the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 15 Sep 2023
| 15 Sep 2023
OH, HO2, and RO2 radical chemistry in a rural forest environment: measurements, model comparisons, and evidence of a missing radical sink
Michelle M. Lew
Youngjun Woo
Pamela Rickly
Matthew D. Rollings
Benjamin Deming
Daniel C. Anderson
Ezra Wood
Hariprasad D. Alwe
Dylan B. Millet
Andrew Weinheimer
Geoff Tyndall
John Ortega
Sebastien Dusanter
Thierry Leonardis
James Flynn
Matt Erickson
Sergio Alvarez
Jean C. Rivera-Rios
Joshua D. Shutter
Frank Keutsch
Detlev Helmig
Hannah M. Allen
Johnathan H. Slade
Paul B. Shepson
Steven Bertman
The hydroxyl (OH), hydroperoxy (HO2), and organic peroxy (RO2) radicals play important roles in atmospheric chemistry. In the presence of nitrogen oxides (NOx), reactions between OH and volatile organic compounds (VOCs) can initiate a radical propagation cycle that leads to the production of ozone and secondary organic aerosols. Previous measurements of these radicals under low-NOx conditions in forested environments characterized by emissions of biogenic VOCs, including isoprene and monoterpenes, have shown discrepancies with modeled concentrations.
During the summer of 2016, OH, HO2, and RO2 radical concentrations were measured as part of the Program for Research on Oxidants: Photochemistry, Emissions, and Transport – Atmospheric Measurements of Oxidants in Summer (PROPHET-AMOS) campaign in a midlatitude deciduous broadleaf forest. Measurements of OH and HO2 were made by laser-induced fluorescence–fluorescence assay by gas expansion (LIF-FAGE) techniques, and total peroxy radical (XO2) mixing ratios were measured by the Ethane CHemical AMPlifier (ECHAMP) instrument. Supporting measurements of photolysis frequencies, VOCs, NOx, O3, and meteorological data were used to constrain a zero-dimensional box model utilizing either the Regional Atmospheric Chemical Mechanism (RACM2) or the Master Chemical Mechanism (MCM). Model simulations tested the influence of HOx regeneration reactions within the isoprene oxidation scheme from the Leuven Isoprene Mechanism (LIM1). On average, the LIM1 models overestimated daytime maximum measurements by approximately 40 % for OH, 65 % for HO2, and more than a factor of 2 for XO2. Modeled XO2 mixing ratios were also significantly higher than measured at night. Addition of RO2 + RO2 accretion reactions for terpene-derived RO2 radicals to the model can partially explain the discrepancy between measurements and modeled peroxy radical concentrations at night but cannot explain the daytime discrepancies when OH reactivity is dominated by isoprene. The models also overestimated measured concentrations of isoprene-derived hydroxyhydroperoxides (ISOPOOH) by a factor of 10 during the daytime, consistent with the model overestimation of peroxy radical concentrations. Constraining the model to the measured concentration of peroxy radicals improves the agreement with the measured ISOPOOH concentrations, suggesting that the measured radical concentrations are more consistent with the measured ISOPOOH concentrations. These results suggest that the models may be missing an important daytime radical sink and could be overestimating the rate of ozone and secondary product formation in this forest.
- Article
(5759 KB) - Full-text XML
-
Supplement
(931 KB) - BibTeX
- EndNote





As a dominant oxidant in the lower troposphere, the hydroxyl radical (OH) initiates reactions with volatile organic compounds (VOCs), leading to the production of hydroperoxy radicals (HO2) and organic peroxy radicals (RO2). In the presence of nitrogen oxides (NOx= NO + NO2), reactions of these radicals establish a fast cycle that can produce ozone and secondary organic aerosol (SOA). Given their central role in atmospheric chemistry, an accurate understanding of radical chemistry is important to address current issues of air quality and climate change. Because of their short atmospheric lifetimes, measurements of these radicals can provide a test of our understanding of this complex chemistry, including our knowledge of radical sources, sinks, and propagation pathways (Heard and Pilling, 2003).
Several field campaigns have been conducted to investigate radical concentrations in both urban and forested environments. Although measurements of OH concentrations in urban areas have been generally consistent with model predictions (Ren et al., 2003; Shirley et al., 2006; Kanaya et al., 2007a; Dusanter et al., 2009b; Lu et al., 2013; Griffith et al., 2016; Tan et al., 2017, 2018, 2019; Whalley et al., 2021), measurements of peroxy radicals in such environments have generally been underpredicted by atmospheric models (Griffith et al., 2016; Baier et al., 2017; Tan et al., 2017; Whalley et al., 2021). Measurements in forested regions characterized by low NOx mixing ratios and elevated emissions of biogenic VOCs, such as isoprene and monoterpenes, have indicated discrepancies with modeled results, with several observations of higher-than-expected OH concentrations in isoprene-rich environments (Tan et al., 2001; Lelieveld et al., 2008; Hofzumahaus et al., 2009; Whalley et al., 2011; Lu et al., 2012; Rohrer et al., 2014). However, several recent studies have revealed potential interferences with measurements of OH radicals in forested environments (Mao et al., 2012; Novelli et al., 2014b; Feiner et al., 2016; Lew et al., 2020). Accounting for these interferences resulted in measured OH concentrations that were in good agreement with model predictions in these forested areas.
In contrast, measurements of HO2 and RO2 radical concentrations in forested areas have shown variable agreement with model predictions. In these environments, measured HO2 concentrations were sometimes found to agree with model predictions (Tan et al., 2001, 2017; Ren et al., 2006; Feiner et al., 2016) but were sometimes lower (Carslaw et al., 2001; Kanaya et al., 2007b; Whalley et al., 2011; Kanaya et al., 2012; Mao et al., 2012; Griffith et al., 2013; Mallik et al., 2018) or higher than model predictions (Carslaw et al., 2001; Kubistin et al., 2010; Kim et al., 2013; Hens et al., 2014). Part of this variability may be due to measurement interferences from certain RO2 radicals in systems that detect HO2 through the conversion to OH using the HO2+ NO → OH + NO2 reaction (Fuchs et al., 2011; Whalley et al., 2013; Hens et al., 2014; Crowley et al., 2018; Lew et al., 2018). However, the extent of RO2 radical contributions to HO2 measurements in many of the earlier campaigns mentioned above is not clear. While accounting for this interference would improve agreement when the model underestimates HO2, it would worsen agreement in the case of an overestimation.
The discrepancies between measured and modeled radical concentrations in forest environments bring into question our understanding of the chemistry of biogenic VOCs (BVOCs) and their contribution to the production of ozone and SOA in the atmosphere. Isoprene is of particular importance due to its global abundance and high reactivity with the OH radical (Wennberg et al., 2018). Current models suggest that emissions of isoprene alone account for half of global non-methane VOC emissions (Guenther et al., 2012; Wennberg et al., 2018). Several theoretical and laboratory studies have investigated the atmospheric chemistry of isoprene and its oxidation products, revealing that isomerization of isoprene-based peroxy radicals and subsequent product pathways could recycle OH and HO2 radicals, resulting in higher radical concentrations under low-NOx conditions (Fuchs et al., 2013; Peeters et al., 2014; Liu et al., 2017; Wennberg et al., 2018; Medeiros et al., 2022).
In addition to isoprene, other biogenic VOCs, including monoterpenes, can play a significant role in the overall oxidative capacity of some environments. Globally, monoterpene emissions are estimated to be more than 100 Tg yr−1 and constitute as much as 10 % of BVOC emissions (Sindelarova et al., 2014). While emissions of isoprene are strongly dependent on photosynthetic photon flux as well as temperature, several plant species also emit monoterpenes under dark conditions (Harley et al., 1996; Owen et al., 2002). Similar to the chemical mechanism of isoprene oxidation, peroxy radicals produced from the oxidation of monoterpenes can undergo isomerization reactions as part of autooxidation mechanisms, leading to the production of highly oxidized peroxy radical products (Jokinen et al., 2014). Under low-NOx conditions, these reactions can compete with reaction with NO as well as with peroxy radical self- and cross-reactions.
While it is known that self- and cross-reactions of RO2 can form either alkoxy radicals (Reaction R1) or an alcohol and a carbonyl species (Reaction R2) (Orlando and Tyndall, 2012), a third pathway that leads to the formation of a dimeric dialkyl peroxide (Reaction R3) has been proposed but previously regarded as less significant due to low yields for small RO2 species (Lightfoot et al., 1992; Tyndall et al., 2001; Noell et al., 2010). However, recent studies have observed the formation of gas-phase C19–C20 dimer compounds and suggest that autoxidation and RO2+ RO2 reactions between terpene-derived peroxy radicals may form low-volatility accretion products (Reaction R3) (Crounse et al., 2013; Ehn et al., 2014; Berndt et al., 2018a, b; Bianchi et al., 2019).
In addition to significantly affecting SOA formation, these reactions could be relevant alongside reactions with NOx or HO2 as radical termination reactions and should be considered when modeling radical concentrations in low-NOx regions characterized by significant biogenic VOC emissions.
This study presents measurements of OH, HO2, and total peroxy radical (XO2= RO2+ HO2) concentrations made within a remote forested region during the PROPHET-AMOS 2016 (Program for Research on Oxidants: Photochemistry, Emissions, and Transport – Atmospheric Measurements of Oxidants in Summer) field campaign. The measurements are compared to predicted radical concentrations from zero-dimensional box models constrained to a wide range of trace gases and meteorological conditions. Additional model simulations that incorporate the Leuven Isoprene Mechanism (LIM1) for isoprene degradation and a series of RO2 + R'O2 reactions that form accretion products are accompanied by a radical budget analysis to test current atmospheric chemistry mechanisms and investigate the fate of isoprene- and monoterpene-derived peroxy radicals in this low-NOx environment.
2.1 PROPHET-AMOS measurement site
All measurements described below were performed as part of the PROPHET-AMOS 2016 field campaign. Measurements were conducted throughout the month of July at the PROPHET facility at the University of Michigan Biological Station (UMBS) in northern Michigan (45.5588∘ N, 84.7145∘ W). The mixed deciduous and coniferous forest site consists primarily of isoprene-emitting species such as big-tooth aspen and red oak but also monoterpene-emitting species such as red maple, white pine, and paper birch (Ortega et al., 2007; Bryan et al., 2015). The site has been described in more detail elsewhere (Carroll et al., 2001; Ortega et al., 2007; Griffith et al., 2013). The majority of the measurements described below were performed near the top of the 31 m tower, approximately 10 m above the forest canopy by placing the instrument directly on the top of the tower, sampling from a glass manifold in the laboratory that pulled air from the top of the tower, or sampling from individual inlets from the top of the tower. Measurements of ozone were taken from the top of the nearby Ameriflux tower, which is 100 m to the north of the PROPHET tower. Table 1 summarizes the measurements used in this study.
Table 1Measured species used for data analysis and model calculations along with respective instruments, measurement techniques, and detection limits.
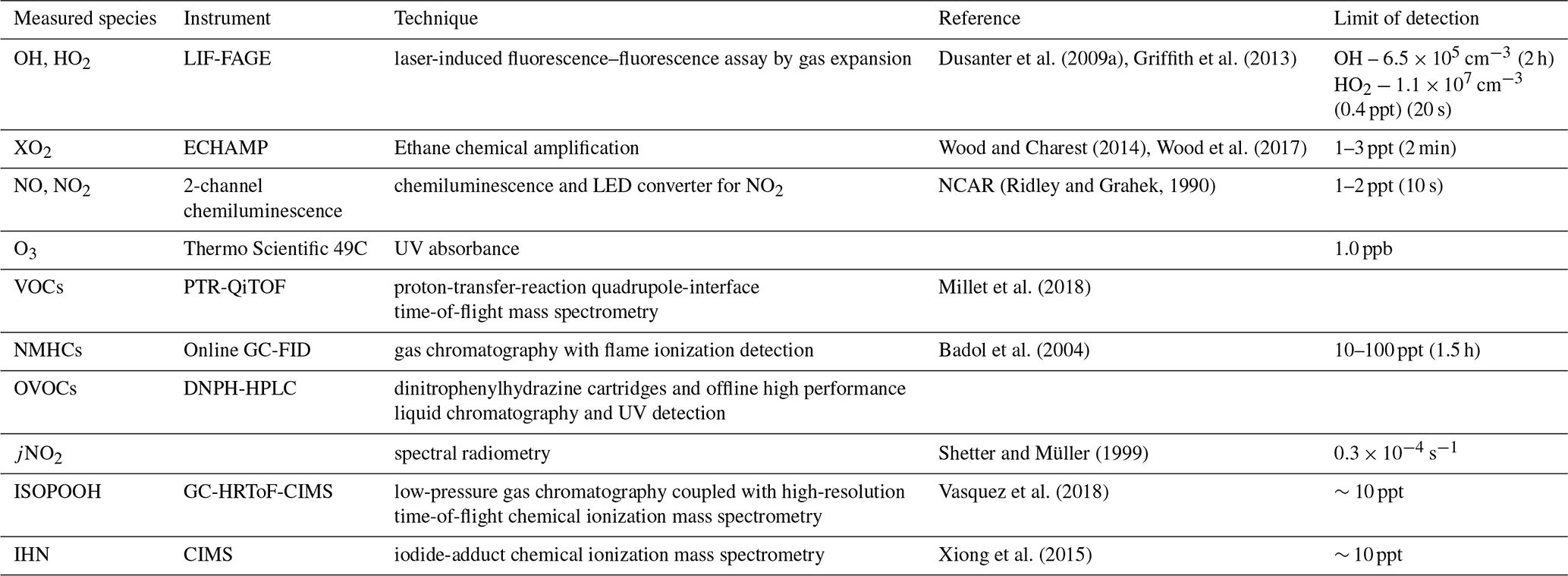
During the campaign, isoprene, the sum of methyl vinyl ketone and methacrolein, monoterpenes, acetaldehyde, and other VOCs and oxygenated VOCs (OVOCs) were measured by the University of Minnesota using proton-transfer- reaction quadrupole-interface time-of-flight mass spectrometry (PTR-QiTOF) (Millet et al., 2018). In addition, C2–C10 alkanes and alkenes, butadiene, C6–C9 aromatic compounds, and isoprene were measured by IMT Nord Europe using a thermal desorption gas chromatography with flame ionization detection (GC-FID) instrument with a 1.5 h time resolution, while C2–C10 aldehydes, C2–C6 ketones, and C2–C4 alcohols were measured by thermal desorption GC-FID with mass spectrometry (GC-FID-MS) with a 1.5 h time resolution (Badol et al., 2004; Roukos et al., 2009). NO and NO2 were measured by the NCAR single-channel chemiluminescence instrument (Ridley and Grahek, 1990), ozone was measured by UV absorption by the University of Colorado and CO by laser-based off-axis integrated cavity output spectroscopy (Los Gatos Research) by the University of Houston and Rice University groups. Isoprene hydroxy hydroperoxides (ISOPOOH) were measured using a gas chromatograph chemical ionization mass spectrometry (GC-ToF-CIMS) instrument by Caltech (Vasquez et al., 2018). Isoprene hydroxy nitrates were measured by an iodine-adduct chemical ionization mass spectrometer by Purdue University (Xiong et al., 2015). Photolysis frequencies were measured using spectral radiometry (Shetter and Müller, 1999) by the University of Houston. Measurements of OH, HO2, and XO2 radicals are described in detail below.
2.2 Measurements of HOx concentrations
Both OH and HO2 were measured using the Indiana University laser-induced fluorescence–fluorescence assay by gas expansion (IU-FAGE) instrument that has been described in more detail previously (Dusanter et al., 2009a; Griffith et al., 2013; Lew et al., 2020). Briefly, OH radicals are detected by laser excitation at 308 nm and subsequent resonant fluorescence detection. The sampled air expands into a low-pressure cell, which extends the OH fluorescence lifetime by reducing the concentration of species that may quench OH fluorescence and allows temporal filtering of OH fluorescence from more intense scattered laser light (Heard and Pilling, 2003).
The IU-FAGE laser system used in this study consisted of a Spectra Physics Navigator II YHP40-532Q that produced approximately 7.5 W of 532 nm radiation (10 kHz repetition rate) to pump a Sirah Credo dye laser (255 mg L−1 of Rhodamine 610 and 80 mg L−1 of Rhodamine 101 in ethanol), resulting in approximately 40 mW of radiation that is tunable near 308 nm. This laser system was housed in the laboratory at the bottom of the PROPHET measurement tower, and 308 nm radiation was focused onto the entrance of a 50 m optical fiber to transmit the laser emission to the sampling cell.
The IU-FAGE sampling cell was located atop the 31 m measurement tower, approximately 10 m above the forest canopy. Ambient air was drawn into the detection cell through a pinhole inlet (0.64 mm diameter) by means of three scroll pumps (Edwards XDS 35i) connected in parallel. The pumps were located at the bottom of the tower and connected to the sampling cell by two parallel 3.8 cm inner-diameter vacuum hoses, which resulted in a sampling cell pressure of 0.6 kPa (4.5 torr) and a flow of 3 SLPM through the sampling inlet.
