the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 May 2021
| 25 May 2021
SO2 and NH3 emissions enhance organosulfur compounds and fine particle formation from the photooxidation of a typical aromatic hydrocarbon
Zhaomin Yang
Li Xu
Narcisse T. Tsona
Jianlong Li
Xin Luo
Aromatic hydrocarbons can dominate the volatile organic compound budget in the urban atmosphere. Among them, 1,2,4-trimethylbenzene (TMB), mainly emitted from solvent use, is one of the most important secondary organic aerosol (SOA) precursors. Although atmospheric SO2 and NH3 levels can affect secondary aerosol formation, the influenced extent of their impact and their detailed driving mechanisms are not well understood. The focus of the present study is to examine the chemical compositions and formation mechanisms of SOA from TMB photooxidation influenced by SO2 and/or NH3. Here, we show that SO2 emission could considerably enhance aerosol particle formation due to SO2-induced sulfate generation and acid-catalyzed heterogeneous reactions. Orbitrap mass spectrometry measurements revealed the generation of not only typical TMB products but also hitherto unidentified organosulfates (OSs) in SO2-added experiments. The OSs designated as being of unknown origin in earlier field measurements were also detected in TMB SOA, indicating that atmospheric OSs might also be originated from TMB photooxidation. For NH3-involved experiments, results demonstrated a positive correlation between NH3 levels and particle volume as well as number concentrations. The effects of NH3 on SOA composition were slight under SO2-free conditions but stronger in the presence of SO2. A series of multifunctional products with carbonyl, alcohols, and nitrate functional groups were tentatively characterized in NH3-involved experiments based on infrared spectra and mass spectrometry analysis. Plausible formation pathways were proposed for detected products in the particle phase. The volatility distributions of products, estimated using parameterization methods, suggested that the detected products gradually condense onto the nucleation particles to contribute to aerosol formation and growth. Our results suggest that strict control of SO2 and NH3 emissions might remarkably reduce organosulfates and secondary aerosol burden in the atmosphere. Updating the aromatic oxidation mechanism in models could result in more accurate treatment of particle formation for urban regions with considerable SO2, NH3, and aromatics emissions.
- Article
(2965 KB) - Full-text XML
-
Supplement
(1758 KB) - BibTeX
- EndNote





Secondary organic and inorganic aerosols have been observed to account for a considerable fraction of fine particulate matter (aerosol particles ≤ 2.5 µm in aerodynamic diameter, PM2.5) during PM2.5 pollution events which have frequently occurred and lasted for days or even weeks in China during the last decade (Huang et al., 2014; Guo et al., 2014). These particles can directly and indirectly impact regional and global climate (Kanakidou et al., 2005), air quality (Zhang et al., 2015), and human health (Lelieveld et al., 2015).
Secondary organic aerosols (SOAs) arise predominantly from the oxidation of volatile organic compounds (VOCs) in the atmosphere. Early atmospheric models underestimated the measured SOA mass concentrations in field studies by 1–2 orders of magnitude (Volkamer et al., 2006; Heald et al., 2010). Although recent efforts such as updating missing SOA precursors, accounting for unknown processes of gas-to-particle conversion, and improving emission inventories have narrowed the observed gap between the modeled and measured SOA mass, uncertainties still exist in organic aerosol estimates (Shrivastava et al., 2011; Cheng et al., 2021). Inorganic perturbations on SOA formation (Shrivastava et al., 2017) are partly responsible for these uncertainties, and they include the addition of mineral particles (Yu and Jang, 2019), nitrogen oxide (NOx) (Zhao et al., 2018), ammonia (NH3) (Hao et al., 2020), and sulfur dioxide (SO2) (Yang et al., 2020), which can engage in the gas- or particle-phase chemistry and subsequently influence SOA formation and growth (Friedman et al., 2016; Ng et al., 2007; Na et al., 2006). NOx effects on particle formation are generally known to be pronounced. High levels of SO2, NH3, and VOCs have been reported in certain regions such as Guangzhou (Zou et al., 2015), Beijing (Meng et al., 2020), and Handan (Li et al., 2017) in China. During haze pollution episodes, Li et al. (2017) observed that SO2 levels can be up to 200 ppb in Handan, China. A recent study also showed significant increasing NH3 levels in the atmosphere over the United States and the European Union (Warner et al., 2017). However, less focus has been placed on the SO2 and NH3 perturbations on SOA formation and chemical composition. Aerosol particles contain a multitude of compounds with different physicochemical properties. Previous laboratory studies examined the photooxidation of cyclohexene, fuel, and 1,3,5-trimethylbenzene in the presence of SO2 and reported organosulfate (OS) formation (Yang et al., 2020; Liu et al., 2017; Blair et al., 2017). The atmospheric oxidation of SO2 can generate sulfuric acid that is critical for the increase of particle acidity. SO2-induced acidic sulfate plays an active role in the production of OSs, which have been recognized as significant SOA tracers describing the enhancement in SOA by SO2 emission (Xu et al., 2015).
OSs are ubiquitous in ambient aerosol particles, and they are estimated to account for 3 %–30 % of the organic mass in fine aerosol particles (Surratt et al., 2008; Tolocka and Turpin, 2012). The presence of OSs could alter aerosol morphology (Riva et al., 2019), viscosity (Riva et al., 2019; Zhang et al., 2019), particle acidity (Riva et al., 2019), phase state (Zhang et al., 2019), hygroscopicity (Estillore et al., 2016; Hansen et al., 2015), and optical properties (Fleming et al., 2019), thereby resulting in large climate effects. A large number of OSs have previously been observed in field measurements, but only a few biogenic VOCs can be clearly designated as OS precursors (Wang et al., 2019b; Shalamzari et al., 2014). Recent field studies reported that some unidentified OSs with C2–C25 skeletons may not be originated from biogenic VOCs and suggested that anthropogenic VOCs may contribute to the formation of these OSs (Wang et al., 2016; Blair et al., 2017). In addition, Ma et al. (2014) demonstrated that OSs derived from aromatic hydrocarbons contribute up to 67 % of the total OS mass in Shanghai, China, highlighting the potentially significant role of anthropogenic aromatics in organosulfate formation. While several studies have shown that SO2 emissions have implications for the SOA burden and OS formation, detailed characterizations of OS formation from anthropogenic monocyclic aromatic photooxidation are poorly performed.
NH3 is the most abundant form of reduced nitrogen, and it is ubiquitous in the ambient environment. NH3 levels have increased substantially in recent years and are estimated to continue to increase in the future (Warner et al., 2017). It is established that the increased NH3 emissions could reduce the effectiveness of PM2.5 control by controlling SO2 and NOx (Wu et al., 2016; Fu et al., 2017). However, the effects of NH3 on the formation of aerosol particles have not been well understood. NH3 has a promoting effect on the formation of new particles (Wang et al., 2020a), where low-volatility organic compounds could condense to form SOA in the atmosphere. A previous chamber study reported that the addition of NH3 could lead to the enhancement in the volume and number concentrations of SOA from the α-pinene ozone system (Na et al., 2007). Another study by Babar et al. (2017) utilized a newly developed flow reactor and confirmed that the presence of NH3 can enhance SOA formation from both ozonolysis and photooxidation of α-pinene. Besides α-pinene, the promoting effects of NH3 on particle formation were also discovered in the photooxidation of aromatics (Chu et al., 2016) and vehicle exhaust (Chen et al., 2019). In contrast, addition of NH3 decreased aerosol particle formation from the reaction of styrene with ozone, owing to the decomposition of products by NH3 nucleophilic attack (Na et al., 2006). Laboratory evidence suggests that NH3 can influence SOA composition via the neutralization of organic acids (Hao et al., 2020) and via the NH3 uptake by carbonyl-containing compounds (Flores et al., 2014). The reaction of organic compounds in particle phase with NH3 can decrease gaseous NH3 concentrations and can enhance the formation of nitrogen-containing organic compounds (Liu et al., 2015b), which are a class of brown carbon and could modify SOA optical properties. The neglect of NH3 effects on SOA formation might increase the model–measurement disagreement in SOA mass and can lead to an overprediction of NH3 concentration in the gas phase, especially in a complex urban environment. Consequently, it is necessary to explicitly explore the influence of NH3 on aerosol particles formation.
The complex mixture of ozone and fine particles is an emerging environmental problem affecting regional and urban air quality in China (Song et al., 2017), and investigating the chemistry of aromatic hydrocarbons has become greatly important for ozone and PM2.5 control because aromatics have high ozone- and SOA-forming potential (Chu et al., 2020). Aromatic hydrocarbons comprise a substantial fraction of the total VOCs at urban locations and even in rural areas (Guo et al., 2006; Ran et al., 2009), and evidence shows that global SOA formation from aromatic hydrocarbons lies in the range of 2 to 12 Tg yr−1 (Henze et al., 2008). 1,2,4-Trimethylbenene (TMB) is a small monocyclic aromatic emitted primarily from industrial solvent evaporation (Mo et al., 2021). In the troposphere, TMB is mainly oxidized via the hydroxyl radical (OH), producing multigenerational oxidized compounds that can contribute to SOA formation and growth (Zaytsev et al., 2019; Mehra et al., 2020). The OH oxidation of TMB can also generate small dicarbonyls glyoxal and methylglyoxal (Zaytsev et al., 2019), which are significant precursors for light-absorbing SOA formation. In addition, the Master Chemical Mechanism (MCM; http://mcm.leeds.ac.uk/MCMv3.2/, last access: 23 February 2021) is a near-explicit chemical mechanism that can describe, in detail, the tropospheric degradation of TMB. A recent study reported that identified autoxidation pathways during OH oxidation of TMB are not included in the current MCM, and the detected TMB products are more diverse than the products shown in the MCM (Wang et al., 2020b). The updates for the OH-initiated oxidation mechanism of TMB can be achieved only when the rate constants, branching ratios, and product distributions can be explicitly obtained. However, TMB photooxidation is highly complex and sensitive to environmental conditions. To better understand TMB SOA formation and growth, investigating chemical processes of TMB photooxidation with inorganic perturbation is required.
The mechanisms leading to secondary aerosol formation in the urban environment remain highly elusive and controversial, particularly for the processes related to changes in secondary aerosol mass and chemical composition. Recent studies have suggested that inorganic pollution emissions could perturb SOA formation, yet very little is known about the SO2 and NH3 effects on SOA formation. Given the ubiquity of SO2, NH3, and TMB in the atmosphere, a key goal of this work is to determine the detailed chemical compositions and formation mechanisms of secondary aerosol from TMB photooxidation with SO2 and/or NH3. We investigated the effects of SO2 and NH3 on the growth of particles from TMB photooxidation for the first time and discussed the role of inorganic species in TMB chemistry. The chemical composition of TMB SOA was rigorously characterized based on laboratory measurements. We also revealed some hitherto unidentified organosulfates and tentatively proposed relevant formation pathways of products.
2.1 Particle generation
Aerosol particles were produced from TMB photooxidation in the presence of NOx in a new indoor smog chamber, which consists of a 1.1 m3 Teflon reactor (0.6 mm Teflon film) housed in a temperature-controlled room. For photooxidation, a panel of black light lamps (F40BLB, GE) were used to provide ultraviolet (UV) irradiation centered at 365 nm. Before each run, the chamber was continually purged with dry and purified air prepared by zero air supply (Model 111, Thermo Scientific, USA) and simultaneously irradiated with UV lights until the concentrations of background contaminants (i.e., NO, NO2, SO2, and O3) were lower than 1 ppb, and the particle number concentration was below 5 cm−3.
The TMB photooxidation experiments were carried out by the following steps. First, a known volume of TMB liquid (98 %, Aladdin) was transported into the chamber through a heated (80 ∘C) Teflon tube carried by a flow of zero air. Second, according to experimental design, different quantities of NO (504 ppm in N2, Qingdao Yuyan Gas Company, China), SO2 (1013 ppm in N2, Qingdao Yuyan Gas Company, China), and NH3 (497 ppm in N2, Qingdao Yuyan Gas Company, China) were introduced into the chamber from corresponding high-pressure cylinders using calibrated mass flow controllers (D07-7, Beijing Sevenstar Electronics Co., Ltd, China). Note that before the injection of NH3, the inlet tubes were flushed with NH3 flow for 30 min to minimize the adsorption losses of NH3 in the tubes. After all species in the chamber were well mixed (initial concentrations of TMB, NO, and/or SO2 were constant), black light lamps were turned on, marking the beginning of photooxidation experiments. The chamber was operated in batch mode with a reaction time of approximately 300–360 min. Seed particles were not introduced into the chamber over the course of particle formation experiments. Temperature and relative humidity (RH) inside the chamber were (299 ± 4) K and (25 ± 1) %, respectively. Detailed experimental conditions and results for each experiment are provided in Table 1. A total of 12 experiments were conducted under four different scenarios. In the first set of experiments (Exps. 1–4, Table 1), SO2 levels in the chamber varied from 0 to 200 ppb, while the initial ratio of [TMB] to [NOx] was kept higher than 10 ppb C ppb−1 (low-NOx conditions). The second set of experiments (Exps. 5–8, Table 1) were performed under high-NOx conditions ([TMB]0 [NOx]0<10 ppb C ppb−1), with SO2 concentration being the only variable (ranged from 0 to 228 ppb). The third set of experiments (Exps. 9–10, Table 1) consisted of an irradiation of TMB, NOx, and NH3, while the subsequent set of experiments (Exps. 11–12, Table 1) consisted of an irradiation of TMB, NOx, SO2, and NH3.
Different physicochemical parameters were measured over the course of photooxidation experiments. A digital thermo-hydrometer (Model 645, Testo AG, Lenzkirch, Germany) was used to measure temperature and RH inside the chamber. The concentrations of NO and NOx were measured with a NO–NO2–NOx analyzer (model 42i, Thermo Scientific, USA), while a Thermo Scientific model 43i-TLE pulsed fluorescence SO2 analyzer was used to measure SO2 levels throughout the experiments. The O3 level was monitored with a Thermo Scientific model 49i O3 analyzer. The initial concentration of NH3 was calculated based on the introduced amount of NH3 and the reactor volume. The decay of TMB was measured by a gas chromatograph (GC, 7890B, Agilent Technologies, USA) equipped with a DB-624 column (30 m × 0.32 mm, 1.8 µm film thickness, Agilent Technologies, USA) and a flame ionization detector (FID). The GC temperature was programmed to increase from 80 to 200 ∘C at a rate of 20 ∘C min−1. Particle size distributions and volume concentrations in all experiments were recorded in situ using a scanning mobility particle sizer (SMPS), which consists of a long differential mobility analyzer (Long DMA, Model 3082, TSI, USA) and a condensation particle counter (CPC, Model 3776, TSI, USA). The Long DMA was available for measuring the particle size distribution in the range of 13.8–723.4 nm, while smaller particles between 4.5–162.5 nm were measured with a nano differential mobility analyzer (Nano DMA, Model 3085, TSI, USA). The measured volume concentration in each experiment was converted into particle mass concentration with an estimated particle density of 1.4 g cm−3.
