the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Jun 2021
| 04 Jun 2021
Importance of secondary organic aerosol formation of α-pinene, limonene, and m-cresol comparing day- and nighttime radical chemistry
Anke Mutzel
Yanli Zhang
Olaf Böge
Maria Rodigast
Agata Kolodziejczyk
Xinming Wang
The oxidation of biogenic and anthropogenic compounds leads to the formation of secondary organic aerosol mass (SOA). The present study aims to investigate α-pinene, limonene, and m-cresol with regards to their SOA formation potential dependent on relative humidity (RH) under night- (NO3 radicals) and daytime conditions (OH radicals) and the resulting chemical composition. It was found that SOA formation potential of limonene with NO3 under dry conditions significantly exceeds that of the OH-radical reaction, with SOA yields of 15–30 % and 10–21 %, respectively. Additionally, the nocturnal SOA yield was found to be very sensitive towards RH, yielding more SOA under dry conditions. In contrast, the SOA formation potential of α-pinene with NO3 slightly exceeds that of the OH-radical reaction, independent from RH. On average, α-pinene yielded SOA with about 6–7 % from NO3 radicals and 3–4 % from OH-radical reaction. Surprisingly, unexpectedly high SOA yields were found for m-cresol oxidation with OH radicals (3–9 %), with the highest yield under elevated RH (9 %), which is most likely attributable to a higher fraction of 3-methyl-6-nitro-catechol (MNC). While α-pinene and m-cresol SOA was found to be mainly composed of water-soluble compounds, 50–68 % of nocturnal SOA and 22–39 % of daytime limonene SOA are water-insoluble. The fraction of SOA-bound peroxides which originated from α-pinene varied between 2 and 80 % as a function of RH.
Furthermore, SOA from α-pinene revealed pinonic acid as the most important particle-phase constituent under day- and nighttime conditions with a fraction of 1–4 %. Other compounds detected are norpinonic acid (0.05–1.1 % mass fraction), terpenylic acid (0.1–1.1 % mass fraction), pinic acid (0.1–1.8 % mass fraction), and 3-methyl-1,2,3-tricarboxylic acid (0.05–0.5 % mass fraction). All marker compounds showed higher fractions under dry conditions when formed during daytime and showed almost no RH effect when formed during night.
- Article
(1594 KB) - Full-text XML
- BibTeX
- EndNote





Large amounts of volatile organic compounds (VOCs) are emitted into the atmosphere from both biogenic and anthropogenic sources with estimated source strengths of about 1300 Tg C yr−1 (Goldstein and Galbally, 2007). Once emitted, VOCs undergo gas-phase reactions with ozone (O3), hydroxyl (OH), or nitrate (NO3) radicals (Atkinson and Arey, 2003). Those reactions result in the formation of oxygenated products, with a lower vapor pressure than the parent hydrocarbons, which are subject to partitioning into the particle phase, leading to the formation of secondary organic aerosol (SOA). The atmospheric degradation of biogenic volatile organic compounds (BVOCs) and subsequent SOA formation have been the subject of numerous studies during the last decades (Hallquist et al., 2009; Glasius and Goldstein, 2016; Shrivastava et al., 2017). The majority of these studies examined the reaction initiated by the OH radical or ozone as they are considered as most dominating VOC sinks, although measurements indicated NO3-radical reaction is the most important sink for several VOCs during nighttime (Geyer et al., 2001). It was demonstrated that NO3-radical-initiated oxidation contributes 28 % of the overall VOC conversion compared to 55 % for OH-radical reaction and 17 % for the ozonolysis (Geyer et al., 2001; Kurtenbach et al., 2002; McLaren et al., 2010; Liebmann et al., 2018a, b). While NO2 and O3 serve as a precursor for nitrate radicals, NO3 is most dominating at night due to the fast photolysis and degradation with NO (Wayne et al., 1991; Brown and Stutz, 2012). The number of studies interconnecting NOx and BVOC emissions (Fry et al., 2009; Xu et al., 2015) are increasing, because the reaction with NO3 is often considered to be more important for BVOCs than for anthropogenic VOCs (Brown and Stutz, 2012).
Even though the number of studies investigating the rise of NO3-radical-initiated SOA formation has increased during the last few years (e.g., Pye et al., 2010; Fry et al., 2014, 2018; Boyd et al., 2015; Qin et al., 2018; Joo et al., 2019), there is still an enormous lack of data with respect to SOA yields, the influence of relative humidity (RH) on SOA formation, and the product distribution in the gas and particle phase. Kinetic studies have shown rate constants for α-pinene and limonene with NO3 in the range of 1.1–6.5 and 1.1–94 cm3 molecule−1 s−1 (Atkinson et al., 1984; Dlugokencky and Howard 1989; Barnes et al., 1990; Kind et al., 1998; Martinez et al., 1998, 1999; Stewart et al., 2013). For m-cresol only two rate constants are reported in the range of 7.0–9.2 cm3 molecule−1 s−1 (Carter et al., 1981; Atkinson et al., 1984). Accordingly, at least for nighttime and on a regional scale, NO3 reaction might lead to important contributions to VOC degradation and SOA formation. According to the comprehensive review by Ng et al. (2017), NO3 + VOC is worth investigating because (i) it can lead to anthropogenically influenced biogenic secondary organic aerosol (BSOA; Hoyle et al., 2007), (ii) SOA yields might be higher than from OH and ozone (Ng et al., 2017), (iii) it compromises an important source for organonitrates that serve as NOx and NOy reservoirs (von Kuhlmann et al., 2004; Horowitz et al., 2007), and (iv) in a few regions it was identified as the most dominating SOA contributor (Hoyle et al., 2007; Pye et al., 2010; Chung et al., 2012; Kiendler-Scharr et al., 2016).
This study aims to investigate three selected precursor compounds, namely α-pinene and limonene as biogenic VOCs and m-cresol as aromatic VOC with regards to their SOA formation potential under nighttime (NO3 radicals) and daytime conditions (OH radicals). While α-pinene and limonene are important BVOCs, m-cresol is often related to biomass burning. The chemical composition of formed SOA was characterized by their fraction of organic material (OM), water-soluble organic material (WSOM), SOA-bound peroxides, and SOA marker compounds. For quantification of marker compounds, well-known BSOA marker compounds (pinic acid, pinonic acid, etc.) were used, while SOA that originated from m-cresol was characterized using a SOA mix that contains mostly anthropogenic SOA compounds that are often related to biomass burning (Hoffmann et al., 2007). Furthermore, SOA yield and SOA growth will be discussed in detail as well as the influence of the relative humidity.
2.1 Chamber experiments
Experiments were conducted in the aerosol chamber under batch mode conditions at the Atmospheric Chemistry Department (ACD) of the Leibniz Institute for Tropospheric Research (TROPOS) in Leipzig. A brief description of the chamber will be given here because a complete description of the chamber can be found elsewhere (Mutzel et al., 2016). The aerosol chamber is made of PTFE and is of cylindrical geometry with a total volume of 19 m3 and a surface to volume ratio of 2 m−1. The chamber is equipped with a humidifier to enable reactions at elevated RH and a temperature-controlled housing to keep the temperature stable at T= 298 K throughout the experimental run. The humidifier is connected to the inlet air stream to enable humidification of air entering the chamber. Experiments were conducted using ammonium sulfate sulfuric acid seed ((NH4)2SO4 H2SO4 particles of pH = 4 at 50 % RH). The seed particles were injected via a nebulizer without a dryer. Their RH-dependent pH value was calculated by E-AIM (Clegg et al., 1998). All experiments were done with an initial hydrocarbon mixing ratio of 60 ppbv.
Table 1Experiments conducted for the NO3- and OH-radical-initiated oxidation of α-pinene, limonene, and m-cresol. All reactions were conducted with 60 ppbv initial hydrocarbon concentration at T= 293 K and in the presence of (NH4)2SO4 H2SO4 (pH = 4 at 50 % RH). OH-radical experiments were done in the presence of 10 ppbv NO.
a Only those studies are reported for OH-radical reaction of limonene and α-pinene that also apply H2O2 NO as an OH source. b Due to the lack of data all available literature is shown.
OH-radical reactions were initialized by photolysis of hydrogen peroxide (H2O2) in the presence of NO (10 ppb). H2O2 was continuously injected into the chamber with a peristaltic pump at 100 µL h−1 and was photolyzed with UV-A lamps (Osram Eversun Super). When the method developed by Barmet et al. (2012) is applied, the average OH-radical mixing ratio in the chamber is about 3–5 ×106 molecules cm−3.
NO3 radicals were produced in a pre-reactor (operated as a flow tube) by the reaction of NO2 and O3. A fraction of the air flow (10 L min−1) out of the total air flow in the flow tube (30 L min−1) was directed to the chamber (Iinuma et al., 2010). When the kinetic box model developed by Fry et al. (2014) is included into the COPASI (COmplex PAthway SImulator), the mixing ratio of NO3 radicals is calculated for the present study to be 7.5 ×107 molecules cm−3. We implemented the reaction mechanism provided by Fry and co-workers in COPASI, and the model was utilized in the aerosol chamber.
After a reaction time of 90 min the reaction was stopped, and samples were taken, passing chamber air over a 47 mm PTFE filter (borosilicate glass fiber filter coated with fluorocarbon, 47 mm in diameter, Pallflex T60A20, Pall, NY, USA) and QF filter (micro-quartz fiber filter, 47 mm in diameter, MK 360, Munktell, Bärenstein, Germany) for 3 min at 30 L min−1. During sampling time no additional air stream was added to the chamber to avoid dilution. PTFE filters were quantified afterwards for biogenic and anthropogenic SOA marker compounds and QF to determine organic elemental carbon (OC EC), non-purgeable organic carbon (NPOC, formerly known as water-soluble organic carbon), and for select experiments also concentration of inorganic nitrate (NO).
Experiments were conducted either under nighttime conditions with NO3 radicals or with OH radicals to represent daytime chemistry. A complete overview of all experiments can be found in Table 1.
Dilution rates and wall losses were considered as follows: NO3 radicals and H2O2 were injected into the chamber with a bypass air of 10 and 5 L min−1. Based on a reaction time of 90 min, a dilution of 4.7 % (NO3) and 2.4 % (OH) can be estimated. These values are within the measurement uncertainty of the proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS). According to the study by Romano and Hanna (2018), an uncertainty of ±10 % can be assumed. Wall losses of VOCs were determined to be 2.5 s−1 (α-pinene), 7.9 s−1 (limonene), and 2.2 s−1 (m-cresol). The consumption recorded by PTR-TOF-MS is corrected for those additional sinks. Particle wall losses were determined from the blank experiments at RH = 50 %. Time-dependent particle losses were used to correct the scanning mobility particle sizer (SMPS) measurements. For blank experiments all compounds were injected into the chamber, except the hydrocarbon. Notably, wall losses at RH = 50 % were also used as an approximation for 0 and 75 % RH, although losses might change under those condition according to their phase state. According to previous studies, wall loss might be small due to the short reaction time. According to McMurry and Grosjean, a 90 min reaction time would result in a 10 % loss of particles, which is within the measurement uncertainty of the SMPS (McMurry and Grosjean, 1985). Additionally, seed particles were injected without a dryer. Consequently, they can be regarded as wet particles when they enter the chamber. Thus at RH = 75 % no additional loss is expected.
