the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Oct 2021
| 07 Oct 2021
Photolytically induced changes in composition and volatility of biogenic secondary organic aerosol from nitrate radical oxidation during night-to-day transition
Emelie L. Graham
Sophie Haslett
Ilona Riipinen
Urs Baltensperger
Amelie Bertrand
Stamatios Giannoukos
Janne Schoonbaert
Imad El Haddad
Andre S. H. Prevot
Wei Huang
Night-time reactions of biogenic volatile organic compounds (BVOCs) and nitrate radicals (NO3) can lead to the formation of NO3-initiated biogenic secondary organic aerosol (BSOA). Here, we study the impacts of light exposure on the chemical composition and volatility of BSOA formed in the dark from three precursors (isoprene, α-pinene, and β-caryophyllene) in atmospheric simulation chamber experiments. Our study represents BSOA formation conditions where reactions between peroxy radicals (RO2 + RO2) and between RO2 and NO3 are favoured. The emphasis here is on the identification of particle-phase organonitrates (ONs) formed in the dark and their changes during photolytic ageing on timescales of ∼ 1 h. The chemical composition of particle-phase compounds was measured with a chemical ionization mass spectrometer with a filter inlet for gases and aerosols (FIGAERO-CIMS) and an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF). Volatility information on BSOA was derived from FIGAERO-CIMS desorption profiles (thermograms) and a volatility tandem differential mobility analyser (VTDMA). During photolytic ageing, there was a relatively small change in mass due to evaporation (< 5 % for the isoprene and α-pinene BSOA, and 12 % for the β-caryophyllene BSOA), but we observed significant changes in the chemical composition of the BSOA. Overall, 48 %, 44 %, and 60 % of the respective total signal for the isoprene, α-pinene, and β-caryophyllene BSOA was sensitive to photolytic ageing and exhibited decay. The photolabile compounds include both monomers and oligomers. Oligomers can decompose into their monomer units through photolysis of the bonds (e.g. likely O–O) between them. Fragmentation of both oligomers and monomers also happened at other positions, causing the formation of compounds with shorter carbon skeletons. The cleavage of the nitrate functional group from the carbon chain was likely not a main degradation pathway in our experiments. In addition, photolytic degradation of compounds changes their volatility and can lead to evaporation. We use different methods to assess bulk volatilities and discuss their changes during both dark ageing and photolysis in the context of the chemical changes that we observed. We also reveal large uncertainties in saturation vapour pressure estimated from parameterizations for the ON oligomers with multiple nitrate groups. Overall, our results suggest that photolysis causes photodegradation of a substantial fraction of BSOA, changes both the chemical composition and the bulk volatility of the particles, and might be a potentially important loss pathway of BSOA during the night-to-day transition.
- Article
(4715 KB) - Full-text XML
- Companion paper
-
Supplement
(1418 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA), formed via the oxidation of volatile organic compounds (VOCs) emitted from human activities (anthropogenic) and vegetation (biogenic), has important impacts on climate (Shrivastava et al., 2017) and human health (Daellenbach et al., 2020). Biogenic VOCs, such as isoprene (C5H8), monoterpenes (C10H16), and sesquiterpenes (C15H24), are key precursors for global SOA formation due to their larger emissions (Guenther et al., 2006; Guenther et al., 1995) and higher reactivity towards atmospheric oxidants compared with the VOCs from anthropogenic emissions. While oxidation initiated by ozone (O3) and hydroxyl radicals (OH) dominates during daytime, nitrate radicals (NO3), generated at night by the reaction of nitrogen dioxide (NO2) with O3, are the major nocturnal oxidant. Modelling studies estimate that 5 %–21 % of SOA is produced by NO3 chemistry at the global scale (Hoyle et al., 2007; Pye et al., 2010).
The reaction of NO3 with VOCs is a major pathway for the production of organonitrates (ONs, e.g. RONO2), which represent a substantial fraction of submicron aerosol nitrate at both urban and rural sites (Kiendler-Scharr et al., 2016). ONs also play an important role in the removal and transport of nitrogen oxides (NOx), and impact NOx cycling and O3 formation (Perring et al., 2013). While the lifetime of aerosols in the atmosphere typically spans over multiple day–night cycles, the lifetime of ONs within particles is much shorter (Lee et al., 2016; Zare et al., 2018). Reactions influencing particulate lifetime of ONs include oxidation, hydrolysis (Pye et al., 2015; Takeuchi and Ng, 2019), and photolysis (Müller et al., 2014; Suarez-Bertoa et al., 2012). They change both chemical and physical properties of SOA and ONs, and have different impacts on the atmospheric NOx budget.
Compared with OH and O3, there are few laboratory studies on NO3-initiated biogenic SOA (BSOA); thus, very little is known about the chemical composition and volatility of BSOA. While there are extensive studies on photolytic/photochemical (photolysis + OH) ageing of OH- and O3-initiated SOA under low-NOx conditions (Surratt et al., 2006; Walser et al., 2007; Mang et al., 2008; Pan et al., 2009; Henry and Donahue, 2012; Presto et al., 2005; Zawadowicz et al., 2020), studies on photolytic/photochemical ageing of NO3-initiated SOA and ONs are limited. The few studies that have examined optical properties of SOA show that there is a significant difference in SOA produced from NO3 oxidation compared with other oxidation pathways (Nakayama et al., 2015; Peng et al., 2018), as well as in BSOA from different precursors (He et al., 2021). Nah et al. (2016) reported that SOA from β-pinene + NO3 is generally resilient to photochemical ageing and does not exhibit significant changes in its chemical composition, whereas most of the SOA from α-pinene + NO3 evaporates during photochemical ageing (photolysis + OH). However, little is known about SOA from other VOC precursors and SOA formed in chemical regimes with different fates of the peroxy radical (RO2), such as RO2 + HO2, RO2 + NO3, and RO2 + RO2. The knowledge gaps related to the effects of photolysis on lifetime and physicochemical properties of ONs currently hinder a quantitative understanding of their impacts on the atmospheric nitrogen budget, chemical interactions between anthropogenic and biogenic compounds, and climate.
In this study, we performed chamber experiments where we formed SOA in the dark and photolytically aged the NO3-initiated SOA and ONs from three biogenic VOCs, namely isoprene, α-pinene, and β-caryophyllene. This article is a companion paper to “Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark”, published in the same journal (Bell et al., 2021). Bell et al. (2021) present the evolution of the composition of NO3-initiated SOA from α-pinene during dark ageing. Here, we focus on the impacts of photolytic ageing on both the chemical composition and volatility of BSOA. We compare the chemical composition of particle-phase compounds before and after 1 h of ultraviolet (UV) irradiation using advanced mass spectrometric techniques, determine photolabile fractions for each BSOA system, and investigate the potential chromophores that photolyze. In addition, we also examine the changes in the bulk volatility with both parameterization and experimental methods.
2.1 Chamber and experimental description
Experiments were conducted in the Paul Scherrer Institute Simulation Chamber for Atmospheric Chemistry (PSI-SCAC, Platt et al., 2013), which is an 8 m3 flexible Teflon chamber suspended in a temperature-controlled shipping container. The chamber is surrounded by a bank of black lights (40×100 W Cleo Performance solarium lamps, Philips; Krapf et al., 2016). The experiments were performed under humid conditions (RH ≈ 60 %) and at a temperature of 21 ± 3 ∘C (with an increase of 3–4 ∘C during photolysis). A list of the experiments is given in Table 1.
Table 1List of experiments and summary of experimental conditions.
SOA was formed in the chamber by the reaction of the precursors with NO3 radicals. Each experiment followed a similar protocol. First, the chamber was cooled from 30 to ∼20 ∘C, and the humidity was then increased by boiling Milli-Q water (18 Mohm) until the desired relative humidity (RH) was reached. After the chamber conditions were stabilized, the desired VOC was injected into the chamber volumetrically, and its concentration was monitored with a proton transfer reaction mass spectrometer (quadrupole-PTR-MS, IONICON). β-Caryophyllene could not be monitored with the PTR-MS because its mass-to-charge () ratio of 204 Th is outside of the mass transmission range for quantitative measurements, although trace amounts of the molecule were detected in the experiment. After the VOC concentration had been stable for 5–15 min, N2O5 was injected into the chamber, which acted as the source of NO3 radicals via decomposition. N2O5 was synthesized by reacting O3 (∼1 %–2 %, generated with an Innovatec ozone generator using PSI-provided O2) with a pure source of NO2 (99 %). N2O5 crystals were collected by passing the gaseous components into a cooled flask (−70 ∘C). Each day, the sample was brought to the chamber in a dry-ice ethanol bath and warmed for 1–2 min prior to injection (5 L min−1 for ∼10 s). A 0-D box model (F0AM) (Wolfe et al., 2016) utilizing the Master Chemical Mechanism (MCM) (Jenkin et al., 1997) was used to model the VOC precursor decay to determine a range of initial N2O5 concentrations. This was performed when the VOC was measurable, which was not the case for the β-caryophyllene experiment (Exp. 4), where the estimate of >200 ppb (Table 1) comes from the average N2O5 concentrations in the other experiments performed. After the injection was complete, the chamber was stirred by injecting zero air into the chamber (100 L min−1 for ∼10 min). Following chamber stirring, the volume of the chamber started to decrease due to all of the instruments sampling air from the chamber. Experiments started under dark conditions to probe NO3-initiated SOA formation and to follow its chemical transformation with no external stimulus. After ∼3–5 h of dark ageing, the chamber was irradiated with UV lights (λmax= 350 nm; see Platt et al., 2013) for about 1 h to test the impacts of photolytic ageing. After each experiment, ∼1000 ppb O3 was added, the chamber was continuously flushed with zero air (∼100 L min−1), and the temperature of the enclosure was increased to 30 ∘C overnight to promote the evaporation of HNO3 from the walls of the chamber.
O3 was measured with an O3 analyser (Thermo 49C), and NOx and NO2 were measured with a chemiluminescence NOx monitor (Thermo 42C). Aerosol number size distributions were measured using a scanning mobility particle sizer (SMPS, TSI Model 3938). Both a chemical ionization mass spectrometer with a filter inlet for gases and aerosols (FIGAERO-CIMS, Aerodyne Research, Inc.) with iodide as the reagent ion and an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF, TOFWERK) were used to measure the molecular composition of organic compounds. The FIGAERO-CIMS performed semi-continuous online measurements and shifted analysis between the particle phase and the gas phase, and the EESI-TOF continuously measured the particle phase with rapid response. In a subset of the experiments (Exp. 1, Exp. 2, and Exp. 4), the evaporation of particles was measured with a custom-built volatility tandem differential mobility analyser (VTDMA) (Tritscher et al., 2011).