Table 1Experimental conditions and results for the TMB photooxidation experiments.
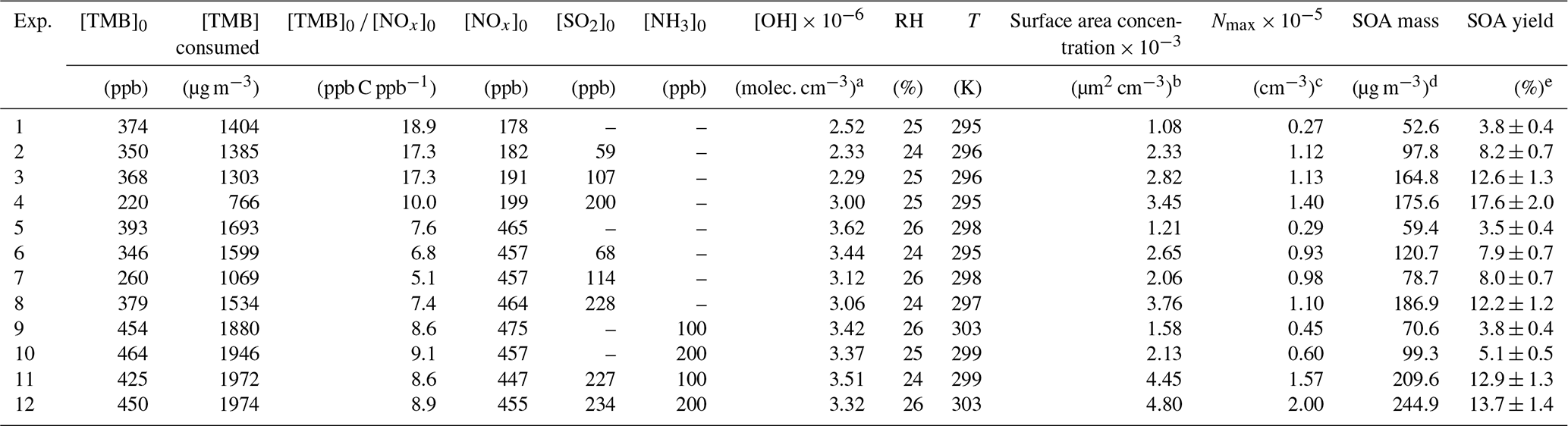
a The average OH concentration was determined from the measured TMB decay. b The particle surface area concentration measured by SMPS at 300 min of each experiment. c Aerosol particle maximum number concentration.d SOA mass concentrations have been wall-loss-corrected. For SOA mass calculation, the inorganic mass concentration has been subtracted from the particle mass concentration. e Errors in SOA yield were calculated from error propagation using the sum of the uncertainties in TMB data and the systematic error of SMPS.
2.2 Particle collection and analysis
2.2.1 Attenuated total reflectance–Fourier transform infrared spectroscopy analysis
Following 300–360 min of reaction, aerosol samples were collected onto aluminum foils (25 mm, Jowin Technology Co. Ltd.) by a low-pressure impactor (DLPI+, DeKati Ltd, Finland) and were stored at −20 ∘C thereafter until analysis to reduce evaporative losses of aerosol. The chemical functional groups of aerosols were characterized by an attenuated total reflectance–Fourier transform infrared (ATR-FTIR) spectrophotometer (Vertex 70, Bruker, Germany) with a mercury cadmium telluride detector. The ATR-FTIR spectra of aerosol particles in each run were recorded by averaging 64 scans from 4000–600 cm−1 with a resolution of 4 cm−1. Prior to each measurement with ATR-FTIR, the surface of the diamond crystal was thoroughly cleaned with ethanol and ultrapure water to rule out interferences of other sources of contamination. The ATR-FTIR spectra of blank aluminum foils were also acquired to confirm the absence of IR absorption by the aluminum foil on which aerosols are collected.
2.2.2 Ion chromatography analysis
Following the ATR-FTIR measurements, aerosol samples were extracted in 3 mL of ultrapure water (Milli-Q water, 18 M Ω) under sonication in an ice bath for 30 min. The extracted samples were filtered through polyethersulfone syringe filters (0.22 µm pore size) and subsequently analyzed for their ionic concentrations using an ion chromatography (Dionex ICS-600, Thermo Fisher Scientific, USA) with electrical conductivity detection. A Dionex IonPac™ AS19 column (4 × 250 mm) connected with AG19 guard column (4 × 50 mm, Dionex Ionpac) was used to separate anions. An aqueous solution of 20 mM potassium hydroxide (KOH) prepared by a reagent-free controller (Dionex, Thermo Scientific, USA) was used as the anion eluent. Cation analysis was carried out with a pair consisting of a CG12A guard column (4 × 50 mm, Dionex Ionpac) and analytical column (4 × 250 mm, CS12A, Dionex IonPac™) and an isocratic 20 mM methanesulfonic acid (CH4O3S). The same volume of extract was injected into the ion chromatograph by a six-way valve mounted with a loop of 250 µL. The elution flow rates of KOH and CH4O3S were both set to 1 mL min−1.
2.2.3 Ultra-high-performance liquid chromatography high-resolution mass spectrometry analysis
Laboratory-generated aerosols were also collected on 47 mm polytetrafluoroethylene (PTFE) filters (0.22 µm pore size, Tianjin Jinteng Experimental Equipment, China) using a stainless steel inline filter holder (Sartorius 16254, Sartorius Stedim Biotech GmbH, Germany) with a flow rate of 10 L min−1. The collected samples were wrapped in foil and stored in the freezer (−20 ∘C) until mass spectrometry analysis. Filter samples were extracted twice with 5 mL of high-purity methanol (Optima® LC-MS grade, Fisher Scientific) under sonication in ice for 30 min. The extracts were mixed, filtered with a 0.2 µm pore size PTFE syringe filter (Millipore), and concentrated to near dryness under a gentle stream of high-purity nitrogen. The concentrated samples were reconstituted with ultrapure water (Milli-Q water, 18.2 M Ω) and methanol (Optima® LC-MS grade, Fisher Scientific) with a volume ratio of 50:50. Control mass spectrometry measurements of solvent and extracts from blank PTFE filters were performed to remove the interferences of solvent and handling protocols. The chemical compositions of aerosols were characterized using an ultra-high-performance liquid chromatograph (UPLC; Ultimate 3000, Thermo Scientific, USA) coupled to a Q-Exactive Focus Hybrid Quadrupole-Orbitrap mass spectrometer (MS; Thermo Scientific, USA) with electrospray ionization (ESI). The ESI source was operated in both positive (+) and negative (−) ionization mode. Product molecules could be detected as [M + H]+ in the positive ion mode, while products could be ionized via deprotonation and were detected as [M − H]− in the negative ion mode. The following parameters were set for the optimal operation of LC/ESI-MS: spray voltage (+), 3.5 kV; spray voltage (−), −3.0 kV; S-lens radio frequency (RF) level (+), 50 V; S-lens RF level (−), 50 V; capillary temperature, 320 ∘C; sheath gas (nitrogen) pressure, 2.76 × 105 Pa; auxiliary gas (nitrogen) flow, 3.33 L min−1. MS spectra were recorded in the range of 50 to 750 in a full MS scan with a mass-resolving power of 70 000 (full width at half maximum, FWHM, at 200). The full MS scan was followed by data-dependent tandem mass spectrometry (MS/MS; dd-MS2) scans using stepped collision energies of 20, 40, and 60 eV via high-energy collisional dissociation. The resolution was 17 500, and an isolation width of 2 units was applied for the dd-MS2 scan. The other parameters for MS2 experiments were as follows: automatic gain control (AGC) target, 2 × 105; maximum IT, 50 ms; loop count, 3; minimum AGC target, 1 × 105; apex trigger, 2–6 s; dynamic exclusion, 6 s. The MS instrument was calibrated every 5 d with standard calibration solutions provided by the manufacturer. The separation of analytes was carried out on an Atlantis T3 C18 column (100 Å, 3 mm particle size, 2.1 mm × 150 mm, Waters, USA) at 35 ∘C. The mobile phases consisted of (A) 0.1 % formic acid (Optima 18 M Ω LC-MS grade, Fisher Scientific) in ultra-pure water (Milli-Q water, 18.2 M Ω) and (B) 0.1 % formic acid in methanol (Optima® LC-MS grade, Fisher Scientific). The injected volume of samples was 2 µL in this study. Samplers were eluted using a 60-min gradient elution program with a flow rate of 200 µL min−1: initially set to 3 % B over the first 3 min, the concentration of eluent B was increased linearly to 50 % in 22 min and from 50 % to 90 % from 25 to 43 min, then it was decreased from 90 % to 3 % from 43 to 48 min and finally kept at 3 % for 12 min. The chemical formulas of observed ions were proposed based on reaction pathways, chemical consideration, and measured value with a mass tolerance of ± 5 ppm. All data were recorded and processed using the Xcalibur V4.2.47 software package.
2.3 Wall losses of vapors and particles
The SOA yield, usually used to quantify the propensity of a parent hydrocarbon to form SOA, could be determined as the ratio of the generated particle mass to the amount of consumed parent hydrocarbon. The particle and vapor wall depositions in chambers can lead to the underestimation of the SOA yield. In order to determine the particles wall loss rates, we carried out independent wall loss experiments using ammonium sulfate particles. An aqueous solution of ammonium sulfate was fed to a constant output atomizer (Model 3706, TSI, USA) to produce droplets, which passed simultaneously through a silica gel diffusion dryer to introduce dry particles into the chamber. The size distributions of ammonium sulfate particles were measured by the SMPS for 480 min. An almost identical humidity (25 ± 1 %) condition was achieved for each experiment. The wall losses of particles are size-dependent, and, thus, we used a size-dependent particle wall loss correction approach, which is described in detail in the Supplement. The size-dependent loss rate (k) of ammonium sulfate particles could be expressed as and was applied to correct the aerosol particle concentrations. The wall loss rates of NO, NO2, SO2, and TMB were determined to be 2.0 × 10−6, 3.9 × 10−6, 4.0 × 10−7, and 2.3 × 10−6 s−1, respectively, indicating that the wall losses of these species were negligible over the course of the experiment. However, gas-phase species that could deposit to the chamber walls include not just the parent hydrocarbon, in this case TMB, but also the oxidation products, which in general are not all totally monitored and characterized. It is difficult to correct directly and accurately for the impact of vapor wall losses on the SOA yield. Therefore, the SOA yield in this work is only the lower limit. Here, the underestimation of SOA yields due to vapor wall losses was determined using the method in the study of Zhang et al. (2014). In brief, the effect of vapor wall losses on SOA yield significantly arose from the competition between vapors condensation onto aerosol particles versus vapor depositions to chamber walls. The extent to which vapor wall depositions influence the SOA yield could be estimated by the ratio of the average timescale of gas–particle partitioning during the photooxidation experiments to the timescale of vapor wall deposition. The evaluated results suggested that the SOA yield could be underestimated by a factor of 1.8 to 8.4 without accounting for vapor losses.
3.1 Effects of SO2
3.1.1 Particle formation and growth in SO2-added photooxidation
To evaluate the impacts of SO2 on aerosol formation and growth from TMB photooxidation, a series of experiments were conducted with various initial SO2 levels under both low- and high-NOx conditions. Evolutions of the number distributions of secondary aerosols with particle size within 4.5–162.5 nm are provided in Fig. 1. There was no new secondary aerosol formation in the beginning of photooxidation. After a period of time, particles were burst-produced, and the number concentration of particles increased rapidly. At the same time, the particles continuously grew via condensation and coagulation mechanisms, consistent with a previous study (Jorga et al., 2020). After 300 min UV irradiation without SO2 introduction, the total maximum number concentration of aerosol particles was only 2.7 × 104 and 2.9 × 104 cm−3 under low-NOx (Exp. 1) and high-NOx (Exp. 5) experiments, respectively. Interestingly, the particle maximum number concentration considerably increased with increasing SO2 levels, regardless of low- or high-NOx conditions (Table 1). As shown in Fig. 1, although the unimodal particle size distribution in the presence of SO2 was similar to that in its absence, particles with mobility diameter ranging from 10 to 80 nm, especially, dominated particle number concentrations with SO2 addition. The time series of total ultrafine particles (diameter < 100 nm) number concentration are shown in Fig. S2, where it is seen that SO2 considerably enhanced ultrafine particle formation. Ultrafine particles are more harmful to humans than larger particles because they can more easily penetrate deep into the lungs and blood circulation (Terzano et al., 2010). Our results indicate that SO2 concentration is a key parameter for ultrafine particle formation. It has been suggested that reducing the number concentration of ultrafine particles can decrease mortality, highlighting again the need to continue to implement strict SO2 emission standards (Fuzzi et al., 2015).