As the chamber is allowed to equilibrate for at least 10 min after seed injection, a dramatic wall loss under dry conditions would be directly observable in SMPS by a drastic decrease of particle volume with a constant particle number. As this was not observed, it can be assumed that wall loss at RH = 0 % is in the same manner as at 50 %.
An ozone monitor was connected for all experiments. Specific conditions in the pre-reactor were set to avoid ozone entering the chamber during NO3-radical reaction. Therefore for this reaction type ozonolysis as a side reaction can be excluded. During H2O2 photolysis small amounts of O3 are always formed, which might lead to ozonolysis. It should be noted that OH-radical reaction was conducted in the presence of NOx. Thus formed O3 will rapidly react with NO rather than with α-pinene and limonene. Due to the low reaction rate constant and low concentration, ozonolysis occurs to a very small extent and cannot be excluded. A maximum O3 concentration of 5 ppb was observed.
2.2 Online instrumentation
The consumption of precursor compounds (ΔHC) was monitored by a PTR-TOF-MS (Ionicon, Lindinger et al., 1998). The particle size distribution was measured by an SMPS (Wiedensohler et al., 2012). In absence of a reliable density estimation, an average density of 1 g cm−3 was used to convert the SMPS measurement data into the increase in organic mass (ΔM). This assumption was also made for OM and non-purgeable organic material (NPOM) measurement. The assumed density was not changed with RH. As the ft between OM and ΔM stays almost constant, the RH seems not to affect the density in the conducted experiments. The particle growth by water uptake was taken into account by collecting particles on the filter and determining the content of OM. For most of the experiments it was found that both values (ΔM and OM) fit well, indicating that particle growth is mainly caused by organics rather than water. Monitors for ozone (49C Ozone Analyzer, Thermo Scientific, USA) and NOx (42i TL Trace Level NOx Analyzer, Thermo Scientific, USA) were connected to the chamber as well.
2.3 Offline measurements
2.3.1 OC EC, NPOC, inorganic nitrate, and SOA-bound peroxides
The quartz filter was cut into halves. One half was used for OC EC quantification, and the second was used for water-soluble organic carbon. The content of OC EC was determined with a C-mat 5500 carbon analyzer, applying a two-step thermographic method (Neusüß et al., 2002). The fraction of water-soluble organic carbon was determined as non-purgeable organic carbon with a TOC-VCPH analyzer (van Pinxteren et al., 2009). To do so, the second half of the QF was extracted in 25 mL ultrapure water for 30 min with an orbital shaker. The resulting extract was filtered through a 0.45 µm syringe filter (Acrodisc 13, Pall, NY, USA). Two hundred fifty microliters of the extract was used for NO analysis. After acidification and sparging with N2, the remaining extract was injected into the TOC analyzer. The amount of NO was determined by ion chromatography coupled with conductivity detection (IC–CD) using an AS18 column combined with AG18 guard column.
For SOA-bound peroxides, half of the PTFE filter was used. One quarter of the filter was used for the peroxide test, and the second quarter filter to determine the blank value. The method is described in detail elsewhere (Mutzel et al., 2013).
2.3.2 Sample preparation for liquid chromatography–mass spectrometry
The sample preparation follows the method described in the literature (Hoffmann et al., 2007; Mutzel et al., 2015). Briefly, half of the PTFE filter was cut into small pieces and transferred into an extraction vial. Five hundred microliters of methanol was added, and the vial was placed in an orbital shaker for 15 min at 1000 rotations min−1. Insoluble material was removed by a syringe filter (0.2 mm, Acrodisc, Pall, NY, USA). Afterwards, the extraction was repeated with 500 mL of MeOH. The combined extracts were dried under a gentle stream of nitrogen and reconstituted in 250 mL of CH3OH H2O (, ).
2.3.3 Analysis with liquid chromatography–mass spectrometry
A high-performance liquid chromatography (HPLC, Agilent, 1100 Series, Santa Clara, CA, USA) connected to a electrospray ionization time-of-flight mass spectrometer (microTOF, Bruker Daltonics, Bremen, Germany) was used for separation and quantification of marker compounds. For the separation an Agilent ZORBAX C18 column (3.0 × 150 mm, 5 µm particle size) was used at a temperature of 25 ∘C and a flow rate of 0.5 mL min−1 with 0.1 % acetic acid in ultrapure water (A) and 100 % methanol (B) as eluents. The gradient was as follows: 10 % B for 2 min, increased from 10 % B to 100 % B in 20 min, and then held constant for 3 min and re-equilibrated for 5 min back to the initial conditions. The quantification was done in the negative ionization mode with a mass range between 50 and 1000, applying a series of sodium acetate clusters to calibrate mass accuracy. Quantification was done using authentic standard solutions within a seven-point calibration with three repetitions of each calibration point.
For anthropogenic SOA compounds, the separation was done as described above at 15 ∘C and with 0.2 % acetic acid in water.
The yield of the single compounds was calculated by taking the quantified amount from the filter, correcting for sampling volume. The numbers are given as a fraction in formed organic mass.
2.3.4 Chemicals
The following chemicals were used as received: α-pinene, limonene, and m-cresol (Sigma-Aldrich, St. Louis, USA; purity: 99, 97, and 99 %, respectively); terebic acid (Sigma-Aldrich, St. Louis, USA; purity: 99 %); and pinic acid (Sigma-Aldrich, St. Louis, USA; purity: 99 %).
The following compounds were synthesized according to procedures given in the literature: norpinonic acid, terpenylic acid (Claeys et al., 2009), 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA; Szmigielski et al., 2007), and diaterpenylic acid acetate (DTAA; Iinuma et al., 2009). The composition of the anthropogenic SOA mix is described in detail in Hoffmann et al. (2007).
3.1 SOA formation and yield
The SOA formation from the reaction of α-pinene, limonene, and m-cresol with NO3 radicals has been investigated within this study with emphasis on SOA yields, the chemical composition in the particle phase, the influence of the RH, and a final comparison to daytime chemistry with OH radicals. The SOA yields were calculated according to Odum et al. (1996) by calculating the amount of produced organic mass in relation to the amount of reacted hydrocarbon according to
where ΔM is the produced organic mass (µg m−3) and ΔHC is the reacted amount of hydrocarbon (µg m−3).
A complete overview on all experiments, the obtained results, and the comparison to literature values is given in Table 1. In general, the discussion is mainly focused on the amount of SOA mass produced after 90 min reaction time. Only the differences in curve shape of growth curves are discussed in detail in the respective section.
In general, α-pinene yielded higher SOA with NO3 radicals ( 6 %) than with OH (YOH≈ 3.5 %). In the case of limonene the difference is not as well discerned but can still be observed ( 15–30 %; YOH≈ 10–21 %). In contrast, m-cresol yielded a dismissable amount of SOA with NO3 radicals and moderate amounts with OH radicals (YOH ≈ 3–9 %). Therefore, the highest SOA formation potential for NO3-radical reaction was found for limonene, and the lowest for m-cresol.
The SOA yield curves were parameterized according to Odum et al. (1996) following
where α is the mass yield of compound i, KOM,i is the partitioning coefficient of compound i, and M0 is the absorbing organic mass.
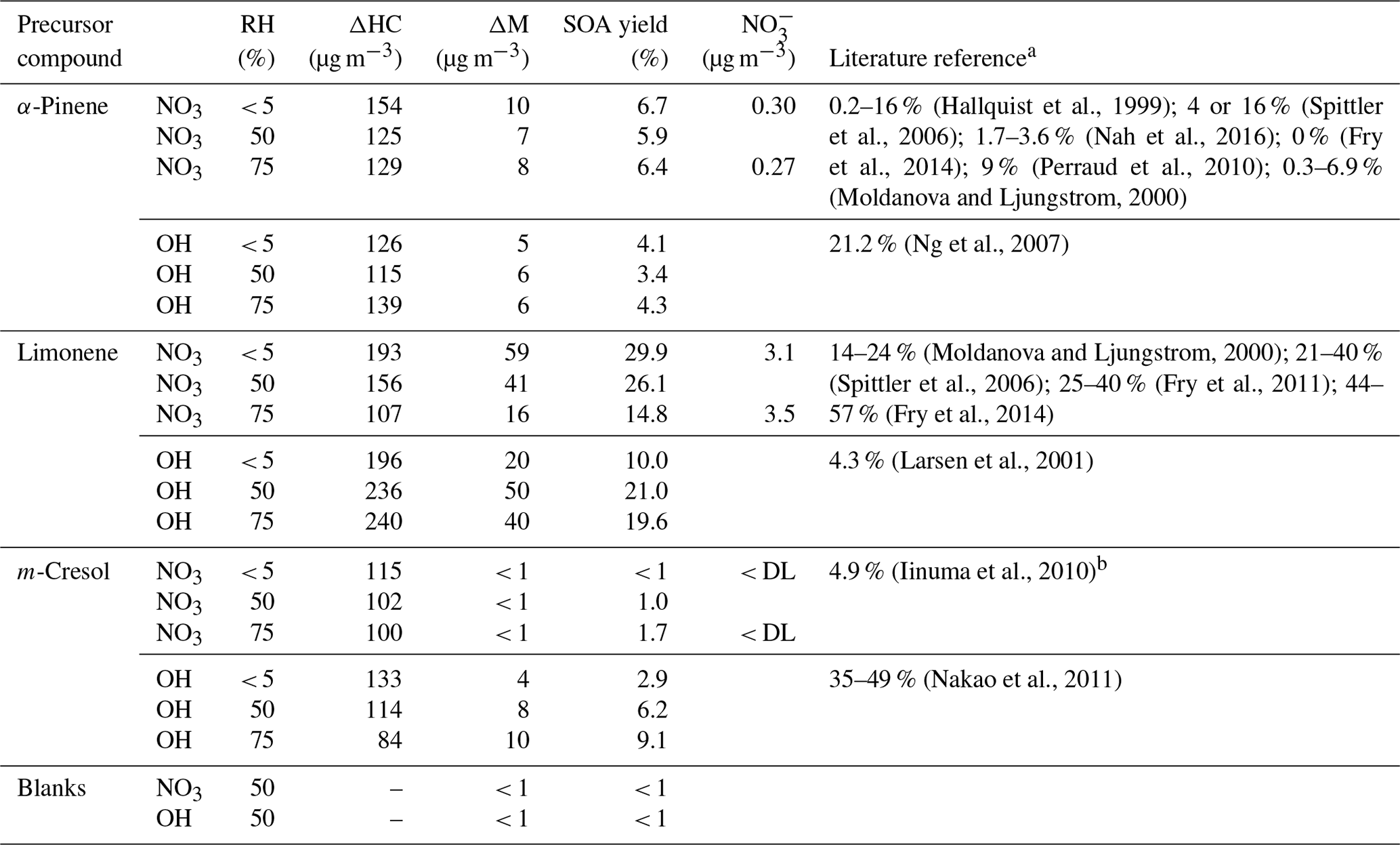
By applying the one-product model approach, the fit produced very good results, with R2>0.99. The applicability of one-product models was also demonstrated by Friedmann and Farmer (2018). Yield curves without any effect of RH result in comparable α and K values. Yield curves with a distinct RH influence show a higher partitioning coefficient for higher SOA yields together with increasing mass yields. All α and K values are depicted in the respective yield curves (Fig. 1).
