the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Dec 2021
| 17 Dec 2021
Development and evaluation of a new compact mechanism for aromatic oxidation in atmospheric models
Kelvin H. Bates
Daniel J. Jacob
Peter D. Ivatt
Mat J. Evans
Yingying Yan
Jintai Lin
Aromatic hydrocarbons, including benzene, toluene, and xylenes, play an important role in atmospheric chemistry, but the associated chemical mechanisms are complex and uncertain. Sparing representation of this chemistry in models is needed for computational tractability. Here, we develop a new compact mechanism for aromatic chemistry (GC13) that captures current knowledge from laboratory and computational studies with only 17 unique species and 44 reactions. We compare GC13 to six other currently used mechanisms of varying complexity in box model simulations of environmental chamber data and diurnal boundary layer chemistry, and show that GC13 provides results consistent with or better than more complex mechanisms for oxygenated products (alcohols, carbonyls, dicarbonyls), ozone, and hydrogen oxide (HO) radicals. Specifically, GC13 features increased radical recycling and increased ozone destruction from phenoxy–phenylperoxy radical cycling relative to other mechanisms. We implement GC13 into the GEOS-Chem global chemical transport model and find higher glyoxal yields and net ozone loss from aromatic chemistry compared with other mechanisms. Aromatic oxidation in the model contributes 23 %, 5 %, and 8 % of global glyoxal, methylglyoxal, and formic acid production, respectively, and has mixed effects on formaldehyde. It drives small decreases in global tropospheric OH (−2.2 %), NOx (; −3.7 %), and ozone (−0.8 %), but a large increase in NO3 (+22 %) from phenoxy–phenylperoxy radical cycling. Regional effects in polluted environments can be substantially larger, especially from the photolysis of carbonyls produced by aromatic oxidation, which drives large wintertime increases in OH and ozone concentrations.
- Article
(12159 KB) - Full-text XML
-
Supplement
(40954 KB) - BibTeX
- EndNote





Aromatic hydrocarbons are a major class of volatile organic compounds (VOCs) emitted to the atmosphere, with important effects on oxidant chemistry and secondary organic aerosol (SOA) formation. They are emitted from anthropogenic sources, including incomplete combustion, industrial processes, solvent evaporation, and fuel leakage (Na et al., 2004; Reimann and Lewis, 2007), and have additional sources from biomass burning and vegetation (Misztal et al., 2015; Cabrera-Perez et al., 2016). Aromatic compounds in the gas phase can be directly harmful to human health (Duarte-Davidson et al., 2001; Sarigiannis and Gotti, 2008; Manuela et al., 2012), and their secondary chemistry can play a dominant role in ozone and SOA production in urban air (Barletta et al., 2005; Tan et al., 2012; Khan et al., 2018; Nault et al., 2018; Oak et al., 2019; Schroeder et al., 2020).
The simplest emitted aromatic hydrocarbons – benzene (C6H6), toluene (C7H8), and xylenes (C8H10), referred to collectively as BTX – together make up over 20 % of global anthropogenic non-methane VOC emissions on a carbon basis (Yan et al., 2019) and up to 60 % of urban emissions (Lee et al., 2002; Ran et al., 2009). The oxidative chemistry of BTX, initiated by reaction with the hydroxyl radical (OH), is unique among VOCs due to the stability of the aromatic ring and to their low hydrogen / carbon (HC) ratios. Thus, standard chemical mechanisms for the fates of the peroxy radicals (RO2) and alkoxy radicals (RO) produced in VOC oxidation may not apply to aromatics (Vereecken, 2018; Xu et al., 2020). Low-volatility oxygenated organics produced from aromatic oxidation contribute to SOA formation (Ng et al., 2007; Hildebrandt et al., 2009; Schwantes et al., 2017) and may lead to new particle formation (Wang et al., 2017; Molteni et al., 2018; Garmash et al., 2020). Efficient production of peroxyacyl nitrates (PANs) from BTX oxidation provides a reservoir of nitrogen oxide radicals (NO), increasing ozone and OH on a global scale (Fischer et al., 2014). Interest in using satellite observations of formaldehyde (CH2O) and glyoxal (C2H2O2) as proxies of VOC emissions has further motivated the need to quantify yields of these species from aromatics (Chance et al., 2000; Wittrock et al., 2006; Liu et al., 2012; Chan Miller et al., 2016). Aromatics are particularly important for glyoxal and methylglyoxal production, providing another avenue for SOA formation (Fu et al., 2008; Lin et al., 2012).
The importance of aromatics for ozone formation spurred the initial development of BTX oxidation mechanisms for air quality models (Carter, 1990; Stockwell et al., 1997). The Master Chemical Mechanism (MCM) gives a quasi-explicit representation of BTX atmospheric chemistry with thousands of reactions (Jenkin et al., 2003) but is computationally intractable for 3-D models. A range of simplified mechanisms are presently used in models (Jenkin et al., 2008; Goliff et al., 2013; Carter and Heo, 2013; Emmons et al., 2020) but can differ greatly in their results. Two recent studies implementing BTX chemistry into global models found opposite effects on global tropospheric ozone (Yan et al., 2019; Taraborrelli et al., 2021). Differences between mechanisms reflect the evolution of knowledge as well as remaining uncertainties and parameterization choices (Schwantes et al., 2017; Xu et al., 2020). Because of the high computational cost of chemical evolution and transport in 3-D models (Nielsen et al., 2017; Hu et al., 2018), it is imperative to minimize the number of species uniquely needed to describe aromatic chemistry (Stockwell et al., 2012; Brown-Steiner et al., 2018; Shen et al., 2020).
Here, we present a new compact mechanism for BTX oxidation, GEOS-Chem version 13 (GC13), that is sufficiently simple for use in 3-D models but retains the accuracy of far more complex mechanisms and successfully fits laboratory data for BTX oxidation products. GC13 incorporates new knowledge on phenoxy–phenylperoxy radical cycling (Taraborrelli et al., 2021), later-generation chemistry of hydroxylated aromatics (Schwantes et al., 2017) and fragmentation products (Newland et al., 2019; Wang et al., 2020), and increased radical cycling in the reactions of first-generation aromatic peroxy radicals (Xu et al., 2020). We evaluate GC13 in box model simulations of laboratory chamber experiments and the continental boundary layer and compare it to six other mechanisms used in atmospheric models: the Common Representative Intermediates (CRI) mechanism, the Master Chemical Mechanism (MCM), the Module Efficiently Calculating the Chemistry of the Atmosphere (MECCA), the Model for Ozone and Related chemical Tracers (MOZART), the Regional Atmospheric Chemistry Mechanism (RACM2), and the Statewide Air Pollution Research Center (SAPRC) mechanism. We implement GC13 into GEOS-Chem, a global chemical transport model (CTM), to diagnose the effects of aromatic chemistry in the troposphere on oxygenated organics and oxidant chemistry.
GC13 includes 17 unique species and 44 unique reactions to describe BTX chemistry. These are listed in Sect. S1 in the Supplement. Figures 1–3 show the dominant routes of BTX oxidation. Initial branching ratios in GC13 are shown in red, and species treated explicitly are shown in blue. Starting from current knowledge on reaction pathways, we reduce the mechanism to be as simple as possible for atmospheric modeling while accurately representing important outcomes, including the following: (a) ozone formation; (b) yields of major first-generation products; (c) short- and long-term yields of formaldehyde, glyoxal, and methylglyoxal; (d) effects on hydrogen oxide (HO) radical budgets; and (e) closure of the total carbon budget. While we do not yet represent SOA formation with GC13, we include intermediates and pathways by which SOA formation is known to occur, facilitating future implementation of an SOA module. We find that separately representing individual xylene isomers provides negligible benefits and, therefore, represent them as a single lumped species. We also combine many xylene oxidation products with toluene products, scaling product yields to retain mass balance.
The first steps of BTX oxidation are summarized in Fig. 1. OH is the only significant oxidant; lifetimes with respect to oxidation at 298 K and are 9.6 d for benzene, 2.1 d for toluene, and 16 h for xylene (Mellouki et al., 2021). Reactions of BTX with NO3 radicals are at least a factor of 105 slower than their reactions with OH (Atkinson et al., 1984).
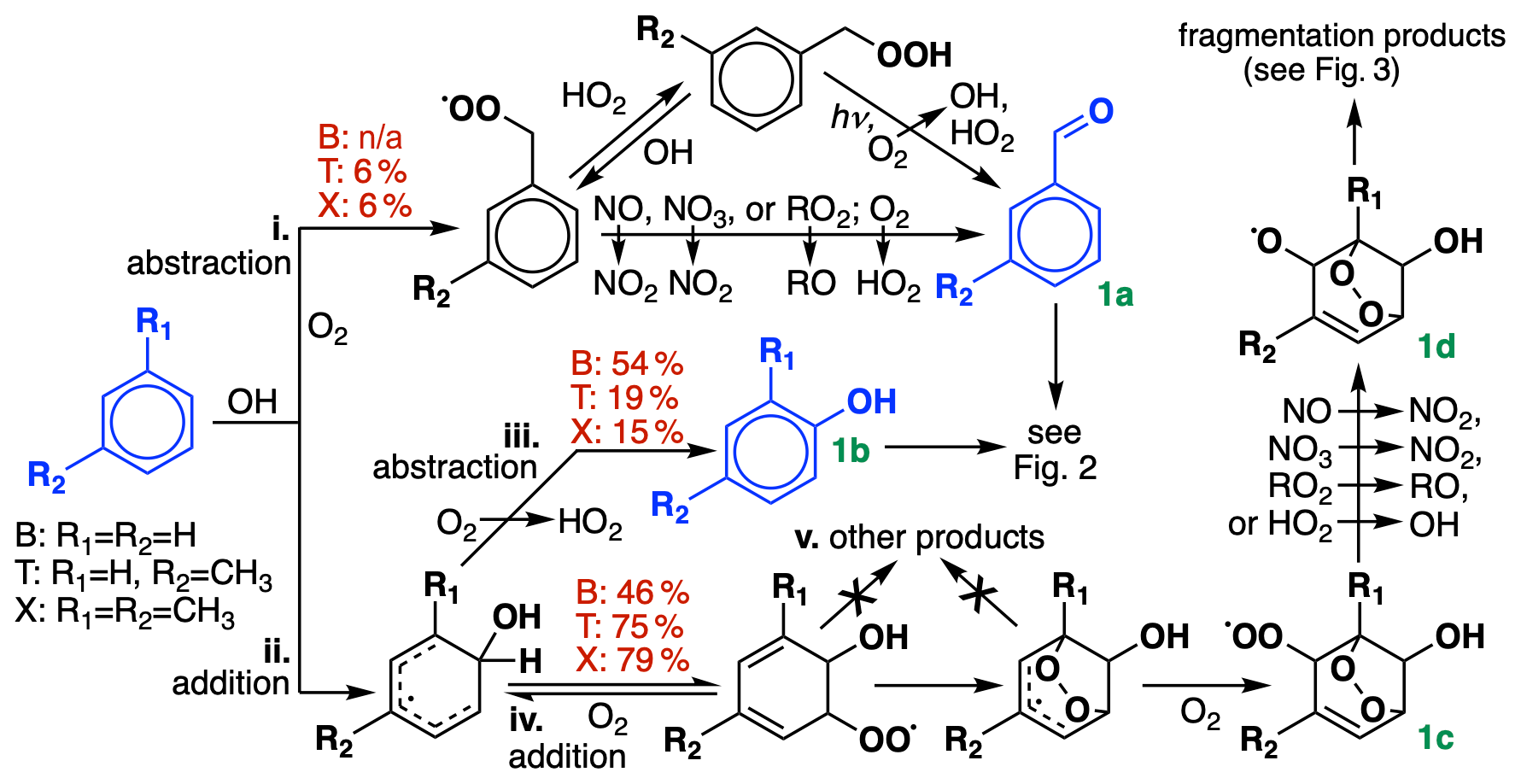
Figure 1Initial oxidation chemistry of benzene, toluene, and xylenes. R1 and R2 denote either H or CH3, depending on the species. For simplicity, only m-xylene is shown; in GC13, the xylene isomers are lumped together. Initial branching ratios in GC13 are shown in red, and species treated explicitly and uniquely in the aromatic oxidation mechanism are highlighted in blue. n/a represents not applicable.
BTX oxidation by OH can proceed via either of two pathways: (i) hydrogen abstraction from an alkyl substituent, only available to toluene and xylene), or (ii) OH addition to the aromatic ring. Route i leads eventually to benzaldehyde (from toluene) or tolualdehyde (from xylene) (Fig. 1a); as described in Sect. 4.1, we find that experimental benzaldehyde yields are best fit by skipping the intermediate peroxy radical and proceeding directly to aldehyde formation. Route ii can either be followed by H abstraction (iii), leading to a stable hydroxylated compound (Fig. 1b, e.g., phenol), or by O2 addition (iv), leading to the formation of a bridged bicyclic peroxy radical (Fig. 1c). (For a more detailed description of the dynamic system of reversible O2 addition, see Xu et al., 2020.) While other mechanisms and past studies have suggested the intermediate formation of other products (pathway v) preceding Fig. 1c, such as C6 epoxides, these remain speculative or observed only under high-NO or low-pressure conditions that are unrepresentative of ambient BTX oxidation (Yu and Jeffries, 1997; Berndt and Böge, 2001; Birdsall and Elrod, 2011). Recent experimental and theoretical evidence suggests that they do not form under ambient conditions (Wang et al., 2013; Zaytsev et al., 2019; Xu et al., 2020), so we assume that Fig. 1c is the only product from route iv.
The bridged bicyclic peroxy radical in Fig. 1c would traditionally be treated as a mechanistic branching point, potentially leading to the formation of a hydroperoxide, an organonitrate, an alcohol, or an alkoxy radical depending on the bimolecular reaction partner. However, recent work by Xu et al. (2020) found that nitrate and hydroperoxide yields from Fig. 1c never exceeded 0.06 % and 1 % for benzene, respectively. Instead, they suggest that reaction with either NO or HO2 leads exclusively to alkoxy radical formation (Fig. 1d, with NO2 and OH as respective coproducts) and subsequent fragmentation and that this pattern should hold for larger aromatics as well. These higher-than-expected radical recycling rates are consistent with other recent results and hypotheses (Orlando and Tyndall, 2012; Praske et al., 2015; Zaytsev et al., 2019) and help to bring OH radical concentrations in line with values inferred from BTX decay rates in chamber experiments (Bloss et al., 2005a). In our mechanism, this means that we can bypass representation of the individual peroxy radical species and proceed straight to ring-opening products, using a single generic peroxy radical species to accomplish conversion of bimolecular reaction partners (e.g., NO to NO2).
The secondary chemistry of the ring-retaining aldehyde (Fig. 1a) and alcohol (Fig. 1b) products is shown in Fig. 2. We lump benzaldehyde with tolualdehyde, given their similar chemistries. Benzaldehyde oxidation by either OH or NO3 generates a peroxyacyl radical (Fig. 2a) that can react with NO2 to form peroxybenzoyl nitrate (Fig. 2b), a PAN-like species that we represent explicitly for its ability to sequester and transport NOx. Reaction of the product in Fig. 2a with HO2 yields perbenzoic acid (Fig. 2c), benzoic acid and ozone, or a benzoyloxy radical and OH, in a ratio (Roth et al., 2010); subsequent chemistry eventually leads to formation of the phenylperoxy radical (Fig. 2d). Benzaldehyde photolysis is also implemented with recently reevaluated rates (Mellouki et al., 2021), producing OH, CO2, and the phenylperoxy radical (Fig. 2d).
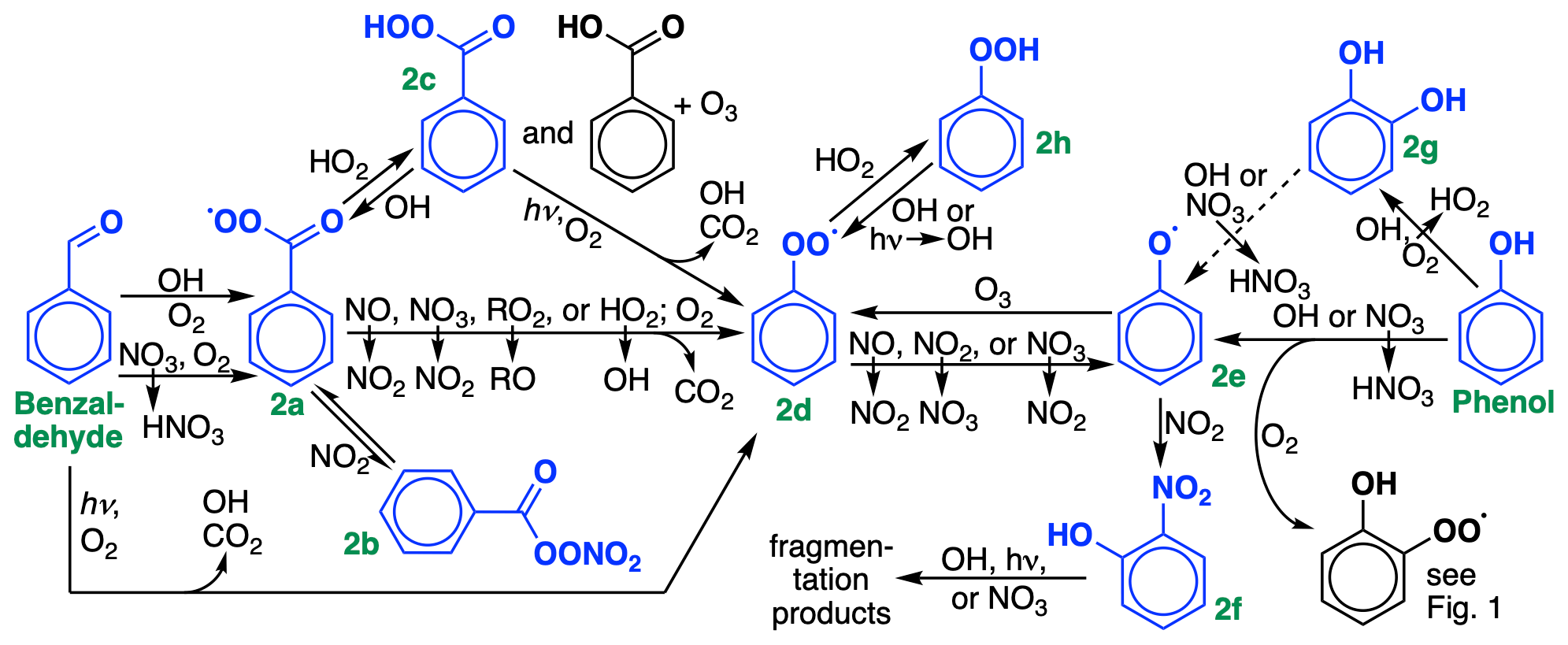
Figure 2Oxidation chemistry of stable ring-retaining aldehydes and alcohols from benzene, toluene, and xylene. For simplicity, only benzaldehyde and phenol are shown; cresol chemistry is not shown but is similar to phenol. GC13 lumps tolualdehyde with benzaldehyde, xylenols with cresol, and methylcatechols with catechol. Species treated explicitly and uniquely in the GC13 mechanism are highlighted in blue. The reaction of catechols (Fig. 2g) with OH produces substituted phenoxy radicals, lumped here with the phenoxy radical (Fig. 2e).