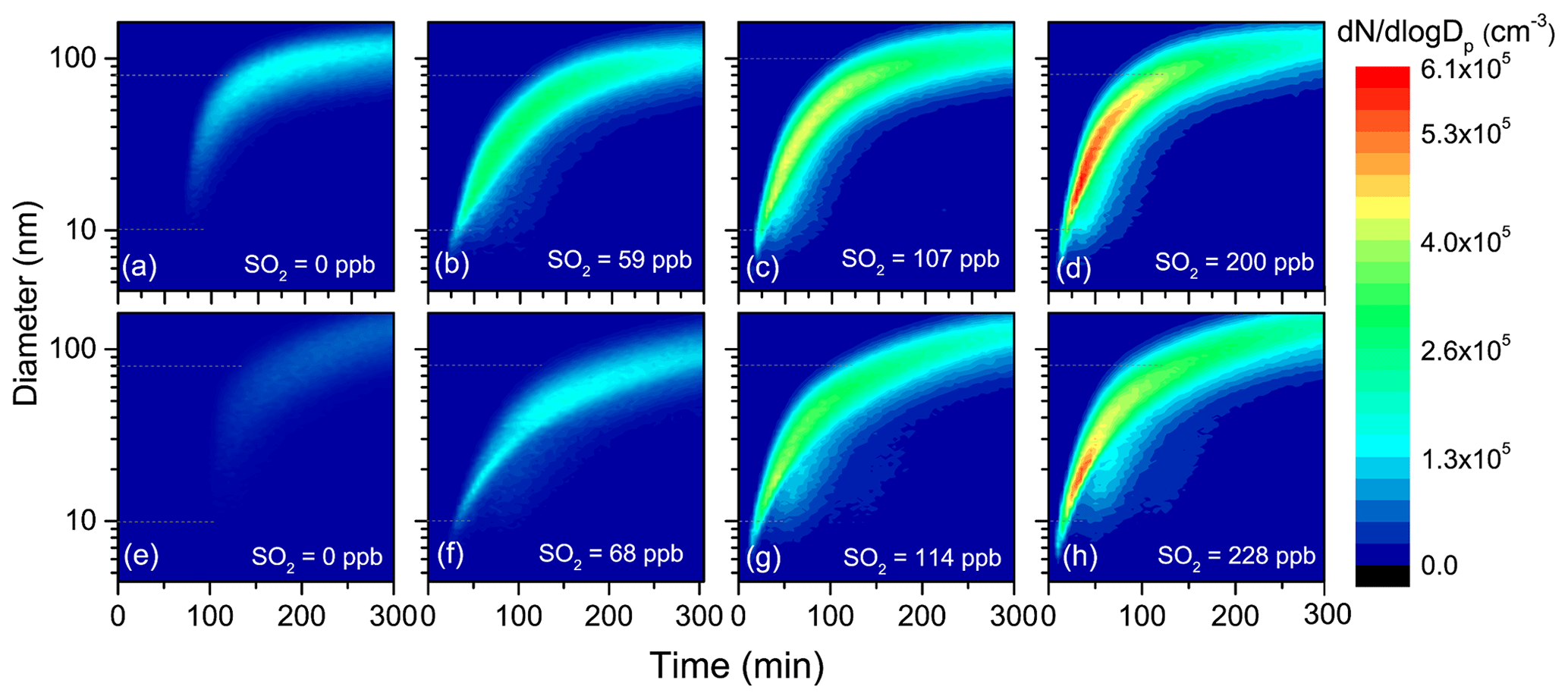
Figure 1Evolutions of the number distributions of aerosol particles generated from TMB photooxidation in low-NOx (a–d) and high-NOx (e–h) experiments.
To fully give an account for the effects of changes in SO2 emissions on aerosol formation, the key particle-phase parameters (i.e., nucleation time, initial growth rate, and particle mean diameter) as a function of SO2 levels are further compared in Fig. 2. In the present study, the SMPS instrument can measure particles larger than 4.5 nm, and, therefore, the nucleation time here is defined as the time required for new secondary aerosols to grow to 4.5 nm after the lights have been turned on (Wyche et al., 2009). The particle size rapidly increased within 30 min after nucleation and gradually reached a stable level within 300 min of photooxidation (Fig. 1). Consequently, the initial growth rate (GRinitial), calculated based on the method of Kulmala et al. (2012), is defined as the particle growth rate within 30 min after nucleation (Li et al., 2018). The mean diameter reported in this work represents the particle mean diameter measured at 300 min in each experiment. From Fig. 2, a significant negative correlation was found between the nucleation time and the initial SO2 level. Furthermore, when similar amounts of SO2 were introduced to the reaction mixture, the gap between the nucleation time of low-NOx and high-NOx conditions would be reduced, which is in agreement with a previous study (Zhao et al., 2018). Under SO2-free conditions, new secondary aerosol could be generated by homogeneous nucleation involving key intermediate products of TMB oxidation. The delay time for particle formation largely corresponds to the time required for intermediate products to build to sufficient concentrations in such a way that their saturation vapor pressure relative to the particle phase is exceeded. New secondary aerosol consists of later stage oxidation products, which might be also responsible for the delayed occurrence. It was demonstrated that higher OH concentration in the chamber could result in faster particle formation (Sarrafzadeh et al., 2016). However, the average OH concentration in SO2-free experiments is comparable with that in SO2-added experiments (Table 1). There has been a gradual fall in the mixing ratio of SO2 (Fig. S3) that could be oxidized to sulfuric acid (H2SO4) during TMB photooxidation. The formed H2SO4 could induce nucleation and increase the nucleation rate (Zhao et al., 2018; Blair et al., 2017), and these processes are responsible for the short nucleation time observed in the TMB NOx SO2 regime (Wyche et al., 2009). The mean diameter of secondary aerosol decreased by 4–10 nm when 60–70 ppb of SO2 was included in the matrix (Fig. 2). In contrast, at high-SO2 levels ([SO2]0> 100 ppb), an increase in the initial SO2 concentration led to an increase in particle mean diameter, regardless of low- or high-NOx conditions. The initial growth rate also showed a similar dependence on the SO2 level as presented in Fig. 2. The nonlinear response of the particle mean diameter to SO2 initial level is similar to the findings of Julin et al. (2018), who found the response of particle size distribution to NH3 emissions to also be nonlinear. Aerosol particles can grow in different ways such as gas–particle partitioning of semi-volatile organic compounds (SVOCs). Since the evaporation of SVOCs is important after partitioning to the particle phase, the rate at which SVOCs participate in the particle growth is lower than their condensation rate. However, recent advances give an insight that the particle-phase chemistry, such as heterogeneous reactions of SVOCs, is substantially pronounced for the particle growth (Shiraiwa et al., 2013; Paasonen et al., 2018; Apsokardu and Johnston, 2018). Organosulfates can be produced by particle-phase reactions involving interactions between organics and inorganics. In this work, organosulfates were only detected in SO2-involved photooxidation, indicating that additional particle-phase reactions can occur under SO2-involved conditions. Increasing the initial SO2 level could induce the formation of more sulfate (Fig. S4) and the enhancement in the particle acidity during photooxidation (Liu et al., 2016; Kroll et al., 2006). The elevated particle acidity can promote more SVOCs to transform into low-volatility products such as organosulfates in the particle phase, thereby promoting the particle growth (Lin et al., 2014; Lal et al., 2012). Then, additional SVOCs could be further transferred from the gas phase to the particle phase to increase the particle size. However, the particle mean diameter in low-SO2 ([SO2]0<100 ppb) experiments is smaller than that in SO2-free experiments. Our result is in line with the study of Wyche et al. (2009), who attributed this phenomenon to the larger number of particles produced under SO2-involved conditions. The presence of 59 ppb SO2 caused the maximum number concentration of particles to increase by 8.5 × 104 cm−3 under low-NOx conditions. When the SO2 level increased from 0 to 68 ppb in high-NOx experiments, the corresponding particle number concentration increased from 2.9 × 104 to 9.3×104 cm−3. Therefore, the amounts of products that condensed onto each aerosol particle significantly decreased in low-SO2 experiments, which could result in the decrease in particle diameter (Liu et al., 2015a). The promoting effect of particle-phase chemistry on the particle size growth may not offset the inhibiting effect of the emergence of a large number of particles on the particle size growth, thereby leading to the low particle diameter in low-SO2 experiments.
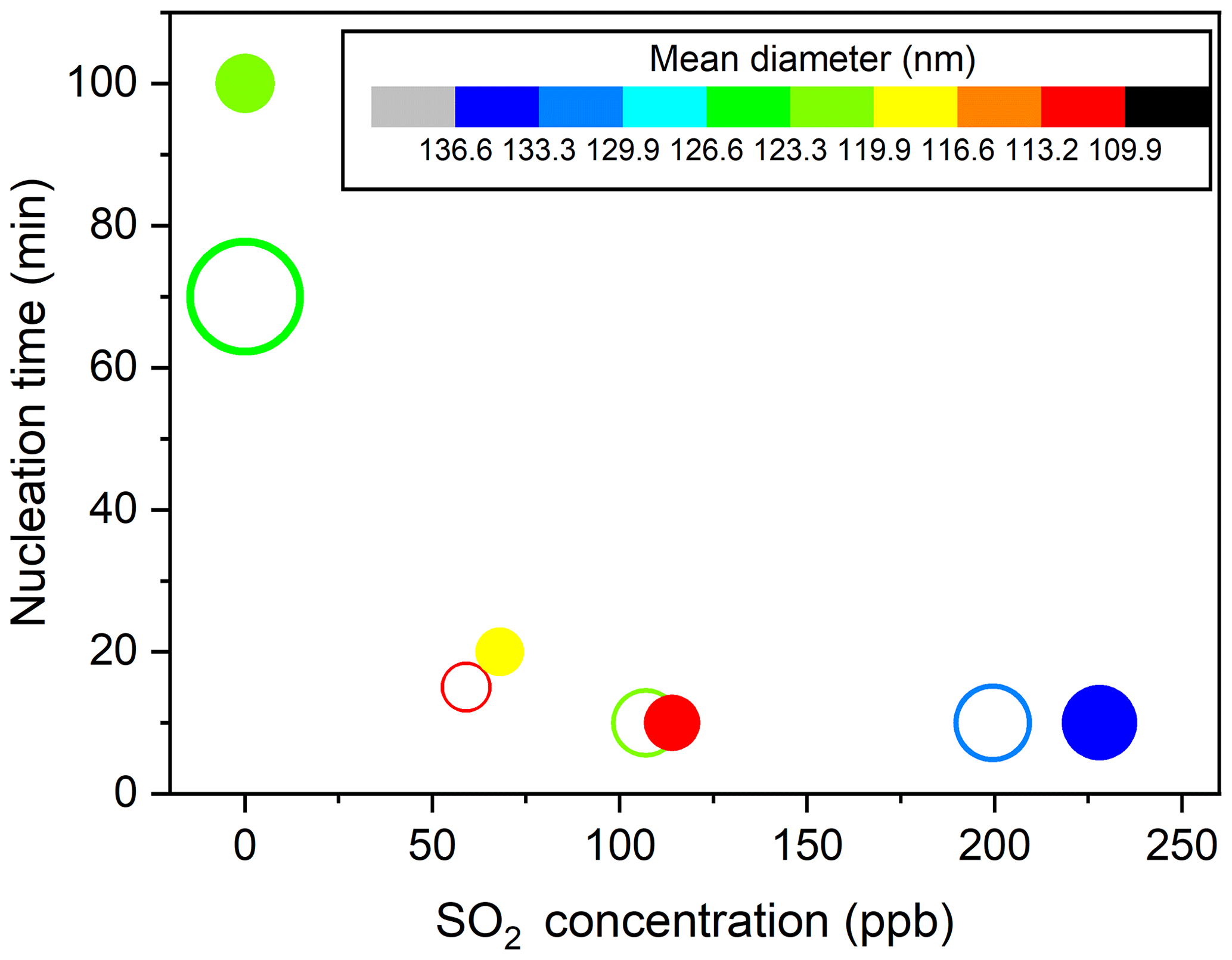
Figure 2Particle nucleation time as a function of initial SO2 concentration under low-NOx (open circles) and high-NOx (solid circles) conditions (Exps. 1–8). The symbol color indicates the particle mean diameter, and the symbol size represents the particle initial growth rate. Values of the particle parameters are listed in Table S1.
3.1.2 SOA yield in SO2-added photooxidation
The particle volume concentration as a function of TMB consumption is presented in Fig. 3, where it can be observed that the particle formation increased with increasing initial SO2 levels, regardless of low- or high-NOx conditions. The present result is consistent with a previous research study, which found that limonene SOA formation is significantly promoted with SO2 addition (Ye et al., 2018). SO2 was found to perturb particle formation by inducing chemical reactions in the gas and particle phase (Wang et al., 2019a; Friedman et al., 2016). We are unable to fully rule out the impacts of SO2 on gas-phase chemistry. However, the decay of TMB was essentially unchanged when SO2 was introduced into the chamber (Fig. S5), which suggests the unlikeliness of SO2 addition to affect the gas-phase chemistry of TMB photooxidation (Kleindienst et al., 2006). Instead, it is more likely attributed to the formation and condensation of H2SO4 and/or the enhancement of organic aerosol formation. We assumed full conversion of the consumed SO2 into H2SO4 aerosol particles and found that the contribution of the formed H2SO4 to the increase in particle volume concentration was less than 100 % (See Sect. S2). In addition, pure SO2 oxidation experiments without TMB addition also indicated that the enhancement in aerosol particles by SO2 introduction cannot be solely attributed to inorganic aerosol formation (See Sect. S2). To calculate the net SOA yield, the inorganic mass concentration was subtracted from the particle mass concentration based on ion chromatography measurements of generated particles. The influence of the SO2 initial level on the SOA yield can be seen in Table 1 as high SO2 levels contribute to produce somewhat high SOA yields. Figure 4 compares the SOA yields obtained from the present work with those found in previous studies with similar experimental conditions. The SOA yields from the two SO2-free experiments are comparable to that reported from the study of Liu et al. (2012) and fit quite well with the yield curve of Odum et al. (1996). The SOA yields in our TMB NOx photooxidation experiments were 3.8 % and 3.5 %, which were close to 3.9 % and 4.2 % derived from the yield curve of Odum et al. (1996) under the same mass concentration. In contrast, with similar mass concentration, the SOA yields in SO2-added regimes were higher than those in previous studies (Odum et al., 1996; Liu et al., 2012). Here the neutralization degree of particle, which was calculated as the molar ratio of to the sum of and (Lin et al., 2013), was used as a tool to roughly estimate the aerosol acidity of collected particle samples. The values of the neutralization degree were lower than 1 in this work, indicating acidic aerosols (Lin et al., 2013). The increase in aerosol acidity could be largely responsible for the observed enhancements in SOA formation in SO2-involved experiments. The OH oxidation of TMB can result in the formation of multifunctional carbonyl compounds (Liu et al., 2012; Zaytsev et al., 2019), which could promote SOA formation via acid-catalyzed heterogeneous reactions. In addition, the particle surface area concentrations significantly increased with increasing SO2 initial concentrations in both low-NOx and high-NOx conditions (Table 1), which might also result in the enhancement in the SOA yield. Besides gas–particle partitioning of SVOCs, the fates of SVOCs in the chamber also include chemical reactions and chamber wall losses. Therefore, in the batch-mode chamber experiments, the gas–particle partitioning of SVOCs has a great sensitivity to particle surface areas (Zhang et al., 2015; Han et al., 2019). Recently, Zhao et al. (2018) examined the SO2 effects on the SOA formation and suggested that providing additional particle surfaces by SO2-induced new particle formation leads to an increase in SOA yield. The effects of the particle surface area concentration on organic aerosol formation were explored by Han et al. (2019), who also found that increasing the particle surface area concentrations can significantly increase the organic aerosol mass yield due to greater partitioning of semi-volatile organic products to the particle phase. Increasing the particle surface area can limit the gas–wall interactions of organic vapors and is favorable for the movement of more SVOCs from the gas phase to the particle side (Han et al., 2019). These additional SVOCs can also undergo further particle chemistry such as acid-catalyzed heterogenous reactions to strongly enhance aerosol particle formation in TMB NOx SO2 photooxidation (Apsokardu and Johnston, 2018).
