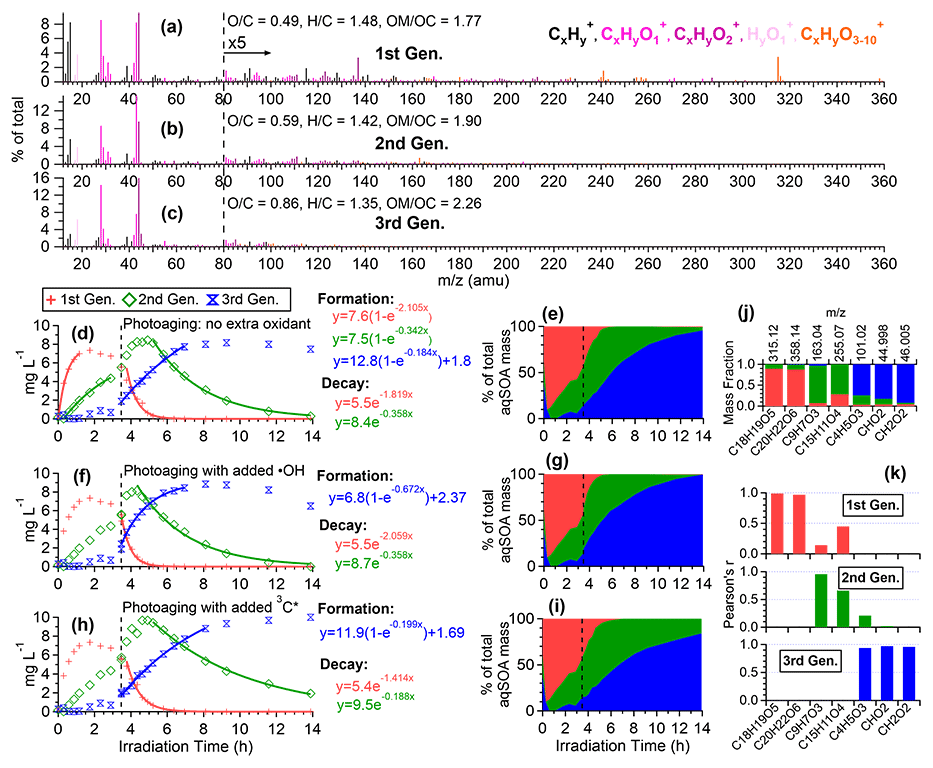
Figure 7Characteristics of the three generations of the 3C∗-aqSOA products resolved by PMF: (a–c) MS profiles, (d, f, and h) mass concentration time series, and (e, g, and i) fractional contribution time series of the PMF factors. (j) Mass fraction of selected AMS tracer ions attributed to each PMF factor. (k) Correlation between PMF factors and selected AMS tracer ions.
The third-generation factor of 3C∗-aqSOA is the most oxidized ( = 0.86 and = 1.36), and its mass spectrum shows negligible high signals but elevated small, oxygenated ions such as CHO, CH2O, and C4H5O (Figs. 7c, j, k and S11a–c). The increase of this factor is observed when the first-generation factor starts to decline. It continues to increase but shows a slight decrease towards the end of prolonged photoaging. These findings agree with our previous studies, which demonstrate that oligomerization and functionalization play a more significant role in the initial formation of phenolic aqSOA, while fragmentation and ring-opening reactions to produce more oxidized compounds become more important later (Jiang et al., 2021; Yu et al., 2016). Further, the observed decay of the third-generation aqSOA indicates that prolonged aging leads to the formation of volatile compounds that evaporate from the condensed phase, resulting in mass loss of aqSOA. This implies that photochemical aging can remove aqSOA from the atmosphere, in addition to wet and dry deposition (Hodzic et al., 2016).
The mass spectral features of the •OH-aqSOA factors (Fig. 6a–c) are generally similar to those of the 3C∗-aqSOA factors (Fig. 7a–c). However, in •OH-aqSOA, we observed that the second generation ( = 0.73 and = 1.36) is the most oxidized factor and shows strong correlations not only with the tracer ions representing functionalized GA monomers (e.g., C9H7O and C15H11O but also with a group of small, oxygenated ions (e.g., CHO, CH2O, and C4H5O that are enriched in the third-generation 3C*-aqSOA. One possible reason for the observed difference is that •OH reaction tends to form highly oxidized products that degrade over long aging times, whereas the 3C∗ reaction can generate highly oxidized SOA products that are more resistant to degradation. Another possible explanation for the observed difference in the evolution of 3C∗-aqSOA and •OH-aqSOA is that the highly oxidized species in the aqSOA exhibit different reactivity with 3C∗ and •OH due to their electron availability (Walling and Gibian, 1965). In general, 3C∗ is known to be less reactive with electron-poor compounds, whereas •OH can rapidly react with a wide range of organic compounds in the aerosol at diffusion-controlled rates (Herrmann et al., 2010). As a result, electron-poor products may persist in 3C∗-aqSOA, while •OH has the capability to further oxidize these products, eventually transforming them into volatile species. This explanation is supported by the more significant decay of the third-generation 3C∗-aqSOA, when extra H2O2 is added during prolonged photoaging (Fig. 7d and f). In addition, compared to 3C∗-aqSOA, •OH-aqSOA exhibits much lower production of first-generation products but higher second generation (Fig. 7d–e vs. Fig. 6d–e), suggesting that oligomerization is more pronounced in 3C∗-aqSOA, while functionalization plays a more important role in •OH-aqSOA.