Rates and branching ratios for phenol and cresol oxidation by OH and NO3 are taken directly from the MCM. We lump xylenols with cresols, given their similar reactivity, and lump all products of cresolxylenol oxidation with those of phenol. Both OH and NO3 can abstract hydrogens, leading either to bridged peroxy radicals (Fig. 1c) or to phenoxy radicals (Fig. 2e), or undergo addition. NO3 addition leads to ring-retaining organonitrates, which we lump with nitrophenols (Fig. 2f), while OH addition forms catechols (Fig. 2g). While other mechanisms including the MCM typically assume that the reaction of catechols with OH proceeds primarily by abstraction from the hydroxyl group, leading to functionalized phenoxy radicals, Schwantes et al. (2017) found that addition pathways dominate, leading to a proliferation of oxidized species that may act as SOA precursors. We cannot represent these species in detail in a condensed mechanism; future work will focus on parameterizing SOA yields from these pathways. Instead, we adopt branching ratios from Schwantes et al. (2017), lump polyhydroxylated aromatics with cresols and other oxidized products with analogs, and adjust the cresol + OH reaction rate such that it represents only the abstraction pathways. H abstraction from cresols would make functionalized phenoxy radicals, which we combine with the phenoxy radical in GC13. We also include the reactions of catechols with NO3 and O3, with rates from the MCM and products lumped in with those from direct phenol and cresol oxidation.
Both benzaldehyde and phenol/cresol oxidation eventually lead to the formation of phenylperoxy and/or phenoxy radicals (Fig. 2d and 2e), the chemistry of which can have important implications for radical and ozone budgets via their cycling and formation of nitrophenols (Fig. 2f; see Sect. 4.3). Due to its aromatic stability, the phenoxy radical is far more stable than a typical alkoxy radical, and rather than fragmenting, tends to react with either NO2 (forming nitrophenols) or ozone (forming the phenylperoxy radical). Thus, the phenoxy and phenylperoxy radicals cycle until interrupted by phenyl hydroperoxide (Fig. 2h) formation from phenylperoxy + HO2, which is only a temporary radical reservoir, or by nitrophenol formation from phenoxy + NO2, which provides a terminal sink and the only route to fragmentation products from this radical system. Here, we treat the phenoxy–phenylperoxy system similarly to their representation in recent work by Taraborrelli et al. (2021), including the phenylperoxy + NO2 reaction (Jagiella and Zabel, 2007), the phenoxy + O3 reaction (Tao and Li, 1999), explicit treatment of phenyl hydroperoxide and nitrophenols (lumped with nitrocatechols), and HONO formation from nitrophenol photolysis (Bejan et al., 2006; Chen et al., 2011).
Finally, Fig. 3 shows the chemical pathways of the bridged alkoxy radical in Fig. 1d produced following OH addition to BTX. These pathways remain uncertain, particularly for toluene and xylene, but represent the main source of glyoxal and methylglyoxal from BTX oxidation. A theoretical study by Wang et al. (2013) suggested ring closure (vi) as the dominant fate of the bicyclic alkoxy radical from benzene, forming an epoxide (Fig. 3a). However, follow-up calculations suggested instead that ring breaking (vii) dominates (98 %+; Wang et al., 2020), resulting in an acyclic alkoxy radical (Fig. 3b), which was confirmed experimentally by Xu et al. (2020). The acyclic alkoxy radical can then decompose directly, forming a conjugated γ-dicarbonyl (Fig. 3c) and glyoxal or methylglyoxal. Conjugated γ-dicarbonyls are highly photolabile, with typical daytime lifetimes of 10–15 min (Newland et al., 2019); however, their photolysis does not result in substantial HOx production, instead leading primarily to ketene-enol formation (Fig. 3e), with a minor route decomposing to CO and an acrolein derivative (Newland et al., 2019).
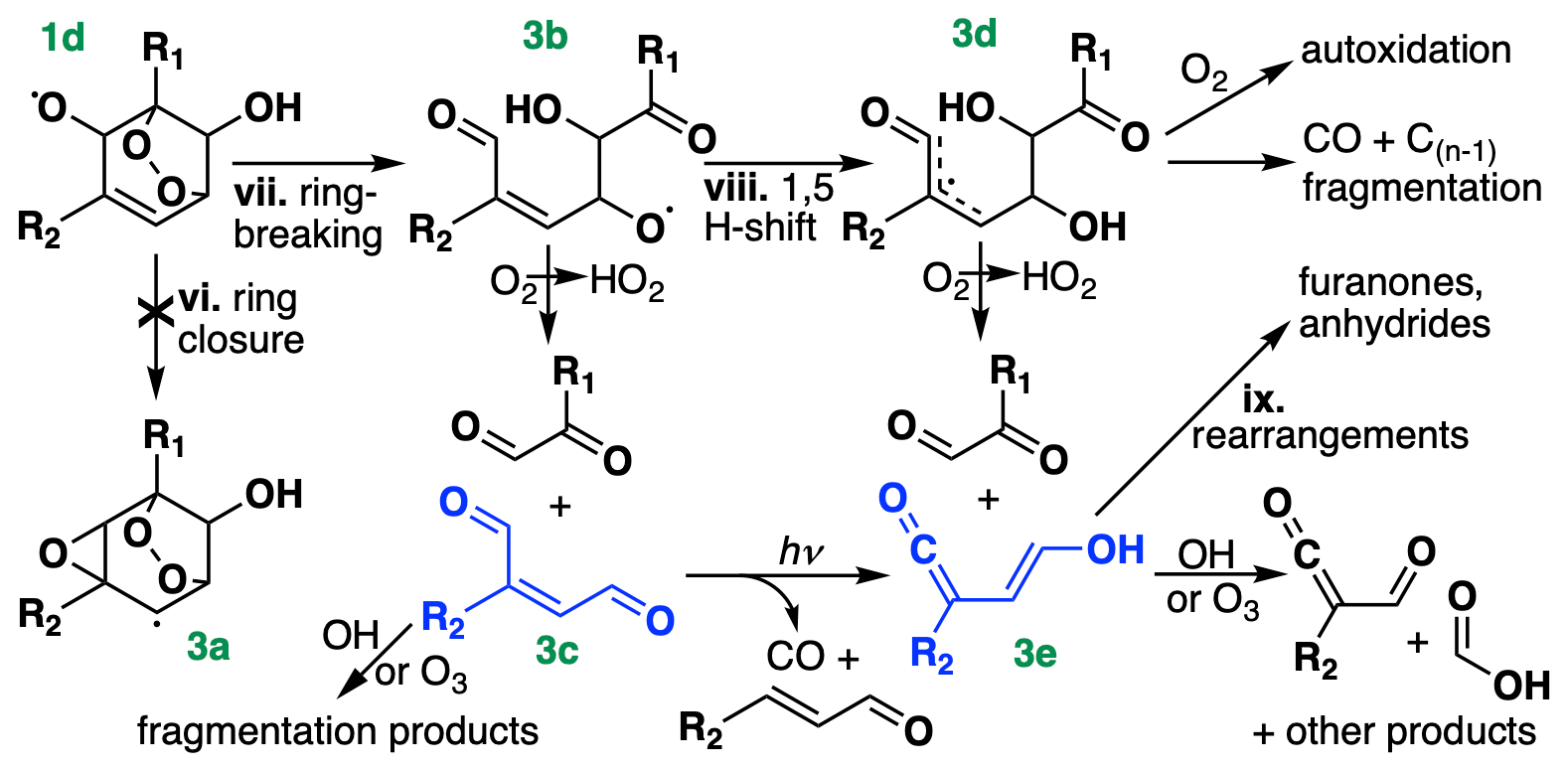
Figure 3Chemical pathways following OH addition to BTX, continued from Fig. 1. R1 and R2 denote either H or CH3, depending on the species. For toluene and xylene, only OH addition ortho to R1 is shown, and for xylene, only the meta isomer is shown; isomeric differences could lead to differing placement of R1 and R2 on the stable products, including the formation of acetic rather than formic acid (bottom right). Compounds in blue (Fig. 3c and 3e) are treated as lumped stable intermediates in GC13.
Recent theoretical (Wang et al., 2020) and experimental (Xu et al., 2020) studies have shown that instead of directly decomposing, the acyclic alkoxy radical (Fig. 3b) can undergo a 1,5 aldehydic H shift (viii), yielding a resonance-stabilized allyl radical (Fig. 3d). This allyl radical represents a mechanistic branching point with many possible fates, dominated by decomposition to an α-dicarbonyl and a ketene-enol (Fig. 3e). Between this direct production and secondary formation from γ-dicarbonyl photolysis, Wang et al. (2020) and Xu et al. (2020) suggest that ketene-enols are the primary product of the ring-opening pathway; however, Xu et al. (2020) note that they do not observe unity yields from benzene, and they suggest that other reactive pathways from Fig. 3d may account for the missing carbon. Such pathways include CO elimination and O2 addition, the latter of which could lead to autoxidation and the formation of highly oxidized SOA precursors from aromatics (Wang et al., 2017; Molteni et al., 2018; Garmash et al., 2020).
Subsequent chemistry of the ketene-enols (Fig. 3e) was studied by Newland et al. (2019) and Wang et al. (2020), and includes unimolecular cyclization to furanones as well as direct formation of formic and acetic acids via reaction with OH and O3. Newland et al. (2019) also observed tautomerization of the ketene-enols to ketene-carbonyls, possibly mediated by chamber surfaces, followed by cyclization to form anhydrides. Further studies are needed to constrain the contributions of these pathways, particularly for ring-opening products from toluene and xylene.
Because the branching ratios of the ring-opening pathways in Fig. 3 remain poorly constrained, particularly for toluene and xylene, we do not speciate them in detail in GC13. Instead, we tune the yields of glyoxal and methylglyoxal (coproduced with Fig. 3c and 3e) to match observed yields from chamber experiments and then lump larger products into two representative intermediates, which stand in for the C4+ stable products in Fig. 3 (primarily Fig. 3c and 3e). One, treated as a C4 compound and produced from all three BTX precursors, yields glyoxal and other compounds lacking methyl groups upon its subsequent oxidation, whereas the other, treated as a C5 compound and produced only from toluene and xylene, also yields methylglyoxal and other methylated products. The subsequent chemistry of these representative C4–C5 intermediates is a weighted combination of the reactive fates of the conjugated dicarbonyls (Fig. 3c) and the ketene-enols (Fig. 3e), drawn from Newland et al. (2019) and the MCM for Fig. 3c and from Newland et al. (2019) and Wang et al. (2020) for Fig. 3e, adjusted slightly to tune the later-generation yields of glyoxal and methylglyoxal. We do not explicitly track the furanones and anhydrides produced in unimolecular rearrangements of the ketene-enols (ix), allocating this carbon instead to other oxygenated intermediates with similar functionalities and lifetimes while maintaining carbon balance. We include a route to direct CO formation from Fig. 3d, using a branching ratio from Xu et al. (2020) for benzene, but do not include the minor pathway to highly oxidized molecule (HOM) formation; subsequent updates to the mechanism focused on SOA formation could represent these pathways explicitly.
In the following sections, we will compare GC13 to a suite of commonly used aromatic mechanisms. Here, we briefly describe each mechanism, in order from most to least complex. The number of species and reactions in each mechanism, excluding inorganic species and generic C1–C3 compounds, are given in Table 1. The mechanisms span a wide range of complexity, with 2 orders of magnitude separating the numbers of species and reactions in the largest and smallest mechanisms.
Bloss et al. (2005b)Taraborrelli et al. (2021)Carter and Heo (2013)Watson et al. (2008)Goliff et al. (2013)Emmons et al. (2020)Porter et al. (2017)Table 1Sizes of the aromatic schemes in common mechanisms.
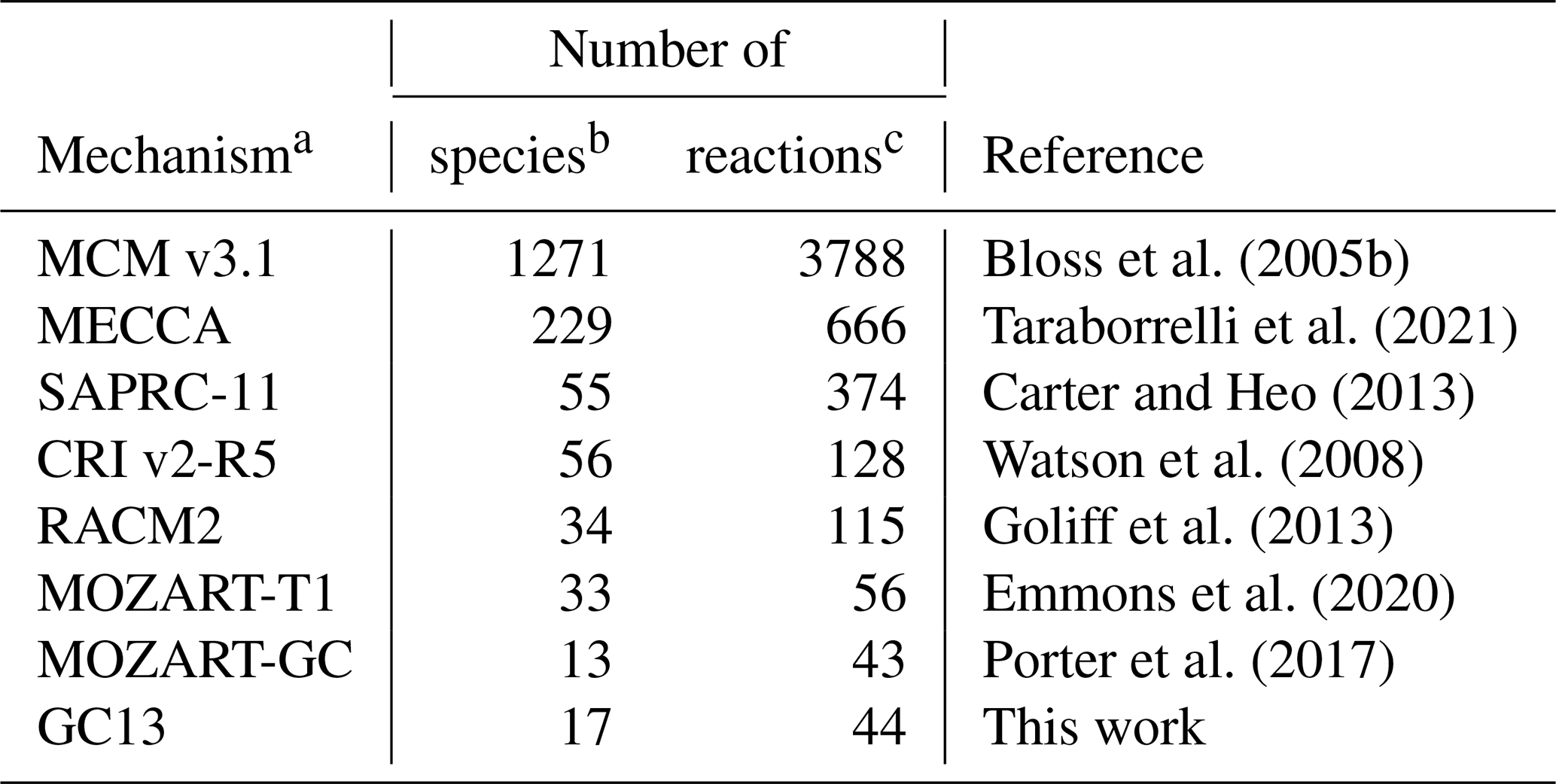
a Mechanisms used in intercomparison to GC13. b The species count includes both stable and radical species, but it does not include inorganic reactants and common/generic C1–C4 species. c The reaction count does not include reactions that only contain these excluded species.
MCM. The Master Chemical Mechanism (MCM) is a near-explicit mechanism that treats the full oxidative degradation of benzene, toluene, and each xylene isomer. The mechanism was developed by Jenkin et al. (2003) and updated to version 3.1 in Bloss et al. (2005b) based on an assessment of experimental work. Bloss et al. (2005a, b) compared the mechanism to a series of chamber experiments and noted that, while ozone was well simulated in benzene oxidation, simulations typically overestimated ozone formation from the larger aromatics while underestimating OH concentrations in all experiments.
MECCA. The Module Efficiently Calculating the Chemistry of the Atmosphere (MECCA; Sander et al., 2011) includes detailed aromatic chemistry described by Cabrera-Perez et al. (2016). The chemistry of toluene and benzene is taken from the MCM without simplification but with some important updates (notably to phenoxy–phenylperoxy radical cycling) described in detail by Cabrera-Perez et al. (2016) and Taraborrelli et al. (2021). MECCA lumps the xylene isomers and sets their chemistry identical to that of toluene, aside from the branching ratios in their initial reaction with OH. Despite its large number of species and reactions, MECCA has been used in a global 3-D model study by Taraborrelli et al. (2021).
SAPRC. The Statewide Air Pollution Research Center (SAPRC) mechanisms are a family of moderately reduced mechanisms that are widely used in airshed models for the prediction of ozone formation and the representation of organic pollutants. Here, we use SAPRC-11 (Carter and Heo, 2013), which was specifically designed to optimize aromatic oxidation and ozone formation rates in comparison to environmental chamber experiments. SAPRC-11 represents the xylene isomers individually and includes many early-generation BTX oxidation products, including phenol, cresol, xylenols, catechol, dicarbonyl compounds, nitrophenols, and benzaldehyde. SAPRC-11 was previously implemented in GEOS-Chem (v9-02) by Yan et al. (2019). Versions of SAPRC have also been implemented in the Weather Research and Forecasting model coupled with Chemistry (WRF-Chem), the Community Multiscale Air Quality (CMAQ) model, and other regional 3-D models (Yu et al., 2010; Cai et al., 2011; Zhang et al., 2012; Kitayama et al., 2019; Shareef et al., 2019).