On average, approximately 1.25 mW of 308 nm radiation exited the 50 m fiber and entered the sampling cell during the campaign. The laser emission enters the sampling cell perpendicular to the sampled air mass and intersects the expanded air in a White cell configuration with approximately 24 passes. The OH fluorescence is collected along an axis that is orthogonal to both the laser beam and sampled air mass and detected using a microchannel plate photomultiplier tube (MCP-PMT) detector (Hamamatsu R5946U), a preamplifier (Stanford Research Systems SR445), and a photon counter (Stanford Research Systems SR400). The MCP-PMT is turned off, and the photon counter is inactive during the laser pulse by means of a delay generator (Berkley Nucleonics 565) to allow the OH fluorescence to be temporally filtered from scattered laser light.
The net OH fluorescence signal is determined through successive spectral-modulation cycles in which the dye laser emission wavelength is tuned on and off resonance, with the Q1(3) transition of OH near 308 nm. A background signal, which primarily consists of scattered laser light that extends into the detection window, is established by tuning the laser emission off resonance with the OH transition and therefore not exciting OH radicals. This background signal is subtracted from the on-resonance signal. A reference cell in which OH is generated by the thermal dissociation of water vapor is used to ensure maximum overlap between dye-laser emission and the OH transition wavelength.
The IU-FAGE measurements of OH are subject to potential interferences when OH radicals are generated inside the detection cell. In the presence of water vapor, the photolysis of ozone by the laser can produce hydroxyl radicals through Reactions (R4) and (R5) (Davis et al., 1981a, b).
To characterize this and any other interference, a chemical scrubbing technique is used to remove ambient OH prior to entering the detection cell (Griffith et al., 2016; Rickly and Stevens, 2018; Lew et al., 2020). This chemical modulation technique is used to monitor levels of the laser-generated ozone-water interference and any other interference that may produce OH radicals inside the detection cell. Hexafluoropropylene (C3F6, 95.5 % in N2; Matheson Gas) was added through a circular injector 1 cm above the inlet with a flow rate of approximately 3.5 sccm to remove 95 % of external OH radicals (Rickly and Stevens, 2018). The differences between the measured OH during C3F6 addition and OH measurements including the interference represent the net ambient OH concentration in the atmosphere. The addition of C3F6 is modulated in between ambient OH measurements every 15 min for a duration of 10 min.
Measurements of HO2 were made indirectly after addition of NO to the sampled air mass to convert ambient HO2 to OH through the fast HO2 + NO → OH + NO2 reaction. A small flow (approximately 2 sccm) of NO (Matheson, 1 % in nitrogen) was added to the sampled air mass through a Teflon loop injector that was positioned directly below the sampling inlet, resulting in an added NO concentration of approximately 9×1011 cm−3. The fraction of HO2 converted into OH was measured during calibration experiments performed during and after the campaign and was 14.0±3.2 %. This low NO concentration minimized the impact of interferences from RO2 radicals derived from the OH-initiated oxidation of alkenes and aromatics that can be quickly converted to HO2 (Fuchs et al., 2011; Lew et al., 2018). The high conversion efficiencies reported for these RO2 radicals are due to the rapid decomposition of β-hydroxyalkoxy radicals that are formed from the RO2+ NO reaction. This decomposition forms a hydroxyalkyl radical that reacts rapidly with O2 to produce HO2 in the detection cell. This can lead to the detection of both HO2 and a fraction of RO2 radicals denoted as HO (HO HO2+αRO2, ). Calibrations before and after the campaign similar to those described in Lew et al. (2018) indicated that the low NO concentration injected into the detection cell (approximately 9×1011 cm−3) resulted in an RO2-to-HO2 conversion efficiency of approximately 10 % for isoprene-based peroxy radicals and an RO2-to-OH conversion efficiency of less than 2 % (Fig. S1 in the Supplement). As a result, the HO2 measurements were performed at the low NO flow that effectively minimized the impact of any potential interference from isoprene-derived RO2 species that are dominant during the day at the PROPHET site (Griffith et al., 2013).
The instrument was calibrated by producing known concentrations of OH and HO2 from the photolysis of water vapor in air as described by Dusanter et al. (2008). The calibration source consists of an aluminum flow reactor with quartz windows on two opposite sides. Aluminum cartridges adjacent to each window house a low-pressure mercury pen lamp and a photodiode detector, both of which are continuously purged with dry nitrogen to stabilize the lamp temperature and prevent light absorption by atmospheric gases. Radiation from the mercury lamp passes through a bandpass filter centered at 185 nm prior to illuminating the flow reactor and detector. The location of the mercury lamp and photodiode is adjustable along the length of the calibration source to allow for the measurement of radical surface loss between the illuminated region and the exit of the calibrator. For calibrations during PROPHET, zero air was delivered to the calibration source at a flow rate of 50 L min−1. A variable fraction of the flow (5 %–40 %) was diverted through a set of custom bubblers containing high-purity water at the base of the tower. This humidified fraction of air was mixed back with the initial flow in approximately 35 m of PTFE tubing (1.25 cm i.d.) before entering the calibration source. Calibrations were performed before, after, and intermittently during the campaign to track changes in sensitivity (Dusanter et al., 2008). The uncertainty associated with this calibration technique is approximately 18 % (1σ) for both OH and HO2.
As previously mentioned, a 50 m fiber optic cable was used to transmit laser radiation to the sampling cell for the above-canopy measurements. The long fiber presented technical challenges that impacted the performance of the IU-FAGE instrument. Due to the length of the fiber, the laser pulse was temporally broadened prior to entering the detection cell and resulted in an increase of background laser scatter of the instrument. This broadened pulse made temporal filtering of scattered laser light difficult and ultimately led to a lower sensitivity and a higher limit of detection for OH. In addition, the length of the fiber corresponded to a decrease in transmission of radiation through the fiber. An average transmission of 8 % led to 0.76–2.15 mW of 308 nm radiation in the detection cell over the course of the campaign. Due to low laser power and high background signal, long averaging times were necessary for OH measurements. The limit of detection for OH was 6.5×105 molecules cm−3 (1σ, 2 h average). Measurements of HO2 were performed approximately once per hour with a limit of detection of 1.1×107 cm−3 (0.4 ppt) (1σ, 20 s average).
2.3 Measurements of total peroxy radicals (XO2)
Total peroxy radicals were measured by the Ethane CHemical AMPlifier (ECHAMP) instrument that has been previously described in detail (Wood et al., 2017). This instrument is similar to traditional chemical amplifiers that mix ambient air with excess CO and NO (Cantrell and Stedman, 1982; Hastie et al., 1991; Cantrell et al., 1996) but instead utilizes chemical amplification by ethane (C2H6) and NO followed by detection of NO2 using cavity-attenuated phase-shift (CAPS) spectroscopy.
The ECHAMP inlet box was positioned on the top platform of the tower at a height of 31 m. Ambient air was sampled at a flow rate of 7.3 SLPM through a 0.4 cm inner diameter (ID) glass inlet that was internally coated with halocarbon wax to minimize radical loss on surfaces. A small flow (0.35 SLPM) of pure O2 was added through a side port to this main flow. The O2 addition increases the O2 mixing ratio in the sampled air to 24.6 % and reduces both the value and the variability of the relative humidity in the sampled air. The sampled air finally entered two reaction chambers at individual flow rates of 1.0 L min−1, with the remaining sampled air used to monitor temperature and RH. In the amplification chamber, the sampled air was immediately mixed with 20 sccm of 50 ppm NO and 20 sccm of 50 % C2H6 through an upstream reagent addition port, leading to final mixing ratios for NO and C2H6 of 1 ppm and 1 % respectively. A flow of 20 sccm N2 was added downstream, 100 ms later. In this chamber, ROx species are converted to HO2 and OH through reactions with NO (R6–R8). Reactions (R8)–(R12) repeat several times, leading to the formation of NO2 that is subsequently measured by a CAPS monitor.
In the background chamber, the sampled air was first mixed with NO and N2, and then C2H6 was added 100 ms later. In this mode, ambient radicals are removed by successive reactions with NO (R6–R8) until they form HONO via the OH + NO → HONO reaction, and therefore amplification chemistry does not occur. After reagent addition, air from each chamber enters identical CAPS monitors. The CAPS NO2 measurements from the background chamber represent ambient NO2, NO2 from the reaction of ambient O3 with added NO, and NO2 from reactions of ambient peroxy radicals with NO but not from ethane amplification reactions. The CAPS NO2 monitor following the amplification chamber measures the sum of that observed from the background chamber and NO2 produced from amplification chemistry. The amount of NO2 produced from amplification reactions (ΔNO2) is determined from the difference between the amplification and background chambers. The concentration of peroxy radicals is calculated by dividing [ΔNO2] by the experimentally determined amplification factor, F.
The amplification factor was determined as a function of relative humidity by producing known concentrations of peroxy radicals with two different calibration sources. The first source relies on the photolysis of water vapor method which is similar to that described above for the IU-FAGE instrument and is commonly used to calibrate other chemical amplifiers (Mihele and Hastie, 2000; Horstjann et al., 2014) and LIF-FAGE instruments (Heard and Pilling, 2003; Dusanter et al., 2008). This method produces equivalent concentrations of OH and HO2 that are quantified by O2 actinometry and measured concentrations of H2O and O3 in the calibration gas, and OH can be quantitatively converted to HO2 or isoprene peroxy radicals through the addition of H2 or isoprene, respectively. The second calibration source was based on the photolysis of methyl iodide (CH3I) at 254 nm to produce CH3O2 radicals (Anderson et al., 2019). The radical concentration is quantified by reaction with NO, in the absence of ethane, to produce NO2 that is measured by CAPS. During PROPHET the ECHAMP limit of detection was 1–3 ppt (2σ, 2 min average). Throughout the campaign, the CH3I calibration method was used as the primary source, and the water vapor photolysis method was used less frequently to quantify the relative response of ECHAMP to HO2 and CH3O2 radicals (see below).
As described in Wood et al. (2017) and Kundu et al. (2019), ECHAMP does not detect all peroxy radicals with equal sensitivity. A portion of RO2 radicals are converted to alkyl nitrites (RONO) and alkyl nitrates (RONO2) via association reactions with NO, and a portion of all sampled radicals are lost to wall reactions. Wall loss rate constants measured in the laboratory for halocarbon-coated 0.4 cm ID glass were typically 1.6 s−1 for HO2 at 60 % RH and less than 0.2 s−1 for CH3O2, with isoprene peroxy radical wall loss rate constants between those two values (Kundu et al., 2019). For the sampling conditions during PROPHET (7.3 SLPM flow rate, 13 cm inlet length), this suggests only 2 % of HO2 was lost to wall reactions. Furthermore, an expected 8 % of isoprene peroxy radicals are lost to formation of organic nitrates. The relative sensitivity of ECHAMP to HO2 radicals and CH3O2 radicals was quantified after the campaign by comparing its response to both types of radicals prepared at equal concentrations using the water vapor photolysis method. These measurements showed that the response to HO2 was 2 % lower than the instrument response to CH3O2. In the absence of sampling losses we would expect that the response to CH3O2 would be 10 % lower than the response to HO2 due to formation of CH3ONO (Wood et al., 2017). These results indicate that sampling losses of HO2 were more likely 10 % and almost equal to the loss of CH3O2 due to CH3ONO formation. Further details of a calibration source comparison between the LIF and ECHAMP instruments are provided in the Supplement.
2.4 Modeling concentrations of OH, HO2, and XO2
Ambient concentrations of OH, HO2, and XO2 were modeled with the Master Chemical Mechanism (MCM) (Jenkin et al., 1997, 2015) and the Regional Atmospheric Chemistry Mechanism version 2 (RACM2) (Goliff et al., 2013). The RACM2 mechanism groups several species according to their reactivity and includes more than 350 reactions. While the near-explicit MCM is expected to better represent the complex oxidation chemistry of this environment, the grouped RACM2 model is more computationally efficient and simpler to use in a radical budget analysis. Due to the limited isoprene oxidation mechanism in the base RACM2 model, a series of reactions described by Tan et al. (2017) was incorporated based on the LIM1 mechanism proposed by Peeters et al. (2009, 2014). The resulting condensed version of LIM1 includes updated bulk reaction rate constants for the 1,6-H shift isomerization reactions of the isoprene peroxy radicals as parameterized by Peeters et al. (2014). These isomerization reactions lead to the formation of HO2 and hydroperoxyaldehydes (HPALDs), which can photolyze leading to OH production, and dihydroperoxy-carbonyl peroxy radicals (di-HPCARPs), which can rapidly decompose to produce additional OH radicals (Teng et al., 2017; Wennberg et al., 2018).
The Master Chemical Mechanism provides a near-explicit mechanism that describes the gas-phase chemical processes involved in the degradation of over 140 VOCs. Model simulations utilized both MCM version 3.2 and MCM version 3.3.1, the latter of which incorporates the explicit LIM1 mechanism and includes the equilibrium between different isoprene peroxy radical isomers and the H-shift isomerization reactions of specific isomers resulting in HOx radical recycling through the production of HPALDs as well as di-HPCARPs (Jenkin et al., 2015). In this mechanism, the equilibrium rate coefficients between different peroxy radical isomers were increased, and the 1,6 H-shift isomerization rate constants were decreased in order to match early experimental results of Crounse et al. (2014) (Peeters, 2015). These changes resulted in effective bulk 1,6 H-shift peroxy radical isomerization rate constants in MCM v3.3.1 that are approximately a factor of 5 lower than the original LIM1 recommended rates (Novelli et al., 2020).
Each of the chemical mechanisms were embedded in the Framework for 0-D Atmospheric Modeling (F0AM) (Wolfe et al., 2016) to calculate radical concentrations predicted by each mechanism. Modeled chemistry for both mechanisms was constrained to measurements of meteorological data and a wide variety of trace gas mixing ratios that were measured during the campaign (Tables S1 and S2). Model simulations were performed with a 30 min integration time and a 5 d spin-up to allow sufficient time to generate unmeasured secondary oxidation products. A 24 h lifetime was assumed for all calculated species to simulate loss via dry deposition and to prevent unexpected accumulation of some unmeasured species. Similar to Ren et al. (2013) and Lu et al. (2012), model sensitivity runs indicate that increasing this depositional loss by a factor of 2 results in changes of the modeled HO2 concentration of less than 4 %. Measurement constraints were synchronized to 30 min time intervals. Species that were measured more frequently were averaged to 30 min intervals, and linear interpolation was used for species measured with lower time resolution.
In cases when speciated measurements or complete measurement sets were not available for species important to radical chemistry, an appropriate correlation analysis or an average of previous measurements conducted at the PROPHET location was used to constrain the model. For example, measurements of the sum of methyl vinyl ketone and methacrolein (MVK + MACR) were available throughout the campaign from the University of Minnesota's PTR-QiTOF instrument, but speciated MACR was measured on some days by the IMT Nord Europe online GC-FID. An MVK:MACR ratio of 0.65:0.35 was derived from a correlation of the available measurements and used to constrain the model when speciated measurements were not available. Similarly, as the sum of monoterpenes was measured by PTR-QiTOF, the mixing ratio of total monoterpenes was constrained as α-pinene in model simulations. Because measurements of HONO concentrations at the top of the tower were unavailable, the model was constrained to the campaign average of previous measurements at this site (Griffith et al., 2013). Photolysis frequencies were calculated using a trigonometric parameterization based on solar zenith angle (Jenkin et al., 1997; Wolfe et al., 2016) and scaled according to measured values of J(NO2) to account for cloud coverage. The uncertainty of modeled radical concentrations is estimated to be 30 % based on uncertainties from model constraint inputs and the measured rate constants for each reaction (Griffith et al., 2013; Wolfe et al., 2016).
In addition to the standard RACM2 and MCM v3.2 models, and the expanded isoprene chemistry in RACM2-LIM1 and MCM v3.3.1, a third set of model simulations were conducted to investigate the influence of RO2+ RO2 accretion reactions and dimer formation on overall radical concentrations. A set of reactions were added to both RACM2-LIM1 and MCM v3.3.1 to create overall mechanisms (RACM-ACC and MCM-ACC) that incorporate RO2+ RO2 accretion reactions for both isoprene- and monoterpene-based peroxy radicals. Several studies have reported observations of highly oxidized C19−20H28−32O10−18 dimer products in chambers (Ehn et al., 2014) and in field measurements (Yan et al., 2016; Zha et al., 2018), suggesting that RO2 reactivity in the process of dimer formation increases along with functionalization and size of the RO2 radical (Berndt et al., 2018a, b). Rate constants for the added reactions were based on measurements from Berndt et al. (2018a, b) and are intended to represent complex autoxidation and dimer formation chemistry into a model process that results in net radical termination.
