the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 15 Jul 2022
| 15 Jul 2022
A novel pathway of atmospheric sulfate formation through carbonate radicals
Yangyang Liu
Yue Deng
Jiarong Liu
Xiaozhong Fang
Tao Wang
Kejian Li
Kedong Gong
Aziz U. Bacha
Iqra Nabi
Qiuyue Ge
Xiuhui Zhang
Christian George
Carbon dioxide is considered an inert gas that rarely participates in atmospheric chemical reactions. Nonetheless, we show here that CO2 is involved in some important photo-oxidation reactions in the atmosphere through the formation of carbonate radicals (CO). This potentially active intermediate CO is routinely overlooked in atmospheric chemistry concerning its effect on sulfate formation. The present work demonstrates that the SO2 uptake coefficient is enhanced by 17 times on mineral dust particles driven by CO. Importantly, upon irradiation, mineral dust particles are speculated to produce gas-phase carbonate radical ions when the atmospherically relevant concentration of CO2 presents, thereby potentially promoting external sulfate aerosol formation and oxidative potential in the atmosphere. Employing a suite of laboratory investigations of sulfate formation in the presence of carbonate radicals on the model and authentic dust particles, ground-based field measurements of sulfate and (bi)carbonate ions within ambient PM, together with density functional theory (DFT) calculations for single electron transfer processes in terms of CO-initiated S(IV) oxidation, a novel role of carbonate radical in atmospheric chemistry is elucidated.
- Article
(9607 KB) - Full-text XML
-
Supplement
(2084 KB) - BibTeX
- EndNote





Atmospheric composition changes are subjected to highly reactive light-induced radicals, such as hydroxyl (•OH), hydroperoxyl (HO2•), or nitrate radicals (NO3•), which are able to alter not only compositions but also the physical and chemical properties of particulate matter (T. Liu et al., 2022; Stevenson et al., 2020; Mahajan et al., 2021). However, when atmospheric chemical reactions occur over humified particles at ambient conditions, which creates a locally enriched aqueous medium of unique chemical activity, other radicals might likewise gain importance. The carbonate radical (CO) is typically such an active radical. The lifetime of CO ranges from a microsecond to even a few milliseconds, and its concentration can be 2 orders of magnitude higher than that of hydroxyl radicals over the water surface (Chandrasekaran and Thomas, 1983; Goldstein et al., 2001; Shafirovich et al., 2001; Sulzberger et al., 1997). In addition, the one electron reduction potential of the E0(CO CO) coupling is 1.78 V vs. a normal hydrogen electrode (NHE) at neutral pH, leaving CO a strong oxidant in aquatic chemistry (Cope et al., 1973; Bisby et al., 1998; Merouani et al., 2010). Previous studies concerning carbonate radical in aqueous media demonstrate that it reacts rapidly with some organic compounds (Merouani et al., 2010), especially for those electron-rich amine compounds (Stenman et al., 2003; Yan et al., 2019). Also, it has been pointed out that the scavenging of hydroxyl radicals by (bi)carbonate species leads to the formation of CO ions (Graedel and Weschler, 1981; Xiong et al., 2016). Besides, a high second-order rate constant, lying at 109 M−1 s−1, has been reported for the reaction of CO with porphyrins (Ferrer-Sueta et al., 2003), indicating that this radical ion has great oxidation capability that may trigger atmospherically relevant chemical reactions, i.e., secondary inorganic species formation. However, it is only regarded as a marginal intermediate in tropospheric anion chemistry so far (Lehtipalo et al., 2016; Beig and Brasseur, 2000; Herrmann et al., 2000; Ge et al., 2021), and its underlying role as an active oxidant for heterogeneous reaction in the atmosphere is barely explored. Very recently, our group observed the promotional effect of CO on atmospheric nitrate formation (Fang et al., 2021). Motivated by this finding, attempts were made to further explore its role in other important, atmospherically relevant reactions.
It is well documented that sulfate (SO) is also a key constituent of aerosols in the atmosphere (Huang et al., 2015; Su et al., 2016). It is able to serve as the precursor of efficient cloud condensation nuclei, with optical properties leading to a cooling effect (Wang et al., 2011). As a consequence, the mechanism aspect of secondary sulfate formation was the focus of numerous studies over the past decades (Hung et al., 2018; Stone, 2002; Zheng et al., 2015). There is a consensus that high-valence sulfur (VI), produced from the oxidation of anthropogenic SO2, is the dominant source of atmospheric secondary sulfate. However, a remarkable missing sulfate budget emerges for the atmospheric modeling (Huang et al., 2019; Liu et al., 2021; Itahashi et al., 2018), which significantly underpredicts SO with respect to observational results when heterogeneous aerosol chemistry is not considered (Zheng et al., 2015; Feng et al., 2018; Wu et al., 2021). This indicates that the heterogeneous sulfate production pathway is a crucial process, and exploring the unrecognized heterogeneous mechanism is very likely to narrow the gap between observations in lab studies, field measurements, and numerical modeling. Due to the missing chemical mechanism that initiated a fast SO2 oxidation, atmospheric models fail to capture the key feature of atmospheric observations of high sulfate production during dust storm episodes in the troposphere (Yu et al., 2020; Dong et al., 2016; Huang et al., 2014), where an evident increase in Ca2+ (Wang et al., 2005; Li et al., 2013), carbonate-containing particles with high alkalinity (Tang et al., 2016; Li et al., 2014; Abou-Ghanem et al., 2020), and photoactive mineral components (Nie et al., 2012; Ta et al., 2003) are prevalent. Air mass is usually in low relative humidity, reportedly being 25 %–35 % (Al-Salihi and Mohammed, 2015; Csavina et al., 2014; Najafpour et al., 2020) in these events, during which the photochemical process is able to alter atmospheric constituents (X. C. Liu et al., 2022). Consequently, there are unknown heterogeneous reaction pathways and previously unconsidered promoters that have great potential to accelerate sulfate formation in the dust-storm-relevant conditions.
Due to the high stability of CO2 under ambient conditions (Hossain et al., 2020), there are rare studies concerning the influence of CO2 in atmospheric chemical processes (Deng et al., 2020; Y. Liu et al., 2020; Xia et al., 2021). CO2 is demonstrated to form (bi)carbonate species over humidified dust particles (Baltrusaitis et al., 2011; Nanayakkara et al., 2014) and is reduced to CO under solar illumination (Deng et al., 2020). Nonetheless, its impact on atmospheric heterogeneous reactions remains poorly characterized. Our early laboratory study illustrates that CO2 decreases the sulfate formation on aluminum oxide particles in the dark (Y. Liu et al., 2020), while upon solar illumination its role in SO2 oxidation over mineral dust surfaces is still an open question. In addition, carbonate salt is abundant in authentic dust particles (Cao et al., 2005) and is reported to reach over 10 wt % of Asian dust particles (Mcnaughton et al., 2009). It is generally accepted that CO affects atmospheric chemistry and aerosol characteristics mainly through its intrinsic alkalinity, which buffers aerosol acidity and increases SO2 adsorption and corresponding sulfate production in the presence of oxidants (Bao et al., 2010; Kerminen et al., 2001; Yu et al., 2018; Li et al., 2007; Al-Hosney and Grassian, 2005). In fact, either CO2 or carbonate salt is able to produce the active CO under the ambient circumstance (Graedel and Weschler, 1981; Ervens et al., 2003) and is prone to increase the oxidative capacity in the atmosphere. Our early study shows that carbonate radicals serve as an active oxidant to accelerate NO2 oxidation over mineral dust particles (Fang et al., 2021), allowing us to consider the possibility that fast heterogeneous SO2 oxidation can be triggered by this active intermediate as well. Nevertheless, to the best of our knowledge, no work has ever considered how and to what extent the carbonate radical influences SO2 heterogeneous oxidation in the atmosphere.
In the current study, through laboratory studies, we present that carbon dioxide and calcium carbonate, working as the precursor of carbonate radicals, have the ability to accelerate sulfate formation over authentic particles in the atmosphere. Together with quantum chemistry calculations, a detailed molecular mechanism regarding a single electron transfer (SET) process between carbonate radical and sulfite ions is elucidated. Furthermore, ground-based observations validate some findings from the laboratory-based simulations.
2.1 Laboratory studies
2.1.1 Methodology for uptake coefficient estimation
The reaction uptake coefficient was estimated by the following Eqs. (1)–(3), as suggested by the previous work (Kong et al., 2014):
where ν is the mean molecular velocity of SO2, As is the effective sample surface, R is the gas constant, T is the temperature, is the molecular weight of SO2, and Z is a total number of surface collisions per unit of time. To be precise, the formation rates () in the equation were determined by ion chromatography (IC) measurements, followed by a conversion factor calculation through linear regression analysis for the integrated absorbance of sulfate bands and corresponding sulfate concentrations. By employing this method, a conversion factor of f = 6.34 × 1015 ions per integrated absorption unit was obtained, and the corresponding SO2 uptake coefficients in the TiO2 + (CO2) + SO2 system were thus estimated using seven time points during the heterogeneous reaction. For SO2 uptake in the TiO2 + (CaCO3) + SO2 system, we estimated SO2 uptake coefficients using three conversion curves established for various types of dust particles. For this purpose, we mixed the known proportion of K2SO4 and dust particles of concern and thus obtained relationship curves between the integrated absorbance of DRIFTS (i.e., diffuse reflectance Fourier transformed infrared spectroscopy) sulfate bands and corresponding theoretical sulfate content through linear fitting (Fig. S4). In total, 10 time points during the heterogeneous reaction were applied for these kinetic calculations.
2.1.2 Preparation for clay and dust membranes and investigation of sulfate formation on those authentic particles during the daytime and nighttime
Each particle suspension for Arizona test dust (ATD; 2.5 mg per 0.5 mL), IMt-2 (Illite, Montana, USA; 10 mg per 0.5 mL) and K-GA (Kaolin, Georgia, USA; 10 mg per 0.5 mL) was first dispersed into water through an ultrasonic bath for 5 min. After that, sample suspensions were transformed onto the cleansed round quartz films (d = 2 cm) using a pipette and subsequently sent to the infrared drying oven for 10 min to prepare the dust membranes. Once taken out from the oven, samples were quickly sealed into a desiccator and cooled down to room temperature before starting the experiments. A membrane sample was then mounted at the center of the reaction chamber (the top half is made of quartz, and the bottom half is made of Teflon). Before each set of experiments, a gas flow (dry air) of 300 mL min−1 was introduced to the chamber for 5 min where a prepared membrane sample was installed. Afterward, samples were exposed to a 4.91 × 1014 molec. cm−3 SO2 (+2.46 × 1018 CO2 when necessary) N2 + O2 mixture in the absence and presence of irradiation (light intensity (I) of 30 mW cm−2) for 15 min before the sample were transferred to a beaker (scale of 10 mL), with 2 mL of 2 % vol. isopropanol leaching solution, then ions were extracted in the ultrasonic tank for 5 min using 0.22 µm PTFE membrane filter, followed by sending the sample into IC. Noting that dust and clay particles possess a considerable volume of sulfate ion in the background, we thus measured the background ions for each batch of synthesized dust particle membranes following the procedures described above. All data demonstrated in Fig. 6 were obtained after the subtraction of background ions.
2.1.3 Determination of gas-phase reactive oxygen species (ROS) production in the flow cell reactor
To measure the concentration of ROS released from TiO2 particles in various reaction systems, an experimental approach using the probe molecule aniline was applied in this study. This is because compound aniline is reported to react rapidly with •OH radicals and CO radicals, which are also evidenced to be two major active ROS species that are responsible for the SO2 oxidation over mineral dust particles. The method applied in this study was almost implemented in the same way as that of the previous study (Behrman, 2018), with a slight modification. Briefly, the degradation rate of aniline in various reaction systems was monitored through high-performance liquid chromatography (HPLC; LC-10AD, Shimadzu, Japan). A Zorbax StableBond (SB) C18 (4.6 mm × 150 mm, 5 µm) reverse-phase column at 25∘ was used with a UV detector at 236 nm to measure the aniline concentration. The mobile phase consisted of an acetonitrile/water ratio of 55 : 45 (), with a flow rate of 1 mL min−1.
The TiO2 suspension (5 mg TiO2 per 100 µL deionized water) was deposited onto the glass substrate (0.13–0.17 mm in thickness) using a pipette and then dried in an oven for 10 min to obtain a TiO2-coated film. Dilute aniline solution, using a 67 mM phosphate buffer solution (pH of 7.0), was prepared and placed below the TiO2-coated film, with an intervening gap between the TiO2 film and solution surface around 2 mm (Fig. 1). This short distance essentially guarantees that gaseous ROS (e.g., •OH radicals or CO radicals) will diffuse and react with aniline molecule (Rodriguez et al., 2013).
In the reaction system containing a TiO2 film upon irradiation (the UV wavelength of 310 nm) in the presence of humidified air (RH of 30 %), when operated in a continuous mode, the overall degradation rate of the probe molecules in the presence of the TiO2 film can be described by Eq. (4), as follows (Wang et al., 2004):
where kobs is the observed degradation rate of aniline, [An] is the concentration of aniline, denoted as [An] hereafter, and rA, rU, and rROS stand for aniline removal rates resulting from air stripping, UV photolysis, and ROS oxidation. kROS, An are the overall second-order reaction rate constants for An with ROS.
Reference experiments without the introduction of ROS were also conducted to measure rA+rU in each reaction system. Upon irradiation, the dust proxy TiO2 produces hole–electron pairs, further forming •OH radicals and superoxide radicals (O) in the presence of absorbed water and oxygen molecules. Thus, an experiment using N2 was adopted to investigate the role of O in consuming aniline. As illustrated in Fig. 7c, a slight change in the degradation rate of aniline after stripping oxygen from the air, indicating that O shows quite a smaller contribution than •OH. This result agrees well with the finding reported by Durán et al. (2019), where the removal of O by adding benzoquinone (BQ) into TiO2 suspension results in a negligible change in the An degradation rate.
Taken above, •OH radicals are assumed to be the only active ROS that accounts for the An degradation. Hence, the maximum steady concentration of •OH radicals can be given by the following equation:
The integration of Eq. (5) yields the following:
Together with the reported second-order rate constant (k•OH, An = 6.5 × 109 M−1 s−1) (Samuni et al., 2002), the steady-state OH radical concentration [•OH]ss-max in a buffered An solution can be calculated from Eq. (7). The observed degradation rate constant of kexp can be obtained from the slope of the semi-log plot of an An degradation, as illustrated in Eq. (6). The maximum steady-state aqueous concentration of •OH supplied by the partitioning process from gas-phase •OH was thus estimated to be 2.15 × 10−15 M for the TiO2 + Air system.
When CO2 (400 ppm, parts per million; atmospherically relevant concentration) is introduced into a flow cell chamber, an increased degradation rate of An is seen, which is very likely to be the generation of active carbonate radical ions (Fig. 9). Similar to the method we adopted for the estimation of [•OH]ss-max, reference experiments were conducted to determine the rates for air stripping and UV photolysis processes in the TiO2 + Air+CO2 system. In the next step, we quenched the hydroxyl radicals by adding tertiary butanol (TBA). This is because it reacts rapidly with hydroxyl radicals (Li et al., 2020) k•OH,TBA = (6 × 108 M−1 s−1), while showing a rather low reaction rate with carbonate radicals (Liu et al., 2015) ( < 1.6 × 102 M−1 s−1). Subsequently, we determined using the previous protocol (Huang and Mabury, 2000) with known (5.4 × 108 M−1 s−1) (Wojnarovits et al., 2020). In the extreme case, assuming that all hydroxyl radical ions were fully trapped by absorbed and dissolved HCO CO, the maximum steady-state CO concentration was determined to be 1.39 × 10−13 M for the TiO2 + Air+CO2 system, matching well with an earlier study where the concentration of CO is reported to be 2 orders of magnitudes more than •OH over the water surface (Sulzberger et al., 1997).
2.2 Quantum chemical calculation
We employed density functional theory (DFT) calculations in the term of the single electron transfer (SET) process using the Gaussian 09 package to investigate this novel route (detailed in Texts S11 and S20 in the Supplement).
2.3 Field observations
2.3.1 Sampling
Our sampling for atmospheric particle matter was launched on the roof of the Department of Environmental Science and Engineering, Fudan University (Jiangwan Campus, 31.340661∘ N, 121.506747∘ E, 16 km away from the city downtown). More geographical information for sampling has been described in detail elsewhere (Y. Liu et al., 2020). Observations for the water-soluble ionic components of particulate matter were performed using an eight-stage non-viable cascade-impactor-type sampler (Tisch Environmental Inc., USA), the size gradients of which are in the sequence of < 0.43, 0.43–0.65, 0.65–1.1, 1.1–2.1, 2.1–.3, 3.3–4.7, 4.7–5.8, 5.8–9.0, and ∼ 9.0 µm. These sizes represent the effective cutoff diameter at each level for unit density spherical particles. In our sampling, the estimations for (bi)carbonate ions in atmospheric particulate matter is conducted for the initial four stages (∼ 3.3 µm). Atmospheric airflow from the head was maintained at the constant rate of 28.3 L min−1 to meet the operation criterion required for the Anderson-type sampler. Quartz filters (81 mm in diameter, Whatman, GE Healthcare, UK) were applied for the sampling, and the membranes were rinsed with ultrapure water (electrical resistivity of 18.2 MΩ) no fewer than three times; that is, they were kept in the ultrasonic cleaning tank for 40 min, then rinsed with ultrapure water twice, before being sent into the infrared drying oven, followed by packing them in aluminum foil prior to the field sampling. We carried out this procedure to eliminate the water-soluble background ions as much as possible and to ensure the balance calculation for (bi)carbonate ions. Additionally, following the same aforementioned pretreatments, we measured the background concentrations of ions in blank membranes. We separated our daily samplings into two periods of 11 h each to give an insight into the influence of potential photoinduced reactions on secondary sulfate formation in the atmosphere.
