the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Oct 2021
| 05 Oct 2021
Reduced volatility of aerosols from surface emissions to the top of the planetary boundary layer
Yangzhou Wu
Kai Bi
Wenkang Gao
Ping Tian
Delong Zhao
Siyuan Li
Chenjie Yu
Guiqian Tang
Yunfei Wu
Kang Hu
Shuo Ding
Qian Gao
Fei Wang
Shaofei Kong
Hui He
Mengyu Huang
Deping Ding
Aerosols from surface emission can be transported upwards through convective mixing in the planetary boundary layer (PBL), which subsequently interact with clouds, serving as important sources to nucleate droplets or ice particles. However, the evolution of aerosol composition during this vertical transport has yet to be explicitly understood. In this study, simultaneous measurements of detailed aerosol compositions were conducted at two sites, namely urban Beijing (50 m above sea level – a.s.l.) and Haituo mountain (1344 m a.s.l.) during wintertime, representing the anthropogenically polluted surface environment and the top of the PBL, respectively. The pollutants from surface emissions were observed to reach the mountain site on daily basis through daytime PBL convective mixing. From the surface to the top of PBL, we found efficient transport or formation of lower-volatility species (black carbon, sulfate, and low-volatile organic aerosol, OA); however, a notable reduction in semivolatile substances, such as the fractions of nitrate and semivolatile OA reduced by 74 % and 76 %, respectively, during the upward transport. This implies that the mass loss of these semivolatile species was driven by the evaporation process, which repartitioned the condensed semivolatile substances to the gas phase when aerosols were transported and exposed to a cleaner environment. In combination with the oxidation processes, these led to an enhanced oxidation state of OA at the top of the PBL compared to surface environment, with an increase of oxygen to carbon atomic ratio by 0.2. Such a reduction in aerosol volatility during vertical transport may be important in modifying its viscosity, nucleation activity, and atmospheric lifetime.
- Article
(3441 KB) - Full-text XML
-
Supplement
(1696 KB) - BibTeX
- EndNote




Substances in the atmosphere, present as aerosol and gas phases, are subject to phase transformation during their lifetime (Pankow, 1994, 1987). Gas-to-particle partition processes thermodynamically determine the production of secondary aerosol mass and the constituents of gases through the condensation or evaporation process. The condensation process leads to gas molecular partitioning in the condensed phase, while the evaporation process occurs when aerosols were diluted in an environment with lower concentration (Donahue et al., 2006). Hereby, their physiochemical properties could be modified, such as the condensation results in enlarging aerosol size (Riipinen et al., 2011, 2012) or the production of new particles (Zhang et al., 2004; Kulmala et al., 2013); the evaporation led to a loss of particulate mass (May et al., 2015; Cubison et al., 2011). These factors have important impacts on altering the radiative interactions of aerosols by changing the mass of aerosols and, thus, direct radiative impacts (Tsigaridis et al., 2014; Wang et al., 2014) or by changing the number concentration and the ability of cloud condensation nuclei for the indirect radiative impacts (D'Andrea et al., 2013; Kuang et al., 2009). The repartitioned gases from aerosols during dilution could experience chemical evolution and further contribute to the modification of aerosol properties (Zhang et al., 2007; Robinson et al., 2007).
In regions with intense anthropogenic activities, such as megacities, nitrate and sulfate are dominant inorganic aerosol chemical components due to intensive emissions of gaseous precursors (anthropogenic NOx and SO2), with secondary formation through photochemical and heterogenous/aqueous reactions (Zhang et al., 2015; Huang et al., 2014; Guo et al., 2014; Sun et al., 2016). Although the formation mechanisms of secondary inorganic aerosol are relatively well understood, low-volatile organic aerosol (OA) processing in atmospheric transport process remains poorly characterized (Shrivastava et al., 2017). Organic compounds are ubiquitous in ambient aerosol, with a large contribution (20 %–90 %) to submicron aerosol mass loadings, including primary OA (POA) and secondary OA (SOA; Zhang et al., 2007). During the atmospheric aging processes, OA physiochemical properties, such as volatility, hygroscopicity, viscosity, and oxidation state, have been significantly transformed. For example, semivolatile organic materials are repartitioned to the gas phase when ambient vapor partial pressure is lower than that in particle phase, and then those vapors can be further photo-oxidized and produce SOA with lower volatility (Robinson et al., 2007). In addition, SOA tends to dominate the OA mass along with atmospheric aging processes, which modulate OA properties to be more oxidized and hygroscopic (Jimenez et al., 2009).
