the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Feb 2021
| 12 Feb 2021
Global trends and European emissions of tetrafluoromethane (CF4), hexafluoroethane (C2F6) and octafluoropropane (C3F8)
Alistair J. Manning
Luke M. Western
Dickon Young
Adam Wisher
Matthew Rigby
Stefan Reimann
Martin K. Vollmer
Michela Maione
Jgor Arduini
Paul B. Krummel
Jens Mühle
Christina M. Harth
Brendan Evans
Ray F. Weiss
Ronald G. Prinn
Simon O'Doherty
Perfluorocarbons (PFCs) are amongst the most potent greenhouse gases listed under the United Nations Framework Convention on Climate Change (UNFCCC). With atmospheric lifetimes on the order of thousands to tens of thousands of years, PFC emissions represent a permanent alteration to the global atmosphere on human timescales. While the industries responsible for the vast majority of these emissions – aluminium smelting and semi-conductor manufacturing – have made efficiency improvements and introduced abatement measures, the global mean mole fractions of three PFCs, namely tetrafluoromethane (CF4, PFC-14), hexafluoroethane (C2F6, PFC-116) and octafluoropropane (C3F8, PFC-218), continue to grow. In this study, we update baseline growth rates using in situ high-frequency measurements from the Advanced Global Atmospheric Gases Experiment (AGAGE) and, using data from four European stations, estimate PFC emissions for northwest Europe. The global growth rate of CF4 decreased from 1.3 ppt yr−1 in 1979 to 0.6 ppt yr−1 around 2010 followed by a renewed steady increase to 0.9 ppt yr−1 in 2019. For C2F6, the growth rate grew to a maximum of 0.125 ppt yr−1 around 1999, followed by a decline to a minimum of 0.075 ppt yr−1 in 2009, followed by weak growth thereafter. The C3F8 growth rate was around 0.007 ppt yr−1 until the early 1990s and then quickly grew to a maximum of 0.03 ppt yr−1 in 2003–2004. Following a period of decline until 2012 to 0.015 ppt yr−1, the growth rate slowly increased again to ∼ 0.017 ppt yr−1 in 2019. We used an inverse modelling framework to infer PFC emissions for northwest Europe. No statistically significant trend in regional emissions was observed for any of the PFCs assessed. For CF4, European emissions in early years were linked predominantly to the aluminium industry. However, we link large emissions in recent years to a chemical manufacturer in northwest Italy. Emissions of C2F6 are linked to a range of sources, including a semi-conductor manufacturer in Ireland and a cluster of smelters in Germany's Ruhr valley. In contrast, northwest European emissions of C3F8 are dominated by a single source in northwest England, raising the possibility of using emissions from this site for a tracer release experiment.
- Article
(6526 KB) - Full-text XML
-
Supplement
(168 KB) - BibTeX
- EndNote





Perfluorocarbons (PFCs) – fully fluorinated hydrocarbons – are a group of extremely potent greenhouse gases that are used extensively in the electronics and semi-conductor industry and are also emitted as a byproduct of aluminium and rare-earth metal smelting. As a consequence of their high global warming potentials (GWPs), PFCs were listed under the Kyoto “basket”, a collection of gases for which regulation on their emissions was introduced under the Kyoto Protocol (UNFCCC, 2009). Countries listed under Annex 1 of the protocol are required to report annual PFC emissions to the United Nations Framework Convention on Climate Change (UNFCCC).
Tetrafluoromethane (CF4, PFC-14) is the simplest and most abundant PFC in the atmosphere, with a reported global mean background mole fraction of 82.7 pmol mol−1 (dry-air mole fraction in parts per trillion; ppt) in 2016 (Prinn et al., 2018). CF4 has a GWP of 6500 over a 100-year time horizon (GWP100) (Burkholder et al., 2019) and, with an estimated atmospheric lifetime of 50 000 years (Burkholder et al., 2019), it is the longest-lived greenhouse gas known. Background atmospheric mole fractions of CF4 have risen sharply since the beginning of the industrial revolution. However, unlike other PFCs, which are solely anthropogenic in origin, CF4 is also emitted naturally from calcium fluorites (CaF2) present in continental crust (Trudinger et al., 2016). Harnisch and Eisenhauer (1998) estimated that the fluorite reservoir is sufficient to maintain an atmospheric background mole fraction of approximately 40 ppt. More recently, this was refined to 34.05 ± 0.33 ppt (Trudinger et al., 2016).
The predominant anthropogenic source of CF4 to the atmosphere is primary aluminium production. Kim et al. (2014) estimated that aluminium smelting accounted for ∼ 68 % of global emissions in 2010. Emissions from the smelting of aluminium ore occur predominantly during “anode effects”, when the feed of alumina to the cell is restricted resulting in formation of CF4 but also during routine operation (Wong et al., 2015). CF4 is also used commercially in the semi-conductor industry as an etchant gas for plasma etching and as a cleaning agent in chemical vapour deposition (CVD) tool chambers. In recent years, the smelting of rare-earth metals, such as neodymium, has also been cited as a small but growing source of CF4 (Cai et al., 2018; Zhang et al., 2018). Firn air measurements show that the atmospheric abundance of CF4 has increased rapidly since 1960 (Trudinger et al., 2016). While emissions peaked in 1980 and have since declined, Engel and Rigby (2019) report renewed growth in recent years.
Hexafluoroethane (C2F6, PFC-116) has an atmospheric lifetime of 10 000 years and a GWP100 of 9200 (Burkholder et al., 2019), making it the most potent PFC, and fourth most potent greenhouse gas, listed under the Kyoto basket in terms of its GWP100. With a global mean background mole fraction of 4.56 ppt in 2016 (Prinn et al., 2018), it is the second most abundant PFC in the atmosphere. The presence of C2F6 has not been detected in the pre-industrial atmosphere (Trudinger et al., 2016), and its sources are very similar to the anthropogenic sources of CF4, with aluminium production and the semi-conductor industry accounting for approximately one-third and two-thirds of the global budget in 2010, respectively (Kim et al., 2014). C2F6 is also a component of the refrigerant blend R-508 (50 %–70 % C2F6 by weight, 30 %–50 % trifluoromethane (CHF3) by weight), which is used in very low temperature refrigeration, though this is thought to be a minor source (Kim et al., 2014). Trudinger et al. (2016) reported peak global emissions of ∼ 3.6 Gg yr−1 in 2000, which was followed by a decline until 2010 and stabilisation thereafter (Engel and Rigby, 2019).
Octafluoropropane (C3F8, PFC-218) has an atmospheric lifetime of 2600 years and a GWP100 of 7000 (Burkholder et al., 2019). It is the fourth most abundant PFC, after perfluorocyclobutane (PFC-318, c-C4F8; see Mühle et al., 2019), with a global mean mole fraction of 0.63 ppt in 2016 (Prinn et al., 2018). The Emission Database for Global Atmospheric Research v4 (EDGAR, 2009) attributes C3F8 emissions to refrigeration/air-conditioning use and semi-conductor manufacture. While the aluminium industry is not thought to be a major source, low concentrations of C3F8 have been detected from smelter stacks (Fraser et al., 2003). At present, the aluminium industry does not account for C3F8 in their emissions reporting, and its low concentration means that it may not even be detectable by the instruments employed by the industry to monitor PFC emissions (Trudinger et al., 2016).
As a result of their exceedingly long lifetimes, PFC emissions represent a permanent (on human timescales) alteration to the atmosphere. PFC sinks are dominated by decomposition during high temperature combustion (Cicerone, 1979; Ravishankara et al., 1993; Morris et al., 1995). Both aluminium and semi-conductor industries have targeted PFCs for emissions reductions in an effort to curb greenhouse gas emissions (Trudinger et al., 2016).
In this study, we present atmospheric PFC measurements from the Advanced Global Atmospheric Gases Experiment (AGAGE) network. These data are used to update baseline mole fraction data and growth rates. The atmospheric measurements from European stations are used, in conjunction with an inverse modelling framework, to estimate PFC emissions for northwest Europe. We compare our emissions maps to the European Pollutant Release and Transfer Register (E-PRTR), which contains a record of PFC emissions from industrial facilities. Finally, we explore potential of using one such PFC-emitting facility as a release location for a tracer experiment.
2.1 Instrumentation
Long-term in situ PFC measurements were made by the AGAGE (Prinn et al., 2018) network. With the exception of Monte Cimone (CMN), all stations were equipped with a Medusa pre-concentration system coupled with a gas chromatograph (GC, Agilent) and quadrupole mass selective detector (MSD) (Miller et al., 2008). At CMN, measurements of C3F8 (neither CF4 or C2F6 were measured) were made using an in-line auto sampler and pre-concentration device coupled to a GC mass spectrometer (Maione et al., 2013). Observations from the five “core” AGAGE stations – Mace Head (MHD), Ireland; Trinidad Head (THD), USA; Ragged Point (RPB), Barbados; Cape Matatula (SMO), American Samoa; and Cape Grim (CGO), Tasmania – were used to infer global trends. Observations from MHD; Jungfraujoch (JFJ), Switzerland; Tacolneston (TAC), United Kingdom; and CMN, Italy were used to infer northwest European emissions (Fig. 1). Station details are given in Table 1.
Table 1Station names, locations and inlet heights for the instruments used to quantify global and regional PFC emissions. Note that for the mountain site, JFJ, the inlet is situated below the instrument.
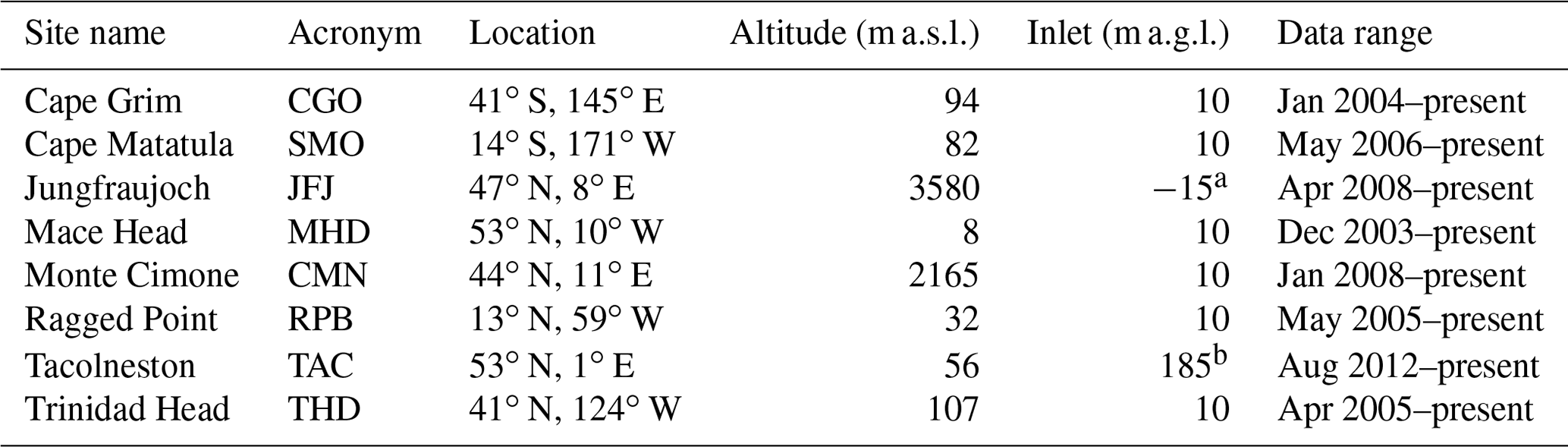
a The original JFJ inlet was situated at 10 m a.g.l. The instrument began sampling from the −15 m a.g.l. inlet on 15 August 2012. b The original TAC inlet was situated at 100 m a.g.l. The instrument began sampling from the 185 m a.g.l. inlet on 10 March 2017.