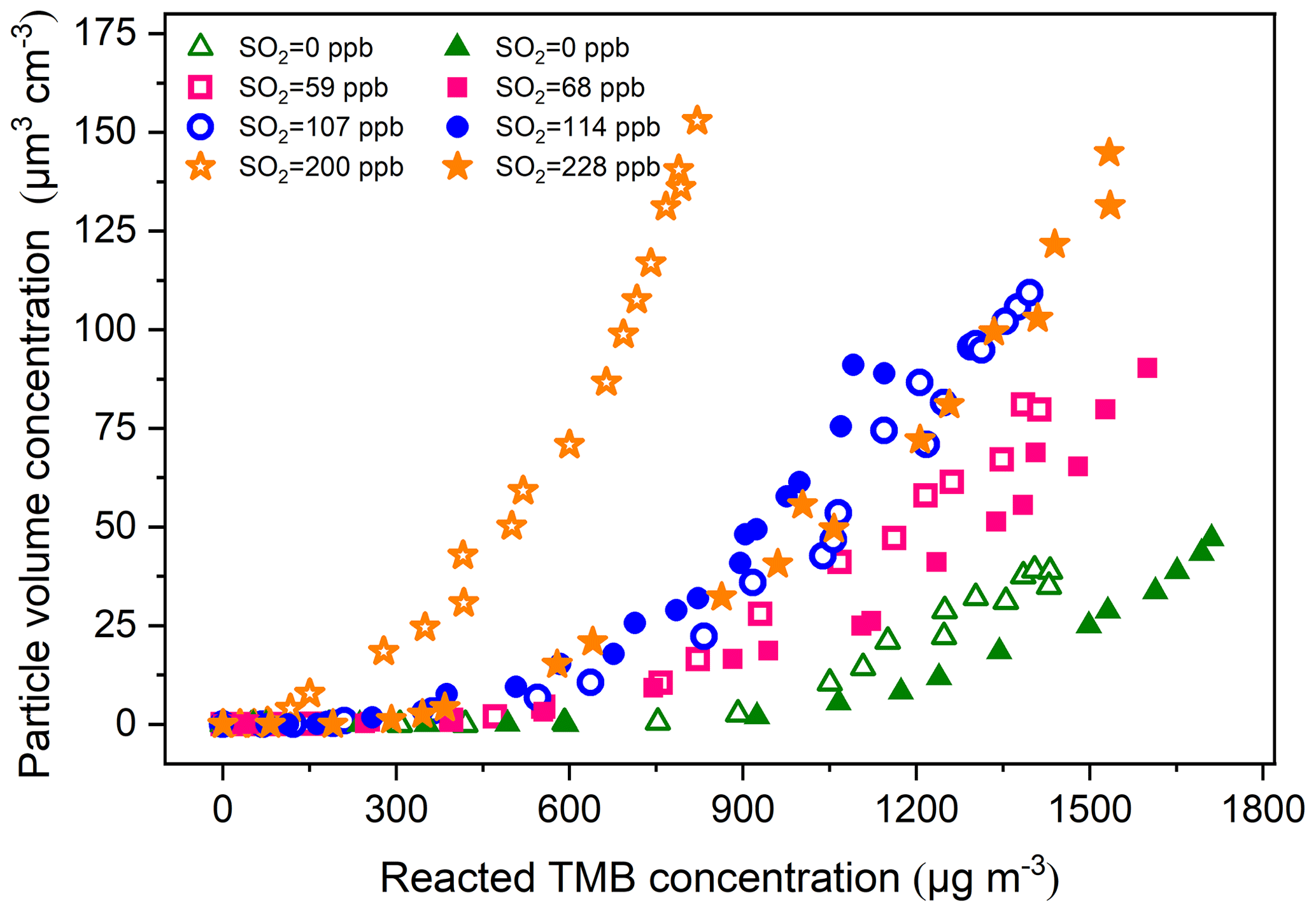
Figure 3Growth of particle volume concentrations from TMB photooxidation as a function of TMB consumption for eight experiments with different initial SO2 concentrations (Exps. 1–8). The open symbols and solid symbols represent low- and high-NOx experiments, respectively.
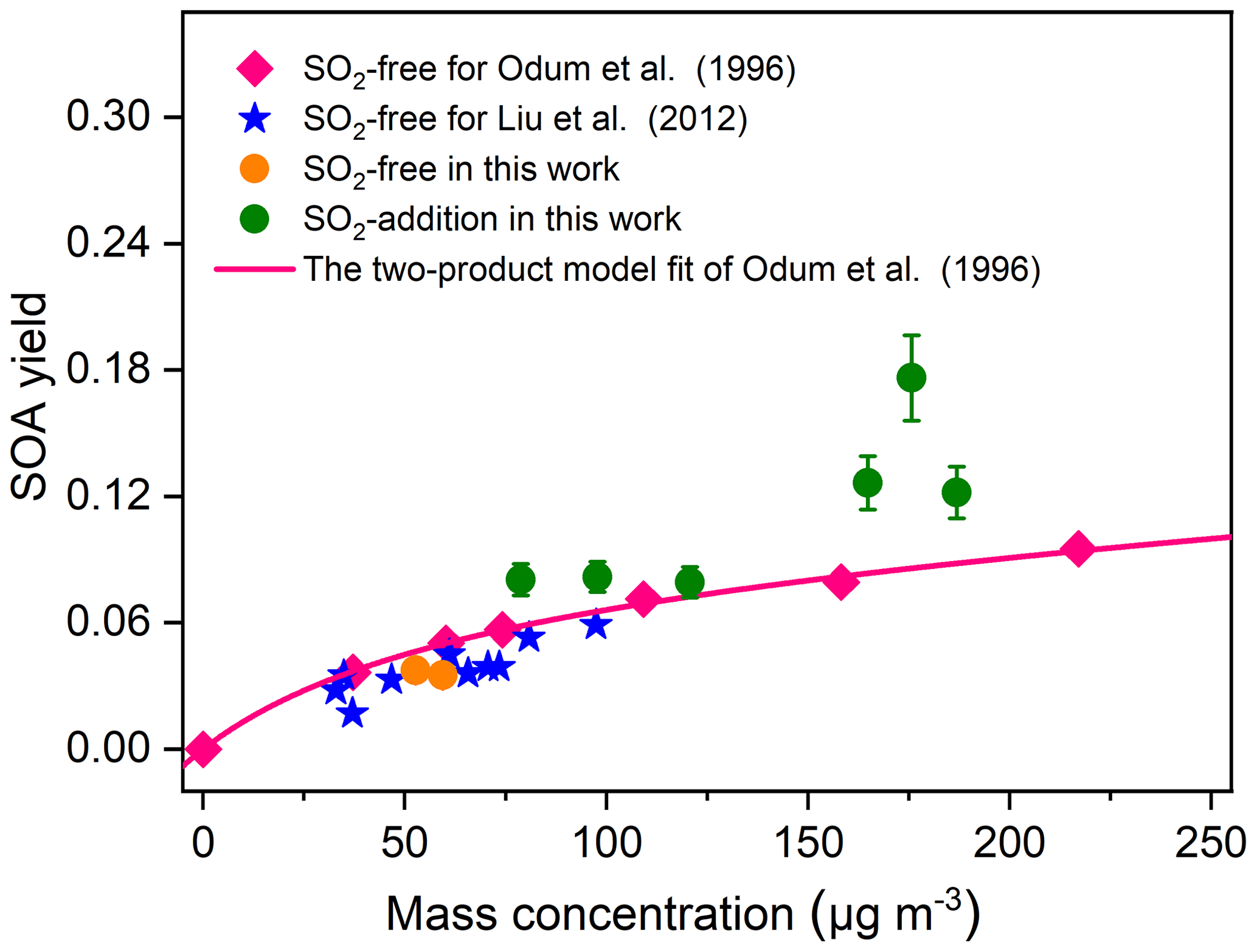
Figure 4Comparison of SOA yields from TMB photooxidation as a function of SOA mass concentration with data reported from literature. The SOA density in the whole study was assumed to be 1.4 g cm−3. The error bars represent errors in the SOA yield results, and the errors were calculated from error propagation using the sum of the uncertainties in TMB data and the systematic error of SMPS. The SOA yields for Liu et al. (2012) were extracted from Table 1 in the study. The pink line is the best fit two-product model for the SOA yields and was extracted from Fig. 2 in the work of Odum et al. (1996).
3.1.3 Particle chemical composition in SO2-added photooxidation
In order to investigate the effects of SO2 on the chemical composition of aerosol particles, the particles were first characterized by ATR-FTIR. Figure 5 compares the characteristic ATR-FTIR spectra of particles formed from the photooxidation of TMB under different conditions, and detailed information on the assignment of absorption peaks is given in Table S2. For the samples collected from TMB NOx photooxidation, the particles exhibited an O–H stretch at 3600–3000 cm−1 as shown in Fig. 5a and b. The C = O stretch of carbonyl at 1720 cm−1 suggests that aldehydes, ketones, and carboxylic acids are significant particle components, while the absorptions at 844 cm−1 (NO symmetric stretch), 1284 cm−1 (NO2 symmetric stretch), and 1647 cm−1 (NO2 asymmetric stretch) are the characteristic peaks of organic nitrates. The slight absorbance at 941 cm−1 indicates the presence of peroxides containing O–O groups. Interestingly, compared with the particles from TMB NOx experiments, there was a new peak at 615 cm−1 for the particles generated from TMB NOx SO2 experiments, regardless of low- or high-NOx conditions. The 615 cm−1 peak is characteristic of inorganic sulfates and further highlights that the presence of SO2 promotes the formation of inorganic sulfates as pointed out in previous studies (Chen et al., 2019; Liu et al., 2016). The strong absorption at 1081 cm−1 arises from the S = O bands in the particle components. Field and laboratory studies have reported that inorganic sulfate could convert into organosulfur compounds in the atmosphere (Riva et al., 2019; Nestorowicz et al., 2018). Therefore, the peak at 1081 cm−1 may be mainly from the absorption of both inorganic sulfates and organosulfur compounds.
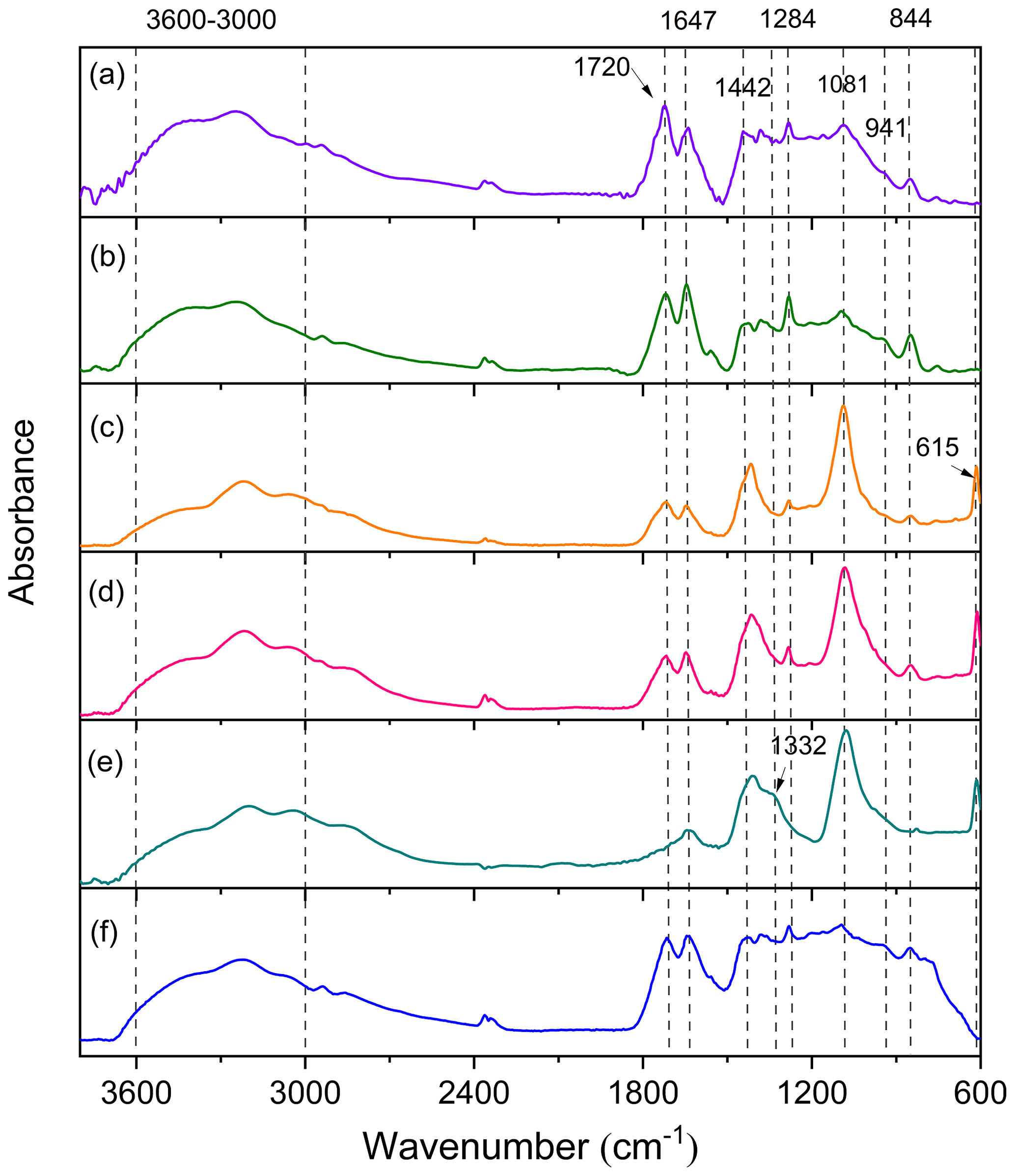
Figure 5ATR-FTIR spectra of aerosol particles generated from TMB NOx (a, Exp. 1; b, Exp. 5), TMB NOx SO2 (c, Exp. 4; d, Exp. 8), TMB NOx NH3 SO2 (e, Exp. 12), and TMB NOx NH3 (f, Exp. 10) photooxidation.