3.4 Evolution of aqSOA optical properties during prolonged aging
Figures 3, S3, and S4 illustrate the evolution of the light absorption properties of the aqSOA during formation and aging. The aqSOA experiences photobleaching during prolonged aging, with the MAC365 nm value of the •OH-aqSOA decreasing from 0.41 (the maximum) to 0.14 m2 g−1 and that of 3C∗-aqSOA decreasing from 0.62 (the maximum) to 0.13 m2 g−1. The rates of sunlight absorption, both normalized and un-normalized by aqSOA mass, also decrease during prolonged aging. Figure 8 displays the absorption Ångström exponent (AAE) of the aqSOA as a function of log10 (MAC405) and an optical-based classification of BrC (Saleh, 2020; Zhai et al., 2022). As a result of prolonged photoaging, the GA aqSOA shifts from being classified as weak BrC to very weak BrC. The changes in the light absorption properties of the GA aqSOA are also influenced by elevated oxidant concentrations (see Sect. 3.4 for further discussions).
Figure 9 presents the mass absorption coefficient spectra resolved by PMF for the three generations of GA aqSOA resulting from •OH and 3C∗ reactions. In general, the 3C∗-aqSOA factors are more light absorbing than the •OH-aqSOA factors, which is consistent with the higher abundance of oligomers and conjugated high molecular weight products in the 3C∗-aqSOA. The first-generation aqSOA factor exhibits a hump in the MAC spectra between 340 and 400 nm, a feature observed previously in phenolic aqSOA (Smith et al., 2016) and attributed to the high conjugation present in oligomeric products. For both •OH- and 3C∗-mediated reactions, the intermediate, second-generation aqSOA are the most light absorbing compared to the fresher (i.e., first generation) and more aged (i.e., third generation) aqSOA. Nevertheless, the second-generation •OH-aqSOA shows relatively lower MAC values (MAC365 nm=0.47 m2 g−1) than the second-generation 3C∗-aqSOA (MAC365 nm=0.89 m2 g−1). This difference could be attributed to the more pronounced oxidative ring-opening reactions that cause the destruction of conjugation in •OH-aqSOA, resulting in the breakdown of chromophores. The third-generation aqSOA factors are the least absorbing (MAC365 nm=0.070 m2 g−1 for the •OH-aqSOA and 0.018 m2 g−1 for the 3C∗-aqSOA), consistent with the dominance of fragmented and ring-opening products in prolonged aging.
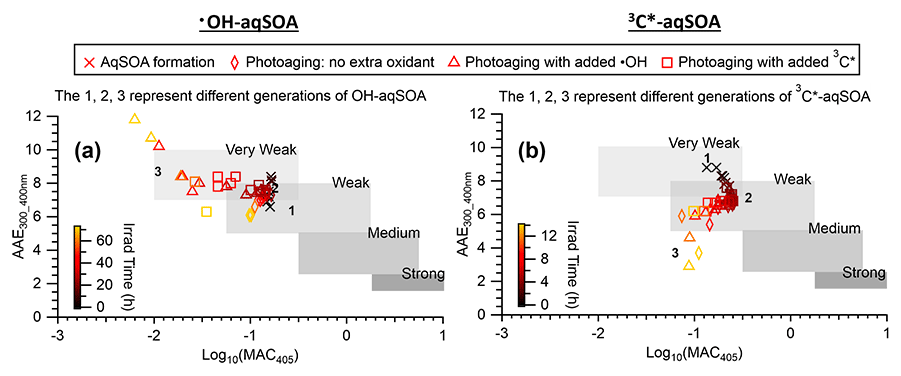
Figure 8The light absorption properties of (a) •OH-aqSOA and (b) 3C∗-aqSOA as shown in the AAE vs. log10(MAC405) space. The shaded areas in each plot represent very weakly, weakly, moderately, and strongly absorbing BrC denoted based on the optical-based BrC classification scheme (Saleh, 2020; Zhai et al., 2022). The numbers 1, 2, and 3 represent the different generations of the •OH-aqSOA and the 3C∗-aqSOA products obtained from PMF.
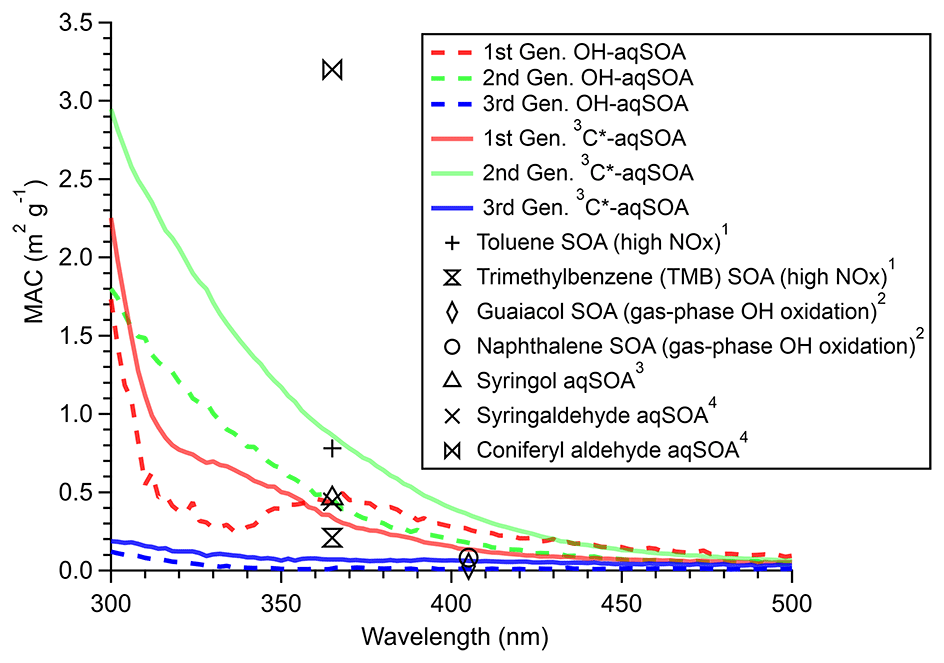
Figure 9Mass absorption coefficient spectra of the PMF resolved three generations of the OH-aqSOA and the 3C∗-aqSOA, comparing with previously reported MAC values of SOA produced from aromatic precursors by 1 Liu et al. (2016), 2 Lambe et al. (2013),3 Yu et al. (2014), and 4 Smith et al. (2016).