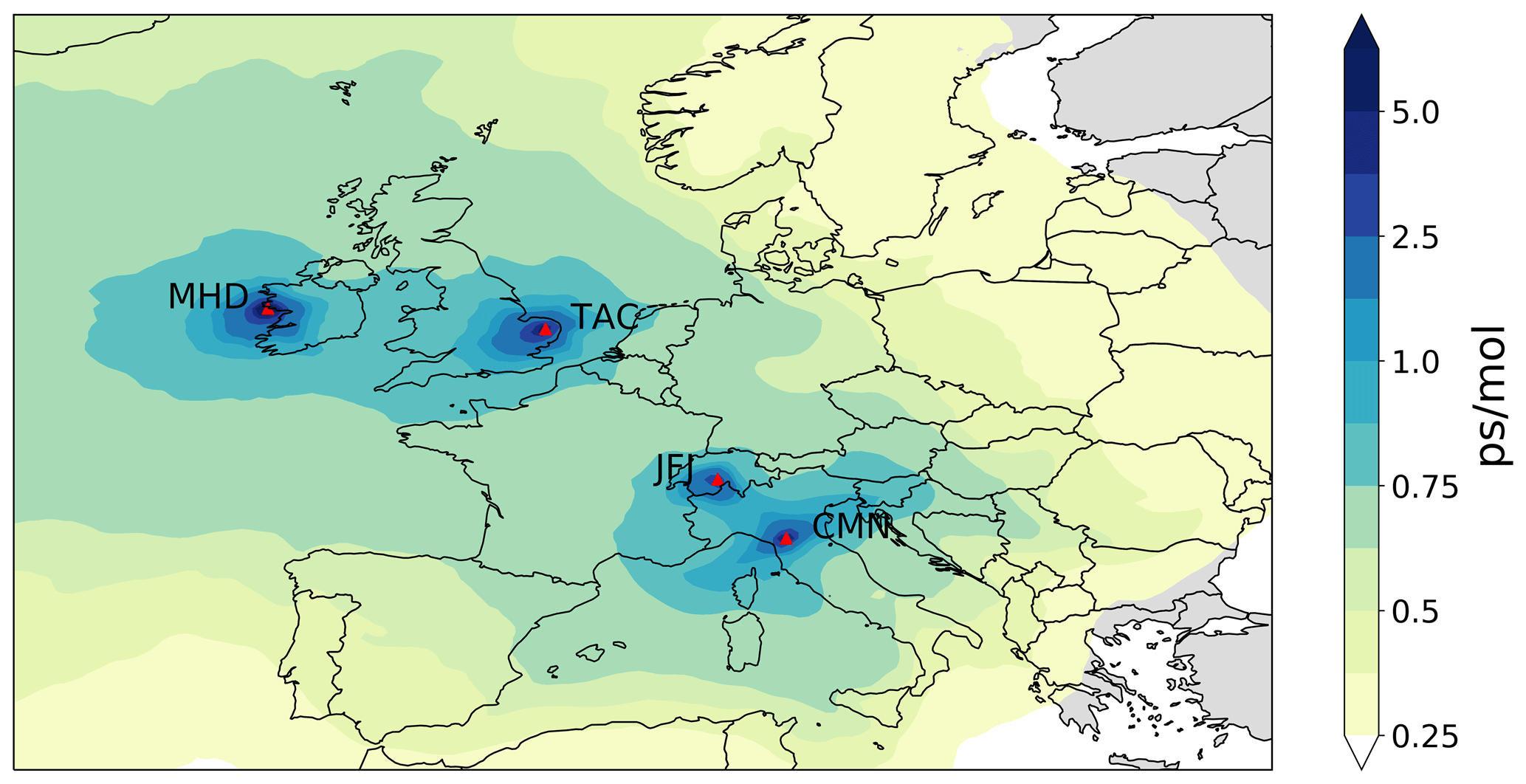
Figure 1Average footprint emission sensitivity in picoseconds per mole (ps mol−1) obtained from NAME 30 d backward calculations for the four measurement sites – Mace Head (MHD), Tacolneston (TAC), Jungfraujoch (JFJ) and Mt. Cimone (CMN) – during 2015. Measurement sites are marked as red triangles.
Each air measurement was bracketed by analysis of a working (quaternary) standard to account for short-term drifts in detector sensitivity. At JFJ and TAC, every two air measurements were bracketed. Quaternary standards were linked to a set of primary calibration scales – SIO-05 for CF4 and SIO-07 for C2F6 and C3F8 – via a hierarchy of compressed real-air standards held in 34 L internally electro-polished stainless-steel canisters (Essex Industries, Missouri, USA). The estimated absolute accuracy of the calibration scales is ∼ 1.5 %, ∼ 2 % and ∼ 4 % for CF4, C2F6 and C3F8, respectively. Average measurement precisions, estimated as the standard deviation of the bracketing standards for all sites across all years, were estimated to be ∼ 0.18 %, ∼ 0.77 % and ∼ 2.70 % for CF4, C2F6 and C3F8, respectively. Mass spectrometers were run in selective ion mode (SIM). CF4 was detected on its base (most abundant) ion, with a mass over charge ratio () of 69, and 50 was used as a qualifier. C2F6 was monitored on 119 with a qualifier ion of 69, and C3F8 was monitored on 169 with 119 as a qualifier. For each gas, the ratio of target to qualifying ion was continuously monitored to ensure that co-eluting species did not interfere with the analysis. Weekly system blanks were conducted to test for system leaks and/or carrier gas impurities. MHD and TAC showed small blanks for all three PFCs. These were carefully assessed for each carrier gas cylinder. Where the blank variability was negligible, all measurements made using that cylinder were blank corrected. Measurements coinciding with high and/or variable blanks were rejected.
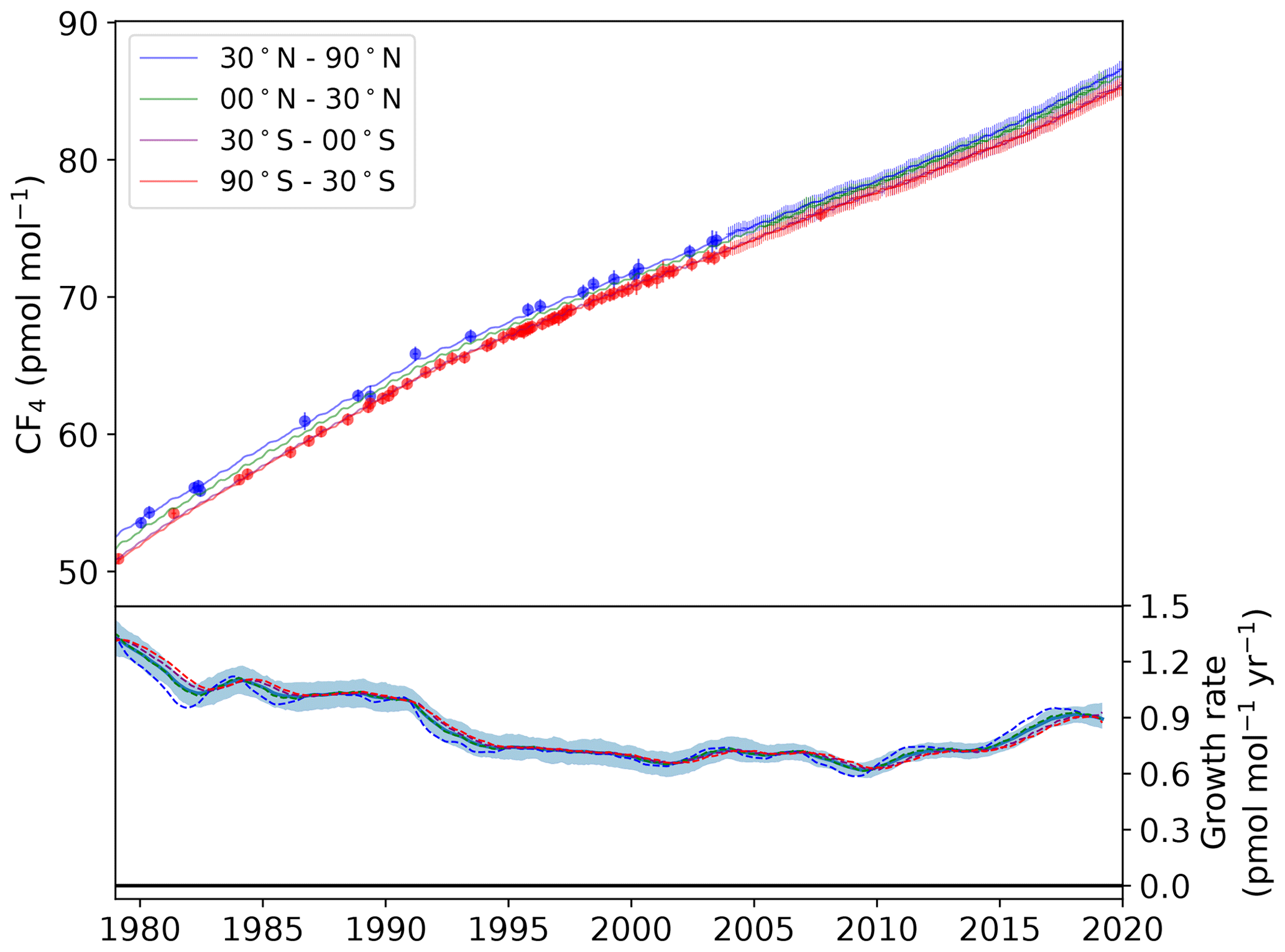
Figure 2Modelled semi-hemispheric monthly average CF4 mole fractions (30–90∘ N: blue; 0–30∘ N: green; 30–0∘ S: purple; 90–30∘ S: red). Monthly averaged baseline observations are shown as data points with 1σ error bars. The more sparse filled circles represent northern (blue) and southern (red) hemispheric air archive samples. The solid trend lines were calculated using the AGAGE 12-box model with emissions from the inversion as input. The lower plot shows the model-derived mole fraction growth rate, smoothed with an approximate 1-year filter, for each semi-hemisphere (dashed lines) and the global mean with 1σ uncertainty (solid line and shading).
2.2 Archived air samples
The atmospheric histories of CF4, C2F6 and C3F8 were extended backwards in time via analysis of northern and southern hemispheric archived air samples (Figs.2–4). A full description of the collection and analysis of these samples can be found in Mühle et al. (2010) and Trudinger et al. (2016). In short, northern hemispheric archive samples, which were provided by a range of laboratories, were filled under baseline conditions using a range of filling techniques and for different purposes. The southern hemisphere archive samples are part of the Cape Grim air archive (CGAA; Fraser et al., 2003) and were cryogenically filled into electro-polished stainless-steel cylinders during baseline conditions. Northern and southern hemispheric samples were analysed at the Scripps Institution of Oceanography and the Commonwealth Scientific and Industrial Research Organisation (CSIRO), Aspendale, Australia, using Medusa gas chromatography – mass spectrometry (GCMS) instruments. Each archive sample was analysed in replicate. Non-linearity data were collected prior to, during and after each sample analysis. Blank runs were also conducted regularly, with blank corrections applied where needed.
2.3 Global emissions estimation
We estimate global emissions using the well-established two-dimensional AGAGE 12-box model, which simulates seasonally varying but annually repeating transport (Cunnold et al., 1983, 2002; Rigby et al., 2013). The model simulates monthly background semi-hemispheric abundances of trace gases given the emissions from the surface into the semi-hemispheres. The model is split into four lower and upper tropospheric boxes and four stratospheric boxes. These boxes are divided zonally at and the Equator, and vertically at 1000, 500, 200 and 0 hPa. The model performs well when the lifetime of the gases are long compared to the interhemispheric exchange time, which makes it well suited to our application due to the long lifetimes of the PFCs, which is considered within the model.