CRI. The Common Representative Intermediates (CRI) mechanism, first developed by Jenkin et al. (2002), is a reduced mechanism based on the MCM that uses a series of generalized intermediates, rather than explicitly simulating multigenerational chemistry, to simulate ozone formation rates. Optimization and comparisons to the MCM for the most recent version, v2, are described by Jenkin et al. (2008). Here, we use CRI v2-R5, a further reduction that lumps together the xylene isomers (Watson et al., 2008). CRI v2-R5 has been widely implemented in regional and global models, including WRF-Chem (Archer-Nicholls et al., 2014), STOCHEM (Utembe et al., 2010; Khan et al., 2015), and UKCA (Archer-Nicholls et al., 2020).
RACM. The Regional Atmospheric Chemistry Mechanism (RACM) is a reduced mechanism first presented by Stockwell et al. (1997) and intended for use in modeling gas-phase chemistry under a wide range of ambient conditions. Here, we use RACM2 as described by Goliff et al. (2013). RACM2 is based on the MCM for benzene and on Calvert (2002) for toluene and the individual xylene isomers. It represents major first-generation products of BTX oxidation, including benzaldehyde, phenol, cresol, an epoxide, and photolabile dicarbonyls. RACM is implemented in both WRF-Chem and CMAQ (Sarwar et al., 2013; Kitayama et al., 2019; Shareef et al., 2019).
MOZART. The Model for Ozone and Related chemical Tracers (MOZART) chemical mechanisms are designed for implementation in the National Center for Atmospheric Research (NCAR) Community Earth System Model (CESM). The most recent iteration, MOZART-T1 (Emmons et al., 2020), is the first to differentiate between the BTX compounds. With an oxidation scheme based on the MCM, it represents some major early-generation products explicitly, including benzaldehyde, photolabile dicarbonyls, and peroxybenzoyl nitrate, but ignores some later-generation products assumed to undergo efficient deposition or aerosol uptake. An earlier version of the aromatic chemistry in MOZART-T1, first described by Knote et al. (2014), was implemented into GEOS-Chem by Porter et al. (2017); for the sake of comparison and contextualization of past studies, we also implement the Porter et al. (2017) mechanism here, which we label “MOZART-GC”.
4.1 Methods
We implemented GC13 and the other mechanisms outlined in Sect. 3 into box model simulations for comparisons to environmental chamber data and for mechanism intercomparisons under a range of boundary layer conditions. The simulations use a fourth-order Rosenbrock kinetic solver implemented with the Kinetic PreProcessor tool (KPP; Damian et al., 2002; Daescu et al., 2003; Sandu et al., 2003). We standardize the inorganic and C1–C3 chemistry to that of the MCM in all mechanisms so that the only differences between the mechanisms are in their BTX oxidation chemistry. For mechanisms with speciated xylenes, we assume equal contributions from the three isomers, and comparisons of xylene product yields with literature values are only conducted for experimental studies that targeted all three isomers.
Environmental chamber simulations. To quantify product yields and compare them to environmental chamber data, we simulate BTX chemistry in a box model representative of chamber experimental conditions. For each mechanism, we initialize the box model with fixed mixing ratios of one aromatic precursor, NO, and H2O2 as a photolytic OH source; we then run the simulation forward in time with a fixed light intensity and temperature until the aromatic precursor is 99 % depleted. Next, we vary all initial settings (temperature, light intensity, and each reactant concentration) individually and rerun the simulation to sample the full range of possible experimental conditions. Result shown in Sect. 4.2 and 4.3 are for simulations with initial [NO]0=5 ppt–2.5 ppm, P=1 atm, T=298 K, [VOC]0=100 ppb, [H2O2]0=2.5 ppm, and an NO2 photolysis rate (jNO2) of s−1 (other photolysis rates, given in Sect. S1 in the Supplement, are scaled to jNO2). “initial” yields shown in Figs. 4–6 are after 10 min of photooxidation; additional results showing long-term yields after 24 h of oxidation are provided in Sect. S2 in the Supplement. Sensitivities to temperature and initial VOC concentrations are generally small, but additional results showing the effects of these parameters, as well as the effects of light intensity and oxidant source, can be found in the Supplement. Wall losses of semi-volatile gases are not represented in these simulations, as we do not seek to model SOA formation or the yields of direct SOA precursors. An improved representation of SOA from aromatics in GEOS-Chem will be the subject of future work. While it is possible that wall losses of semi-volatile intermediates affect experimental yields of oxidized VOCs (OVOCs), this multigenerational chemistry is expected to play only a minor role at the short timescales isolated here.
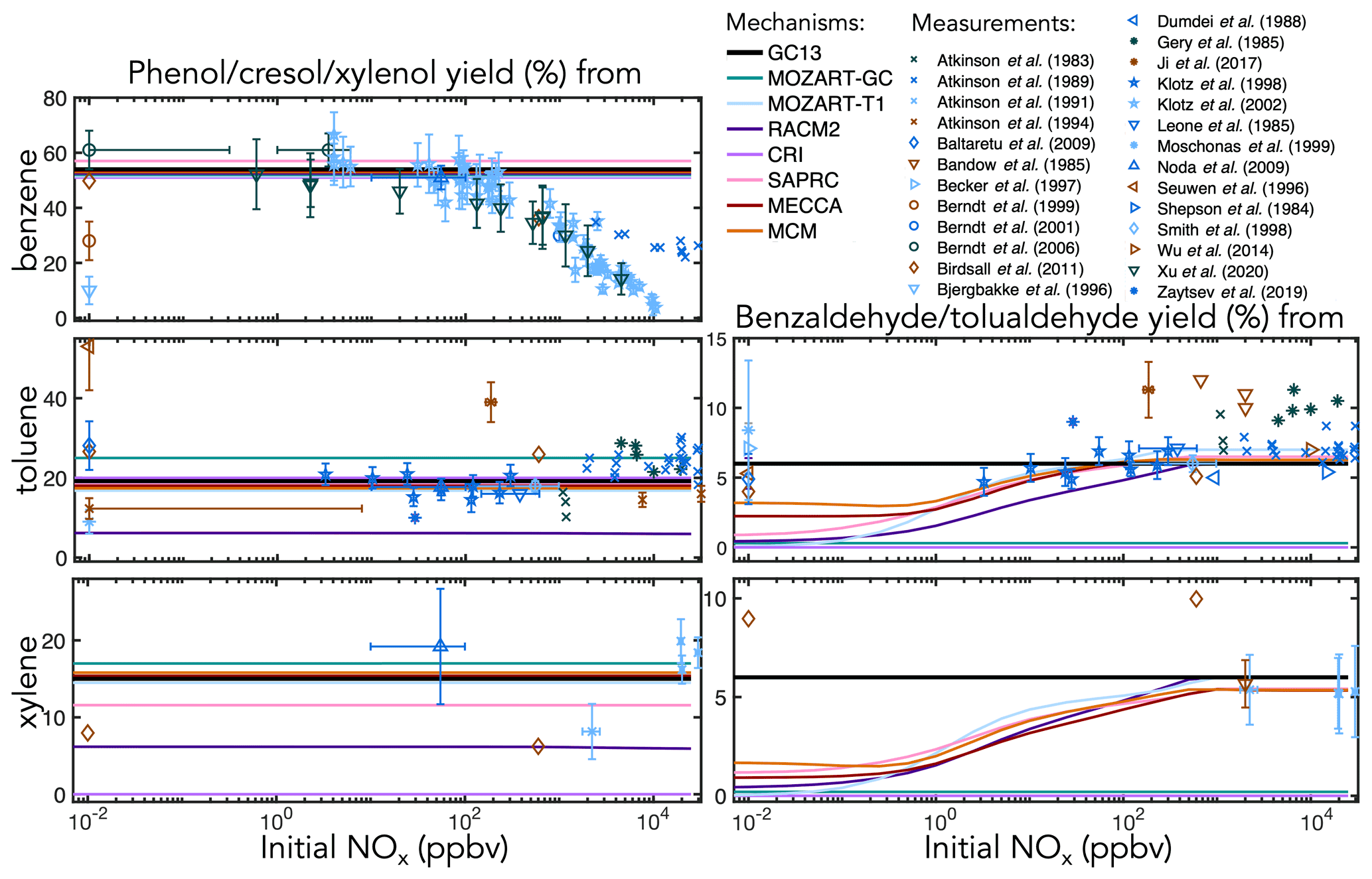
Figure 4Experimental (points) and simulated (lines) prompt molar yields of aromatic alcohols and aldehydes from BTX oxidation in environmental chambers as a function of initial NOx mixing ratio. Overlapping simulation lines are offset slightly for visibility.
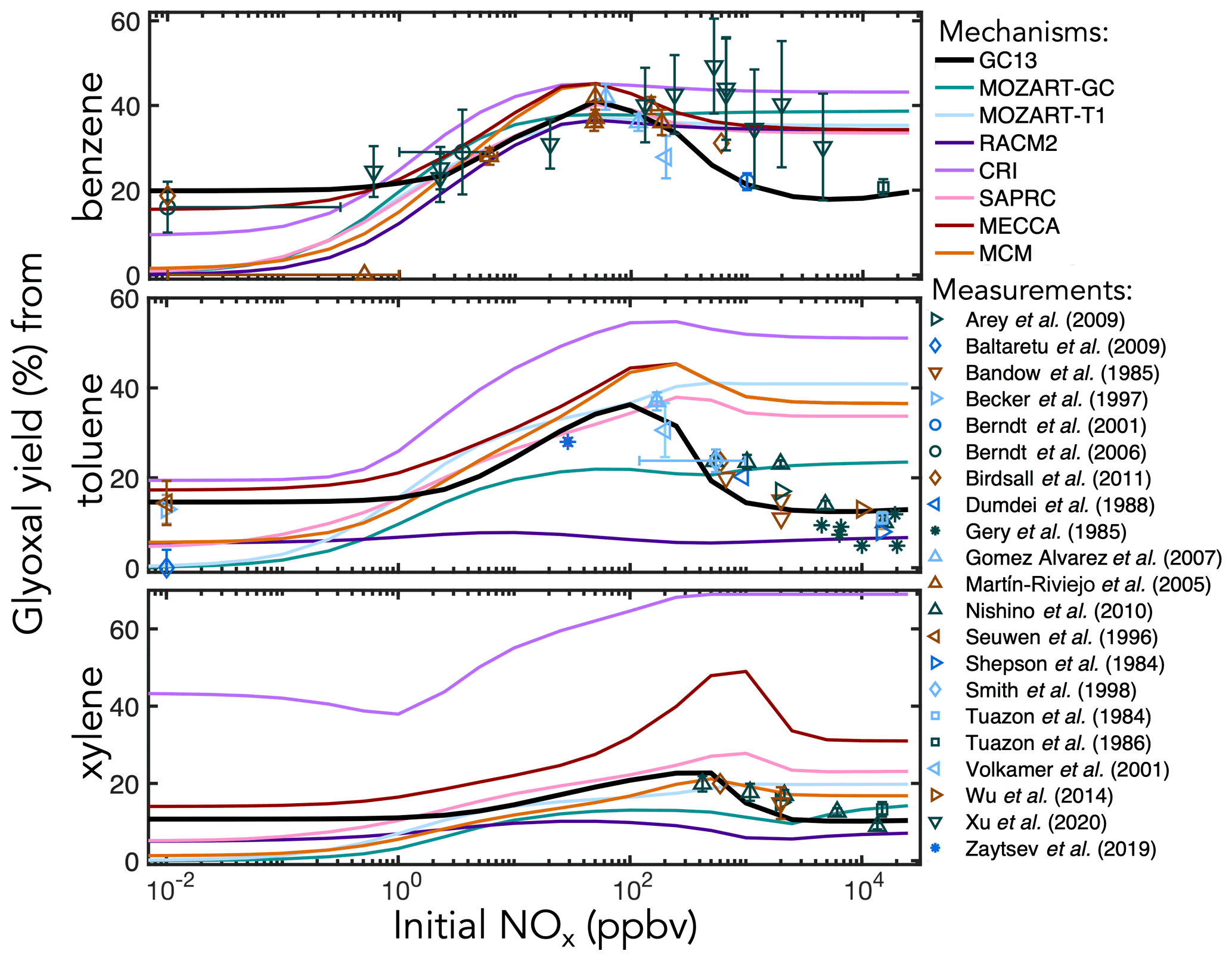
Figure 5Experimental (points) and simulated (lines) prompt molar yields of glyoxal from BTX oxidation in environmental chambers as a function of initial NOx mixing ratio.
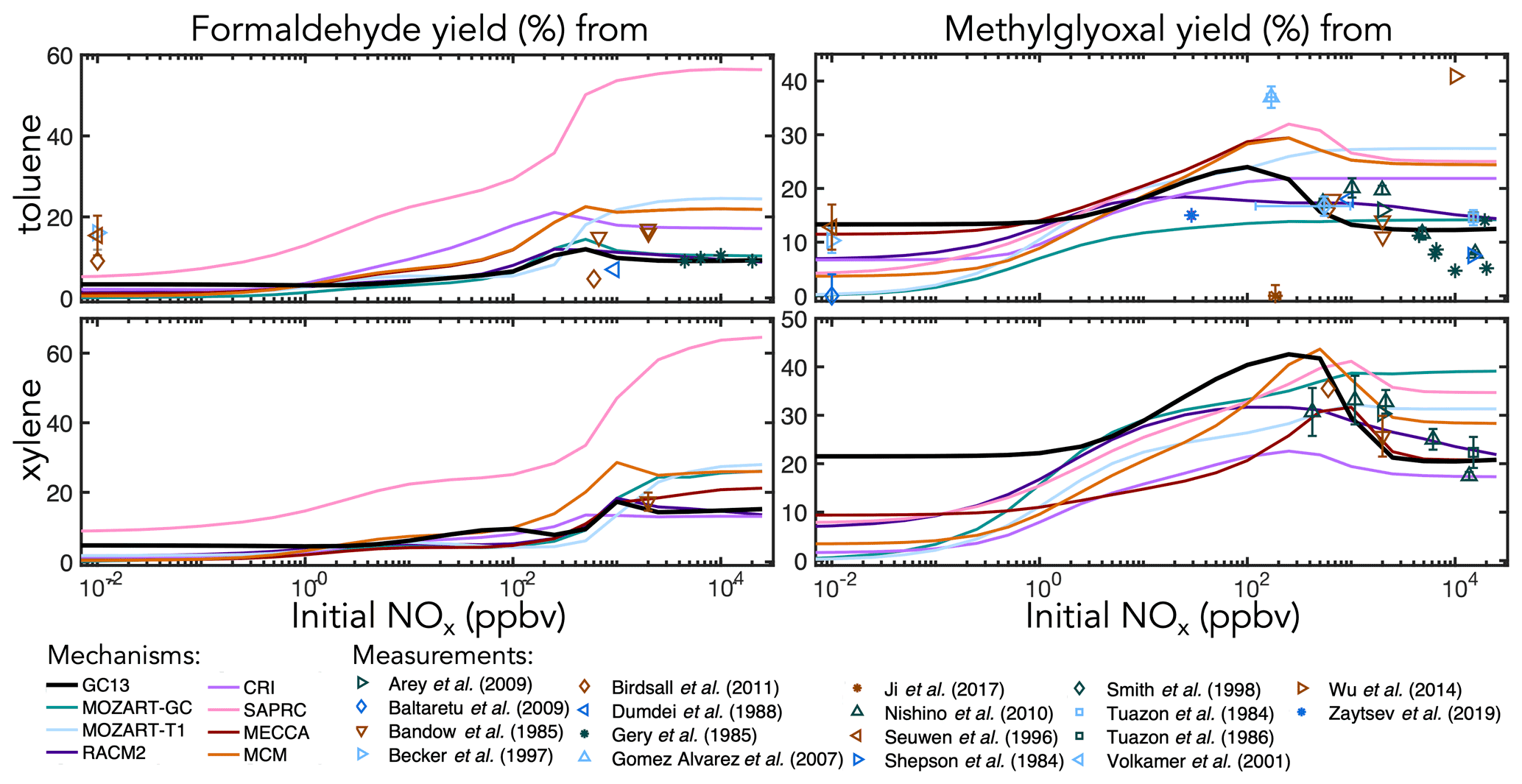
Figure 6Experimental (points) and simulated (lines) prompt molar yields of methylglyoxal and formaldehyde from toluene and xylene oxidation in environmental chambers as a function of initial NOx mixing ratio.
Continental boundary layer simulations. To examine longer-term product yields and effects of BTX oxidation on the ambient atmosphere, the same box model described above is also run under conditions meant to simulate a continental boundary layer like that of the heavily studied Seoul metropolitan area (Oak et al., 2019; Schroeder et al., 2020) with constant NO and aromatic emissions. The well-mixed boundary layer exchanges with the background free troposphere with a fixed ventilation timescale of 1 d for all species. Simulations are initialized with 75 ppb O3, 1.8 ppm CH4, 200 ppb CO, 300 ppt CH2O, and 1 % H2O, and these species are also present in the same concentrations in the background free troposphere with which the boundary layer box exchanges. Photolysis rates follow a clear-sky diurnal profile at 45∘ latitude at the summer solstice with an ozone column of 350 DU, while temperature varies sinusoidally with an amplitude of 4 ∘C, centered at 25 ∘C, peaking at 13:00 solar time, and a period of 1 d. Results shown in Sect. 4.2 and 4.3 are for a total aromatic VOC emission rate of 120 ppt h−1, distributed between benzene, toluene, and xylene in a molar ratio or for a single precursor, and for fixed NO emission rates between 1 ppt h−1 and 10 ppb h−1. Additional results showing sensitivities to VOC emission rates can be found in the Supplement. The model does not represent deposition or aerosol uptake processes, except to impose a 1 h loss rate on N2O5 for conversion to HNO3. We apply 7 d of initialization to reach diurnal steady state; the results shown in Sect. 4.3 are from the eighth simulated day.
4.2 Oxygenated VOC yields
We use environmental chamber simulations to determine prompt product yields from each mechanism and compare them to experimental data; we also use both chamber and boundary layer simulations to investigate the differences in long-term yields of later-generation and multigenerational products between mechanisms. In both cases, we present our results as a function of initial NOx mixing ratio ([NOx]0) imposed in the model either directly (chamber simulations) or through emissions (boundary layer simulations). In some cases, yields from organic peroxy radical reactions may depend on the branching between bimolecular and unimolecular reactions in addition to the branching between reaction with NO and other reactions, and comparisons would be more appropriately made as a function of peroxy radical lifetime against all bimolecular reactions rather than just NO (see, e.g., Xu et al., 2020); however, it is difficult to approximate these bimolecular lifetime conditions for many past experimental results, so we opt for the more straightforward comparisons as a function of initial NOx.
In comparisons with experimental yields (Figs. 4–6), simulated yields are shown after 20 min of oxidation, whereas experimental yields are the earliest reported value. Because the experimental yields shown here come from a range of chamber studies with differing setups (e.g., chamber size, oxidant source, light spectrum and intensity, duration, and initial concentrations), care should be taken when comparing observed yields as a function of [NOx]0 to each other or to simulated yields. Experiments conducted with no added NOx are shown at [NOx]0=10 ppt (below which modeled yields are invariant with [NOx]0) regardless of whether NOx concentrations were monitored during the experiment. In some cases, NOx may off-gas from chamber walls (Carter et al., 2005). When reported, instrumental uncertainties on measured NOx mixing ratios are shown as horizontal error bars on experimental points.