2.2 Chemical ionization mass spectrometer with a filter inlet for gases and aerosols
The design and operation of the FIGAERO-CIMS were similar to those described in previous studies (Lopez-Hilfiker et al., 2014; Lee et al., 2014; Huang et al., 2018). In this study, the FIGAERO inlet was coupled to a high-resolution time-of-flight chemical-ionization mass spectrometer (HR-TOF-CIMS) ( 5000–6000), and I− was used as the reagent ion. An X-ray generator was used to ionize methyl iodide and produce the reagent ion in a nitrogen flow. Particles were collected on a 25 mm Zefluor® polytetrafluoroethylene (PTFE) filter (Pall Corp.) inside the FIGAERO via a sampling port (stainless-steel tube of ca. 1.5 m length, flow rate 5 L min−1). The duration of particle-phase sampling depended on the mass concentrations in the chamber and was 10–20 min for most of the experiments. For each experiment, three to four filters were sampled during dark ageing, and one filter was collected after about 1 h of photolytic ageing. In addition, one particle blank was performed for each experiment during dark ageing (for details on the background subtraction, see Fig. S1 and the supporting information in the Supplement). During particle-phase collection, gases were measured via a 6 mm Teflon tube of ∼1 m length at 5 L min−1. When the particle-phase sampling was completed, the gas-phase measurement was switched off and particles on the filter were desorbed by a flow of ultra-high-purity (99.999 %) nitrogen. A FIGAERO desorption round lasted about 55 min: 20 min of ramping the temperature of the nitrogen flow from ambient temperature up to 200 ∘C was followed by a 20 min “soak period” at a constant temperature of 200 ∘C and 15 min of cooling down to room temperature. The mass spectral signal evolutions as a function of desorption temperature are termed thermograms (Lopez-Hilfiker et al., 2014). The integration of thermograms of individual compounds (cooling period excluded) yields their total signal in counts per deposition. Here, we did not convert the counts per deposition into chamber concentrations, as we only show normalized signal (either signal normalized to the dominating compound or to the sum signal of all compounds). We observed modest or negligible fragmentation due to thermal desorption in the FIGAERO inlet (5 %–27 %, 1 %–4 %, and 10 %–23 % of the total organic signal of the isoprene, α-pinene, and β-caryophyllene SOA respectively). As shown in Fig. S3, most of the thermograms of individual compounds for all systems exhibited unimodal and sharp peaks. Thermal fragmentation products were detected either through thermograms of individual compounds with multiple peaks (normally double peaks) or one peak with Tmax (desorption temperature at which a compound's signal exhibits a maximum) much higher than the estimated Tmax (e.g. for α-pinene SOA, a C10 monomer had a Tmax≈ 140 ∘C which is in the range of Tmax for C20 dimers; Faxon et al., 2018). Artefacts resulting from thermal fragmentation products were corrected for (see Sect. S1.2 and Fig. S2 in the Supplement). In addition, only a small fraction (< 1 % of the total mass) of the compounds were deprotonated. In this study, we only report the molecules clustered with I−. Further information concerning the correction of thermal decomposition and the data analysis is provided in the supporting information in the Supplement.
2.3 Extractive electrospray ionization time-of-flight mass spectrometer
A detailed description of the EESI-TOF can be found in previous work (Lopez-Hilfiker et al., 2019; Pospisilova et al., 2020) and in the companion paper to this publication (Bell et al., 2021). In brief, the EESI-TOF samples the aerosol formed in the chamber via a sampling line (∼3.5 m long stainless-steel tube) and removes the gaseous components by passing the sampled volume through a multichannel charcoal denuder. The aerosol sample then intersects a spray of droplets emanating from a fused silica electrospray capillary (50 : 50 H2O : acetonitrile doped with 100 ppm NaI). The droplets envelope the aerosol sample and extract the water-soluble fraction of the aerosol. During droplet evaporation, a majority of the extracted molecules bind to Na+, creating positively charged adducts. The ions are guided to the time-of-flight mass spectrometer ( 5500–7000) where their ratio is determined. Background measurements were conducted every 4 min for 1 min by passing the sample air through a particle filter. The reported signal throughout the work is the difference spectrum of consecutive 4 min average signal and 1 min average background signal. The EESI-TOF signal (Hz) is scaled by the molecular weight of each ion in order to represent the EESI-TOF signal as a mass-based measurement (ag s−1). Artefacts associated with these measurements are discussed in Bell et al. (2021). The EESI-TOF data for most experiments had similar bulk sensitivities, except for the α-pinene experiment (Exp. 3), which was about a factor of 10 lower because the EESI-TOF was still being optimized and the capillary position and TOF settings were being altered.
2.4 Volatility tandem differential mobility analyser
The custom-built VTDMA (see e.g. Tritscher et al., 2011) sampled aerosols through the same inlet as the EESI-TOF, the SMPS, and a silica gel diffusion drier downstream of the chamber. The VTDMA combines two differential mobility particle sizer (DMPS) systems, coupled in series with a heating unit in between. Both DMPS systems consist of a custom-made DMA (Stockholm University, operated with closed-loop sheath air) and a condensation particle counter (CPC, TSI Model 3010). Before entering each DMPS system, the particles were brought into charge equilibrium using a Ni-63 source. The first DMA selected a nearly monodisperse aerosol with the diameter set to the geometric mean diameter measured in situ by the SMPS upstream (127, 87, and 219 nm) during both dark ageing and photolysis for Exp. 1, Exp. 2, and Exp. 4 respectively). The sample flow was then split into two, with one half going to the CPC measuring the selected particle concentration and the other half entering a 35 cm long custom-built thermodenuder (TD) with a residence time of 1.9 s, which heated the aerosol to 150 ∘C in Exp. 1 and Exp. 2 and to 175 ∘C in Exp. 4. The size distribution (over 15 bins) of the heated aerosol was measured in the second DMPS system with a time resolution of approximately 5 min, and the change in size was converted to an estimate of the volume fraction remaining (VFR):
where Dinit. is the initial diameter, and D(T) denotes the heated (temperature, T) diameters, represented by the geometric mean mode diameter estimated using a Gaussian fit of the whole heated size distribution.
2.5 Wall loss correction
The particle mass concentrations (in µg m−3) were derived from integrated number size distributions and their conversions to mass using an assumed organic aerosol density (1.19 g cm−3; Vaden et al., 2011). They were corrected for wall losses using a uniform dynamic wall loss rate kwall for the whole size range. kwall was determined from the observed exponential decay of the particle number concentration (taking coagulation into account) using Eq. (2), where N corresponds to the particle number concentration, and kcoag corresponds to the coagulation coefficient ( s−1; Pospisilova et al., 2020):
The wall-loss-corrected particle mass concentration was divided by the uncorrected mass concentration to obtain the wall loss correction factor applied to the EESI-TOF and FIGAERO-CIMS signal. In the experiments performed here, the total number concentration (in cm−3) varied between 106 and 104 at the beginning of the experiment to 104 and 103 at the end of the experiment. In experiments with large number concentrations (e.g. > 5×104 cm−3 in Exp. 4), coagulation was the dominant particle sink, which resulted in a mobility diameter that increased during the experiment, even though the mass was decreasing due to evaporation. The particle number concentration was constrained to be constant throughout the lifetime in the chamber, based upon kwall and kcoag. Evaporation of semi-volatile compounds will not affect the particle wall loss correction because this does not change the particle number concentration, only the particle volume (or mass).
2.6 Estimating bulk saturation mass concentration C* based on FIGAERO-CIMS and VTDMA
The saturation mass concentration (C*) of individual compounds was calculated based on their molecular composition using three different, previously published parameterizations:
-
an updated version of the parameterization by Donahue et al. (2011), modified based on the saturation concentrations of highly oxygenated molecules (HOMs) detected by Tröstl et al. (2016) and published by Mohr et al. (2019):
where n0= 25, bC= 0.475, bO= 0.2, bCO= 0.9, and bN= 2.5. nC, nO, and nN are the number of carbon, oxygen, and nitrogen atoms in the compound respectively. For ONs, it is assumed that a nitrate group reduces a compound's vapour pressure by about 2.5 orders of magnitude (Donahue et al., 2011; Pankow and Asher, 2008).
-
an updated version of Li et al. (2016) with a modified nitrogen coefficient for organic nitrates (Isaacman-VanWertz and Aumont, 2021):
where n0= 22.66, bC= 0.4481, bO= 1.656, and bCO= −0.7790 for CHO compounds, and n0= 24.13, bC= 0.3667, bO= 0.7732, bCO= −0.07790, and bN= −1.5464 for CHON compounds. nC, nO, and nN are the number of carbon, oxygen, and nitrogen atoms in the compound respectively. The modification of the parameterization is based on the observation that a nitrate group has a similar impact on vapour pressure as a hydroxyl; thus, one nitrate group (one nitrogen atom and three oxygen atoms) reduces a compound's vapour pressure by 0.7732 orders of magnitude (Isaacman-VanWertz and Aumont, 2021).
-
a parameterization based on highly oxygenated organic molecules (HOMs) from α-pinene ozonolysis (Peräkylä et al., 2020):
where nC, nH, nO, and nN are the number of carbon, hydrogen, oxygen, and nitrogen atoms in the compound respectively. With this parameterization, a nitrate group reduces a compound's vapour pressure by 0.34 orders of magnitude.
We note here that more such parameterizations exist in literature (Donahue et al., 2011; Daumit et al., 2013). We choose these three parameterizations as they have explicit formulations for nitrate groups/nitrogen. The parameterizations calculate C* at a temperature of 300 K. The temperature dependence of C* is considered as follows:
where T is the temperature in kelvin, C*(300 K) is the saturation concentration at 300 K, R is the gas constant, and ΔHVAP is the vaporization enthalpy. The latter is calculated as follows (Epstein et al., 2010):
For each filter sample from the FIGAERO-CIMS, the mass-weighted average log10C* for the entire particle population deposited on the filter (bulk) was calculated and adjusted to the same T (298.3 K) as that used by the kinetic model (see the description below) for further comparison.
The Tmax, the desorption temperature at which a compound's signal exhibits a maximum in the FIGAERO-CIMS, has previously been shown to be qualitatively connected to the compound's volatility (log10C* or enthalpy of vaporization) (Lopez-Hilfiker et al., 2014; Thornton et al., 2020; Bannan et al., 2019). For each filter sample from the FIGAERO-CIMS, we calculated the mass-weighted average Tmax of the entire particle population deposited on the filter (bulk).
As an additional method to constrain the bulk volatility, VTDMA measurements of the VFR were used to determine the bulk log10C* with a kinetic model that simulates the evaporation of a monodisperse aerosol in the heated part of the VTDMA (Riipinen et al., 2010). The bulk log10C* (at 298.3 K) was determined manually so that the calculated VFR matches the measurements. Additional model input parameters include the settings of the VTDMA (i.e. initial particle diameter and concentration), the residence time and temperature in the heated section, and the ambient temperature at the time of the measurement.
3.1 Mass yields of isoprene, α-pinene, and β-caryophyllene BSOA
As shown in Table 1, the wall-loss-corrected SOA yields and maximal mass concentrations formed from isoprene, α-pinene, and β-caryophyllene are 4 %–9 % at 11–24 µg m−3, 3 % at 18 µg m−3, and 110 % at 464 µg m−3 respectively. The SOA yields observed here are comparable to previously reported values: for isoprene + NO3, yields between 2 % and 24 % have been presented (Rollins et al., 2009; Ng et al., 2008); for α-pinene + NO3, yields between 0 % and 16 % have been presented (Hallquist et al., 2009; Nah et al., 2016; Fry et al., 2014); and for β-caryophyllene + NO3, yields between 86 % and 146 % have been presented (Fry et al., 2014; Jaoui et al., 2013). The yields at the lower end of the range of reported values of the isoprene and α-pinene SOA in our study can likely be explained by the lack of seed aerosol in those experiments. For β-caryophyllene, most of the products are of sufficiently low volatility (see Sect. 3.5), and the SOA yields are similar to those from the seeded experiments. The variation in SOA yields can also be caused by different chemical regimes, resulting, for example, in different branching ratios for the peroxy radicals (RO2) + hydroperoxyl radical (HO2), RO2 + NO3, and RO2 + RO2 pathways after the initial RO2 formation via NO3 + VOCs. The RO2 + RO2 pathway is a more effective channel for forming SOA than the RO2 + HO2 and RO2 + NO3 pathways (Ng et al., 2008). In Ng et al. (2008), the SOA yield from the isoprene + NO3 experiments (seeded), where the RO2 + RO2 pathway dominated, was much higher (23.8 %) than that from experiments where the RO2 + NO3 pathway dominated (4.3 %), as RO2 + RO2 chemistry led to the formation of oligomers. In the study of Nah et al. (2016), the branching ratios for the RO2 + HO2, RO2 + NO3, and RO2 + RO2 pathways were 5 : 4 : 1, and an SOA yield of 1.7 % at 1.2 µg m−3 (no seed) was observed for α-pinene + NO3 SOA. In our experiments, we achieved higher SOA yields from α-pinene + NO3, due to N2O5 : VOC ratios between 1 and 5 and the radical balance being dominated by either RO2 + RO2 (at low N2O5 : VOC) or RO2 + NO3 (at high N2O5 : VOC) chemistry and no significant source of HO2 radicals. As discussed in Bell et al. (2021), even in experiments where the N2O5 : VOC ratio is high (∼3) and the MCM model predicts the RO2 + NO3 pathway to be dominant, the initial composition of BSOA formed here in the dark is still dominated by RO2 + RO2 reactions, resulting in substantial fractions of oligomers and indicating that the rate of the RO2 + RO2 pathway is likely underestimated by the MCM model.
