the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Jul 2021
| 02 Jul 2021
Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling
Robin Wollesen de Jonge
Jonas Elm
Bernadette Rosati
Sigurd Christiansen
Noora Hyttinen
Dana Lüdemann
Merete Bilde
Pontus Roldin
Dimethyl sulfide (DMS) is the dominant biogenic sulfur compound in the ambient marine atmosphere. Low-volatility acids from DMS oxidation promote the formation and growth of sulfur aerosols and ultimately alter cloud properties and Earth's climate. We studied the OH-initiated oxidation of DMS in the Aarhus University Research on Aerosol (AURA) smog chamber and the marine boundary layer (MBL) with the aerosol dynamics and gas- and particle-phase chemistry kinetic multilayer model ADCHAM. Our work involved the development of a revised and comprehensive multiphase DMS oxidation mechanism, capable of both reproducing smog chamber and atmospheric relevant conditions. The secondary aerosol mass yield in the AURA chamber was found to have a strong dependence on the reaction of methyl sulfinic acid (MSIA) and OH, causing a 82.8 % increase in the total PM at low relative humidity (RH), while the autoxidation of the intermediate radical CH3SCH2OO forming hydroperoxymethyl thioformate (HPMTF) proved important at high temperature and RH, decreasing the total PM by 55.8 %. The observations and modelling strongly support the finding that a liquid water film existed on the Teflon surface of the chamber bag, which enhanced the wall loss of water-soluble intermediates and oxidants dimethyl sulfoxide (DMSO), MSIA, HPMTF, SO2, methanesulfonic acid (MSA), sulfuric acid (SA) and H2O2. The effect caused a 64.8 % and 91.7 % decrease in the secondary aerosol mass yield obtained at both dry (0 % RH–12 % RH) and humid (50 % RH–80 % RH) conditions, respectively. Model runs reproducing the ambient marine atmosphere indicate that OH comprises a strong sink of DMS in the MBL (accounting for 31.1 % of the total sink flux of DMS) although less important than the combined effect of halogen species Cl and BrO (accounting for 24.3 % and 38.7 %, respectively). Cloudy conditions promote the production of particular mass (PM) from SO2 accumulated in the gas phase, while cloud-free periods facilitate MSA formation in the deliquesced particles. The exclusion of aqueous-phase chemistry lowers the DMS sink as no halogens are activated in the sea spray particles and underestimates the secondary aerosol mass yield by neglecting and MSA PM production in the particle phase. Overall, this study demonstrated that the current DMS oxidation mechanisms reported in literature are inadequate in reproducing the results obtained in the AURA chamber, whereas the revised chemistry captured the formation, growth and chemical composition of the formed aerosol particles well. Furthermore, we emphasize the importance of OH-initiated oxidation of DMS in the ambient marine atmosphere during conditions with low sea spray emissions.
- Article
(2674 KB) - Full-text XML
-
Supplement
(6142 KB) - BibTeX
- EndNote





Dimethyl sulfide (DMS: CH3SCH3) from biogenic ocean emissions is the largest source of natural sulfur in the ambient atmosphere (Lovelock et al., 1972; Andreae, 1990; Seinfeld and Pandis, 2016). DMS is oxidized mainly in the gas phase by OH (66 %), NO3 (16 %) and various halogen species (Hoffmann et al., 2016; Chen et al., 2018) globally, either by OH-, NO3- or Cl-initiated H abstraction or OH and BrO addition.
Each reaction defines a distinct abstraction and addition pathway, leading to the formation of low-volatility acids, methanesulfonic acid (MSA: CH3SO3H) and sulfuric acid (SA: H2SO4) (Yin et al., 1990; Barnes et al., 2006). SA is known to undergo binary or ternary nucleation in the presence of H2O and NH3, while MSA is believed to nucleate only under certain favourable conditions (Korhonen et al., 1999; Chen et al., 2015). New particle formation (NPF) from SA is an important initial step in the production of cloud condensation nuclei (CCN) and has the potential to alter the properties of clouds and hence climate. MSA condenses on the new particles and promotes their growth into the CCN size range (> 100 nm in particle diameter) (Saltzman et al., 1983). Although the chemistry of DMS oxidation and subsequent formation of SA and MSA has been studied in great detail, the current mechanisms remain uncertain (Barnes et al., 2006; Hoffmann et al., 2016). Large-scale models simplify the process by assuming fixed yields of SO2 and MSA from DMS oxidation (Berglen et al., 2004; Kloster et al., 2006), leaving out important intermediates such as dimethyl sulfoxide (DMSO: CH3SOCH3), methane sulfinic acid (MSIA: CH3SO2H) and the recently discovered autoxidation product hydroperoxymethyl thioformate (HPMTF: HOOCH2SCHO) (Wu et al., 2014; Berndt et al., 2019; Veres et al., 2020). A study by Hoffmann et al. (2016) pointed out how model studies often exclude or simplify the effect of multiphase DMS chemistry. Soluble products and intermediates DMS, SO2, DMSO, MSIA, MSA and SA will readily partition to the aqueous phase (Campolongo et al., 1999) and transform in various reaction pathways. Berresheim et al. (2014) noted that SA field measurements could not be explained when considering the oxidation of SO2 by OH as the only source of SA and questioned the importance of SO2 in SA production and NPF. Taking into account the aqueous-phase uptake of SO2 during cloud cover, the authors theorized the rapid formation of SA from SO3 (produced from thermal decomposition of CH3SO3) as a possible explanation to close the gap between measured and modelled SA concentrations. Multiphase DMS chemistry is also essential to explain the MSA to ratios found in atmospheric aerosol samples (Lucas and Prinn, 2002; Hoffmann et al., 2016). While MSA production in the gas phase is scarce (Lucas and Prinn, 2002), aqueous-phase reactions almost predominantly form MSA (Hoffmann et al., 2016). However, MSA formation in the gas phase remains uncertain, and earlier studies have suggested an alternative pathway via the MSIA intermediate (Yin et al., 1990; von Glasow and Crutzen, 2004). In pristine marine environments, halogen compounds from sea spray aerosols comprise an important addition to the otherwise OH dominated oxidation of DMS (von Glasow and Crutzen, 2004). A modelling study by Braeuer et al. (2013) examined the activation and subsequent transfer of chlorine and bromine species from the particle phase to the gas phase. Cl and BrO radicals in particular were found to exert a strong increase in the DMS sink flux (Hoffmann et al., 2016).
Few have studied the gas-phase and multiphase DMS chemistry in controlled smog chamber experiments, and prior to Rosati et al. (2021b) and this work, none have tested model predictions on the formation, growth and chemical composition of the aerosol particles being produced. Therefore, this study and the companion paper by Rosati et al. (2021b) investigate the oxidation of DMS by OH radicals performed in the Aarhus University Research on Aerosol (AURA) smog chamber, using the state-of-the-art gas- and particle-phase chemistry model for laboratory chamber studies (ADCHAM) (Roldin et al., 2014). A comprehensive multiphase mechanism was developed based on the Master Chemical Mechanism version 3.3.1 (MCMv3.3.1) (Jenkin et al., 1997, 2003), the CAPRAM DMS module 1.0 (DM1.0) (Hoffmann et al., 2016) and the multiphase halogen chemistry mechanism CAPRAM Halogen Module 2.0 (HM2.0) (Braeuer et al., 2013). Additional reactions and rate constants were implemented from individual studies (Turnipseed et al., 1995; Kukui et al., 2003; Wu et al., 2014; Cao et al., 2013; Berndt et al., 2019, 2020). A stand-alone experiment examining butanol oxidation by OH was used to quantify the effect of high and low relative humidity (RH) on the OH concentration in the AURA chamber (Sect. 2.2). The results were used to estimate the formation of a RH-dependent water film forming on the Teflon bag surface and implemented as a mean to explain the difference in DMS experiments performed at dry and humid conditions (Sect. 3.3.1–3.3.3). Finally, we performed several sensitivity runs investigating how the new revised DMS multiphase chemistry mechanism compares with previous DMS mechanisms under different relevant atmospheric conditions (Sect. 3.3.4).
2.1 Smog chamber experiments
The experiments studied in this work were performed in the Aarhus University Research on Aerosol (AURA) smog chamber (Kristensen et al., 2017). Here we only give a brief overview of the smog chamber setup. For a detailed description of the setup, e.g. different instrumentations as well as results from experiments performed under dry conditions, the reader is referred to Rosati et al. (2021b).
AURA consists of a 5 m3 Teflon bag mounted inside a temperature-controlled enclosure. All experiments studied in this work were performed in batch sampling mode. The chamber was first filled with clean particle-free air. After this, the UV lights were turned on, followed by addition of H2O2, which served as an OH source. For the modelled dry (low RH) and humid experiments, the injected H2O2 amounts were equivalent to a H2O2 mixing ratio of 20 and 70 ppmv, respectively. The reference time zero, which marks the start of the experiments and model simulations, was set to the time when DMS was injected into the chamber. DMS was injected after the H2O2. The total instrument sampling rates ranged between 4.2 and 6.9 L min−1. During the sampling, the Teflon bag was partially compressed. However, since the Teflon bag was mounted inside a fixed metal frame (Kristensen et al., 2017), a fraction of the sampled air must have been replaced with air leaking in from outside the Teflon bag, resulting in gradual dilution. Based on observations of the chamber volume, it was estimated that the chamber volume shrank to a minimum of 3 m3. In the model, we considered the gradual compaction and dilution of the chamber volume due to the instrument sampling (see description in the Supplement Sect. S2.1) (Rosati et al., 2021a). The inflow of air from outside the chamber is a likely source for the particle contamination (nitrate and organics) observed with the high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) (Rosati et al., 2021b). However, since this source of contamination cannot explicitly be quantified, the inflow of air was assumed to be free of particles in the model simulations. For the humid experiments, an additional inflow of humidified particle-free air with a rate of 2.0 L min−1 was used to keep the RH stable. This resulted in additional dilution of the chamber.
In addition to the DMS oxidation experiments, several butanol OH oxidation experiments were used to investigate how the humidity and temperature influenced the OH concentration in the chamber. Table 1 provides an overview of the conditions for all AURA experiments modelled with ADCHAM. Experiments DMS1-5 and BUT-1 are described in detail in Rosati et al. (2021b), and experimental data for these experiments are adopted from Rosati et al. (2021b); see also Sect. S2 for model details.
Table 1Experimental (model) conditions. Experiments DMS2, DMS6 and DMS7 marked in bold are referred to as “dry”, “humid” and “cold” in Sect. 3. Experiments DMS1-5 and BUT-1 are described in detail in Rosati et al. (2021b), and experimental data for these experiments are adopted from Rosati et al. (2021b).
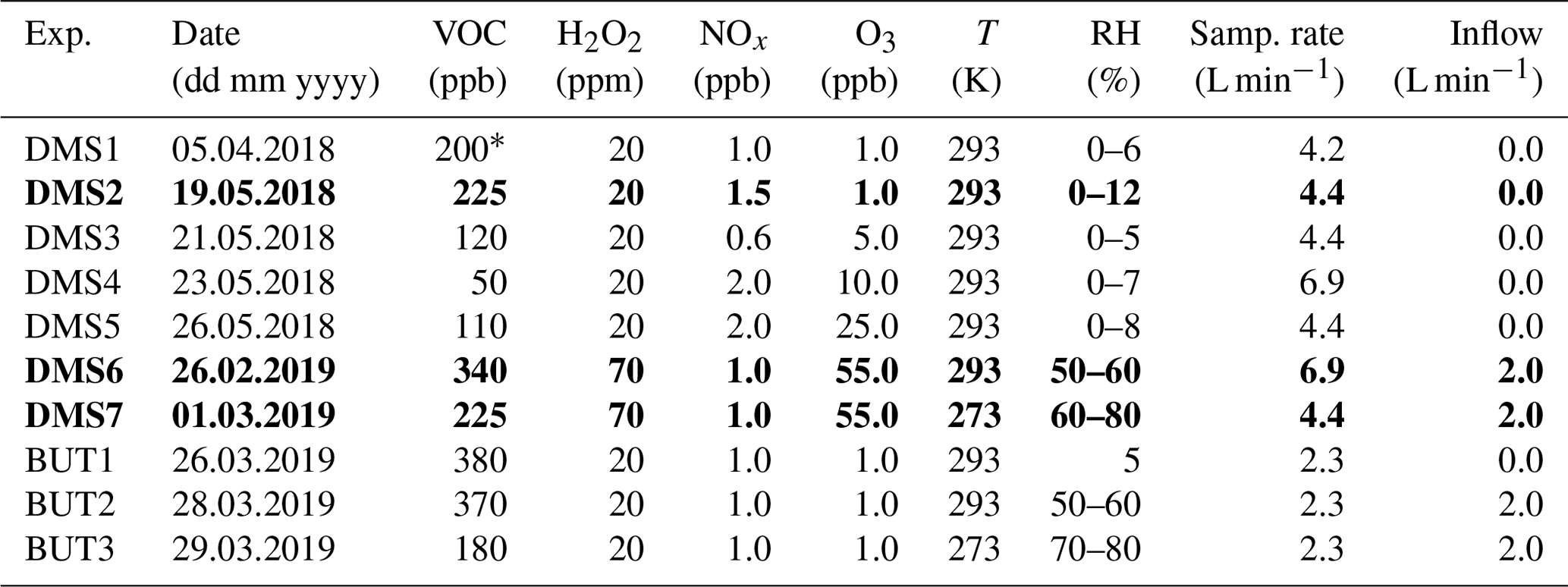
∗ Estimated value based on the injected DMS sample volume (no proton-transfer-reaction mass spectrometry (PTR-MS) measurements).
Table 2Default model setup. Experiments DMS2, DMS6 and DMS7 marked in bold are referred to as “dry”, “humid” and “cold” in Sect. 3.