The chemical compositions of particles generated in TMB NOx and TMB NOx SO2 photooxidation were further measured with a high-resolution mass spectrometer (HRMS), to determine whether organosulfur compounds were formed in SO2-added experiments. The formation of organosulfur compounds was recognized by the oxidation mechanism and the loss of characteristic fragment ions at 79.95 (SO3), 80.96 (), and 96.96 () in MS/MS spectra (Figs. S7–S8). The list of compounds observed in this work, along with molecular weights (MWs), measured masses, and proposed structures, is presented in Table S3. In SO2-free experiments, the major aerosol components were multifunctional alcohols, peroxides, organic nitrates, ketoaldehydes, and ketocarboxylic acids. With the same analytical methods, the same products were also observed in SO2-added photooxidation. The most striking result to emerge from Table S3 is that 10 organosulfates (OS-214, OS-226, OS-228, OS-240, OS-242, OS-244, OS-268, OS-300, OS-316, and OS-345) and two organic sulfonates were only detected in filter samples collected from the TMB NOx SO2 photooxidation experiment, indicating that SO2 emissions in the atmosphere can alter the aerosol formation chemistry and thus influence the aerosol chemical composition. To the best of our knowledge, this is the first time the 10 organosulfates are identified in TMB NOx SO2 photooxidation experiments. Recently, some sulfur-containing compounds from field measurements were designated as being compounds of unknown origin. For example, the MW 214 organosulfate has been detected in PM2.5 collected from the highly polluted megacity Shanghai (Cai et al., 2020). However, the VOC precursor for this organosulfate formation was not reported. The MW 242 organosulfate found in Baengnyeong Island was also classified as organosulfur of unknown origin (Boris et al., 2016). O'Brien et al. (2014) found the formation of MW 228 organosulfate in ambient aerosol particles, but its specific source was not pointed out. Evidence from this study suggests that TMB photooxidation in the presence of SO2 might contribute to the formation of these organosulfates (MW = 214, 228, and 242) in the ambient air. Importantly, an organosulfur compound with a formula of C7H12O7S (MW = 240) was observed in ambient fine aerosols and was tentatively assigned to an oxidation product of anthropogenic 1,3,5-trimethylbenzene (Boris et al., 2016). The finding of the current study suggests that the organosulfur compound (MW = 240) may also be produced from the photooxidation of 1,2,4-trimethylbenzene in the presence of SO2. In addition, the MW 226, 240, and 268 organosulfur compounds were designated as biogenic-derived organosulfates in previous field studies (Cai et al., 2020; Boris et al., 2016). More recently, Chen et al. (2020b) suggested that heterogeneous OH oxidation of isoprene-derived SOA can contribute to the formation of an organosulfate with a molecular weight of 228. Our results show the detection of OS-226, OS-228, OS-240, and OS-268 organosulfates, which are isomers of organosulfates derived from isoprene (Cai et al., 2020), isoprene (Chen et al., 2020b), limonene (Cai et al., 2020), and limonene (Boris et al., 2016), respectively. More studies need to be undertaken to differentiate various sources of organosulfates in the ambient aerosols through chemical synthesis of authentic organosulfates standards.
The mechanisms describing the formation of OS-226, OS-228, OS-240, OS-242, OS-244, OS-300, OS-316, and OS-345 are proposed in Fig. 6. Following analogous mechanisms for toluene photooxidation, the oxidation of TMB is dominantly initiated by OH addition to the benzene ring to form TMB-OH adduct, which can react with O2 through recombination to produce the bicyclic peroxy radical. It has been established that a series of ring-retaining (product A in Fig. 6) and ring-opening products (products B, C, and D in Fig. 6) can be generated by the further reaction of the bicyclic peroxy radical in the presence of NO (Zaytsev et al., 2019; Li and Wang, 2014). Both ring-opening and ring-retaining compounds are expected to contribute significantly to organosulfate production. Here, we take unsaturated ketoaldehyde (product C in Fig. 6) as an example to describe the possible formation mechanism of organosulfate observed in the present study. The reaction of OH with compound C involves OH addition to unsaturated C = C bonds to form an alkyl radical, which can react subsequently with O2 to yield an organic peroxy radical. Further reactions of the organic peroxy radical can follow two different pathways. One pathway is that the organic peroxy radical undergoes a 1,5-H shift isomerization to form a new acyl radical. Insight from a previous review suggested that the acyl radical reacts with O2 to yield an acyl peroxy radical, which could further react with HO2 to form multifunctional hydroperoxide (Ziemann and Atkinson, 2012). The second channel is the reaction of the organic peroxy radical with HO2 to produce hydroperoxide, terminating the radical chain directly. The acid-driven heterogeneous chemistry of hydroperoxide has been previously adopted to explain the generation of certain OSs (Riva et al., 2016a, b). With the presence of sulfuric acid formed by the oxidation of SO2 by OH, TMB-derived hydroperoxides can be hydrolyzed by H+ and then react with inorganic to form organosulfates. The conversion of inorganic sulfates to organosulfates could cause changes in aerosol growth, multiphase chemistry, and acidity (Zhang et al., 2019; Riva et al., 2019).
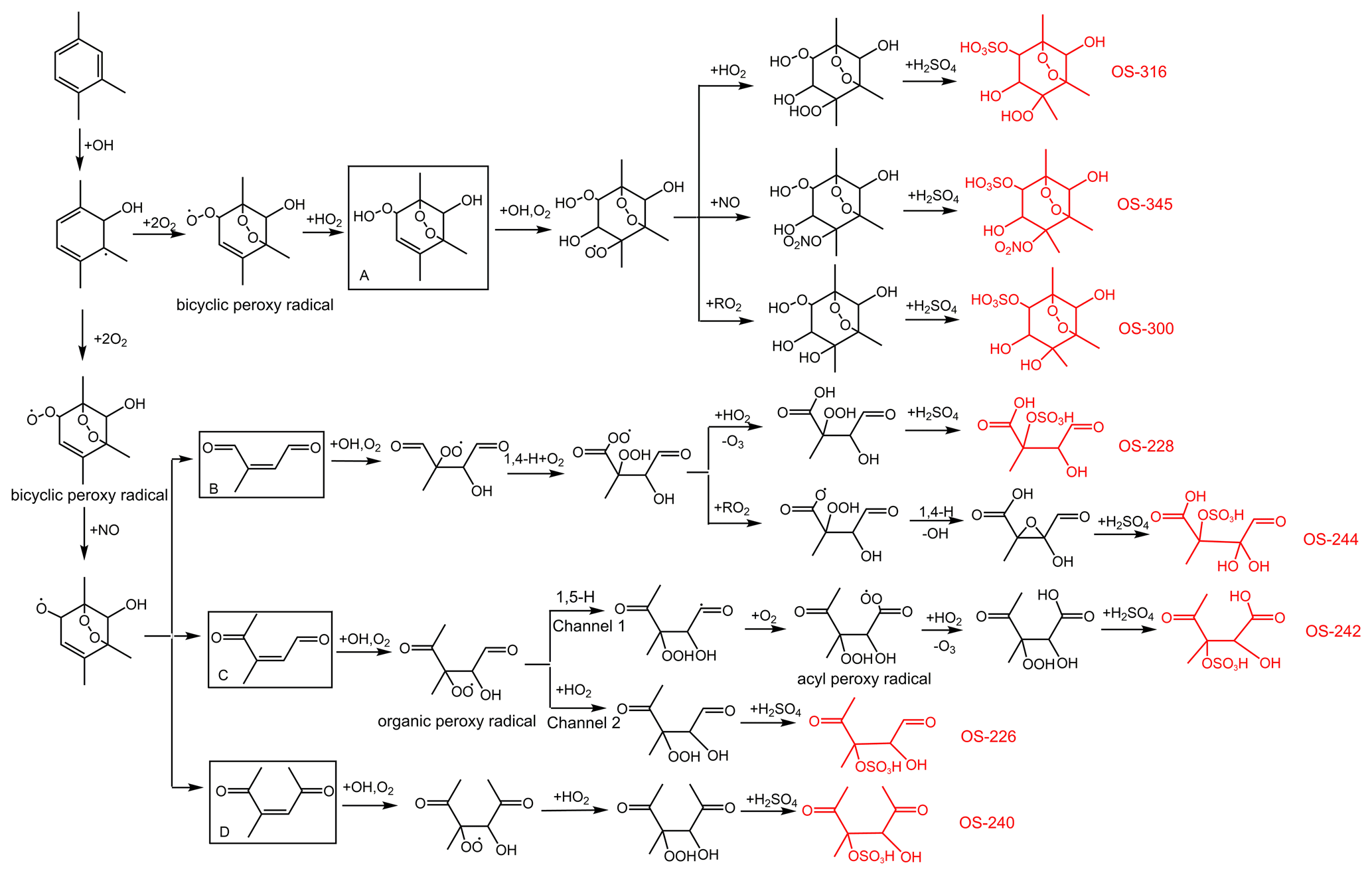
Figure 6Proposed mechanisms for organosulfate formation from the photooxidation of TMB in the presence of SO2. The black boxes mark the ring-opening and ring-retaining products suggested in previous studies (Li and Wang, 2014; Zaytsev et al., 2019). The compounds in red are organosulfates detected by the UPLC-HRMS in this work.
The MW 228 and 230 organic sulfonates were assigned as sulfonates containing an aromatic ring based on accurate mass measurements and comparison of mass fragmentation patterns with other aromatic sulfonates (Riva et al., 2015a). MS/MS spectra and the proposed fragmentation schemes of sulfonates are reported in Fig. S8. For MW 228 sulfonate, the MS/MS spectra showed the fragment ions at 79.95721 (SO, 118.96625 (C8H7O, M-SO-CO), and 163.04025 (C9H7O, M-SO2), as presented in Fig. S8. For the MW 230 sulfonate, the fragment of parent ion at 229.01706 could occur by the loss of 44 mass units to give the product ion at 185.02777 (Fig. S8). The loss of 79.95747 mass units as a sulfite radical is in accord with the MS/MS spectra of aromatic sulfonates generated from the photooxidation of polycyclic aromatic hydrocarbons (Riva et al., 2015b). A recent field measurement demonstrated that aromatic organosulfur compounds account for a substantial fraction of total organosulfur compounds in Shanghai, China, highlighting the importance of aromatic organosulfur compounds (Ma et al., 2014). Aromatic sulfonate formation from the photooxidation of TMB in the presence of SO2 was unexpected, and the exact formation pathway of the aromatic sulfonates warrants further investigation.
3.2 Effects of NH3
3.2.1 Particle formation and growth in NH3-involved photooxidation
High-NOx photooxidation experiments were carried out in the presence and absence of NH3. The average OH concentrations were similar for each experiment within 300 min of irradiation (Table 1). Figure 7 displays the volume and number concentrations of aerosols as a function of time in different photooxidation (Exps. 5, 8–12). TMB was oxidized to produce many secondary aerosols under continuous UV irradiation. The volume concentrations of aerosol particles have a clear positive correlation with NH3 initial level for all conditions. However, the effect of NH3 on particle formation was not as pronounced as that of SO2 with similar concentration (Fig. 7). In TMB NOx NH3 photooxidation, the net SOA yield increased slightly from 3.5 % to 5.1 % as the NH3 initial level increased from 0 to 200 ppb (Table 1). Our result is consistent with the finding of Chen et al. (2020a), who showed that NH3 did not significantly affect SOA formation from toluene NOx photooxidation under dry conditions. Interestingly, SMPS measurements demonstrated that the coexistence of SO2 and NH3 can considerably promote secondary aerosol formation (Fig. 7). After subtracting the inorganic components, it was seen that the net SOA yield could increase to 13.7 % with the introduction of 200 ppb NH3 and 234 ppb SO2, indicating the synergetic effects of NH3 and SO2 (Chu et al., 2016). The flux of the gas-phase products diffusing to a particle partly depends on the surface area of the particle. The coexistence of SO2 and NH3 promoted the increase in particle surface area concentrations (Table 1). The ability of particle formation originating from gas-to-particle conversion may be significantly stronger with SO2 and NH3 introduction, leading to the enhancement in particle formation. The total number concentrations of aerosol particles increased rapidly in all experiments after nucleation was initiated as shown in Fig. 7b. After reaching the maximum concentration, the number density of particles declined gradually because of particles' coagulation and deposition to the chamber walls. Increasing the NH3 level to 200 ppb enhanced the particle maximum number concentration by factors of 2.0 and 1.7 under SO2-free and SO2-involved conditions, respectively. Our results are valuable in terms of the potential of NH3 emission reductions to improve air quality by decreasing total particle number concentrations.
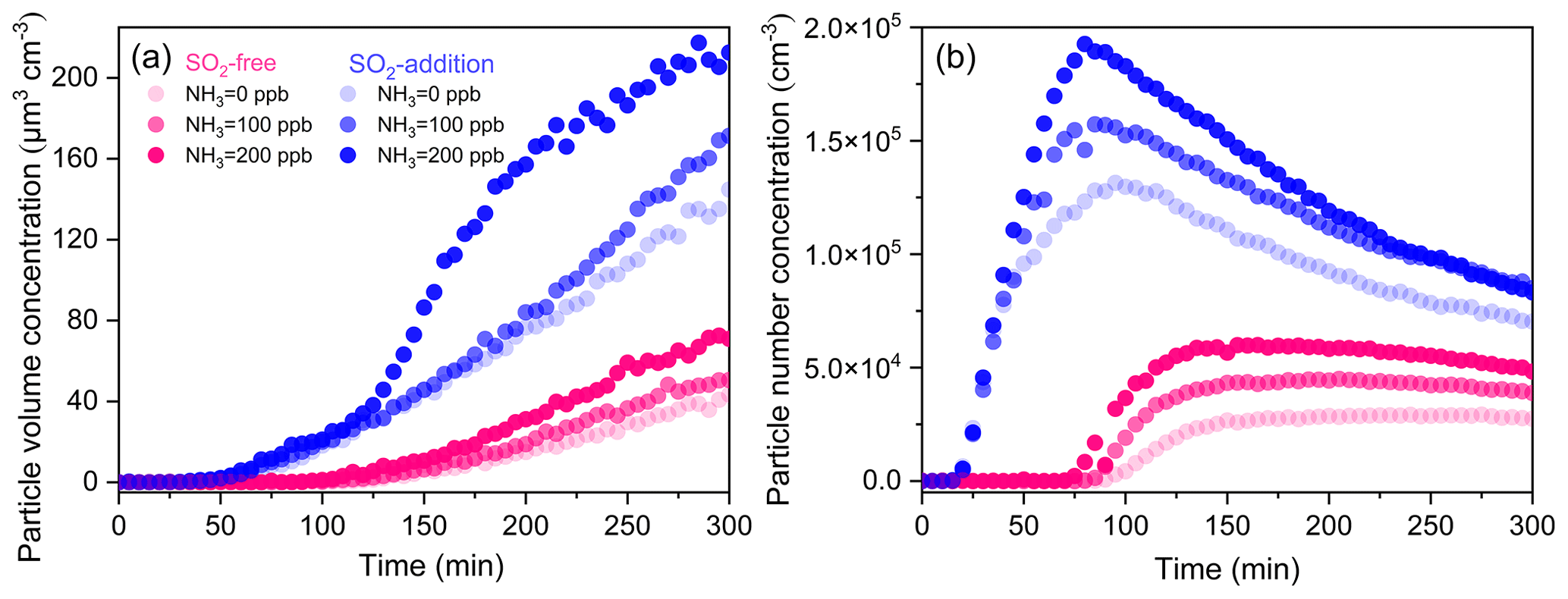
Figure 7Time evolutions of the volume (a) and number (b) concentrations of aerosol particles from TMB photooxidation with different initial NH3 levels under SO2-free and SO2-addition (∼ 230 ppb) conditions.