The evolution of the SOA mass concentrations and particle size during both dark ageing (2 h before photolysis) and photolysis are shown in Fig. 1 for all systems. Before photolysis, for the α-pinene system (Exp. 3), steady evaporation was observed from the particles, which is consistent with a shrinking mean diameter (from 240 to 233 nm) resulting in a 15 % loss of particle mass (corrected for wall loss) or volume. For the two isoprene experiments (Exp. 1 and Exp. 2), ∼6 % of the mass was lost during dark ageing. The wall-loss-corrected mass concentrations of the β-caryophyllene SOA are stable throughout the dark ageing period, while the geometric mean diameter of the particles increases from 229 nm (at time −2 h in Fig. 1) to 266 nm (at time 0 h), as a result of particle coagulation following the large number concentrations (∼106 cm−3). The differences in evaporation can likely be attributed to the lower volatility of β-caryophyllene monomers (C15) compared with isoprene dimers (C10) and α-pinene monomers (C10) (for details, see Sect. 3.5). Overall, the BSOA formed during dark ageing exhibits slight to moderate shrinkage of its mass and size (following correction for wall loss) after the initial production.

Figure 1Time series of the normalized mass concentration (a) and geometric mean diameter (b) of the SOA measured by the SMPS. SOA mass loadings were corrected for particle wall losses based on the decay of the particle number concentration. The mass concentration is normalized to the mass concentration value when the lights were turned on (time = 0 in the figure). The area shaded in yellow corresponds to the period of UV irradiation. The expected curvature of the mass concentration and geometric mean diameter without UV irradiation is represented by the dashed lines.
3.2 Chemical composition of isoprene, α-pinene, and β-caryophyllene BSOA
In total, 478, 425, and 366 organic molecular compositions (including 436, 332, and 348 ONs) were identified with the FIGAERO-CIMS, and 359, 158, and 441 organic molecular compositions (including 273, 134, and 268 ONs) were identified with the EESI-TOF, for the isoprene, α-pinene, and β-caryophyllene SOA respectively. In the α-pinene + NO3 experiment (Exp. 3), due to the lower sensitivity of the EESI-TOF compared with other experiments (described in Sect. 2.3), the compounds detected by the EESI-TOF were fewer than those detected in other experiments. Due to the dominating RO2 + RO2 pathway, oligomers make a substantial contribution to the total mass formed in the dark. The mass fractions of oligomers are ∼100 %, 86–88 %, and 60–63 % with the FIGAERO-CIMS and 97 %–98 %, 85 %–99 %, and 13 %–22 % with the EESI-TOF, for the isoprene, α-pinene, and β-caryophyllene SOA respectively. The dimer-to-monomer ratio varies across the three systems, and it increases with decreasing carbon number of the precursors.
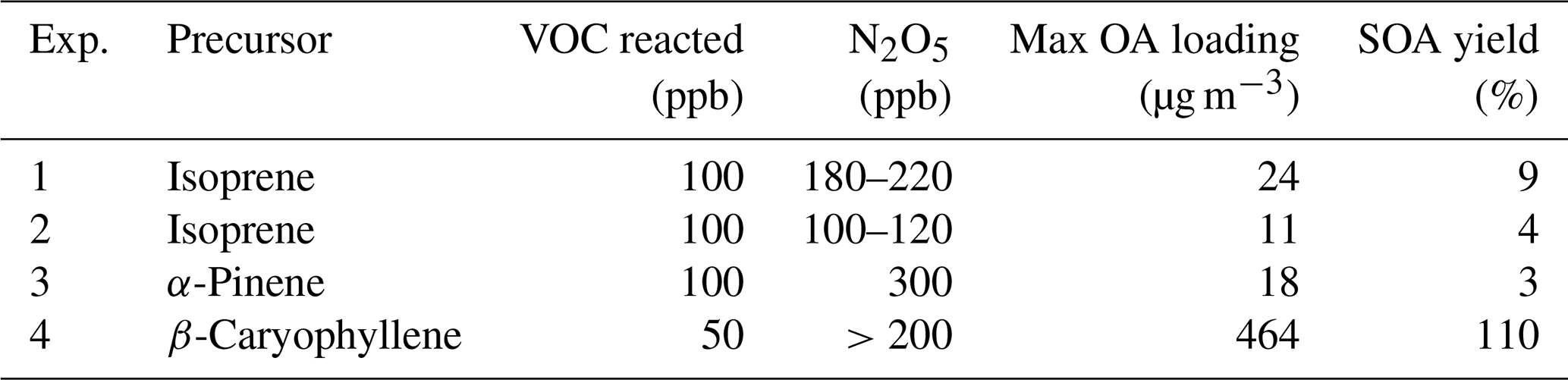
The mass spectra of the particle-phase species of the three systems obtained from the FIGAERO-CIMS and the EESI-TOF shortly before switching on the lights are shown in Fig. 2. For the isoprene system, Exp. 1 and Exp. 2 are similar; thus, only Exp. 1 is presented here, and Exp. 2 is presented in the Supplement (Fig. S6). For the isoprene SOA, monomers are negligible (C1–5 compounds are less likely to partition into the particle phase due to their relatively high volatility), and dimers with three to four nitrate groups (e.g. C10H17N3O13 and C10H18N4O16, each contributing 15 % to the total mass) dominate the mass measured by the FIGAERO-CIMS. Dimers with one, two, and five nitrate groups (44 %), and trimers (21 %) also contribute a substantial fraction to the total mass. The major compounds observed in our study are similar to those observed by Ng et al. (2008), who used a Waters ACQUITY ultra-performance liquid chromatography system coupled to a Waters LCT Premier XT time-of-flight mass spectrometer equipped with an ESI source that is operated in the negative (–) ionization mode (UPLC/(–)ESI-TOFMS) and investigated N2O5 : isoprene ratios from 0.6 to 5. For the EESI-TOF, the major compounds are similar, with the same highest peak (C10H17N3O13) as measured by the FIGAERO-CIMS. However, the relative intensities are different. Compared with the FIGAERO-CIMS, the EESI-TOF measures higher signals of C10H16N2O10, C15H24N4O18, and C20H25N3O19, but lower signals of C10H17N3O12, C10H18N4O16, and C10H17N5O18. There is a high signal from C4H6O2, which comes from the degradation of oligomeric species C10H16N2O9,10 and C10H17N3O13.
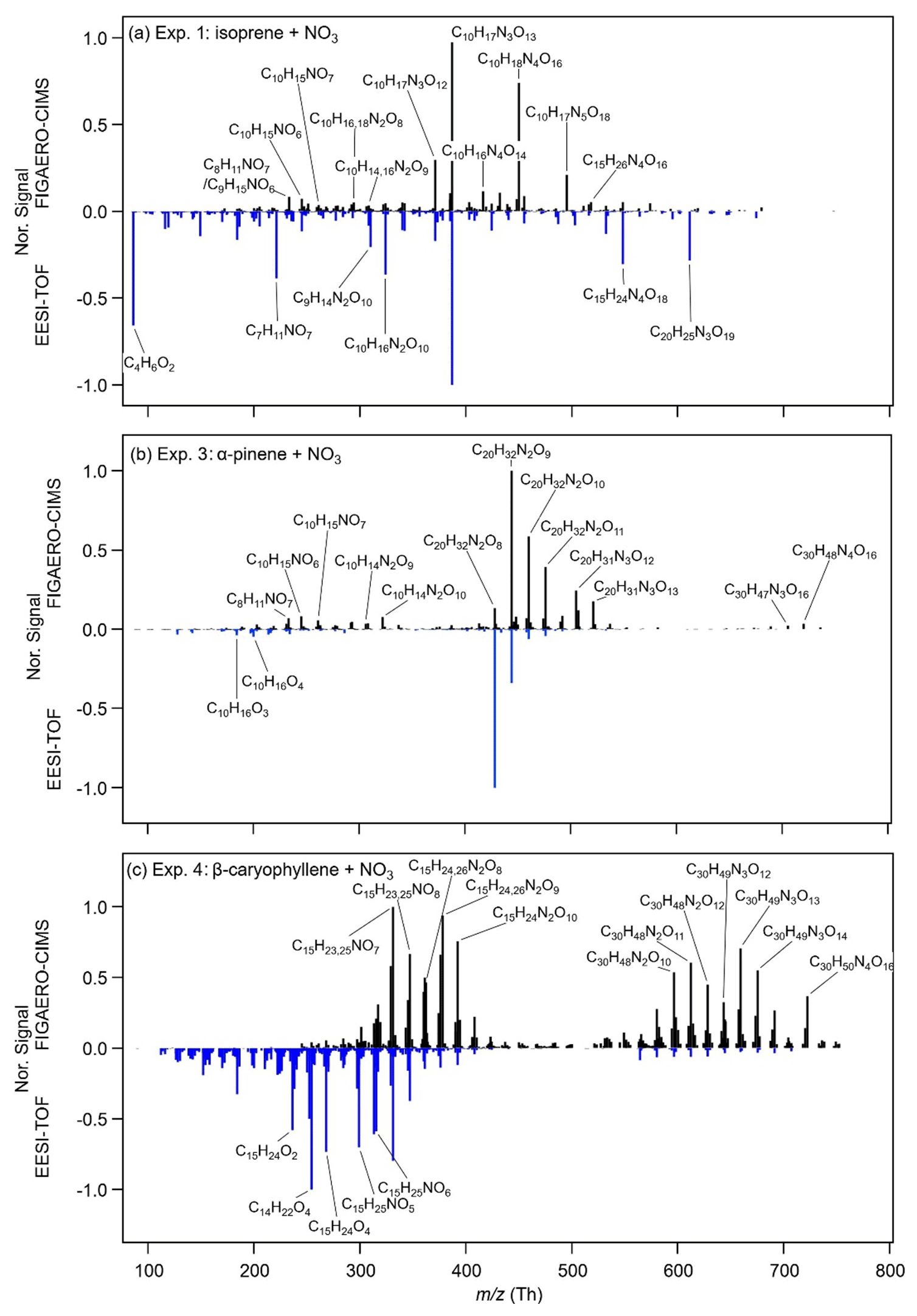
Figure 2FIGAERO-CIMS (positive axis) and EESI-TOF (negative axis) mass spectra of particle-phase CxHyOzN0–4 formed during (a) Exp. 1 (isoprene + NO3), (b) Exp. 3 (α-pinene + NO3), and (c) Exp. 4 (β-caryophyllene + NO3) shortly before photolysis. For FIGAERO-CIMS, the last filter before photolysis is chosen (Pre 2 in Fig. 3). The EESI-TOF mass spectra are averaged mass spectra during the same sampling time as that of FIGAERO-CIMS. All mass spectra are normalized to the corresponding maximal signal.