The vertical transport of aerosol and gases in the planetary boundary layer (PBL) from the surface to the lower free troposphere (FT) importantly determines the influence of anthropogenic emissions to the upper level of the atmosphere; for example, aerosols in the upper level of the PBL may have more important feedback effects in influencing the boundary layer dynamics (Li et al., 2017). The aerosol could be upward transported from surface to upper level through daytime convective mixing in the PBL (Garratt, 1994). Previous studies have used aircraft (Liu et al., 2020b; Zhao et al., 2019) or balloon platforms (Ran et al., 2016; Li et al., 2015) to measure the vertical profiles of PBL in situ; however, these lack sufficient temporal coverage or full chemical components. The evolution of aerosol properties during this vertical transport in the PBL on a daily basis has not been fully understood. In this study, simultaneous and continuous measurements of detailed aerosol compositions were performed at both surface and surface-influenced mountain sites using advanced instrumentation, which provides an opportunity to realize the high time resolution variations at different altitudes. The relative location of the mountain site with respect to the top of the PBL varies with the diurnal variation of PBL height (PBLH), which leaves the mountain site in the free troposphere most of the day and allows it to be influenced by PBL air masses around midday. Through comparing the difference in aerosol chemical compositions between the two sites, we aim to investigate the modification of compositions during the upward transport in the PBL and explore the generic mechanisms in driving the evolution of chemical composition.
2.1 Experimental sites
Simultaneous measurements of detailed aerosol compositions (organics, nitrate, sulfate, ammonium, chloride, and black carbon) and gaseous pollutants (NOx, SO2, CO, and O3) were conducted during 6 and 22 January 2019 at both the surface and surface-influenced mountain sites, representing the anthropogenically polluted surface urban environment and the top of the PBL, respectively. All aerosol measurements were performed downstream of a PM2.5 impactor (BGI SCC1.829) at both sites and dried by Nafion tubing before splitting to the sample inlets of instruments.

Figure 1Experimental overview. (a) Spatial distribution of mean aerosol optical depth for the experimental month, with locations of surface and mountain sites, colored by terrain height. (b) A typical example of westerly dominated air mass from the Numerical Atmospheric–dispersion Modelling Environment (NAME) dispersion model outputs. (c) Diurnal cycles of the planetary boundary layer height (PBLH) and local air mass fraction contributing to the mountain site during the experimental period, with whiskers showing the 25th and 75th percentiles, and the dashed line denoting the altitude of the mountain site. (d) Time series of air mass fractions at the mountain and PBLH. (e–h) Time series of black carbon (BC), nitrate (NO3), sulfate (SO4), and organics (Org), with the black and red lines showing the surface and mountain site, respectively.
Figure 1a shows the locations of surface and mountain sites and the spatial distribution of mean aerosol optical depth during the observation period. The surface site locates in the Institute of Atmospheric Physics (IAP), Chinese Academy of Science (39.97∘ N, 116.37∘ E; 50 m above sea level – a.s.l.), which represents the urban environment in Beijing influenced by intense surface anthropogenic emissions, such as local traffic and cooking emissions (Sun et al., 2016; Zhang et al., 2014). The mountain site (Haituo Mountain; 40.52∘ N, 115.78∘ E, 1344 m a.s.l.) is located in the northwestern Beijing area, with a straight line distance of ∼ 85 km from downtown Beijing. The Haituo Mountain belongs to the Taihang Mountains and connects to the continental plateau extending to the west. The surroundings of this site are covered with broadleaf forest and are without distinct anthropogenic emissions, except for a few villages at the foot of the mountain. Thus, it can be considered an ideal receptor site for regional transport influences and/or local influences by vertical transport under certain conditions.
2.2 Instrumentation and data analysis
Non-refractory aerosol chemical components, including nitrate (NO3), sulfate (SO4), ammonium (NH4), chloride (Chl), and organics (Org), were measured by the high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Research, Inc.) at each site. The applied collection efficiency (CE) for the AMS measurement at the surface and mountain site is shown in Fig. S1 in the Supplement, following the principle by Middlebrook et al. (2012). Ionization efficiency (IE) calibrations were performed by using size-selected (300 nm) ammonium nitrate particles, and default relative IE (RIE) values were used for Org (1.4), NO3 (1.1), SO4 (1.2), NH4 (4.0), and Chl (1.3). The IE calibration was conducted before and after the experiment at both sites.
