the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 Jun 2021
| 29 Jun 2021
Highly oxygenated organic molecule (HOM) formation in the isoprene oxidation by NO3 radical
Iida Pullinen
Hendrik Fuchs
Stephanie Schrade
Rongrong Wu
Ismail-Hakki Acir
Ralf Tillmann
Franz Rohrer
Jürgen Wildt
Yindong Guo
Astrid Kiendler-Scharr
Andreas Wahner
Sungah Kang
Luc Vereecken
Highly oxygenated organic molecules (HOM) are found to play an important role in the formation and growth of secondary organic aerosol (SOA). SOA is an important type of aerosol with significant impact on air quality and climate. Compared with the oxidation of volatile organic compounds by ozone (O3) and hydroxyl radical (OH), HOM formation in the oxidation by nitrate radical (NO3), an important oxidant at nighttime and dawn, has received less attention. In this study, HOM formation in the reaction of isoprene with NO3 was investigated in the SAPHIR chamber (Simulation of Atmospheric PHotochemistry In a large Reaction chamber). A large number of HOM, including monomers (C5), dimers (C10), and trimers (C15), both closed-shell compounds and open-shell peroxy radicals (RO2), were identified and were classified into various series according to their formula. Their formation pathways were proposed based on the peroxy radicals observed and known mechanisms in the literature, which were further constrained by the time profiles of HOM after sequential isoprene addition to differentiate first- and second-generation products. HOM monomers containing one to three N atoms (1–3N-monomers) were formed, starting with NO3 addition to carbon double bond, forming peroxy radicals, followed by autoxidation. 1N-monomers were formed by both the direct reaction of NO3 with isoprene and of NO3 with first-generation products. 2N-monomers (e.g., C5H8N2On(n=7–13), C5H10N2On(n=8–14)) were likely the termination products of C5H9N2On•, which was formed by the addition of NO3 to C5-hydroxynitrate (C5H9NO4), a first-generation product containing one carbon double bond. 2N-monomers, which were second-generation products, dominated in monomers and accounted for ∼34 % of all HOM, indicating the important role of second-generation oxidation in HOM formation in the isoprene + NO3 reaction under our experimental conditions. H shift of alkoxy radicals to form peroxy radicals and subsequent autoxidation (“alkoxy–peroxy” pathway) was found to be an important pathway of HOM formation. HOM dimers were mostly formed by the accretion reaction of various HOM monomer RO2 and via the termination reactions of dimer RO2 formed by further reaction of closed-shell dimers with NO3 and possibly by the reaction of C5–RO2 with isoprene. HOM trimers were likely formed by the accretion reaction of dimer RO2 with monomer RO2. The concentrations of different HOM showed distinct time profiles during the reaction, which was linked to their formation pathway. HOM concentrations either showed a typical time profile of first-generation products, second-generation products, or a combination of both, indicating multiple formation pathways and/or multiple isomers. Total HOM molar yield was estimated to be 1.2 %, which corresponded to a SOA yield of ∼3.6 % assuming the molecular weight of C5H9NO6 as the lower limit. This yield suggests that HOM may contribute a significant fraction to SOA yield in the reaction of isoprene with NO3.
- Article
(5895 KB) - Full-text XML
-
Supplement
(1011 KB) - BibTeX
- EndNote





Highly oxygenated organic molecules (HOM) are an important class of compound formed in the oxidation of volatile organic compounds (VOC), including biogenic VOC (BVOC) and anthropogenic VOC (Crounse et al., 2013; Ehn et al., 2014; Jokinen et al., 2014; Rissanen et al., 2014; Jokinen et al., 2015; Krechmer et al., 2015; Mentel et al., 2015; Rissanen et al., 2015; Kenseth et al., 2018; Molteni et al., 2018; Garmash et al., 2020; McFiggans et al., 2019; Molteni et al., 2019; Quéléver et al., 2019). A number of recent studies have demonstrated that HOM play a pivotal role in both nucleation and also particle growth of pre-existing particles, thus contributing to secondary organic aerosol (SOA) (Ehn et al., 2014; Kirkby et al., 2016; Tröstl et al., 2016). Particularly, in the early stage of aerosol growth, HOM may contribute a significant fraction of SOA mass (Tröstl et al., 2016).
HOM are formed by the autoxidation of peroxy radicals (RO2), which means they undergo intramolecular H shift forming alkyl radicals, followed by O2 addition leading to formation of new RO2 as shown below (Vereecken et al., 2007; Crounse et al., 2013; Ehn et al., 2017; Bianchi et al., 2019; Møller et al., 2019; Nozière and Vereecken, 2019; Vereecken and Nozière, 2020).
Besides autoxidation, the RO2 can also react with HO2, RO2, and NO3, either forming a series of termination products (Reactions R1–3), including organic hydroxyperoxide, alcohol, and carbonyl, or forming alkoxy radicals (RO, Reactions R4–5) via the following reactions.
The termination products are detected in the mass spectra at masses M+1, M−15, and M−17, respectively, with M being the molecular mass of the parent RO2 (Ehn et al., 2014; Mentel et al., 2015). In case RO2 is an acyl peroxy radical, percarboxylic acids and carboxylic acids are formed instead of hydroperoxides and alcohols in Reactions (R3) and (R1), respectively (Atkinson et al., 2006; Mentel et al., 2015). RO2 can also form HOM dimers by the accretion reaction of two RO2: Reaction (R6) (Berndt et al., 2018a, b; Valiev et al., 2019). Additionally, HOM can be formed via H shift in RO followed by O2 addition (referred to as the “alkoxy-proxy” pathway) (Finlayson-Pitts and Pitts, 2000; Vereecken and Peeters, 2010; Vereecken and Francisco, 2012; Mentel et al., 2015). These pathways are summarized in a recent comprehensive review (Bianchi et al., 2019), which also further clarifies HOM definition.
Currently, most laboratory studies of HOM formation focus on the VOC oxidation by OH and O3 (Crounse et al., 2013; Ehn et al., 2014; Jokinen et al., 2014; Rissanen et al., 2014; Jokinen et al., 2015; Krechmer et al., 2015; Mentel et al., 2015; Rissanen et al., 2015; Kirkby et al., 2016; Tröstl et al., 2016; Kenseth et al., 2018; Molteni et al., 2018; Garmash et al., 2020; McFiggans et al., 2019; Molteni et al., 2019; Quéléver et al., 2019; Wang et al., 2020; Yan et al., 2020). HOM formation in the oxidation of VOC with NO3 has received much less attention. NO3 is another important oxidant of VOC mainly operating during nighttime. Particularly, NO3 has high reactivity with unsaturated BVOC such as monoterpene and isoprene. It is often the dominant oxidant of these compounds at night, especially in regions where biogenic and anthropogenic emissions mix (Geyer et al., 2001; Brown et al., 2009, 2011). The reaction products contribute to SOA formation (Xu et al., 2015; Lee et al., 2016). Also, the organic nitrates produced in these reactions play an important role in nitrogen chemistry by altering NOx concentration, which further influences photochemical recycling and ozone formation on the next day. Among these reaction products, HOM can also be formed (Xu et al., 2015; Lee et al., 2016; Yan et al., 2016). Despite the potential importance, studies of HOM formation in the oxidation of BVOC by NO3 are still limited compared with the HOM formation via oxidation by O3 and OH. Although a number of laboratory studies have investigated the reaction of NO3 with BVOC (Ng et al., 2008; Fry et al., 2009; Rollins et al., 2009; Fry et al., 2011; Kwan et al., 2012; Fry et al., 2014; Boyd et al., 2015; Schwantes et al., 2015; Nah et al., 2016; Boyd et al., 2017; Claflin and Ziemann, 2018; Faxon et al., 2018; Draper et al., 2019; Takeuchi and Ng, 2019; Novelli et al., 2021; Vereecken et al., 2021), these studies mostly focus on either SOA yield and composition or on the gas-phase chemistry mechanism mainly for “traditional” oxidation products that stem from a few oxidation steps.
Importantly, HOM formation in the reaction of NO3 with isoprene, the most abundant BVOC accounting for more than half of the global BVOC emissions, has not been explicitly addressed yet, to the best of our knowledge. Although isoprene from plants is mainly emitted under light conditions, i.e., in the daytime, isoprene can remain high after sunset in significant concentrations (Starn et al., 1998; Stroud et al., 2002; Brown et al., 2009) because of the reduced consumption by OH and is found to decay rapidly. A substantial fraction of isoprene can then be oxidized by NO3 (Brown et al., 2009). Regarding the budget of NO3, the reaction of isoprene with NO3 can contribute a significant or even dominant fraction of NO3 loss at night in regions where VOC is dominated by isoprene such as the northeastern US (Brown et al., 2009). Under some circumstances, the reaction of isoprene with NO3 can contribute a significant fraction during the afternoon and afterwards (Ayres et al., 2015; Hamilton et al., 2021). The reaction of isoprene with NO3 is the subject of a number of studies (Ng et al., 2008; Perring et al., 2009; Rollins et al., 2009; Kwan et al., 2012; Schwantes et al., 2015; Vereecken et al., 2021). These studies focus on the oxidation mechanism and “traditional” oxidation products, as well as SOA yields. The initial step is the NO3 addition to one of the C = C double bounds, preferentially to the carbon C1 (Schwantes et al., 2015), followed by O2 addition forming a nitrooxyalkyl peroxy radical (RO2). This RO2 can undergo the reactions described above, forming a series of products such as C5-nitrooxyhydroperoxide, C5-nitrooxycarbonyl, and C5-hydroxynitrate (Ng et al., 2008; Kwan et al., 2012) as well as methyl vinyl ketone (MVK), potentially methacrolein (MACR), formaldehyde, OH radical, and NO2 as minor products (Schwantes et al., 2015). A high nitrate yield (57 %–95 %) was found (Perring et al., 2009; Rollins et al., 2009; Kwan et al., 2012; Schwantes et al., 2015). Products in the particle phase such as C10 dimers were also detected (Ng et al., 2008; Kwan et al., 2012; Schwantes et al., 2015). The SOA yield varies from 2 % to 23.8 % depending on the organic aerosol concentration (Ng et al., 2008; Rollins et al., 2009). These studies have provided valuable insights into oxidation mechanism, particle yield, and composition. However, because HOM formation was not the focus of these studies, only a limited number of products, mainly moderately oxygenated ones (oxygen number ≤2 in addition to NO3 functional groups), were detected in the gas phase. The detailed mechanism of HOM formation and their yields in the reaction of BVOC+NO3 are still unclear.
In this study, we investigated the HOM formation in the oxidation of isoprene by NO3. We report the identification of HOM, including HOM monomers, dimers, and trimers. According to the reaction products and the literature, we discuss the formation mechanism of these HOM. The formation mechanism of various HOM is further constrained with time series of HOM upon repeated isoprene additions. We also provide an estimate of HOM yield in the isoprene + NO3 reaction and assess their roles in SOA formation.
2.1 Chamber setup and experiments
Experiments investigating the reaction of isoprene with NO3 were conducted in the SAPHIR chamber (Simulation of Atmospheric PHotochemistry In a large Reaction chamber) at the Forschungszentrum Jülich, Germany. The details of the chamber have been described before (Rohrer et al., 2005; Zhao et al., 2015a, b, 2018). Briefly, SAPHIR is a Teflon chamber with a volume of 270 m3. It can utilize natural sunlight for illumination and is equipped with a louvre system to switch between light and dark conditions. In this study, the experiments were conducted in the dark with the louvres closed.
Temperature and relative humidity were continuously measured. Gas- and particle-phase species were characterized using a comprehensive set of instruments with the details described before (Zhao et al., 2015b). VOC were characterized using a Proton Transfer Reaction Time-of-Flight Mass Spectrometer (PTR-ToF-MS, Ionicon Analytik, Austria). NOx and O3 concentrations were measured using a chemiluminescence NOx analyzer (ECO PHYSICS TR480) and a UV photometer O3 analyzer (ANSYCO, model O341M), respectively. OH, HO2, and RO2 concentrations were measured using a laser-induced fluorescence system (LIF) (Fuchs et al., 2012). NO3 and N2O5 were detected by a custom-built instrument based on cavity ring-down spectroscopy. The design of the instrument is similar to that described by Wagner et al. (2011). NO3 was directly detected in one cavity by its absorption at 662 nm and the sum of NO3 and N2O5 in a second, heated cavity, which had a heated inlet to thermally decompose N2O5 to NO3. The sampling flow rate was 3 to 4 L/min. The detection by cavity ring-down spectroscopy was achieved by a diode laser that was periodically switched on and off with a repetition rate of 200 Hz. Ring-down events were observed by a digital oscilloscope PC card during the time when the laser was switched off and were averaged over 1 s. The zero-decay time that is needed to calculate the concentration of NO3 was measured every 20 s by chemically removing NO3 in the reaction with excess nitric oxide (NO) in the inlet system. The accuracy of measurements was limited by the uncertainty in the correction for inlet losses of NO3 and N2O5. In the case of N2O5 a transmission of (85 ± 10) % was achieved and, in the case of NO3, (50 ± 30) %.
Before an experiment, the chamber was flushed with high-purity synthetic air (purity >99.9999 % O2 and N2). Experiments were conducted under dry conditions (RH < 2 %), and temperature was at 302 ± 3 K. NO2 and O3 were added to the chamber first to form N2O5 and NO3, reaching concentrations of ∼60 ppb for NO2 and ∼100 ppb for O3. After around half an hour, isoprene was sequentially added into the chamber for three times at intervals of ∼1 h. Around 40 min after the third isoprene injection, NO2 was added to compensate for the loss of NO3 and N2O5. Afterwards, three isoprene additions were repeated in the same way as before. O3 was added before the fifth and sixth isoprene additions to compensate for its loss by reaction. The schematic for the experimental procedure is shown in Fig. S1. Experiments were designed such that the chemical system was dominated by the reaction of isoprene with NO3 and the reaction of isoprene with O3 did not play a major role (<3 % of the isoprene consumption). Figure S2 shows the relative contributions of the reaction of O3 and NO3 with isoprene to the total chemical loss of isoprene using the NO3 and O3 concentrations measured. The reaction with NO3 accounted for >95 % of the isoprene consumption for the whole experiment periods. The contribution of the reaction of isoprene with a trace amount of OH, mainly produced in the reaction of isoprene + O3 via Criegee intermediates (Nguyen et al., 2016), is negligible as the OH yield is less than one (Malkin et al., 2010), and thus its contribution is less than that of isoprene + O3. This is consistent with the contribution determined using measured OH concentration, despite some uncertainty in measured OH concentration due to the interference from NO3. In these experiments, RO2 fate is estimated to be dominated by its reaction with NO3 according to the measured NO3, RO2, and HO2 concentrations and their rate constants for the reactions with RO2 (MCM v3.2; Jenkin et al., 1997, 2003; Saunders et al., 2003; Jenkin et al., 2015, via the website: http://mcm.york.ac.uk/, last access: 28 June 2021) despite uncertainties in the measured RO2 and HO2 concentration due to interference from NO3. As a large portion of RO2 is not measured by LIF (Vereecken et al., 2021) and thus RO2 is underestimated, we expected the reaction of RO2+RO2 to also be important. Overall, we estimate that the RO2 fate is dominated by the reaction RO2+NO3 with a significant contribution of RO2+RO2.