The α-pinene system according to the FIGAERO-CIMS measurements is dominated by C20 dimers with two nitrate groups, i.e. C20H32N2O8–13 make up 39 % of the total mass, while the monomers and trimers contribute 12 %–14 % and 3.7 %–4.4 % respectively. Our mass spectra are similar to those reported in Takeuchi and Ng (2019) (measured with a FIGAERO-CIMS), where the α-pinene + NO3 SOA was dominated by C20 dimers with two dominating compounds, C20H32N2O9 and C20H32N2O10. The dominating monomer in their study was C10H15NO6, also identical to our study. Their ratio of C20H32N2O9 to C10H15NO6 was about 5, which was ∼2 times higher (about 10) in our study. In Takeuchi and Ng (2019), reactions between peroxy radicals (RO2 + RO2) and RO2 + NO3 are also favoured. The presence of large concentrations of RO2 in both studies likely leads to the large fraction of oligomers. The EESI-TOF detected similar dimers and monomers as those measured by the FIGAERO-CIMS. However, the highest peak measured is C20H32N2O9 for the FIGAERO-CIMS and C20H32N2O8 for the EESI-TOF. It is also worth noting that the EESI-TOF measured a few CHO compounds without nitrate groups, such as C10H16O3 and C10H16O4.
The β-caryophyllene SOA mass spectrum from the FIGAERO-CIMS is dominated by C15 monomers with one to two nitrate groups (C15H23,25NO7,8 and C15H24,26N2O8−10) and C30 dimers with two to three nitrate groups (C30H48N2O10–12 and C30H49N3O12−14). The mass spectrum from the EESI-TOF is dominated by C15 monomers with zero and one nitrate group. The contribution from the dimers is substantially smaller compared with FIGAERO-CIMS, which may result from a decreased sensitivity of larger components, as observed previously (Lopez-Hilfiker et al., 2019), and/or from decomposition of ON dimers. The higher contribution from the monomers, compared with the isoprene and α-pinene systems, is likely due to the larger number of carbon atoms of β-caryophyllene, and the two double bonds (one more than α-pinene), which both lead to the low volatility of the monomers (for details, see Sect. 3.5).
Overall, both instruments are able to cover the major compounds from both monomer and dimer regions, although the sensitivity towards different compounds varies. The I− CIMS is not sensitive towards non-oxygenated compounds, monoalcohols, monoketones, or monoaldehydes (Lee et al., 2014), whereas the EESI-TOF is sensitive to nearly all compounds present in SOA but is not sensitive to non-oxygenated compounds (Lopez-Hilfiker et al., 2019). Differences are also caused by the fragmentation of oligomers into smaller compounds (CxHyOz) inside the EESI-TOF, such as C4H6O2 in the isoprene + NO3 system, and a few CHO compounds in the α-pinene and β-caryophyllene systems. These compounds are likely related to artefacts from the electrospray ionization (Goracci et al., 2017; James et al., 2006; Keith-Roach, 2010; Maire and Lange, 2010; Kourtchev et al., 2020; Rovelli et al., 2020). They may occur due to the proximity of –ONO2 functional groups next to peroxy linkages, which results in fragmentation of molecules when particles are exposed to water in the electrospray (Bell et al., 2021) as well as the loss of a HNO3 fragment during ionization (Liu et al., 2019).
3.3 Photolytic ageing of isoprene, α-pinene, and β-caryophyllene BSOA
As can be observed in Fig. 1, switching on the lights affects particle mass and size for all BSOA systems, although to a different extent. For the isoprene SOA, the particle size and mass show a slightly steeper decrease compared with the situation expected without UV irradiation (dashed lines). For the α-pinene SOA, both the particle size and mass decrease compared with the dark, from 229 nm and a normalized mass concentration of 0.9 to 219 nm and 0.87 after 1 h photolysis respectively. The β-caryophyllene SOA, although the least volatile, shows a decrease of 12 % (normalized mass concentration of 0.83 vs. 0.95 expected without UV irradiation) and a reduction of the mean particle size from 273 to 230 nm. These results indicate that there are slight to moderate losses of particle-phase compounds due to UV irradiation. The possible reasons for the differences in the three systems will be discussed in this section and in Sect. 3.5.
In Fig. 3, the relative changes in the chemical composition of the SOA before (pre) and after (post) photolytic ageing measured by the FIGAERO-CIMS are illustrated. During irradiation, processes/reactions occurring in the dark may also continue with the lights on – for example, consumption/decomposition of reactive oxygen species (Pospisilova et al., 2020), evaporation of semi-volatile compounds, and hydrolysis of nitrate functional groups can alter the chemical composition of SOA particles. To account for this, Fig. 3 compares the chemical composition of two particle filter samples collected during dark ageing (Pre 1 and Pre 2) and one filter sample collected after 1 h of photolytic ageing (Post). The time interval of the sampling was about 1.5–2 h or about 3 h if a particle blank was performed between two filters. The difference between the two filters during dark ageing (Pre 2 – Pre 1) shows the changes in the chemical composition caused by all processes other than photolysis, whereas the difference between the last filter during dark ageing and the one after 1 h photolysis (Post – Pre 2) shows the impacts of the above-mentioned processes plus the impact of UV irradiation.
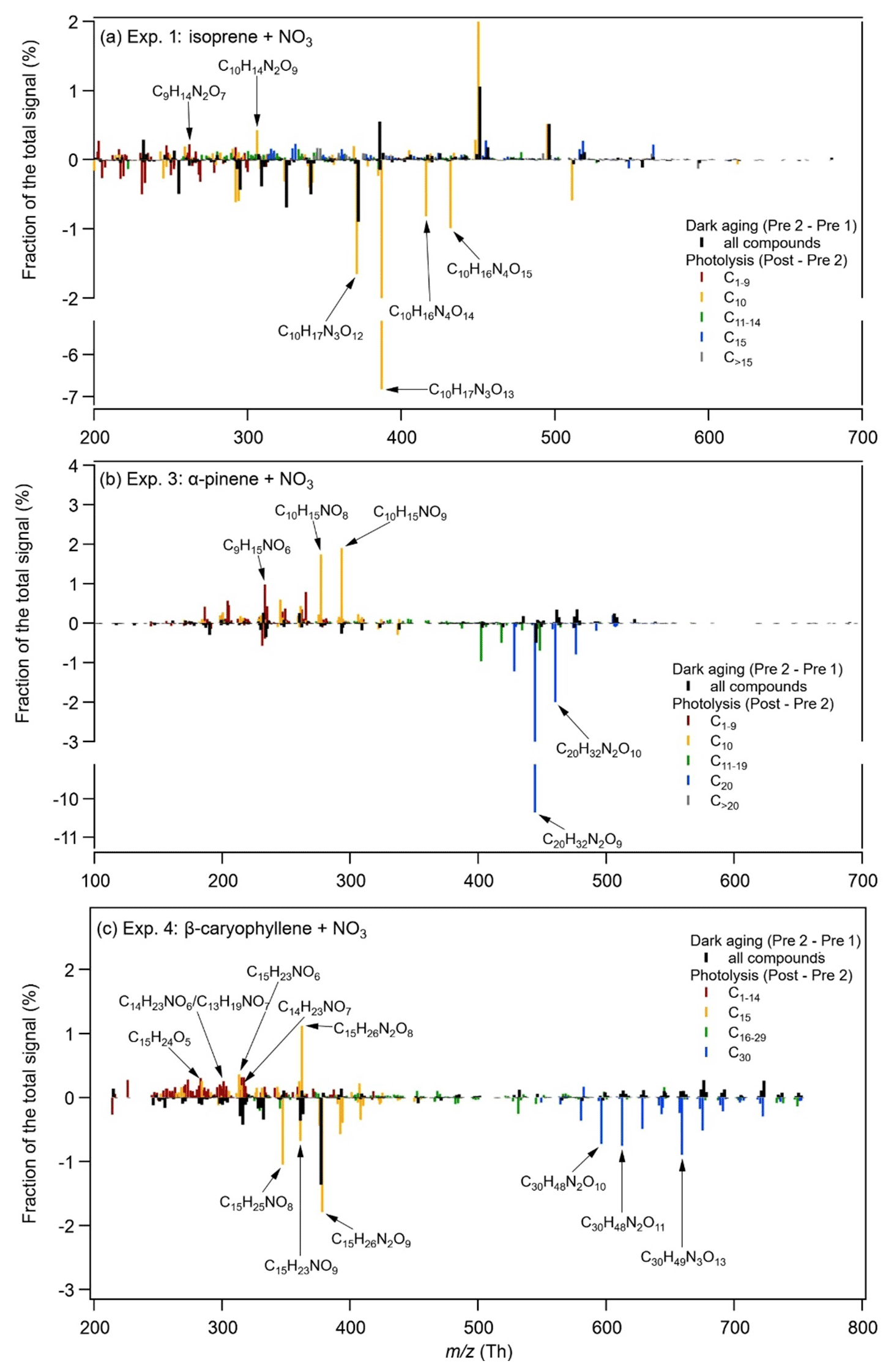
Figure 3Relative changes in the fraction of the total signal between two filters during dark ageing (Pre 2 – Pre 1) and between the last filter during dark ageing and the filter after 1 h of photolysis (Post – Pre 2) for (a) Exp. 1 (isoprene + NO3), (b) Exp. 3 (α-pinene + NO3), and (c) Exp. 4 (β-caryophyllene + NO3).
The comparison between the two filters during dark ageing (Pre 2 – Pre 1 in Fig. 3) shows relatively small changes in the chemical composition that are similar for all three systems: the signal fractions of high-molecular-weight compounds increase and the signal fractions of low-molecular-weight compounds decrease. The signal fraction changes in individual compounds are less than 2 %. Such change is mainly due to the evaporation of semi-volatile compounds, as gas-phase wall losses act as a sink for semi-volatile compounds, resulting in the repartitioning of the particle phase (Bertrand et al., 2018). Bell et al. (2021) show that most of the changes in chemical composition in the α-pinene SOA system occur over the first 2 h of dark ageing, meaning that the chemical composition has mostly stabilized at the time directly prior to photolysis. Figure S7 in the Supplement shows the changes in absolute signal fraction for the filter samples pre- and post-photolysis for Exp. 3.
During photolysis, the changes observed in the mass spectral patterns (Post – Pre 2 in Fig. 3) differ from those during dark ageing and also have a larger magnitude. For the isoprene system, the compounds exhibiting the largest decays in signal fraction are C10H17N3O12–13 and C10H16N4O14–15 (8 % and 2 % of the total signal respectively). At the same time, there are a few compounds (e.g. C9H14N2O7 and C10H14N2O9) whose signal fractions increase (< 1 %). For the α-pinene system, the signal fraction of the main dimer compounds (C20H32N2O8−10) decreases by 13.6 % during photolysis, whereas the signal fractions of a few monomers (e.g. C9H15NO6−8 and C10H15NO8−9) increase by up to 2 %. The β-caryophyllene SOA system shows decreases in the signal fractions of both monomers and dimers, mainly C15H25NO8, C15H23NO9, C15H26N2O9, and C30H48N2O11–13, whereas the signal fractions of quite a few compounds, including C12–14 compounds (e.g. C14H23NO6,7), C15 compounds (e.g. C15H23NO6, C15H25N2O8), C16–29 compounds (e.g. C29H46N2O9–11), and C30 compounds (e.g. C30H46N2O12), increase. Overall, our results show that the major compounds decomposing during photolysis are both monomers and oligomers and that there are also compounds formed that contain lower carbon numbers and/or fewer nitrate groups.