Ring-retaining products. Figure 4 shows environmental chamber molar yields of ring-retaining alcohols and aldehydes from BTX oxidation in the mechanisms and in the experimental literature as functions of initial NOx mixing ratio. For the ring-retaining alcohols formed via OH addition followed by H abstraction (Fig. 1iii), all mechanisms implement fixed direct yields from the BTX precursors, which therefore do not vary with NO. GC13 uses a phenol yield from benzene of 54 %, derived from an error-weighted average of the yields measured by Klotz et al. (2002) (53±7 %), Berndt and Böge (2006) (61±7 %), and Xu et al. (2020) (49±13 %) at atmospherically relevant NOx levels. Among the other mechanisms implemented here, SAPRC uses a fixed phenol yield of 57 % from benzene, whereas all others use 53 %. Fixed cresol and xylenol yields of 19 % and 15 % in GC13, from toluene and xylene, respectively, are taken from MECCA and are consistent with observations.
For phenol, the mechanisms' fixed yields of 53 %–57 % fit most data under low-to-moderate NO conditions; however, when [NOx]0 exceeds 100 ppb, the observed yield declines, as bimolecular reactions become too fast to allow equilibration to occur. This behavior is not exhibited by any of the mechanisms studied here. Such high NO concentrations are also rarely seen in ambient environments; therefore, the fixed yields are suitable for atmospheric simulations. Experimental yields of cresol (from toluene) and xylenol (from xylene) exhibit little correlation with NOx and are, therefore, adequately represented by fixed branching ratios, although the ratios implemented vary widely between mechanisms, with low yields in RACM2 (6 % from toluene and xylene) and no representation of xylenols in CRI v2-R5.
The ring-retaining aldehydes, benzaldehyde and tolualdehyde, are formed following H abstraction from the methyl groups of toluene and xylene, respectively (Fig. 1i). Most mechanisms that include benzaldehyde chemistry (CRI and MOZART-GC do not) explicitly treat the benzyl peroxy intermediate in this reaction pathway, resulting in peak aldehyde yields of ∼6 % when [NOx]0 exceeds 100 ppb, dropping to 1 %–3 % at (NOx]0<1 ppb due to the competing formation of benzyl hydroperoxide. However, this results in lower benzaldehyde yields than observed under NOx-free conditions. The lack of observed NOx dependence in the benzaldehyde yield may reflect a short lifetime of benzyl hydroperoxide, a high incidence of RO2+RO2 reactions (Moschonas et al., 1999), a non-hydroperoxide-forming channel in the benzyl peroxy + HO2 reaction (Baltaretu et al., 2009), or another pathway altogether (Salta et al., 2020). Regardless, to fit observations and further simplify the mechanism, we bypass the benzyl peroxy intermediate and form benzaldehyde directly from the reaction of toluene and xylene with OH with a fixed yield of 6 %, consistent with most experimental results at both high and low NOx.
C1–C3 carbonyl products. Figure 5 shows prompt environmental chamber yields of glyoxal from BTX oxidation in both the mechanisms and the experimental literature as functions of initial NOx mixing ratio (initialized as NO in simulations and in most chamber experiments, with some NO2 in chamber experiments). Initial glyoxal yields from BTX oxidation generally range between 10 % and 40 %, with the highest yields from benzene and the lowest from xylenes. With a fixed NOx-independent first-generation glyoxal yield and secondary yields from the representative C4–C5 intermediates (Fig. 3), GC13 accurately simulates experimentally measured glyoxal yields across a wide range of [NOx]0 for all three aromatic precursors, whereas most other mechanisms exhibit excessive glyoxal formation at [NOx]0>100 ppb and insufficient production at [NOx]0<1 ppb. Due to the long lifetime of benzene relative to its intermediate oxidation products, it is difficult to isolate prompt vs. multigenerational glyoxal yields, which partially explains the range of experimental results; the prompt yield that we implement in GC13 (18 %) is able to match the lower limit of observed yields at both high and low NOx. We also find in GC13 simulations that the glyoxal yield from benzene is sensitive to the initial benzene concentration in chamber experiments (see Sect. S4 in the Supplement), which may further explain the spread of measured yields.
Figure 6 shows simulated and observed environmental chamber yields of formaldehyde and methylglyoxal from toluene and xylene oxidation as functions of initial NOx mixing ratio. Because it lacks alkyl substituents, benzene is unable to produce either formaldehyde or methylglyoxal. As with glyoxal, GC13 is consistent with experimental yields across the full spectrum of [NOx]0 conditions, although some observed methylglyoxal yields from toluene deviate considerably from the general trend. Most other mechanisms exhibit lower prompt formaldehyde and methylglyoxal yields at [NOx]0<1 ppb and higher yields at [NOx]0>100 ppb than GC13 or observed values, although observational evidence is sparse for xylene. In particular, SAPRC and the MCM exhibit high prompt formaldehyde and methylglyoxal yields at [NOx]0>100 ppb, respectively.
In addition to these prompt yields, later-generation formation of C1–C3 carbonyl species can be important for HOx radical and carbon budgets. Data on long-term yields are sparse, so here we rely mostly on model intercomparisons of boundary layer simulations with the MCM taken as a reference. Results are shown in Fig. 7 for mixed aromatic emissions and in Fig. S6 in the Supplement for individual aromatic precursors. The maxima in yields at intermediate NOx, particularly evident for formaldehyde, reflect the corresponding maxima in OH concentrations. Generally, the MCM is able to produce higher late-generation yields of the C1–C3 carbonyls than more reduced mechanisms, but GC13 exhibits high yields similar to the MCM. GC13 also simulates similar midday glyoxal-to-formaldehyde concentration ratios from aromatic oxidation to the MCM, ranging from 0.3 when NOx exceeds 1 ppb to >0.7 at NOx<10 ppt (Fig. S8 in the Supplement). Results speciated by BTX precursor reveal a similar trend. MOZART-GC and MOZART-T1, the most reduced of the previous mechanisms, tend to simulate the lowest long-term carbonyl yields. MECCA is generally able to reproduce the MCM's high long-term carbonyl yields except from xylene, which MECCA lumps as toluene; this results in an overprediction of glyoxal yields and underprediction of formaldehyde and methylglyoxal yields relative to the MCM. Long-term yields from simulated chamber experiments (Figs. S3 and S4 in the Supplement) show results similar to the boundary layer simulations.
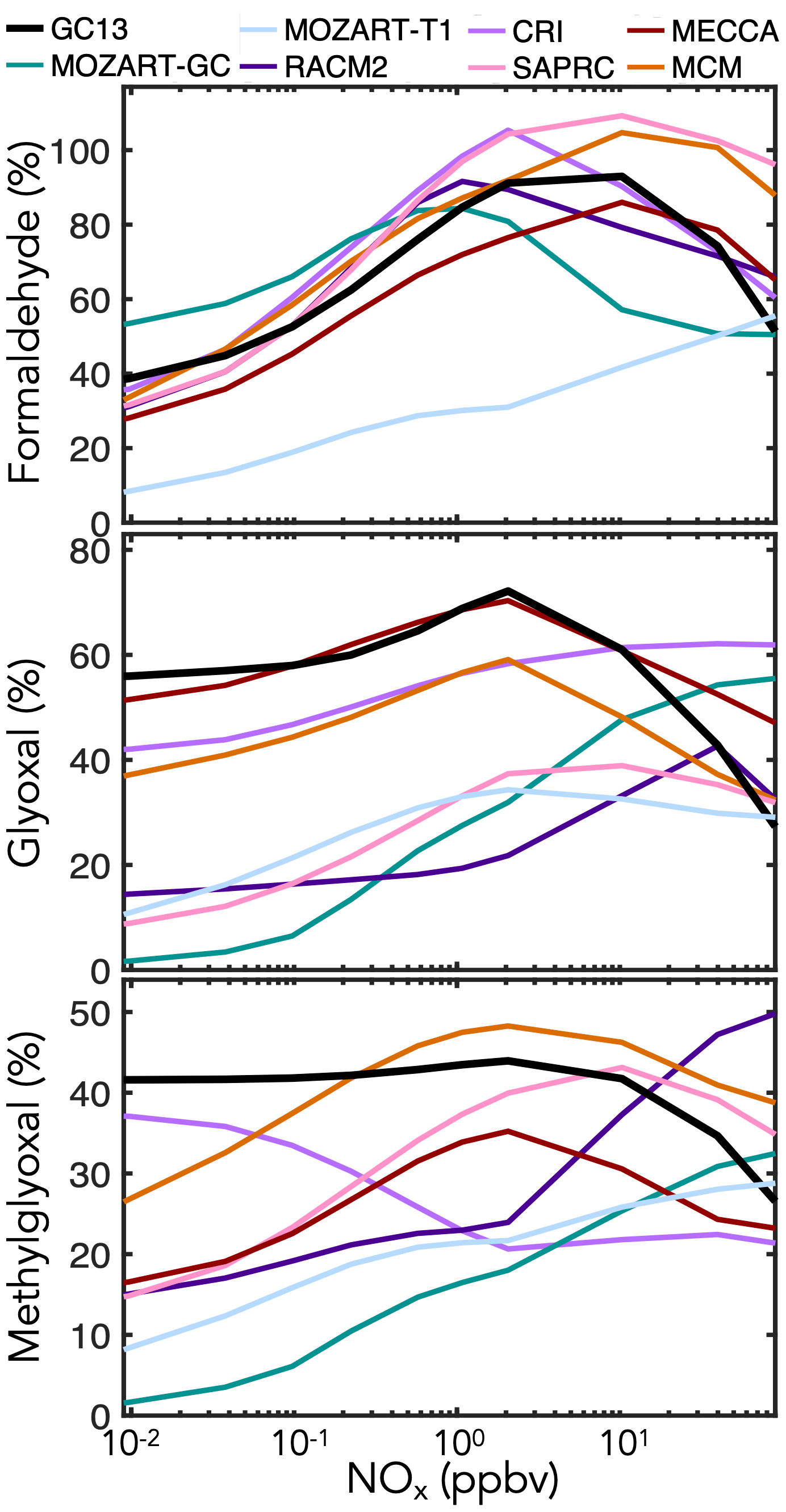
Figure 7Simulated molar yields of formaldehyde, glyoxal, and methylglyoxal from BTX oxidation in a continental boundary layer as a function of midday NOx mixing ratio. The model setup is described in Sect. 4.1. Molar yields are averaged over the eighth simulated day.
Other VOC products. Some experimental and theoretical evidence exists for formation pathways of fumaraldehydic, formic, and acetic acids from BTX oxidation (Berndt et al., 1999; Dumdei and Kenny, 1988; Wang et al., 2020; Xu et al., 2020), but this is generally not included in mechanisms. Global models tend to underestimate ambient concentrations of these compounds, and additional formation pathways could help alleviate this discrepancy (Millet et al., 2015; Khan et al., 2018). Here, we include the formation of formic and acetic acids as described in Newland et al. (2019) and Wang et al. (2020) via the ketene-enols, represented as part of the lumped C4 and C5 products. Few chamber data are available for comparison; Berndt et al. (1999) measured a 13 % yield of formic acid from benzene under nominally NOx-free conditions, while Dumdei and Kenny (1988) measured a 6 % yield of acetic acid from toluene under high-NO (940 ppb) conditions. Both are consistent with prompt yields in GC13. Long-term yields of formic acid in the mechanism can reach 32 % from benzene, 28 % from toluene, and 16 % from xylene, while acetic acid yields can reach 12 %–13 % from toluene and xylene (See Figs. S2 and S5 in the Supplement).
Most mechanisms also represent the formation of larger (C4+) dicarbonyls, including biacetyl, photolabile conjugated dialdehydes, and less-reactive ketones, with varying degrees of complexity. In GC13, these compounds are not treated explicitly, and are instead grouped into the two representative C4–C5 reactive intermediates. For this reason, we do not optimize any branching ratios in GC13 with environmental chamber yields of C4+ dicarbonyls, but Fig. S1 in the Supplement provides a comparison between measured and simulated yields of photolabile dicarbonyls in each mechanism.
Finally, as described in Sect. 2, most mechanisms include the direct formation of a ring-opened C6+ epoxide from BTX oxidation. These yields are NOx-independent and span a wide range, from 0 % from all precursors in GC13 to >70 % from xylene in RACM. While little evidence for this pathway exists under atmospherically relevant conditions, it may be useful as an intermediate, particularly given that other known pathways cannot achieve carbon closure in many experimental studies (e.g., Xu et al., 2020). Although these epoxides are not included in GC13, Fig. S1 provides a comparison between measured yields in environmental chambers and simulated yields in other mechanisms.
4.3 Effects on oxidants
Figure 8 shows the effects of BTX oxidation on daytime abundances of ozone, OH, and HO2 in the continental boundary layer simulations. Results speciated by aromatic precursor are shown in Fig. S7. GC13 and MECCA have less ozone production than the MCM, RACM, and SAPRC, all of which are known to overestimate peak ozone from chamber experiments (Bloss et al., 2005a; Goliff et al., 2013; Carter and Heo, 2013). This is primarily due to phenoxy–phenylperoxy radical cycling chemistry, represented only in GC13 and MECCA (Sander et al., 2019; Taraborrelli et al., 2021). The dominant phenoxy–phenylperoxy cycle converts NO2 to NO3 and consumes one ozone molecule; during the day, this cycle largely balances via NO3 photolysis (although the minor channel to NO + O2 represents an odd oxygen sink), but at night, conversion of NO3 to N2O5 and on to HNO3 amplifies ozone loss. MECCA and GC13 both exhibit a surge in NO3 (and corresponding reduction in NO2) at sunset, which propagates to lower NO2 and ozone levels throughout the following day (Fig. S30 in the Supplement). Flux through this phenoxy–phenylperoxy system is highest from benzene because of the high phenol yields (Fig. 1), so the ozone differences between mechanisms are strongest for benzene (see Fig. S7). The reduced ozone formation in GC13 and MECCA relative to other mechanisms may improve model biases relative to chamber experiments (Bloss et al., 2005a; Goliff et al., 2013; Carter and Heo, 2013) and would likely reduce the high simulated contribution of aromatics to ambient ozone formation in box model analyses of polluted environments (Luecken et al., 2018; Oak et al., 2019; Schroeder et al., 2020).
GC13 also simulates higher HOx concentrations than other mechanisms, especially under low-NOx (<1 ppb) conditions. This effect is due primarily to increased radical propagation from the bimolecular reactions of the bridged bicyclic peroxy radicals (Fig. 1c), which do not form radical-terminating hydroperoxides or organonitrates in GC13. Higher radical recycling from subsequent reactions of the representative C4 and C5 intermediates also contributes. It has been reported that other mechanisms tend to underpredict HOx concentrations in simulations of chamber experiments (Chen, 2008), and both Bloss et al. (2005a) and Carter and Heo (2013) comment on the need to increase HOx recycling from aromatic oxidation; thus, GC13 brings HOx concentrations into improved alignment with chamber results. These effects are stronger for toluene and xylene because of their higher bridged bicyclic peroxy radical and C4–C5 intermediate yields relative to benzene (see Fig. S7).
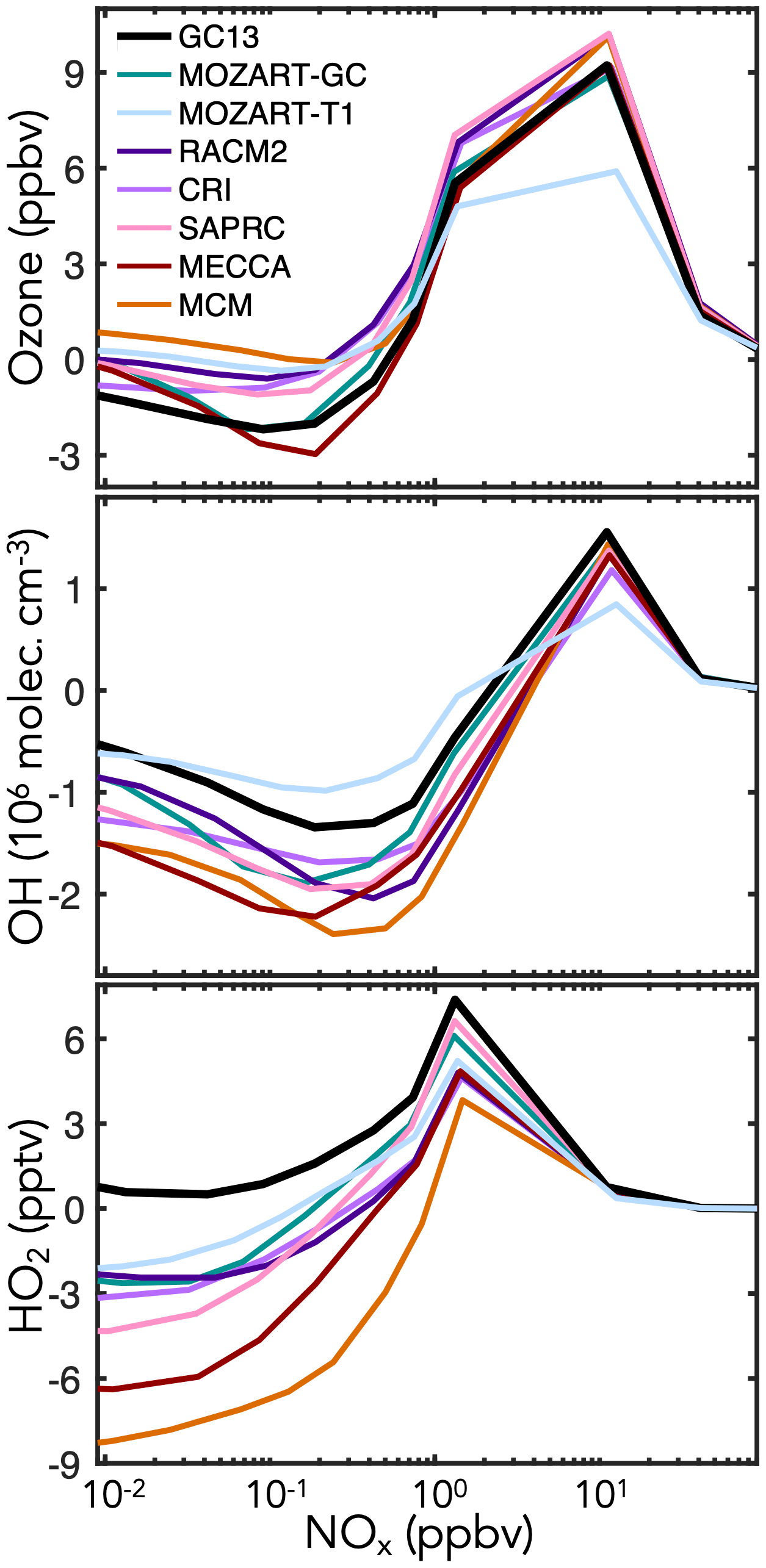
Figure 8Changes in midday ozone, OH, and HO2 concentrations due to aromatic chemistry in box model simulations of the continental boundary layer with different aromatic oxidation mechanisms. Changes in species concentrations are calculated by subtracting their midday (10:00–14:00 LT) mean values in a simulation without aromatic emissions from those in an equivalent simulation with aromatic emissions, and these values are plotted against the midday NOx mixing ratio. Aromatic emissions are 120 ppt h−1 with molar ratios for benzene : toluene : xylenes. Additional model setup is described in Sect. 4.1.
