the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 May 2021
| 18 May 2021
Formation kinetics and mechanisms of ozone and secondary organic aerosols from photochemical oxidation of different aromatic hydrocarbons: dependence on NOx and organic substituents
Hao Luo
Jiangyao Chen
Guiying Li
Taicheng An
Aromatic hydrocarbons (AHs) contribute significantly to ozone and secondary organic aerosol (SOA) formation in the atmosphere, but their formation mechanisms are still unclear. Herein, the photochemical oxidation of nine AHs was investigated in a chamber. Only a small amount of ozone was produced from the direct photochemical oxidation of AHs, while a lower number of AH substituents resulted in higher concentrated ozone. Addition of NOx increased ozone and SOA production. The synergetic effect of accelerated NO2 conversion and NO reaction with AHs boosted ozone and volatile intermediate formation. Promoting AH concentration in the VOC NOx ratio further increased formation rates and concentrations of both ozone and SOA. Additionally, ozone formation was enhanced with increasing AH substituent number but negligibly affected by their substituent position. Differently, SOA yield decreased with an increased substituent number of AHs but increased with ortho-methyl-group-substituted AHs. Model fitting and intermediates consistently confirmed that increasing the substituent number on the phenyl ring inhibited generation of dicarbonyl intermediates, which however were preferentially produced from oxidation of ortho-methyl-group-substituted AHs, resulting in different changing trends of the SOA yield. The restrained oligomerization by increased substituent number was another main cause for decreased SOA yield. These results are helpful to understand the photochemical transformation of AHs to secondary pollutants in the real atmosphere.
- Article
(3693 KB) - Full-text XML
-
Supplement
(2295 KB) - BibTeX
- EndNote





As an abundant group of volatile organic compounds (VOCs), aromatic hydrocarbons (AHs) are important precursors of ozone (O3) and secondary organic aerosols (SOAs) in the atmospheric environment (Peng et al., 2017; Tong et al., 2020), directly or indirectly threatening air quality and public health (Henze et al., 2008; Lane et al., 2008; Yang et al., 2016). Atmospheric AHs mainly come from anthropogenic sources, such as industrial emission and motor vehicle emission (Luo et al., 2020a; Sun et al., 2018; Chen et al., 2020; An et al., 2014; He et al., 2015), and these emitted AHs are commonly composed of a single phenyl ring with fewer than four methyl groups (e.g., toluene, xylene) or ethyl groups (e.g., ethylbenzene) (Han et al., 2019; Hu et al., 2015; Chen et al., 2019). It is also found that photochemical oxidation of these AHs is sensitive to reaction conditions (e.g., VOC NOx ratio; Odum et al., 1996; Metzger et al., 2008; Bloss et al., 2005a; Carter and Heo, 2013), ultimately influencing the formation kinetics and mechanisms of O3 and SOA from AH oxidation (Borrás and Tortajada-Genaro, 2012; Sato et al., 2010; Cocker et al., 2001; Ji et al., 2017; Jia and Xu, 2018).
Previous experimental simulation studies have confirmed that a relatively high VOC NOx ratio had an inhibitory effect on SOA productivity (Wang et al., 2015), while the VOC NOx ratio might also influence O3 formation (Wang et al., 2016). From field observations, the actual ratio of VOC NOx in the atmosphere always changes with the variation of the seasons (Geng et al., 2008; Zou et al., 2015; Li et al., 2013; Seinfeld, 1989). Nevertheless, although it is of important environmental significance, studies about the influence of VOC NOx ratio on photochemical oxidation of AHs to form O3 and SOA are mostly focused on the varied NOx concentration. The effects of AH content as well as substitute groups of AHs to the formation kinetics and mechanisms of O3 and SOA have still not been studied. Therefore, in view of the complexity of the real atmosphere, it is very necessary to effectively simulate atmospheric photochemical reactions at different VOC NOx ratios in the laboratory smog chamber and explore the formation kinetics and mechanisms of O3 and SOA from AHs at different concentrations.
Usually, OH-initiated reactions have been confirmed to dominate in AH photochemical oxidation (Ji et al., 2018), in which the reaction rate constant increases with increased substituent number of AHs (Atkinson and Arey, 2003; Aschmann et al., 2013; Glasson and Tuesday, 1970). The important role of substituent position has also been observed in the OH-initiated alkane and alkene oxidation (Atkinson, 2007; Ziemann, 2011). All these previous studies inspire us that the influence of substituents, including the number and position, on the photochemical transformation of AHs to O3 and SOA cannot be ignored. However, although both O3 and SOA generated from different AHs have been studied in laboratory smog chamber simulations, the role of AH substituents in the formation kinetics and mechanisms of O3 and SOA, as well as their relationship with the oxidation intermediates, has not been systematically investigated and established.
In this work, nine AHs with different substituent numbers and positions (e.g., benzene, toluene, ethylbenzene, m-xylene, o-xylene, p-xylene, 123-trimethylbenzene, 124-trimethylbenzene, 135-trimethylbenzene) were chosen to study their photochemical oxidation behavior in an indoor smog chamber system to compare the formation activity in O3 and SOA. The influences of NOx, AH concentration, and AH substituent on the formation kinetics of O3 and SOA were studied in detail. All volatile intermediates were qualitatively and quantitatively analyzed online to propose their potential contribution to the formation of O3 and SOA. The relationship between AH structure, intermediates, and production of O3 and SOA was established to reveal the transformation mechanisms of AHs to O3 and SOA. The results of this work will further elucidate the photochemical behavior of AHs in the atmosphere and provide reliable experimental data for modeling and prediction in the future.
2.1 Photochemical oxidation experiment
All experiments were conducted in a GDUT-DRC dual-reactor chamber with two 2 m3 pillow-shaped reactors; a detailed description of the reactor is reported in our earlier work (Luo et al., 2020b). The experimental relative humidity (RH) and temperature were set to < 5 % and around 303 ± 1 K, respectively. No inorganic seed aerosol was supplied in this work. A total of 60 black lamps (40 W, F40BL, GE, USA) were equipped to provide the UV light source, and the light intensity in the dual reactor was determined to be 0.161 min−1 (left) and 0.169 min−1 (right) using the NO2 photolysis rate constant (Luo et al., 2020b). The center of the UV lamp wavelength was 360 nm.
Nine AHs (benzene, 99.5 %; toluene, 99.0 %; ethylbenzene, 99.8 %; m-xylene, 99.0 %; o-xylene, 99.0 %; p-xylene, 99.0 %; 1,2,3-trimethylbenzene (123-TMB), 90.0 %; 1,2,4-trimethylbenzene (124-TMB), 98.0 %; 1,3,5-trimethylbenzene (135-TMB), 97.0 %) purchased from Aladdin Industrial Co., Ltd. (USA) and a certain amount of NO2 were directly injected into the reactor to conduct the photochemical oxidation experiments. Before turning on the light, all AHs and NO2 were injected with the background gas flow and adjusted to stabilize for 1 h. Typical experimental conditions (e.g., concentrations of AHs and NOx, VOC NOx ratio, RH, and temperature) of this study for nine AHs are supplied in the Supplement in Table S1.
2.2 Organic gas measurement
The concentrations of AHs and their oxidation products were all measured online using a proton-transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS; Ionicon Analytik Inc., Austria). In the setting model, all gaseous organics with proton affinity greater than H2O, including AHs, hydrocarbons, acids, and carbonyl groups, were all measured quantitatively and qualitatively. The processing software TOF-DAQ (Tofwerk AG, Switzerland) recorded the material with ≤ 240, and the original signal strength was converted into parts per billion (ppb) concentration by a formula (Lindinger et al., 1998). Before sampling and measurement, PTR-ToF-MS was calibrated once a week throughout the measurement period (Han et al., 2019). The detection limit of PTR-ToF-MS was < 20 ppt for 79 and < 10 ppt for 181 averaged over 1 min. Detailed parameters of the instrument and the yield calculation formula of products are given in the Supplement.
2.3 Inorganic gas measurement
The real-time concentrations of NO, NO2, and NOx were all spontaneously monitored with an NOx analyzer (Model 42i, Thermo Scientific Inc., USA), and the real-time concentration of O3 was monitored with an O3 analyzer (Model 49i, Thermo Scientific Inc., USA). The detection limits of the NOx and O3 analyzers were < 0.4 ppb averaged over 1 min and 0.5 ppb, respectively. All devices were calibrated weekly using a gas calibrator (Model 146i, Thermo Scientific Inc., USA).
2.4 Particle measurement
Particle size distribution was measured by a scanning mobility particle sizer spectrometer (SMPS; TSI Inc., USA) equipped with electrostatic classifiers (ECs; Model 3082, TSI Inc., USA) and a long differential mobility analyzer (DMA) (Model 3081, TSI Inc., USA) or an optional nano-DMA (Model 3085, TSI Inc., USA) and a condensation particle counter (CPC; Model 3776, TSI Inc., USA). The velocities of sheath gas and aerosol flows were set at 3.0 and 0.3 L min−1, respectively. Under this setting, the particle size range was observed from 13.8 to 723.4 nm. The yield calculation formula of SOA is given in the Supplement.
3.1 Formation kinetics and mechanisms of O3 and SOA without NOx
The direct photochemical oxidation of nine AHs was first conducted to evaluate the formation potential of O3 and SOA. No SOA was detected within 480 min reaction duration. The O3 concentration increased steadily from 0 to 16 ppb for 135-TMB and 28 ppb for ethylbenzene within 420 min (Fig. 1a). The peak concentration of O3 for these nine AHs followed the trend of (ethylbenzene, toluene, benzene, 23–28 ppb) > (m-, o-, p-xylene, 18–21 ppb) > (123-, 124-, 135-TMB, 16–20 ppb). Clearly, a lower number of AH substituents resulted in the generation of higher concentrated O3. This may be because the substituent number of AHs determined their reactivity, while the reaction rate constants with •OH could be applied to evaluate the reactivity. Previous studies indicated that the rate constants of AHs with •OH were found to follow the order of toluene (5.61 ± 0.08) × 10−12 cm3 molecule−1 s−1 < xylene (7.4–14) × 10−12 cm3 molecule−1 s−1 < TMB (14–31) × 10−12 cm3 molecule−1 s−1 at 304 ± 1 K (Doyle et al., 1975; Anderson and Hites, 1996). Combining this with our results, it was solidly concluded that AHs with a lower number of substituents showed lower reactivity, which resulted in the formation of higher concentrated O3.