3.5 Effects of additional oxidant exposure on aqSOA aging
To investigate the effect of the concentrations of condensed-phase oxidants on the photoaging of phenolic aqSOA, we added either 100 µM H2O2 or 5 µM 3,4-DMB into the solution after the majority (∼95 %) of GA had reacted. Since GA decay follows first-order kinetics, we assumed that the steady-state concentration of oxidants remained constant during initial aqSOA formation. By introducing additional H2O2 or 3,4-DMB, we increased the •OH or 3C∗ concentration, as well as the overall oxidant concentration in the solution during the photoaging of the aqSOA.
As shown in Figs. 1c and 4 and Table S3, compared to continued photoaging without addition of extra oxidant (k=0.017 h−1), the photodegradation rates of •OH-aqSOA are substantially faster when extra •OH or 3C∗ are introduced (k=0.11 and 0.057 h−1, respectively). Likewise, the addition of extra •OH or 3C∗ results in more extensive mass loss of the •OH-aqSOA with reductions of 88 % or 79 % of the aqSOA mass observed at the end of the photoaging, respectively. These levels of mass loss were significantly higher than without extra oxidant (i.e., 62 %). These findings suggest that the presence of additional •OH or 3C∗ accelerates the photochemical aging process and leads to increased formation of volatile and semivolatile products that subsequently evaporate. As shown in Fig. 6d, f, and h, the decay of the first- and second-generation •OH-aqSOA is increased, and concurrently, the formation of the third-generation factor is accelerated when extra oxidants are introduced, suggesting a faster transformation from the first to second to third generation. In addition, in the later stage of photoaging, we observed a more significant decay of the third-generation •OH-aqSOA when extra oxidants are added, which suggests that higher concentrations of oxidants also facilitate the ultimate breakdown of the third-generation •OH-aqSOA. The and the average oxidation state of carbon (OSc) of •OH-aqSOA exhibit a slightly faster increase upon the addition of extra •OH or 3C∗ but eventually decrease more significantly by the end of the photoaging (Fig. 1f and g), indicating accelerated formation of highly oxidized species and enhanced production of volatile compounds under high oxidant concentrations. Furthermore, increased oxidant concentrations also have significant impacts on the photobleaching of •OH-aqSOA. Specifically, the MAC values of the aqSOA decrease faster when extra oxidants are added (Fig. 3c–f). It is noteworthy that the addition of 100 µM of H2O2, the source of •OH, has a greater impact on the degradation of •OH-aqSOA mass and light absorption than the addition of 5 µM of 3,4-DMB, the source of 3C∗, despite the fact that the GA reaction with 3C∗ is much faster than that with •OH in this study. This result may reflect the reactivity differences between GA and the aqSOA towards the oxidants, i.e., while both oxidants react quickly with GA, for the electron-poor aqSOA products, 3C∗ may be much less reactive, whereas •OH can oxidize them rapidly. Another possible interpretation is that the addition of 3,4-DMB into the solution during aqSOA aging produces unique low-volatility, light-absorbing products, which cannot be generated in •OH reactions. These products may counteract some of the mass and absorption loss due to fragmentation and evaporation.
Elevated oxidant concentrations also affect the photoaging of 3C∗-aqSOA. Unlike •OH-aqSOA, the addition of •OH during aging only slightly accelerates the decrease of mass and light absorption of the 3C∗-aqSOA (Figs. 1j–k and 3g–l). One possible explanation is that the added •OH only accounts for a small fraction of the total oxidant amount in the 3C∗-initiated reaction system and thus shows little impact on the aqSOA aging compared to the preexisting oxidants (e.g., 3C∗ and •OH generated from 3C∗). Another possible explanation is that the added •OH reacts with 3,4-DMB in the solution, resulting in a decrease in the amount of 3C∗ source. Additionally, the reaction of •OH with 3,4-DMB may also generate low-volatility products that balance out the increased decay of the aqSOA. This interpretation is consistent with the fast decay of 3,4-DMB after the addition of extra •OH (Fig. 1i). However, when extra 3C∗ (i.e., 3,4-DMB) is added to the 3C∗-aqSOA solution during extended aging, it slows down the decay of 3C∗-aqSOA mass and light absorption due to the enhanced formation of second-generation products (Fig. 7h vs. d). This suggests that an increase in 3C∗ concentration during aging promotes the formation of low-volatility functionalized products.
This study investigates the evolution of the composition and optical properties of phenolic aqSOA during prolonged photoaging, including the effects of increased oxidant concentrations. The aqSOA was generated by reacting GA with •OH or 3C∗ under relatively dilute cloud/fog water conditions. Compared to the •OH reaction, the 3C∗ reaction is significantly faster and leads to higher mass yields of aqSOA, more oligomers, and increased high-molecular-weight species when the same fraction of initial GA has reacted. On the other hand, the •OH reaction generates aqSOA that is more oxidized and more enriched in small, highly oxygenated species. Consistent with their compositional differences, the 3C∗-aqSOA is more light absorbing than the •OH-aqSOA.
The chemical composition of the aqSOA evolves during photoaging, with oligomerization and functionalization being the dominant mechanisms during initial aqSOA formation, while fragmentation and volatile product formation become more important during prolonged aging. This leads to a loss of 62 %–88 % of the •OH-aqSOA mass after 48 h of prolonged aging under simulated sunlight with or without added oxidants, while the 3C∗-aqSOA experienced a loss of 25 %–54 % of its mass after 10.5 h of extended photoaging. These results indicate that aqueous-phase photochemical aging can significantly reduce atmospheric aqSOA, in addition to wet and dry deposition. In this study, the rate of loss for phenolic aqSOA during photochemical aging was found to be in the range of 0.017–0.11 h−1 (i.e., 5– s−1). The photochemical kinetics in our RPR-200 photoreactor system were ∼7 times faster than those experienced under ambient winter solstice sunlight in northern California (George et al., 2015). Consequently, these observations indicate a photochemical lifetime of 3–17 d for phenolic aqSOA in ambient conditions. The deposition loss rate constant of submicron particles in the atmosphere, assuming wet deposition is the dominant process, is approximately s−1 (resulting in a lifetime of approximately 5 d) (Henry and Donahue, 2012; Molina et al., 2004). These findings suggest that the contribution of photochemical aging to the removal of phenolic aqSOA can be comparable to that of wet deposition.