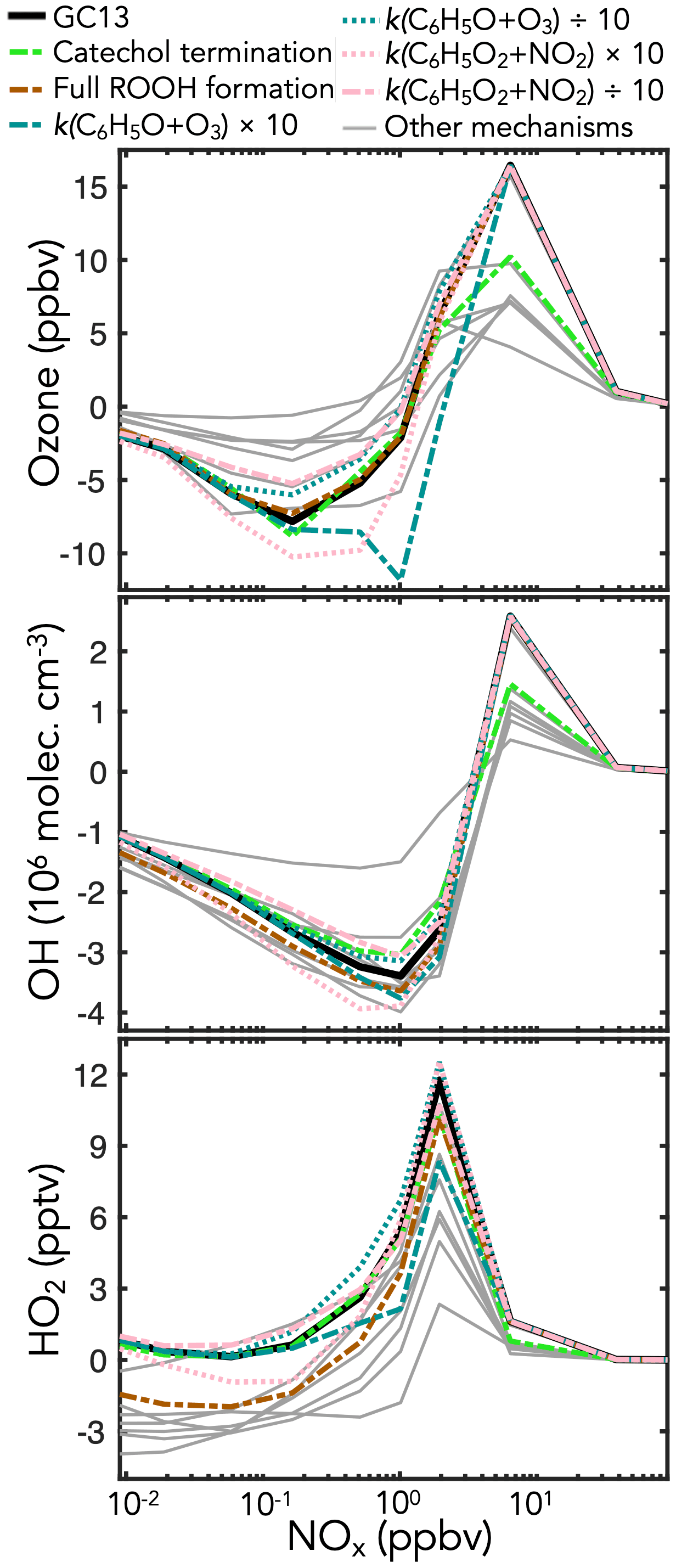
Figure 9Sensitivity of midday ozone, OH, and HO2 concentrations in the GC13 aromatic mechanism to changes in individual reaction rate constants and yields. The black lines show the standard GC13 box model simulation for the effect of mixed aromatic emissions in the continental boundary layer at 10:00–14:00 LT, relative to a simulation with no aromatic emission. The colored and patterned lines show the effects of individual changes in the GC13 mechanism. Gray lines show the results from the other mechanisms in Table 1. Results shown for the GC13 standard simulation and other mechanisms are the same as for Fig. 8 except that BTX emissions were doubled to better show the changes from the individual reactions.
To test the sensitivities of these outcomes to specific aspects of the GC13 mechanism, we conduct additional simulations with individually perturbed reaction rate constants and yields. Results from the sensitivity simulations with the most prominent changes are shown in Fig. 9. We find that ozone is most sensitive to changes in the rates of the key reactions in the phenoxy–phenylperoxy system. These rates remain uncertain; Tao and Li (1999) measured a rate constant of 2.86 (±0.35) at 298 K for the C6H5O+O3 reaction, which we use in GC13, but noted that it might be a lower limit, whereas Jagiella and Zabel (2007) did not set uncertainty bounds on their best fit rate constant of for C6H5O2+NO2 (used in GC13), instead only specifying a minimum of consistent with their results. Increasing either rate by a factor of 10 substantially increases ozone loss due to phenoxy–phenylperoxy cycling, highlighting the importance of better constraints on these rates.
Ozone and OH are both sensitive to changes in the fates of catechols and methylcatechols. While most mechanisms assume that the reactions of catechols and methylcatechols with OH proceed by abstraction to form functionalized phenoxy radicals, Schwantes et al. (2017) showed that addition pathways dominate, leading to heavily substituted low-volatility products that may contribute to SOA formation. In a sensitivity simulation with the product channels from catechols + OH turned off (representing complete loss of products to aerosols), the effects of aromatic oxidation on ozone production and OH are strongly diminished, as this represents a major loss of later-generation gas-phase products such as the C1–C3 carbonyls and C4–C5 intermediates. For both catechol and phenoxy–phenylperoxy perturbations, benzene is the most sensitive of the primary aromatics, due to its higher yields of the phenolic pathway than toluene or xylene (Fig. S25 in the Supplement).
HOx concentrations are also highly sensitive to the relative contributions of the radical propagation and termination pathways from the reactions of HO2 with the initial bridged bicyclic peroxy radicals from BTX + OH under low-NOx (<1 ppb) conditions. A perturbation simulation with 100 % radical termination (i.e., hydroperoxide formation), as in most mechanisms, reduces HO2 in GC13 to levels similar to the other mechanisms, and causes a smaller reduction in OH, highlighting the importance of this branching ratio. However, Xu et al. (2020) showed that hydroperoxide formation is minimal from the benzene-derived bridged bicyclic peroxy radical, motivating our treatment in GC13.
Results from additional sensitivity simulations are shown in Sect. S5 in the Supplement (Figs. S25–S29 in the Supplement). Changes to other reactions in the phenoxy–phenylperoxy system can be important – perturbations to the C6H5O+NO2 rate have similar effects to those of C6H5O+O3 because the two reactions are in direct competition, whereas increasing the C6H5O2+NO3 rate increases ozone loss in a manner similar to increasing the C6H5O2+NO2 rate, although peaking at slightly lower ambient NOx concentrations. Mechanism outcomes are only mildly sensitive to changes in the reactions of nitrophenols, a pathway implemented in GC13 and MECCA. Other novel aspects of GC13, including the 0 % yield of organonitrates from the reactions of NO with the initial bridged bicyclic peroxy radicals (following Xu et al., 2020) and increased HOx recycling from the benzoylperoxy radical + HO2 reaction, have minimal effects on ambient ozone and HOx.
5.1 Methods
To more deeply investigate the effects of BTX oxidation on atmospheric chemistry, we implement the GC13 mechanism into the GEOS-Chem CTM and compare it to other selected mechanisms in the GEOS-Chem environment. GEOS-Chem is driven by meteorology from the Modern-Era Retrospective analysis for Research and Applications, Version 2 (MERRA-2) assimilation product of the NASA Global Modeling and Assimilation Office (GMAO). We use GEOS-Chem version 12.3 (DOI https://doi.org/10.5281/zenodo.2658178) with added C2H4 and C2H2 chemistry (Kwon et al., 2021) as a base, which includes 196 species in its chemical mechanism (not including aromatic chemistry), of which 149 are transported. We run global simulations at a horizontal resolution with 47 vertical layers. For each simulation, we perform an initial 8 month spin-up (1 March–1 December 2015), followed by 1 year of simulation from which seasonal and annual averages are output. We conduct one simulation with no aromatic emissions as a base case as well as one simulation with GC13 for comparison. For simulations with GC13 chemistry, Henry's law coefficients of newly included species (Table S1 in the Supplement) are taken from Sander (2015) and Cabrera-Perez et al. (2016) for use in GEOS-Chem dry and wet deposition modules. The added aromatic chemistry in the GC13 simulation increases overall CPU time by an average of 1.7 % relative to the base simulation, attributable predominantly to gas-phase chemistry (64 %) and transport (26 %).
Anthropogenic VOC emissions in our GEOS-Chem simulations are from the Community Emissions Data System (CEDS) (Hoesly et al., 2018), overwritten with the Multi-resolution Emission Inventory for China (MEIC; Zheng et al., 2018) and with the KORUS v5 inventory for the rest of East Asia (Woo et al., 2012; Jang et al., 2020). Biogenic emissions in GEOS-Chem are from the Model of Emissions of Gases and Aerosols from Nature (MEGAN) version 2.1 (Guenther et al., 2012), and open-fire emissions are from the Global Fire Emissions Database (GFED) version 4 (van der Werf et al., 2010). Global annual emissions are 7.23, 10.42, and 7.30 Tg for benzene, toluene, and xylene from anthropogenic sources, respectively, and the corresponding values from open fires are 1.67, 0.88, and 0.26 Tg, respectively. No emissions of other gas-phase aromatics are included. Total BTX emissions are 60 % higher than in the global model simulation of Taraborrelli et al. (2021) but only 4 % higher (in carbon mass) than their total emissions of C6–C9 aromatics (including phenol, benzaldehyde, ethyl benzene, and lumped C9 aromatics).
We also implement the two simplest alternative mechanisms, RACM2 and MOZART-T1, in the GEOS-Chem environment. MOZART-GC and SAPRC-11 were previously implemented in GEOS-Chem by Porter et al. (2017) and Yan et al. (2019), respectively, but neither were incorporated into the standard version of GEOS-Chem. Instead, aromatic chemistry previously implemented in the standard version of GEOS-Chem was simply parameterized to achieve reasonable glyoxal and methylglyoxal yields with fixed branching ratios for SOA and peroxyacetyl nitrate (PAN) formation (Fu et al., 2008; Fischer et al., 2014). Comparison of GC13 to this parameterized GEOS-Chem aromatic chemistry and to MOZART-GC is shown in the Supplement (Sects. S7 and S8) for reference to past GEOS-Chem studies.
5.2 Effects on oxygenated VOCs
Figure 10 shows the impact of GC13 aromatic chemistry on concentrations of glyoxal, methylglyoxal, and formic acid in the lowest 1 km of the atmosphere. Aromatic oxidation increases the tropospheric production of these three oxygenated VOCs by 30 %, 5 %, and 9 %, respectively. Although absolute changes are strongest in source regions, the relative contribution of aromatic chemistry to these concentrations extends globally, due both to later-generation production and to the longer lifetimes of aromatics relative to other precursors (e.g., anthropogenic alkenes and isoprene). Changes to gas-phase acetic acid are similar but smaller in magnitude than those of formic acid, with aromatic chemistry increasing production by 5 %. Because these oxygenated VOCs are also formed in isoprene oxidation, the relative contribution of aromatics is much lower in high-isoprene regions, especially the tropics. In the Middle East, aromatics are responsible for >80 % of glyoxal because anthropogenic VOC emissions are high while biogenic emissions are low.
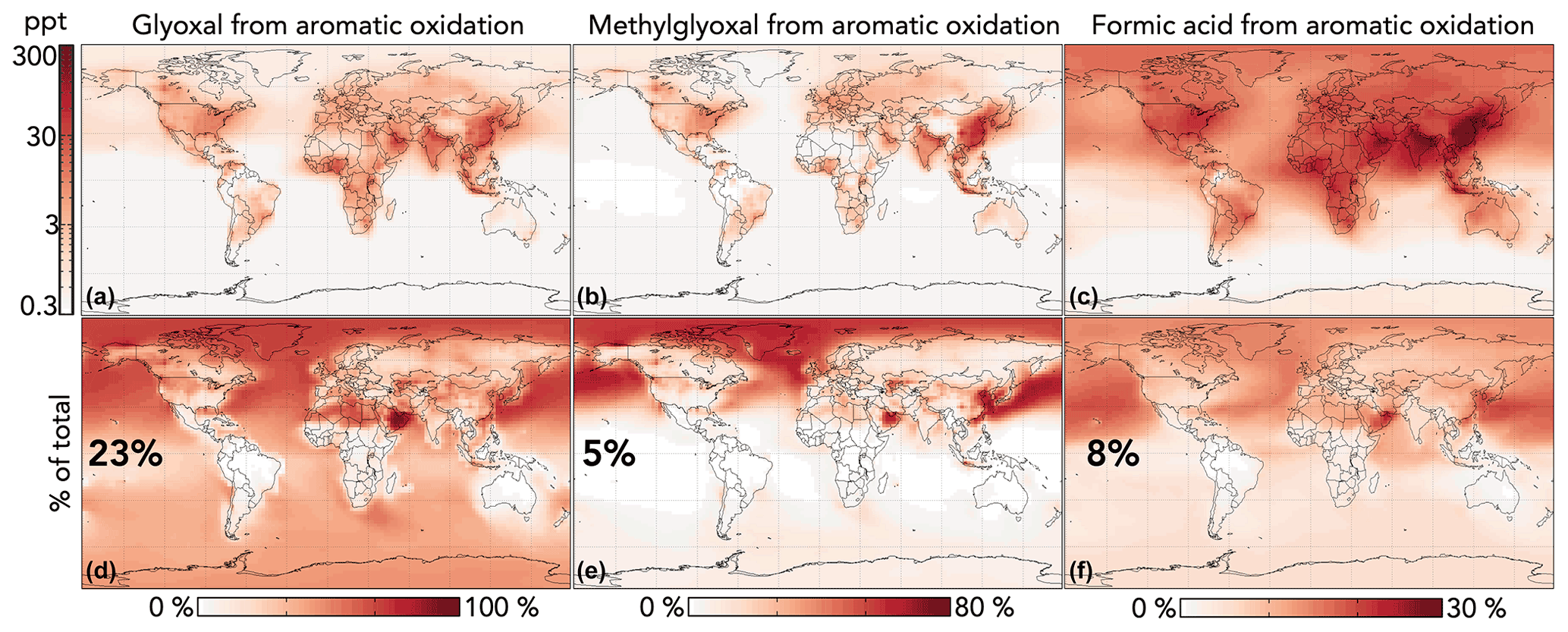
Figure 10Effects of GC13 aromatic chemistry on glyoxal, methylglyoxal, and formic acid concentrations in GEOS-Chem. Panels (a)–(c) show absolute differences in annual mean concentrations below 1 km altitude between GEOS-Chem simulations with GC13 aromatic chemistry and with no aromatic chemistry. Panels (d)–(f) show the percentage contributions of aromatic chemistry to the total model concentrations of the three species. Inset numbers are global percentages. The color scales are logarithmic for panels (a)–(c) and linear for panels (d)–(f).
Our simulated contributions of aromatic oxidation to the budgets of glyoxal and methylglyoxal are substantially larger than in previous GEOS-Chem studies using simplified mechanisms with lower yields (Fu et al., 2008; Silva et al., 2018), but they are more consistent with results from the detailed mechanisms in Taraborrelli et al. (2021) and Yan et al. (2019). The increased glyoxal yields in GC13 align with general model findings of negative biases relative to satellite observations of glyoxal columns in regions with strong anthropogenic influence (Chan Miller et al., 2016; Liu et al., 2012; Myriokefalitakis et al., 2008; Silva et al., 2018; Stavrakou et al., 2009). Taraborrelli et al. (2021) simulated even higher aromatic contributions to the global glyoxal budget, due in part to differences in the mechanisms (higher long-term yields of glyoxal and methylglyoxal from MECCA; see Figs. S3 and S4 in the Supplement) and potentially to differences in non-aromatic glyoxal and methylglyoxal sources between the models. Neither Taraborrelli et al. (2021) nor Yan et al. (2019) discuss the contribution of aromatic chemistry to gas-phase formic or acetic acid budgets.
Additional effects from aromatic chemistry on the global distribution of oxygenated VOCs are shown in Fig. 11. PAN is subject to competing influences: methylglyoxal formation from aromatic oxidation increases the source strength of the acyl peroxy radical, the organic precursor to PAN, whereas lower NO2 due to phenoxy–phenylperoxy cycling tends to decrease PAN production. The former effect dominates in source regions; aromatic oxidation with the GC13 mechanism increases PAN mixing ratios over Northern Hemisphere continents by up to 40 %. Downwind, particularly over oceans, PAN decreases due to phenoxy–phenylperoxy consumption of NO2.