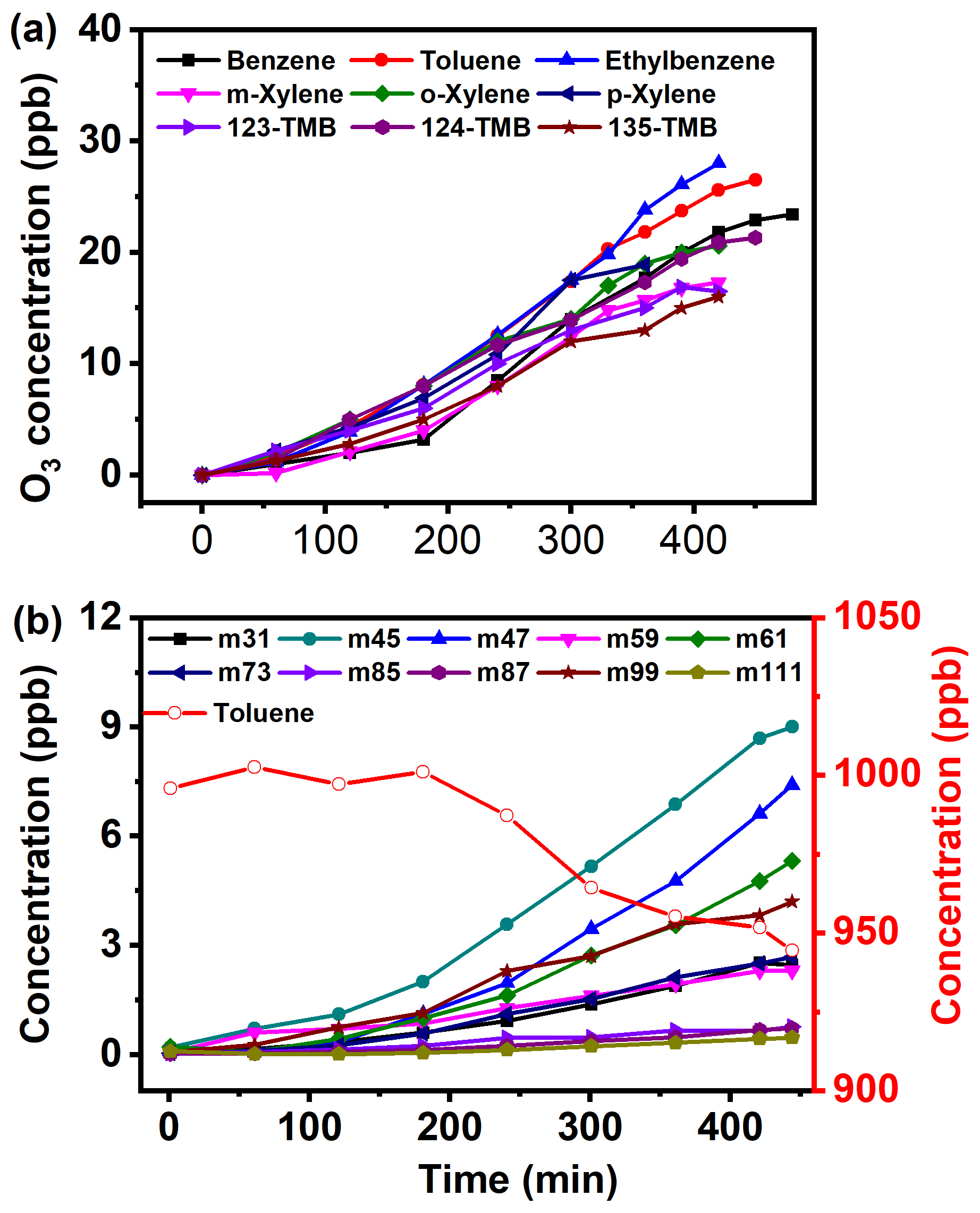
Figure 1(a) O3 formation curve from AH photochemical oxidation (2110 ppb of benzene, 996 ppb of toluene, 1060 ppb of ethylbenzene, 889 ppb of m-xylene, 1160 ppb of o-xylene, 1040 ppb of p-xylene, 824 ppb of 123-TMB, 935 ppb of 124-TMB, 913 ppb of 135-TMB) without NOx. (b) The concentration variation of intermediates formed from 996 ppb of toluene photochemical oxidation without NOx.
Furthermore, to figure out the formation mechanisms of O3 from direct AH oxidation, the corresponding volatile intermediates were also monitored. Toluene was taken as an example to illustrate the concentration variation of volatile intermediates. As Fig. 1b shows, with a decrease of toluene's concentration from 996 to 944.5 ppb, the concentrations of nine intermediates increased at different degrees. The concentrations of = 45 (m45, acetaldehyde), = 47 (m47, formic acid), and = 61 (m61, acetic acid or glycolaldehyde) increased faster than others and peaked at 5–9 ppb within 450 min, indicating easy oxidation of toluene to small molecular carbonyl products. The peak concentration of = 99 (m99, 3-methyl-2(5H)-furanone or 4-keto-2-pentenal) reached 4.2 ppb, while the production of = 31 (m31, formaldehyde), = 59 (m59, glyoxal), = 73 (m73, methylglyoxal) was at the same level (ca. 2.4 ppb). The concentrations of some intermediates including 85 (m85, 2-butenedial), 87 (m87, butanedione), and 111 (m111, hexa-2,4-dienedial) were lower than 0.8 ppb within 450 min reaction duration. Similar variation trends of volatile intermediates were observed for the other eight AHs (Fig. S1 in the Supplement).
It is worth mentioning that most of the above intermediates are well-known precursors of O3 and SOA (Li et al., 2016; Ji et al., 2017; Nishino et al., 2010). However, the formation of SOA was not observed in this study. Two reasons might be involved. Since this study was carried out at low RH (< 5 %) and without seed particles, no SOA precursor oligomers existed. Furthermore, the concentrations of intermediates produced were too low to trigger the initial nucleation reaction and then generate SOA under low RH conditions. Therefore, SOA formation could not be observed in the NOx-free photochemical oxidation of these nine AHs. In general, tropospheric O3 is mainly from NO2 photolysis, and the existence of AHs could enhance O3 formation. However, with the absence of NOx in this study, the low concentrated O3 was observed from AH photochemical oxidation. The possible contributors of these O3 might be intermediates such as carbonyl compounds. In all, our results indicated that direct photochemical transformation of AHs to O3 actually occurred and should be taken into consideration in the atmospheric environment.
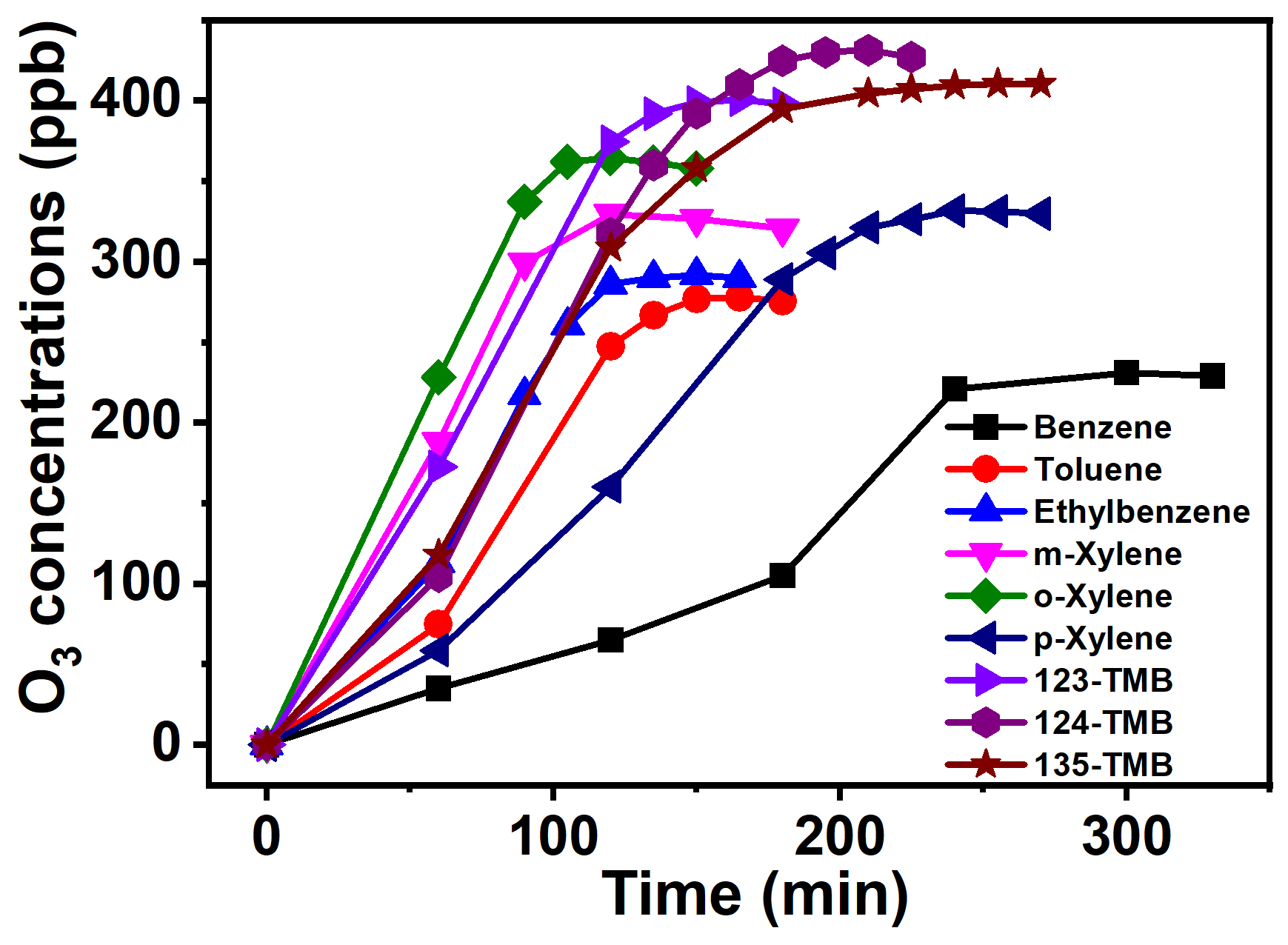
Figure 2O3 formation curve from AH photochemical oxidation with the presence of NOx (2000 ppb of benzene and 160.2 ppb of NOx, 1048 ppb of toluene and 162.0 ppb of NOx, 1050 ppb of ethylbenzene and 162.4 ppb of NOx, 889 ppb of m-xylene and 172.1 ppb of NOx, 1052 ppb of o-xylene and 159.8 ppb of NOx, 1040 ppb of p-xylene and 157.2 ppb of NOx, 956 ppb of 123-TMB and 171.4 ppb of NOx, 1010 ppb of 124-TMB and 169.5 ppb of NOx, 1040 ppb of 135-TMB and 164.2 ppb of NOx).
3.2 Formation kinetics and mechanisms of O3 in the presence of NOx
To further explore the role of NOx in O3 formation during photochemical oxidation of AHs, about 160 ± 10 ppb of NO2 was added into the reactor. The concentration of NO2 was selected based on previous works (Luo et al., 2019; Chen et al., 2018). Under this condition, the generated O3 was found to significantly increase, and the O3 peak concentrations ranged from 230 to 440 ppb within 100 to 250 min (Fig. 2). All these data were about 200–400 ppb higher than those obtained in the absence of NOx (Fig. 1a), indicating the quick enhancement of NOx to O3 formation. In this work, the added NO2 was firstly photolyzed under 360 nm light irradiation to form NO and O(3P) (Eq. 1). Then, the latter was oxidized to form O3 (Eq. 2), which further reacted with NO to form NO2 (Eq. 3). Meanwhile, AHs were photochemically oxidized to form RO2 and HO2, both of which then reacted with NO to form NO2 (Eqs. 4 and 5). Clearly, the presence of AHs could compete with O3 for the NO reaction and reduce the consumption of O3. The synergetic effect of direct NO2 conversion and AH competition reaction led to the boosting of the formation of O3 in the presence of both AHs and NOx.
Furthermore, the effect of AH content on the O3 formation in the presence of NOx (e.g., VOC NOx ratio) was investigated. Here, the concentration of NOx was maintained constantly and that of AH was gradually increased. For toluene (Fig. 3a), O3 peak concentration of 250 ppb was achieved after reaction for 420 min under a VOC NOx ratio of 2.47. When increasing the VOC NOx ratio to 6.29, the time needed to achieve a higher peak O3 concentration of 280 ppb was shortened to 150 min. All these data confirmed that O3 formation rate and concentration were both accelerated with increased AH concentration. Similar results of shorter reaction time leading to higher O3 concentration were observed for the rest of the AHs (Fig. S2). Increasing AH concentration would result in the enhanced formation of RO2 and HO2, both of which reacted with NO to save the O3 consumption. Meanwhile, the photolysis of NO2 to form NO and then O3 was also accelerated. Both of these reasons were responsible for the fast-enhanced formation of O3 with the increased AH concentration in the VOC NOx ratio.
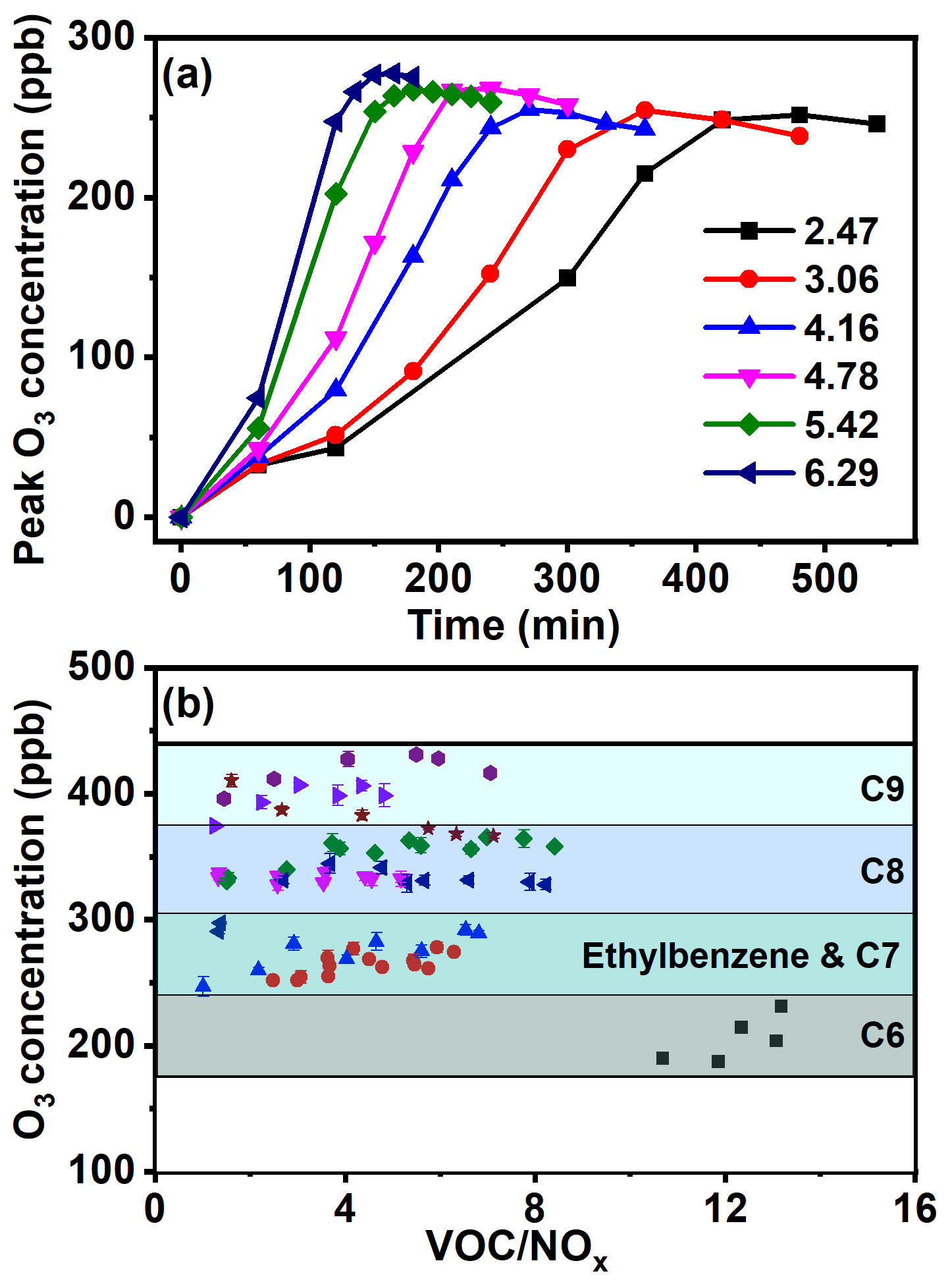
Figure 3(a) O3 formation curve of toluene photochemical oxidation at different VOC NOx ratios and (b) the changing trend of the peak O3 generated by photochemical oxidation of AHs at different VOC NOx ratios (square: benzene; circle: toluene; upper triangle: ethylbenzene; lower triangle: m-xylene; diamond: o-xylene; left triangle: p-xylene; right triangle: 123-TMB; hexagon: 124-TMB; pentacle: 135-TMB).
To study the effect of AH substituents on O3 formation, the O3 peak concentrations of nine AHs obtained at the same VOC NOx ratio were compared. As Fig. 3b shows, the O3 peak concentrations of nine AHs followed the order of TMB (366.4–431.2 ppb) > xylene (290.6–365.7 ppb) > toluene and ethylbenzene (246.7–291.7 ppb) > benzene (187.3–231.2 ppb). Clearly, the O3 peak value was positively correlated with the number of AH substituents, suggesting AHs with more substituents possessed higher O3 production potential at the same VOC NOx ratio. In previous studies, O3 concentrations from AH oxidation with the presence of NOx were reported as follows: 160–300 ppb for toluene, 400 ppb for m-xylene, and 340–470 ppb for 123-TMB (Luo et al., 2019; Li et al., 2018; Xu et al., 2015). However, these studies only focused on one or several AHs, and the relationship between AH substituents and O3 formation was still not understood. Meanwhile, the VOC NOx ratio ranging from 1.0 to 13.0 was selected for its effect on O3 formation. The range of VOC NOx ratio in the above research studies was close to that in our study (Table S2). Then, our results of O3 concentration were comparable to those in the previous studies under a similar range of the VOC NOx ratio. Furthermore, the results obtained in this study clearly confirmed that increasing the substituent number of AHs correspondingly increased O3 concentration. It was also noticed that the O3 peak concentrations of xylene or TMB isomers were in the same range, suggesting the negligible effect of substituent position of AHs on their O3 formation.
3.3 Accelerated formation of SOA in the presence of NOx
Besides O3, the effect of AH concentration on the formation kinetics of SOA with the presence of NOx was also investigated. As Fig. 4 shows, from the photochemical oxidation of toluene, the peak number concentration of SOA increased from 2.0 × 104 to 5.5 × 104 particle cm−3 with an increase of the VOC NOx ratio from 2.37 to 5.58. The time to achieve the above concentration was shortened from 250 to 120 min, while the median particle size range also increased from 300–400 to 400–500 nm. Similar results of shorter time leading to higher concentration and larger particle size for SOA were observed for the other eight AHs with the increasing VOC NOx ratio (Figs. S3–S10).
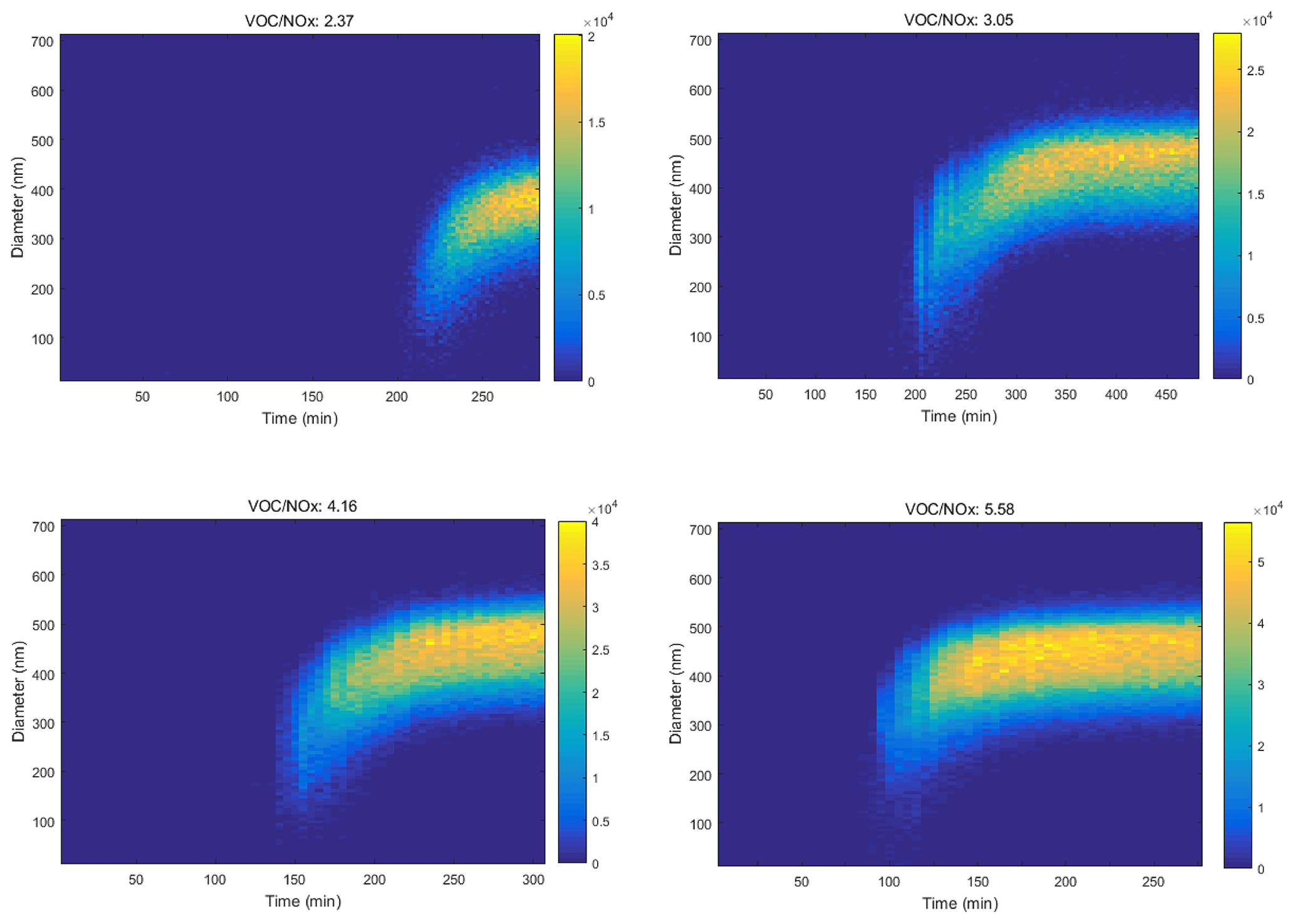
Figure 4Nanoparticle distribution from toluene photochemical oxidation varied with time at different VOC NOx ratios (the initial concentrations of NOx were in the range of 158.2 to 179.7 ppb).
Previous studies reported the enhanced SOA yield with increased NOx concentration (Zhao et al., 2018; Hurley et al., 2001; Song et al., 2007; Sarrafzadeh et al., 2016). This was because NOx mainly influenced the distribution of oxidation products by affecting the RO2 reaction equilibrium, where RO2 easily converted to low-volatility ROOH or ROOR and thus resulted in the nucleation of new particles (Sarrafzadeh et al., 2016). However, in this study, we kept the NOx concentration unchanged and modified the initial concentration of AHs. The increased AHs could lead to promoted RO2 formation, resulting in more low-volatility products' formation. The accumulation of low-volatility products promoted the nucleation of particulate matter and finally increased the yield of SOA.
The particle number and mass concentrations of SOA generated from nine AHs were further compared to evaluate the effect of AH substituents on the SOA formation. The particle number concentration of SOA was obtained at the endpoint of each reaction. With the increase of substituent number, the number concentration of SOA decreased (e.g., from 6.9 × 103 particle m−3 for 135-TMB to 7.8 × 104 particle m−3 for toluene) (Fig. 5a). With the progress of the reaction, the mass concentration of SOA increased, and the increase of substituent number shortened the time achieving the peak mass concentration (Fig. 5b). These results revealed that the increase of substituent number of AHs increased SOA mass concentration but decreased its particle number. AHs with different substituent position also showed different SOA formation characteristics. For xylene, the peak mass concentration of o-xylene (88.6 µg m−3) was higher than that of m-xylene and p-xylene, while the peak mass concentration of 123-TMB (82.0 µg m−3) was significantly higher than that of 124-TMB (31.8 µg m−3) and 135-TMB (27.6 µg m−3) (Fig. 5b). These phenomena indicated that xylene and TMB with ortho-methyl substituents facilitated SOA formation. Further, the ortho-methyl group of isomers (e.g., o-xylene, 123-TMB) could more thoroughly be oxidized, producing more particles (Sato et al., 2010).
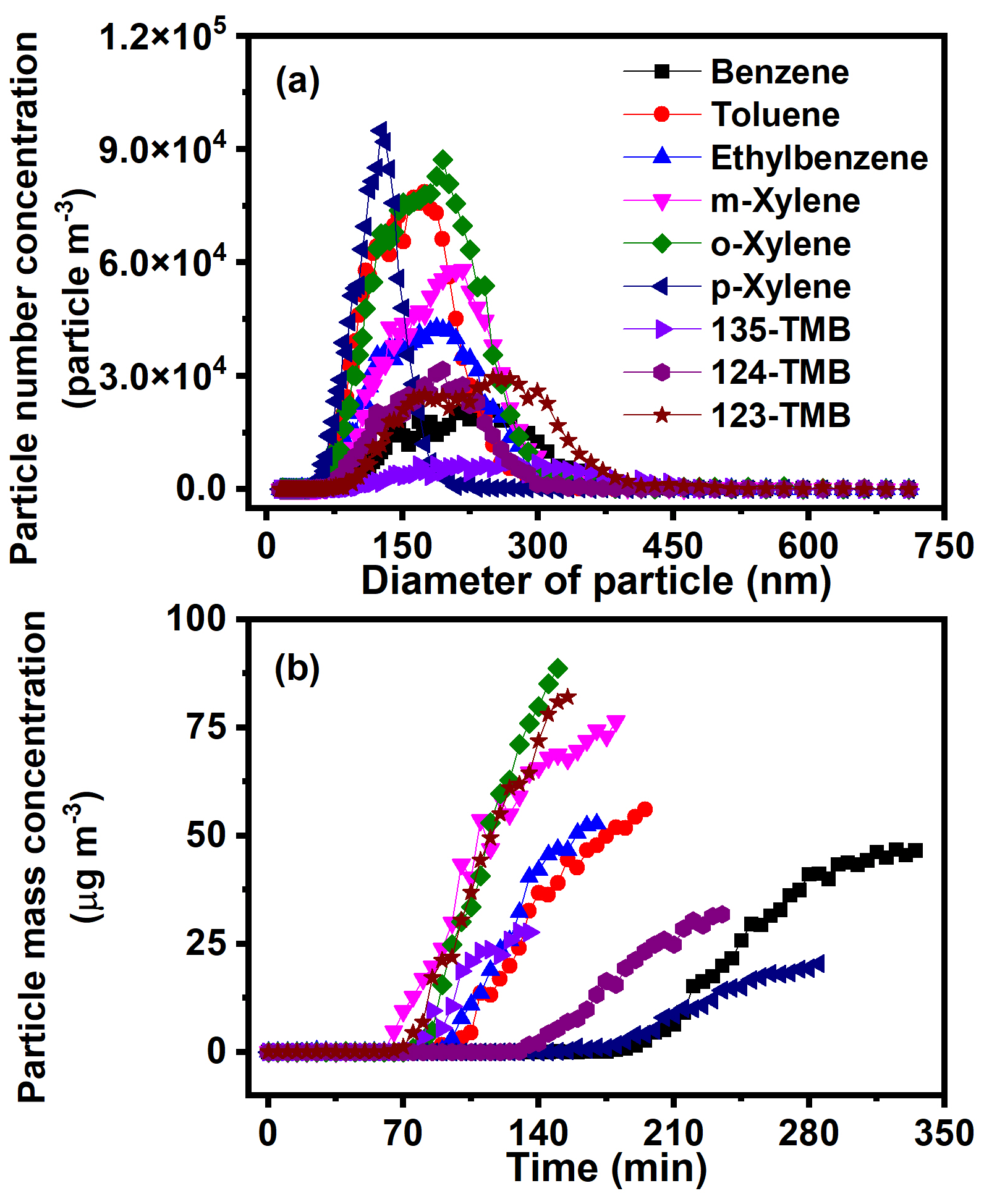
Figure 5(a) Number concentration and (b) mass concentration of SOA produced from AH photochemical oxidation with the presence of NOx (2000 ppb of benzene and 160.2 ppb of NOx, 1048 ppb of toluene and 162.0 ppb of NOx, 1050 ppb of ethylbenzene and 162.4 ppb of NOx, 889 ppb of m-xylene and 172.1 ppb of NOx, 1052 ppb of o-xylene and 159.8 ppb of NOx, 1040 ppb of p-xylene and 157.2 ppb of NOx, 956 ppb of 123-TMB and 171.4 ppb of NOx, 1010 ppb of 124-TMB and 169.5 ppb of NOx, 1040 ppb of 135-TMB and 164.2 ppb of NOx).
It has also been reported that seed particles (e.g., NaCl) and highly relative humidity (up to 90 %) can significantly increase the yield of SOA (Wang et al., 2016; Luo et al., 2019; Jia and Xu, 2018). However, in this study, the maximum SOA yield of 25 % (Fig. S11) was produced with increasing AH concentration, which was consistent with that from previous research studies (Sato et al., 2012; Li et al., 2016; Song et al., 2007; Odum et al., 1997). Further considering the oxidation conditions of low RH (less than 5 %) and seedless particles in this study, our results indicated that AH concentration should also be paid much attention in relation to SOA formation, although the addition of NOx, seed particles, and high RH are also all very important.
To further investigate the effect of AH substituents on SOA yield, a two-product semi-empirical model was employed. As Fig. S12 shows, the model fitted the SOA yield of nine AHs well, and the corresponding fitting parameters are listed in Table 1. Similarly, high-volatility components were assumed from the photochemical oxidation of AHs and the same Kom,2 of 0.005 m3 µg−1 was assigned. As seen from the table, benzene (0.242 m3 µg−1), toluene (0.162 m3 µg−1), and ethylbenzene (0.422 m3 µg−1) showed higher α2 than that of xylenes (e.g., 0.086 m3 µg−1 for m-xylene) and TMBs (e.g., 0.082 m3 µg−1 for 123-TMB), indicating the production of more high-volatility products from AHs with a lower number of substituents. Meanwhile, benzene (0.022 m3 µg−1), toluene (0.027 m3 µg−1), and ethylbenzene (0.023 m3 µg−1) displayed lower Kom,1 in comparison with xylenes (e.g., 0.074 m3 µg−1 for p-xylene) and TMBs (e.g., 0.085 m3 µg−1 for 135-TMB), and the corresponding α1 decreased with increasing substituent number. All these results demonstrated that the increase of substituent number on the phenyl ring inhibited the generation of low-volatility products, thus reducing the generation of SOA particles and finally leading to the decrease of SOA yield. The results also indicated that the oxidation degree became lower and lower for AHs with increased substituent number, since the oxidation of methyl carbon was more difficult than that of carbon of the phenyl ring. Li et al. (2016) reported a similar phenomenon previously, which is consistent with our results. However, they did not further investigate the relationship of isomer AHs with the SOA yield.
Table 1Fitting yield curve parameters of two-product semi-empirical models.
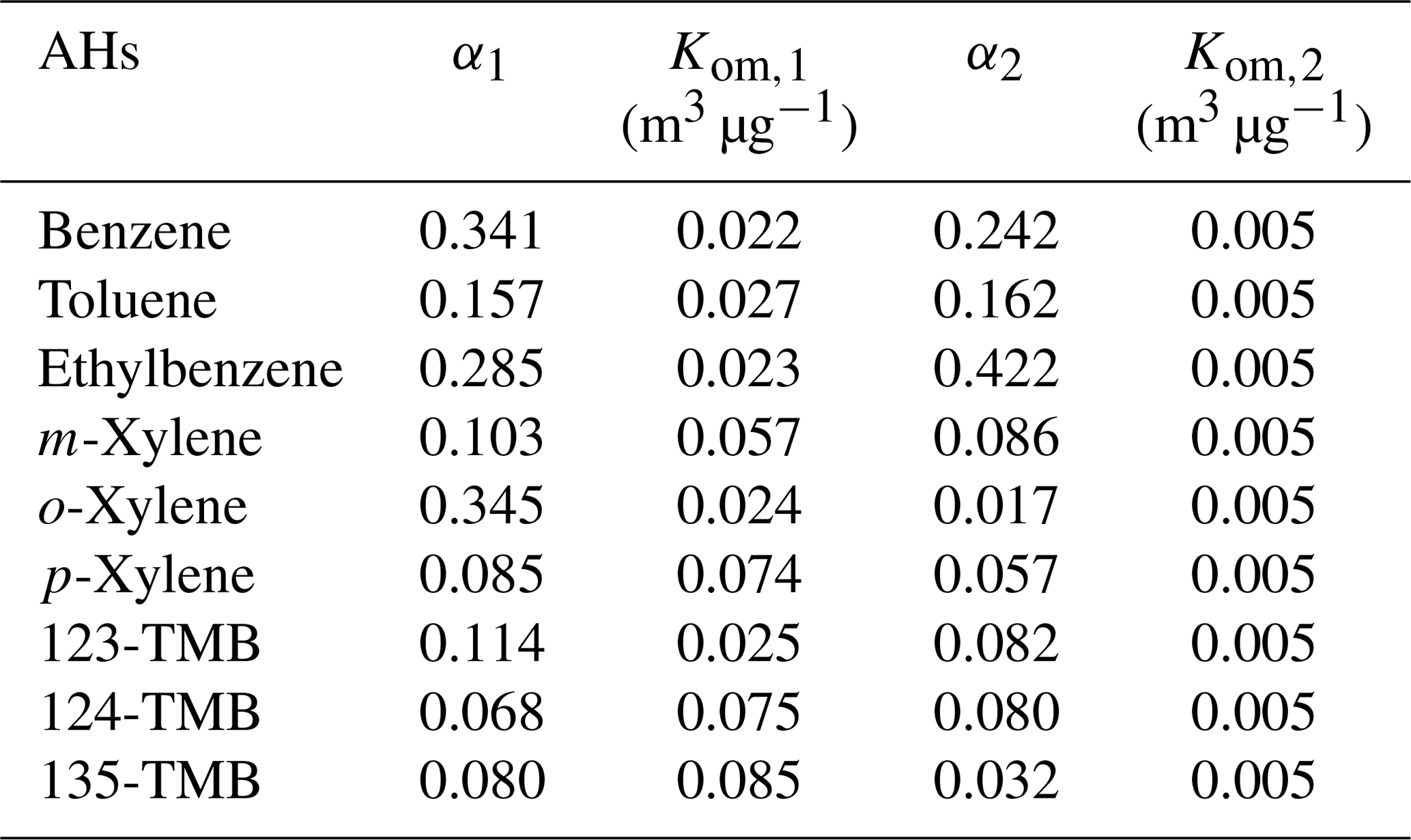
In the present study, SOA yield of o-xylene was found higher than that of m-xylene and p-xylene, consistent with the SOA number and mass results. The fitting results showed that the Kom,1 of o-xylene (0.024 m3 µg−1) was much lower than that of m-xylene (0.057 m3 µg−1) and p-xylene (0.074 m3 µg−1), indicating the production of more low-volatility products from o-xylene. Similarly, 123-TMB showed the highest SOA yield and lowest Kom,1 among three TMBs. These results further confirmed that AHs with an ortho-methyl substituent favored the yield of SOA. This might be because these AHs were more susceptible to being oxidized and forming ring-opening products and finally producing more RO2 than other isomers. Some previous studies have also obtained results consistent with this (Zhou et al., 2011; Song et al., 2007). In addition, ethylbenzene was also isomeric to xylene, and its SOA yield was higher than that of xylenes. Recent studies have found that the SOA yield during the oxidation of alkanes and alkenes by •OH increased with the carbon chain length (Lim and Ziemann, 2009; Tkacik et al., 2012). Clearly, the length of carbon chain also affected the oxidation degree of AHs. Then, the longer ethyl group led to a higher degree of photochemical oxidation for ethylbenzene than xylenes, promoting the formation of more SOA precursors and finally higher SOA yield.
3.4 Enhanced formation mechanisms of SOA with NOx
In order to further reveal the enhanced formation mechanisms of SOA from the oxidation of AHs with the presence of NOx, the corresponding volatile intermediates were all identified and quantified comparably. As Fig. 6a shows, with a gradual decrease of toluene concentration, the concentrations of small molecule carbonyl products, such as m31, m45, m47, and m61, quickly increased to 16.0, 45.3, 31.0, and 17.0 ppb within 200 min. Acetaldehyde showed the highest concentration, which was by far higher than that obtained without NOx (9 ppb). The ones that followed were m85, m87, and m111, with a peak concentration below 2.8 ppb. The increase of concentration of m59, m73, and m99 began to slow down after 120 min, and this trend was consistent with the trend of SOA formation (Fig. 5b). A similar variation trend of volatile intermediates for other AHs was also measured (Fig. S13). All these results demonstrated that there was a specific window period, and the intermediates in the gaseous phase were transformed into the particulate phase. The significant increase of SOA occurred after breaking through the window period.
.
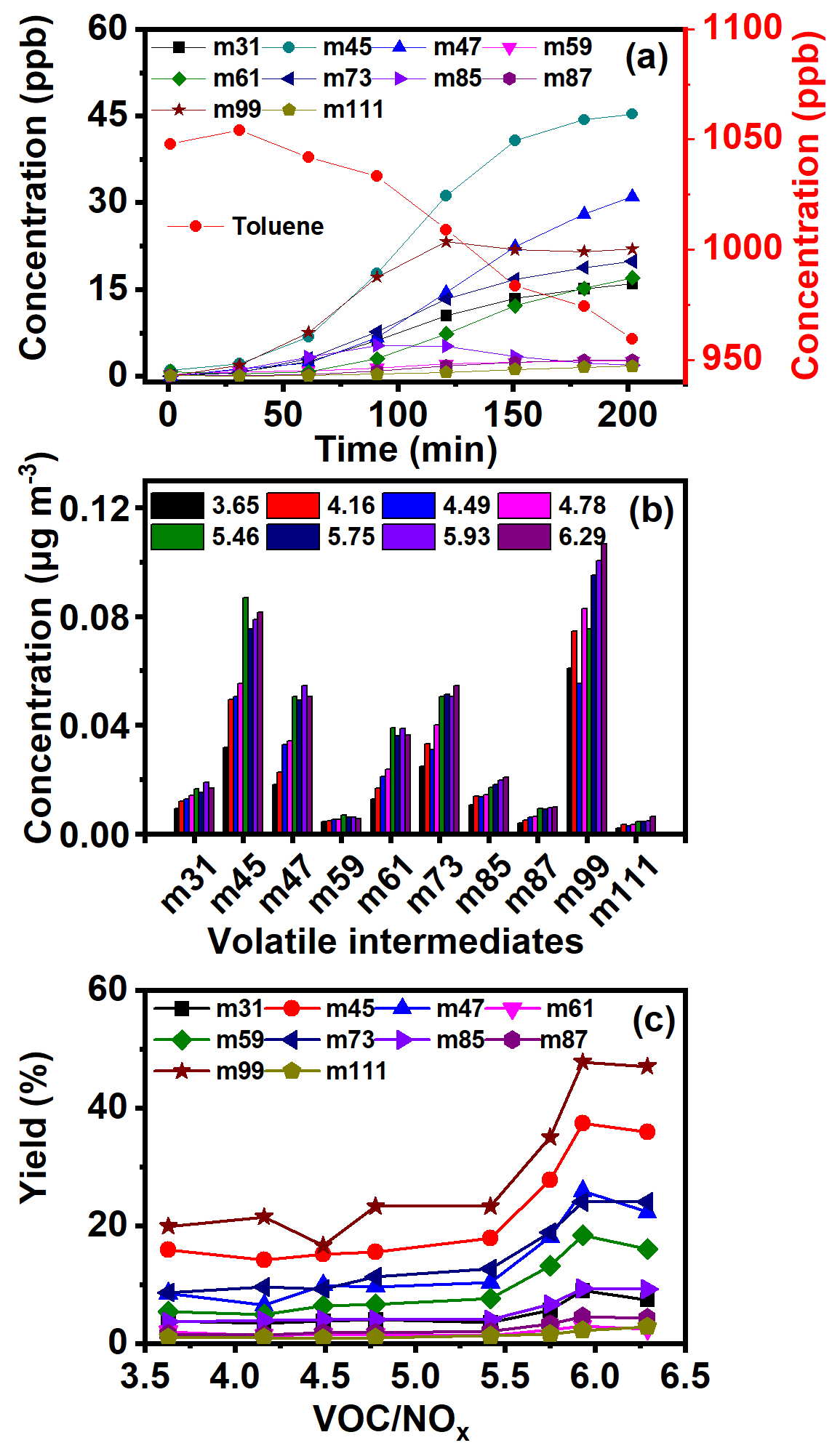
Figure 6(a) Intermediate concentration variation along with the reaction time under 1048 ppb of toluene and 162.0 ppb of NOx, (b) intermediate concentration, and (c) yield from toluene photochemical oxidation with the increase of the VOC NOx ratio
Further comparison of volatile intermediates during photochemical oxidation of AHs with different VOC NOx ratios was also carried out. For toluene oxidation (Fig. 6b), the concentrations of all intermediates increased with the increase of the VOC NOx ratio. The carbonyl intermediates such as m59 and m73 were believed to play an important role in the photochemical oxidation of AHs to form SOA (Li et al., 2016; Ji et al., 2017; Nishino et al., 2010). Bloss et al. (2005b) also found glyoxal and methylglyoxal produced from toluene photochemical oxidation to be the main precursor of SOA. In previous studies, maximum yields of glyoxal and methylglyoxal of 20 % and 17 % were obtained during toluene photochemical oxidation (Baltaretu et al., 2009; Volkamer et al., 2001; Nishino et al., 2010), which were lower than those of the present study (24 % in Fig. 6c). Meanwhile, the yields of glyoxal and methylglyoxal during photochemical oxidation of toluene, xylenes, and TMBs increased with increasing AH concentration. Therefore, the increase of AH content in the reaction system promoted the photochemical oxidation of AHs to produce more volatile carbonyl intermediates, finally leading to higher SOA yield in this study. Moreover, the yield of m59 and m73 from benzene photochemical oxidation was found to be the lowest among all AHs (Figs. 6b, c and S14–S22), indicating that the presence of the branch chain on the phenyl ring did not favor the production of unsaturated carbonyl compounds. This was because the increasing methyl group number of AHs weakened their oxidation reactivity, resulting in the inhibition of the ring-opening reaction (Li et al., 2016). Further, the formation of glyoxal and methylglyoxal from RO2 was also subsequently suppressed. Furthermore, the yields of m85 from AHs containing an ortho-methyl group (e.g., o-xylene, 123-TMB, and 124-TMB) were found higher to be than those of their isomers, due to ortho-methyl groups of the phenyl ring preferring the ring-opening reaction and the subsequent generation of ketone products (Li et al., 2016).
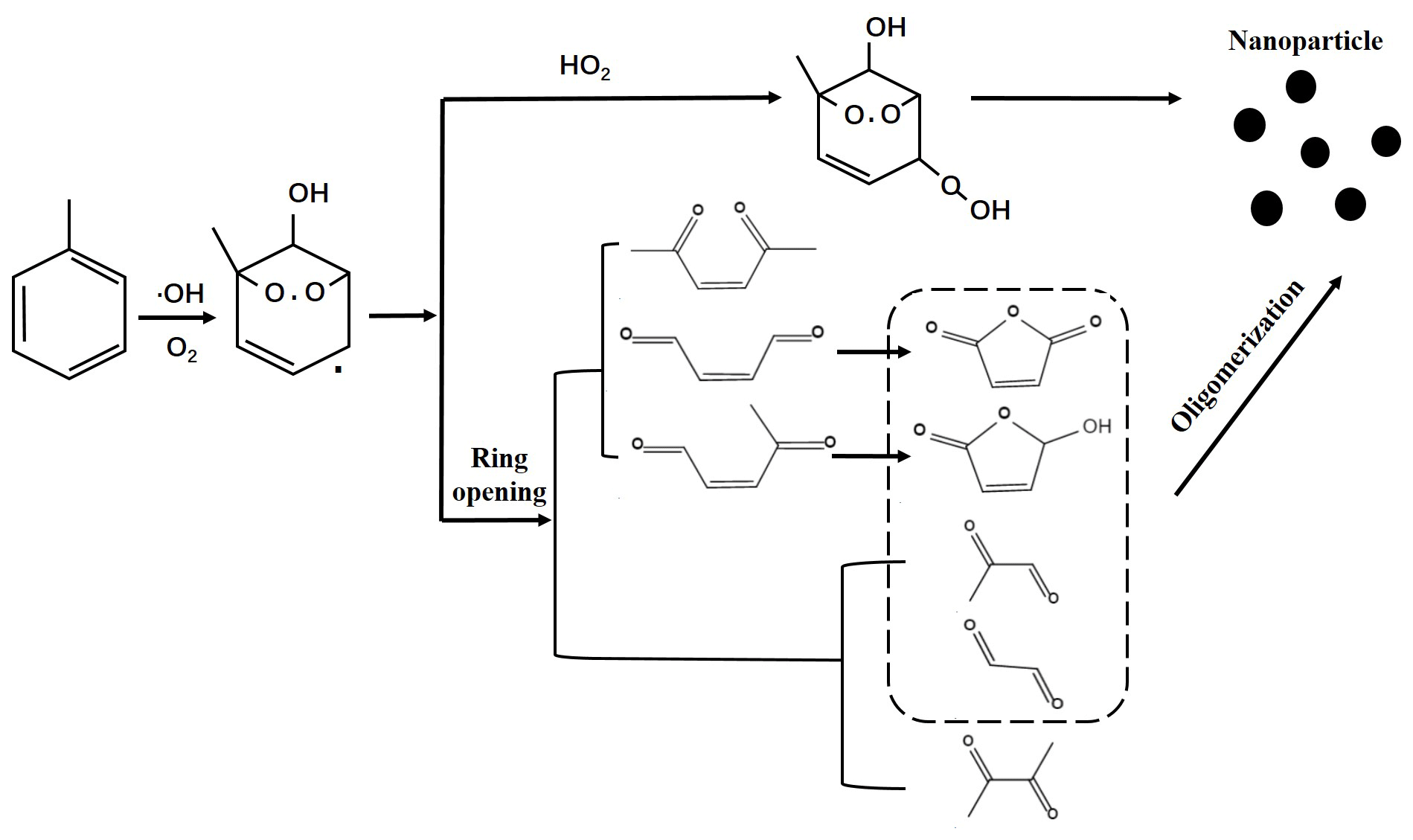
Based on the above results, the possible photochemical oxidation mechanism from AH to SOA was proposed. Toluene was selected as an example. As Fig. 7 shows, the phenyl ring of toluene firstly reacted with •OH to produce cresol (Ziemann, 2011; Atkinson, 2007) and then further oxidized to form an intermediate, which could cause a ring-opening reaction or react with HO2 radicals to form bicyclic peroxide compounds. The latter has been suggested as an important SOA precursor of AH photochemical oxidation (Song et al., 2005; Wyche et al., 2009; Nakao et al., 2011; Johnson et al., 2005). The ring-opening intermediates were consisted of saturated and unsaturated dicarbonyl compounds (Jang and Kamens, 2001; Birdsall and Elrod, 2011). However, the possibility of these dicarbonyl intermediates directly partitioning into the particulate phase was very small (Jang and Kamens, 2001), but they could oligomerize to form low-volatility compounds (Forstner et al., 1997; Jang and Kamens, 2001). The oligomerization was an important pathway for SOA formation from AH photochemical oxidation (Sato et al., 2012; Li et al., 2016; Hu et al., 2007). In our study, the detection of m85 and m99 proved the formation of ring-opening products. These unsaturated 1,4-dicarbonyls were observed to form small cyclic furanone compounds (Jang and Kamens, 2001; Bloss et al., 2005b). Therefore, the ring-opening products with saturated or unsaturated dicarbonyl groups finally transformed into SOA through the oligomerization process.
As mentioned above, with the increase of the substituent number on AHs, the yield of SOA decreased. The enhanced ring-opening products and restrained oligomerization reactions by the increased methyl group number were supposed to be the main cause. The methyl group was found to stabilize the ring-opening radicals (Ziemann, 2011). When the phenyl ring contained a methyl group, the oxidation pathway was prone to ring-opening. The concentrations of m87 and m111 increased with the increase of the methyl group number (Fig. S23), meaning that these two intermediates were dominant in the ring-opening products. However, they could not oligomerize to further partition into SOA formation (Fig. 7). Both non-cyclic dicarbonyls and cyclic compounds formed by unsaturated dicarbonyls were deemed to have a small probability of oligomerizing (Li et al., 2016; Kalberer et al., 2004). In previous study, no m85 and m111 were detected in particulate-phase SOA under 300 ± 1 K and dry conditions (RH < 0.1 %) in the absence of inorganic seed aerosol (Li et al., 2016). However, m85 was measured in the gas phase of this study, indicating that it was not the precursor of SOA for the oligomerization reaction. The presence of methyl groups would inhibit the oligomerization to prevent the formation of ring compounds by unsaturated dicarbonyl groups and finally decrease SOA formation.
In this study, no SOA formation was observed from the direct photochemical oxidation of AHs, while a small amount of O3 was produced without NOx addition. The presence of NOx significantly increased the production of O3 and SOA, due to the synergetic effect of accelerated NO2 conversion and AH reaction with NO as well as enhanced formation of volatile intermediates. Further increased formation of both O3 and SOA was observed by promoted AH concentration. In addition, an increase of AH substituent number could enhance O3 formation but decrease SOA yield. The ortho-methyl-group-substituted AHs exhibited a higher SOA yield. The preferential formation of variation of dicarbonyl intermediates and restrained oligomerization reaction were responsible for the above differences. These results showed more clear understanding of the effect of NOx and organic molecule structures on photochemical oxidation of AHs to form O3 and SOA, which could provide a solid experimental basis for studying the transformation of AHs to secondary pollutants in the real atmospheric environment.
The code used in this study is available upon request from Hao Luo (luohao6@foxmail.com).
The data that support the results are available from the corresponding author upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-7567-2021-supplement.
TA designed research. HL and JC conducted experiments. HL, JC, and GL analyzed data. HL prepared the manuscript with contributions from all coauthors.
The authors declare that they have no conflict of interest.
This research has been supported by the National Natural Science Foundation of China (grant nos. 42020104001 and 41731279), the Local Innovative and Research Team Project of Guangdong Pearl River Talents Program (2017BT01Z032), the Guangdong Provincial Key Research and Development Program (2019B110206002), and the National Key R&D Program of China (2019YFC0214402).
This paper was edited by Jianping Huang and reviewed by two anonymous referees.
An, T. C., Huang, Y., Li, G. Y., He, Z. G., Chen, J. Y., and Zhang, C. S.: Pollution profiles and health risk assessment of VOCs emitted during e-waste dismantling processes associated with different dismantling methods, Environ. Int., 73, 186–194, https://doi.org/10.1016/j.envint.2014.07.019, 2014.
Anderson, P. N. and Hites, R. A.: OH radical reactions: the major removal pathway for polychlorinated biphenyls from the atmosphere, Environ. Sci. Technol., 30, 1756–1763, 1996.
Aschmann, S. M., Arey, J., and Atkinson, R.: Rate constants for the reactions of OH radicals with 1,2,4,5-tetramethylbenzene, pentamethylbenzene, 2,4,5-trimethylbenzaldehyde, 2,4,5-trimethylphenol, and 3-methyl-3-hexene-2,5-dione and products of OH + 1,2,4,5-tetramethylbenzene, J. Phys. Chem. A, 117, 2556–2568, https://doi.org/10.1021/jp400323n, 2013.
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/s1352-2310(03)00391-1, 2003.
Baltaretu, C. O., Lichtman, E. I., Hadler, A. B., and Elrod, M. J.: Primary atmospheric oxidation mechanism for toluene, J. Phys. Chem. A, 113, 221–230, 2009.
Birdsall, A. W. and Elrod, M. J.: Comprehensive NO-dependent study of the products of the oxidation of atmospherically relevant aromatic compounds, J. Phys. Chem. A, 115, 5397–5407, https://doi.org/10.1021/jp2010327, 2011.
Bloss, C., Wagner, V., Bonzanini, A., Jenkin, M. E., Wirtz, K., Martin-Reviejo, M., and Pilling, M. J.: Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data, Atmos. Chem. Phys., 5, 623–639, https://doi.org/10.5194/acp-5-623-2005, 2005a.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005b.
Borrás, E. and Tortajada-Genaro, L. A.: Secondary organic aerosol formation from the photo-oxidation of benzene, Atmos. Environ., 47, 154–163, https://doi.org/10.1016/j.atmosenv.2011.11.020, 2012.
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013.
Chen, J. Y., He, Z. G., Ji, Y. M., Li, G. Y., An, T. C., and Choi, W. Y.: (OH)-O-center dot radicals determined photocatalytic degradation mechanisms of gaseous styrene in TiO2 system under 254 nm versus 185 nm irradiation: Combined experimental and theoretical studies, Appl. Catal. B-Environ., 257, 117912, https://doi.org/10.1016/J.Apcatb.2019.117912, 2019.
Chen, J. Y., Yi, J. J., Ji, Y. M., Zhao, B. C., Ji, Y. P., Li, G. Y., and An, T. C.: Enhanced H-abstraction contribution for oxidation of xylenes via mineral particles: Implications for particulate matter formation and human health, Environ. Res., 186, 109568, https://doi.org/10.1016/J.Envres.2020.109568, 2020.
Chen, Y., Tong, S. R., Wang, J., Peng, C., Ge, M. F., Xie, X. F., and Sun, J.: Effect of titanium dioxide on secondary organic aerosol formation, Environ. Sci. Technol., 52, 11612–11620, https://doi.org/10.1021/acs.est.8b02466, 2018.
Cocker III, D. R., Mader, B. T., Kalberer, M., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas-particle partitioning of secondary organic aerosol: II. m-xylene and 1,3,5-trimethylbenzene photooxidation systems, Atmos. Environ., 35, 6073–6085, 2001.
Doyle, G. J., Lloyd, A. C., Darnall, K. R., Winer, A. M., and Pitts Jr., J. N.: Gas phase kinetic study of relative rates of reaction of selected aromatic compounds with hydroxyl radicals in an environmental chamber, Environ. Sci. Technol., 9, 237–241, 1975.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: molecular composition, Environ. Sci. Technol., 31, 1345–1358, 1997.
Geng, F., Tie, X., Xu, J., Zhou, G., Peng, L., Gao, W., Tang, X., and Zhao, C.: Characterizations of ozone, NOx, and VOCs measured in Shanghai, China, Atmos. Environ., 42, 6873–6883, https://doi.org/10.1016/j.atmosenv.2008.05.045, 2008.
Glasson, W. A. and Tuesday, C. S.: Hydrocarbon reactivities in the atmospheric photooxidation of nitric oxide, Environ. Sci. Technol., 4, 916–924, 1970.
Han, C., Liu, R., Luo, H., Li, G., Ma, S., Chen, J., and An, T.: Pollution profiles of volatile organic compounds from different urban functional areas in Guangzhou China based on GC/MS and PTR-TOF-MS: Atmospheric environmental implications, Atmos. Environ., 214, 116843, https://doi.org/10.1016/j.atmosenv.2019.116843, 2019.
He, Z. G., Li, G. Y., Chen, J. Y., Huang, Y., An, T. C., and Zhang, C. S.: Pollution characteristics and health risk assessment of volatile organic compounds emitted from different plastic solid waste recycling workshops, Environ. Int., 77, 85–94, https://doi.org/10.1016/j.envint.2015.01.004, 2015.
Henze, D. K., Seinfeld, J. H., Ng, N. L., Kroll, J. H., Fu, T.-M., Jacob, D. J., and Heald, C. L.: Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 8, 2405–2420, https://doi.org/10.5194/acp-8-2405-2008, 2008.
Hu, D., Tolocka, M., Li, Q., and Kamens, R. M.: A kinetic mechanism for predicting secondary organic aerosol formation from toluene oxidation in the presence of NOx and natural sunlight, Atmos. Environ., 41, 6478–6496, https://doi.org/10.1016/j.atmosenv.2007.04.025, 2007.
Hu, L., Millet, D. B., Baasandorj, M., Griffis, T. J., Travis, K. R., Tessum, C. W., Marshall, J. D., Reinhart, W. F., Mikoviny, T., Müller, M., Wisthaler, A., Graus, M., Warneke, C., and de Gouw, J.: Emissions of C6−C8 aromatic compounds in the United States: Constraints from tall tower and aircraft measurements, J. Geophys. Res.-Atmos., 120, 826–842, https://doi.org/10.1002/2014JD022627, 2015.
Hurley, M. D., Sokolov, O., Wallington, T. J., Takekawa, H., Karasawa, M., Klotz, B., Barnes, I., and Becker, K. H.: Organic aerosol formation during the atmospheric degradation of toluene, Environ. Sci. Technol., 35, 1358–1366, https://doi.org/10.1021/es0013733, 2001.
Jang, M. and Kamens, R. M.: Characterization of Secondary Aerosol from the Photooxidation of Toluene in the Presence of NOx and 1-Propene, Environ. Sci. Technol., 35, 3626–3639, https://doi.org/10.1021/es010676+, 2001.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Ji, Y., Zheng, J., Qin, D., Li, Y., Gao, Y., Yao, M., Chen, X., Li, G., An, T., and Zhang, R.: OH-Initiated Oxidation of Acetylacetone: Implications for Ozone and Secondary Organic Aerosol Formation, Environ. Sci. Technol., 52, 11169–11177, https://doi.org/10.1021/acs.est.8b03972, 2018.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Johnson, D., Jenkin, M. E., Wirtz, K., and Martin-Reviejo, M.: Simulating the formation of secondary organic aerosol from the photooxidation of toluene, Environ. Chem., 2, 35–48, https://doi.org/10.1071/en04069, 2005.