The FIGAERO-CIMS offers insight into the pre- vs. post-photolysis SOA composition, and the EESI-TOF allows one to monitor the evolution of individual compounds at high time resolution. Using a linear fit to the 2 h pre-photolysis time series of the individual compounds measured by the EESI-TOF and then comparing a predicted value based on this fit at 1 h post-photolysis (with 95 % confidence interval) with the actual measured signal (see Fig. S8 in the Supplement), we find that 54 (the average value from Exp. 1 and Exp. 2) out of 359, 24 out of 158, and 104 out of 441 compounds decreased significantly during photolysis, i.e. by 44 % ± 20 %, 64 % ± 24 %, and 24 % ± 18 % for the isoprene, α-pinene, and β-caryophyllene SOA respectively. These compounds contributed 48 %, 44 %, and 60 % to the total pre-photolysis signal for these three respective systems. Meanwhile, there were 116, 53, and 164 compounds that increased during photolysis for the isoprene, α-pinene, and β-caryophyllene SOA respectively. We note, however, that the fraction appearing to be resistant to photolysis or even showing increase may undergo functional group changes during photodegradation. For example, the decay of molecules CxHyOzN may be compensated for by the photodegradation of other molecules which produces products with the same molecule formula CxHyOzN but different structures. Here, we cannot fully exclude particle-phase compound volatilization resulting from an increase in chamber temperature (3–4 ∘C, caused by heating from the chamber lights). However, because most of the compounds showing decay are oligomers with very low predicted volatilities, such an increase in temperature is unlikely to play a major role in the change of composition.
Figure 4 shows the time series of the main compounds that degrade during photolysis for each system, measured by both the EESI-TOF and the FIGAERO-CIMS. Within the error tolerances, the time series of the compounds measured by the two instruments have a good agreement. The decay rates of these compounds are compound dependent. Even though 1 h of photolytic ageing might be too short to observe full decay trajectories (Zawadowicz et al., 2020; O'Brien and Kroll, 2019), it is long enough to distinguish the non-photolabile compounds and the photolabile compounds and to compare the decay rates of the photolabile compounds (Henry and Donahue, 2012; Wong et al., 2015). As also shown by Nah et al. (2016), most of the changes in α-pinene + NO3 SOA due to photochemical ageing happened in the first 1 h of light exposure.
Figure 4Time series of the isoprene tracers C10H17N3O12,13 (Exp. 1 and Exp. 2), the α-pinene tracers C20H32N2O8−10 (Exp. 3), and the β-caryophyllene tracers C30H48N2O8–10 (Exp. 4) from the EESI-TOF and the FIGAERO-CIMS.

Figure 5Mass-weighted number of nitrate groups per monomer (number of carbon atoms/number of the carbon atoms in precursors – 5, 10, and 15 for the isoprene, α-pinene, and β-caryophyllene SOA, respectively) and mass-weighted number of non-nitrate oxygen atoms per monomer for three filters (Pre 1, Pre 2, and Post) for all experiments.
Photolysis requires chromophores, which absorb particular wavelengths of
visible light. Earlier studies on the photodegradation of OH- and
O3-initiated SOA under low-NOx conditions have demonstrated that
photolysis fragments molecules with functional groups like carbonyls (C=O)
and peroxides (R–O–O–R) (Surratt et al., 2006; Walser et al., 2007; Mang
et al., 2008; Pan et al., 2009; Krapf et al., 2016). In contrast to these
studies, the BSOA in our study is rich in nitrate groups and
oligomers. In Fig. 5a, we plot the average number of nitrate groups per
monomer (monomer unit) for the three FIGAERO-CIMS filter samples (the same
as in Fig. 3) selected before and after photolysis for all three SOA
systems. Overall, the particle-phase compounds of all three SOA types
contain a similar number of nitrate groups per monomer on average (1–1.5).
During the last 2–3 h of dark ageing, the nitrate group fraction is stable,
but photolysis causes a slight decrease in the nitrate-to-monomer ratio for
all systems, consistent with the decrease of 1 %–3 % of the mass fraction of ONs of total organic compounds for all systems. It is clear from this that UV light fragmentation of nitrate groups only plays a minor role in
changing the chemical composition of SOA when transitioning from dark to
light conditions. It is known that organic nitrates can undergo photolysis
and release NO2 via RONO2 + hv−>
RO• + NO2 (Barnes et al., 1993). Measurements of the absorption cross-sections of a number of alkyl nitrates, such as methyl nitrate, and some bifunctional organic nitrates have shown lifetimes of up to between 14 h and 6 d (Barnes et al., 1993). The absorption cross-sections of organic nitrates normally have an absorption maximum at λ < 290 nm and start to decrease when λ > 310 nm (Barnes et al., 1993; He et al., 2021). The black lights in the chamber reproduce the solar spectrum well in the range of 320–400 nm, but the intensity at lower wavelengths (< 320 nm) falls off faster than the solar spectrum.
Thus, the photolysis of nitrate groups observed in our study represents a
lower limit of what would be expected in the atmosphere. More recent studies have shown that the presence of a nitrate group can enhance the absorption cross-sections and make the photolysis of carbonyl nitrates faster than their reaction with OH (Müller et al., 2014; Suarez-Bertoa et al., 2012). However, based on the stability of the nitrate group fraction during photolytic ageing as described above (compare also Fig. 5), the cleavage of the nitrate functional group from the carbon chain is not the main loss pathway in our study and on the timescales of our experiments.
In terms of oligomers, for the α-pinene and β-caryophyllene systems, the major degrading compounds are C20 and C30 dimers, while the major compounds formed are their corresponding C10 and C15 monomers (the isoprene system is a mix of C10 dimers, C15 trimers, and C20 tetramers, which makes it challenging to identify the potential parent compounds of newly formed compounds). This indicates that for some fraction of the oligomers, the linkage between two monomer blocks is photolabile. Oligomers may be formed through both gas- and particle-phase processes. As RO2 + R'O2−> ROOR' + O2 is the main reaction channel for RO2 self- and cross-reactions in the gas phase, a large fraction of peroxides can be expected, and such mechanisms of dimer formation were proposed for isoprene + NO3 SOA (Ng et al., 2008; Zhao et al., 2021; Wu et al., 2021). Recent studies also showed multiple pathways of dimerization in the particle phase or the gas–particle interface (Zhao et al., 2019; Claflin and Ziemann, 2018). All the studies mentioned above show that the formation of dimers, in both the gas- and particle-phases, changes the functional groups of the resulting molecules compared with their building blocks (monomers). It is likely that peroxide functional groups are formed during RO2 self-/cross-reactions, which could make some dimers photolabile, whereas the formation of potentially photolabile carbonyl functional groups is less certain from NO3 oxidation.
Some of the compounds being formed during photolytic ageing are molecules with one to three fewer carbon atoms than their potential parent compounds (e.g. C13, C14, and C29 compounds in the β-caryophyllene system), indicating fragmentation of the carbon skeleton. On average, each monomer (or monomer unit) of the isoprene, α-pinene, and β-caryophyllene SOA contains ca. 2, 2.5, and 3 oxygen atoms pre-photolysis (disregarding the oxygen atoms in the nitrate groups) respectively (Fig. 5b), suggesting that, in addition to nitrate groups, these compounds also possess other oxygenated functional groups and other possible chromophores. The β-caryophyllene SOA exhibited higher losses of the particle mass and reduction in size (Fig. 1), followed by the α-pinene SOA, which could partly be explained by its higher number of functional groups per monomer (or monomer unit), i.e. larger photolabile fraction, compared with the other two systems.
Overall, the particle mass loss of nitrate-initiated SOA after 1 h photolysis in our study is less than that reported for the OH- and O3-initiated SOA under low-NOx conditions (Zawadowicz et al., 2020; Wong et al., 2015; Henry and Donahue, 2012; Epstein et al., 2014), despite observing larger changes in the chemical composition. One possible reason could be that our systems have a large fraction of oligomers, and photolysis-derived products may not directly evaporate, meaning that a substantial fraction of these products retain low enough volatility to remain in the particle phase. Moreover, in terms of different functional groups, the addition of a nitrate group in a given molecule is estimated to reduce its vapour pressure by about 2.5 orders of magnitude, which is comparable to other oxygenated functional groups often observed in the OH- and O3-initiated BSOA, for example, the hydroxyl group (−OH) and hydroperoxy group (−OOH) (2.4–2.5 orders of magnitude), or the carbonyl group (=O) (1 order of magnitude) (Pankow and Asher, 2008; Donahue et al., 2011). Thus, the gas-phase compounds formed in our systems could contain fewer functional groups (disregarding nitrate groups) than those in OH- and O3-initiated BSOA but could still be of low enough volatility to condense into the particle phase. The elemental oxygen-to-carbon (O : C) ratio (disregarding the oxygen atoms in the nitrate groups) of the α-pinene SOA was calculated to be 0.26 ± 0.01 before photolysis in Exp. 3, which is lower than the reported O : C ratios of 0.34–0.36 for BSOA from ozonolysis of α-pinene measured by the FIGAERO-CIMS in laboratory experiments (Huang et al., 2018). As shown above, nitrate groups might be less photolabile than carbonyls and peroxides; thus, different sensitivities towards photolysis can be expected for the systems initiated by different oxidants.
3.4 Changes in the gas phase during photolysis
The primary focus of this study is on the changes in condensed-phase chemical composition of BSOA due to photolytic ageing. However, we cannot decouple processes happening in both the particle and gas phases. For example, photodegradation of semi-volatile compounds in the gas phase could lead to a decrease in compounds in the particle phase, as they will evaporate to re-establish equilibrium. If gas-phase photodegradation was the dominant cause of mass loss in all systems, the largest decays in the particle phase would be expected from the most volatile species. Further, large non-volatile molecules (e.g. dimers in the β-caryophyllene SOA) should be non-responsive to such a pathway. Because there is a systematic degradation of dimers in all systems, it is unlikely that repartitioning derived from gas-phase photodegradation is driving the change in SOA composition during UV ageing.
For the experiments shown here, the FIGAERO-CIMS using I− as the reagent ion (and an X-ray generator as the ion source) also measured the gas phase. However, in systems with a high abundance of NO3 radicals, charge transfer can cause losses of I− and formation of NO (Lee et al., 2014). Additionally, the reaction of N2O5 and I− may partly result in NO + INO2, and H transfer reactions can occur between I− and HNO3 (HNO3 can come from the hydrolysis of N2O5 in our injection inlet or on chamber walls), both providing other routes to the formation of NO. As the amount of NO introduced in our systems was very high, the signal of NO was comparable to and sometimes even higher than the signal of I−. Thus, gas-phase compounds were detected clustered with both NO and I−, with changing ratios of I− NO during the experiments (higher during dark ageing and lower during photolysis). This was not an issue for the particle phase, as particles are desorbed from the filter with pure nitrogen. We probed the uncertainties of detected signals due to the changing I− NO ratio (from ∼0.1 to 1.6) after Exp. 2 (for details, see the description in the supporting information and Fig. S4 in the Supplement). In our system, for those compounds clustered with I−, the normalized signal did not change significantly with decreasing I− NO as long as I− NO was higher than ∼0.3, but it decreased by about 20 %–40 % when I− NO decreased further to 0.1, as more molecules were clustered with NO.