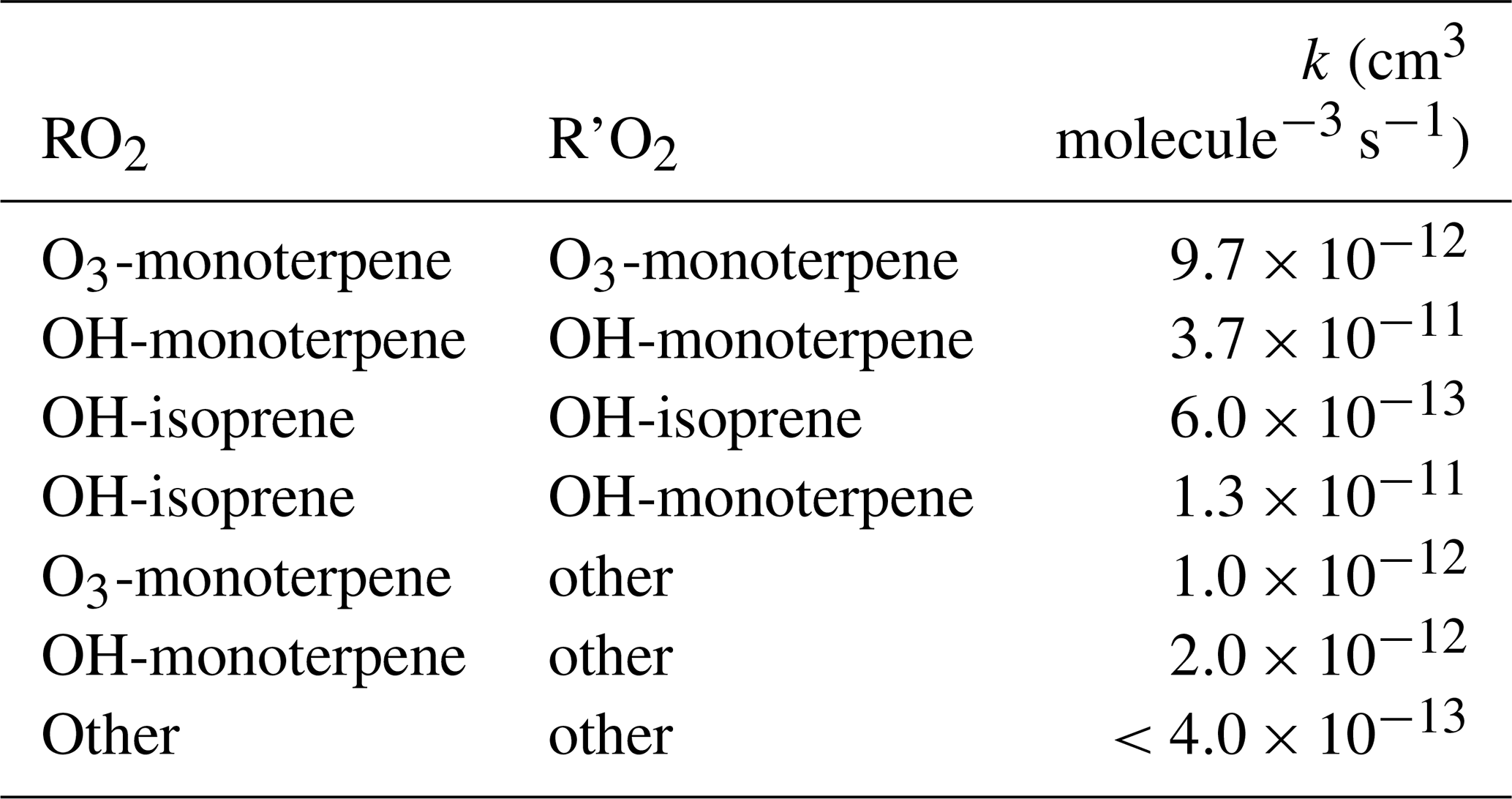
Rate constants for several RO2+ RO2 reactions used in this study are shown in Table 2. As described above, measurements of the sum of all monoterpenes were interpreted as α-pinene in the model, and thus rate constants measured in an exclusively α-pinene system (Berndt et al., 2018a) were used and provide only an estimation of the terpene chemistry at the PROPHET site that also includes emissions of β-pinene, limonene, and others (Carroll et al., 2001; Ortega et al., 2007). In addition to C10–RO2 radicals derived from monoterpenes, measured rate constants for self- and cross-reactions of C5-RO2 radicals derived from isoprene are also included, as well as more general, slower reactions between C10-RO2 and other smaller RO2 species. For the purposes of this study, rate constants are based on measurements of the least oxidized C10-RO2 species described in Berndt et al. (2018a) and thus may represent a lower limit in terms of autooxidation and dimer reactions as a radical sink. As such, the goal of this model was not to provide a detailed mechanism or exact representation of chemistry but instead to investigate the plausibility of autooxidation and dimer formation and the relative importance that the process may have as a radical termination process in a low-NOx, high-BVOC environment.
Table 2Summary of RO2+ R'O2→ ROOR' rate constants added to RACM-ACC and MCM-ACC based on Berndt et al. (2018a, b).
3.1 Meteorological and chemical conditions
A complete suite of supporting measurements, including meteorological conditions and important chemical species that were used as model constraints is shown in Fig. 1, and campaign average measured values of important model constraints are shown in Fig. 2. In general, weather during the campaign was sunny with intermittent clouds, with some exceptions of more overcast days (8, 15, 17, and 24 July). Mixing ratios of NO, O3, and photolysis rate constants were similar to those observed during previous field campaigns at the same site (Griffith et al., 2013). The maximum observed NO mixing ratio was 480 ppt on 11 July, and the average peak mixing ratio of NO was approximately 115 ppt at 09:00 local time. NO mixing ratios at night were typically less than 0.5 ppt. Average ozone mixing ratios were between 25 and 35 ppb. Maximum average daytime temperatures of 24 ∘C were similar to measurements at this site in 2008 but warmer than measurements at this site in 2009, resulting in average mixing ratios of isoprene that peaked near 3 ppb at approximately 18:00, similar to that measured in 2008 but greater than that measured in 2009 (Griffith et al., 2013). These measurements are summarized along with those from previous campaigns at the PROPHET site in Table S3. Mixing ratios of anthropogenic VOCs were low, with average mixing ratios of toluene and benzene below 65 and 40 ppt, respectively.
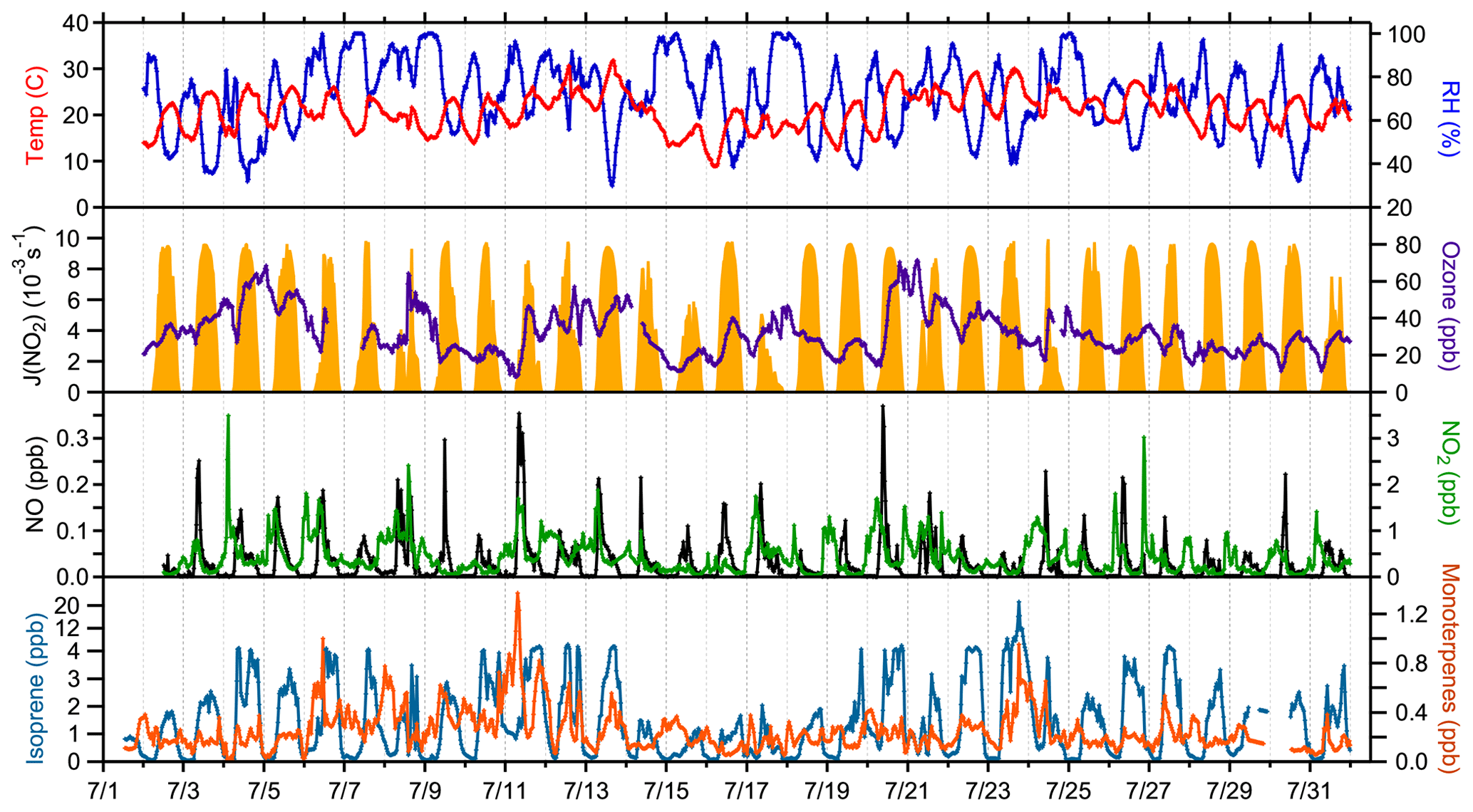
Figure 1Time series of measured meteorological and chemical conditions used as constraints for model calculations.
3.2 OH measurements and model predictions
Measured and modeled OH, HO2, and XO2 concentrations from 2 July through 31 July are shown in Fig. 3 with correlation plots shown in Fig. S2. Measurements of OH were hampered by high background signals and limited laser power. Diurnal profiles with a 2 h time resolution of the OH measurements are shown in Fig. 4, in addition to the model results. An average of all OH measurements performed during the campaign shows a peak of 1.25×106 molecules cm−3 at 13:00. Measurements of OH during the morning hours were significantly lower than all model calculations. An experimental OH budget based on measured concentrations of OH, HO2, and other species that contribute to OH production and loss is shown in Fig. S3. The imbalance between 07:00 and 12:00 suggests either a missing OH sink or errors with the OH measurement during this time. The reason for this discrepancy is not clear but may be the result of participant activity on the top of the tower near the detection cell in the mornings during the campaign which may have influenced the OH measurements, although a systematic measurement error during this time cannot be ruled out.
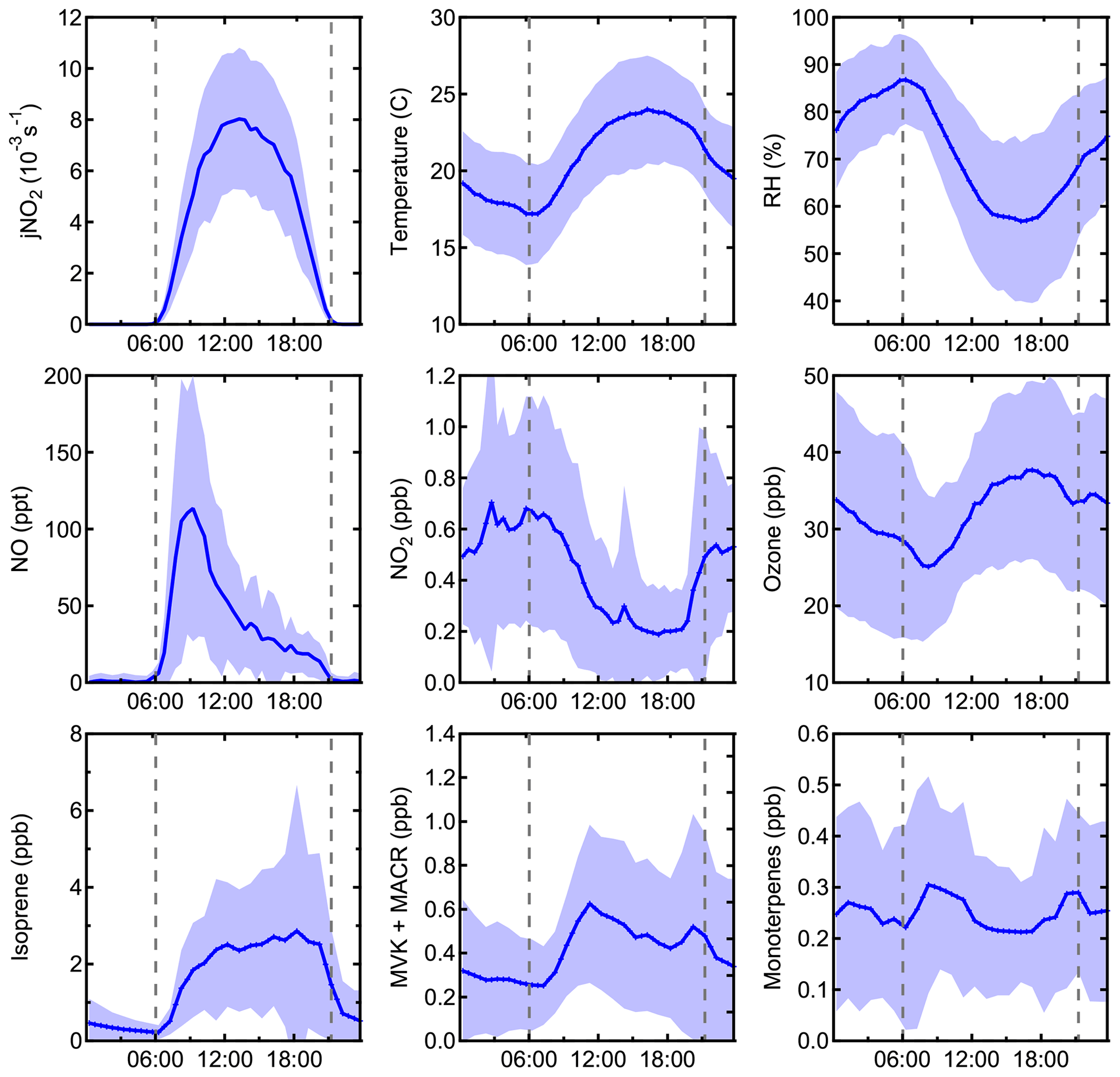
Figure 2Campaign average measurements of jNO2, temperature, relative humidity, NO, NO2, O3, isoprene, methyl vinyl ketone and methacrolein, and monoterpenes. Shaded areas represent the 1σ variability.
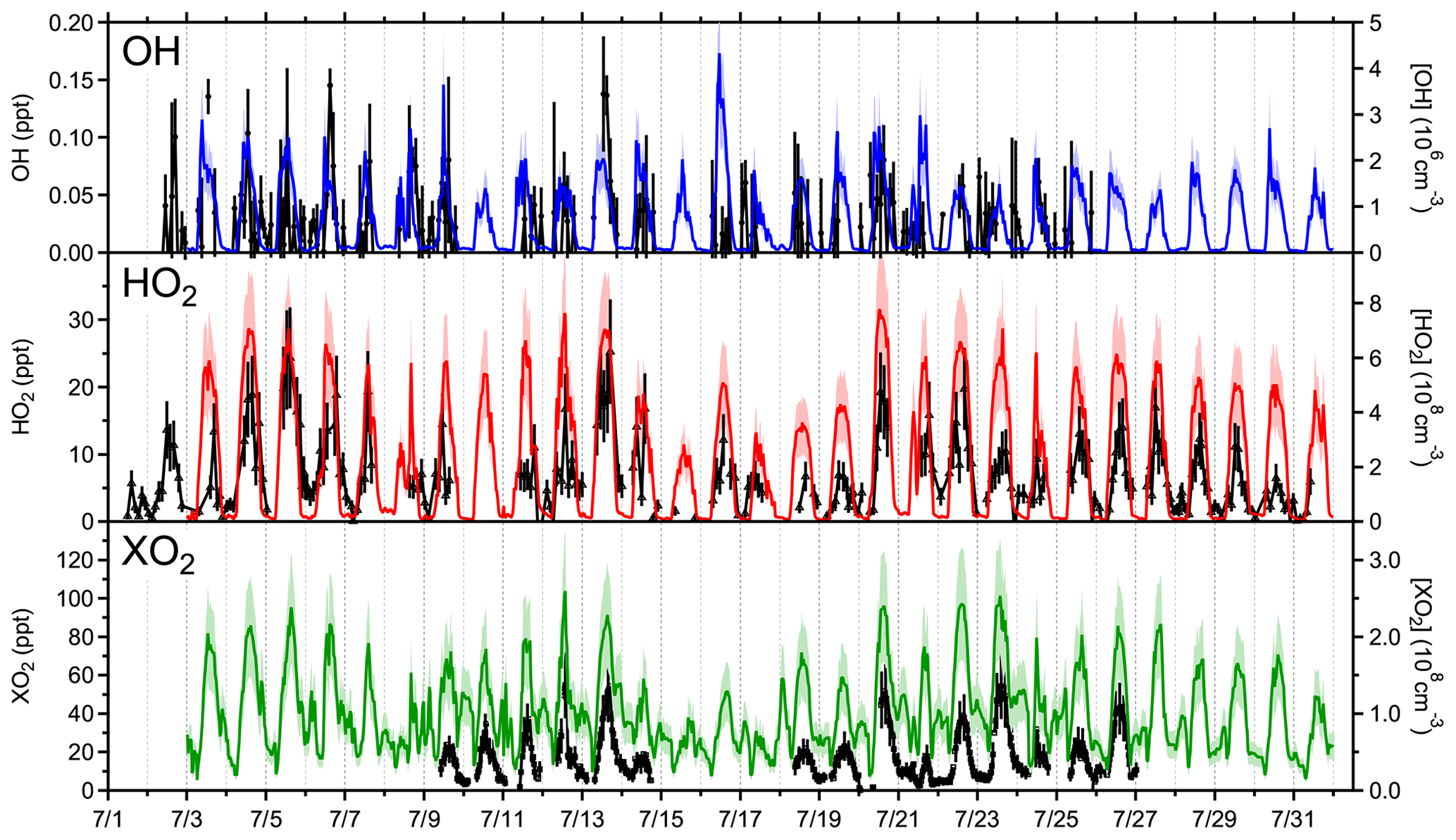
Figure 3Time series of radical measurements (black) and MCM v3.3.1 model predictions of OH (blue), HO2 (red), and XO2 (green) from 2 to 31 July. Measurements of any potential interferences in the OH measurements have been subtracted, and only positive OH measurements are shown for simplicity.