2.2 Characterization of HOM
In this study we refer to a similar definition for HOM by Bianchi et al. (2019); i.e., HOM typically contain six or more oxygen atoms formed via autoxidation and related chemistry of peroxy radicals. HOM were detected using a Chemical Ionization time-of-flight Mass Spectrometer (Aerodyne Research Inc., USA) with nitrate as the reagent ion (CIMS) (Eisele and Tanner, 1993; Jokinen et al., 2012). 15N nitric acid was used to produce 15NO in order to distinguish the NO3 group into target molecules formed in the reaction from the reagent ion. The details of the instrument are described in our previous publications (Ehn et al., 2014; Mentel et al., 2015; Pullinen et al., 2020). The CIMS has a mass resolution of ∼4000 (m/dm). Examples of peak fitting are shown in Fig. S3. HOM concentrations were estimated using the calibration coefficient of H2SO4 as described by Pullinen et al. (2020) because the charge efficiency of HOM and H2SO4 can be assumed to be equal and close to the collision limit (Ehn et al., 2014; Pullinen et al., 2020). The details of the calibration with H2SO4 are provided in Supplement Sect. S1. Since HOM contain more than six oxygen atoms and their clusters with nitrate ions are quite stable (Ehn et al., 2014), the charge efficiency of HOM is thus assumed to be equal to that of H2SO4, which is close to the collision limit (Viggiano et al., 1997). If HOM do not charge with nitrate ions at their collision limit or the clusters formed break during the short residence time in the charger, its concentration would be underestimated as pointed out by Ehn et al. (2014). Thus, our assumption provides a lower limit of the HOM concentration. The HOM yield was derived using the concentration of the HOM produced, divided by the concentration of isoprene that was consumed by NO3. The uncertainty of HOM yield was estimated to −55 %/+103 %. The loss of HOM to the chamber was corrected using a wall loss rate of 6 × 10−4 s−1 as quantified previously (Zhao et al., 2018). HOM concentrations were also corrected for dilution due to the replenishment flow needed to maintain a constant overpressure of the chamber (loss rate ∼1 × 10−6 s−1) (Zhao et al., 2015b). The influence of wall loss correction and dilution correction on HOM yield was ∼12 % and <1 %, respectively. Although the wall loss rate of vapors in this study might not be exactly the same as in our previous photooxidation experiments (Zhao et al., 2018), HOM yield is not sensitive to the vapor wall loss rate. An increase in wall loss rate by 100 % or a decrease by 50 % only changes the HOM yield by 11 % and −6 %, respectively.
3.1 Overview of HOM
The mass spectra of HOM in the gas phase formed in the oxidation of isoprene by NO3 are shown in Fig. 1. A large number of HOM were detected. Almost all peaks are assigned HOM containing nitrogen atoms, with possibly few exceptions such as C5H10O8 and C5H8O11 with very minor peaks (≲1 % of the maximum peak). The reaction products can be roughly divided into three classes: monomers (C5, ∼200–400 Th), dimers (C10, ∼400–600 Th), and trimers (C15, ≳600 Th), according to their mass-to-charge ratio (). The detailed peak assignment of monomers, dimers, and trimers is discussed in the following sections.
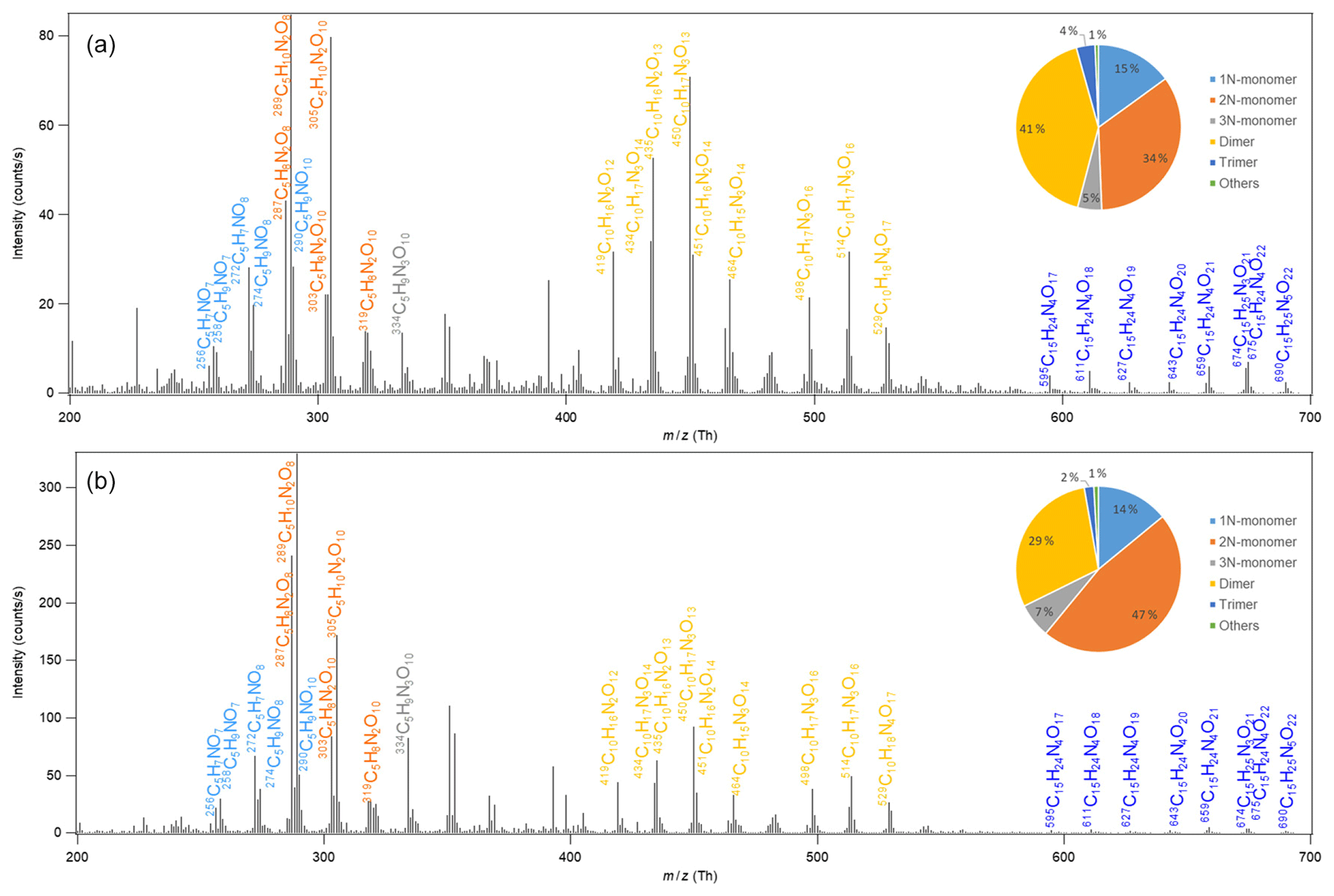
Figure 1Mass spectrum of the HOM formed in the oxidation of isoprene by NO3. HOM are detected as clusters with the reagent ion 15NO, which is not shown in the molecular formula in the figure for simplicity. Panels (a) and (b) show the average spectrum during the first isoprene addition period (P1) and for the whole period of six isoprene additions (P1–6), respectively. The insets show the contributions of different classes of HOM. 1–3N-monomer refers to the monomers containing one to three nitrogen atoms in the molecular formula.
3.2 HOM monomers and their formation
3.2.1 Overview of HOM monomers
HOM monomers showed a roughly repeating pattern in the mass spectrum at every 16 Th (corresponding to the mass of oxygen) (Fig. 1a). Here a number of series of HOM monomers with continuously increasing oxygenation were found, such as C5H9NOn, C5H7NOn, C5H8N2On, and C5H10N2On (Tables 1 and S1–2 and Fig. 2). These monomers included both stable closed-shell molecules and open-shell radicals, such as C5H8NOn• and C5H9N2On•. The open-shell molecules were likely RO2 radicals because of their much longer lifetime and hence higher concentrations compared with alkoxy radicals (RO) and alkyl radicals (R). Since the observed stable products were mostly termination products of RO2 reactions, we describe the stable products in a RO2-oriented approach. It is worth noting that some of the termination products may contain multiple isomers formed from different pathways.
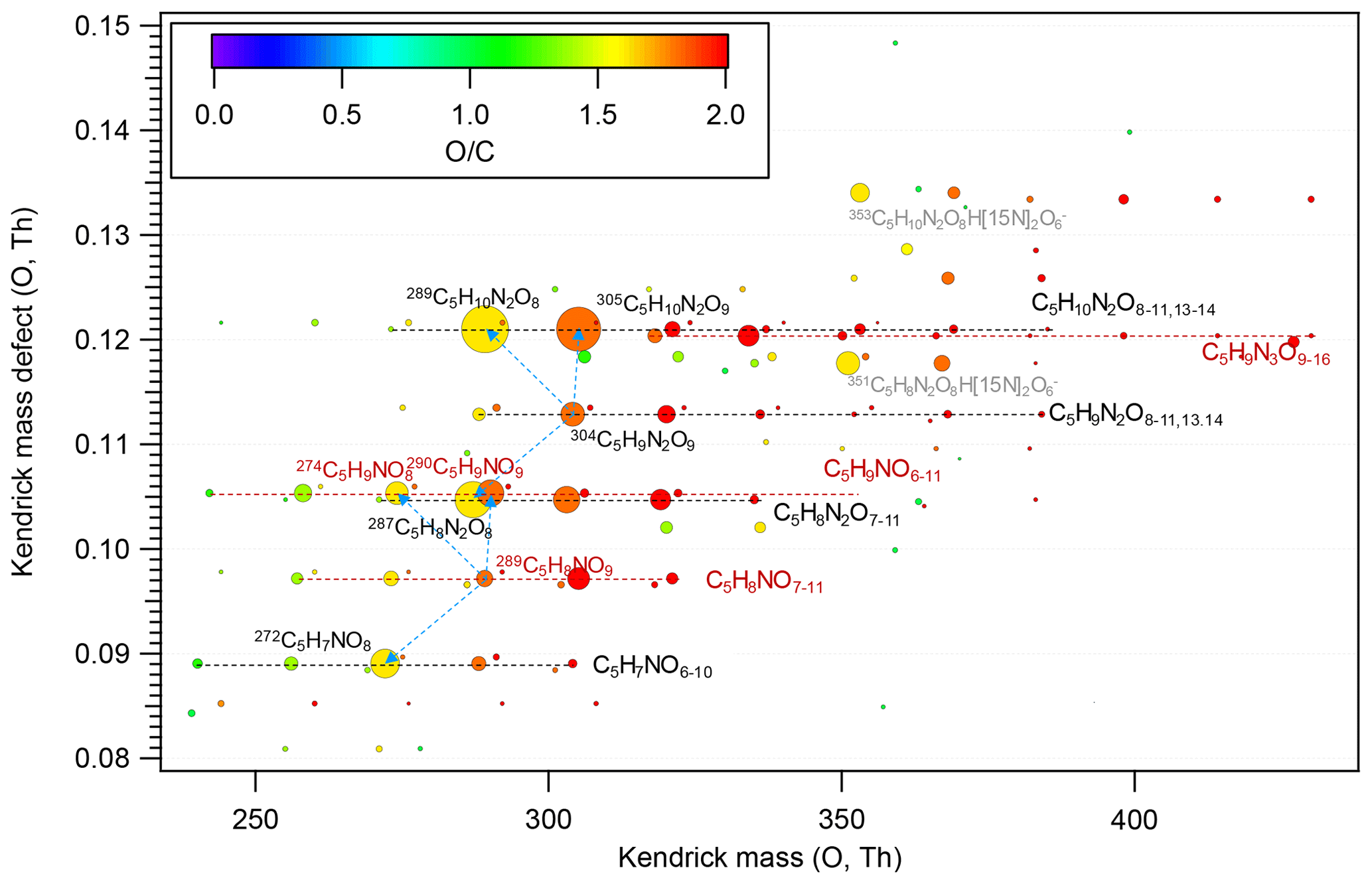
Figure 2Kendrick mass defect plot for O of HOM monomers. The in the molecular formula includes the reagent ion 15NO, which is not shown for simplicity. The size (area) of circles is set to be proportional to the average peak intensity of each molecular formula during the first isoprene addition period (P1). The species at 351 and 353 (labeled in grey) are the adducts of C5H8N2O8 and C5H10N2O8 with H[15N]2O, respectively. The blue dashed lines with arrows indicate the termination products hydroperoxide (M+H), alcohol (), and ketone (), with M the molecular formula of a HOM RO2.
Table 1HOM monomers formed in the oxidation of isoprene by NO3.
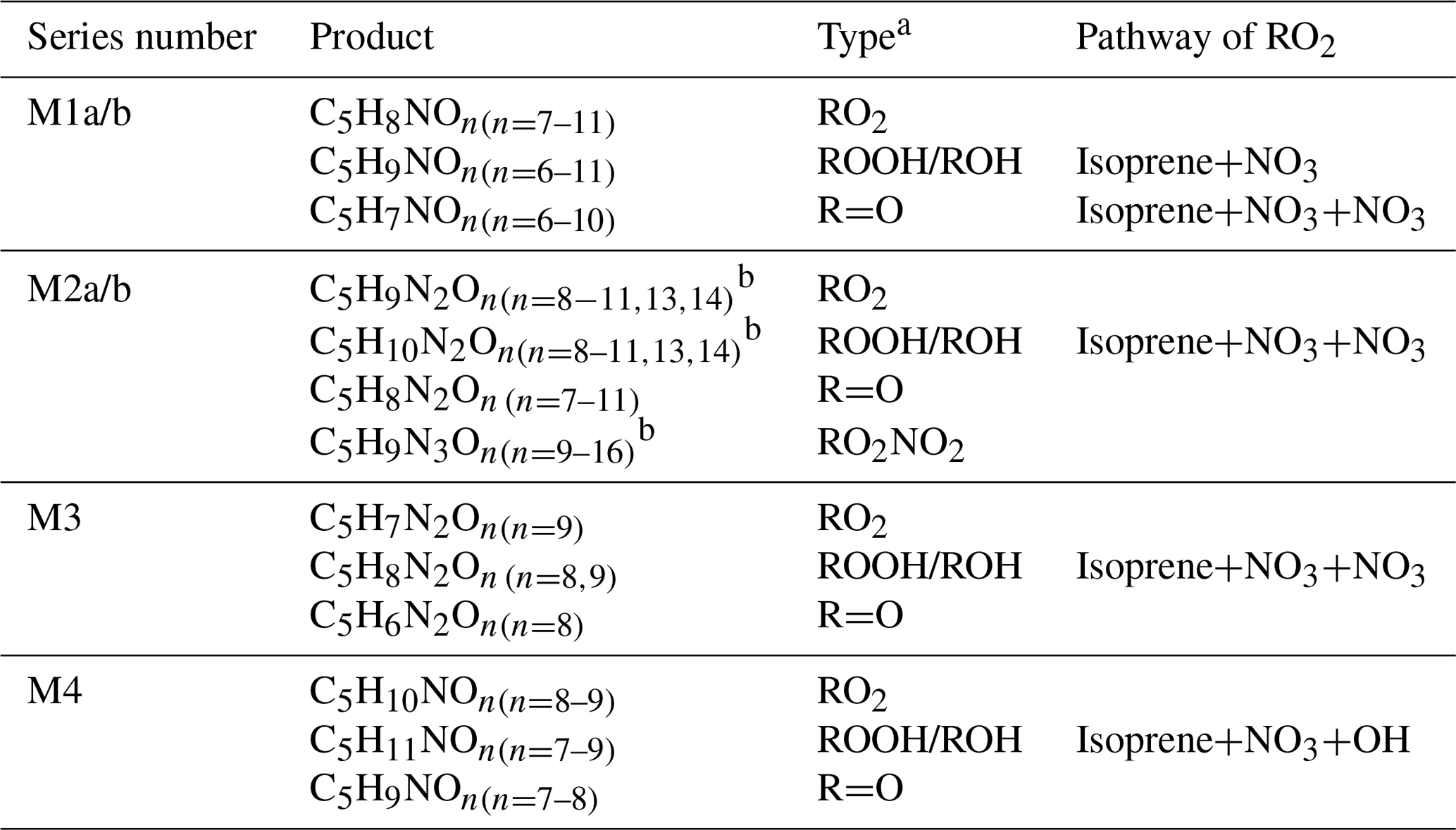
a RO2 denotes peroxy radical, and ROOH, ROH, R=O, and RO2NO2 denote the termination products containing hydroperoxy, hydroxyl, carbonyl group, and peroxynitrate, respectively. b Peak assignment of compounds with may be subject to uncertainties.
