the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 May 2021
| 04 May 2021
Heterogeneous interactions between SO2 and organic peroxides in submicron aerosol
Shunyao Wang
Tengyu Liu
Jinmyung Jang
Jonathan P. D. Abbatt
Arthur W. H. Chan
Atmospheric models often underestimate particulate sulfate, a major component in ambient aerosol, suggesting missing sulfate formation mechanisms in the models. Heterogeneous reactions between SO2 and aerosol play an important role in particulate sulfate formation and its physicochemical evolution. Here we study the reactive uptake kinetics of SO2 onto aerosol containing organic peroxides. We present chamber studies of SO2 reactive uptake performed under different relative humidity (RH), particulate peroxide contents, peroxide types, and aerosol acidities. Using different model organic peroxides mixed with ammonium sulfate particles, the SO2 uptake coefficient () was found to be exponentially dependent on RH. increases from 10−3 at RH 25 % to 10−2 at RH 71 % as measured for an organic peroxide with multiple O–O groups. Under similar conditions, the kinetics in this study were found to be structurally dependent: organic peroxides with multiple peroxide groups have a higher than those with only one peroxide group, consistent with the reactivity trend previously observed in the aqueous phase. In addition, is linearly related to particle-phase peroxide content, which in turn depends on gas–particle partitioning of organic peroxides. Aerosol acidity plays a complex role in determining SO2 uptake rate, influenced by the effective Henry's Law constant of SO2 and the condensed-phase kinetics of the peroxide–SO2 reaction in the highly concentrated aerosol phase. These uptake coefficients are consistently higher than those calculated from the reaction kinetics in the bulk aqueous phase, and we show experimental evidence suggesting that other factors, such as particle-phase ionic strength, can play an essential role in determining the uptake kinetics. values for different types of secondary organic aerosol (SOA) were measured to be on the order of 10−4. Overall, this study provides quantitative evidence of the multiphase reactions between SO2 and organic peroxides, highlighting the important factors that govern the uptake kinetics.
- Article
(1414 KB) - Full-text XML
-
Supplement
(1259 KB) - BibTeX
- EndNote





Sulfate and organic compounds are ubiquitous particulate components in both polluted and pristine environments (Chen et al., 2009; Andreae et al., 2018; He et al., 2011; Sun et al., 2013; Huang et al., 2014), with important implications for public health and global climate (Hallquist et al., 2009). Particulate sulfate can form via S(IV) oxidation by OH radicals in the gas phase and via oxidation in cloud water, fog droplets, or the aerosol aqueous phase, by H2O2, O2 (catalyzed by transition metals), O3, NO2, and small organic peroxides (methyl hydroperoxide and peroxyacetic acid) (Seinfeld and Pandis, 2012). However, atmospheric models tend to underestimate particulate sulfate production on both global (Tie et al., 2001; Yang et al., 2017; Fairlie et al., 2010) and regional scales, especially during heavy haze episodes (Wang et al., 2014; B. Zheng et al., 2015; Sha et al., 2019; Gao et al., 2016; Li et al., 2017; Huang et al., 2019), suggesting that the overall kinetics may be underestimated and/or important mechanisms may be missing in models.
To reconcile these differences, studies have investigated novel reaction mechanisms of sulfate formation. Stabilized Criegee intermediates (sCIs) were hypothesized to oxidize SO2 rapidly and potentially serve as an important source of ambient sulfate (Mauldin et al., 2012). In the work by Newland et al. (2015) and Nguyen et al. (2016), this sCI pathway was shown to play a minor role in sulfate formation. More recently, when L. Liu et al. (2019) applied this mechanism and kinetics to a source-oriented WRF-Chem model, the sCI pathway was found to only account for at most 9 % of the total particulate sulfate. Reactive nitrogen species (such as NO2) have also been proposed as a dominant sulfate formation pathway when aerosol pH was estimated to be 5–6 in Cheng et al. (2016) and close to 7 in Wang et al. (2016) under severe haze scenarios. While such high aerosol pH is not substantiated by some thermodynamic modeling results, which concluded that pH ranges between 4 and 5 in polluted regions (Song et al., 2018; Guo et al., 2017), other studies that highlighted the roles of ammonia and dust found aerosol pH could be higher than 6 (Shi et al., 2017; Ding et al., 2019). Furthermore, higher aerosol water content and PM mass concentration in polluted areas have been shown to enhance aerosol pH via a multiphase buffering process (G. J. Zheng et al., 2020). Meanwhile, a recent modeling study incorporating this heterogeneous NOx mechanism still exhibited a discrepancy of 20 % between the predicted and observed sulfate, indicating the possibility of unknown mechanisms (Huang et al., 2019). Other factors may play a role in enhancing particle-phase sulfate formation rates. Chen et al. (2019) investigated the synergistic effects of NO2 and NH3 on sulfate formation and found that the rate of this reaction can be enhanced by high ionic strength in the particle phase. This enhancement effect by solute strength on sulfate formation was also investigated for the H2O2 pathway in aerosol liquid water. Liu et al. (2020) found that ionic strength and general acid-catalyzed mechanisms can cause the S(VI) formation rate to be nearly 50 times faster in the aerosol phase than in dilute solutions. On the other hand, during the severe haze episodes in China (Li et al., 2020; Guo et al., 2017), transition metal ion (TMI) catalysis of SO2 oxidation by O2 can be significantly suppressed in the aerosol phase due to high ionic strength (Liu et al., 2020; Cheng et al., 2016; Su et al., 2020).
In addition to high solute strength, submicron aerosol is also rich in organic compounds (Jimenez et al., 2009; Hallquist et al., 2009). In recent years, many studies have investigated the potential role of heterogeneous interactions between SO2 and organic aerosol in particulate sulfate formation. Song et al. (2019) found that heterogeneous oxidation of hydroxymethanesulfonate (HMS) by OH can trigger rapid sulfate formation. Wang et al. (2020) studied photosensitizers in ambient particles and found that this pathway could be essential under specific light conditions. Recent studies found that reactive intermediates from isoprene oxidation (L. B. Huang et al., 2019) and benzoic acid (L. B. Huang et al., 2020) can yield a variety of organosulfur species upon catalysis by TMI. Other studies have also investigated the interactions between secondary organic aerosol (SOA) and SO2. Field observations found that ambient sulfate abundance is highly correlated with SOA formation (Yee et al., 2020; Xu et al., 2015). C. Liu et al. (2019) found that SO2 enhances SOA formation and the average carbon oxidation state during methoxyphenol photooxidation. By performing chamber experiments with limonene SOA formation in the presence of SO2, Ye et al. (2018) also observed significant SO2 decay along with increased SOA yields and carbon oxidation state, proposing that organic peroxides in SOA may be the key reactive intermediates for SO2 oxidation.
Organic peroxides are key intermediates for aerosol formation and ubiquitously exist in many SOA systems (Hallquist et al., 2009; Bianchi et al., 2019). Numerous studies have reported peroxide content of 20 %–60 % for isoprene- and monoterpene-derived SOA (Surratt et al., 2006; Ng et al., 2008; Ye et al., 2018; Epstein et al., 2014). A significant fraction of organic peroxide (30 %–50 %) has also been found in naphthalene-derived SOA under low and high NOx conditions (Kautzman et al., 2009). Using model simulations, Bonn et al. (2004) found that organic hydroperoxides can account for up to 60 % of global SOA. The aqueous-phase reaction kinetics between organic peroxides and dissolved SO2 have been explored in previous studies (Lind et al., 1987; Gunz and Hoffmann, 1990; Wang et al., 2019; Dovrou et al., 2019; Yao et al., 2019). The second-order reaction rate constants for organic peroxides in SOA (Dovrou et al., 2019; Yao et al., 2019) and S(IV) were measured to be on the order of 102–103 M−1 s−1, which is within the range of those measured for commercially available organic peroxides (Wang et al., 2019) and small organic peroxides (Lind et al., 1987). Yao et al. (2019) quantified the reactive uptake coefficient of SO2 () onto α-pinene SOA to be on the order of 10−4–10−3, which is positively dependent on RH and inferred particle-phase peroxide content. These reactions are also linked to the formation of organosulfates (Wang et al., 2019). Both inorganic sulfate (85 %–90 %) and organosulfates (10 %–15 %) were observed as products of SO2 reactive uptake onto SOA (Yao et al., 2019).
Given the potential significance of SO2 reactive uptake in particulate sulfate formation, a more in-depth study is needed to determine the important factors that govern the heterogeneous kinetics of SO2 onto organic-peroxide-containing aerosol. In this study, we measured for two categories of aerosol: (1) model organic peroxides mixed with ammonium sulfate or malonic acid and (2) SOA from a few representative biogenic and anthropogenic precursors. The impacts of RH, peroxide type, peroxide content, and condensed-phase pH on SO2 reactive uptake were systematically evaluated with the goal of better understanding atmospheric multiphase sulfate formation.
The reactive uptake of SO2 onto peroxide-containing particles was studied in a 1 m3 Teflon chamber under ambient temperature and pressure. In brief, generated particles and SO2 were introduced into the chamber separately. The consumption of SO2, changes in particle size distribution, and chemical composition were monitored to estimate the reactive uptake coefficients. Particles were also collected on filters for offline chemical characterization.
2.1 Seed aerosol generation
In this work, two types of aerosol were used to investigate the uptake of SO2. The first is ammonium sulfate or malonic acid mixed with model organic peroxides (Fig. S1 in the Supplement). In this first set of experiments, an aerosol atomizer (model 3076, TSI Inc., USA) was used to generate aqueous particles from dilute solution. Each solution consists of ammonium sulfate (≥ 99 %, Sigma-Aldrich) or malonic acid (99 %, Sigma-Aldrich) and a model organic peroxide in ultrapure water (HPLC-grade, Fisher Chemical). For the experiments investigating the relationship between and peroxide type (Expts. 2–14; Table S1 in the Supplement), different commercially available organic peroxides were used, including tert-butyl hydroperoxide (70 wt % in water, Sigma-Aldrich), cumene hydroperoxide (80 wt % in water, Sigma-Aldrich), and 2-butanone peroxide (35 wt %, Sigma-Aldrich). The molar ratio of organic peroxide to ammonium sulfate in the atomizing solution was 2:1, with the aim of being atmospherically relevant (corresponding to a maximum particulate peroxide molar fraction of 66 % and a mass fraction of approximately 50 %–70 % if all the organic peroxides were assumed to remain in the particle phase). This ratio was used as a proxy for total peroxide content in both the gas and particle phase relative to that of ammonium sulfate upon atomization. For experiments studying the relationship between and particle-phase peroxide content, the molar ratio of organic peroxide to ammonium sulfate (Expts. 10–12, 15–18; Table S1) in the solution was adjusted to be 0.02, 0.2, 1, 2, and 4, respectively. For experiments in which malonic acid was used (Expts. 19–22; Table S1), molar ratios of 0.2, 1, 2, and 4 were adopted. For measuring with different aerosol pH (Expts. 17, 23–25; Table S1), different amounts of HCl (37 %, Sigma-Aldrich) were added into the solution (0 M, 0.00002 M, 0.0001 M, 0.001 M HCl) prior to atomization. The initial pH values of aerosol (2.5, 2.2, 1.6, 1, respectively) were modeled using the E-AIM III model (Clegg et al., 1998) based on the initial molar ratios of inorganic species (H+, NH, SO, Cl−) in the atomizing solution and measured RH (around 50 %). The atomized particles were flowed into the chamber without drying and therefore assumed to remain deliquesced under the range of RH we studied. Expts. 2–14 (Table S1) also represent those in which the relationship between and RH conditions was studied.