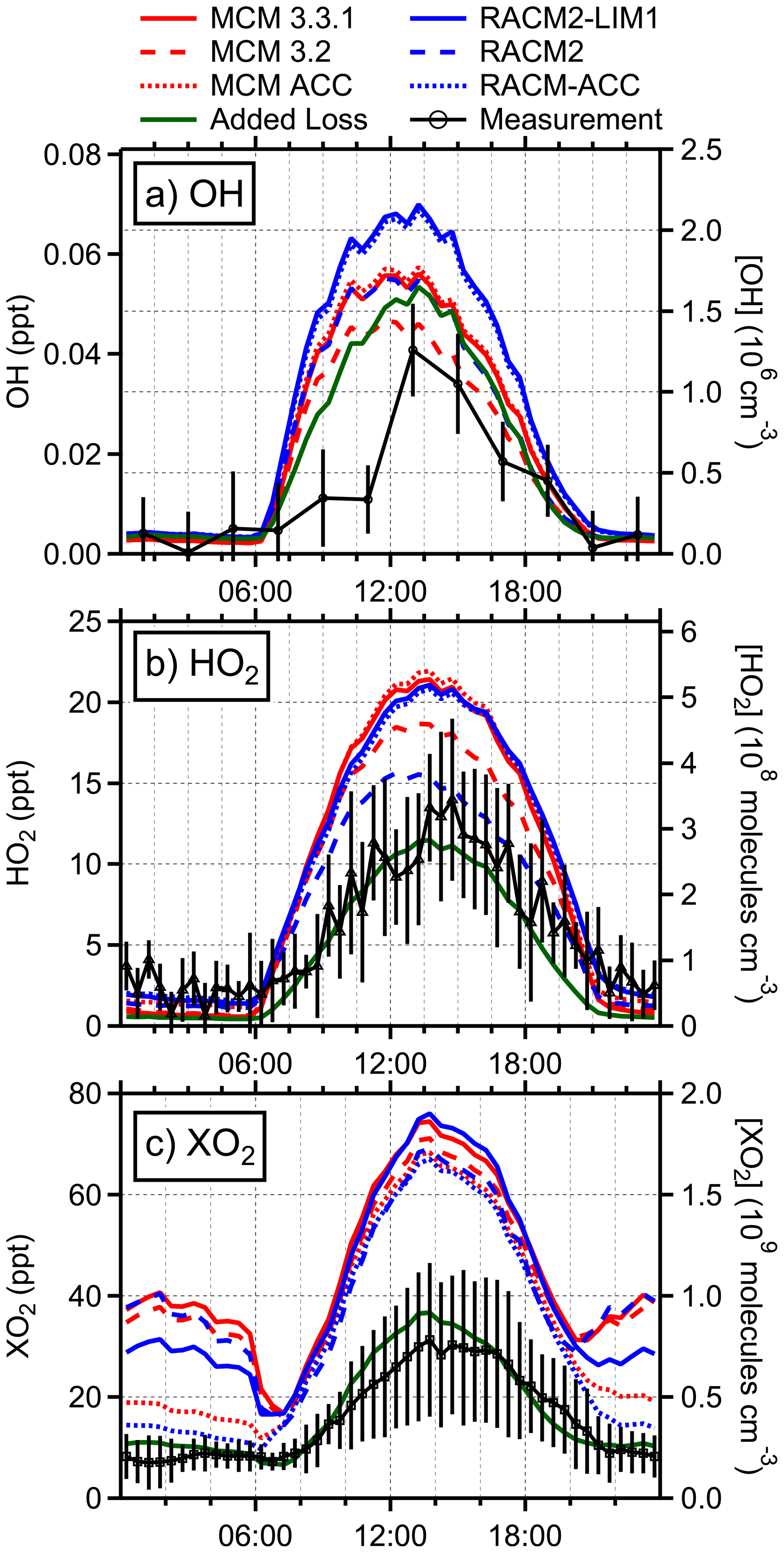
Figure 4Diurnal average measured (black) and modeled concentrations of (a) OH, (b) HO2, and (c) XO2. MCM models are shown in red and RACM2 in blue. The green line represents an additional version of the RACM-ACC model with added sinks for HO2 and isoprene peroxy radicals. The colored lines represent an average of individual daily model runs from only the days that each respective species was measured (OH: 7/3–7/9, 7/11–7/14, and 7/16–7/25; HO2: 7/3–7/9 and 7/11–7/31; XO2: 7/9–7/14 and 7/18–7/26). Error bars represent the 1σ measurement precision.
Measurements of potential interferences by chemical modulation through addition of C3F6 as described above did not reveal any significant unknown interferences, similar to that observed previously at this site (Griffith et al., 2013) but in contrast to measurements by the IU FAGE instrument during the IRRONIC (Indiana Radical, Reactivity and Ozone Production Intercomparison) campaign (Lew et al., 2020). Lew et al. (2020) found that the measured interference increased with both ozone and temperature, similar to that observed by Mao et al. (2012), who also measured a similar interference that increased with both temperature and total OH reactivity. Laboratory studies suggest that the interference could be due to the decomposition of Criegee intermediates inside the low-pressure detection cell leading to OH production (Novelli et al., 2014a, 2017; Fuchs et al., 2016; Rickly and Stevens, 2018), although estimated concentrations of Criegee intermediates in similar environments are too low to explain the observed interference (Novelli et al., 2017). Another proposed source of the interference is the decomposition of ROOOH molecules inside the FAGE detection cell formed from the reaction of OH with RO2 radicals (Fittschen et al., 2019). While the sources of these interferences are still unknown, one possible explanation for the absence of a measurable interference during PROPHET-AMOS is the lower measured mixing ratios of ozone and lower temperatures compared to those measured during IRRONIC, resulting in lower mixing ratios of isoprene and other BVOCs. Based on the observed correlation of the interference with ozone and temperature highlighted in Lew et al. (2020), it is likely that a similar interference was undetectable during PROPHET-AMOS.
The measured OH concentrations reported here are similar to previous measurements made by the IU-FAGE instrument at the PROPHET site in 2009 but are lower than those measured at the site in 2008, although the latter measurements suffered from poor precision (Griffith et al., 2013). The results reported here are also in contrast to measurements of OH at this site in 1998 as reported by Tan et al. (2001), who reported maximum daytime concentrations of approximately 4×106 cm−3 that were approximately a factor of 3 greater than model predictions (Table S3). While the mixing ratios of NOx and isoprene in 1998 were similar to those observed during PROPHET-AMOS, mixing ratios of ozone were higher in 1998, with the average maximum of approximately 45 ppb similar to that observed during the IRRONIC campaign (Lew et al., 2020). In addition, anomalously elevated concentrations of OH were observed at night in 1998 (Faloona et al., 2001; Tan et al., 2001). These results suggest that the OH measurements in 1998 at the PROPHET site may have been influenced by interferences similar to those observed by Mao et al. (2012), Feiner et al. (2016), and Lew et al. (2020). As illustrated in Fig. 4, the base RACM2 and MCM v3.2 models are able to reproduce the maximum observed OH concentrations to within the combined measurement precision and uncertainty of the models. The addition of LIM1 chemistry to the models increased the predicted average maximum OH concentration by approximately 20 % between MCM v3.2 and MCM v3.3.1 and by 30 % between RACM2 and RACM2-LIM1, with the MCM v3.3.1 maximum modeled OH concentrations approximately 30 % greater than the measured concentrations and the RACM2-LIM1 maximum modeled OH concentrations approximately 60 % greater than the measured concentrations. These results are in contrast to several previous LIF measurements in forested environments (Rohrer et al., 2014), in which measured OH concentrations were significantly higher than modeled predictions. However, the results reported here are similar to those found by Feiner et al. (2016) in an Alabama forest during SOAS (Southern Oxidant and Aerosol Study) where isoprene was the dominant BVOC. In that study, the modeled OH concentrations using MCM v3.3.1 were in good agreement with the measured concentrations when interferences were subtracted from the measurements.
While the predictions by both the RACM2 and MCM models are within the combined uncertainty of the measurements and the models, the MCM v3.3.1 results are in better agreement with the measurements (Fig. 4), which could suggest that the measurements are consistent with the lower effective bulk 1,6-H shift peroxy radical isomerization rate constants in MCM v3.3.1 compared to the original LIM1 recommended rates (Novelli et al., 2020). This is in contrast to the results of from the IRRONIC campaign discussed above, where the MCM v3.3.1 model underpredicted the measured concentrations by approximately a factor of 2, with the RACM2-LIM1 model predictions in better agreement with the measurements (Lew et al., 2020). Similarly, Novelli et al. (2020) reported that the MCM v3.3.1 mechanism underpredicted measurements of OH by a factor of approximately 1.4 during isoprene oxidation experiments in the SAPHIR (Simulation of Atmospheric Photochemistry In a large Reaction) chamber when mixing ratios of NO were less than 0.2 ppb. Unfortunately, the poor precision of the OH measurements reported here do not allow a robust test of the two mechanisms.
3.3 HO2 and XO2 measurements and model predictions
The time series of measured and modeled HO2 and total peroxy radical (XO2) concentrations from 2 July through 31 July are shown in Fig. 3, and correlation plots of the measured concentrations and MCM v3.3.1 model predictions are shown in Fig. S2. Daily maxima were typically observed between 13:30 and 15:30 local time and ranged from 1.7×108 cm−3 (6.7 ppt) to 7.0×108 cm−3 (28.2 ppt) for HO2 and 2.7×108 cm−3 (10.8 ppt) to 1.3×109 cm−3 (52.1 ppt) for XO2. Measured diurnal average profiles are shown in Fig. 4 along with average model results that consider only the days on which each respective species was measured (7/3–7/9 and 7/11–7/31 for HO2; 7/9–7/14 and 7/18–7/26 for XO2). In addition, measured RO2 mixing ratios (HO2 measured by LIF subtracted from XO2 measured by ECHAMP) are compared with modeled RO2 mixing ratios in Fig. S4. The average maximum (12:00–15:00) of HO2 measurements performed during the campaign was 2.85×108 cm−3 (11.6 ppt), while the maximum daytime average of the XO2 measurements was approximately 7.7×108 cm−3 (29.0 ppt).
The measured HO2 concentrations were similar to previous measurements at this site. Median daytime maximum concentrations of HO2* measured in 2008 were approximately 7×108 cm−3 (28 ppt), while median daytime maximum concentrations of HO2* measured in 2009 were approximately 5×108 cm−3 (20 ppt), with nighttime concentrations below 1×108 cm−3 (4 ppt) during both years (Griffith et al., 2013). The conversion efficiency of isoprene peroxy radicals to the measured HO2* concentrations during these studies was estimated to be approximately 90 %, suggesting that the measured HO2* concentrations reflected the sum of HO2+ isoprene peroxy radicals. Given that isoprene peroxy radicals contribute to approximately 33 % of the total peroxy radical concentrations during the daytime, the measured HO2* concentrations in 2008 and 2009 were greater than HO2 but less than XO2 concentrations (Table S3). When compared to 2009, the higher HO2* concentrations measured in 2008 were likely due to the higher mixing ratios of HCHO observed in 2008, leading to greater rates of radical production (Griffith et al., 2013). The higher mixing ratios of HCHO may be a result of the higher mixing ratios of isoprene leading to a greater production of HCHO during the warmer temperatures observed in 2008 (Griffith et al., 2013).
Average daytime maximum concentrations of HO2 measured at this site in 1998 were reported to be approximately 16 ppt 3.9×108 cm−3 (16 ppt), with nighttime concentrations less than 1.2×108 cm−3 (5 ppt) (Tan et al., 2001), similar to the measurements in this study. However, it is not clear whether the 1998 HO2 measurements were influenced by interferences from isoprene-based peroxy radicals, as discussed above. As a result, these measurements may be an upper limit to the actual HO2 concentrations. The measured XO2 concentrations are similar to the total RO2+ HO2 concentrations measured at this site in 1997 by Mihele and Hastie (2003), who reported daytime maximum mixing ratios between 20 and 65 ppt using a radical chemical amplifier technique, and nighttime mixing ratios of 3–6 ppt. Although not measured at the site during the 1997 campaign, monoterpene mixing ratios observed in 1998, 2008, and 2009 were similar to measurements from 2016 (Table S3).
As illustrated in Fig. 4, the base RACM2 and MCM v3.2 models overpredict both the measured HO2 and XO2 concentrations during the daytime, although the agreement with the measured HO2 concentrations is within the combined uncertainty of the measurements and the model. The base RACM2 model overpredicts the measured average maximum HO2 concentrations by approximately 30 %, while the MCM v3.2 overpredicts the measured daytime maximum HO2 by approximately 60 %. However, including the LIM1 isoprene oxidation mechanism increases the daytime HO2 concentrations predicted by the base models by approximately 15 % and 35 % for MCM and RACM2 models, respectively (Fig. 4). Overall, both the RACM2-LIM1 and MCM v3.3.1 models overpredict the measured daytime maximum HO2 concentrations by approximately 80 %, which is outside of the combined measurement and model uncertainties. Similarly, the base RACM2 and MCM v3.2 models as well as the updated RACM2-LIM1 and the MCM v3.3.1 models overpredict the daytime XO2 concentrations by more than a factor of 2, with predicted daytime maximum XO2 mixing ratios ranging from 65.5 (RACM2) to 72.6 (RACM2-LIM1) ppt (1.6–1.8×109 cm−3).
The model overprediction of the daytime measured HO2 concentrations is consistent with model simulations of the measured HO2* concentrations at this site in 2008 and 2009 (Griffith et al., 2013). In 2008, a base RACM model overpredicted the measured HO2* concentrations by approximately 30 % on average, while the same model overpredicted the HO2* concentrations measured in 2009 by approximately 50 %. Similar to the results presented here, addition of the LIM1 mechanism for isoprene oxidation to the RACM model likely would have increased the discrepancy between the 2008 and 2009 measurements. However, these model results are in contrast to those observed at this site in 1998, where a RACM-based model was able to reproduce the reported measured HO2 concentrations (Tan et al., 2001). As discussed above, these measurements likely represent an upper limit to the actual HO2 concentrations as it is not clear whether the measurements of HO2 were free from interferences from isoprene-based and other alkene-based peroxy radicals (Fuchs et al., 2011; Lew et al., 2018). As a result, it is likely that this RACM-based model of Tan et al. (2001) overestimated the actual HO2 concentrations in 1998. The results reported here are also in contrast to the results of Mihele and Hastie (2003), who found that a 0-D MCM-based model could reproduce the measured daytime XO2 concentrations on several days. Similarly, these results are in contrast to the IRRONIC campaign, where the MCM and RACM2 models were able to reproduce the measured HO2* concentrations to within 30 % (Lew et al., 2020), and the results from SOAS, where the MCM v3.3.1 model was able to reproduce the measured HO2 concentrations to within the combined uncertainties of the measurement and the model (Feiner et al., 2016). The ability of the models to reproduce the measured peroxy radical concentrations in these studies may reflect the higher mixing ratios of NOx observed at the PROPHET site in 1997, the SOAS site, and at the IRRONIC site, resulting in a greater contribution of the RO2+ NOx reactions to the fate of peroxy radicals during these campaigns (Mihele and Hastie, 2003; Sanchez et al., 2018; Lew et al., 2020). An analysis of the discrepancies between the PROPHET and IRRONIC campaigns will be presented in a subsequent paper.
The composition of the total peroxy radical concentration (XO2) in the RACM2-LIM1 model is shown in Fig. 5. As illustrated in this figure, the model predicts that HO2 radicals comprise approximately 33 % of the total daytime maximum XO2 concentration, with isoprene peroxy radicals accounting for approximately 33 %, methyl peroxy radicals for approximately 12 %, and acyl peroxy radicals for approximately 7 % and with peroxy radicals from alkane, alkene, and terpene oxidation comprising the remaining 15 %. Given that the model agreement with the measurements is better for HO2, the majority of the discrepancy between the modeled and measured XO2 concentrations is likely due to a greater overestimation of RO2 radicals, including isoprene-based peroxy radicals. These results are in contrast to that reported by Kundu et al. (2019), who found that their measurements of XO2 concentrations by the ECHAMP instrument during the IRRONIC campaign could be reproduced on several days by a model incorporating the MCM v3.2. As discussed above, the ability of the models to reproduce the measured concentrations during IRRONIC may reflect the higher mixing ratios of NO observed during this campaign, resulting in a greater contribution of RO2+ NOx reactions to the fate of peroxy radicals at this site.