The average oxidation state of the 3C∗-aqSOA increases continuously during photoaging, while that of •OH-aqSOA exhibits a slight decrease towards the end of photoaging when additional oxidants are introduced; this is likely due to the formation and evaporation of highly oxidized volatile products. This finding indicates that photoaging does not necessarily increase the average oxidation state of condensed-phase organics, as the evaporation of highly oxidized products may decrease the average of aqSOA. As photoaging continues, photobleaching becomes more pronounced, causing the aqSOA to shift from weakly absorbing BrC to very weak BrC. We also observed through PMF analysis that the second-generation aqSOA, enriched in functionalized phenolic compounds, is the most light absorbing. This suggests that intermediately aged phenolic aqSOA is more light absorbing than other generations, and that the light absorptivity of phenolic aqSOA is the result of a competition between BrC formation and photobleaching. Elevated oxidant concentrations during photoaging promote fragmentation reactions over oligomerization and functionalization reactions and can ultimately promote the breakdown and evaporation of the aqSOA products, resulting in a faster decline in aqSOA mass and light absorption.
The HR-AMS data analysis toolkits (SQUIRREL and PIKA) can be downloaded from https://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/ (University of Colorado, 2023). The PMF Evaluation Tool (PET v3.08) can be downloaded from http://cires1.colorado.edu/jimenez-group/wiki/index.php/PMF-AMS_Analysis_Guide (MediaWiki, 2023).
The data used in this paper can be made available upon request to the corresponding author (Qi Zhang, dkwzhang@ucdavis.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-7103-2023-supplement.
QZ and WJ developed the research goals and designed the experiments. WJ and CN performed the experiments. WJ and QZ analyzed the data and prepared the paper with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research was supported by grants from the National Science Foundation (NSF) (grant no. AGS-2220307), the U.S. Department of Energy (DOE) Atmospheric System Research program (grant no. DESC0022140), and the California Agricultural Experiment Station (projects CA-D-ETX-2102-H and CA-D-LAW-6403-RR). Wenqing Jiang also acknowledges funding from the Fumio Matsumura Memorial Fellowship by the Department of Environmental Toxicology at the University of California at Davis.
This research has been supported by the National Science Foundation (grant no. AGS-2220307), the U.S. Department of Energy (grant no. DESC0022140), and the California Agricultural Experiment Station (projects CA-D-ETX-2102-H and 15 CA-D-LAW-6403-RR).
This paper was edited by Barbara Ervens and reviewed by two anonymous referees.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Al-Nu'airat, J., Dlugogorski, B. Z., Gao, X., Zeinali, N., Skut, J., Westmoreland, P. R., Oluwoye, I., and Altarawneh, M.: Reaction of phenol with singlet oxygen, Phys. Chem. Chem. Phys., 21, 171–183, https://doi.org/10.1039/C8CP04852E, 2019.
Anastasio, C. and McGregor, K. G.: Chemistry of fog waters in California's Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen, Atmos. Environ., 35, 1079–1089, https://doi.org/10.1016/S1352-2310(00)00281-8, 2001.
Anastasio, C., Faust, B. C., and Rao, C. J.: Aromatic Carbonyl Compounds as Aqueous-Phase Photochemical Sources of Hydrogen Peroxide in Acidic Sulfate Aerosols, Fogs, and Clouds. 1. Non-Phenolic Methoxybenzaldehydes and Methoxyacetophenones with Reductants (Phenols), Environ. Sci. Technol., 31, 218–232, https://doi.org/10.1021/es960359g, 1997.
Arciva, S., Niedek, C., Mavis, C., Yoon, M., Sanchez, M. E., Zhang, Q., and Anastasio, C.: Aqueous • OH Oxidation of Highly Substituted Phenols as a Source of Secondary Organic Aerosol, Environ. Sci. Technol., 56, 9959–9967, https://doi.org/10.1021/acs.est.2c02225, 2022.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions, Chem. Rev., 86, 69–201, https://doi.org/10.1021/cr00071a004, 1986.
Barzaghi, P. and Herrmann, H.: A mechanistic study of the oxidation of phenol by OH/NO2/NO3 in aqueous solution, Phys. Chem. Chem. Phys., 4, 3669–3675, https://doi.org/10.1039/B201652D, 2002.
Berndt, T. and Böge, O.: Formation of phenol and carbonyls from the atmospheric reaction of OH radicals with benzene, Phys. Chem. Chem. Phys., 8, 1205–1214, https://doi.org/10.1039/B514148F, 2006.
Brege, M., Paglione, M., Gilardoni, S., Decesari, S., Facchini, M. C., and Mazzoleni, L. R.: Molecular insights on aging and aqueous-phase processing from ambient biomass burning emissions-influenced Po Valley fog and aerosol, Atmos. Chem. Phys., 18, 13197–13214, https://doi.org/10.5194/acp-18-13197-2018, 2018.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Canonica, S., Hellrung, B., and Wirz, J.: Oxidation of Phenols by Triplet Aromatic Ketones in Aqueous Solution, J. Phys. Chem. A, 104, 1226–1232, https://doi.org/10.1021/jp9930550, 2000.
Chang, J. L. and Thompson, J. E.: Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds, Atmos. Environ., 44, 541–551, https://doi.org/10.1016/j.atmosenv.2009.10.042, 2010.