In the second set of experiments, the uptake of SO2 onto SOA was investigated (Expts. 26–28; Fig. S2, Table S1). A custom-built 10 L quartz oxidation flow reactor was used to produce SOA (Ye et al., 2016) from different hydrocarbon precursors. In this work, we studied SOA formed from toluene photooxidation, limonene ozonolysis, and α-pinene ozonolysis, which are three of the most commonly studied SOA systems (Ng et al., 2007; Hildebrandt et al., 2009; Hartz et al., 2005; Varutbangkul et al., 2006). Toluene (analytical standard, Sigma Aldrich) was continuously injected into zero air flow by a syringe (1000 mL, Hamilton) installed on a syringe pump (KDS Legato100) to achieve an initial concentration of 0.5 ppm. Limonene (Sigma-Aldrich, 97 %) and α-pinene (Sigma-Aldrich, 98 %) were pre-dissolved in cyclohexane (Sigma-Aldrich, 99.5 %) with a volumetric ratio of 1:1500 and 1:500 to ensure that OH formed from limonene or α-pinene ozonolysis was scavenged by cyclohexane, which was estimated based on the rate constants (Atkinson and Arey, 2003). The initial steady-state concentrations of limonene and α-pinene were controlled to be around 2 and 1 ppm entering the flow tube. O3, used as the oxidant (for limonene and α-pinene) or the OH precursor (for toluene), was generated by passing 0.5 L min−1 of pure oxygen (99.6 %, Linde, Mississauga, Canada) through an O3 generator (no. 97006601, UVP, Cambridge, UK). Humidified air was produced by bubbling zero air through a custom-made humidifier at a flow rate of 1 L min−1. The photolysis of O3 produces O (1D), which reacts with water vapor to produce •OH with illumination from the 254 nm UV lamps (UVP, Cambridge, UK) to initiate the photooxidation of toluene. The average residence time inside the flow tube was controlled to be around 5 min. A gas chromatography–flame ionization detector (GC-FID, model 8610C, SRI Instruments Inc., LV, USA) equipped with a Tenax® TA trap was used to monitor the concentration of hydrocarbon precursors at the inlet and outlet of the flow reactor. In all cases, the O3 concentration was maintained to be at least 10 times higher than that of the hydrocarbon. Temperature and relative humidity were monitored by an Omega HX94C RH transmitter. The particle size distribution and volume concentration were monitored using a custom-built scanning mobility particle sizer (SMPS), which is a combination of a differential mobility analyzer column (DMA, model 3081, TSI, Shoreview, MN, USA) with flow controls and a condensation particle counter (CPC, model 3772, TSI, Shoreview, MN, USA).
2.2 Quantification of
Prior to each experiment, the chamber was flushed by purified air overnight with a flow rate of 25 L min−1 until the particle number concentration was less than 5 cm−3 and SO2 was less than 1 ppb. To adjust RH, the chamber was humidified by passing purified air through a custom-built humidifier filled with ultrapure water. For experiments with atomized ammonium sulfate or malonic acid, SO2 was injected into the chamber prior to the introduction of particles. For experiments studying onto SOA, aerosol generated from the flow tube was injected into the Teflon chamber continuously after passing through an O3 denuder (Ozone Solutions, Iowa, USA) to achieve a specific aerosol concentration inside the chamber prior to SO2 addition. The SO2 mixing ratio in the chamber during each experiment was continuously monitored using an SO2 analyzer (model 43i, Thermo Scientific). The initial mixing ratio of SO2 in each experiment was controlled to be around 200 ppb. Aerosol size distribution was monitored by SMPS. The reactive uptake coefficient of SO2 was calculated by integrating the following equation:
where [SO2] is the SO2 mixing ratio (ppb) monitored by the SO2 analyzer, A is the average surface area concentration (µm2 cm−3) derived from the particle size distribution measured by SMPS, and represents the mean molecular velocity (cm s−1) of SO2. d[SO2]dt is solved over the initial SO2 decay such that the peroxide concentration in the aerosol liquid phase is assumed to be constant and pseudo-first-order kinetics can be applied (Abbatt et al., 2012; Thornton et al., 2003). A summary of all the measured can be found in Table S1. The typical evolution of monitored species can be seen in Fig. 1. Control experiments were performed in order to rule out other potential factors (e.g., SO2 loss in the in-line filter in front of the SO2 analyzer, interferences inside the SO2 analyzer, chamber wall losses, SO2 uptake onto wet ammonium sulfate, gas-phase reaction of SO2 with peroxide vapor) that may contribute to the SO2 decay observed during the measurement inside the chamber (Figs. S3–S6). The measurement uncertainty and precision of in this study can be found in Table S1. Also, we observed that there was SO2 repartitioning from the humid chamber wall in the presence of organic peroxide under high RH (Fig. S6b, RH 74 %). The observed SO2 repartitioning rate was then applied to correct the measured under high RH conditions (above 70 %, Expt. 14), and this correction amounts to a 40 % increase in calculated .
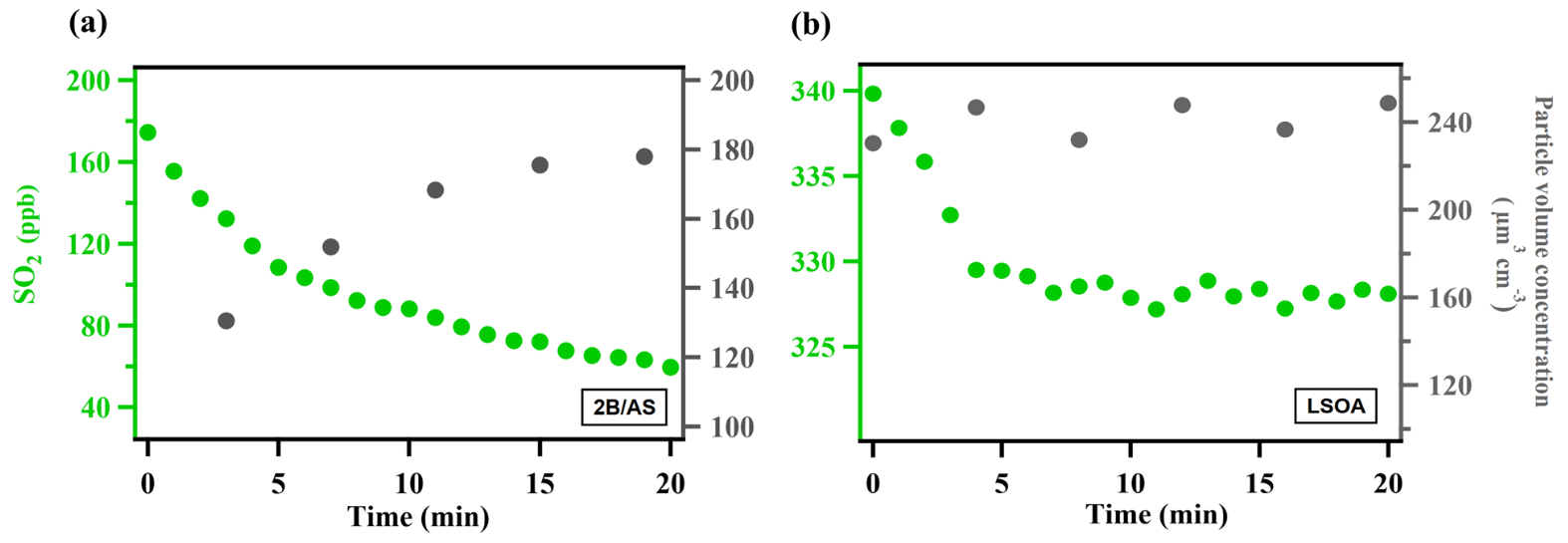
Figure 1Typical evolution of the species monitored during measurement for (a) ammonium sulfate mixed with 2-butanone organic peroxide (2B/AS, Expt. 16) and (b) limonene SOA (LSOA, Expt. 27). Particle volume concentrations measured by SMPS have been corrected for wall loss assuming a pseudo-first-order loss rate (Ye et al., 2016). was calculated for the initial portion of the decay (first 7 min).
2.3 Offline peroxide quantification
Aerosol was collected onto 47 mm PTFE (polytetrafluoroethylene) filters with 0.2 µm pore size (Whatman®, GE Healthcare) from the chamber by a diaphragm pump (KNF Neuberger Inc., USA) for offline chemical analysis. The total particulate peroxide content (H2O2, ROOH, and ROOR) in these samples prior to SO2 uptake was quantified using the iodometric–spectrophotometric assay (Docherty et al., 2005). I2 produced from the reaction between I− and peroxides can further quickly combine with the excess amount of I− to form I, which has a brown color and absorbs UV–visible (UV–Vis) light at 470 nm. The SOA extraction was then aliquoted into a 96-well UV plate (Greiner Bio-One, Kremsmünster, AT) with 160 µL per well. 20 µL of formic acid (≥ 95 %, Sigma-Aldrich) was added into each well, followed by 20 µL of potassium iodide (BioUltra, ≥ 99.5 %, Sigma-Aldrich) solution (dissolved in DI water). The plate was then immediately covered by an adhesive plate sealer (EdgeBio, Gaithersburg, USA) in order to avoid reagent evaporation and O2 oxidation. After incubation for an hour in the dark, the absorption at 470 nm was measured using a UV–Vis spectrophotometer (Spectramax 190, Molecular Devices Corporation, Sunnyvale, CA) and then converted to a peroxide concentration using the calibration curve made by tert-butyl hydroperoxide (70 wt % in H2O, Sigma-Aldrich) with a series of concentrations (0–10 mM). An average molecular mass for seed particles (organics + ammonium sulfate) was assumed based on the chemical composition in order to calculate the molar fraction of total peroxides using the following equation:
where maerosol is the weighed aerosol mass on the filter, and Mperoxide are the molecular mass of ammonium sulfate and peroxide, respectively, and fperoxide are the initial molar fraction of ammonium sulfate and peroxide, and Nperoxide and Naerosol are the measured peroxide molar and calculated aerosol molar, respectively. More details about the iodometric–spectrophotometric procedures were described in previous work (Wang et al., 2018).
3.1 SO2 uptake and RH
A positive relationship between and RH (between 25 %–71 %) was observed for all types of organic peroxides studied (Fig. 2). The positive dependence of the reactive uptake coefficient of water-soluble gaseous species on RH has also been observed in other studies (Thornton et al., 2003; Griffiths et al., 2009; Zhao et al., 2017; Zhang et al., 2019). Recently, the uptake behavior of SO2 onto soot, mineral dust, and SOA was also shown to positively depend on RH (Zhang et al., 2019; Zhao et al., 2017; Yao et al., 2019).