HOM monomers were classified into 1N-, 2N-, and 3N-monomers according to the number of nitrogen atoms that they contain. HOM without nitrogen atoms were barely observed except for very minor peaks (≲1 % of the maximum peak) possibly assigned to C5H10O8 and C5H8O11. The contribution of 2N-monomers such as C5H10N2On and C5H8N2On was higher than that of the 1N-HOM monomers, and that of 3N-monomers was the lowest (Fig. 1, inset). The most abundant monomers were C5H10N2O8, C5H10N2O9, and C5H8N2O8. The termination products of C5H9NO8, C5H9NO9, and C5H7NO8 also showed relatively high abundance. These limited numbers of compounds dominated the HOM monomers. Since 2N-monomers were second-generation products as discussed below, the higher-abundance 2N-monomers indicate that the second-generation HOM play an important role in the reaction of NO3 with isoprene in the reaction conditions of our study, as also seen by Wu et al. (2020). This is more evident for the mass spectrum averaged over six isoprene addition periods (Fig. 1b), where the abundances of C5H10N2On and C5H8N2On were more dominant. This observation is in contrast with the finding for the reaction of O3 with BVOC, which contains only one double bond such as α-pinene (Ehn et al., 2014), where HOM are mainly first-generation products formed via autoxidation. The higher abundance of HOM 2N-monomers than 1N-monomers is likely because HOM production rate via the autoxidation of 1N-monomer RO2 following the reaction of isoprene with NO3 may be slower than that of the reaction of 1N-monomers (including both HOM and non-HOM monomers) with NO3. We would like to note that some less oxygenated 1N-monomers such as C5H9NO and C5H7NO4 may have high abundance but are not detected by NO-CIMS and are not HOM and thus not included in HOM 1N-monomers.
3.2.2 1N-monomers
In our experiments we observed a C5H8NOn• (n=7–12) series (series M1) as well as its corresponding termination products C5H7NOn−1, C5H9NOn−1, and C5H9NOn via the reactions with RO2 and HO2, which contain carbonyl, hydroxyl, and hydroperoxy group, respectively. Overall, the peak intensities of C5H9NOn and C5H7NOn series first increased and then decreased as oxygen number increased (Fig. 2), with the peak intensity of C5H9NO8 and C5H7NO8 being the highest within their respective series when averaged over the whole experiment period.
C5H8NOn• with odd numbers of oxygen atoms (n=7, 9, 11, series M1a) were possibly formed by the attack of NO3 to one double bond, preferentially to C1 according to previous studies (Skov et al., 1992; Berndt and Böge, 1997; Schwantes et al., 2015) and followed by autoxidation (Scheme 1a). We would like to note that NO-CIMS only observed HOM with oxygen numbers ≥6 in this study due to its selectivity of detection. C5H8NOn• with even numbers of oxygen atoms (n=8, 10, series M1b in Table 1) were possibly formed after H shift of an alkoxy radical formed in Reactions (R4) or (R5) and subsequent O2 addition (“alkoxy–peroxy” channel) (Scheme 1b), where the alkoxy radicals can be formed from both the RO2+NO3 and RO2+RO2 reactions. The hydroxy RO2 formed can undergo further autoxidation, adding two oxygen atoms after each H shift. We would like to note that the scheme and other schemes in this study only show example isomers and pathways to form these molecules. It is likely that many of the reactions occurring are not the dominant channels as otherwise there would be much higher HOM yield as discussed below.
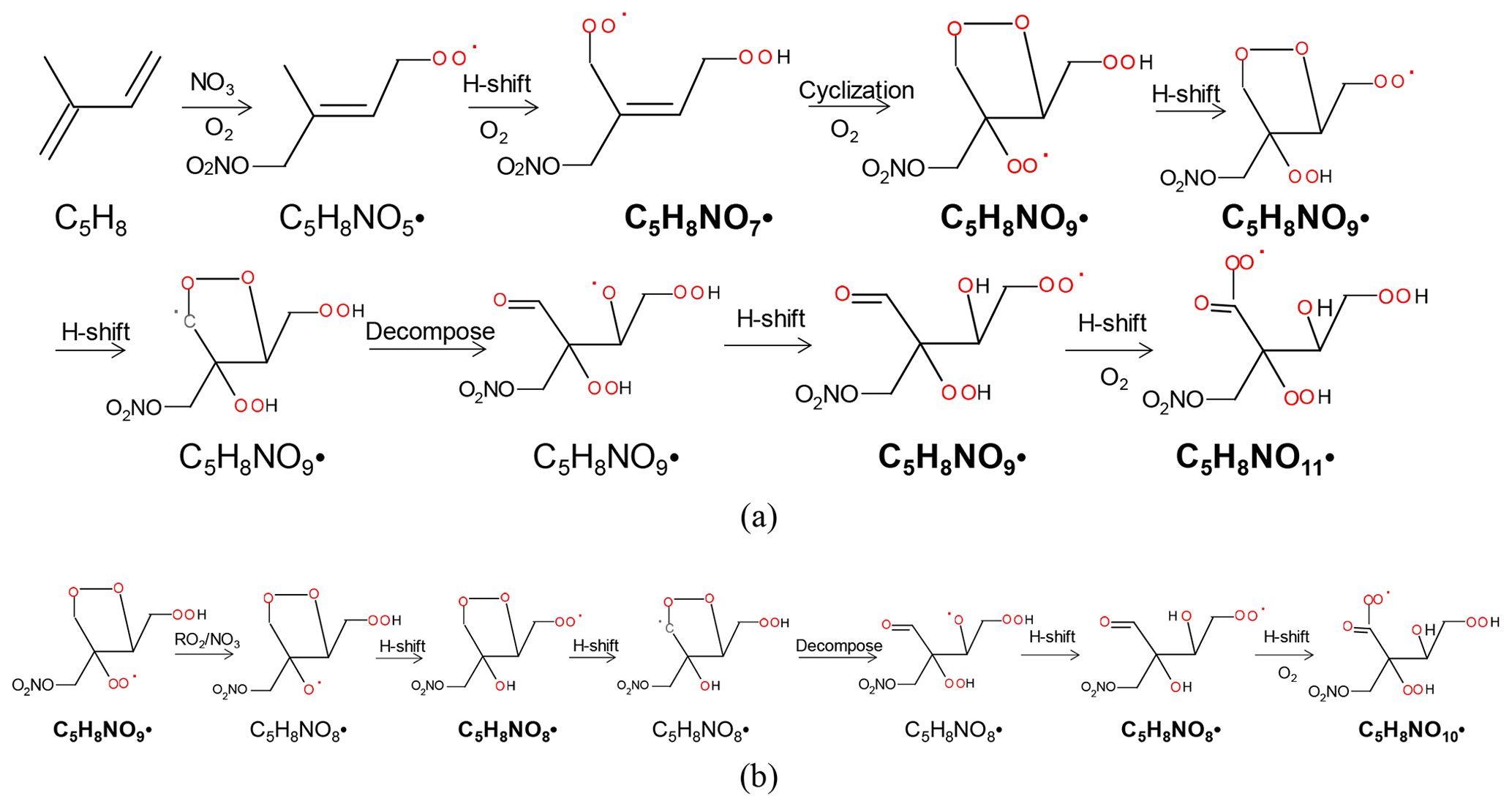
Scheme 1The example pathways to form HOM RO2 C5H8NOn• (n=7, 9, 11) series (a) and C5H8NOn• (n=8, 10) series (b) in the reaction of isoprene with NO3. The detected products are in bold.
Some HOM monomers may contain multiple isomers and be formed via different pathways. For example, C5H9NOn can contain alcohols derived from RO2 C5H8NOn+1•, hydroperoxides derived from RO2 C5H8NOn•, or the ketones from RO2C5H10NOn+1•. Some RO2 C5H8NOn• may be formed via the reaction of first-generation products with NO3 in addition to direct reaction of isoprene with NO3. For example, C5H8NO7• can be formed by the reaction of NO3 with C5H8O2, which is a first-generation product observed previously in the reaction of isoprene with NO3 or OH (Scheme S1b) (Kwan et al., 2012). Moreover, RO2 C5H8NOn• can be formed from C5-carbonylnitrate, a first-generation product, with OH (Scheme S1a). Trace amount of OH can be produced in the reaction of isoprene with NO3 (Kwan et al., 2012; Wennberg et al., 2018). OH can also be formed via Criegee intermediates formed in the isoprene + O3 reaction (Nguyen et al., 2016), but this OH source was likely minor because the contribution of the isoprene + O3 reaction to total isoprene loss was negligible (<5 %, Fig. S2). In addition, C5H8NO8• may also be formed by the reaction of NO3 with C5H8O3, which is a first-generation product observed in the reaction of isoprene with OH (Kwan et al., 2012). The C5H8NOn• formed via direct reaction of isoprene with NO3 is a first-generation RO2, while that formed via other indirect pathways is a second-generation RO2. The time profiles of the isomers from these two pathways, however, are expected to be different, as will be discussed below.
Time series of HOM can shed light on their formation mechanisms. It is expected that first-generation products will increase quickly with isoprene addition and reach a maximum earlier in the presence of wall loss of organic vapor, while second-generation products will reach a maximum in the later stage or increase continuously if the production rate is higher than the loss rate. As a reference to analyze the time profiles of HOM, the time profiles of isoprene, NO3, and N2O5 are also shown (Fig. S4). After isoprene was added in each period, NO3 and N2O5 dropped dramatically and then gradually increased. We found that termination products within the same M1 series showed different time profiles. For example, in C5H9NOn series, C5H9NO8 clearly increased instantaneously with isoprene addition and decreased quickly afterwards (Fig. 3a), indicating that it was a first-generation product, which was expected according to mechanism Scheme 1. C5H9NO6 and C5H9NO10 had a general increasing trend with time. While C5H9NO6 increased continuously with time, C5H9NO10 reached maximum intensity in the late phase of each isoprene addition period and then decreased naturally or after isoprene addition. The faster loss of C5H9NO10 than C5H9NO6 may result from the faster wall loss due to its lower volatility. C5H9NO7 and C5H9NO9 showed a mixing time profile with features of the former two kinds of time profiles, increasing almost instantaneously with isoprene additions, especially in the first two periods, while increasing continuously or decreasing first with isoprene additions and then increasing later in each period. This kind of time series indicates that there were significant contributions from both first- and second-generation products.
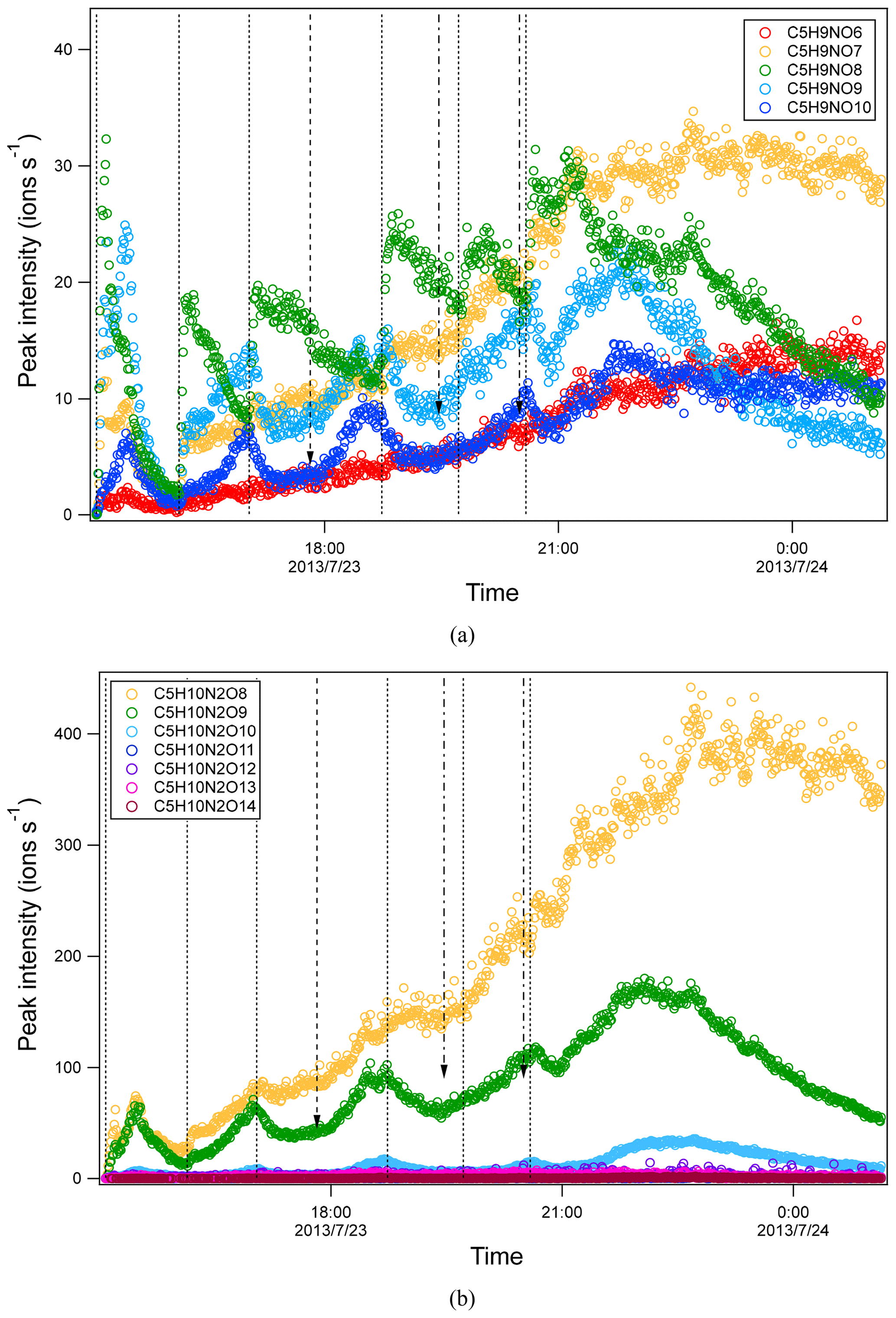
Figure 3Time series of peak intensity of several HOM monomers of C5H9NOn series (a) and of C5H10N2On series (b). They are likely the termination products of RO2 C5H8NOn• and C5H9N2On•, respectively. The dashed lines indicate the time of isoprene additions. The long-dashed arrow indicates the time of NO2 addition. The dash-dotted arrows indicate the time of O3 additions.