Once generated, aerosol particles need to grow to a larger size (> 50–100 nm) before they exert a significant impact on global climate and public health. To explore the effects of NH3 on particle growth, the initial growth rate of aerosol particles from different experiments is also compared in Fig. S9. In SO2-free experiments, the increase in NH3 initial concentrations led to a remarkable increase of the initial growth rate of aerosol particles. High initial growth rates were also found in the photooxidation of other aromatics such as toluene and -xylene with NH3 addition (Li et al., 2018). This result may be explained by the fact that under SO2-free conditions, NH3 mainly reacts with acids to produce ammonium salts. Previous studies have reported that ammonium salts could partition into the initial growth process of new secondary aerosol particles and thus increase the particle initial growth rate (Li et al., 2018; Zhu et al., 2014). More interestingly, the NH3 level did not substantially affect the initial growth rate of particles in the presence of SO2 (Fig. S9). The results in Fig. 7 have demonstrated that the synergetic effects of NH3 and SO2 can promote new particle formation (Lehtipalo et al., 2018). In SO2-involved experiments, NH3 molecules tend to promote new particle formation rather than particle growth. Note that these effects of NH3 on particle initial growth might be more complex in the ambient air with high levels of SO2, NH3, NOx, and VOCs than current smog chamber experiments.
3.2.2 Particle chemical composition in NH3-involved photooxidation
The ATR-FTIR spectra of aerosol particles from the NH3-added experiment are also given in Fig. 5. A previous study suggested that the absorbances at 3310–3360 and 1550–1650 cm−1 can be assigned to N–H stretch and C–N–H bend in secondary amine molecules, respectively (Babar et al., 2017). However, no evidence of secondary amine formation was detected, and no clear FTIR spectra differences between aerosols from the TMB NOx experiment and aerosols from TMB NOx NH3 photooxidation were found in this work (Fig. 5). It has been recently discovered that the conversion of oxidized organics to nitrogen-containing compounds in the presence of NH3 is more likely to occur in high RH conditions (Zhang et al., 2020). NH3 uptake by TMB-derived aerosol particles may be limited to the aerosol surface under low RH conditions (RH < 20 %) (Bell et al., 2017). The amounts of secondary amine compounds formed from the NH3 uptake by aerosol may be small, resulting in no characteristic peaks of secondary amine in ATR-FTIR spectra. Surprisingly, the effects of NH3 on aerosol chemical compositions were increasingly important under SO2-rich conditions. As displayed in Fig. 5e, the strong C = O band at 1720 cm−1 converted to a shoulder upon NH3 addition to the TMB NOx SO2 reaction system. In addition, the C–N stretch at 1332 cm−1 was observed in TMB NOx SO2 NH3 photooxidation (Fig. 5). The particle-phase C–N stretch has been also characterized in m-xylene photooxidation experiments with NH3 addition and was suggested to account for nitrogen-containing compounds (Liu et al., 2015b). A smog chamber study also found that the coexistence of SO2 and NH3 substantially enhanced the formation of nitrogen-containing compounds from the photooxidation of toluene (Chu et al., 2016). In the current work, the presence of SO2 could promote the increase in particle acidity. Elevating particle acidity could facilitate the reaction of with carbonyl-containing compounds, leading to the formation of nitrogen-containing organic compounds (Liu et al., 2015b).
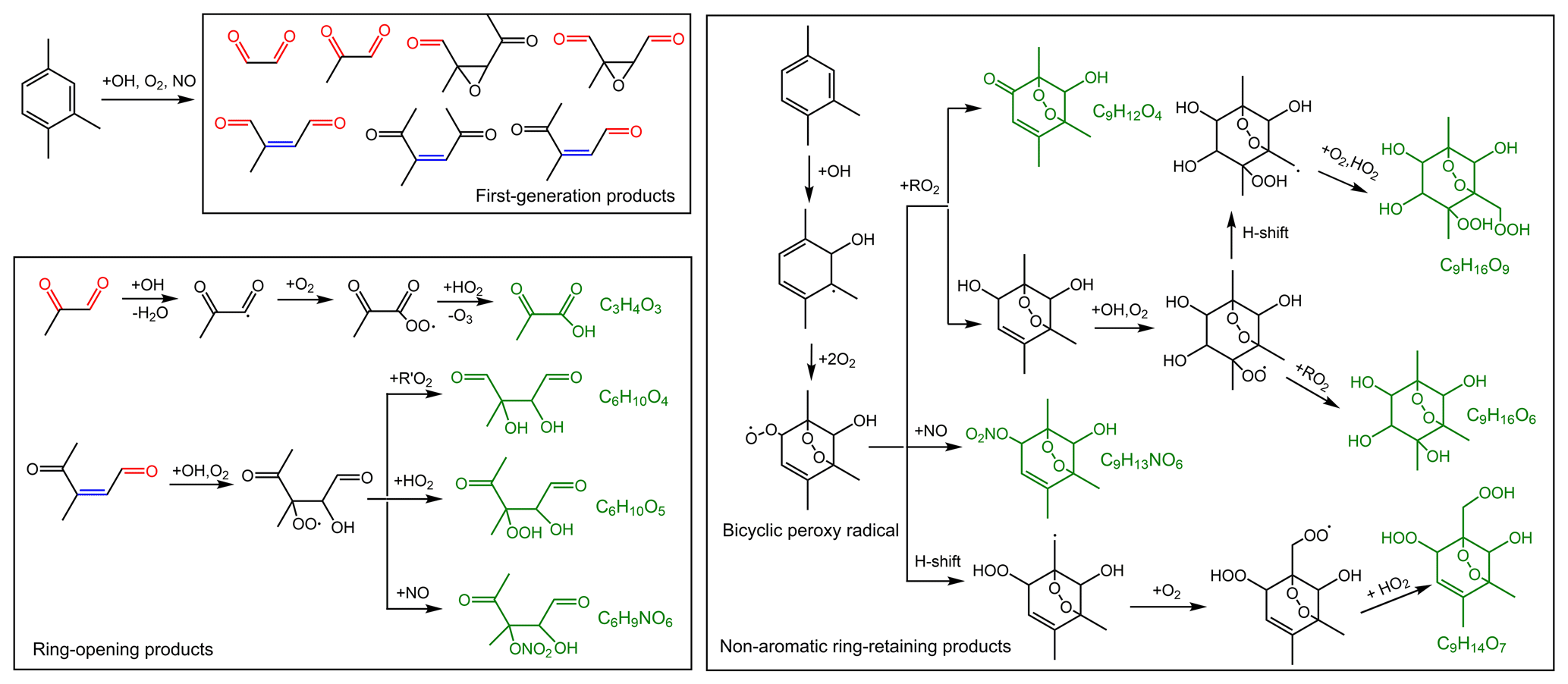
Figure 8Simplified formation mechanism of the detected products in SOA formed from NH3-involved photooxidation. Some observed products are produced through the same chemical mechanism, and for simplicity, only one product is drawn in this figure as an example.
The MS measurement results are consistent with FTIR analysis. As shown in the MS spectra of aerosol samples (Fig. S10), under SO2-free conditions, the presence of NH3 did not result in considerable changes in peak numbers and abundance for both positive ion mode and negative ion mode. NH3 could slightly enhance SOA formation in SO2-free experiment as mentioned in Sect. 3.2.1. Therefore, the NH3-induced changes in the absolute concentrations of organic components might be small in SO2-free experiments, leading to similar mass spectra for Fig. S10a and b. In addition, the major products (Table S4) are likely generated by similar chemical mechanisms (Fig. 8), which are not sensitive to the change in initial NH3 levels under current experimental conditions. First, the photooxidation of TMB in the presence of NO can result in the formation of bicyclic oxy radicals that could decompose to a series of ring-opening products including biacetyl, epoxy-dicarbonyl, and carbonylic products (Li and Wang, 2014). As depicted in Fig. 8, the first-generation products could be further oxidized by OH. For example, oxidation of methylglyoxal by OH can proceed through abstraction of the aldehydic hydrogen to form the CH3C(O)C(O)• radical, which may react with O2 and then with HO2 to form pyruvic acid. Second, the bicyclic peroxy radical (BPR) is a key intermediate for the formation of non-aromatic ring-retaining products. Reactions of BPR with RO2 can form either bicyclic carbonyl or bicyclic alcohol, which further undergoes OH oxidation to yield the C9H16O6 and C9H16O9 compounds. BPR can produce bicyclic organonitrates by reaction with NO and can also undergo an intramolecular H shift followed by O2 addition to form a new bicyclic peroxy radical. The new bicyclic peroxy radical reacts with HO2 to generate highly oxygenated organic molecules, consistent with a recent study (Wang et al., 2020b). To the best of our knowledge, NH3 does not basically affect the reaction of free radicals in gas phase during the photooxidation of TMB. Generally, NH3 levels play a negligible role in the aerosol organic composition in TMB photooxidation without SO2 addition. In contrast, under SO2-rich conditions, the increase in NH3 level led to a significant increase in the abundance of organic compounds (especially for compounds with ) in both positive and negative ion modes (Fig. S11). The introduction of SO2 and NH3 led to the formation of ammonium sulfate (Fig. S12), which is an attractive condensation sink for organic vapors. High particle surface area concentration in TMB NOx SO2 NH3 experiments may increase the abundance of organic compounds in the bulk phase. To better explain this effect, the saturation mass concentrations of detected products were predicted based on a previous method (Li et al., 2016), and the calculated results are shown in Fig. 9 and Table S4. During the photooxidation of TMB, the fates of organic compounds are mainly governed by the competition between fragmentation and functionalization. Losing carbon atoms increases product volatility, which could be partly compensated for by functionalization. Among the compounds present in aerosol particles formed from NH3-added systems, 14 products were C9 or smaller multifunctional oxidation products. From Fig. 9, the range of product saturation mass concentration spanned approximately 8 orders of magnitude, indicating that the measured particle-phase products are considerably different regarding volatility. The products in the particle phase are classified into three classes in Fig. 9: low-volatility organic compounds, intermediate-volatility organic compounds, and semi-volatile organic compounds. The measured products might not be responsible for homogenous nucleation, but these compounds can gradually condense onto nucleation particles (i.e., ammonium sulfate) to contribute to aerosol formation and growth, which highlights the role of ammonium sulfate in this case.
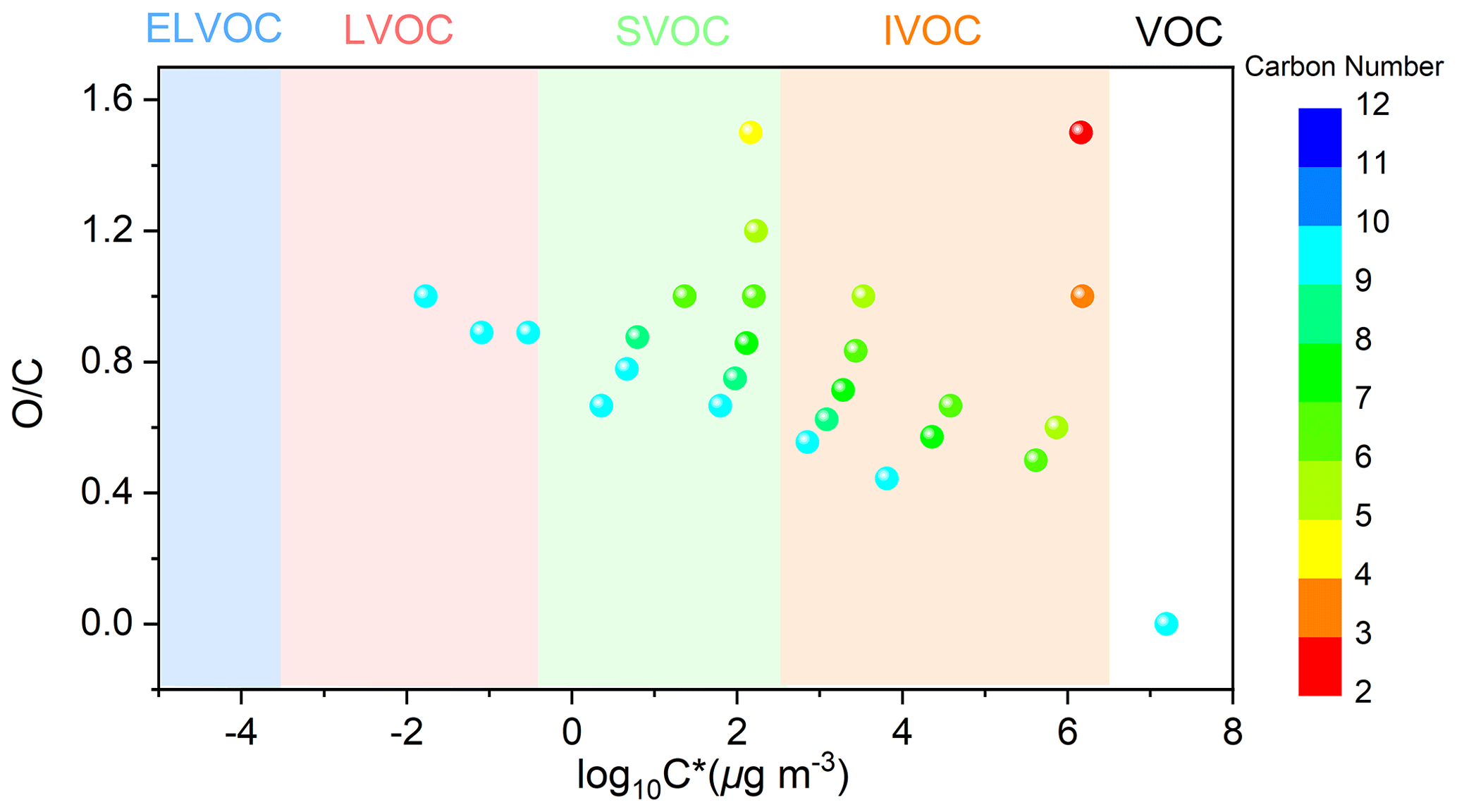
Figure 9TMB and detected products (Table S4) displayed in the two-dimensional volatility-oxidation space. Based on the method of Li et al. (2016), we grouped the detected compounds in the classes of extremely low-volatility organic compound (ELVOC), low-volatility organic compound (LVOC), semi-volatile organic compound (SVOC), intermediate-volatility organic compound (IVOC), and volatile organic compound (VOC).