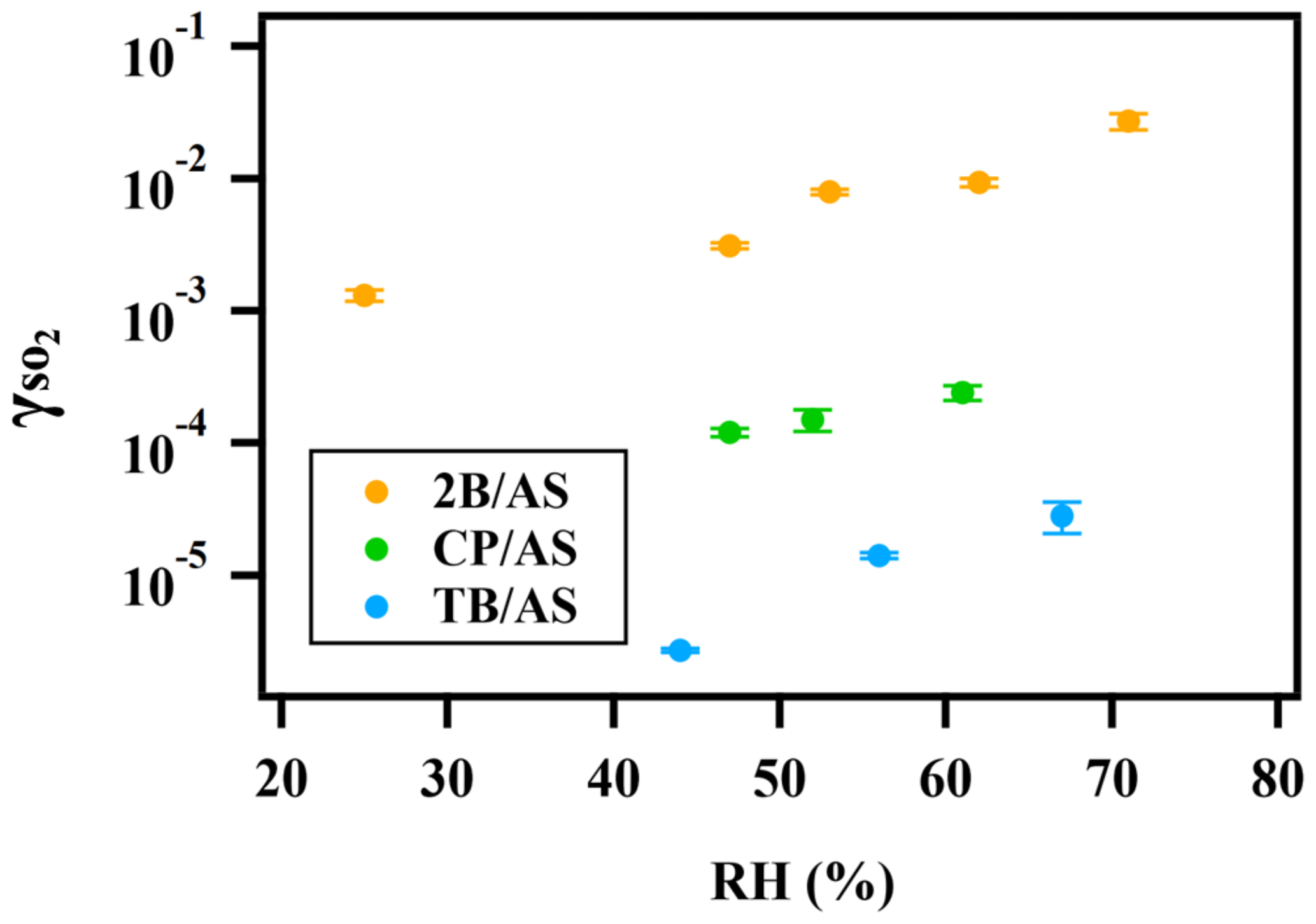
Figure 2Exponential relationship between and RH for ammonium sulfate aerosol containing 2-butanone peroxide (2B), cumene hydroperoxide (CP), and tert-butyl hydroperoxide (TB).
It is also noteworthy that an exponential dependence of the SO2 reactive uptake coefficient on RH was observed in our study. increases with increased relative humidity, which could be even more significant under a high-RH regime. This is consistent with previous laboratory studies that measured the reactive uptake coefficient of SO2 onto aerosol to be exponentially dependent on RH (Zhang et al., 2019; Yao et al., 2019). Additionally, multiple field campaigns have observed a significant correlation between particulate sulfate formation and ambient RH (Song et al., 2019; Sun et al., 2013; Huang et al., 2020). Sun et al. (2013) observed a faster sulfate formation rate under humid conditions, proposing a significant impact of aerosol liquid water on sulfate production during wintertime in Beijing. G. J. Zheng et al. (2015) reported a notably higher SOR (molar ratio of sulfate to the sum of sulfate and SO2) during wet periods (RH > 50 %), indicating the importance of heterogeneous reactions for secondary sulfur transformation with abundant aerosol water content under humid conditions. In a recent study by Song et al. (2019), rapid sulfate formation rate observed under high-RH conditions was found to be significantly higher than atmospheric modeling results implemented with homogeneous SO2 oxidation pathways, which was later attributed to heterogeneous sulfate formation mechanisms. Multiple mechanisms can potentially explain this observed –RH dependence. An enhanced relative humidity would result in a nonlinear increase in aerosol water content, which can lead to more SO2 dissolved in the aerosol aqueous phase (Seinfeld and Pandis, 2012). It should be noted that while the relative humidity is varied systematically in these experiments, the relationship is more complex since RH also affects other aerosol properties, which can affect the uptake kinetics in turn. For example, a higher aerosol liquid water content could dilute protons and thus lower the aerosol acidity. In a study by Laskin et al. (2003), an enhanced uptake of SO2 onto sea-salt particles was observed with an increased aerosol alkalinity at a high pH range.
3.2 Dependence of SO2 uptake on peroxide content and type
As expected, the measured uptake rate of SO2 is dependent on the particulate peroxide content in the current study. Figure 3 shows that is linearly proportional to the amount of particulate peroxide for aerosol with similar volume-to-surface ratios and containing the same type of organic peroxides. This positive relationship between and condensed-phase peroxide content has also been inferred from experiments with SO2 uptake onto α-pinene SOA (Yao et al., 2019), in which the peroxide content in α-pinene SOA was varied indirectly by introducing NO and adjusting the branching ratio of the peroxide-yielding RO2+HORO2 pathway.
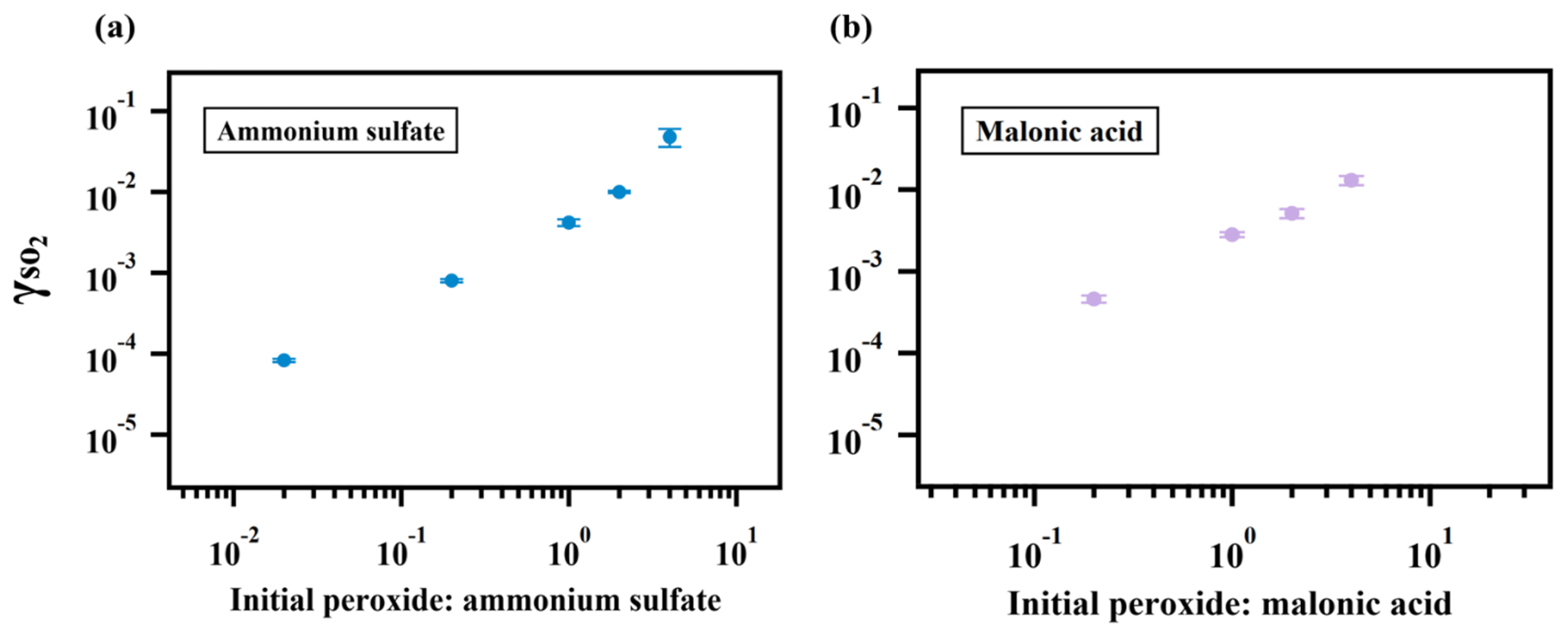
Figure 3Relationship between and particulate peroxide content. values for ammonium sulfate (a) and malonic acid aerosol (b) containing different amounts of 2-butanone peroxide are shown here. The observed dependence of on the amount of peroxide injected is linear since the slopes of the relationship are both nearly 1 in (a) and (b).
In addition to the amount of peroxide injected, the particulate fraction of organic peroxide available for heterogeneous reaction is also influenced by gas–particle partitioning. As indicated in Fig. 2, the reactive uptake coefficients of different organic peroxides vary amongst each other by about an order of magnitude in the range of RH studied, despite the same amounts of peroxide relative to ammonium sulfate initially being in the atomizing solution. Based on our previous work (Wang et al., 2019), the aqueous-phase rate constants for these organic peroxides with dissolved S(IV) only vary by a factor of 2–3 and therefore cannot fully explain the observed difference in uptake rates. Since vapor pressure varies considerably among the different peroxides in the present study, gas–particle partitioning is likely to influence the amount of peroxide in the particle phase that reacts with dissolved SO2. The relative particulate peroxide content on filters for the three peroxides collected from chamber experiments under RH 50 % without SO2 uptake was measured by the offline KI method (Fig. S7). Although the initial ratio of organic peroxide to ammonium sulfate in the atomizing solution was nominally the same, we measured the highest amount of particulate peroxide with 2-butanone peroxide (16.7 %), followed by cumene hydroperoxide (12.7 %) and then tert-butyl hydroperoxide (3.8 %) using the offline iodometric method. This trend in particulate peroxide content is consistent with the vapor pressures calculated using the SIMPOL group contribution method (Pankow et al., 2008), with 2-butanone peroxide being the least volatile and tert-butyl hydroperoxide being the most volatile. Also, the order of particle-phase peroxide content is consistent with the order of observed, as shown in Fig. 2. A simple visualization of these relationships between different peroxide characteristics (number of peroxide groups, vapor pressure, and aqueous-phase rate constants) and measured (at RH = 50 %) is illustrated in Fig. S7, which indicates that higher can be expected for organic peroxides with multiple O–O groups, lower vapor pressures, and higher aqueous-phase reactivities. It should be noted that the order-of-magnitude difference in experimentally measured among various organic peroxides (Fig. 2) is still not fully explained when both volatility and reaction kinetics are taken into account (Fig. S7), suggesting that the reactive uptake may be influenced by other factors. In summary, for our current experiments in which we nominally maintained the total injected amount of organic peroxide as constant, measured depends on both the reactivity and gas–particle partitioning of the organic peroxides.
3.3 SO2 uptake and aqueous-phase kinetics
Since the aqueous-phase reaction rate constants between S(IV) and these model organic peroxides have been measured previously (Wang et al., 2019), we can test our understanding of the measured using a simple model. By assuming the amount of SO2 dissolved in the aerosol is in equilibrium with the gas phase, the overall can be expressed using the simplified resistor model (Hanson et al., 1994):
where α is the mass accommodation coefficient, is the mean molecular speed of SO2 (cm s−1), H is the effective Henry's law constant that includes both the dissolution of SO2 and the dissociation of H2SO3 (M atm−1), R is the ideal gas constant (atm L mol−1 K−1), T is the temperature (K), and the parameter q is used to describe the competition between the reaction and diffusion of the dissolved gaseous species within a particle, which is further calculated as
where r is the radius (cm) of a given particle, Dl is the aqueous-phase diffusion coefficient (cm2 s−1), and kI is the first-order rate constant (s−1) for the reaction. For experiments in the current study, the calculated q values were consistently found to be far less than 1, which indicates a volume-limited reaction regime. Combined with the assumption of a relatively fast mass accommodation process compared with the bulk-phase reaction, Eq. (3) can be further simplified to describe reactive uptake in the volume-limited regime:
Here, we assume that all the peroxides remain in the condensed phase upon atomizing and reaction inside the chamber for the upper-bound prediction of . The notation [peroxide] represents the particle-phase concentration of total organic peroxide (M) based on the initial ratio between organic peroxide and ammonium sulfate in the atomizing solution and based on the aerosol water content output by E-AIM III (Clegg et al., 1998). kII is the second-order reaction rate constant (M−1 s−1), which we have previously measured in the bulk phase at dilute concentrations (Wang et al., 2019), and is the ratio between the particle volume concentration (µm3 cm−3) and particle surface area concentration (µm2 cm−3) derived from SMPS measurements. As a result, the observed reactive uptake coefficient of SO2 can be compared to that predicted from the bulk-phase reaction rate constant, and the results are shown in Figs. 4 and S8. Overall, we noticed that this model captures the dependence of on peroxide content, but the modeled results were found to be generally 15–50 times lower than the experimentally measured values (Fig. S8). The current predictions are likely upper-bound estimates since all the peroxides were assumed to stay in the condensed phase without partitioning. As a result, this observed discrepancy of 15–50 times could even be larger if the particulate peroxide content during the chamber experiments were lower due to partitioning.