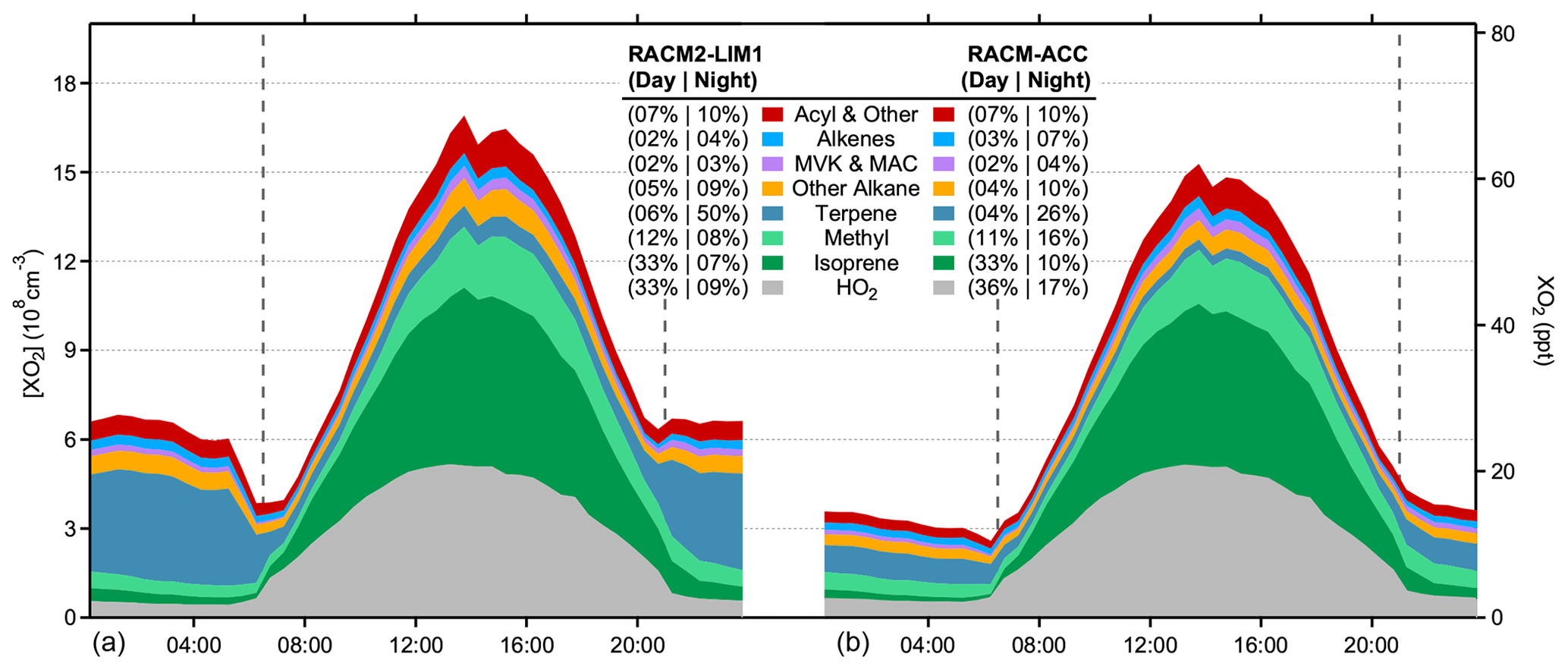
Figure 5Modeled XO2 composition from RACM2-LIM1 (a) and RACM-ACC (b). Colors represent peroxy radicals derived from the listed VOCs and numbers represent the percentage contribution of each species to the total concentration of XO2 during the day (06:30 to 21:00) and at night (21:00 to 06:30), respectively.
During the nighttime, the models reproduce the measured HO2 concentrations but overestimate the measured XO2 radical concentrations (Fig. 4). The RACM2 and MCM models overpredict the nighttime XO2 concentrations by a factor of approximately 4, with the RACM2-LIM1 model predicting mixing ratios of total peroxy radicals of approximately 27 ppt between 21:00 and 06:00 and the MCM v3.3.1 model predicting XO2 mixing ratios of approximately 36 ppt during the night (Fig. 4) compared to the measured concentrations of less than 10 ppt. These results are similar to those from the 1997 PROPHET campaign in which measured XO2 mixing ratios of 3–6 ppt were overpredicted by more than a factor of 10 by a model that included reactive terpene emissions (Mihele and Hastie, 2003; Sillman et al., 2002). This is in contrast to the results of Kundu et al. (2019), who found that the MCM v3.2 could reproduce measured nighttime mixing ratios of less than 10 ppt during the IRRONIC campaign, which is likely a result of the elevated NOx mixing ratios at that site.
The RACM2-LIM1 model predicts that approximately 50 % of the nighttime total XO2 radical concentration is composed of peroxy radicals derived from the ozonolysis of monoterpenes (Fig. 5). As mentioned above, the measured sum of monoterpenes was constrained as α-pinene in all model simulations, resulting in an average monoterpene ozonolysis rate constant that is likely similar to that expected from previous speciated measurements of monoterpenes, including limonene and β-pinene, at this site (Ortega et al., 2007; Kim et al., 2011). However, the average ozonolysis rate constant assumed in the model could represent an upper limit if the monoterpene composition was dominated by species less reactive with ozone (e.g., camphene, cymene) or a lower limit if more reactive terpene species were present (e.g., ocimene, limonene) (Atkinson et al., 1990; Khamaganov and Hites, 2001; Atkinson and Arey, 2003).
The addition of the RO2+ RO2 accretion reactions described above to the RACM2-LIM1 and MCM v3.3.1 models (RACM-ACC and MCM-ACC) significantly reduces the predicted XO2 radical concentrations at night by 50 %, lowering the measurement–model discrepancy to less than 5 ppt for the RACM-ACC model. As shown in Fig. 5, this is largely due to a reduction in the concentration of organic peroxy radicals derived from monoterpenes due to the relatively large rate constants for the associated RO2+ RO2 accretion reactions (Table 2).
3.4 Radical budget analysis
A radical budget analysis for OH, HO2, isoprene-based peroxy radicals (ISOP) and total ROx was conducted to provide information about the processes that drive radical production and the radical loss pathways in this environment and also to highlight the relative importance of the changes in radical chemistry upon the addition of the LIM1 mechanism and accretion reactions. Figure 6a illustrates the campaign average production and loss pathways of OH for the RACM2-LIM1 model. This includes both initiation reactions and propagation steps that produce OH in blue, while termination pathways are shown alongside propagation steps that convert OH to HO2 or RO2 in red. The addition of the LIM1 reactions increases the maximum OH production rate at 13:45 by 35 % from 2.01 ppb h−1 in RACM2 to 2.71 ppb h−1 in RACM2-LIM1, primarily due to the isomerization of isoprene peroxy radicals to form HPALDs, which readily photolyze to form OH, and also di-HPCARPs, which rapidly decompose to produce additional OH radicals (Peeters et al., 2014; Teng et al., 2017; Wennberg et al., 2018). In the morning (06:45–13:15), RACM2-LIM1 suggests the HO2+ NO reaction is the dominant source of OH radicals, accounting for 41 % of total OH production and as much as 53 % when NO mixing ratios are the highest. This decreases to 21 % in the afternoon and evening as the NO concentration decreases. Photolytic processes are significant throughout the day, with ozone and HONO photolysis contributing up to 28% and 13 % respectively during the day. Ozonolysis of alkenes, primarily monoterpenes, is a minor contributor of up to 6 % during the day but is the dominant source of OH at night.
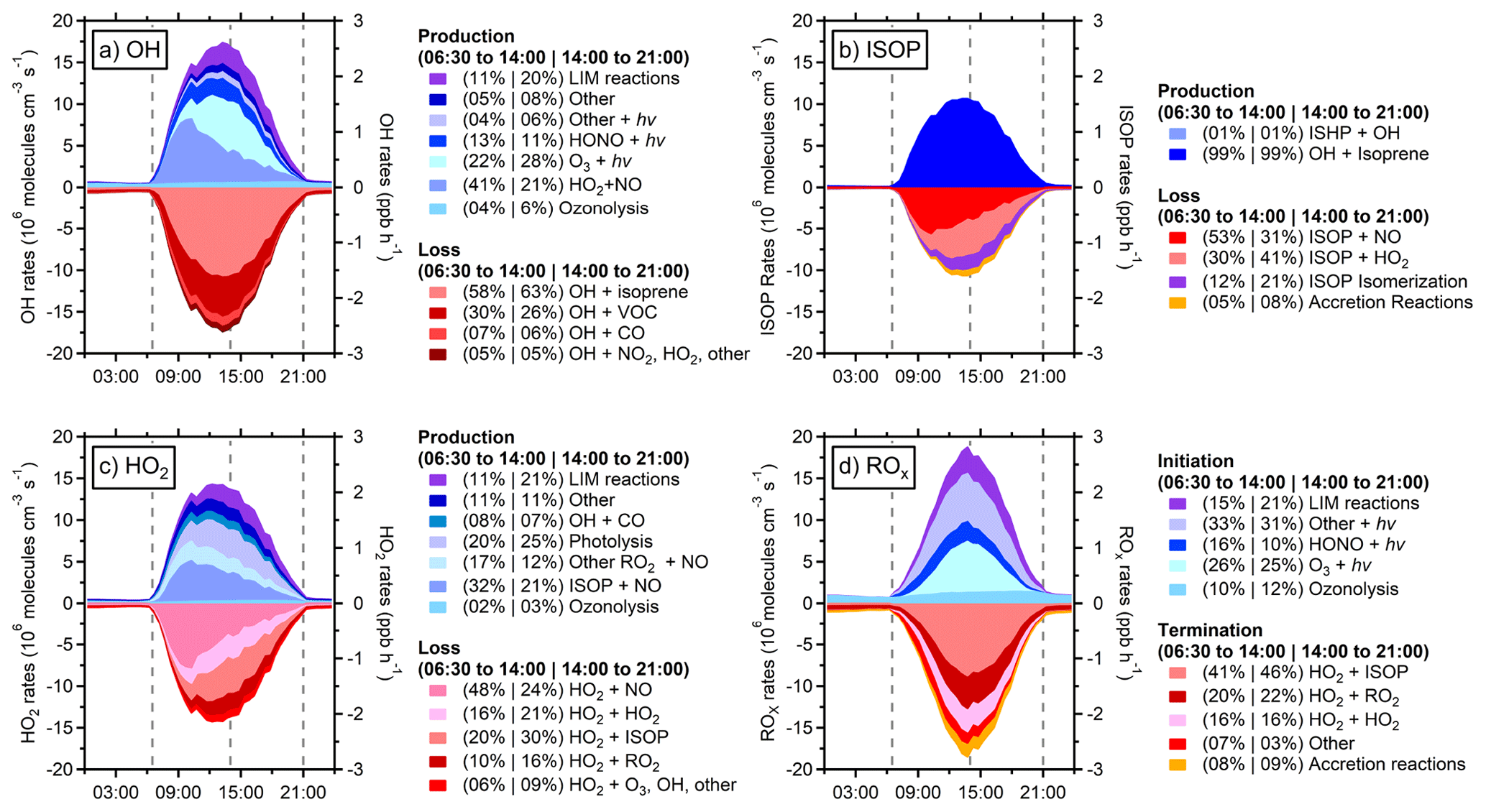
Figure 6Radical budgets from the RACM2-LIM1 model with additional accretion reactions (RACM-ACC) for (a) OH, (b) isoprene-based peroxy radicals (ISOP), (c) HO2, and (d) total ROx. Shades of blue represent reactions that produce/initiate radicals, and shades of red represent radical loss/termination reactions. LIM reactions (purple) include reactions added as part of the Leuven Isoprene Mechanism. Percentages represent the relative initiation or termination rates of each respective process in the morning (06:30 to 14:00) and during the evening (14:00 to 21:00), which are indicated by the vertical dashed lines.
Reaction with isoprene is the dominant loss pathway for OH radicals accounting for approximately 60 % of daytime OH reactivity. Other VOCs (16 %), namely monoterpenes, and OVOCs (10 %), such as formaldehyde, methyl vinyl ketone, and methacrolein, make up the majority of the remaining daytime OH reactivity. Propagation through reaction with CO is minor (6 %), and termination through the OH + NO2 reaction is not significant (< 2 %). Consistent with the OH radical budget, the OH + isoprene reaction is the dominant source of isoprene-based peroxy radicals (ISOP; Fig. 6b), with the ISOP + NO reaction accounting for approximately 53 % of their total loss in the morning, while the ISOP + HO2 reaction and peroxy radical isomerization reactions in the LIM1 mechanism account for 62 % of isoprene-based peroxy radical loss in the afternoon. The ISOP accretion reaction accounts for only 8 % of the loss of isoprene-based peroxy radicals in the afternoon.
Figure 6c illustrates the campaign average HO2 radical production and loss pathways for RACM2-LIM1. The production of HO2 in the RACM2-LIM1 model is largely due to turnover from the RO2+ NO reaction. During the morning, when NO concentrations are greatest, 32 % of HO2 is produced from the reaction of NO and peroxy radicals derived from isoprene, while 17 % is produced from the reaction of NO with other RO2 species. In addition to reactions with NO, the photolysis of formaldehyde can account for up to 15 % of daytime HO2 production and turnover from the OH + CO reaction near 8 %. HO2 loss is primarily due to reaction with NO in the morning (48 %) but dominated by the HO2 self-reaction, reaction with isoprene RO2 to form ISOPOOH, and reaction with other peroxy radicals in the afternoon and evening (21 %, 30 %, and 16 % respectively).
The total ROx radical budget is illustrated in Fig. 6d. The addition of LIM1 reactions increases the maximum radical initiation rate by 28 % from 2.11 to 2.69 ppb h−1, again primarily due to fast photolysis of HPALDs and decomposition of di-HPCARPs. Overall radical initiation in RACM2-LIM1 is largely due to photolytic processes, with a combined 51 % from ozone photolysis (26 %), HONO (13 %), and HPALDs (18 %) and 32 % from the photolysis of other species such as hydrogen peroxide, aldehydes, organic peroxides, and nitric acid. Ozonolysis is a consistent radical initiation source of approximately 0.21 ppb h−1 throughout the day, which dominates ROx initiation at night and is a significant contributor (11 %) throughout the day when photolysis sources are dominant.
Daytime termination of radicals in RACM2-LIM1 is dominated by peroxy radical self- and cross-reactions, primarily the reaction of isoprene peroxy radicals with HO2 to form ISOPOOH (44 %) but also the HO2 self-reaction (16 %) and HO2+ other RO2 species (21 %). Radical reactions with NOx were less significant due to the low NOx concentrations and accounted for at most 0.13 ppb h−1 or less than 8 % of the RACM2-LIM1 termination budget when NOx mixing ratios were highest and less than 3 % total during the daytime. The addition of RO2+ RO2 accretion reactions in the RACM-ACC model provides an alternative pathway that results in a termination rate equivalent to half that of HO2+ RO2 reactions at night (4.0×105 molecules cm−3 s−1) and accounts for 30 % of total ROx termination during this time. During the day, when isoprene and NO mixing ratios are higher, these reactions only contribute to 9 % of the overall termination due to the lower rate constants for reactions of C5-RO2 from isoprene (Table 2). As shown in Fig. 4, this results in better agreement between the measurement and model at night, but model overprediction during the day remains.
As illustrated in Fig. 4, including the accretion reactions shown in Table 2 in both the RACM2-LIM1 and MCM v3.3.1 models improves the agreement between the model and measured XO2 concentrations at night to within the combined uncertainty of the model and the measurements, although the agreement of the RACM2 model is better. However, including these accretion reactions in the model only decreases the modeled XO2 concentrations by 9 % during the daytime when isoprene-based peroxy radicals dominate the total XO2 composition (Fig. 5), as the RO2+ RO2 accretion rate constants for isoprene-based peroxy radicals are smaller compared to those for monoterpene-based peroxy radicals (Table 2).
One possible explanation for the model discrepancies with the measured HO2 and XO2 concentrations during the daytime is the errors associated with the measurements of these radicals, such as a systematic error in the calibration of HO2 or XO2 radicals. However, as discussed above, measurements of XO2 concentrations during the IRRONIC campaign were in good agreement with model predictions by the RACM2 and MCM mechanisms, where isoprene dominated OH reactivity during the daytime and isoprene-based peroxy radicals likely contributed to approximately 30 % of the total XO2 concentrations, similar to that observed during PROPHET-AMOS (Kundu et al., 2019). While measurements of HO2 were not conducted during IRRONIC, the measured HO2* concentrations were also found to be in good agreement with the model predictions (Lew et al., 2020). In addition, the measured XO2 HO2* ratio was found to be in good agreement with the modeled ratio (Kundu et al., 2019). While these results do not rule out the possibility of errors associated with the calibration of the ECHAMP and IU-FAGE instruments, they suggest that the discrepancy between the measurement and model predictions during PROHET-AMOS may not be due to a systematic error in the measurements. As noted in Sect. 2.3, ECHAMP is expected to be 8 % less sensitive to isoprene RO2 than other peroxy radicals. As the modeled isoprene RO2 mixing ratio accounted for approximately 33 % of modeled XO2 during the daytime (Fig. 5), this suggests that the measured XO2 represents a lower limit and could be as much as 3 % higher than reported. Given the large differences between modeled and measured XO2 of more than a factor of 2 during midday, this difference cannot account for the discrepancy with the modeled concentrations.
Measurements of isoprene hydroxy hydroperoxides (ISOPOOH) produced from the reaction of isoprene-based RO2 radicals with HO2 can provide an additional test of the model chemistry at this site. Figure 7a shows the average ISOPOOH mixing ratio measured during PROPHET-AMOS between 22 and 27 July by the Caltech low-pressure GC-CIMS instrument (Vasquez et al., 2018) along with MCM model results. The measured mixing ratios were similar to those observed during the SOAS campaign (Kaiser et al., 2016). The measurements shown include both the 1,2- and 4,3-ISOPOOH isomers, although the 1,2-ISOPOOH constitutes the dominant fraction (Vasquez et al., 2018). In order to achieve a more realistic comparison, a measurement-based deposition term for ISOPOOH and isoprene hydroxy nitrates (IHN) (Nguyen et al., 2015; Wei et al., 2021) was included in the mechanism for all model runs shown in this figure. Still, as illustrated in Fig. 7a, the RACM2-ACC and MCM-ACC models overpredict the measured ISOPOOH concentrations by approximately a factor of 8–10 during the daytime, consistent with the overprediction of peroxy radicals by the models. Constraining the model to the measured concentrations of HO2 and isoprene-RO2 (assuming the same relative distribution of RO2 radicals predicted by the models) improves the agreement (Fig. 7a), although the model still overestimates the measured concentrations. This overestimate of the measured ISOPOOH is similar to that observed during the SOAS campaign (Kaiser et al., 2016), where a large dilution rate was needed to bring the modeled ISOPOOH into agreement with the measurements. Similarly, the model also overestimates the concentrations of IHN produced from the reaction of isoprene peroxy radicals with NO and measured using iodine-adduct CIMS (Xiong et al., 2015). Constraining the model to the measured peroxy radical concentrations improves the agreement with the measurements (Fig. 7b). It is also worth noting that the model does not account for losses of IHN due to reactive uptake onto aerosol and subsequent hydrolysis in the aerosol phase (Jacobs et al., 2014; Morales et al., 2021; Wang et al., 2021). Knowledge and incorporation of such loss rates in the model could better constrain the modeled IHN concentrations, but the effect is expected to be small in comparison to the adjustment in the modeled output when constrained to measured RO2 (Wei et al., 2021; Mayhew et al., 2022). These results suggest that the measured HO2 and XO2 concentrations are consistent with the measured ISOPOOH and IHN concentrations and that the models are overpredicting the concentrations of HO2 and isoprene-based peroxy radicals, either through an overestimation of their production or an underestimation of their loss.
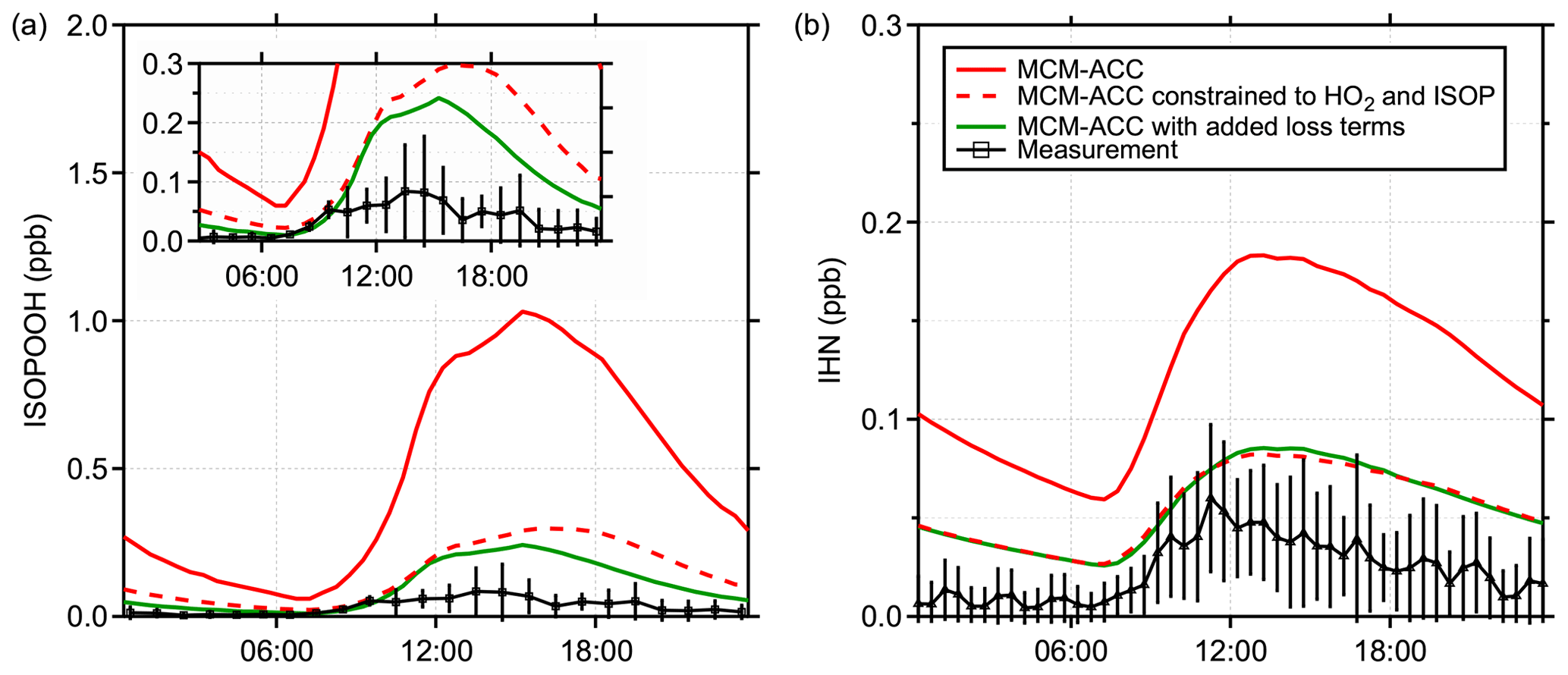
Figure 7Measured and modeled mixing ratios of (a) isoprene hydroxy hydroperoxides (ISOPOOH) and (b) isoprene hydroxy nitrates (IHN). Measurements of ISOPOOH are an average from 23–27 July (Vasquez et al., 2018), and measurements of IHN are an average from 6–31 July. The solid lines represent modeled mixing ratios from MCM-ACC models. The dashed line represents predictions of the same model constrained to measured values of HO2 and measurements of XO2 scaled to the modeled isoprene RO2 composition.
The radical budget analysis suggests that the OH + isoprene reaction is the main source of isoprene-based peroxy radicals during PROPHET-AMOS (Fig. 6b). Measurements of the total OH reactivity together with the measurement of the concentration of OH can provide an estimate of the rate of peroxy radical production from reactions of VOCs with OH. Measurements of total OH reactivity were also conducted during PROPHET-AMOS using both the Indiana University Total OH Loss Method (IU-TOHLM) instrument (Hansen et al., 2014) and the IMT Nord Europe Comparative Reactivity Measurement (CRM) instrument (Hansen et al., 2015), and an analysis of the results and the instrument intercomparison will be presented in a subsequent paper. Figure 8 shows the diurnal averaged total OH reactivity as measured by the IU-TOHLM instrument along with that predicted by the MCM v3.3.1 model. As illustrated in this figure, the measured OH reactivity agreed with that calculated from measured and modeled OH sinks, including the reactivity of some unmeasured oxidation products, suggesting that the loss of OH is well represented by the models. Reaction with isoprene is the dominant daytime OH radical sink, accounting for approximately 60 % of the total OH reactivity during the day, in both the MCM v3.3.1 (Fig. 8) and RACM2-LIM1 (Fig. S6) models.
Figure 8Diurnal average of the measured (IU-TOHLM instrument) and modeled total OH reactivity at the top of the tower during PROPHET-AMOS. Modeled reactivity is largely based on measured species that are used as constraints in the model but also includes contributions from unmeasured oxidation products in the MCM v.3.3.1 model.
The reasonable agreement between the measured and modeled OH concentrations and total OH reactivity suggests that the rate of production of peroxy radicals by the reaction of OH with isoprene and other VOCs is not overestimated by the model given that these reactions are the dominant source of peroxy radicals during PROPHET-AMOS. In addition, because radical propagation by the RO2+ NO reaction is a major source of HO2 radicals, it is unlikely that the model is overestimating the production of HO2 radicals, although the photolysis of HCHO and other aldehydes is also predicted to be a significant source of HO2 radicals, contributing up to 20 %–25 % of total HO2 production. However, HCHO and other aldehydes were measured during the campaign, providing a constraint on radical production by the photolysis of these compounds.
Reactant segregation, where unevenly distributed surface flux leads to incomplete mixing in the convective boundary layer, could lead to an effective reduction in the rate of isoprene oxidation by OH, resulting in an overestimation of the reaction rate by the models. While it has been suggested that segregation between OH and isoprene could effectively reduce the rate of the OH + isoprene reaction by 60 %% (Butler et al., 2008), recent studies have suggested that segregation of OH and isoprene may result in an effective reduction in the rate of the OH + isoprene reaction of less than 15 % (Ouwersloot et al., 2011; Pugh et al., 2011). As a result, it is unlikely that reactant segregation is responsible for the discrepancy between the measured and modeled HO2 and XO2 concentrations described above, and the overprediction of these peroxy radicals by the models is likely due to an underestimation of radical termination rather than an overestimation of the production of these radicals. An additional loss of HO2 and isoprene-based RO2 radicals on the order of the rate of these peroxy radicals with NO is needed in order to resolve the daytime discrepancy between the model and the measurements. Figure 4 includes the results of an additional model that features the RACM2-ACC chemical mechanism but also includes additional sinks for HO2 and isoprene peroxy radicals (green line in Fig. 4). The added HO2 sink corresponds to a first-order loss rate of 0.012 s−1, which is approximately 40 % of the daytime HO2 loss, while the added isoprene-based RO2 sink corresponds to a first-order loss rate of 0.024 s−1, which is approximately 60 % of the daytime loss of isoprene-based peroxy radicals (Fig. S5). The addition of these peroxy radical loss mechanisms reduces the predicted daytime maximum OH concentration by 25 % to 1.65×106 cm−3, which is within the combined uncertainties of the measurement and the model (Fig. 4a). These loss processes could potentially include several components, such as uptake of radicals and important precursors to aerosols or the forest canopy, faster self- and cross-reactions between C5-RO2 and other RO2 species that serve as RO2 radical sinks, or reaction of RO2 radicals with isoprene or other unsaturated VOCs.
The first-order loss of HO2 on aerosols can be estimated assuming a first-order loss to aerosol surfaces (Ravishankara, 1997; Whalley et al., 2010) (Eq. 1), where A is the aerosol surface area per volume (cm2 cm−3); γ is the uptake coefficient; and cg is the mean molecular speed of a gas (cm s−1) given by Eq. (2), where R is the gas constant, T is the temperature, and Mw is the molecular weight of the gas. Aerosol uptake coefficients for HO2 radicals have been measured in both laboratory and field studies, with most values ranging from less than 0.1 to 0.4 (Taketani et al., 2008; Thornton et al., 2008; Taketani et al., 2012; George et al., 2013; Zhou et al., 2021). Assuming values of A=100 µm2 cm−3 typical of rural aerosols (Cai et al., 2017) and γ=0.1 results in an estimated first-order loss rate of approximately 0.001 s−1, while assuming values of A=200 µm2 cm−3 and γ=0.4 results in an estimated first-order loss of approximately 0.008 s−1. Similar assumptions for isoprene-based peroxy radicals result in an estimated first-order loss of approximately – s−1. Assuming an uptake coefficient of γ=1 for isoprene-based peroxy radicals would lead to estimated first-order loss rates of approximately – s−1. These results suggest that while heterogeneous loss of peroxy radicals on aerosols may contribute to the model overestimation of the measurements, they may not be the only loss mechanism missing in the model.
Recent studies have detected products of the reaction of RO2 radicals with unsaturated VOCs under atmospheric conditions and suggested that the reaction of isoprene-based peroxy radicals with isoprene could be a significant radical termination reaction in low-NOx regions (NO ≤ 0.05 ppb) (Nozière et al., 2023). Assuming a rate constant of 10−14 cm3 s−1 for this reaction based on measurements of the rate of the reaction of acyl peroxy radicals with 2,3-dimethyl-2-butene, the reaction of isoprene-based peroxy radicals with isoprene would result in an estimated first-order loss of approximately s−1. Although this RO2+ alkene rate coefficient is not large enough to resolve the discrepancy between the measured and modeled XO2 mixing ratios at the PROPHET site, RO2 radicals derived from the OH oxidation of isoprene and monoterpenes could exhibit enhanced reactivity to alkenes and constitute a more significant portion of the missing radical sink (Nozière and Fache, 2021).
An underestimation of radical termination by the reactions of isoprene-based RO2 radicals could also be responsible for the discrepancies between the modeled and measured peroxy radical concentrations. The overestimation of the measured ISOPOOH concentration by the model (Fig. 7a) suggests that the model is not underestimating the rate of radical termination by the reaction of HO2 with isoprene-based RO2 radicals. To account for the missing loss of isoprene-based RO2 radicals, an accretion rate constant for the self-reaction of isoprene-based RO2 radicals of approximately cm3 molecule−1 s−1, similar to that for the self-reaction of monoterpene RO2 radicals (Table 2), would bring the modeled peroxy radical concentrations into agreement with the measurements. While this is greater than the factor of 2–3 uncertainty associated with the measured rate constant for this reaction (Berndt et al., 2018b), a combination of loss rates from aerosol uptake, RO2 reactions with alkenes, and accretion reactions would require a smaller accretion rate constant for isoprene-based RO2 radicals. An analysis of the experimental radical budgets including the impact of potential additional loss rates will be presented in a subsequent publication.
Another potential loss process in the 0-D model includes vertical and/or horizontal transport of peroxy radicals given their relatively longer modeled lifetimes under the low NOx conditions at the PROPHET site. The average chemical lifetimes of HO2 and isoprene-based peroxy radicals during the daytime range from 35–135 s and 40–160 s, respectively. These calculated lifetimes depend primarily on the reactions of HO2 and isoprene-based RO2 with the measured radical concentrations and the measured concentration of NO but also on the reactions of HO2 with O3 and the isoprene RO2 isomerization reactions included in the LIM1 mechanism. These lifetimes are on the order of the expected canopy mixing timescale in forested environments (∼ 2 min) (Wolfe et al., 2011; Wei et al., 2021), suggesting that deposition to the canopy surface could constitute a portion of the missing radical loss process and could be more significant on well-mixed days. Similar to the above discussion, radical loss to surfaces within the forest canopy can be estimated using Eq. (2), where A now represents the ratio of the canopy surface area to the height of the mixing layer. Previous measurements at the PROPHET site reported a leaf area index (LAI) of approximately 3.8 m2 m−2 (Ortega et al., 2007). Assuming a mixing layer height of 1500 m, this suggests that an HO2 uptake coefficient of would result in a first-order loss rate of 0.013 s−1, which could account for the proposed missing HO2 sink. This uptake coefficient is lower than that measured for many atmospheric aerosols but is similar to measurements of HO2 uptake on organic aerosols (Lakey et al., 2015). Similarly, an uptake coefficient of for isoprene peroxy radicals would result in a first-order loss rate of 0.024 s−1 and could account for the missing radical sink. These results imply that loss to surfaces within the canopy could be a substantial radical loss mechanism in dense forests where low NOx mixing ratios result in longer peroxy radical lifetimes that are on the order of the transport time through the canopy.
The daytime maximum measured OH radical concentrations during the PROPHET-AMOS campaign were generally in good agreement with model simulations using both the RACM2 and MCM v3.2, though both models overestimated the measured values in the morning. In contrast to previous measurements by the IU-FAGE instrument, no significant OH interferences were measured during the campaign, perhaps due to the lower temperatures and ozone concentrations, which seem to be correlated with unknown interferences associated with the LIF-FAGE technique (Lew et al., 2020). Including the LIM1 isoprene chemical mechanism into the RACM2-LIM1 and MCM v3.3.1 models increases the maximum modeled OH concentration by approximately 30 %, with the MCM v3.3.1 mechanism in better agreement with the measurements. These results are in contrast to previous measurements in forest environments, where the measurements were found to be significantly greater than model predictions (Rohrer et al., 2014).
Both the RACM2 and MCM models overpredict the measured daytime concentration of HO2 by approximately 50 % and the measured XO2 concentrations by approximately a factor of 2, similar to previous measurements at this site (Griffith et al., 2013). During the nighttime, the models are able to reproduce the measured HO2 concentrations but overestimate the measured XO2 radical concentrations by factors of approximately 3–5, with approximately 50 % of the nighttime total XO2 radical concentration composed of peroxy radicals derived from the ozonolysis of monoterpenes. The addition of the RO2+ RO2 accretion reactions to the models significantly reduces the predicted XO2 radical concentrations at night by up to 60 % due to the relatively large rate constants for the RO2 + RO2 accretion reactions of monoterpene-derived peroxy radicals. However, including these RO2+ RO2 accretion reactions does not significantly impact the modeled daytime peroxy radical concentrations when isoprene-based peroxy radicals dominate the total XO2 composition, as the reported RO2+ RO2 accretion rate constants for isoprene-based peroxy radicals are smaller compared to that for monoterpene-based peroxy radicals.