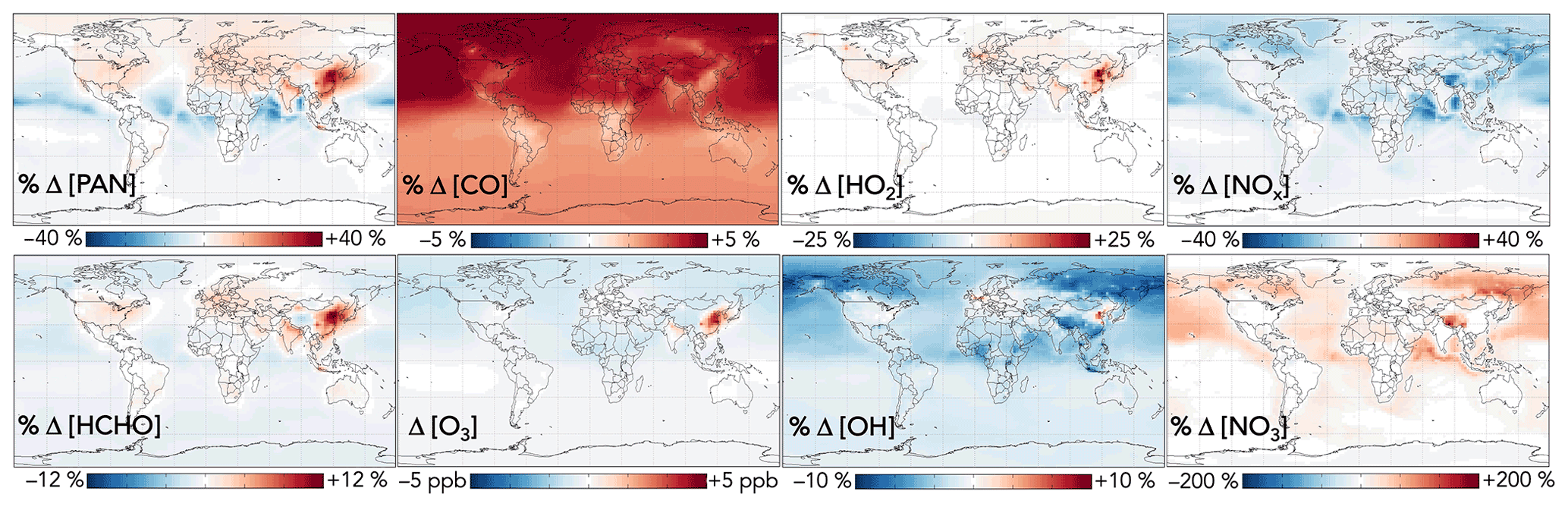
Figure 11Effects of GC13 aromatic chemistry on PAN, formaldehyde, CO, ozone, HO2, OH, NOx, and NO3 concentrations. Panels show the absolute (for ozone) and relative (for others) differences in annual mean concentrations below 1 km altitude between GEOS-Chem simulations with GC13 aromatic chemistry and with no aromatic chemistry. The color scales are linear.
Formaldehyde exhibits a similar spatial pattern to PAN, with competing effects from its direct secondary production via aromatic oxidation, leading to locally increased mixing ratios of up to 12 % from aromatic oxidation, and indirect decreases due to reduced OH, which dominates downwind. Global formaldehyde production changes by just −0.1 % from aromatic oxidation. GC13 increases the tropospheric CO burden relative to the simulation without aromatic chemistry by 3 %, due to both direct production and the decreased OH sink, with less spatial heterogeneity than other effects. The changes to both formaldehyde and CO are spatially consistent with the findings of Taraborrelli et al. (2021) using MECCA but are smaller in magnitude, driven primarily by the smaller change in tropospheric OH in GC13.
Figure 12 compares the effects of aromatic chemistry on oxygenated VOC concentrations in GEOS-Chem with GC13 to simulations with the MOZART-T1 and RACM2 mechanisms. The most prominent difference between the simulations is the higher overall yield of glyoxal from aromatic oxidation in GC13, especially in later-generation chemistry. This results in increases of up to 60 % in surface glyoxal when switching from either MOZART-T1 or RACM2 to GC13, with the strongest effects over the Middle East (where a lack of biogenic emissions renders aromatics the dominant glyoxal source) and in remote areas where later-generation chemistry dominates and decreases in OH increase VOC lifetimes. Overall tropospheric glyoxal loadings are 10 % lower in MOZART-T1 and 13 % lower in RACM2 than in GC13, whereas tropospheric glyoxal production from aromatics is 38 % lower in MOZART-T1 and 61 % lower in RACM2 than in GC13.
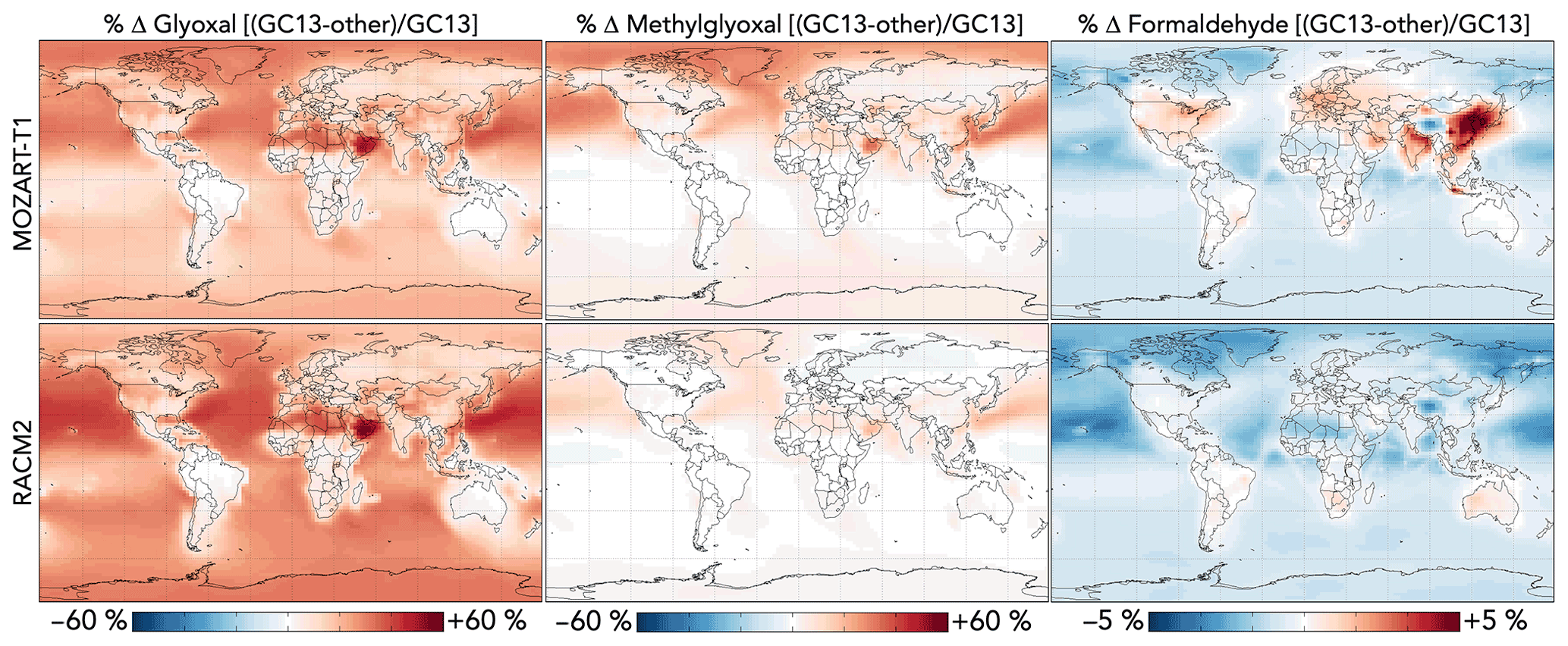
Figure 12Differences in glyoxal, methylglyoxal, and formaldehyde concentrations between the GC13 mechanism and the MOZART-T1 and RACM2 mechanisms for aromatic chemistry. Values are percent differences in annual mean concentrations below 1 km altitude between GEOS-Chem simulations with the GC13 mechanism and with the MOZART-T1 or RACM2 mechanism. The color scales are linear.
Methylglyoxal exhibits similar, although less pronounced, differences between the mechanisms, confined mostly to the Northern Hemisphere where its production is greatest. The strongest differences are seen for the MOZART-T1 mechanism, which produces only 62 % of the methylglyoxal from BTX that GC13 produces in global simulations, resulting in decreases in the surface methylglyoxal mixing ratio of up to 30 % (2 % globally). The RACM2 mechanism produces more methylglyoxal than MOZART-T1 (74 % of the amount produced by GC13 globally) and, therefore, exhibits smaller changes (up to 10 % decreases locally and 1 % decreases globally) and even some local increases. Differences in formaldehyde between the mechanisms are minor; global tropospheric formaldehyde is 0.4 % higher with MOZART-T1 and 0.8 % higher with RACM2 relative to GC13. The most prominent change is an increase of 5 % in boundary layer formaldehyde over Northeast China with GC13 relative to MOZART-T1.
5.3 Effects on oxidants
The effects of aromatic chemistry on radical and ozone budgets are shown in Fig. 11. Impacts on HOx and ozone are consistent with the results of the continental boundary layer simulations in Sect. 4.3. HO2 is increased by aromatic chemistry by up to 20 % annually averaged in high-NOx aromatic source regions, but it exhibits little change (<0.1 %) globally. OH and ozone both increase in high-NOx aromatic source regions, by up to 6 % and 5 ppb, respectively, on annual averages but decrease elsewhere, largely due to phenoxy–phenylperoxy radical cycling. On a global scale, these decreases slightly dominate; aromatic chemistry reduces tropospheric OH and ozone by 2.2 % and <1 % (0.37 ppb) on annual average. The effect on OH has a strong seasonal variation (Fig. 13), with increases in source regions in the Northern Hemisphere winter – up to 24 % over Northeast China – but small effects and even slight decreases in the summer. This is due to the importance of carbonyl photolysis as a wintertime OH source (Li et al., 2021).

Figure 13Seasonal effects of aromatic chemistry on OH and ozone concentrations. Panels show the relative (for OH) and absolute (for ozone) differences in mean concentrations below 1 km altitude between GEOS-Chem simulations with GC13 aromatic chemistry and with no aromatic chemistry for 1 December 2015–1 March 2016 (a, c) and 1 June 2016–1 September 2016 (b, d). The color scales are linear.
NOx concentrations decrease everywhere as a result of aromatic chemistry, most notably in regions downwind of aromatic emissions in the Northern Hemisphere (Fig. 11). Although PAN and peroxybenzoyl nitrate can act as NOx reservoirs, releasing NOx in remote air, there are additional NOx sinks from phenoxy–phenylperoxy cycling and nitrophenol formation. The impacts of NO3 production from phenylperoxy + NO2 are particularly pronounced in downwind regions with very low NOx concentrations, where this pathway can increase annual average NO3 concentrations by up to 200 %.
Generally, these effects of aromatic chemistry on oxidants in GC13 are consistent with those from MECCA in Taraborrelli et al. (2021), who also showed global decreases in NOx, OH, and ozone from aromatic chemistry, along with local increases and seasonal cycles for OH and ozone in areas of strong aromatic emissions, and strong increases in NO3. Global average changes tend to be stronger in Taraborrelli et al. (2021), especially for OH and ozone, consistent with the sharper decreases in these compounds due to aromatic chemistry in continental boundary layer simulations (Fig. 8). By contrast, SAPRC implemented in GEOS-Chem by Yan et al. (2019) showed increases in ozone, OH, and NOx concentrations as well as decreases in NO3. We attribute this primarily to the absence of phenoxy–phenylperoxy cycling in SAPRC.

Figure 14Differences in ozone, OH, and NO3 concentrations between the GC13 mechanism and the MOZART-T1 and RACM2 mechanisms for aromatic chemistry. Values are absolute (for OH) or percent (for other species) differences in annual mean concentrations below 1 km altitude between GEOS-Chem simulations with the GC13 mechanism and with the MOZART-T1 or RACM2 mechanism. The color scales are linear.
Differences in oxidant concentrations between global simulations with GC13 and with the MOZART-T1 and RACM2 mechanisms are shown in Fig. 14. Changes to ozone, HOx, and NOx between the mechanisms can be attributed primarily to the inclusion of phenoxy–phenylperoxy cycling and increased OH recycling in GC13. Surface ozone and OH are reduced in GC13 relative to MOZART-T1 and RACM2, consistent with findings in box model simulations (Sect. 4.3). Both MOZART-T1 and RACM2 cause increases in the tropospheric ozone burden relative to a simulation without aromatic chemistry, as Yan et al. (2019) also showed for SAPRC, whereas aromatic chemistry in GC13, like MECCA (Taraborrelli et al., 2021), causes a global ozone decrease. As an additional consequence of their lack of phenoxy–phenylperoxy cycling, the MOZART-T1 and RACM2 mechanisms simulate much lower tropospheric burdens of NO3 (by 18 % and 19 %, respectively) and slightly higher NOx burdens (by 3 % and 2 %, respectively) than GC13. Finally, higher OH recycling in GC13 leads to local increases in OH relative to MOZART-T1 (up to 4 %) in source regions, but globally, differences in tropospheric and surface OH are <2 %.
Our GC13 mechanism and its implementation in GEOS-Chem focused on the effect of BTX emissions. Taraborrelli et al. (2021) estimated that non-BTX aromatics contribute 54 % of global aromatic emissions by carbon mass with C6–C8 oxygenates, C8–C9 aromatic hydrocarbons, and higher aromatics each contributing approximately one-third of this non-BTX fraction. The contributions from C6–C8 oxygenate emissions including benzaldehyde, phenol, cresols, and catechols can be readily accounted for in GC13, as they are already included explicitly as secondary species. C8–C9 aromatic hydrocarbons including ethylbenzene, styrene, and trimethylbenzenes can be added as independent species using simplified initial oxidation reactions to convert them directly to products already included in GC13. We have provided such reactions, simplified from the MCM (Jenkin et al., 2003), in Sect. S1 in the Supplement, and this would add three species and seven reactions to GC13. Higher aromatics such as polycyclic aromatic hydrocarbons (PAHs) are semi-volatile, and their oxidation products partition heavily into the aerosol phase (Chan et al., 2009; Chen et al., 2016), so their simulation is more relevant to SOA formation than to oxidant chemistry.
We developed a new compact mechanism (GC13) for fast and accurate simulation of benzene, toluene, and xylene (BTX) oxidation chemistry in atmospheric models. GC13 includes only 17 unique species to describe the aromatic oxidation chain and 44 unique reactions. It includes recent information from experimental and computational studies, and captures the important features of much more complex and computationally costly mechanisms. In particular, it incorporates recent evidence for efficient radical recycling from the bimolecular reactions of bridged bicyclic peroxy radicals, and it explicitly treats phenoxy–phenylperoxy radical cycling as a sink for ozone and NOx. Ethylbenzene, styrene, and trimethylbenzene can be added to GC13 with just three additional species and seven reactions. GC13 presently focuses on gas-phase chemistry, but future application to secondary organic aerosol (SOA) formation could build on the current treatment of methylcatechols and nitrophenols as explicit species in GC13.
We compared outcomes of GC13 to other aromatic oxidation mechanisms in box model simulations of environmental chamber observations and of the continental boundary layer. Product yields from the mechanism exhibit good agreement with environmental chamber observations and result in increased glyoxal and methylglyoxal yields from aromatic oxidation relative to previous reduced mechanisms. Radical cycling in GC13 tends to increase simulated HOx radical concentrations, which past mechanisms have tended to underestimate (Bloss et al., 2005a; Carter and Heo, 2013). Phenoxy–phenylperoxy radical cycling decreases ozone production, which past mechanisms have tended to overestimate (Bloss et al., 2005a). We find that the effects of aromatic chemistry on HOx, NOx, and ozone are strongly sensitive to uncertainties in the chemistry of the phenoxy–phenylperoxy system, with smaller but significant sensitivities to radical recycling from bridged bicyclic peroxy radicals and the oxidative fates of catechols.
We implemented the GC13 mechanism in the GEOS-Chem global atmospheric chemistry model and compared results to simulations without aromatic chemistry and with alternative mechanisms (MOZART-T1, RACM2). GC13 increases the computational cost of an annual simulation by 1.7 % relative to a simulation without aromatic chemistry. Aromatic oxidation plays a particularly important role in the tropospheric budgets of small dicarbonyl species, contributing 23 % and 5 % to the global production of glyoxal and methylglyoxal, respectively. These values are substantially higher than those simulated with the MOZART-T1 and RACM2 mechanisms. Formaldehyde concentrations over Northeast China increase by 12 % due to aromatic chemistry. Formic and acetic acids increase globally by 9 % and 5 %, respectively, through oxidation of ketene-enols generated by aromatic ring-breaking. Aromatic chemistry decreases global tropospheric OH by 2.2 % and ozone by less than 1 % but increases them in polluted environments in winter, such as in Northeast China where wintertime OH increases by 24 % and surface ozone increases by 5 ppb.
GEOS-Chem and box model output, as well as code files for replication, can be found in the Harvard Dataverse repository (https://doi.org/10.7910/DVN/0UQYOI; Bates, 2021). The GEOS-Chem version used here (12.3) can be found at https://doi.org/10.5281/zenodo.2658178 (Yantosca, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-18351-2021-supplement.
KHB compiled the GC13 mechanism and designed and carried out the modeling described herein. YY, PDI, KL, KHB, MJE, and JL prepared the other mechanisms for implementation in KPP. KHB prepared the paper with substantial assistance from DJJ.
The contact author has declared that neither they nor their co-authors have any competing interests.
The views and opinions expressed in this article are those of the authors and do not represent the official views of the US EPA.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kelvin H. Bates acknowledges the support of the Harvard University Center for the Environment and the National Oceanic and Atmospheric Administration's Climate and Global Change fellowship programs.
This work was supported by the U.S. Environmental Protection Agency (EPA) Science To Achieve Results (STAR) program (grant no. 840014 to Harvard University).
This paper was edited by Nga Lee Ng and reviewed by two anonymous referees.
Archer-Nicholls, S., Lowe, D., Utembe, S., Allan, J., Zaveri, R. A., Fast, J. D., Hodnebrog, Ø., Denier van der Gon, H., and McFiggans, G.: Gaseous chemistry and aerosol mechanism developments for version 3.5.1 of the online regional model, WRF-Chem, Geosci. Model Dev., 7, 2557–2579, https://doi.org/10.5194/gmd-7-2557-2014, 2014. a
Archer-Nicholls, S., Abraham, N. L., Shin, Y. M., Weber, J., Russo, M. R., Lowe, D., Utembe, S., O'Connor, F. M., Kerridge, B., Latter, B., Siddans, R., Jenkin, M., Wild, O., and Archibald, A. T.: The Common Representative Intermediates Mechanism version 2 in the United Kingdom Chemistry and Aerosols Model, Earth and Space Science Open Archive, 49 pp., https://doi.org/10.1002/essoar.10505092.1, 2020. a
Arey, J., Obermeyer, G., Aschmann, S. M., Chattopadhyay, S., Cusick, R. D., and Atkinson, R.: Dicarbonyl products of the OH radical-initiated reaction of a series of aromatic hydrocarbons, Environ. Sci. Technol., 43, 683–689, https://doi.org/10.1021/es8019098, 2009.
Atkinson, R. and Aschmann, S. M.: Products of the gas-phase reactions of aromatic hydrocarbons: effect of NO2 concentration, Int. J. Chem. Kinet., 26, 929–944, https://doi.org/10.1002/kin.550260907, 1994.