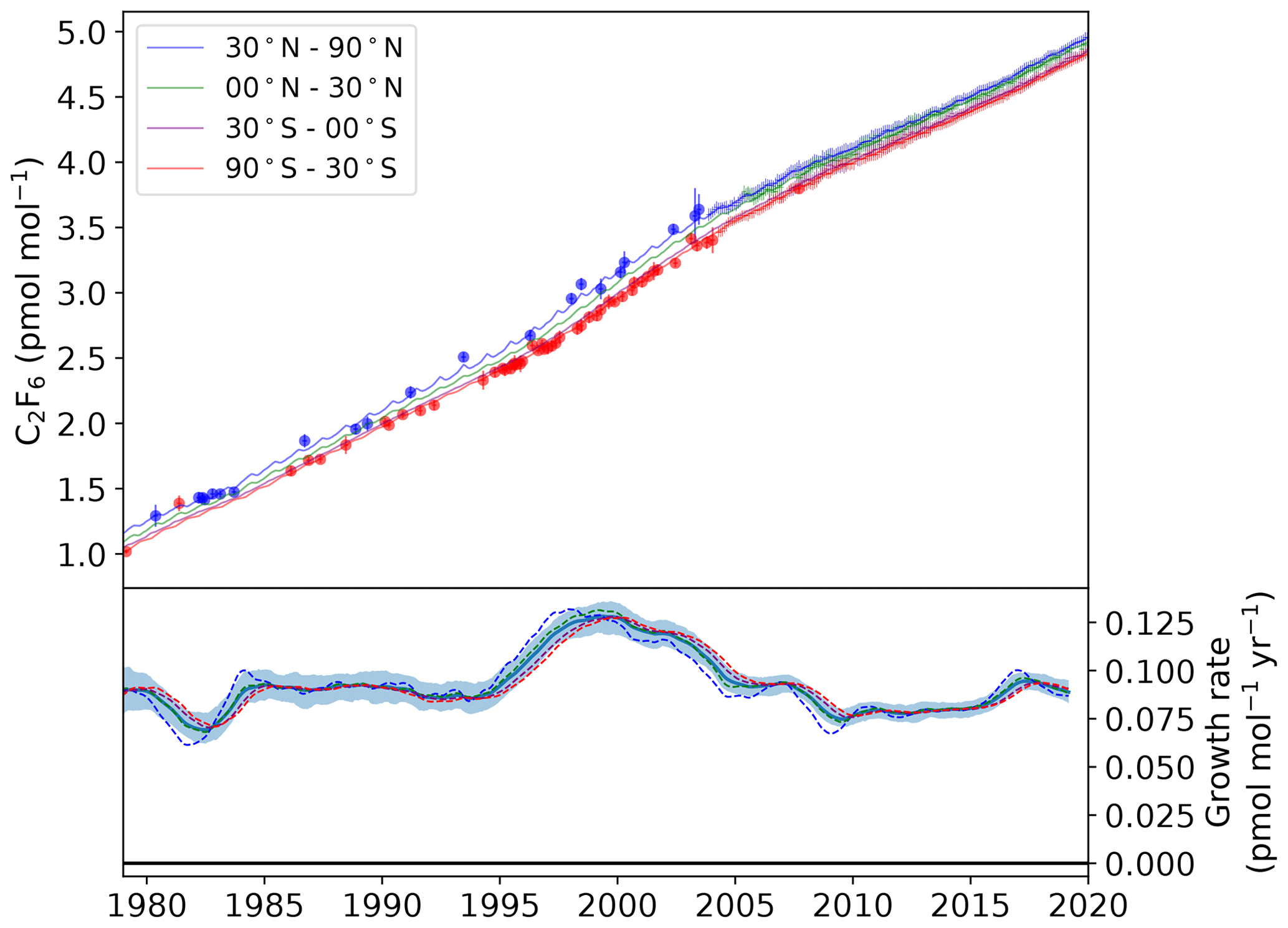
Figure 3Modelled semi-hemispheric monthly average C2F6 mole fractions (30–90∘ N: blue; 0–30∘ N: green; 30–0∘ S: purple; 90–30∘ S: red). Monthly averaged baseline observations are shown as data points with 1σ error bars. The more sparse filled circles represent northern (blue) and southern (red) hemispheric air archive samples. The solid trend lines were calculated using the AGAGE 12-box model with emissions from the inversion as input. The lower plot shows the model-derived mole fraction growth rate, smoothed with an approximate 1-year filter, for each semi-hemisphere (dashed lines) and the global mean with 1σ uncertainty (solid line and shading).
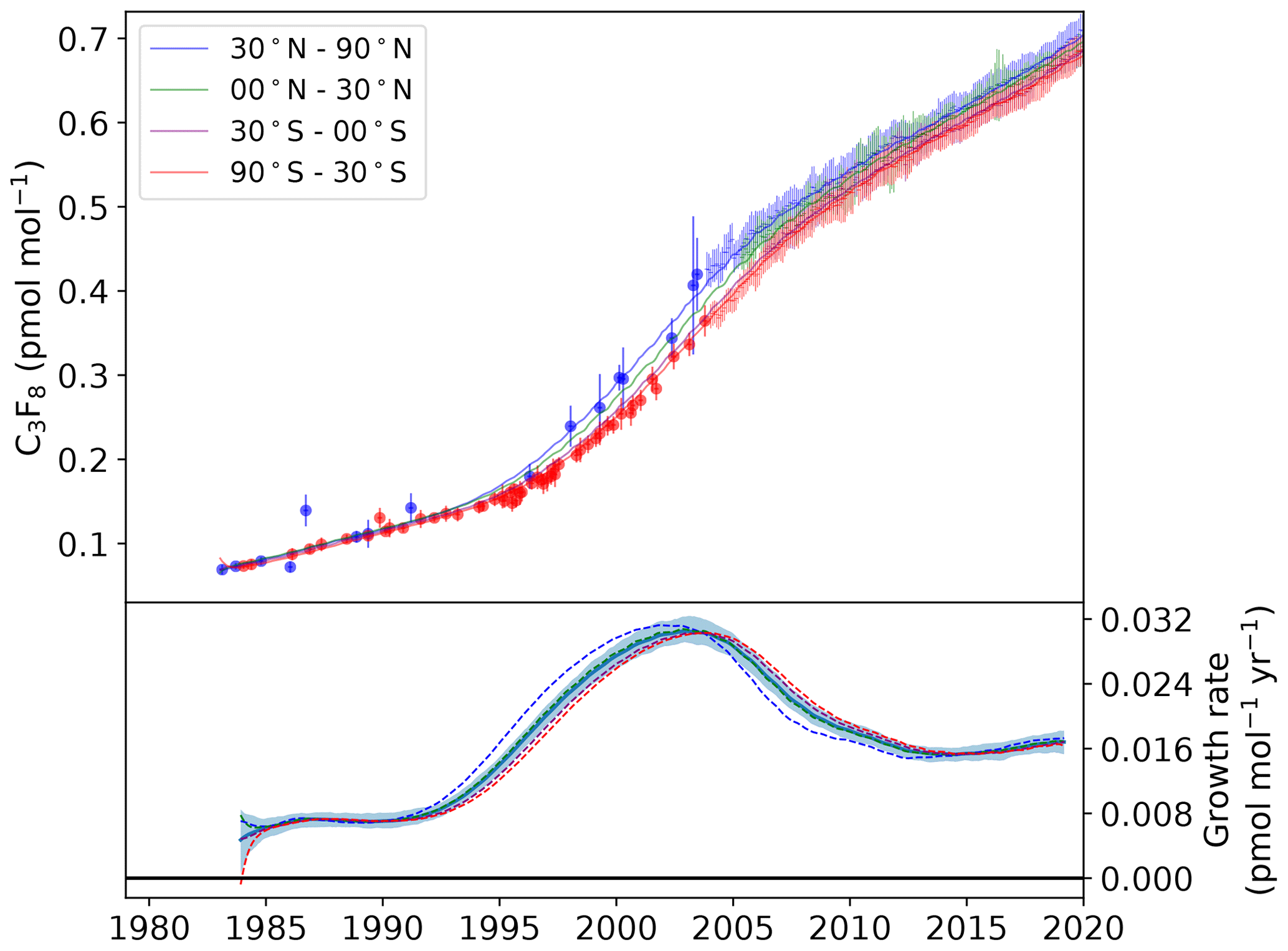
Figure 4Modelled semi-hemispheric monthly average C3F8 mole fractions (30–90∘ N: blue; 0–30∘ N: green; 30–0∘ S: purple; 90–30∘ S: red). Monthly averaged baseline observations are shown as data points with 1σ error bars. The more sparse filled circles represent northern (blue) and southern (red) hemispheric air archive samples. The solid trend lines were calculated using the AGAGE 12-box model with emissions from the inversion as input. The lower plot shows the model-derived mole fraction growth rate, smoothed with an approximate 1-year filter, for each semi-hemisphere (dashed lines) and the global mean with 1σ uncertainty (solid line and shading).
Estimates of global emissions are derived using a Bayesian method in which global emission growth rates are constrained a priori (following Rigby et al., 2014). Here, we assumed that, in the absence of observations, the annual emissions growth rate would be zero plus or minus 20 % of the maximum emissions from the Emissions Database for Global Atmospheric Research v4.2 (EDGAR, 2009). The derived emissions were not found to be sensitive to reasonable changes to this constraint. The estimate is informed using monthly mean baseline-filtered observations from the five background AGAGE stations (MHD, THD, RPB, SMO and CGO), averaged into semi-hemispheric monthly means (see Rigby et al., 2013). Prior to 2004, the inversion is constrained by archived air samples from the CGAA (Fraser et al., 2003) and various archived northern hemispheric samples (Mühle et al., 2010). Model–measurement uncertainties are assumed to be equal to the monthly baseline variability. For archived air samples, this term is assumed to be equal to the mean variability in the high-frequency baseline data, scaled by the mole fraction difference between the high-frequency mean and archived air sample. For the archived samples, the repeatability of each measurement was included in the model–measurement uncertainty. These uncertainties are propagated through the inversion, along with the uncertainty due to the prior constraint, to calculate the posterior emissions uncertainty.
2.4 Estimating European emissions using a regional inverse modelling technique
We infer regional PFC emissions by combining atmospheric measurements (Sect. 2.1) and air histories, derived using the atmospheric-dispersion model NAME (Jones et al., 2007), within the Met Office's Inversion Technique for Emissions Modelling (InTEM) framework. Numerous examples exist within the literature that describe the use of InTEM for the estimation of long-lived greenhouse gas emissions (e.g. Manning et al., 2011; Say et al., 2016; Arnold et al., 2018; Rigby et al., 2019; Mühle et al., 2019). In short, the simulated transport of gas in the NAME atmospheric transport model creates a sensitivity matrix that maps the spatial surface emissions to a modelled measurement. Meteorology from the UK Met Office Unified Model (UM) (Walters et al., 2019), including the nested high-resolution UK model (UKV) from 2014 onwards, drives the transport through advection, diffusion and turbulence in NAME. The global UM and UKV are available at 3- and 1-hourly resolutions, respectively. The horizontal resolution of the global UM has increased from ∼ 40 km in 2003 to ∼ 12 km in 2020; the UKV is at ∼ 1.5 km over the UK and Ireland. The NAME output spatial latitudinal by longitudinal resolution is 0.234∘ by 0.352∘. We regard measurements as being sensitive to emissions from the surface when simulated particles in the model fall below 40 m above ground level (a.g.l.) within a 30 d period prior to the measurement. NAME simulates the release of 20 000 particles per hourly measurement interval within the computational domain −98 to 40∘ E and 11 to 79∘ N. At MHD and TAC, these particles were released from a 20 m vertical line centred on the sample inlet. At JFJ and CMN, the mountain meteorology presents additional challenges for the model. At these stations, we assumed a release point of 1000 m a.g.l. for JFJ and 500 m a.g.l. for CMN, representing a compromise between the model surface altitude and the actual station height. The NAME model was run offline prior to the inversion and tailored to create a sensitivity matrix for a specific gas following Arnold et al. (2018).
InTEM estimates the spatial emissions of CF4, C2F6 and C3F8 by combining measurements, the sensitivities generated using NAME, any prior knowledge available about the emissions sources and magnitudes, and the uncertainty in these quantities. InTEM is a Bayesian inversion system that uses a non-negative least-squares solver. This is described in detail in Arnold et al. (2018). The baseline, or background, mole fraction is solved for within the inversion system. Prior baselines were estimated for MHD, JFJ and CMN using the measurements at those sites. The prior baseline for TAC was assumed to be equivalent to the prior MHD baseline, since they occupy similar latitudes.
The measurement uncertainty was estimated to be a combination of the precision described in Sect. 2.1 and the variability of three consecutive measurements centred around the measurement point. The observations were averaged into 4 h windows. The model uncertainty was a combination of the prior baseline uncertainty and the magnitude of the median pollution event at the measurement location per year. Observations recorded at TAC were selected when the difference between CH4 observations (made using a Picarro G2301) at different heights on the mast during the 4 h period were less than 20 ppb; i.e. the air was well mixed in the vertical. Analysis of the meteorology during these selected times provided thresholds that were applied at MHD which only has one measurement height. Off-diagonal elements of the model–observation uncertainty matrix were calculated by assuming a temporal correlation timescale of 12 h. The sensitivity of the results to this arbitrary value is low for reasonable changes – tests with 8 and 16 h windows showed that there was little discernible impact on the annually estimated national emissions.
For each inversion, the system used an a priori estimate of emissions which were distributed uniformly over land, on which a prior distribution with very large uncertainty was placed, ensuring that the posterior solution was informed entirely by the atmospheric measurements. Only two constraints were applied a priori: (1) all grid cells were forced to have a non-negative emission; (2) the nine grid cells centred on the locations of known PFC emitters, as reported to the E-PRTR, were specifically solved for. The E-PRTR only reports total PFC emissions (e.g. the sum total of all individual PFCs). Therefore, we define all E-PRTR locations for all three PFCs. Elsewhere in the domain, the spatial resolution of the underlying grid was allowed to vary within each country, with finer resolution in areas found to have high emissions and high sensitivity to the measurements.
3.1 Atmospheric trends and global emissions
The modelled and measured baseline atmospheric trends (1979–2019) of CF4, C2F6 and C3F8 are shown in Figs. 2–4. Prior to the calculation of monthly baseline estimates, the AGAGE pollution algorithm (Cunnold et al., 2002) was used to remove regional pollution effects observed at each station.