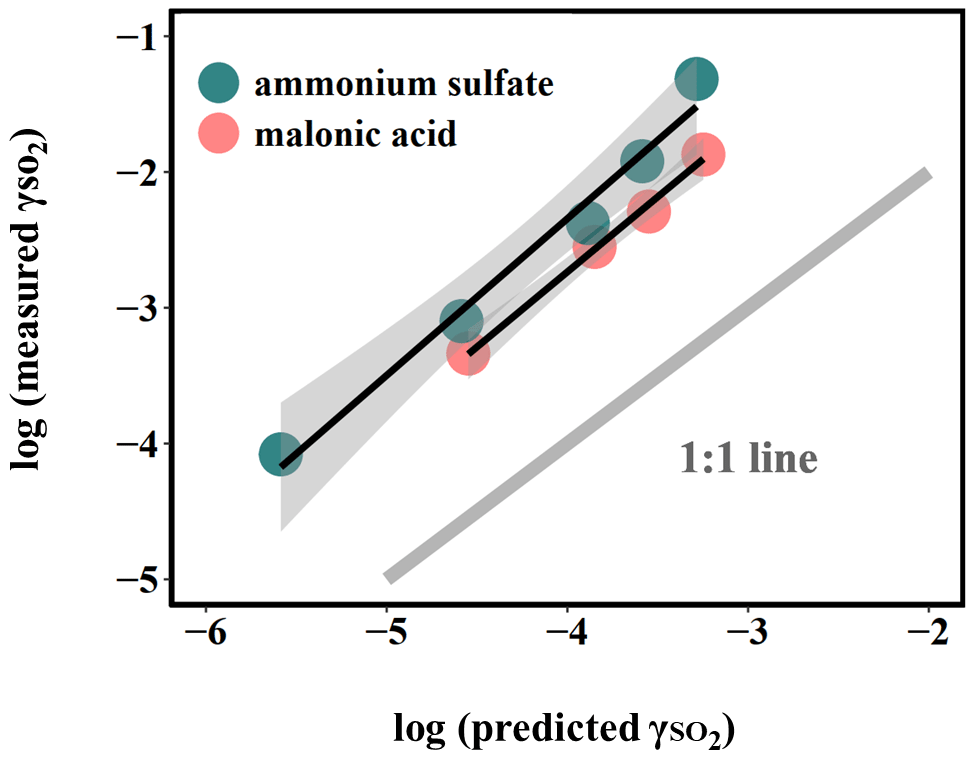
Figure 4Relationship between measured and predicted by Eq. (5). The large deviation from the 1:1 line, which represents the difference between the measured uptake coefficient and predicted values based on kinetics in the dilute aqueous phase, indicates that aerosol reactive uptake is significantly faster than reactions in the dilute aqueous phase. This enhancement is likely driven in part by high ionic strengths, as the difference between measured and predicted is consistently higher for organic-peroxide-containing ammonium sulfate (high ionic strength) than for that mixed with malonic acid (lower ionic strength).
It should be noted that the calculated was based on reaction kinetics measured in dilute solutions, while the experimental values were measured directly from suspended particles. This large difference in kinetics between those in aerosol and in dilute bulk solution suggests that this multiphase interaction is strongly favored in a highly concentrated aerosol environment. One of the potential explanations for this discrepancy could be liquid–liquid phase separation (LLPS) in aerosol between organic peroxide and ammonium sulfate (Ciobanu et al., 2009; O'Brien et al., 2015) such that SO2 can directly interact with the acidic organic phase, wherein the concentration of peroxides can be higher and the kinetics can be different from what we measured in dilute solution (Wang et al., 2019). However, LLPS is generally governed by the chemical composition of the hydrophobic phase (Freedman, 2017). A higher level of oxygenation in organic aerosol is related to higher hydrophilicity, which would favor a homogeneous particle instead of phase separation. Previous studies showed that LLPS did not occur for organic coating with O:C above 0.8 (You et al., 2013; You et al., 2014). The LLPS phenomenon in simple organic–inorganic mixtures can also be affected by the functional groups. The maximum O:C for LLPS can be 0.71 for organics with multiple carboxylic and hydroxyl groups but low aromatic content (Song et al., 2012), while the 2-butanone peroxide we used for both measurement and prediction in the present study has multiple peroxide groups with an O:C value of 0.75. Particle size could also have impacts on phase separation (Cheng et al., 2015). Particle diameters in the current study are mainly under 200 nm, while a previous study showed that particles smaller than this size are less likely to experience LLPS (Veghte et al., 2013). We therefore believe that LLPS is not likely to be responsible for the enhanced uptake rate observed under these experimental conditions.
Another explanation is the high solute strength in the concentrated aerosol phase. Although the aerosol water content for ammonium sulfate aerosol was found to be higher than that of malonic acid aerosol under RH 50 %, as indicated in Figs. 4 and S8, the difference between the measured and predicted is larger for ammonium sulfate aerosol than for malonic acid. Meanwhile, the calculated ionic strength in the aerosol liquid phase under RH 50 % for ammonium sulfate (40 mol kg−1) is significantly larger than that of malonic acid (0.45 mol kg−1). It has been previously reported that the reaction rate between sulfite and hydrogen peroxide in the aqueous phase increases with ionic strength (Maaß et al., 1999). Based on the reaction mechanisms proposed for dissolved SO2 and hydrogen peroxide (Halperin and Taube, 1952), we speculate that the reaction between aqueous-phase S(IV) and organic peroxides follows a similar mechanism.
Here, the overall rate constant is equal to , assuming fast equilibrium steps for reactions (Eq. 6) and (Eq. 7). Dissociated solutes are surrounded by an extended solvation shell, which could affect the reaction rates (Herrmann, 2003). Fewer available free water molecules would therefore shift the equilibrium to the right in Eq. (6). Additionally, higher ionic strength also corresponds to an increased concentration of electrolytes in the aqueous phase, which could hinder the dissociation of peroxymonosulfurous acid and shift the equilibrium in Eq. (7) to the right. In recent work by Liu et al. (2020), the rate of S(IV) oxidation by H2O2 was found to be enhanced by up to a factor of 50 in the aerosol aqueous phase compared to that of dilute solution. The highest ionic strength at which such an enhancement was measured for the H2O2 oxidation pathway was 15 mol kg−1 (Liu et al., 2020).
Whereas the above analysis is based on the assumption that all the chemistry occurs in the bulk component of the particle, it is also possible that some component of the reaction occurs at the gas–particle interface, and the overall kinetics can be affected by interfacial characteristics. For example, an enhanced ionic strength in the aerosol phase can also impact the interfacial reaction mechanisms. A previous study has shown evidence that interfacial chemistry is important for SO2 oxidation in the aerosol phase (Laskin et al., 2003). With higher ionic strength, anions partitioning in the air–liquid interface can promote the overall reaction kinetics via proton transfer and thus accelerate the interfacial chemistry (Knipping et al., 2000; Mishra et al., 2012; Mekic et al., 2018; Mekic et al., 2020; Wei et al., 2018; Ruiz-Lopez et al., 2020). In addition to the catalytic effects of protons indicated in Eqs. (6)–(8), Hung et al. (2015, 2018) observed a significant SO signal at the acidic microdroplet surface, which can promote sulfate formation via a radical propagation chain initiated by surrounding radicals and molecular oxygen (Eqs. 9–12).
Here, the hydroxy radical can potentially be produced from decomposition of the labile organic peroxide in our system (Tong et al., 2016). However, we cannot distinguish whether the interfacial protons promote sulfate formation by catalyzing the peroxide S(IV) oxidation pathway or the sulfur radical pathway at the current stage. In the recent study by Wei et al. (2018), a pH gradient was observed for phosphate-buffered aerosol droplets with the proton accumulated at the interface. Based on the pH-dependent aqueous-phase kinetics measured in our previous work (Wang et al., 2019), such interfacial proton accumulation could potentially explain the enhanced kinetics we observed for aerosol in the current study. However, the chemical compositions are quite different. While phosphate-buffered particles were studied in Wei et al. (2018), acidic ammonium sulfate aerosol was used in our study. Also, the particle size in Wei et al. (2018) is significantly larger (20 µm) than in the current study (200 nm). Thus, it should be noted that there is no direct evidence from the current study showing the relationship between the interfacial properties and , and future studies are warranted.
Therefore, while more studies are needed to clearly delineate the roles of ionic strength, interfacial activity, bulk reactivity, and the particle-phase state quantitatively, the enhancement of SO2 oxidation kinetics by highly concentrated aerosol particles compared to dilute aqueous solutions is concluded to be large (factor of 15–50) for the experimental conditions in the current study.
3.4 SO2 uptake and aerosol pH
As indicated by the proposed reaction mechanisms (Eqs. 6–8), protons are important reaction intermediates for this SO2 oxidation pathway. Previously, the aqueous-phase reaction rate constants between organic peroxides and dissolved SO2 were measured to be pH-dependent (Wang et al., 2019). Moreover, the dissolution equilibrium of SO2 into the aqueous phase is also pH-sensitive (Seinfeld and Pandis, 2012). Many studies have also shown that the uptake kinetics for gaseous species can be affected by the condensed-phase pH (Shi et al., 1999; Gaston et al., 2014; Drozd et al., 2013; Jang and Kamens, 2001; Liu et al., 2015). Reactive uptake of ammonia was observed to depend on condensed-phase acidity (Shi et al., 1999). Heterogeneous condensation of isoprene-derived epoxydiol onto seed aerosol was found to increase with proton concentration (Gaston et al., 2014). In the current study, the potential impact of particle-phase pH on was explored by adding HCl into the atomizing solution. To estimate the particle-phase pH, two different methods associated with two different assumptions were used. In the first scenario, the aerosol pH in each experiment was estimated using the E-AIM III model (Clegg et al., 1998) based on the initial molar ratios of inorganic species (H+, NH, SO, Cl−) in the atomizing solution and measured RH (around 50 %). In the second scenario, the additional sulfate formed from reactive uptake of SO2 was taken into consideration. The partitioning of HCl was allowed in the model simulation for both scenarios. The formation of sulfate can enhance the proton concentration in the aerosol liquid phase, thus lowering the aerosol pH. The average pH during the SO2 uptake process is likely between these two extremes.
Figure 5 shows the measured reactive uptake coefficients of SO2 as a function of the calculated pH. The reactive uptake coefficient was found to weakly increase with decreasing pH, which is consistent with acid-catalyzed reactions between peroxides and dissolved SO2 as measured in the bulk phase (Lind et al., 1987; Wang et al., 2019). was also predicted for the same range of pH based on Eq. (5) and the pH-dependent bulk-phase reaction rate constants measured previously (Wang et al., 2019). Indicated by Fig. 5, the measured again exceeds the predicted by about a factor of 50, which is consistent with what we reported earlier and is likely due to the effects of aerosol ionic strength.