Figure 1Yield curves for α-pinene, limonene, and m-cresol with NO3 and OH radicals for 0, 50, and 75 % RH. Yield curves were parameterized with the one-product approach (Odum et al., 1996). The obtained values for α (mass yield) and KOM (partitioning coefficient) are included as well. Please note that the SOA yield of m-cresol NO3 was below 0.01 %. Therefore, no parameterization can be provided. Each fit presents a single chamber experiment. SOA yield was calculated by deviating the produced organic mass by the consumed amount of hydrocarbon.
Only a limited number of studies provided parameterization of yield curves for VOC OH NOx and VOC NO3 according to Odum et al. (1996), which highlights the need for the present data set. Spittler et al. (2006) reported based on a two-product model for limonene NO3 and values of 0.1249/0.3128 and 0.0348/0.0181. The reported values for α correspond well to values obtained in this study, whereas K values are by 1 order of magnitude smaller. This variation could be caused by the different seed particles used, because Spittler and co-workers employed a pure organic seed and in this study an inorganic seed was utilized.
Iinuma et al. (2010) reported based on a two-product model for cresol OH and K values of 0.1231/0.0004 and 0.0753. These values are not in agreement with reported values, which might be caused by different OH sources used.

Figure 2Growth curves for α-pinene, limonene, and m-cresol with NO3 and OH radicals for 0, 50, and 75 % RH.
3.1.1 α-Pinene
SOA yields for α-pinene with NO3 radicals ranged from ≈ 5.9 to 6.4 % in reasonable agreement with the literature data (0–16 %; Table 1). However, comparing the SOA formation from nighttime chemistry with daytime, the yields from NO3 radical chemistry are higher. The SOA yield in this study is very close to those that have been reported by Moldanova and Ljungstrom (2000) ( 0.3–6.9 %) and Nah et al. (2016) ( 1.7–3.6 %). Although the values agree very well with the majority of the studies, it is still unclear why Fry and co-workers reported no SOA formation from α-pinene NO3 in the presence of seed particles (Fry et al., 2014). Even so, small SOA yields were observed within our present investigation; α-pinene NO3 always yielded SOA. The initial conditions in the study of Fry et al., and this study are very similar, with the exception of the workflow. Fry and co-workers injected the BVOCs into a chamber that was filled with NO3 radicals, whereas for the present study the BVOC was injected at first and afterwards the reaction was initialized. Further studies are needed to reveal the reasons for the discrepancies in the SOA yields from NO3-radical reaction.
Furthermore, when comparing the growth curves for OH- and NO3-radical reaction with α-pinene and limonene, a clear difference in the curve shapes can be seen (Fig. 2). The SOA formation from the OH-radical-initiated reaction starts later than in the case of NO3 for both systems, α-pinene and limonene. Such a long induction period is most likely caused by further reaction of first-generation oxidation products leading to SOA formation as has been demonstrated in previous studies (Ng et al., 2006; Mutzel et al., 2016). As reported by Mutzel et al. (2016), the SOA formation of α-pinene OH and limonene OH is partly controlled via further reaction of myrtenal and limonaketone/endolim, respectively. The reaction of these first-generation oxidation products is the limiting factor for SOA formation and explains the delay in SOA growth (Mutzel et al., 2016). In contrast, SOA originating from NO3 starts immediately after 30 µg m−3 is consumed. Thus, condensable oxidation products are directly formed and partitioned into the particle phase. Potential candidates of those products might be organonitrates.
3.1.2 Limonene
Limonene for both oxidation regimes (day and night) yielded the highest SOA yields compared to α-pinene and m-cresol. The SOA yield from limonene ( 16–29 %) is by a factor of 3–5 higher than α-pinene and by a factor of 10 higher than m-cresol. Those values are close to the lowest values reported for limonene ozonolysis (Northcross and Jang, 2007; Chen and Hopke, 2010; Gong et al., 2018). Furthermore, according to the present data set, limonene with NO3 ( 16–29 %) is more efficient in SOA production than the OH-radical reaction (YOH≈ 10–21 %).
Consequently, nocturnal oxidation of limonene with NO3 yields more SOA than ozonolysis and OH-radical reaction. This additional SOA source should be considered in future studies, in particular under less humid conditions. In addition to the strong SOA formation potential, the organic mass production of limonene + NO3 seems to be highly dependent on humidity. This will be discussed separately in the corresponding section below.
3.1.3 m-Cresol
In contrast to α-pinene and limonene, m-cresol yielded only negligible amounts of SOA with NO3 radicals, while the OH-radical reaction seems to be more efficient than α-pinene. This observation was unexpected because anthropogenic VOCs are often suggested to form less SOA than biogenic ones. SOA production from anthropogenic VOCs has often been investigated but usually led to inconsistent results and very low yields (e.g., Izumi and Fukuyama, 1990; Healy et al., 2009; Emanuelsson et al., 2013). A study by Hildebrandt et al. (2009) raised the question about the low SOA yields and observed much higher yields by using artificial sunlight. The present study also demonstrates higher SOA yields than expected and supports the hypothesis of Hildebrandt and co-workers about a higher importance of SOA production from anthropogenic VOCs.
It should be noted that, due to the low SOA yields from NO3-radical reaction, no parameterization of the yield curves can be provided (Fig. 1). In general, the SOA yields (YOH≈ 2.9–9.1 %) for OH-radical reaction with m-cresol are in good agreement with Iinuma and co-workers (YOH≈ 4.9 %), although the photolysis of methyl nitrite was used to generate OH radicals. Compared to the study by Nakao et al. (2011) (YOH≈ 35–49 %) the values are much lower. This is not surprising, as Nakao and co-workers conducted experiments in the absence of NOx, whereas in this study NOx was always present. The effect of NOx lowering SOA yields has often been described in the literature (e.g., Presto et al., 2005; Ng et al., 2007; Zhao et al., 2018).
Furthermore, the yield curves clearly indicated a strong effect of relative humidity, which can also be seen from the growth curves (Fig. 2). This effect will be discussed in the following section.
3.2 Influence of RH on SOA yield and growth
Within this study, experiments were conducted at RH < 5 %, RH = 50 %, and RH = 75 % to investigate the effect of humidity on SOA yield, growth and composition. As discussed in the section above, relative humidity has been suggested to influence SOA formation and yield for the NO3-radical-initiated reaction of VOCs. The observed humidity dependencies could be caused by four main factors: (i) the uptake of the SOA marker compounds or their precursor compounds changes as a function of the experimental conditions, (ii) the formation process of SOA marker compounds is directly affected by the experimental conditions, (iii) further reactions take place within the particle phase, and/or (iv) the uptake behavior of the first-generation oxidation products might change with liquid water content (LWC). It remains a challenge to differentiate between all these factors because the observed dependency is most likely the result of a combination of all four factors. A discussion of factor (i)–(iii) is provided in the respective subsection (Sect. 3.3). The influence of RH on the uptake behavior of first-generation oxidation products cannot be excluded. At has been demonstrated in previous studies, the uptake coefficient of first-generation oxidation products, in particular carbonyl compounds, might depend on RH (Healy et al., 2009)
Furthermore, the limited number of studies investigating the effect of RH on the OH-radical reaction often contradict each other (Cocker et al., 2001; Bonn and Moortgat, 2002). Only a very limited number of studies are available that investigate the influence of RH on SOA formation originating from VOCs + NO3 (the only ones, to the authors' knowledge, are as follows: Spittler et al., 2006; Fry et al., 2009; Bonn and Moortgat, 2002; Boyd et al., 2015). According to Fig. 2, a significant effect can be observed for two systems – i.e., limonene NO3 and m-cresol OH – while α-pinene NO3 and m-cresol NO3 were not affected by RH, which is in good agreement with the literature studies (by Bonn and Moortgat, 2002; Fry et al., 2009; Boyd et al., 2015). Only Spittler and co-workers observed lower SOA yields under humid conditions (20 vs. 40 % RH). Notably, in the case of limonene NO3, the SOA yield varies by a factor of 2 between 29.1 % (at RH < 5 %) and 14.8 % (at RH = 75 %). In the literature, higher SOA yields for other cresol isomers have been reported. Due to the different volatilities of the cresol isomers, different SOA formation potentials are expected (Ramasamy et al., 2019).
In the case of m-cresol OH the SOA yield increases with humidity by a factor of 5. Thus far, no study has investigated the role of RH on the SOA formation from limonene NO3 and m-cresol OH, and the data set for α-pinene NO3 is small and inconsistent. Since an effect was only observed for limonene NO3 and m-cresol OH, these systems will be discussed in more detail.
3.2.1 Limonene + NO3
The SOA yield was found to decrease with increasing relative humidity from 29 down to 14.8 %. This pronounced effect could be a result of a direct effect of RH on the partitioning of condensable products. Organonitrates (ONs) are well-known oxidation products of VOC with NO3 and are often related to SOA formation and growth (e.g., Day et al., 2010; Rollins et al., 2010; Zaveri et al., 2010). ONs are reported to be very prone to hydrolysis, which might explain the lower SOA yields under humid conditions (e.g., Darer et al., 2011; Hu et al., 2011; Jacobs et al., 2014; Rindelaub et al., 2015). As a contribution of ONs is excluded, the formation of the hydrophobic compounds that partition into the organic phase needs to be considered as a potential explanation. As depicted in Fig. 4, limonene yields the highest fraction of water-insoluble OM. Although this process needs to be considered, it is unlikely that this explains the current observation; Fig. 2 clearly indicates a decreasing consumption when RH increases as a potential reason for lower SOA yields at higher RH. One might assume that lower consumptions are caused by an enhanced partitioning of NO3 radicals into the particle phase due to an enhanced aerosol liquid water content (ALWC). This seems to be supported by the quantification of particulate inorganic nitrate as this shows a higher fraction in SOA under elevated RH (Table 1). Under dry conditions the ratio of produced organic mass : particulate inorganic nitrate is around 9.65, whereas under elevated RH this ratio decreases to 4.65. Therefore, a stronger contribution of particulate inorganic NO can be inferred.
A second aspect to be considered is the contribution of first-generation oxidation products. According to theoretical investigations, endolim is the most favored product formed during limonene + NO3 (Jiang et al., 2009). It could be speculated that endolim reacts faster with NO3 than limonene, scavenging NO3 radicals. As no rate constants are available for endolim with NO3, the values from the Master Chemical Mechanism (MCM, version 3.3.1; Jenkin et al., 1997; Saunders et al., 2003) were taken for comparison. For the reaction of endolim with NO3 (LIMAL) a rate constant of 2.6 cm3 molecule−1 s−1 can be found. According to k values taken from MCM and kinetic studies, limonene with NO3 is by 2 orders of magnitude (: 1.2 6 cm3 molecule−1 s−1) faster compared to endolim. Consequently, a competition between limonene and the respective first-generation oxidation product can be excluded.