2.3.2 (Bi)carbonate estimation
The concentration of bi(carbonate) ions were estimated following the protocol reported in the early works (Zhang et al., 2011; Palmer and Cherry, 1984; Fang et al., 2021; Y. Liu et al., 2020). The following two assumptions were made to simplify the estimation for bi(carbonate) concentrations within the system: (a) dominating cations in each system are H+, Li+, Na+, NH, K+, Mg2+, and Ca2+, whereas those transition and heavy metal ions were out of consideration, considering their limited contents of dissolve cations in the atmospheric particulate matter. (b) There were three typical organic acid ions (CH3COO−, COOH−, and C2O, which are major soluble organic acid ions in the atmosphere) taken into account for the ionization balance and the rest of the charge gap in each system was assumed to originate from (bi)carbonate ions. Then we established balance equations (Eqs. 8–11) for each sample on the basis of charge conservation and ionization equilibrium constant of carbonic acid (K1 = 4.47 × 10−7 and K2 = 4.69 × 10−11 at 273 K).
It is worth mentioning that [X] is referring to the charge concentration for ions (coulomb M). Additionally, the temperature factor was also considered to correct the equilibrium constant for (bi)carbonate ions using the following equation:
where ΔH is the temperature variation (K), R is the ideal gas constant (8.31451 J mol−1 K−1), and T is the temperature (K) during pH measurements. We then solved those equations to obtain a series of , which were eventually corrected by subtracting blank values.
2.4 Other measurements and analysis
In addition to the above descriptions of measurements and calculation methodologies, more information, including the heterogenous reaction setup, kinetics reaction order determination, pretreatment of mineral dust, DRIFTS, IC, Raman measurements and analysis, etc., is available in Texts S1–S13 in the Supplement.
3.1 Accelerated sulfate production in the presence of carbonate
The physicochemical properties of the employed mineral dust proxies, including Brunauer–Emmett–Teller (BET) surface area, crystal phase, and structure, were first characterized (Fig. S1), consistent with early studies (Su et al., 2008; Balachandran and Eror, 1982; Shang et al., 2010), with an additional discussion in Text S14. The spectral irradiance of the solar simulator applied in the present study is well covered by natural sunlight (Fig. S2), and we tried as much as possible to have the experimental results from the lab simulate the real atmosphere. Upon solar irradiation under RH of 30 % SO2 N2 + O2 flow ([SO2] = 2.21 × 1014 molec. cm−3), the sulfate production on TiO2 + CaCO3 mixture particles (50 wt % TiO2 and 50 wt % CaCO3), measured by IC, is significantly enhanced by 7 times and 23 times compared to that of pristine TiO2 and CaCO3 (Fig. 2a), respectively. In stark contrast, there is a negligible increase in sulfate production detected in the TiO2 + CaCO3 mixture relative to that of pristine CaCO3 and TiO2 in dark experiments (Fig. S3). Great discrepancies in sulfate production over TiO2 + CaCO3 particles between dark and light experiments suggest that carbonate salt may play a different role in these two scenarios. However, the alkalinity of carbonate salt favors SO2 adsorption (Al-Hosney and Grassian, 2005; Yu et al., 2018), and the photo-oxidation process assisted by TiO2 particles is able to strengthen the oxidation efficiency of adsorbed SO2 (Shang et al., 2010; Chen et al., 2012), which is a plausible explanation for the increased sulfate production over TiO2 + CaCO3 particles. Following this speculation, two types of mixtures, TiO2 + CaCO3 and TiO2 + CaO, were employed. In the dark experiments (Fig. 2b), both TiO2 + CaO and TiO2 + CaCO3 almost yield an identical concentration of sulfite and sulfate. On the contrary, TiO2 + CaCO3 particles produce nearly 2 times more sulfate than TiO2 + CaO particles once irradiated, along with a sharp decrease in S(IV) species on the surface of TiO2 + CaCO3 surfaces. Besides, CaCO3 tends to show relatively humble physical properties, including BET surface area, surface pH, and solubility, etc., relative to CaO (see the detailed discussion in Text S15). The above results allow us to assert that the carbonate-containing system contains an alternative important mechanism for sulfate formation beyond the production of an alkaline environment (additional discussion available in Text S16). Figure 2c and d illustrate that the DRIFTS features of S(IV) and S(VI) species (Nanayakkara et al., 2014; Wu et al., 2011) increase over time on theoretical and experimental TiO2 + CaCO3 mixtures (wt wt = 50 50) upon irradiation. The theoretical mixture is calculated based on the DRIFTS experiments of pristine TiO2 and CaCO3 particles through a simple linear superposition, whereas the experimental mixture is directly derived from the DRIFTS experiment of TiO2 + CaCO3 (wt wt = 50 50) particles. These results suggest a synergistic effect presented in this mixture for sulfate formation under solar irradiation.
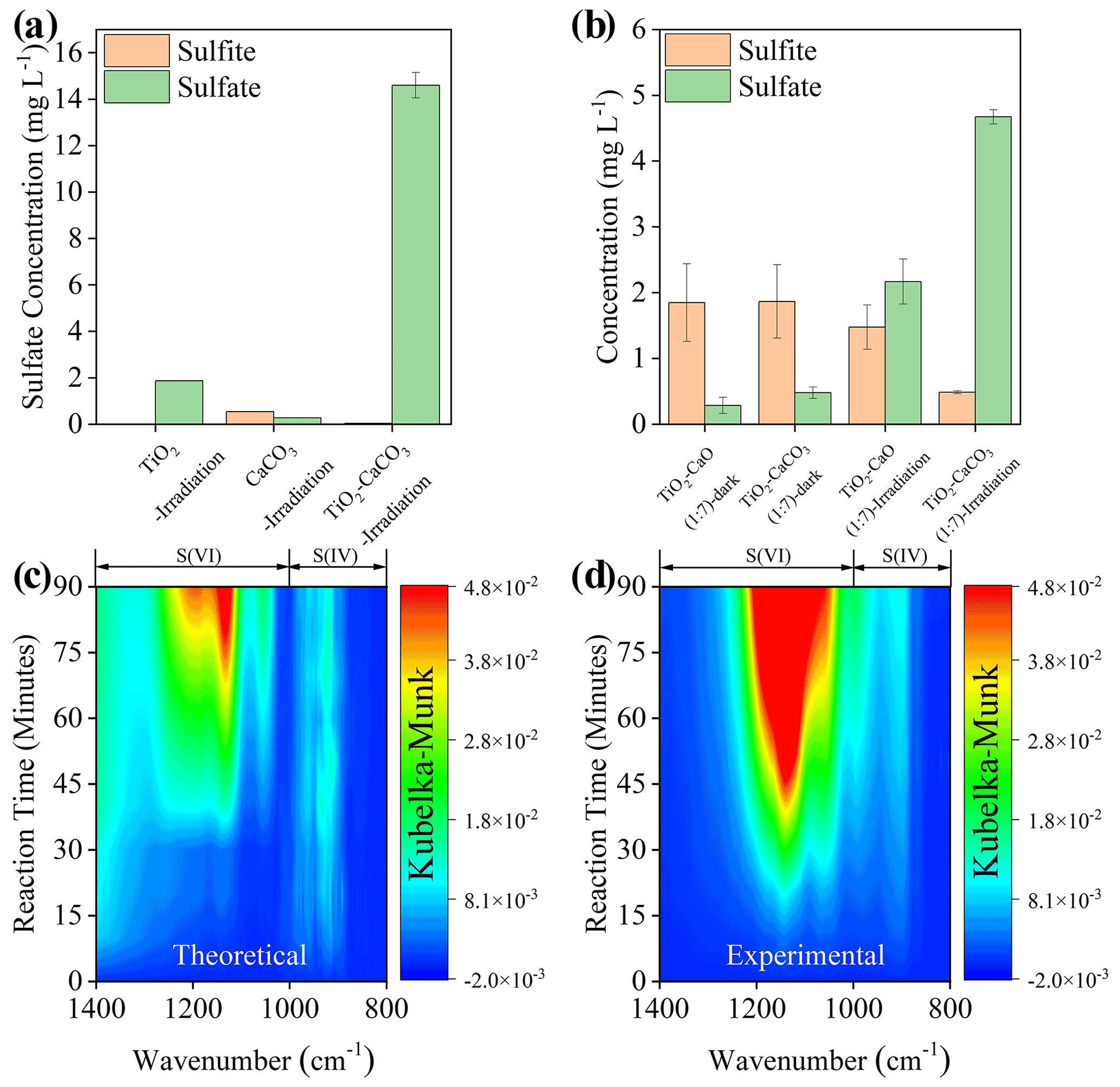
Figure 2(a) Sulfate or sulfite concentration quantified by IC on TiO2, CaCO3, and TiO2 + CaCO3 particles (wt wt = 50 50) after exposure to gaseous SO2 under irradiation for 60 min. (b) Sulfate and sulfite concentration quantified by IC on mineral dust particles of concern after exposure to SO2 under dark or irradiation for 20 min. In situ DRIFTS of S(IV) and S(VI) production on theoretical (c) and (d) experimental TiO2 + CaCO3 mixtures (wt wt = 50 50) upon irradiation for 90 min. All spectra were processed by the Kubelka–Munk (K–M) algorithm. Note that the production of sulfur species in theoretical TiO2 + CaCO3 mixtures refers to 0.5 × K–M bands of sulfur species of TiO2− + 0.5 × K–M bands of sulfur species of CaCO3, while those for experimental TiO2 + CaCO3 mixtures refer to 1 × K–M bands of sulfur species of TiO2 + CaCO3 mixtures (wt wt = 50 50). Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, total flow rate of 52.5 mL min−1, and SO2 of 2.21 × 1014 molec. cm−3.
Combining DRIFTS experiments with the obtained calibration curve (Fig. S4), we estimated that the uptake coefficient of TiO2 + CaCO3 mixture (50 wt % CaCO3) increased by about a factor of 17 as compared to that of pure CaCO3 or TiO2 (Table S1). More importantly, upon irradiation, SO2 uptake coefficients for these dust mixtures lie at the 10−4 order of magnitude, which is proven to gain importance in overall sulfate production by numerical modeling investigations (Wang et al., 2014; Zhang and Carmichael, 1999). Hence, the photochemical pathway associated with carbonate species is likely a potential driving force to increase sulfate production in the atmosphere. Meanwhile, the reaction order of SO2 in the TiO2 + CaCO3 + SO2 reaction system in the range of 400–20 000 ppb (parts per billion) is determined to be 0.80 (Fig. S5), indicating that the uptake coefficients obtained at the ppm level of SO2 would somewhat overestimate the real one obtained at atmosphere-relevant SO2 concentration level (several or a few tens of ppb). While we note that the difference between the lab and atmospheric conditions regarding the SO2 concentration remains even after considering the 400 ppb case, employing hundreds of ppb SO2 in the laboratory simulation to obtain the kinetic parameter of sulfate formation is acceptable (T. Y. Liu et al., 2020; Liu and Abbatt, 2021). Therefore, we tentatively believe that uptake coefficients estimated in this work would be valid after being calibrated.
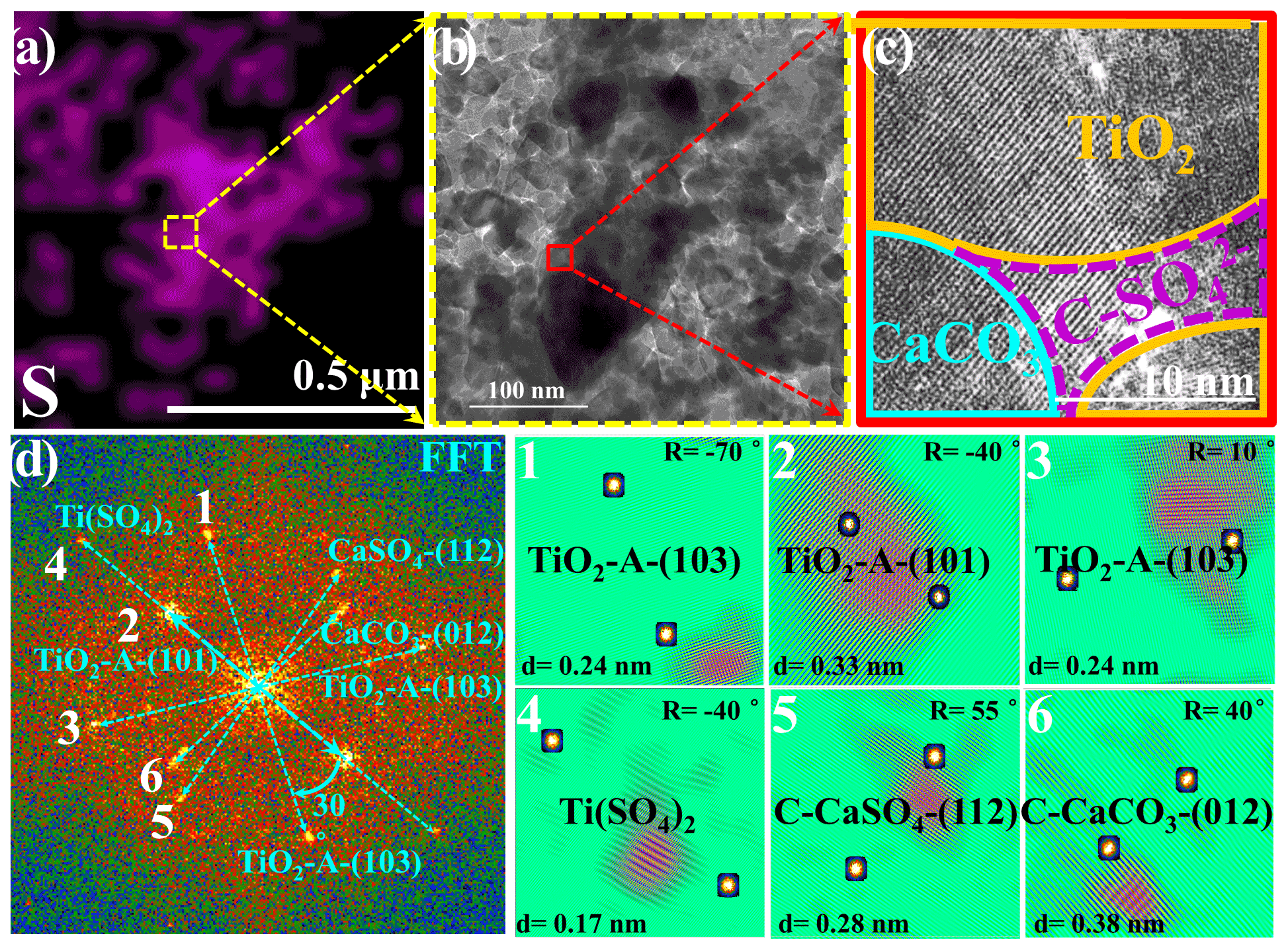
Figure 3(a) Energy dispersive spectroscopy (EDS) mapping of sulfur. (b) A selected HRTEM (high-resolution transmission electron microscopy) region containing a high density of sulfur for further observation is shown, and the red rectangle refers to the region shown in panel (c). (c) The HRTEM image in high resolution with lattice fringes and (d) corresponding fast Fourier transform (FFT) power spectra, lattice indexing, and (1–6) the inverse FFT analysis of lattice signal is shown in panel (d). In panel (c), the term C-SO stands for crystalline SO, i.e., CaSO4 and Ti(SO4)2. Particles employed for the HRTEM measurement refer to TiO2 (50 wt %) + CaCO3 (50 wt %) mixture particles upon exposure to the 4.42 × 1014 molec. cm−3 SO2 N2 + O2 for 60 min, while other reaction conditions are as same as those of the above sulfate quantification experiments in Fig. 1. Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, and total flow rate of 52.5 mL min−1.
A high-resolution transmission electron microscopy (HRTEM) analysis of TiO2 (50 wt %) + CaCO3 (50 wt %) particles after reaction, in combination with energy dispersive spectrometer mapping measurements of the sulfur component, was conducted to investigate the synergistic effect between TiO2 and carbonate ions (Figs. 3a–d and S6). A region with a relatively high density of sulfur species was selected for further observation (Fig. 3a and b), and the distribution of each component was determined by fast Fourier transformation (FFT) and inverse FFT analyses (Fig. 3d) of the selected HRTEM image in high resolution with lattice fringes, as shown in Fig. 3c. The observation of crystalline Ti(SO4)2 and CaSO4 on the interface of TiO2 and CaCO3 components implies that the synergistic effect on sulfate production likely originates from interplays of those two types of components under solar illumination.