For CF4, a large increase in baseline mole fraction is evident across all semi-hemispheres. The global mean increased from 52.1 ppt in 1979 to 85.5 ppt in 2019, representing an approximate 3-fold increase following subtraction of the natural background. We observed significant variations in growth rate across the measurement window. In 1979, the growth rate was estimated to be 1.3 ppt yr−1 but declined steadily to a minimum of 0.6 ppt yr−1 by 2009. The drop in growth rate observed around 2009 is probably due to a fall in aluminium production following the 2008 financial crisis. Production of primary aluminium dropped by roughly 6 % (∼ 200 000 metric tonnes) between 2008 and 2009 (IAI, 2020). Global emissions declined by 0.6 Gg over the same period (Table 2). However, more recent years have seen a renewed increase in the growth rate of CF4, rising from 0.7 ppt yr−1 in 2010 to 0.9 ppt yr−1 in 2019. At 14.1 Gg yr−1, emissions in 2018 are the largest observed since high-frequency measurements began in 2003. Our work is consistent with Trudinger et al. (2016) and Engel and Rigby (2019), who showed the first increase in global CF4 emissions since the 1980s. Despite this, given global primary aluminium production has increased ∼ 5-fold over the last 40 years (IAI, 2020), our estimates highlight the success of efficiency improvements and abatement technology in reducing emissions of CF4 per tonne of aluminium produced.
Table 2Global annual PFC emissions, estimated using the AGAGE 12-box model – an extension of the work by Rigby et al. (2014), Trudinger et al. (2016) and Engel and Rigby (2019) – in gigagrams per year (Gg yr−1). Lower and upper uncertainty bounds correspond to the 16th and 84th percentiles of the posterior model distribution, respectively.

The global C2F6 baseline mole fraction grew from 1.1 ppt in 1979 to 4.8 ppt in 2019, an increase of more than 4-fold. The relative increase is larger than that of CF4, suggesting that there are major sources of C2F6 not linked to the aluminium industry. The growth rate peaked in 1999 at an estimated 0.13 ppt yr−1, followed by a sustained period of decline to a minimum of 0.07 ppt yr−1 in 2009. As with CF4, the minimum rate of growth in 2009 is probably a result, at least in part, of the reduced demand for aluminium following the 2008 financial crisis. The effect of the crisis on demand for electronics, and therefore the consumption of C2F6 as an etchant gas, is not known. The 2009 minimum was followed by a period of stagnation, with a near constant growth rate between 2009 and 2013. Annual global emissions did not vary significantly during this period, remaining stable at ∼ 1.9 Gg yr−1. However, there is some evidence for a resurgence in C2F6 emissions post-2013. The global growth rate in 2017 was estimated to be 0.09 ppt yr−1, with corresponding global emissions of 2.3 Gg yr−1, the largest observed since prior to the financial crisis.
The global C3F8 baseline mole fraction grew from an estimated 0.07 ppt in 1983 to 0.68 ppt in 2019. The large almost 10-fold increase shows that, in terms of relative growth, emissions of C3F8 have increased sharply when compared to CF4 and C2F6. Unlike CF4 and C2F6, the aluminium industry is not a major contributor of global emissions of C3F8, though detectable concentrations have been observed in the outflow from smelter stacks (Fraser et al., 2003). Following a period of relative stability between 1985 and 1992, the growth rate increased rapidly, reaching a maximum of 0.03 ppt yr−1 in 2003. Thereafter, a period of steady decline saw the growth rate fall to 0.015 ppt yr−1 in 2014, with corresponding global emissions of 0.51 (0.48–0.55) Gg yr−1. Of the three PFCs discussed, C3F8 is the only gas for which a pronounced “dip” in growth rate was not observed around the time of the financial crisis, perhaps indicative of the resilience of the semi-conductor industry to the crisis relative to aluminium producers. Since 2015, the global growth rate has remained comparatively stable, with no statistically significant trend in global emissions. Emissions in 2019 were estimated to be 0.56 (0.51–0.60) Gg yr−1.
3.2 Northwest European PFC emissions
We present estimates of PFC emissions in northwest Europe (the United Kingdom; Ireland; France; Belgium, the Netherlands and Luxembourg (collectively termed Benelux); and Germany) using the procedure described in Sect. 2.4, based on the sensitivity of the measurements to emissions from these countries (see Fig. 1).
From 2005 to 2007 (2005 to 2010 for CF4), we only report emissions for the UK, Ireland and Benelux, due to the lack of atmospheric measurements from continental Europe and therefore sensitivity to southern France and eastern Germany during this period. Reported estimates for France and Germany (and the northwest Europe total) begin in 2008 (C2F6 and C3F8) and 2010 (CF4), corresponding with the availability of measurements from JFJ (Table 1).
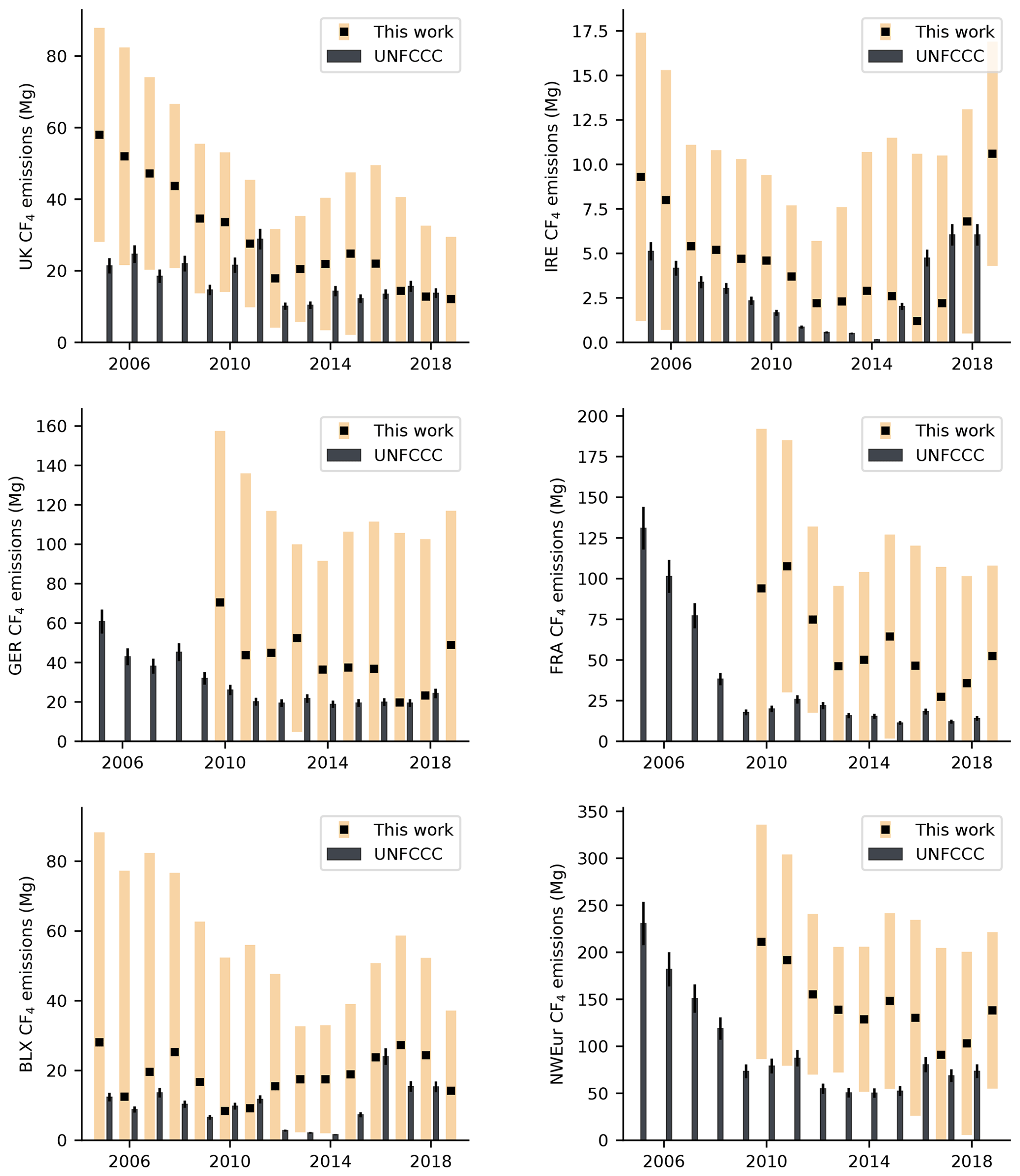
Figure 5Annual CF4 emissions (2005–2019) for northwest European countries in Mg yr−1. InTEM estimates are shown as black squares with pale orange uncertainty bounds. Emissions reported to the UNFCCC (sum of individual reporting countries) are shown as black bars, with an assumed uncertainty (black error bars) of 10 %. Note that UNFCCC data are only available up until 2018 inclusive.
3.2.1 CF4
Annual CF4 emissions for northwest Europe are shown in Fig. 5 and Table S1. Due to the considerable uncertainties, we find no statistically significant trend in emissions from the region over the measurement period (based on the 95 % uncertainty in the trend).
European aluminium production dropped significantly during the measurement period, declining from 3.2 MT in 2004, to just 2.2 MT in 2016 (IAI, 2020). In that time, the number of active European aluminium smelters also declined, falling from 25 to 16. On average, northwest Europe accounted for 2.1 % of global emissions in 2010 (0.21 Gg in northwest Europe; 10.2 Gg globally) but only 0.7 % in 2018 (0.1 Gg in northwest Europe; 14.1 Gg globally). Despite no significant trend in northwest European emissions, global emissions increased considerably over the same period (Table 2), indicating that emissions from other regions have increased. China was the largest producer of primary aluminium in 2019 (IAI, 2020). Comparison of our northwest European estimates with compiled emissions reported to the UNFCCC indicates a discrepancy between reporting methods (Fig. 5) throughout most of the reporting window. Our estimates are typically larger than the inventory, but some agreement is observed from 2016 onwards.
Of the individual countries/regions examined, Ireland is the only country whose reported emissions to the UNFCCC have increased in recent years. The InTEM estimates mirror this trend, though the uncertainties are considerable. Our estimates increased from 2.2 (0.0–5.7) Mg yr−1 in 2012 to 10.6 (4.3–16.9) Mg yr−1 in 2019. Ireland is not a producer of aluminium metal, though it does have a bauxite refinery. Therefore, its emissions of CF4 are probably due to consumption by semi-conductor manufacturers. Across all years of the E-PRTR (2007–2017), Ireland only reported emissions from a single facility, a semi-conductor factory to the west of Dublin. From 2017 onwards, this source is evident in our spatial maps. It is therefore likely that emissions from this site are the main driver of the recent observed increase in Ireland's CF4 emissions.