Che, H., Segal-Rozenhaimer, M., Zhang, L., Dang, C., Zuidema, P., Dobracki, A., Sedlacek, A. J., Coe, H., Wu, H., Taylor, J., Zhang, X., Redemann, J., and Haywood, J.: Cloud processing and weeklong ageing affect biomass burning aerosol properties over the south-eastern Atlantic, Commun. Earth Environ., 3, 182, https://doi.org/10.1038/s43247-022-00517-3, 2022.
Chen, L.-W. A., Chow, J. C., Wang, X., Cao, J., Mao, J., and Watson, J. G.: Brownness of Organic Aerosol over the United States: Evidence for Seasonal Biomass Burning and Photobleaching Effects, Environ. Sci. Technol., 55, 8561–8572, https://doi.org/10.1021/acs.est.0c08706, 2021.
Corral Arroyo, P., Bartels-Rausch, T., Alpert, P. A., Dumas, S., Perrier, S., George, C., and Ammann, M.: Particle-Phase Photosensitized Radical Production and Aerosol Aging, Environ. Sci. Technol., 52, 7680–7688, https://doi.org/10.1021/acs.est.8b00329, 2018.
D'Alessandro, N., Bianchi, G., Fang, X., Jin, F., Schuchmann, H.-P., and von Sonntag, C.: Reaction of superoxide with phenoxyl-type radicals, J. Chem. Soc. Perkin Trans., 2, 1862–1867, https://doi.org/10.1039/B003346O, 2000.
Dalrymple, R. M., Carfagno, A. K., and Sharpless, C. M.: Correlations between Dissolved Organic Matter Optical Properties and Quantum Yields of Singlet Oxygen and Hydrogen Peroxide, Environ. Sci. Technol., 44, 5824–5829, https://doi.org/10.1021/es101005u, 2010.
Daumit, K. E., Carrasquillo, A. J., Sugrue, R. A., and Kroll, J. H.: Effects of Condensed-Phase Oxidants on Secondary Organic Aerosol Formation, J. Phys. Chem. A, 120, 1386–1394, https://doi.org/10.1021/acs.jpca.5b06160, 2016.
Deng, J., Ma, H., Wang, X., Zhong, S., Zhang, Z., Zhu, J., Fan, Y., Hu, W., Wu, L., Li, X., Ren, L., Pavuluri, C. M., Pan, X., Sun, Y., Wang, Z., Kawamura, K., and Fu, P.: Measurement report: Optical properties and sources of water-soluble brown carbon in Tianjin, North China – insights from organic molecular compositions, Atmos. Chem. Phys., 22, 6449–6470, https://doi.org/10.5194/acp-22-6449-2022, 2022.
Dong, P., Chen, Z., Qin, X., and Gong, Y.: Water Significantly Changes the Ring-Cleavage Process During Aqueous Photooxidation of Toluene, Environ. Sci. Technol., 55, 16316–16325, https://doi.org/10.1021/acs.est.1c04770, 2021.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Farley, R., Bernays, N., Jaffe, D. A., Ketcherside, D., Hu, L., Zhou, S., Collier, S., and Zhang, Q.: Persistent Influence of Wildfire Emissions in the Western United States and Characteristics of Aged Biomass Burning Organic Aerosols under Clean Air Conditions, Environ. Sci. Technol., 56, 3645–3657, https://doi.org/10.1021/acs.est.1c07301, 2022.
García, N. A.: New trends in photobiology: Singlet-molecular-oxygen-mediated photodegradation of aquatic phenolic pollutants. A kinetic and mechanistic overview, J. Photochem. Photobiol. B Biol., 22, 185–196, https://doi.org/10.1016/1011-1344(93)06932-S, 1994.
Garofalo, L. A., Pothier, M. A., Levin, E. J. T., Campos, T., Kreidenweis, S. M., and Farmer, D. K.: Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Sp. Chem., 3, 1237–1247, https://doi.org/10.1021/acsearthspacechem.9b00125, 2019.
George, K. M., Ruthenburg, T. C., Smith, J., Yu, L., Zhang, Q., Anastasio, C., and Dillner, A. M.: FT-IR quantification of the carbonyl functional group in aqueous-phase secondary organic aerosol from phenols, Atmos. Environ., 100, 230–237, https://doi.org/10.1016/j.atmosenv.2014.11.011, 2015.
Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Heal, M. R., Harrison, M. A. J., and Neil Cape, J.: Aqueous-phase nitration of phenol by N2O5 and ClNO2, Atmos. Environ., 41, 3515–3520, https://doi.org/10.1016/j.atmosenv.2007.02.003, 2007.
Henry, K. M. and Donahue, N. M.: Photochemical Aging of α-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 116, 5932–5940, https://doi.org/10.1021/jp210288s, 2012.
Herrmann, H., Hoffmann, D., Schaefer, T., Bräuer, P., and Tilgner, A.: Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, Chem. Phys. Chem., 11, 3796–3822, https://doi.org/10.1002/cphc.201000533, 2010.
Herrmann, H., Schaefer, T., Tilgner, A., Styler, S. A., Weller, C., Teich, M., and Otto, T.: Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 115, 4259–4334, https://doi.org/10.1021/cr500447k, 2015.
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016.
Huang, D. D., Zhang, Q., Cheung, H. H. Y., Yu, L., Zhou, S., Anastasio, C., Smith, J. D., and Chan, C. K.: Formation and Evolution of aqSOA from Aqueous-Phase Reactions of Phenolic Carbonyls: Comparison between Ammonium Sulfate and Ammonium Nitrate Solutions, Environ. Sci. Technol., 52, 9215–9224, https://doi.org/10.1021/acs.est.8b03441, 2018.
Jiang, W., Misovich, M. V, Hettiyadura, A. P. S., Laskin, A., McFall, A. S., Anastasio, C., and Zhang, Q.: Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase – Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 55, 5199–5211, https://doi.org/10.1021/acs.est.0c07581, 2021.