We further assessed the importance of interfacial contact between TiO2 and CaCO3 in sulfate production by two synthetic approaches in which the interface abundance is modulated for comparison. Typically, a grinding method was used to make a TiO2 + CaCO3 mixture with compact contact between those two components, thus leading to abundant interfaces. Meanwhile, the shaking method is designed to create a TiO2 + CaCO3 mixture of loose contact, leaving relatively lower numbers of interfaces within the mixtures. The resulting mixing statuses of the two samples meet our expectations, evidenced by the scanning electron microscope (SEM) technique (Fig. S7). IC quantification analysis suggests that particles with considerable junctions exhibit a more pronounced promotion for sulfate production than those having relatively few junctions (Fig. S8). These results emphasize the importance of an indispensable interface contact between TiO2 and CaCO3 in a quick production upon irradiation.
Table 1Chemical compositions of mineral dust simulants.
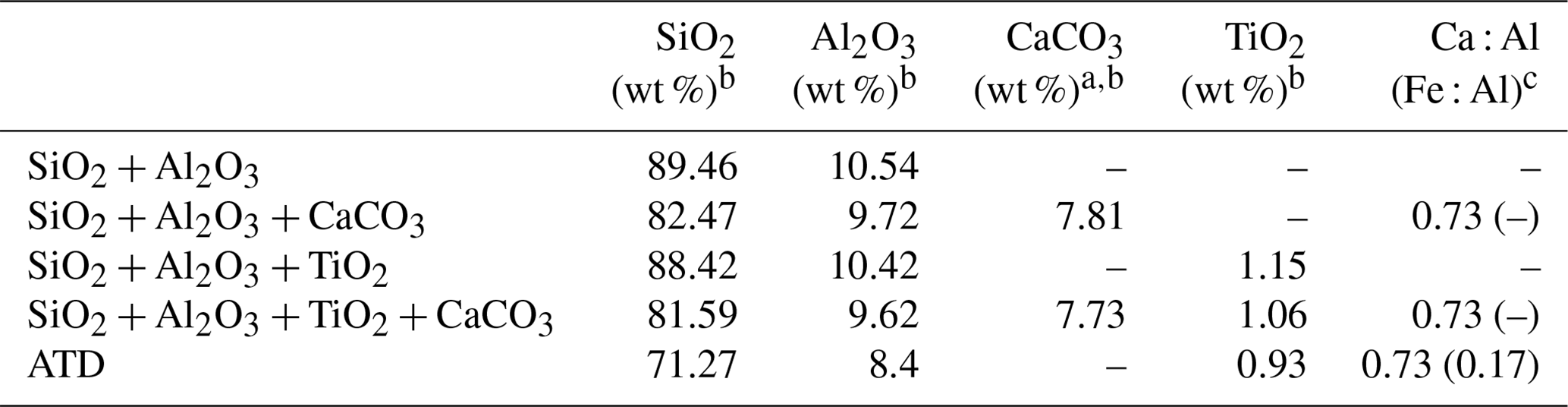
a In the present study, CaCO3 was taken as representative of alkaline earth metal oxide in our proxies for the authentic dust. b The mass ratio of four components (if any) in the simulants were controlled in the ratio of SiO2 : Al2O3 : CaCO3 : TiO2 = 81.59 : 9.62 : 7.73 : 1.06. For instance, the mass ratio of SiO2 to Al2O3 is 81.59 : 9.62 in the SiO2 + Al2O3 simulant, whereas it is 81.59 : 9.62 : 7.73 in the SiO2 + Al2O3 + CaCO3 mixture. The ratios of each component are derived from the EDS mapping analysis of ATD dust particles. c This column refers to the molar ratio.
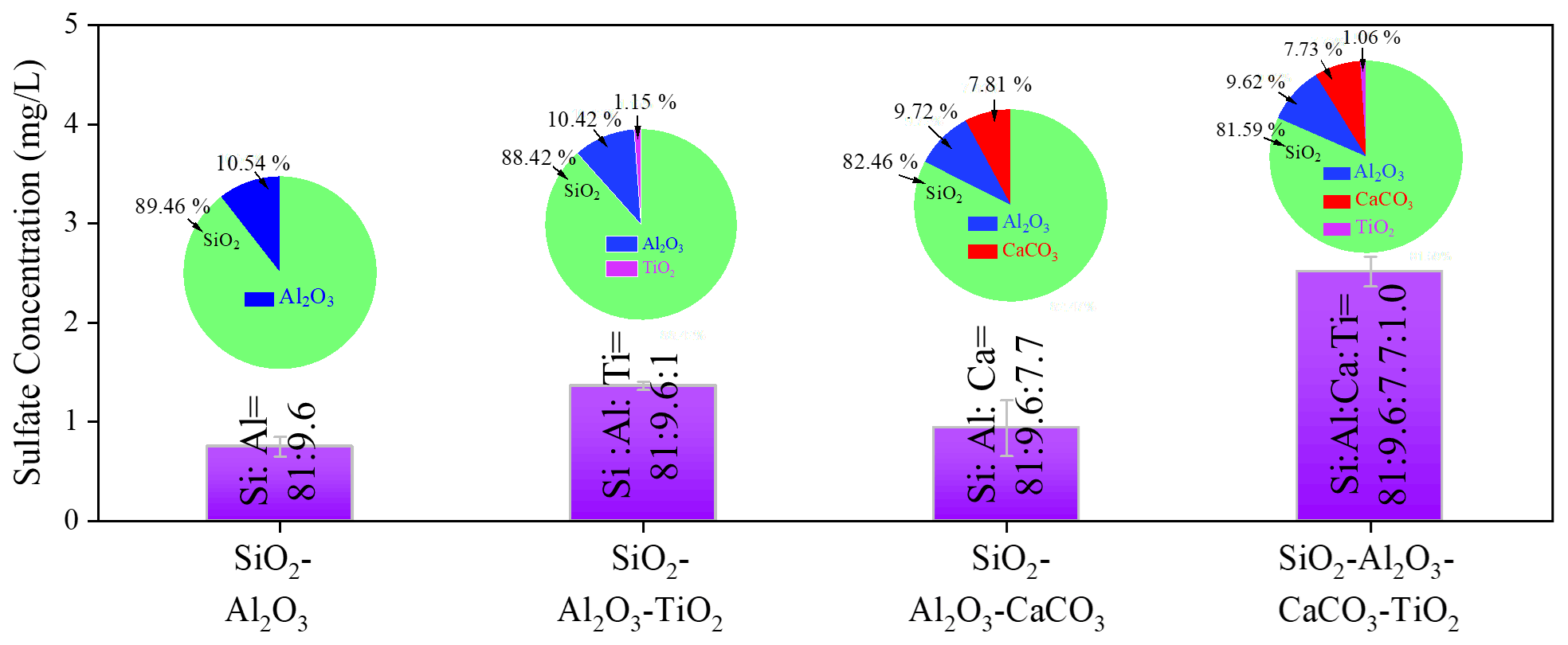
Figure 4Sulfate concentration quantified by IC. Sulfate concentration was measured by IC on mineral dust simulants after exposure to gaseous SO2 (2.46 × 1014 molec. cm−3) under irradiation. Noting that SiO2 : Al2O3 : CaCO3 : TiO2 refers to the mass fraction ratios of the components in simulants. Experiments were all conducted at an RH of 30 % and light intensity (I) of 30 mW cm−2.
The increased sulfate production was further probed by employing mineral dust simulants where two dominant crust constituents SiO2 and Al2O3 were introduced into TiO2 + CaCO3 particles to mimic the authentic mineral dust particles in the atmosphere, with the specific component and corresponding ratio information shown in Table 1. It is worth mentioning that the determination of the ratio of each component in the simulants relies upon the EDS mapping results of ATD particles. In Fig. 4, the introduction of TiO2 components (1 wt %) into SiO2 + Al2O3 leads to 81.6 % enhancement of sulfate production because of photolabile ROS. On the other hand, a mere 24.8 wt % increase in the sulfate yield was observed once ∼ 8 wt % of CaCO3 was incorporated into SiO2 + Al2O3 dust particles. This can be attributed to the alkaline environment created by CaCO3, which is thought to increase SO2 adsorption (Al-Hosney and Grassian, 2005) and sulfate production accordingly. Surprisingly, mixing a ∼ 1 % mass fraction of TiO2 and ∼ 8 wt % of CaCO3 into SiO2 + Al2O3 gives rise to a 235 % increase in sulfate formation relative to that of SiO2 + Al2O3. It represents nearly an extra 100 % enhancement of sulfate production due to the presence of TiO2 and CaCO3 of an atmospherically relevant mass fraction. These results lead to the hypothesis that the observed synergistic effect on the heterogeneous oxidation of SO2 is likely to take effect in the atmosphere.
Fe2O3 is also one of the crucial components found in authentic mineral dust (El Zein et al., 2013), and it has been reported to produce electron–hole pairs under solar irradiation (Li et al., 2019), thus likely involving the reaction channel considered in this work (see a detailed discussion in a later section). Similar to the protocol applied for synthesizing the TiO2 + CaCO3 mixture, αFe2O3 + CaCO3 are prepared accordingly. In Fig. S9a, our results show that αFe2O3 cannot trigger fast SO2 oxidation in the presence of carbonate ions upon irradiation, which is distinguished from the results we derived from the TiO2 + CaCO3 mixture. This can be explained by the fact that Fe2O3 shows a lower redox activity relative to TiO2 (Fig. S9b), where its strong redox capability essentially enables photoinduced electrons and holes to produce O and •OH radical ions. In stark contrast, the valence band and conduct band of Fe2O3 lie at −0.18 and at 1.68 V vs. NHE (pH = 7), lower than the redox potential required for generating an O, •OH and carbonate-containing ROS (Li et al., 2016). Hence, no promoted sulfate production is seen for αFe2O3 + CaCO3 particles under irradiation. We also note the inconsistency between our study and the previous literature with regard to the response of SO2 oxidation to solar irradiation over αFe2O3 particles, which has been interpreted in Text S17.
Overall, we show that, upon irradiation, the atmospherically relevant content of TiO2 (nearly 1 %) in mineral dust simulants is able to interact with carbonate ions to launch an increased sulfate production, which is beyond the conventional regime of alkaline neutralization of H2SO4. Unlike TiO2, αFe2O3 lacks the ability to initiate fast SO2 oxidation by generating carbonate-containing ROS due to its limited photochemical activity, although ferric chemistry is important in secondary sulfate formation in the atmosphere (Sullivan et al., 2007; Yermakov and Purmal, 2003).
3.2 Accelerated sulfate production in the presence of CO2
Atmospheric CO2 is also an important source of (bi)carbonate. Its influence on photochemical SO2 uptake on mineral dust was thus studied. Distinguishing S(VI) from S(IV) species over TiO2 particles relies upon the position of the infrared (IR) bands, according to the assignment of the previous literature (Nanayakkara et al., 2014), and S(VI) and S(IV) build up as heterogeneous reactions occur in all cases (Fig. 5). In the presence of atmospherically relevant CO2 (9.83 × 1015 molec. cm−3), the sulfate yield was increased under irradiation as compared to the CO2-free case (Fig. 5a and b). We cautiously examined the net effect of formed (bi)carbonate on sulfate production by time-resolved DRIFTS spectra (Fig. 5c and d) using dark experiments. CO2 suppresses both S(IV) and S(VI) products in the dark probably because of the competitive adsorption effect, as we observed over Al2O3 particles (Y. Liu et al., 2020).
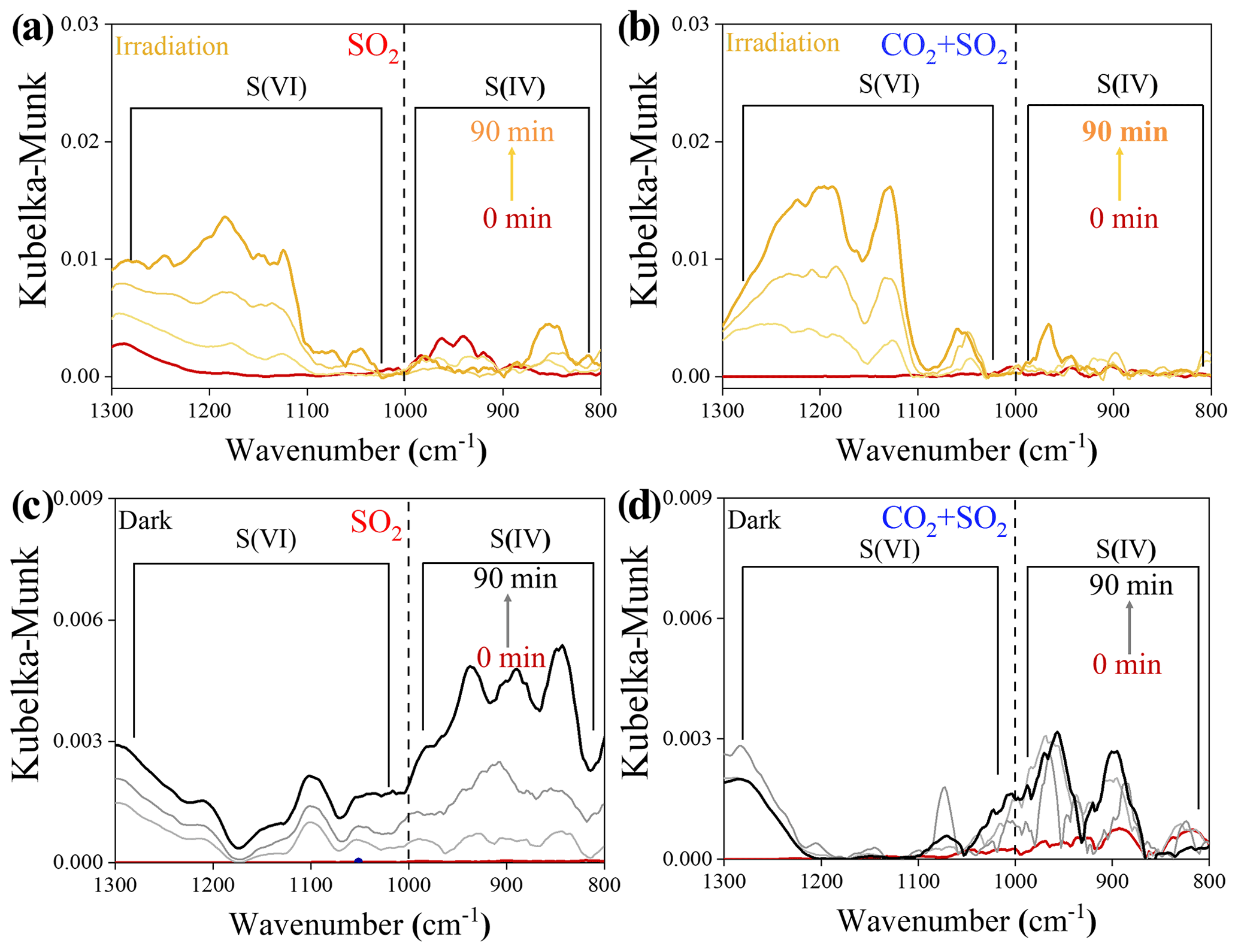
Figure 5Time-resolved DRIFTS of S(IV) and S(VI) products over TiO2 particles after exposure to SO2 N2 + O2 in the absence and presence of CO2 upon irradiation (a, b) and those reactions under dark experiments (c, d). Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, total flow rate of 52.5 mL min−1, and SO2 of 7.37 × 1013 molec. cm−3.
The above observation leads us to speculate that the active intermediate derived from (bi)carbonate species upon irradiation is a plausible force to drive rapid sulfate formation. Besides, a nearly 50 % increase in the SO2 uptake coefficient is observed for the mineral dust proxy TiO2 after being exposed to the 9.83 × 1015 molec. cm−3 (400 ppm) CO2 + SO2 N2 + O2 mixture (Table S1). The pseudo first reaction order (1.13) was also determined in the selected concentration range of 400–20 000 ppb (Fig. S5b), which satisfies the prerequisite for the uptake coefficients derived from laboratory chambers potentially being generalized to the atmosphere condition, as we expounded in the early context.

Figure 6Laboratory studies of sulfate production on authentic dust and clay membranes (a) K-Ga-2, (b) ATD, and (c) IMt-2 under the dark and irradiation (30 mW cm−2) upon exposure to 4.91 × 1014 molec. cm−3 SO2 N2 + O2 and 2.46 × 1018 molec. cm−3 CO2 + 4.91 × 1014 molec. cm−3 and SO2 N2 + O2 at RH of 30 %. Note that the sulfate yield in three cases was normalized by the mass of dust particles employed for a heterogeneous reaction.