Taking these uncertainties into account, we found that the signal of the major gas-phase compounds either stayed stable (e.g. C15H25NO) or increased significantly (e.g. C14H23NO4, C15H23NO6, and C15H26N2O8, with increases in the normalized signals of more than 5 times) during photolytic ageing for the β-caryophyllene SOA. For the isoprene and α-pinene SOA, although the low I− NO ratio (< 0.1) hindered a quantitative comparison, we still observed increases of several major compounds (e.g. C5H10N2O8 for the isoprene SOA, and C10H15NO8, C10H17NO7 for the α-pinene SOA). These are likely fragmentation products from oligomers and/or monomers. As the volatility of these small compounds is much higher than their parent oligomer compounds, they would tend to entirely or partly (depending on their volatility) evaporate to the gas phase. Different from the photolytic ageing of O3- or OH- initiated SOA, we did not, however, observe a significant increase in the signals of small acids in the gas phase, such as formic acid and acetic acid (Pan et al., 2009), despite observations of fragmentation reactions in the particle phase. Their minor importance seems not to significantly impact the measured formic or acetic acid in the gas phase.
In addition to photolytic ageing of gas-phase compounds, there are other gas-phase reactions initiated by UV irradiation, which may impact the chemical composition, e.g. OH oxidation and the reactions with O3 or NO3 radicals. Under our experimental conditions (>100 ppb NO2 from decomposition of N2O5), if there are any OH radicals formed, they will nearly exclusively react with NO2 given the high concentrations. However, we did not observe a significant increase in the gas-phase HNO3 signal. For the reactions with O3 or NO3, if there were such appreciable reactions in the experiments with isoprene and β-caryophyllene, where double bonds are still present, we would expect to observe the significant formation of more highly oxygenated molecules, which are absent in Fig. 3. Additionally, Fig. 3 shows that there is a consistent degradation of dimers in all systems, including α-pinene, where double bonds are not retained, thereby suggesting that the dominant photolytic degradation pathways are independent of gas-phase chemistry.
3.5 Impacts of photolysis on particle volatility
Considering photolytic ageing results in the degradation of molecules, there is a shift in the volatility distribution towards higher volatility species, which can cause evaporation of particulate mass. Here, we discuss how the apparent volatility of remaining particles changes during photolytic ageing as well as the potential influence that the previously discussed chemical processes have on these changes.
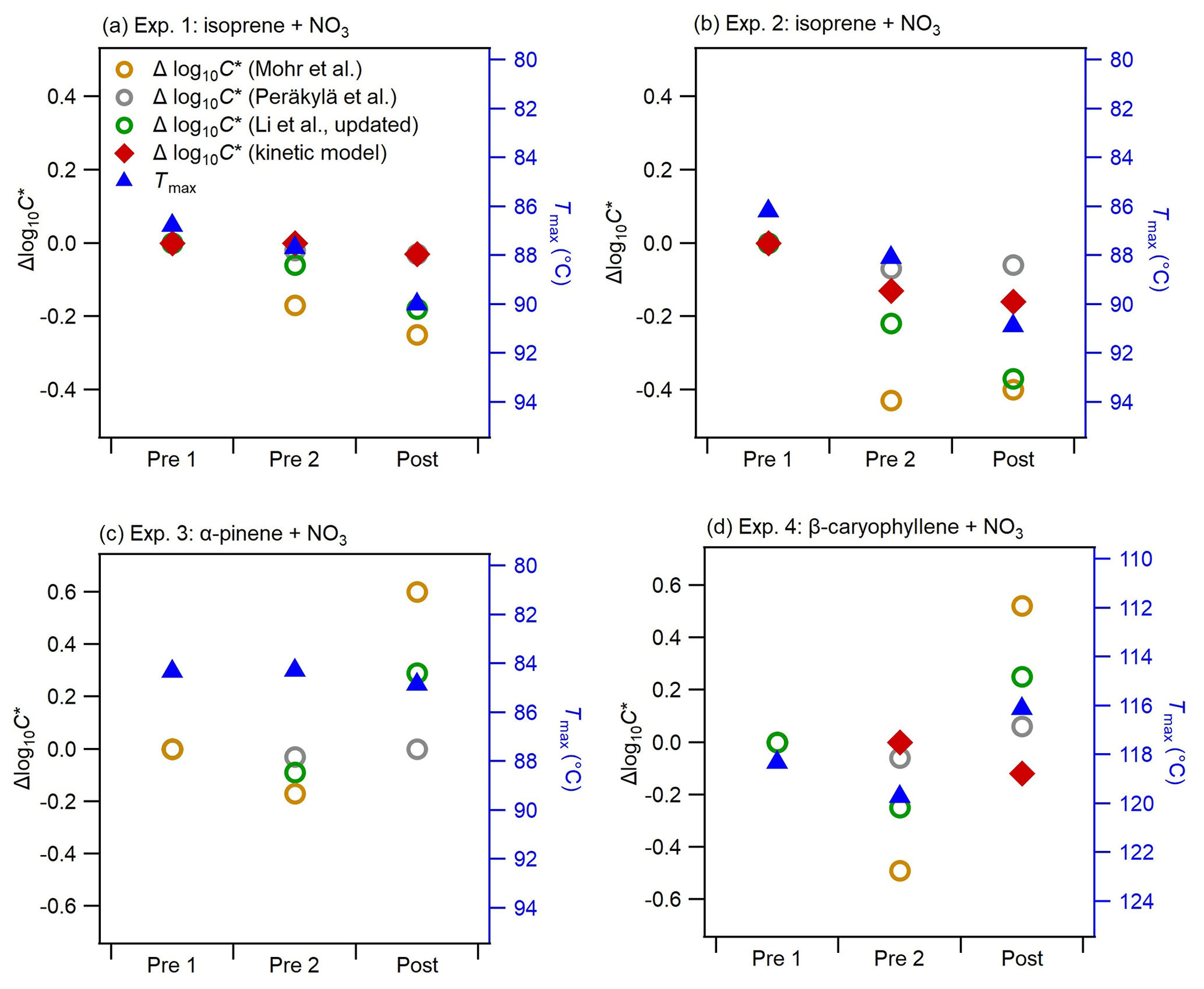
Figure 6Comparison of changes in the bulk volatility measured with three methods before and after photolysis (Pre 1, Pre 2, and Post) for all experiments: Method 1 was Δlog10C* based on the parameterizations of Mohr et al. (2019), Li et al. (2016) (updated by Isaacman-VanWertz and Aumont, 2021), and Peräkylä et al. (2020); Method 2 was mass-weighted average Tmax (note the right axes are swapped, and the range in panel d is different from that in the other panels); and Method 3 was Δlog10C* from the kinetic model (data available only for Pre 2 and Post in Exp. 4).
Figure 6 shows the changes in the bulk volatility derived from different methods (bulk log10C* derived from molecular composition using different parameterizations, bulk log10C* derived from the kinetic model using the VTDMA measurements, and mass-weighted average Tmax from the FIGAERO-CIMS thermograms) during dark ageing and photolysis for all BSOA systems. Note that we show the Δlog10C* (calculated by subtracting the bulk log10C* of the filter Pre 1 from the bulk log10C* of each filter), as the variabilities in the absolute bulk log10C* calculated by different parameterizations span several orders of magnitude: the bulk log10C* for all three systems is in the range of −11.9 to −7.6 based on the parameterization by Mohr et al. (2019), −3.7 to −0.3 by Li et al. (2016) and Isaacman-VanWertz and Aumont (2021), and −0.5 to 0 by Peräkylä et al. (2020) (see Fig. S9 in the Supplement). As Fig. S10 in the Supplement shows, the discrepancy in log10C* from the three parameterizations becomes larger with increasing nitrogen number. While a detailed discussion on these discrepancies is beyond the scope of this paper, they clearly reflect the uncertainties related to volatility estimates of complex organic particles (O'Meara et al., 2014; Wu et al., 2021). Tmax is a qualitative measure of volatility and is influenced by filter mass loadings, temperature ramp rate, and FIGAERO geometry (Huang et al., 2018; Schobesberger et al., 2018; Thornton et al., 2020). However, Tmax variation due to these factors is generally smaller than the difference in monomers and dimers. We compared the shape of thermograms and Tmax of several monomers and dimers observed in our study to those of the same compounds from a few field campaigns (see Fig. S11 in the Supplement). Overall, the thermal desorption behaviour observed in our chamber experiments is similar to that from field measurements. By further comparing the log10C* of individual compounds, based on studies such as Mohr et al. (2019), with their corresponding Tmax (Fig. S12), we found good qualitative agreement with a clear trend of increasing Tmax with decreasing log10C*, and higher Tmax for dimers than corresponding monomers for all systems. We also noticed large uncertainties in the calculated log10C*, especially for ON oligomers with multiple nitrate groups. For example, the dominating dimer C20H32N2O9 has a Tmax of about 70 ∘C, which is in the range of Tmax of C10 monomers such as C10H15NO9. However, based on the parameterization of Mohr et al. (2019), the log10C* of the dimer C20H32N2O9(−7.9) is much lower than the log10C* of the C10 monomers (log).
Overall, for the two isoprene experiments (Exp. 1 and Exp. 2), almost all methods show a decrease in volatility as the experiment progresses, with no change in trend during the transition from dark to light conditions. This agrees well with the changes in the chemical composition as shown in Fig. 3: during dark ageing, the chemical composition shifts to higher-mass (lower-volatility) compounds and causes a decrease in the bulk volatility; during photolysis, there is additional decay of some oligomers such as C10H17N3O13, whose volatility is in the intermediate range of all compounds (Fig. S12). As a consequence, despite clear changes in the chemical composition, the bulk volatility does not change substantially due to photolysis. In Exp. 2, the Δlog10C* by both Peräkylä et al. (2020) and Mohr et al. (2019) decreased during the dark ageing but increased slightly during photolysis, which is different from other methods. The difference between Exp. 1 and Exp. 2 is probably due to the differences in the chemical composition (Fig. 2 vs. Fig. S6), as Exp. 1 had a higher NO3 isoprene ratio and SOA with more nitrate groups (Fig. 5), and also due to different mechanisms to treat nitrate groups in the different parameterizations. It again reflects uncertainties related to volatility estimates by different parameterizations.
In Exp. 3 (α-pinene + NO3), no clear trend in volatility can be discerned. Tmax does not exhibit significant changes during both dark ageing and photolysis. However, the Δlog10C* by the parameterizations shows a slight decrease during the dark ageing and an increase during photolysis. As shown in Fig. 3, we observed decreases in a few dimers and increases in a few monomers, which, based on the parameterizations, causes a shift from low-volatility compounds to high-volatility compounds. However, as mentioned above, the Tmax of these dimers is not necessarily higher than those monomers (i.e. the volatility of these dimers is not necessarily lower than that of those monomers). Thus, the mass-weighted average Tmax does not decrease. Unfortunately, no VTDMA data are available for this experiment.