Jiang, W., Ma, L., Niedek, C., Anastasio, C., and Zhang, Q.: Chemical and Light-Absorption Properties of Water-Soluble Organic Aerosols in Northern California and Photooxidant Production by Brown Carbon Components, ACS Earth Sp. Chem., 7, 1107–1119, https://doi.org/10.1021/acsearthspacechem.3c00022, 2023.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 80, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kamath, D., Mezyk, S. P., and Minakata, D.: Elucidating the Elementary Reaction Pathways and Kinetics of Hydroxyl Radical-Induced Acetone Degradation in Aqueous Phase Advanced Oxidation Processes, Environ. Sci. Technol., 52, 7763–7774, https://doi.org/10.1021/acs.est.8b00582, 2018.
Kang, E., Toohey, D. W., and Brune, W. H.: Dependence of SOA oxidation on organic aerosol mass concentration and OH exposure: experimental PAM chamber studies, Atmos. Chem. Phys., 11, 1837–1852, https://doi.org/10.5194/acp-11-1837-2011, 2011.
Kaur, R. and Anastasio, C.: Light absorption and the photoformation of hydroxyl radical and singlet oxygen in fog waters, Atmos. Environ., 164, 387–397, https://doi.org/10.1016/j.atmosenv.2017.06.006, 2017.
Kaur, R. and Anastasio, C.: First Measurements of Organic Triplet Excited States in Atmospheric Waters, Environ. Sci. Technol., 52, 5218–5226, https://doi.org/10.1021/acs.est.7b06699, 2018.
Kaur, R., Labins, J. R., Helbock, S. S., Jiang, W., Bein, K. J., Zhang, Q., and Anastasio, C.: Photooxidants from brown carbon and other chromophores in illuminated particle extracts, Atmos. Chem. Phys., 19, 6579–6594, https://doi.org/10.5194/acp-19-6579-2019, 2019.
Kim, H., Collier, S., Ge, X., Xu, J., Sun, Y., Jiang, W., Wang, Y., Herckes, P., and Zhang, Q.: Chemical processing of water-soluble species and formation of secondary organic aerosol in fogs, Atmos. Environ., 200, 158–166, https://doi.org/10.1016/j.atmosenv.2018.11.062, 2019.
Kılıç, M., Koçtürk, G., San, N., and Çınar, Z.: A model for prediction of product distributions for the reactions of phenol derivatives with hydroxyl radicals, Chemosphere, 69, 1396–1408, https://doi.org/10.1016/j.chemosphere.2007.05.002, 2007.
Kleinman, L. I., Sedlacek III, A. J., Adachi, K., Buseck, P. R., Collier, S., Dubey, M. K., Hodshire, A. L., Lewis, E., Onasch, T. B., Pierce, J. R., Shilling, J., Springston, S. R., Wang, J., Zhang, Q., Zhou, S., and Yokelson, R. J.: Rapid evolution of aerosol particles and their optical properties downwind of wildfires in the western US, Atmos. Chem. Phys., 20, 13319–13341, https://doi.org/10.5194/acp-20-13319-2020, 2020.
Kobayashi, S. and Higashimura, H.: Oxidative polymerization of phenols revisited, Prog. Polym. Sci., 28, 1015–1048, https://doi.org/10.1016/S0079-6700(03)00014-5, 2003.
Kroll, J. H., Smith, J. D., Che, D. L., Kessler, S. H., Worsnop, D. R., and Wilson, K. R.: Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol, Phys. Chem. Chem. Phys., 11, 8005–8014, https://doi.org/10.1039/B905289E, 2009.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, J. Phys. Chem. A, 119, 10767–10783, https://doi.org/10.1021/acs.jpca.5b06946, 2015.
Lambe, A. T., Onasch, T. B., Croasdale, D. R., Wright, J. P., Martin, A. T., Franklin, J. P., Massoli, P., Kroll, J. H., Canagaratna, M. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Transitions from Functionalization to Fragmentation Reactions of Laboratory Secondary Organic Aerosol (SOA) Generated from the OH Oxidation of Alkane Precursors, Environ. Sci. Technol., 46, 5430–5437, https://doi.org/10.1021/es300274t, 2012.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between Oxidation Level and Optical Properties of Secondary Organic Aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Lee, H. J. (Julie), Aiona, P. K., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 48, 10217–10226, https://doi.org/10.1021/es502515r, 2014.
Leresche, F., Salazar, J. R., Pfotenhauer, D. J., Hannigan, M. P., Majestic, B. J., and Rosario-Ortiz, F. L.: Photochemical Aging of Atmospheric Particulate Matter in the Aqueous Phase, Environ. Sci. Technol., 55, 13152–13163, https://doi.org/10.1021/acs.est.1c00978, 2021.
Li, M., Wang, X., Zhao, Y., Du, P., Li, H., Li, J., Shen, H., Liu, Z., Jiang, Y., Chen, J., Bi, Y., Zhao, Y., Xue, L., Wang, Y., Chen, J., and Wang, W.: Atmospheric Nitrated Phenolic Compounds in Particle, Gaseous, and Aqueous Phases During Cloud Events at a Mountain Site in North China: Distribution Characteristics and Aqueous-Phase Formation, J. Geophys. Res.-Atmos., 127, e2022JD037130, https://doi.org/10.1029/2022JD037130, 2022a.
Li, X., Tao, Y., Zhu, L., Ma, S., Luo, S., Zhao, Z., Sun, N., Ge, X., and Ye, Z.: Optical and chemical properties and oxidative potential of aqueous-phase products from OH and 3C∗-initiated photooxidation of eugenol, Atmos. Chem. Phys., 22, 7793–7814, https://doi.org/10.5194/acp-22-7793-2022, 2022b.
Lim, Y. B., Tan, Y., and Turpin, B. J.: Chemical insights, explicit chemistry, and yields of secondary organic aerosol from OH radical oxidation of methylglyoxal and glyoxal in the aqueous phase, Atmos. Chem. Phys., 13, 8651–8667, https://doi.org/10.5194/acp-13-8651-2013, 2013.