The second-generation products may be different isomers formed in pathways other than shown in Scheme 1. Second-generation C5H9NO6 can be formed via C5H8NO7•, which can also be formed by the reaction of NO3 and O2 with C5H8O2 as mentioned above (Scheme S2b) or by the reaction of OH with C5H7NO4 (Scheme S2a). The time profiles of C5H8NO7• did show more contribution of second-generation processes because C5H8NO7• continuously increased with time in general. If the pathways via the reaction of NO3 and O2 with C5H8O2 and the reaction of OH with C5H7NO4 contribute most to C5H9NO6, C5H9NO6 would show mostly a time profile of second-generation products. Similarly, second-generation C5H9NO7 can be formed via C5H8NO7• or C5H8NO8•. The time series of C5H8NO8• did show the contribution of both the first- and second-generation processes, which generally increased with time while also responding to isoprene addition (Fig. S5). Similarly to C5H9NO6, the second-generation pathways for C5H9NO7, C5H9NO9, and C5H9NO10 are shown in Schemes S1, S3, and S4. For the RO2 in C5H8NOn• series other than C5H8NO, the peak of C5H8NOn• overlaps with C5H10N2On in the mass spectra, which is a much larger peak and thus cannot be differentiated from C5H10N2On. Therefore, it is not possible to obtain reliable separate time profiles in order to differentiate their major sources. It is worth noting that nitrate CIMS may not be able to detect all isomers of C5H9NO6 due to the sensitivity limitation. Therefore, we cannot exclude the possibility that the absence of some first-generation isomers of C5H9NO6 was due to the low sensitivity of these isomers.
Among the termination products of the 1N-monomer RO2, carbonyl and hydroxyl/hydroperoxide species had comparable abundance in general (Table S1), suggesting that disproportionation reactions between RO2 and RO2 forming hydroxy and carbonyl species (R1–2) were likely an important RO2 termination pathway. However, dependence of the exact ratio of carbonyl species to hydroxyl/hydroperoxide species on the number of oxygen atoms did not show a clear trend (Table S1), suggesting that the reactions of HOM RO2 depended on their specific structure. There was no clear difference in the abundance between the termination products from C5H8NOn• with odd and even numbers of oxygen atoms in general, although the most abundant termination product of C5H8NOn•, i.e., C5H7NO8, was likely formed from C5H8NO9• in series M1a. This fact indicates that both the peroxy pathway and alkoxy–peroxy pathway were important for the HOM formation in the isoprene + NO3 reaction under our conditions, in agreement with the significant formation of alkoxy radicals from the reaction of RO2 with NO3 and RO2.
In addition to the termination products of RO2 M1, minor peaks of the RO2 series C5H10NOn• (n=8–9) (M4, Table 1) and their corresponding termination products including hydroperoxide, alcohol, and carbonyl species were detected (Table S3). C5H10NOn were likely formed by sequential addition of NO3 and OH to two double bonds of isoprene (Scheme S5). OH can react quickly with isoprene or with the first-generation products of the reaction of isoprene with NO3, thus forming C5H10NOn•. In addition, a few very minor but noticeable peaks of C5H9On• and their corresponding termination products C5H10On and C5H8On were also observed. These HOM may be formed by the reactions of isoprene with trace amounts of OH and with O3, although their contributions to reacted isoprene were negligible. These HOM were also observed in the reaction of isoprene with O3 with and without OH scavengers (Jokinen et al., 2015).
Among 1N-monomer HOM, C5H9NO7 has been observed in the particle phase using ESI-TOFMS by Ng et al. (2008), while others have not been observed in previous laboratory studies of the reaction of isoprene with NO3, to our knowledge. A number of C5 organic nitrates have been observed in field studies. For example, C5H7–11NO6–8 and C5H7–11NO4–9 have been observed in the gas phase (Massoli et al., 2018) and the particle phase (Lee et al., 2016; Chen et al., 2020), respectively, in a rural area of the southeastern US, where isoprene is abundant. Xu et al. (2021) observed a number of C5 1N-HOM such as C5HNO6,7 in the polluted megacities of Nanjing and Shanghai of eastern China during summer. While many of these HOM have daytime sources and are attributed to photooxidation in the presence of NOx, nighttime oxidation with NO3 also contributed to their formation (Lee et al., 2016; Chen et al., 2020; Xu et al., 2021). C5H7–11NO4–9 were also observed in chamber experiments of the reaction of isoprene with OH in the presence of NOx (Lee et al., 2016). C5HxNO4–9 and C5HxNO4–10 have also been observed in the gas phase and particle phase, respectively, in a monoterpene-dominating rural area in southwestern Germany (Huang et al., 2019).
3.2.3 2N-mononmers
The 2N-monomer RO2 series C5H9N2On• (n=8–14) were observed, as well as its likely termination products, C5H8N2On and C5H10N2On, which contain a carbonyl and hydroxyl or hydroperoxide functional group, respectively. The RO2 series C5H9N2On• with an odd number of oxygen atoms (n=9, 11) (M2a in Table 1) were likely formed from the first-generation product C5H9NO4 (C5-hydroxynitrate) by adding NO3 to the remaining double bond, forming C5H9N2O9•, followed by autoxidation (Scheme 2a). This RO2 series can also be formed by the addition of NO3 to the double bond of first-generation products (e.g., C5H9NO5, C5-nitrooxyhydroperoxide) and a subsequent alkoxy–peroxy step (Scheme 2b). C5H9N2On• with an even number of oxygen atoms (n=8, 10, 12) (M2b in Table 1) can be formed by the addition of NO3 to the double bond of C5H9NO5 followed by autoxidation (Scheme 3a) or of C5H9NO4 followed by an alkoxy–peroxy step (Scheme 3b). The formation pathways of C5H9N2O and C5H9N2O8• cannot be well explained, as they contain too many or too few oxygen atoms to be formed via the pathways in Scheme 2 or 3. In Schemes 2 and 3, we show the reactions starting from 1-NO3-isoprene-4-OO as an example. In the Supplement, we have also shown the pathways starting from 1-NO3-isoprene-2-OO peroxy radicals, which is indicated in a recent study by Vereecken et al. (2021) to be the dominant RO2 in the reaction of isoprene with NO3.
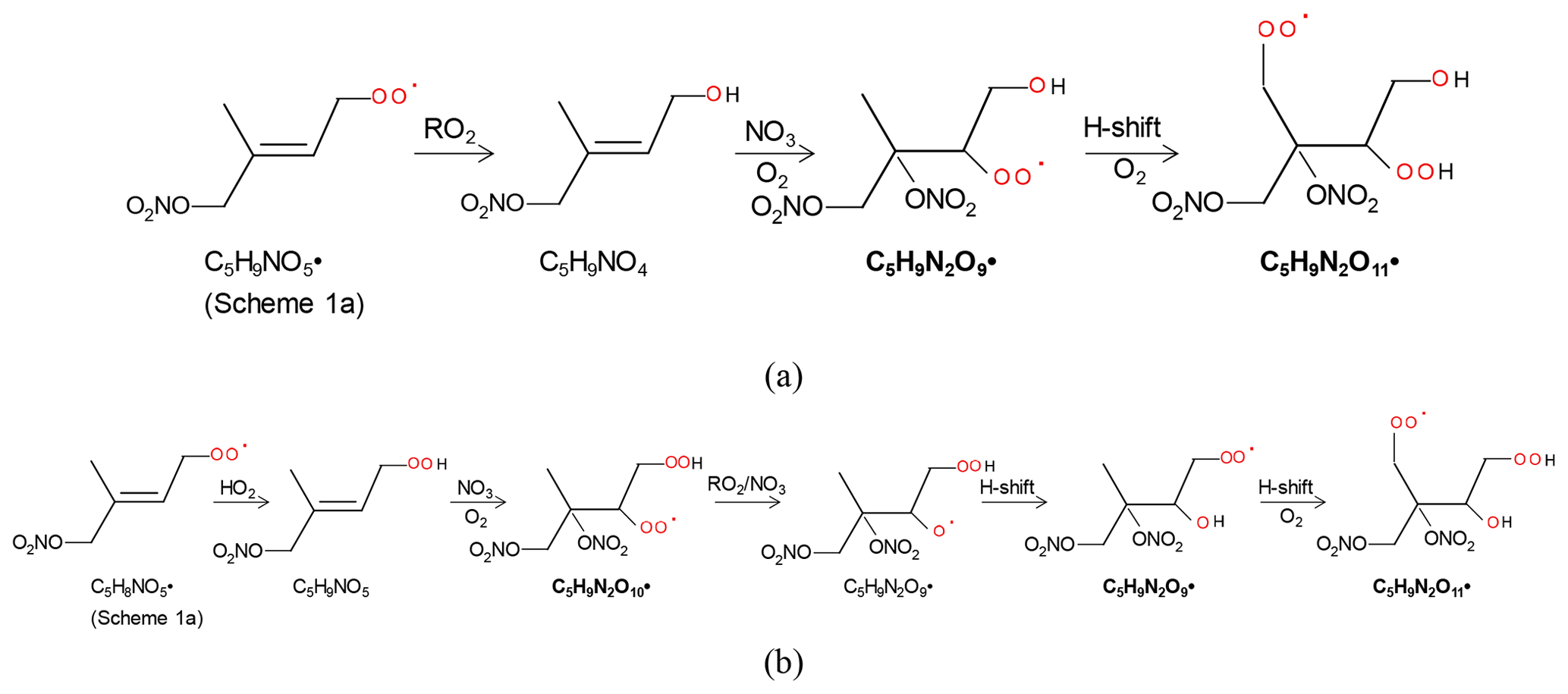
Scheme 2The example pathways to form C5H9N2On (n=9, 11) HOM RO2 series by RO2 channel (a) and alkoxy–peroxy channel. The detected products are in bold.
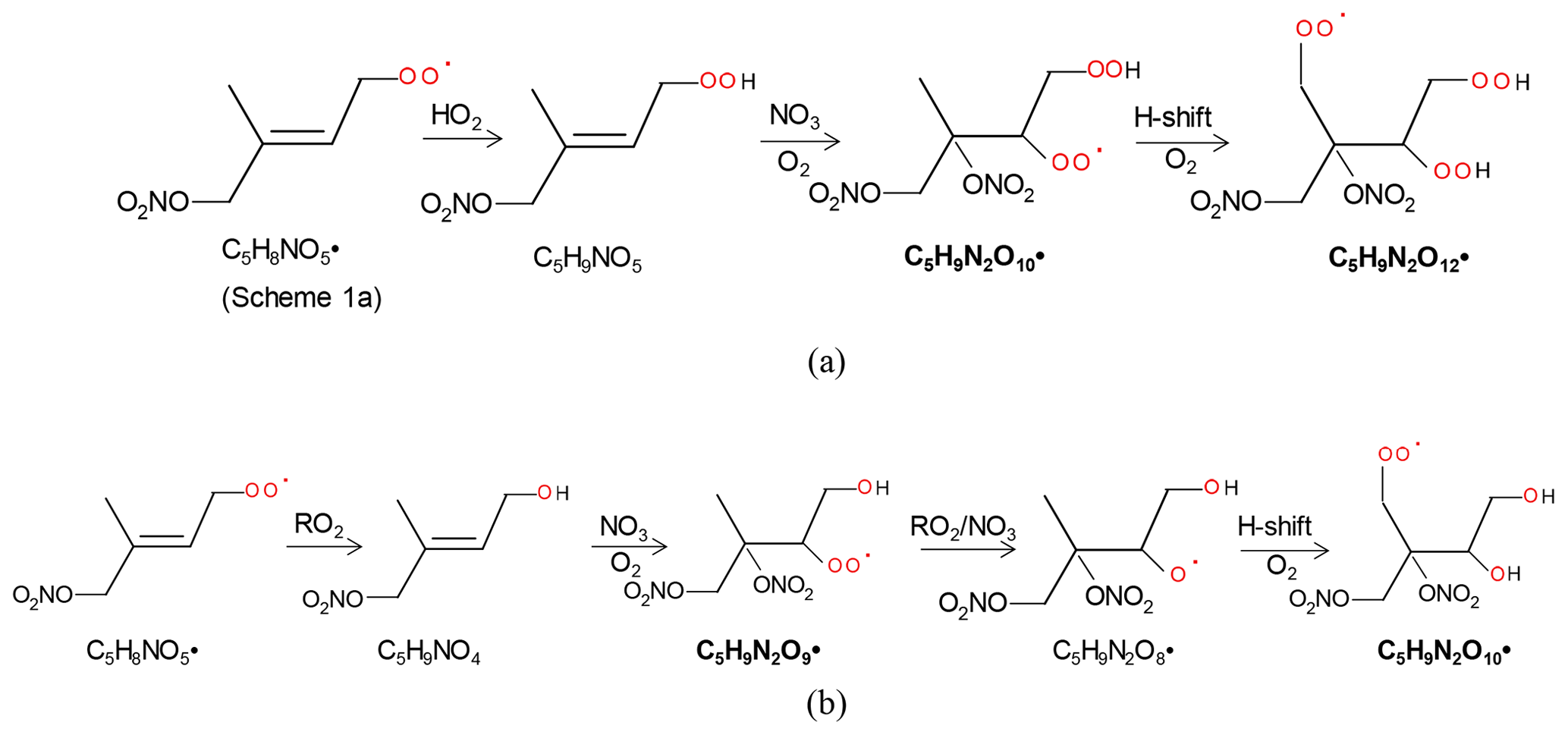
Scheme 3The example pathways to form C5H9N2On (n=10, 12) HOM RO2 series by RO2 channel (a) and alkoxy–peroxy channel (b). The detected products are in bold.
Formation through either Scheme 2 or 3 means that C5H8N2On and C5H10N2On were second-generation products. The time series of C5H10N2On species clearly indicates that they were indeed second-generation products. C5H10N2On species generally did not increase immediately with isoprene addition (Fig. 3b) but increased gradually with time and reached its maximum in the later stage of each period before decreasing with time (in periods 1 and 6) or decreasing after the next isoprene addition (periods 2–5). This time profile can be explained by the time series of the precursor of C5H10N2On, C5H9N2On• (RO2) (Fig. S6). The changing rate (production rate minus destruction rate) of C5H10N2On concentration was dictated by the concentration of C5H9N2On• and the wall loss rate. During periods 2 to 5, C5H9N2On• gradually increased but decreased sharply after the isoprene additions, resulting from chemical reactions of C5H9N2On• and additionally from wall loss. When the rate of change of the C5H10N2On concentration was positive, the concentration of C5H10N2On increased with time. After isoprene additions, the rate of change of the C5H10N2On concentration decreased dramatically to even negative, leading to decreasing concentrations. Similarly to C5H10N2On, the C5H8N2On series did not respond immediately to isoprene additions (Fig. S7), which is expected for second-generation products according to the mechanism discussed above (Schemes 2–3). Particularly, the continuing increase in C5H8N2On even after isoprene was completely depleted (at ∼ 21:40, Fig. S7) clearly indicates that these compounds were second-generation products, although in the end they decreased due to wall loss.