In Exp. 4 (β-caryophyllene + NO3), the different methods yield different information on the relative changes in volatility from dark to photolytic ageing. Both Tmax and Δlog10C* from the parameterizations show a bulk volatility decrease during dark ageing and an increase during photolysis, which would agree with changes in chemical composition. The shift from small (high-volatility) compounds to large (low-volatility) compounds during dark ageing causes the decrease in the bulk volatility, whereas the major compounds that decay during photolysis cover both monomers and oligomers with a wide range of volatility, and there are a few newly formed C< 15 compounds that remain in the particle phase (Fig. 3); thus, the bulk volatility increases. However, the Δlog10C* from the VTDMA data does not agree with the trend during photolysis. We note that the particle size distribution and mean particle size in Exp. 1, 2, and 3, as shown in Fig. 1, did not change substantially, whereas the particles size in Exp. 4 (β-caryophyllene + NO3) became significantly smaller during photolysis (Fig. 1). In Fig. 7, the evolution of the volume distribution of the β-caryophyllene SOA in Exp. 4 shows that the size distribution slowly shifts to larger sizes during dark ageing, but during photolysis, the decrease in particles with diameters larger than 300 nm is much greater than the decrease in small particles, indicating that the photodegradation is dependent upon particle size. The size dependence can be explained by calculating the mass absorption efficiency as a function of particle size (see Fig. S5 in the Supplement), which shows that particles with a diameter of 300 nm are 1.5–1.7 times more efficient at absorbing the chamber lights (λ= 350 nm) than 100 nm particles. Other processes, such as coagulation and evaporation, tend to lead to larger decreases in smaller particles, which is the opposite of what we found here, while the wall loss rate (which is tested in another experiment during the same campaign) was nearly constant for the particle size range up to 400 nm; thus, the latter was unlikely the reason for the size dependence of light absorption. In comparison, the isoprene and α-pinene systems have a smaller and narrower size distribution and, consequently, do not have an equally important size dependence in their response to UV light. This could also partly explain why the β-caryophyllene SOA had the largest decay in particle mass and size among the three systems. As the particles with different sizes will not have a uniform change in their chemical composition, the volatility measured for one selected size by the VTDMA may not be able to represent the change in the bulk volatility for experiments with a wide distribution of particle sizes; similarly, caution should be applied when interpreting the representativeness of measurements based on the bulk mass for the whole particle size distribution.
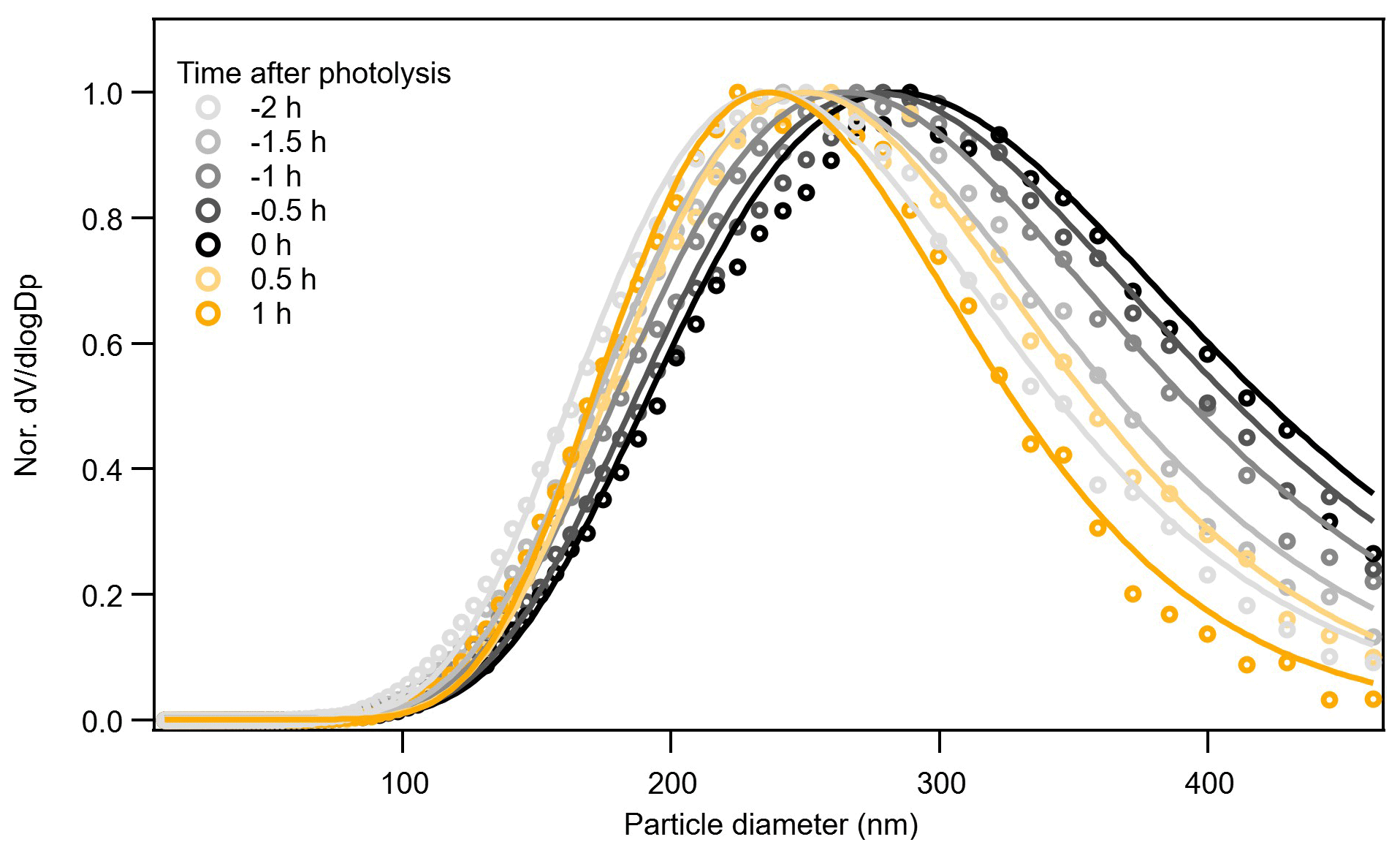
Figure 7Evolution of the particle size distribution of the β-caryophyllene SOA during dark ageing and photolysis. The solid lines are log-normal fits.
Our experimental observations on three BSOA systems show that the dark-to-light transition (1 h photolytic ageing) causes slight to moderate evaporation in BSOA (∼0, 3.5, and 12 % of the total mass for the isoprene, α-pinene, and β-caryophyllene SOA respectively). Evaporation is due to the fragmentation of photolabile compounds and the formation of volatile fragmentation products. Despite finding 0 %–12 % of the mass evaporating during photolysis, 48 %, 44 %, and 60 % of the total signal of the respective isoprene, α-pinene, and β-caryophyllene SOA is sensitive to photolytic ageing, representing a majority of the pre-photolysis composition.
Fragmentation of nitrate groups is not the main loss pathway on the timescale of our experiments. For all BSOA studied here, UV light fragments oligomers at the linkage between the monomer units (likely peroxides), as well as functional groups at other positions, causing the formation of monomers and compounds with a shorter carbon skeleton (e.g. C10→ C9). The newly formed compounds are more volatile than their parent compounds and are, thus, entirely/partly released back into the gas phase.
The changes in bulk volatility vary with individual experiments and depend on (1) whether the volatility of the photolabile fractions is higher/lower than that of non-photolabile fractions and (2) how the fragment products remain in the particle phase. The comparisons of different methods to assess the bulk volatility reveal the complexity of volatility assessments. There is generally a good agreement with the trends between the VTDMA and thermal desorption behaviour from the FIGAERO-CIMS (both desorption-based methods). However, the uncertainties related to molecular-formula-derived parameterization, especially for the compounds with multiple nitrate groups, likely cause disagreement when assessing the changes in the absolute bulk volatility. It is also worth noting that the particle size dependence of light absorption should be considered when studying the changes in the bulk volatility for systems with broad size distributions.
Our systems represent conditions in which RO2–RO2 and RO2–NO3 reactions are favoured and ON oligomers contribute to the high SOA yields we observed. Different changes in particle mass/size due to photolysis could be expected for the BSOA formed under different conditions, as they have different chemical composition and, thus, different amounts and/or types of chromophores. In addition, the differences among the three BVOC systems also indicate that results obtained for specific BVOCs cannot be directly generalized to SOA present in the atmosphere, and it is necessary to study different BVOC SOA systems.
To our knowledge, the photolysis of SOA has mainly been studied under low-NOx conditions to date. The sensitivity of ONs to UV light is poorly understood. As ONs are important in high-NOx environments and make substantial contributions to the total organic aerosols (Farmer et al., 2010; Lee et al., 2016), future studies that probe the photolytic ageing of ONs under different ambient conditions and for different SOA precursors are needed to improve our understanding of the life cycle of ONs.
The datasets are available upon request from the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-14907-2021-supplement.
CW, DMB, and CM designed the study. Chamber experiments were carried out by CW, DMB, ELG, SH, AB, SG, and CM. Data analysis and interpretation were performed by CW, DMB, ELG, IR, JS, and CM. CW wrote the manuscript, with input from all co-authors. All co-authors read and commented on the manuscript.
Some authors are members of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors have also no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
We thank Taina Yli-Juuti for helpful discussion on photolytically induced changes in BSOA volatility and Joel A. Thornton for his inspiring discussion and comments on this study.
This work was supported by the Swiss National Science Foundation (grant nos. 200020_172602 and 200021_169787), the European Union's Horizon 2020 Research and Innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997, the Knut and Alice Wallenberg Foundation (WAF project20 CLOUDFORM, grant no. 2017.0165), and the European Research Council H2020 programme (CHAPAs, grant no. 850614).
The article processing charges for this open-access publication were covered by Stockholm University.
This paper was edited by Jason Surratt and reviewed by two anonymous referees.
Bannan, T. J., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Mehra, A., Hammes, J., Hallquist, M., Alfarra, M. R., Krieger, U. K., Reid, J. P., Jayne, J., Robinson, W., McFiggans, G., Coe, H., Percival, C. J., and Topping, D.: A method for extracting calibrated volatility information from the FIGAERO-HR-ToF-CIMS and its experimental application, Atmos. Meas. Tech., 12, 1429–1439, https://doi.org/10.5194/amt-12-1429-2019, 2019.
Barnes, I., Becker, K. H., and Zhu, T.: Near UV absorption spectra and photolysis products of difunctional organic nitrates: Possible importance as NOx reservoirs, J. Atmos. Chem., 17, 353–373, https://doi.org/10.1007/BF00696854, 1993.
Bell, D. M., Wu, C., Bertrand, A., Graham, E., Schoonbaert, J., Giannoukos, S., Baltensperger, U., Prevot, A. S. H., Riipinen, I., El Haddad, I., and Mohr, C.: Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-379, in review, 2021.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
Daellenbach, K. R., Uzu, G., Jiang, J., Cassagnes, L.-E., Leni, Z., Vlachou, A., Stefenelli, G., Canonaco, F., Weber, S., Segers, A., Kuenen, J. J. P., Schaap, M., Favez, O., Albinet, A., Aksoyoglu, S., Dommen, J., Baltensperger, U., Geiser, M., El Haddad, I., Jaffrezo, J.-L., and Prévôt, A. S. H.: Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 587, 414–419, https://doi.org/10.1038/s41586-020-2902-8, 2020.
Daumit, K. E., Kessler, S. H., and Kroll, J. H.: Average chemical properties and potential formation pathways of highly oxidized organic aerosol, Faraday Discuss., 165, 181–202, https://doi.org/10.1039/C3FD00045A, 2013.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A Semiempirical Correlation between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Epstein, S. A., Blair, S. L., and Nizkorodov, S. A.: Direct Photolysis of α-Pinene Ozonolysis Secondary Organic Aerosol: Effect on Particle Mass and Peroxide Content, Environ. Sci. Technol., 48, 11251–11258, https://doi.org/10.1021/es502350u, 2014.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Goracci, L., Tortorella, S., Tiberi, P., Pellegrino, R. M., Di Veroli, A., Valeri, A., and Cruciani, G.: Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics, Anal. Chem., 89, 6257–6264, https://doi.org/10.1021/acs.analchem.7b01259, 2017.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., and McKay, W.: A global model of natural volatile organic compound emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, 1995.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
He, Q., Tomaz, S., Li, C., Zhu, M., Meidan, D., Riva, M., Laskin, A., Brown, S. S., George, C., Wang, X., and Rudich, Y.: Optical Properties of Secondary Organic Aerosol Produced by Nitrate Radical Oxidation of Biogenic Volatile Organic Compounds, Environ. Sci. Technol., 55, 2878–2889, https://doi.org/10.1021/acs.est.0c06838, 2021.
Henry, K. M. and Donahue, N. M.: Photochemical Aging of α-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 116, 5932–5940, https://doi.org/10.1021/jp210288s, 2012.
Hoyle, C. R., Berntsen, T., Myhre, G., and Isaksen, I. S. A.: Secondary organic aerosol in the global aerosol – chemical transport model Oslo CTM2, Atmos. Chem. Phys., 7, 5675–5694, https://doi.org/10.5194/acp-7-5675-2007, 2007.
Huang, W., Saathoff, H., Pajunoja, A., Shen, X., Naumann, K.-H., Wagner, R., Virtanen, A., Leisner, T., and Mohr, C.: α-Pinene secondary organic aerosol at low temperature: chemical composition and implications for particle viscosity, Atmos. Chem. Phys., 18, 2883–2898, https://doi.org/10.5194/acp-18-2883-2018, 2018.
Isaacman-VanWertz, G. and Aumont, B.: Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 21, 6541–6563, https://doi.org/10.5194/acp-21-6541-2021, 2021.
James, P. F., Perugini, M. A., and O'Hair, R. A. J.: Sources of artefacts in the electrospray ionization mass spectra of saturated diacylglycerophosphocholines: From condensed phase hydrolysis reactions through to gas phase intercluster reactions, J. Am. Soc. Mass Spectrom., 17, 384–394, https://doi.org/10.1021/jasms.8b02630, 2006.
Jaoui, M., Kleindienst, T. E., Docherty, K. S., Lewandowski, M., and Offenberg, J. H.: Secondary organic aerosol formation from the oxidation of a series of sesquiterpenes: α-cedrene, β-caryophyllene, α-humulene and α-farnesene with O3, OH and NO3 radicals, Environ. Chem., 10, 178–193, https://doi.org/10.1071/EN13025, 2013.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Keith-Roach, M. J.: A review of recent trends in electrospray ionisation–mass spectrometry for the analysis of metal–organic ligand complexes, Anal. Chim. Acta, 678, 140–148, https://doi.org/10.1016/j.aca.2010.08.023, 2010.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kourtchev, I., Szeto, P., O'Connor, I., Popoola, O. A. M., Maenhaut, W., Wenger, J., and Kalberer, M.: Comparison of Heated Electrospray Ionization and Nanoelectrospray Ionization Sources Coupled to Ultra-High-Resolution Mass Spectrometry for Analysis of Highly Complex Atmospheric Aerosol Samples, Anal. Chem., 92, 8396–8403, https://doi.org/10.1021/acs.analchem.0c00971, 2020.
Krapf, M., El Haddad, I., Bruns, Emily A., Molteni, U., Daellenbach, Kaspar R., Prévôt, André S. H., Baltensperger, U., and Dommen, J.: Labile Peroxides in Secondary Organic Aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Liu, X., Day, D. A., Krechmer, J. E., Brown, W., Peng, Z., Ziemann, P. J., and Jimenez, J. L.: Direct measurements of semi-volatile organic compound dynamics show near-unity mass accommodation coefficients for diverse aerosols, Commun. Chem., 2, 98, https://doi.org/10.1038/s42004-019-0200-x, 2019.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Pospisilova, V., Huang, W., Kalberer, M., Mohr, C., Stefenelli, G., Thornton, J. A., Baltensperger, U., Prevot, A. S. H., and Slowik, J. G.: An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 12, 4867–4886, https://doi.org/10.5194/amt-12-4867-2019, 2019.
Maire, F. and Lange, C. M.: Formation of unexpected ions from a first-generation polyamidoamine dendrimer by use of methanol: an artefact due to electrospray emitter corrosion?, Rapid Commun. Mass Spectrom., 24, 995–1000, https://doi.org/10.1002/rcm.4475, 2010.
Mang, S. A., Henricksen, D. K., Bateman, A. P., Andersen, M. P. S., Blake, D. R., and Nizkorodov, S. A.: Contribution of Carbonyl Photochemistry to Aging of Atmospheric Secondary Organic Aerosol, J. Phys. Chem. A, 112, 8337–8344, https://doi.org/10.1021/jp804376c, 2008.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petäjä, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 4442, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Nakayama, T., Sato, K., Tsuge, M., Imamura, T., and Matsumi, Y.: Complex refractive index of secondary organic aerosol generated from isoprene/NOx photooxidation in the presence and absence of SO2, J. Geophys. Res.-Atmos., 120, 7777–7787, https://doi.org/10.1002/2015JD023522, 2015.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
O'Brien, R. E. and Kroll, J. H.: Photolytic Aging of Secondary Organic Aerosol: Evidence for a Substantial Photo-Recalcitrant Fraction, J. Phys. Chem. Lett., 10, 4003–4009, https://doi.org/10.1021/acs.jpclett.9b01417, 2019.
O'Meara, S., Booth, A. M., Barley, M. H., Topping, D., and McFiggans, G.: An assessment of vapour pressure estimation methods, Phys. Chem. Chem. Phys., 16, 19453–19469, https://doi.org/10.1039/C4CP00857J, 2014.
Pan, X., Underwood, J. S., Xing, J.-H., Mang, S. A., and Nizkorodov, S. A.: Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry, Atmos. Chem. Phys., 9, 3851–3865, https://doi.org/10.5194/acp-9-3851-2009, 2009.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Peng, C., Wang, W., Li, K., Li, J., Zhou, L., Wang, L., and Ge, M.: The Optical Properties of Limonene Secondary Organic Aerosols: The Role of NO3, OH, and O3 in the Oxidation Processes, J. Geophys. Res.-.Atmos., 123, 3292–3303, https://doi.org/10.1002/2017JD028090, 2018.
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Science Advances, 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922, 2020.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary Organic Aerosol Production from Terpene Ozonolysis. 1. Effect of UV Radiation, Environ. Sci. Technol., 39, 7036–7045, https://doi.org/10.1021/es050174m, 2005.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Riipinen, I., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Equilibration time scales of organic aerosol inside thermodenuders: Evaporation kinetics versus thermodynamics, Atmos. Environ., 44, 597–607, https://doi.org/10.1016/j.atmosenv.2009.11.022, 2010.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Rovelli, G., Jacobs, M. I., Willis, M. D., Rapf, R. J., Prophet, A. M., and Wilson, K. R.: A critical analysis of electrospray techniques for the determination of accelerated rates and mechanisms of chemical reactions in droplets, Chem. Sci., 11, 13026–13043, https://doi.org/10.1039/D0SC04611F, 2020.
Schobesberger, S., D'Ambro, E. L., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: A model framework to retrieve thermodynamic and kinetic properties of organic aerosol from composition-resolved thermal desorption measurements, Atmos. Chem. Phys., 18, 14757–14785, https://doi.org/10.5194/acp-18-14757-2018, 2018.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Suarez-Bertoa, R., Picquet-Varrault, B., Tamas, W., Pangui, E., and Doussin, J. F.: Atmospheric Fate of a Series of Carbonyl Nitrates: Photolysis Frequencies and OH-Oxidation Rate Constants, Environ. Sci. Technol., 46, 12502–12509, https://doi.org/10.1021/es302613x, 2012.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Thornton, J. A., Mohr, C., Schobesberger, S., D'Ambro, E. L., Lee, B. H., and Lopez-Hilfiker, F. D.: Evaluating Organic Aerosol Sources and Evolution with a Combined Molecular Composition and Volatility Framework Using the Filter Inlet for Gases and Aerosols (FIGAERO), Accounts Chem. Res., 53, 1415–1426, https://doi.org/10.1021/acs.accounts.0c00259, 2020.
Tritscher, T., Dommen, J., DeCarlo, P. F., Gysel, M., Barmet, P. B., Praplan, A. P., Weingartner, E., Prévôt, A. S. H., Riipinen, I., Donahue, N. M., and Baltensperger, U.: Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber, Atmos. Chem. Phys., 11, 11477–11496, https://doi.org/10.5194/acp-11-11477-2011, 2011.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vaden, T. D., Imre, D., Beránek, J., Shrivastava, M., and Zelenyuk, A.: Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol, P. Natl. Acad. Sci. USA, 108, 2190–2195, https://doi.org/10.1073/pnas.1013391108, 2011.
Walser, M. L., Park, J., Gomez, A. L., Russell, A. R., and Nizkorodov, S. A.: Photochemical Aging of Secondary Organic Aerosol Particles Generated from the Oxidation of d-Limonene, J. Phys. Chem. A, 111, 1907–1913, https://doi.org/10.1021/jp066293l, 2007.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wong, J. P. S., Zhou, S., and Abbatt, J. P. D.: Changes in Secondary Organic Aerosol Composition and Mass due to Photolysis: Relative Humidity Dependence, J. Phys. Chem. A, 119, 4309–4316, https://doi.org/10.1021/jp506898c, 2015.
Wu, R., Vereecken, L., Tsiligiannis, E., Kang, S., Albrecht, S. R., Hantschke, L., Zhao, D., Novelli, A., Fuchs, H., Tillmann, R., Hohaus, T., Carlsson, P. T. M., Shenolikar, J., Bernard, F., Crowley, J. N., Fry, J. L., Brownwood, B., Thornton, J. A., Brown, S. S., Kiendler-Scharr, A., Wahner, A., Hallquist, M., and Mentel, T. F.: Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical, Atmos. Chem. Phys., 21, 10799–10824, https://doi.org/10.5194/acp-21-10799-2021, 2021.
Zare, A., Romer, P. S., Nguyen, T., Keutsch, F. N., Skog, K., and Cohen, R. C.: A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates, Atmos. Chem. Phys., 18, 15419–15436, https://doi.org/10.5194/acp-18-15419-2018, 2018.
Zawadowicz, M. A., Lee, B. H., Shrivastava, M., Zelenyuk, A., Zaveri, R. A., Flynn, C., Thornton, J. A., and Shilling, J. E.: Photolysis Controls Atmospheric Budgets of Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 54, 3861–3870, https://doi.org/10.1021/acs.est.9b07051, 2020.
Zhao, D., Pullinen, I., Fuchs, H., Schrade, S., Wu, R., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Guo, Y., Kiendler-Scharr, A., Wahner, A., Kang, S., Vereecken, L., and Mentel, T. F.: Highly oxygenated organic molecule (HOM) formation in the isoprene oxidation by NO3 radical, Atmos. Chem. Phys., 21, 9681–9704, https://doi.org/10.5194/acp-21-9681-2021, 2021.
Zhao, Z., Tolentino, R., Lee, J., Vuong, A., Yang, X., and Zhang, H.: Interfacial Dimerization by Organic Radical Reactions during Heterogeneous Oxidative Aging of Oxygenated Organic Aerosols, J. Phys. Chem. A, 123, 10782–10792, https://doi.org/10.1021/acs.jpca.9b10779, 2019.