Liu, J., Lin, P., Laskin, A., Laskin, J., Kathmann, S. M., Wise, M., Caylor, R., Imholt, F., Selimovic, V., and Shilling, J. E.: Optical properties and aging of light-absorbing secondary organic aerosol, Atmos. Chem. Phys., 16, 12815–12827, https://doi.org/10.5194/acp-16-12815-2016, 2016.
Ma, L., Guzman, C., Niedek, C., Tran, T., Zhang, Q., and Anastasio, C.: Kinetics and Mass Yields of Aqueous Secondary Organic Aerosol from Highly Substituted Phenols Reacting with a Triplet Excited State, Environ. Sci. Technol., 55, 5772–5781, https://doi.org/10.1021/acs.est.1c00575, 2021.
Mabato, B. R. G., Lyu, Y., Ji, Y., Li, Y. J., Huang, D. D., Li, X., Nah, T., Lam, C. H., and Chan, C. K.: Aqueous secondary organic aerosol formation from the direct photosensitized oxidation of vanillin in the absence and presence of ammonium nitrate, Atmos. Chem. Phys., 22, 273–293, https://doi.org/10.5194/acp-22-273-2022, 2022.
Mabato, B. R. G., Li, Y. J., Huang, D. D., Wang, Y., and Chan, C. K.: Comparison of aqueous secondary organic aerosol (aqSOA) product distributions from guaiacol oxidation by non-phenolic and phenolic methoxybenzaldehydes as photosensitizers in the absence and presence of ammonium nitrate, Atmos. Chem. Phys., 23, 2859–2875, https://doi.org/10.5194/acp-23-2859-2023, 2023.
McNeill, K. and Canonica, S.: Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci. Process. Impacts, 18, 1381–1399, https://doi.org/10.1039/C6EM00408C, 2016.
MediaWiki: PMF-AMS Analysis Guide, MediaWiki [data set], http://cires1.colorado.edu/jimenez-group/wiki/index.php/PMF-AMS_Analysis_Guide (last access: May 2023), 2023.
Misovich, M. V, Hettiyadura, A. P. S., Jiang, W., Zhang, Q., and Laskin, A.: Molecular-Level Study of the Photo-Oxidation of Aqueous-Phase Guaiacyl Acetone in the Presence of 3C∗: Formation of Brown Carbon Products, ACS Earth Sp. Chem., 5, 1983–1996, https://doi.org/10.1021/acsearthspacechem.1c00103, 2021.
Molina, M. J., Ivanov, A. V, Trakhtenberg, S., and Molina, L. T.: Atmospheric evolution of organic aerosol, Geophys. Res. Lett., 31, 1–5, https://doi.org/10.1029/2004GL020910, 2004.
Morgan, W. T., Allan, J. D., Bower, K. N., Highwood, E. J., Liu, D., McMeeking, G. R., Northway, M. J., Williams, P. I., Krejci, R., and Coe, H.: Airborne measurements of the spatial distribution of aerosol chemical composition across Europe and evolution of the organic fraction, Atmos. Chem. Phys., 10, 4065–4083, https://doi.org/10.5194/acp-10-4065-2010, 2010.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
O'Brien, R. E. and Kroll, J. H.: Photolytic Aging of Secondary Organic Aerosol: Evidence for a Substantial Photo-Recalcitrant Fraction, J. Phys. Chem. Lett., 10, 4003–4009, https://doi.org/10.1021/acs.jpclett.9b01417, 2019.
Olariu, R. I., Klotz, B., Barnes, I., Becker, K. H., and Mocanu, R.: FT–IR study of the ring-retaining products from the reaction of OH radicals with phenol, o-, m-, and p-cresol, Atmos. Environ., 36, 3685–3697, https://doi.org/10.1016/S1352-2310(02)00202-9, 2002.
Palm, B. B., Peng, Q., Fredrickson, C. D., Lee, B. H., Garofalo, L. A., Pothier, M. A., Kreidenweis, S. M., Farmer, D. K., Pokhrel, R. P., Shen, Y., Murphy, S. M., Permar, W., Hu, L., Campos, T. L., Hall, S. R., Ullmann, K., Zhang, X., Flocke, F., Fischer, E. V., and Thornton, J. A.: Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, P. Natl. Acad. Sci. USA, 117, 29469–29477, https://doi.org/10.1073/pnas.2012218117, 2020.
Pang, H., Zhang, Q., Lu, X., Li, K., Chen, H., Chen, J., Yang, X., Ma, Y., Ma, J., and Huang, C.: Nitrite-Mediated Photooxidation of Vanillin in the Atmospheric Aqueous Phase, Environ. Sci. Technol., 53, 14253–14263, https://doi.org/10.1021/acs.est.9b03649, 2019.
Parandaman, A., Kumar, M., Francisco, J. S., and Sinha, A.: Organic Acid Formation from the Atmospheric Oxidation of Gem Diols: Reaction Mechanism, Energetics, and Rates, J. Phys. Chem. A, 122, 6266–6276, https://doi.org/10.1021/acs.jpca.8b01773, 2018.
Saleh, R.: From Measurements to Models: Toward Accurate Representation of Brown Carbon in Climate Calculations, Curr. Pollut. Reports, 6, 90–104, https://doi.org/10.1007/s40726-020-00139-3, 2020.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of Emissions from Air Pollution Sources. 3. C1-C29 Organic Compounds from Fireplace Combustion of Wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Sedlacek, A. J. I. I. I., Lewis, E. R., Onasch, T. B., Zuidema, P., Redemann, J., Jaffe, D., and Kleinman, L. I.: Using the Black Carbon Particle Mixing State to Characterize the Lifecycle of Biomass Burning Aerosols, Environ. Sci. Technol., 56, 14315–14325, https://doi.org/10.1021/acs.est.2c03851, 2022.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Smith, J. D., Sio, V., Yu, L., Zhang, Q., and Anastasio, C.: Secondary Organic Aerosol Production from Aqueous Reactions of Atmospheric Phenols with an Organic Triplet Excited State, Environ. Sci. Technol., 48, 1049–1057, https://doi.org/10.1021/es4045715, 2014.