According to the finding by Ng et al. (2008), C5-hydroxynitrate decays much more quickly than C5-nitrooxyhydroperoxides. Additionally, C5-hydroxynitrate concentration is expected to be higher than that of nitrooxyhydroperoxides because RO2+RO2 forming alcohol is likely more important than RO2+HO2 forming hydroperoxide in this study. Therefore, it is likely that C5H9N2On• M2a series were mainly formed from C5H9NO4 instead of C5H9NO5, while C5H9N2On• M2b were formed from C5H9NO4 followed by an alkoxy–peroxy step. That is, Schemes 2a and 3b appear more likely.
Similarly to C5H8NOn•, the intensity of carbonyl species from C5H9N2On• was also comparable with that of hydroxyl/hydroperoxide species, suggesting that RO2+RO2 reaction forming ketone and alcohol was likely an important pathway of HOM formation in the isoprene + NO3 reaction. In general, the intensities of the termination products from C5H9N2On• with both even and odd oxygen numbers were comparable. This again suggests that both peroxy and alkoxy–peroxy pathways were important for HOM formation in the isoprene + NO3 reaction. The intensity of C5H8N2On first increased and then decreased with oxygen number, while C5H10N2On decreased with oxygen number, with C5H10N2O8 and C5H8N2O8 being the most abundant within their respective series.
Some 2N-monomers have been detected in previous studies of the reaction of isoprene with NO3. C5H10N2O8 was detected in the particle phase by Ng et al. (2008), and C5H8N2O7 was detected in the gas phase by Kwan et al. (2012). C5H9N2O9• has been proposed to be formed via the pathway as in Scheme 2a (Ng et al., 2008), and it was directly detected in our study. C5H8N2O7 species has been proposed as a dinitrooxy epoxide formed by the oxidation of nitrooxyhydroperoxide (Kwan et al., 2012) instead of being a dinitrooxy ketone proposed in our study, a termination product of C5H9N2O8•. Admittedly, C5H8N2O7 may contain both isomers. In addition, Ng et al. (2008) detected C5H8N2O6 in the gas phase, which was not detected in this study, likely due to the selectivity of NO-CIMS. 2N-monomers have also been observed in previous field studies. For example, Massoli et al. (2018) observed C5H10N2O8–10 in rural Alabama, US, during the SOAS campaign. Xu et al. (2021) observed C5H8,10N2O8 and C5H10N2O8 in the polluted megacities of Nanjing and Shanghai during summer.
One could suppose that C5H7N2On• should also be formed since C5-nitrooxycarbonyl (C5H7NO4) also contains one double bond that can be attacked by NO3 in a second oxidation step. However, concentrations of C5H7N2On were too low to assign molecular formulas with confidence except for C5H7N2O9•, clearly showing that C5H7N2On• was not important. This fact is consistent with the finding by Ng et al. (2008) that C5-nitrooxycarbonyls react slowly with NO3. Additionally, the peroxy radical formed in the reaction of C5-nitrooxycarbonyls with NO3 likely leads to more fragmentation in H shift as found in the OH oxidation of methacrolein (Crounse et al., 2012), which may also contribute to the low abundance of C5H7N2On. The presence of HOM containing two N atoms is in line with the finding by Faxon et al. (2018), who detected products containing two N atoms in the reaction of NO3 with limonene, which also contain two carbon double bonds. It is anticipated that for VOC with more than one double bond, NO3 can add to all the double bonds as for isoprene and limonene.
3.2.4 3N-monomers
HOM containing three nitrogen atoms, C5H9N3On (n=9–16), were observed. These compounds were possibly peroxynitrates formed by the reaction of RO2 (C5H9N2On•) with NO2. The time series of C5H9N3On was examined to check whether they match such a mechanism. If C5H9N3On were formed by the reaction of C5H9N2On−2• with NO2, the concentration would be a function of the concentrations of C5H9N2On−2• and NO2 as follows:
where [C5H9N3On], [C5H9N2On−2•], and [NO2] are the concentrations of these species, k is the rate constant, and kwall is the wall loss rate. Because the products of C5H9N2On−2• and NO2 were at their maximum at the end of each period and decreased rapidly after isoprene addition (Fig. S8), the concentration should have its maximum increasing rate at the end of each isoprene addition period. However, we found that only C5H9N3O showed such a time profile (Fig. S9), while C5H9N3O generally increased with time, different from what one would expect based on the proposed pathway. Therefore, it is likely that C5H9N3O were mainly formed via the reaction of C5H9N2On• with NO2, whereas C5H9N3O were not. Moreover, C5H9N3O9 cannot be explained by the reaction C5H9N2On• (n≥9) with NO2 or NO3, because these reactions would add at least one more oxygen atom. One possible pathway to form C5H9N3O9 was the direct addition of N2O5 to the carbon double bond of C5-hydroxynitrate, forming a nitronitrate. Such a mechanism was proposed previously in the heterogeneous reaction of N2O5 with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) because -NO2 and -NO3 groups were detected (Lai and Finlayson-Pitts, 1991). This pathway generally matched the time series of C5H9N3O typical of second-generation products since C5-hydroxynitrate was a first-generation product. It is possible that the main pathway of C5H9N3O was the reaction of C5H9NO with N2O5, although the reaction of N2O5 with C=C double bonds in common alkenes and unsaturated alcohols is believed to be unimportant (Japar and Niki, 1975; Pfrang et al., 2006).
3N-monomer, C5H9N3O10, has been observed in the particles formed in the isoprene + NO3 reaction by Ng et al. (2008). Here a complete series of C5H9N3On was observed. C5H9N3O10 was previously proposed to be formed by another pathway, i.e., the reaction of RO2 (C5H9N2O9•) and NO3 (Ng et al., 2008). We further examined the possibility of such a pathway in our study. Similarly to NO2, if C5H9N3On were formed by the reaction of C5H9N2On−2• with NO3, the concentration would have its maximum increasing rate at the end of each isoprene addition period. Among C5H9N2On•, the precursors of C5H9N3On, C5H9N2O showed a maximum increasing rate and a subsequent decrease after isoprene addition. The difference in oxygen number between C5H9N3O, the termination products, and C5H9N2O, the corresponding RO2 with the consistent time profile, is mostly two. Since the reaction of C5H9N2On with NO2 and NO3 results in increased oxygen numbers by two and by one, respectively, we infer that it is more likely that C5H9N3O were formed by the reaction of C5H9N2O with NO2 rather than NO3, and thus they were likely peroxynitrates rather than nitrates formed by the reaction of RO2 with NO3. Since alkyl peroxynitrates decompose rapidly (Finlayson-Pitts and Pitts, 2000; Ziemann and Atkinson, 2012), it is possible that these compounds contained peroxyacylnitrates.
Little attention has been paid to the RO2+NO2 pathway in nighttime chemistry of isoprene in the literature (Wennberg et al., 2018), which is likely due to the instability of the products. According to this pathway, C5H8N2On, which was proposed as a ketone formed via C5H9N2O9• in the M2 series (Table 1) as discussed above, can also comprise peroxynitrate formed by the reaction of C5H8NOn• (M1a RO2) with NO2. 3N dimers such as C5H9N3O10 have been observed in a recent field study in polluted cities in eastern China (Xu et al., 2021).
3.3 HOM dimers and their formation
A number of HOM dimer series were observed, including C10H16N2On (n=10–17), C10H17N3On (n=11–19), and C10H18N4On (n=15–18), C10H18N2On (n=10–16), C10H15N3On (n=13–17), and C10H19N3On (n=14–15) series (Tables 2, S3). C10H16N2On series (dimer 1, Table 2) was likely formed by the accretion reaction of two monomer RO2 of M1a/b (Reaction R7).
Similarly, C10H18N4On series (dimer 2, Table 2) were likely formed by the accretion reaction of two monomer RO2 of M2 (Reaction R8). As n1 and n2 are ≥9, the number of oxygen in C10H18N4On is expected to be ≥16. This is consistent with our observation that only C10H18N4On with n≥16 had significant concentrations.
Table 2HOM dimers and trimers formed in the oxidation of isoprene by NO3.

a ROOR denotes organic peroxide. b The numbering refers to Table 1.
C10H17N3On series (dimer 3, Table 2) were likely formed by the cross-accretion reaction of one M1 RO2 and one M2 RO2 (Reaction R9). Since n1 is ≥5 and n2 is ≥9, the number of oxygen atoms in C10H17N3On is expected to be ≥12, which is also roughly consistent with our observation that only C10H17N3On with n≥11 were detected.
Similarly, C10H18N2On(n=10–16) and C10H15N3On(n=13–17) series (dimer 4, dimer 5, Table 2) were likely formed from the accretion reaction between one M1 RO2 and one M4 RO2 and between one M1 RO2 and one M3 RO2 (C5H7N2O9•). Other dimer series than dimers 1–5 were also present. However, they had quite low intensity (Fig. 4), which was consistent with the low abundance of their parent monomer RO2. They can be formed from various accretion reactions of monomer RO2. For example, C10H19N3On can be formed by the accretion reaction of C5H9N2On• and C5H10NOn• (Table 2).
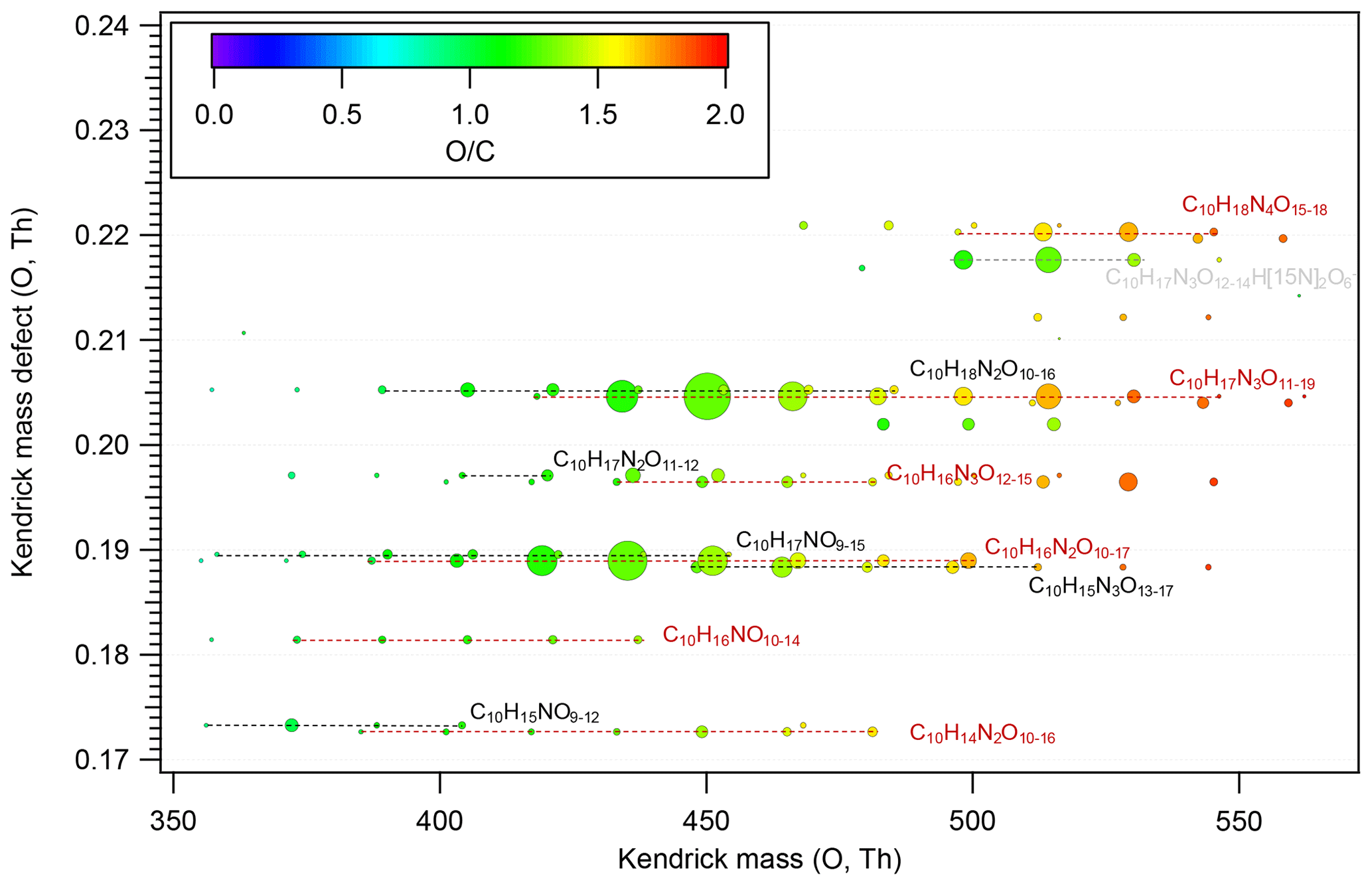
Figure 4Kendrick mass defect plot for O of HOM dimers formed in the isoprene + NO3 reaction. The size (area) of circles is set to be proportional to the average peak intensity of each molecular formula during the first isoprene addition period (P1). The molecular formula includes the reagent ion 15NO, which is not shown for simplicity. The species labeled in grey (C10H17N3O12–14 H[15N]2O) are the adducts of C10H17N3O12–14 with H[15N]2O.
Similarly to monomers, a few species dominated in the HOM dimer spectrum. The dominant dimer series were C10H17N3Ox and C10H16N2Ox series, with C10H17N3O12–14 and C10H16N2O12–14 showing the highest intensity among each series (Fig. 4). In addition, the O C ratio or oxidation state of HOM dimers was generally lower than that of monomers (Figs. 2, 4), which resulted from the loss of two oxygen atoms in the accretion reaction of two monomer RO2.
According to the mechanism above (Reactions R7–R9), we attempt to explain the relative intensities of the dimers using the signal intensities of monomer RO2. Assuming that the rate constant for each of the HOM-RO2 + HOM-RO2 reactions forming dimers is the same considering that all HOM-RO2 are highly oxygenated with a number of functional groups, it is expected that the dimer formed by the recombination between the most abundant RO2 has the highest intensity. The most abundant monomers RO2 were C5H9N2O9• and C5H9N2O10•, and thus the most abundant dimers are expected to be C10H16N4O16, C10H16N4O17, and C10H16N4O18. This expected result is in contrast with our observation showing that the most abundant dimers were C10H17N3O12–14 and C10H16N2O12–14 (Fig. 4). The discrepancy is possibly attributed to the presence of less oxygenated RO2 (with O ≤ 5) that have a low detection sensitivity in the NO3-CIMS (Riva et al., 2019) due to their lower oxygenation compared with other HOM RO2 shown above. These RO2 may react with C5H9N2O9• and C5H9N2O10•. For example, C5H8NO5• (RO2) is proposed as an important first-generation RO2 in the oxidation of isoprene by NO3 (Ng et al., 2008; Rollins et al., 2009; Kwan et al., 2012; Schwantes et al., 2015). Although C5H8NO5• showed a very low signal in our mass spectra, it was likely to have high abundance since it was the first RO2 formed in the reaction of isoprene with NO3. Indeed, we found that the termination products of C5H8NO5• such as C5H9NO5, C5H7NO4, and C5H9NO4 had high abundance in another study, indicating the high abundance of C5H8NO5•. The accretion reaction of C5H8NO5• with C5H9N2O9–10• and C5H8NO9–10• can explain the high abundance of C10H17N3O12–14 and C10H16N2O12–14 among all dimers.