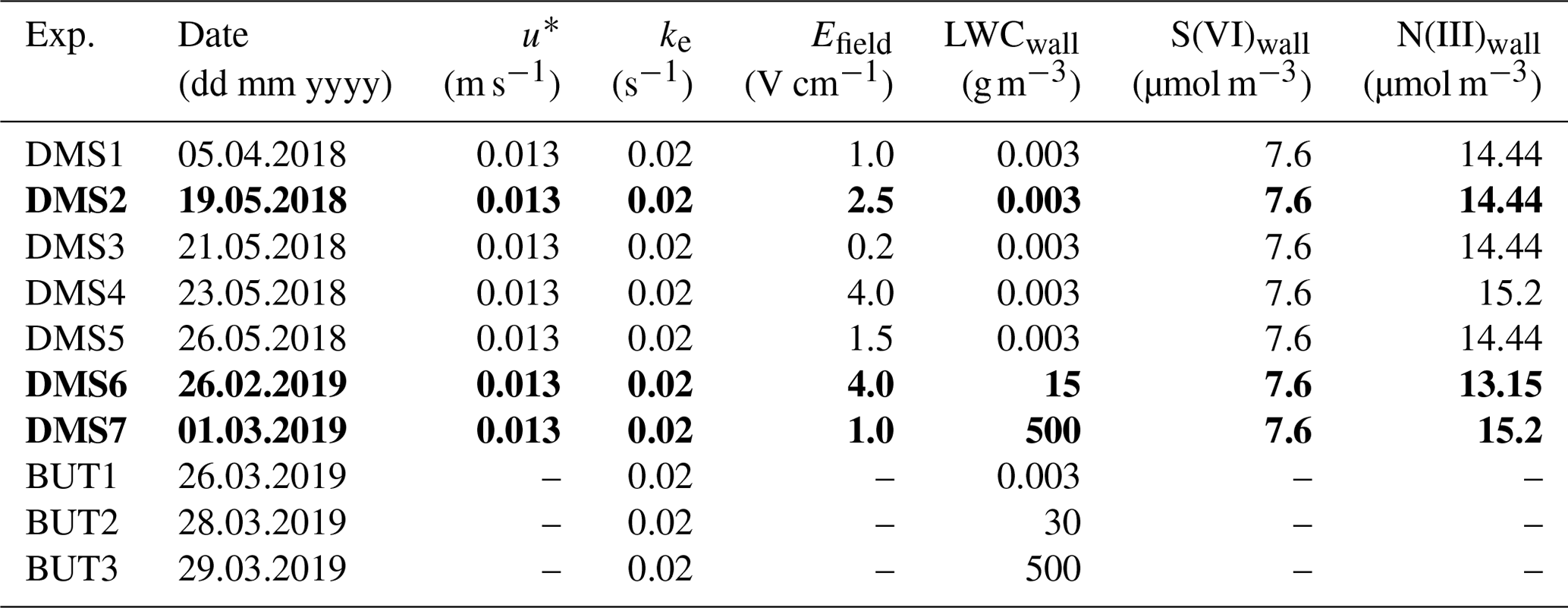
u∗ denotes the friction velocity, ke the coefficient of eddy diffusion and LWCwall the liquid water content on the Teflon wall.
2.2 ADCHAM – AURA model setup
ADCHAM was set up and used to simulate seven DMS oxidation experiments (DMS1-7) and three butanol oxidation experiments (BUT1-3) in total at different environmental conditions (T and RH) (Table 1). The primary aim was to evaluate and constrain the relative role of different DMS gas-phase oxidation pathways leading to atmospheric secondary aerosol formation. The model simulations were started 1 h before the DMS injection with initially particle-free air and NOx, O3 and H2O2 concentrations as specified in Table 1. The measured temperature and relative humidity (RH) time series with 1 min resolution were read in as input to the model. The modelled particle number size distribution was represented by 200 fixed size bins between 1.07 and 500 nm in diameter.
2.2.1 Chamber wall effects – gas to wall partitioning
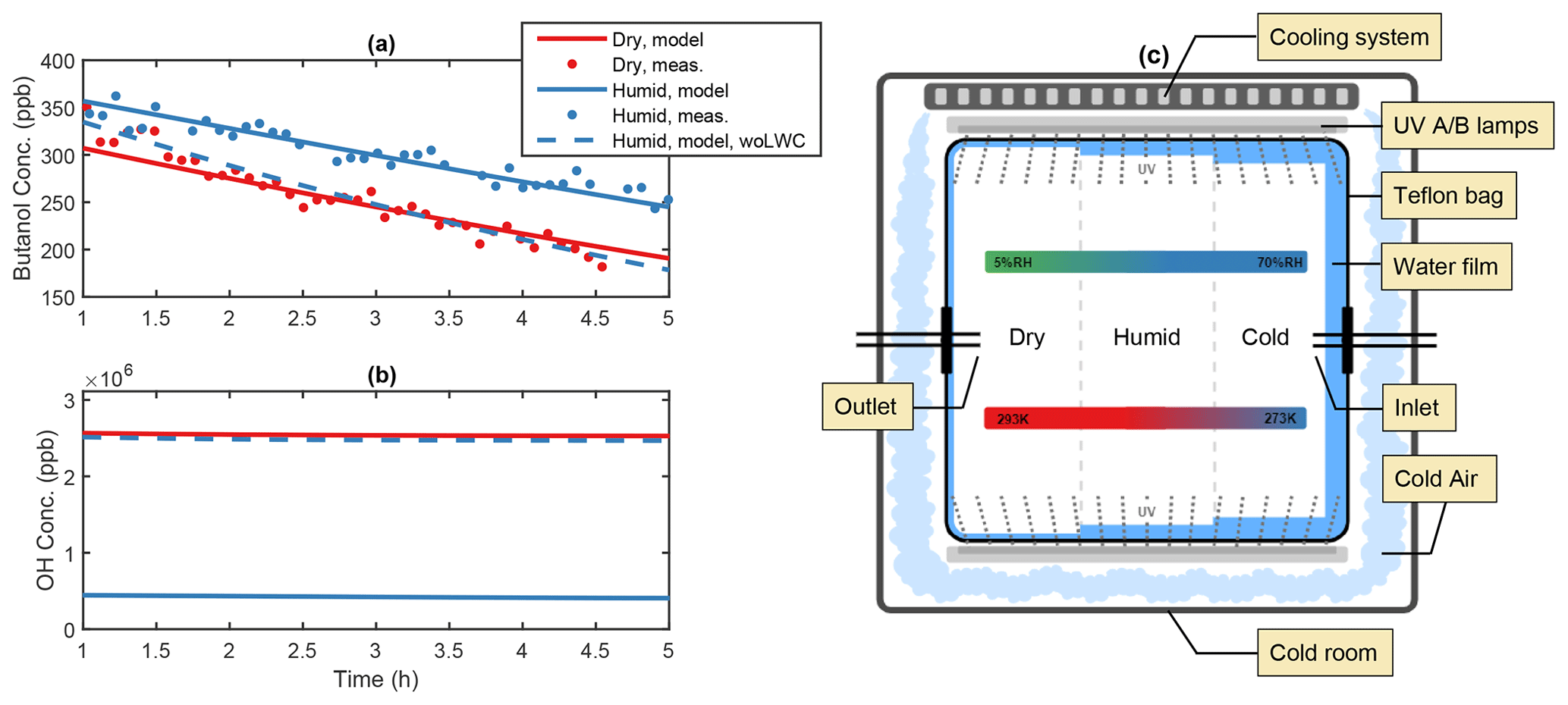
Figure 1Illustration of the water film effect in the AURA chamber. Panel (a) depicts the PTR-MS butanol decay from experiments BUT1 (red) and BUT2 (blue) and (b) the OH concentration at varying RH with and without the influence of a liquid water film (woLWC: without liquid water content). The water content changed in accordance with varying temperature and RH (c). Outside cooling of the chamber may affect the water adsorption on the Teflon surface.
Comparing secondary aerosol yields from chamber experiments performed at high and low RH revealed a significant decrease in the overall PM during humid conditions. The effect may be caused by the formation of a liquid film on the chamber walls (Fig. 1c), lowering the gas-phase concentration of oxidants and water-soluble products from the DMS oxidation mechanism. Adsorption of water onto Teflon bag surfaces is known to occur in smog chamber experiments (Sumner et al., 2004) and may lead to condensation during high RH conditions (Svensson et al., 1987). Wall contamination including HONO and HNO3 from NOx exposure increases the uptake by fixating the adsorbed water molecules (Sumner et al., 2004). Separate experiments examining 1-butanol oxidation by OH were used to quantify the thickness of the Teflon bag water film. Analogous to the method by Song et al. (2019), the OH concentration was estimated based on the decay of 1-butanol. Humid chamber conditions decreased OH production and slowed down the butanol oxidation (Fig. 1a). The effect likely arose from the enhanced water uptake to the Teflon walls, which lowered the gas-phase concentration of H2O2 and thus the production of OH (Fig. 1b). The water content needed to suppress OH formation to match the experimental observations at ∼ 60 % RH and 293 K, in the centre of the chamber, corresponded to approximately 30 g m−3 or a liquid film layer of ∼10 µm on the chamber surface (see Fig. S1). The predicted concentration requires water to condensate on the Teflon bag. While Sumner et al. (2004) reject the idea that condensation is possible, chamber experiments by Svensson et al. (1987) showed that water condensates on both polluted and clean Teflon surfaces at RH conditions beyond 70 %. The authors also observed the formation of a few monolayers of water during low RH (< 5 %) conditions. During dry experiments in the AURA chamber, a small increase in RH was observed, caused by water contamination from instrument sampling (Table 1).
We performed several sensitivity tests with different water film thickness for the modelled DMS experiments (see Figs. S2–S4). From this, we concluded that with an effective water film concentration of 3 mg m−3, ∼ three water monolayers, the model generally captures the observed PM mass formation and SA to MSA PM mass ratio during the dry experiments. For the humid DMS experiments, the water concentration on the walls needs to be ∼ 15 and ∼ 500 g m−3 for the 293 and 273 K experiments, respectively. The optimal values found for the wall liquid water content are on the same order of magnitude as the values estimated based on the butanol experiments performed at similar RH and temperature conditions (Table 2).
On average the temperature proved smaller at the chamber bag surface as opposed to the chamber bag centre. The effect was caused by the temperature regulation setup cooling the chamber bag from the outside (Fig. 1c). These results are of interest, as the local decrease in temperature facilitates a local increase in RH. Consequently, the outside cooling of the chamber bag contributed to the condensation of water vapour and formation of a relatively thick liquid film on the Teflon surface during the humid experiments. The temperature observations at the vertical facing chamber walls together with the observed RH in the centre of the chamber indicate that the RH next to the chamber walls reached up to ∼ 80 % during the 293 K experiments and that the air may have been supersaturated with respect to water (RH > 100 %) during the 273 K experiments. This motivates the use of a substantially thicker liquid water film during the 273 K experiments.
In all experiments, the AMS measurements reveal that the formed PM contains a substantial mass fraction of ammonium. We expect that the ammonium mass mainly originates from NH3(g), gradually evaporating from the chamber walls and possibly to a smaller extent from NH3(g) leaking into the chamber. To be able to capture the observed secondary ammonium mass formation and the new particle formation (Sect. 2.4) in all experiments, we assumed that an initial pool of 1 mg m−3 (mass per air volume) dissolved ammonium sulfate was present on the chamber walls and that no NH3(g) was leaking in with air from outside the chamber. The pool of ammonium and sulfate on the chamber walls can be motivated by previous experiments in the AURA chamber with ammonium sulfate seed particles, SA(g) and NH3(g) depositing on the walls. While the sulfate (S(VI)) on the walls can be considered to be non-volatile, the semi-volatile ammonium (N(III)) will partition between the liquid film on the walls and the gas phase to different extents, which depend on the wall liquid water content, the acidity and the temperature. Hence, the N(III) concentration on the chamber walls most likely differs from one experiment to another. This motivates why we decided to use the initial N(III) wall concentration as an unknown model fitting parameter, when comparing the modelled and observed new particle and secondary ammonium mass formation. This resulted in initial N(III):S(VI) on the walls ranging between 1.7 and 2.0 for the different experiments (Table 2). The mass transfer of NH3(g) to and from the chamber walls was represented by the thermodynamics and multiphase chemistry module in ADCHAM (Sect. 2.3). Figure S5 shows the modelled NH3(g) concentration and the NH3 concentration on the chamber walls for all modelled experiments. The modelled NH3(g) concentration gradually decreased during most of the experiments because of the NH3 uptake to the formed aerosol particles and the SA and MSA deposition on the chamber walls, which resulted in a gradually more acidic liquid water film. However, in DMS6 the modelled NH3 concentration increases slightly in the end of the experiments. This is because the evaporation of NH3(g) from the chamber walls becomes larger than the sink of NH3(g) to the particle phase.
The first-order wall loss rates of gases were modelled using the theory proposed by McMurry and Grosjean (1985) (Eq. 1).
is the chamber surface area A to volume V ratio, αw the vapour wall mass accommodation coefficient, the mean thermal speed of the molecules, ke the coefficient of eddy diffusion and D the molecular diffusivity. ke and αw are the two key unknown parameters in Eq. (1). In the AURA smog chamber, ke has been estimated to ∼ 10−3 s−1 for low-volatility highly oxygenated organic molecules (HOMs) formed from α-pinene ozonolysis (Quéléver et al., 2019). For a typical α-pinene HOM monomer or SA and , this corresponds to ke≈0.02 s−1. This estimated value of ke can, e.g., be compared with the value reported by Zhang et al. (2014) of 0.015 s−1, for a Teflon chambers with a volume of 28 m3.
Figure S6 illustrates how ke varies as a function of ke and αw for a compound with similar molecular properties as MSA and SA. For , the vapour wall losses are entirely governed by the chamber mixing and the molecular diffusion through a thin surface layer next to the chamber walls, while for smaller values of αw, the surface reactivity also limits the wall uptake (McMurry and Grosjean, 1985). By default, the chamber wall loss of the important condensable vapours (SA, MSA, HNO3, NH3) and the highly water-soluble H2O2 were assumed to be limited exclusively by the chamber mixing and molecular diffusion across the surface layer next to the chamber walls (i.e. using αw=1.0). For the important intermediate DMS oxidation products, i.e. DMSO, DMSO2, MSIA and HPMTF, the wall mass accommodation coefficients were by default set to 10−5. Wall mass accommodation coefficients ∼ 10−5 have previously been suggested when modelling wall losses of volatile and semi-volatile organic molecules in Teflon smog chambers (Matsunaga and Ziemann, 2010; Zhang et al., 2014). For DMS, O3, SO2 and NO2, we used a default αw of 10−7. This αw value is approximately 1 order of magnitude greater than the value reported by McMurry and Grosjean (1985) for O3, SO2 and NO2, during dry chamber conditions. The relatively low αw values used for DMS, compared to the DMS oxidation products, was motivated by the reported low mass accommodation coefficients for DMS uptake to particles and cloud droplets (Hoffmann et al., 2016; Zhu et al., 2006; Kreidenweis et al., 2003) and the observed and modelled DMS trends during the humid experiments. When using the default αw values, ke becomes ∼ 10−3 s−1 for MSA and SA, ∼ 10−4 s−1 for the intermediate DMS oxidation products, and ∼ s−1 for DMS, O3, SO2 and NO2, for a fully inflated chamber (V=5 m3).
We performed several model sensitivity simulations with αw values ± 1 order of magnitude from the default values for DMS, DMSO, DMSO2, MSIA, HPMTF, O3 and SO2, for all experiments listed in Tables 1–2 (see Figs. S7–S21). From this, we conclude that the modelled secondary aerosol formation is especially sensitive to the rate at which O3 partitions to the liquid water film on the chamber walls. The uptake of O3 in the liquid water film governs the uptake and oxidation of MSIA(g) on the walls, both during the dry and humid experiments and the oxidation of DMS during the humid experiments (Figs. S7–S11). Also lower wall loss rates (i.e. lower αw) for the intermediate oxidation products, especially MSIA, have relatively large impact on the modelled secondary aerosol mass formation. Decreasing αw with 1 order of magnitude for the intermediate oxidation products results in increasing particle mass formation with a factor of 1.5–2.5 in experiment DMS1-6 but has very minor impact on the modelled particle mass (PM) formation during the humid and cold experiment (DMS7) (Figs. S17–S21). In DMS7, the PM formation is instead to a large extent influenced by the rate at which O3 is dissolved into the liquid water film (Figs. S8–S10). The wall losses of O3 and NO2 have been shown to increase during humid chamber conditions (Grosjean, 1985), and it is possible that the liquid water content on the chamber walls influence αw. However, for the base case model simulations, we chose to use the same αw values for both the dry and humid experiments.
The calculated ke were used as input parameters to the multiphase chemistry module (Sect. 2.1.2). This module solves a set of coupled ordinary differential equations which describe how the gas-phase and wall aqueous-phase concentration of all compounds ([c(g)] and [c(aq)]) evolve over time:
Equations (1) and (2) take into account the mass transfer between the gas phase and aqueous phase for all compounds ci and all relevant reactions (R1 to Rn) in the gas (R(g)) and aqueous phase (R(aq)), respectively. kaq is a first-order mass transfer rate (s−1), which in the case of the gas–wall partitioning is equal to ke and in the case of particles or cloud droplets is equal to the particle and cloud droplet condensation sink. ([c(g)] and [c(aq)]) are both given in units of molecules per cubic centimetre (molec. cm−3), and H∗ is the dimensionless Henry solubility (Jacobson, 2005) which is given by
Hcp denotes Henry's law solubility in units of kilograms per atmosphere per mole (kg atm−1 mol−1), R∗ is the universal gas constant (82.06 cm3 atm mol−1 K−1), T is the temperature in kelvin (K) and LWC is the aqueous-phase liquid water content, which in the case of gas–wall partitioning is equal to the effective wall water concentration given in units of kilograms per cubic centimetre (kg cm−3).
Table S2 gives the values of Hcp and αw for all species that were considered to undergo phase transfer between the gas and aqueous phase in the multiphase chemistry module.
2.2.2 Particle wall losses
The particle wall losses of particles with zero to three elemental charges were calculated using the particle wall loss parameterizations described in Roldin et al. (2014). For this, the model takes into account the initial fraction of neutral and charged new particles, which was calculated with ACDC, and the evolution of the aerosol particle charge distribution over time. The key unknown parameters which govern the particle wall losses of neutral and charged particles are the friction velocity (u∗) and the electric field strength Efield. We used a fixed value of (u∗) of 0.013 m s−1 for all experiments. This value was chosen in order for the particle wall losses to be consistent with the gas–wall loss rates calculated with Eq. (1); i.e., for a hypothetical non-charged particle or gas molecule of 0.6 nm in diameter, both parameterizations give first-order wall loss rates of ∼ 10−3 s−1. Efield (Table 2) was set to different values in the range of 0.2–4 V cm−1, depending on the observed and modelled particle number and volume concentration loss rates. The air ion concentration in the chamber was calculated from the steady-state solution of the ion balance equation, taking into account the ion formation rate, ion–ion recombination, condensation sink and wall losses of air ions (Kirkby et al., 2016). The steady-state air ion concentration of positively and negatively charged ions ([]) was used to derive the particle charge distribution by solving a system of differential equations:
In the above equations, and [N3]i denote the number concentration of particles with zero, one, two or three elemental charges in each size bin (i). kqi denotes the aerosol particle ion attachment coefficients (unit cm3 s−1), which depend on the size and sign of the particle charge (q) and air ions (Fuchs, 1963; Hoppel and Frick, 1986). For example, k−2i represents the attractive air ion attachment coefficients for an air ion approaching a particle, in size bin i, with two elemental charges when the sign of the particle and ion charge are different, while k1i represents the repellent air ion attachment coefficient for an air ion approaching a particle with one elemental charge, when the signs of the charge of the ion and particle are the same. Equations (5)–(8) assume an even distribution of positive and negative charged air ions (i.e. . Particles with more than three elemental charges were not considered in the model. Thus, half of all particles with three elemental charges which in reality would have gained four elemental charges upon collision with air ions were assumed to keep their three elemental charges. Figure S25 illustrates how the modelled particle charge distribution and particle wall losses evolve during experiment DMS2.
2.2.3 Chamber contaminants
Smog chamber experiments have the advantage of elucidating atmospheric phenomena in controlled temperature, RH, UV light and volatile organic compound (VOC) concentration conditions. Even so, contamination from walls and instrument sampling affects the outcome (Sumner et al., 2004). NOx pollutants have a high impact on DMS chemistry (Barnes et al., 2006), both by direct oxidation of intermediate species in the DMS oxidation mechanisms and indirectly by the formation of ozone. Chambers exposed regularly build up HONO on the walls from the heterogeneous reaction of NO2 and water on the Teflon bag surface (Svensson et al., 1987). The HONO wall pool comprises an additional source of NOx during chamber experiments. When exposed to UV lights, NO2 photodissociates to form NO and ground-state atomic oxygen O(3P) that combine rapidly with molecular oxygen to yield ozone. In the gas phase, ozone facilitates the formation of SO2 via the CH3SO intermediate, thereby reducing SA production and NPF. The effect is dominant in high RH conditions, during which SO2 is taken up by the water film on the Teflon bag. In the aqueous phase, ozone drives the uptake of DMS by rapid oxidation (see Sect. 3.2). NOx concentrations were estimated based on the observed ozone formation in the chamber experiments.
Matsunaga and Ziemann (2010) described the direct uptake of gas-phase molecules and particulate matter to Teflon surfaces. During long-term usage, the concentration of certain sticky species may be built up on the chamber bag in spite of thorough cleaning. Previous experiments performed in the AURA chamber have used (NH4)2SO4 as seed particles. Therefore, we motivate the presence of a (NH4)2SO4 coating from (NH4)2SO4 seed particles deposited on the chamber walls.
Depending on the pH of the aqueous solution, a fraction of the dissolved will deprotonate and form NH3(aq), which evaporates and forms a continuous source of NH3(g). Different smog chamber studies have examined NPF from ternary nucleation involving SA, water and NH3 (Benson et al., 2011; Kürten et al., 2016), and all demonstrate that NH3 enhances SA nucleation. The evaporation of NH3 from the walls to the gas phase was essential to reproduce the particle number (PN) concentration and PM observed by PSM/WCPC and HR-ToF-AMS measurements, respectively. The increasing LWC on the chamber walls during humid and cold conditions resulted in lower NH3(g) concentrations (Fig. S5), which enables the model to capture the observed lower particle number concentrations during the humid and cold experiment (DMS7) (Fig. S32). Furthermore, a study by Kirkby et al. (2011) performed in the Cosmics Leaving Outdoor Droplets (CLOUD) chamber revealed that trace amounts of NH3 were always present during experiments, even under extremely clean conditions. Consequently, NH3 most likely was present, albeit at low concentration, during all chamber experiments.
2.3 Multiphase chemistry
The multiphase chemistry mechanism was developed based on the DMS gas-phase oxidation scheme in the Master Chemical Mechanism version 3.3.1 (MCMv3.3.1) (Jenkin et al., 1997, 2003, 2015; Saunders et al., 2003), the CAPRAM DMS module 1.0 (DM1.0) (Hoffmann et al., 2016) and a subset of reactions from the multiphase halogen chemistry mechanism CAPRAM Halogen Module 2.0 (HM2.0) (Braeuer et al., 2013). Additional reactions and rate constants were implemented from individual studies (Turnipseed et al., 1995; Kukui et al., 2003; Wu et al., 2014; Cao et al., 2013; Berndt et al., 2019, 2020). A complete list of all reactions can be found in Table S1. In total, the mechanism includes 922 species and 2946 reactions, of which 100 reactions are phase transfer reactions (forward and backward), 2542 are gas-phase reactions and 304 are aqueous-phase reactions. However, the majority of the reactions are only relevant for the atmospheric model simulations (Sect. 3), including 1900 gas-phase reactions which are part of the MCMv3.3.1 isoprene chemistry scheme (Jenkin et al., 2015) (not listed in Table S1), 411 halogen gas-phase reactions, 216 halogen aqueous-phase reactions and 58 halogen phase transfer reactions. The concentrations of H+ and OH− which are involved in many of the aqueous-phase reactions were updated outside the multiphase chemistry mechanism in the ADCHAM thermodynamics module (Roldin et al., 2014) and were not considered to be influenced by the irreversible aqueous-phase chemistry. Hence, [H+] and [OH−] were multiplied directly into the reaction rate expressions in the multiphase chemistry mechanism and are not explicitly included as reactants and products in the reactions listed in Table S1. The multiphase chemistry mechanism was generated with the Kinetic PreProcessor (KPP) (Damian et al., 2002) and solved with the ordinary differential equation solver DLSODE (Hindmarsh, 1983). Figures S22–S24 show examples of the modelled concentrations of the intermediate DMS oxidation products, O3, OH, HO2 and H2O2 in the gas phase and wall aqueous phase for the experiments DMS2, DMS6 and DMS7. The figures also show the calculated pH in the liquid water film.
2.4 New particle formation
The new particle formation was modelled with the Atmospheric Cluster Dynamics Code (ACDC) (McGrath et al., 2012; Olenius et al., 2013) using the same ACDC version and methodology as described in Roldin et al. (2019). With this approach, both neutral and ion-induced nucleation of ammonia–sulfuric acid clusters of a size up to five ammonia and five sulfuric acid molecules are simulated explicitly in ACDC. For this, we used an estimated galactic cosmic ray ionization rate of 1.7 cm−3 s−1. The model considered the losses of clusters by coagulation with the existing aerosol particles and their losses to the chamber walls. The number of clusters that are able to grow to the upper size range in ACDC are defined as new particles in the model and are added to the lowest particle number size bin in ADCHAM. Quantum chemical calculations suggest that MSA may be involved already in the initial molecule cluster formation steps (Rosati et al., 2021b). However, ions have been shown to be very important for the formation of clusters involving sulfuric acid and ammonia, and presently the SA–MSA–NH3 clusters from Rosati et al. (2021b) only consider electrically neutral species and thus were not used in the model simulations performed in this work.
2.5 Atmospheric model runs
To evaluate the updated multiphase DMS chemistry for atmospheric realistic conditions, ADCHAM was set up to reproduce the pristine marine environments of the open ocean. For this purpose an emission estimate of relevant gas-phase and particle-phase species was implemented based on the model scenarios in the work by Braeuer et al. (2013) and Hoffmann et al. (2016). While halogens are of insignificant importance in the AURA smog chamber, they comprise an important oxidant in the ambient atmosphere. Bromine and chlorine released from sea salt aerosols interact strongly with sulfur compounds including DMS (Braeuer et al., 2013) and alter the mechanism presented in Fig. 2. Consequently, the CAPRAM Halogen Module was implemented to address both the gas-phase and aqueous-phase oxidation of DMS by halogen compounds. A base run scenario (named AtmMain) reproduced the movement of an air parcel along a marine environment trajectory for 120 h (Fig. 5). Starting at midnight, the simulation included eight in-cloud periods, four during the day and four at night. ADCHAM clouds formed and evaporated during 75 min adiabatic cooling and warming periods, respectively, in which the RH changed slowly over time. The in-cloud residence time was chosen in accordance with the study by Pruppacher and Jaenicke (1995). The maximum cloud liquid water content was assumed to be 0.3 g m−3. The last cloud period included a rain event, with below-cloud particle scavenging described by the parameterization by Laakso et al. (2003) and gas scavenging described by a parameterization from Simpson et al. (2003). Cloud conditions were introduced to illustrate the effect of multiphase DMS chemistry and varying UV light intensity to reproduce the oxidation capacity of the marine atmosphere during both daytime and night-time conditions. The wet deposition of particles and gases by rain was introduced to spark a NPF event. Between in-cloud periods, the RH was kept at 90 %. Consequently, the aerosols formed were treated as deliquesced particles receptive to aqueous-phase chemistry. This setup was essential to capture the gas-phase concentrations of halogens bromide and chloride mainly formed by halogen activation inside the particles. Sea spray emissions were based on a temperature- and wind-speed-dependent parameterization by Salter et al. (2015). Wind speed was kept fixed at 8 m s−1 in accordance with the global annual average marine wind speed (Kent et al., 2013). Besides the base setup, four sensitivity runs were performed to validate the effect of varying atmospheric conditions. The first (named “PolAtm”) represented a polluted marine environment with high NOx and O3 gas-phase concentrations. The second (named “woCloudAtm”) did not include any in-cloud periods. The third run (named “woAqAtm”) disregarded all aqueous-phase chemistry. The final run (named “lowWindAtm”) kept the wind speed at 2 m s−1.
For the atmospheric model simulations, the particle number size distribution was represented by 200 fixed size bins in the diameter range 1.07 nm to 10 µm.