Smith, J. D., Kinney, H., and Anastasio, C.: Aqueous benzene-diols react with an organic triplet excited state and hydroxyl radical to form secondary organic aerosol, Phys. Chem. Chem. Phys., 17, 10227–10237, https://doi.org/10.1039/c4cp06095d, 2015.
Smith, J. D., Kinney, H., and Anastasio, C.: Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products, Atmos. Environ., 126, 36–44, https://doi.org/10.1016/j.atmosenv.2015.11.035, 2016.
Stefania, G., Paola, M., Marco, P., Lara, G., Claudio, C., Matteo, R., Stefano, D., Silvia, S., Francesca, C., Paolo, G. G., Chiara, P. M., Marco, V., Fabiana, S., Sandro, F., and Cristina, F. M.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Suh, I., Zhang, R., Molina, L. T., and Molina, M. J.: Oxidation Mechanism of Aromatic Peroxy and Bicyclic Radicals from OH-Toluene Reactions, J. Am. Chem. Soc., 125, 12655–12665, https://doi.org/10.1021/ja0350280, 2003.
Sun, Y., Zhang, Q., Zheng, M., Ding, X., Edgerton, E. S., and Wang, X.: Characterization and Source Apportionment of Water-Soluble Organic Matter in Atmospheric Fine Particles (PM2.5) with High-Resolution Aerosol Mass Spectrometry and GC–MS, Environ. Sci. Technol., 45, 4854–4861, https://doi.org/10.1021/es200162h, 2011.
Sun, Y. L., Zhang, Q., Anastasio, C., and Sun, J.: Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 10, 4809–4822, https://doi.org/10.5194/acp-10-4809-2010, 2010.
Talukdar, R. K., Gierczak, T., McCabe, D. C., and Ravishankara, A. R.: Reaction of Hydroxyl Radical with Acetone. 2. Products and Reaction Mechanism, J. Phys. Chem. A, 107, 5021–5032, https://doi.org/10.1021/jp0273023, 2003.
Tan, Y., Carlton, A. G., Seitzinger, S. P., and Turpin, B. J.: SOA from methylglyoxal in clouds and wet aerosols: Measurement and prediction of key products, Atmos. Environ., 44, 5218–5226, https://doi.org/10.1016/j.atmosenv.2010.08.045, 2010.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
University of Colorado: TOF-AMS software downloads, University of Colorado [data set], https://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/ (last access: May 2023), 2023.
Vione, D., Minella, M., Maurino, V., and Minero, C.: Indirect Photochemistry in Sunlit Surface Waters: Photoinduced Production of Reactive Transient Species, Chem. – A Eur. J., 20, 10590–10606, https://doi.org/10.1002/chem.201400413, 2014.
Wagner, P. and Park, B.-S.: Photoinduced hydrogen atom abstraction by carbonyl compounds, in Organic photochemistry, 227–366, CRC Press, https://doi.org/10.1201/9780203744826, 2017.
Wagstrom, K. M. and Pandis, S. N.: Determination of the age distribution of primary and secondary aerosol species using a chemical transport model, J. Geophys. Res.-Atmos., 114, D14, https://doi.org/10.1029/2009JD011784, 2009.
Walling, C. and Gibian, M. J.: Hydrogen abstraction reactions by the triplet states of Ketones1, J. Am. Chem. Soc., 87, 3361–3364, 1965.
Yang, J., Au, W. C., Law, H., Lam, C. H., and Nah, T.: Formation and evolution of brown carbon during aqueous-phase nitrate-mediated photooxidation of guaiacol and 5-nitroguaiacol, Atmos. Environ., 254, 118401, https://doi.org/10.1016/j.atmosenv.2021.118401, 2021.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Yu, L., Smith, J., Laskin, A., George, K. M., Anastasio, C., Laskin, J., Dillner, A. M., and Zhang, Q.: Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: competition among oligomerization, functionalization, and fragmentation, Atmos. Chem. Phys., 16, 4511–4527, https://doi.org/10.5194/acp-16-4511-2016, 2016.
Zepp, R. G., Wolfe, N. L., Baughman, G. L., and Hollis, R. C.: Singlet oxygen in natural waters, Nature, 267, 421–423, https://doi.org/10.1038/267421a0, 1977.
Zhai, L., An, Y., Feng, L., Qin, X., and Xu, J.: Contrasting the physical and chemical characteristics of dissolved organic matter between glacier and glacial runoff from a mountain glacier on the Tibetan Plateau, Sci. Total Environ., 848, 157784, https://doi.org/10.1016/j.scitotenv.2022.157784, 2022.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, Y., He, L., Sun, X., Ventura, O. N., and Herrmann, H.: Theoretical Investigation on the Oligomerization of Methylglyoxal and Glyoxal in Aqueous Atmospheric Aerosol Particles, ACS Earth Sp. Chem., 6, 1031–1043, https://doi.org/10.1021/acsearthspacechem.1c00422, 2022.
Zhou, S., Collier, S., Jaffe, D. A., Briggs, N. L., Hee, J., Sedlacek III, A. J., Kleinman, L., Onasch, T. B., and Zhang, Q.: Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 17, 2477–2493, https://doi.org/10.5194/acp-17-2477-2017, 2017.
Zhou, S., Collier, S., Jaffe, D. A., and Zhang, Q.: Free tropospheric aerosols at the Mt. Bachelor Observatory: more oxidized and higher sulfate content compared to boundary layer aerosols, Atmos. Chem. Phys., 19, 1571–1585, https://doi.org/10.5194/acp-19-1571-2019, 2019.