A last sink of NO3 radicals is the reaction of RO2 radicals with NO3, as has been investigated by Boyd et al. (2015) for α-pinene. In conducting two different sets of reactions with “RO2 + NO3 dominant” and “RO2 + HO2 dominant”, no effect on SOA yield of α-pinene was found. Nevertheless, taking into account that α-pinene contains only one double bond, formed RO2 radicals are saturated, whereas limonene as a diene forms RO2 radicals with one remaining double bond, which could be expected to be more reactive than saturated RO2 radicals. Therefore, limonene-originated RO2 radicals are more reactive and might represent an important sink for NO3, which is in competition to limonene + NO3. Thus far only one study exists that investigates this reaction channel. Thus, this competitor for limonene with NO3 seems to be likely and should be systematically investigated in the future.
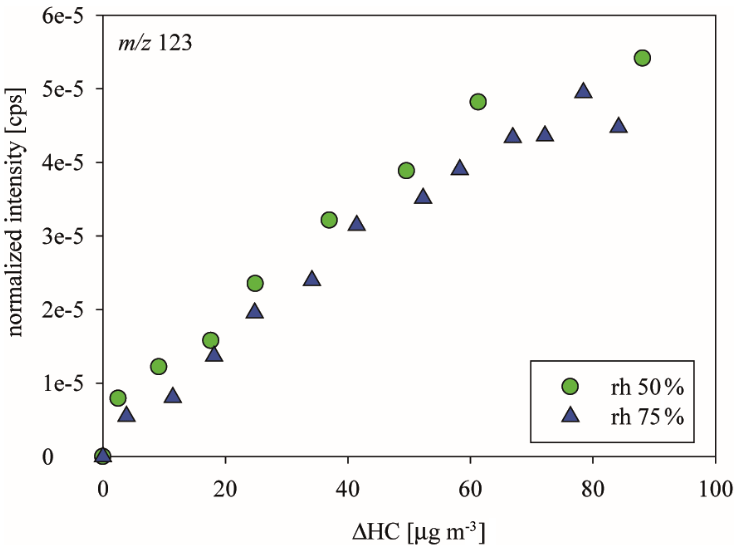
Figure 3Evolution of 123 as a function of consumption of m-cresol (ΔHC). The signal at 123 can be attributed to methyl-1,4-benzoquinone.
3.2.2 m-Cresol + OH
In contrast to limonene NO3, the OH-radical-initiated oxidation of m-cresol showed higher SOA yields with increasing RH (Fig. 1). Even though the consumption also decreases under humid conditions, the particulate OM increases (Fig. 2). When analyzing the respective growth curves, a delay in aerosol production can be seen. At 0 and 50 % RH conditions, aerosol production starts at ΔHC ≈ 80–90 µg m−3. At more elevated RH the SOA production starts immediately after initialization of the reaction (ΔHC ≈ 5–10 µg m−3). According to Ng et al. (2006), such a difference in mass production can be caused by one or both of two factors, the first being a delay in mass transfer from the gas into particle phase and the second being the fact that condensable products are only formed from second-generation oxidation products and thus the formation of those products is the limiting parameter for SOA formation.
According to a study by Coeur-Tourneur et al. (2006), methyl-1,4-benzoquinone (MBQ) is the most dominating oxidation product, with up to 12 % molar yield. MBQ was also detected in this study by means of the PTR-TOF-MS at 123. Nevertheless, the increase of the signal in relation to consumption does not show a significant effect of RH on the formation (Fig. 3). Therefore, a strong contribution due to further reactions of MBQ can be excluded. Thus, the delay might be caused by the effect of relative humidity on the partitioning of condensable products, such as methyl-nitro-catechol. This hypothesis is supported by the comprehensive characterization of the particle phase as discussed in Sect. 3.4.
Figure 4Comparison of organic mass calculated from SMPS with an offline-determined concentration of organic material (OM) and non-purgeable OM. Measurement uncertainties are given as follows: 10 % for SMPS measurements (Wiedensohler et al., 2012), 5 % for OC EC measurements (Spindler et al., 2004), and 10 % for WSOM measurements (Timonen et al., 2010). Please note that no values can be given for m-cresol NO3 as the produced organic mass was too small to be within the detection limits of the different techniques. The values of OM and WSOM were also illustrated as a fraction of the produced organic mass ΔM (expressed as percent above to the corresponding bar).
3.3 Characterization of particle-phase chemical composition
The filters collected after each experiment were analyzed with regards to their content of OC, WSOM, SOA-bound peroxides, and SOA marker compounds. The results are summarized in Figs. 4–7.
3.3.1 Organic carbon and water-soluble organic carbon
Pre-heated quartz fiber filters were analyzed for OM and WSOM content. Please note that WSOM was determined as NPOM. The obtained results were compared to the increase in organic mass (ΔM) obtained from the SMPS (Fig. 4). In general, the values agree, meaning that the increase in organic mass corresponds to organic carbon and, secondly, that the majority of this mass is water-soluble, except in the limonene experiments.
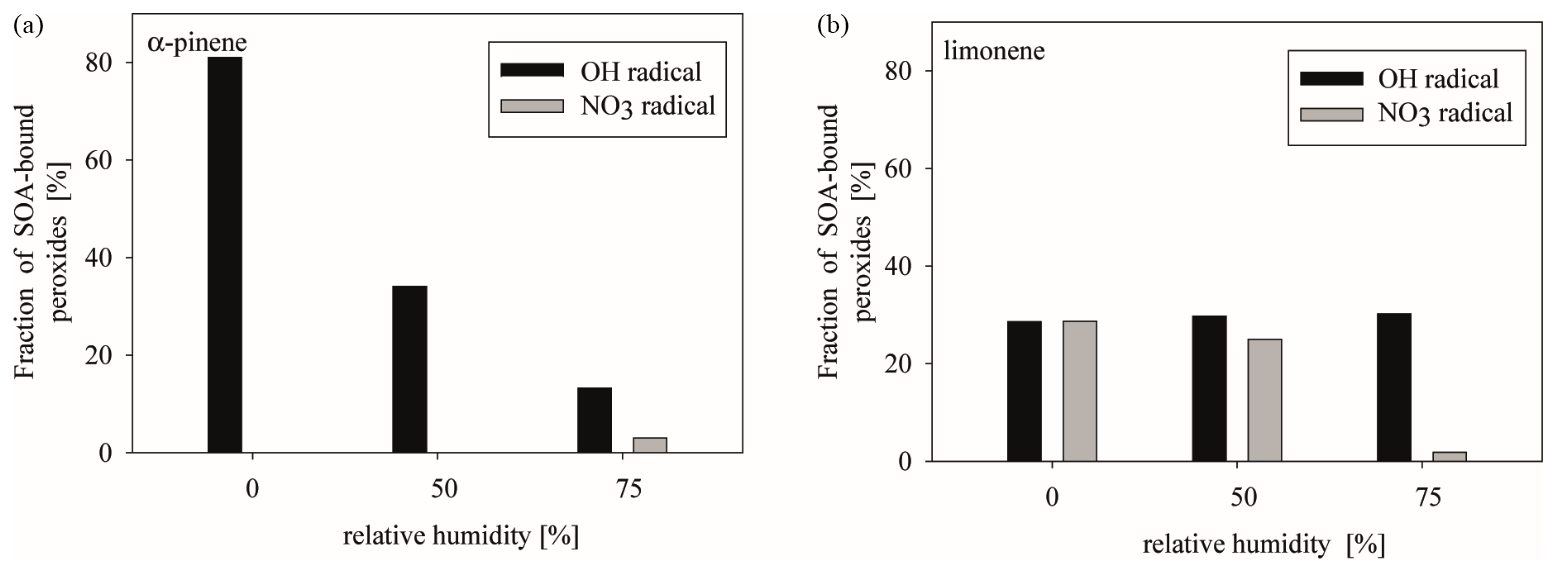
Figure 5Fraction of SOA-bound peroxides from α-pinene (a) and limonene (b) oxidation with OH radicals (black) and NO3 radicals (grey). Quantification was done following the method described by Mutzel et al. (2013) assuming a molar mass of 300 g mol−1 (Docherty et al., 2005).
In general, limonene with 22–36 % organic mass is the only precursor hinting at water-insoluble material. From both systems, limonene NO3 and limonene OH, the WSOM fraction ranges between 32–50 and 61–78 %, respectively. In the case of limonene NO3 the fraction of water-soluble carbon decreases dramatically when the relative humidity is reduced. Only one-third (32 %) of ΔM is composed of water-soluble carbon, although the SOA yield was highest under dry conditions. Thus, under reduced RH more water-insoluble compounds partition into the particle phase, leading to enhanced SOA growth. Potential candidates might be higher-molecular-weight compounds (HMWCs), which seem to be involved in SOA growth for the NO3 and OH system.
To further investigate the fraction of organic material found in the formed SOA, discussions about SOA-bound peroxides and single compounds are provided in the following sections.
3.3.2 SOA-bound peroxides
Organic peroxides in SOA were quantified according to a method published by our laboratory (Mutzel et al., 2013), assuming a molar mass of 300 g mol−1 (Fig. 5), as is recommended by Docherty et al. (2005), presuming that the majority of organic peroxides are higher-molecular-weight compounds (e.g., dimers). Notably, the assumed molar mass has a significant influence of the calculated amount of SOA-bound peroxides. This might cause some uncertainties. The method applies an iodometric detection by UV–Vis spectroscopy. Although earlier studies demonstrated that H2O2 injected into an aerosol chamber does not cause artifacts, blank experiments were also conducted to exclude them. The blank run was conducted by injecting seed, H2O2, and NO. After a reaction time of 90 min, filter samples were taken and analyzed for their SOA-bound peroxide content. These experiments also show no peroxide content. Thus for pure inorganic seed particles an effect of H2O2 originating from the injected H2O2 can be excluded. Additionally, mixed organic and inorganic seed particles might be prone to partitioning of injected H2O2. If the organic content controlled the H2O2 uptake, it could be expected that the detected amount of peroxides is the same for particles containing the same amount of OM. In the case of α-pinene OH the content of OM is almost constant (around 5–6 mg m−3), whereby the peroxide constant differs from 80 to 20 %. With a contribution of mixed organic and inorganic particles an H2O2 uptake cannot be excluded, but based on the present data set it can be regarded to be very small.
The fraction of SOA-bound peroxides is always expressed as a fraction of organic mass formed during the experiment and was calculated as follows:
where
-
nperox is amount of substance in micromoles (calculated from the iodometric peroxide test)
-
mperox is mass of organic peroxides in micrograms
-
Mperox is mass concentration of organic peroxides in micrograms per cubic meter
-
Vsampling is sampling volume of the filter in cubic meters
-
Fperox is mass fraction of SOA-bound peroxides in percent
-
Morg is amount of organic mass formed during experiment in micrograms per cubic meter.