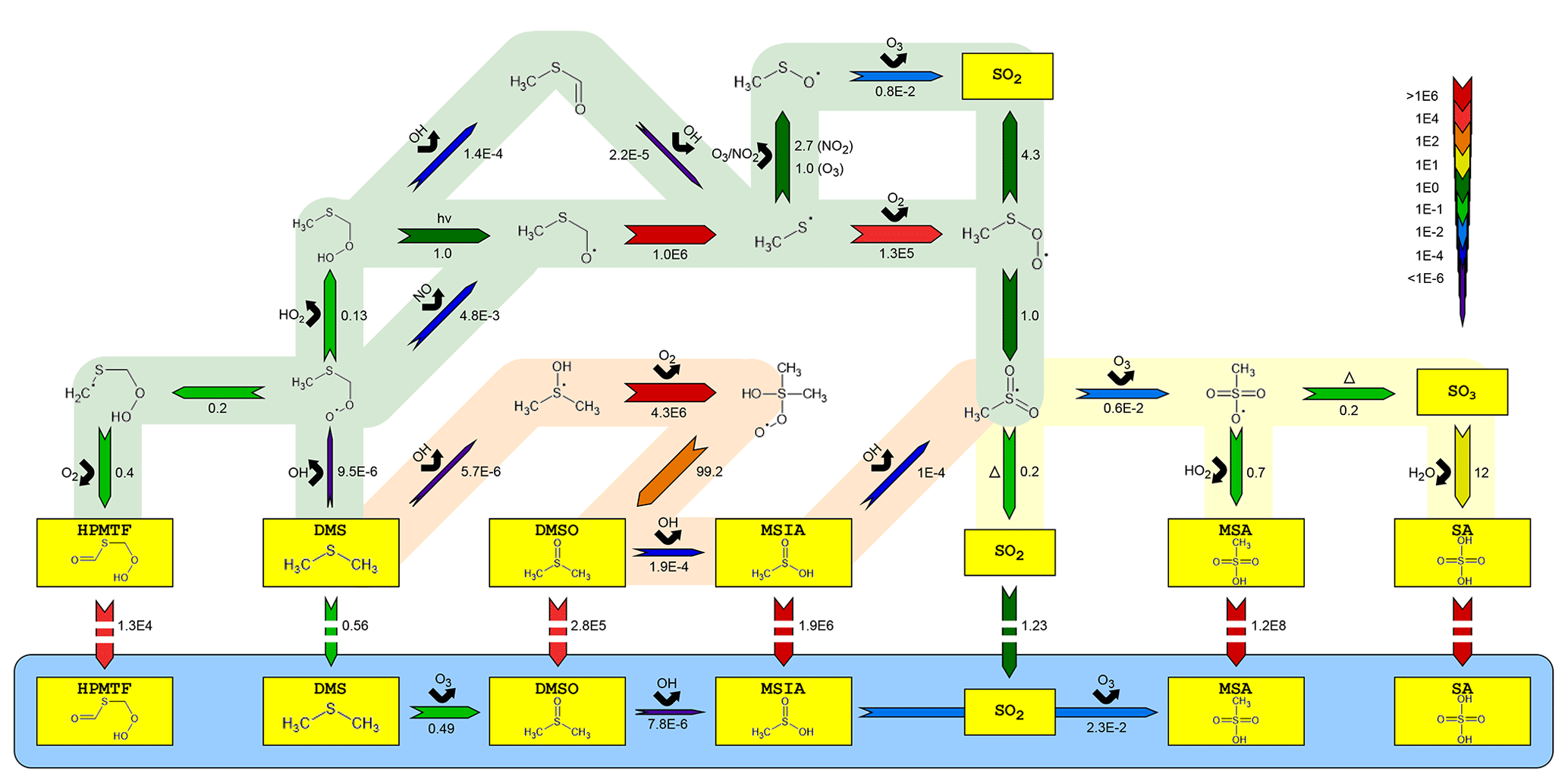
Figure 2Simplified mechanism of the OH-initiated oxidation of DMS. Reaction rates are reported per second s−1 for gas-phase and aqueous-phase chemistry and in units of per atmosphere, mol / (kg atm), for phase transfers (marked with broken arrows). All have been estimated at 293 K, 5 % RH and relevant OH, O3, HO2, NO and NO2 concentrations in the AURA smog chamber. The blue area denotes the aqueous-phase oxidation of important water-soluble intermediates and products (marked in yellow). Red and green shaded areas represent the OH addition and H abstraction pathways, respectively, while the yellow area illustrates the remaining mechanism after the two previous pathways intercept. The number beneath each arrow denotes the rate of the said reaction and the chemical species above its reactant.
3.1 Gas-phase DMS oxidation
The presented gas-phase mechanism has predominantly been based on the MCMv3.3.1 reaction scheme, although several pathways were added or modified to explain the observations in the AURA chamber. Figure 2 presents an outline of the most important multiphase DMS chemistry. Utilizing the MCMv3.3.1 chemistry mechanism alone caused the model to slightly overestimate the sink flux of DMS by OH addition compared to proton-transfer-reaction mass spectrometry (PTR-MS) measurement made in the chamber (R2=0.92 between the measured and modelled DMS concentration in experiment DMS2). At the same time, MSA PM concentrations were significantly underestimated in proportion to those measured by the HR-ToF-AMS (49.5 % on average in experiment DMS2). Therefore, efforts were made to reduce the rate of DMS oxidation in the addition pathway while promoting MSA production, by revising the MCMv3.3.1 mechanism. Analogous to the DM1.0 mechanism (Hoffmann et al., 2016), the initial addition of OH to DMS was modified by implementing the DMS–OH adduct as an intermediate product, either decomposing back to DMS (Lucas and Prinn, 2002), reacting with O2 to form a stabilized peroxyl radical (RO2), or fragmenting into CH3SOH. Most important was the decomposition back to DMS (not considered in MCM) that lowered the initial rate of DMS oxidation to improve the fit between the measured and modelled DMS concentration (R2=0.95, experiment DMS2).
MSA, often believed to be formed solely in the abstraction pathway (MCMv3.3.1), is produced almost exclusively from the reaction of CH3SO3 with HO2. However, this reaction and the pathway leading to it did not explain the yields obtained in the chamber experiments. Therefore, an alternative pathway considering oxidation of the stable intermediate MSIA by OH was implemented to increase SA and MSA production. The importance of MSIA oxidation by OH was discussed by von Glasow and Crutzen (2004) and mentioned in several other studies (Yin et al., 1990; Lucas and Prinn, 2002; Kukui et al., 2003; Ghahremaninezhad et al., 2019). While MCM assumes unity production of SO2, there are strong indications that intermediate CH3SO2 in the abstraction pathway comprises the main product. As a consequence, the MSIA+OH reaction creates a link between the addition and abstraction pathway. The increased production of CH3SO2 promotes the formation of SA and MSA in the gas phase along with PM yields in the smog chamber experiments (82.8 % increase in the total PM during experiment DMS2). It should be noted that during high RH chamber conditions (or cloudy MBL in the atmosphere), the MSIA+OH reaction becomes less important, since MSIA and its precursor DMSO partition almost exclusively to the aqueous phase (Campolongo et al., 1999; Barnes et al., 2006).
In the abstraction pathway, the large increase in MSA and SA production by the MSIA+OH pathway was counterbalanced to match observations by implementing a newly discovered DMS autoxidation pathway (Veres et al., 2020). The pathway proceeds via the CH3SCH2OO radical (the main product of the initial DMS+OH reaction in the abstraction pathway) and undergoes two unimolecular hydrogen shifts (H shifts) and O2 addition to form the stable intermediate product HPMTF. The mechanism was propounded in theory by Wu et al. (2014) and confirmed experimentally by Berndt et al. (2019). Wu et al. (2014) estimated an upper limit of 2.48 s−1 at 295 K for the rate-limiting H shift in CH3SCH2OO, considerably larger than the laboratory study by Berndt et al. (2019) (0.23 s−1) and the multiconformer transition state theory (MC-TST) calculation by Veres et al. (2020) (0.047 s−1). While Veres et al. (2020) argue that the slow rate coincides with autoxidation rates of similar peroxides (Crounse et al., 2013), a combination of the intermediate rate proposed by Berndt et al. (2019) and temperature dependence suggested by Veres et al. (2020) offered a good agreement with observations from the AURA chamber experiments.
SO2 comprised the main product in the abstraction pathway, formed mainly through the thermal decomposition of CH3SO2. While SO2 is often mentioned as the most important source of SA (Barnes et al., 2006), the slow oxidation by OH and uptake to the aqueous phase during high RH chamber conditions suggested that its contribution was less important than other sources. SO2 comprised 30.8 % of the SA source flux during the first 5 h of the humid experiment DMS6, while SO3 formed by thermal decomposition of CH3SO3 made up 69.2 %. In pristine marine atmospheric conditions (see Sect. 3.3.4), SO2 comprised 0.8 % of the SA source flux in the gas phase throughout the entire simulation. SO3 reacts rapidly with water to form SA and is not affected by multiphase DMS chemistry (Hoffmann et al., 2016). Consequently, SO3 from thermal decomposition of CH3SO3 may be the main driver of NPF from DMS oxidation in the chamber experiments and possibly the ambient marine atmosphere. A similar theory was presented in a field study by Berresheim et al. (2014), in which the authors suggested that the said reaction may be able to explain the gap between modelled and measured SA concentrations. Although very fast thermal decay rates for CH3SO3 have been proposed in literature (Cao et al., 2013), rates close to the value given in MCM provide the best match between the observed and modelled SA and MSA PM. Since SO2 has little effect on the gas-phase SA production in the chamber experiments, the fate of CH3SO3 may be the factor controlling the MSA/SA formation, ratio and temperature dependence. The critical factor concerns the branching between thermal decay and reaction with HO2.
Isomerization of CH3S(OO) to CH3SO2 was added to the MCM mechanism to increase the rate of SA production and hence the onset of NPF (previously too slow compared to observations in the chamber). CH3SO2 is the main precursor of CH3SO3. Consequently, the rate of CH3S(OO) isomerization and CH3SO2 production affects the formation of both SA and MSA (11.6 % and 15.8 % increase in SA and MSA PM, respectively, during experiment DMS6). Turnipseed et al. (1995) reported an upper limit for the said reaction of 20–25 s−1, while other studies (Campolongo et al., 1999; Lucas and Prinn, 2002; Hoffmann et al., 2016) found the isomerization rate to be substantially slower (1 s−1). The latter was sufficient to match experimental results.
3.2 Aqueous-phase DMS oxidation
During high RH conditions in the chamber experiments or cloud cover in the MBL, water affects the gas-phase oxidation of DMS (Hoffmann et al., 2016). In the atmosphere the aqueous-phase chemistry proceeds in cloud droplets and deliquesced particles (Seinfeld and Pandis, 2016). In the chamber the water adsorbs to the Teflon bag and forms a thin liquid film (see Sect. 2.2.1). In either case the presence of water can significantly lower the gas-phase concentration of oxidants H2O2, OH, O3, HO2 and water-soluble intermediates and products in the DMS oxidation mechanism. These include DMS, DMSO, MSIA, MSA, SA and SO2 (Campolongo et al., 1999) (Figs. S22–S24). The dominant pathway in the multiphase DMS oxidation mechanism is initiated by the oxidation of DMS by O3. Although DMS has a small Henry law solubility, the constant turnover by O3 ensures a steady flux of DMS from the gas phase to the aqueous phase. The mechanism proceeds via the DMSO and MSIA intermediates with MSA as the main product (von Glasow and Crutzen, 2004; Hoffmann et al., 2016). Henry's law solubility for DMS was based on a study by Campolongo et al. (1999), while COSMOtherm (COSMOtherm, 2019) calculations (see Sect. S2.3) were used to quantify Henry's law solubility and acid dissociation (pKa) of DMSO, DMSO2, MSIA, MSA and hydrated and non-hydrated HPMTF. Henry's law solubility of hydrated HPMTF was found to be comparable to that of MSA, while that of non-hydrated HPMTF proved to be similar to DMSO. Although Veres et al. (2020) suggested that HPMTF may contribute directly to the growth of aerosol particles, the increase in PM from hydrated HPMTF did not coincide with observations in the chamber experiments. Consequently, HPMTF likely played no (or an insignificant) direct part in the particle growth but remained in the gas phase as a sulfur reservoir, reducing the overall secondary aerosol yield from DMS oxidation (55.8 % decrease in the total PM during experiment DMS6). In atmospheric relevant conditions HPMTF may be oxidized in cloud droplets analogous to compounds with similar functional groups (Doussin and Monod, 2013). While this reaction pathway has not been validated in the literature, we propose a mechanism that incorporates the aqueous-phase OH-initiated oxidation of HPMTF to HOOCH2SCO. The subsequent transfer of HOOCH2SCO to the gas phase strongly increases the HPMTF-derived production of SO2 and thus .
3.3 Model simulations
Yin et al. (1990)Yin et al. (1990)Veres et al. (2020)Veres et al. (2020)Yin et al. (1990)Yin et al. (1990)Lucas and Prinn (2002)Lucas and Prinn (2002)Table 3Overview of conditions used in the model base runs (rows in bold) and sensitivity runs (rows in roman) in both chamber and atmospheric relevant simulations.

Table 4Summary of modelled DMS conversion yields to HPMTF, SO2, MSA and S(IV), total PM yields and PM ratios in humid, dry and cold chamber plus atmospheric relevant conditions. Rows in bold denote base runs, while rows in roman denote sensitivity runs.
Conversion yields, PM yields and ratios in atmospheric relevant simulations obtained before the precipitation event at max PM. PM yields in atmospheric relevant simulations exclude Cl− and Na+ from sea salt aerosols. PM yields in smog chamber simulations obtained at maximum SMPS PM concentration.
The ADCHAM model was constrained to conditions specific for the experiments performed in the AURA smog chamber. Figures S26–S32 compare the modelled and measured gas and particle concentrations for all AURA DMS experiments listed in Tables 1–2. Different sensitivity runs were performed for three representative experiments (DMS2, DMS6 and DMS7) to highlight the effects of our revised DMS multiphase chemistry mechanism, compared to previous studies. An additional run examined the implemented mechanism in atmospheric relevant conditions. Table 3 provides an outline of each simulation, while results are given in Table 4.