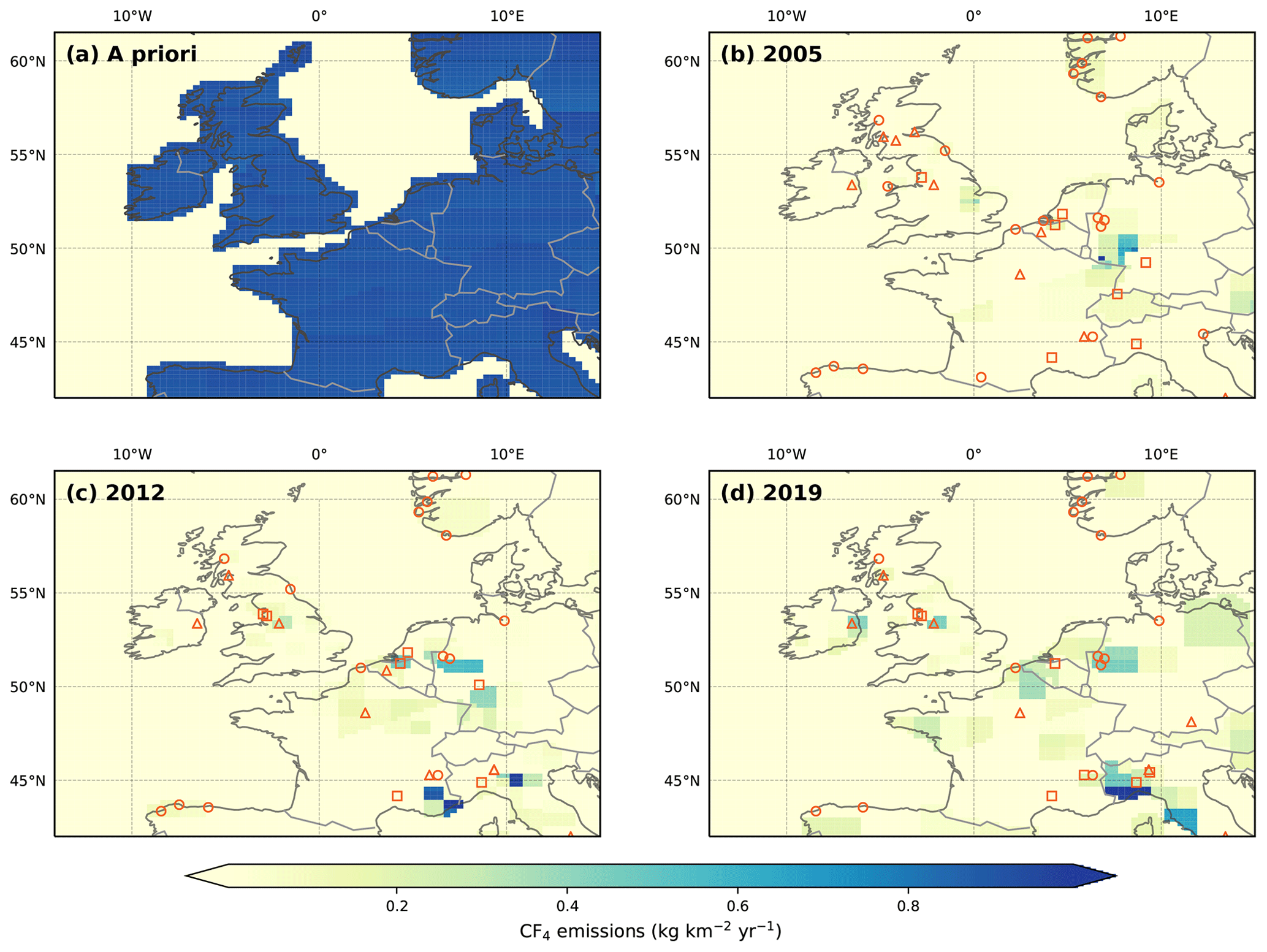
Figure 6Northwest European CF4 emissions in kg km−2 yr−1. (a) The a priori emissions field. With the exception of the oceans, emissions were distributed uniformly across the model domain. A posteriori emissions are shown for (b) 2005, (c) 2012 and (d) 2019. Facilities that reported PFC emissions to the E-PRTR in the selected year are shown as orange circles (aluminium smelters), triangles (electronics manufacturers) and squares (chemical manufacturers, including petroleum products). Since the reporting period for the E-PRTR is shorter than that of our measurements, 2005 and 2019 emissions are compared to the earliest (2007) and latest (2017) years of the E-PRTR database, respectively. Note that the E-PRTR database reports cumulative PFC emissions.
The inferred spatial distribution of northwest European CF4 emissions is shown in Fig. 6. In 2005, regional emissions were dominated by those from western Germany, roughly consistent (accounting for transport errors) with the location of three aluminium smelters in the Ruhr valley. Aluminium production is also the probable source of smaller emissions from southwest Norway and northern Denmark, although we do not report national estimates for these countries. Aluminium production in the Ruhr valley in Germany remains a significant source of CF4 in later years, though emissions from other areas and industries become apparent. Starting in 2010, strong emissions were found for southeast France and northwest Italy. Southeast France has previously been linked to emissions of other halogenated species (Maione et al., 2014). Our results show that emissions from this region diminished after 2015. In contrast, emissions from northwest Italy continued to grow until the end of the record. The E-PRTR lists two PFC emitters in the region. The largest of these sources, a chemicals manufacturer located near the Italian city of Alessandria, is consistent with our emissions maps after 2010. This manufacturer reported total PFC emissions of 185 Mg in 2012, making it one of the largest emitters in Europe. Interestingly, this region has previously been linked to considerable emissions of the hydrofluorocarbon, trifluoromethane (CHF3, HFC-23) (Keller et al., 2011), though we are unable to ascertain whether theses gases share a common source.
In comparison to other large European countries, emissions from the UK and Ireland are small throughout the reporting window. Between 2005 and 2012, emissions from northern England are consistent with the location of the Lynemouth aluminium smelter, which was closed in March 2012. After this date, the predominant UK source is located near the city of Manchester, consistent with the location of an electronics manufacturer.
Emissions from the Benelux region are small and the uncertainties are large. In later years, the majority of these emissions are consistent with the location of chemical manufacturers. These emissions are in a similar location to a strong source of c-C4F8 reported by Mühle et al. (2019), which the authors attributed to the consumption of c-C4F8 as an intermediate feed stock in the manufacture of polytetrafluoroethylene (PTFE). There is no available evidence supporting the use of CF4 in PTFE manufacture, though the production of CF4, perhaps as a byproduct, cannot be ruled out.
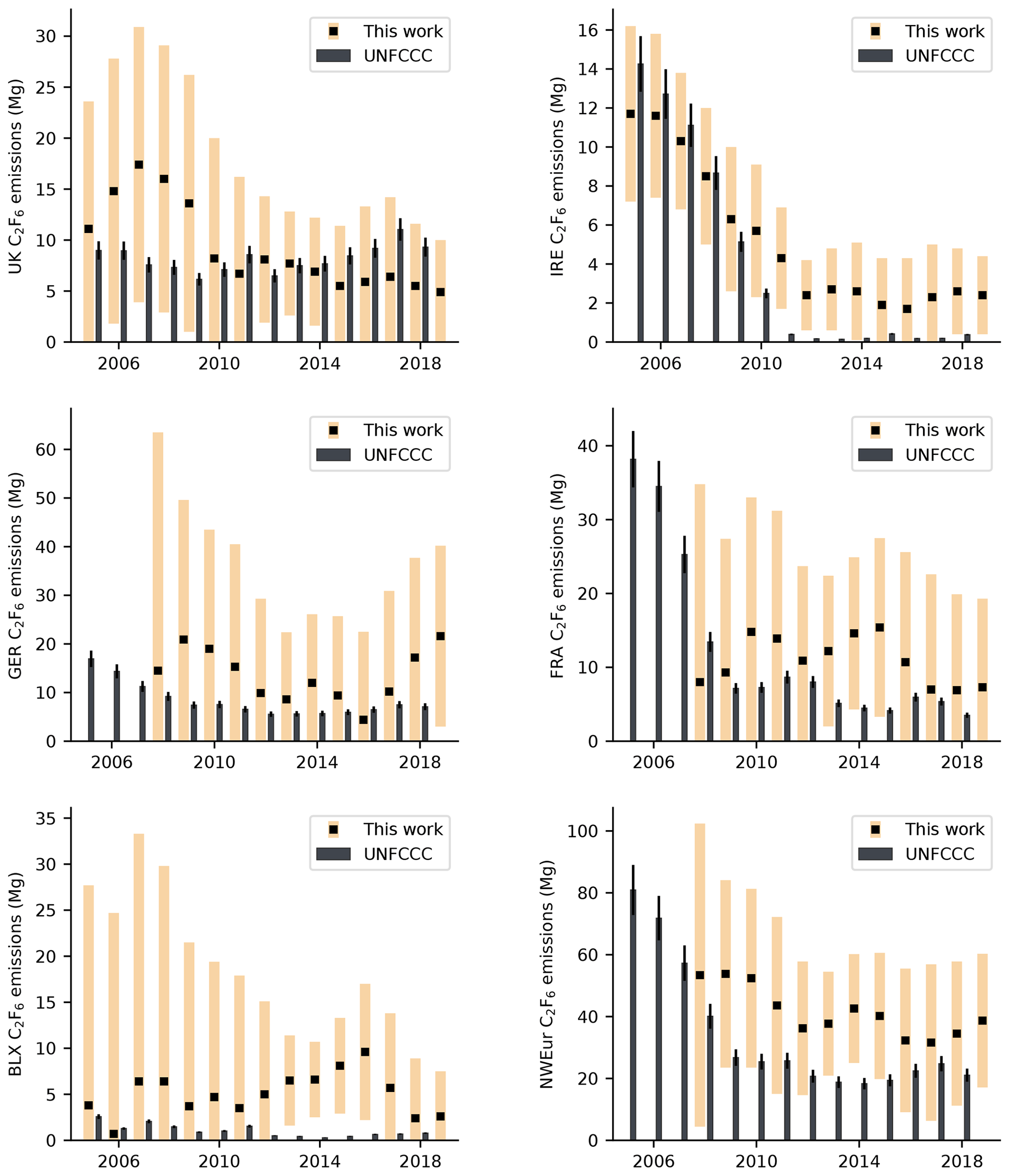
Figure 7Annual C2F6 emissions (2005–2019) for northwest European countries in Mg yr−1. InTEM estimates are shown as black squares with pale orange uncertainty bounds. Emissions reported to the UNFCCC (sum of individual reporting countries) are shown as black bars, with an assumed uncertainty (black error bars) of 10 %. Note that UNFCCC data are only available up until 2018 inclusive.
3.2.2 C2F6
Annual C2F6 emissions are shown in Fig. 7 and Table S2. Like CF4, the uncertainty of our estimates for Germany and France, particularly in early years, is large and overall, there is no statistically significant trend in emissions from northwest Europe over the measurement period (based on the 95 % uncertainty in the trend). When compared to global emissions, the average contribution of northwest Europe declined from 2.4 % in 2008 (0.05 Gg in northwest Europe; 2.1 Gg globally) to 1.6 % in 2018 (0.03 Gg in northwest Europe; 2.2 Gg globally).
Our work is in reasonable agreement with C2F6 emissions reported to the UNFCCC, particularly for Ireland between 2006 and 2011, where our top-down estimates captured the significant fall in emissions reported in Ireland's national inventory. Elsewhere, our uncertainties typically overlap the average UNFCCC estimates, with the exception of Benelux, where our emissions for 2013–2016 are significantly larger than the inventory, suggesting an under-reporting of emissions during this period.
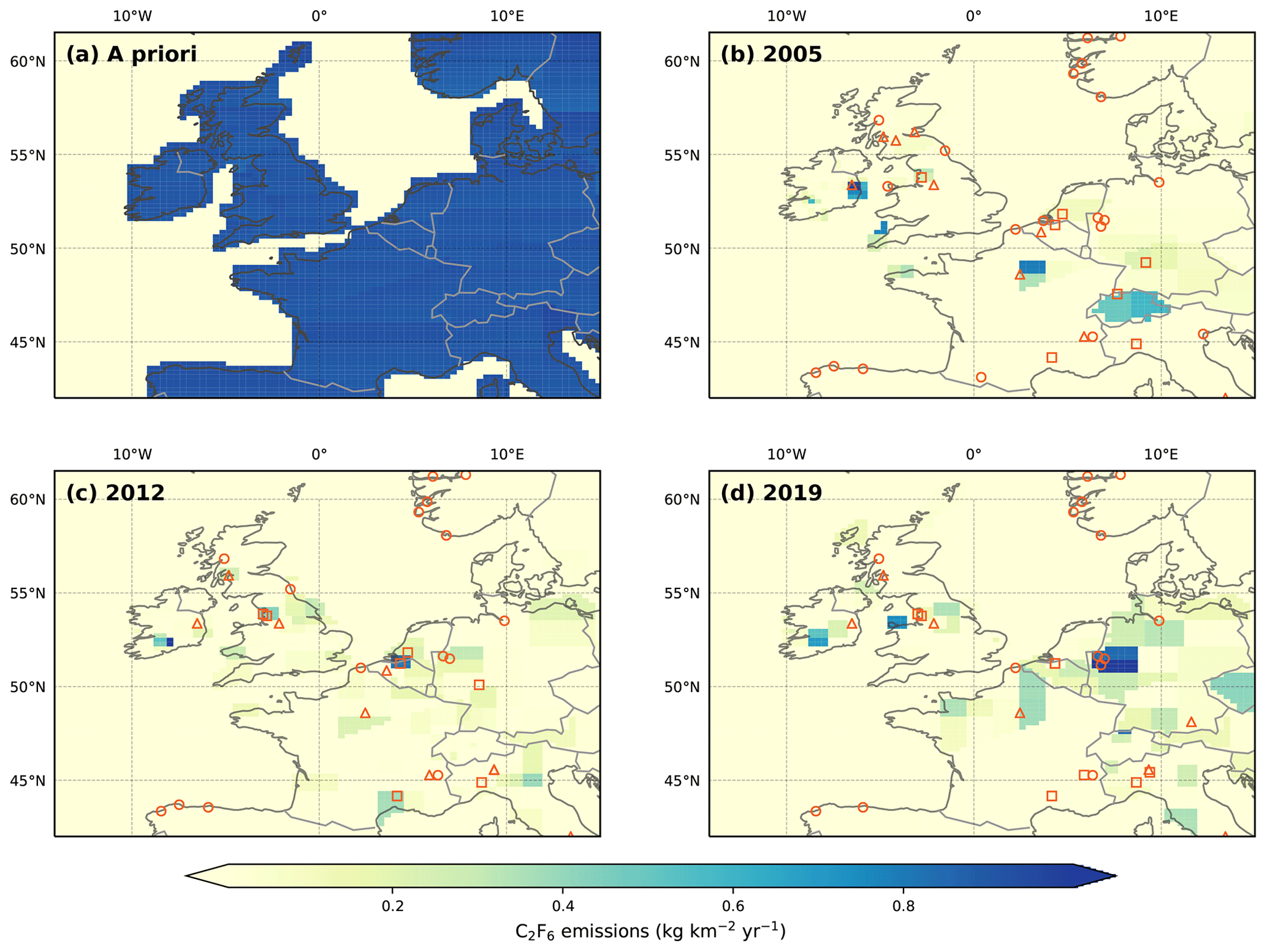
Figure 8Northwest European C2F6 emissions in kg km−2 yr−1. (a) The a priori emissions field. With the exception of the oceans, emissions were distributed uniformly across the model domain. A posteriori emissions are shown for (b) 2005, (c) 2012 and (d) 2019. Facilities that reported PFC emissions to the E-PRTR in the selected year are shown as orange circles (aluminium smelters), triangles (electronics manufacturers) and squares (chemical manufacturers, including petroleum products). Since the reporting period for the E-PRTR is shorter than that of our measurements, 2005 and 2019 emissions are compared to the earliest (2007) and latest (2017) years of the E-PRTR database, respectively. Note that the E-PRTR database reports cumulative PFC emissions.