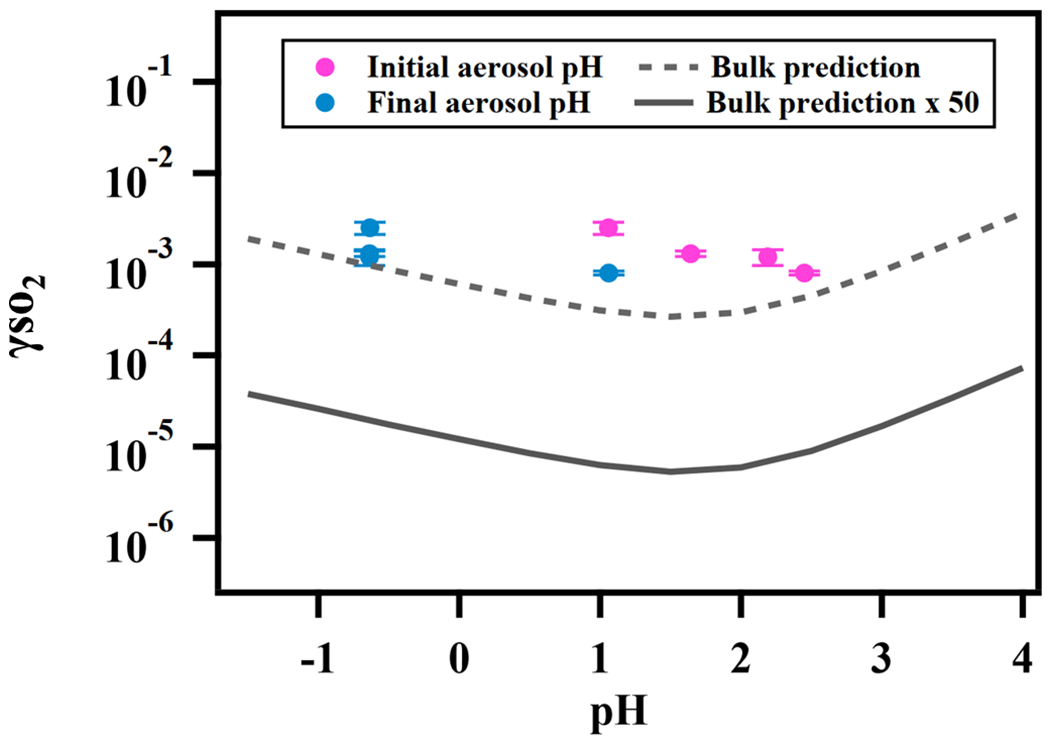
Figure 5Relationship between and aerosol-phase pH for ammonium sulfate aerosol containing 2-butanone peroxide.
Unlike the observed , however, the predicted does not exhibit a monotonic trend. is expected to decrease with decreasing pH at high pH (> 2) as the effective Henry's law constant of SO2 decreases with higher acidity (Seinfeld and Pandis, 2012). is not expected to increase with decreasing pH until pH is below 2, at which the acidity enhancement in the reaction rate constant exceeds the decrease in SO2 solubility. As illustrated earlier, extrapolating dilute aqueous-phase kinetics to highly concentrated aerosol requires considering effects from high solute strength. Solute strength may change the pH dependence of in two ways. First, the solubility of SO2 may decrease and become less dependent on pH as ionic strength increases (Rodríguez-Sevilla et al., 2002). A former study (Leng et al., 2015) has shown that the effective Henry's law of triethylamine decreases with increased ionic strength. Another potential explanation is that the aqueous-phase reaction rate constant can be more pH-dependent at high ionic strengths than what we measured previously in dilute solutions. In either case, the inflection of the predicted would change and could become more negatively dependent on pH (d[]d[pH] becomes less positive in the high pH range and/or more negative in the low pH range), which would more closely match the observed dependence. It should also be noted that there are substantial uncertainties in estimating pH values originating from the partitioning of organics, organic–inorganic phase separations, the mixing state of specific ions, uncertain activity coefficients, and the propagation of RH uncertainties (Clegg et al., 2008; Fountoukis et al., 2009; Guo et al., 2016). Also, the reactive uptake is a dynamic process and will influence aerosol pH in turn upon sulfate formation. In summary, while the magnitude of predicted is consistent with our expected values (after accounting for enhancement by high aerosol solute strength), we cannot fully explain the dependence of on aerosol pH at the current stage. Future studies should investigate how the effective Henry's law of SO2 and the pH dependence of reaction rate constants vary in the aerosol liquid phase with high solute strength in order to have a more comprehensive understanding of the relationship between and aerosol pH.
3.5 SO2 uptake onto SOA
was measured for a few model SOA systems, as organic peroxides are abundant in SOA (Surratt et al., 2006; Kautzman et al., 2009; Krapf et al., 2016; Bonn et al., 2004). Here we studied SOA formed from monoterpene ozonolysis and toluene photooxidation. It should be noted that for the measurements of toluene SOA, a strong hydrocarbon interference was observed with the SO2 analyzer, likely stemming from the high concentrations of gas-phase aromatic compounds. A rough estimate of the uptake rate for toluene SOA from aerosol mass spectrometer sulfate measurements is provided in the Supplement (Sect. S1). The reactive uptake coefficient of SO2 onto Saharan mineral dust was reported to be on the order of 10−5 (Adams et al., 2005). onto dust with the coexistence of NO2 and NH3 under various RH conditions was measured to be 10−7 to 10−5 (Zhang et al., 2019). For a variety of metal oxides, SO2 reactive uptake coefficients were quantified to be between 10−6 and 10−4 (Usher et al., 2002; Fu et al., 2007; Shang et al., 2010). More recently, studied for heterogeneous sulfate formation by photolysis of particulate nitrate was reported to be in the range of 10−6 to 10−5 (Gen et al., 2019). As shown in Fig. 6, for all SOA systems was measured to be on the order of 10−4. Similar values on the order of 10−4 were measured for α-pinene SOA by Yao et al. (2019), and 10−5 for limonene SOA was estimated from the chamber study by Ye et al. (2018). The reaction products from this SOA and SO2 interaction will be reported in a separate study.
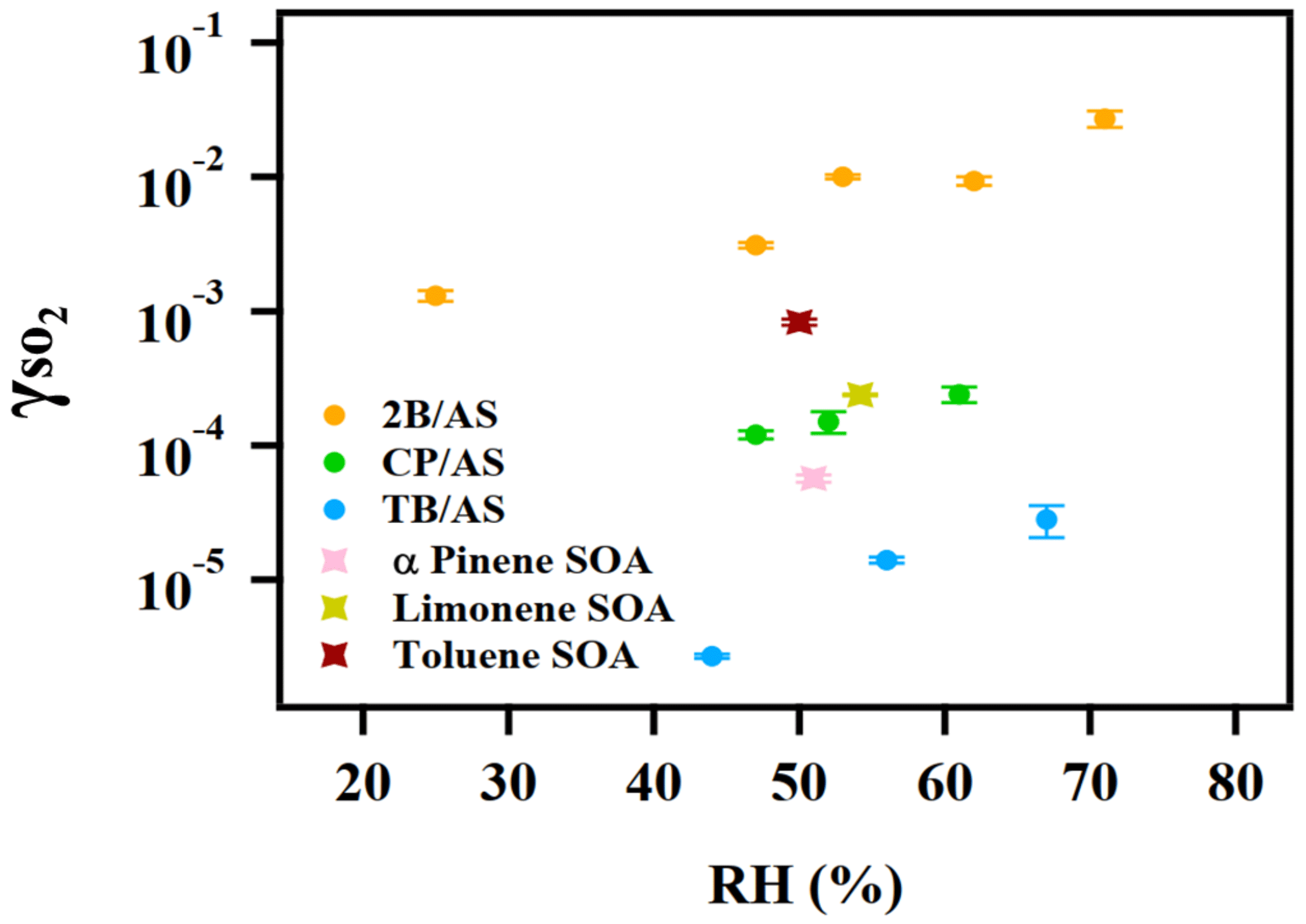
Figure 6 measured for different types of organic aerosol. The reactive uptake coefficient of SO2 onto SOA is on the order of 10−4.
Oxidation of atmospheric hydrocarbons produces reactive intermediates that can potentially interact with SO2 and form particulate sulfate, contributing to PM formation and growth (Berndt et al., 2015; Mauldin et al., 2012; Yao et al., 2019). Organic peroxides generated from both biogenic and anthropogenic hydrocarbon emissions are abundant in submicron aerosol. Given that they are highly reactive with relatively short lifetimes (Bonn et al., 2004; Krapf et al., 2016; Qiu et al., 2020), these species could serve as important condensed-phase oxidants for gas-phase SO2. Combining laboratory measurements and model predictions, the current study investigated heterogeneous reactions between SO2 and particulate organic peroxide. The measured for organic-peroxide-containing aerosol ranges from 10−5 to 10−2 in this study. Based on the modeling work by Wang et al. (2014), adding an SO2 uptake pathway to GEOS-Chem with a reactive uptake coefficient of 10−4 could improve the surface sulfate prediction by more than 50 % during severe haze episodes over northern China (RH 50 %), suggesting the potential importance of this multiphase reaction pathway, especially when SOA is the dominant component in particulate matter.
The dependence of heterogeneous kinetics on RH, aerosol pH, peroxide type, and peroxide content was also evaluated. The experimentally measured was found to be consistently higher than that predicted from reaction kinetics with organic peroxides in the dilute aqueous phase. This discrepancy can be potentially explained by the effects of high ionic strength presented in the aerosol, suggesting that the impact of highly concentrated solutes needs to be taken into consideration when applying aqueous-phase kinetics to aerosol multiphase chemistry, especially for particles containing strong electrolytes. We also observed that the kinetics of this multiphase reaction exhibit a weak dependence on pH. Increasing the condensed-phase acidity may enhance the heterogeneous rate constant at low pH, and while this pH dependence is consistent with that of the aqueous-phase reaction rate constant measured previously, it is not consistent with the decrease in the effective Henry's law constant of SO2 along with enhanced acidity. Also, it is likely that within the uncertainties, there may not be an observable –pH trend. Currently, we are not able to fully explain the pH dependence, and further studies are warranted. Particle-phase peroxide content was observed to be linearly correlated with . Moreover, measured for 2-butanone peroxide was found to be orders of magnitude higher than that of cumene hydroperoxide and tert-butyl hydroperoxide. The difference in among various types of organic peroxides can be partially explained by their condensed-phase reactivity and gas–particle partitioning.