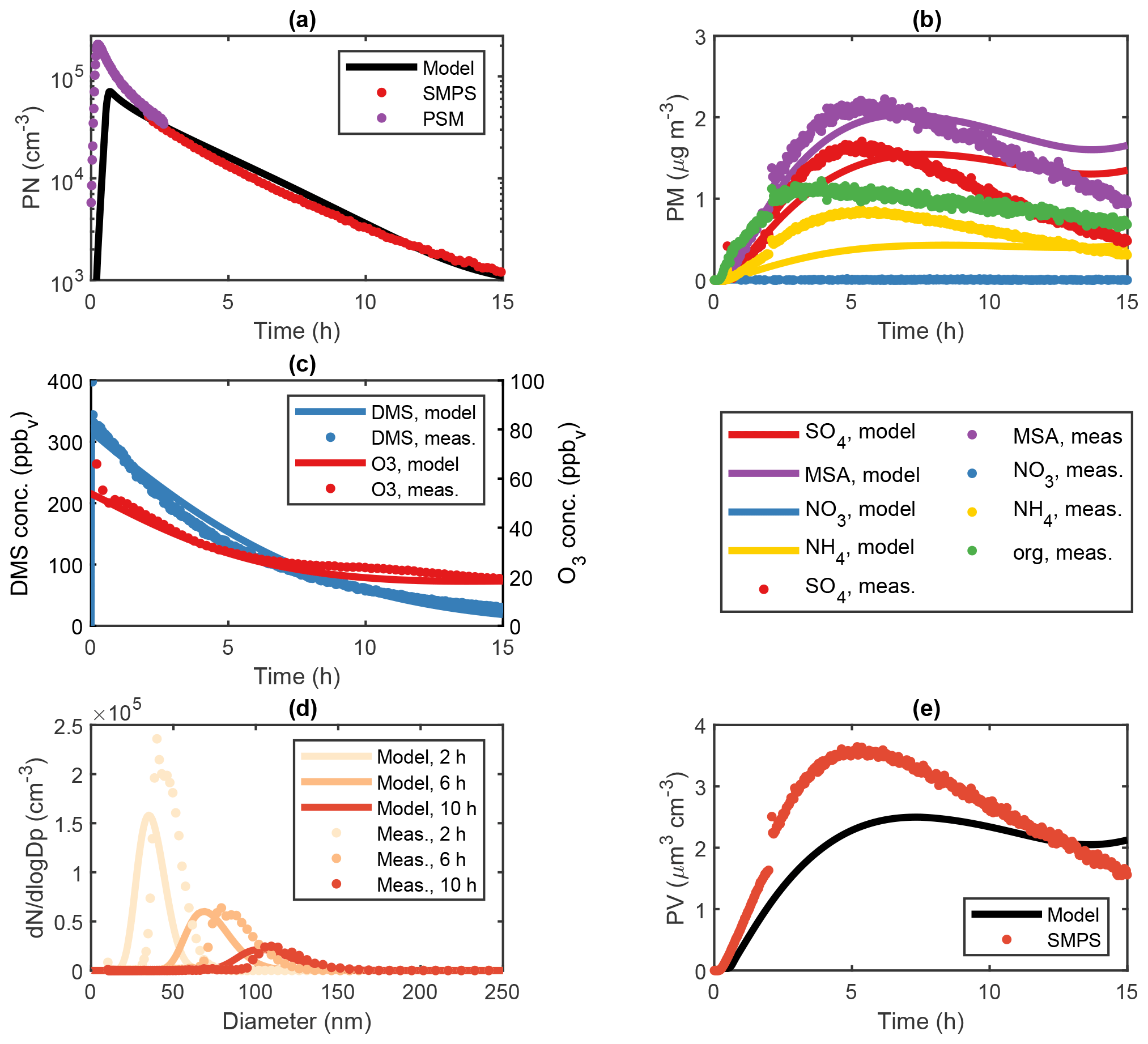
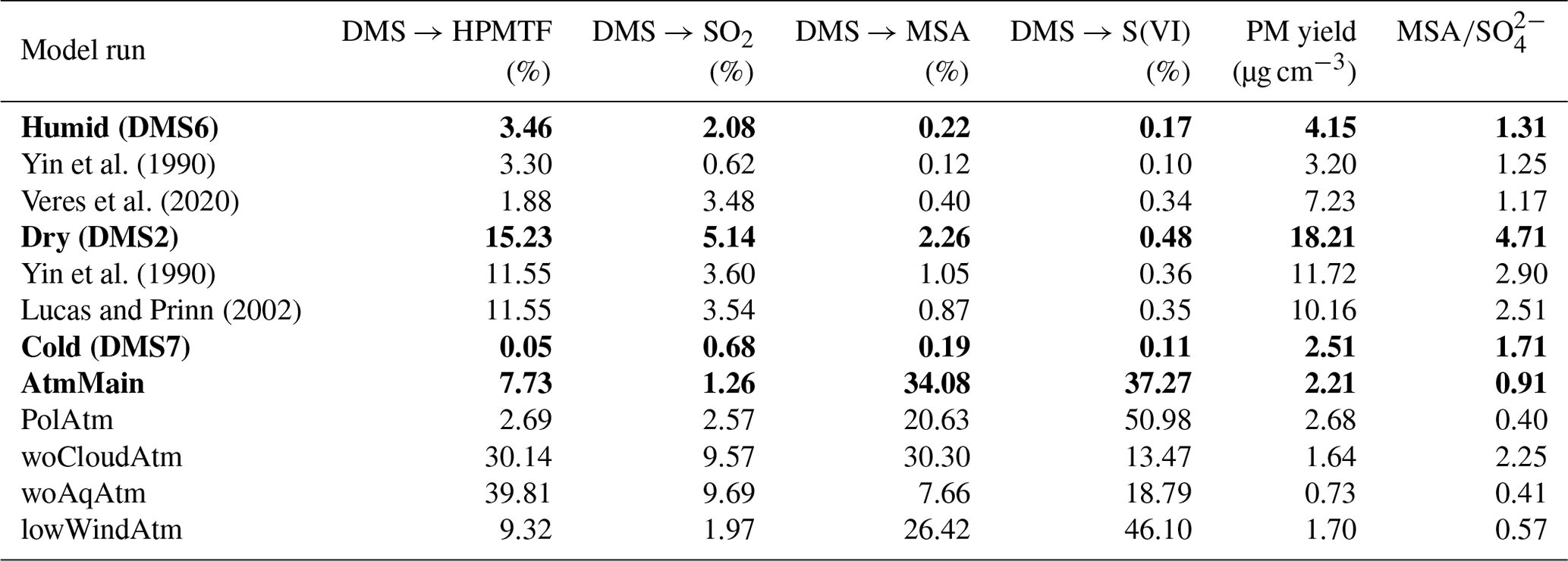
Figure 3Modelled and measured results from an OH-initiated DMS oxidation experiment (exp. DMS6) performed at 293 K and 70 % RH in the AURA smog chamber. Panel (a) illustrates PN concentrations at 1.7 nm cut-off obtained from PSM and SMPS measurements, (b) the HR-ToF-AMS PM composition, (c) the DMS and O3 gas-phase concentration decay, (d) the SMPS number size distribution and (e) the SMPS PV concentration.
3.3.1 Humid chamber
ADCHAM reproduced the MSA (R2=0.81), (R2=0.49) and (R2=0.46) PM from HR-ToF-AMS measurements along with DMS (R2=0.93) and O3 (R2=0.96) concentrations in the AURA smog chamber at 293 K and high RH conditions (Fig. 3b, c). In this context, it should be noted that the HR-ToF-AMS PM concentration was corrected using the SMPS particle volume concentration (PV) (Fig. 3e) and HR-ToF-AMS aerosol density (Rosati et al., 2021b) analogous to the method by Bahreini et al. (2009) to account for the uncertainties in the HR-ToF-AMS measurements. The model also captured trends in the PN concentration from PSM and SMPS measurements reasonably well (R2=0.32 for the combined PSM and SMPS measurements and R2 = 0.99 for SMPS measurements alone) (Fig. 3a, d). We motivate the PN concentration underestimate in the first 2 h of the experiment by the presence of organic contamination (Fig. 3b). A similar effect is seen in the modelled PV (Fig. 3e), which likewise does not consider the influence of organics in the aerosol particle formation. A water concentration of 15 g m−3 corresponding to a ∼ 5 µm water film was implemented based on model sensitivity runs (Fig. S3). The optimal LWC found is within the same order of magnitude as the value predicted by the butanol experiment (see Sect. 2.2.1). Consequently, H2O2 partitioned strongly to the aqueous phase, reducing gas-phase concentration of HO2 by 78.0 % compared to the dry experiments (Figs. S22–S23). With the reduction in HO2, MSA production from the CH3SO3+HO2 reaction decreased correspondingly – a change from 4.06×106 to 1.04×106 cm−3 s−1 in the average MSA source flux. The reduced conversion from CH3SO3 to MSA favoured the decomposition of CH3SO3 to SO3 and lowered the ratio from 4.71 to 1.31 compared to experiments performed in dry conditions (Table 4). The overall mass yield was strongly influenced by the uptake of DMSO and MSIA to the water film. Since the oxidation of MSIA by OH was implemented as an alternative source of CH3SO2 (an important precursor of SA and MSA) in the gas-phase mechanism (Kukui et al., 2003), SA and MSA production and concentrations in the gas phase were reduced in accordance with the DMSO and MSIA dissolution in the wall aqueous film (Figs. S22–S23). Thus, the presence of a water film on the chamber bag surface strongly altered the DMS oxidation product ratio and total PM yield (91.7 % total PM decrease in experiment DMS6).
An organic PM signal was observed consistently in all chamber experiments (Fig. 3b). The effect could not be replicated by considering condensation of DMSO and MSIA onto pre-existing particles and may originate from organic wall contaminants, inflow of air from outside the chamber caused by the continuous instrument sampling or from deposits and related memory effects in the AMS instrument, as explained in Rosati et al. (2021b). Considering the proportion between organic, MSA and PM, we cannot exclude that such contamination may have affected both particle formation and growth. ADCHAM reproduced the PM (Fig. 3b) by considering an influx of NH3 from (NH4)2SO4 wall contamination (see Sect. 2.2.3). High amounts of water ensured a more dilute solution, which allowed more NH3 to be dissolved in the aqueous phase on the chamber walls, resulting in lower NH3(g) concentration in the chamber (Fig. S5).
PTR-MS measurements indicated a strong DMS oxidation in the first few hours of the experiment (Fig. 3c). While OH-initiated gas-phase oxidation alone could not explain the observed trend, the aqueous-phase oxidation by O3 improved the fit (Fig. S8). DMS has a small Henry law solubility, and the majority resides in the gas phase (Campolongo et al., 1999). However, the O3 present in the water film ensured a steady conversion of DMS to DMSO and hence a flux of DMS between the gas phase and aqueous phase. The uptake of O3 to the water film was apparent from the decrease in O3 concentration observed in the experiment (Fig. 3c). In dry conditions, O3 was found to increase gradually from NOx contamination (see Sect. 2.2.3). These results strongly advocate the presence of a chamber wall water film. Despite the aqueous-phase uptake, O3 remained abundant in the gas phase, thus favouring the CH3SO+O3 reaction and promoting SO2 production. The reduced importance of SO2 in gas-phase SA formation worked to lower the overall SA and MSA production, since SO2 was taken up by the water film. Consequently, the gas-phase O3 abundance decreased SA production and hence NPF.
MSIA oxidation by OH was essential to capture the observed onset in NPF from PSM and SMPS measurements (Fig. 3a). The reaction increases the rate of CH2SO2, CH3SO3 and hence SA production and caused a 41.6 % increase in the maximum gas-phase SA concentration during experiment DMS6. The effect was also achieved by implementing the isomerization of CH3SOO to form CH3SO2. This is an important result, since neither of the said reactions are implemented in MCM. The strong uptake of H2O2 to the water film (96.5 % decrease in the gas phase, concentrating during experiment DMS6) likewise increased SA production and NPF by lowering HO2 concentrations in the gas phase, which led to a 82.9 % decrease in the rate of the CH3SO3+HO2 reaction and promoted SO3 production from the thermal decomposition of CH3SO3. Consequently, SA production and hence NPF occurred faster during high RH conditions, reaching a PN concentration of 1000 molec. cm−3 at a 1.7 nm cut-off diameter 11 min earlier in experiment DMS6 compared to experiment DMS2. The total PN concentrations remained low due to the uptake of DMSO and MSIA to the water film.
The autoxidation of the CH3SCH2OO radical leading to the formation of HPMTF exerted a strong influence on the PM yield in the humid chamber experiments, lowering the total PM by 55.8 % in experiment DMS6. The effect was evident when implementing the rate-limiting H-shift reaction constant as suggested by Yin et al. (1990) and Veres et al. (2020), respectively (Table 4), as opposed to the rate proposed by Berndt et al. (2019). PM yields decreased by 22.9 % in accordance with the study by Yin et al. (1990) and increased by 74.2 % in accordance with the study by Veres et al. (2020). The difference coincides with the production and accumulation of HPMTF in the gas phase. In consequence of the high sensitivity of the reaction rate to the model outcome, more detailed studies of the HPMTF autoxidation pathway are needed to overcome the uncertainty related to the current mechanism.
Overall, modelling the OH-initiated DMS oxidation at high RH strongly indicated the presence of a water film on the Teflon bag. This crucial finding comprises an important consideration when performing smog chamber experiments and could help to explain the complex interaction between gaseous species and chamber walls. The effect, however, remains uncertain (Svensson et al., 1987; Sumner et al., 2004) and requires further investigation to be validated.
3.3.2 Dry chamber
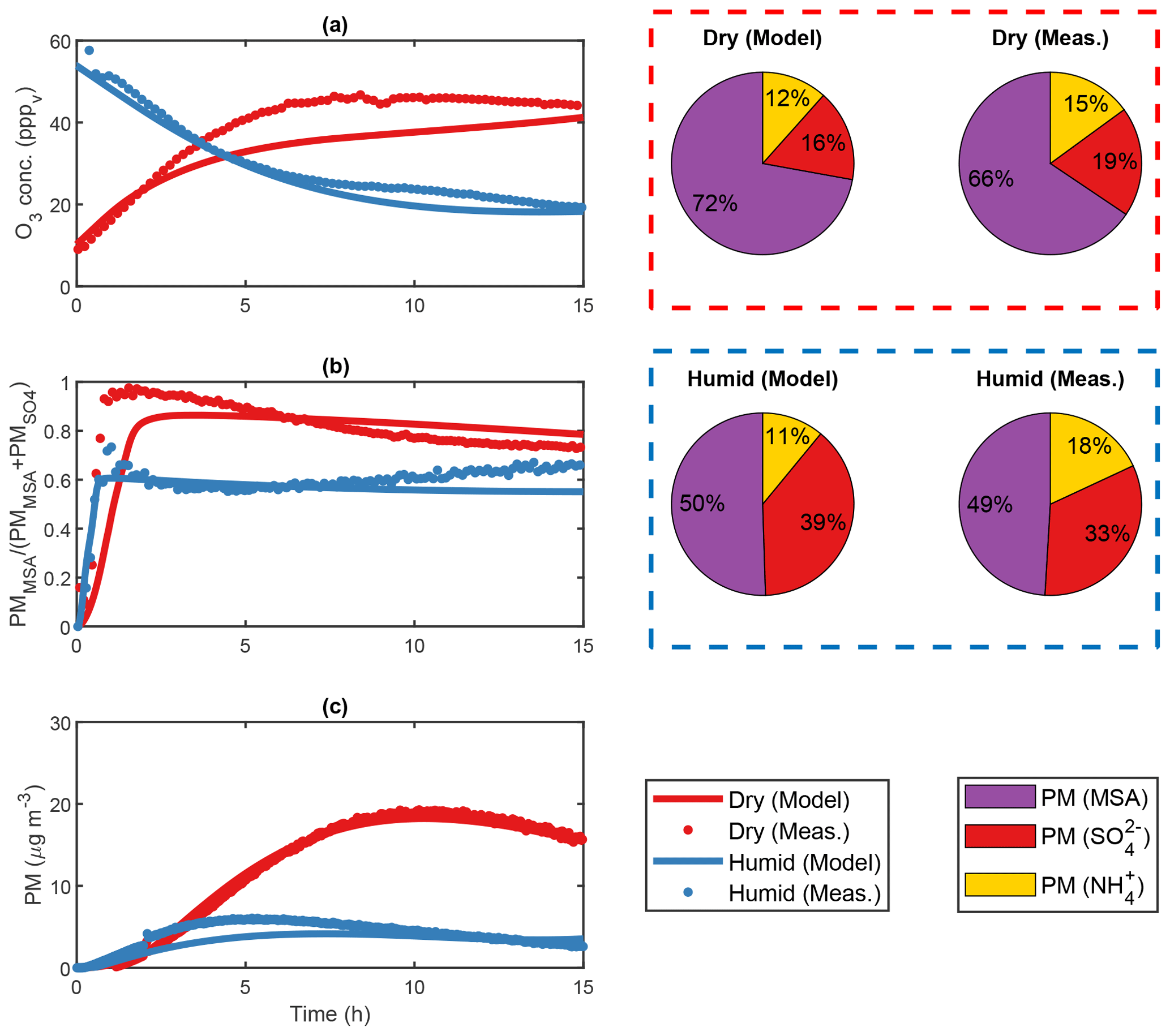
Figure 4Modelled and measured results from two separate DMS experiments (exp. DMS2 and DMS6) performed in the AURA smog chamber at dry (red) and humid (blue) conditions, respectively. Panel (a) shows the PTR-MS progression in O3 concentration, (b) the HR-ToF-AMS MSA to ratio and (c) the SMPS total secondary aerosol mass concentration. The HR-ToF-AMS chemical composition is illustrated by pie charts.
ADCHAM captures the difference in secondary aerosol PM concentration between dry and humid experiments performed in the AURA smog chamber (Fig. 4c). The water film concentration was kept low at 3 mg m−3 in accordance with the study by Svensson et al. (1987) and had less effect on the DMS oxidation compared to the humid experiments (64.8 % and 91.7 % decrease in the total PM yield obtained in dry and humid conditions, respectively). Consequently, almost all DMSO and a large fraction of MSIA remained in the gas phase (Figs. S17–S18, S22) and increased the total MSA and SA PM. The limited uptake of MSIA to the water film made the MSIA+OH reaction essential to reproduce the MSA PM production. By contrast, the said reaction had less impact on the overall PM yield in high RH conditions since MSIA dissolves and reacts almost exclusively in the aqueous phase. While different rates have been reported for the oxidation of MSIA by OH in literature, cm3 molec.−1 (Lucas and Prinn, 2002) and cm3 molec.−1 (Yin et al., 1990), the increased rate of cm3 molec.−1 proposed by Kukui et al. (2003) offered a good agreement with the observed PM concentration (R2 = 0.99) (see Fig. S33). This result entails that the main pathway leading to gas-phase MSA production in low RH chamber or cloud-free atmospheric conditions proceeds not by the abstraction pathway as assumed in previous studies but via the addition pathway. A similar conclusion was drawn in a study by von Glasow and Crutzen (2004). The effect became more profound when incorporating the production of HPMTF by autoxidation (Veres et al., 2020). In this case, the rate proposed by Berndt et al. (2019) ensured a strong decrease in SA and MSA production formed via the abstraction pathway (36.0 % PM decrease during experiment DMS2). Consequently, the OH-initiated addition to DMS and subsequent oxidation pathway proved to be the main source of secondary aerosol PM in the AURA chamber at low RH conditions. The effect of the MSIA+OH reaction rate on the PM yield is evident from the results in Table 4. The reaction rate proposed by Yin et al. (1990) and Lucas and Prinn (2002) decreased the PM yield by 35.6 % and 44.2 %, respectively, as opposed to the rate suggested by Kukui et al. (2003). A better understanding of this reaction is needed to improve model predictions of the production of MSA and SA in the gas phase.
The H2O2 (and hence HO2) partitioned readily to the aqueous phase in the humid experiments but not in the dry ones. The difference is evident from the PM ratio of 4.71 and 1.31 observed in dry and humid experiments, respectively (Fig. 4b). In the dry experiments, the high HO2 gas-phase concentration favoured the CH3SO3+HO2 reaction and therefore MSA as opposed to SA production. Thus, MSA dominated the observed PM mass in dry conditions. In the humid experiments, the substantially lower H2O2 and hence HO2 gas-phase concentration limited the CH3SO3+HO2 reaction and favoured the decomposition of CH3SO3 to SO3 and hence SA. Consequently, PM production matched that of MSA in humid conditions. The initial O3 concentration appeared low during dry conditions but increased consistently as each experiment progressed (Fig. 4a). This response incites a constant influx of NOx which may arise from a HONO wall pool (see Sect. 2.2.3) and NO2 contamination from air leaking in from outside the Teflon bag. The decrease in the gas-phase O3 concentration observed in humid conditions was associated with the uptake of O3 to the Teflon bag water film. Our model results indicate that such uptake was of no or insignificant importance during dry conditions (Fig. S7).
3.3.3 Cold chamber
At 273 K, the rate of OH addition surpassed that of H abstraction in the initial step of DMS oxidation. Production rates of DMSO and MSIA increased accordingly, but gas-phase concentrations remained low as both species partitioned strongly to the water film (94.0 % and 99.7 % decrease in DMSO and MSIA gas-phase concentrations, respectively, during experiment DMS7) (Fig. S24). The autoxidation pathway leading to HPMTF formation proved less significant at 273 K due to the temperature dependence on the rate-determining H shift in the CH3SCH2OO radical intermediate (77.2 % decrease in rate compared to 293 K). Thus, the strong decrease in secondary aerosol formation from the increased formation and uptake of DMSO and MSIA was counterbalanced by the decrease in HPMTF production. The rate of SO3 formation from thermal decomposition of CH3SO3 decreased by 91.7 % in accordance with the decrease in chamber temperature. This response induced a substantial increase in MSA gas-phase and PM production that did not reflect observations in the AURA chamber – Table 4 illustrates how the ratio remains independent of changes in temperature (1.31 and 1.71 in experiment DMS6 and DMS7, respectively). To solve the gap between model and measurements, the water film concentration was increased from 15 g m−3 to 500 g m−3 (Fig. S4). Consequently, the gas-phase concentration of H2O2 and hence HO2 decreased along with the CH3SO3+HO2 reaction rate, thereby reducing MSA formation while promoting SO3 and thus SA production. Sumner et al. (2004) applied Brunauer–Emmett–Teller (BET) theory to describe the adsorption of water on a Teflon bag surface and argued that a decrease in temperature would enhance the uptake of water. Furthermore, RH calculations based on temperature measurements outside the chamber bag suggest that supersaturation may have occurred along the bag surface due to strong outside cooling. These findings, together with the observed slow 1-butanol decay during similar cold humid conditions (Fig. S1), advocate the increase in water film concentration needed to reproduce PM observations (Fig. S32).
The uptake of gaseous species to the aqueous phase increased in accordance with the water film concentration and Henry's law theory (Fig. S24). Most important was the decrease in the gas-phase NH3 that worked to suppress the onset and total concentration of NPF (Fig. S5).
3.3.4 Atmospheric implication
Figure 5Modelled DMS oxidation and subsequent PM production in pristine marine environment conditions. Panel (a) illustrates the evolution of DMS, SA, MSA, HPMTF, MSIA and DMSO gas-phase concentrations, (b) the sink fluxes of DMS due to Cl, OH, BrO, NO3 and O3 and (c) the number size distribution and secondary aerosol PM production. Light blue areas denote in-cloud period, in which rain events are represented as dark blue. Night-time and daytime periods are represented by the normalized UV intensity and marked by grey and white areas, respectively.
DMS decay dominates during the day when the UV light intensity and thus the gas-phase concentration of oxidant species OH, Cl and BrO is high (Fig. 5b). BrO- and Cl-initiated oxidation of DMS accounted for 38.7 % and 24.3 % of the total sink flux over the entire simulation, respectively, thus highlighting the importance of halogen chemistry in the DMS oxidation mechanism. In this context, it is important to note that the presence of sea spray aerosols and consequently halogen compounds is highly dependent on wind speed. While Cl and BrO radicals comprise the main sink flux of DMS at 8 m s−1, their influence decrease substantially at 2 m s−1 (21.8 % and 13.2 % mean DMS sink flux in lowWindAtm, respectively). This effect is apparent from the lowWindAtm sensitivity run, in which OH dominates the oxidation of DMS at 55.5 % mean DMS sink flux (see Fig. S37). Furthermore, other studies (Kristensen et al., 2016) regard sea salt particles as unimportant to the submicron aerosol number concentrations in the marine atmosphere. This statement stands in stark contrast to the sea spray parameterization by Salter et al. (2015) utilized in this work. Therefore, it is plausible that the emission of the sea salt aerosols and thus the halogen radical concentration may be overestimated in the model.
Halogen activation inside the sea salt aerosols comprised the main source of both Br and Cl in the gas phase. Analogous to the work by Braeuer et al. (2013), chloride ions were activated by the iodine species HOI in the deliquesced sea-salt particles, forming ICl capable of transferring to the gas phase. BrCl formed by bromide activation also partitioned to the gas phase. Both species photodissociated proportional to the UV light intensity, causing Br and Cl concentrations to peak at midday in non-cloud conditions. Br and Cl gas-phase radicals reacted strongly with ozone to form BrO and ClO. While ClO had little effect on the DMS decay, BrO comprised the main oxidant and dominated the sink flux of DMS (Fig. 5b). During in-cloud periods, the halogen activation terminated, and both bromide and chloride stayed in the aqueous phase. Consequently, neither BrCl nor ICl was released to the gas phase, thus decreasing the sink flux of DMS by halogen species (16.9 % and 9.9 % mean DMS sink flux by Cl and BrO during cloud periods).
The net effect of the OH-initiated oxidation of DMS accounted for 31.1 % of the total sink flux over the entire simulation. Although the oxidation capacity of OH differed from the capacities of halogens BrO and Cl, it still comprised a considerable part in the decay of DMS in atmospheric relevant conditions. During in-cloud periods, the phase transfer of OH and its precursor O3 decreased the gas-phase oxidation of DMS. Despite the uptake a significant amount remained in the gas phase. As a result, the sink flux of DMS by OH radicals exceeded that of halogen species during in-cloud periods. Besides OH, the aqueous-phase oxidation of DMS by O3 proved to be the main sink of DMS, at 32.0 % during in-cloud periods. The importance of the said reaction increased as the UV light intensity decreased, and hence the Cl, BrO and OH gas-phase concentrations decreased. Consequently, the night-time in-cloud oxidation of DMS was solely due to aqueous-phase O3 (4.1 % of the total sink flux over the entire simulation). During nighttime non-cloud periods, the NO3 radical proved to be a small but stand-alone sink of DMS, comprising 1.8 % of the total DMS sink flux throughout the entire simulation.
The main gas-phase products comprised HPMTF (7.7×108 molec. cm−3), DMSO (6.4×108 molec. cm−3), MSIA (1.1×108 molec. cm−3) and SO2 (1.0×108 molec. cm−3), whereas MSA (3.2×106 molec. cm−3) and SA (2.0×106 molec. cm−3) were present only in small quantities (Fig. 5a). HPMTF, DMSO and SO2 accumulated in the gas phase between cloud periods, while MSIA, SA and MSA condensed onto the pre-existing sea salt aerosols. The large surface area ensured a strong condensation sink and prevented NPF from SA and NH3 nucleation. SA in the gas phase formed predominantly via the SO3 intermediate from the thermal decomposition of CH3SO3, comprising 99.2 % of the SA source flux in AtmMain. The oxidation of SO2 by OH proved to be a small but insignificant source of SA (0.8 %). Consequently, NPF involving SA from marine DMS emissions may solely stem from SA production via the CH3SO3 intermediate. This finding is consistent with the study by Hoffmann et al. (2016). Although essential for the formation of new particles, SA produced in the gas phase proved insignificant to the overall formation of PM. During in-cloud periods, SO2 partitioned strongly to the aqueous phase. The subsequent oxidation by O3 and H2O2 comprised the main source of PM throughout the simulation. MSA in the gas phase formed almost exclusively via the CH3SO3 intermediate. However, the strong uptake of MSIA to the particles made the system less sensitive to the MSIA+OH reaction. As a results, the gas-phase MSA production proved insignificant to the total MSA PM concentration. Instead, MSA was almost exclusively formed via the aqueous-phase oxidation by O3 in both the deliquesced particles and cloud droplets between and during in-cloud periods, respectively (see Fig. S38). The uptake of MSIA to the aerosol particles, and thus the importance of the MSIA+OH reaction, does, however, depend strongly on Henry's law solubility of MSIA, which is highly uncertain. Henry's law solubility utilized in this work was based on COSMOtherm calculations and was found to be lower than that suggested in previous studies (Barnes et al., 2006; Hoffmann et al., 2016) by more than 2 orders of magnitude. In this context, it is important to note that Henry's law solubility values of MSIA utilized by Barnes et al. (2006) and Hoffmann et al. (2016) were assumptions based on Henry's law solubility of DMSO and MSA. The extent of the aqueous-phase MSA production increased in accordance with the uptake of MSIA to the particle phase. This finding contradicts the work by von Glasow and Crutzen (2004), in which the gas-phase production of MSA comprised more than half of the total MSA yield. The difference was unexpected, since von Glasow and Crutzen (2004) implemented the MSIA+OH reaction and rate based on the experimental work by Kukui et al. (2003) also utilized in this paper. The effect was explained by the aqueous-phase oxidation of MSIA by O3, not considered in the study by von Glasow and Crutzen (2004). Without this reaction, MSA in the aqueous phase formed primarily by OH oxidation and thus solely during in-cloud periods when the aqueous OH concentration was high.
DMSO resided in the gas phase between cloud periods but partitioned strongly to the aqueous phase when clouds were present (80.8 % decrease in gas-phase DMSO going into the first cloud period in AtmMain). OH radicals facilitated the conversion of DMSO to MSIA in the cloud droplets during the day. At night, the OH oxidation terminated, and DMSO was released back to the gas phase in accordance with the cloud evaporation. The uptake of DMSO and subsequent oxidation to MSIA and thus MSA increased the MSA production during cloud periods (3.0×104 and 1.2×104 cm−3 s−1 MSA source flux during and between cloud periods, respectively) (see Fig. S38).
Due to its high stability, HPMTF accumulated in the gas phase during clear conditions, an insignificant fraction being oxidized by OH. Analogous to the field measurements by Veres et al. (2020), HPMTF was taken up by aqueous cloud particles during in-cloud periods. Veres et al. (2020) claimed that HPMTF may likely contribute directly to particle formation and growth but failed to provide adequate proof for this finding. COSMOtherm calculations quantifying Henry's law solubility of HPMTF render its contribution to particle growth very insignificant, and neither ADCHAM nor the AURA chamber experiments suggest that HPMTF resides in the aerosol phase. Furthermore, HPMTF is unlikely to contribute to NPF. This is caused by the fact that the functional groups in the HPMTF molecule are a carbonyl group and a hydroperoxide group. Based on COSMOtherm calculations (Kurtén et al., 2016), it has been well established that highly oxygenated organic molecules (HOMs) consisting of multiple carbonyl, hydroperoxy and peroxy acid functional groups may not have low vapour pressures, despite their high O:C ratios, especially if the HOM contains only few H-bond donors (Hyttinen et al., 2021). Furthermore, quantum chemical calculations have shown that in order for a given organic highly oxidized organic molecule to be involved in sulfuric-acid-based new particle formation, it must contain several strong binding moieties such as carboxylic acid groups (Elm et al., 2017). The importance of HPMTF in new particle formation is further diminished by the fact that the monomer can be stabilized by an intramolecular hydrogen bond between the –OOH and –C=O groups. This implies that the intramolecular hydrogen bond needs to be broken before HPMTF can cluster with another molecule. Using quantum chemical calculations, it has been shown that such intramolecular hydrogen bonds hinder strong cluster formation between sulfuric acid and highly oxidized C6H8O7 peroxy acid products formed from cyclohexene ozonolysis (Elm et al., 2015, 2016). HPMTF may, however, indirectly contribute to particle growth via the aqueous-phase oxidation by OH forming SO2 and hence . At present, no data on the aqueous-phase oxidation of HPMTF are available in the literature. Instead, a rate constant of the OH-initiated oxidation of HPMTF was estimated based on experimental data of compounds with similar functional groups (Doussin and Monod, 2013). Unlike the slow gas-phase oxidation, the implemented rate ensured a strong sink flux of HPMTF in the aqueous phase ( and molec. cm−3 s−1 mean HPMTF sink flux in the gas phase and aqueous phase, respectively). The formed intermediate HOOCH2SCO partitioned readily to the gas phase and oxidized to form SO2. Consequently, the aqueous-phase oxidation of HPMTF may increase the conversion of HPMTF to SO2 and thus the PM production. However, further investigations are required to overcome the uncertainties linked to the said reaction. The weak oxidation of HPMTF by OH and consequent accumulation in the gas phase could potentially reduce the local effect of DMS-derived aerosol formation, as HPMTF may be transported large distances before partitioning to and oxidizing in aqueous cloud particles.
Previous model studies (von Glasow and Crutzen, 2004; Braeuer et al., 2013; Hoffmann et al., 2016) did not consider the effect of precipitation on the gas-phase concentration of aerosol particles and gaseous species. Therefore, a 5 mm h−1 intensity rain event and below-cloud scavenging of particles and gases according to the parameterizations by Laakso et al. (2003) and Simpson et al. (2003) were implemented in the base model run (Fig. 5c). The wet deposition of aerosol particles efficiently lowered the available surface area and thus decreased the condensation sink of SA and NH3. Following the rain event, a distinct growth mode occurred, initialized by NPF of said gaseous species. Unlike the main growth mode dominated by the presence of sea spray aerosols, the new mode grew solely by SA, MSA and produced in the gas phase and particle phase, respectively. A distinct Hoppel minimum formed in the main growth mode at 140 nm proceeding the first cloud period (Fig. 5c) (Hoppel and Frick, 1990), becoming more profound by each cloud passage. The effect was most distinct during the day, in which the activated cloud particles grew considerably by the ample SO2 concentration. The absence of a Hoppel minimum in both the woCloudAtm (see Fig. S33) and woAqAtm (see Fig. S34) sensitivity run demonstrates the contribution of in-cloud and aqueous-phase chemistry to the total PM concentration.
The woCloudAtm sensitivity run demonstrated the importance of cloud chemistry in the ambient atmosphere. Semi-soluble products HPMTF and SO2 did not partition to the deliquesced particles, and thus their conversion yields increased by a factor of 3.9 and 7.6, respectively, compared to AtmMain, as both species accumulated in the gas phase (Table 4). PM production decreased by 66.4 % in consequence of the reduction in aqueous SO2. Unlike AtmMain, SA proved an important source of secondary aerosol mass. The increase in the SO2 gas-phase concentration promoted SA production (23.2 % SO2-derived SA source flux in woCloudAtm) and thus SA-derived PM. MSA formation remained indifferent from AtmMain and dominated in the deliquesced aerosol particles. Consequently, the MSA to PM ratio proved higher in woCloudAtm than AtmMain (2.25 and 0.91, respectively). The absence of a rain event in woCloudAtm increased the final secondary aerosol mass yield by 95 % compared to AtmMain. This effect was caused by the decrease in the wet deposition of aerosol particles. The PN concentration in the NPF event that followed decreased accordingly, as the condensation sink of species SA and NH3 capable of forming new particles remained high. By comparison, the PN concentration in the NPF event following the rain event in AtmMain was more than 1 order of magnitude higher. Unlike AtmMain, NPF events in woCloudAtm occurred consistently throughout the simulation (see Fig. S35). Each event initiated in the morning in accordance with the increase in OH, BrO and Cl concentrations and thus DMS oxidation and SA production. The lack of clouds allowed the particles to grow to approximately 30 nm in particle mode diameter towards the end of the simulation. In AtmMain, morning NPF events were terminated by the uptake of SA and NH3 to the cloud particles. Overall, cloud-free conditions promoted the gas-phase SA production and thus NPF but impeded SO2- derived PM formation in the aqueous particle phase as species SO2 and HPMTF accumulated in the gas phase.
The effects of aqueous-phase chemistry in both the deliquesced particles and cloud droplets were validated in the woAqAtm sensitivity run. Bromide and chloride ions did not activate as no iodine chemistry took place in the aerosol particles. Consequently, neither HOBr nor ICl entered the gas phase, and thus no BrO or Cl radicals were formed. The lack of reactive halogen species lowered the sink flux of DMS by 37.2 %, causing it to accumulate in the gas phase throughout the simulation (see Fig. S36). The total secondary aerosol yield decreased from 2.21 to 0.73 µg cm−3 in accordance with the decrease in DMS decay (Table 4). MSA PM production proved strongly reduced in woAqAtm (0.06 µg cm−3 at the end of the simulation) relative to AtmMain (0.27 µg cm−3). While woAqAtm still considered the phase transfer of soluble species, no reactions took place in the particle phase. As a result, MSIA did not oxidize by O3 to form MSA in the deliquesced particles. Instead, MSA formed solely in the gas phase via the CH3SO3 intermediate. The decrease in MSA production is evident from the decrease in the PM ratio (0.91 and 0.41 in AtmMain and woAqAtm, respectively) (Table 4). Analogous to woCloudAtm, the PM production was strongly reduced (67.0 % compared to AtmMain) as SO2 did not transform in the particle phase. SO2- and CH3SO3-derived SA formation in the gas phase therefore comprised the only source of PM throughout the simulation. In conclusion, the absence of aqueous-phase chemistry led to an underestimation of both and in particular MSA PM production and thus the total secondary aerosol yield.
The PolAtm sensitivity run reproduced DMS chemistry in polluted marine environments of high initial O3 and NO2 gas-phase concentrations. Model inputs were based on measurements in the marine boundary layer at Tudor Hill, Bermuda, and the English Channel (Boylan et al., 2015; Leser et al., 2003). Both locations experience high O3 and NOx pollution from the USA and Europe, respectively. The daytime and night-time DMS sink flux increased by 21.8 % in accordance with increased O3 and NO2 pollution from the increase in oxidants OH, NO3, Cl and Cl2 (see Fig. S34). The effect is evident from the increase in the total secondary aerosol mass yield (2.21 and 2.68 µg cm−3 in AtmMain and PolAtm, respectively) (Table 4). Elevated NO3 concentrations favoured the abstraction pathway and thus the formation of HPMTF and suppressed the gas-phase production of DMSO and MSIA in the addition pathway. The effect was counterbalanced by the increase in aqueous-phase production of DMSO and MSIA from the increase in O3-initiated DMS oxidation in the aerosol cloud droplets. Elevated O3 concentrations promoted SO2 production via the CH3SO and HOOCH2SO intermediates and increased the PM yield by 60.4 % compared with AtmMain. However, analogous to HPMTF, the increase was not reflected in the conversion yields (Table 4). The increased OH and O3 oxidation capacity of the aqueous phase ensured a high uptake and turn-over rate of species SO2 and HPMTF, thus lowering their respective gas-phase concentrations.
We have presented ADCHAM simulation of the OH-derived oxidation of DMS and subsequent particle formation and growth in the AURA smog chamber and under relevant atmospheric conditions. New particles in the chamber experiments formed primarily by nucleation of SA (produced in the gas phase via the CH3SO3 intermediate) and NH3 and grew by condensation of MSA. The total production of secondary aerosol mass and the MSA to SA ratio were strongly influenced by the formation of a liquid water film on the Teflon bag chamber wall, the effect of which increased in accordance with RH. Water-soluble reaction products and intermediates DMS, DMSO, DMSO2, MSIA, HPMTF, MSA and SA were taken up by the water film. Consequently, the secondary aerosol PM production decreased significantly during experiments performed at humid conditions (50 % RH–80 % RH) compared to experiments performed at dry conditions (0 % RH–12 % RH). The recently discovered autoxidation product HPMTF comprised a large fraction of the gas-phase products produced in both smog chamber and atmospheric relevant simulations but proved insignificant to the direct formation and growth of aerosol particles. HPMTF may, however, contribute indirectly to the particle growth by oxidizing in the aqueous phase to form SO2 and thus SA and . At high RH, the rate of CH3SCH3OO autoxidation leading to the formation of HPMTF had a strong impact on the secondary mass yield in the chamber experiments. At low RH, the oxidation of MSIA by OH proved essential to the total particle mass. Overall, a significant revision of the DMS oxidation mechanisms presented in literature was needed to reproduce the measurements obtained in the AURA smog chamber.
OH-derived oxidation of DMS in an atmospheric relevant context comprised a significant DMS sink but proved less important than the equivalent oxidation by halogen species BrO and Cl. The relative importance of OH oxidation increased in accordance with a decrease in wind speed, which lowered sea spray emissions and thus the gas-phase concentration of reactive halogen species. The large surface area of the sea spray aerosols induced a strong condensation sink, thus impeding NPF. By introducing precipitation in the model, the decrease in the condensation sink was sufficient to allow new particles to form by nucleation of SA and NH3.
Smog chamber studies are able to elucidate atmospheric phenomena in a controlled environment but rarely represent actual atmospheric conditions. Therefore, future studies will focus on implementing the revised DMS chemistry in the chemistry transport model ADCHEM and test the setup on field measurements from the marine Arctic environment.
All source codes, including the complete ADCHAM model version and plotting programs used to conduct the analysis presented in this paper, can be obtained by contacting the corresponding author Robin Wollesen de Jonge. All results presented in the paper and Supplement and the complete DMS multiphase chemistry mechanism (Table S1) written in a format compatible with the Kinetic PreProcessor (KPP) (Damian et al., 2002) can be downloaded from an open archive provided by the data publisher Zenodo (https://doi.org/10.5281/zenodo.5016758, Roldin and Wollesen de Jonge, 2021).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-9955-2021-supplement.
BR and SC conceived and planned the DMS experiments, while RWdJ and PR conceived the modelling of these experiments. RWdJ, PR, JE and NH took part in the development of code related to ADCHAM, ACDC and COSMOtherm, respectively. RWdJ, PR and DL utilized ADCHAM to analyze the DMS experiments, which were performed by RWdJ, BR and SC. Resources were provided by PR and MB. RWdJ, PR, JE and NH wrote the original draft, which included visualizations by RWdJ and PR. The project was supervised by PR, JE and MB. All the authors discussed the results and contributed to the final manuscript.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors would like to thank Tinja Olenius from the Swedish Meteorological and Hydrological Institute (SMHI) for help with the implementation of ADCD in ADCHAM and Shamjad P. Moosakutty and Mads Mørk Jensen for the calibration and interpretation of the AMS measurements. We would also like to thank Anders Feilberg from the Department of Biological and Chemical Engineering, Aarhus University, for making the PTR-MS instrument available, and we would like to thank him and Jon Bjarke Valbæk Mygind for help with the PTR-MS data. We thank the Swedish Strategic Research Program MERGE, the Centre for Scientific and Technical Computing at Lund University, LUNARC, the Swedish National Infrastructure for Computing, SNIC and CSC – IT Center for Science, Finland, for computational resources.
This project has received funding from the Swedish Research Council Formas project no. 2018-01745-COBACCA, Swedish Research Council VR project no. 2019-05006, the Austrian Science Fund (FWF: J 3970-N36), Aarhus University, the Independent Research Fund Denmark grant no. 9064-00001B and the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation programme, Project SURFACE (grant agreement no. 717022).
This paper was edited by Harald Saathoff and reviewed by two anonymous referees.