As another step toward a real scenario in the atmosphere, experimental trials employing authentic mineral dust particles, i.e., K-Ga-2 (Kaolin, Georgia, USA), Arizona test dust (ATD), and IMt-2 (Illite, Montana, USA) clays were implemented, with the component analysis results shown in Table 2. In Fig. 6, the pronounced increase in sulfate yield (a nearly 100 % increase in sulfate production in the CO2-involved case under irradiation) is best seen in K-Ga-2 clay (Fig. 6a). The promotional effect of CO2 on sulfate formation under irradiation, nonetheless, is less evident for IMt-2 (the content of TiO2 ≈ 0.99 %) and ATD (the content of TiO2 ≈ 0.46 %) as compared to K-Ga-2 particles. This may correlate to their higher mass fraction of alkaline earth metal oxide (denoted as AE), which enables dust particles to possess a substantial number of (bi)carbonate species within the natural environment where they have experienced long-term exposure to atmospheric CO2 during regional transport. Therefore, the aforementioned synergetic effect takes effect over IMt-2 and ATD particles even without exposure to CO2, presumably due to the presence of the abundant alkaline carbonate formed, and a relatively moderate increase in sulfate production was thus observed. On the other hand, TiO2 content is not necessarily an accurate predictor of photoreactivity; the content and proportion of the active phase of TiO2 in K-Ga-2 altogether contribute to a more pronounced increase in sulfate production relative to the other two clays (see the detailed discussion in Text S18).
Table 2Chemical compositions of the ATD dust and standard clays.
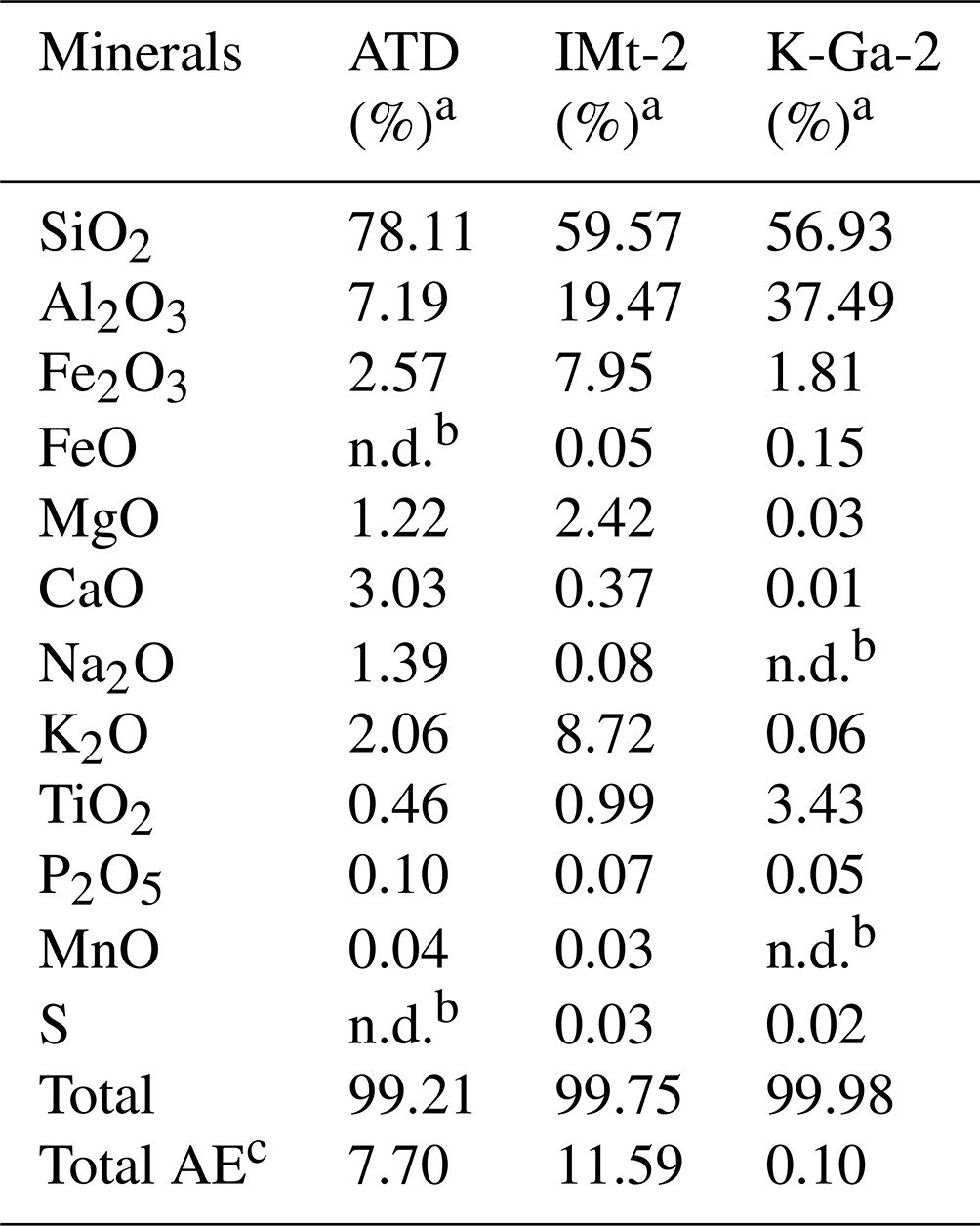
a Chemical compositions of the dust and clays were determined by XRF (X-ray fluorescence) results. Note that b n.d. refers to not detected and that c AE refers to alkaline earth metal oxide.
3.3 Reaction mechanism
The heterogeneous reaction of SO2 on dust particles in the atmosphere is a complicated process, covering a series of reactions taking place in both homogeneous and heterogeneous ways. At a sufficiently low RH condition (normally below 10 % RH), water readily dissociates on the surface of metal oxide under ambient atmospheric conditions, where the metal oxide surface is terminated by hydroxyl groups that hydrogen bond to adsorbed water molecules (Cwiertny et al., 2008). In this case, SO2 oxidation over dust particles is dominated by the reaction regime where the resulting hydroxyl groups react with gaseous SO2 to form adsorbed S(IV)ad species. Afterward, S(IV)ad will be oxidized by oxidants in the atmosphere or photoinduced active intermediates produced from the dust surface upon irradiation. As the RH increases beyond 10 %–15 %, multilayer water coverage occurs, reaching approximately two monolayers at RH of 30 % (Mogili et al., 2006). Under these circumstances, the amount of water adsorbed onto the surface of the dust particles is believed to be sufficiently large so that it is liquid-like in the corresponding physical and chemical properties (Cwiertny et al., 2008; Peters and Ewing, 1997). In this work, heterogenous SO2 oxidation over mineral dust proxies proceeds at the RH of 30 %, and two water layers are likely to attach to dust particles. Thus, radical ions are anticipated to play a key role in fast SO2 oxidation over humidified mineral dust, and mechanism studies performed in the aqueous phase are persuasive to some extent.

Figure 7(a) Single-wavelength transient absorption spectra of various aqueous solutions. (b) The removal rate of aniline after exposure to airflow under irradiation in the absence and presence of mineral dust particles for 300 s. Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, and a total flow rate of 52.5 mL min−1. Note that an adequate amount of NaOH was introduced into the TiO2 suspension system to achieve a pH environment condition identical to that of the TiO2 + Na2CO3 suspension system. (c) Determination of sulfite and sulfate concentration after exposure to air flow under irradiation in the absence and presence of mineral dust particles for 20 min. Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, and a total flow rate of 52.5 mL min−1. (d) Sulfate formation change Δ(SO) is determined by different sulfate concentrations with and without the addition of isopropanol as hydroxyl radical scavenger. (e) The difference in transient absorption kinetics of the sulfite radical and carbonate radical at the various aqueous solutions and their corresponding growth–decay fit curves. A ΔA signal was recorded at 255 and 600 nm after a pulsed 355 nm laser excitation. (f) Electron spin resonance (ESR) spectrometry of [DMPO–SO] intermediary formed in a solution of TiO2 (3 mg per 4 mL) + 0.1 M Na2SO3 and TiO2 (3 mg per 4 mL) + 0.5 M Na2CO3 + 0.1 M Na2SO3. For clarity, the integrated areas of the ESR profiles were also presented for direct comparison. Note that Exp. and Sti. stand for experimental results and corresponding fitting results using isotropic radicals software.
Our preliminary sulfate quantification results (Fig. 2a and b) suggest that the presence of (bi)carbonate ions under solar light contributes to increased sulfate yield. In this carbonate-containing reaction system, a plausible intermediate is the active CO. It is readily produced via the following two pathways. First of all, a carbonate anion can be directly oxidized by photoinduced holes produced from typical mineral dust upon solar irradiation (Reactions R1 and R2), as the redox potential of CO CO is 1.78 V (vs. NHE at pH = 7), which is lower than the TiO2 valence band (VB) potential of 2.67 V (vs. NHE at pH = 7), as follows (Li et al., 2016; Xiong et al., 2016):
In the second pathway, carbonate radicals evolve through the reaction of (bi)carbonate anion with hydroxyl radicals •OH forming over mineral dust surfaces (Zhang et al., 2015; Reaction R3 and R4).
The above assumptions are supported by nanosecond transient absorption spectra (NTAS), in which a signal (ΔOD) of a carbonate radical CO at 600 nm (Bhattacharya et al., 1998) only emerges for dust suspension containing (bi)carbonate species (Fig. 7a). An increased degradation rate of aniline observed in TiO2 suspension due to the presence of carbonate ions produces additional evidence of the formation of active CO ions and the strengthened oxidation capability of TiO2 (Fig. 7b; see the additional discussion in Text S19). The CO-induced chemistry was further evidenced by •OH scavenging experiments using tertiary butyl alcohol (TBA) and isopropanol (i-PrOH), as they show lower reaction rates with CO ( < 4.0 × 104 M−1 s−1 and < 1.6 × 102 M−1 s−1) relative to those with •OH (ki-PrOH∼•OH < 1.9 × 109 M−1 s−1 and (k•OH,TBA = 6 × 108 M−1 s−1; Buxton et al., 2009; Liu et al., 2015; Li et al., 2020). TBA dramatically decreases the yield of sulfate on the TiO2 surface by nearly 70 %, with sulfite ions being the dominant sulfur species (Fig. 7c). Meanwhile, a great loss of sulfate yield when TiO2 suspension was added with i-PrOH (Fig. 7d). This is in strong contrast to the result of a carbonate-involved system, where the reactivity is sustained as carbonate radicals offer an alternative reaction pathway for SO2 oxidation. This is plausible since the carbonate ions are an excellent •OH scavenger, and CO becomes the predominant species in a relatively strong alkaline aqueous-like environment in the presence of carbonate salt. This is supported by a previous work (Sun et al., 2016) in which adding 0.1 M of bicarbonate salt into the UV H2O2 system (H2O2 = 0.3 mM) was sufficient to suppress •OH concentration to around 10−15 M, creating a carbonate-radical-dominated regime ([CO] = 8.64 × 1012 M). In our experiments (Fig. 7b), 0.2 M of carbonate salt was employed, and the reaction rate of CO with •OH is nearly 2 orders of magnitude higher than that of HCO, thus giving rise to carbonate radical being the substitute for the hydroxyl radical in the reaction. The above results suggest that •OH is a major contributor to the sulfate yield on TiO2 particles in the absence of carbonate ions, while CO ions dominate the SO2 oxidation over humidified carbonate-containing TiO2 particles upon irradiation. In addition to experimental investigations, the carbonate radical formation process is proved to be thermodynamically favorable and supported by density functional theory (DFT) calculations (Fig. S10 and Text S20).
On the other hand, early studies (Das, 2001; Neta and Huie, 1985; Chameides and Davis, 1982) agree with the key role of the sulfite radical (SO) in rapid sulfate production in an aqueous medium, and the present reaction system creates a localized environment in which the SO can be readily produced from the TiO2 and S(IV) species upon solar illumination (Salama et al., 1995). Consequently, a probe light of NTAS at wavelength 255 nm (ascribed to the sulfite radical) and 600 nm (ascribed to carbonate radical) were simultaneously monitored (Hayon et al., 1972; Ghalei et al., 2016; Goldstein et al., 2001). A weak signal of sulfite radical was observed in the system of the TiO2 + Na2SO3 suspension under irradiation (Fig. 7e). On the contrary, the sulfite radical signal is strengthened after the introduction of carbonate ions into the TiO2 + Na2SO3 suspension, along with a significant decrease in the signal for carbonate radical. Electron spin resonance (ESR) data (Fig. 7f) further confirm the increase in the SO after 2 min UV irradiation in the presence of a carbonate ion. Based on the above results, one may deduce that the interplay between carbonate radical and sulfite ions is a crucial step giving rise to the increased SO, which is reported to account for rapid atmospheric sulfate formation through chain propagation reactions that involve SO and SO intermediates (Hung and Hoffmann, 2015; Hung et al., 2018). Additionally, this sulfite radical ion chemistry is believed to drive fast sulfate formation over mineral dust particles as well (Gankanda et al., 2016; Rubasinghege et al., 2010). Nevertheless, there are two possibilities that might explain the aforementioned interaction. One way is through the oxygen transfer and the other route is electron transfer, which needs further clarification.
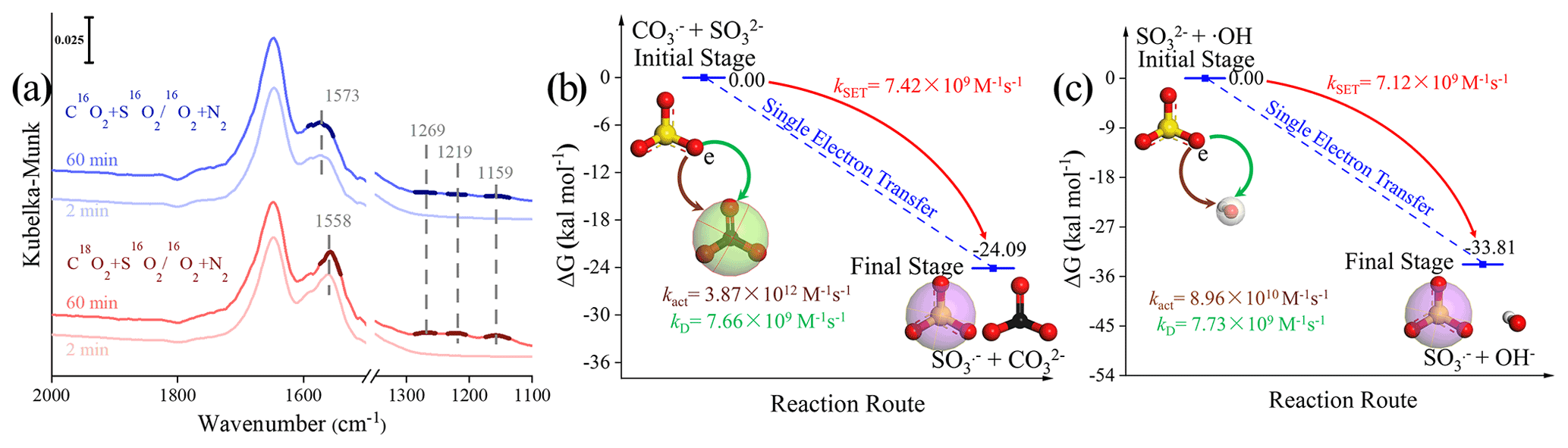
Figure 8(a) In situ DRIFTS of heterogeneous reaction of SO2 on the TiO2 particles for 2 and 60 min after being exposed to C16(18)O2 N2 for 20 min under irradiation. (b) Reaction pathway of the interaction between the carbonate radical (CO) and sulfite (SO) and the (c) interaction between the hydroxyl radical (•OH) and sulfite (SO) through the SET process at the coupled cluster calculations with all single and double excitations (CCSDs) (T)-F12/cc-PVDZ-F12//M06-2X/6-311G (3df, 3pd) level (where polarized valence double zeta is PVDZ), and represents the difference in the Gibbs free energy between the reactant and product. The white, black, yellow, and red spheres represent H, C, S, and O atoms, respectively. In order to visualize the variation in the surface products in oxygen isotope experiments (a), DRIFTS features of these concerned species are highlighted in dark colors.
We first examined the oxygen transfer path through 18O isotope labeling experiments. TiO2 particles were initially exposed to C16O2 N2 and C18O2 N2, followed by the exposure of SO2 N2 + O2 under irradiation (Fig. 8a). A bidentate carbonate band centered at 1573 cm−1 appears after the introduction of C16O2 N2, while this band shifts to 1558 cm−1 when C18O2 N2 is introduced, indicating the incorporation of 18O into bidentate carbonate species, in accordance with a previous report (Liao et al., 2002). However, no shift of IR features at 1269, 1219, and 1159 cm−1, assigned to (bi)sulfate species on TiO2 particles, was observed throughout the reaction. This implies that the oxygen transfer path does not account for the rapid SO2 oxidation on particles of concern.
In light of the above analysis, the electron transfer might be a plausible pathway to explain the fast oxidation within the reaction system. DFT calculations provide an accessible approach to studying the electron transfer pathway. The result in Fig. 8b illustrates SO formation is a SET process of CO and SO, where the O atom in SO transfers an electron to the O atom in CO to form SO and CO. This SET reaction is a thermodynamically favorable process, with the difference in the Gibbs free energy between reactant and product lying at −24.09 kcal mol−1. We note that the insufficient O2 supply in aqueous media may be an underlying constraint to the proposed CO-initiated SO2 oxidation pathway. Therefore, a careful estimation of both the oxygen consumption and supply rates were conducted, revealing that oxygen supply flux can be sufficiently larger than corresponding consumption (see additional details in Text S21). This enables us to deduce that considered chain reactions can continually proceed. Taking the above results and discussions altogether, the following reactions are proposed accordingly (Reactions R5–R8):
Another important pathway needs to be considered as well; that is, SO can also be formed via the conventional reaction of •OH and SO (Reaction R9).
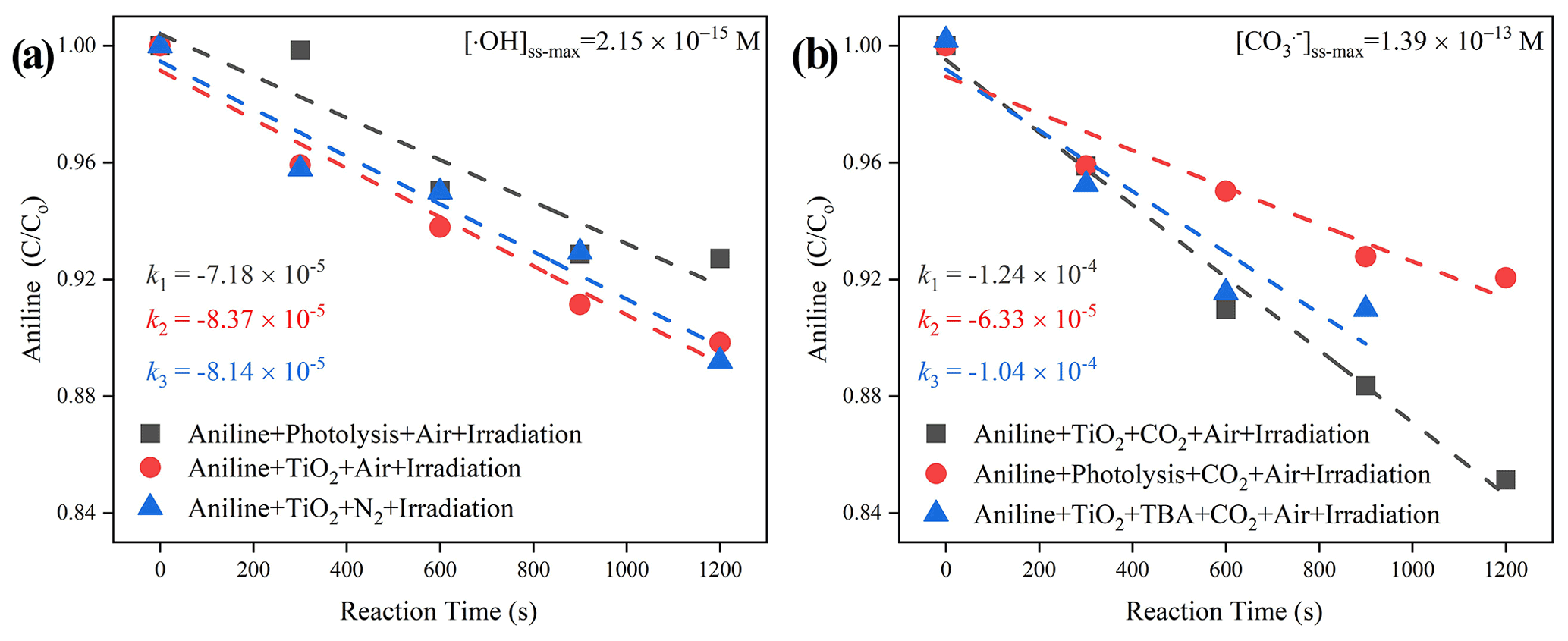
Figure 9The degradation rate of aniline after exposure to air flow under irradiation in the absence (a) and presence (b) of CO2 over mineral dust proxy particles TiO2 as a function of the reaction time. Reaction conditions are RH of 30 %, light intensity (I) of 30 mW cm−2, and a total flow rate of 52.5 mL min−1.
In this SET process, the electron donor SO reacts spontaneously with electron acceptor •OH (Fig. 8c), and the calculated activation free energy barrier for this SET reaction is 2.50 kcal mol−1. Hence, the reaction process of •OH with SOis diffusion controlled, and the total rate constant kSET−2 was calculated to be 7.12 × 109 M−1 s−1. In comparison, the rate constant kSET−1 of the diffusion-controlled SET process for CO and SO was estimated to be 7.42 × 109 M−1 s−1. Despite a slight net increase in the rate, the distinguishable concentration of CO and •OH should also be taken into account for the rate comparison in varied reaction paths. To visualize the difference, relative rates were calculated according to Eq. (12), as follows:
where r is the ratio of two reaction rates, and [CO], [SO], and [•OH] refer to the concentration of corresponding reactants. In the literature, it is suggested that the concentration of carbonate radicals is able to show 2 orders of magnitude higher than that of hydroxyl radicals at the surface of the water under solar irradiation (Chandrasekaran and Thomas, 1983; Goldstein et al., 2001; Sulzberger et al., 1997), which is consistent with the concentration gap between carbonate radicals and hydroxyl radicals through a partitioning process from the gas phase determined in our reaction system (Fig. 9). While the net concentrations of carbonate and hydroxyl radicals in the water layers of humified particles are very likely to be different from those found in the bulk aqueous media, the concentration inputs of two radicals with the gap of 2 orders of magnitude could somehow reflect the relative contribution of carbonate radicals and hydroxyl radicals to sulfate production, based on the literature results and our experimental trails. The concentrations of CO and •OH were set in the range from 1.0 × 10−10 to 1 × 10−12 mol L−1 and from 1.0 × 10−12 to 1 × 10−14 mol L−1 (Sulzberger et al., 1997), and the r value could thus reach to 1.04 × 104 at most (Fig. S11). As a result, we speculate that the formation pathway of SO via an interaction between CO and SO is a more efficient route that corresponds well with our experimental results.
In addition to the pathway launched by photo-generated holes, the sink of photo-generated electrons is also considered. In our reaction system, O2 is thought to be an electron trap and to produce the superoxide radical ions (O), which are reported to play a non-negligible role in sulfate formation (Shang et al., 2010) and should be taken into account to give a whole picture of reaction scheme in triggering sulfate formation on the surface of TiO2-containing mineral dust particles. p-benzoquinone is a commonly used O scavenger for trapping the O radical ions (Yan et al., 2018). Our data show that adding an excess amount of p-benzoquinone into TiO2 particles reduces the sulfate yield by 32 %, along with the appearance of sulfite ions over TiO2 particles upon exposure to SO2 (Fig. 7c). Notably, the decrease in sulfate yield by around 30 % in the presence of O scavenger p-benzoquinone is almost complementary to that added with •OH scavenger using TBA (70 %), pointing toward a minor sulfate formation pathway contributed by O relative to the major pathway by CO when carbonate ions are presented to efficiently capture •OH ions. Following the work of Shang et al. (2010), O-involved SO2 oxidation can be given as Reactions (R10)–(R12), as follows:
where intermediate SO3, formed via the interaction between SO2 and O, subsequently couples with water molecules to produce sulfate species as a final product. pH is an important factor within aqueous chemical reaction processes and is preferred to alter the dominant regime for sulfate production. Yet, so far, adjusting the pH of particle surfaces is quite tough, and exploring the role of dust surface pH in the reactivity of CO is not easily achieved. Notwithstanding, the increase in pH in TiO2 suspension was observed to promote the production of CO, further strengthening the oxidation capability of dust particles (Fig. 7b). In contrast, decreasing pH is expected to reduce the yield of CO since the reaction rate of CO with •OH is nearly 2 orders of magnitude higher than that with HCO. On this basis, a question arises as to whether the surface pH of mineral dust can be sustained to maintain the fast SO2 oxidation triggered by CO in the typical lifespan of mineral dust.
Considering that the SO2 concentration employed in this work is higher than that in the real atmosphere, the concept of equivalent exposure time is introduced to evaluate the influence of pH on the SO2 oxidation pathway initiated by CO (see a more detailed discussion on determining equivalent exposure time in Text S22). The heterogeneous sulfate production over TiO2 and TiO2 + CaCO3 particles versus equivalent exposure time were plotted (Fig. S12). Clearly, the sulfate yield builds up steadily during the 2-week equivalent exposure time, suggesting that the regime of CO-initiated SO2 oxidation over TiO2 and TiO2 + CaCO3 particles is slightly affected by the possible decrease in surface pH because of the accumulation of sulfate production over entire reaction course. In the atmosphere, the lifetime of mineral dust particles ranges from several days to weeks (Bauer and Koch, 2005), and the equivalent exposure time considered in this study (nearly 2 weeks) falls right within the characteristic lifespan range of mineral dust particles. Besides, 20 ppb is assumed to be an atmospherically relevant concentration to calculate equivalent exposure time in this study, whereas even low SO2 concentrations (several or a few tens ppb of SO2) were monitored in the field observation (He et al., 2014; Watanabe et al., 2020). Therefore, the reduction of dust surfaces pH would be more moderate than what we now consider, and even a little influence of surface pH on our proposed reaction scheme would have an impact. Therefore, persistent growth of sulfate shows a negligible effect on the CO-initiated SO2 oxidation scheme proposed in this work.
Additionally, dust particles are reported to eject the radical ions from the surface under solar light irradiation, severing as an underlying pathway for sulfate aerosol formation in the atmosphere (Chen et al., 2021; Dupart et al., 2012) and is described as follows:
where ROS (g) stands for the active intermediates in the gas phase. Over 400 ppm of CO2 is universal in the atmosphere, and it is expected to form (bi)carbonate ions once enters the quasi-liquid layer of humified particles. Bi(carbonate) ions are then prone to reacting with hydroxyl radical ions to form carbonate radicals. Following this line of reasoning, we attempt to monitor the plausible gas ROS species that are formed in the presence of CO2 (see the detailed discussion in Sect. 2).
When CO2 (atmospherically relevant concentration) is introduced into the homemade flow cell chamber, with the intervening gap between the TiO2-coated film and probe molecule solution fixing at nearly 2 mm, the short distance guarantees that gaseous ROS will diffuse and react with aniline (Rodriguez et al., 2013). An increased degradation rate of this probe molecule was seen, which can be speculated to be the generation of active carbonate radical ions (Fig. 9). Aqueous CO is believed to be supplied by the partitioning processes from CO (g) that come from humified dust particles, and its maximum steady-state concentration was determined to be 1.39 × 10−13 M for TiO2 + Air + CO2 system, which is over 1.8 orders of magnitudes higher than that of •OH for the TiO2 + Air system (2.15 × 10−15 M). This observation matches with an earlier study, where the concentration of a carbonate radical can be nearly 2 orders of magnitudes greater than •OH over the water surface (Sulzberger et al., 1997).
Overall, the above results suggest that the photochemistry that involves carbonate ions, more precisely CO radicals, increases sulfate production. This finding broadens the prevailing view that the acceleration of SO2 oxidation over the carbonate salt is merely due to the favorable neutralization of H2SO4 over an alkaline surface. Worth noting is that upon irradiation the active component TiO2 in mineral dust produces carbonate radicals in the gas phase when CO2 presents, therefore potentially promoting sulfate aerosol formation in the atmosphere. Overall, it could be speculated that (bi)carbonate species strengthen the oxidative capacity of TiO2-containing dust particles with regard to SO2 oxidation.
3.4 Field measurements of sulfate and (bi)carbonate ions
Complementary field sampling and analysis were further conducted to examine our hypothesis that intermediate CO may play role in secondary sulfate formation in the atmosphere. We first considered the meteorological condition of wind speed, which is an important parameter determining whether the local chemical process gains importance in affecting secondary sulfate formation. Meteorological information was collected from an open-access database (https://www.aqistudy.cn/, last access: 7 September 2021). During the sampling period, the wind scale mainly varies from 0 to 1, corresponding to the wind speed ranging from 0 to 1.5 m s−1 (Fig. S13). All plots shown in Fig. S13 give rise to a statistical wind speed of 0.76 ± 0.73, which represents the weak dispersion of pollutants at a low wind speed (not exceeding 2.5 m s−1) (Witkowska et al., 2016; P. Liu et al., 2020), indicating that a local source is a dominant contributor to the air pollution.
Under stagnant meteorological conditions (wind speed < 1.5 m s−1), for the coarse mode (2.5–10 µm) of sulfate, the heterogeneous reaction of SO2 on the dust surfaces is thought to be a major contributor (Liu et al., 2017). This correlates to the fact that a large mass fraction of mineral dust is abundant in the coarse-mode particulate matter (PM; Fang et al., 2017; Miller-Schulze et al., 2015) in which TiO2 was found at mass mixing ratios ranging from 0.1 % to 10 % depending on the exact location where particles were lifted up (Hanisch and Crowley, 2003; Chen et al., 2012). Therefore, PM with relatively larger size dimensions are expected to contribute to secondary sulfate formation via heterogeneous reactions, which is supported by the recent field study in which the carbonate fraction of coarse PM is evidenced to promote secondary sulfate production (Song et al., 2018). Considering this, rather than determining the concentration of water-soluble ions in all stages, special attention is focused on PM collected in stages 1–4 (particles with their dimension of ≥ 3.3 µm). As (bi)carbonate ions are known as key precursors in producing CO and accelerating sulfate formation, quantifications of those relevant water-soluble ions were thus conducted (see details in Sect. 2 and Text S13).

Figure 10Field observation for the relationship between carbonate and sulfate ions during daytime and nighttime hours. Linear relationship analyses for measured sulfate ions and estimated carbonate ions (a) and for measured sulfate ions and estimated bicarbonate ions (b) during the daytime and nighttime hours, with particle sizes of PM ranging from 3.3 to 9 µm.
We further consider the relationships between sulfate ions and (bi)carbonate ions by means of linear regression analysis. However, under the low wind speed (0.76 ± 0.73), correlation coefficients R2 obtained for the relationship between bi(carbonate) and sulfate ions are not promising, with 0.56 (sulfate vs. carbonate) and 0.61 (sulfate vs. bicarbonate) for PM3.3-PM9.0 during daytime hours (Fig. 10). A plausible explanation is that in spite of having little significance, a local primary emission source also brings bias and uncertainty to the correlation results. Shanghai is a coastal city, and sulfate species such as K2SO4 and Na2SO4 from the sea salt contribute to the local sulfate emission as well (Long et al., 2014). On the other hand, this novel SO2 oxidation channel is yet to be in the infant stage, and only active mineral dust components have been considered in this work, whereas other components found in the coarse mode of PM such as organic matter, elemental carbon, and sea salt (Cheung et al., 2011) are likely to involve this mechanism and alter the response of sulfate yield to SO2 heterogeneous uptake. In addition, the water-soluble ions determined in these samples may not come from the net contribution of heterogenous reaction processes in absolute daytime and nighttime periods. In other words, some of the collected samples, experiencing the heterogeneous reaction that occurs during the day(night)–night(day) shifts periods, will inevitably be assigned to the sulfate ions measured in separate sampling hours, thus reducing the correlation coefficients.
For those large particles (LPs), which refer to the particles with a diameter large than 9 µm in this work, sulfate ions show a rather weak or even no correlation to (bi)carbonate ions during the nighttime or daytime hours (Fig. S14). This is likely due to the short lifetime of the LPs. Generally, the aerosol lifetime is of the order of less than an hour to days (Koelemeijer et al., 2006), highly depending on the particle size. For example, the lifetime of PM10 ranges from minutes to hours, and its travel distance, in general, is less than 10 km (Agustine et al., 2018). As a consequence, secondary sulfate formation through chemical reaction over LPs is not significant with respect to in situ emissions. When PM downsizes to 2.5 µm, then PM2.5 has a lifetime prolonged to nearly 1 d or longer (Wu et al., 2020). Therefore, PM3.3–PM9.0 are expected to have a relatively long lifetime, of the order of several hours on average, which enables the heterogeneous reaction process to become a more important contributor to overall sulfate measured in PM3.3–PM9.0 than that in PM≥9.0. This is supported by our observations where, during the daytime hours, the correlation coefficients for PM3.3–PM9.0, i.e., 0.56 (sulfate vs. carbonate) and 0.61 (sulfate vs. bicarbonate), are higher than that of PM≥9.0, i.e., 0.489 (sulfate vs. carbonate) and 0.36 (sulfate vs. bicarbonate), respectively. Likewise, higher correlation coefficients are also observed for PM3.3–PM9.0 than PM≥9.0 in the sample collected during the nighttime periods.
While we note that the correlations between sulfate and (bi)carbonate are not high in this work, ground-based field measurements of sulfate and (bi)carbonate ions shed light on their distinct correlations during the daytime and nighttime hours. In Figs. 10 and S14, the negative correlations between the mass concentrations of sulfate ions and (bi)carbonate ions are observed in the nighttime hours, consistent with the suppression of sulfate formation by CO2 in the dark experiments. This is also supported by lab-based observations where CO2-derived (bi)carbonate species are demonstrated to suppress sulfate production over two dominant mineral dust components, i.e., aluminum oxide (Y. Liu et al., 2020) and silicon dioxide (Fig. S15 and Text S23). Alternatively, while CO2-derived (bi)carbonate may slightly affect sulfate accumulation over PM with high water content in the dark scenario, fresh PM is usually dry when emitted into the atmosphere. Due to the competitive adsorption, the occurrence of the suppression of SO2 adsorption and subsequent sulfate formation is possible in the early emission stage before PM becomes wet, thus contributing to the overall negative correlation.
Instead, positive correlations were seen for those ions within PM sampled during the daytime hours, regardless of the size ranges and carbonate types (HCO CO). This matches with the scenarios in which sulfate production, upon irradiation in the presence of (bi)carbonate ions, is increased over both model and authentic dust particles. Except for this case (nighttime period, size larger than 9 µm), most of the significance P values for their correlations were smaller than 0.1, specifically with significant P values below 0.5 determined for bicarbonate vs. sulfate, implying the plausible underlying connection between sulfate and (bi)carbonate ions. In fact, preceding ground-based observations of a highly correlated relationship between Ca2+ and SO water-soluble ions (Wu et al., 2020) during the carbonate-enriched dust storm episodes, together with persistent reports on the significant role of photochemical channels in elevating the sulfate concentration level during the daytime hours (Wei et al., 2019; Kim et al., 2018; Wu et al., 2017), indirectly reflects the possibility of accelerated SO2 oxidation triggered by photo-generated active intermediates associated with carbonate species.
Overall, this is the first time that relationships between those ions have been explored separately in these two periods. Taken together, carbonate radicals are likely to promote sulfate production in the atmosphere during daytime hours. A detailed and systematic SO2 oxidation channel triggered by CO needs further investigation to enable a better interpretation of the correlations between these inorganic ions at the given meteorological conditions of sampling and physicochemical properties of PM.
On the basis of the experimental and theoretical results derived from this work, we, for the first time, propose a novel reaction channel for fast SO2 oxidation over mineral dust particles due to the formation of carbonate radical ions. A schematic chart for the sulfate formation in the presence of carbonate radicals upon solar light or bi(carbonate) ions under dark conditions is summarized and elucidated in Fig. 11. During the nighttime hours at 298 K (ambient temperature) CO2-derived (bi)carbonate species are prone to having a slightly negative effect on sulfate formation, presumably due to the competitive adsorption between CO2 and SO2. For alkaline carbonate salt, it favors sulfate formation through the neutralization process. On the other hand, in the daytime, both CO2-derived (bi)carbonate species and carbonate salt work as the precursors of CO, which promotes sulfate formation. In particular, uptake coefficients for carbonate salt containing mineral dust can be increased 17 times, which is more pronounced than the increase due to the neutralization regime in the dark condition. Consistent with the findings reported in the early studies (Chen et al., 2021; Dupart et al., 2012), we speculate the production of gas-phase CO ions when mineral dust particles are irradiated in the presence of CO2 (atmospherically relevant concentration 400 ppm). This observation potentially implies that the increased sulfate yield in part comes from increased external secondary sulfate aerosol triggered by CO (g).
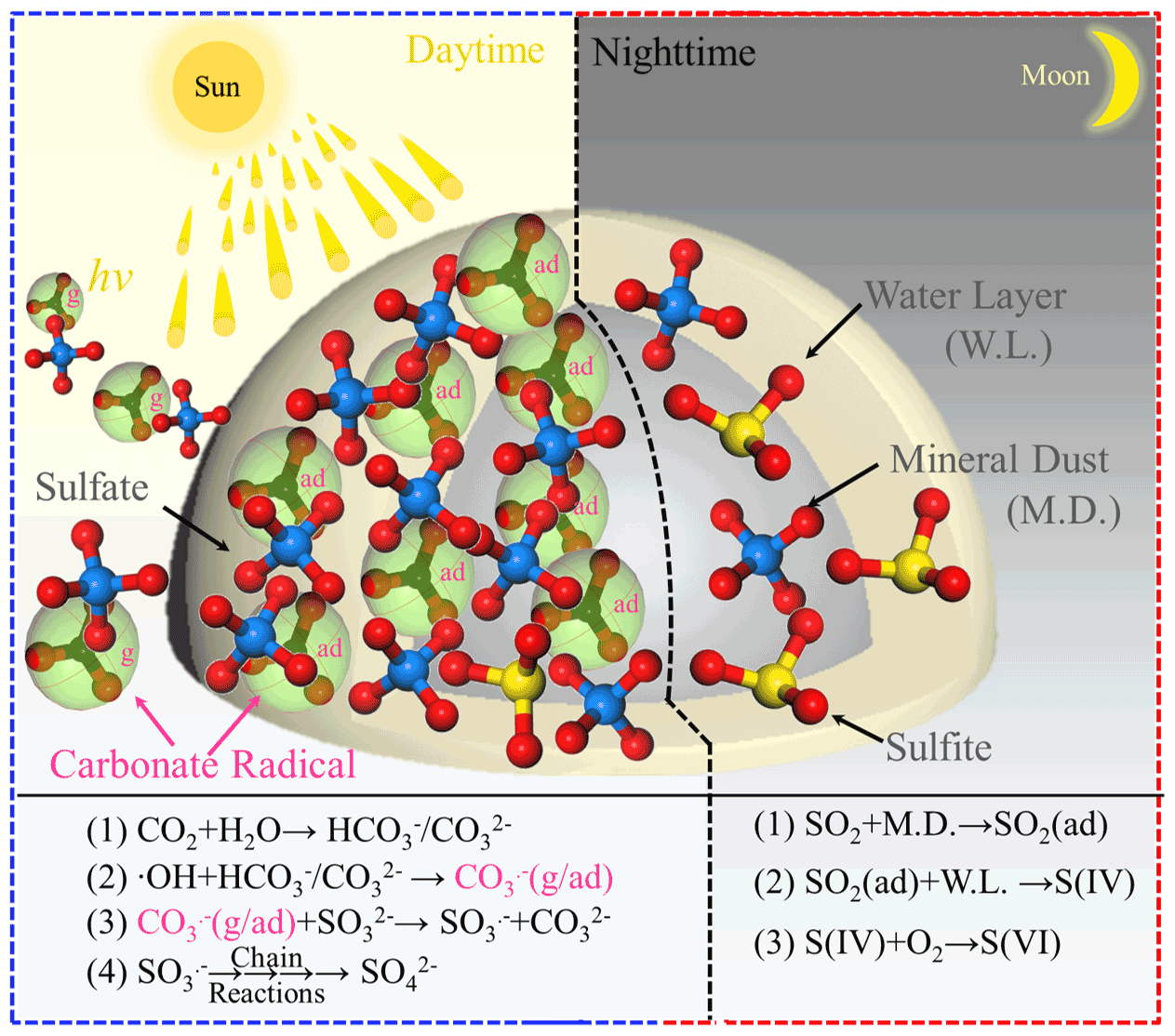
Figure 11Schematic of the sulfate formation in the presence and absence of a carbonate radical. Note that g and ad represent gas phase and adsorbed carbonate radical ions, respectively.
By means of ROS scavenger experiments, direct observation of carbonate radicals using NTAS analysis, oxygen isotope assay, ESR measurement, and DFT calculations, CO-initiated S(IV) oxidation involving single electron transfer process is elucidated. While carbonate radical ions are mainly responsible for rapid sulfate formation, superoxide radical ions are likely to serve as a minor pathway over TiO2-containing mineral dust particles (not shown in the schematic chart to direct readers' focus on CO). In addition, a weak correlation between sulfate ions and (bi)carbonate ions observed for PM3.3–PM9.0 in this work reasonably correlates to non-chemical primary emission and the complicated nature of CO regime of sulfate production in the atmosphere. Nonetheless, complementary field sampling of ambient PM and analysis of sulfate and (bi)carbonate ions in this study unfold their distinct correlations during the daytime and nighttime hours; these two tendencies agree with the experimental observations.
In this work, only atmospheric secondary sulfate formation was considered, whereas the oxidation of primary organic species has not yet been investigated. In fact, carbonate radical ions are prone to reacting rapidly with electron-rich organic amines (Stenman et al., 2003; Yan et al., 2019) and phenol (Xiong et al., 2016; Busset et al., 2007), and they may potentially serve as the key oxidants that drive the fast formation of secondary organic aerosol in the atmosphere. Besides, an observation of the strengthened photochemistry launched by carbonate radicals suggests that such chemistry may be amplified on atmospherically relevant reactions that occur in cloud droplets and fog water, where they often contain hydroxyl radicals and water-soluble (bi)carbonate ions.
Importantly, gas-phase carbonate radical ions are speculated to be formed in the atmospherically relevant CO2 concentration (400 ppm) when mineral dust is irradiated. This will help in the formation of external sulfate aerosol formation. Since both sulfate aerosol and CO2 are known to regulate the radiation budget and solar energy balance on the Earth (Cheung et al., 2011; Möller, 1964), coupled with the CO2-initiated promoted sulfate pathway found here, their overall influence on the global climate needs further investigation. Therefore, our study highlights the necessity for a comprehensive understanding of the CO-relevant chemistry in the underlying impacts of fine PM concentration, human health, and climate. All these assumptions need to be investigated in further detail. This study provides the indication that carbonate radicals not only play a role as a marginal intermediate in tropospheric anion chemistry but also as a strong oxidant for the surface processing of trace gas in the atmosphere.
The data that support the results are available from the corresponding author upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-9175-2022-supplement.
YL, YD, and LZ initially proposed the idea. YL and YD designed the experiments together, and YL performed the most of the experiments. JL performed DFT calculations. YL, XF, and TW contributed to field samplings and data analysis. KL, KG, AUB, IN, QG, XZ, CG, and LZ provided suggestions on the experiments and writing of the draft. All authors wrote the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We greatly appreciate Yang Yang and Keli Han from the Dalian institute of chemical physics, for NTAS and some helpful discussions.
This research has been supported by the National Natural Science Foundation of China (grant nos. 22176036, 21976030, and 21677037), the National Key Research and Development Program of China (grant nos. 2016YFE0112200 and 2016YFC0202700), and the Natural Science Foundation of Shanghai (grant nos. 19ZR1471200 and 17ZR1440200).
This paper was edited by Ryan Sullivan and reviewed by three anonymous referees.
Abou-Ghanem, M., Oliynyk, A. O., Chen, Z. H., Matchett, L. C., McGrath, D. T., Katz, M. J., Locock, A. J., and Styler, S. A.: Significant Variability in the Photocatalytic Activity of Natural Titanium-Containing Minerals: Implications for Understanding and Predicting Atmospheric Mineral Dust Photochemistry, Environ. Sci. Technol., 54, 13509–13516, https://doi.org/10.1021/acs.est.0c05861, 2020.
Agustine, I., Yulinawati, H., Gunawan, D., and Suswantoro, E.: Potential impact of particulate matter less than 10 micron (PM10) to ambient air quality of Jakarta and Palembang, Iop C Ser. Earth Env., 106, 012057, https://doi.org/10.1088/1755-1315/106/1/012057, 2018.
Al-Hosney, H. A. and Grassian, V. H.: Water, sulfur dioxide and nitric acid adsorption on calcium carbonate: A transmission and ATR-FTIR study, Phys. Chem. Chem. Phys., 7, 1266–1276, https://doi.org/10.1039/b417872f, 2005.
Al-Salihi, A. M. and Mohammed, T. H.: The effect of dust storms on some meteorological elements over Baghdad, Iraq: Study Cases, IOSR Journal of Applied Physics, 7, Ver. II PP 01-07, https://iosrjournals.org/iosr-jap/papers/Vol7-issue2/Version-2/A07220107.pdf (last access: 12 July 2022), 2015.
Balachandran, U. and Eror, N. G.: Raman-Spectra Of Titanium-Dioxide, J. Sol. State Chem., 42, 276–282, https://doi.org/10.1016/0022-4596(82)90006-8, 1982.
Baltrusaitis, J., Schuttlefield, J., Zeitler, E., and Grassian, V. H.: Carbon dioxide adsorption on oxide nanoparticle surfaces, Chem. Eng. J., 170, 471–481, https://doi.org/10.1016/j.cej.2010.12.041, 2011.
Bao, H., Yu, S., and Tong, D. Q.: Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America, Nature, 465, 909–912, https://doi.org/10.1038/nature09100, 2010.
Bauer, S. E. and Koch, D.: Impact of heterogeneous sulfate formation at mineral dust surfaces on aerosol loads and radiative forcing in the Goddard Institute for Space Studies general circulation model, J. Geophys. Res., 110, D17202, https://doi.org/10.1029/2005jd005870, 2005.
Behrman, E. J.: Degradation kinetics and mechanism of aniline by heat-assisted persulfate oxidation Comment, J. Environ. Sci. China, 64, 352–352, https://doi.org/10.1016/j.jes.2018.02.008, 2018.
Beig, G. and Brasseur, G. P.: Model of tropospheric ion composition: A first attempt, J. Geophys. Res., 105, 22671–22684, https://doi.org/10.1029/2000JD900119, 2000.
Bhattacharya, A., Amitabha, D., and Mandal, P. C.: Carbonate radical induced polymerisation of pyrrole: A steady state and flash photolysis study, J. Radioanal. Nucl. Ch., 230, 91–95, https://doi.org/10.1007/BF02387452, 1998.
Bisby, R. H., Johnson, S. A., Parker, A. W., and Tavender, S. M.: Time-resolved resonance Raman spectroscopy of the carbonate radical, J. Chem. Soc. Faraday Trans., 94, 2069–2072, https://doi.org/10.1039/A801239C, 1998.
Busset, C., Mazellier, P., Sarakha, M., and De Laat, J.: Photochemical generation of carbonate radicals and their reactivity with phenol, J. Photoch. Photobio. A, 185, 127–132, https://doi.org/10.1016/j.jphotochem.2006.04.045, 2007.
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B.: Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O− in aqueous solution, J. Phys. Chem. Ref. Data, 17, 513–886, https://doi.org/10.1063/1.555805, 2009.
Cao, J. J., Lee, S. C., Zhang, X. Y., Chow, J. C., An, Z. S., Ho, K. F., Watson, J. G., Fung, K., Wang, Y. Q., and Shen, Z. X.: Characterization of airborne carbonate over a site near Asian dust source regions during spring 2002 and its climatic and environmental significance, J. Geophys. Res., 110, 1–8, https://doi.org/10.1029/2004JD005244, 2005.
Chameides, W. L. and Davis, D. D.: The Free-Radical chemistry of cloud droplets and its impact upon the composition of rain, J. Geophs Res.-Oceans, 87, 4863–4877, https://doi.org/10.1029/JC087iC07p04863, 1982.
Chandrasekaran, K. and Thomas, J. K.: Photochemical reduction of carbonate to formaldehyde on TiO2 powder, Chem. Phys. Lett., 99, 7–10, https://doi.org/10.1016/0009-2614(83)80259-0, 1983.
Chen, H. H., Nanayakkara, C. E., and Grassian, V. H.: Titanium Dioxide Photocatalysis in Atmospheric Chemistry, Chem. Rev., 112, 5919–5948, https://doi.org/10.1021/cr3002092, 2012.
Chen, Y., Tong, S. R., Li, W. R., Liu, Y. P., Tan, F., Ge, M. F., Xie, X. F., and Sun, J.: Photocatalytic Oxidation of SO2 by TiO2: Aerosol Formation and the Key Role of Gaseous Reactive Oxygen Species, Environ. Sci. Technol., 55, 9784–9793, https://doi.org/10.1021/acs.est.1c01608, 2021.
Cheung, K., Daher, N., Kam, W., Shafer, M. M., Ning, Z., Schauer, J. J., and Sioutas, C.: Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area, Atmos. Environ., 45, 2651–2662, https://doi.org/10.1016/j.atmosenv.2011.02.066, 2011.
Cope, V. W., Chen, S.-N., and Hoffman, M. Z.: Intermediates in the photochemistry of of carbonato-amine complexes of cobalt(III). carbonate(-) radicals and the aquocarbonato complex, J. Am. Chem. Soc., 95, 3116–3121, https://doi.org/10.1021/ja00791a005, 1973
Csavina, J., Field, J., Felix, O., Corral-Avitia, A. Y., Saez, A. E., and Betterton, E. A.: Effect of wind speed and relative humidity on atmospheric dust concentrations in semi-arid climates, Sci. Total Envrion., 487, 82–90, https://doi.org/10.1016/j.scitotenv.2014.03.138, 2014.
Cwiertny, D. M., Young, M. A., and Grassian, V. H.: Chemistry and photochemistry of mineral dust aerosol, Annu. Rev. Phys. Chem., 59, 27–51, https://doi.org/10.1146/annurev.physchem.59.032607.093630, 2008.
Das, T. N.: Reactivity and role of SO radical in aqueous medium chain oxidation of sulfite to sulfate and atmospheric sulfuric acid generation, J. Phys. Chem. A, 105, 9142–9155, https://doi.org/10.1021/jp011255h, 2001.
Deng, Y., Liu, Y., Wang, T., Cheng, H., Feng, Y., Yang, Y., and Zhang, L.: Photochemical reaction of CO2 on atmospheric mineral dusts, Atmos. Environ., 223, 117222, https://doi.org/10.1016/j.atmosenv.2019.117222, 2020.
Dong, X., Fu, J. S., Huang, K., Tong, D., and Zhuang, G.: Model development of dust emission and heterogeneous chemistry within the Community Multiscale Air Quality modeling system and its application over East Asia, Atmos. Chem. Phys., 16, 8157–8180, https://doi.org/10.5194/acp-16-8157-2016, 2016.
Dupart, Y., King, S. M., Nekat, B., Nowak, A., Wiedensohler, A., Herrmann, H., David, G., Thomas, B., Miffre, A., Rairoux, P., D'Anna, B., and George, C.: Mineral dust photochemistry induces nucleation events in the presence of SO2, P. Natl. Acad. Sci. USA, 109, 20842–20847, https://doi.org/10.1073/pnas.1212297109, 2012.
Duran, A., Monteagudo, J. M., Martin, I. S., Merino, S., Chen, X., and Shi, X.: Solar photo-degradation of aniline with rGO/TiO2 composites and persulfate, Sci. Total Envrion., 697, 134086, https://doi.org/10.1016/j.scitotenv.2019.134086, 2019.
El Zein, A., Romanias, M. N., and Bedjanian, Y.: Kinetics and Products of Heterogeneous Reaction of HONO with Fe2O3 and Arizona Test Dust, Environ. Sci. Technol., 47, 6325–6331, https://doi.org/10.1021/es400794c, 2013.
Ervens, B., George, C., Williams, J. E., Buxton, G. V., Salmon, G. A., Bydder, M., Wilkinson, F., Dentener, F., Mirabel, P., Wolke, R., and Herrmann, H.: CAPRAM 2.4 (MODAC mechanism): An extended and condensed tropospheric aqueous phase mechanism and its application, J. Geophys. Res., 108, 4426, https://doi.org/10.1029/2002jd002202, 2003.
Fang, T., Guo, H., Zeng, L., Verma, V., Nenes, A., and Weber, R. J.: Highly Acidic Ambient Particles, Soluble Metals, and Oxidative Potential: A Link between Sulfate and Aerosol Toxicity, Environ. Sci. Technol., 51, 2611–2620, https://doi.org/10.1021/acs.est.6b06151, 2017.
Fang, X., Liu, Y., Kejian, Tao, W., Yue, D., Yiqing, F., Yang, Y., Cheng, H., Chen, J., and liwu, Z.: Atmospheric Nitrate Formation through Oxidation by carbonate radical, ACS Earth Space Chem., 5, 1801–1811, https://doi.org/10.1021/acsearthspacechem.1c00169, 2021.
Feng, T., Bei, N. F., Zhao, S. Y., Wu, J. R., Li, X., Zhang, T., Cao, J. J., Zhou, W. J., and Li, G. H.: Wintertime nitrate formation during haze days in the Guanzhong basin, China: A case study, Environ. Pollut., 243, 1057–1067, https://doi.org/10.1016/j.envpol.2018.09.069, 2018.
Ferrer-Sueta, G., Vitturi, D., Batinic-Haberle, I., Fridovich, I., Goldstein, S., Czapski, G., and Radi, R.: Reactions of manganese porphyrins with peroxynitrite and carbonate radical anion, J. Biol. Chem., 278, 27432–27438, https://doi.org/10.1074/jbc.M213302200, 2003.
Gankanda, A., Coddens, E. M., Zhang, Y. P., Cwiertny, D. M., and Grassian, V. H.: Sulfate formation catalyzed by coal fly ash, mineral dust and iron(III) oxide: variable influence of temperature and light, Environ. Sci.-Proc. Imp., 18, 1484–1491, https://doi.org/10.1039/c6em00430j, 2016.
Ge, W., Liu, J., Yi, K., Xu, J., Zhang, Y., Hu, X., Ma, J., Wang, X., Wan, Y., Hu, J., Zhang, Z., Wang, X., and Tao, S.: Influence of atmospheric in-cloud aqueous-phase chemistry on the global simulation of SO2 in CESM2, Atmos. Chem. Phys., 21, 16093–16120, https://doi.org/10.5194/acp-21-16093-2021, 2021.
Ghalei, M., Ma, J., Schmidhammer, U., Vandenborre, J., Fattahi, M., and Mostafavi, M.: Picosecond pulse radiolysis of highly concentrated carbonate solutions, J. Phys. Chem. B, 120, 2434–2439, https://doi.org/10.1021/acs.jpcb.5b12405, 2016.
Goldstein, S., Czapski, G., Lind, J., and Merényi, G.: Carbonate radical ion is the only observable intermediate in the reaction of peroxynitrite with CO2, Chem. Res. Toxicol., 14, 1273–1276, https://doi.org/10.1021/tx0100845, 2001.
Graedel, T. E. and Weschler, C. J.: Chemistry within Aqueous Atmospheric Aerosols And Raindrops, J. Geophys. Res., 19, 505–539, https://doi.org/10.1029/RG019i004p00505, 1981.
Hanisch, F. and Crowley, J. N.: Ozone decomposition on Saharan dust: an experimental investigation, Atmos. Chem. Phys., 3, 119–130, https://doi.org/10.5194/acp-3-119-2003, 2003.
Hayon, E., Treinin, A., and Wilf, J.: Electronic spectra, photochemistry, and autoxidation mechanism of the sulfite-bisulfite-pyrosulfite systems. SO, SO, SO, and SO radicals, J. Am. Chem. Soc., 94, 47–57, https://doi.org/10.1021/ja00756a009, 1972.
He, J., Xu, H. H., Balasubramanian, R., Chan, C. Y., and Wang, C. J.: Comparison of NO2 and SO2 Measurements Using Different Passive Samplers in Tropical Environment, Aerosol Air Qual. Res., 14, 355–363, https://doi.org/10.4209/aaqr.2013.02.0055, 2014.
Herrmann, H., Ervens, B., Jacobi, H. W., Wolke, R., Nowacki, P., and Zellner, R.: CAPRAM2.3: A chemical aqueous phase radical mechanism for tropospheric chemistry, J. Atmos. Chem., 36, 231–284, https://doi.org/10.1023/A:1006318622743, 2000.
Hossain, M. D., Huang, Y., Yu, T. H., Goddard Iii, W. A., and Luo, Z.: Reaction mechanism and kinetics for CO2 reduction on nickel single atom catalysts from quantum mechanics, Nat. Commun., 11, 2256, https://doi.org/10.1038/s41467-020-16119-6, 2020.
Huang, H. L., Chao, W., and Lin, J. J. M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
Huang, J. P. and Mabury, S. A.: Steady-state concentrations of carbonate radicals in field waters, Environ. Toxicol. Chem., 19, 2181–2188, https://doi.org/10.1002/etc.5620190906, 2000.
Huang, L., An, J., Koo, B., Yarwood, G., Yan, R., Wang, Y., Huang, C., and Li, L.: Sulfate formation during heavy winter haze events and the potential contribution from heterogeneous SO2 + NO2 reactions in the Yangtze River Delta region, China, Atmos. Chem. Phys., 19, 14311–14328, https://doi.org/10.5194/acp-19-14311-2019, 2019.
Huang, X., Song, Y., Zhao, C., Li, M. M., Zhu, T., Zhang, Q., and Zhang, X. Y.: Pathways of sulfate enhancement by natural and anthropogenic mineral aerosols in China, J. Geophys. Res., 119, 14165–14179, https://doi.org/10.1002/2014jd022301, 2014.
Hung, H. M. and Hoffmann, M. R.: Oxidation of Gas-Phase SO2 on the Surfaces of Acidic Microdroplets: Implications for Sulfate and Sulfate Radical Anion Formation in the Atmospheric Liquid Phase, Environ. Sci. Technol., 49, 13768–13776, https://doi.org/10.1021/acs.est.5b01658, 2015.
Hung, H. M., Hsu, M. N., and Hoffmann, M. R.: Quantification of SO2 oxidation on interfacial surfaces of acidic micro-droplets: Implication for ambient sulfate formation, Environ. Sci. Technol., 52, 9079–9086, https://doi.org/10.1021/acs.est.8b01391, 2018.
Itahashi, S., Yamaji, K., Chatani, S., and Hayami, H.: Refinement of Modeled Aqueous-Phase Sulfate Production via the Fe- and Mn-Catalyzed Oxidation Pathway, Atmosphere, 9, 132, https://doi.org/10.3390/atmos9040132, 2018.
Kerminen, V. M., Hillamo, R., Teinilä, K., Pakkanen, T., Allegrini, I., and Sparapani, R.: Ion balances of size-resolved tropospheric aerosol samples: implications for the acidity and atmospheric processing of aerosols, Atmos. Environ., 35, 5255–5265, https://doi.org/10.1016/S1352-2310(01)00345-4, 2001.
Kim, H., Zhang, Q., and Heo, J.: Influence of intense secondary aerosol formation and long-range transport on aerosol chemistry and properties in the Seoul Metropolitan Area during spring time: results from KORUS-AQ, Atmos. Chem. Phys., 18, 7149–7168, https://doi.org/10.5194/acp-18-7149-2018, 2018.
Koelemeijer, R., Homan, C. D., and Matthijsen, J.: Comparison of spatial and temporal variations of aerosol optical thickness and particulate matter over Europe, Atmos. Environ., 40, 5304–5315, https://doi.org/10.1016/j.atmosenv.2006.04.044, 2006.
Kong, L. D., Zhao, X., Sun, Z. Y., Yang, Y. W., Fu, H. B., Zhang, S. C., Cheng, T. T., Yang, X., Wang, L., and Chen, J. M.: The effects of nitrate on the heterogeneous uptake of sulfur dioxide on hematite, Atmos. Chem. Phys., 14, 9451–9467, https://doi.org/10.5194/acp-14-9451-2014, 2014.
Lehtipalo, K., Rondo, L., Kontkanen, J., Schobesberger, S., Jokinen, T., Sarnela, N., Kürten, A., Ehrhart, S., Franchin, A., Nieminen, T., Riccobono, F., Sipilä, M., Yli-Juuti, T., Duplissy, J., Adamov, A., Ahlm, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Downard, A. J., Dunne, E. M., Flagan, R. C., Guida, R., Hakala, J., Hansel, A., Jud, W., Kangasluoma, J., Kerminen, V.-M., Keskinen, H., Kim, J., Kirkby, J., Kupc, A., Kupiainen-Määttä, O., Laaksonen, A., Lawler, M. J., Leiminger, M., Mathot, S., Olenius, T., Ortega, I. K., Onnela, A., Petäjä, T., Praplan, A., Rissanen, M. P., Ruuskanen, T., Santos, F. D., Schallhart, S., Schnitzhofer, R., Simon, M., Smith, J. N., Tröstl, J., Tsagkogeorgas, G., Tomé, A., Vaattovaara, P., Vehkamäki, H., Vrtala, A. E., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Virtanen, A., Donahue, N. M., Carslaw, K. S., Baltensperger, U., Riipinen, I., Curtius, J., Worsnop, D. R., and Kulmala, M.: The effect of acid-base clustering and ions on the growth of atmospheric nano-particles, Nat. Commun., 7, 11594, https://doi.org/10.1038/ncomms11594, 2016.
Li, B. Q., Ma, X. Y., Li, Q. S., Chen, W. Z., Deng, J., Li, G. X., Chen, G. Y., and Liao, W. C.: Factor affecting the role of radicals contribution at different wavelengths, degradation pathways and toxicity during UV-LED/chlorine process, Chem. Eng. J., 392, 124552, https://doi.org/10.1016/j.cej.2020.124552, 2020.
Li, K. J., Kong, L. D., Zhanzakova, A., Tong, S. Y., Shen, J. D., Wang, T., Chen, L., Li, Q., Fu, H. B., and Zhang, L. W.: Heterogeneous conversion of SO2 on nano alpha-Fe2O3: the effects of morphology, light illumination and relative humidity, Environ. Sci. Nano., 6, 1838–1851, https://doi.org/10.1039/c9en00097f, 2019.
Li, L., Chen, Z. M., Zhang, Y. H., Zhu, T., Li, S., Li, H. J., Zhu, L. H., and Xu, B. Y.: Heterogeneous oxidation of sulfur dioxide by ozone on the surface of sodium chloride and its mixtures with other components, J. Geophys. Res., 112, D18301, https://doi.org/10.1029/2006jd008207, 2007.
Li, W. J., Shao, L. Y., Shi, Z. B., Chen, J. M., Yang, L. X., Yuan, Q., Yan, C., Zhang, X. Y., Wang, Y. Q., Sun, J. Y., Zhang, Y. M., Shen, X. J., Wang, Z. F., and Wang, W. X.: Mixing state and hygroscopicity of dust and haze particles before leaving Asian continent, J. Geophys. Res., 119, 1044–1059, https://doi.org/10.1002/2013jd021003, 2014.
Li, X., Yu, J., and Jaroniec, M.: Hierarchical photocatalysts, Chem. Soc. Rev., 45, 2603–2636, https://doi.org/10.1039/c5cs00838g, 2016.
Li, X. R., Wang, L. L., Ji, D. S., Wen, T. X., Pan, Y. P., Sun, Y., and Wang, Y. S.: Characterization of the size-segregated water-soluble inorganic ions in the Jing-Jin-Ji urban agglomeration: Spatial/temporal variability, size distribution and sources, Atmos. Environ., 77, 250–259, https://doi.org/10.1016/j.atmosenv.2013.03.042, 2013.
Liao, L. F., Lien, C. F., Shieh, D. L., Chen, M. T., and Lin, J. L.: FTIR study of adsorption and photoassisted oxygen isotopic exchange of carbon monoxide, carbon dioxide, carbonate, and formate on TiO2, J. Phys. Chem. B, 106, 11240–11245, https://doi.org/10.1021/jp0211988, 2002.
Liu, J. R., Ning, A., Liu, L., Wang, H. X., Kurten, T., and Zhang, X. H.: A pH dependent sulfate formation mechanism caused by hypochlorous acid in the marine atmosphere, Sci. Total Envrion., 787, 147551, https://doi.org/10.1016/j.scitotenv.2021.147551, 2021.
Liu, P., Ye, C., Xue, C., Zhang, C., Mu, Y., and Sun, X.: Formation mechanisms of atmospheric nitrate and sulfate during the winter haze pollution periods in Beijing: gas-phase, heterogeneous and aqueous-phase chemistry, Atmos. Chem. Phys., 20, 4153–4165, https://doi.org/10.5194/acp-20-4153-2020, 2020.
Liu, T., Hong, Y., Li, M., Xu, L., Chen, J., Bian, Y., Yang, C., Dan, Y., Zhang, Y., Xue, L., Zhao, M., Huang, Z., and Wang, H.: Atmospheric oxidation capacity and ozone pollution mechanism in a coastal city of southeastern China: analysis of a typical photochemical episode by an observation-based model, Atmos. Chem. Phys., 22, 2173–2190, https://doi.org/10.5194/acp-22-2173-2022, 2022.
Liu, T. Y. and Abbatt, J. P. D.: Oxidation of sulfur dioxide by nitrogen dioxide accelerated at the interface of deliquesced aerosol particles, Nat. Chem., 13, 1173–1177, https://doi.org/10.1038/s41557-021-00777-0, 2021.
Liu, T. Y., Clegg, S. L., and Abbatt, J. P. D.: Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles, P. Natl. Acad. Sci. USA, 117, 1354–1359, https://doi.org/10.1073/pnas.1916401117, 2020.
Liu, X. C., Tang, W. J., Chen, H. N., Guo, J. M., Tripathee, L., and Huang, J.: Observational Study of Ground-Level Ozone in the Desert Atmosphere, B. Environ. Contam. Tox., 108, 219–224, https://doi.org/10.1007/s00128-021-03444-9, 2022.
Liu, Y., Wang, T., Fang, X., Deng, Y., Cheng, H., Fu, H., and Zhang, L.: Impact of greenhouse gas CO2 on the heterogeneous reaction of SO2 on Alpha-Al2O3, Chinese Chem. Lett., 31, 2712–2716, https://doi.org/10.1016/j.cclet.2020.04.037, 2020.
Liu, Y. Q., He, X. X., Duan, X. D., Fu, Y. S., and Dionysiou, D. D.: Photochemical degradation of oxytetracycline: Influence of pH and role of carbonate radical, Chem. Eng. J., 276, 113–121, https://doi.org/10.1016/j.cej.2015.04.048, 2015.
Liu, Z. R., Xie, Y. Z., Hu, B., Wen, T. X., Xin, J. Y., Li, X. R., and Wang, Y. S.: Size-resolved aerosol water-soluble ions during the summer and winter seasons in Beijing: Formation mechanisms of secondary inorganic aerosols, Chemosphere, 183, 119–131, https://doi.org/10.1016/j.chemosphere.2017.05.095, 2017.