In general, we found that the observed in this study can be summarized using the following semi-empirical multilinear relationship:
where γ is the reactive uptake coefficient, kII is the aqueous-phase S(IV) oxidation rate constant (M−1 s−1), PAS is the molar ratio between particulate peroxide and ammonium sulfate in the atomizing solution, which is a proxy for the amount of peroxide in both the gas and particle phases applied in the current study, RH is the relative humidity (%), and Vp is the vapor pressure (kPa) of peroxide. Figure 7 illustrates the degree to which this semi-empirical expression describes the experimental data for ammonium sulfate aerosol mixed with the three types of organic peroxides. Residual evaluations of this multilinear regression can be found Fig. S9. We caution that this equation is not directly applicable to atmospheric models in its current form, especially since the particle-phase peroxide content (PAS) value we applied as input is a calculated value rather than a measurement. However, it illustrates the internal consistency of our experimental results across a range of RH, peroxide contents, and aqueous-phase reactivities, which are the key variables for uptake rates. A better understanding of ionic strengths and pH in aerosol, either through modeling or direct measurements of these variables, is needed to establish the coefficient dependence.
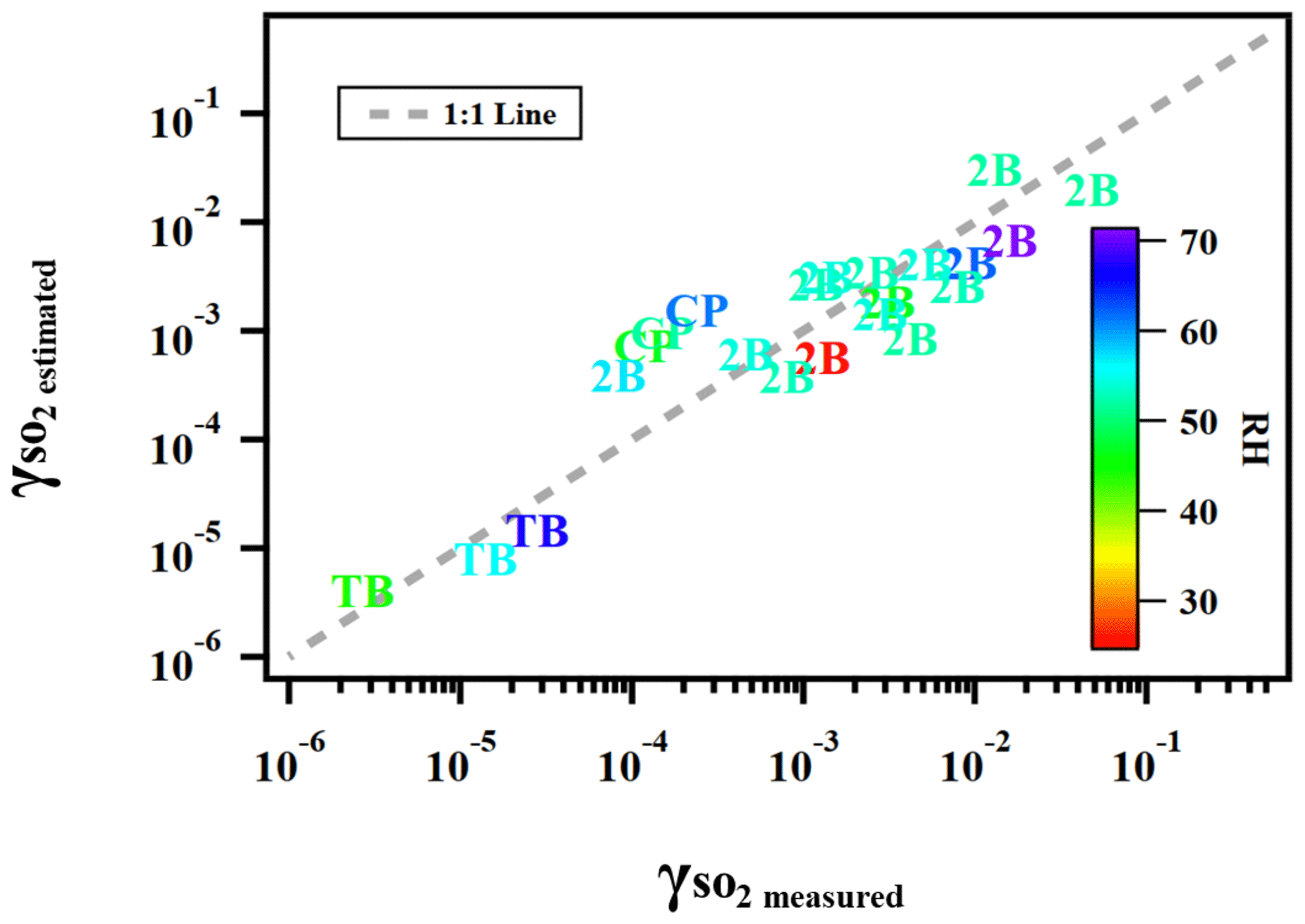
Figure 7Predicted using Eq. (13) versus measured for ammonium sulfate or malonic acid aerosol containing 2-butanone peroxide (2B), cumene hydroperoxide (CP), and tert-butyl hydroperoxide (TB) under different experimental conditions.
Future studies should be focused on exploring and the reaction products for various types of SOA as well as ambient particles under atmospherically relevant conditions by evaluating the underlying impacts of photochemical condition, chemical composition, particle morphology, ionic strength, and interfacial properties on this multiphase physicochemical process. Overall, presented in our study and its relationship with ambient RH, aerosol pH, ionic strength, and particulate peroxide content and type could provide a framework for the implementation of this heterogeneous mechanism in atmospheric models to have a better understanding of ambient sulfate formation and particle growth.
All data presented in this study are available in the Supplement and have been deposited in figshare (https://doi.org/10.6084/m9.figshare.13078112.v1 Wang, 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-6647-2021-supplement.
AWHC and SW designed the study. SW, TL, and JJ performed the experiments. SW, AWHC, TL, and JJ analyzed data. SW and AWHC wrote the paper with input from all co-authors.
The authors declare that they have no conflict of interest.
The authors would like to thank Greg Evans, Yue Zhao, and Christopher Lim for helpful comments and discussions. Special thanks are owed to SOCAAR for providing the SO2 analyzer.
This research has been supported by the Natural Sciences and Engineering Research Council Discovery Grant (RGPIN-2019-06936) and funding from the Canada Research Chairs Program (950-232582).
This paper was edited by Hang Su and reviewed by two anonymous referees.
Abbatt, J. P. D., Lee, A. K. Y., and Thornton, J. A.: Quantifying trace gas uptake to tropospheric aerosol: recent advances and remaining challenges, Chem. Soc. Rev., 41, 6555–6581, https://doi.org/10.1039/c2cs35052a, 2012.
Adams, J. W., Rodriguez, D., and Cox, R. A.: The uptake of SO2 on Saharan dust: a flow tube study, Atmos. Chem. Phys., 5, 2679–2689, https://doi.org/10.5194/acp-5-2679-2005, 2005.
Andreae, M. O., Afchine, A., Albrecht, R., Holanda, B. A., Artaxo, P., Barbosa, H. M. J., Borrmann, S., Cecchini, M. A., Costa, A., Dollner, M., Fütterer, D., Järvinen, E., Jurkat, T., Klimach, T., Konemann, T., Knote, C., Krämer, M., Krisna, T., Machado, L. A. T., Mertes, S., Minikin, A., Pöhlker, C., Pöhlker, M. L., Pöschl, U., Rosenfeld, D., Sauer, D., Schlager, H., Schnaiter, M., Schneider, J., Schulz, C., Spanu, A., Sperling, V. B., Voigt, C., Walser, A., Wang, J., Weinzierl, B., Wendisch, M., and Ziereis, H.: Aerosol characteristics and particle production in the upper troposphere over the Amazon Basin, Atmos. Chem. Phys., 18, 921–961, https://doi.org/10.5194/acp-18-921-2018, 2018.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, 2003.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: Formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: A key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bonn, B., von Kuhlmann, R., and Lawrence, M. G.: High contribution of biogenic hydroperoxides to secondary organic aerosol formation, Geophys. Res. Lett., 31, L10108, https://doi.org/10.1029/2003GL019172, 2004.
Chen, Q., Farmer, D. K., Schneider, J., Zorn, S. R., Heald, C. L., Karl, T. G., Guenther, A., Allan, J. D., Robinson, N., Coe, H., Kimmel, J. R., Pauliquevis, T., Borrmann, S., Pöschl, U., Andreae, M. O., Artaxo, P., Jimenez, J. L., and Martin, S. T.: Mass spectral characterization of submicron biogenic organic particles in the Amazon Basin, Geophys. Res. Lett., 36, L20806, https://doi.org/10.1029/2009GL039880, 2009.
Chen, T., Chu, B., Ge, Y., Zhang, S., Ma, Q., He, H., and Li, S.-M.: Enhancement of aqueous sulfate formation by the coexistence of NO2/NH3 under high ionic strengths in aerosol water, Environ. Pollut., 252, 236–244, https://doi.org/10.1016/j.envpol.2019.05.119, 2019.
Cheng, Y. F., Su, H., Koop, T., Mikhailov, E., and Pöschl, U.: Size dependence of phase transitions in aerosol nanoparticles, Nat. Commun., 6, 5923, https://doi.org/10.1038/ncomms6923, 2015.
Cheng, Y. F., Zheng, G. J., Wei, C., Mu, Q., Zheng, B., Wang, Z. B., Gao, M., Zhang, Q., He, K. B., Carmichael, G., Pöschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Ciobanu, V. G., Marcolli, C., Krieger, U. K., Weers, U., and Peter, T.: Liquid-liquid phase separation in mixed organic/inorganic aerosol particles, J. Phys. Chem. A, 113, 10966–10978, 2009.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic model of the system HNHNa SONOClH2O at 298.15 K, J. Phys. Chem. A, 102, 2155–2171, https://doi.org/10.1021/jp973043j, 1998.
Clegg, S. L., Kleeman, M. J., Griffin, R. J., and Seinfeld, J. H.: Effects of uncertainties in the thermodynamic properties of aerosol components in an air quality model – Part 1: Treatment of inorganic electrolytes and organic compounds in the condensed phase, Atmos. Chem. Phys., 8, 1057–1085, https://doi.org/10.5194/acp-8-1057-2008, 2008.
Ding, J., Zhao, P., Su, J., Dong, Q., Du, X., and Zhang, Y.: Aerosol pH and its driving factors in Beijing, Atmos. Chem. Phys., 19, 7939–7954, https://doi.org/10.5194/acp-19-7939-2019, 2019.
Docherty, K. S., Wu, W., Lim, Y. B., and Ziemann, P. J.: Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O3, Environ. Sci. Technol., 39, 4049–4059, 2005.
Dovrou, E., Rivera-Rios, J. C., Bates, K. H., and Keutsch, F. N.: Sulfate formation via cloud processing from isoprene hydroxyl hydroperoxides (ISOPOOH), Environ. Sci. Technol., 53, 12476-12484, https://doi.org/10.1021/acs.est.9b04645, 2019.
Drozd, G. T., Woo, J. L., and McNeill, V. F.: Self-limited uptake of α-pinene oxide to acidic aerosol: the effects of liquid–liquid phase separation and implications for the formation of secondary organic aerosol and organosulfates from epoxides, Atmos. Chem. Phys., 13, 8255–8263, https://doi.org/10.5194/acp-13-8255-2013, 2013.