The models also overpredict the daytime measured concentrations of isoprene hydroxy hydroperoxide and isoprene hydroxy nitrates, consistent with an overprediction of the concentration of isoprene-based peroxy radicals. Constraining the model to the measured peroxy radical concentrations improves the agreement with the measured ISOPOOH and IHN concentrations. These results suggest that the measured radical concentrations are more consistent with the measured ISOPOOH and IHN concentrations, providing additional confidence in the accuracy of the HO2 and XO2 radical measurements, and suggest that the model is either overestimating the production of peroxy radicals or, more likely, underestimating their loss. The modeled OH concentrations and total OH reactivity were in good agreement with the measurements, suggesting that the model is not overestimating the production of peroxy radicals, including isoprene-based peroxy radicals.
To reproduce the measured peroxy radical concentrations, an additional loss process equivalent to the reaction of peroxy radicals with NO must be added to the model, accounting for approximately 60 % of the total rate of radical termination in the model. The additional loss processes could potentially include several components, such as direct surface deposition of radicals and important precursors to aerosols or the forest canopy, faster self- and cross-reactions between C5-RO2 and other RO2 species, reactions of peroxy radicals with isoprene and other alkenes, or vertical transport of peroxy radicals given their longer lifetime under the low NOx conditions at the PROPHET site. The overestimation of peroxy radical concentrations suggests that current atmospheric chemistry models may be overestimating the rate of production of ozone and other secondary products in similar low NOx areas impacted by isoprene emissions. Additional measurements and modeling studies are needed to resolve these discrepancies.
Data presented in this study can be obtained from the authors upon request (pstevens@indiana.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-10287-2023-supplement.
BB, ML, YW, PR, and PSS were responsible for the LIF-FAGE measurements of OH, HO2, and OH reactivity. BD, MDR, DCA, and EW were responsible for the ECHAMP measurements of XO2. HDA and DBM were responsible for the PTR-MS measurements of VOCs and OVOCs. SD and TL were responsible for the GC measurements of VOCs and OVOCs. AW, GT, JO, and DM were responsible for the measurements of NO and NO2. WW and DH were responsible for the measurements of O3 at the Ameriflux tower. JF, ME, and SA were responsible for the measurements of photolysis frequencies and CO. JR, JS, and FK were responsible for the measurements of formaldehyde. HMA was responsible for the measurements of ISOPOOH. JHS and PBS were responsible for the IHN measurements. SB was responsible for coordination and preparation of the PROPHET site. BB, ML, YW, PR, and PSS conducted the analysis and photochemical modeling and wrote the paper with feedback from all co-authors.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Joe Sakowski for his assistance with the OH, HO2, and OH reactivity measurements. We also thank Krystal Vasquez, Eric Praske, John Crounse, and Paul Wennberg for their hard effort obtaining the ISOPOOH measurements; Deedee Montzka for assistance in obtaining the NOx measurements; all PROPHET-AMOS participants for making this work possible; and the University of Michigan Biological Station for hosting the field study.
This research has been supported by the National Science Foundation (grant nos. AGS-1440834, AGS-1827450, AGS-1443842, AGS-1719918, AGS-1561755, AGS-1643306, AGS-1932771, and AGS-1428257).
This paper was edited by Lisa Whalley and reviewed by two anonymous referees.
Anderson, D. C., Pavelec, J., Daube, C., Herndon, S. C., Knighton, W. B., Lerner, B. M., Roscioli, J. R., Yacovitch, T. I., and Wood, E. C.: Characterization of ozone production in San Antonio, Texas, using measurements of total peroxy radicals, Atmos. Chem. Phys., 19, 2845–2860, https://doi.org/10.5194/acp-19-2845-2019, 2019.
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Atkinson, R., Hasegawa, D., and Aschmann, S. M.: Rate constants for the gas-phase reactions of O3 with a series of monoterpenes and related compounds at 296 ± 2 K, Int. J. Chem. Kinet., 22, 871–887, https://doi.org/10.1002/kin.550220807, 1990.
Badol, C., Borbon, A., Locoge, N., Léonardis, T., and Galloo, J. C.: An automated monitoring system for VOC ozone precursors in ambient air: development, implementation and data analysis, Anal. Bioanal. Chem, 378, 1815–1827, https://doi.org/10.1007/s00216-003-2474-0, 2004.
Baier, B. C., Brune, W. H., Miller, D. O., Blake, D., Long, R., Wisthaler, A., Cantrell, C., Fried, A., Heikes, B., Brown, S., McDuffie, E., Flocke, F., Apel, E., Kaser, L., and Weinheimer, A.: Higher measured than modeled ozone production at increased NOx levels in the Colorado Front Range, Atmos. Chem. Phys., 17, 11273–11292, https://doi.org/10.5194/acp-17-11273-2017, 2017.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bryan, A. M., Cheng, S. J., Ashworth, K., Guenther, A. B., Hardiman, B. S., Bohrer, G., and Steiner, A. L.: Forest-atmosphere BVOC exchange in diverse and structurally complex canopies: 1-D modeling of a mid-successional forest in northern Michigan, Atmos. Environ., 120, 217–226, https://doi.org/10.1016/j.atmosenv.2015.08.094, 2015.
Butler, T. M., Taraborrelli, D., Brühl, C., Fischer, H., Harder, H., Martinez, M., Williams, J., Lawrence, M. G., and Lelieveld, J.: Improved simulation of isoprene oxidation chemistry with the ECHAM5/MESSy chemistry-climate model: lessons from the GABRIEL airborne field campaign, Atmos. Chem. Phys., 8, 4529–4546, https://doi.org/10.5194/acp-8-4529-2008, 2008.
Cai, R., Yang, D., Fu, Y., Wang, X., Li, X., Ma, Y., Hao, J., Zheng, J., and Jiang, J.: Aerosol surface area concentration: a governing factor in new particle formation in Beijing, Atmos. Chem. Phys., 17, 12327–12340, https://doi.org/10.5194/acp-17-12327-2017, 2017.
Cantrell, C. A. and Stedman, D. H.: A possible technique for the measurement of atmospheric peroxy radicals, Geophys. Res. Lett., 9, 846–849, https://doi.org/10.1029/GL009i008p00846, 1982.
Cantrell, C. A., Shetter, R. E., and Calvert, J. G.: Dual-Inlet Chemical Amplifier for Atmospheric Peroxy Radical Measurements, Anal. Chem., 68, 4194–4199, https://doi.org/10.1021/ac960639e, 1996.
Carroll, M. A., Bertman, S. B., and Shepson, P. B.: Overview of the Program for Research on Oxidants: PHotochemistry, Emissions, and Transport (PROPHET) summer 1998 measurements intensive, J. Geophys. Res.-Atmos., 106, 24275–24288, https://doi.org/10.1029/2001JD900189, 2001.
Carslaw, N., Creasey, D. J., Harrison, D., Heard, D. E., Hunter, M. C., Jacobs, P. J., Jenkin, M. E., Lee, J. D., Lewis, A. C., Pilling, M. J., Saunders, S. M., and Seakins, P. W.: OH and HO2 radical chemistry in a forested region of north-western Greece, Atmos. Environ., 35, 4725–4737, https://doi.org/10.1016/S1352-2310(01)00089-9, 2001.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Crounse, J. D., Teng, A., and Wennberg, P. O.: Experimental constraints on the distribution and fate of peroxy radicals formed in reactions of isoprene + OH + O2, presented at Atmospheric Chemical Mechanisms: Simple Models – Real World Complexities, University of California, Davis, USA, 10–12 December, 2014.
Crowley, J. N., Pouvesle, N., Phillips, G. J., Axinte, R., Fischer, H., Petäjä, T., Nölscher, A., Williams, J., Hens, K., Harder, H., Martinez-Harder, M., Novelli, A., Kubistin, D., Bohn, B., and Lelieveld, J.: Insights into HOx and ROx chemistry in the boreal forest via measurement of peroxyacetic acid, peroxyacetic nitric anhydride (PAN) and hydrogen peroxide, Atmos. Chem. Phys., 18, 13457–13479, https://doi.org/10.5194/acp-18-13457-2018, 2018.
Davis, D. D., Rodgers, M. O., Fischer, S. D., and Asai, K.: An experimental assessment of the O3/H2O interference problem in the detection of natural levels of OH via laser induced fluorescence, Geophys. Res. Lett., 8, 69–72, https://doi.org/10.1029/GL008i001p00069, 1981a.
Davis, D. D., Rodgers, M. O., Fischer, S. D., and Heaps, W. S.: A theoretical assessment of the O3/H2O interference problem in the detection of natural levels of OH via laser induced fluorescence, Geophys. Res. Lett., 8, 73–76, https://doi.org/10.1029/GL008i001p00073, 1981b.
Dusanter, S., Vimal, D., and Stevens, P. S.: Technical note: Measuring tropospheric OH and HO2 by laser-induced fluorescence at low pressure. A comparison of calibration techniques, Atmos. Chem. Phys., 8, 321–340, https://doi.org/10.5194/acp-8-321-2008, 2008.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., and Molina, L. T.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 1: Deployment of the Indiana University laser-induced fluorescence instrument, Atmos. Chem. Phys., 9, 1665–1685, https://doi.org/10.5194/acp-9-1665-2009, 2009a.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009b.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Faloona, I., Tan, D., Brune, W., Hurst, J., Barket Jr, D., Couch, T. L., Shepson, P., Apel, E., Riemer, D., Thornberry, T., Carroll, M. A., Sillman, S., Keeler, G. J., Sagady, J., Hooper, D., and Paterson, K.: Nighttime observations of anomalously high levels of hydroxyl radicals above a deciduous forest canopy, J. Geophys. Res.-Atmos., 106, 24315–24333, https://doi.org/10.1029/2000JD900691, 2001.
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., DeGouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing Atmospheric Oxidation in an Alabama Forest, J. Atmos. Sci., 73, 4699–4710, https://doi.org/10.1175/JAS-D-16-0044.1, 2016.
Fittschen, C., Al Ajami, M., Batut, S., Ferracci, V., Archer-Nicholls, S., Archibald, A. T., and Schoemaecker, C.: ROOOH: a missing piece of the puzzle for OH measurements in low-NO environments?, Atmos. Chem. Phys., 19, 349–362, https://doi.org/10.5194/acp-19-349-2019, 2019.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013.
Fuchs, H., Tan, Z., Hofzumahaus, A., Broch, S., Dorn, H.-P., Holland, F., Künstler, C., Gomm, S., Rohrer, F., Schrade, S., Tillmann, R., and Wahner, A.: Investigation of potential interferences in the detection of atmospheric ROx radicals by laser-induced fluorescence under dark conditions, Atmos. Meas. Tech., 9, 1431–1447, https://doi.org/10.5194/amt-9-1431-2016, 2016.
George, I. J., Matthews, P. S. J., Whalley, L. K., Brooks, B., Goddard, A., Baeza-Romero, M. T., and Heard, D. E.: Measurements of uptake coefficients for heterogeneous loss of HO2 onto submicron inorganic salt aerosols, Phys. Chem. Chem. Phys., 15, 12829–12845, https://doi.org/10.1039/C3CP51831K, 2013.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Stevens, P. S., Alaghmand, M., Bertman, S. B., Carroll, M. A., Erickson, M., Galloway, M., Grossberg, N., Hottle, J., Hou, J., Jobson, B. T., Kammrath, A., Keutsch, F. N., Lefer, B. L., Mielke, L. H., O'Brien, A., Shepson, P. B., Thurlow, M., Wallace, W., Zhang, N., and Zhou, X. L.: OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 13, 5403–5423, https://doi.org/10.5194/acp-13-5403-2013, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res.-Atmos., 121, 4211–4232, https://doi.org/10.1002/2015JD024358, 2016.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hansen, R. F., Griffith, S. M., Dusanter, S., Rickly, P. S., Stevens, P. S., Bertman, S. B., Carroll, M. A., Erickson, M. H., Flynn, J. H., Grossberg, N., Jobson, B. T., Lefer, B. L., and Wallace, H. W.: Measurements of total hydroxyl radical reactivity during CABINEX 2009 – Part 1: field measurements, Atmos. Chem. Phys., 14, 2923–2937, https://doi.org/10.5194/acp-14-2923-2014, 2014.
Hansen, R. F., Blocquet, M., Schoemaecker, C., Léonardis, T., Locoge, N., Fittschen, C., Hanoune, B., Stevens, P. S., Sinha, V., and Dusanter, S.: Intercomparison of the comparative reactivity method (CRM) and pump–probe technique for measuring total OH reactivity in an urban environment, Atmos. Meas. Tech., 8, 4243–4264, https://doi.org/10.5194/amt-8-4243-2015, 2015.
Harley, P., Guenther, A., and Zimmerman, P.: Effects of light, temperature and canopy position on net photosynthesis and isoprene emission from sweetgum (Liquidambar styraciflua) leaves, Tree Physiol., 16, 25–32, https://doi.org/10.1093/treephys/16.1-2.25, 1996.
Hastie, D. R., Weissenmayer, M., Burrows, J. P., and Harris, G. W.: Calibrated chemical amplifier for atmospheric ROx measurements, Anal. Chem., 63, 2048–2057, https://doi.org/10.1021/ac00018a029, 1991.
Heard, D. E. and Pilling, M. J.: Measurement of OH and HO2 in the Troposphere, Chem. Rev., 103, 5163–5198, https://doi.org/10.1021/cr020522s, 2003.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified Trace Gas Removal in the Troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Horstjann, M., Andrés Hernández, M. D., Nenakhov, V., Chrobry, A., and Burrows, J. P.: Peroxy radical detection for airborne atmospheric measurements using absorption spectroscopy of NO2, Atmos. Meas. Tech., 7, 1245–1257, https://doi.org/10.5194/amt-7-1245-2014, 2014.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016.
Kanaya, Y., Cao, R., Akimoto, H., Fukuda, M., Komazaki, Y., Yokouchi, Y., Koike, M., Tanimoto, H., Takegawa, N., and Kondo, Y.: Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res.-Atmos., 112, D21312, https://doi.org/10.1029/2007JD008670, 2007a.
Kanaya, Y., Cao, R., Kato, S., Miyakawa, Y., Kajii, Y., Tanimoto, H., Yokouchi, Y., Mochida, M., Kawamura, K., and Akimoto, H.: Chemistry of OH and HO2 radicals observed at Rishiri Island, Japan, in September 2003: Missing daytime sink of HO2 and positive nighttime correlations with monoterpenes, J. Geophys. Res.-Atmos., 112, D11308, https://doi.org/10.1029/2006JD007987, 2007b.
Kanaya, Y., Hofzumahaus, A., Dorn, H.-P., Brauers, T., Fuchs, H., Holland, F., Rohrer, F., Bohn, B., Tillmann, R., Wegener, R., Wahner, A., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., Kleffmann, J., Elshorbany, Y., and Schurath, U.: Comparisons of observed and modeled OH and HO2 concentrations during the ambient measurement period of the HOxComp field campaign, Atmos. Chem. Phys., 12, 2567–2585, https://doi.org/10.5194/acp-12-2567-2012, 2012.
Khamaganov, V. G. and Hites, R. A.: Rate Constants for the Gas-Phase Reactions of Ozone with Isoprene, α- and β-Pinene, and Limonene as a Function of Temperature, J. Phys. Chem. A, 105, 815–822, https://doi.org/10.1021/jp002730z, 2001.
Kim, S., Guenther, A., Karl, T., and Greenberg, J.: Contributions of primary and secondary biogenic VOC tototal OH reactivity during the CABINEX (Community Atmosphere-Biosphere INteractions Experiments)-09 field campaign, Atmos. Chem. Phys., 11, 8613–8623, https://doi.org/10.5194/acp-11-8613-2011, 2011.
Kim, S., Wolfe, G. M., Mauldin, L., Cantrell, C., Guenther, A., Karl, T., Turnipseed, A., Greenberg, J., Hall, S. R., Ullmann, K., Apel, E., Hornbrook, R., Kajii, Y., Nakashima, Y., Keutsch, F. N., DiGangi, J. P., Henry, S. B., Kaser, L., Schnitzhofer, R., Graus, M., Hansel, A., Zheng, W., and Flocke, F. F.: Evaluation of HOx sources and cycling using measurement-constrained model calculations in a 2-methyl-3-butene-2-ol (MBO) and monoterpene (MT) dominated ecosystem, Atmos. Chem. Phys., 13, 2031–2044, https://doi.org/10.5194/acp-13-2031-2013, 2013.
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010.
Kundu, S., Deming, B. L., Lew, M. M., Bottorff, B. P., Rickly, P., Stevens, P. S., Dusanter, S., Sklaveniti, S., Leonardis, T., Locoge, N., and Wood, E. C.: Peroxy radical measurements by ethane – nitric oxide chemical amplification and laser-induced fluorescence during the IRRONIC field campaign in a forest in Indiana, Atmos. Chem. Phys., 19, 9563–9579, https://doi.org/10.5194/acp-19-9563-2019, 2019.
Lakey, P. S. J., George, I. J., Whalley, L. K., Baeza-Romero, M. T., and Heard, D. E.: Measurements of the HO2 Uptake Coefficients onto Single Component Organic Aerosols, Environ. Sci. Technol., 49, 4878–4885, https://doi.org/10.1021/acs.est.5b00948, 2015.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Lew, M. M., Dusanter, S., and Stevens, P. S.: Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 11, 95–109, https://doi.org/10.5194/amt-11-95-2018, 2018.
Lew, M. M., Rickly, P. S., Bottorff, B. P., Reidy, E., Sklaveniti, S., Léonardis, T., Locoge, N., Dusanter, S., Kundu, S., Wood, E., and Stevens, P. S.: OH and HO2 radical chemistry in a midlatitude forest: measurements and model comparisons, Atmos. Chem. Phys., 20, 9209–9230, https://doi.org/10.5194/acp-20-9209-2020, 2020.