C2F6 emissions maps are shown in Fig. 8. Unlike CF4, whose emissions appear to be dominated by the aluminium industry, the C2F6 distribution is consistent with a greater contribution from the electronics industry (Kim et al., 2014). For instance, in 2005, the two largest sources (on the outskirts of Dublin, Ireland, and Paris, France) correspond with the locations of electronics manufacturers, suggesting these emissions are linked with the consumption of C2F6 as an etching gas. In contrast, emissions from the Ruhr valley in Germany are small. By 2012, a source in northern Belgium, which is concurrent with the location of a basic chemicals manufacturer, dominates European emissions. The manufacture of basic chemicals also appear to be a source of C2F6 in southern France and northwest England. In contrast, the emissions “hot-spot” located near Dublin in 2005 is greatly diminished by 2012, in line with the planned phase-out of C2F6 in favour of NF3 by the manufacturer.
By 2019, the spatial distribution of emissions is more varied. Large emissions are found for the Ruhr valley in Germany and likely originate from three aluminium smelters in the region that were also found to emit CF4, which is known to be co-emitted during the smelting process at a ratio of around 0.1 kg kg−1 CF4 C2F6 (Kim et al., 2014). In Ireland, emissions associated with electronics manufacture, located on the outskirts of Dublin, have ceased. However, significant emissions are now found further to the southwest. This source region does not appear to be listed under the E-PRTR and may be a contributing factor in the small discrepancy between our work and the UNFCCC, from 2011 onwards. These emissions are situated in a comparable location to the Irish city of Limerick. While several electronics manufacturers are based here, these sources cannot be confirmed without further information from individual companies.
3.2.3 C3F8
Northwest European C3F8 emissions are shown in Fig. 9 and Table S3. As with CF4 and C2F6, northwest European emissions of C3F8 exhibited no statistically significant trend over the measurements period. The contribution of northwest Europe to global emissions is considerably greater than other PFCs −4.8 % in 2008 (0.03 Gg in northwest Europe; 0.69 Gg globally) and 5.1 % by 2018 (0.03 Gg in northwest Europe; 0.55 Gg globally). Of the gases studied, C3F8 is the only PFC for which northwest Europe contributed a greater fraction of the global total in 2018 than it did in 2008. Several countries, including France and Ireland, reported no emissions of C3F8 to the UNFCCC across the measurement window. In general, our work is in agreement with these reports, with the uncertainty bounds of our estimates typically encapsulating 0 Mg yr−1. Our work shows the UK to be the largest emitter of C3F8 in northwest Europe. In the early years of the record, and again after 2016, there is a significant discrepancy between reporting methods, potentially indicative of under-reporting by UK emitters.
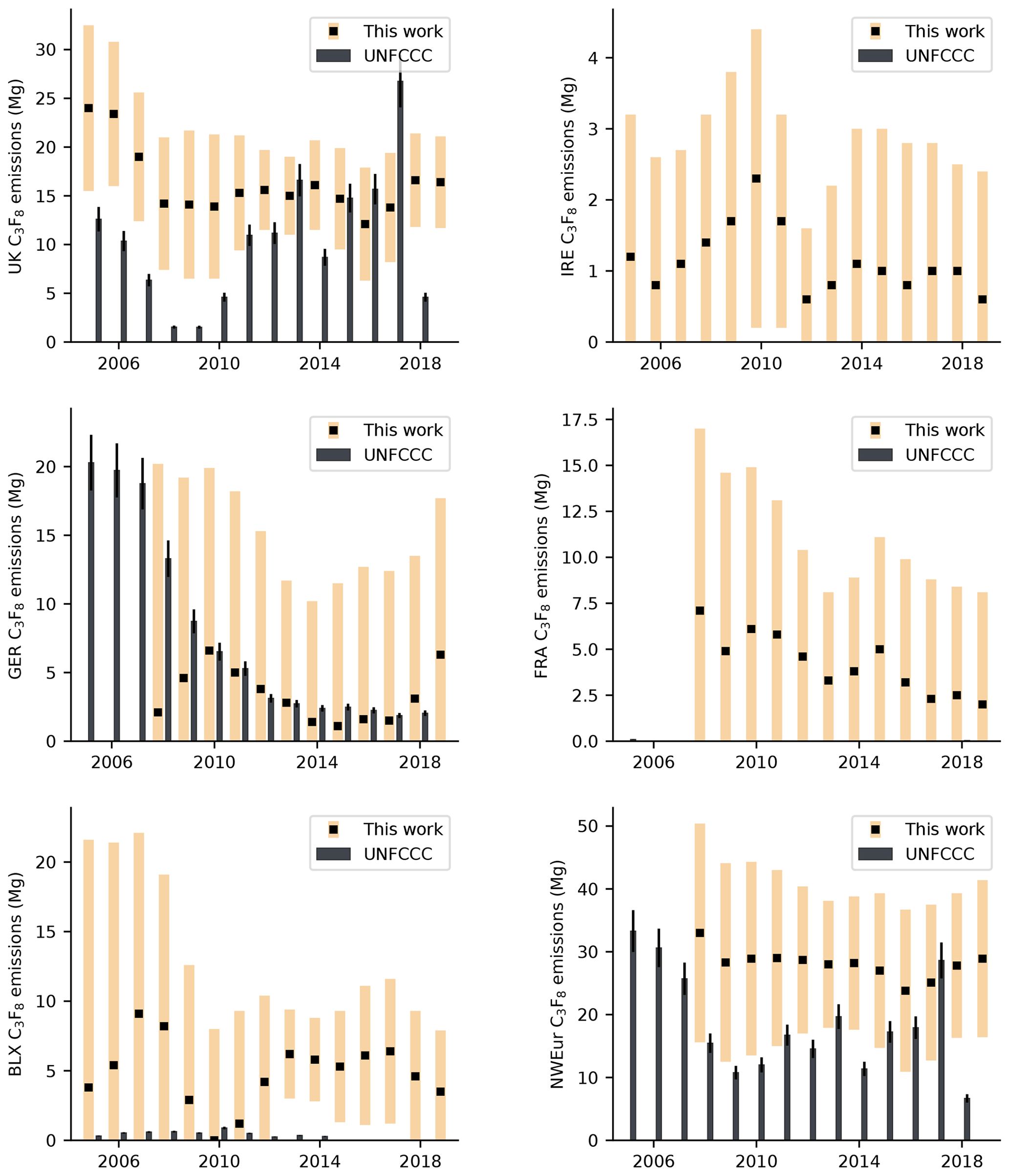
Figure 9Annual C3F8 emissions (2005–2019) for northwest European countries in Mg yr−1. InTEM estimates are shown as black squares with pale orange uncertainty bounds. Emissions reported to the UNFCCC (sum of individual reporting countries) are shown as black bars, with an assumed uncertainty (black error bars) of 10 %. Note that UNFCCC data are only available up until 2018 inclusive.
The spatial maps reveal few notable sources of C3F8 across continental Europe. The apparent reduction of continental sources in later years may be due to the substitution of C3F8 in the semi-conductor industry by lower GWP alternatives. However, it is also likely that in the absence of European measurements prior to 2008 (when JFJ came online), the inversion has less skill in inferring point source emissions at such a distance from the receptor (MHD). In 2012, the spatial maximum in emissions from the Benelux region are consistent with that found for C2F6 and c-C4F8 (Mühle et al., 2019), probably due to close vicinity of chemical industries or perhaps due to a common PFC source.
UK emissions of C3F8 appear to be dominated by a single source located in northwest England (Fig. 10). Facility listings from the E-PRTR show a PFC manufacturer whose location is consistent with this source (Fig. 10). This company is the only known manufacturer of C3F8 in the UK and possibly in all of Europe.
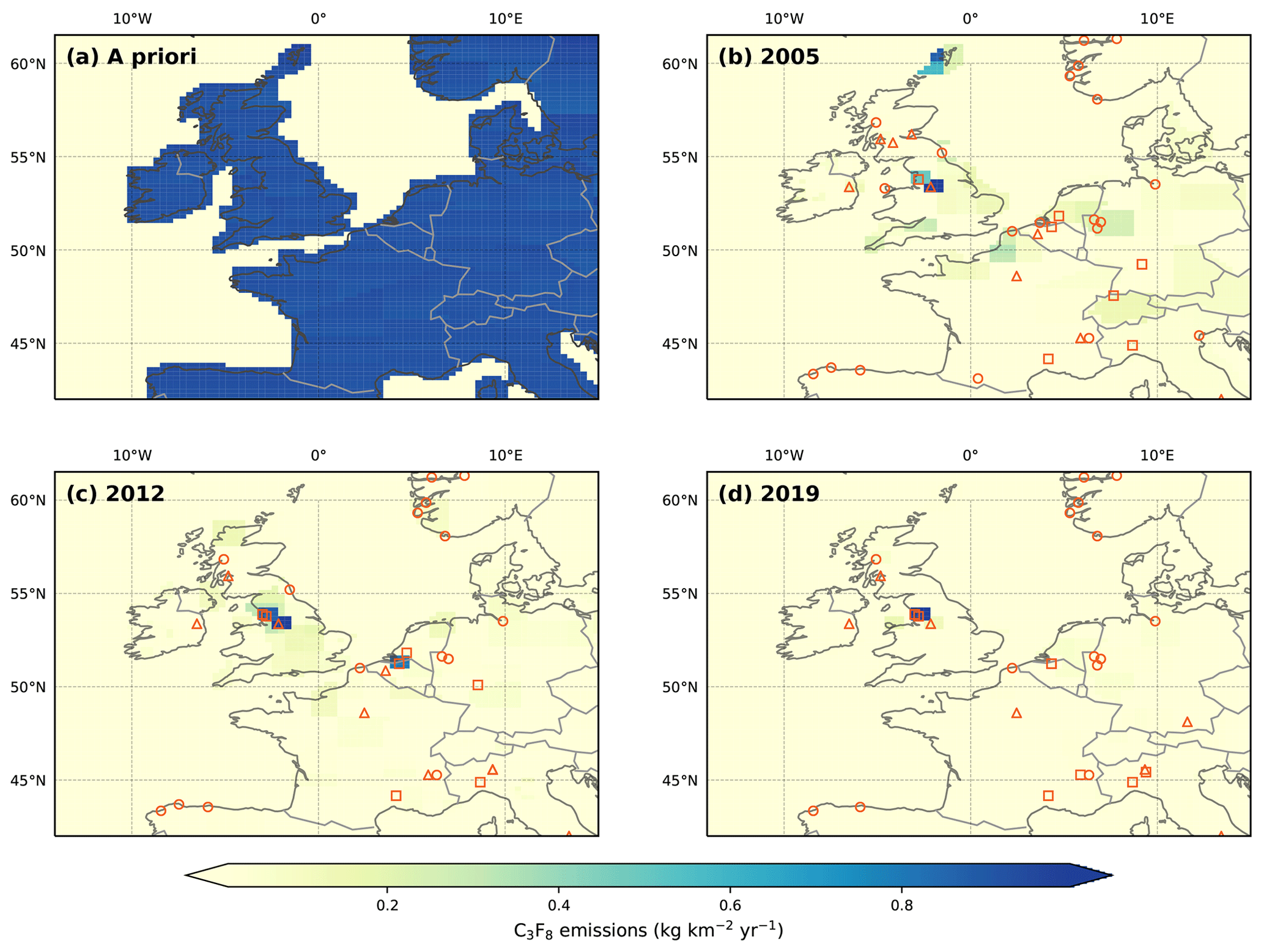
Figure 10Northwest European C3F8 emissions in kg km−2 yr−1. (a) The a priori emissions field. With the exception of the oceans, emissions were distributed uniformly across the model domain. A posteriori emissions are shown for (b) 2005, (c) 2012 and (d) 2019. Facilities that reported PFC emissions to the E-PRTR in the selected year are shown as orange circles (aluminium smelters), triangles (electronics manufacturers) and squares (chemical manufacturers, including petroleum products). Since the reporting period for the E-PRTR is shorter than that of our measurements, 2005 and 2019 emissions are compared to the earliest (2007) and latest (2017) years of the E-PRTR database, respectively. Note that the E-PRTR database reports cumulative PFC emissions.