Atkinson, R., Carter, W. P. L., and Winer, A. M.: Effects of pressure on product yields in the NOx photooxidations of selected aromatic hydrocarbons, J. Phys. Chem., 87, 1605–1610, https://doi.org/10.1021/j100232a029, 1983.
Atkinson, R., Carter, W. P. L., Plum, C. N., Winer, A. M., and Pitts Jr., J. N.: Kinetics of the gas-phase reactions of NO3 radicals with a series of aromatics at 296 ± 2 K, Int. J. Chem. Kinet., 16, 887–898, https://doi.org/10.1002/kin.550160709, 1984. a
Atkinson, R., Aschmann, S. M., Arey, J., and Carter, W. P. L.: Formation of ring-retaining products from the OH radical-initiated reactions of benzene and toluene, Int. J. Chem. Kinet., 21, 801–827, https://doi.org/10.1002/kin.550210907, 1989.
Atkinson, R., Aschmann, S. M., and Arey, J.: Formation of ring-retaining products from the OH radical-initiated reactions of o-, m-, and p-xylene, Int. J. Chem. Kinet., 23, 77–97, https://doi.org/10.1002/kin.550210907, 1991.
Baltaretu, C. O., Lichtman, E. I., Hadler, A. B., and Elrod, M. J.: Primary atmospheric oxidation mechanism for toluene, J. Phys. Chem. A, 113, 221–230, https://doi.org/10.1021/jp806841t, 2009. a
Bandow, H., Washida, N., and Akimoto, H.: Ring-cleavage reactions of aromatic hydrocarbons studied by FT-IR spectroscopy. I. Photooxidation of toluene and benzene in the NOx-air system, B. Chem. Soc. Jpn., 58, 2531–2540, https://doi.org/10.1246/bcsj.58.2531, 1985.
Barletta, B., Meinardi, S., Rowland, F. S., Chan, C.-Y., Wang, X., Zou, S., Chan, L. Y., and Blake, D. R.: Volatile organic compounds in 43 Chinese cities, Atmos. Environ., 39, 5979–5990, https://doi.org/10.1016/j.atmosenv.2005.06.029, 2005. a
Bates, K.: Code and files for “Development and evaluation of a new reduced mechanism for aromatic oxidation in atmospheric models”, V2, Harvard Dataverse [code], https://doi.org/10.7910/DVN/0UQYOI, 2021. a
Becker, K.-H., Barnes, I., Bierbach, A., Brockmann, K. J., and Kirchner, F.: Chemical Processes in Atmospheric Oxidation, Springer, Berlin, 1997.
Bejan, I., Abd El Aal, Y., Barnes, I., Benter, T., Bohn, B., Wiesen, P., and Kleffmann, J.: The photolysis of ortho-nitrophenols: a new gas phase source of HONO, Phys. Chem. Chem. Phys., 8, 2028–2035, https://doi.org/10.1039/B516590C, 2006. a
Berndt, T. and Böge, O.: Gas-phase reaction of OH radicals with benzene: products and mechanism, Phys. Chem. Chem. Phys., 3, 4946–4956, https://doi.org/10.1039/b106667f, 2001. a
Berndt, T. and Böge, O.: Formation of phenol and carbonyls from the atmospheric reaction of OH radicals with benzene, Phys. Chem. Chem. Phys., 8, 1205–1214, https://doi.org/10.1039/b514148f, 2006. a
Berndt, T., Böge, O., and Herrmann, H.: On the formation of benzene oxide/oxepin in the gas-phase reaction of OH radicals with benzene, Chem. Phys. Lett., 314, 435–442, https://doi.org/10.1016/S0009-2614(99)01041-6, 1999. a, b
Birdsall, A. W. and Elrod, M. J.: Comprehensive NO-dependent study of the products of the oxidation of atmospherically relevant aromatic compounds, J. Phys. Chem. A, 115, 5397–5407, https://doi.org/10.1021/jp2010327, 2011. a
Bjergbakke, E., Sillesen, A., and Pagsberg, P.: UV spectrum and kinetics of hydroxycyclohexadienyl radicals, J. Phys. Chem., 100, 5729–5736, https://doi.org/10.1021/jp951588c, 1996.
Bloss, C., Wagner, V., Bonzanini, A., Jenkin, M. E., Wirtz, K., Martin-Reviejo, M., and Pilling, M. J.: Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data, Atmos. Chem. Phys., 5, 623–639, https://doi.org/10.5194/acp-5-623-2005, 2005a. a, b, c, d, e, f, g
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005b. a, b, c
Brown-Steiner, B., Selin, N. E., Prinn, R., Tilmes, S., Emmons, L., Lamarque, J.-F., and Cameron-Smith, P.: Evaluating simplified chemical mechanisms within present-day simulations of the Community Earth System Model version 1.2 with CAM4 (CESM1.2 CAM-chem): MOZART-4 vs. Reduced Hydrocarbon vs. Super-Fast chemistry, Geosci. Model Dev., 11, 4155–4174, https://doi.org/10.5194/gmd-11-4155-2018, 2018. a
Cabrera-Perez, D., Taraborrelli, D., Sander, R., and Pozzer, A.: Global atmospheric budget of simple monocyclic aromatic compounds, Atmos. Chem. Phys., 16, 6931–6947, https://doi.org/10.5194/acp-16-6931-2016, 2016. a, b, c, d
Cai, C., Kelly, J. T., Avise, J. C., Kaduwela, A. P., and Stockwell, W. R.: Photochemical Modeling in California with Two Chemical Mechanisms: Model Intercomparison and Response to Emission Reductions, J. Air Waste Manage., 61, 559–572, https://doi.org/10.3155/1047-3289.61.5.559, 2011. a
Calvert, J. G.: The mechanisms of atmospheric oxidation of aromatic hydrocarbons, Oxford University Press, Oxford, 2002. a
Carter, W. P.: A detailed mechanism for the gas-phase atmospheric reactions of organic compounds, Atmos. Environ., 24, 481–518, https://doi.org/10.1016/0960-1686(90)90005-8, 1990. a
Carter, W. P., Cocker, D. R., Fitz, D. R., Malkina, I. L., Bumiller, K., Sauer, C. G., Pisano, J. T., Bufalino, C., and Song, C.: A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation, Atmos. Environ., 39, 7768–7788, https://doi.org/10.1016/j.atmosenv.2005.08.040, 2005. a
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013. a, b, c, d, e, f, g
Chan, A. W. H., Kautzman, K. E., Chhabra, P. S., Surratt, J. D., Chan, M. N., Crounse, J. D., Kürten, A., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 9, 3049–3060, https://doi.org/10.5194/acp-9-3049-2009, 2009. a
Chan Miller, C., Jacob, D. J., González Abad, G., and Chance, K.: Hotspot of glyoxal over the Pearl River delta seen from the OMI satellite instrument: implications for emissions of aromatic hydrocarbons, Atmos. Chem. Phys., 16, 4631–4639, https://doi.org/10.5194/acp-16-4631-2016, 2016. a, b
Chance, K., Palmer, P. I., Spurr, R. J. D., Martin, R. V., Kurosu, T. P., and Jacob, D. J.: Satellite observations of formaldehyde over North America from GOME, Geophys. Res. Lett., 27, 3461–3464, https://doi.org/10.1029/2000GL011857, 2000. a
Chen, C.-L., Kacarab, M., Tang, P., and Cocker, D. R.: SOA formation from naphthalene, 1-methylnaphthalene, and 2-methylnaphthalene photooxidation, Atmos. Environ., 131, 424–433, https://doi.org/10.1016/j.atmosenv.2016.02.007, 2016. a
Chen, J., Wenger, J. C., and Venables, D. S.: Near-Ultraviolet Absorption Cross Sections of Nitrophenols and Their Potential Influence on Tropospheric Oxidation Capacity, J. Phys. Chem. A, 115, 12235–12242, https://doi.org/10.1021/jp206929r, 2011. a
Chen, S.: A comparison of chemical mechanisms based on TexAQS-2006 field data, Master's thesis, Pennsylvania State University, State College, PA, USA, 2008. a
Daescu, D., Sandu, A., and Carmichael, G.: Direct and adjoint sensitivity analysis of chemical kinetic systems with KPP: II – validation and numerical experiments, Atmos. Environ., 37, 5097–5114, https://doi.org/10.1016/j.atmosenv.2003.08.020, 2003. a
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G.: The Kinetic PreProcessor (KPP) – a software environment for solving chemical kinetics, Comput. Chem. Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002. a
Duarte-Davidson, R., Courage, C., Rushton, L., and Levy, L.: Benzene in the environment: an assessment of the potential risks to the health of the population, Occup. Environ. Med., 58, 2–13, https://doi.org/10.1136/oem.58.1.2, 2001. a
Dumdei, B. E. and Kenny, D. V.: MS/MS analysis of the products of toluene photooxidation and measurement of their mutagenic activity, Environ. Sci. Technol., 22, 1493–1498, https://doi.org/10.1021/es00177a017, 1988. a, b
Emmons, L. K., Schwantes, R. H., Orlando, J. J., Tyndall, G., Kinnison, D., Lamarque, J.-F., Marsh, D., Mills, M. J., Tilmes, S., Bardeen, C., Buchholz, R. R., Conley, A., Gettelman, A., Garcia, R., Simpson, I., Blake, D. R., Meinardi, S., and Pétron, G.: The chemistry mechanism in the Community Earth System Model Version 2 (CESM2), J. Adv. Model. Earth Sy., 12, e2019MS001882, https://doi.org/10.1029/2019MS001882, 2020. a, b, c
Fischer, E. V., Jacob, D. J., Yantosca, R. M., Sulprizio, M. P., Millet, D. B., Mao, J., Paulot, F., Singh, H. B., Roiger, A., Ries, L., Talbot, R. W., Dzepina, K., and Pandey Deolal, S.: Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 14, 2679–2698, https://doi.org/10.5194/acp-14-2679-2014, 2014. a, b
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.-Atmos., 113, D15303, https://doi.org/10.1029/2007JD009505, 2008. a, b, c
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020. a, b
Gery, M. W., Fox, D. L., Jeffries, H. E., Stockburger, L., and Weathers, W. S.: A continuous stirred tank reactor investigation of the gas-phase reaction of hydroxyl radicals and toluene, Int. J. Chem. Kinet., 17, 931–955, https://doi.org/10.1002/kin.550170903, 1985.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013. a, b, c, d, e
Gómez Alvarez, E., Viidanoja, J., Muñoz, A., Wirtz, K., and Hjorth, J.: Experimental confirmation of the dicarbonyl route in the photo-oxidation of toluene and benzene, Environ. Sci. Technol., 41, 8362–8369, https://doi.org/10.1021/es0713274, 2007.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009. a
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018. a
Hu, L., Keller, C. A., Long, M. S., Sherwen, T., Auer, B., Da Silva, A., Nielsen, J. E., Pawson, S., Thompson, M. A., Trayanov, A. L., Travis, K. R., Grange, S. K., Evans, M. J., and Jacob, D. J.: Global simulation of tropospheric chemistry at 12.5 km resolution: performance and evaluation of the GEOS-Chem chemical module (v10-1) within the NASA GEOS Earth system model (GEOS-5 ESM), Geosci. Model Dev., 11, 4603–4620, https://doi.org/10.5194/gmd-11-4603-2018, 2018. a
Jagiella, S. and Zabel, F.: Reaction of phenylperoxy radicals with NO2 at 298 K, Phys. Chem. Chem. Phys., 9, 5036–5051, https://doi.org/10.1039/B705193J, 2007. a, b
Jang, Y., Lee, Y., Kim, J., Kim, Y., and Woo, J.-H.: Improvement China point source for improving bottom-up emission inventory, Asia-Pac. J. Atmos. Sci., 56, 107–118, https://doi.org/10.1007/s13143-019-00115-y, 2020. a
Jenkin, M., Watson, L., Utembe, S., and Shallcross, D.: A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 1: Gas phase mechanism development, Atmos. Environ., 42, 7185–7195, https://doi.org/10.1016/j.atmosenv.2008.07.028, 2008. a, b
Jenkin, M. E., Saunders, S. M., Derwent, R. G., and Pilling, M. J.: Development of a reduced speciated VOC degradation mechanism for use in ozone models, Atmos. Environ., 36, 4725–4734, https://doi.org/10.1016/S1352-2310(02)00563-0, 2002. a
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003. a, b, c
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Khan, A., Szulejko, J. E., Kim, K.-H., and Brown, R. J. C.: Airborne volatile aromatic hydrocarbons at an urban monitoring station in Korea from 2013 to 2015, J. Environ. Manage., 209, 525–538, https://doi.org/10.1016/j.jenvman.2017.12.055, 2018. a, b
Khan, M., Cooke, M. C., Utembe, S. R., Archibald, A. T., Maxwell, P., Morris, W. C., Xiao, P., Derwent, R. G., Jenkin, M. E., Percival, C. J., Walsh, R. C., Young, T. D. S., Simmonds, P. G., Nickless, G., O'Doherty, S., and Shallcross, D. E.: A study of global atmospheric budget and distribution of acetone using global atmospheric model STOCHEM-CRI, Atmos. Environ., 112, 269–277, https://doi.org/10.1016/j.atmosenv.2015.04.056, 2015. a
Kitayama, K., Morino, Y., Yamaji, K., and Chatani, S.: Uncertainties in O3 concentrations simulated by CMAQ over Japan using four chemical mechanisms, Atmos. Environ., 198, 448–462, https://doi.org/10.1016/j.atmosenv.2018.11.003, 2019. a, b
Klotz, B., Sorensen, S., Barnes, I., Becker, K.-H., Etzkorn, T., Volkamer, R., Platt, U., Wirtz, K., and Martín-Reviejo, M.: Atmospheric oxidation of toluene in a large-volume outdoor photoreactor: in situ determination of ring-retaining product yields, J. Phys. Chem. A, 102, 10289–10299, https://doi.org/10.1021/jp982719n, 1998.
Klotz, B., Volkamer, R., Hurley, M. D., Sulbaek Andersen, M. P., Nielsen, O. J., Barnes, I., Imamura, T., Wirtz, K., Becker, K.-H., Platt, U., Wallington, T. J., and Washida, N.: OH-initiated oxidation of benzene. Part I I. Influence of elevated NOx concentrations, Phys. Chem. Chem. Phys., 4, 4399–4411, https://doi.org/10.1039/b204398j, 2002. a
Knote, C., Hodzic, A., Jimenez, J. L., Volkamer, R., Orlando, J. J., Baidar, S., Brioude, J., Fast, J., Gentner, D. R., Goldstein, A. H., Hayes, P. L., Knighton, W. B., Oetjen, H., Setyan, A., Stark, H., Thalman, R., Tyndall, G., Washenfelder, R., Waxman, E., and Zhang, Q.: Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model, Atmos. Chem. Phys., 14, 6213–6239, https://doi.org/10.5194/acp-14-6213-2014, 2014. a
Kwon, H.-A., Park, R. J., Oak, Y. J., Nowlan, C. R., Janz, S. J., Kowalewski, M. G., Fried, A., Walega, J., Bates, K. H., Choi, J., Blake, D. R., Wisthaler, A., and Woo, J.-H.: Top-down estimates of anthropogenic VOC emissions in South Korea using formaldehyde vertical column densities from aircraft during the KORUS-AQ campaign, Elementa: Science of the Anthropocene, 9, 00109, https://doi.org/10.1525/elementa.2021.00109, 2021. a
Lee, S. C., Chiu, M. Y., Ho, K. F., Zou, S. C., and Wang, X.: Volatile organic compounds (VOCs) in urban atmosphere of Hong Kong, Chemosphere, 48, 375–382, https://doi.org/10.1016/S0045-6535(02)00040-1, 2002. a
Leone, J. A., Flagan, R. C., Grosjean, D., and Seinfeld, J. H.: An outdoor smog chamber and modeling study of toluene-NOx photooxidation, Int. J. Chem. Kinet., 17, 177–216, https://doi.org/10.1002/kin.550170206, 1985.