Lightfoot, P. D., Cox, R. A., Crowley, J. N., Destriau, M., Hayman, G. D., Jenkin, M. E., Moortgat, G. K., and Zabel, F.: Organic peroxy radicals: Kinetics, spectroscopy and tropospheric chemistry, Atmos. Environ. A-Gen., 26, 1805–1961, https://doi.org/10.1016/0960-1686(92)90423-I, 1992.
Liu, Z., Nguyen, V. S., Harvey, J., Müller, J.-F., and Peeters, J.: Theoretically derived mechanisms of HPALD photolysis in isoprene oxidation, Phys. Chem. Chem. Phys., 19, 9096–9106, https://doi.org/10.1039/C7CP00288B, 2017.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Häseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Mallik, C., Tomsche, L., Bourtsoukidis, E., Crowley, J. N., Derstroff, B., Fischer, H., Hafermann, S., Hüser, I., Javed, U., Keßel, S., Lelieveld, J., Martinez, M., Meusel, H., Novelli, A., Phillips, G. J., Pozzer, A., Reiffs, A., Sander, R., Taraborrelli, D., Sauvage, C., Schuladen, J., Su, H., Williams, J., and Harder, H.: Oxidation processes in the eastern Mediterranean atmosphere: evidence from the modelling of HOx measurements over Cyprus, Atmos. Chem. Phys., 18, 10825–10847, https://doi.org/10.5194/acp-18-10825-2018, 2018.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Mayhew, A. W., Lee, B. H., Thornton, J. A., Bannan, T. J., Brean, J., Hopkins, J. R., Lee, J. D., Nelson, B. S., Percival, C., Rickard, A. R., Shaw, M. D., Edwards, P. M., and Hamilton, J. F.: Evaluation of isoprene nitrate chemistry in detailed chemical mechanisms, Atmos. Chem. Phys., 22, 14783–14798, https://doi.org/10.5194/acp-22-14783-2022, 2022.
Medeiros, D. J., Blitz, M. A., Seakins, P. W., and Whalley, L. K.: Direct Measurements of Isoprene Autoxidation: Pinpointing Atmospheric Oxidation in Tropical Forests, JACS Au, 2, 809–818, https://doi.org/10.1021/jacsau.1c00525, 2022.
Mihele, C. M. and Hastie, D. R.: Optimized Operation and Calibration Procedures for Radical Amplifier-Type Detectors, J. Atmos. Ocean. Technol., 17, 788–794, https://doi.org/10.1175/1520-0426(2000)017<0788:OOACPF>2.0.CO;2, 2000.
Mihele, C. M. and Hastie, D. R.: Radical chemistry at a forested continental site: Results from the PROPHET 1997 campaign, J. Geophys. Res.-Atmos., 108, 4450, https://doi.org/10.1029/2002JD002888, 2003.
Millet, D. B., Alwe, H. D., Chen, X., Deventer, M. J., Griffis, T. J., Holzinger, R., Bertman, S. B., Rickly, P. S., Stevens, P. S., Léonardis, T., Locoge, N., Dusanter, S., Tyndall, G. S., Alvarez, S. L., Erickson, M. H., and Flynn, J. H.: Bidirectional Ecosystem–Atmosphere Fluxes of Volatile Organic Compounds Across the Mass Spectrum: How Many Matter?, ACS Earth Space Chem., 2, 764–777, https://doi.org/10.1021/acsearthspacechem.8b00061, 2018.
Morales, A. C., Jayarathne, T., Slade, J. H., Laskin, A., and Shepson, P. B.: The production and hydrolysis of organic nitrates from OH radical oxidation of β-ocimene, Atmos. Chem. Phys., 21, 129–145, https://doi.org/10.5194/acp-21-129-2021, 2021.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Noell, A. C., Alconcel, L. S., Robichaud, D. J., Okumura, M., and Sander, S. P.: Near-Infrared Kinetic Spectroscopy of the HO2 and C2H5O2 Self-Reactions and Cross Reactions, J. Phys. Chem. A, 114, 6983–6995, https://doi.org/10.1021/jp912129j, 2010.
Novelli, A., Vereecken, L., Lelieveld, J., and Harder, H.: Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 16, 19941–19951, https://doi.org/10.1039/C4CP02719A, 2014a.
Novelli, A., Hens, K., Tatum Ernest, C., Kubistin, D., Regelin, E., Elste, T., Plass-Dülmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014b.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020.
Nozière, B. and Fache, F.: Reactions of organic peroxy radicals, RO2, with substituted and biogenic alkenes at room temperature: unsuspected sinks for some RO2 in the atmosphere?, Chem. Sci., 12, 11676–11683, https://doi.org/10.1039/D1SC02263F, 2021.
Nozière, B., Durif, O., Dubus, E., Kylington, S., Emmer, Å., Fache, F., Piel, F., and Wisthaler, A.: The Reaction of Organic Peroxy Radicals with Unsaturated Compounds Controlled by a non-Epoxide Pathway under Atmospheric Conditions, Phys. Chem. Chem. Phys., 25, 7772–7782, https://doi.org/10.1039/D2CP05166D, 2023.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/C2CS35166H, 2012.
Ortega, J., Helmig, D., Guenther, A., Harley, P., Pressley, S., and Vogel, C.: Flux estimates and OH reaction potential of reactive biogenic volatile organic compounds (BVOCs) from a mixed northern hardwood forest, Atmos. Environ., 41, 5479–5495, https://doi.org/10.1016/j.atmosenv.2006.12.033, 2007.
Ouwersloot, H. G., Vilà-Guerau de Arellano, J., van Heerwaarden, C. C., Ganzeveld, L. N., Krol, M. C., and Lelieveld, J.: On the segregation of chemical species in a clear boundary layer over heterogeneous land surfaces, Atmos. Chem. Phys., 11, 10681–10704, https://doi.org/10.5194/acp-11-10681-2011, 2011.
Owen, S. M., Harley, P., Guenther, A., and Hewitt, C. N.: Light dependency of VOC emissions from selected Mediterranean plant species, Atmos. Environ., 36, 3147–3159, https://doi.org/10.1016/S1352-2310(02)00235-2, 2002.
Peeters, J.: Interactive comment on “The MCM v3.3 degradation scheme for isoprene” by M.E. Jenkin et al., Atmospheric Chemistry and Physics Discussions, 15, C2486, https://acp.copernicus.org/preprints/15/C2486/2015/acpd-15-C2486-2015-supplement.pdf (last access: 1 June 2023), 2015.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/B908511D, 2009.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Pugh, T. A. M., MacKenzie, A. R., Langford, B., Nemitz, E., Misztal, P. K., and Hewitt, C. N.: The influence of small-scale variations in isoprene concentrations on atmospheric chemistry over a tropical rainforest, Atmos. Chem. Phys., 11, 4121–4134, https://doi.org/10.5194/acp-11-4121-2011, 2011.
Ravishankara, A. R.: Heterogeneous and Multiphase Chemistry in the Troposphere, Science, 276, 1058–1065, https://doi.org/10.1126/science.276.5315.1058, 1997.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X., and Gao, H.: OH and HO2 Chemistry in the urban atmosphere of New York City, Atmos. Environ., 37, 3639–3651, https://doi.org/10.1016/S1352-2310(03)00459-X, 2003.
Ren, X., Brune, W. H., Oliger, A., Metcalf, A. R., Simpas, J. B., Shirley, T., Schwab, J. J., Bai, C., Roychowdhury, U., Li, Y., Cai, C., Demerjian, K. L., He, Y., Zhou, X., Gao, H., and Hou, J.: OH, HO2, and OH reactivity during the PMTACS–NY Whiteface Mountain 2002 campaign: Observations and model comparison, J. Geophys. Res.-Atmos., 111, D10S03, https://doi.org/10.1029/2005JD006126, 2006.
Ren, X., van Duin, D., Cazorla, M., Chen, S., Mao, J., Zhang, L., Brune, W. H., Flynn, J. H., Grossberg, N., Lefer, B. L., Rappenglück, B., Wong, K. W., Tsai, C., Stutz, J., Dibb, J. E., Thomas Jobson, B., Luke, W. T., and Kelley, P.: Atmospheric oxidation chemistry and ozone production: Results from SHARP 2009 in Houston, Texas, J. Geophys. Res.-Atmos., 118, 5770–5780, https://doi.org/10.1002/jgrd.50342, 2013.
Rickly, P. and Stevens, P. S.: Measurements of a potential interference with laser-induced fluorescence measurements of ambient OH from the ozonolysis of biogenic alkenes, Atmos. Meas. Tech., 11, 1–16, https://doi.org/10.5194/amt-11-1-2018, 2018.
Ridley, B. and Grahek, F.: A Small, Low Flow, High Sensitivity Reaction Vessel for NO Chemiluminescence Detectors, J. Atmos. Ocean. Technol., 7, 307-311, https://doi.org/10.1175/1520-0426(1990)007<0307:ASLFHS>2.0.CO;2, 1990.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Häseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559–563, https://doi.org/10.1038/ngeo2199, 2014.
Roukos, J., Plaisance, H., Leonardis, T., Bates, M., and Locoge, N.: Development and validation of an automated monitoring system for oxygenated volatile organic compounds and nitrile compounds in ambient air, J. Chromatogr. A, 1216, 8642–8651, https://doi.org/10.1016/j.chroma.2009.10.018, 2009.
Sanchez, D., Jeong, D., Seco, R., Wrangham, I., Park, J.-H., Brune, W. H., Koss, A., Gilman, J., de Gouw, J., Misztal, P., Goldstein, A., Baumann, K., Wennberg, P. O., Keutsch, F. N., Guenther, A., and Kim, S.: Intercomparison of OH and OH reactivity measurements in a high isoprene and low NO environment during the Southern Oxidant and Aerosol Study (SOAS), Atmos. Environ., 174, 227–236, https://doi.org/10.1016/j.atmosenv.2017.10.056, 2018.
Shetter, R. E. and Müller, M.: Photolysis frequency measurements using actinic flux spectroradiometry during the PEM-Tropics mission: Instrumentation description and some results, J. Geophys. Res.-Atmos., 104, 5647–5661, https://doi.org/10.1029/98JD01381, 1999.
Shirley, T. R., Brune, W. H., Ren, X., Mao, J., Lesher, R., Cardenas, B., Volkamer, R., Molina, L. T., Molina, M. J., Lamb, B., Velasco, E., Jobson, T., and Alexander, M.: Atmospheric oxidation in the Mexico City Metropolitan Area (MCMA) during April 2003, Atmos. Chem. Phys., 6, 2753–2765, https://doi.org/10.5194/acp-6-2753-2006, 2006.
Sillman, S., Carroll, M. A., Thornberry, T., Lamb, B. K., Westberg, H., Brune, W. H., Faloona, I., Tan, D., Shepson, P. B., Sumner, A. L., Hastie, D. R., Mihele, C. M., Apel, E. C., Riemer, D. D., and Zika, R. G.: Loss of isoprene and sources of nighttime OH radicals at a rural site in the United States: Results from photochemical models, J. Geophys. Res.-Atmos., 107, ACH 2-1–ACH 2-14, https://doi.org/10.1029/2001JD000449, 2002.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Taketani, F., Kanaya, Y., and Akimoto, H.: Kinetics of Heterogeneous Reactions of HO2 Radical at Ambient Concentration Levels with (NH4)2SO4 and NaCl Aerosol Particles, J. Phys. Chem. A, 112, 2370–2377, https://doi.org/10.1021/jp0769936, 2008.
Taketani, F., Kanaya, Y., Pochanart, P., Liu, Y., Li, J., Okuzawa, K., Kawamura, K., Wang, Z., and Akimoto, H.: Measurement of overall uptake coefficients for HO2 radicals by aerosol particles sampled from ambient air at Mts. Tai and Mang (China), Atmos. Chem. Phys., 12, 11907–11916, https://doi.org/10.5194/acp-12-11907-2012, 2012.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOx budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign, J. Geophys. Res.-Atmos., 106, 24407–24427, https://doi.org/10.1029/2001JD900016, 2001.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Thornton, J. A., Jaeglé, L., and McNeill, V. F.: Assessing known pathways for HO2 loss in aqueous atmospheric aerosols: Regional and global impacts on tropospheric oxidants, J. Geophys. Res.-Atmos., 113, D05303, https://doi.org/10.1029/2007JD009236, 2008.
Tyndall, G. S., Cox, R. A., Granier, C., Lesclaux, R., Moortgat, G. K., Pilling, M. J., Ravishankara, A. R., and Wallington, T. J.: Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res.-Atmos., 106, 12157–12182, https://doi.org/10.1029/2000JD900746, 2001.
Vasquez, K. T., Allen, H. M., Crounse, J. D., Praske, E., Xu, L., Noelscher, A. C., and Wennberg, P. O.: Low-pressure gas chromatography with chemical ionization mass spectrometry for quantification of multifunctional organic compounds in the atmosphere, Atmos. Meas. Tech., 11, 6815–6832, https://doi.org/10.5194/amt-11-6815-2018, 2018.
Wang, Y., Piletic, I. R., Takeuchi, M., Xu, T., France, S., and Ng, N. L.: Synthesis and Hydrolysis of Atmospherically Relevant Monoterpene-Derived Organic Nitrates, Environ. Sci. Technol., 55, 14595–14606, https://doi.org/10.1021/acs.est.1c05310, 2021.
Wei, D., Alwe, H. D., Millet, D. B., Bottorff, B., Lew, M., Stevens, P. S., Shutter, J. D., Cox, J. L., Keutsch, F. N., Shi, Q., Kavassalis, S. C., Murphy, J. G., Vasquez, K. T., Allen, H. M., Praske, E., Crounse, J. D., Wennberg, P. O., Shepson, P. B., Bui, A. A. T., Wallace, H. W., Griffin, R. J., May, N. W., Connor, M., Slade, J. H., Pratt, K. A., Wood, E. C., Rollings, M., Deming, B. L., Anderson, D. C., and Steiner, A. L.: FORest Canopy Atmosphere Transfer (FORCAsT) 2.0: model updates and evaluation with observations at a mixed forest site, Geosci. Model Dev., 14, 6309–6329, https://doi.org/10.5194/gmd-14-6309-2021, 2021.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L. K., Furneaux, K. L., Goddard, A., Lee, J. D., Mahajan, A., Oetjen, H., Read, K. A., Kaaden, N., Carpenter, L. J., Lewis, A. C., Plane, J. M. C., Saltzman, E. S., Wiedensohler, A., and Heard, D. E.: The chemistry of OH and HO2 radicals in the boundary layer over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 1555–1576, https://doi.org/10.5194/acp-10-1555-2010, 2010.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Percival, C. J., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys., 21, 2125–2147, https://doi.org/10.5194/acp-21-2125-2021, 2021.
Wolfe, G. M., Thornton, J. A., Bouvier-Brown, N. C., Goldstein, A. H., Park, J.-H., McKay, M., Matross, D. M., Mao, J., Brune, W. H., LaFranchi, B. W., Browne, E. C., Min, K.-E., Wooldridge, P. J., Cohen, R. C., Crounse, J. D., Faloona, I. C., Gilman, J. B., Kuster, W. C., de Gouw, J. A., Huisman, A., and Keutsch, F. N.: The Chemistry of Atmosphere-Forest Exchange (CAFE) Model – Part 2: Application to BEARPEX-2007 observations, Atmos. Chem. Phys., 11, 1269–1294, https://doi.org/10.5194/acp-11-1269-2011, 2011.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wood, E. C. and Charest, J. R.: Chemical Amplification – Cavity Attenuated Phase Shift Spectroscopy Measurements of Atmospheric Peroxy Radicals, Anal. Chem., 86, 10266–10273, https://doi.org/10.1021/ac502451m, 2014.
Wood, E. C., Deming, B. L., and Kundu, S.: Ethane-Based Chemical Amplification Measurement Technique for Atmospheric Peroxy Radicals, Environ. Sci. Tech. Lett., 4, 15–19, https://doi.org/10.1021/acs.estlett.6b00438, 2017.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Zha, Q., Yan, C., Junninen, H., Riva, M., Sarnela, N., Aalto, J., Quéléver, L., Schallhart, S., Dada, L., Heikkinen, L., Peräkylä, O., Zou, J., Rose, C., Wang, Y., Mammarella, I., Katul, G., Vesala, T., Worsnop, D. R., Kulmala, M., Petäjä, T., Bianchi, F., and Ehn, M.: Vertical characterization of highly oxygenated molecules (HOMs) below and above a boreal forest canopy, Atmos. Chem. Phys., 18, 17437–17450, https://doi.org/10.5194/acp-18-17437-2018, 2018.
Zhou, J., Sato, K., Bai, Y., Fukusaki, Y., Kousa, Y., Ramasamy, S., Takami, A., Yoshino, A., Nakayama, T., Sadanaga, Y., Nakashima, Y., Li, J., Murano, K., Kohno, N., Sakamoto, Y., and Kajii, Y.: Kinetics and impacting factors of HO2 uptake onto submicron atmospheric aerosols during the 2019 Air QUAlity Study (AQUAS) in Yokohama, Japan , Atmos. Chem. Phys., 21, 12243–12260, https://doi.org/10.5194/acp-21-12243-2021, 2021.