Provided that C5H8NO5• is abundant, we still cannot explain the relative intensities of C10H17N3O12, C10H17N3O13, and C10H17N3O14 that were all formed by the accretion reaction with C5H8NO5•. C10H17N3O12 should have the highest intensity among C10H17N3O12–14 as its precursor RO2, C5H9N2O9•, is the most abundant. This suggests that accretion reactions other than those of C5H8NO5• with C5H9N2O9–10• also contributed to C10H17N3O12–14. Admittedly, the assumption of different RO2 having similar rate constants in accretion reactions may not be valid. For example, self-reaction of tertiary RO2 is slower than secondary and primary RO2 (Jenkin et al., 1998; Finlayson-Pitts and Pitts, 2000). Different rate constants may also lead to the observation that the most abundant dimers could not be explained by the most abundant RO2.
The time profiles of C10H16N2On indicate contributions of both the first- and second-generation products. The dominance of the first- or second-generation products depended on the specific compounds. Most C10H16N2On compounds increased instantaneously after isoprene additions, indicating significant contributions of first-generation products. Since the formation of C10H16N2On likely involved C5H8NO5• as discussed above, the instantaneous increase may result from the increase in C5H8NO5• as well as other first-generation RO2. After the initial increase, C10H16N2O10–12 then decayed with time (Fig. 5), while C10H16N2O13–15 increased again in the later phase of a period and when NO2 and O3 were added. The second increase indicated that C10H16N2O13–15 may contain more than one isomer, which had different production pathways. As discussed above, C5H8NOn• can be either a first-generation RO2 formed directly via the reaction of isoprene with NO3 and autoxidation or a second-generation RO2, e.g., formed via the reaction of C5H8O2 with NO3. Therefore, the second increase in C10H16N2O13–15 may result from the reaction of two first-generation RO2 and of two second-generation RO2 or between one first-generation and one second-generation RO2. The increase in C10H16N2O14–15 after isoprene addition was not large, indicating the larger contributions from second-generation products compared with other C10H16N2On. Overall, as the number of oxygen increased, the contribution of second-generation products to C10H16N2On increased.
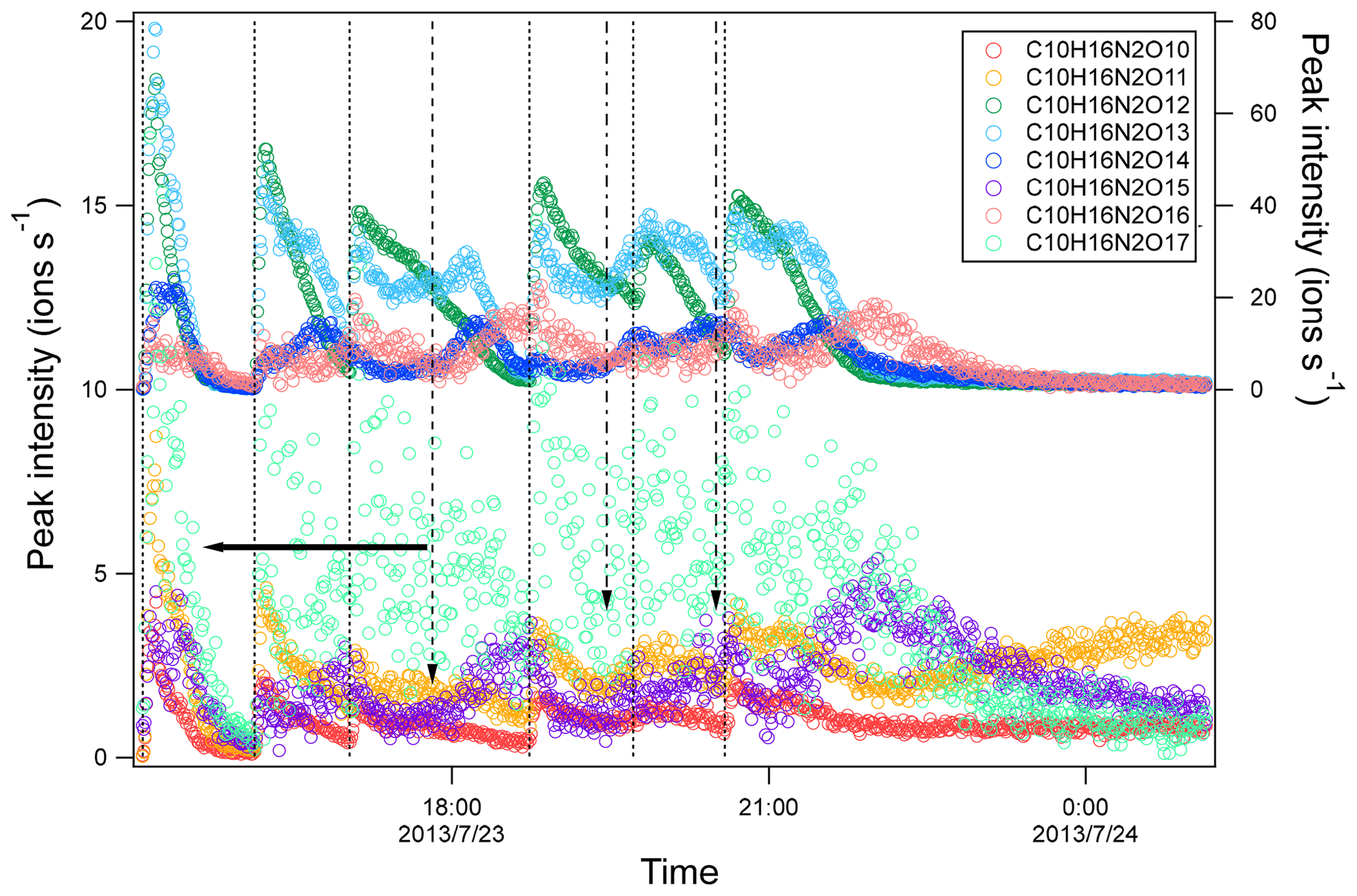
Figure 5Time series of peak intensity of several HOM dimers of C10H16N2On series. The dashed lines indicate the time of isoprene additions. The long-dashed arrow indicates the time of NO2 addition. The dash-dotted arrows indicate the time of O3 additions. The horizontal arrows indicate y-axis scales for different markers.
In contrast to C10H16N2On series, C10H18N4On increased gradually after each isoprene addition and then decreased afterward (Fig. 6), either naturally or after isoprene additions, which is typical for second-generation products. Since C10H18N4On was likely formed by the accretion reaction of C5H9N2On• (RO2), the time profile of C10H18N4On was as expected since C5H9N2On• was formed via the reaction of NO3 with first-generation products C5H9NOn. The C10H18N4On concentration depended on the product of the concentrations of two C5H9N2On•. Taking C10H18N4O16 as an example, its concentration can be expressed as follows:
When the concentration of C5H9N2O9• increased, the changing rate of C10H18N4O16 was positive and increased, and thus the concentration of C10H18N4O16 increased. When the concentration C5H9N2O9• decreased sharply after isoprene additions, the changing rate of C10H18N4O16 decreased and even became negative values, and thus the concentration of C10H18N4O16 decreased after isoprene addition.
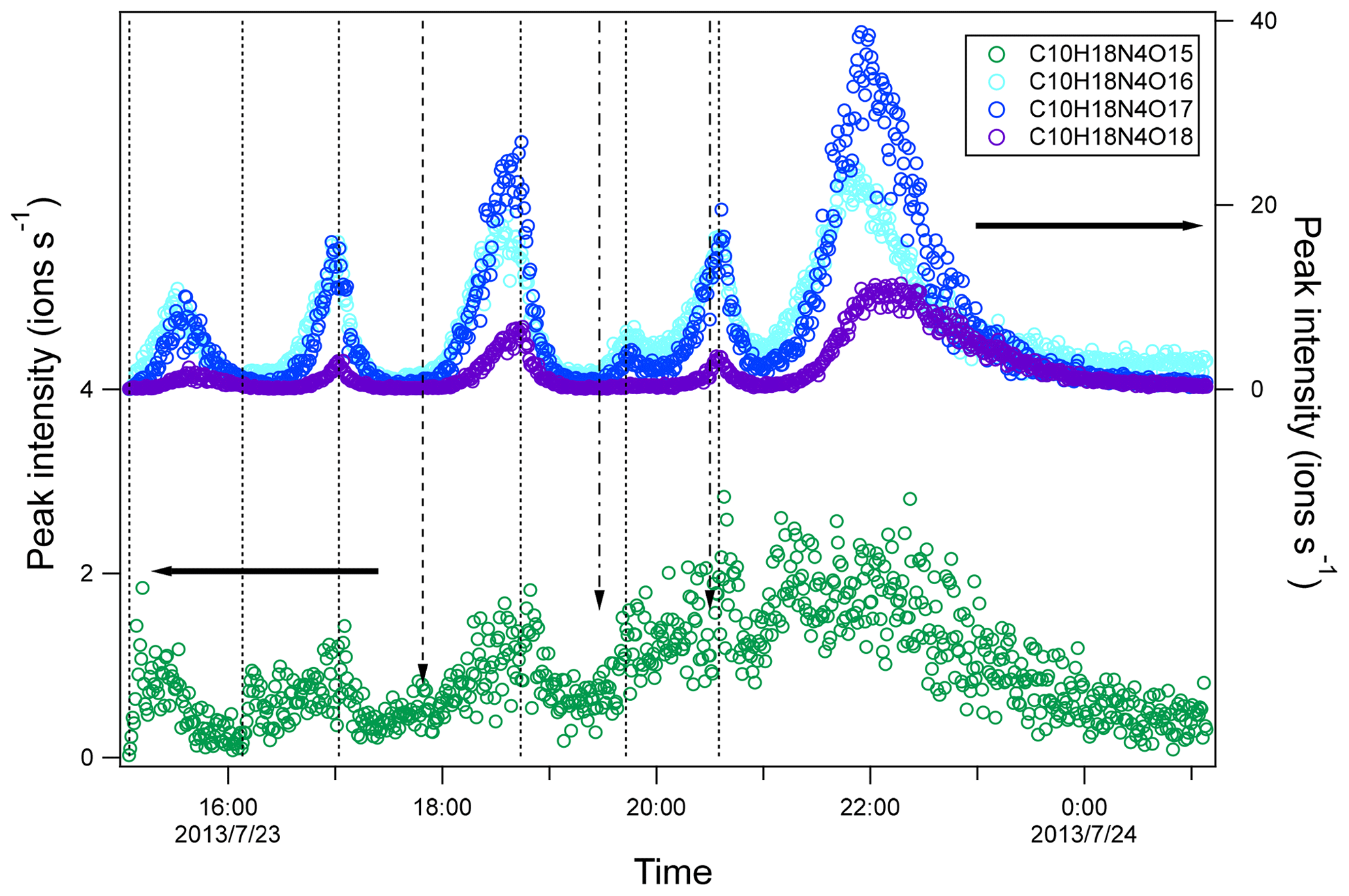
Figure 6Time series of peak intensity of several HOM dimers of C10H18N4On series. The dashed lines indicate the time of isoprene additions. The long-dashed arrow indicates the time of NO2 addition. The dash-dotted arrows indicate the time of O3 additions. The horizontal arrows indicate y-axis scales for different markers.
Similarly to the C10H16N2On series, while C10H17N3On first increased instantaneously with isoprene addition, it increased again during the later stage of each period (Fig. S10), showing a mixed behavior of the first-generation products and second-generation products. The time series of C10H17N3On was as expected in general because C10H17N3On was likely formed via the accretion reaction of C5H8NOn• (M1 RO2) and C5H9N2On• (M2 RO2), which were first- or second-generation, and second-generation RO2, respectively.
Some dimers that cannot be explained by accretion reactions, such as C10H16N3On(n=12–15)•, C10H17N2On(n=11–12)•, C10H16NOn(n=10–14)•, C10H15NOn(n=9–12), and C10H17NOn(n=9–15), were also observed. These dimers had low abundance. We note that due to their low signals in the mass spectra, their assignment and thus range of n may be subject to uncertainties. Since C10H16NOn(n=10–16)•, C10H16N3On(n=12–15)•, and C10H17N2On• contain unpaired electrons, they cannot be formed via the direct accretion reaction of two RO2. Instead, C10H16N3On(n=12–15)• (dimer R1) and C10H17N2On• (dimer R2) were likely RO2 formed by the reaction of HOM dimers containing a double bond (dimer 1) with NO3 and with OH, respectively, followed by the reaction with O2.
The corresponding termination products of C10H16N3On• RO2 series such as C10H15N3On (ketone) and C10H17N3On (hydroperoxide/alcohol) were also observed, although these compounds can also be formed via reactions between two RO2 radicals (Reaction R9 and Table 1). Among the termination products, C10H15N3On had low intensity. Reaction (R13) and the termination reaction of C10H17N2On• with HO2 provided an additional pathway to C10H17N3On besides the Reaction (R9) pathway discussed above. Similarly, other dimers may also be formed by the termination reactions of dimer RO2 with RO2 or HO2. For example, C10H18N4On can be formed via termination reaction of C10H17N4On• with another RO2 wherein C10H17N4On• can be formed as follows.
C10H16NOn(n=10–14)• could be explained by the reaction of monomer RO2 with isoprene.
Only C10H16NOn• with n≥10 were detected, while according to the mechanism of self-reaction between C5H8NOn•, the n range of C10H16NOn• is expected to be 7–14. The absence of C10H16NOn(n<10)• is likely attributed to their low abundance, which might result from low precursor concentrations, low reaction rates with isoprene, and/or faster reactive losses with other radicals. Such a reaction of RO2 with isoprene has been proposed by Ng et al. (2008) and Kwan et al. (2012). The corresponding termination products of C10H16NOn• are C10H15NOn (ketone) and C10H17NOn species (hydroperoxide/alcohol). C10H17NOn species showed a time profile of typical first-generation products (Fig. S11), i.e., increasing immediately with isoprene addition and then decaying with time. This behavior further supports the possibility of Reaction (R13). Yet the reaction rate of alkene with RO2 is likely low due to the high activation energy (Stark, 1997, 2000). It is worth noting that to our knowledge no experimental kinetic data on the addition of RO2 to alkenes in the gas phase in atmospheric relevant conditions are available, though fast, low-barrier ring closure reactions in unsaturated RO2 radicals have been reported (Vereecken and Peeters, 2004, 2012; Kaminski et al., 2017; Richters et al., 2017; Chen et al., 2021). We would like to note that there is unlikely interference to C10-HOM from monoterpenes, which has been reported previously (Bernhammer et al., 2018), as the concentration of monoterpenes in the chamber during this study was below the limit of detection, which was ∼50 ppt (3σ).