3.2.4 UK C3F8 emissions as a tracer for atmospheric transport
If the facility in northwest England is the only source of C3F8 in northwest Europe, this gas could potentially be used as a tracer species for the validation of atmospheric transport models, assuming that emissions are well defined. To test the validity of UK C3F8 emissions as a potential tracer, the mole fraction at MHD was modelled by multiplying the NAME sensitivity matrices (see Sect. 2.4) with an emissions grid, where the UK's total reported emissions of C3F8 in 2014 (UNFCCC, 8.69 Mg yr−1) were placed in the grid cell corresponding with the location of the PFC manufacturer. The resulting time series was then compared to the MHD observations. Figure 11 shows the comparison of the forward model with MHD C3F8 observations for July–August 2014 (time frame chosen for illustrative purposes). In general, pollution events observed at MHD coincide with modelled events, though in the example shown, the magnitude of the modelled events is significantly smaller than the observed mole fraction. This indicates that the assumed emissions rate is too small, or there are errors in the transport model (NAME). Alternatively, the current assumption, that emissions from the site are released at a constant rate throughout the year, may also be an oversimplification – emissions are likely to vary depending on the rate of production.
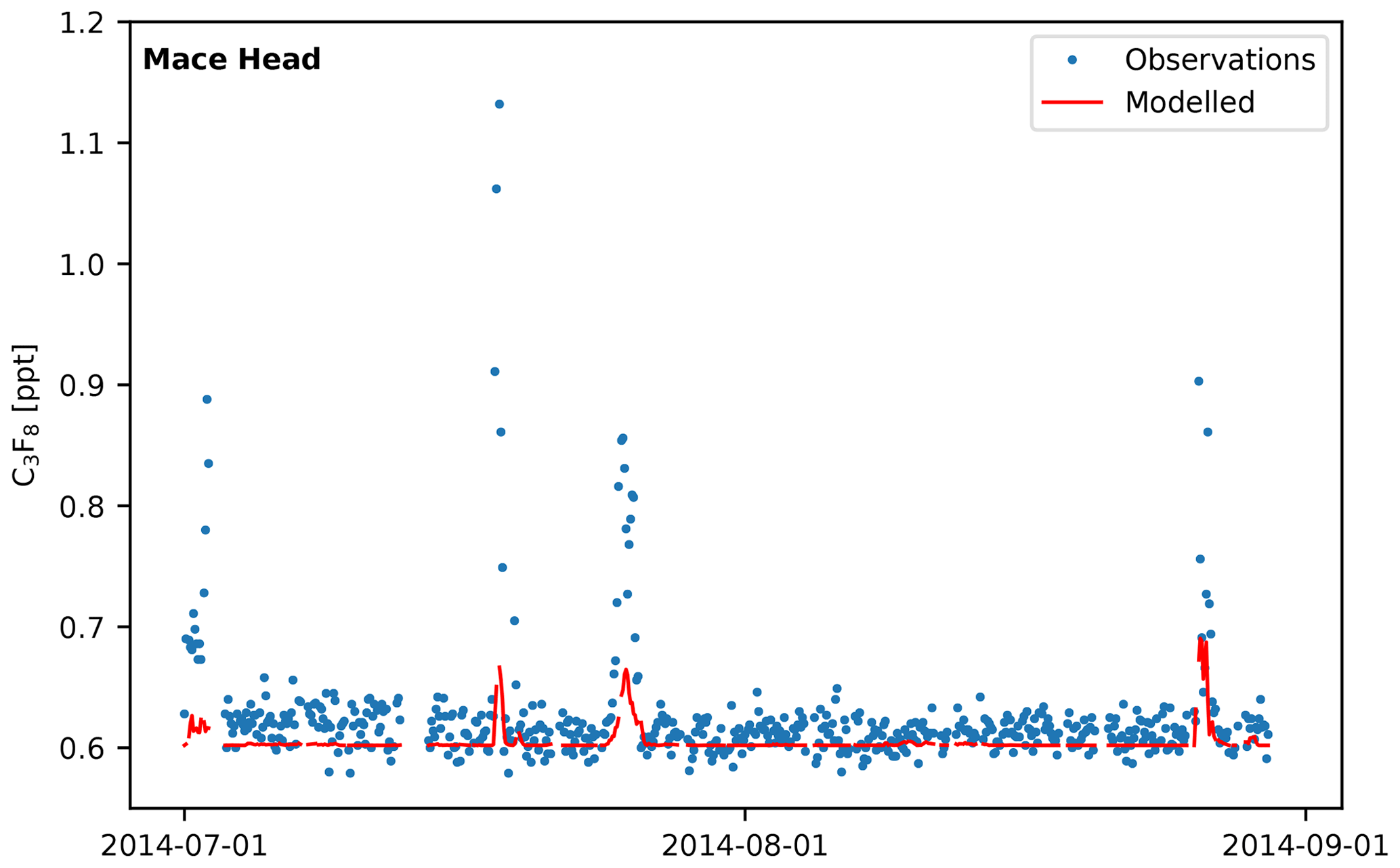
Figure 11Comparison of measured (blue dots) and modelled (red line) C3F8 mole fractions at MHD for July–August 2014. Dates are chosen for illustrative purposes only.
While atmospheric dispersion models, such as NAME, are used extensively to simulate atmospheric transport, they rely on simulated atmospheric dynamics that are subject to considerable uncertainties. If emissions from this source in northwest England were known better (e.g. by collaboration with the company), then differences between the forward model and observed mole fractions might be used to improve model transport or make estimates of transport uncertainties. Due to the long atmospheric lifetime of C3F8, photochemical loss processes do not need to be taken into account, thus further reducing uncertainty. Quantifying these differences would be a useful means by which to assess and perhaps improve the performance of individual model simulations in future inverse modelling work.
We have presented measurements of tetrafluoromethane (CF4, PFC-14), hexafluoroethane (C2F6, PFC-116) and octafluoropropane (C3F8, PFC-218) from the AGAGE network. We combined measurements from five background stations, in conjunction with a box model, to infer global trends. For CF4, the global mean baseline mole fraction increased by ∼ 33 ppt between 1979 and 2019. The global growth rate declined across much of the measurement period, falling to a minimum of 0.6 ppt yr−1 around the time of the 2008 financial crisis. However, the growth rate began to rise again after 2011, consistent with increasing global emissions. For C2F6, a maximum growth rate of 0.125 ppt yr−1 was observed around 1999, followed by a period of steady decline. Like with CF4, we found a renewed increase in the global growth rate after 2011. C3F8 exhibited a rapid increase in growth rate, starting in the early 1990s and ending in the early 2000s, followed by steady decline until 2013. In recent years, a small increase in growth rate was observed.
We used observations from four European observatories to infer PFC emissions from northwest Europe. Between 2010 and 2019, northwest European emissions of CF4 exhibited no statistically significant trend, despite an increase in global aluminium production and continued demand for electronic components, consistent with growth in emissions from other regions. In early years, emissions were predominantly consistent with the locations of aluminium smelters. However, in more recent years, the largest source of CF4 was northwest Italy. These emissions might be linked to a chemicals manufacturer that reports substantial PFC emissions to the E-PRTR.
Likewise for C2F6, no significant trend was observed for northwest European emissions from 2008 until 2019. A notable fall in emissions from Ireland was observed between 2007 and 2012. This trend was mirrored by Ireland's national inventory report and appears to be linked with an electronics manufacture on the outskirts Dublin. By 2019, the largest source of C2F6 in northwest Europe was western Germany, most notably the Ruhr valley region in which three aluminium smelters are found. Northwest European C3F8 emissions were stable over the measurement period. Several countries, including France and Ireland, reported no emissions of C3F8, which our results are consistent with. With the exception of a small source in Benelux, we found northwest European emissions of C3F8 were dominated by a single source in northwest England, consistent with the location of a PFC manufacturer. We explored the potential of using this facility in a tracer release experiment and showed that, if accurate high-frequency emissions data were made available, this site could be used to provide useful information related to transport model performance.
Atmospheric measurement data from AGAGE stations are available from the AGAGE website (http://agage.mit.edu/data/agage-data, Prinn et al., 2020). Data from the Tacolneston observatory are available from the Centre for Environmental Data Analysis (CEDA) data archive (https://catalogue.ceda.ac.uk/uuid/a18f43456c364789aac726ed365e41d1, O'Doherty et al., 2019). AGAGE 12-box model code will be made available upon request by contacting Matt Rigby. Licenses to use NAME and InTEM are available for research purposes via a request to the UK Met Office or on request from Alistair Manning.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-2149-2021-supplement.
Measurement data were collected by SO'D, DY, AW, DS, SR, MKV, MM, JA, PBK, JM, RFW and RGP. CMH produced and maintained the gravimetric SIO calibration scales for these gases. AJM conducted the NAME runs and ran the InTEM inverse model. MR performed the global 12-box model inversions. BE performed the C3F8 forward model runs. DS and LW analysed InTEM and forward model output. DS and LW wrote the manuscript, with contributions from all co-authors.
The authors declare that they have no conflict of interest.
The authors would like to thank the on-site technicians across the AGAGE network for their cooperation and efforts in maintaining AGAGE instrumentation.
The operations of Mace Head and Tacolneston were funded by the UK Department of Business, Energy and Industrial Strategy (BEIS) through contract no. 1537/06/2018 to the University of Bristol. The operations of Mace Head and Ragged Point were also partly funded under NASA contract no. NNX16AC98G to MIT with a sub-award (no. 5710002970) to the University of Bristol. Ragged Point was also partly funded by NOAA grant no. RA133R15CN0008 to the University of Bristol. Support for the observations at Jungfraujoch comes through Swiss national programmes HALCLIM and CLIMGAS-CH (Swiss Federal Office for the Environment, FOEN), the International Foundation High Altitude Research Stations Jungfraujoch and Gornergrat (HFSJG) and ICOS-CH (Integrated Carbon Observation System Research Infrastructure). Observations at Cape Grim are supported largely by the Australian Bureau of Meteorology, CSIRO and NASA contract NNX16AC98G to MIT with sub-award no. 5710004055 to CSIRO. Operations at the O. Vittori station (Monte Cimone) are supported by the National Research Council of Italy. Trinidad Head, Cape Matatula and data processing and calibration across the AGAGE network were funded by NASA grant nos. NNX16AC96G and NNX16AC97G to the Scripps Institution of Oceanography. Luke M. Western and Matthew Rigby were funded by NERC grant nos. NE/N016548/1 and NE/S004211/1, and Matthew Rigby was funded under NERC grant no. NE/M014851/1. Inverse analysis was carried out on hardware supported by NERC grant no. NE/L013088/1.