Kalberer, M., Paulsen, D., Sax, M., Steinbacher, M., Dommen, J., Prevot, A. S. H., Fisseha, R., Weingartner, E., Frankevich, V., and Zenobi, R.: Identification of polymers as major components of atmospheric organic aerosols, Science, 303, 1659–1662, 2004.
Lane, T. E., Donahue, N. M., and Pandis, S. N.: Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model, Atmos. Environ., 42, 7439–7451, https://doi.org/10.1016/j.atmosenv.2008.06.026, 2008.
Li, K., Chen, L., White, S. J., Yu, H., Wu, X., Gao, X., Azzi, M., and Cen, K.: Smog chamber study of the role of NH3 in new particle formation from photo-oxidation of aromatic hydrocarbons, Sci. Total Environ., 619–620, 927–937, https://doi.org/10.1016/j.scitotenv.2017.11.180, 2018.
Li, L., Tang, P., Nakao, S., Chen, C.-L., and Cocker III, D. R.: Role of methyl group number on SOA formation from monocyclic aromatic hydrocarbons photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 2255–2272, https://doi.org/10.5194/acp-16-2255-2016, 2016.
Li, Y., Lau, A. K. H., Fung, J. C. H., Zheng, J., and Liu, S.: Importance of NOx control for peak ozone reduction in the Pearl River Delta region, J. Geophys. Res.-Atmos., 118, 9428–9443, https://doi.org/10.1002/jgrd.50659, 2013.
Lim, Y. B. and Ziemann, P. J.: Effects of molecular structure on aerosol yields from OH radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of NOx, Environ. Sci. Technol., 43, 2328–2334, https://doi.org/10.1021/es803389s, 2009.
Lindinger, W., Hansel, A., and Jordan, A.: Proton-transfer-reaction mass spectrometry (PTR–MS): on-line monitoring of volatile organic compounds at pptv levels, Chem. Soc. Rev., 27, 347–354, 1998.
Luo, H., Jia, L., Wan, Q., An, T., and Wang, Y.: Role of liquid water in the formation of O3 and SOA particles from 1,2,3-trimethylbenzene, Atmos. Environ., 217, 116955, https://doi.org/10.1016/j.atmosenv.2019.116955, 2019.
Luo, H., Li, G., Chen, J., Ma, S., Wang, Y., and An, T.: Spatial and temporal distribution characteristics and ozone formation potentials of volatile organic compounds from three typical functional areas in China, Environ. Res., 183, 109141, https://doi.org/10.1016/j.envres.2020.109141, 2020a.
Luo, H., Li, G., Chen, J., Wang, Y., and An, T.: Reactor characterization and primary application of a state of art dual-reactor chamber in the investigation of atmospheric photochemical processes, J. Environ. Sci., 98, 161–168, 2020b.
Metzger, A., Dommen, J., Gaeggeler, K., Duplissy, J., Prevot, A. S. H., Kleffmann, J., Elshorbany, Y., Wisthaler, A., and Baltensperger, U.: Evaluation of 1,3,5 trimethylbenzene degradation in the detailed tropospheric chemistry mechanism, MCMv3.1, using environmental chamber data, Atmos. Chem. Phys., 8, 6453–6468, https://doi.org/10.5194/acp-8-6453-2008, 2008.
Nakao, S., Clark, C., Tang, P., Sato, K., and Cocker III, D.: Secondary organic aerosol formation from phenolic compounds in the absence of NOx, Atmos. Chem. Phys., 11, 10649–10660, https://doi.org/10.5194/acp-11-10649-2011, 2011.
Nishino, N., Arey, J., and Atkinson, R.: Formation yields of glyoxal and methylglyoxal from the gas-phase OH radical-initiated reactions of toluene, xylenes, and trimethylbenzenes as a function of NO2 concentration, J. Phys. Chem. A, 114, 10140–10147, 2010.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, 1996.
Odum, J. R., Jungkamp, T. P., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: The atmospheric aerosol-forming potential of whole gasoline vapor, Science, 276, 96–99, 1997.
Peng, J., Hu, M., Du, Z., Wang, Y., Zheng, J., Zhang, W., Yang, Y., Qin, Y., Zheng, R., Xiao, Y., Wu, Y., Lu, S., Wu, Z., Guo, S., Mao, H., and Shuai, S.: Gasoline aromatics: a critical determinant of urban secondary organic aerosol formation, Atmos. Chem. Phys., 17, 10743–10752, https://doi.org/10.5194/acp-17-10743-2017, 2017.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Sato, K., Takami, A., Isozaki, T., Hikida, T., Shimono, A., and Imamura, T.: Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons, Atmos. Environ., 44, 1080–1087, https://doi.org/10.1016/j.atmosenv.2009.12.013, 2010.
Sato, K., Takami, A., Kato, Y., Seta, T., Fujitani, Y., Hikida, T., Shimono, A., and Imamura, T.: AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: effects of chemical structure on SOA aging, Atmos. Chem. Phys., 12, 4667–4682, https://doi.org/10.5194/acp-12-4667-2012, 2012.
Seinfeld, J. H.: Urban air pollution: State of the science, Science, 243, 745–752, 1989.
Song, C., Na, K., and Cocker III, D. R.: Impact of the hydrocarbon to NOx ratio on secondary organic aerosol formation, Environ. Sci. Technol., 39, 3143–3149, https://doi.org/10.1021/es0493244, 2005.
Song, C., Na, K., Warren, B., Malloy, Q., and Cocker III, D. R.: Secondary organic aerosol formation from the photooxidation of p- and o-xylene, Environ. Sci. Technol., 41, 7403–7408, https://doi.org/10.1021/es0621041, 2007.
Sun, J., Wang, Y., Wu, F., Tang, G., Wang, L., Wang, Y., and Yang, Y.: Vertical characteristics of VOCs in the lower troposphere over the North China Plain during pollution periods, Environ. Pollut., 236, 907–915, https://doi.org/10.1016/j.envpol.2017.10.051, 2018.
Tkacik, D. S., Presto, A. A., Donahue, N. M., and Robinson, A. L.: Secondary organic aerosol formation from intermediate-volatility organic compounds: cyclic, linear, and branched alkanes, Environ. Sci. Technol., 46, 8773–8781, https://doi.org/10.1021/es301112c, 2012.
Tong, D., Chen, J. Y., Qin, D. D., Ji, Y. M., Li, G. Y., and An, T. C.: Mechanism of atmospheric organic amines reacted with ozone and implications for the formation of secondary organic aerosols, Sci. Total Environ., 737, 139830, https://doi.org/10.1016/j.scitotenv.2020.139830, 2020.
Volkamer, R., Platt, U., and Wirtz, K.: Primary and secondary glyoxal formation from aromatics: Experimental evidence for the bicycloalkyl-radical pathway from benzene, toluene, and p-xylene, J. Phys. Chem. A, 105, 7865–7874, https://doi.org/10.1021/jp010152w, 2001.
Wang, W. G., Li, K., Zhou, L., Ge, M. F., Hou, S. Q., Tong, S. R., Mu, Y. J., and Jia, L.: Evaluation and Application of Dual-Reactor Chamber for Studying, Acta Phys.-Chim. Sin., 31, 1251–1259, https://doi.org/10.3866/PKU.WHXB201504161, 2015.
Wang, Y., Luo, H., Jia, L., and Ge, S.: Effect of particle water on ozone and secondary organic aerosol formation from benzene-NO2-NaCl irradiations, Atmos. Environ., 140, 386–394, https://doi.org/10.1016/j.atmosenv.2016.06.022, 2016.
Wyche, K. P., Monks, P. S., Ellis, A. M., Cordell, R. L., Parker, A. E., Whyte, C., Metzger, A., Dommen, J., Duplissy, J., Prevot, A. S. H., Baltensperger, U., Rickard, A. R., and Wulfert, F.: Gas phase precursors to anthropogenic secondary organic aerosol: detailed observations of 1,3,5-trimethylbenzene photooxidation, Atmos. Chem. Phys., 9, 635–665, https://doi.org/10.5194/acp-9-635-2009, 2009.
Xu, J., Griffin, R. J., Liu, Y., Nakao, S., and Cocker III, D. R.: Simulated impact of NOx on SOA formation from oxidation of toluene and m-xylene, Atmos. Environ., 101, 217–225, https://doi.org/10.1016/j.atmosenv.2014.11.008, 2015.
Yang, Y., Vance, M., Tou, F., Tiwari, A., Liu, M., and Hochella, M. F.: Nanoparticles in road dust from impervious urban surfaces: distribution, identification, and environmental implications, Environ. Sci.-Nano, 3, 534–544, https://doi.org/10.1039/c6en00056h, 2016.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zhou, Y., Zhang, H., Parikh, H. M., Chen, E. H., Rattanavaraha, W., Rosen, E. P., Wang, W., and Kamens, R. M.: Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: Water and particle seed effects (II), Atmos. Environ., 45, 3882–3890, https://doi.org/10.1016/j.atmosenv.2010.12.048, 2011.
Ziemann, P. J.: Effects of molecular structure on the chemistry of aerosol formation from the OH-radical-initiated oxidation of alkanes and alkenes, Int. Rev. Phys. Chem., 30, 161–195, https://doi.org/10.1080/0144235x.2010.550728, 2011.
Zou, Y., Deng, X. J., Zhu, D., Gong, D. C., Wang, H., Li, F., Tan, H. B., Deng, T., Mai, B. R., Liu, X. T., and Wang, B. G.: Characteristics of 1 year of observational data of VOCs, NOx and O3 at a suburban site in Guangzhou, China, Atmos. Chem. Phys., 15, 6625–6636, https://doi.org/10.5194/acp-15-6625-2015, 2015.