Some of the dimers discussed above have been observed in previous laboratory studies. Ng et al. (2008) found C10H16N2O8 and C10H16N2O9 in the gas phase and C10H17N3O12, C10H17N3O13, C10H18N4O16, and C10H17N5O18 in the particle phase. C10H16N2O8 and C10H16N2O9 were also observed in our study, but their intensity in the MS was too low to assign molecular formulas with high confidence. The low intensity may be due to the low sensitivity of C10H16N2O8,9 in NO-CIMS. According to modeling results of the products formed in cyclohexene ozonolysis by Hyttinen et al. (2015), at least two hydrogen bond donor functional groups are needed for a compound to be detected in a nitrate CIMS. As C10H16N2O8 and C10H16N2O9 have no and only one H-bond donor function groups, respectively, they are expected to have low sensitivity in NO-CIMS. Moreover, the low intensity can be partly attributed to the much lower isoprene concentrations used in this study compared to previous studies, leading to the low concentration of C10H16N2O8 and C10H16N2O9 (Ng et al., 2008). C10H17N3O12, C10H17N3O13, C10H18N4O16, and C10H17N5O18 were all observed in the gas phase in this study, wherein the concentration of C10H17N5O18 was very low. The formation pathways of C10H17N3O12, C10H17N3O13, and C10H18N4O16 (Reaction R8) were generally similar to those proposed by Ng et al. (2008), except that the products from H shift of RO2 were involved in the formation of C10H17N3O13. Among the two pathways of C10H18N4O16 formation (Reaction R8 and via Reaction R12), our results indicate that Reaction (R8) was the main pathway, based on the low concentrations of C10H17N4O and other termination products of them, C10H16N4O. That the time profile of C10H18N4O16 was consistent with what is expected from Reaction (R8) as discussed above offers additional evidence for that conclusion.
Few field studies have reported HOM dimers formed via the reaction of NO3 with isoprene. This might be because NO3 + isoprene-HOM dimers can have the identical molecular formula to the HOM monomers from monoterpene oxidation. Possible contribution of dimer formation in the isoprene oxidation to C6–10 HOM in the particle phase observed at a rural site, Yorkville, US, is reported by Chen et al. (2020), although these HOM are thought to be more likely from monoterpene oxidation.
3.4 HOM trimers and their formation
A series of HOM trimers were observed, such as C15H24N4On(n=17–22), C15H25N5On(n=20–22), C15H25N3On(n=13–20), C15H26N4On(n=17–21), and C15H24N2On(n=12–16). Among the trimers, C15H24N4On was the most abundant series (Fig. S12). The C15H24N4On series can be explained by the accretion reaction of one monomer HOM RO2 and one dimer HOM RO2.
The formation pathways of dimer RO2 C10H16N3On (n=12–15) and C10H17N2On are shown above (Reactions R10 and R11).
The other trimers were likely formed via similar pathways (Table 2 and Supplement Sect. S2). Since NO-CIMS cannot provide the structural information of these HOM trimers, we cannot elucidate the major pathways. However, in all these pathways, dimer-RO2 is necessary to form a trimer, and most of the dimer-RO2 formation pathways require at least one double bond in the dimer molecule except for the reaction of RO2 with isoprene. Since one double bond has already reacted in the monomer-RO2 formation, we anticipate that in the reaction with NO3 it will be more favorable for precursors (VOC) containing more than one double bond to form trimer molecules than precursors containing only one double bond, as it is easier to generate new RO2 radicals from these dimers by attack on the remaining double bond(s).
The time profile of C15H24N4On showed the mixed behavior of first- and second-generation products (Fig. S13), consistent with the mechanism discussed above since C5H8NOn• and C10H16N3On• were of first- or second-generation and second-generation, respectively. The contributions of the second-generation products became larger as the number of oxygen atoms increased. In contrast, C15H25N3On showed instantaneous increase with isoprene addition (Fig. S14), which was typical for time profiles of first-generation products. Both proposed formation pathways of C15H25N3On (Reactions RS6 and RS7) contained a second-generation RO2, which was not in line with the time profile observed. The observation cannot be well explained unless we assume molecular adducts of a dimer with one monomer. It is also possible that some C10H17N2On• were formed very, very quickly or that there were other formation pathways of C15H25N3On not accounted for here.
We are not aware of field studies reporting NO3 + isoprene-HOM trimers, which is likely for the same reason for dimers discussed above. It is challenging to distinguish HOM trimers formed in the reaction NO3 with isoprene from the dimers formed by cross-reaction of the RO2 from monoterpene oxidation (C10-RO2) with that from isoprene oxidation (C5-RO2), as their molecular formulas can be identical.
3.5 Contributions of monomers, dimers, and trimers to HOM
The concentration (represented by peak intensity) of monomers was higher than that of dimers, but overall their concentrations remained on the same order of magnitude (Fig. 1a, inset). The concentration of trimers was much lower than that of monomers and dimers. The relative contributions of monomers, dimers, and trimers evolved in time due to the changing concentration of each HOM species. Comparing the contributions of various classes of HOM in period 1 with those in periods 1–6 reveals that the relative contribution of monomers increased with time, especially that of 2N-monomers, while the contribution of dimers decreased. This trend is attributed to the larger wall loss of dimers compared to monomers because of their lower volatility and also to the continuous formation of second-generation monomers, mostly 2N-monomers. Overall, the relative contribution of total HOM monomers decreased immediately after isoprene addition, while the contribution of HOM dimers increased rapidly (Fig. S15), which was attributed to the faster increase in dimer intensity due to their rapid formation. Afterwards, the contribution of monomers to total HOM gradually increased and that of dimers decreased, which was partly due to the faster wall loss rate of dimers and to the continuous formation of second-generation monomers.
3.6 Yield of HOM
The HOM yield in the oxidation of isoprene by NO3 was estimated using the sensitivity of H2SO4. It was derived for the first isoprene addition period to minimize the contribution of multi-generation products and to better compare with the data in the literature, thus denoted as primary HOM yield (Pullinen et al., 2020), and was estimated to be 1.2 %. The uncertainty was estimated as shown in Supplement Sect. S1. Despite the uncertainty, the primary HOM yield here was much higher than the HOM yield from the ozonolysis and photooxidation of isoprene (Jokinen et al., 2015). The difference may be attributed to the more efficient oxygenation in the addition of NO3 to carbon double bonds. Compared with the reaction with O3 or OH, the initial peroxy radicals contain five oxygen atoms when isoprene reacts with NO3, while the initial peroxy radicals contain only three oxygen atoms when reacting with OH, and the ozonide contains three oxygen atoms in the case of O3.
HOM formation in the reaction of isoprene with NO3 was investigated in the SAPHIR chamber. A number of HOM monomers, dimers, and trimers containing one to five nitrogen atoms were detected, and their time-dependent concentration profiles were tracked throughout the experiment. Some formation mechanisms for various HOM were proposed according to the molecular formula identified and the available literature. HOM showed a variety of time profiles with multiple isoprene additions during the reaction. First-generation HOM increased instantaneously after isoprene addition and then decreased, while second-generation HOM increased gradually and then decreased with time, reaching a maximum concentration at the later stage of each period. The time profiles provide additional constraints on their formation mechanism besides the molecular formula, suggesting whether they were first-generation products or second-generation products or a combination of both. 1N-monomers (mostly C5) were likely formed by NO3 addition to a double bond of isoprene, forming monomer RO2, followed by autoxidation and termination via the reaction with HO2, RO2, and NO3. Time series suggest that some 1N-monomer could also be formed by the reaction of first-generation products with NO3 and thus be of second generation. 2N-monomers were likely formed via the reaction of first-generation products such as C5-hydroxynitrate with NO3 and are thus second-generation products. 3N-monomers likely comprised peroxy/peroxyacyl nitrates formed by the reaction of 2N-monomer RO2 with NO2 and possibly nitronitrates formed via the direct addition of N2O5 to the first-generation products. HOM dimers were mostly formed by the accretion reactions between various HOM monomers RO2, either first-generation or second-generation or with the contributions of both, and thus showed time profiles typical of either first-generation products, second-generation products, or a combination of both. Additionally, some dimer peroxy radicals (dimer RO2) were formed by the reaction of NO3 with dimers containing a C=C double bond. HOM trimers were proposed to be formed by accretion reactions between the monomer RO2 and dimer RO2.
Overall, both HOM monomers and dimers contribute significantly to total HOM, while trimers only contributed a minor fraction. Within both the monomer and dimer compounds, a limited set of compounds dominated the abundance, such as C5H8N2On, C5H10N2On, C10H17N3On, and C10H16N2On series. 2N-monomers, which were second-generation products, dominated in monomers and accounted for ∼34 % of all HOM, indicating the important role of second-generation oxidation in HOM formation in the isoprene + NO3 reaction. Both RO2 autoxidation and “alkoxy–peroxy” pathways were found to be important for 1N- and 2N-HOM formation. In total, the yield of HOM monomers, dimers, and trimers accounted for 1.2 % of the isoprene reacted, which was much higher than the HOM yield in the oxidation of isoprene by OH and O3 reported in the literature (Jokinen et al., 2015). This means that the reaction of isoprene with NO3 is a competitive pathway of HOM formation from isoprene.
The HOM in the reaction of isoprene with NO3 may account for a significant fraction of SOA. If all the HOM condense on particles, using the molecular weight of the HOM with the least molecular weight observed in this study (C5H9NO6), the HOM yield corresponds to a SOA yield of 3.6 %. Although SOA concentrations were not measured in this study, Ng et al. (2008) reported a SOA yield of the isoprene + NO3 reaction of 4.3 %–23.8 %. Rollins et al. (2009) reported a SOA yield of 2 % at low organic aerosol loading (∼0.52 µg m−3) and 14 % if the further oxidation of the first-generation products is considered in the isoprene + NO3 reaction. Comparing the potential SOA yield produced by HOM with SOA yields in the literature suggests that HOM may play an important role in the SOA formation in the isoprene + NO3 reaction.
The RO2 lifetime is approximately 20–50 s in our experiments, which is generally comparable to or shorter than the lifetime of RO2 in the ambient atmosphere at night, varying from several tens to several hundreds of seconds (Fry et al., 2018), depending on the NO3, HO2, and RO2 concentrations. Assuming a HO2, RO2, and NO3 concentration of 5 ppt, 5 ppt (Tan et al., 2019), and 300 ppt (Brown and Stutz, 2012), respectively, the RO2 lifetime in our study is comparable to the nighttime RO2 lifetime (50 s) found in urban locations and areas influenced by urban plume. In areas with longer RO2 lifetime such as remote areas, the autoxidation is expected to be more important relative to bimolecular reactions. This may enhance HOM yield and thus enhance SOA yield. However, on the other hand, at lower RO2 concentration and thus longer RO2 lifetime, reduced rates of RO2+RO2 reactions producing low-volatility dimers can reduce the SOA yield by reducing dimer yield (McFiggans et al., 2019; Pullinen et al., 2020). The RO2 fate in our experiments is dominated the reaction RO2+NO3 with significant contribution of RO2+RO2, which can also represent the RO2 fate in the urban areas and areas influenced by urban plume. Our experiment condition cannot represent the chemistry in HO2-dominated regions such as clean forest environments (Schwantes et al., 2015).
We observed the second-generation products formed by the reaction of first-generation products. The lifetime of first-generation nitrates in the ambient atmosphere, according their rate constants with OH and NO3 (Wennberg et al., 2018), are ∼5 and ∼1.3–4 h, respectively, with respect to the reaction with OH and NO3 assuming a typical OH concentration of 2 × 106 molec. cm−3 (Lu et al., 2014; Tan et al., 2019) and a NO3 concentration of 100–300 ppt in urban areas (Brown and Stutz, 2012). Therefore, they have the chance to react further with OH and NO3 at dawn. In our experiments, the lifetimes of these first-generation nitrates with respect to OH and NO3 are comparable to the aforementioned lifetime due to comparable OH and NO3 concentrations with these ambient conditions. Therefore, our findings on the second-generation products are relevant to the ambient urban atmosphere and areas influenced by urban plumes. Some of these products such as C5H8,10N2O8 and multi-generation nitrooxyorganosulfates have been observed in recent field studies in polluted megacities in eastern China (Hamilton et al., 2021; Xu et al., 2021).
All the data in the figures of this study are available upon request to the corresponding author (t.mentel@fz-juelich.de or dfzhao@fudan.edu.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-9681-2021-supplement.
TFM, HF, StS, DZ, IP, AW, and AKS designed the experiments. Instrument deployment and operation were carried out by IP, HF, StS, IHA, RT, FR, DZ, and RW. Data analysis was done by DZ, HF, StS, RW, IHA, RT, FR, YG, SK, DZ, TFM, RW, JW, and SK, and LV interpreted the compiled data set. DZ and TFM wrote the paper. All the co-authors discussed the results and commented on the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
We thank the SAPHIR team for supporting our measurements and providing helpful data. Defeng Zhao and Yindong Guo would like to thank the National Natural Science Foundation of China (41875145). We would like to thank three anonymous reviewers and Kristian Møller for their helpful comments.
This research has been supported by the National Natural Science Foundation of China (grant no. 41875145).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Nga Lee Ng and reviewed by three anonymous referees.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Berndt, T. and Böge, O.: Gas-phase reaction of NO3 radicals with isoprene: a kinetic and mechanistic study, Int. J. Chem. Kinet., 29, 755–765, https://doi.org/10.1002/(sici)1097-4601(1997)29:10<755::Aid-kin4>3.0.Co;2-l, 1997.
Berndt, T., Mender, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of alpha-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem.-Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bernhammer, A.-K., Fischer, L., Mentler, B., Heinritzi, M., Simon, M., and Hansel, A.: Production of highly oxygenated organic molecules (HOMs) from trace contaminants during isoprene oxidation, Atmos. Meas. Tech., 11, 4763–4773, https://doi.org/10.5194/amt-11-4763-2018, 2018.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Boyd, C. M., Nah, T., Xu, L., Berkemeier, T., and Ng, N. L.: Secondary Organic Aerosol (SOA) from Nitrate Radical Oxidation of Monoterpenes: Effects of Temperature, Dilution, and Humidity on Aerosol Formation, Mixing, and Evaporation, Environ. Sci. Technol., 51, 7831–7841, https://doi.org/10.1021/acs.est.7b01460, 2017.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Brown, S. S., deGouw, J. A., Warneke, C., Ryerson, T. B., Dubé, W. P., Atlas, E., Weber, R. J., Peltier, R. E., Neuman, J. A., Roberts, J. M., Swanson, A., Flocke, F., McKeen, S. A., Brioude, J., Sommariva, R., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 9, 3027–3042, https://doi.org/10.5194/acp-9-3027-2009, 2009.
Brown, S. S., Dube, W. P., Peischl, J., Ryerson, T. B., Atlas, E., Warneke, C., de Gouw, J. A., Hekkert, S. t. L., Brock, C. A., Flocke, F., Trainer, M., Parrish, D. D., Feshenfeld, F. C., and Ravishankara, A. R.: Budgets for nocturnal VOC oxidation by nitrate radicals aloft during the 2006 Texas Air Quality Study, J. Geophys. Res.-Atmos., 116, D24305, https://doi.org/10.1029/2011jd016544, 2011.
Chen, J., Møller, K. H., Wennberg, P. O., and Kjaergaard, H. G.: Unimolecular Reactions Following Indoor and Outdoor Limonene Ozonolysis, J. Phys. Chem. A, 125, 669–680, 10.1021/acs.jpca.0c09882, 2021.
Chen, Y., Takeuchi, M., Nah, T., Xu, L., Canagaratna, M. R., Stark, H., Baumann, K., Canonaco, F., Prévôt, A. S. H., Huey, L. G., Weber, R. J., and Ng, N. L.: Chemical characterization of secondary organic aerosol at a rural site in the southeastern US: insights from simultaneous high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) and FIGAERO chemical ionization mass spectrometer (CIMS) measurements, Atmos. Chem. Phys., 20, 8421–8440, https://doi.org/10.5194/acp-20-8421-2020, 2020.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Draper, D. C., Myllys, N., Hyttinen, N., Møller, K. H., Kjaergaard, H. G., Fry, J. L., Smith, J. N., and Kurten, T.: Formation of Highly Oxidized Molecules from NO3 Radical Initiated Oxidation of Δ-3-Carene: A Mechanistic Study, ACS Earth and Space Chemistry, 3, 1460–1470, https://doi.org/10.1021/acsearthspacechem.9b00143, 2019.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, https://doi.org/10.1029/93jd00031, 1993.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Finlayson-Pitts, B. and Pitts, J.: Chemistry of the upper and lower atmosphere, Academic Press, San Diego, 2000.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C.: SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 11, 3879–3894, https://doi.org/10.5194/acp-11-3879-2011, 2011.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkle, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Fuchs, H., Dorn, H.-P., Bachner, M., Bohn, B., Brauers, T., Gomm, S., Hofzumahaus, A., Holland, F., Nehr, S., Rohrer, F., Tillmann, R., and Wahner, A.: Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Meas. Tech., 5, 1611–1626, https://doi.org/10.5194/amt-5-1611-2012, 2012.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Geyer, A., Alicke, B., Konrad, S., Schmitz, T., Stutz, J., and Platt, U.: Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.-Atmos., 106, 8013–8025, https://doi.org/10.1029/2000jd900681, 2001.
Hamilton, J. F., Bryant, D. J., Edwards, P. M., Ouyang, B., Bannan, T. J., Mehra, A., Mayhew, A. W., Hopkins, J. R., Dunmore, R. E., Squires, F. A., Lee, J. D., Newland, M. J., Worrall, S. D., Bacak, A., Coe, H., Percival, C., Whalley, L. K., Heard, D. E., Slater, E. J., Jones, R. L., Cui, T., Surratt, J. D., Reeves, C. E., Mills, G. P., Grimmond, S., Sun, Y., Xu, W., Shi, Z., and Rickard, A. R.: Key Role of NO3 Radicals in the Production of Isoprene Nitrates and Nitrooxyorganosulfates in Beijing, Environ. Sci. Technol., 55, 842–853, https://doi.org/10.1021/acs.est.0c05689, 2021.
Huang, W., Saathoff, H., Shen, X. L., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015.
Japar, S. M. and Niki, H.: Gas-phase reactions of the nitrate radical with olefins, J. Phys. Chem., 79, 1629–1632, https://doi.org/10.1021/j100583a002, 1975.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/s1352-2310(96)00105-7, 1997.
Jenkin, M. E., Boyd, A. A., and Lesclaux, R.: Peroxy Radical Kinetics Resulting from the OH-Initiated Oxidation of 1,3-Butadiene, 2,3-Dimethyl-1,3-Butadiene and Isoprene, J. Atmos. Chem., 29, 267–298, https://doi.org/10.1023/A:1005940332441, 1998.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipila, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem.-Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V. M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipila, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kaminski, M., Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017.
Kenseth, C. M., Huang, Y. L., Zhao, R., Dalleska, N. F., Hethcox, C., Stoltz, B. M., and Seinfeld, J. H.: Synergistic O3 + OH oxidation pathway to extremely low-volatility dimers revealed in beta-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 115, 8301–8306, https://doi.org/10.1073/pnas.1804671115, 2018.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W. W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipila, M., Junninen, H., St Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Lai, C. C. and Finlayson-Pitts, B. J.: Reactions of dinitrogen pentoxide and nitrogen-dioxide with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, Lipids, 26, 306–314, https://doi.org/10.1007/bf02537142, 1991.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Brauers, T., Oebel, A., Dlugi, R., Hu, M., Li, X., Lou, S. R., Shao, M., Zhu, T., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Nighttime observation and chemistry of HOx in the Pearl River Delta and Beijing in summer 2006, Atmos. Chem. Phys., 14, 4979–4999, https://doi.org/10.5194/acp-14-4979-2014, 2014.
Malkin, T. L., Goddard, A., Heard, D. E., and Seakins, P. W.: Measurements of OH and HO2 yields from the gas phase ozonolysis of isoprene, Atmos. Chem. Phys., 10, 1441–1459, https://doi.org/10.5194/acp-10-1441-2010, 2010.
Massoli, P., Stark, H., Canagaratna, M. R., Krechmer, J. E., Xu, L., Ng, N. L., Mauldin, R. L., Yan, C., Kimmel, J., Misztal, P. K., Jimenez, J. L., Jayne, J. T., and Worsnop, D. R.: Ambient Measurements of Highly Oxidized Gas-Phase Molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013, ACS Earth and Space Chemistry, 2, 653–672, https://doi.org/10.1021/acsearthspacechem.8b00028, 2018.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The Importance of Peroxy Radical Hydrogen-Shift Reactions in Atmospheric Isoprene Oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kurten, A., Lehtipalo, K., Makhmutov, V., Petaja, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tome, A., Trostl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from alpha-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, Acs Earth and Space Chemistry, 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of alpha-pinene and beta-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/c6cp00053c, 2016.
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/D0CP05555G, 2021.
Nozière, B. and Vereecken, L.: Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem.-Int. Edit., 58, 13976–13982, https://doi.org/10.1002/anie.201907981, 2019.
Perring, A. E., Wisthaler, A., Graus, M., Wooldridge, P. J., Lockwood, A. L., Mielke, L. H., Shepson, P. B., Hansel, A., and Cohen, R. C.: A product study of the isoprene+NO3 reaction, Atmos. Chem. Phys., 9, 4945–4956, https://doi.org/10.5194/acp-9-4945-2009, 2009.
Pfrang, C., Martin, R. S., Canosa-Mas, C. E., and Wayne, R. P.: Gas-phase reactions of NO3 and N2O5 with (Z)-hex-4-en-1-ol, (Z)-hex-3-en-1-ol (`leaf alcohol'), (E)-hex-3-en-1-ol, (Z)-hex-2-en-1-ol and (E)-hex-2-en-1-ol, Phys. Chem. Chem. Phys., 8, 354–363, https://doi.org/10.1039/b510835g, 2006.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Quéléver, L. L. J., Kristensen, K., Normann Jensen, L., Rosati, B., Teiwes, R., Daellenbach, K. R., Peräkylä, O., Roldin, P., Bossi, R., Pedersen, H. B., Glasius, M., Bilde, M., and Ehn, M.: Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis, Atmos. Chem. Phys., 19, 7609–7625, https://doi.org/10.5194/acp-19-7609-2019, 2019.
Richters, S., Pfeifle, M., Olzmann, M., and Berndt, T.: endo-Cyclization of unsaturated RO2 radicals from the gas-phase ozonolysis of cyclohexadienes, Chem. Commun., 53, 4132–4135, https://doi.org/10.1039/c7cc01350g, 2017.
Rissanen, M. P., Kurten, T., Sipila, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petaja, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Rissanen, M. P., Kurten, T., Sipila, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petaja, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding alpha-Pinene, J. Phys. Chem. A, 119, 4633–4650, https://doi.org/10.1021/jp510966g, 2015.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2+HO2 Pathway, J. Phys. Chem. A 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Skov, H., Hjorth, J., Lohse, C., Jensen, N. R., and Restelli, G.: Products and mechanisms of the reactions of the nitrate radical (NO3) with isoprene, 1,3-butadiene and 2,3-dimethyl-1,3-butadiene in air, Atmos. Environ. A.-Gen., 26, 2771–2783, https://doi.org/10.1016/0960-1686(92)90015-D, 1992.
Stark, M. S.: Epoxidation of Alkenes by Peroxyl Radicals in the Gas Phase: Structure-Activity Relationships, J. Phys. Chem. A, 101, 8296–8301, https://doi.org/10.1021/jp972054+, 1997.
Stark, M. S.: Addition of Peroxyl Radicals to Alkenes and the Reaction of Oxygen with Alkyl Radicals, J. Am. Chem. Soc., 122, 4162–4170, https://doi.org/10.1021/ja993760m, 2000.
Starn, T. K., Shepson, P. B., Bertman, S. B., Riemer, D. D., Zika, R. G., and Olszyna, K.: Nighttime isoprene chemistry at an urban-impacted forest site, 103, 22437–22447, https://doi.org/10.1029/98JD01201, 1998.
Stroud, C. A., Roberts, J. M., Williams, E. J., Hereid, D., Angevine, W. M., Fehsenfeld, F. C., Wisthaler, A., Hansel, A., Martinez-Harder, M., Harder, H., Brune, W. H., Hoenninger, G., Stutz, J., and White, A. B.: Nighttime isoprene trends at an urban forested site during the 1999 Southern Oxidant Study, 107, ACH 7-1–ACH 7-14, https://doi.org/10.1029/2001JD000959, 2002.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Valiev, R. R., Hasan, G., Salo, V.-T., Kubecka, J., and Kurten, T.: Intersystem Crossings Drive Atmospheric Gas-Phase Dimer Formation, J. Phys. Chem. A, 123, 6596–6604, https://doi.org/10.1021/acs.jpca.9b02559, 2019.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, https://doi.org/10.1039/c2cs35070j, 2012.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L. and Peeters, J.: Nontraditional (per)oxy ring-closure paths in the atmospheric oxidation of isoprene and monoterpenes, J. Phys. Chem. A, 108, 5197–5204, https://doi.org/10.1021/jp049219g, 2004.
Vereecken, L. and Peeters, J.: A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/c0cp00387e, 2010.
Vereecken, L. and Peeters, J.: A theoretical study of the OH-initiated gas-phase oxidation mechanism of beta-pinene (C10H16): first generation products, Phys. Chem. Chem. Phys., 14, 3802–3815, https://doi.org/10.1039/c2cp23711c, 2012.
Vereecken, L., Mueller, J. F., and Peeters, J.: Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of alpha-pinene: impact of non-traditional peroxyl radical chemistry, Phys. Chem. Chem. Phys., 9, 5241–5248, https://doi.org/10.1039/b708023a, 2007.
Vereecken, L., Carlsson, P. T. M., Novelli, A., Bernard, F., Brown, S. S., Cho, C., Crowley, J. N., Fuchs, H., Mellouki, W., Reimer, D., Shenolikar, J., Tillmann, R., Zhou, L., Kiendler-Scharr, A., and Wahner, A.: Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 23, 5496–5515, https://doi.org/10.1039/d0cp06267g, 2021.
Viggiano, A. A., Seeley, J. V., Mundis, P. L., Williamson, J. S., and Morris, R. A.: Rate Constants for the Reactions of XO3−(H2O)n (X = C, HC, and N) and NO3−(HNO3)n with H2SO4: Implications for Atmospheric Detection of H2SO4, J. Phys. Chem. A, 101, 8275–8278, https://doi.org/10.1021/jp971768h, 1997.
Wagner, N. L., Dubé, W. P., Washenfelder, R. A., Young, C. J., Pollack, I. B., Ryerson, T. B., and Brown, S. S.: Diode laser-based cavity ring-down instrument for NO3, N2O5, NO, NO2 and O3 from aircraft, Atmos. Meas. Tech., 4, 1227–1240, https://doi.org/10.5194/amt-4-1227-2011, 2011.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Wu, R., Vereecken, L., Tsiligiannis, E., Kang, S., Albrecht, S. R., Hantschke, L., Zhao, D., Novelli, A., Fuchs, H., Tillmann, R., Hohaus, T., Carlsson, P. T. M., Shenolikar, J., Bernard, F., Crowley, J. N., Fry, J. L., Brownwood, B., Thornton, J. A., Brown, S. S., Kiendler-Scharr, A., Wahner, A., Hallquist, M., and Mentel, T. F.: Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2020-1180, in review, 2020.
Xu, L., Guo, H. Y., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Xu, Z. N., Nie, W., Liu, Y. L., Sun, P., Huang, D. D., Yan, C., Krechmer, J., Ye, P. L., Xu, Z., Qi, X. M., Zhu, C. J., Li, Y. Y., Wang, T. Y., Wang, L., Huang, X., Tang, R. Z., Guo, S., Xiu, G. L., Fu, Q. Y., Worsnop, D., Chi, X. G., and Ding, A. J.: Multifunctional Products of Isoprene Oxidation in Polluted Atmosphere and Their Contribution to SOA, Geophys. Res. Lett., 48, e2020GL089276, https://doi.org/10.1029/2020GL089276, 2021.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Yan, C., Nie, W., Vogel, A. L., Dada, L., Lehtipalo, K., Stolzenburg, D., Wagner, R., Rissanen, M. P., Xiao, M., Ahonen, L., Fischer, L., Rose, C., Bianchi, F., Gordon, H., Simon, M., Heinritzi, M., Garmash, O., Roldin, P., Dias, A., Ye, P., Hofbauer, V., Amorim, A., Bauer, P. S., Bergen, A., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Canagaratna, M. R., Chen, X., Ding, A., Dommen, J., Draper, D. C., Duplissy, J., Frege, C., Heyn, C., Guida, R., Hakala, J., Heikkinen, L., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kirkby, J., Kontkanen, J., Kurten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nieminen, T., Nowak, J., Ojdanic, A., Onnela, A., Pajunoja, A., Petaja, T., Piel, F., Quelever, L. L. J., Sarnela, N., Schallhart, S., Sengupta, K., Sipila, M., Tome, A., Trostl, J., Vaisanen, O., Wagner, A. C., Ylisirnio, A., Zha, Q., Baltensperger, U., Carslaw, K. S., Curtius, J., Flagan, R. C., Hansel, A., Riipinen, I., Smith, J. N., Virtanen, A., Winkler, P. M., Donahue, N. M., Kerminen, V. M., Kulmala, M., Ehn, M., and Worsnop, D. R.: Size-dependent influence of NOx on the growth rates of organic aerosol particles, Science Advances, 6, eaay4945, https://doi.org/10.1126/sciadv.aay4945, 2020.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zhao, D. F., Buchholz, A., Kortner, B., Schlag, P., Rubach, F., Kiendler-Scharr, A., Tillmann, R., Wahner, A., Flores, J. M., Rudich, Y., Watne, Å. K., Hallquist, M., Wildt, J., and Mentel, T. F.: Size-dependent hygroscopicity parameter (κ) and chemical composition of secondary organic cloud condensation nuclei, Geophys. Res. Lett., 42, 10920–10928, https://doi.org/10.1002/2015gl066497, 2015a.
Zhao, D. F., Kaminski, M., Schlag, P., Fuchs, H., Acir, I.-H., Bohn, B., Häseler, R., Kiendler-Scharr, A., Rohrer, F., Tillmann, R., Wang, M. J., Wegener, R., Wildt, J., Wahner, A., and Mentel, Th. F.: Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 15, 991–1012, https://doi.org/10.5194/acp-15-991-2015, 2015b.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.