In summary, we explored the detailed effects of SO2 and NH3 on secondary aerosol formation from TMB photooxidation. Our results demonstrate a substantial increase in ultrafine particle (< 100 nm) number concentrations resulting from SO2 addition. Significant increases in SOA yields were found in TMB NOx SO2 photooxidation, due to acid-driven heterogeneous reactions. The laboratory characterization of SOA composition confirmed the formation of new organosulfates at MW 214, 226, 228, 240, 242, 244, 268, 300, 316, and 345. The MS data give experimental evidence that the MW 214 and 242 organosulfates could account for organosulfates previously designated as being of unknown origin in ambient PM2.5, while some of them that were observed in TMB NOx SO2 photooxidation are isomers of recognized biogenic organosulfates. This indicates that care must be taken in the identification of TMB-derived organosulfates in ambient aerosols. More laboratory and field studies with organosulfate authentic standards should be carried out to determine the accurate yield of the measured organosulfates and the contribution of TMB-derived organosulfate to total atmospheric organosulfate. In addition, the composition of secondary aerosol could determine the physicochemical properties of aerosol particles (e.g., viscosity and phase state). Changes in SO2 emissions in different regions all over the world have great implications for the physicochemical properties of aromatics-derived SOA and, thus, highly influence the global climate.
The presence of NH3 also increased the number and volume concentration of secondary aerosol particles, especially under SO2-rich conditions. We characterized a series of multifunctional ring-retaining and ring-opening organic compounds containing one and even more carbonyl and alcohol functional groups. The predicted volatility distributions of products suggested that the measured later generation products progressively condense onto nucleation particles to enhance particle formation in NH3-added photooxidation. Current models that are used to assess aerosol–climate interactions should fully take into account the influence of NH3 on secondary aerosol formation, which is especially significant in regions with strong NH3 emissions.
Experimental data are available upon request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-7963-2021-supplement.
LD and ZY designed the experiments, and ZY carried them out. ZY performed data analysis with assistance from LX, XL, and LD. ZY prepared the paper with contributions from all co-authors. NTT, JL, and LD commented on the paper.
The authors declare that they have no conflict of interest.
We thank Jingyao Qu and Zhifeng Li from the State Key Laboratory of Microbial Technology of Shandong University for help and guidance with MS measurements.
This research has been supported by the National Natural Science Foundation of China (grant no. 91644214), the Department of Education of Shandong Province (grant no. 2019KJD007), and the Fundamental Research Fund of Shandong University (grant no. 2020QNQT012).
This paper was edited by Jason Surratt and reviewed by three anonymous referees.
Apsokardu, M. J. and Johnston, M. V.: Nanoparticle growth by particle-phase chemistry, Atmos. Chem. Phys., 18, 1895–1907, https://doi.org/10.5194/acp-18-1895-2018, 2018.
Babar, Z. B., Park, J.-H., and Lim, H.-J.: Influence of NH3 on secondary organic aerosols from the ozonolysis and photooxidation of α-pinene in a flow reactor, Atmos. Environ., 164, 71–84, https://doi.org/10.1016/j.atmosenv.2017.05.034, 2017.
Bell, D. M., Imre, D., S, T. M., and Zelenyuk, A.: The properties and behavior of alpha-pinene secondary organic aerosol particles exposed to ammonia under dry conditions, Phys. Chem. Chem. Phys., 19, 6497–6507, https://doi.org/10.1039/c6cp08839b, 2017.
Blair, S. L., MacMillan, A. C., Drozd, G. T., Goldstein, A. H., Chu, R. K., Pasa-Tolic, L., Shaw, J. B., Tolic, N., Lin, P., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Molecular characterization of organosulfur compounds in biodiesel and diesel fuel secondary organic aerosol, Environ. Sci. Technol., 51, 119–127, https://doi.org/10.1021/acs.est.6b03304, 2017.
Boris, A. J., Lee, T., Park, T., Choi, J., Seo, S. J., and Collett Jr., J. L.: Fog composition at Baengnyeong Island in the eastern Yellow Sea: detecting markers of aqueous atmospheric oxidations, Atmos. Chem. Phys., 16, 437–453, https://doi.org/10.5194/acp-16-437-2016, 2016.
Cai, D., Wang, X., Chen, J., and Li, X.: Molecular characterization of organosulfates in highly polluted atmosphere using ultra-high-resolution mass spectrometry, J. Geophys. Res.-Atmos., 125, e2019JD032253, https://doi.org/10.1029/2019jd032253, 2020.
Chen, L., Bao, Z., Wu, X., Li, K., Han, L., Zhao, X., Zhang, X., Wang, Z., Azzi, M., and Cen, K.: The effects of humidity and ammonia on the chemical composition of secondary aerosols from toluene/NOx photo-oxidation, Sci. Total Environ., 728, 138671, https://doi.org/10.1016/j.scitotenv.2020.138671, 2020a.
Chen, T., Liu, Y., Ma, Q., Chu, B., Zhang, P., Liu, C., Liu, J., and He, H.: Significant source of secondary aerosol: formation from gasoline evaporative emissions in the presence of SO2 and NH3, Atmos. Chem. Phys., 19, 8063–8081, https://doi.org/10.5194/acp-19-8063-2019, 2019.
Chen, Y., Zhang, Y., Lambe, A. T., Xu, R., Lei, Z., Olson, N. E., Zhang, Z., Szalkowski, T., Cui, T., Vizuete, W., Gold, A., Turpin, B. J., Ault, A. P., Chan, M. N., and Surratt, J. D.: Heterogeneous Hydroxyl Radical Oxidation of Isoprene-Epoxydiol-Derived Methyltetrol Sulfates: Plausible Formation Mechanisms of Previously Unexplained Organosulfates in Ambient Fine Aerosols, Environ. Sci. Tech. Let., 7, 460–468, https://doi.org/10.1021/acs.estlett.0c00276, 2020b.
Cheng, Y., Yu, Q.-Q., Liu, J.-M., Zhu, S., Zhang, M., Zhang, H., Zheng, B., and He, K.-B.: Model vs. observation discrepancy in aerosol characteristics during a half-year long campaign in Northeast China: The role of biomass burning, Environ. Pollut., 269, 116167–116167, https://doi.org/10.1016/j.envpol.2020.116167, 2021.
Chu, B., Zhang, X., Liu, Y., He, H., Sun, Y., Jiang, J., Li, J., and Hao, J.: Synergetic formation of secondary inorganic and organic aerosol: effect of SO2 and NH3 on particle formation and growth, Atmos. Chem. Phys., 16, 14219–14230, https://doi.org/10.5194/acp-16-14219-2016, 2016.
Chu, B., Ma, Q., Liu, J., Ma, J., Zhang, P., Chen, T., Feng, Q., Wang, C., Yang, N., Ma, H., Ma, J., Russell, A. G., and He, H.: Air pollutant correlations in china: Secondary air pollutant responses to NOx and SO2 control, Environ. Sci. Tech. Let., 7, 695–700, https://doi.org/10.1021/acs.estlett.0c00403, 2020.
Estillore, A. D., Hettiyadura, A. P. S., Qin, Z., Leckrone, E., Wombacher, B., Humphry, T., Stone, E. A., and Grassian, V. H.: Water Uptake and Hygroscopic Growth of Organosulfate Aerosol, Environ. Sci. Technol., 50, 4259–4268, https://doi.org/10.1021/acs.est.5b05014, 2016.
Fleming, L. T., Ali, N. N., Blair, S. L., Roveretto, M., George, C., and Nizkorodov, S. A.: Formation of Light-Absorbing Organosulfates during Evaporation of Secondary Organic Material Extracts in the Presence of Sulfuric Acid, ACS Earth Space Chem., 3, 947–957, https://doi.org/10.1021/acsearthspacechem.9b00036, 2019.
Flores, J. M., Washenfelder, R. A., Adler, G., Lee, H. J., Segev, L., Laskin, J., Laskin, A., Nizkorodov, S. A., Brown, S. S., and Rudich, Y.: Complex refractive indices in the near-ultraviolet spectral region of biogenic secondary organic aerosol aged with ammonia, Phys. Chem. Chem. Phys., 16, 10629–10642, https://doi.org/10.1039/c4cp01009d, 2014.
Friedman, B., Brophy, P., Brune, W. H., and Farmer, D. K.: Anthropogenic Sulfur Perturbations on Biogenic Oxidation: SO2 Additions Impact Gas-Phase OH Oxidation Products of α- and β-Pinene, Environ. Sci. Technol., 50, 1269-1279, https://doi.org/10.1021/acs.est.5b05010, 2016.
Fu, X., Wang, S., Xing, J., Zhang, X., Wang, T., and Hao, J.: Increasing Ammonia Concentrations Reduce the Effectiveness of Particle Pollution Control Achieved via SO2 and NOx Emissions Reduction in East China, Environ. Sci. Tech. Let., 4, 221–227, https://doi.org/10.1021/acs.estlett.7b00143, 2017.
Fuzzi, S., Baltensperger, U., Carslaw, K., Decesari, S., Denier van der Gon, H., Facchini, M. C., Fowler, D., Koren, I., Langford, B., Lohmann, U., Nemitz, E., Pandis, S., Riipinen, I., Rudich, Y., Schaap, M., Slowik, J. G., Spracklen, D. V., Vignati, E., Wild, M., Williams, M., and Gilardoni, S.: Particulate matter, air quality and climate: lessons learned and future needs, Atmos. Chem. Phys., 15, 8217–8299, https://doi.org/10.5194/acp-15-8217-2015, 2015.
Guo, H., Wang, T., Blake, D., Simpson, I., Kwok, Y., and Li, Y.: Regional and local contributions to ambient non-methane volatile organic compounds at a polluted rural/coastal site in Pearl River Delta, China, Atmos. Environ., 40, 2345–2359, 2006.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Han, Y., Gong, Z., Liu, P., de Sá, S. S., McKinney, K. A., and Martin, S. T.: Influence of Particle Surface Area Concentration on the Production of Organic Particulate Matter in a Continuously Mixed Flow Reactor, Environ. Sci. Technol., 53, 4968–4976, https://doi.org/10.1021/acs.est.8b07302, 2019.
Hansen, A. M. K., Hong, J., Raatikainen, T., Kristensen, K., Ylisirniö, A., Virtanen, A., Petäjä, T., Glasius, M., and Prisle, N. L.: Hygroscopic properties and cloud condensation nuclei activation of limonene-derived organosulfates and their mixtures with ammonium sulfate, Atmos. Chem. Phys., 15, 14071–14089, https://doi.org/10.5194/acp-15-14071-2015, 2015.
Hao, L., Kari, E., Leskinen, A., Worsnop, D. R., and Virtanen, A.: Direct contribution of ammonia to α-pinene secondary organic aerosol formation, Atmos. Chem. Phys., 20, 14393–14405, https://doi.org/10.5194/acp-20-14393-2020, 2020.
Heald, C. L., Kroll, J. H., Jimenez, J. L., Docherty, K. S., DeCarlo, P. F., Aiken, A. C., Chen, Q., Martin, S. T., Farmer, D. K., and Artaxo, P.: A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 37, L08803, https://doi.org/10.1029/2010gl042737, 2010.
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., El Haddad, I., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Jorga, S. D., Kaltsonoudis, C., Liangou, A., and Pandis, S. N.: Measurement of Formation Rates of Secondary Aerosol in the Ambient Urban Atmosphere Using a Dual Smog Chamber System, Environ. Sci. Technol., 54, 1336–1343, https://doi.org/10.1021/acs.est.9b03479, 2020.
Julin, J., Murphy, B. N., Patoulias, D., Fountoukis, C., Olenius, T., Pandis, S. N., and Riipinen, I.: Impacts of Future European Emission Reductions on Aerosol Particle Number Concentrations Accounting for Effects of Ammonia, Amines, and Organic Species, Environ. Sci. Technol., 52, 692–700, https://doi.org/10.1021/acs.est.7b05122, 2018.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Kulmala, M., Petaja, T., Nieminen, T., Sipila, M., Manninen, H. E., Lehtipalo, K., Dal Maso, M., Aalto, P. P., Junninen, H., Paasonen, P., Riipinen, I., Lehtinen, K. E., Laaksonen, A., and Kerminen, V. M.: Measurement of the nucleation of atmospheric aerosol particles, Nat. Protoc., 7, 1651–1667, https://doi.org/10.1038/nprot.2012.091, 2012.
Lal, V., Khalizov, A. F., Lin, Y., Galvan, M. D., Connell, B. T., and Zhang, R.: Heterogeneous reactions of epoxides in acidic media, J. Phys. Chem. A, 116, 6078–6090, https://doi.org/10.1021/jp2112704, 2012.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D. X., Chen, X. M., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X. C., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V. M., Kim, C., Kirkby, J., Kontkanen, J., Kurten, A., Lawler, M. J., Mai, H. J., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petaja, T., Piel, F., Pospisilova, V., Quelever, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipila, M., Tauber, C., Tome, A., Trostl, J., Vaisanen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M. Y., Weitz, L., Wimmer, D., Ye, P. L., Ylisirnio, A., Zha, Q. Z., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, Sci. Adv., 4, 9, https://doi.org/10.1126/sciadv.aau5363, 2018.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Li, H., Zhang, Q., Zhang, Q., Chen, C., Wang, L., Wei, Z., Zhou, S., Parworth, C., Zheng, B., Canonaco, F., Prévôt, A. S. H., Chen, P., Zhang, H., Wallington, T. J., and He, K.: Wintertime aerosol chemistry and haze evolution in an extremely polluted city of the North China Plain: significant contribution from coal and biomass combustion, Atmos. Chem. Phys., 17, 4751–4768, https://doi.org/10.5194/acp-17-4751-2017, 2017.
Li, K., Chen, L., White, S. J., Yu, H., Wu, X., Gao, X., Azzi, M., and Cen, K.: Smog chamber study of the role of NH3 in new particle formation from photo-oxidation of aromatic hydrocarbons, Sci. Total Environ., 619–620, 927-937, https://doi.org/10.1016/j.scitotenv.2017.11.180, 2018.
Li, Y. and Wang, L.: The atmospheric oxidation mechanism of 1,2,4-trimethylbenzene initiated by OH radicals, Phys. Chem. Chem. Phys., 16, 17908–17917, https://doi.org/10.1039/c4cp02027h, 2014.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Lin, Y.-H., Knipping, E. M., Edgerton, E. S., Shaw, S. L., and Surratt, J. D.: Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013, 2013.
Lin, Y. H., Budisulistiorini, H., Chu, K., Siejack, R. A., Zhang, H. F., Riva, M., Zhang, Z. F., Gold, A., Kautzman, K. E., and Surratt, J. D.: Light-Absorbing Oligomer Formation in Secondary Organic Aerosol from Reactive Uptake of Isoprene Epoxydiols, Environ. Sci. Technol., 48, 12012–12021, https://doi.org/10.1021/es503142b, 2014.
Liu, C., Ma, Q., Chu, B., Liu, Y., He, H., Zhang, X., Li, J., and Hao, J.: Effect of aluminium dust on secondary organic aerosol formation in m-xylene/NOx photo-oxidation, Sci. China Earth Sci., 58, 245–254, https://doi.org/10.1007/s11430-014-5023-0, 2015a.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015b.
Liu, S., Shilling, J. E., Song, C., Hiranuma, N., Zaveri, R. A., and Russell, L. M.: Hydrolysis of Organonitrate Functional Groups in Aerosol Particles, Aerosol Sci. Technol., 46, 1359–1369, 10.1080/02786826.2012.716175, 2012.
Liu, S., Jia, L., Xu, Y., Tsona, N. T., Ge, S., and Du, L.: Photooxidation of cyclohexene in the presence of SO2: SOA yield and chemical composition, Atmos. Chem. Phys., 17, 13329–13343, https://doi.org/10.5194/acp-17-13329-2017, 2017.
Liu, T., Wang, X., Hu, Q., Deng, W., Zhang, Y., Ding, X., Fu, X., Bernard, F., Zhang, Z., Lü, S., He, Q., Bi, X., Chen, J., Sun, Y., Yu, J., Peng, P., Sheng, G., and Fu, J.: Formation of secondary aerosols from gasoline vehicle exhaust when mixing with SO2, Atmos. Chem. Phys., 16, 675–689, https://doi.org/10.5194/acp-16-675-2016, 2016.
Ma, Y., Xu, X., Song, W., Geng, F., and Wang, L.: Seasonal and diurnal variations of particulate organosulfates in urban Shanghai, China, Atmos. Environ., 85, 152–160, https://doi.org/10.1016/j.atmosenv.2013.12.017, 2014.
Mehra, A., Wang, Y., Krechmer, J. E., Lambe, A., Majluf, F., Morris, M. A., Priestley, M., Bannan, T. J., Bryant, D. J., Pereira, K. L., Hamilton, J. F., Rickard, A. R., Newland, M. J., Stark, H., Croteau, P., Jayne, J. T., Worsnop, D. R., Canagaratna, M. R., Wang, L., and Coe, H.: Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation, Atmos. Chem. Phys., 20, 9783–9803, https://doi.org/10.5194/acp-20-9783-2020, 2020.
Meng, Z., Wu, L., Xu, X., Xu, W., Zhang, R., Jia, X., Liang, L., Miao, Y., Cheng, H., Xie, Y., He, J., and Zhong, J.: Changes in ammonia and its effects on PM2.5 chemical property in three winter seasons in Beijing, China, Sci. Total Environ., 749, 142208, https://doi.org/10.1016/j.scitotenv.2020.142208, 2020.
Mo, Z., Lu, S., and Shao, M.: Volatile organic compound (VOC) emissions and health risk assessment in paint and coatings industry in the Yangtze River Delta, China, Environ. Pollut., 269, 115740, https://doi.org/10.1016/j.envpol.2020.115740, 2021.
Na, K., Song, C., and Cockeriii, D.: Formation of secondary organic aerosol from the reaction of styrene with ozone in the presence and absence of ammonia and water, Atmos. Environ., 40, 1889–1900, https://doi.org/10.1016/j.atmosenv.2005.10.063, 2006.
Na, K., Song, C., Switzer, C., and Cocker, D. R., III: Effect of ammonia on secondary organic aerosol formation from alpha-Pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, https://doi.org/10.1021/es061956y, 2007.
Nestorowicz, K., Jaoui, M., Rudzinski, K. J., Lewandowski, M., Kleindienst, T. E., Spólnik, G., Danikiewicz, W., and Szmigielski, R.: Chemical composition of isoprene SOA under acidic and non-acidic conditions: effect of relative humidity, Atmos. Chem. Phys., 18, 18101–18121, https://doi.org/10.5194/acp-18-18101-2018, 2018.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
O'Brien, R. E., Laskin, A., Laskin, J., Rubitschun, C. L., Surratt, J. D., and Goldstein, A. H.: Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass Spectrometry: CalNex 2010 field study, J. Geophys. Res.-Atmos., 119, 12706–12720, https://doi.org/10.1002/2014jd021955, 2014.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Paasonen, P., Peltola, M., Kontkanen, J., Junninen, H., Kerminen, V.-M., and Kulmala, M.: Comprehensive analysis of particle growth rates from nucleation mode to cloud condensation nuclei in boreal forest, Atmos. Chem. Phys., 18, 12085–12103, https://doi.org/10.5194/acp-18-12085-2018, 2018.
Ran, L., Zhao, C., Geng, F., Tie, X., Tang, X., Peng, L., Zhou, G., Yu, Q., Xu, J., and Guenther, A.: Ozone photochemical production in urban Shanghai, China: Analysis based on ground level observations, J. Geophys. Res.-Atmos., 114, D15301, https://doi.org/10.1029/2008jd010752, 2009.
Riva, M., Tomaz, S., Cui, T., Lin, Y.-H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an Unrecognized Secondary Anthropogenic Source of Organosulfates and Sulfonates: Gas-Phase Oxidation of Polycyclic Aromatic Hydrocarbons in the Presence of Sulfate Aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015a.
Riva, M., Tomaz, S., Cui, T. Q., Lin, Y. H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015b.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016a.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., Canagaratna, M. R., and Surratt, J. D.: Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016b.
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Ribeiro, I. O., RL, E. O., Dos Santos, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sa, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin Junior, S., de Souza, R. A. F., Palm, B. B., Jimenez, J. L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing Isoprene Epoxydiol-to-Inorganic Sulfate Aerosol Ratio Results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Shalamzari, M. S., Kahnt, A., Vermeylen, R., Kleindienst, T. E., Lewandowski, M., Cuyckens, F., Maenhaut, W., and Claeys, M.: Characterization of polar organosulfates in secondary organic aerosol from the green leaf volatile 3-Z-hexenal, Environ. Sci. Technol., 48, 12671–12678, https://doi.org/10.1021/es503226b, 2014.
Shiraiwa, M., Yee, L. D., Schilling, K. A., Loza, C. L., Craven, J. S., Zuend, A., Ziemann, P. J., and Seinfeld, J. H.: Size distribution dynamics reveal particle-phase chemistry in organic aerosol formation, P. Natl. Acad. Sci. USA, 110, 11746–11750, https://doi.org/10.1073/pnas.1307501110, 2013.
Shrivastava, M., Fast, J., Easter, R., Gustafson Jr., W. I., Zaveri, R. A., Jimenez, J. L., Saide, P., and Hodzic, A.: Modeling organic aerosols in a megacity: comparison of simple and complex representations of the volatility basis set approach, Atmos. Chem. Phys., 11, 6639–6662, https://doi.org/10.5194/acp-11-6639-2011, 2011.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Song, C., Wu, L., Xie, Y., He, J., Chen, X., Wang, T., Lin, Y., Jin, T., Wang, A., Liu, Y., Dai, Q., Liu, B., Wang, Y. N., and Mao, H.: Air pollution in China: Status and spatiotemporal variations, Environ. Pollut., 227, 334–347, https://doi.org/10.1016/j.envpol.2017.04.075, 2017.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Terzano, C., Di Stefano, F., Conti, V., Graziani, E., and Petroianni, A.: Air pollution ultrafine particles: toxicity beyond the lung, Eur. Rev. Med. Pharmacol. Sci., 14, 809–821, 2010.
Tolocka, M. P. and Turpin, B.: Contribution of organosulfur compounds to organic aerosol mass, Environ. Sci. Technol., 46, 7978–7983, https://doi.org/10.1021/es300651v, 2012.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006gl026899, 2006.
Wang, M., Kong, W., Marten, R., He, X. C., Chen, D., Pfeifer, J., Heitto, A., Kontkanen, J., Dada, L., Kurten, A., Yli-Juuti, T., Manninen, H. E., Amanatidis, S., Amorim, A., Baalbaki, R., Baccarini, A., Bell, D. M., Bertozzi, B., Brakling, S., Brilke, S., Murillo, L. C., Chiu, R., Chu, B., De Menezes, L. P., Duplissy, J., Finkenzeller, H., Carracedo, L. G., Granzin, M., Guida, R., Hansel, A., Hofbauer, V., Krechmer, J., Lehtipalo, K., Lamkaddam, H., Lampimaki, M., Lee, C. P., Makhmutov, V., Marie, G., Mathot, S., Mauldin, R. L., Mentler, B., Muller, T., Onnela, A., Partoll, E., Petaja, T., Philippov, M., Pospisilova, V., Ranjithkumar, A., Rissanen, M., Rorup, B., Scholz, W., Shen, J., Simon, M., Sipila, M., Steiner, G., Stolzenburg, D., Tham, Y. J., Tome, A., Wagner, A. C., Wang, D. S., Wang, Y., Weber, S. K., Winkler, P. M., Wlasits, P. J., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Zhou, X., Volkamer, R., Riipinen, I., Dommen, J., Curtius, J., Baltensperger, U., Kulmala, M., Worsnop, D. R., Kirkby, J., Seinfeld, J. H., El-Haddad, I., Flagan, R. C., and Donahue, N. M.: Rapid growth of new atmospheric particles by nitric acid and ammonia condensation, Nature, 581, 184–189, https://doi.org/10.1038/s41586-020-2270-4, 2020a.
Wang, S., Zhou, S., Tao, Y., Tsui, W. G., Ye, J., Yu, J. Z., Murphy, J. G., McNeill, V. F., Abbatt, J. P. D., and Chan, A. W. H.: Organic Peroxides and Sulfur Dioxide in Aerosol: Source of Particulate Sulfate, Environ. Sci. Technol., 53, 10695–10704, https://doi.org/10.1021/acs.est.9b02591, 2019a.
Wang, X. K., Rossignol, S., Ma, Y., Yao, L., Wang, M. Y., Chen, J. M., George, C., and Wang, L.: Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River, Atmos. Chem. Phys., 16, 2285–2298, https://doi.org/10.5194/acp-16-2285-2016, 2016.
Wang, Y., Ma, Y., Li, X., Kuang, B. Y., Huang, C., Tong, R., and Yu, J. Z.: Monoterpene and Sesquiterpene alpha-Hydroxy Organosulfates: Synthesis, MS/MS Characteristics, and Ambient Presence, Environ. Sci. Technol., 53, 12278–12290, https://doi.org/10.1021/acs.est.9b04703, 2019b.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020b.
Warner, J. X., Dickerson, R. R., Wei, Z., Strow, L. L., Wang, Y., and Liang, Q.: Increased atmospheric ammonia over the world's major agricultural areas detected from space, Geophys. Res. Lett., 44, 2875–2884, https://doi.org/10.1002/2016gl072305, 2017.
Wu, Y., Gu, B., Erisman, J. W., Reis, S., Fang, Y., Lu, X., and Zhang, X.: PM2.5 pollution is substantially affected by ammonia emissions in China, Environ. Pollut., 218, 86–94, https://doi.org/10.1016/j.envpol.2016.08.027, 2016.
Wyche, K. P., Monks, P. S., Ellis, A. M., Cordell, R. L., Parker, A. E., Whyte, C., Metzger, A., Dommen, J., Duplissy, J., Prevot, A. S. H., Baltensperger, U., Rickard, A. R., and Wulfert, F.: Gas phase precursors to anthropogenic secondary organic aerosol: detailed observations of 1,3,5-trimethylbenzene photooxidation, Atmos. Chem. Phys., 9, 635–665, https://doi.org/10.5194/acp-9-635-2009, 2009.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Yang, Z., Tsona, N. T., Li, J., Wang, S., Xu, L., You, B., and Du, L.: Effects of NOx and SO2 on the secondary organic aerosol formation from the photooxidation of 1,3,5-trimethylbenzene: A new source of organosulfates, Environ. Pollut., 264, 114742, https://doi.org/10.1016/j.envpol.2020.114742, 2020.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Yu, Z. and Jang, M.: Atmospheric Processes of Aromatic Hydrocarbons in the Presence of Mineral Dust Particles in an Urban Environment, ACS Earth Space Chem., 3, 2404–2414, https://doi.org/10.1021/acsearthspacechem.9b00195, 2019.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhang, G., Lian, X., Fu, Y., Lin, Q., Li, L., Song, W., Wang, Z., Tang, M., Chen, D., Bi, X., Wang, X., and Sheng, G.: High secondary formation of nitrogen-containing organics (NOCs) and its possible link to oxidized organics and ammonium, Atmos. Chem. Phys., 20, 1469–1481, https://doi.org/10.5194/acp-20-1469-2020, 2020.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of urban fine particulate matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensber, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, 2014.
Zhang, Y., Chen, Y., Lei, Z., Olson, N. E., Riva, M., Koss, A. R., Zhang, Z., Gold, A., Jayne, J. T., Worsnop, D. R., Onasch, T. B., Kroll, J. H., Turpin, B. J., Ault, A. P., and Surratt, J. D.: Joint Impacts of Acidity and Viscosity on the Formation of Secondary Organic Aerosol from Isoprene Epoxydiols (IEPOX) in Phase Separated Particles, ACS Earth Space Chem., 3, 2646–2658, https://doi.org/10.1021/acsearthspacechem.9b00209, 2019.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zhu, Y., Sabaliauskas, K., Liu, X., Meng, H., Gao, H., Jeong, C.-H., Evans, G. J., and Yao, X.: Comparative analysis of new particle formation events in less and severely polluted urban atmosphere, Atmos. Environ., 98, 655–664, https://doi.org/10.1016/j.atmosenv.2014.09.043, 2014.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.
Zou, Y., Deng, X. J., Zhu, D., Gong, D. C., Wang, H., Li, F., Tan, H. B., Deng, T., Mai, B. R., Liu, X. T., and Wang, B. G.: Characteristics of 1 year of observational data of VOCs, NOx and O3 at a suburban site in Guangzhou, China, Atmos. Chem. Phys., 15, 6625–6636, https://doi.org/10.5194/acp-15-6625-2015, 2015.