Andreae, M. O.: Ocean–atmosphere Interactions in the Global Biogeochemical Sulfur Cycle, Mar. Chem., 30, 1–29, 1990. a
Bahreini, R., Ervens, B., Middlebrook, A., Warneke, C., de Gouw, J., DeCarlo, P., Jimenez, J., Brock, C., Neuman, J., Ryerson, T., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J., Peischl, J., Richter, D., Walega, J., Weibring, P., and Fehsenfeld, F.: Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas, J. Geophys. Res., 114, D00F16, https://doi.org/10.1029/2008JD011493, 2009. a
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006. a, b, c, d, e, f, g
Benson, D. R., Yu, J. H., Markovich, A., and Lee, S.-H.: Ternary homogeneous nucleation of H2SO4, NH3, and H2O under conditions relevant to the lower troposphere, Atmos. Chem. Phys., 11, 4755–4766, https://doi.org/10.5194/acp-11-4755-2011, 2011. a
Berglen, T. F., Berntsen, T. K., Isaksen, I. S. A., and Sundet, J. K.: A global model of the coupled sulfur/oxidant chemistry in the troposphere: The sulfur cycle, J. Geophys. Res.-Atmos., 109, https://doi.org/10.1029/2003JD003948, 2004. a
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 2019, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019. a, b, c, d, e, f, g, h
Berndt, T., Chen, J., Møller, K. H., Hyttinen, N., Prisle, N. L., Tilgner, A., Hoffmann, E. H., Herrmann, H., and Kjaergaard, H. G.: SO2 formation and peroxy radical isomerization in the atmospheric reaction of OH radicals with dimethyl disulfide, Chem. Commun., 56, 13634–13637, https://doi.org/10.1039/D0CC05783E, 2020. a, b
Berresheim, H., Adam, M., Monahan, C., O'Dowd, C., Plane, J. M. C., Bohn, B., and Rohrer, F.: Missing SO2 oxidant in the coastal atmosphere? – observations from high-resolution measurements of OH and atmospheric sulfur compounds, Atmos. Chem. Phys., 14, 12209–12223, https://doi.org/10.5194/acp-14-12209-2014, 2014. a, b
Boylan, P., Helmig, D., and Oltmans, S.: Ozone in the Atlantic Ocean marine boundary layer, Elementa, 3, 000045, https://doi.org/10.12952/journal.elementa.000045, 2015. a
Braeuer, P., Tilgner, A., Wolke, R., and Herrmann, H.: Mechanism development and modelling of tropospheric multiphase halogen chemistry: The CAPRAM Halogen Module 2.0 (HM2), J. Atmos. Chem., 70, 19–52, https://doi.org/10.1007/s10874-013-9249-6, 2013. a, b, c, d, e, f, g
Campolongo, F., Saltelli, A., Jensen, N. R., Wilson, J., and Hjorth, J.: The Role of Multiphase Chemistry in the Oxidation of Dimethylsulphide (DMS), A Latitude Dependent Analysis, J. Atmos. Chem., 32, 327–356, https://doi.org/10.1023/A:1006154618511, 1999. a, b, c, d, e, f
Cao, J., Wang, W.-L., Gao, L.-J., and Fu, F.: Mechanism and Thermodynamic Properties of CH3SO3 Decomposition, Acta Phys.-Chim. Sin., 29, 1161–1167, https://doi.org/10.3866/PKU.WHXB201304021, 2013. a, b, c
Chen, H., Varner, M., Gerber, R., and Finlayson-Pitts, B.: Reactions of Methanesulfonic Acid with Amines and Ammonia as a Source of New Particles in Air, J. Phys. Chem. B, 120, 1526–1536, https://doi.org/10.1021/acs.jpcb.5b07433, 2015. a
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018. a
COSMOtherm: version C3.0, Release 19, COSMOlogic GmbH & Co. KG., Leverkusen, Germany, 2019. a
Crounse, J., Nielsen, L., Jørgensen, S., Kjaergaard, H., and Wennberg, P.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G. R.: The kinetic preprocessor KPP-a software environment for solving chemical kinetics, Comput. Chem. Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002. a, b
Doussin, J.-F. and Monod, A.: Structure–activity relationship for the estimation of OH-oxidation rate constants of carbonyl compounds in the aqueous phase, Atmos. Chem. Phys., 13, 11625–11641, https://doi.org/10.5194/acp-13-11625-2013, 2013. a, b
Elm, J., Myllys, N., Hyttinen, N., and Kurtén, T.: Computational Study of the Clustering of a Cyclohexene Autoxidation Product C6H8O7 with Itself and Sulfuric Acid, J. Phys. Chem. A, 119, 8414–8421, 2015. a
Elm, J., Myllys, N., Luy, J., Kurtén, T., and Vehkamäki, H.: The Effect of Water and Bases on the Clustering of a Cyclohexene Autoxidation Product C6H8O7 with Sulfuric Acid, J. Phys. Chem. A, 120, 2240–2249, 2016. a
Elm, J., Myllys, N., and Kurtén, T.: What is Required for Highly Oxidized Molecules to Form Clusters with Sulfuric Acid?, J. Phys. Chem. A., 121, 4578–4587, 2017. a
Fuchs, N. A.: On the stationary charge distribution on aerosol particles in a bipolar ionic atmosphere, Geofisica Pura e Applicata, 56, 185–193, https://doi.org/10.1007/BF01993343, 1963. a
Ghahremaninezhad, R., Gong, W., Galí, M., Norman, A.-L., Beagley, S. R., Akingunola, A., Zheng, Q., Lupu, A., Lizotte, M., Levasseur, M., and Leaitch, W. R.: Dimethyl sulfide and its role in aerosol formation and growth in the Arctic summer – a modelling study, Atmos. Chem. Phys., 19, 14455–14476, https://doi.org/10.5194/acp-19-14455-2019, 2019. a
Grosjean, D.: Wall Loss of Gaseous Pollutants in Outdoor Teflon Chambers, Environ. Sci. Technol, 19, 1059–1065, 1985. a
Hindmarsh, A. C.: ODEPACK, A Systematized Collection of ODE Solvers in Scientific Computing, edited by: Stepleman, R. S., Carver, M., Peskin, R., Ames, W. F., and Vichnevetsky, R., Vol. 1, North-Holland, Amsterdam, 55–64, 1983. a
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s
Hoppel, W. A. and Frick, G. M.: Ion-Aerosol Attachment Coefficients and the Steady-State Charge Distribution on Aerosols in a Bipolar Ion Environment, Aerosol Sci. Tech., 5, 1–21, https://doi.org/10.1080/02786828608959073, 1986. a
Hoppel, W. A. and Frick, G. M.: The Nonequilibrium Character of the Aerosol Charge Distributions Produced by Neutralizes, Aerosol Sci. Tech., 12, 471–496, https://doi.org/10.1080/02786829008959363, 1990. a
Hyttinen, N., Wolf, M., Rissanen, M. P., Ehn, M., Peräkylä, O., Kurtén, T., and Prisle, N. L.: Gas-to-Particle Partitioning of Cyclohexene-and α-Pinene-Derived Highly Oxygenated Dimers Evaluated Using COSMOtherm, J. Phys. Chem. A, 125, 3726–3738, https://doi.org/10.1021/acs.jpca.0c11328, 2021. a
Jacobson, M. Z.: Fundamentals of Atmospheric Modelling (2nd edition), Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 828 pp., 2005. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a, b
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003. a, b
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a, b
Kent, E. C., Fangohr, S., and Berry, D. I.: A comparative assessment of monthly mean wind speed products over the global ocean, Int. J. Clim., 33, 2520–2541, https://doi.org/10.1002/joc.3606, 2013. a
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–33, https://doi.org/10.1038/nature10343, 2011. a
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., and Heinritzi, M.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016. a
Kloster, S., Feichter, J., Maier-Reimer, E., Six, K. D., Stier, P., and Wetzel, P.: DMS cycle in the marine ocean-atmosphere system – a global model study, Biogeosciences, 3, 29–51, https://doi.org/10.5194/bg-3-29-2006, 2006. a
Korhonen, P., Kulmala, M., Laaksonen, A., Viisanen, Y., McGraw, R., and Seinfeld, J. H.: Ternary nucleation of H2SO4, NH3, and H2O in the atmosphere, J. Geophys. Res.-Atmos., 104, 26349–26353, https://doi.org/10.1029/1999JD900784, 1999. a
Kreidenweis, S. M., Walcek, C. J., Feingold, G., Gong, W., Jacobson, M. Z., Kim, C., Liu, X., Penner, J. E., Nenes, A., and Seinfeld, J. H.: Modification of aerosol mass and size distributiondue to aqueous-phase SO2 oxidation in clouds: Comparisons of several models, J. Geophys. Res.-Atmos., 108, 4213, https://doi.org/10.1029/2002JD002697, 2003. a
Kristensen, K., Jensen, L. N., Glasius, M., and Bilde, M.: The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alpha-pinene, Environ. Sci., 19, 1220–1234, https://doi.org/10.1039/C7EM00231A, 2017. a, b
Kristensen, T. B., Müller, T., Kandler, K., Benker, N., Hartmann, M., Prospero, J. M., Wiedensohler, A., and Stratmann, F.: Properties of cloud condensation nuclei (CCN) in the trade wind marine boundary layer of the western North Atlantic, Atmos. Chem. Phys., 16, 2675–2688, https://doi.org/10.5194/acp-16-2675-2016, 2016. a
Kukui, A., Borissenko, D., Laverdet, G., and Bras, G.: Gas-Phase Reactions of OH Radicals with Dimethyl Sulfoxide and Methane Sulfinic Acid Using Turbulent Flow Reactor and Chemical Ionization Mass Spectrometry, J. Phys. Chem. A, 107, 5732–5742, https://doi.org/10.1021/jp0276911, 2003. a, b, c, d, e, f, g
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene Autoxidation Products May Not Have Extremely Low Saturation Vapor Pressures Despite High O:C Ratios, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016. a
Kürten, A., Bianchi, F., Almeida, J., Kupiainen-Määttä, O., Dunne, E., Duplissy, J., Williamson, C., Barmet, P., Breitenlechner, M., Dommen, J., Donahue, N., Flagan, R., Franchin, A., Gordon, H., Hakala, J., Hansel, A., Heinritzi, M., Ickes, L., Jokinen, T., and Curtius, J.: Experimental particle formation rates spanning tropospheric sulfuric acid and ammonia abundances, ion production rates, and temperatures, J. Geophys. Res.-Atmos., 121, 12377–12400, https://doi.org/10.1002/2015JD023908, 2016. a
Laakso, L., Grönholm, T., Üllar Rannik, Kosmale, M., Fiedler, V., Vehkamäki, H., and Kulmala, M.: Ultrafine particle scavenging coefficients calculated from 6 years field measurements, Atmos. Environ., 37, 3605–3613, https://doi.org/10.1016/S1352-2310(03)00326-1, 2003. a, b
Leser, H., Honninger, G., and Platt, U.: MAX-DOAS measurements of BrO and NO2 in the marine boundary layer, Geophys. Res. Lett., 30, 101029, https://doi.org/10.1029/2002GL015811, 2003. a
Lovelock, J. E., Maggs, R. J., and Rasmussen, R. A.: Atmospheric Dimethyl Sulfide and the Natural Sulfur Cycle, Nature, 237, 452–453, 1972. a
Lucas, D. D. and Prinn, R. G.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res.-Atmos., 107, 1–26, https://doi.org/10.1029/2001JD000843, 2002. a, b, c, d, e, f, g, h, i, j
Matsunaga, A. and Ziemann, P.: Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Technol., 44, 881–892, 2010. a, b
McGrath, M. J., Olenius, T., Ortega, I. K., Loukonen, V., Paasonen, P., Kurtén, T., Kulmala, M., and Vehkamäki, H.: Atmospheric Cluster Dynamics Code: a flexible method for solution of the birth-death equations, Atmos. Chem. Phys., 12, 2345–2355, https://doi.org/10.5194/acp-12-2345-2012, 2012. a
McMurry, P. H. and Grosjean, D.: Gas and Aerosol Wall Losses in Teflon Film Smog Chambers, Environ. Sci. Technol., 19, 1176–1182, 1985. a, b, c
Olenius, T., Kupiainen-Määttä, O., Ortega, I., Kurtén, T., and Vehkamäki, H.: Free energy barrier in the growth of sulfuric acid-ammonia and sulfuric acid-dimethylamine clusters, J. Chem. Phys., 139, 084312, https://doi.org/10.1063/1.4819024, 2013. a
Pruppacher, H. and Jaenicke, R.: The processing of water vapor and aerosols by atmospheric clouds, a global estimate, Atmos. Res., 38, 283–295, https://doi.org/10.1016/0169-8095(94)00098-X, 1995. a
Quéléver, L. L. J., Kristensen, K., Normann Jensen, L., Rosati, B., Teiwes, R., Daellenbach, K. R., Peräkylä, O., Roldin, P., Bossi, R., Pedersen, H. B., Glasius, M., Bilde, M., and Ehn, M.: Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis, Atmos. Chem. Phys., 19, 7609–7625, https://doi.org/10.5194/acp-19-7609-2019, 2019. a
Roldin, P. and Wollesen de Jonge, R.: DMS multiphase chemistry mechanism and model results, Zenodo [Dataset], https://doi.org/10.5281/zenodo.5016758, 2021. a
Roldin, P., Eriksson, A. C., Nordin, E. Z., Hermansson, E., Mogensen, D., Rusanen, A., Boy, M., Swietlicki, E., Svenningsson, B., Zelenyuk, A., and Pagels, J.: Modelling non-equilibrium secondary organic aerosol formation and evaporation with the aerosol dynamics, gas- and particle-phase chemistry kinetic multilayer model ADCHAM, Atmos. Chem. Phys., 14, 7953–7993, https://doi.org/10.5194/acp-14-7953-2014, 2014. a, b, c
Roldin, P., Ehn, M., Kurtén, T., Olenius, T., Rissanen, M. P., Sarnela, N., Elm, J., Rantala, P., Hao, L., Hyttinen, N., Heikkinen, L., Worsnop, D. R., Pichelstorfer, L., Xavier, C., Clusius, P., Öström, E., Petäjä, T., Kulmala, M., Vehkamäki, H., Virtanen, A., Riipinen, I., and Boy, M.: The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system, Nat. Commun., 10, 1–15, 2019. a
Rosati, B., Christiansen, S., Dinesen, A., Roldin, P., Massling, A., Nilsson, E. D., and Bilde, M.: The impact of atmospheric oxidation on hygroscopicity and cloud droplet activation of inorganic sea spray aerosol, Sci. Rep., 11, 10008, https://doi.org/10.1038/s41598-021-89346-6, 2021a. a
Rosati, B., Christiansen, S., Wollesen de Jonge, R., Roldin, P., Jensen, M. M., Wang, K., Moosakutty, S. P., Thomsen, D., Salomonsen, C., Hyttinen, N., Elm, J., Feilberg, A., Glasius, M., and Bilde, M.: New Particle Formation and Growth from Dimethyl Sulfide Oxidation by Hydroxyl Radicals, ACS Earth Space Chem., 5, 801–811, https://doi.org/10.1021/acsearthspacechem.0c00333, 2021b. a, b, c, d, e, f, g, h, i, j, k, l
Salter, M. E., Zieger, P., Acosta Navarro, J. C., Grythe, H., Kirkevåg, A., Rosati, B., Riipinen, I., and Nilsson, E. D.: An empirically derived inorganic sea spray source function incorporating sea surface temperature, Atmos. Chem. Phys., 15, 11047–11066, https://doi.org/10.5194/acp-15-11047-2015, 2015. a, b
Saltzman, E. S., Savoie, D. L., Zika, R. G., and Prospero, J. M.: Methane sulfonic acid in the marine atmosphere, J. Geophys. Res.-Oceans, 88, 10897–10902, https://doi.org/10.1029/JC088iC15p10897, 1983. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, NJ, 3 Edn., 1152 pp., 2016. a, b
Simpson, D., Fagerli, H., Jonson, J. E., Tsyro, S., Wind, P., and Tuovinen, J.-P.: Transboundary Acidification, Eutrophication and Groud Level Ozone in Europe, Part I, Unified EMEP Model Description, EMEP Status Report 2003, 104 pp., 2003. a, b
Song, M., Zhang, C., Wu, H., Mu, Y., Ma, Z., Zhang, Y., Liu, J., and Li, X.: The influence of OH concentration on SOA formation from isoprene photooxidation, Sci. Total Environ., 650, 951–957, https://doi.org/10.1016/j.scitotenv.2018.09.084, 2019. a
Sumner, A. L., Menke, E. J., Dubowski, Y., Newberg, J. T., Penner, R. M., Hemminger, J. C., Wingen, L. M., Brauers, T., and Finlayson-Pitts, B. J.: The nature of water on surfaces of laboratory systems and implications for heterogeneous chemistry in the troposphere, Phys. Chem. Chem. Phys., 6, 604–613, https://doi.org/10.1039/B308125G, 2004. a, b, c, d, e, f
Svensson, R., Ljungström, E., and Lindqvist, O.: Kinetics of the reaction between nitrogen dioxide and water vapour, Atmos. Environ., 21, 1529–1539, https://doi.org/10.1016/0004-6981(87)90315-5, 1987. a, b, c, d, e
Turnipseed, A. A., Barone, S. B., Jensen, N. R., Hanson, D. R., Howard, C. J., and Ravishankara, A. R.: Kinetics of the Reactions of CF3O Radicals with CO and H2O, J. Phys. Chem., 99, 6000–6009, https://doi.org/10.1021/j100016a041, 1995. a, b, c
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020. a, b, c, d, e, f, g, h, i, j, k, l, m, n
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004. a, b, c, d, e, f, g, h, i
Wu, R., Wang, S., and Wang, L.: A New Mechanism for The Atmospheric Oxidation of Dimethyl Sulfide, The Importance of Intramolecular Hydrogen Shift in CH3SCH2OO Radical., J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2014. a, b, c, d, e
Yin, F., Grosjean, D., Flagan, R. C., and Seinfeld, J. H.: Photooxidation of dimethyl sulfide and dimethyl disulfide, II: Mechanism evaluation, J. Atmos. Chem., 11, 365–399, https://doi.org/10.1007/BF00053781, 1990. a, b, c, d, e, f, g, h, i, j, k, l, m
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014. a, b
Zhu, L., Nenes, A., Wine, P. H., and Nicovich, J. M.: Effects of aqueous organosulfur chemistry on particulate methanesulfonate to non–sea salt sulfate ratios in the marine atmosphere, J. Geophys. Res.-Atmos., 111, D05316, https://doi.org/10.1029/2005JD006326, 2006. a