Li, K., Jacob, D. J., Liao, H., Qiu, Y., Shen, L., Zhai, S., Bates, K. H., Sulprizio, M. P., Song, S., Lu, X., Zhang, Q., Zheng, B., Zhang, Y., Zhang, J., Lee, H. C., and Kuk, S. K.: Ozone pollution in the North China Plain spreading into the late-winter haze season, P. Natl. Acad. Sci. USA, 118, e2015797118, https://doi.org/10.1073/pnas.2015797118, 2021. a
Lin, G., Penner, J. E., Sillman, S., Taraborrelli, D., and Lelieveld, J.: Global modeling of SOA formation from dicarbonyls, epoxides, organic nitrates and peroxides, Atmos. Chem. Phys., 12, 4743–4774, https://doi.org/10.5194/acp-12-4743-2012, 2012. a
Liu, Z., Wang, Y., Vrekoussis, M., Richter, A., Wittrock, F., Burrows, J. P., Shao, M., Chang, C.-C., Liu, S.-C., Wang, H., and Chen, C.: Exploring the missing source of glyoxal (CHOCHO) over China, Geophys. Res. Lett., 39, L10812, https://doi.org/10.1029/2012GL051645, 2012. a, b
Luecken, D. J., Napelenok, S. L., Strum, M., Scheffe, R., and Phillips, S.: Sensitivity of ambient atmospheric formaldehyde and ozone to precursor species and source types across the U.S, Environ. Sci. Technol., 52, 4668–4675, https://doi.org/10.1021/acs.est.7b05509, 2018. a
Manuela, C., Gianfranco, T., Maria, F., Tiziana, C., Carlotta, C., Giorgia, A., Assunta, C., Pia, S. M., Jean-Claude, A., Francesco, T., and Angela, S.: Assessment of occupational exposure to benzene, toluene and xylenes in urban and rural female workers, Chemosphere, 87, 813–819, https://doi.org/10.1016/j.chemosphere.2012.01.008, 2012. a
Martín-Reviejo, M. and Wirtz, K.: Is benzene a precursor for secondary organic aerosol?, Environ. Sci. Technol., 39, 1045–1054, https://doi.org/10.1021/es049802a, 2005.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021. a, b
Millet, D. B., Baasandorj, M., Farmer, D. K., Thornton, J. A., Baumann, K., Brophy, P., Chaliyakunnel, S., de Gouw, J. A., Graus, M., Hu, L., Koss, A., Lee, B. H., Lopez-Hilfiker, F. D., Neuman, J. A., Paulot, F., Peischl, J., Pollack, I. B., Ryerson, T. B., Warneke, C., Williams, B. J., and Xu, J.: A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 15, 6283–6304, https://doi.org/10.5194/acp-15-6283-2015, 2015. a
Misztal, P. K., Hewitt, C. N., Wildt, J., Blande, J. D., Eller, A. S. D., Fares, S., Gentner, D. R., Gilman, J. B., Graus, M., Greenberg, J., Guenther, A. B., Hansel, A., Harley, P., Huang, M., Jardine, K., Karl, T., Kaser, L., Keutsch, F. N., Kiendler-Scharr, A., Kleist, E., Lerner, B. M., Li, T., Mak, J., Nölscher, A. C., Schnitzhofer, R., Sinha, V., Thornton, B., Warneke, C., Wegener, F., Werner, C., Williams, J., Worton, D. R., Yassaa, N., and Goldstein, A. H.: Atmospheric benzenoid emissions from plants rival those from fossil fuels, Sci. Rep.-UK, 5, 12064, https://doi.org/10.1038/srep12064, 2015. a
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018. a, b
Moschonas, N., Danalatos, D., and Glavas, S.: The effect of O2 and NO2 on the ring retaining products of the reaction of toluene with hydroxyl radicals, Atmos. Environ., 33, 111–116, https://doi.org/10.1016/S1352-2310(98)00134-4, 1999. a
Myriokefalitakis, S., Vrekoussis, M., Tsigaridis, K., Wittrock, F., Richter, A., Brühl, C., Volkamer, R., Burrows, J. P., and Kanakidou, M.: The influence of natural and anthropogenic secondary sources on the glyoxal global distribution, Atmos. Chem. Phys., 8, 4965–4981, https://doi.org/10.5194/acp-8-4965-2008, 2008. a
Na, K., Kim, Y. P., Moon, I., and Moon, K.-C.: Chemical composition of major VOC emission sources in the Seoul atmosphere, Chemosphere, 55, 585–594, https://doi.org/10.1016/j.chemosphere.2004.01.010, 2004. a
Nault, B. A., Campuzano-Jost, P., Day, D. A., Schroder, J. C., Anderson, B., Beyersdorf, A. J., Blake, D. R., Brune, W. H., Choi, Y., Corr, C. A., de Gouw, J. A., Dibb, J., DiGangi, J. P., Diskin, G. S., Fried, A., Huey, L. G., Kim, M. J., Knote, C. J., Lamb, K. D., Lee, T., Park, T., Pusede, S. E., Scheuer, E., Thornhill, K. L., Woo, J.-H., and Jimenez, J. L.: Secondary organic aerosol production from local emissions dominates the organic aerosol budget over Seoul, South Korea, during KORUS-AQ, Atmos. Chem. Phys., 18, 17769–17800, https://doi.org/10.5194/acp-18-17769-2018, 2018. a
Newland, M. J., Rea, G. J., Thüner, L. P., Henderson, A. P., Golding, B. T., Rickard, A. R., Barnes, I., and Wenger, J.: Photochemistry of 2-butenedial and 4-oxo-2- pentenal under atmospheric boundary layer conditions, Phys. Chem. Chem. Phys., 21, 1160–1171, https://doi.org/10.1039/c8cp06437g, 2019. a, b, c, d, e, f, g, h
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007. a
Nielsen, J. E., Pawson, S., Molod, A., Auer, B., da Silva, A. M., Douglass, A. R., Duncan, B., Liang, Q., Manyin, M., Oman, L. D., Putman, W., Strahan, S. E., and Wargan, K.: Chemical Mechanisms and Their Applications in the Goddard Earth Observing System (GEOS) Earth System Model, J. Adv. Model. Earth Sy., 9, 3019–3044, https://doi.org/10.1002/2017MS001011, 2017. a
Nishino, N., Arey, J., and Atkinson, R.: Formation yields of glyoxal and methylglyoxal from the gas-phase OH radical-initiated reactions of toluene, xylenes, and trimethylbenzenes as a function of NO2 concentration, J. Phys. Chem. A, 114, 10140–10147, https://doi.org/10.1021/jp105112h, 2010.
Noda, J., Volkamer, R., and Molina, M. J.: Dealkylation of alkylbenzenes: a significant pathway in the toluene, o-, m-, p-xylene + OH reaction, J. Phys. Chem. A, 113, 9658–9666, https://doi.org/10.1021/jp901529k, 2009.
Oak, Y., Park, R., Schroeder, J., Crawford, J., Blake, D., Weinheimer, A., Woo, J.-H., Kim, S.-W., Yeo, H., Fried, A., Wisthaler, A., and Brune, W.: Evaluation of simulated O3 production efficiency during the KORUS-AQ campaign: implications for anthropogenic NOx emissions in Korea, Elementa: Science of the Anthropocene, 7, 56, https://doi.org/10.1525/elementa.394, 2019. a, b, c
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, 2012. a
Porter, W. C., Safieddine, S. A., and Heald, C. L.: Impact of aromatics and monoterpenes on simulated tropospheric ozone and total OH reactivity, Atmos. Environ., 169, 250–257, https://doi.org/10.1016/j.atmosenv.2017.08.048, 2017. a, b, c, d
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric fate of methyl vinyl ketone: Peroxy radical reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, 2015. a
Ran, L., Zhao, C., Geng, F., Tie, X., Tang, X., Peng, L., Zhou, G., Yu, Q., Xu, J., and Guenther, A.: Ozone photochemical production in urban Shanghai, China: analysis based on ground level observations, J. Geophys. Res., 114, D15301, https://doi.org/10.1029/2008JD010752, 2009. a
Reimann, S. and Lewis, A. C.: Anthropogenic VOCs, in: Volatile Organic Compounds in the Atmosphere, chap. 2, edited by: Koppmann, R., Blackwell Publishing, Oxford, UK, 33–81, https://doi.org/10.1002/9780470988657.ch2, 2007. a
Roth, E., Chakir, A., and Ferhati, A.: Study of a benzoylperoxy radical in the gas phase: ultraviolet spectrum and C6H5C(O)O2+HO2 reaction between 295 and 357 K, J. Phys. Chem. A, 114, 10367–10379, https://doi.org/10.1021/jp1021467, 2010. a
Salta, Z., Kosmas, A. M., Segovia, M. E., Kieninger, M., Tasinato, N., Barone, V., and Ventura, O. N.: Reinvestigation of the Deceptively Simple Reaction of Toluene with OH and the Fate of the Benzyl Radical: The “Hidden” Routes to Cresols and Benzaldehyde, J. Phys. Chem. A, 124, 5917–5930, https://doi.org/10.1021/acs.jpca.0c03727, 2020. a
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015. a
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011. a
Sander, R., Baumgaertner, A., Cabrera-Perez, D., Frank, F., Gromov, S., Grooß, J.-U., Harder, H., Huijnen, V., Jöckel, P., Karydis, V. A., Niemeyer, K. E., Pozzer, A., Riede, H., Schultz, M. G., Taraborrelli, D., and Tauer, S.: The community atmospheric chemistry box model CAABA/MECCA-4.0, Geosci. Model Dev., 12, 1365–1385, https://doi.org/10.5194/gmd-12-1365-2019, 2019. a
Sandu, A., Daescu, D., and Carmichael, G.: Direct and adjoint sensitivity analysis of chemical kinetic systems with KPP: I – theory and software tools, Atmos. Environ., 37, 5083–5096, https://doi.org/10.1016/j.atmosenv.2003.08.019, 2003. a
Sarigiannis, D. and Gotti, A.: Biology-based dose-response models for health risk assessment of chemical mixtures, Fresen. Environ. Bull., 17, 1439–1451, available ata: http://search.proquest.com/docview/19629308/ (last access: 12 December 2021), 2008. a
Sarwar, G., Godowitch, J., Henderson, B. H., Fahey, K., Pouliot, G., Hutzell, W. T., Mathur, R., Kang, D., Goliff, W. S., and Stockwell, W. R.: A comparison of atmospheric composition using the Carbon Bond and Regional Atmospheric Chemistry Mechanisms, Atmos. Chem. Phys., 13, 9695–9712, https://doi.org/10.5194/acp-13-9695-2013, 2013. a
Schroeder, J., Crawford, J., Ahn, J.-Y., Chang, L., Fried, A., Walega, J., Weinheimer, A., Montzka, D., Hall, S., Ullmann, K., Wisthaler, A., Mikoviny, T., Chen, G., Blake, D., Blake, N., Hughes, S., Meinardi, S., Diskin, G., Digangi, J., Choi, Y., Pusede, S., Huey, G., Tanner, D., Kim, M., and Wennberg, P.: Observation-based modeling of ozone chemistry in the Seoul metropolitan area during the Korea-United States Air Quality Study (KORUS-AQ), Elementa: Science of the Anthropocene, 8, 3, https://doi.org/10.1525/elementa.400, 2020. a, b, c
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017. a, b, c, d, e, f
Seuwen, R. and Warneck, P.: Oxidation of toluene in NOx-free air: product distribution and mechanism, Int. J. Chem. Kinet., 28, 315–332, https://doi.org/10.1002/(SICI)1097-4601(1996)28:5<315::AID-KIN1>3.0.CO;2-Y, 1996.
Shareef, M. M., Husain, T., and Alharbi, B.: Studying the Effect of Different Gas-Phase Chemical Kinetic Mechanisms on the Formation of Oxidants, Nitrogen Compounds and Ozone in Arid Regions, J. Environ. Protection, 10, 1006–1031, https://doi.org/10.4236/jep.2019.108060, 2019. a, b
Shen, L., Jacob, D. J., Santillana, M., Wang, X., and Chen, W.: An adaptive method for speeding up the numerical integration of chemical mechanisms in atmospheric chemistry models: application to GEOS-Chem version 12.0.0, Geosci. Model Dev., 13, 2475–2486, https://doi.org/10.5194/gmd-13-2475-2020, 2020. a
Shepson, P. B., Edney, E. O., and Corse, E. W.: Ring fragmentation reactions on the photooxidations of toluene and o-xylene, J. Phys. Chem., 88, 4122–4126, https://doi.org/10.1021/j150662a053, 1984.
Silva, S. J., Heald, C. L., and Li, M.: Space-Based Constraints on Terrestrial Glyoxal Production, J. Geophys. Res.-Atmos., 123, 13583–13594, https://doi.org/10.1029/2018JD029311, 2018. a, b
Smith, D. F., McIver, C. D., and Kleindienst, T. E.: Primary product distribution from the reaction of hydroxyl radicals with toluene at ppb NOx mixing ratios, J. Atmos. Chem., 30, 209–228, 1998.
Stavrakou, T., Müller, J.-F., De Smedt, I., Van Roozendael, M., Kanakidou, M., Vrekoussis, M., Wittrock, F., Richter, A., and Burrows, J. P.: The continental source of glyoxal estimated by the synergistic use of spaceborne measurements and inverse modelling, Atmos. Chem. Phys., 9, 8431–8446, https://doi.org/10.5194/acp-9-8431-2009, 2009. a
Stockwell, W. R., Kirchner, F., Kuhn, M., and Seefeld, S.: A new mechanism for regional atmospheric chemistry modeling, J. Geophys. Res.-Atmos., 102, 25847–25879, https://doi.org/10.1029/97JD00849, 1997. a, b
Stockwell, W. R., Lawson, C. V., Saunders, E., and Goliff, W. S.: A Review of Tropospheric Atmospheric Chemistry and Gas-Phase Chemical Mechanisms for Air Quality Modeling, Atmosphere, 3, 1–32, https://doi.org/10.3390/atmos3010001, 2012. a
Tan, J.-H., Guo, S.-J., Ma, Y.-L., Yang, F.-M., He, K.-B., Yu, Y.-C., Wang, J.-W., Shi, Z.-B., and Chen, G.-C.: Non-methane hydrocarbons and their ozone formation potentials in Foshan, China, Aerosol Air Qual. Res., 12, 387–398, https://doi.org/10.4209/aaqr.2011.08.0127, 2012. a
Tao, Z. and Li, Z.: A kinetics study on reactions of C6H5O with C6H5O and O3 at 298 K, Int. J. Chem. Kinet., 31, 65–72, https://doi.org/10.1002/(SICI)1097-4601(1999)31:1<65::AID-KIN8>3.0.CO;2-J, 1999. a, b
Taraborrelli, D., Cabrera-Perez, D., Bacer, S., Gromov, S., Lelieveld, J., Sander, R., and Pozzer, A.: Influence of aromatics on tropospheric gas-phase composition, Atmos. Chem. Phys., 21, 2615–2636, https://doi.org/10.5194/acp-21-2615-2021, 2021. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p
Tuazon, E. C., Atkinson, R., MacLeod, H., Biermann, H. W., Winer, A. M., Carter, W. P. L., and Pitts Jr., J. N.: Yields of glyoxal and methylglyoxal from the NOx-air photooxidations of toluene and m- and p-xylene, Environ. Sci. Technol., 18, 981–984, https://doi.org/10.1021/es00130a017, 1984.
Tuazon, E. C., MacLeod, H., Atkinson, R., and Carter, W. P. L.: α-Dicarbonyl yields from the NOx-air photooxidations of a series of aromatic hydrocarbons in air, Environ. Sci. Technol., 20, 383–387, https://doi.org/10.1021/es00146a010, 1986.
Utembe, S., Cooke, M., Archibald, A., Jenkin, M., Derwent, R., and Shallcross, D.: Using a reduced Common Representative Intermediates (CRIv2-R5) mechanism to simulate tropospheric ozone in a 3-D Lagrangian chemistry transport model, Atmos. Environ., 44, 1609–1622, https://doi.org/10.1016/j.atmosenv.2010.01.044, 2010. a
van der Werf, G. R., Randerson, J. T., Giglio, L., Collatz, G. J., Mu, M., Kasibhatla, P. S., Morton, D. C., DeFries, R. S., Jin, Y., and van Leeuwen, T. T.: Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009), Atmos. Chem. Phys., 10, 11707–11735, https://doi.org/10.5194/acp-10-11707-2010, 2010. a
Vereecken, L.: Advances in Atmospheric Chemistry, World Scientific, Singapore, 377–527, 2018. a
Volkamer, R., Platt, U., and Wirtz, K.: Primary and secondary glyoxal formation from aromatics: experimental evidence for the bicycloalkyl-radical pathway from benzene, toluene, and p-xylene, J. Phys. Chem. A, 105, 7865–7874, https://doi.org/10.1021/jp010152w, 2001.
Wang, L., Wu, R., and Xu, C.: Atmospheric oxidation mechanism of benzene: fates of alkoxy radical intermediates and revised mechanism, J. Phys. Chem. A, 117, 14163–14168, https://doi.org/10.1021/jp4101762, 2013. a, b
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017. a, b
Wang, S., Newland, M. J., Deng, W., Rickard, A. R., Hamilton, J. F., Muñoz, A., Rádenas, M., Vázquez, M. M., Wang, L., and Wang, X.: Aromatic Photo-oxidation, A New Source of Atmospheric Acidity, Environ. Sci. Technol., 54, 7798–7806, https://doi.org/10.1021/acs.est.0c00526, 2020. a, b, c, d, e, f, g, h
Watson, L., Shallcross, D., Utembe, S., and Jenkin, M.: A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 2: Gas phase mechanism reduction, Atmos. Environ., 42, 7196–7204, https://doi.org/10.1016/j.atmosenv.2008.07.034, 2008. a, b
Wittrock, F., Richter, A., Oetjen, H., Burrows, J. P., Kanakidou, M., Myriokefalitakis, S., Volkamer, R., Beirle, S., Platt, U., and Wagner, T.: Simultaneous global observations of glyoxal and formaldehyde from space, Geophys. Res. Lett., 33, L16804, https://doi.org/10.1029/2006GL026310, 2006. a
Woo, J.-H., Choi, K.-C., Kim, H. K., Baek, B. H., Jang, M., Eum, J.-H., Song, C. H., Ma, Y.-I., Sunwoo, Y., Chang, L.-S., and Yoo, S. H.: Development of an anthropogenic emissions processing system for Asia using SMOKE, Atmos. Environ., 58, 5–13, https://doi.org/10.1016/j.atmosenv.2011.10.042, air Quality Modeling in Asia (AQMA) 2011, 2012. a
Wu, R., Pan, S., Li, Y., and Wang, L.: Atmospheric oxidation mechanism of toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/jp500077f, 2014.
Xu, L., Möller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New Insights into the Radical Chemistry and Product Distribution in the OH-Initiated Oxidation of Benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q
Yan, Y., Cabrera-Perez, D., Lin, J., Pozzer, A., Hu, L., Millet, D. B., Porter, W. C., and Lelieveld, J.: Global tropospheric effects of aromatic chemistry with the SAPRC-11 mechanism implemented in GEOS-Chem version 9-02, Geosci. Model Dev., 12, 111–130, https://doi.org/10.5194/gmd-12-111-2019, 2019. a, b, c, d, e, f, g, h
Yantosca, B.: geoschem/geos-chem: GEOS-Chem 12.3.2 (12.3.2), Zenodo [data set], https://doi.org/10.5281/zenodo.2658178, 2019. a
Yu, J. and Jeffries, H. E.: Atmospheric photooxidation of alkylbenzenes: I I. Evidence of formation of epoxide intermediates, Atmos. Environ., 31, 2281–2287, https://doi.org/10.1016/S1352-2310(97)88637-2, 1997. a
Yu, S., Mathur, R., Sarwar, G., Kang, D., Tong, D., Pouliot, G., and Pleim, J.: Eta-CMAQ air quality forecasts for O3 and related species using three different photochemical mechanisms (CB4, CB05, SAPRC-99): comparisons with measurements during the 2004 ICARTT study, Atmos. Chem. Phys., 10, 3001–3025, https://doi.org/10.5194/acp-10-3001-2010, 2010. a
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019. a, b
Zhang, Y., Chen, Y., Sarwar, G., and Schere, K.: Impact of gas-phase mechanisms on Weather Research Forecasting Model with Chemistry (WRF/Chem) predictions: Mechanism implementation and comparative evaluation, J. Geophys. Res.-Atmos., 117, D01301, https://doi.org/10.1029/2011JD015775, 2012. a
Zheng, B., Tong, D., Li, M., Liu, F., Hong, C., Geng, G., Li, H., Li, X., Peng, L., Qi, J., Yan, L., Zhang, Y., Zhao, H., Zheng, Y., He, K., and Zhang, Q.: Trends in China's anthropogenic emissions since 2010 as the consequence of clean air actions, Atmos. Chem. Phys., 18, 14095–14111, https://doi.org/10.5194/acp-18-14095-2018, 2018. a
- Abstract
- Introduction
- GC13: a new compact aromatic mechanism for atmospheric models
- Previous aromatic mechanisms
- Mechanism evaluation and intercomparison
- Global effects of aromatic oxidation
- Accounting for primary non-BTX aromatics
- Conclusions
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- GC13: a new compact aromatic mechanism for atmospheric models
- Previous aromatic mechanisms
- Mechanism evaluation and intercomparison
- Global effects of aromatic oxidation
- Accounting for primary non-BTX aromatics
- Conclusions
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement