the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Oct 2021
| 27 Oct 2021
Winter ClNO2 formation in the region of fresh anthropogenic emissions: seasonal variability and insights into daytime peaks in northern China
Xiang Peng
Weihao Wang
Chuan Yu
Yee Jun Tham
Jianmin Chen
Yujing Mu
Chenglong Zhang
Pengfei Liu
Likun Xue
Xinfeng Wang
Jian Gao
Hong Li
Nitryl chloride (ClNO2) is an important chlorine reservoir in the atmosphere that affects the oxidation of volatile organic compounds (VOCs) and the production of ROx radicals and ozone (O3). This study presents measurements of ClNO2 and related compounds at urban, polluted rural, and polluted lower tropospheric (mountaintop) sites in the winter of 2017–2018 over the North China Plain (NCP). The nocturnal concentrations of ClNO2 were lower at the urban and polluted rural sites but higher at the polluted lower tropospheric site. The winter concentrations of ClNO2 were generally lower than the summer concentrations that were previously observed at these sites, which was due to the lower nitrate radical (NO3) production rate (P(NO3)) and the smaller N2O5 uptake coefficients (γ(N2O5)) in winter, despite the higher ratios of dinitrogen pentoxide (N2O5) to NO3 in winter. Significant daytime peaks of ClNO2 were observed at all the sites during the winter campaigns, with ClNO2 mixing ratios of up to 1.3 ppbv. Vertical transport of ClNO2 from the residual layers and prolonged photochemical lifetime of ClNO2 in winter may explain the elevated daytime concentrations. The daytime-averaged chlorine radical (Cl) production rates (P(Cl)) from the daytime ClNO2 were 0.17, 0.11, and 0.12 ppbv h−1 at the polluted rural, urban, and polluted lower tropospheric sites, respectively, which were approximately 3–4 times higher than the campaign-averaged conditions. Box model calculations showed that the Cl atoms liberated during the daytime peaks of ClNO2 increased the ROx levels by up to 27 %–37 % and increased the daily O3 productions by up to 13 %–18 %. Our results provide new insights into the ClNO2 processes in the lower troposphere impacted by fresh and intense anthropogenic emissions and reveal that ClNO2 can be an important daytime source of Cl radicals under certain conditions in winter.
- Article
(8163 KB) - Full-text XML
-
Supplement
(6855 KB) - BibTeX
- EndNote




Cl is a potent atmospheric oxidant that reacts analogously to hydroxyl radicals (OH) with hydrocarbons (Simpson et al., 2015). Cl is highly reactive toward alkanes, with the rate constants of its reactions with alkanes being approximately 10–200 times greater than some of the OH + VOC reactions (Atkinson and Arey, 2003; Young et al., 2014; Burkholder et al., 2015). Consequently, Cl enhances the production of ROx (= OH + HO2 + RO2) via Reactions (R1)–(R4), which promotes O3 formation by converting nitric oxide (NO) to nitrogen dioxide (NO2) (Reactions R3 and R5). Cl also consumes O3 via Reaction (R8). The net effect of the Cl chemistry is typically the depletion of O3 in the remote atmosphere, such as the stratosphere (Molina and Rowland, 1974) and remote oceans (Simpson et al., 2015; Wang et al., 2019), and an increase in O3 production in the polluted troposphere (Riedel et al., 2014; Xue et al., 2015).
Here M denotes the third body in ambient air.
The production of Cl is determined by the formation and decomposition of Cl precursors such as ClNO2 (Chang et al., 2011; Simpson et al., 2015). ClNO2 is produced mostly in dark conditions by the heterogeneous uptake of N2O5 on chloride (Cl−)-laden aerosols (Reactions R9–R13) and undergoes photolysis during the day (Reaction R14) (Finlayson-Pitts et al., 1989). ClNO2 formation is constrained by the NO3 production rate (P(NO3), Reaction R9). NO3 is in thermal equilibrium with N2O5 (Reaction R10), and the equilibrium constant (Keq) depends on temperature and NO2 concentrations. N2O5 formation is suppressed by NO and VOCs as they consume NO3 (Reactions R11–12). The N2O5 uptake probability (γ(N2O5)) and ClNO2 production yield (φ(ClNO2)) are kinetic parameters with values between 0 and 1, which can be derived from the observation data of N2O5, ClNO2, and related species (Brown et al., 2006; Phillips et al., 2016). Previous laboratory studies have demonstrated that γ(N2O5) is enhanced by higher relative humidity (RH) and particulate Cl− concentrations but suppressed by higher temperature and concentrations of aerosol nitrate (NO) and organic species (Behnke et al., 1997; Hallquist et al., 2003; Bertram et al., 2009; Griffiths and Anthony Cox, 2009).
Field observations of ClNO2 were first reported in the marine boundary layer off the coast of the Houston–Galveston area in the United States (Osthoff et al., 2008). Subsequent studies demonstrated the worldwide ubiquity of ClNO2 and confirmed its significant role in photochemistry (Thornton et al., 2010; Mielke et al., 2011; Phillips et al., 2012; Edwards et al., 2013; Bannan et al., 2015; Wild et al., 2016; Wang et al., 2016; Bannan et al., 2019; Eger et al., 2019). The role of ClNO2 in the radical budget could be more important than that of OH in winter, because OH production is reduced in winter owing to lower concentrations of O3 and H2O vapor in this season. A limited number of winter observations of ClNO2 have been conducted on various platforms, including on aircraft over northern Europe (Priestley et al., 2018) and the eastern United States (Haskins et al., 2018, 2019); on a tall tower in Boulder, United States (Riedel et al., 2013); on a mountaintop in Hong Kong (Wang et al., 2016); and at ground sites in Alberta, Canada (Mielke et al., 2016), and Heshan, China (Yun et al., 2018). These studies found high ClNO2 mixing ratios of up to 7.7 ppbv (Yun et al., 2018) in winter and a contribution of ClNO2 to Cl liberation of up to 83 % (Priestley et al., 2018) in urban Manchester, and that ClNO2 was a more dominant radical source than OH in both the early morning and the whole day in the polluted marine boundary layer downwind of the northeast United States (Haskins et al., 2019). ClNO2 usually exhibits higher concentrations in aged and polluted air masses than in clean air and in regions subject to significant fresh NO emissions (Wang et al., 2016; Z. Wang et al., 2017; Osthoff et al., 2018).
The chemical production of ClNO2 in winter has some unique features compared with that in warmer seasons. Long winter nights provide more time for ClNO2 production and accumulation. Lower temperatures in winter shift the N2O5–NO3 equilibrium to the N2O5 side (Brown et al., 2003) and increase the γ(N2O5) on aerosols (Bertram and Thornton, 2009). In addition, NOx has longer lifetimes in winter compared with summer due to less abundant OH radicals in winter and its slower reaction rate with OH (Kenagy et al., 2018). However, P(NO3) might be lower in winter due to reduced O3 concentrations. The availability of aerosol Cl− also varies in winter and summer. More Cl− is emitted due to coal burning in winter (McCulloch et al., 1999; Fu et al., 2018). In places like East Asia, the winter monsoon brings air masses from the interior of the continent, thereby suppressing the transport of sea salt to inland areas. Because of the contrasts in the availability of aerosol chloride and the variability in meteorology and NOx emissions that affect the N2O5 chemistry, it is not clear whether ClNO2 formation is more prevalent in winter.
The North China Plain (NCP) – home to Beijing and several other megacities – is one of the most industrialized and populous regions of China and frequently suffers from severe haze pollution in winter (An et al., 2019; Fu et al., 2020). ClNO2 concentrations have been measured over the NCP (Le Breton et al., 2018; Zhou et al., 2018), but only one study was conducted in winter (Le Breton et al., 2018). The present study presents recent field observations of ClNO2 concentrations from three campaigns conducted in winter and early spring at three sites in the NCP. The results were compared with those obtained in the previous summer campaigns at the same locations. We examined the factors controlling ClNO2 formation, i.e., P(NO3), which is the nocturnal reactivity of NO3, and N2O5, γ(N2O5), and φ(ClNO2). We then focused on the unexpected daytime peaks of ClNO2 concentrations that were observed at the sites and evaluated their impact on the daytime atmospheric oxidative capacity using a chemical box model.
2.1 Observation sites
Field campaigns were performed in Wangdu, Beijing, and Mt. Tai in sequence during the winter–early spring of 2017–2018 (Table 1). The locations of the three sites are shown in Supplement Fig. S1. The sites were selected for investigation of ClNO2 in urban, rural, and mountain areas of the NCP. The winter indoor-heating period lasts from early November to 15 March of the following year (Ran et al., 2016), and thus the observations were made mostly during the heating period during which coal is intensively used. Detailed descriptions of the measurement sites are available in previous studies (Tham et al., 2016; Z. Wang et al., 2017; Xia et al., 2019), and a brief introduction is given here.
Table 1Locations and periods of the field campaigns relevant to this study.

1 Observations from this study. 2−4 Observations from previous studies, i.e., Tham et al. (2016), Xia et al. (2019), and Z. Wang et al. (2017), respectively.
Our observations at the Wangdu site were part of the Campaign of Oxidation Potential Research for air Pollution in winter (COPPER). The Wangdu site is located in Dongbaituo Village, Hebei Province. Local villagers use coal stoves for cooking and heating during winter. National road G4 and provincial road S335 are 1 and 3 km to the west of the sampling site, respectively. Many heavy-duty trucks passed through G4 and S335 every night during the study period, emitting a large amount of NOx and particulate matter. Therefore, the site experienced heavy pollution from coal burning and road traffic (Peng et al., 2020).
The Beijing site is located at the Chinese Research Academy of Environmental Science (CRAES), which is 15 km northeast of the city center. The sampling site is surrounded by intra-city roads, commercial buildings, and residential buildings with few industrial facilities. When the prevailing wind originates from the north (i.e., remote mountainous regions), the site is upwind from the Beijing downtown area and thus is less polluted. However, when the wind originates from the south, the site receives pollutants from Beijing's urban areas in the NCP (Xia et al., 2019).
Mt. Tai is located approximately 40 km south of Jinan City (population: 8.9 million) and 15 km north of Tai'an City (population: 5.6 million) (Wen et al., 2018). Measurements were taken at Mt. Tai meteorological station (1534 m a.s.l.). The site is isolated from the anthropogenic emissions of tourist areas and is not affected by local emissions. The observation period, i.e., March to April, was in early spring in the NCP. However, considering the low temperature (4.6 ± 6.3∘) observed on top of Mt. Tai, this study considered the observation period to be winter to early spring.
2.2 Measurements of N2O5 and ClNO2 concentrations
N2O5 and ClNO2 were simultaneously measured by a chemical ionization mass spectrometer with a quadrupole mass analyzer (Q-CIMS; THS Instruments). The principles and calibrations of the Q-CIMS measurements are available in previous studies (Wang et al., 2016; Tham et al., 2016; Z. Wang et al., 2017; Xia et al., 2019). Briefly, the primary ions used in the Q-CIMS were iodide (I−) and its water clusters, which were generated using CH3I with an inline ionizer (210Po). The iodide adducts, namely IN2O and IClNO, were then detected by the mass spectrometer. An example of the mass spectrum is shown in Fig. S2. The integration time of the signals recorded by the Q-CIMS is shown in Table S1. The isotopic ratios of I35ClNO and I37ClNO in the ambient data were used to confirm the identity of ClNO2 (Fig. S3). Gas-phase mixtures of NO2 and O3 produced N2O5 for N2O5 calibration. The synthetic N2O5 was converted to ClNO2 by passage through a humidified NaCl slurry for ClNO2 calibration. The dependence of the N2O5 sensitivities on ambient RH was tested once in each campaign and used to calibrate the N2O5 data (Fig. S4a). ClNO2 sensitivities were found to be not affected by RH (Fig. S4b). Single-concentration calibrations of N2O5 and ClNO2 were performed every 1–2 d, which showed stable sensitivities of N2O5 and ClNO2 (Text S1 and Fig. S5). And the linearity of the N2O5 and ClNO2 signals to concentrations was checked via multi-concentration calibrations in the Mt. Tai campaign (Fig. S6). Backgrounds of N2O5 and ClNO2 were determined by passing the ambient air through glass wool once a day at different time. The background signals of N2O5 (3.3–7.7 pptv) and ClNO2 (1.0–7.5 pptv) were stable and independent of the time of the day (Fig. S7). The detection limits of N2O5 and ClNO2 were 6.9–7.3 and 3.8–5.3 pptv, which is defined here as 3 times the standard deviation of the background signals in 5 min (Table S2 in the Supplement). A virtual-impactor design (Peng et al., 2020) was adopted, and the sampling tube was replaced daily to minimize inlet artifacts.
2.3 Other measurements
The trace gases, particle number size distribution (PNSD), and ionic composition of aerosols and other species were simultaneously measured (Table S3). Online non-methane hydrocarbons were measured by gas chromatography–flame ionization detection–mass spectrometry (GC-FID/MS; Chromatotec Group) at the Beijing site (Zhang et al., 2017) and Wangdu site (Zhang et al., 2020). At Mt. Tai, we used canisters to collect air samples, which were analyzed using GC-FID/MS. In Wangdu and Mt. Tai, oxygenated volatile organic compound (OVOC) samples were collected on DNPH-coated sorbent cartridges followed by post-campaign analysis using high-performance liquid chromatography. The ionic compositions of PM2.5 (e.g., NH, NO, SO, and Cl−) were quantified by the Monitor for AeRosols and GAses in ambient air (MARGA, Metrohm) at the Beijing and Mt. Tai sites (Wen et al., 2018). An aerosol chemical speciation monitor (ACSM, Aerodyne Research Inc.) was utilized at the Wangdu site to monitor the non-refractory components of these ions in PM2.5. The concentrations of the NO, SO, and NH measured simultaneously by the MARGA and ACSM were in good agreement, whereas the concentration of Cl− measured by the ACSM was slightly lower than that measured by the MARGA, which was possibly due to the significant proportion of refractory chloride, e.g., NaCl, present in the aerosols (Xia et al., 2020). We assumed that the particles sampled by a wide-range particle spectrometer (WPS) were spherical in shape and calculated the aerosol surface area density (Sa) and volume density (Va). A parameterization was adopted to consider the hygroscopic growth factor (GF) of aerosol sizes, as follows: (Lewis, 2008), where the parameters a and b were derived as 0.582 and 8.460, respectively, in a previous field study over the NCP (Achtert et al., 2009).
2.4 Calculation of N2O5 loss and ClNO2 production
Some analytical metrics were calculated from the observation data. P(NO3) was calculated using Eq. (1), where k1 represents the rate constant of Reaction (R9) (Atkinson and Lloyd, 1984).
k(NO3) during the night was calculated using the measured mixing ratios of NO and non-methane hydrocarbons that were measured by GC (Sect. 2.3). As most OVOCs react with NO3 at much slower rates compared to those with hydrocarbons, especially alkenes (Atkinson and Arey, 2003), the OVOCs were not included in the calculation of k(NO3). Nonetheless, the k(NO3) might be slightly underestimated here.
where ki is the rate constant for a specific VOC + NO3 reaction and is adopted from Atkinson and Arey (2003), and represents the rate constant for Reaction (R11) (DeMore et al., 1997). The ambient concentrations of NO3 were estimated by assuming that NO3 and N2O5 were in dynamic equilibrium (DeMore et al., 1997).
The loss rates of NO3 due to NO and VOCs were then calculated by [NO][NO3] and ∑ki[VOCi][NO3], respectively.
The loss rate coefficient of N2O5 on the aerosol surface (k(N2O5)) is expressed as follows.
where c(N2O5) represents the average molecular velocity of N2O5. The rate constants (k1, ki, and ) and equilibrium constant (Keq) are calculated as temperature-dependent parameters.
γ(N2O5) and φ(ClNO2) were estimated using steady-state analysis in applicable cases (Brown et al., 2006). This method assumes a steady state of N2O5, which means that the production rate of N2O5 is equal to its loss rate. We adopted the criteria described by Xia et al. (2020) to select the cases, namely low concentrations of NO, an increasing trend of ClNO2 concentrations, and stable air masses. Equation (5) was then established by plotting against , with γ(N2O5) as the slope and k(NO3) as the intercept in the linear regression (Brown et al., 2003). Here, the derived γ(N2O5) was accepted when the regression had R2 > 0.5 and k(NO3) > 0.
φ(ClNO2) was then calculated using the following equation:
where d[ClNO2] dt and [N2O5] represent the increasing rate of ClNO2 production and the average concentration of N2O5, respectively, within the selected cases.
2.5 Box model
An observation-based chemical box model was utilized to simulate the concentrations of Cl and ROx radicals and the production and loss pathways of O3. The detailed model description is available in Peng et al. (2020). Based on Master Chemical Mechanism (MCM) v3.3.1 (Jenkin et al., 2015), Peng et al. (2020) modified the chemical mechanisms to include up-to-date gas-phase chlorine and bromine chemistry. The observed N2O5, ClNO2, NOx, HONO, O3, jNO2, and related species were constrained in the model for every 10 min of model time, after interpolating or averaging the data (Table S4). The mixing ratios of non-methane hydrocarbons (NMHCs) and OVOCs (Sect. 2.3) were constrained every hour. As OVOCs were not measured in Beijing in this study, we adopted the concentrations of OVOCs measured in previous studies in winter Beijing (Gu et al., 2019; Qian et al., 2019). We also assumed the CH4 mixing ratio to be 2000 ppbv, which was the mean value in summer at Wangdu (Tan et al., 2017), for our Wangdu and Beijing sites. We acknowledge that the adoption of the summer CH4 concentrations for our winter studies may underestimate the CH4 level, but the effect on the ROx budget is expected to be insignificant due to much smaller contribution of CH4 to ROx than NMHCs and OVOCs in polluted environments. The photolysis frequencies of ClNO2, O3, and other species were simulated according to the solar zenith angle using the tropospheric ultraviolet and visible (TUV) radiation model and scaled by the observed jNO2 values. Numerical experiments were conducted by constraining (Case 1) and not constraining ClNO2 data (Case 2) at each site. The differences in the radical concentrations and O3 budgets between Cases 1 and 2 represented the effect of ClNO2. For example, the increase in ROx (%) due to ClNO2 was calculated by (ROx_w – ROx_wo) ROx_wo, where ROx_w represents the concentration of ROx in Case 1 with ClNO2 constrained in the model, and ROx_wo represents the concentration of ROx in Case 2 without ClNO2 constrained.
3.1 Overall measurements, diurnal patterns, and comparison with other studies
The time series of N2O5 and ClNO2 levels in the three campaigns are displayed in Fig. 1. Overall, elevated levels of N2O5 and ClNO2 were observed with different patterns at each site. The ground sites (Wangdu and Beijing) were characterized by high NOx levels (83.2 ± 81.3 ppbv and 35.6 ± 27.3 ppbv, respectively) and low O3 levels (8.5 ± 8.8 ppbv and 17.3 ± 11.4 ppbv, respectively), whereas the mountain site, Mt. Tai, was marked by relatively lower NOx levels (2.4 ± 2.0 ppbv) and higher O3 levels (64.6 ± 14.7 ppbv) (Fig. S8). The campaign-averaged mixing ratios of ClNO2 were similar at the ground sites (71 ± 132 pptv and 76 ± 103 pptv in Wangdu and Beijing, respectively) and were significantly lower than that at Mt. Tai (179 ± 247 pptv). The nocturnal ratio of ClNO2 N2O5 at each site displayed large day-to-day variability, which was positively dependent on the ambient RH (Fig. S9) and, to a lesser extent, positively correlated with Sa (figure not shown).
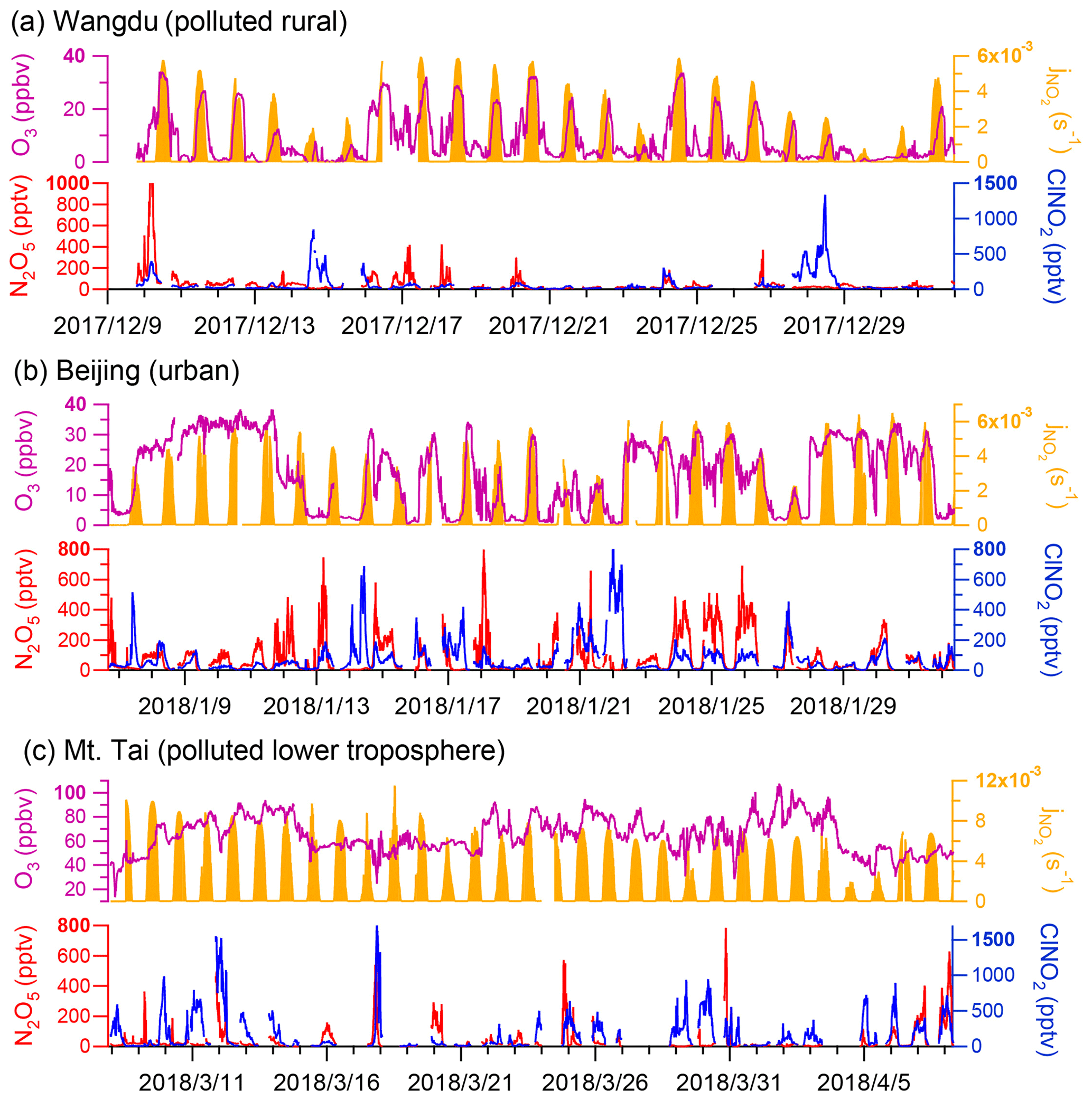
Figure 1Overall observations of N2O5, ClNO2, and related species at the (a) Wangdu, (b) Beijing, and (c) Mt. Tai sites.

Figure 2Diurnal average levels of N2O5, ClNO2, O3, and jNO2 observed at the Wangdu, Beijing, and Mt. Tai sites throughout the campaign in winter (this study) and previous summer field studies (Table 1). The shaded areas indicate the 10th and 90th percentiles.
The campaign-averaged diurnal patterns of the mixing ratios of N2O5, ClNO2, and related species are depicted in Fig. 2. ClNO2 levels typically exhibited a daily cycle, peaking at night and decreasing during the day. The diurnal pattern of ClNO2 at the Wangdu site in winter was an exception, with elevated concentrations (10th–90th percentiles) around midday (12:00–14:00 local time, LT), which resulted from a noontime peak in ClNO2 concentrations during a few days at Wangdu. The detailed observation results from each site are separately introduced as follows.
The nocturnal production of ClNO2 was insignificant in Wangdu despite the presence of abundant Cl− (3.3 ± 3.2 µg m−3 throughout the observation), which likely originated from the intensive residential coal combustion in the area (Peng et al., 2020). The Wangdu site experienced high mass concentrations of PM2.5 (a maximum of approximately 450 µg m−3) and very large mixing ratios of NO (a maximum of approximately 350 ppbv). The wind rose analysis showed that the high concentrations of NO originated from the west of the sampling site where two major roads were located. Numerous heavy-duty trucks on these roads were responsible for high NO concentrations. The presence of abundant NO inhibited N2O5 formation by consuming O3 and NO3 at the Wangdu site. When the ambient concentrations of NO substantially decreased, e.g., on 10 December, the N2O5 mixing ratios increased to 1 ppbv. The mixing ratios of ClNO2 were mostly low (< 200 pptv) during the night. The relationship between nighttime levels of ClNO2 and grouped NO and NOx concentrations is shown in Fig. 3. ClNO2 showed higher levels when the NO mixing ratios were below 10 ppbv and NOx mixing ratios ranged from 10–20 ppbv (Fig. 3a, d). However, significant daytime peaks in ClNO2 mixing ratios were observed on 14 and 28 December, reaching approximately 0.8 and 1.3 ppbv, respectively. The daytime peaks in ClNO2 concentrations at the three sites are discussed in detail in Sect. 3.3. For comparison, the ambient mixing ratios of NO in the summer campaign at Wangdu were much lower (mostly 0–10 ppbv), and O3 mixing ratios were much higher (i.e., exceeded 90 ppbv on most days), which favored the production of N2O5 and ClNO2 (Tham et al., 2016).
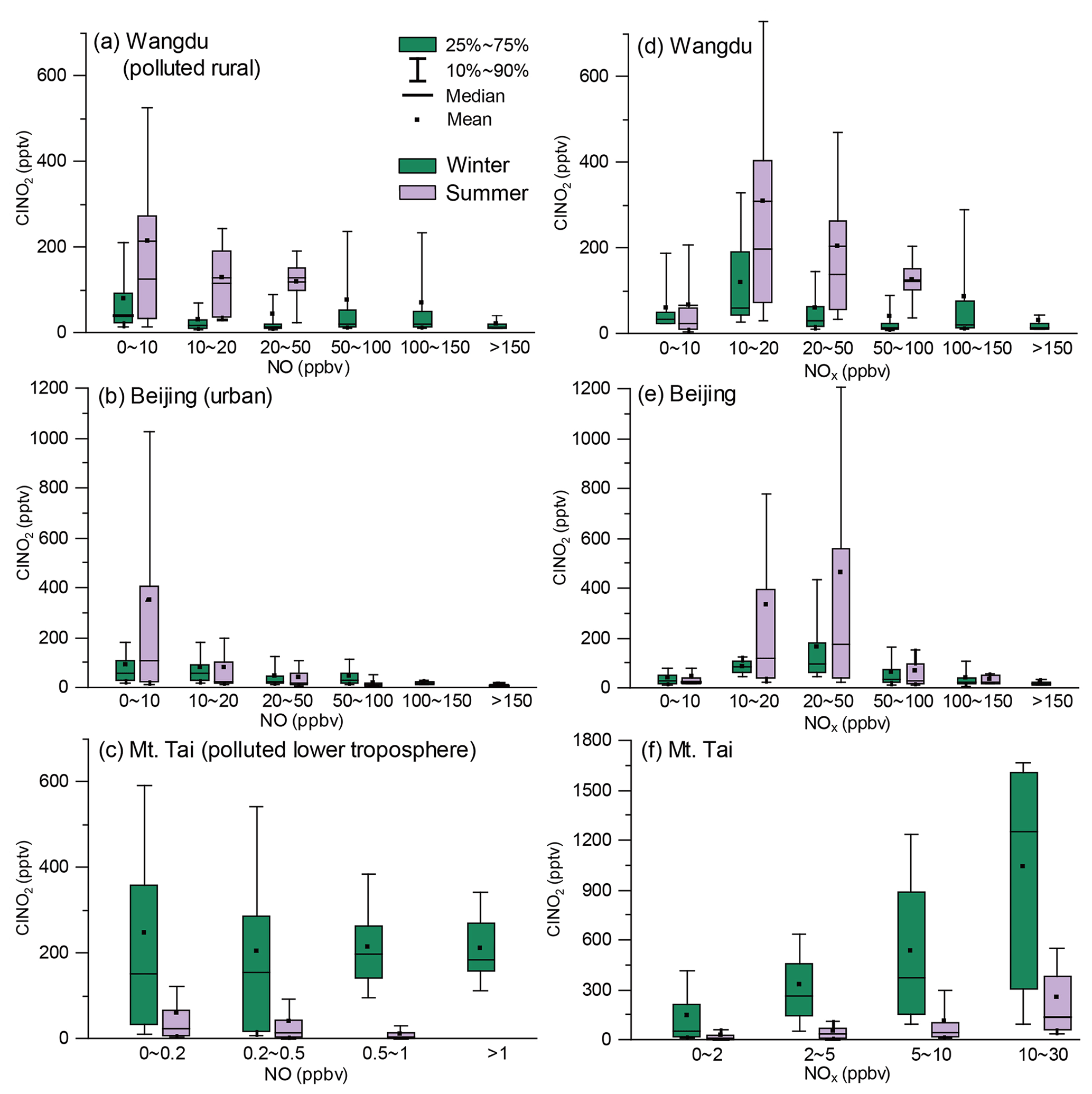
Figure 3The relationship between nighttime levels of ClNO2 and grouped NO (a, b, c) and NOx (d, e, f) mixing ratios in the winter (green color) and summer (purple color) campaigns. The difference in the scale of ClNO2 in (c) and (f) is caused by statistic factors, since only the 10th to 90th percentiles of ClNO2 data are shown here.
The winter Beijing observations showed that there was significant production of N2O5 but limited conversion of N2O5 to ClNO2 in dry conditions. The observation period in Beijing was divided into polluted days (24 h PM2.5 > 75 µg m−3; China's Grade II air quality standard for PM2.5) and clean days (24 h PM2.5 < 35 µg m−3; Grade I standard). The polluted periods were characterized by simultaneous high levels of PM2.5 and NO, e.g., on 19 January. The clean periods were marked by relatively high mixing ratios of O3 and low levels of PM2.5 and NOx, e.g., from 8 to 11 January. Both polluted and clean conditions were unfavorable for ClNO2 formation owing to the high concentrations of NO on the polluted days and the low concentrations of NO2 and aerosols on the clean days. Moreover, the RH observed in Beijing was typically below 40 %, which indicated relatively slow heterogeneous loss of N2O5 and slow ClNO2 formation. Consequently, N2O5 mixing ratios frequently accumulated to elevated levels, exceeding 0.4 ppbv on 10 of the 26 observation nights, and the mixing ratio of ClNO2 was mostly below 0.4 ppbv. Nighttime levels of ClNO2 in winter Beijing were higher when NO mixing ratios ranged from 0–10 ppbv and NOx mixing ratios ranged 20–50 ppbv (Fig. 3b, d). The highest mixing ratios of ClNO2 were observed (up to approximately 0.8 ppbv) when the site occasionally intercepted air masses with a higher RH (approximately 75 %), e.g., on the night of 22 January. This result is similar to the previous observation in Beijing (Xia et al., 2019), in which the ratio of ClNO2 to N2O5 increased significantly from late spring with a low RH (10 %–30 %) to early summer with a higher RH (20 %–80 %). The overall mixing ratios of ClNO2 in the present Beijing study in winter were also significantly lower than those reported in summer (maximum of 1.4 to 2.9 ppbv) in other studies (Le Breton et al., 2018; Zhou et al., 2018).
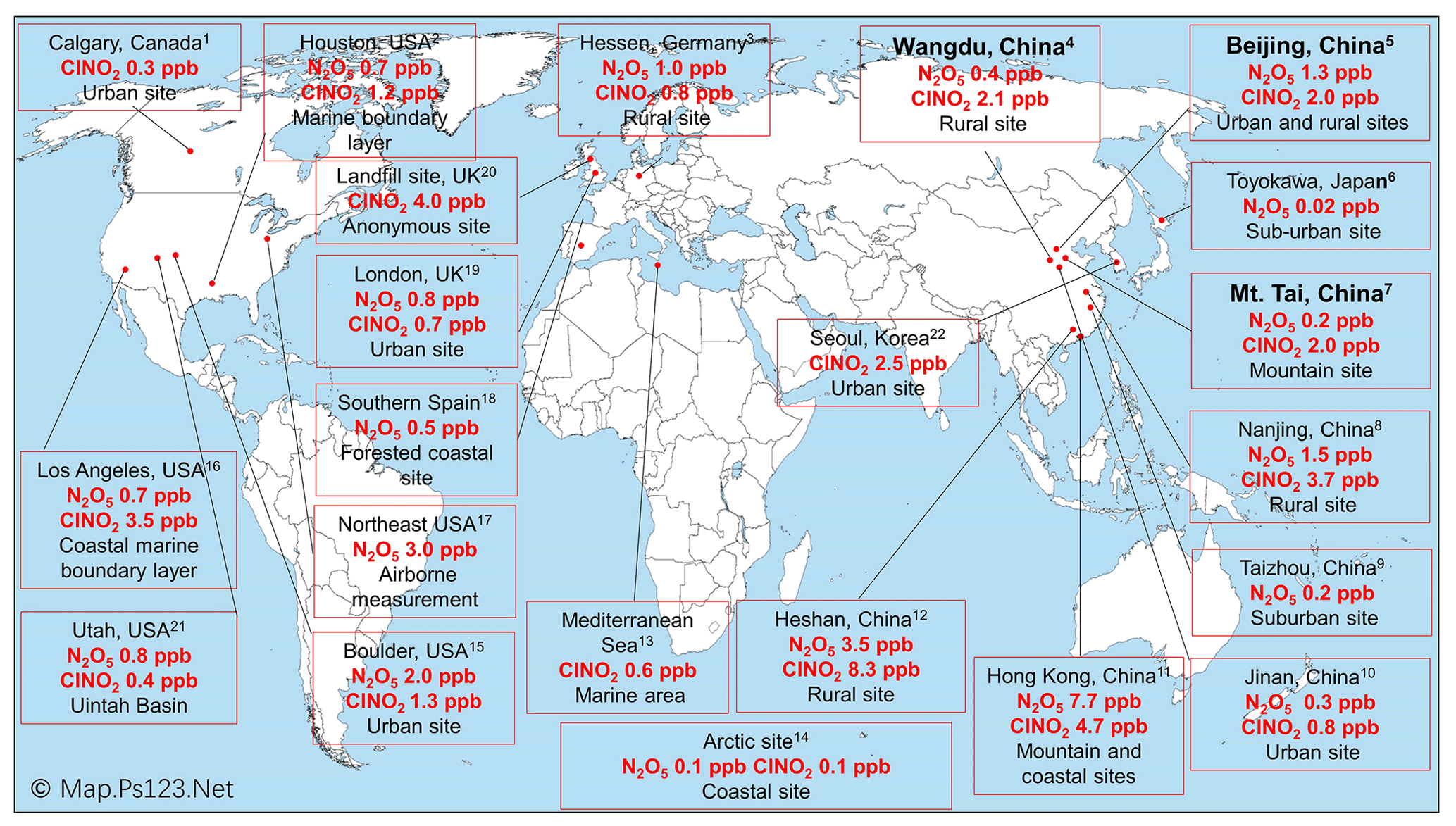
Figure 4Previous observations of ClNO2 and N2O5 levels worldwide. Observation sites in this study are shown in bold. The ClNO2 and N2O5 levels shown are the highest that were measured at these sites. Footnotes associated with the locations refer to the references as follows. (1) Mielke et al. (2011), (2016); Osthoff et al. (2018). (2) Osthoff et al. (2008); Faxon et al. (2015). (3) Phillips et al. (2012). (4) Tham et al. (2016); Liu et al. (2017). (5) H. Wang et al. (2017); Le Breton et al. (2018); Wang et al. (2018); Zhou et al. (2018); Xia et al. (2019). (6) Nakayama et al. (2008). (7) Z. Wang et al. (2017). (8) Xia et al. (2020). (9) H. Wang et al. (2019). (10) X. Wang et al. (2017). (11) Wang et al. (2016); Yun et al. (2017); Yan et al. (2019). (12) Yun et al. (2018). (13) Eger et al. (2019). (14) Apodaca et al. (2008); McNamara et al. (2019). (15) Thornton et al. (2010); Riedel et al. (2013). (16) Riedel et al. (2012); Mielke et al. (2013). (17) Brown et al. (2006); Brown et al. (2007); Haskins et al. (2018). (18) Crowley et al. (2011). (19) Bannan et al. (2015); Sommariva et al. (2018). (20) Bannan et al. (2019). (21) Edwards et al. (2013); Wild et al. (2016); McDuffie et al. (2019). (22) Jeong et al. (2019).
Elevated mixing ratios of ClNO2 (i.e., above 0.5 ppbv) were frequently recorded at the Mt. Tai station in winter. Nighttime levels of ClNO2 were slightly higher when NO levels were below 0.5 ppbv (Fig. 3c) and showed a positive correlation with NOx levels (Fig. 3f). High concentrations of PM2.5 (34.5 ± 27.3 µg m−3) and high RH (63.6 ± 27.1 %) favored the ClNO2 formation at Mt. Tai. The maximum level of ClNO2 (approximately 1.7 ppbv) was observed just before midnight on 18 March, which was slightly lower than the highest concentration observed at Mt. Tai in the summer of 2014 (Z. Wang et al., 2017). The elevated concentrations of ClNO2 observed in the previous summer study at Mt. Tai were due to emissions from distinct coal-fired power plants, whereas this winter study found that coal burning had less effect on concentrations of ClNO2. The campaign-averaged levels of SO2 and particulate SO were 1.6 ± 1.6 ppbv and 3.6 ± 2.9 µg m−3, respectively, during the winter observations, which were significantly lower than those observed in the summer campaigns (2.9 ± 3.7 ppbv and 14.8 ± 9.0 µg m−3, respectively). The decreases in SO2 and sulfate were attributed to strengthened emission control for coal-fired power generation during 2014–2018 and also less transport of emissions from the ground to the Mt. Tai site (1534 m a.s.l.) in late winter and early spring compared with that in summer.
We compared the observed winter concentrations of ClNO2 with those reported in previous studies in Asia, North America, and Europe (Fig. 4). The highest winter concentrations of ClNO2 to date were observed in southern China, with a maximum level of 4.7 ppbv at a mountain top in Hong Kong in aged urban/industrial plumes from the Pearl River Delta (PRD) (Wang et al., 2016) and 8.3 ppbv during a severe pollution episode within the PRD (Yun et al., 2018). The high-concentration ClNO2 events in southern China were due to concurrent high levels of PM2.5 and O3 (e.g., 400 µg m−3 and 160 ppbv found by Yun et al., 2018), which contrast the high concentrations of PM2.5 and low concentrations of O3 over northern China during the cold winter. The winter mixing ratios of ClNO2 in the United States and Europe range from approximately 0.3 ppbv in urban California (Mielke et al., 2016) and urban Manchester (Priestley et al., 2018), respectively, to 1.3 ppbv in the outflow of coastal urban areas (Riedel et al., 2013; Haskins et al., 2019). In general, the winter concentrations of ClNO2 over northern China were comparable to or slightly higher than those observed in the United States and Europe.
3.2 NO3 production and loss pathways
To gain insight into the processes controlling the variability in concentrations of ClNO2, nocturnal P(NO3) and NO3 loss pathways were compared using Eqs. (1–5) in Sect. 2.4. The average P(NO3) was comparable at the three sites in winter, ranging from 0.15 to 0.25 ppbv h−1, and these rates were significantly lower than the respective summer values (Fig. 5a). The lower P(NO3) in winter was caused by both lower k1 and lower [NO2] × [O3] in winter (see Eq. 1). The lower k1 in winter is caused by lower temperature in winter, while the lower [NO2] × [O3] in winter is mainly caused by less photochemical production of O3 and more NO that consumes the available O3 in winter (Table S5). Nighttime NO3 removal through NO3 and N2O5 was estimated by comparing k(NO3) × [NO3] (Eqs. 2–3) and k(N2O5) × [N2O5] (Eqs. 4–5). The average γ(N2O5) values derived from each campaign (Table S6 and Fig. S10) were used in Eq. (4). The nighttime NO3 loss via NO titration and VOC oxidation was greater than the N2O5 heterogeneous loss in all the winter and summer campaigns (Fig. 5b). These were the campaign average results. In contrast, the N2O5 loss was greater than the NO3 loss in selected cases in summer at Mt. Tai (Wang et al., 2017c). To determine the nocturnal loss of NO3, we further compared the N2O5 NO3 ratio and γ(N2O5) at the three sites.
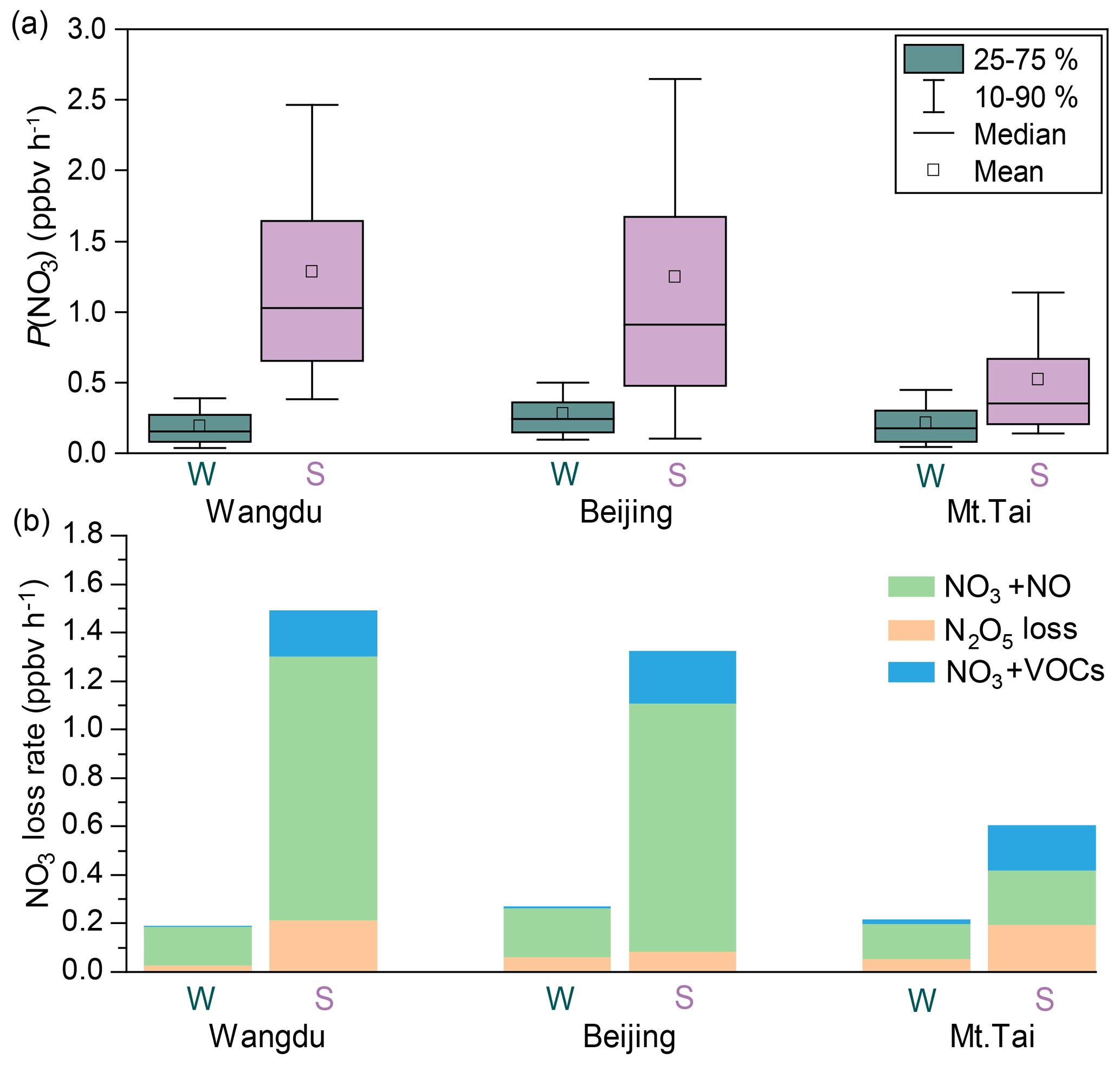
Figure 5Comparison of P(NO3) and loss pathways of NO3 during the winter and summer observations over the NCP. W and S are abbreviations for winter and summer, respectively.
The thermal decomposition of N2O5 was suppressed in winter and resulted in high ratios of N2O5 NO3 (Fig. 6a; up to approximately 1000), which favored N2O5 loss over NO3 loss. However, the γ(N2O5) in winter was systematically lower than that in summer (Fig. 6b), which indicated slower N2O5 loss in winter. A previous field study in winter Beijing also reported small values of γ(N2O5), ranging from < 0.001 to 0.02 (Wang et al., 2020). This result differs from previous laboratory studies, which reported larger γ(N2O5) on (NH4)2SO4 aerosols at lower temperatures (Hallquist et al., 2003; Griffiths and Anthony Cox, 2009). It is possible that other factors, such as RH and aerosol composition (aside from (NH4)2SO4), had a large influence on γ(N2O5). The limited number (two to four) of γ(N2O5) values obtained in each winter campaign (Table S6) may have also caused a bias in the estimation of the overall γ(N2O5). The opposite effects – a higher N2O5 NO3 ratio and lower γ(N2O5) in winter – offset each other in Wangdu (Fig. 6b) but favored N2O5 loss in Beijing and NO3 loss at Mt. Tai compared with those in the respective summer campaigns. The higher concentrations of ClNO2 at Mt. Tai during the winter campaigns may be attributable to higher φ(ClNO2) values in Mt. Tai (Fig. 6c).
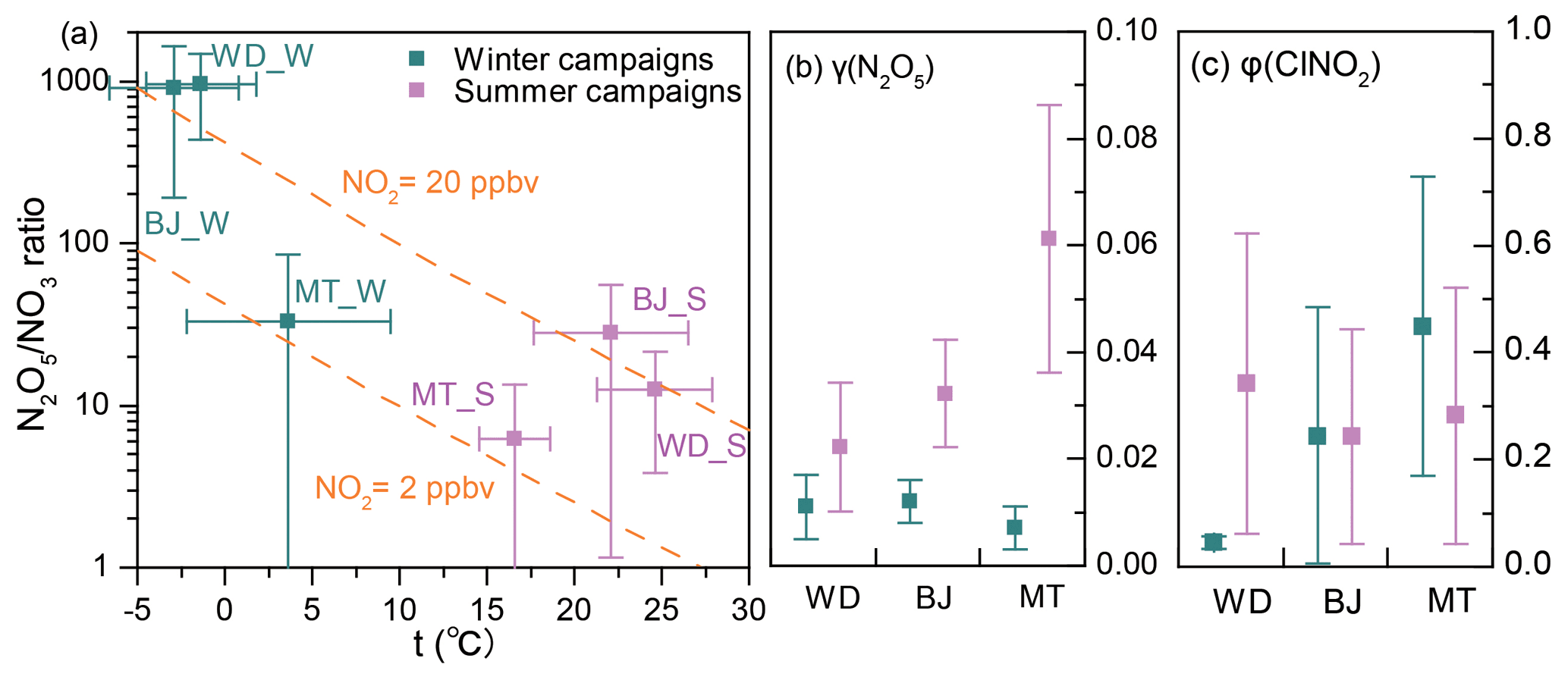
Figure 6Comparison of the (a) N2O5 NO3 ratio, (b) γ(N2O5), and (c) φ(ClNO2) during the winter and summer campaigns. Square dots and error bars indicate the average values and standard deviations, respectively.
3.3 Daytime peaks in ClNO2 concentrations
In the winter campaigns, high concentrations of ClNO2 were sustained after sunrise. Distinct peaks in ClNO2 concentrations were observed on 3–4 d in each campaign, as shown in Fig. 7 displaying one case at each site. Other daytime cases from the three sites are shown in Figs. S11–13. The validity of the daytime peaks was checked by performing isotopic analysis of ClNO2, background detection, and on-site calibration. The signals of I35ClNO and I37ClNO were well correlated (R2 > 0.99) during daytime peaks in ClNO2 concentrations (Fig. S3a–c) and calibrations (Fig. S3d–f). The ratio of I37ClNO to I35ClNO (0.32–0.35) was consistent with the natural isotopic ratio of 37Cl to 35Cl. The background signals of ClNO2 were checked when its daytime peaks in concentrations were observed, and no increase in the background was found. We also checked the signal of primary ions (IH2O−) and found no abnormal changes when ClNO2 concentrations showed daytime peaks. These results confirmed that the daytime peaks in ClNO2 concentrations were real atmospheric phenomena.
The daytime ClNO2 episodes usually occurred from 10:00 to 11:00 LT at each site. The highest daytime mixing ratio of ClNO2 was 1.3 ppbv (5 min average) observed at 11:30 on 28 December 2017 in Wangdu. In comparison, the daytime ClNO2 concentration observed in the previous summer study at Wangdu (Tham et al., 2016) reached a maximum in the early morning (08:00 LT) and declined to several parts per trillion by volume at 11:00. Attenuated solar radiation was observed during the days with daytime peaks in ClNO2 concentrations. For example, the daily maximum rates of jNO2 (1 min average) for the Wangdu case shown in Fig. 7a (2.5 × 10−3 s−1) was significantly lower than the highest rate observed during this campaign (6.0 × 10−3 s−1). The attenuated solar radiation reduced the photolysis of ClNO2, which allowed it to persist for a longer period during the day. The chemical data showed contrasting features during the daytime peaks in ClNO2 concentrations at the three sites. At Wangdu, ClNO2 concentrations showed a sharp peak while the concentrations of other pollutants decreased (Fig. 7a); in Beijing, the daytime peak in ClNO2 concentrations appeared with little simultaneous change in the NO, NOx, and O3 levels after sunrise (Fig. S12a). In two cases, daytime peaks of ClNO2 concentrations at Mt. Tai (Figs. 7c and S13c) occurred together with significant increases in NO, NOx, and PM2.5 levels, whereas O3 concentrations decreased after sunrise and resumed previous levels.
The daytime peaks in ClNO2 concentrations were likely caused by the transport of air masses to the respective sites. In situ production of ClNO2 was limited during the days on which significant daytime ClNO2 occurred, because the mixing ratios of N2O5 were near the detection limit of the instrument (several parts per trillion by volume). The photochemical lifetime of ClNO2 at 10:00 am LT was estimated to be 1–2 h, based on the inverse of jClNO2, which allowed the transport of ClNO2 produced elsewhere to the observation sites. As daytime peaks in ClNO2 concentrations appeared at both the ground and mountain sites, the high-ClNO2 region may exist in the residual layer above the nocturnal mixing layers. At sunrise, ClNO2-rich air masses may be transported downward to the ground sites (Wangdu and Beijing) and upward to the mountaintop site (Mt. Tai). The downward transport of ClNO2 at Wangdu in summer has been illustrated by Tham et al. (2016), and the upward transport to the top of Mt. Tai has also been implicated by the increasing daytime concentrations of O3 and other pollutants (e.g., Gao et al., 2005; Zhou et al., 2009; Jiang et al., 2020). Measurements in the residual layers are needed to further investigate the transport of ClNO2 within the entire boundary layer.
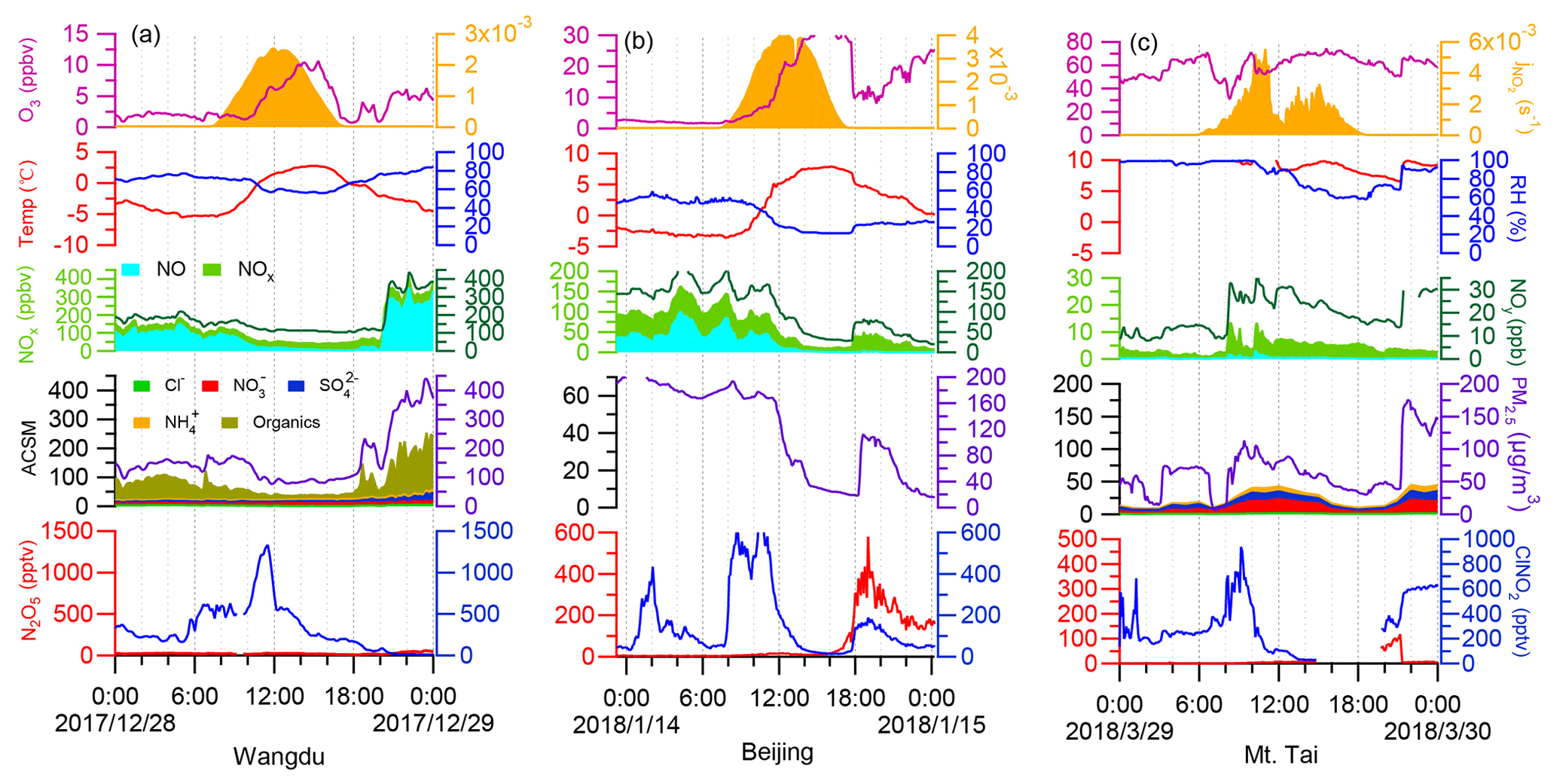
Figure 7Examples of daytime peaks of ClNO2 levels observed at (a) Wangdu, (b) Beijing, and (c) Mt. Tai in the winter campaigns. These examples show the highest levels of daytime ClNO2 at each site. The ionic composition of aerosols was not available on 14 January 2018, owing to an instrument problem.
3.4 Impact of daytime ClNO2 on atmospheric oxidation capacity
We used the box model (Sect. 2.5) to show the impact of ClNO2 on photochemical oxidation at the three sites (Fig. 7a–c). In campaign-averaged conditions, the impact of ClNO2 was minor, owing to the low daytime concentrations of ClNO2. The daytime-averaged P(Cl) (06:00–18:00 LT) from ClNO2 photolysis was in the range of 0.03–0.06 ppbv h−1, with the peak values of 0.07–0.12 ppbv h−1, and the photolysis of ClNO2 enhanced the daytime ROx concentrations by 1.3 %–3.8 % and net O3 production by 1.3 %–6.2 % at the three sites (figures not shown). Such impacts were lower than those during summer at Wangdu (Tham et al., 2016).
However, the impact of ClNO2 increased considerably in the cases of daytime-peak concentrations, as shown in Fig. 8. The daytime-averaged P(Cl) values from ClNO2 photolysis were 0.15 ± 0.13 (maximum of 0.46), 0.11 ± 0.09 (maximum of 0.32), and 0.19 ± 0.20 (maximum of 0.74) ppbv h−1 at Wangdu, Beijing, and Mt. Tai, respectively (Fig. 8a–c). The winter P(Cl) peak in Wangdu (Fig. 8a, 0.46 ppbv h−1) was twice the summer average value (0.24 ppbv h−1) (Tham et al., 2016). The P(Cl) during the daytime peaks of ClNO2 in this study is significantly higher than that in Riedel et al. (2012) (maximum ∼ 0.08 ppbv h−1) but slightly lower than that in Haskins et al. (2019) (maximum ∼ 1.3 ppbv h−1). P(Cl) from other sources (e.g., the HCl + OH reaction) was minor (8.8 %–14.5 %) during these cases. The relative importance of ClNO2 in primary radical production varied among these sites. ClNO2 had a minor contribution in Beijing but became increasingly important in Wangdu and Mt. Tai (Fig. 8b, c). HONO photolysis was the most important source of OH at the two ground sites, whereas O3 was also important at Mt. Tai.
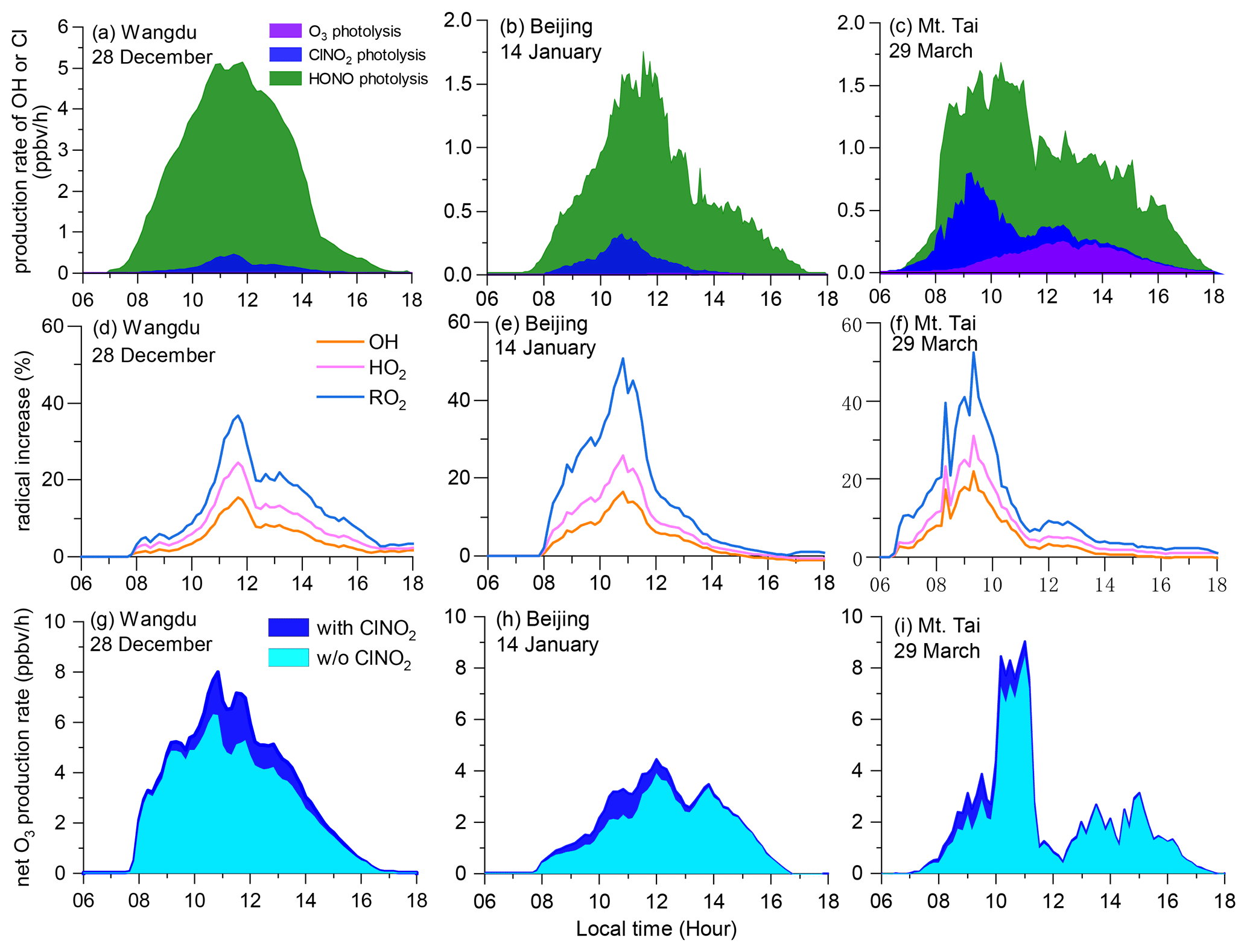
Figure 8The impact of ClNO2 photolysis on atmospheric oxidation during daytime-ClNO2 episodes: (a) primary radical production from the photolysis of O3, ClNO2, and HONO; (b) percentage increase in OH, HO2, and RO2 due to ClNO2 photolysis (Sect. 2.5); and (c) enhancement of net O3 production rates due to ClNO2 photolysis.
The liberated Cl (mostly from ClNO2 photolysis) accounted for 28.5 %–57.7 % of the daytime (06:00–18:00 LT) oxidation of alkanes, 6.1 %–13.7 % of that of alkenes, 5.3 %–14.2 % of that of aromatics, and 4.6 %–6.0 % of that of aldehydes in the cases of high levels of daytime ClNO2. The Cl + VOC reactions enhanced the production of OH, HO2, and RO2 by up to 15 %–22 %, 24 %–31 %, and 36 %–52 %, respectively (Fig. 8d–f). The photolysis of ClNO2 increased the daytime net O3 production by 5.4 ppbv (18 %), 2.8 ppbv (17 %), and 2.6 ppbv (13 %) at Wangdu, Beijing, and Mt. Tai, respectively (Fig. 8g–i). These results indicate the considerable impact of daytime ClNO2 on the atmospheric oxidative capacity and production of secondary pollutants.
The impact of Cl in the NCP is likely larger than the result shown above. Our model calculations considered photolysis of ClNO2 (and HCl + OH) as the source of Cl, but not other photolabile Cl-containing gases. However, in the Wangdu field campaign, we frequently observed elevated daytime concentrations of bromine chloride (BrCl) and molecular chlorine (Cl2), which dominated the Cl production (Peng et al., 2020). In addition, our ClNO2 measurements were conducted at polluted ground-level sites and at a high mountain site (1534 m a.s.l.), which are not in the nocturnal residual layer where strong ClNO2 production is expected to occur (Zhang et al., 2017). It is thus highly desirable to measure ClNO2 in the residual layer in future studies to comprehensively assess the role of ClNO2 in the lower part of the atmosphere.
Observations of ClNO2 and related species were conducted at urban, polluted rural, and polluted lower tropospheric sites in the winter of 2017–2018 in the NCP, which suffers from severe winter haze pollution. The winter measurements showed lower concentrations of ClNO2 compared with those in previous summer observations. The campaign-averaged NO3 loss via reaction with NO at night dominated over the N2O5 loss at all the sites due to high NO concentrations, and in situ ClNO2 formation was generally insignificant. However, high levels of daytime ClNO2 (exceeding 1 ppbv) were observed at the three sites. We suggest that ClNO2 was efficiently produced in the nocturnal residual layer and was transported to ground-level and high-elevation sites. The daytime concentrations of ClNO2 had great effects on the production of Cl, ROx, and O3. Vertical measurements of the concentrations of ClNO2 and related compounds are needed to better understand the distribution and impact of these species in the lower troposphere. Compared to the previous studies in the clean troposphere or in more aged air masses, our results provide new insights into ClNO2 formation in the region affected by fresh and intense anthropogenic emissions.
The datasets described in this study are available by contacting the corresponding author (cetwang@polyu.edu.hk).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-15985-2021-supplement.
TW designed this study. JC, YM, LX, JG, and HL provided field measurement sites. MX, XP, and WW conducted the CIMS measurements. CY, ZW, YJT, HC, CZ, PL, and XW provided supporting data. XP and WW performed the box model simulation. MX analyzed and virtualized the research data. MX and TW wrote the manuscript with discussions and comments from all co-authors.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors are grateful to Yujie Zhang, Fang Bi, Zhenhai Wu, and Xi Cheng for providing supporting data in Beijing. The authors acknowledge helpful discussions and opinions from Peng Wang and Xiao Fu and logistics support from Liwei Guan in Wangdu, and they thank the meteorological observatory at Mt. Tai for providing experiment platforms.
This work was funded by National Natural Science Foundation of China (grant nos. 91544213, 91844301, and 41922051), the Hong Kong Research Grants Council (grant nos. T24-504/17-N and A-PolyU502/16), and the National Key Research and Development Program of China (grant no. 2016YFC0200500).
This paper was edited by Gabriele Stiller and reviewed by Jessica Haskins and one anonymous referee.
Achtert, P., Birmili, W., Nowak, A., Wehner, B., Wiedensohler, A., Takegawa, N., Kondo, Y., Miyazaki, Y., Hu, M., and Zhu, T.: Hygroscopic growth of tropospheric particle number size distributions over the North China Plain, J. Geophys. Res.-Atmos., 114, D00G07, https://doi.org/10.1029/2008JD010921, 2009.
An, Z., Huang, R.-J., Zhang, R., Tie, X., Li, G., Cao, J., Zhou, W., Shi, Z., Han, Y., Gu, Z., and Ji, Y.: Severe haze in northern China: A synergy of anthropogenic emissions and atmospheric processes, P. Natl. Acad. Sci. USA, 116, 8657–8666, 2019.
Apodaca, R. L., Huff, D. M., and Simpson, W. R.: The role of ice in N2O5 heterogeneous hydrolysis at high latitudes, Atmos. Chem. Phys., 8, 7451–7463, https://doi.org/10.5194/acp-8-7451-2008, 2008.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, 2003.
Atkinson, R. and Lloyd, A. C.: Evaluation of kinetic and mechanistic data for modeling of photochemical smog, J. Phys. Chem, Ref. Data, 13, 315-444, 1984.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B. A., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prévôt, A. S. H., Lee, J., Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp, T., Stone, D., Heard, D. E., Fleming, Z. L., Leigh, R., Shallcross, D. E., and Percival, C. J.: The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.-Atmos., 120, 5638–5657, 2015.
Bannan, T. J., Khan, M. A. H., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Lowe, D., Pitt, J., and Allen, G.: A large source of atomic chlorine from ClNO2 photolysis at a UK landfill site, Geophys. Res. Lett., 46, 8508–8516, 2019.
Behnke, W., George, C., Scheer, V., and Zetzsch, C.: Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments, J. Geophys. Res.-Atmos., 102, 3795–3804, 1997.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Bertram, T. H., Thornton, J. A., Riedel, T. P., Middlebrook, A. M., Bahreini, R., Bates, T. S., Quinn, P. K., and Coffman, D. J.: Direct observations of N2O5 reactivity on ambient aerosol particles, Geophys. Res. Lett., 36, L19803, https://doi.org/10.1029/2009GL040248, 2009.
Brown, S., Ryerson, T., Wollny, A., Brock, C., Peltier, R., Sullivan, A., Weber, R., Dube, W., Trainer, M., and Meagher, J.: Variability in nocturnal nitrogen oxide processing and its role in regional air quality, Science, 311, 67–70, 2006.
Brown, S. S., Stark, H., Ryerson, T. B., Williams, E. J., Nicks, D. K., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A.: Nitrogen oxides in the nocturnal boundary layer: Simultaneous in situ measurements of NO3, N2O5, NO2, NO, and O3, J. Geophys. Res.-Atmos., 108, 4299, https://doi.org/10.1029/2002JD002917, 2003.
Brown, S. S., Dubé, W. P., Osthoff, H. D., Stutz, J., Ryerson, T. B., Wollny, A. G., Brock, C. A., Warneke, C., De Gouw, J. A., and Atlas, E.: Vertical profiles in NO3 and N2O5 measured from an aircraft: Results from the NOAA P-3 and surface platforms during the New England Air Quality Study 2004, J. Geophys. Res.-Atmos., 112, D22304, https://doi.org/10.1029/2007JD008883, 2007.
Burkholder, J., Sander, S., Abbatt, J., Barker, J., Huie, R., Kolb, C., Kurylo, M., Orkin, V., Wilmouth, D., and Wine, P.: Chemical kinetics and photochemical data for use in atmospheric studies: evaluation number 18, Pasadena, CA, Jet Propulsion Laboratory, National Aeronautics and Space, 2015.
Chang, W. L., Bhave, P. V., Brown, S. S., Riemer, N., Stutz, J., and Dabdub, D.: Heterogeneous atmospheric chemistry, ambient measurements, and model calculations of N2O5: A review, Aerosol Sci. Tech., 45, 665–695, 2011.
Crowley, J. N., Thieser, J., Tang, M. J., Schuster, G., Bozem, H., Beygi, Z. H., Fischer, H., Diesch, J.-M., Drewnick, F., Borrmann, S., Song, W., Yassaa, N., Williams, J., Pöhler, D., Platt, U., and Lelieveld, J.: Variable lifetimes and loss mechanisms for NO3 and N2O5 during the DOMINO campaign: contrasts between marine, urban and continental air, Atmos. Chem. Phys., 11, 10853–10870, https://doi.org/10.5194/acp-11-10853-2011, 2011.
DeMore, W. B., Sander, S. P., Golden, D. M., Hampson, R. F., Kurylo, M. J., Howard, C. J., Ravishankara, A. R., Kolb, C. E., and Molina, M. J.: Chemical Kinetics and Photochemistry data for use in Stratospheric Modeling. Evaluation number 12, JPL Publication 97-4, JPL, Pasadena, CA, 1997.
Edwards, P. M., Young, C. J., Aikin, K., deGouw, J., Dubé, W. P., Geiger, F., Gilman, J., Helmig, D., Holloway, J. S., Kercher, J., Lerner, B., Martin, R., McLaren, R., Parrish, D. D., Peischl, J., Roberts, J. M., Ryerson, T. B., Thornton, J., Warneke, C., Williams, E. J., and Brown, S. S.: Ozone photochemistry in an oil and natural gas extraction region during winter: simulations of a snow-free season in the Uintah Basin, Utah, Atmos. Chem. Phys., 13, 8955–8971, https://doi.org/10.5194/acp-13-8955-2013, 2013.
Eger, P. G., Friedrich, N., Schuladen, J., Shenolikar, J., Fischer, H., Tadic, I., Harder, H., Martinez, M., Rohloff, R., Tauer, S., Drewnick, F., Fachinger, F., Brooks, J., Darbyshire, E., Sciare, J., Pikridas, M., Lelieveld, J., and Crowley, J. N.: Shipborne measurements of ClNO2 in the Mediterranean Sea and around the Arabian Peninsula during summer, Atmos. Chem. Phys., 19, 12121–12140, https://doi.org/10.5194/acp-19-12121-2019, 2019.
Faxon, C., Bean, J., and Ruiz, L.: Inland Concentrations of Cl2 and ClNO2 in Southeast Texas suggest chlorine chemistry significantly contributes to atmospheric reactivity, Atmosphere, 6, 1487–1506, 2015.
Finlayson-Pitts, B., Ezell, M., and Pitts, J.: Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2, Nature, 337, 241–244, 1989.
Fu, X., Wang, T., Wang, S., Zhang, L., Cai, S., Xing, J., and Hao, J.: Anthropogenic emissions of hydrogen chloride and fine particulate chloride in China, Environ. Sci. Technol., 52, 1644–1654, 2018.
Fu, X., Wang, T., Gao, J., Wang, P., Liu, Y., Wang, S., Zhao, B., and Xue, L.: Persistent Heavy Winter Nitrate Pollution Driven by Increased Photochemical Oxidants in Northern China, Environ. Sci. Technol., 54, 3881–3889, 2020.
Gao, J., Wang, T., Ding, A., and Liu, C.: Observational study of ozone and carbon monoxide at the summit of mount Tai (1534 m asl) in central-eastern China, Atmos. Environ., 39, 4779–4791, 2005.
Griffiths, P. T. and Anthony Cox, R.: Temperature dependence of heterogeneous uptake of N2O5 by ammonium sulfate aerosol, Atmos. Sci. Lett., 10, 159–163, 2009.
Gu, Y., Li, Q., Wei, D., Gao, L., Tan, L., Su, G., Liu, G., Liu, W., Li, C., and Wang, Q.: Emission characteristics of 99 NMVOCs in different seasonal days and the relationship with air quality parameters in Beijing, China, Ecotox. Environ. Safe., 169, 797–806, 2019.
Hallquist, M., Stewart, D. J., Stephenson, S. K., and Cox, R. A.: Hydrolysis of N2O5 on sub-micron sulfate aerosols, Phys. Chem. Chem. Phys., 5, 3453–3463, 2003.
Haskins, J., Lopez-Hilfiker, F., Lee, B., Shah, V., Wolfe, G., DiGangi, J., Fibiger, D., McDuffie, E., Veres, P., and Schroder, J.: Anthropogenic control over wintertime oxidation of atmospheric pollutants, Geophys. Res. Lett., 46, 14826–14835, 2019.
Haskins, J. D., Jaeglé, L., Shah, V., Lee, B. H., Lopez-Hilfiker, F. D., Campuzano-Jost, P., Schroder, J. C., Day, D. A., Guo, H., and Sullivan, A. P.: Wintertime Gas-Particle Partitioning and Speciation of Inorganic Chlorine in the Lower Troposphere Over the Northeast United States and Coastal Ocean, J. Geophys. Res.-Atmos., 123, 12897–812916, 2018.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jeong, D., Seco, R., Gu, D., Lee, Y., Nault, B. A., Knote, C. J., Mcgee, T., Sullivan, J. T., Jimenez, J. L., Campuzano-Jost, P., Blake, D. R., Sanchez, D., Guenther, A. B., Tanner, D., Huey, L. G., Long, R., Anderson, B. E., Hall, S. R., Ullmann, K., Shin, H., Herndon, S. C., Lee, Y., Kim, D., Ahn, J., and Kim, S.: Integration of airborne and ground observations of nitryl chloride in the Seoul metropolitan area and the implications on regional oxidation capacity during KORUS-AQ 2016, Atmos. Chem. Phys., 19, 12779–12795, https://doi.org/10.5194/acp-19-12779-2019, 2019.
Jiang, Y., Xue, L., Gu, R., Jia, M., Zhang, Y., Wen, L., Zheng, P., Chen, T., Li, H., Shan, Y., Zhao, Y., Guo, Z., Bi, Y., Liu, H., Ding, A., Zhang, Q., and Wang, W.: Sources of nitrous acid (HONO) in the upper boundary layer and lower free troposphere of the North China Plain: insights from the Mount Tai Observatory, Atmos. Chem. Phys., 20, 12115–12131, https://doi.org/10.5194/acp-20-12115-2020, 2020.
Kenagy, H. S., Sparks, T. L., Ebben, C. J., Wooldrige, P. J., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., McDuffie, E. E., Fibiger, D. L., and Brown, S. S.: NOx Lifetime and NOy Partitioning During WINTER, J. Geophys. Res.-Atmos., 123, D028736 https://doi.org/10.1029/2018JD028736, 2018.
Le Breton, M., Hallquist, Å. M., Pathak, R. K., Simpson, D., Wang, Y., Johansson, J., Zheng, J., Yang, Y., Shang, D., Wang, H., Liu, Q., Chan, C., Wang, T., Bannan, T. J., Priestley, M., Percival, C. J., Shallcross, D. E., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Chlorine oxidation of VOCs at a semi-rural site in Beijing: significant chlorine liberation from ClNO2 and subsequent gas- and particle-phase Cl–VOC production, Atmos. Chem. Phys., 18, 13013–13030, https://doi.org/10.5194/acp-18-13013-2018, 2018.
Lewis, E. R.: An examination of Köhler theory resulting in an accurate expression for the equilibrium radius ratio of a hygroscopic aerosol particle valid up to and including relative humidity 100 %, J. Geophys. Res.-Atmos., 113, D03205, https://doi.org/10.1029/2007JD008590, 2008.
Liu, X., Qu, H., Huey, L. G., Wang, Y., Sjostedt, S., Zeng, L., Lu, K., Wu, Y., Hu, M., and Shao, M.: High levels of daytime molecular chlorine and nitryl chloride at a rural site on the North China Plain, Environ. Sci. Technol., 51, 9588–9595, 2017.
McCulloch, A., Aucott, M. L., Benkovitz, C. M., Graedel, T. E., Kleiman, G., Midgley, P. M., and Li, Y. F.: Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8391–8403, 1999.
McDuffie, E. E., Womack, C. C., Fibiger, D. L., Dube, W. P., Franchin, A., Middlebrook, A. M., Goldberger, L., Lee, B. H., Thornton, J. A., Moravek, A., Murphy, J. G., Baasandorj, M., and Brown, S. S.: On the contribution of nocturnal heterogeneous reactive nitrogen chemistry to particulate matter formation during wintertime pollution events in Northern Utah, Atmos. Chem. Phys., 19, 9287–9308, https://doi.org/10.5194/acp-19-9287-2019, 2019.
McNamara, S. M., Raso, A. R., Wang, S., Thanekar, S., Boone, E. J., Kolesar, K. R., Peterson, P. K., Simpson, W. R., Fuentes, J. D., and Shepson, P. B.: Springtime Nitrogen Oxide-Influenced Chlorine Chemistry in the Coastal Arctic, Environ. Sci. Technol., 53, 8057–8067, 2019.
Mielke, L., Stutz, J., Tsai, C., Hurlock, S., Roberts, J., Veres, P., Froyd, K., Hayes, P., Cubison, M., and Jimenez, J.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res.-Atmos., 118, 10638–10652, 2013.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of ClNO2 in a mid-continental urban environment, Environ. Sci. Technol., 45, 8889–8896, 2011.
Mielke, L. H., Furgeson, A., Odame-Ankrah, C. A., and Osthoff, H. D.: Ubiquity of ClNO2 in the urban boundary layer of Calgary, Alberta, Canada, Can. J. Chem., 94, 414–423, 2016.
Molina, M. J. and Rowland, F. S.: Stratospheric sink for chlorofluoromethanes: chlorine atom-catalysed destruction of ozone, Nature, 249, 810–812, 1974.
Nakayama, T., Ide, T., Taketani, F., Kawai, M., Takahashi, K., and Matsumi, Y.: Nighttime measurements of ambient N2O5, NO2, NO and O3 in a sub-urban area, Toyokawa, Japan, Atmospheric Environment, 42, 1995-2006, 2008.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, 2008.
Osthoff, H. D., Odame-Ankrah, C. A., Taha, Y. M., Tokarek, T. W., Schiller, C. L., Haga, D., Jones, K., and Vingarzan, R.: Low levels of nitryl chloride at ground level: nocturnal nitrogen oxides in the Lower Fraser Valley of British Columbia, Atmos. Chem. Phys., 18, 6293–6315, https://doi.org/10.5194/acp-18-6293-2018, 2018.
Peng, X., W. Wang, M. Xia, H. Chen, A. R. Ravishankara, Q. Li, A. Saiz-Lopez, P. Liu, F. Zhang, C. Zhang, L. Xue, X. Wang, C. George, J. Wang, Y. Mu, J. Chen, and T. Wang, An unexpected large continental source of reactive bromine and chlorine with significant impact on wintertime air quality, Natl. Sci. Rev., 8, nwaa304, https://doi.org/10.1093/nsr/nwaa304, 2020.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012GL051912, 2012.
Phillips, G. J., Thieser, J., Tang, M., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Priestley, M., le Breton, M., Bannan, T. J., Worrall, S. D., Bacak, A., Smedley, A. R. D., Reyes-Villegas, E., Mehra, A., Allan, J., Webb, A. R., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of organic and inorganic chlorinated compounds and their contribution to chlorine radical concentrations in an urban environment in northern Europe during the wintertime, Atmos. Chem. Phys., 18, 13481–13493, https://doi.org/10.5194/acp-18-13481-2018, 2018.
Qian, X., Shen, H., and Chen, Z.: Characterizing summer and winter carbonyl compounds in Beijing atmosphere, Atmos. Environ., 214, 116845, https://doi.org/10.1016/j.atmosenv.2019.116845, 2019.
Ran, L., Deng, Z., Wang, P., and Xia, X.: Black carbon and wavelength-dependent aerosol absorption in the North China Plain based on two-year aethalometer measurements, Atmos. Environ., 142, 132–144, 2016.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl chloride and molecular chlorine in the coastal marine boundary layer, Environ. Sci. Technol., 46, 10463–10470, 2012.
Riedel, T. P., Wagner, N. L., Dubé, W. P., Middlebrook, A. M., Young, C. J., Öztürk, F., Bahreini, R., VandenBoer, T. C., Wolfe, D. E., and Williams, E. J.: Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl2 measurements from a tall tower in a polluted continental setting, J. Geophys. Res.-Atmos., 118, 8702–8715, 2013.
Riedel, T. P., Wolfe, G. M., Danas, K. T., Gilman, J. B., Kuster, W. C., Bon, D. M., Vlasenko, A., Li, S.-M., Williams, E. J., Lerner, B. M., Veres, P. R., Roberts, J. M., Holloway, J. S., Lefer, B., Brown, S. S., and Thornton, J. A.: An MCM modeling study of nitryl chloride (ClNO2) impacts on oxidation, ozone production and nitrogen oxide partitioning in polluted continental outflow, Atmos. Chem. Phys., 14, 3789–3800, https://doi.org/10.5194/acp-14-3789-2014, 2014.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric halogen chemistry: Sources, cycling, and impacts, Chem. Rev., 115, 4035–4062, 2015.
Sommariva, R., Hollis, L. D., Sherwen, T., Baker, A. R., Ball, S. M., Bandy, B. J., Bell, T. G., Chowdhury, M. N., Cordell, R. L., and Evans, M. J.: Seasonal and geographical variability of nitryl chloride and its precursors in Northern Europe, Atmos. Sci. Lett., 19, e844, https://doi.org/10.1002/asl.844, 2018.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, 2010.
Wang, H., Lu, K., Chen, X., Zhu, Q., Chen, Q., Guo, S., Jiang, M., Li, X., Shang, D., and Tan, Z.: High N2O5 Concentrations Observed in Urban Beijing: Implications of a Large Nitrate Formation Pathway, Environ. Sci. Technol. Lett., 4, 416–420, 2017.
Wang, H., Lu, K., Guo, S., Wu, Z., Shang, D., Tan, Z., Wang, Y., Le Breton, M., Lou, S., Tang, M., Wu, Y., Zhu, W., Zheng, J., Zeng, L., Hallquist, M., Hu, M., and Zhang, Y.: Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing, Atmos. Chem. Phys., 18, 9705–9721, https://doi.org/10.5194/acp-18-9705-2018, 2018.
Wang, H., Chen, X., Lu, K., Hu, R., Li, Z., Wang, H., Ma, X., Yang, X., Chen, S., and Dong, H.: NO3 and N2O5 chemistry at a suburban site during the EXPLORE-YRD campaign in 2018, Atmos. Environ., 224, 117180, https://doi.org/10.1016/j.atmosenv.2019.117180, 2019.
Wang, H., Chen, X., Lu, K., Tan, Z., Ma, X., Wu, Z., Li, X., Liu, Y., Shang, D., and Wu, Y.: Wintertime N2O5 uptake coefficients over the North China Plain, Sci. Bull., 65, 765–774, 2020.
Wang, T., Tham, Y. J., Xue, L., Li, Q., Zha, Q., Wang, Z., Poon, S. C., Dubé, W. P., Blake, D. R., and Louie, P. K.: Observations of nitryl chloride and modeling its source and effect on ozone in the planetary boundary layer of southern China, J. Geophys. Res.-Atmos., 121, 2476–2489, 2016.
Wang, X., Wang, H., Xue, L., Wang, T., Wang, L., Gu, R., Wang, W., Tham, Y. J., Wang, Z., Yang, L., Chen, J., and Wang, W.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Environ., 156, 125–134, 2017.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Wen, L., Xue, L., Wang, X., Xu, C., Chen, T., Yang, L., Wang, T., Zhang, Q., and Wang, W.: Summertime fine particulate nitrate pollution in the North China Plain: increasing trends, formation mechanisms and implications for control policy, Atmos. Chem. Phys., 18, 11261–11275, https://doi.org/10.5194/acp-18-11261-2018, 2018.
Wild, R. J., Edwards, P. M., Bates, T. S., Cohen, R. C., de Gouw, J. A., Dubé, W. P., Gilman, J. B., Holloway, J., Kercher, J., Koss, A. R., Lee, L., Lerner, B. M., McLaren, R., Quinn, P. K., Roberts, J. M., Stutz, J., Thornton, J. A., Veres, P. R., Warneke, C., Williams, E., Young, C. J., Yuan, B., Zarzana, K. J., and Brown, S. S.: Reactive nitrogen partitioning and its relationship to winter ozone events in Utah, Atmos. Chem. Phys., 16, 573–583, https://doi.org/10.5194/acp-16-573-2016, 2016.
Xia, M., Wang, W., Wang, Z., Gao, J., Li, H., Liang, Y., Yu, C., Zhang, Y., Wang, P., Zhang, Y., Bi, F., Cheng, X., and Wang, T.: Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing, Atmosphere, 10, 204, https://doi.org/10.3390/atmos10040204, 2019.
Xia, M., Peng, X., Wang, W., Yu, C., Sun, P., Li, Y., Liu, Y., Xu, Z., Wang, Z., Xu, Z., Nie, W., Ding, A., and Wang, T.: Significant production of ClNO2 and possible source of Cl2 from N2O5 uptake at a suburban site in eastern China, Atmos. Chem. Phys., 20, 6147–6158, https://doi.org/10.5194/acp-20-6147-2020, 2020.
Xue, L. K., Saunders, S. M., Wang, T., Gao, R., Wang, X. F., Zhang, Q. Z., and Wang, W. X.: Development of a chlorine chemistry module for the Master Chemical Mechanism, Geosci. Model Dev., 8, 3151–3162, https://doi.org/10.5194/gmd-8-3151-2015, 2015.
Yan, C., Tham, Y. J., Zha, Q., Wang, X., Xue, L., Dai, J., Wang, Z., and Wang, T.: Fast heterogeneous loss of N2O5 leads to significant nighttime NOx removal and nitrate aerosol formation at a coastal background environment of southern China, Sci. Total Environ., 677, 637–647, 2019.
Young, C. J., Washenfelder, R. A., Edwards, P. M., Parrish, D. D., Gilman, J. B., Kuster, W. C., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., Roberts, J. M., Griffith, S., Dusanter, S., Stevens, P. S., Flynn, J., Grossberg, N., Lefer, B., Holloway, J. S., Peischl, J., Ryerson, T. B., Atlas, E. L., Blake, D. R., and Brown, S. S.: Chlorine as a primary radical: evaluation of methods to understand its role in initiation of oxidative cycles, Atmos. Chem. Phys., 14, 3427–3440, https://doi.org/10.5194/acp-14-3427-2014, 2014.
Yun, H., Wang, T., Wang, W., Tham, Y. J., Li, Q., Wang, Z., and Poon, S.: Nighttime NOx loss and ClNO2 formation in the residual layer of a polluted region: Insights from field measurements and an iterative box model, Sci. Total Environ., 622, 727–734, 2017.
Yun, H., Wang, W., Wang, T., Xia, M., Yu, C., Wang, Z., Poon, S. C. N., Yue, D., and Zhou, Y.: Nitrate formation from heterogeneous uptake of dinitrogen pentoxide during a severe winter haze in southern China, Atmos. Chem. Phys., 18, 17515–17527, https://doi.org/10.5194/acp-18-17515-2018, 2018.
Zhang, F., Shang, X., Chen, H., Xie, G., Fu, Y., Wu, D., Sun, W., Liu, P., Zhang, C., Mu, Y., Zeng, L., Wan, M., Wang, Y., Xiao, H., Wang, G., and Chen, J.: Significant impact of coal combustion on VOCs emissions in winter in a North China rural site, Sci. Total Environ., 720, 137617, https://doi.org/10.1016/j.scitotenv.2020.137617, 2020.
Zhang, H., Li, H., Zhang, Q., Zhang, Y., Zhang, W., Wang, X., Bi, F., Chai, F., Gao, J., and Meng, L.: Atmospheric Volatile Organic Compounds in a Typical Urban Area of Beijing: Pollution Characterization, Health Risk Assessment and Source Apportionment, Atmosphere, 8, https://doi.org/10.3390/atmos8030061, 2017.
Zhou, W., Zhao, J., Ouyang, B., Mehra, A., Xu, W., Wang, Y., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Chen, Q., Xie, C., Wang, Q., Wang, J., Du, W., Zhang, Y., Ge, X., Ye, P., Lee, J. D., Fu, P., Wang, Z., Worsnop, D., Jones, R., Percival, C. J., Coe, H., and Sun, Y.: Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmos. Chem. Phys., 18, 11581–11597, https://doi.org/10.5194/acp-18-11581-2018, 2018.
Zhou, Y., Wang, T., Gao, X., Xue, L., Wang, X., Wang, Z., Gao, J., Zhang, Q., and Wang, W.: Continuous observations of water-soluble ions in PM2.5 at Mount Tai (1534 m a.s.l.) in central-eastern China, J. Atmos. Chem., 64, 107–127, 2009.