Epstein, S. A., Blair, S. L., and Nizkorodov, S. A.: Direct photolysis of α-pinene ozonolysis secondary organic aerosol: effect on particle mass and peroxide content, Environ. Sci. Technol., 48, 11251–11258, 2014.
Fairlie, T. D., Jacob, D. J., Dibb, J. E., Alexander, B., Avery, M. A., van Donkelaar, A., and Zhang, L.: Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes, Atmos. Chem. Phys., 10, 3999–4012, https://doi.org/10.5194/acp-10-3999-2010, 2010.
Fountoukis, C., Nenes, A., Sullivan, A., Weber, R., Van Reken, T., Fischer, M., Matías, E., Moya, M., Farmer, D., and Cohen, R. C.: Thermodynamic characterization of Mexico City aerosol during MILAGRO 2006, Atmos. Chem. Phys., 9, 2141–2156, https://doi.org/10.5194/acp-9-2141-2009, 2009.
Freedman, M. A.: Phase separation in organic aerosol, Chem. Soc. Rev., 46, 7694–7705, https://doi.org/10.1039/C6CS00783J, 2017.
Fu, H. B., Wang, X., Wu, H. B., Yin, Y., and Chen, J. M.: Heterogeneous uptake and oxidation of SO2 on iron oxides, J. Phys. Chem. C, 111, 6077–6085, 2007.
Gao, M., Carmichael, G. R., Wang, Y., Saide, P. E., Yu, M., Xin, J., Liu, Z., and Wang, Z.: Modeling study of the 2010 regional haze event in the North China Plain, Atmos. Chem. Phys., 16, 1673–1691, https://doi.org/10.5194/acp-16-1673-2016, 2016.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Gen, M., Zhang, R., Huang, D. D., Li, Y., and Chan, C. K.: Heterogeneous SO2 oxidation in sulfate formation by photolysis of particulate nitrate, Environ. Sci. Technol. Lett., 6, 86–91, https://doi.org/10.1021/acs.estlett.8b00681, 2019.
Griffiths, P. T., Badger, C. L., Cox, R. A., Folkers, M., Henk, H. H., and Mentel, T. F.: Reactive uptake of N2O5 by aerosols containing dicarboxylic acids. Effect of particle phase, composition, and nitrate content, J. Phys. Chem. A, 113, 5082-5090, https://doi.org/10.1021/jp8096814, 2009.
Gunz, D. W. and Hoffmann, M. R.: Atmospheric chemistry of peroxides: A review, Atmos. Environ., 24A, 1601–1633, https://doi.org/10.1016/0960-1686(90)90496-A, 1990.
Guo, H., Sullivan, A. P., Campuzano-Jost, P., Schroder, J. C., LopezHilfiker, F. D., Dibb, J. E., Jimenez, J. L., Thornton, J. A., Brown, S. S., Nenes, A., and Weber, R. J.: Fine particle pH and the partitioning of nitric acid during winter in the northeastern United States, J. Geophys. Res.-Atmos., 121, 10355–10376, https://doi.org/10.1002/2016JD025311, 2016.
Guo, H., Weber, R. J., and Nenes, A.: High levels of ammonia do not raise fine particle pH sufficiently to yield nitrogen oxide-dominated sulfate production, Sci. Rep., 7, 12109, https://doi.org/10.1038/s41598-017-11704-0, 2017.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Halperin, J. and Taube, H.: The transfer of oxygen atoms in oxidation – reduction reactions. IV. The reaction of hydrogen peroxide with sulfite and thiosulfate, and of oxygen, manganese dioxide and of permanganate with sulfite, J. Am. Chem. Soc., 74, 380–382, 1952.
Hanson, D. R., Ravishankara, A. R., and Solomon, S.: Heterogeneous reactions in sulfuric acid aerosols: A framework for model calculations, J. Geophys. Res., 99, 3615, https://doi.org/10.1029/93JD02932, 1994.
Hartz, K. E. H., Rosenorn, T., Ferchak, S. R., Raymond, T. M., Bilde, M., Donahue, N. M., and Pandis, S. N.: Cloud condensation nuclei activation of monoterpene and sesquiterpene secondary organic aerosol, J. Geophys. Res.-Atmos., 110, D14208, https://doi.org/10.1029/2004JD005754, 2005.
He, L.-Y., Huang, X.-F., Xue, L., Hu, M., Lin, Y., Zheng, J., Zhang, R., and Zhang, Y.-H.: Submicron aerosol analysis and organic source apportionment in an urban atmosphere in Pearl River Delta of China using high-resolution aerosol mass spectrometry, J. Geophys. Res. Atmos., 116, D12304, https://doi.org/10.1029/2010JD014566, 2011.
Herrmann, H.: Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 103, 4691–4716, 2003.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Huang, L., An, J., Koo, B., Yarwood, G., Yan, R., Wang, Y., Huang, C., and Li, L.: Sulfate formation during heavy winter haze events and the potential contribution from heterogeneous SO2 + NO2 reactions in the Yangtze River Delta region, China, Atmos. Chem. Phys., 19, 14311–14328, https://doi.org/10.5194/acp-19-14311-2019, 2019a.
Huang, L. B., Coddens, E. M., and Grassian, V. H.: Formation of organosulfur compounds from aqueous phase reactions of S (IV) with methacrolein and methyl vinyl ketone in the presence of transition metal ions, ACS Earth Space Chem., 3, 1749–1755, 2019.
Huang, L. B., Liu, T. and Grassian, V. H.: Radical-initiated formation of aromatic organosulfates and sulfonates in the aqueous phase, Environ. Sci. Technol., 54, 11857–11864, 2020.
Huang, R. J., Zhang, Y. L., Bozzetti, C., Ho, K. F., Cao, J. J., Han, Y. M., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, 2014.
Huang, R. J., He, Y., Duan, J., Li, Y., Chen, Q., Zheng, Y., Chen, Y., Hu, W., Lin, C., Ni, H., Dai, W., Cao, J., Wu, Y., Zhang, R., Xu, W., Ovadnevaite, J., Ceburnis, D., Hoffmann, T., and O'Dowd, C. D.: Contrasting sources and processes of particulate species in haze days with low and high relative humidity in wintertime Beijing, Atmos. Chem. Phys., 20, 9101–9114, https://doi.org/10.5194/acp-20-9101-2020, 2020.
Hung, H. M. and Hoffmann, M. R.: Oxidation of gas-phase SO2 on the surfaces of acidic microdroplets: Implications for sulfate and sulfate radical anion formation in the atmospheric liquid phase, Environ. Sci. Technol., 49, 13768–13776, 2015.
Hung, H. M., Hsu, M. N., and Hoffmann, M. R.: Quantification of SO2 oxidation on interfacial surfaces of acidic micro-droplets: Implication for ambient sulfate formation, Environ. Sci. Technol., 52, 9079–9086, https://doi.org/10.1021/acs.est.8b01391, 2018.
Jang, M. and Kamens, R. M.: Atmospheric secondary aerosol formation by heterogeneous reactions of aldehydes in the presence of a sulfuric acid aerosol catalyst, Environ. Sci. Technol., 35, 4758–4766, https://doi.org/10.1021/es010790s, 2001.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kautzman, K., Surratt, J., Chan, M., Chan, A., Hersey, S., Chhabra, P., Dalleska, N., Wennberg, P., Flagan, R., and Seinfeld, J.: Chemical composition of gas-and aerosol-phase products from the photooxidation of naphthalene, J. Phys. Chem. A, 114, 913–934, 2009.
Knipping, E. M., Lakin, M. J., Foster, K. L., Jungwirth, P., Tobias, D. J., Gerber, R. B., Dabdub, D., and Finlayson-Pitts, B. J.: Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols, Science, 288, p. 301, https://doi.org/10.1126/science.288.5464.301, 2000.
Krapf, M., El Haddad, I., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prévôt, A. S., Baltensperger, U., and Dommen, J.: Labile peroxides in secondary organic aerosol, Chem, 1, 603–616, 2016.
Laskin, A., Gaspar, D. J., Wang, W., Hunt, S. W., Cowin, J. P., Colson, S. D., and Finlayson-Pitts, B. J.: Reactions at interfaces as a source of sulfate formation in sea-salt particles, Science, 301, p. 340, https://doi.org/10.1126/science.1085374, 2003.
Leng, C. B., Roberts, J. E., Zeng, G., Zhang, Y. H., and Liu, Y.: Effects of temperature, pH, and ionic strength on the Henry's law constant of triethylamine, Geophys. Res. Lett., 42, 3569–3575, https://doi.org/10.1002/2015gl063840, 2015.
Li, G., Bei, N., Cao, J., Huang, R., Wu, J., Feng, T., Wang, Y., Liu, S., Zhang, Q., Tie, X., and Molina, L. T.: A possible pathway for rapid growth of sulfate during haze days in China, Atmos. Chem. Phys., 17, 3301–3316, https://doi.org/10.5194/acp-17-3301-2017, 2017.
Li, J., Zhang, Y.L., Cao, F., Zhang, W., Fan, M., Lee, X., and Michalski, G.: Stable sulfur isotopes revealed a major role of transition-metal ion-catalyzed SO2 oxidation in haze episodes, Environ. Sci. Technol., 54, 2626–2634, https://doi.org/10.1021/acs.est.9b07150, 2020.
Lind, J. A., Lazrus, A. L., and Kok, G. L.: Aqueous phase oxidation of sulfur (IV) by hydrogen peroxide, methylhydroperoxide, and peroxyacetic acid, J. Geophys. Res.-Atmos., 92, 4171–4177, 1987.
Liu, C., Chen, T., Liu, Y., Liu, J., He, H., and Zhang, P.: Enhancement of secondary organic aerosol formation and its oxidation state by SO2 during photooxidation of 2-methoxyphenol, Atmos. Chem. Phys., 19, 2687–2700, https://doi.org/10.5194/acp-19-2687-2019, 2019.
Liu, L., Bei, N., Wu, J., Liu, S., Zhou, J., Li, X., Yang, Q., Feng, T., Cao, J., Tie, X., and Li, G.: Effects of stabilized Criegee intermediates (sCIs) on sulfate formation: a sensitivity analysis during summertime in Beijing–Tianjin–Hebei (BTH), China, Atmos. Chem. Phys., 19, 13341–13354, https://doi.org/10.5194/acp-19-13341-2019, 2019.
Liu, T., Clegg, S. L., and Abbatt, J. P. D.: Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles, P. Natl. Acad. Sci. USA, 117, 1354–1359, 2020.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Maaß, F., Elias, H., and Wannowius, K. J.: Kinetics of the oxidation of hydrogen sulfite by hydrogen peroxide in aqueous solution: ionic strength effects and temperature dependence, Atmos. Environ., 33, 4413–4419, https://doi.org/10.1016/S1352-2310(99)00212-5, 1999.
Mauldin, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V. M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
Mekic, M., Loisel, G., Zhou, W., Jiang, B., Vione, D., and Gligorovski, S.: Ionic-strength effects on the reactive uptake of ozone on aqueous pyruvic acid: Implications for air–sea ozone deposition, Environ. Sci. Technol., 52, 12306–12315, https://doi.org/10.1021/acs.est.8b03196, 2018.
Mekic, M., Zeng, J., Zhou, W., Loisel, G., Jin, B., Li, X., Vione, D., and Gligorovski, S.: Ionic strength effect on photochemistry of fluorene and dimethylsulfoxide at the air–sea interface: Alternative formation pathway of organic sulfur compounds in a marine atmosphere, ACS Earth Space Chem., 4, 1029–1038, https://doi.org/10.1021/acsearthspacechem.0c00059, 2020.
Mishra, H., Enami, S., Nielsen, R. J., Hoffmann, M. R., Goddard, W. A., and Colussi, A. J.: Anions dramatically enhance proton transfer through aqueous interfaces, P. Natl. Acad. Sci. USA, 109, 10228–10232, 2012.