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Arnold, T., Manning, A. J., Kim, J., Li, S., Webster, H., Thomson, D., Mühle, J., Weiss, R. F., Park, S., and O'Doherty, S.: Inverse modelling of CF4 and NF3 emissions in East Asia, Atmos. Chem. Phys., 18, 13305–13320, https://doi.org/10.5194/acp-18-13305-2018, 2018. a, b, c
Burkholder, J. B., Hodnebrog, Ø., and Orkin, V. L.: Annex 1: Summary of abundances, lifetimes, ozone depletion potentials (ODPs), radiative efficiences (REs), global warming potentials (GWPs), and global temperature change potentials (GTPs), Vol. 58, Global Ozone Research and Monitoring Project, World Meteorological Organization, Geneva, Switzerland, available at: https://csl.noaa.gov/assessments/ozone/2018/downloads/AppendixA_2018OzoneAssessment.pdf (last access: 6 September 2020), 2019. a, b, c, d
Cai, B., Liu, H., Kou, F., Yang, Y., Yao, B., Chen, X., Wong, D. S., Zhang, L., Li, J., Kuang, G., Chen, L., Zheng, J., Guan, D., and Shan, Y.: Estimating perfluorocarbon emission factors for industrial rare earth metal electrolysis, Resour. Conserv. Recycl., 136, 315–323, 2018. a
Cicerone, R. J.: Atmospheric carbon tetrafluoride: A nearly inert gas, Science, 206, 59–61, 1979. a
Cunnold, D., Prinn, R., Rasmussen, R., Simmonds, P., Alyea, F., Cardelino, C., Crawford, A., Fraser, P., and Rosen, R.: The atmospheric lifetime experiment: 3. Lifetime methodology and application to three years of CFCl3 data, J. Geophys. Res.-Oceans, 88, 8379–8400, 1983. a
Cunnold, D., Steele, L., Fraser, P., Simmonds, P., Prinn, R., Weiss, R., Porter, L., O'Doherty, S., Langenfelds, R., Krummel, P., Wang, H., Emmons, L., Tie, X., and Dlugokencky, E.: In situ measurements of atmospheric methane at GAGE/AGAGE sites during 1985-2000 and resulting source inferences, J. Geophys. Res-Atmos., 107, 4225, https://doi.org/10.1029/2001JD001226, 2002. a, b
EDGAR: Emission Database for Global Atmospheric Research (EDGAR), release version 4.0, available at: https://edgar.jrc.ec.europa.eu/ (last access: 6 September 2020), 2009. a, b
Engel, A. and Rigby, M.: Chapter 1: Update on Ozone Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, in: Scientific Assessment of Ozone Depletion: 2018, Vol. 58 of Global Ozone Research and Monitoring Project, World Meteorological Organization, Geneva, Switzerland, available at: https://csl.noaa.gov/assessments/ozone/2018/downloads/Chapter1_2018OzoneAssessment.pdf (last access: 6 September 2020), 2019. a, b, c, d
Fraser, P., Steele, P., and Cooksey, M.: PFC and carbon dioxide emissions from an Australian aluminium smelter using time-integrated stack sampling and GC-MS, GC-FID analysis, in: Light Metals 2013, Springer US, Boston, MA, 871–876, 2003. a, b, c, d
Harnisch, J. and Eisenhauer, A.: Natural CF4 and SF6 on Earth, Geophys. Res. Lett., 25, 2401–2404, 1998. a
IAI: Primary aluminium production statistics, available at: http://www.world-aluminium.org/statistics/, last access: 6 September 2020. a, b, c, d
Jones, A., Thomson, D., Hort, M., and Devenish, B.: The UK Met Office's next-generation atmospheric dispersion model, NAME III, in: Air pollution modeling and its application XVII, Springer US, Boston, MA, 580–589, 2007. a
Keller, C. A., Brunner, D., Henne, S., Vollmer, M. K., O'Doherty, S., and Reimann, S.: Evidence for under-reported western European emissions of the potent greenhouse gas HFC-23, Geophys. Res. Lett., 38, L15808, https://doi.org/10.1029/2011GL047976, 2011. a
Kim, J., Fraser, P. J., Li, S., Mühle, J., Ganesan, A. L., Krummel, P. B., Steele, L. P., Park, S., Kim, S-K., Park, M.-K., Arnold, T., Harth, C. M., Salameh, P. K., Prinn, R. G., Weiss, R. F., and Kim, K.-R.: Quantifying aluminum and semiconductor industry perfluorocarbon emissions from atmospheric measurements, Geophys. Res. Lett., 41, 4787–4794, 2014. a, b, c, d, e
Maione, M., Giostra, U., Arduini, J., Furlani, F., Graziosi, F., Vullo, E. L., and Bonasoni, P.: Ten years of continuous observations of stratospheric ozone depleting gases at Monte Cimone (Italy) – Comments on the effectiveness of the Montreal Protocol from a regional perspective, Sci. Total Environ., 445, 155–164, 2013. a
Maione, M., Graziosi, F., Arduini, J., Furlani, F., Giostra, U., Blake, D. R., Bonasoni, P., Fang, X., Montzka, S. A., O'Doherty, S. J., Reimann, S., Stohl, A., and Vollmer, M. K.: Estimates of European emissions of methyl chloroform using a Bayesian inversion method, Atmos. Chem. Phys., 14, 9755–9770, https://doi.org/10.5194/acp-14-9755-2014, 2014. a
Manning, A., O'Doherty, S., Jones, A., Simmonds, P., and Derwent, R.: Estimating UK methane and nitrous oxide emissions from 1990 to 2007 using an inversion modeling approach, J. Geophys. Res.-Atmos., 116, D02305, https://doi.org/10.1029/2010JD014763, 2011. a
Miller, B. R., Weiss, R. F., Salameh, P. K., Tanhua, T., Greally, B. R., Mühle, J., and Simmonds, P. G.: Medusa: A sample preconcentration and GC/MS detector system for in situ measurements of atmospheric trace halocarbons, hydrocarbons, and sulfur compounds, Anal. Chem., 80, 1536–1545, 2008. a
Morris, R. A., Miller, T. M., Viggiano, A., Paulson, J. F., Solomon, S., and Reid, G.: Effects of electron and ion reactions on atmospheric lifetimes of fully fluorinated compounds, J. Geophys. Res.-Atmos., 100, 1287–1294, 1995. a
Mühle, J., Ganesan, A. L., Miller, B. R., Salameh, P. K., Harth, C. M., Greally, B. R., Rigby, M., Porter, L. W., Steele, L. P., Trudinger, C. M., Krummel, P. B., O'Doherty, S., Fraser, P. J., Simmonds, P. G., Prinn, R. G., and Weiss, R. F.: Perfluorocarbons in the global atmosphere: tetrafluoromethane, hexafluoroethane, and octafluoropropane, Atmos. Chem. Phys., 10, 5145–5164, https://doi.org/10.5194/acp-10-5145-2010, 2010. a, b
Mühle, J., Trudinger, C. M., Western, L. M., Rigby, M., Vollmer, M. K., Park, S., Manning, A. J., Say, D., Ganesan, A., Steele, L. P., Ivy, D. J., Arnold, T., Li, S., Stohl, A., Harth, C. M., Salameh, P. K., McCulloch, A., O'Doherty, S., Park, M.-K., Jo, C. O., Young, D., Stanley, K. M., Krummel, P. B., Mitrevski, B., Hermansen, O., Lunder, C., Evangeliou, N., Yao, B., Kim, J., Hmiel, B., Buizert, C., Petrenko, V. V., Arduini, J., Maione, M., Etheridge, D. M., Michalopoulou, E., Czerniak, M., Severinghaus, J. P., Reimann, S., Simmonds, P. G., Fraser, P. J., Prinn, R. G., and Weiss, R. F.: Perfluorocyclobutane (PFC-318, c-C4F8) in the global atmosphere, Atmos. Chem. Phys., 19, 10335–10359, https://doi.org/10.5194/acp-19-10335-2019, 2019. a, b, c, d
O'Doherty, S., Say, D., and Stanley, K.: Deriving Emissions related to Climate Change Network: CO2, CH4, N2O, SF6, CO and halocarbon measurements from Tacolneston Tall Tower, Norfolk, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/ae483e02e5c345c59c2b72ac46574103 (last access: 6 September 2020), 2019.
Prinn, R. G., Weiss, R. F., Arduini, J., Arnold, T., DeWitt, H. L., Fraser, P. J., Ganesan, A. L., Gasore, J., Harth, C. M., Hermansen, O., Kim, J., Krummel, P. B., Li, S., Loh, Z. M., Lunder, C. R., Maione, M., Manning, A. J., Miller, B. R., Mitrevski, B., Mühle, J., O'Doherty, S., Park, S., Reimann, S., Rigby, M., Saito, T., Salameh, P. K., Schmidt, R., Simmonds, P. G., Steele, L. P., Vollmer, M. K., Wang, R. H., Yao, B., Yokouchi, Y., Young, D., and Zhou, L.: History of chemically and radiatively important atmospheric gases from the Advanced Global Atmospheric Gases Experiment (AGAGE), Earth Syst. Sci. Data, 10, 985–1018, https://doi.org/10.5194/essd-10-985-2018, 2018. a, b, c, d
Prinn, R. G., Weiss, R. F., Arduini, J., Arnold, T., Fraser, P. J., Ganesan, A. L., Gasore, J., Harth, C. M., Hermansen, O., Kim, J., Krummel, P. B., Li, S., Loh, Z. M., Lunder, C. R., Maione, M., Manning, A. J., Miller, B. R., Mitrevski, B., Mühle, J., O’Doherty, S., Park, S., Reimann, S., Rigby, M., Salameh, P. K., Schmidt, R., Simmonds, P. G., Steele, L. P., Vollmer, M. K., Wang, R. H., and Young, D.: The ALE/GAGE/AGAGE Data Base http://agage.mit.edu/data, last access: 6 September 2020.
Ravishankara, A., Solomon, S., Turnipseed, A. A., and Warren, R.: Atmospheric lifetimes of long-lived halogenated species, Science, 259, 194–199, 1993. a
Rigby, M., Prinn, R., O'Doherty, S., Miller, B., Ivy, D., Mühle, J., Harth, C., Salameh, P., Arnold, T., Weiss, R., et al.: Recent and future trends in synthetic greenhouse gas radiative forcing, Geophys. Res. Lett, 41, 2623–2630, 2014. a, b
Rigby, M., Park, S., Saito, T., Western, L. M., Redington, A. L., Fang, X., Henne, S., Manning, A. J., Prinn, R. G., Dutton, G. S., Fraser, P. J., Ganesan, A. L., Hall, B. D., Harth, C. M., Kim, J., Kim, K.-R., Krummel, P. B., Lee, T., Li, S., Liang, Q., Lunt, M. F., Montzka, S. A., Muhle, J., O'Doherty, S., Park, M.-K., Reimann, S., Salameh, P. K., Simmonds, P., Tunnicliffe, R. L., Weiss, R. F., Yokouchi, Y., and Young, D.: Increase in CFC-11 emissions from eastern China based on atmospheric observations, Nature, 569, 546–550, 2019. a
Rigby, M., Prinn, R. G., O'Doherty, S., Montzka, S. A., McCulloch, A., Harth, C. M., Mühle, J., Salameh, P. K., Weiss, R. F., Young, D., Simmonds, P. G., Hall, B. D., Dutton, G. S., Nance, D., Mondeel, D. J., Elkins, J. W., Krummel, P. B., Steele, L. P., and Fraser, P. J.: Re-evaluation of the lifetimes of the major CFCs and CH3CCl3 using atmospheric trends, Atmos. Chem. Phys., 13, 2691–2702, https://doi.org/10.5194/acp-13-2691-2013, 2013. a, b
Say, D., Manning, A. J., O’Doherty, S., Rigby, M., Young, D., and Grant, A.: Re-Evaluation of the UK’s HFC-134a emissions inventory based on atmospheric observations, Environ. Sci. Tech., 50, 11129–11136, 2016. a
Trudinger, C. M., Fraser, P. J., Etheridge, D. M., Sturges, W. T., Vollmer, M. K., Rigby, M., Martinerie, P., Mühle, J., Worton, D. R., Krummel, P. B., Steele, L. P., Miller, B. R., Laube, J., Mani, F. S., Rayner, P. J., Harth, C. M., Witrant, E., Blunier, T., Schwander, J., O'Doherty, S., and Battle, M.: Atmospheric abundance and global emissions of perfluorocarbons CF4, C2F6 and C3F8 since 1800 inferred from ice core, firn, air archive and in situ measurements, Atmos. Chem. Phys., 16, 11733–11754, https://doi.org/10.5194/acp-16-11733-2016, 2016. a, b, c, d, e, f, g, h, i, j
UNFCCC: Kyoto protocol reference manual on accounting of emissions and assigned amount, Tech. rep., eSocialSciences, available at: https://unfccc.int/resource/docs/publications/08_unfccc_kp_ref_manual.pdf (last access: 6 September 2020), 2009. a
Walters, D., Baran, A. J., Boutle, I., Brooks, M., Earnshaw, P., Edwards, J., Furtado, K., Hill, P., Lock, A., Manners, J., Morcrette, C., Mulcahy, J., Sanchez, C., Smith, C., Stratton, R., Tennant, W., Tomassini, L., Van Weverberg, K., Vosper, S., Willett, M., Browse, J., Bushell, A., Carslaw, K., Dalvi, M., Essery, R., Gedney, N., Hardiman, S., Johnson, B., Johnson, C., Jones, A., Jones, C., Mann, G., Milton, S., Rumbold, H., Sellar, A., Ujiie, M., Whitall, M., Williams, K., and Zerroukat, M.: The Met Office Unified Model Global Atmosphere 7.0/7.1 and JULES Global Land 7.0 configurations, Geosci. Model Dev., 12, 1909–1963, https://doi.org/10.5194/gmd-12-1909-2019, 2019. a
Wong, D. S., Fraser, P., Lavoie, P., and Kim, J.: PFC emissions from detected versus nondetected anode effects in the aluminum industry, JOM-J. min. Met. Mat. S., 67, 342–353, 2015. a
Zhang, L., Wang, X., and Gong, B.: Perfluorocarbon emissions from electrolytic reduction of rare earth metals in fluoride/oxide system, Atmos. Pollut. Res., 9, 61–65, 2018. a