Organic peroxides were detected from α-pinene and limonene, but not from m-cresol. The absence of organic peroxides for m-cresol might be a result of the aromatic structure. Notably, in the case of α-pinene, organic peroxides were only detected from the OH-radical reaction, albeit in very high fractions. This observation was unexpected as NOx was present in the system, usually suppressing ROOH formation. Considering the reaction of alkylperoxy radicals (RO2) with hydroperoxy radicals (HO2) is the most important source for ROOH, this source decreases with rising NOx levels due to the competition with RO2 + NO (Presto et al., 2005). Other processes than RO2 + HO2 should have yielded organic peroxides, and thus other compounds of peroxidic nature are detected by the applied test. As daytime experiments were performed with H2O2 as an OH source, blank filters were carefully checked to exclude the contribution of H2O2 present in the particle phase due to gas-to-particle partitioning of the injected oxidant. In general, peroxide fractions of 10–80 % of the organic mass have been detected from α-pinene OH experiments. The high peroxide fractions of 10–80 % are in contradiction to the small SOA yields from α-pinene OH (YOH≈ 3.5 %). While the organic peroxide formation from the ozonolysis of α-pinene and limonene has been studied in the past (Docherty et al., 2005; Mertes et al., 2012; Epstein et al., 2014; Krapf et al., 2016; Gong et al., 2018), peroxide fractions from OH-radical-induced oxidation are rare (Mertes et al., 2012). Mertes and co-workers reported peroxide fractions between 5 and 17 % (low NOx at 50 % RH) and 5.5–6.4 % (high NOx at 75 % RH). Those values are slightly lower than observed in this study, with 34 % (medium NOx at 50 % RH) and 13 % (medium NOx at 75 % RH). The difference between the two studies is most likely caused by usage of other OH-radical sources. The observed tendency of lower peroxide fractions under elevated RH is consistent in both studies and might be a result of two facts: (i) the uptake of HO2 radicals from the gas phase and (ii) decomposition and/or hydrolysis of hydroperoxides. It has been reported that the gas-phase HO2 radical concentration is significantly suppressed by 3 orders of magnitude when a liquid phase is present (Herrmann et al., 1999). If the HO2 uptake is increased under elevated RH, then HO2, in the gas phase, is only available in a smaller amount to react with RO2 radicals (Herrmann et al., 1999). Furthermore, decomposition and/or hydrolysis occur to a larger extent, lowering the peroxide fraction under high RH (Chen et al., 2011; Wang et al., 2011).
In contrast, limonene yielded SOA-bound peroxides from both oxidation regimes, NO3 and OH. For the OH-radical reaction no difference between the respective fractions can be observed, which leads to an average organic peroxide fraction of about 30 % without a dependency on RH. This therefore indicates that (i) organic peroxides are of a different nature than those formed from α-pinene, (ii) they originate from other reactions, and/or (iii) they are protected from hydrolysis. The latter hypothesis is supported by the high fraction of water-insoluble material, which could hint at a separate organic phase protecting peroxides from hydrolysis. The almost stable content might indicate that those peroxides and their respective formation pathways are not affected by humidity. Thus, it could be speculated that peroxides of higher molecular weight – i.e., dimers with peroxyhemiacetal structure – are formed. Due to the lack of data, no comparison to other studies can be provided.
The reaction of limonene with NO3 radicals yielded peroxides as well, in fractions comparable to those measured for both OH reactions with lower RH. Under higher RH (75 %), the peroxide fraction decreases dramatically to almost 0.5 %. The formation of organic peroxides in NO3-initiated reactions has not been a subject of VOC oxidation studies, with regards to the published information at present. Only a few studies have examined the further processing of nitrooxy-, alkyl-, peroxy-radical formed during NO3-radical reaction. For isoprene it has been shown that the reaction of nitrooxy, alkyl, and peroxy radical with other RO2 and HO2 is able to form peroxidic compounds, such as ROOR C10 dimers and nitrooxyhydroperoxide (Kwan et al., 2012; Schwantes et al., 2015). As the detected fraction of SOA-bound peroxides decreases with RH, the reaction of RO2 + HO2 seems to be the major source of these peroxides.
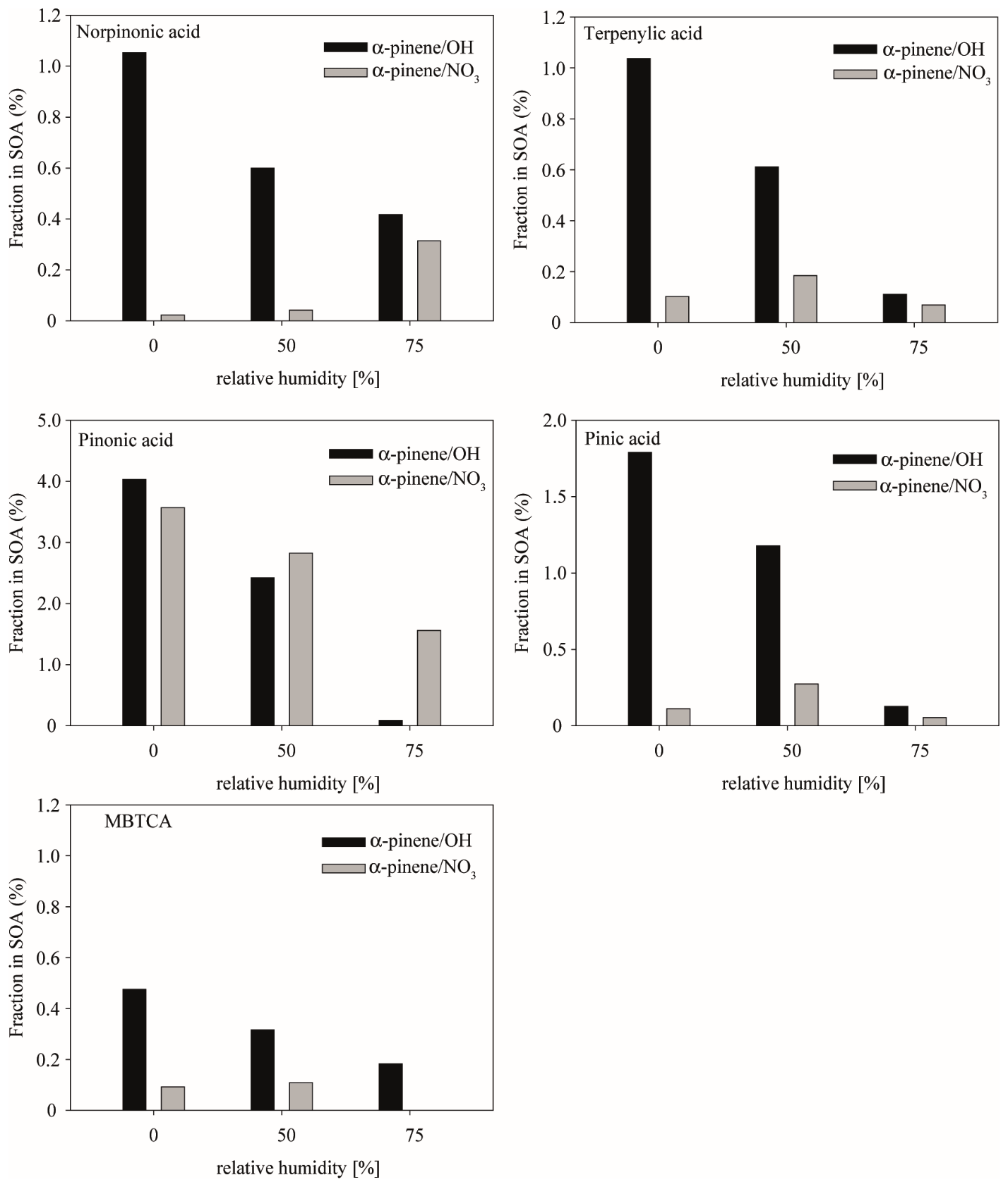
Figure 6Fraction of α-pinene marker compounds norpinonic acid, terpenylic acid, pinonic acid, pinic acid, and MBTCA for OH-radical reaction (black) and NO3-radical reaction (grey).

Figure 7Fraction of 3-methyl-6-nitrocatechol from the oxidation of m-cresol with OH. Please note that other compounds from the ASOA standard (Hoffmann et al., 2007) were not identified, and due to the low SOA yield from NO3-radical reaction the concentration might be below the detection limit.
3.3.3 Biogenic SOA marker compounds
The quantification of biogenic marker compounds was performed using a BSOA standard containing norpinonic acid, terpenylic acid, pinonic acid, pinic acid, and MBTCA (Fig. 6). These compounds were detected from night- and daytime chemistry of α-pinene, while, due to its different structure, limonene does not form these compounds. Therefore, the discussion about the fraction of those BSOA marker compounds refers to those formed within the α-pinene oxidations. Fractions of SOA marker compounds were expressed as mass fraction in formed organic mass (derived from SMPS measurements).
The most important SOA marker compounds formed from the OH-radical reaction of α-pinene are pinonic acid < pinic acid < norpinonic acid and terpenylic acid < MBTCA. A significant dependency on RH was observed in the same manner as with the organic peroxides (Fig. 5) for all compounds: increasing RH leads to decreasing SOA fractional contributions, while the SOA yields themselves remain about constant (Fig. 1).
As stated above, this observation might be caused by four main factors: (i) the uptake of the SOA marker compounds or their precursor compounds change as a function of the experimental conditions, (ii) the formation process of SOA marker compounds is directly affected by the experimental conditions, (iii) further reactions take place within the particle phase, and (iv) the uptake behavior of the first-generation oxidation products might change with LWC. However, it should be noted that earlier studies (Seinfeld et al., 2001; Ma et al., 2007) suggest the ALWC does influence the partitioning behavior of carboxylic acids such as pinic acid and pinonic acid. As a higher RH corresponds to a higher ALWC, enhanced partitioning is expected. However, this hypothesis is not supported by the experimental results of this study due to the observation of higher fractions of the SOA marker compounds under almost dry conditions; factor (i) seems to be unlikely. Factor (ii) may play a partial role in the formation of carboxylic acids. In the literature, the formation of carboxylic acids is often described by OH attacks on a particular precursor compound, and subsequent mechanisms usually involve the reaction of a formed acylperoxy radical with HO2 radicals (Niki et al., 1985; Moortgat et al., 1989; Lightfoot et al., 1992; Larsen et al., 2001). For both reactions, water is most likely to have a direct effect on OH and HO2 radicals, except in opposite directions. Vöhringer-Martinez et al. (2007) suggest that water molecules catalyze the attack of OH radicals (in the gas and particle phases) due to the formation of hydrogen bonds which can lower the reaction barrier. This catalytic effect could lead to higher fractions of specific markers under elevated RH, which was not observed in the data set obtained from this study. On the contrary, the reaction of the acylperoxy radical with HO2 might be RH-dependent because, as discussed before, HO2 tends to partition into the particle phase at elevated RH and thus is not sufficiently available in the gas phase (Herrmann et al., 1999). This might explain the low fractions of marker compounds at elevated RH.
The last factor (iii) to be considered involves further reactions of the SOA marker compounds yielding HMWCs in the particle phase, as has been often described in the literature (e.g., Gao et al., 2004; Tolocka et al., 2004; Müller et al., 2008; Yasmeen et al., 2010). It was suggested that compounds such as terpenylic acid and pinic acid can react further in the particle phase to form dimers (Yasmeen et al., 2010). Thus, under elevated RH, the formation of higher-molecular-weight compounds might be enhanced, lowering the fraction of individual monomeric compounds. Nevertheless, based on the experimental results of this study and the literature data, the combination of two factors appears to be important for the formation of carboxylic acids: first, the suppression of carboxylic acid formation due to enhanced partitioning of HO2 into the particle phase under elevated RH and, second, the further reaction of particulate marker compounds yielding HMWCs.
Notably, pinonic acid was detected in comparable amounts from α-pinene OH and α-pinene NO3 with the same RH dependency (Fig. 6). The reasons for such a pronounced RH dependency have been discussed intensively above. In addition to the described factors, in the case of NO3-radical reactions, the central role of organonitrates needs to be considered. It cannot be ignored that ONs act as potential precursors for the detected marker compounds. With elevated RH conditions and depending on their structure, ONs are prone to undergoing hydrolysis, and the respective products should show higher fractions under elevated RH. This observation seems to be the case for norpinonic acid, leading to the theory that these might be the hydrolysis products of respective organonitrates. More data are needed for the hypothesis of norpinonic acid being a hydrolysis product of ONs, as it remains speculative at this time.
The remaining marker compounds observed after the NO3 reactions – terpenylic acid, pinic acid, and MBTCA – do not show a significant variation with RH and are not affected by water or ALWC. Nevertheless, the fractions of all compounds, except pinonic acid, are significantly lower compared to the OH-radical reaction. This is most likely caused by an enhanced formation of ONs.
3.3.4 Anthropogenic SOA marker compounds
A well-established analytical method was applied to identify and quantify anthropogenic secondary organic aerosol (ASOA) marker compounds from m-cresol oxidation (Hoffmann et al., 2007). Despite the larger number of standards present in the authentic standard mixture, only 3-methyl-6-nitro-catechol (MNC) could be detected and quantified (Fig. 7). This compound was only detected from the OH-radical reaction. Quantification of marker compounds in samples after nighttime processing cannot be provided due to the very small SOA yields.
For MNC, an intense RH dependency was found in higher values under humid conditions and lower values under dry conditions (1.5 %). In particular, with elevated RH the fraction of 3-methyl-6-nitro-catechol reached 6 % of overall formed SOA mass, highlighting the importance of this particular oxidation product. Methyl-nitro-catechols are of special interest because they are important biomass burning tracer compounds, and their ambient concentration can reach up to 29 ng m−3 (Iinuma et al., 2010). Additionally, such a high fraction of MNC could also affect the phase state of the particles. It has been shown that MNC particles adsorb water under elevated RH, leading to a change in the phase state of the particles (Slade and Knopf, 2014). This effect is connected to a lower uptake of OH radicals into the particle phase by a factor of 4 when RH increases from 15 to 30 %. The lower fractions under dry conditions might be a result of a stronger OH uptake into the particle phase, leading to a greater extent of heterogenous reactions of MNC in the particle phase.
3.4 Atmospheric implications and conclusion
The examination of the oxidation of atmospherically relevant compounds and the resulting SOA formation is of large importance for a better understanding of atmospheric chemistry and its response to future climate change. Several studies predict an increase in BVOC emissions as a response to a warmer climate (e.g., Sanderson et al., 2003; Lathière et al., 2005; Heald et al., 2008). The increase in monoterpenes emission is estimated to be up to 50 % (Lathière et al., 2005). Such a dramatic increase might lead to an enhanced formation of reactive oxygen species (ROS) as determined here as peroxides and SOA. ROS are suggested to cause oxidative stress that influences human morbidity and mortality (Squadrito et al., 2001; Schwartz et al., 2002; Xiao et al., 2003; Ayres et al., 2008). The current knowledge is limited, with large uncertainties for predicting the global SOA burden (see review by Hallquist et al., 2009; Glasius and Goldstein, 2016; Shrivastava et al., 2017). With an expected increase in VOC emissions the knowledge about SOA formation process and their response to changes in the parameters investigated in this study will become more important. The present study provides important data concerning SOA formation potential of OH and NO3 radical oxidation of biogenic and anthropogenic VOCs, the influence of relative humidity on the SOA yield, and its resulting chemical composition.
During the night and early morning hours, the RH near the surface is high; NO3 radical chemistry is competitive with that of other oxidants; and, accordingly, RH starts to play a crucial role. The investigation of the effect of RH on the SOA formation and chemical composition shed light on various aspects, especially for NO3-initiated SOA formation. NO3 radical reaction can form SOA more efficiently than OH-radical reaction in the presence of NOx, at least for α-pinene and limonene, highlighting the importance of this atmospherically relevant nighttime sink. Furthermore, pinonic acid was found to contribute significantly, up to 4 %, for NO3- and OH-originated α-pinene-SOA, indicating this compound might play a key role for both day- and nighttime conditions, not just daytime conditions. Pinonic acid might be formed from the oxidation of pinonaldehyde that itself has been found for NO3-radical-initiated reactions of α-pinene (Spittler et al., 2006). It should be also noted that huge amounts of organic peroxides were found from α-pinene OH, which are an important part of ROS and can be associated with oxidative stress after inhalation of such particles. The peroxide fraction was found to be higher under dry conditions and, somewhat surprisingly, decreases with RH.
Relative humidity was found to affect SOA growth and composition, in particular the formation of MNC during m-cresol oxidation. While daytime chemistry of α-pinene and limonene is RH-independent (YOH ≈ 6 % and 20 %), SOA yields from m-cresol + OH increased with elevated RH (YOH≈ 3–9 %). This observed effect is most likely to be attributed to the huge fraction of MNC of up to 6 % under high RH, lowering the uptake of OH radicals and changing the phase state. Additionally, the reaction of limonene + NO3 appeared to be very sensitive towards RH, yielding the highest SOA ( 29 %) under dry conditions. This observation is suggested to be caused by a competitive reaction between limonene and formed RO2 radicals, leading to a lower conversion of limonene. Furthermore, m-cresol was found to yield only insignificant amounts with NO3, thus producing a highly reactive gas phase, since almost all oxidation products stay in the gas phase. The concentration of reactive species in the gas phase could act as a reservoir for compounds with a much higher SOA formation potential.
All data presented in this study are available from the authors upon request (herrmann@tropos.de). In addition data from experiment at RH = 50 % (α-pinene OH, α-pinene NO3, limonene OH, limonene NO3, cresol NO3) are available at the EUROCHAMP web page (https://data.eurochamp.org/data-access/chamber-experiments/#/, last access: 24 January 2020, Herrmann, 2020).
AM, OB, and HH planned the experiments. AM, YZ, MR, AK, and OB performed the chamber experiments. AM, YZ, and AK analyzed the data. AM and HH wrote the manuscript. YZ, MR, AK, XW, and HH edited the manuscript.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
All support received is gratefully acknowledged.
The present study was supported by the German Research Foundation (DFG) under grant number HE 3086/25-1. It received further support through funding from the European Union's Horizon 2020 research and innovation program through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997. Exchange of staff was supported through the EU Marie Skłodowska-Curie Actions AMIS (295132) and MARSU (690958-MARSU-RISE-2015).
This paper was edited by Jean-Francois Doussin and reviewed by three anonymous referees.
Atkinson, R., Aschmann, S. M., Winer, A. M., and Pitts, J. N.: Kinetics of the gas-phase reactions of NO3 radicals with a series of dialkenes, cycloalkenes, and monoterpenes at 295 1 K, Environ. Sci. Technol., 18, 370–375, https://doi.org/10.1021/es00123a016, 1984.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, S197–S219, https://doi.org/10.1016/s1352-2310(03)00391-1, 2003.
Ayres, J. G., Borm, P., Cassee, F. R., Castranova, V., Donaldson, K., Ghio, A., Harrison, R. M., Hider, R., Kelly, F., Kooter, I. M., Marano, F., Maynard, R. L., Mudway, I., Nel, A., Sioutas, C., Smith, S., Baeza-Squiban, A., Cho, A., Duggan, S., and Froines, J.: Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress potential – A workshop report and consensus statement, Inhal. Toxicol., 20, 75–99, https://doi.org/10.1080/08958370701665517, 2008.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Barnes, I., Bastian, V., Becker, K. H., and Tong, Z.: Kinetics and Products of the reactions of NO3 with monoalkene, dialkenes and monoterpenes, J. Phys. Chem., 94, 2413–2419, https://doi.org/10.1021/j100369a041, 1990.
Bonn, B. and Moorgat, G. K.: New particle formation during a- and b-pinene oxidation by O3, OH and NO3, and the influence of water vapour: particle size distribution studies, Atmos. Chem. Phys., 2, 183–196, https://doi.org/10.5194/acp-2-183-2002, 2002.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Carter, W. P. L., Winer, A. M., and Pitts, J. N.: Major atmospheric sink for phenol and the cresols. Reaction with the nitrate radical, Environ. Sci. Technol., 15, 829–831, https://doi.org/10.1021/es00089a009, 1981.
Chen, Q., Liu, Y., Donahue, N. M., Shilling, J. E., and Martin, S. T.: Particle-phase chemistry of secondary Organic material: modeled compared to measured O : C and H : C elemental ratios provide constraints, Environ. Sci. Technol., 45, 4763–4770, https://doi.org/10.1021/es104398s, 2011.
Chen, X. and Hopke, P. K.: A chamber study of secondary organic aerosol formation by limonene ozonolysis, Indoor Air, 20, 320–328, https://doi.org/10.1111/j.1600-0668.2010.00656.x, 2010.
Chung, C. E., Ramanathan, V., and Decremer, D.: Observationally constrained estimates of carbonaceous aerosol radiative forcing, P. Natl. Acad. Sci. USA, 109, 11624–11629, https://doi.org/10.1073/pnas.1203707109, 2012.
Claeys, M., Iinuma, Y., Szmigielski, R., Surratt, J. D., Blockhuys, F., Van Alsenoy, C., Böge, O., Sierau, B., Gómez-González, Y., Vermeylen, R., Van der Veken, P., Shahgholi, M., Chan, A. W. H., Herrmann, H., Seinfeld, J. H., and Maenhaut, W.: Terpenylic acid and related compounds from the oxidation of α-pinene: implications for new particle formation and growth above forests, Environ. Sci. Technol., 43, 6976–6982, https://doi.org/10.1021/es9007596, 2009.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic model of the system HNHSONOH2O at tropospheric temperatures, J. Phys. Chem. A, 102, 2137–2154, https://doi.org/10.1021/jp973042r, 1998.
Cocker, D. R., Clegg, S. L., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas–particle partitioning of secondary organic aerosol. Part I: α-pinene/ozone system, Atmos. Environ., 35, 6049–6072, https://doi.org/10.1016/S1352-2310(01)00404-6, 2001.
Coeur-Tourneur, C., Henry, F., Janquin, M.-A., and Brutier, L.: Gas-phase reaction of hydroxyl radicals with m-, o- and p-cresol, Int. J. Chem. Kin., 38, 553–562, https://doi.org/10.1002/kin.20186, 2006.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Day, D. A., Liu, S., Russell, L. M., and Ziemann, P. J.: Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California, Atmos. Environ., 44, 1970–1979, https://doi.org/10.1016/j.atmosenv.2010.02.045, 2010.
Dlugokencky, E. J. and Howard, C. J.: Studies of NO3 radical reacions with some atmospheric organic-compounds at low pressures, J. Phys. Chem., 93, 1091–1096, https://doi.org/10.1021/j100340a015, 1989.
Docherty, K. S., Wu, W., Lim, Y. B., and Ziemann, P. J.: Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O3, Environ. Sci. Technol., 39, 4049–4059, https://doi.org/10.1021/es050228s, 2005.
Emanuelsson, E. U., Hallquist, M., Kristensen, K., Glasius, M., Bohn, B., Fuchs, H., Kammer, B., Kiendler-Scharr, A., Nehr, S., Rubach, F., Tillmann, R., Wahner, A., Wu, H.-C., and Mentel, Th. F.: Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 13, 2837–2855, https://doi.org/10.5194/acp-13-2837-2013, 2013.
Epstein, S. A., Blair, S. L., and Nizkorodov, S. A.: Direct photolysis of α-pinene ozonolysis secondary organic aerosol: effect on particle mass and peroxide content, Environ. Sci. Technol., 48, 11251–11258, https://doi.org/10.1021/es502350u, 2014.
Friedmann, B. and Farmer, D. K.: SOA and gas phase organic yields from the sequential photooxidation of seven monoterpenes, Atmos. Environ., 187, 335–345, 2018.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C.: SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 11, 3879–3894, https://doi.org/10.5194/acp-11-3879-2011, 2011.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkle, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Gao, S., Ng, N. L., Keywood, M., Varutbangkul, V., Bahreini, R., Nenes, A., He, J., Yoo, K. Y., Beauchamp, J. L., Hodyss, R. P., Flagan, R. C., and Seinfeld, J. H.: Particle phase acidity and oligomer formation in secondary organic aerosol, Environ. Sci. Technol., 38, 6582–6589, https://doi.org/10.1021/es049125k, 2004.
Geyer, A., Alicke, B., Konrad, S., Schmitz, T., Stutz, J., and Platt, U.: Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.-Atmos., 106, 8013–8025, https://doi.org/10.1029/2000jd900681, 2001.
Glasius, M. and Goldstein, A. H.: Recent discoveries and future challenges in atmospheric organic chemistry, Environ. Sci. Technol., 50, 2754–2764, https://doi.org/10.1021/acs.est.5b05105, 2016.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth’s Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Gong, Y., Chen, Z., and Li, H.: The oxidation regime and SOA composition in limonene ozonolysis: roles of different double bonds, radicals, and water, Atmos. Chem. Phys., 18, 15105–15123, https://doi.org/10.5194/acp-18-15105-2018, 2018.
Hallquist, M., Wangberg, I., Ljungstrom, E., Barnes, I., and Becker, K. H.: Aerosol and product yields from NO3 radical-initiated oxidation of selected monoterpenes, Environ. Sci. Technol., 33, 553–559, https://doi.org/10.1021/es980292s, 1999.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heald, C. L., Henze, D. K., Horowitz, L. W., Feddema, J., Lamarque, J.-F., Guenther, A., Hess, P. G., Vitt, F., Seinfeld, J. H., Goldstein, A. H., and Fung, I.: Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res.-Atmos., 113, D05211, https://doi.org/10.1029/2007jd009092, 2008.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of relative humidity on gas/particle partitioning and aerosol mass yield in the photooxidation of p-xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Herrmann, H.: Database of atmospheric simulation chamber studies, available at: https://data.eurochamp.org/data-access/chamber-experiments/#/, last access: 24 January 2020.
Herrmann, H., Ervens, B., Nowacki, P., Wolke, R., and Zellner, R.: A chemical aqueous phase radical mechanism for tropospheric chemistry, Chemosphere, 38, 1223-1232, https://doi.org/10.1016/S0045-6535(98)00520-7, 1999.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Hoffmann, D., Iinuma, Y., and Herrmann, H.: Development of a method for fast analysis of phenolic molecular markers in biomass burning particles using high performance liquid chromatography/atmospheric pressure chemical ionisation mass spectrometry, J. Chromatogr.A, 1143, 168–175, https://doi.org/10.1016/j.chroma.2007.01.035, 2007.
Horowitz, L. W., Fiore, A. M., Milly, G. P., Cohen, R. C., Perring, A., Wooldridge, P. J., Hess, P. G., Emmons, L. K., and Lamarque, J.-F.: Observational constraints on the chemistry of isoprene nitrates over the eastern United States, J. Geophys. Res.-Atmos., 112, D12S08, https://doi.org/10.1029/2006jd007747, 2007.
Hoyle, C. R., Berntsen, T., Myhre, G., and Isaksen, I. S. A.: Secondary organic aerosol in the global aerosol – chemical transport model Oslo CTM2, Atmos. Chem. Phys., 7, 5675–5694, https://doi.org/10.5194/acp-7-5675-2007, 2007.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Iinuma, Y., Böge, O., Keywood, M., Gnauk, T., and Herrmann, H.: Diaterebic acid acetate and diaterpenylic acid acetate: atmospheric tracers for secondary organic aerosol formation from 1,8-cineole oxidation, Environ. Sci. Technol., 43, 280–285, https://doi.org/10.1021/es802141v, 2009.
Iinuma, Y., Böge, O., Gräfe, R., and Herrmann, H.: Methyl-nitrocatechols: atmospheric tracer compounds for biomass burning secondary organic aerosols, Environ. Sci. Technol., 44, 8453–8459, https://doi.org/10.1021/es102938a, 2010.
Izumi, K. and Fukuyama, T.: Photochemical aerosol formation from aromatic hydrocarbons in the presence of NOx, Atmos. Environ. A, 24, 1433–1441, https://doi.org/10.1016/0960-1686(90)90052-O, 1990.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jiang, L., Wang, W., and Xu, Y.-S.: Theoretical investigation of the NO3 radical addition to double bonds of limonene, Int. J. Mol. Sci., 10, 3743–3754, https://doi.org/10.3390/ijms10093743, 2009.
Joo, T., Rivera-Rios, J. C., Takeuchi, M., Alvarado, M. J., and Ng, N. L.: Secondary organic aerosol formation from reaction of 3-methylfuran with nitrate radicals, ACS Earth Space Chem., 3, 922–934, https://doi.org/10.1021/acsearthspacechem.9b00068, 2019.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Aijala, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016gl069239, 2016.
Kind, I., Berndt, T., and Böge, O.: Gas-phase rate constants for the reaction of NO3 radicals with a series of cyclic alkenes, 2-ethyl-1-butene and 2,3-dimethyl-1,3-butadiene, Chem. Phys. Lett., 288, 111–118, https://doi.org/10.1016/s0009-2614(98)00254-1, 1998.
Krapf, M., El Haddad, I., Bruns, Emily A., Molteni, U., Daellenbach, Kaspar R., Prévôt, André S. H., Baltensperger, U., and Dommen, J.: Labile peroxides in secondary organic aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Kurtenbach, R., Ackermann, R., Becker, K. H., Geyer, A., Gomes, J. A. G., Lorzer, J. C., Platt, U., and Wiesen, P.: Verification of the contribution of vehicular traffic to the total NMVOC emissions in Germany and the importance of the NO3 chemistry in the city air, J. Atmos. Chem., 42, 395–411, https://doi.org/10.1023/a:1015778616796, 2002.
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Larsen, B. R., Di Bella, D., Glasius, M., Winterhalter, R., Jensen, N. R., and Hjorth, J.: Gas-phase OH oxidation of monoterpenes: gaseous and particulate products, J. Atmos. Chem., 38, 231–276, https://doi.org/10.1023/A:1006487530903, 2001.
Lathière, J., Hauglustaine, D. A., De Noblet-Ducoudré, N., Krinner, G., and Folberth, G. A.: Past and future changes in biogenic volatile organic compound emissions simulated with a global dynamic vegetation model, Geophys. Res. Lett., 32, L20818, https://doi.org/10.1029/2005gl024164, 2005.
Liebmann, J., Karu, E., Sobanski, N., Schuladen, J., Ehn, M., Schallhart, S., Quéléver, L., Hellen, H., Hakola, H., Hoffmann, T., Williams, J., Fischer, H., Lelieveld, J., and Crowley, J. N.: Direct measurement of NO3 radical reactivity in a boreal forest, Atmos. Chem. Phys., 18, 3799–3815, https://doi.org/10.5194/acp-18-3799-2018, 2018a.
Liebmann, J. M., Muller, J. B. A., Kubistin, D., Claude, A., Holla, R., Plass-Dülmer, C., Lelieveld, J., and Crowley, J. N.: Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: identification of reactive trace gases and comparison with OH reactivity, Atmos. Chem. Phys., 18, 12045–12059, https://doi.org/10.5194/acp-18-12045-2018, 2018b.
Lightfoot, P. D., Cox, R. A., Crowley, J. N., Destriau, M., Hayman, G. D., Jenkin, M. E., Moortgat, G. K., and Zabel, F.: Organic peroxy radicals: kinetics, spectroscopy and tropospheric chemistry, Atmos. Environ. Pt. A, 26, 1805–1961, https://doi.org/10.1016/0960-1686(92)90423-I, 1992.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998.
Ma, Y., Willcox, T. R., Russell, A. T., and Marston, G.: Pinic and pinonic acid formation in the reaction of ozone with α-pinene, Chem. Commun., 1328–1330, https://doi.org/10.1039/B617130C, 2007.
Martinez, E., Cabanas, B., Aranda, A., and Martin, P.: Kinetics of the reactions of NO3 radical with selected monoterpenes: A temperature dependence study, Environ. Sci. Technol., 32, 3730–3734, https://doi.org/10.1021/es970899t, 1998.
Martinez, E., Cabanas, B., Aranda, A., Martin, P., and Salgado, S.: Absolute rate coefficients for the gas-phase reactions of NO3 radical with a series of monoterpenes at T= 298 to 433 K, J. Atmos. Chem., 33, 265–282, https://doi.org/10.1023/a:1006178530211, 1999.
McMurry, P. H. and Grosjean, D.: Gas and Aerosol Wall Losses in Teflon Film Smog Chamber, Environ. Sci. Technol., 19, 1176–1182, 1985.
McLaren, R., Wojtal, P., Majonis, D., McCourt, J., Halla, J. D., and Brook, J.: NO3 radical measurements in a polluted marine environment: links to ozone formation, Atmos. Chem. Phys., 10, 4187–4206, https://doi.org/10.5194/acp-10-4187-2010, 2010.
Mertes, P., Pfaffenberger, L., Dommen, J., Kalberer, M., and Baltensperger, U.: Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP), Atmos. Meas. Tech., 5, 2339–2348, https://doi.org/10.5194/amt-5-2339-2012, 2012.
Moldanova, J. and Ljungstrom, E.: Modelling of particle formation from NO3 oxidation of selected monoterpenes, J. Aerosol Sci., 31, 1317–1333, https://doi.org/10.1016/s0021-8502(00)00041-0, 2000.
Moortgat, G. K., Veyret, B., and Lesclaux, R.: Kinetics of the reaction of HO2 with CH3C(O)O2 in the temperature range 253–368 K, Chem. Phys. Lett., 160, 443–447, https://doi.org/10.1016/0009-2614(89)87624-9, 1989.
Müller, L., Reinnig, M.-C., Warnke, J., and Hoffmann, Th.: Unambiguous identification of esters as oligomers in secondary organic aerosol formed from cyclohexene and cyclohexene/α-pinene ozonolysis, Atmos. Chem. Phys., 8, 1423–1433, https://doi.org/10.5194/acp-8-1423-2008, 2008.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: An improved method for the quantification of SOA bound peroxides, Atmos. Environ., 67, 365–369, https://doi.org/10.1016/j.atmosenv.2012.11.012, 2013.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M., Böge, O., Richters, S., Spindler, G., Sipilä, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly oxidized multifunctional organic compounds observed in tropospheric particles: A field and laboratory study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: Monoterpene SOA – Contribution of first-generation oxidation products to formation and chemical composition, Atmos. Environ., 130, 136–144, https://doi.org/10.1016/j.atmosenv.2015.10.080, 2016.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical aging of alpha-pinene and beta-pinene secondary organic aerosol formed from nitrate radical oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Nakao, S., Clark, C., Tang, P., Sato, K., and Cocker III, D.: Secondary organic aerosol formation from phenolic compounds in the absence of NOx, Atmos. Chem. Phys., 11, 10649–10660, https://doi.org/10.5194/acp-11-10649-2011, 2011.
Neusüß, C., Gnauk, T., Plewka, A., Herrmann, H., and Quinn, P. K.: Carbonaceous aerosol over the Indian Ocean: OC EC fractions and selected specifications from size-segregated onboard samples, J. Geophys. Res.-Atmos., 107, 30–13, https://doi.org/10.1029/2001jd000327, 2002.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of First- versus Second-Generation Products to Secondary Organic Aerosols Formed in the Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Niki, H., Maker, P. D., Savage, C. M., and Breitenbach, L. P.: FTIR study of the kinetics and mechanism for chlorine-atom-initiated reactions of acetaldehyde, J. Phys. Chem., 89, 588–591, https://doi.org/10.1021/j100250a008, 1985.
Northcross, A. L. and Jang, M.: Heterogeneous SOA yield from ozonolysis of monoterpenes in the presence of inorganic acid, Atmos. Environ., 41, 1483–1493, https://doi.org/10.1016/j.atmosenv.2006.10.009, 2007.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Perraud, V., Bruns, E. A., Ezell, M. J., Johnson, S. N., Greaves, J., and Finlayson-Pitts, B. J.: Identification of organic nitrates in the NO3 radical initiated oxidation of alpha-pinene by atmospheric pressure chemical ionization mass spectrometry, Environ. Sci. Technol., 44, 5887–5893, https://doi.org/10.1021/es1005658, 2010.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration, Environ. Sci. Technol., 39, 7046–7054, https://doi.org/10.1021/es050400s, 2005.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Qin, M. M., Hu, Y. T., Wang, X. S., Vasilakos, P., Boyd, C. M., Xu, L., Song, Y., Ng, N. L., Nenes, A., and Russell, A. G.: Modeling biogenic secondary organic aerosol (BSOA) formation from monoterpene reactions with NO3: A case study of the SOAS campaign using CMAQ, Atmos. Environ., 184, 146–155, https://doi.org/10.1016/j.atmosenv.2018.03.042, 2018.
Ramasamy, S., Nakayama, T., Imamura, T., Morino, Y., Kajii, Y., and Sato, K.: Investigation of dark condition nitrate radical-and ozone-initiated aging of toluene secondary organic aerosol: Importance of nitrate radical reactions with phenolic products, Atmos. Environ., 219, 117049, https://doi.org/10.1016/j.atmosenv.2019.117049, 2019.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from alpha-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Rollins, A. W., Smith, J. D., Wilson, K. R., and Cohen, R. C.: Real time in situ detection of organic nitrates in atmospheric aerosols, Environ. Sci. Technol., 44, 5540–5545, https://doi.org/10.1021/es100926x, 2010.
Romano, A. and Hanna, G. B.: Identification and quantification of VOCs by proton transfer reaction time of flight mass spectrometry: An experimental workflow for the optimization of specificity, sensitivity, and accuracy, J. Mass Spectrom., 53, 287–295, https://doi.org/10.1002/jms.4063, 2018.
Sanderson, M. G., Jones, C. D., Collins, W. J., Johnson, C. E., and Derwent, R. G.: Effect of climate change on isoprene emissions and surface ozone levels, Geophys. Res. Lett., 30, 1936, https://doi.org/10.1029/2003gl017642, 2003.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 oxidation products from the RO2 + HO2 pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Schwartz, J., Laden, F., and Zanobetti, A.: The concentration-response relation between PM2.5 and daily deaths, Environ. Health Persp., 110, 1025–1029, https://doi.org/10.1289/ehp.021101025, 2002.
Seinfeld, J., Erdakos, G., Asher, W., and Pankow, J.: Modeling the formation of secondary organic aerosol (SOA). 2. The predicted effects of relative humidity on aerosol formation in the alpha-pinene-, beta-pinene-, sabinene-, delta 3-carene-, and cyclohexene-ozone systems, Environ. Sci. Technol., 35, 1806–1817, 2001.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys. 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Slade, J. H. and Knopf, D. A.: Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity, Geophys. Res. Lett., 41, 5297–5306, https://doi.org/10.1002/2014gl060582, 2014.
Spindler, G., Müller, K., Brüggemann, E., Gnauk, T., and Herrmann, H.: Long-term size-segregated characterization of PM10, PM2.5, and PM1 at the IfT research station Melpitz downwind of Leipzig (Germany) using high and low-volume filter samplers, Atmos. Environ., 38, 5333–5347, https://doi.org/10.1016/j.atmosenv.2003.12.047, 2004.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, T., and Wirtz, K.: Reactions of NO3 radicals with limonene and alpha-pinene: Product and SOA formation, Atmos. Environ., 40, S116–S127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Squadrito, G. L., Cueto, R., Dellinger, B., and Pryor, W. A.: Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter, Free Radical Bio. Med., 31, 1132–1138, https://doi.org/10.1016/S0891-5849(01)00703-1, 2001.
Stewart, D. J., Almabrok, S. H., Lockhart, J. P., Mohamed, O. M., Nutt, D. R., Pfrang, C., and Marston, G.: The kinetics of the gas-phase reactions of selected monoterpenes and cyclo-alkenes with ozone and the NO3 radical, Atmos. Environ., 70, 227–235, https://doi.org/10.1016/j.atmosenv.2013.01.036, 2013.
Szmigielski, R., Surratt, J., Gomez-Gonzalez, Y., Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T., Lewandowski, M., Offenberg, J., Edney, E., Seinfeld, J., Maenhaut, W., and Claeys, M.: 3-Methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007gl031338, 2007.
Timonen, H., Aurela, M., Carbone, S., Saarnio, K., Saarikoski, S., Mäkelä, T., Kulmala, M., Kerminen, V.-M., Worsnop, D. R., and Hillamo, R.: High time-resolution chemical characterization of the water-soluble fraction of ambient aerosols with PILS-TOC-IC and AMS, Atmos. Meas. Tech., 3, 1063–1074, https://doi.org/10.5194/amt-3-1063-2010, 2010.
Tolocka, M. P., Jang, M., Ginter, J. M., Cox, F. J., Kamens, R. M., and Johnston, M. V.: Formation of oligomers in secondary organic aerosol, Environ. Sci. Technol., 38, 1428–1434, https://doi.org/10.1021/es035030r, 2004.
van Pinxteren, D., Brüggemann, E., Gnauk, T., Iinuma, Y., Müller, K., Nowak, A., Achtert, P., Wiedensohler, A., and Herrmann, H.: Size- and time-resolved chemical particle characterization during CAREBeijing-2006: Different pollution regimes and diurnal profiles, J. Geophys. Res.-Atmos., 114, D00G09, https://doi.org/10.1029/2008jd010890, 2009.
Vöhringer-Martinez, E., Hansmann, B., Hernandez, H., Francisco, J. S., Troe, J., and Abel, B.: Water catalysis of a radical-molecule gas-phase reaction, Science, 315, 497–501, https://doi.org/10.1126/science.1134494, 2007.
von Kuhlmann, R., Lawrence, M. G., Pöschl, U., and Crutzen, P. J.: Sensitivities in global scale modeling of isoprene, Atmos. Chem. Phys., 4, 1–17, https://doi.org/10.5194/acp-4-1-2004, 2004.
Wang, Y., Kim, H., and Paulson, S. E.: Hydrogen peroxide generation from α- and β-pinene and toluene secondary organic aerosols, Atmos. Environ., 45, 3149–3156, https://doi.org/10.1016/j.atmosenv.2011.02.060, 2011.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosa-Mas, C. E., Hjorth, J., Lebras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The nitrate radical – physics, chemistry, and the atmosphere, Atmos. Environ. Pt. A, 25, 1–203, https://doi.org/10.1016/0960-1686(91)90192-a, 1991.
Wiedensohler, A., Birmili, W., Nowak, A., Sonntag, A., Weinhold, K., Merkel, M., Wehner, B., Tuch, T., Pfeifer, S., Fiebig, M., Fjäraa, A. M., Asmi, E., Sellegri, K., Depuy, R., Venzac, H., Villani, P., Laj, P., Aalto, P., Ogren, J. A., Swietlicki, E., Williams, P., Roldin, P., Quincey, P., Hüglin, C., Fierz-Schmidhauser, R., Gysel, M., Weingartner, E., Riccobono, F., Santos, S., Grüning, C., Faloon, K., Beddows, D., Harrison, R., Monahan, C., Jennings, S. G., O'Dowd, C. D., Marinoni, A., Horn, H.-G., Keck, L., Jiang, J., Scheckman, J., McMurry, P. H., Deng, Z., Zhao, C. S., Moerman, M., Henzing, B., de Leeuw, G., Löschau, G., and Bastian, S.: Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions, Atmos. Meas. Tech., 5, 657–685, https://doi.org/10.5194/amt-5-657-2012, 2012.
Xiao, G. G., Wang, M. Y., Li, N., Loo, J. A., and Nel, A. E.: Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line, J. Biol. Chem., 278, 50781–50790, https://doi.org/10.1074/jbc.M306423200, 2003.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-Van Wertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Yasmeen, F., Vermeylen, R., Szmigielski, R., Iinuma, Y., Böge, O., Herrmann, H., Maenhaut, W., and Claeys, M.: Terpenylic acid and related compounds: precursors for dimers in secondary organic aerosol from the ozonolysis of α- and β-pinene, Atmos. Chem. Phys., 10, 9383–9392, https://doi.org/10.5194/acp-10-9383-2010, 2010.
Zaveri, R. A., Berkowitz, C. M., Brechtel, F. J., Gilles, M. K., Hubbe, J. M., Jayne, J. T., Kleinman, L. I., Laskin, A., Madronich, S., Onasch, T. B., Pekour, M. S., Springston, S. R., Thornton, J. A., Tivanski, A. V., and Worsnop, D. R.: Nighttime chemical evolution of aerosol and trace gases in a power plant plume: Implications for secondary organic nitrate and organosulfate aerosol formation, NO3 radical chemistry, and N2O5 heterogeneous hydrolysis, J. Geophys. Res.-Atmos., 115, D12304, https://doi.org/10.1029/2009jd013250, 2010.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.