Newland, M. J., Rickard, A. R., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide, Atmos. Chem. Phys., 15, 9521–9536, https://doi.org/10.5194/acp-15-9521-2015, 2015.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/C6CP00053C, 2016.
O'Brien, R. E., Wang, B., Kelly, S. T., Lundt, N., You, Y., Bertram, A. K., Leone, S. R., Laskin, A., and Gilles, M. K.: Liquid–liquid phase separation in aerosol particles: Imaging at the nanometer scale, Environ. Sci. Technol., 49, 4995-5002, https://doi.org/10.1021/acs.est.5b00062, 2015.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Qiu, J., Liang, Z., Tonokura, K., Colussi, A. J., and Enami, S.: Stability of monoterpene-derived α-hydroxyalkyl-hydroperoxides in aqueous qrganic media – relevance to the fate of hydroperoxides in aerosol particle phases, Environ. Sci. Technol., 54, 3890–3899, https://doi.org/10.1021/acs.est.9b07497, 2020.
Rodríguez-Sevilla, J., Álvarez, M., Limiñana, G., Díaz, M. C.: Dilute SO2 absorption equilibria in aqueous HCl and NaCl solutions at 298.15 K, J. Chem. Eng. Data, 47, 1339–1345, 2002.
Ruiz-Lopez, M. F., Francisco, J. S., Martins-Costa, M. T., and Anglada, J. M.: Molecular reactions at aqueous interfaces, Nat. Rev. Chem., 4, 459–475, 2020.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, 2012.
Sha, T., Ma, X., Jia, H., Tian, R., Chang, Y., Cao, F., and Zhang, Y.: Aerosol chemical component: Simulations with WRF-Chem and comparison with observations in Nanjing, Atmos. Environ., 218, 116982, https://doi.org/10.1016/j.atmosenv.2019.116982, 2019.
Shang, J., Li, J., Zhu, T.: Heterogeneous reaction of SO2 on TiO2 particles, Sci. China Chem., 53, 2637–2643, 2010.
Shi, G., Xu, J., Peng, X., Xiao, Z., Chen, K., Tian, Y., Guan, X., Feng, Y., Yu, H., Nenes, A., and Russell, A. G.: pH of aerosols in a polluted atmosphere: Source contributions to highly acidic aerosol, Environ. Sci. Technol., 51, 4289–4296, https://doi.org/10.1021/acs.est.6b05736, 2017.
Shi, Q., Davidovits, P., Jayne, J. T., Worsnop, D. R., and Kolb, C. E.: Uptake of gas-phase ammonia. 1. Uptake by aqueous surfaces as a function of pH, J. Phys. Chem. A, 103, 8812–8823, https://doi.org/10.1021/jp991696p, 1999.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid–liquid phase separation in aerosol particles: dependence on O:C, organic functionalities, and compositional complexity, Geophys. Res. Lett., 39, L19801, https://doi.org/10.1029/2012GL052807, 2012.
Song, S., Gao, M., Xu, W., Shao, J., Shi, G., Wang, S., Wang, Y., Sun, Y., and McElroy, M. B.: Fine-particle pH for Beijing winter haze as inferred from different thermodynamic equilibrium models, Atmos. Chem. Phys., 18, 7423–7438, https://doi.org/10.5194/acp-18-7423-2018, 2018.
Song, S., Gao, M., Xu, W., Sun, Y., Worsnop, D. R., Jayne, J. T., Zhang, Y., Zhu, L., Li, M., Zhou, Z., Cheng, C., Lv, Y., Wang, Y., Peng, W., Xu, X., Lin, N., Wang, Y., Wang, S., Munger, J. W., Jacob, D. J., and McElroy, M. B.: Possible heterogeneous chemistry of hydroxymethanesulfonate (HMS) in northern China winter haze, Atmos. Chem. Phys., 19, 1357–1371, https://doi.org/10.5194/acp-19-1357-2019, 2019.
Su, H., Cheng, Y., and Pöschl, U.: New multiphase chemical processes influencing atmospheric aerosols, air quality, and climate in the anthropocene, Accounts Chem. Res., 53, 2034–2043, https://doi.org/10.1021/acs.accounts.0c00246, 2020.
Sun, Y., Wang, Z., Fu, P., Jiang, Q., Yang, T., Li, J., and Ge, X.: The impact of relative humidity on aerosol composition and evolution processes during wintertime in Beijing, China, Atmos. Environ., 77, 927–934, 2013.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., and Claeys, M.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A, 110, 9665–9690, 2006.
Thornton, J. A., Braban, C. F., and Abbatt, J. P. D.: N2O5 hydrolysis on sub-micron organic aerosols: the effect of relative humidity, particle phase, and particle size, Phys. Chem. Chem Phys., 5, 4593–4603, https://doi.org/10.1039/B307498F, 2003.
Tie, X., Brasseur, G., Emmons, L., Horowitz, I., and Kinnison, D.: Effects of aerosols on tropospheric oxidants: a global model study, J. Geophys. Res.-Atmos., 106, 22931–22964, 2001.
Tong, H., Arangio, A. M., Lakey, P. S. J., Berkemeier, T., Liu, F., Kampf, C. J., Brune, W. H., Pöschl, U., and Shiraiwa, M.: Hydroxyl radicals from secondary organic aerosol decomposition in water, Atmos. Chem. Phys., 16, 1761–1771, https://doi.org/10.5194/acp-16-1761-2016, 2016.
Usher, C. R., Al-Hosney, H., Carlos-Cuellar, S., and Grassian,V. H.: A laboratory study of the heterogeneous uptake and oxidation of sulfur dioxide on mineral dust particles, J. Geophys. Res., 107, 4713, https://doi.org/10.1029/2002JD002051, 2002.
Varutbangkul, V., Brechtel, F. J., Bahreini, R., Ng, N. L., Keywood, M. D., Kroll, J. H., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Hygroscopicity of secondary organic aerosols formed by oxidation of cycloalkenes, monoterpenes, sesquiterpenes, and related compounds, Atmos. Chem. Phys., 6, 2367–2388, https://doi.org/10.5194/acp-6-2367-2006, 2006.
Veghte, D. P., Altaf, M. B., and Freedman, M. A.: Size dependence of the structure of organic aerosol, J. Am. Chem. Soc., 135, 16046–16049, 2013.
Wang, G., Zhang, R., Gomez, M. E., Yang, L., Zamora, M. L., Hu, M., Lin, Y., Peng, J., Guo, S., and Meng, J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, 2016.
Wang, S.: SO2 uptake .csv, figshare, https://doi.org/10.6084/m9.figshare.13078112.v1, 2020.
Wang, S., Ye, J., Soong, R., Wu, B., Yu, L., Simpson, A. J., and Chan, A. W. H.: Relationship between chemical composition and oxidative potential of secondary organic aerosol from polycyclic aromatic hydrocarbons, Atmos. Chem. Phys., 18, 3987–4003, https://doi.org/10.5194/acp-18-3987-2018, 2018.
Wang, S., Zhou, S., Tao, Y., Tsui, W. G., Ye, J., Yu, J. Z., Murphy, J. G., McNeill, V. F., Abbatt, J. P. D., and Chan, A. W. H.: Organic peroxides and sulfur dioxide in aerosol: Source of particulate sulfate, Environ. Sci. Technol., 53, 10695–10704, https://doi.org/10.1021/acs.est.9b02591, 2019.
Wang, X., Gemayel, R., Hayeck, N., Perrier, S., Charbonnel, N., Xu, C., Chen, H., Zhu, C., Zhang, L., Wang, L., Nizkorodov, S. A., Wang, X., Wang, Z., Wang, T., Mellouki, A., Riva, M., Chen, J., and George, C.: Atmospheric photosensitization: A new pathway for sulfate formation, Environ. Sci. Technol., 54, 3114–3120, 2020.
Wang, Y., Zhang, Q., Jiang, J., Zhou, W., Wang, B., He, K., Duan, F., Zhang, Q., Philip, S., and Xie, Y.: Enhanced sulfate formation during China's severe winter haze episode in January 2013 missing from current models, J. Geophys. Res.-Atmos., 119, 10425–10440, 2014.
Wei, H., Vejerano, E. P., Leng, W., Huang, Q., Willner, M. R., Marr, L. C., and Vikesland, P. J.: Aerosol microdroplets exhibit a stable pH gradient, P. Natl. Acad. Sci. USA, 115, 7272, https://doi.org/10.1073/pnas.1720488115, 2018.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., and Knote, C.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States,P. Natl. Acad. Sci. USA, 112, 37–42, 2015.
Yang, Y., Wang, H., Smith, S. J., Easter, R., Ma, P.-L., Qian, Y., Yu, H., Li, C., and Rasch, P. J.: Global source attribution of sulfate concentration and direct and indirect radiative forcing, Atmos. Chem. Phys., 17, 8903–8922, https://doi.org/10.5194/acp-17-8903-2017, 2017.
Yao, M., Zhao, Y., Hu, M., Huang, D., Wang, Y.C., Yu, J. Z., and Yan, N.: Multiphase reactions between secondary organic aerosol and sulfur dioxide: kinetics and contributions to sulfate formation and aerosol aging, Environ. Sci. Technol. Lett. 6, 768–774, 2019.
Ye, J., Gordon, C. A., and Chan, A. W. H: Enhancement in secondary organic aerosol formation in the presence of preexisting organic particle, Environ. Sci. Technol., 50, 3572–3579, 2016.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Yee, L. D., Isaacman-VanWertz, G., Wernis, R. A., Kreisberg, N. M., Glasius, M., Riva, M., Surratt, J. D., de Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Liu, Y., Misztal, P. K., Artaxo, P., Viegas, J., Manzi, A., de Souza, R. A. F., Edgerton, E. S., Baumann, K., and Goldstein, A. H.: Natural and anthropogenically influenced isoprene oxidation in southeastern United States and central Amazon, Environ. Sci. Technol., 54, 5980–5991, https://doi.org/10.1021/acs.est.0c00805, 2020.
You, Y., Renbaum-Wolff, L., and Bertram, A. K.: Liquid–liquid phase separation in particles containing organics mixed with ammonium sulfate, ammonium bisulfate, ammonium nitrate or sodium chloride, Atmos. Chem. Phys., 13, 11723–11734, https://doi.org/10.5194/acp-13-11723-2013, 2013.
You, Y., Smith, M. L., Song, M., Martin, S. T., and Bertram, A. K.: Liquid–liquid phase separation in atmospherically relevant particles consisting of organic species and inorganic salts, Int. Rev. Phys. Chem., 33, 43–77, https://doi.org/10.1080/0144235X.2014.890786, 2014.
Zhang, S., Xing, J., Sarwar, G., Ge, Y., He, H., Duan, F., Zhao, Y., He, K., Zhu, L., and Chu, B.: Parameterization of heterogeneous reaction of SO2 to sulfate on dust with coexistence of NH3 and NO2 under different humidity conditions, Atmos. Environ., 208, 133–140, 2019.
Zhao, Y., Liu, Y., Ma, J., Ma, Q., and He, H.: Heterogeneous reaction of SO2 with soot: The roles of relative humidity and surface composition of soot in surface sulfate formation, Atmos. Environ., 152, 465–476, 2017.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015.
Zheng, G. J., Duan, F. K., Su, H., Ma, Y. L., Cheng, Y., Zheng, B., Zhang, Q., Huang, T., Kimoto, T., Chang, D., Pöschl, U., Cheng, Y. F., and He, K. B.: Exploring the severe winter haze in Beijing: the impact of synoptic weather, regional transport and heterogeneous reactions, Atmos. Chem. Phys., 15, 2969–2983, https://doi.org/10.5194/acp-15-2969-2015, 2015.
Zheng, G. J., Su, H., Wang, S., Andreae, M. O., Pöschl, U., and Cheng, Y.: Multiphase buffer theory explains contrasts in atmospheric aerosol acidity, Science, 369, 1374–1377, https://doi.org/10.1126/science.aba3719, 2020.