the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Mar 2022
| 09 Mar 2022
Secondary organic aerosol formation from camphene oxidation: measurements and modeling
Qi Li
Jia Jiang
Isaac K. Afreh
Kelley C. Barsanti
David R. Cocker III
While camphene is one of the dominant monoterpenes measured in biogenic and pyrogenic emission samples, oxidation of camphene has not been well-studied in environmental chambers and very little is known about its potential to form secondary organic aerosol (SOA). The lack of chamber-derived SOA data for camphene may lead to significant uncertainties in predictions of SOA from oxidation of monoterpenes using existing parameterizations when camphene is a significant contributor to total monoterpenes. Therefore, to advance the understanding of camphene oxidation and SOA formation and to improve representation of camphene in air quality models, a series of experiments was performed in the University of California Riverside environmental chamber to explore camphene SOA mass yields and properties across a range of chemical conditions at atmospherically relevant OH concentrations. The experimental results were compared with modeling simulations obtained using two chemically detailed box models: Statewide Air Pollution Research Center (SAPRC) and Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A). SOA parameterizations were derived from the chamber data using both the two-product and volatility basis set (VBS) approaches. Experiments performed with added nitrogen oxides (NOx) resulted in higher SOA mass yields (up to 64 %) than experiments performed without added NOx (up to 28 %). In addition, camphene SOA mass yields increased with SOA mass (Mo) at lower mass loadings, but a threshold was reached at higher mass loadings in which the SOA mass yields no longer increased with Mo. SAPRC modeling of the chamber studies suggested that the higher SOA mass yields at higher initial NOx levels were primarily due to higher production of peroxy radicals (RO2) and the generation of highly oxygenated organic molecules (HOMs) formed through unimolecular RO2 reactions. SAPRC predicted that in the presence of NOx, camphene RO2 reacts with NO and the resultant RO2 undergoes hydrogen (H)-shift isomerization reactions; as has been documented previously, such reactions rapidly add oxygen and lead to products with very low volatility (i.e., HOMs). The end products formed in the presence of NOx have significantly lower volatilities, and higher O : C ratios, than those formed by initial camphene RO2 reacting with hydroperoxyl radicals (HO2) or other RO2. Further analysis reveals the existence of an extreme NOx regime, wherein the SOA mass yield can be suppressed again due to high NO HO2 ratios. Moreover, particle densities were found to decrease from 1.47 to 1.30 g cm−3 as [HC]0 [NOx]0 increased and O : C decreased. The observed differences in SOA mass yields were largely explained by the gas-phase RO2 chemistry and the competition between RO2+ HO2, RO2+ NO, RO2+ RO2, and RO2 autoxidation reactions.
- Article
(2770 KB) - Full-text XML
-
Supplement
(3517 KB) - BibTeX
- EndNote





On a global scale, biogenic monoterpene emissions are estimated to contribute 14 % of the total reactive volatile organic compound (VOC) flux (Tg C) (Guenther, 1995). Camphene is a ubiquitous monoterpene emitted from biogenic sources (Geron et al., 2000; Hayward et al., 2001; Ludley et al., 2009; Maleknia et al., 2007; White et al., 2008) and pyrogenic sources (Akagi et al., 2013; Gilman et al., 2015; Hatch et al., 2015). Many studies have reported camphene as a top contributor by mass in measured biogenic and pyrogenic monoterpene emissions (Benelli et al., 2018; Hatch et al., 2019; Komenda, 2002; Mazza and Cottrell, 1999; Moukhtar et al., 2006). For example, in measurements of laboratory and prescribed fires reported by Hatch et al. (2019), camphene was among the top two monoterpenes emitted from subalpine and Douglas fir fires based on emission factors (mass of compound emitted to the mass of fuel burned).

Figure 1Camphene chemical structure and reaction rate constants (unit: cm3 molec.−1 s−1) with major atmospheric oxidants.
When emitted to the atmosphere, monoterpenes form oxygenated compounds through reactions with oxidants such as hydroxyl radicals (OH), ozone (O3), and nitrate radicals (NO3); compounds with sufficiently low volatility can then condense to form secondary organic aerosol (SOA). Figure 1 shows the chemical structure of camphene and its reaction rate constants with major atmospheric oxidants. The SOA formation potential of individual monoterpenes can vary greatly based on their molecular structure, atmospheric lifetimes, and the volatility of their oxidation products (Atkinson and Arey, 2003; Griffin et al., 1999; Ng et al., 2007; Zhang et al., 1992). Previous experimental studies of other monoterpenes (such as α-pinene, β-pinene, and d-limonene) have reported SOA mass yields from ∼ 10 % to 50 % through OH oxidation and from ∼ 0 % to 65 % through NO3 oxidation; among the studied monoterpenes, d-limonene often has the highest reported yields (Mutzel et al., 2016; Griffin et al., 1999; Ng et al., 2007; Fry et al., 2014). Few studies have been published regarding camphene SOA formation.
Past experimental studies of camphene have largely been focused on gas-phase reactivity with OH, NO3, and/or O3 and gas-phase product identification (e.g., Atkinson et al., 1990; Gaona-Colmán et al., 2017; Hakola et al., 1994). Baruah et al. (2018) performed a kinetic and mechanism study of the camphene oxidation initiated by OH radicals using density functional theory (DFT), in which the rate constant and atmospheric lifetime were reported. It was also suggested that addition at the terminal double-bond carbon atom could account for 98.4 % of the initial OH addition. A product study by Gaona-Colmán et al. (2017) showed obvious NOx dependence in OH + camphene experiments, in which the molar yield of acetone was enhanced by a factor of 3, or 33 % relative to 10 %, in the presence of NOx (2–2.3 ppmv of NO).
Hatfield and Huff-Hartz studied SOA formation from ozonolysis of VOC mixtures, in which the added camphene was considered a non-reactive VOC and assumed to have little to no effect on SOA mass yields (Hatfield and Huff Hartz, 2011). Mehra et al. (2020) recently published a compositional analysis study of camphene SOA. Although SOA mass yields were not provided, they demonstrated the potential contribution of highly oxygenated organic molecules (HOMs) and oligomers to camphene SOA formed in an oxidation flow reactor (OFR). Afreh et al. (2021) presented the first mechanistic modeling study of camphene SOA formation. While relatively high SOA mass yields were reported (with final SOA mass and yields twice that of α-pinene), no chamber-based SOA data were available for measurement–model comparison at that time.
SOA formation has been shown to be highly dependent on gas-phase NOx concentrations, more precisely the relative ratios of NO : HO2, hydroperoxyl radicals : RO2, and peroxy radicals (Henze et al., 2008; Ng et al., 2007; Presto et al., 2005; Ziemann and Atkinson, 2012; Kroll and Seinfeld, 2008; Song et al., 2005). During chamber experiments, VOCs are subject to oxidation by OH, O3, and/or NO3. For some precursors, NOx levels influence the amount of SOA produced in the initial oxidation steps by controlling the relative proportions of oxidants, the fractional reactivity with those oxidants, and thus the volatility distribution of the products formed (Hurley et al., 2001; Nøjgaard et al., 2006; Kroll and Seinfeld, 2008). For other precursors, NOx levels influence the amount of SOA produced via the fate of RO2. The reactions between RO2 and HO2 form hydroperoxides, which can have sufficiently low volatility to condense into the particle phase. In the presence of NOx, RO2 will react with NO, forming organic nitrate and carbonyl compounds that have higher volatilities than the products formed through the HO2 pathway (Kroll and Seinfeld, 2008; Ziemann and Atkinson, 2012). Previous studies of relatively small compounds (carbon number ≤ 10), including monoterpenes such as α-pinene, have reported that SOA mass yields generally increase as initial NOx decreases, with a proposed mechanism of competitive chemistry between RO2+ HO2 and RO2+ NO pathways, the latter of which would form more volatile products (Kroll et al., 2006; Ng et al., 2007; Song et al., 2005). The NOx dependence of camphene oxidation and SOA formation has been relatively understudied.
The atmospheric gas-phase autoxidation of RO2 has been identified as another key pathway of SOA formation (Crounse et al., 2013; Jokinen et al., 2014; Ehn et al., 2017; Bianchi et al., 2019). The RO2 radical undergoes intramolecular H-atom abstraction reactions to form a hydroperoxide functionality and an alkyl radical (RO), to which a new RO2 will be formed by adding O2. The autoxidation process can repeat several times until terminated by other pathways and will form low-volatility compounds known as highly oxygenated organic molecules (HOMs) (Bianchi et al., 2019). Recent theoretical and experimental studies have been conducted to understand HOM formation from monoterpenes such as α-pinene and β-pinene (Zhang et al., 2017; Quéléver et al., 2019; Xavier et al., 2019; Pullinen et al., 2020; Ye et al., 2019), but the potential importance and mechanisms of HOM formation from camphene have not been well-investigated.
Here, we present the first systematic study of SOA formation from camphene using laboratory-based chamber experiments and chemically detailed box models. The experiments were conducted at varying NOx levels, and the chamber data were used to provide SOA parameterizations based on the two-product (Odum et al., 1996) and volatility basis set (VBS) modeling approaches (Donahue et al., 2006, 2009). Two chemically detailed box models, Statewide Air Pollution Research Center (SAPRC) and Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A), were used to provide mechanistic insights into the chamber observations and to elucidate the connections between the fate of RO2, HOM-forming mechanisms, and camphene SOA formation.
2.1 Chamber facility and instrumentation
The camphene photooxidation experiments were conducted in the University of California Riverside (UCR) dual indoor environmental chamber. Chamber characterization and features have been previously described in detail (Carter et al., 2005). Briefly, the UCR environmental chamber consists of two 90 m3 collapsible Teflon reactors (2MIL (0.0508 mm) FEP film) kept at a positive pressure differential (3.73–4.98 Pa) to the enclosure wherein the reactors are located to minimize contamination during experiments. The enclosure is relative-humidity-controlled (< 0.1 %), temperature-controlled (300 ± 1 K), and continuously flushed with dry purified air (dew point < −40 ∘C). Prior to and between experiments, reactors were collapsed to a volume < 20 m3 for cleaning. The cycle of filling–purging the reactors was repeated until particle number concentrations were < 5 cm−3 and NOx mixing ratios were < 1 ppb. The reactors were then flushed with dry purified air and filled up to 90 m3 overnight. The filling–purging of the reactors is controlled by an “elevator” program in LabView.
NO and NO2 mixing ratios were monitored by a Thermo Environmental Instruments model 42C chemiluminescence NOx analyzer. O3 mixing ratios were monitored by a Dasibi Environmental Corp. model 1003-AH O3 analyzer. An Agilent 6890 gas chromatograph with flame ionization detector (GC-FID) was used to measure the camphene levels during the experiments.
Multiple instruments were used for particle-phase monitoring. Each reactor was equipped with a scanning mobility particle sizer (SMPS), including a TSI 3081 differential mobility analyzer (DMA), to measure the particle mass concentration. Particle effective density was directly measured by an aerosol particle mass analyzer (APM, Kanomax) with an SMPS built in-house (Malloy et al., 2009). Chemical composition of SOA was measured using an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) (DeCarlo et al., 2006) and analyzed to obtain O : C and H : C ratios by applying the method of Canagaratna et al. (2015). Data processing was performed using the ToF-AMS Analysis Toolkit 1.57 and PIKA 1.16 on Igor Pro 6.36. A prior characterization of this UCR chamber system (Li et al., 2016) reported an experimental uncertainty in SOA yields of < 6.65 %.
Particle wall loss corrections were performed following the method described in Cocker et al. (2001). Vapor wall loss of organics has been reported in multiple chambers (e.g., Zhang et al., 2015, 2014; Schwantes et al., 2019) and has been modeled as a function of the mass and volatility of the condensing compounds, condensation sink, and characteristics of the chamber (e.g., La et al., 2016; Zhang et al., 2014; Ye et al., 2016). The extent to which these observations and modeling simulations are relevant in the UCR chamber is unclear given the significant difference in chamber sizes. The UCR chamber is 4.5 times larger (90 m3) than the largest referenced chamber in these studies (20 m3), and most are ∼ 10 m3. In the UCR chamber, the role of vapor wall loss has been investigated in SOA experiments using various precursor compounds (including α-pinene and m-xylene) under seed and no seed conditions (Clark et al., 2016; Li et al., 2015). There has been no evidence of non-negligible vapor wall loss in those experiments, including no measurable differences in SOA formation in experiments with and without seed. In this work, stability tests on camphene demonstrated negligible vapor wall loss of the parent compound. Thus without evidence to suggest otherwise, negligible vapor wall loss was assumed for these experiments. This assumption is further discussed where it may affect the major conclusions regarding the role of gas-phase chemistry in SOA formation.
2.2 Experimental conditions
A series of 13 camphene photooxidation experiments was carried out under varying levels of camphene and NOx (Table 1). Due to the relatively high melting point of camphene (51 ∘C), camphene (Sigma-Aldrich, purity > 96 %, FG) was injected into a glass manifold (heated to 50 ∘C by heating tape) using a preheated (∼ 50–55 ∘C) microliter syringe. As camphene evaporated it was carried to the reactors by dry purified compressed air flowing through a glass manifold at 8 L min−1 for 15 min. Injection lines from the glass manifold to the reactors were also heated to reduce losses of camphene. H2O2 (Sigma Aldrich, 50 wt % in H2O) was injected by adding 200 µL onto glass wool in glass tubing and then placing the tubing in a 56 ∘C oven with 10 L min−1 of dry purified compressed air flowing through the tubing for 15 min and into the reactors. An inert tracer, perfluorohexane (Sigma-Aldrich, 99 %) or perfluorobutane (Sigma-Aldrich, 99 %) was injected to the reactors through the heated glass manifold by a carrier gas of 50 ∘C pure N2. NO (Matheson, UHP) at known volume and pressure was transferred and injected through the same glass manifold as the inert tracer. When gaseous injection of camphene, H2O2, the inert tracer, and NO (when used) was completed, the reactors were internally mixed using built-in blowers to ensure uniform distribution of chemicals and then irradiated using UV black lights (115w Sylvania 350BL) to start photooxidation. No seed aerosol was used in this study. All experiments were conducted under dry conditions (relative humidity < 0.1 %) at 300 K. The initial conditions of the experiments are summarized in Table 1.
Table 1Summary of initial conditions for chamber experiments and box model simulations.

* H2O2 mixing ratio was targeted at 1 ppm but corrected based on tracer (perfluorohexane or perfluorobutane) concentration to offset initial reactor volume bias. Corrected H2O2 mixing ratios were used in SAPRC modeling.
2.3 Model configurations and conditions
The chamber experiments were modeled using two different box models: SAPRC and GECKO-A. The SAPRC model was chosen because it has been designed to evaluate gas-phase chemistry in the UCR chamber. The GECKO-A model was chosen because of the ability to predict both gas- and particle-phase composition, as well as the prior work of Afreh et al. (2021), in which GECKO-A was used to study SOA formation from camphene. The initial conditions of the simulations are summarized in Table 1.
2.3.1 SAPRC
A gas-phase oxidation mechanism was derived using the SAPRC mechanism generation system (MechGen) with modified initial rate constants (camphene with OH, NO3, and O3) based on published literature data (Atkinson and Arey, 2003). MechGen, described elsewhere (Carter, 2020b, 2021; Jiang et al., 2020), is capable of generating fully explicit mechanisms for the atmospheric reactions of many types of organic compounds and the intermediate radicals they form. MechGen uses experimentally derived rate constants and branching ratios if data are available and otherwise uses estimated rate constants and branching ratios based on group additivity and other estimation methods. This system was used to derive reactions of explicit and lumped organic compounds and products in the development of the SAPRC-18 mechanism (Carter, 2020a) and a detailed SAPRC furan mechanism (Jiang et al., 2020).
The MechGen-derived camphene mechanism was implemented into the SAPRC box model to simulate chamber experiments under the same chemical conditions as the chamber experiments, for which the initial hydrocarbon concentrations and NOx levels are as defined in Table 1. The SAPRC box model system has been used for chemical mechanism development, evaluation, and box modeling applications since the mid-1970s (Carter, 1990, 1994, 2000, 2010a, b, 2020a). The initial conditions and relevant chemical parameters for environmental chamber experiments are required inputs; simulations can be performed using multiple versions of the SAPRC gas-phase chemical mechanism. In this work, the recently published version, SAPRC-18 (Carter, 2020a), was selected as the base mechanism because it represents the current state of the science and includes the most up-to-date model species and explicit representation of RO2 chemistry.
2.3.2 GECKO-A
GECKO-A is a nearly explicit mechanism generator and SOA box model. GECKO-A relies on experimental data and structure–activity relationships (SARs) to generate detailed oxidation reaction schemes for organic compounds. The generated reaction schemes are applied in the SOA box model to simulate SOA formation based on the absorptive gas–particle partitioning model of Pankow (1994), wherein thermodynamic equilibrium between the gas and an ideal particle phase is assumed. Detailed descriptions of GECKO-A, including mechanism generation and SOA formation, are provided by Aumont et al. (2005) and Camredon et al. (2007). GECKO-A has been used to predict SOA in a number of studies (e.g., Aumont et al., 2012; Lannuque et al., 2018; McVay et al., 2016), including camphene (Afreh et al., 2021). Details of the camphene mechanism and SOA box modeling are described in Afreh et al. (2021). Briefly, the camphene mechanism includes 1.3×106 reactions and 1.8×105 oxidation products; vapor pressures of products were calculated based on the Nannoolal method (Nannoolal et al., 2008).
The GECKO-A simulations were performed for a predefined set of conditions, prior to the chamber experiments, and thus in some cases differ from the experimental conditions. GECKO-A simulations were performed under two NOx conditions: with 80 ppb of NOx and without NOx (Table 1). For both NOx conditions, the initial hydrocarbon mixing ratios were set at 10, 25, 50, 100, and 150 ppb. All simulations were run under the following initial conditions: 1000 ppb of H2O2, 1 µg m−3 of organic seed with a molecular weight of 250 g mol−1, 298 K temperature, 1 % relative humidity, and 50∘ solar zenith angle (required to compute the photolysis frequencies). Simulation results for camphene were compared with chamber data including SOA mass yields, precursor decay rates, and oxidant levels.
Table 2 summarizes the measured initial NO NO2 mixing ratios, initial camphene concentration ([HC]0), reacted camphene concentration (Δ[HC]), SOA mass (Mo) formed, particle density, final peak particle diameter (dp), photochemical aging time, irradiation time, and SOA mass yield (SOA mass formed, Mo hydrocarbon reacted, ΔHC) for all 13 experiments. Except for Fig. 4, in which SOA mass yields are shown as a function of photochemical age, all SOA mass yields refer to the mass at the end of the experiments (∼ 6 h). Measured and predicted gas-phase species are presented in Sect. 3.1; SOA mass and yields are presented in Sect. 3.2. The predicted fate of RO2 in the context of the initial HC to initial NOx mixing ratio ([HC]0 [NOx]0) is presented in Sect. 3.3.
Table 2Chamber SOA data; WO indicates experiments without added NOx, and W indicates experiments with added NOx.
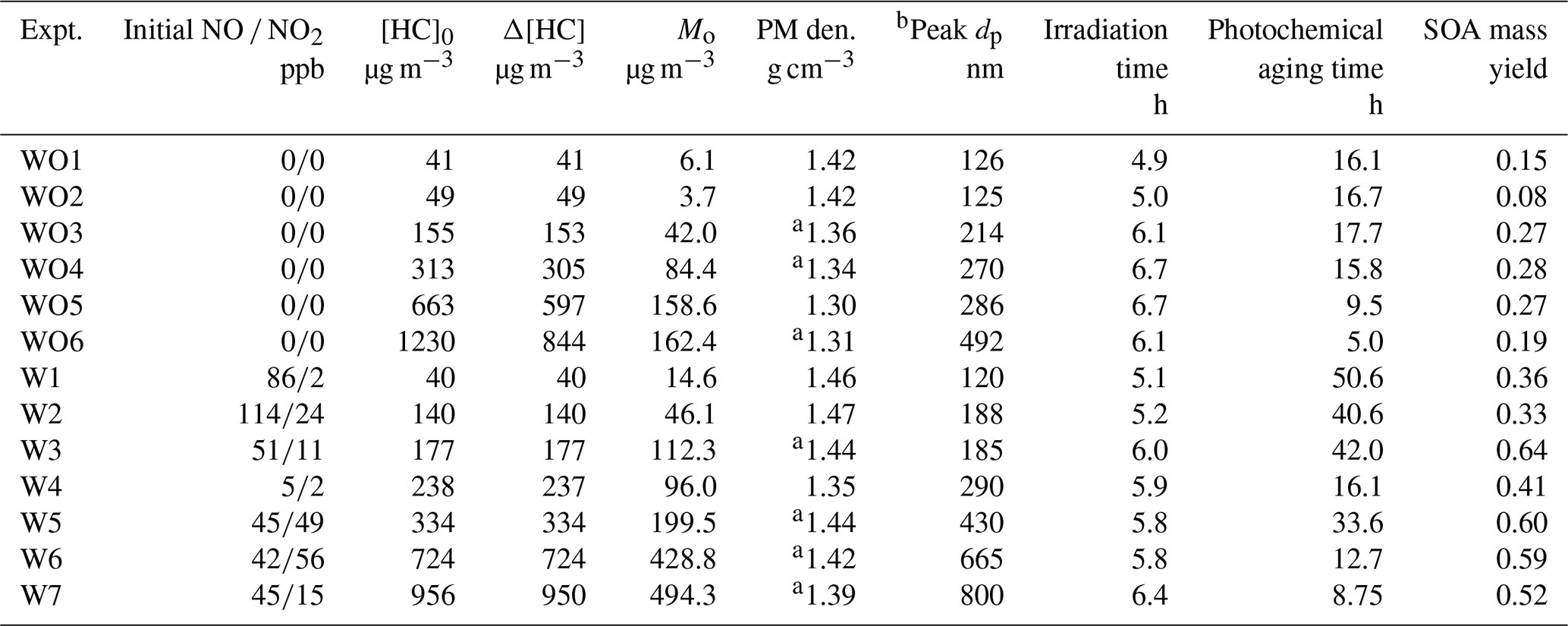
a Estimated using the best-fit line shown in Fig. S6. b Peak dp refers to the diameter of particles at the peak of the size distribution plot at the end of the experiment. The uncertainty of peak dp values is less than 5 %.
3.1 Gas-phase reactivity
Figure 2 shows measured and predicted camphene consumption for the 13 photooxidation experiments and the calculated time-dependent β values (ratio of RO2+ NO to the sum of RO2+ NO and RO2 + HO2) (Henze et al., 2008; Pye et al., 2010) based on SAPRC predictions for each experimental condition. Additional comparisons of measured and predicted gas-phase species are shown in Fig. S1. Higher camphene decay rates and higher OH levels (0.15–0.88 ppt with added NOx; 0.05–0.29 ppt without added NOx) were observed and predicted for experiments with added NOx than without, likely due to the fast recycling of OH when NOx was present (Fig. 2). For all experiments, the β values changed as a function of time due to changing chemical conditions. Note that due to off-gassing of NOx from the Teflon reactor (Carter et al., 2005), β values simulated here were larger than 0 even for experiments without added NOx. Experiments with added NOx have β values from 0.12–1, while experiments without added NOx have values < 0.12. For all parameters (camphene consumption, NOx decay, O3 formation, and OH levels), the SAPRC simulation results were generally in good agreement with the experimental data. The exception to the generally good agreement was O3 predictions in experiments without added NOx, which have a relatively strong dependence on the HONO off-gassing rate. The quantity Δ([O3]–[NO]) has been used to evaluate the rate of NO oxidation by RO2 for VOC–NOx systems in SAPRC mechanism development (Carter and Lurmann, 1989; Carter, 2000, 2007, 2020a). Figure S2 shows the comparison of the Δ([O3] − [NO]) values between chamber measurements and SAPRC simulations for experiments with added NOx. The SAPRC box model captures the rates of RO2+ NO well and supports the use of the SAPRC model to interpret chamber observations, especially in the presence of NOx. Unfortunately, it is hard to quantify how well-constrained the other RO2 reaction rates and product yields are without corresponding measurements, which are not available. In this case, the SAPRC model was largely used to probe the mechanism (diagnostic) and not to predict yields (prognostic).
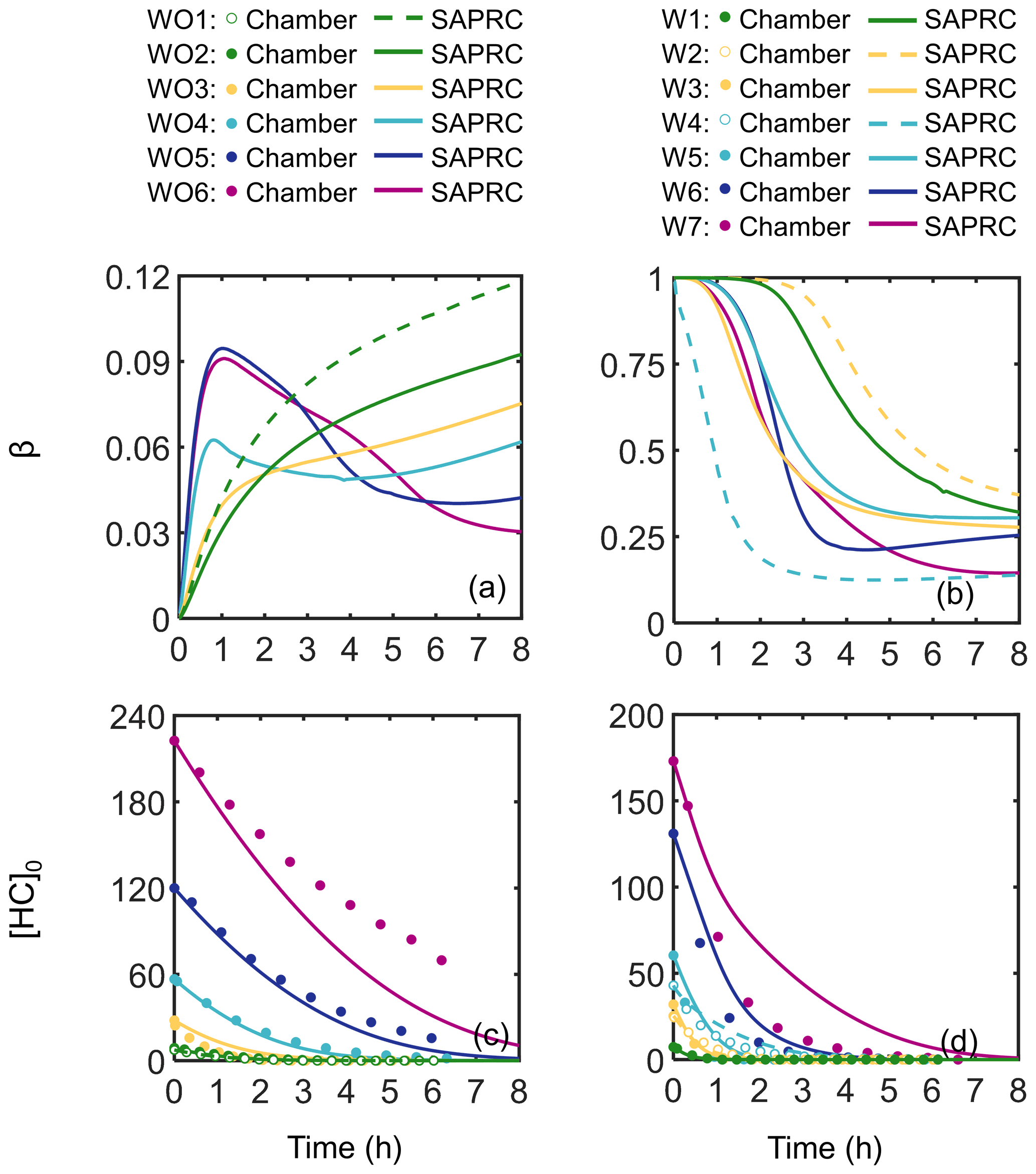
Figure 2SAPRC-predicted β values: (a) without added NOx and (b) with added NOx. Measured (circles) and predicted (lines) camphene consumption as a function of irradiation time: (c) without added NOx and (d) with added NOx. The hollow makers used in (c) and (d) are equivalent to dashed lines defined in the legends.
3.2 SOA mass and yield
Measured SOA mass yields are shown in Fig. 3 as a function of SOA mass (Mo) for experiments with (squares) and without (circles) added NOx. The SOA mass yields were much higher in experiments with added NOx (0.33–0.64) than experiments without added NOx (0.08–0.28). The SOA mass yields measured at the lowest [HC]0 Δ[HC] may be an underestimate due to the assumption of negligible vapor wall loss, which would have the largest effect at low Δ[HC] (Krechmer et al., 2020). The observed trends in SOA mass yields were unexpected based on prior chamber studies of SOA formation from monoterpenes, such as OH oxidation studies of α- and β-pinene, in which SOA mass yields were reported to be suppressed under high-NOx conditions (Eddingsaas et al., 2012; Pullinen et al., 2020; Sarrafzadeh et al., 2016).
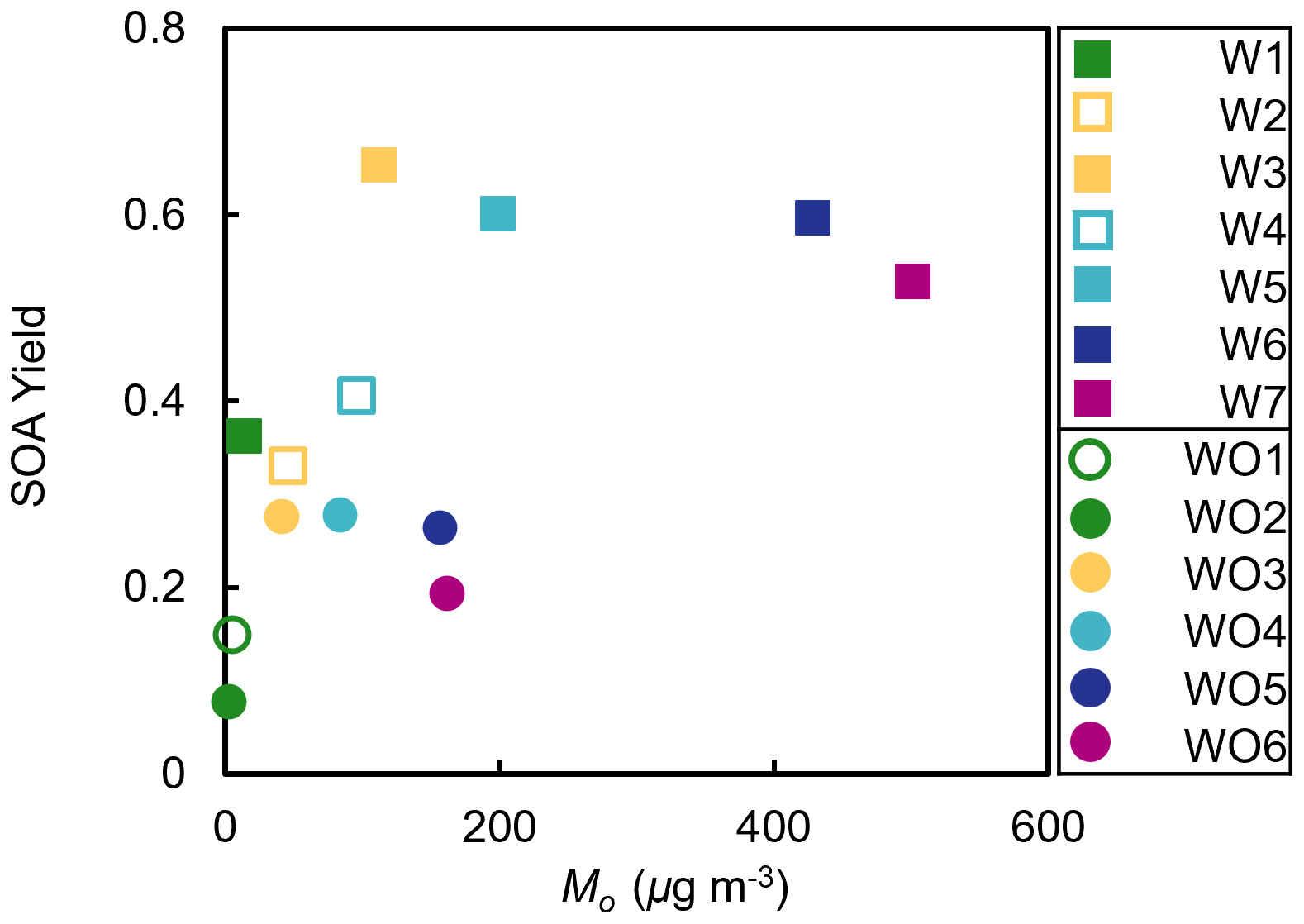
Figure 3Measured camphene SOA mass yields as a function of SOA mass (Mo). Squares indicate experiments with (W) and circles without (WO) added NOx. Initial HC mixing ratios are differentiated by color; open symbols are used to indicate replicate initial HC mixing ratios.
Figure 3 shows another unexpected observation: the SOA mass yields decreased at high SOA mass under both NOx conditions. In the presence of NOx, the observed SOA mass yields increased with Mo for Mo≤ 112 µg m−3, plateaued at 112 µg m−3 < Mo≤ 429 µg m−3, and then decreased for Mo > 429 µg m−3. Without NOx, the observed SOA mass yields increased for Mo≤ 42 µg m−3, plateaued at 42 µg m−3 < Mo≤ 159 µg m−3, and then decreased for Mo > 159 µg m−3. The difference between the peak SOA mass yield and the SOA mass yield at the highest [HC]0 was ∼ 0.12 with added NOx and ∼ 0.08 without added NOx. While the SOA mass yields at the highest [HC]0 may not be statistically different within the uncertainty of the measurements, this trend was also observed in the GECKO-A model simulations (see Sect. 5) and were thus further investigated, and reasonably explained, by the RO2 fate based on box model simulations (see Sects. 4 and 5).
The varying [OH] levels in the chamber experiments led to a wide range of photochemical aging times from hours to days. The irradiation time was converted to equivalent photochemical aging time in the ambient atmosphere using Eq. (1) (Aumont et al., 2012):
where [OH]atm was assumed to be 2×106 molec. cm−3. Figure 4 shows the measured SOA mass yields as a function of photochemical aging time calculated using OH values predicted by SAPRC ([OH]sim). The SOA mass yields are dependent on OH levels and thus photochemical aging time. The yield curves for most experiments plateaued or nearly plateaued by the end of the experiment. Higher [HC]0 generally led to steeper increases in SOA mass yield as a function of aging time. Experiments with added NOx generally had longer photochemical aging times than experiments without added NOx; without added NOx, all experiments may not have fully plateaued and would thus have had higher SOA mass yields at longer irradiation times. However, even at the same aging time (Fig. S8), the SOA yields were higher in the experiments with added NOx. The higher SOA mass yields in experiments with added NOx may partially be attributed to the difference in [OH] levels and extents of aging. Similar NOx effects have been reported in many previous studies (e.g., Ng et al., 2007; Sarrafzadeh et al., 2016). Sarrafzadeh et al. (2016) proposed that in a study of β-pinene the OH level was the main factor that accounted for differences in SOA mass yields under varying [NOx]0. In the camphene experiments presented herein, the aging effects were determined to be less important than RO2 chemistry, since the SOA mass yield curves as a function of photochemical aging already plateau or nearly plateau by the end of experiments (Fig. 4), and the shapes of the growth curves (Figs. S9 and S10) indicate different kinetics and contributions from oxidation products that form slowly among and between experiments with and without added NOx (Ng et al., 2006).
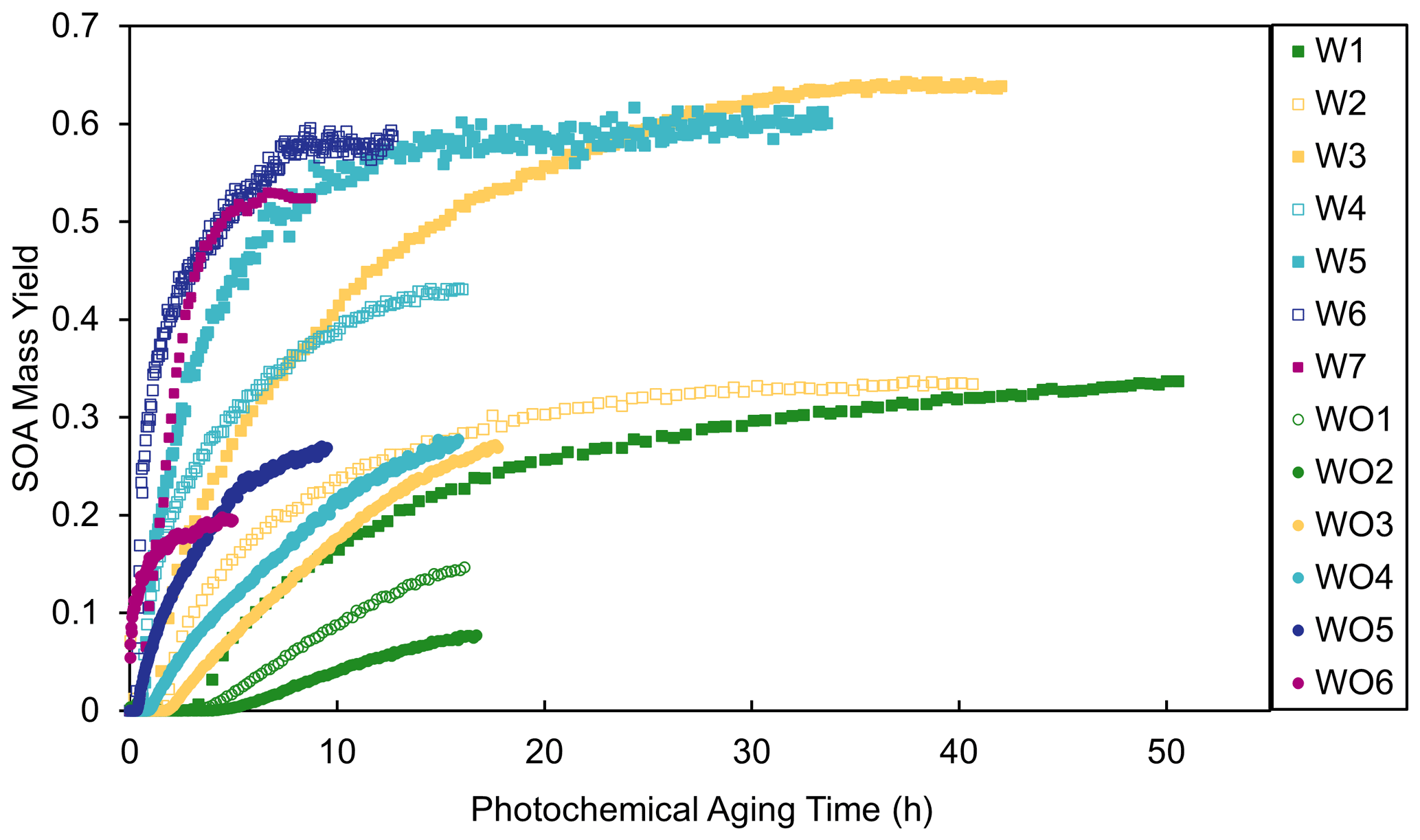
Figure 4Measured SOA mass yields as a function of photochemical aging time in experiments with added NOx (squares) and experiments without added NOx (circles).
SOA mass yields are shown as a function of Δ[HC], [HC]0 [NOx]0, and photochemical aging time in Fig. 5. For the experiments without added NOx, a constant value of 1 ppb was used in the calculations of [HC]0 [NOx]0 to account for NOx off-gassing from the Teflon reactors. Based on recent characterization experiments, the UCR chamber has a NOx off-gassing rate of 2.8 ppt min−1 in the form of HONO; the camphene experiments lasted for ∼ 300 to 360 min. Over low Δ[HC], SOA mass increased in experiments without added NOx due to the dependence of partitioning on Mo (or Δ[HC]). This trend may be exaggerated due to the assumption of negligible vapor wall loss, which could result in an underestimation of SOA mass yield, particularly at low Δ[HC] (Krechmer et al., 2020). The sensitivity of SOA formation to [HC]0 [NOx]0 over the range of [HC] sampled is also shown. At a given Δ[HC] level, a lower [HC]0 [NOx]0 (when within 0.5–200) led to a higher SOA mass yield (decreasing [HC]0 [NOx]0 by ∼ 2 orders of magnitude resulted in a factor of 2 increase in SOA mass yield). The chamber data presented here indicate that the highest SOA mass yields from camphene were observed in a regime of high Δ[HC] and moderate [HC]0 [NOx]0; this regime is distinguished from an extreme [NOx] regime, proposed in Sect. 4.2, in which SOA mass yields are suppressed at the lowest [HC]0 [NOx]0 (also shown in Fig. 5). These observations are different from those in studies of α-pinene, in which lower [HC]0 [NOx]0 generally led to lower SOA mass yield (Eddingsaas et al., 2012). The observed trends are further explored in the following sections, particularly the role of RO2 based on SAPRC simulations.
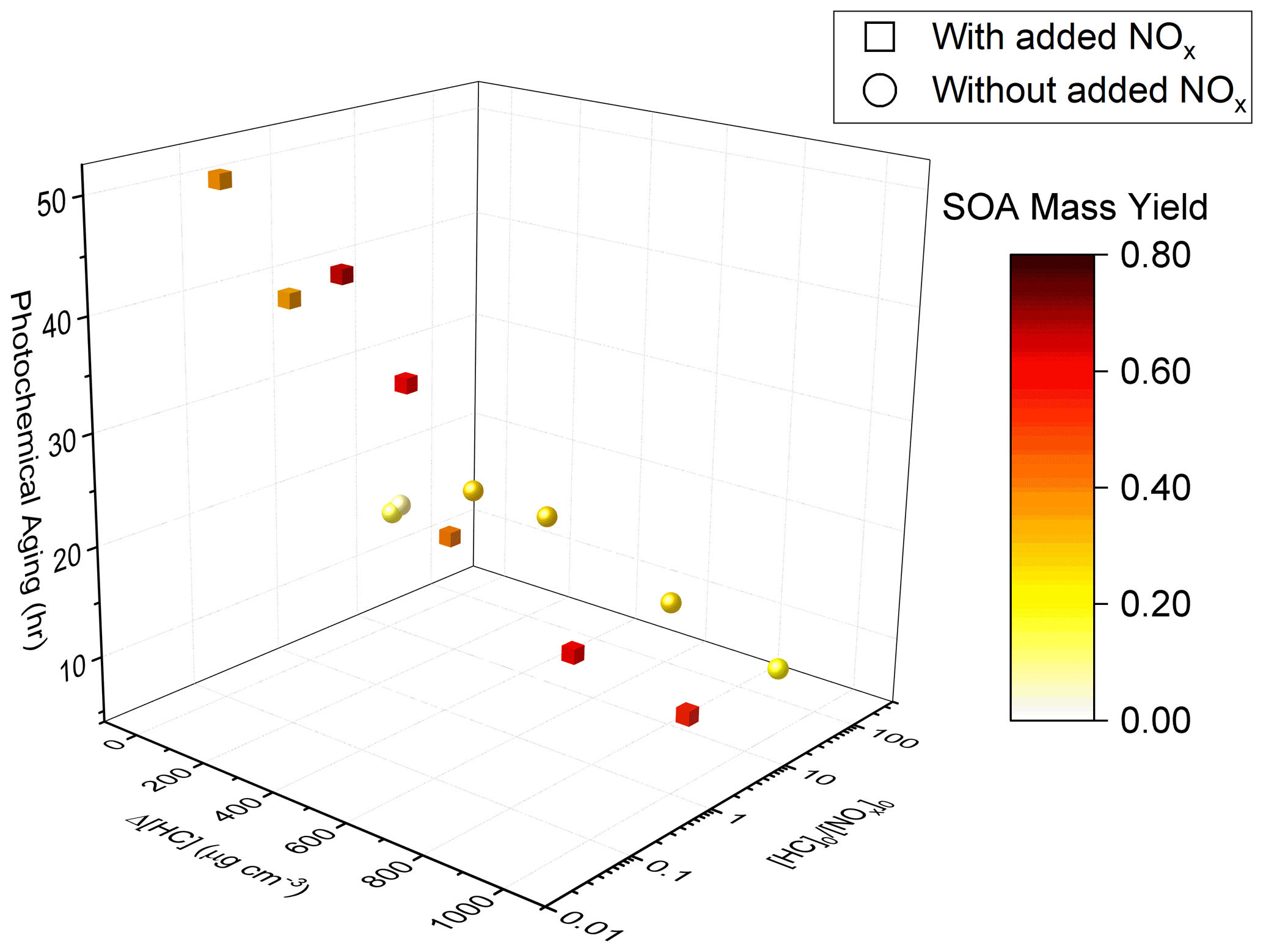
Figure 5SOA mass yield (color bar) as a function of Δ[HC], [HC]0 [NOx]0, and photochemical aging time. Experiments with added NOx are shown as square markers, and experiments without added NOx are shown as round markers.
3.3 [HC]0 [NOx]0 and the fate of peroxy radicals
Table S1 shows the experimental [HC]0 [NOx]0 and the SAPRC-predicted fate of total RO2 (calculated as the summation of RO2 radicals that undergo bimolecular reactions) for all the chamber runs. In Fig. 6, the fate of total RO2 is shown as a function of [HC]0 [NOx]0. The majority of RO2 was predicted to undergo bimolecular reactions with HO2 or NO across the range of [HC]0 [NOx]0 values sampled. At [HC]0 [NOx]0 < 6, > 50 % of the RO2 was predicted to react with NO, and at [HC]0 [NOx]0 > 10, > 50 % of the RO2 was predicted to react with HO2. A roughly 50 : 50 branching of RO2 between NO and HO2 was reached when [HC]0 [NOx]0 was 6 : 1, which is close to the ratio that was suggested in Presto et al. (2005). When [HC]0 [NOx]0 increased over 50, the total fraction of bimolecular RO2+ RO2 increased from 0 % to 30 %. In addition, the normalized total RO2 concentration (total [RO2] [HC]0, ppbv ppbv−1) increased as [HC]0 [NOx]0 decreased (Fig. 7), suggesting that more oxygenated RO2 was formed by the NO pathway than others, which is consistent with the formation of HOMs with added NOx. There is a general trend of increasing SOA mass yield with decreasing [HC]0 [NOx]0 (Figs. 5 and 7), with the exception of four outliers (W1, W2, WO1, and WO2) that have relatively low SOA mass yields. Experiments WO1, WO2, and W1 had the lowest Δ[HC] (49, 41, and 40 µg m−3, respectively, Table 2), indicating that the SOA mass yields were influenced by Δ[HC] and RO2 chemistry. The connections between the fate of RO2 and observed SOA mass yields are further discussed in Sect. 4. Though vapor wall loss has been found to be negligible in previous UCR chamber experiments, such experiments were typically conducted at higher [HC]0. Thus, it is acknowledged that vapor wall loss could affect the measured SOA yields, particularly for experiments W1–2 and WO1–2 with low [HC]0 (or Mo). A vapor wall loss correction for those experiments would increase the measured SOA but would not affect the following discussion or conclusions regarding the role of RO2 chemistry.
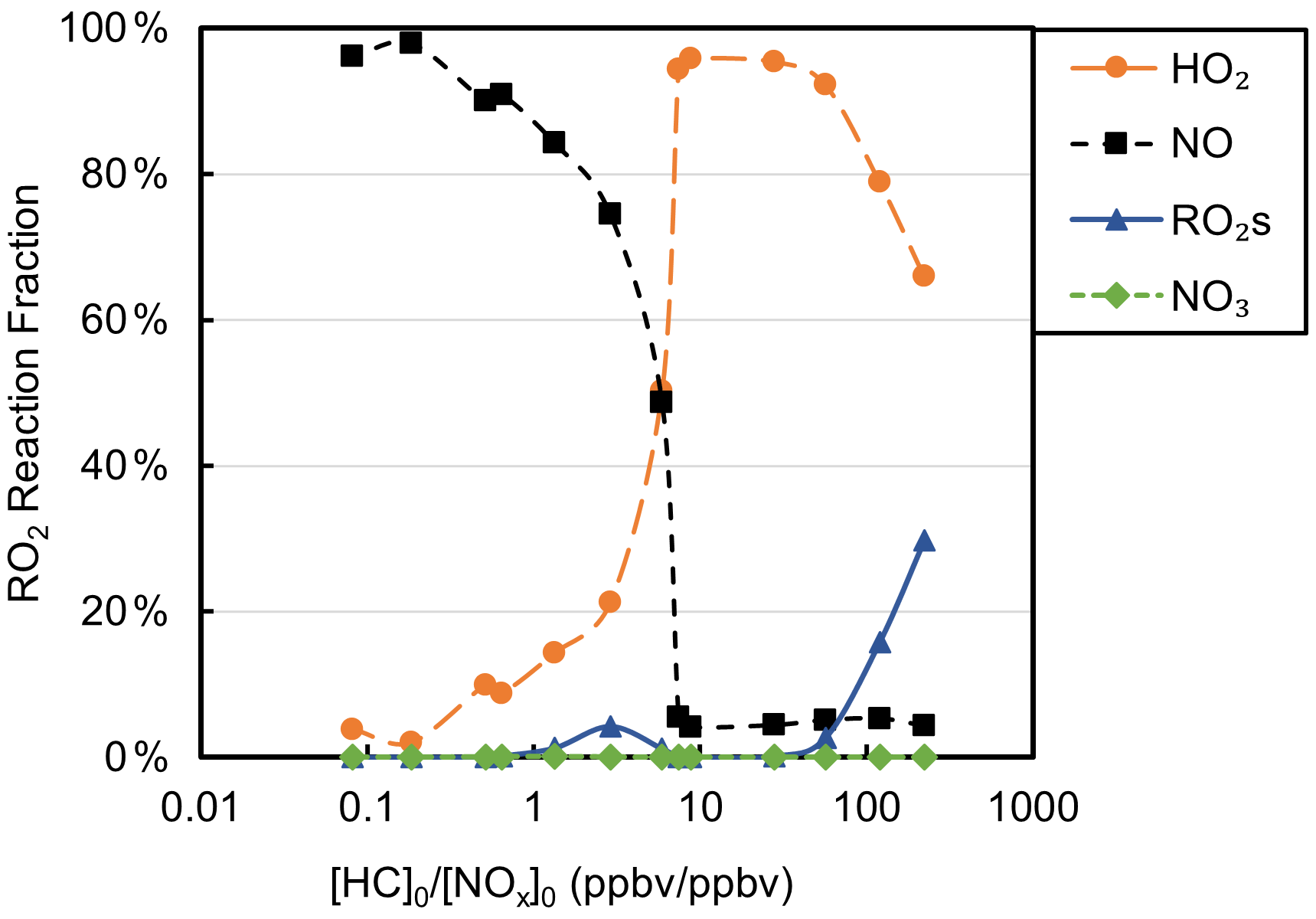
Figure 6Fractions of total RO2 reactions of each type as a function of [HC]0 [NOx]0 based on Table S1.

By assuming the gas-phase chemistry and product distribution were similar when RO2+ NO accounted for more than 80 % of the total RO2 consumption and when RO2+ HO2 accounted for more than 80 % of the total RO2 consumption, experiments with (W1–3, 5–6) and without (WO1–4) added NOx were grouped and used to derive SOA parameters using the two-product (Odum et al., 1996) and VBS approaches (Donahue et al., 2006, 2009). The resultant parameters are shown in Table 3 (two-product) and Table 4 (VBS).
Table 3Two-product model SOA parameters.
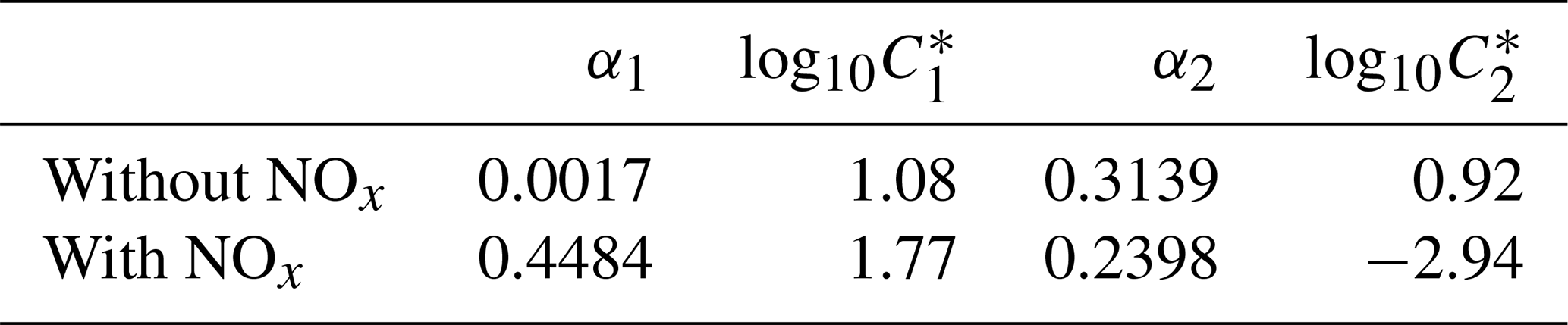
α: gas-phase mass fraction; C*: saturation vapor pressure (µg m−3).
Table 4VBS model SOA parameters.

αwo: gas-phase mass fraction without NOx added; αw: gas-phase mass fraction with NOx added; C*: saturation vapor pressure (µg m−3).
The reaction rate constant of camphene with O3 is relatively low compared to OH, and thus it is expected that OH is the dominant oxidant in the photooxidation of camphene under chamber conditions, especially with the high initial H2O2 (∼ 1 ppm) concentrations. This is supported by SAPRC simulation results (see Fig. S3 in the Supplement), in which O3 accounts for 0 %–3 % and NO3 for 0 %–16 % of camphene oxidation, demonstrating the important role of OH oxidation in these studies.
4.1 Camphene + OH gas-phase mechanism
Figure 8 shows the MechGen-predicted reactions and products of OH-initiated oxidation of camphene in the presence of NOx through one major pathway, which had a yield of 0.83 (a more detailed reaction mechanism schematic is presented in Fig. S4). The reaction starts with OH addition to the CH2= (C) position to form a ring-retaining alkyl radical, which further reacts with O2 to form the camphene peroxy radical, RO2-a. RO2-a can react with oxidants (NO, NO3, HO2, and/or other RO2) to create an alkoxy radical (RO-a) or with NO-to-NO2 conversion, or it can form stable products such as organic nitrate (NO3CAMP1), hydroperoxide (HO2CAMP1), and alcohol (RO2CAMP1) compounds. The cyclic alkoxy radical RO-a can undergo prompt beta (β)-scission ring-opening reaction and then O2 addition to form another peroxy radical, RO2-b. In the presence of NOx, rapid β-scission decomposition or ring-opening reactions of the camphene alkoxy radicals (RO-b and RO-c) occur through the RO2+ NO pathway, leading to the generation of the peroxy radical RO2-d with a lower carbon number and higher O : C ratio (increases from 0.30 for RO2-a to 0.71 for RO2-d).
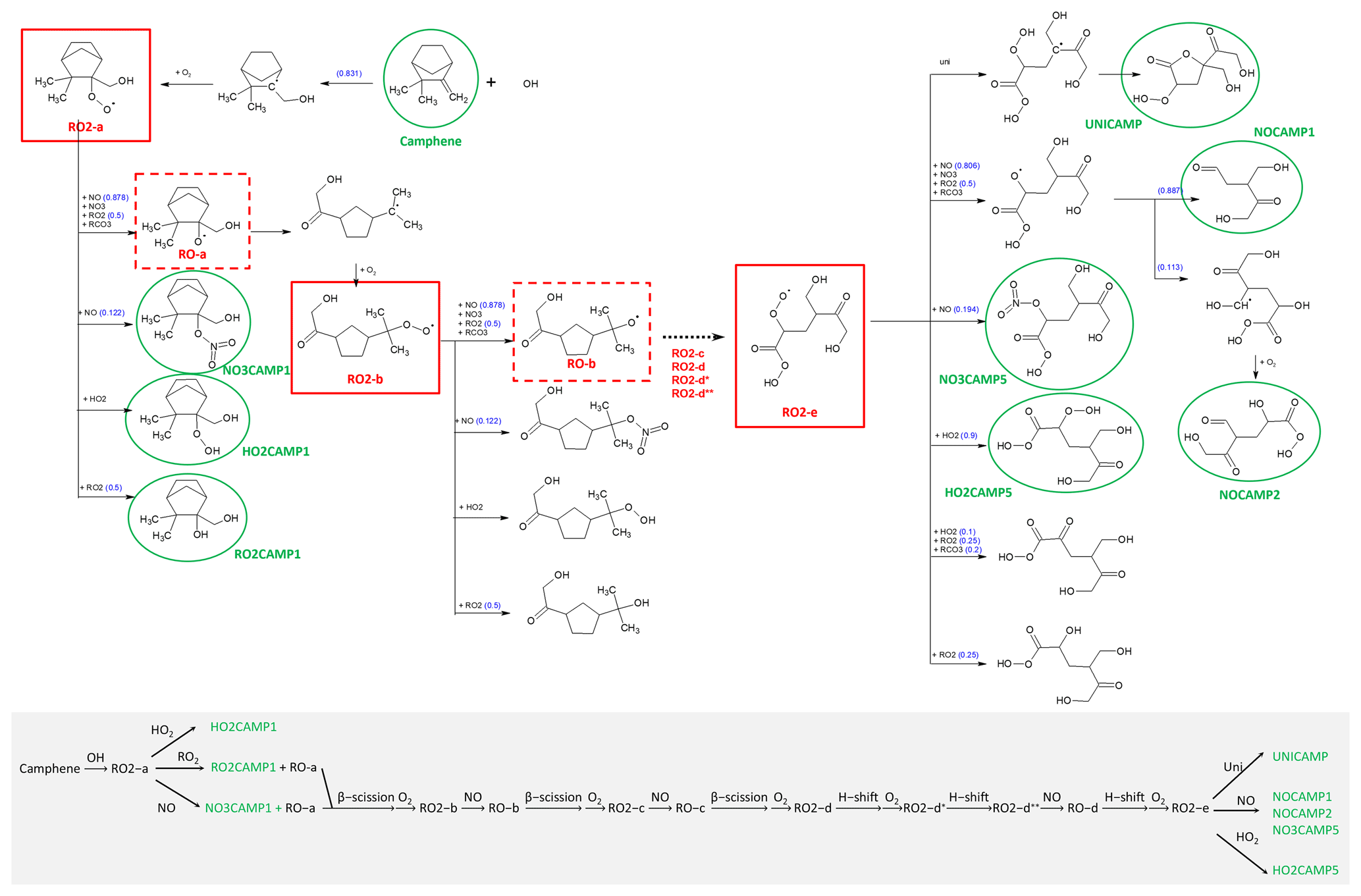
Figure 8Schematic of the OH-initiated oxidation of the camphene mechanism in SAPRC at 298 K and atmospheric pressure in the presence of NOx. See Fig. S4 for more details.
MechGen predicted that RO2-d could undergo 1,5 H-shift isomerization nearly instantaneously, even in the presence of ∼ 100 ppb NOx. Subsequent rapid addition of O2 can form a new peroxy radical RO2-d*, which can undergo 1,7 H-shift isomerization and form the peroxy radical RO2-d. RO2-d can participate in termination reactions with NO and HO2 to form organic nitrate (NO3CAMP4) and hydroperoxide (HO2CAMP4) products, which are known as highly oxygenated organic molecules (HOMs). In the presence of NOx, RO2-d can also react with NO to form the alkoxy radical RO-d that can undergo 1,4 H-shift isomerization and then O2 addition to form the new peroxy radical RO2-e, which will also lead to the formation of HOMs such as NO3CAMP5, HO2CAMP5, and UNICAMP. A recent SOA study by Mehra et al. (2020) demonstrated the formation of HOMs in camphene chamber experiments under both low-NOx (30 ppb camphene, ∼ 0 ppb NOx) and medium-NOx (30 ppb camphene, 2.2 ppb NO, 58.4 ppb NO2) conditions. Based on their observations and analysis, the average molecular formula of the camphene SOA was C7.26H9.85O4.03 for low-NOx and C6.63H9.7N0.12O4.21 for medium-NOx conditions, which also suggests the occurrence of ring-opening and decomposition reactions during camphene photooxidation, as predicted by MechGen.
Table 5Log10C* value for the selected first generation of stable end products formed from camphene reactions with OH.
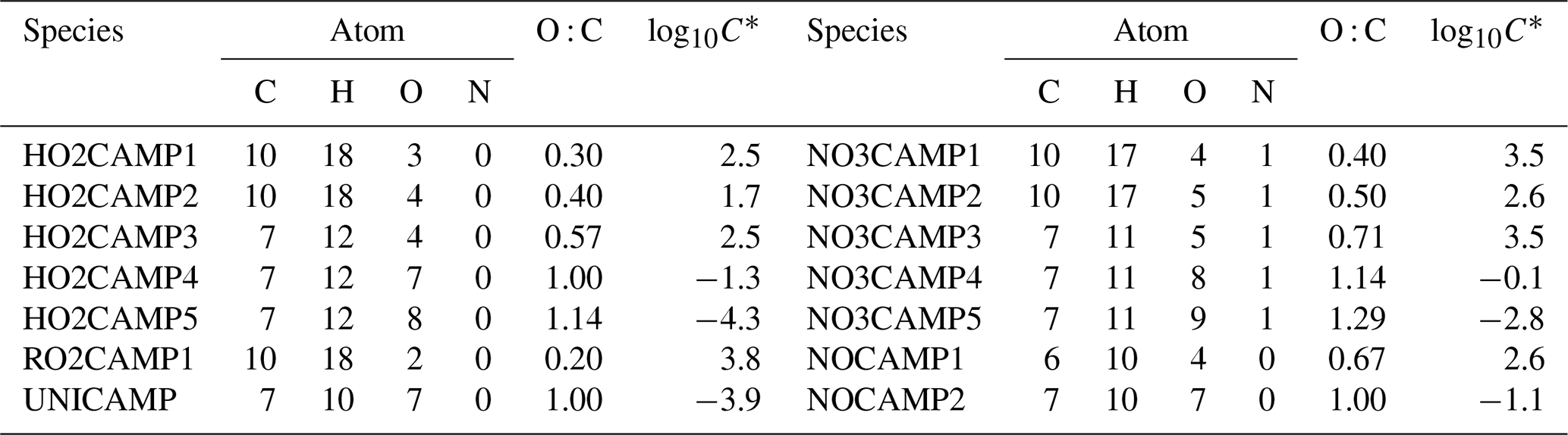
Table 6Fractions of peroxy radical RO2-a reactions of each type, calculated based on SAPRC simulations.
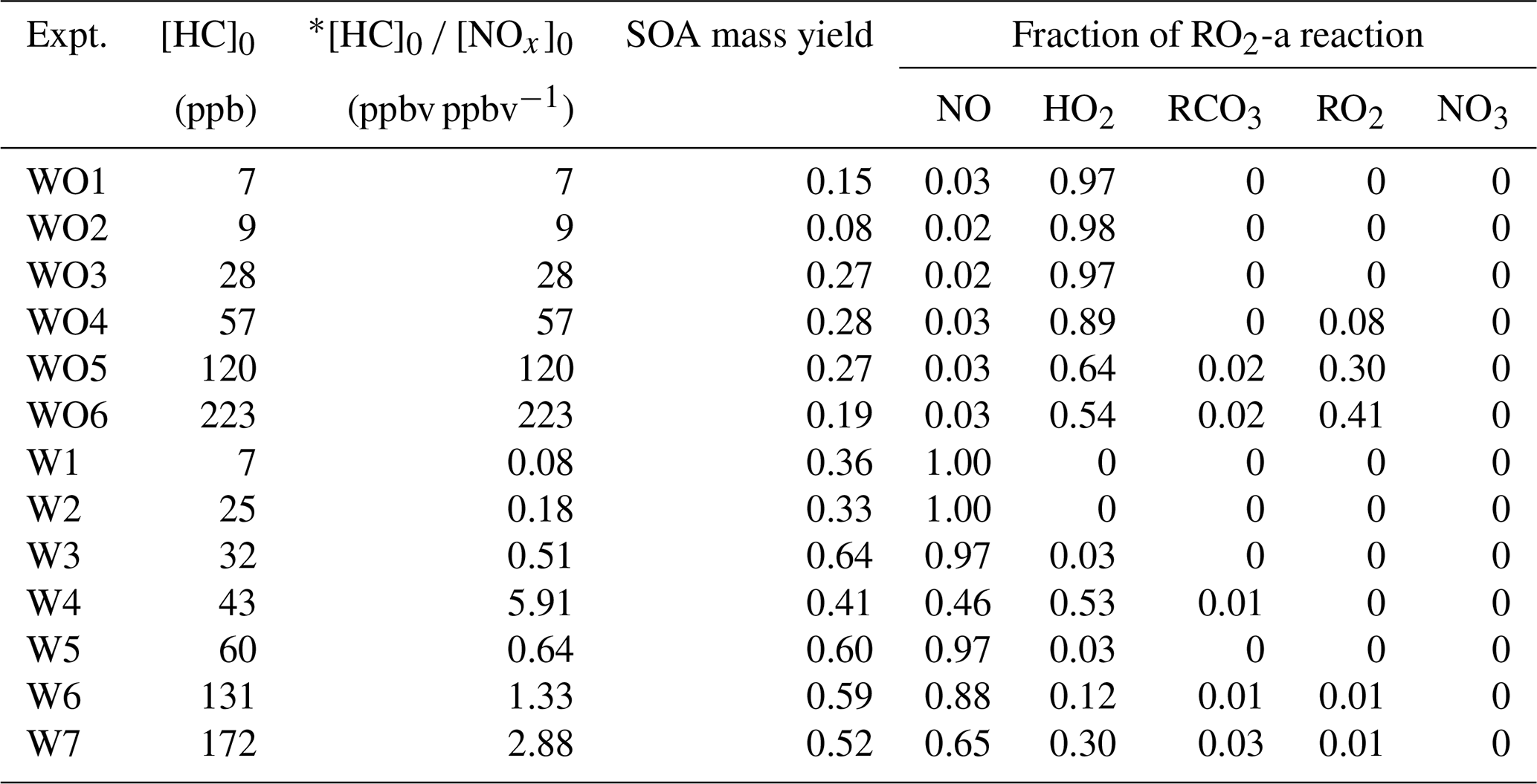
* The [HC]0 [NOx]0 for WO1–6 experiments was estimated assuming 1 ppb of NOx.
4.2 The formation of HOMs and influence on SOA mass yields
Table 5 lists the log C* values and O : C ratios for the major camphene products predicted; vapor pressures of products were calculated based on the Nannoolal method (Nannoolal et al., 2008). HOMs have much lower volatilities than the earlier terminal products such as NO3CAMP1, HO2CAMP1, and RO2CAMP1. HOMs formed by autoxidation steps in camphene radical chain reactions are mediated by the H-shift isomerization of RO2-d and RO-d. Table 6 shows the SAPRC-predicted fate of RO2-a for all chamber runs; the fate of summed RO2 is shown in Table S1, which includes RO2-a–d and all the RO2 radicals formed from other minor pathways. For the experiments without added NOx (WO1–6), once the initial peroxy radical RO2-a was formed, a large fraction of RO2-a (0.54–0.98) quickly reacted with HO2 to form the terminal product HO2CAMP1, while only 2 %–3 % of RO2-a reacted through the NO pathway and led to the generation of HOMs. For the experiments with added NOx (W1–7), much higher RO2-a + NO fractions (0.65–1.00) were predicted by SAPRC. The fates of summed RO2 also suggested that not only RO2-a, but also the other RO2 radical intermediates would tend to favor further reactions through the NO reaction chain to form lower-volatility products.
Based on the predicted fate of RO2 in SAPRC simulations, the higher SOA mass yields in experiments with NOx were due to the formation of HOMs through autoxidation in the presence of NOx. In general, faster RO2 reaction with NO, HO2, or other RO2 limits HOM formation by autoxidation (Bianchi et al., 2019). In previous monoterpene SOA studies, HOM formation was often observed when NOx was absent or under lower-NOx conditions (Pye et al., 2019; Schervish and Donahue, 2020; Zhao et al., 2018). For example, Zhao et al. (2018) demonstrated that autoxidation for some RO2 is competitive with RO2+ NO at parts per billion levels of NO for O3-initiated α-pinene oxidation. They also reported that HOM formation decreased as the initial NO concentration increased from 0 to 30 ppb. In the camphene experiments presented herein, the reverse trend was observed (see experiments WO4, W4, and W5 conducted with ∼ 50 ppb camphene at different NOx levels). This was due to the key RO2 species, RO2-d, which was predicted to form only in the presence of NOx and had a fast enough autoxidation rate constant to effectively compete with bimolecular reactions.
While the decreasing SOA mass yields at high [HC]0 and Mo in experiments with and without added NOx (shown in Fig. 3) may not be statistically different within the uncertainty of the measurements, RO2 chemistry was explored as an explanation for the apparent trends. For experiments with added NOx, a shift in the RO2 reaction pathways from NO to HO2 can explain the decreasing SOA mass yields. The fraction of RO2-a+ NO decreased from 0.97 (W5) to 0.65 (W7), while the fraction of RO2-a+ HO2 increased from 0.03 (W5) to 0.3 (W7). For the experiments without NOx, the shift from RO2+ HO2 to self- and cross-reactions of RO2 at high [HC]0 and Mo can explain the decreasing SOA mass yields. When [HC]0 increased from 57 to 223 ppb, the fractions of RO2-a + HO2 decreased from 0.89 (WO4) to 0.54 (WO6) and the fraction of RO2-a + RO2 increased by a factor of 5 from 0.08 to 0.41. Moreover, this shift from bimolecular reactions with HO2 to RO2 as [HC]0 increased also occurred in the context of the total RO2 (Table S1). Generally, products that were predicted to form from one RO2 reacting with another RO2 in the absence of NOx had relatively higher volatility than those formed from that RO2 reacting with HO2; for example, RO2CAMP1 formed from RO2-a + RO2 was more volatile than HO2CAMP1 formed from RO2-a + HO2 (Table 5). The increasing fraction of self- and cross-reactions of RO2 is therefore one likely explanation for the decreasing SOA mass yields at high ΔHC and Mo in the experiments without NOx.
The relatively low SOA mass yields in experiments W1 and W2 (0.36 and 0.33) can also be explained due to differences in product distribution. An underestimation of the SOA mass yields in these experiments due to the assumption of negligible wall loss is not sufficient to explain these relatively low yields. A comparison of the product distributions between W1, W2, W3, and W5 suggested similar yields of NO3CAMP1–5 and NOCAMP1–2, but major differences in yields of UNICAMP and HO2CAMP1–5 (Fig. S5). Experiments W3 and W5 were selected for comparison because of their closest total RO2 fractional reaction distribution (approximately 90 % RO2+ NO and 10 % RO2+ HO2) to W2 (98 % RO2 + NO and 2 % RO2+ HO2) and W1 (96 % RO2+ NO and 4 % RO2+ HO2) but higher SOA mass yield (0.64 and 0.6). W1 and W2 were predicted to have much smaller SOA mass yield than W3 and W5 in the low-volatility products HO2CAMP1–5 (especially product HO2CAMP5, the lowest volatility among all listed products in Table 5, log10C* = −4.3) and UNICAMP (the second lowest volatility shown in Table 5, log −3.9), which can contribute to the lower SOA mass yield. Further analysis of W1 and W2 revealed a likely cause for the different yields of HO2CAMP1–5 and UNICAMP. W1 and W2 were predicted to have delayed peaks of [OH] (after 3–4 h of irradiation), which was likely due to the high NOx concentrations (Fig. S1b). Correspondingly, the [HO2] was highly suppressed during the first 2 h of irradiation. Under high [NOx], the RO2-e + HO2 pathway shown in Fig. 8 could therefore be suppressed, resulting in a lower yield of HO2CAMP5. This indicates that a second “extreme NOx” regime may exist at high [NOx] and significantly lower [HC]0 [NOx]0.
5.1 SOA mass and yield
The comparison of gas- and particle-phase species between chamber experiments and GECKO-A model simulations is shown in Fig. S1a and S1b. Without added NOx, GECKO-A predicts much smaller camphene consumption rates and no O3 formation, while both the chamber data and SAPRC simulations suggest a final O3 mixing ratio of ∼ 10 ppb (Fig. S1a). This may be due to an underrepresentation of data and relevant pathways for low- to no-NOx conditions in the GECKO-A mechanism generation system, and the incomplete treatment of wall effects in the application of the GECKO-A box model. The simulations without added NOx are therefore not further discussed. With added NOx, GECKO-A shows good agreement with the experimental data and SAPRC simulations in the context of camphene consumption, O3, and OH levels.
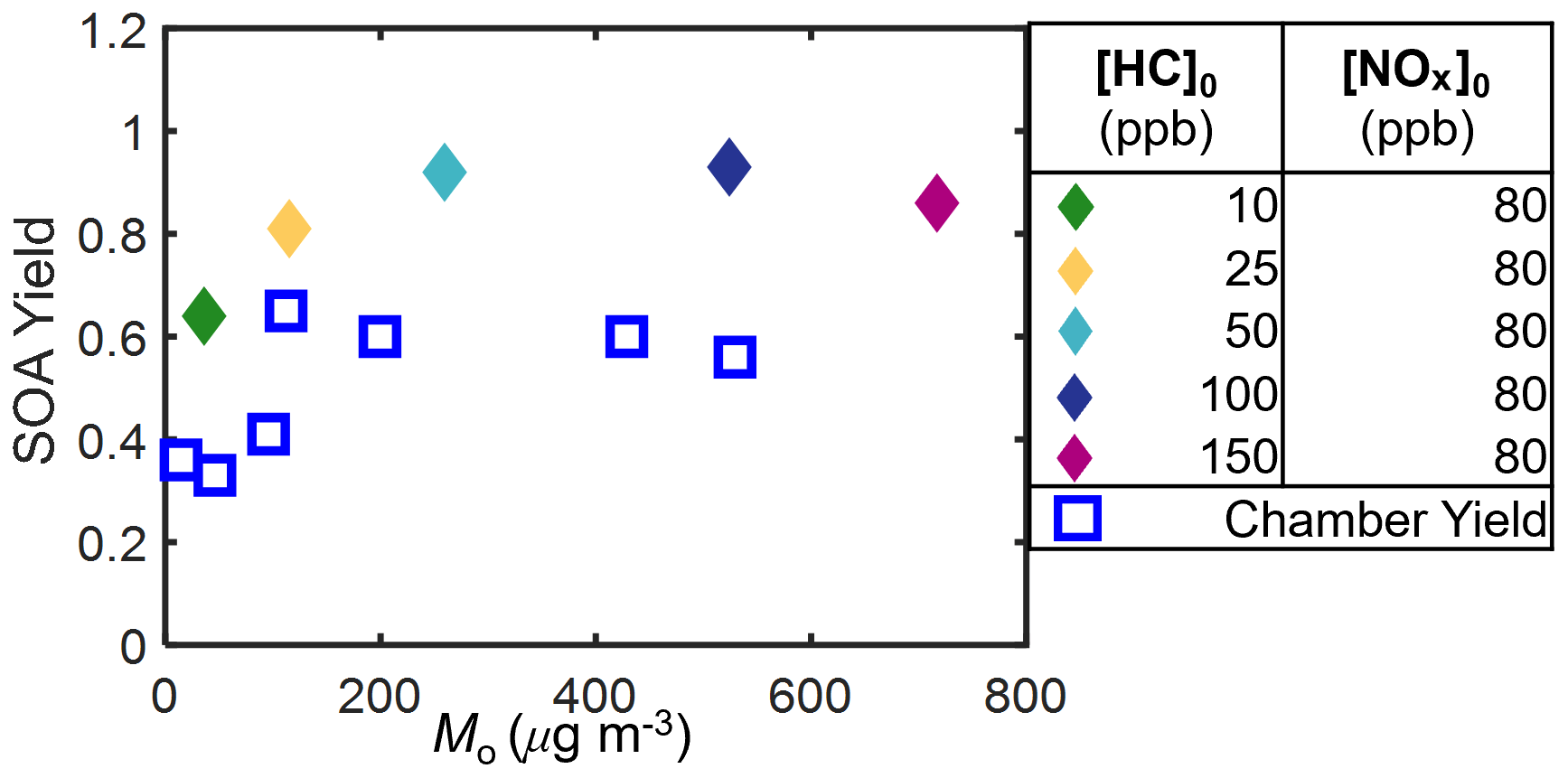
Figure 9 shows the predicted SOA mass yields based on GECKO-A. For simulations with added NOx, while the model predicted higher SOA mass yields (0.64–0.93) than were observed (0.33–0.64), the trends in the SOA mass yields were consistent between chamber observations and simulations. The simulated SOA mass yield increased with SOA mass for SOA mass < 260 µg m−3, plateaued for SOA mass between 260 and 524 µg m−3, and then decreased for SOA mass > 524 µg m−3.
The predicted O : C ratio and average carbon number (Fig. 10), defined as the mole-weighted average carbon number for the main products (∼ 95 % by mass), were consistent with the plateauing and/or decreasing SOA yields at higher [HC]0 (Fig. 9). The average carbon number was calculated using Eq. (2):
where nCi, Mo,i, and MWi are the carbon number, mass, and molecular weight of species i, respectively. With added NOx, the average carbon number of both the gas and particle phases increased as [HC]0 increased, while the O : C ratio decreased. These trends indicate that there is a significant fraction of higher-volatility compounds formed that contribute to SOA at higher [HC]0 (or Mo), resulting in lower SOA mass yields. In addition, only at the highest two [HC]0 were non-negligible fractions of precursor predicted to react with O3 and NO3 (Fig. S7), suggesting a larger fraction of higher-volatility nitrogen-containing products. More detailed comparisons of GECKO-A simulations with chamber experiments are presented by Afreh et al. (2021) for camphene and McVay et al. (2016) for α-pinene.

Figure 10GECKO-A-predicted particle O : C and mole-weighted average carbon number of products with added NOx.
5.2 Particle density and O : C
Figure 11a shows the GECKO-A-predicted O : C ratio and measured O : C ratio as a function of [HC]0 [NOx]0 for all experiments. Good agreement in O : C ratios was observed between the model predictions and chamber data. The O : C ratio decreased from 1.21 to 0.39 as [HC]0 [NOx]0 increased from 0.13 to 223, supporting the idea that more highly oxygenated products were formed at lower [HC]0 [NOx]0.
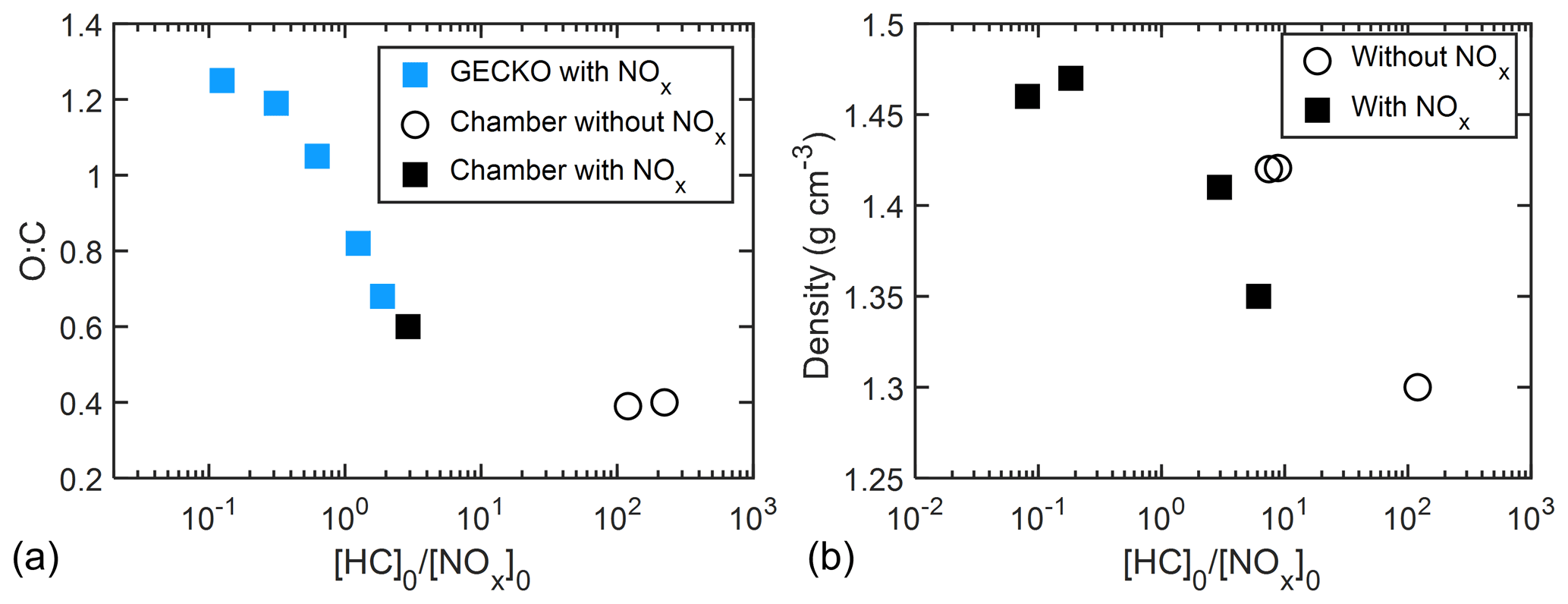
Figure 11(a) O : C ratio as a function of [HC]0 [NOx]0 with AMS data and prediction by GECKO-A simulation. (b) Particle density (directly measured by APM–SMPS) shown as a function of [HC]0 [NOx]0.
A negative correlation was also observed between measured particle density and [HC]0 [NOx]0. The final density of particles decreased from 1.47 to 1.30 g cm−3 as [HC]0 [NOx]0 increased from 0.08 to 120 (Fig. 11b). The change in O : C ratio could account for the change in density. O : C and H : C have been used in semi-empirical SOA density parameterizations (Nakao et al., 2013; Kuwata et al., 2012), in which O : C plays a dominant role in determining organic particle density compared to H : C. Consistent with the semi-empirical formulations, the density of particles formed from oxidation of camphene increased as O : C increased (from 0.39 to 1.21), while H : C varies over a smaller range (from 1.42 to 1.79). The change in density supports the proposed explanation that more oxygenated products were formed under lower [HC]0 [NOx]0. The wide range in final density and the correlation with [HC]0 [NOx]0 shown here have not been previously reported. The SOA mass of each experiment in this study was calculated with its own density of SOA instead of applying an averaged density. A list of particle densities used in this study can be found in Table 2.
The first SOA mass yields from oxidation of camphene based on experiments performed in the UCR environmental chamber with varying [NOx]0 are presented herein. Higher SOA mass yields were measured with added NOx (0.33–0.64) than without added NOx (0.08–0.26) at atmospherically relevant OH concentrations. SOA formation from the oxidation of camphene showed different NOx dependence than what has previously been reported for other monoterpenes (e.g., α-pinene, d-limonene) and n-alkanes (carbon ≤ 10), in which higher SOA mass yields were measured when [NOx] was lower (Nøjgaard et al., 2006; Ng et al., 2007). For camphene oxidation, higher Δ[HC] and lower [HC]0 [NOx]0 (within 0.5–200) generally led to higher SOA mass yields. Similar NOx dependence has been observed for two sesquiterpenes (longifolene and aromadendrene) but was attributed to the production of nonvolatile organic nitrates with no detailed mechanistic analysis provided at that time (Ng et al., 2007).
Although [HC]0 [NOx]0 shows clear correlation with SOA mass yield, this quantity cannot completely represent the underlying RO2 chemistry. The RO2 chemistry and the competition between varying bimolecular RO2 and unimolecular RO2 reaction pathways, explored using SAPRC MechGen, can be used to explain the dependence of SOA mass yields on HC and NOx. The RO2+ NO pathway favored in experiments with added NOx formed HOMs with much lower volatilities than products formed in other pathways. In addition to the regular NOx regime introduced above ([HC]0 [NOx]0 > 0.5), the results suggested an extreme NOx regime wherein high [NOx] may suppress SOA mass yield. High NOx levels may suppress HO2 levels at the beginning of the experiments, causing a subsequent reduction in the yields of low-volatility products such as UNICAMP and HO2CAMP5. This suggests that if the reactions happened in NOx-rich environments with extremely high ratios of NO to HO2 (NO HO2), the SOA mass yield from oxidation of camphene might be significantly suppressed. As demonstrated here, simulations with chemically detailed box models such as SAPRC are useful for identifying SOA formation regimes.
Overall, SOA formation from oxidation of camphene may be larger in polluted environments (e.g., urban environments) than NOx-free environments. This reveals a possible underestimation of SOA formed from oxidation of camphene and potentially other VOCs that are assumed to have lower SOA mass yields at higher NOx levels. Further chamber and modeling studies of other understudied VOCs will be important for identifying other systems in which moderate NOx levels can promote HOM formation.
The experimental and modeling data are available upon request from the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-3131-2022-supplement.
QL and JJ contributed equally to the study and share the first authorship. QL performed chamber experiments, conducted data analysis, and led the first draft of the paper. JJ derived and implemented the camphene mechanism in SAPRC, conducted SAPRC model simulations, and led discussions on the chemistry of camphene SOA formation. IKA carried out GECKO-A model simulations. QL and JJ interpreted the results and wrote the paper with IKA. QL, JJ, KCB, and DRC III finalized the paper. All the listed authors contributed to the revisions of the paper. The project was supervised by KCB and DRC III.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
The views expressed in this document are solely those of the authors and do not necessarily reflect those of the agency. The U.S. Environmental Protection Agency (EPA) does not endorse any products or commercial services mentioned in this publication. This publication has not been formally reviewed by the EPA.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors acknowledge William Carter for helping with the discussions in the MechGen estimation methods.
This research has been supported by NSF Atmospheric and Geospace Sciences (grant no. 1753364). This publication was developed under assistance agreement no. 84000701 awarded by the EPA to the University of California Riverside.
This paper was edited by Nga Lee Ng and reviewed by two anonymous referees.
Afreh, I. K., Aumont, B., Camredon, M., and Barsanti, K. C.: Using GECKO-A to derive mechanistic understanding of secondary organic aerosol formation from the ubiquitous but understudied camphene, Atmos. Chem. Phys., 21, 11467–11487, https://doi.org/10.5194/acp-21-11467-2021, 2021.
Akagi, S. K., Yokelson, R. J., Burling, I. R., Meinardi, S., Simpson, I., Blake, D. R., McMeeking, G. R., Sullivan, A., Lee, T., Kreidenweis, S., Urbanski, S., Reardon, J., Griffith, D. W. T., Johnson, T. J., and Weise, D. R.: Measurements of reactive trace gases and variable O3 formation rates in some South Carolina biomass burning plumes, Atmos. Chem. Phys., 13, 1141–1165, https://doi.org/10.5194/acp-13-1141-2013, 2013.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003.
Atkinson, R., Aschmann, S. M., and Arey, J.: Rate constants for the gas-phase reactions of OH and NO3 radicals and O3 with sabinene and camphene at 296±2 K, Atmos. Environ. A-Gen., 24, 2647–2654, https://doi.org/10.1016/0960-1686(90)90144-C, 1990.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Aumont, B., Valorso, R., Mouchel-Vallon, C., Camredon, M., Lee-Taylor, J., and Madronich, S.: Modeling SOA formation from the oxidation of intermediate volatility n-alkanes, Atmos. Chem. Phys., 12, 7577–7589, https://doi.org/10.5194/acp-12-7577-2012, 2012.
Baruah, S. D., Gour, N. K., Sarma, P. J., and Deka, R. C.: OH-initiated mechanistic pathways and kinetics of camphene and fate of product radical: a DFT approach, Environ. Sci. Pollut. Res., 25, 2147–2156, https://doi.org/10.1007/s11356-017-0646-2, 2018.
Benelli, G., Govindarajan, M., Rajeswary, M., Vaseeharan, B., Alyahya, S. A., Alharbi, N. S., Kadaikunnan, S., Khaled, J. M., and Maggi, F.: Insecticidal activity of camphene, zerumbone and α-humulene from Cheilocostus speciosus rhizome essential oil against the Old-World bollworm, Helicoverpa armigera, Ecotox. Environ. Safe, 148, 781–786, https://doi.org/10.1016/j.ecoenv.2017.11.044, 2018.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Camredon, M., Aumont, B., Lee-Taylor, J., and Madronich, S.: The SOA/VOC/NOx system: an explicit model of secondary organic aerosol formation, Atmos. Chem. Phys., 7, 5599–5610, https://doi.org/10.5194/acp-7-5599-2007, 2007.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Carter, W. P. L.: A detailed mechanism for the gas-phase atmospheric reactions of organic compounds, Atmos. Environ. A-Gen., 24, 481–518, https://doi.org/10.1016/0960-1686(90)90005-8, 1990.
Carter, W. P. L.: Development of ozone reactivity scales for volatile organic compounds, J. Air Waste Manage., 44, 881–899, https://doi.org/10.1080/1073161x.1994.10467290, 1994.
Carter, W. P. L.: Documentation of the SAPRC-99 chemical mechanism for VOC reactivity assessment. Final Report to California Air Resources Board, Contract 92-329 and Contract 95–308, https://intra.engr.ucr.edu/~carter/pubs/s99txt.pdf (last access: December 2021), 2000.
Carter, W. P. L.: Development of the SAPRC-07 chemical mechanism and updated ozone reactivity scales, Final report to the California Air Resources Board, Contract No. 03-318, 06-408, and 07-730, https://intra.engr.ucr.edu/~carter/SAPRC/saprc07.pdf (last access: December 2021), 2007.
Carter, W. P. L.: Development of a condensed SAPRC-07 chemical mechanism, Atmos. Environ., 44, 5336–5345, https://doi.org/10.1016/j.atmosenv.2010.01.024, 2010a.
Carter, W. P. L.: Development of the SAPRC-07 chemical mechanism, Atmos. Environ., 44, 5324–5335, https://doi.org/10.1016/j.atmosenv.2010.01.026, 2010b.
Carter, W. P. L.: Documentation of the SAPRC-18 Mechanism; Report to California Air Resources Board Contract No. 11-761, May 2020, https://intra.engr.ucr.edu/~carter/SAPRC/18/ (last access: December 2021), 2020a.
Carter, W. P. L.: Estimates and Assignments used in the SAPRC-18 Mechanism Generation System; Report to California Air Resources Board Contract No. 11-761, http://intra.engr.ucr.edu/~carter/SAPRC/18 (last access: December 2021), 2020b.
Carter, W. P. L.: Gateway to the SAPRC Mechanism Generation System, http://mechgen.cert.ucr.edu/, last access: 30 March 2021.
Carter, W. P. L. and Lurmann, F. W.: Evaluation of the RADM Gas-phase Chemical Mechanism. U.S. Environmental Protection Agency Cooperative Agreement CR-814558-01-0, Statewide Air Pollution Research Center, University of California, Riverside, 1989.
Carter, W. P. L., Cocker, D. R., Fitz, D. R., Malkina, I. L., Bumiller, K., Sauer, C. G., Pisano, J. T., Bufalino, C., and Song, C.: A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation, Atmos. Environ., 39, 7768–7788, https://doi.org/10.1016/j.atmosenv.2005.08.040, 2005.
Clark, C. H., Kacarab, M., Nakao, S., Asa-Awuku, A., Sato, K., and Cocker, D. R.: Temperature Effects on Secondary Organic Aerosol (SOA) from the Dark Ozonolysis and Photo-Oxidation of Isoprene, Environ. Sci. Technol., 50, 5564–5571, https://doi.org/10.1021/acs.est.5b05524, 2016.
Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: State-of-the-art chamber facility for studying atmospheric aerosol chemistry, Environ. Sci. Technol., 35, 2594–2601, https://doi.org/10.1021/es0019169, 2001.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/JZ4019207, 2013.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Donahue, N. M., Robinson, A. L., and Pandis, S. N.: Atmospheric organic particulate matter: From smoke to secondary organic aerosol, Atmos. Environ., 43, 94–106, https://doi.org/10.1016/j.atmosenv.2008.09.055, 2009.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/KIN.21130, 2017.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons Terms of Use CC-BY, Environ. Sci. Technol, 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Gaona-Colmán, E., Blanco, M. B., Barnes, I., Wiesen, P., and Teruel, M. A.: OH- and O3-initiated atmospheric degradation of camphene: temperature dependent rate coefficients, product yields and mechanisms, RSC Adv., 7, 2733–2744, https://doi.org/10.1039/c6ra26656h, 2017.
Geron, C., Rasmussen, R., Arnts, R. R., and Guenther, A.: A review and synthesis of monoterpene speciation from forests in the United States, Atmos. Environ., 34, 1761–1781, https://doi.org/10.1016/S1352-2310(99)00364-7, 2000.
Gilman, J. B., Lerner, B. M., Kuster, W. C., Goldan, P. D., Warneke, C., Veres, P. R., Roberts, J. M., de Gouw, J. A., Burling, I. R., and Yokelson, R. J.: Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 15, 13915–13938, https://doi.org/10.5194/acp-15-13915-2015, 2015.
Griffin, R. J., Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: Organic aerosol formation from the oxidation of biogenic hydrocarbons, J. Geophys. Res., 104, 3555–3567, https://doi.org/10.1029/1998JD100049, 1999.
Guenther, A.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Hakola, H., Arey, J., Aschmann, S. M., and Atkinson, R.: Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes, J. Atmos. Chem., 18, 75–102, https://doi.org/10.1007/BF00694375, 1994.
Hatch, L. E., Luo, W., Pankow, J. F., Yokelson, R. J., Stockwell, C. E., and Barsanti, K. C.: Identification and quantification of gaseous organic compounds emitted from biomass burning using two-dimensional gas chromatography–time-of-flight mass spectrometry, Atmos. Chem. Phys., 15, 1865–1899, https://doi.org/10.5194/acp-15-1865-2015, 2015.
Hatch, L. E., Jen, C. N., Kreisberg, N. M., Selimovic, V., Yokelson, R. J., Stamatis, C., York, R. A., Foster, D., Stephens, S. L., Goldstein, A. H., and Barsanti, K. C.: Highly Speciated Measurements of Terpenoids Emitted from Laboratory and Mixed-Conifer Forest Prescribed Fires, Environ. Sci. Technol., 53, 9418–9428, https://doi.org/10.1021/acs.est.9b02612, 2019.
Hatfield, M. L. and Huff Hartz, K. E.: Secondary organic aerosol from biogenic volatile organic compound mixtures, Atmos. Environ., 45, 2211–2219, https://doi.org/10.1016/j.atmosenv.2011.01.065, 2011.
Hayward, S., Muncey, R. J., James, A. E., Halsall, C. J., and Hewitt, C. N.: Monoterpene emissions from soil in a Sitka spruce forest, Atmos. Environ., 35, 4081–4087, https://doi.org/10.1016/S1352-2310(01)00213-8, 2001.
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008.
Hurley, M. D., Sokolov, O., and Wallington, T. J.: Organic aerosol formation during the atmospheric degradation of toluene, Environ. Sci. Technol., 35, 1358–1366, https://doi.org/10.1021/es0013733, 2001.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585 https://doi.org/10.1021/ES950943, 1996.
Jiang, J., Carter, W. P. L., Cocker, D. R., and Barsanti, K. C.: Development and Evaluation of a Detailed Mechanism for Gas-Phase Atmospheric Reactions of Furans, ACS Earth Sp. Chem., 4, 1254–1268, https://doi.org/10.1021/acsearthspacechem.0c00058, 2020.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/ANIE.201408566, 2014.
Komenda, M.: Monoterpene emissions from Scots pine (Pinus sylvestris): Field studies of emission rate variabilities, J. Geophys. Res., 107, 4161, https://doi.org/10.1029/2001JD000691, 2002.
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always Lost but Never Forgotten: Gas-Phase Wall Losses Are Important in All Teflon Environmental Chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Kuwata, M., Zorn, S. R., and Martin, S. T.: Using elemental ratios to predict the density of organic material composed of carbon, hydrogen, and oxygen, Environ. Sci. Technol., 46, 787–794, https://doi.org/10.1021/es202525q, 2012.
La, Y. S., Camredon, M., Ziemann, P. J., Valorso, R., Matsunaga, A., Lannuque, V., Lee-Taylor, J., Hodzic, A., Madronich, S., and Aumont, B.: Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation, Atmos. Chem. Phys., 16, 1417–1431, https://doi.org/10.5194/acp-16-1417-2016, 2016.
Lannuque, V., Camredon, M., Couvidat, F., Hodzic, A., Valorso, R., Madronich, S., Bessagnet, B., and Aumont, B.: Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: development of the VBS-GECKO parameterization, Atmos. Chem. Phys., 18, 13411–13428, https://doi.org/10.5194/acp-18-13411-2018, 2018.
Li, L., Tang, P., and Cocker, D. R.: Instantaneous nitric oxide effect on secondary organic aerosol formation from m-xylene photooxidation, Atmos. Environ., 119, 144–155, https://doi.org/10.1016/j.atmosenv.2015.08.010, 2015.
Li, L., Tang, P., Nakao, S., and Cocker III, D. R.: Impact of molecular structure on secondary organic aerosol formation from aromatic hydrocarbon photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 10793–10808, https://doi.org/10.5194/acp-16-10793-2016, 2016.
Ludley, K. E., Jickells, S. M., Chamberlain, P. M., Whitaker, J., and Robinson, C. H.: Distribution of monoterpenes between organic resources in upper soil horizons under monocultures of Picea abies, Picea sitchensis and Pinus sylvestris, Soil Biol. Biochem., 41, 1050–1059, https://doi.org/10.1016/j.soilbio.2009.02.002, 2009.
Maleknia, S. D., Bell, T. L., and Adams, M. A.: PTR-MS analysis of reference and plant-emitted volatile organic compounds, Int. J. Mass Spectrom., 262, 203–210, https://doi.org/10.1016/j.ijms.2006.11.010, 2007.
Malloy, Q. G. J., Nakao, S., Qi, L., Austin, R., Stothers, C., Hagino, H., and Cocker, D. R.: Real-Time aerosol density determination utilizing a modified scanning mobility particle sizer aerosol particle mass analyzer system, Aerosol Sci. Tech., 43, 673–678, https://doi.org/10.1080/02786820902832960, 2009.
Mazza, G. and Cottrell, T.: Volatile components of roots, stems, leaves, and flowers of Echinacea species, J. Agr. Food Chem., 47, 3081–3085, https://doi.org/10.1021/jf981117y, 1999.
McVay, R. C., Zhang, X., Aumont, B., Valorso, R., Camredon, M., La, Y. S., Wennberg, P. O., and Seinfeld, J. H.: SOA formation from the photooxidation of α-pinene: systematic exploration of the simulation of chamber data, Atmos. Chem. Phys., 16, 2785–2802, https://doi.org/10.5194/acp-16-2785-2016, 2016.
Mehra, A., Krechmer, J. E., Lambe, A., Sarkar, C., Williams, L., Khalaj, F., Guenther, A., Jayne, J., Coe, H., Worsnop, D., Faiola, C., and Canagaratna, M.: Oligomer and highly oxygenated organic molecule formation from oxidation of oxygenated monoterpenes emitted by California sage plants, Atmos. Chem. Phys., 20, 10953–10965, https://doi.org/10.5194/acp-20-10953-2020, 2020.
Moukhtar, S., Couret, C., Rouil, L., and Simon, V.: Biogenic Volatile Organic Compounds (BVOCs) emissions from Abies alba in a French forest, Sci. Total Environ., 354, 232–245, https://doi.org/10.1016/j.scitotenv.2005.01.044, 2006.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: Monoterpene SOA – Contribution of first-generation oxidation products to formation and chemical composition, Atmos. Environ., 130, 136–144, https://doi.org/10.1016/j.atmosenv.2015.10.080, 2016.
Nakao, S., Tang, P., Tang, X., Clark, C. H., Qi, L., Seo, E., Asa-Awuku, A., and Cocker, D.: Density and elemental ratios of secondary organic aerosol: Application of a density prediction method, Atmos. Environ., 68, 273–277, https://doi.org/10.1016/j.atmosenv.2012.11.006, 2013.
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contribution and group interactions, Fluid Phase Equilibr., 269, 117–133, https://doi.org/10.1016/j.fluid.2008.04.020, 2008.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of first- versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, 2006.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Nøjgaard, J. K., Bilde, M., Stenby, C., Nielsen, O. J., and Wolkoff, P.: The effect of nitrogen dioxide on particle formation during ozonolysis of two abundant monoterpenes indoors, Atmos. Environ., 40, 1030–1042, https://doi.org/10.1016/j.atmosenv.2005.11.029, 2006.
Pankow, J. F.: An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 41, 75–79, https://doi.org/10.1016/j.atmosenv.2007.10.060, 1994.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration, Environ. Sci. Technol., 39, 7046–7054, https://doi.org/10.1021/es050400s, 2005.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Quéléver, L. L. J., Kristensen, K., Normann Jensen, L., Rosati, B., Teiwes, R., Daellenbach, K. R., Peräkylä, O., Roldin, P., Bossi, R., Pedersen, H. B., Glasius, M., Bilde, M., and Ehn, M.: Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis, Atmos. Chem. Phys., 19, 7609–7625, https://doi.org/10.5194/acp-19-7609-2019, 2019.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Schervish, M. and Donahue, N. M.: Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 20, 1183–1199, https://doi.org/10.5194/acp-20-1183-2020, 2020.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Song, C., Na, K., and Cocker, D. R.: Impact of the hydrocarbon to NOx ratio on secondary organic aerosol formation, Environ. Sci. Technol., 39, 3143–3149, https://doi.org/10.1021/es0493244, 2005.
White, M. L., Russo, R. S., Zhou, Y., Mao, H., Varner, R. K., Ambrose, J., Veres, P., Wingenter, O. W., Haase, K., Stutz, J., Talbot, R., and Sive, B. C.: Volatile organic compounds in northern New England marine and continental environments during the ICARTT 2004 campaign, J. Geophys. Res., 113, D08S90, https://doi.org/10.1029/2007JD009161, 2008.
Xavier, C., Rusanen, A., Zhou, P., Dean, C., Pichelstorfer, L., Roldin, P., and Boy, M.: Aerosol mass yields of selected biogenic volatile organic compounds – a theoretical study with nearly explicit gas-phase chemistry, Atmos. Chem. Phys., 19, 13741–13758, https://doi.org/10.5194/acp-19-13741-2019, 2019.
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Tech., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, 2016.
Ye, Q., Wang, M., Hofbauer, V., Stolzenburg, D., Chen, D., Schervish, M., Vogel, A., Mauldin, R. L., Baalbaki, R., Brilke, S., and Dada, L.: Molecular Composition and Volatility of Nucleated Particles from α-Pinene Oxidation between −50 ∘C and +25 ∘C, Environ. Sci. Technol., 53, 12357–12365, https://doi.org/10.1021/ACS.EST.9B03265, 2019.
Zhang, S.-H., Shaw, M., Seinfeld, J. H., and Flagan, R. C.: Photochemical aerosol formation from α-pinene- and β-pinene, J. Geophys. Res., 97, 20717–20729, https://doi.org/10.1029/92jd02156, 1992.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/PNAS.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Bé, A. G., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z., Gold, A., Graf, S., Cubison, M. J., Groessl, M., Jayne, J. T., Worsnop, D. R., and Canagaratna, M. R.: Highly Oxygenated Multifunctional Compounds in α-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 51, 5932–5940, https://doi.org/10.1021/ACS.EST.6B06588, 2017.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.