Long, S. L., Zeng, J. R., Li, Y., Bao, L. M., Cao, L. L., Liu, K., Xu, L., Lin, J., Liu, W., Wang, G. H., Yao, J., Ma, C. Y., and Zhao, Y. D.: Characteristics of secondary inorganic aerosol and sulfate species in size-fractionated aerosol particles in Shanghai, J. Environ. Sci. China, 26, 1040–1051, https://doi.org/10.1016/S1001-0742(13)60521-5, 2014.
Mahajan, A. S., Li, Q., Inamdar, S., Ram, K., Badia, A., and Saiz-Lopez, A.: Modelling the impacts of iodine chemistry on the northern Indian Ocean marine boundary layer, Atmos. Chem. Phys., 21, 8437–8454, https://doi.org/10.5194/acp-21-8437-2021, 2021.
McNaughton, C. S., Clarke, A. D., Kapustin, V., Shinozuka, Y., Howell, S. G., Anderson, B. E., Winstead, E., Dibb, J., Scheuer, E., Cohen, R. C., Wooldridge, P., Perring, A., Huey, L. G., Kim, S., Jimenez, J. L., Dunlea, E. J., DeCarlo, P. F., Wennberg, P. O., Crounse, J. D., Weinheimer, A. J., and Flocke, F.: Observations of heterogeneous reactions between Asian pollution and mineral dust over the Eastern North Pacific during INTEX-B, Atmos. Chem. Phys., 9, 8283–8308, https://doi.org/10.5194/acp-9-8283-2009, 2009.
Merouani, S., Hamdaoui, O., Saoudi, F., Chiha, M., and Petrier, C.: Influence of bicarbonate and carbonate ions on sonochemical degradation of Rhodamine B in aqueous phase, J. Hazard Mater., 175, 593–599, https://doi.org/10.1016/j.jhazmat.2009.10.046, 2010.
Miller-Schulze, J. P., Shafer, M., Schauer, J. J., Heo, J., Solomon, P. A., Lantz, J., Artamonova, M., Chen, B., Imashev, S., and Sverdlik, L.: Seasonal contribution of mineral dust and other major components to particulate matter at two remote sites in Central Asia, Atmos. Environ., 119, 11–20, https://doi.org/10.1016/j.atmosenv.2015.07.011, 2015.
Mogili, P. K., Kleiber, P. D., Young, M. A., and Grassian, V. H.: Heterogeneous uptake of ozone on reactive components of mineral dust aerosol: an environmental aerosol reaction chamber study, J. Phys. Chem. A, 110, 13799–13807, https://doi.org/10.1021/jp063620g, 2006.
Möller, F.: On the influence of changes in the CO2 concentration in air on the radiation balance of the Earth's surface and on the climate, J. Geophys. Res., 68, 3877–3886, https://doi.org/10.1029/JZ068i013p03877, 1964.
Najafpour, N., Afshin, H., and Firoozabadi, B.: Dust concentration over a semi-arid region: Parametric study and establishment of new empirical models, Atmos. Res., 243, 104995, https://doi.org/10.1016/j.atmosres.2020.104995, 2020.
Nanayakkara, C. E., Larish, W. A., and Grassian, V. H.: Titanium dioxide nanoparticle surface reactivity with atmospheric gases, CO2, SO2, and NO2: roles of surface hydroxyl groups and adsorbed water in the formation and stability of adsorbed products, J. Phys. Chem. C, 118, 23011–23021, https://doi.org/10.1021/jp504402z, 2014.
Neta, P. and Huie, R. E.: Free-radical chemistry of sulfite, Environ. Health Persp., 64, 209–217, https://doi.org/10.1289/ehp.8564209, 1985.
Nie, W., Wang, T., Xue, L. K., Ding, A. J., Wang, X. F., Gao, X. M., Xu, Z., Yu, Y. C., Yuan, C., Zhou, Z. S., Gao, R., Liu, X. H., Wang, Y., Fan, S. J., Poon, S., Zhang, Q. Z., and Wang, W. X.: Asian dust storm observed at a rural mountain site in southern China: chemical evolution and heterogeneous photochemistry, Atmos. Chem. Phys., 12, 11985–11995, https://doi.org/10.5194/acp-12-11985-2012, 2012.
Palmer, C. D. and Cherry, J. A.: Geochemical Evolution of Groundwater in Sequences of Sedimentary-Rocks, J. Hydrol., 75, 27–65, https://doi.org/10.1016/0022-1694(84)90045-3, 1984.
Peters, S. J. and Ewing, G. E.: Water on Salt: An Infrared Study of Adsorbed H2O on NaCl (100) under Ambient Conditions, J. Phys. Chem. B, 101, 10880–10886, https://doi.org/10.1021/jp972810b, 1997.
Rodriguez, J. A., Hanson, J., and Chupas, P.: In-Situ Characterization of Heterogeneous Catalysts, Focus on Catal. 8, Hoboken, New Jersey, John Wiley & Sons, Inc., https://doi.org/10.1002/9781118355923, 2013.
Rubasinghege, G., Elzey, S., Baltrusaitis, J., Jayaweera, P. M., and Grassian, V. H.: Reactions on Atmospheric Dust Particles: Surface Photochemistry and Size-Dependent Nanoscale Redox Chemistry, J. Phys. Chem. Lett., 1, 1729–1737, https://doi.org/10.1021/jz100371d, 2010.
Salama, S. B., Natarajan, C., Nogami, G., and Kennedy, J. H.: The role of reducing agent in oxidation reactions of water on illuminated TiO2 electrodes, J. Electrochem. Soc., 142, 806–810, https://doi.org/10.1149/1.2048539, 1995.
Samuni, A., Goldstein, S., Russo, A., Mitchell, J. B., Krishna, M. C., and Neta, P.: Kinetics and mechanism of hydroxyl radical and OH-adduct radical reactions with nitroxides and with their hydroxylamines, J. Am. Chem. Soc., 124, 8719–8724, https://doi.org/10.1021/ja017587h, 2002.
Shafirovich, V., Dourandin, A., Huang, W., and Geacintov, N. E.: The carbonate radical is a site-selective oxidizing agent of guanine in double-stranded oligonucleotides, J. Biol. Chem., 276, 24621–24626, https://doi.org/10.1074/jbc.M101131200, 2001.
Shang, J., Li, J., and Zhu, T.: Heterogeneous reaction of SO2 on TiO2 particles, Sci. China Chem., 53, 2637–2643, https://doi.org/10.1007/s11426-010-4160-3, 2010.
Song, X., Li, J., Shao, L., Zheng, Q., and Zhang, D.: Inorganic ion chemistry of local particulate matter in a populated city of North China at light, medium, and severe pollution levels, Sci. Total Envrion., 650, 566–574, https://doi.org/10.1016/j.scitotenv.2018.09.033, 2018.
Stenman, D., Carlsson, M., Jonsson, M., and Reitberger, T.: Reactivity of the carbonate radical anion towards carbohydrate and lignin model compounds, J. Wood Chem. Technol., 23, 47–69, https://doi.org/10.1081/Wct-120018615, 2003.
Stevenson, D. S., Zhao, A., Naik, V., O'Connor, F. M., Tilmes, S., Zeng, G., Murray, L. T., Collins, W. J., Griffiths, P. T., Shim, S., Horowitz, L. W., Sentman, L. T., and Emmons, L.: Trends in global tropospheric hydroxyl radical and methane lifetime since 1850 from AerChemMIP, Atmos. Chem. Phys., 20, 12905–12920, https://doi.org/10.5194/acp-20-12905-2020, 2020.
Stone, R.: Air pollution. Counting the cost of London's killer smog, Science, 298, 2106–2107, https://doi.org/10.1126/science.298.5601.2106b, 2002.
Su, H., Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Zhang, Q., He, K., and Carmichael, G.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Su, W. G., Zhang, J., Feng, Z. C., Chen, T., Ying, P. L., and Li, C.: Surface phases of TiO2 nanoparticles studied by UV Raman spectroscopy and FT-IR spectroscopy, J. Phys. Chem. C, 112, 7710–7716, https://doi.org/10.1021/jp7118422, 2008.
Sullivan, R. C., Guazzotti, S. A., Sodeman, D. A., and Prather, K. A.: Direct observations of the atmospheric processing of Asian mineral dust, Atmos. Chem. Phys., 7, 1213–1236, https://doi.org/10.5194/acp-7-1213-2007, 2007.
Sulzberger, B., Canonica, S., Egli, T., Giger, W., Klausen, J., and Gunten, U. v.: Oxidative transformations of contaminants in natural and in technical systems, Chimia, 51, 900–907, https://doi.org/10.1051/epjconf/20101105003, 1997.
Sun, P., Tyree, C., and Huang, C. H.: Inactivation of Escherichia coli, Bacteriophage MS2, and Bacillus Spores under UV H2O2 and UV/Peroxydisulfate Advanced Disinfection Conditions, Environ. Sci. Technol., 50, 4448–4458, https://doi.org/10.1021/acs.est.5b06097, 2016.
Ta, W. Q., Xiao, Z., Qu, J. J., Yang, G. S., and Wang, T.: Characteristics of dust particles from the desert/Gobi area of northwestern China during dust-storm periods, Environ. Geol., 43, 667–679, https://doi.org/10.1007/s00254-002-0673-1, 2003.
Tang, M. J., Cziczo, D. J., and Grassian, V. H.: Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 116, 4205–4259, https://doi.org/10.1021/acs.chemrev.5b00529, 2016.
Wang, X. M., Huang, X., Zuo, C. Y., and Hu, H. Y.: Kinetics of quinoline degradation by O3/UV in aqueous phase, Chemosphere, 55, 733–741, https://doi.org/10.1016/j.chemosphere.2003.11.019, 2004.
Wang, Y., Wan, Q., Meng, W., Liao, F., Tan, H., and Zhang, R.: Long-term impacts of aerosols on precipitation and lightning over the Pearl River Delta megacity area in China, Atmos. Chem. Phys., 11, 12421–12436, https://doi.org/10.5194/acp-11-12421-2011, 2011.
Wang, Y., Zhuang, G. S., Tang, A. H., Yuan, H., Sun, Y. L., Chen, S. A., and Zheng, A. H.: The ion chemistry and the source of PM2.5 aerosol in Beijing, Atmos. Environ., 39, 3771–3784, https://doi.org/10.1016/j.atmosenv.2005.03.013, 2005.
Wang, Y. X., Zhang, Q. Q., Jiang, J. K., Zhou, W., Wang, B. Y., He, K. B., Duan, F. K., Zhang, Q., Philip, S., and Xie, Y. Y.: Enhanced sulfate formation during China's severe winter haze episode in January 2013 missing from current models, J. Geophys. Res., 119, 10425–10440, https://doi.org/10.1002/2013jd021426, 2014.
Watanabe, K., Yang, L., Nakamura, S., Otani, T., and Mori, K.: Volcanic Impact of Nishinoshima Eruptions in Summer 2020 on the Atmosphere over Central Japan: Results from Airborne Measurements of Aerosol and Trace Gases, Sola, 17, 109–112, https://doi.org/10.2151/sola.2021-017, 2020.
Wei, J., Yu, H., Wang, Y., and Verma, V.: Complexation of Iron and Copper in Ambient Particulate Matter and Its Effect on the Oxidative Potential Measured in a Surrogate Lung Fluid, Environ. Sci. Technol., 53, 1661–1671, https://doi.org/10.1021/acs.est.8b05731, 2019.
Witkowska, A., Lewandowska, A. U., Saniewska, D., and Falkowska, L. M.: Effect of agriculture and vegetation on carbonaceous aerosol concentrations (PM2.5 and PM10) in Puszcza Borecka National Nature Reserve (Poland), Air Qual. Atmos. Hlth., 9, 761–773, https://doi.org/10.1007/s11869-015-0378-8, 2016.
Wojnarovits, L., Toth, T., and Takacs, E.: Rate constants of carbonate radical anion reactions with molecules of environmental interest in aqueous solution: A review, Sci. Total Envrion., 717, 137219, https://doi.org/10.1016/j.scitotenv.2020.137219, 2020.
Wu, C., Zhang, S., Wang, G., Lv, S., Li, D., Liu, L., Li, J., Liu, S., Du, W., Meng, J., Qiao, L., Zhou, M., Huang, C., and Wang, H.: Efficient Heterogeneous Formation of Ammonium Nitrate on the Saline Mineral Particle Surface in the Atmosphere of East Asia during Dust Storm Periods, Environ. Sci. Technol., 54, 15622–15630, https://doi.org/10.1021/acs.est.0c04544, 2020.
Wu, D., Fan, Z., Ge, X., Meng, Y., Xia, J., Liu, G., and Li, F.: Chemical and Light Extinction Characteristics of Atmospheric Aerosols in Suburban Nanjing, China, Atmosphere-Basel, 8, 149, https://doi.org/10.3390/atmos8080149, 2017.
Wu, L. Y., Tong, S. R., Wang, W. G., and Ge, M. F.: Effects of temperature on the heterogeneous oxidation of sulfur dioxide by ozone on calcium carbonate, Atmos. Chem. Phys., 11, 6593–6605, https://doi.org/10.5194/acp-11-6593-2011, 2011.
Wu, Q., Tang, X., Kong, L., Dao, X., Lu, M. M., Liu, Z. R., Wang, W., Wang, Q., Chen, D. H., Wu, L., Pan, X. L., Li, J., Zhu, J., and Wang, Z. F.: Evaluation and Bias Correction of the Secondary Inorganic Aerosol Modeling over North China Plain in Autumn and Winter, Atmosphere, 12, 578, https://doi.org/10.3390/atmos12050578, 2021.
Xia, D. M., Zhang, X. R., Chen, J. W., Tong, S. R., Xie, H. B., Wang, Z. Y., Xu, T., Ge, M. F., and Allen, D. T.: Heterogeneous Formation of HONO Catalyzed by CO2, Environ. Sci. Technol., 55, 12215–12222, https://doi.org/10.1021/acs.est.1c02706, 2021.
Xiong, X. Q., Zhang, X., and Xu, Y. M.: Incorporative Effect of Pt and Na2CO3 on TiO2-Photocatalyzed Degradation of Phenol in Water, J. Phys. Chem. C, 120, 25689–25696, https://doi.org/10.1021/acs.jpcc.6b07951, 2016.
Yan, J. F., Peng, J. L., Lai, L. D., Ji, F. Z., Zhang, Y. H., Lai, B., Chen, Q. X., Yao, G., Chen, X., and Song, L. P.: Activation CuFe2O4 by Hydroxylamine for Oxidation of Antibiotic Sulfamethoxazole, Environ. Sci. Technol., 52, 14302–14310, https://doi.org/10.1021/acs.est.8b03340, 2018.
Yan, S. W., Liu, Y. J., Lian, L. S., Li, R., Ma, J. Z., Zhou, H. X., and Song, W. H.: Photochemical formation of carbonate radical and its reaction with dissolved organic matters, Water Res., 161, 288–296, https://doi.org/10.1016/j.watres.2019.06.002, 2019.
Yermakov, A. N. and Purmal, A. P.: Iron-catalyzed oxidation of sulfite: From established results to a new understanding, Prog. React. Kinet. Mec., 28, 189–255, https://doi.org/10.3184/007967403103165503, 2003.
Yu, T., Zhao, D., Song, X., and Zhu, T.: NO2-initiated multiphase oxidation of SO2 by O2 on CaCO3 particles, Atmos. Chem. Phys., 18, 6679–6689, https://doi.org/10.5194/acp-18-6679-2018, 2018.
Yu, Z. C., Jang, M. S., Kim, S., Bae, C., Koo, B. Y., Beardsley, R., Park, J., Chang, L. S., Lee, H. C., Lim, Y. K., and Cho, J. H.: Simulating the Impact of Long-Range-Transported Asian Mineral Dust on the Formation of Sulfate and Nitrate during the KORUS-AQ Campaign, ACS Earth Space Chem., 4, 1039–1049, https://doi.org/10.1021/acsearthspacechem.0c00074, 2020.
Zhang, G. S., He, X. X., Nadagouda, M. N., O'Shea, K. E., and Dionysiou, D. D.: The effect of basic pH and carbonate ion on the mechanism of photocatalytic destruction of cylindrospermopsin, Water Res., 73, 353–361, https://doi.org/10.1016/j.watres.2015.01.011, 2015.
Zhang, T., Cao, J. J., Tie, X. X., Shen, Z. X., Liu, S. X., Ding, H., Han, Y. M., Wang, G. H., Ho, K. F., Qiang, J., and Li, W. T.: Water-soluble ions in atmospheric aerosols measured in Xi'an, China: Seasonal variations and sources, Atmos. Res., 102, 110–119, https://doi.org/10.1016/j.atmosres.2011.06.014, 2011.
Zhang, Y. and Carmichael, G. R.: The role of mineral aerosol in tropospheric chemistry in East Asia – A model study, J Appl Meteorol, 38, 353–366, https://doi.org/10.1175/1520-0450(1999)038<0353:Tromai>2.0.Co;2, 1999.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015.