Standard AMS data analysis software packages (SQUIRREL 1.59D and PIKA 1.19D) were used to deconvolve the mass spectrum and obtain mass concentrations of chemical components. Then, high-resolution mass spectra of OA for 12–150 at both sites were analyzed by using positive matrix factorization (PMF) with the PMF2 algorithm (Paatero and Tapper, 1994), following the data processing and factor selecting steps given by Ulbrich et al. (2009) and Zhang et al. (2011). As a result, five factors were resolved from the surface OA, including three primary OA (POA), that is, hydrocarbon-like OA (HOA) from traffic emissions, cooking-related OA (COA), and coal combustion OA (CCOA), and two secondary OA (SOA), that is, semivolatile oxidized OA (SV-OOA) and low-volatility oxygenated OA (LV-OOA). Note that the COA factor was not resolved from the mountain OA, which has been frequently reported in urban environments (Zhang et al., 2011). The other four analogous factors were also resolved from the mountain OA. Detailed diagnostic plots of the PMF results are presented in Figs. S2 and S3, while the PMF-derived mass spectra and time series of organic components at both sites are shown in Figs. S4 and S5. Elemental ratios, including oxygen to carbon () and hydrogen to carbon (), were determined from analysis of high-resolution mass spectra of OA based on the improved-ambient (I-A) method (Canagaratna et al., 2015), while the elemental ratios were also calculated using the Aiken-ambient (A-A) method (Aiken et al., 2008) for comparison with previous studies. The elemental ratios reported in this study are obtained from the I-A method, unless otherwise stated.
Black carbon (BC) mass was measured by a single particle soot photometer (SP2; DMT Inc.), following the calibration and data analysis processes by Liu et al. (2020). Gaseous pollutants (i.e., NOx, SO2, CO, and O3) were measured by gas analyzers (models 42i, 43i, 48i, and 49i; Thermo Fisher Scientific Inc.). The PBLH was determined by an enhanced single-lens lidar ceilometer (CL51; Vaisala), using the retrieval method by Tang et al. (2015). Meteorology variables (RH and temperature) were measured using WXT510 (Vaisala).
2.3 Air mass history
The three-dimensional air mass histories were calculated by the Numerical Atmospheric-dispersion Modeling Environment (NAME; Jones et al., 2007), which is a Lagrangian dispersion model following 3D trajectories of plume parcels by Monte Carlo methods. The meteorological data source uses the global configuration of UK Met Office's Unified Model. In order to calculate the historical air mass contribution, the model releases tracer particles at a nominal rate of 1 g s−1, with a maximum travel time of 24 h in a backward mode from the target site (i.e., the Haituo Mountain site in this study), and the integrated time was recorded on a horizontal grid from 0 to 1000 m above the ground, aggregating over all particles for a given release period. In this study, the potential source contribution of particles to the mountain site (back to 24 h) from air masses are classified into the following four main regions (Fig. 1b): local (39–41.5∘ N, 115–117∘ E; a square region around central Beijing covering both measurement sites), west (32–41.5∘ N, 104–115∘ E), north (41.5–45∘ N, 104–121∘ E), and South (32–39 ∘ N, 115–121∘ E). Figure 1b presents a typical example of westerly dominated air mass from NAME outputs. The particle flux is integrated in each segregated region, and contributions of each air mass fractions could be obtained.
3.1 Upward transport of aerosols around midday
We aim to classify the source influences on the mountain site from the local surface emission or wider regional area. Air mass history analysis showed a pronounced diurnal pattern of local air mass influence (as determined by the NAME dispersion model), peaking around midday (11:00–14:00 local time). The local air mass faction, as calculated from the dispersion model, is only used to indicate the predominant local air mass influence in the midday. The aerosol concentration contained in the air mass depends on the transport efficiency, reaction, and deposition rate of each aerosol type. The fraction of transported aerosols, even for the inert BC may not be quantitatively comparable with the air mass fraction. The maximum local influence was consistent with the most developed PBLH (Fig. 1c). This suggested the strongest influence of surface emissions to the mountain through midday convective mixing (CM; termed, hereafter, as the CM period). For a certain period (9–12 January), westerly air mass continuously influenced the mountain site (gray bar in Fig. 1d–h), which advected regional pollutants from the polluted high plateau, adding on the persistent local emission (termed the regional advection, RA, period). Note that the RA period was also influenced by the convective mixing of surface sources around midday but was combined with additional sources from other regions besides the surface emission. In this study, the statistical results of the RA period include the whole period marked with the gray bar in Fig. 1d–h, and the rest period is used for the statistics of the CM period.
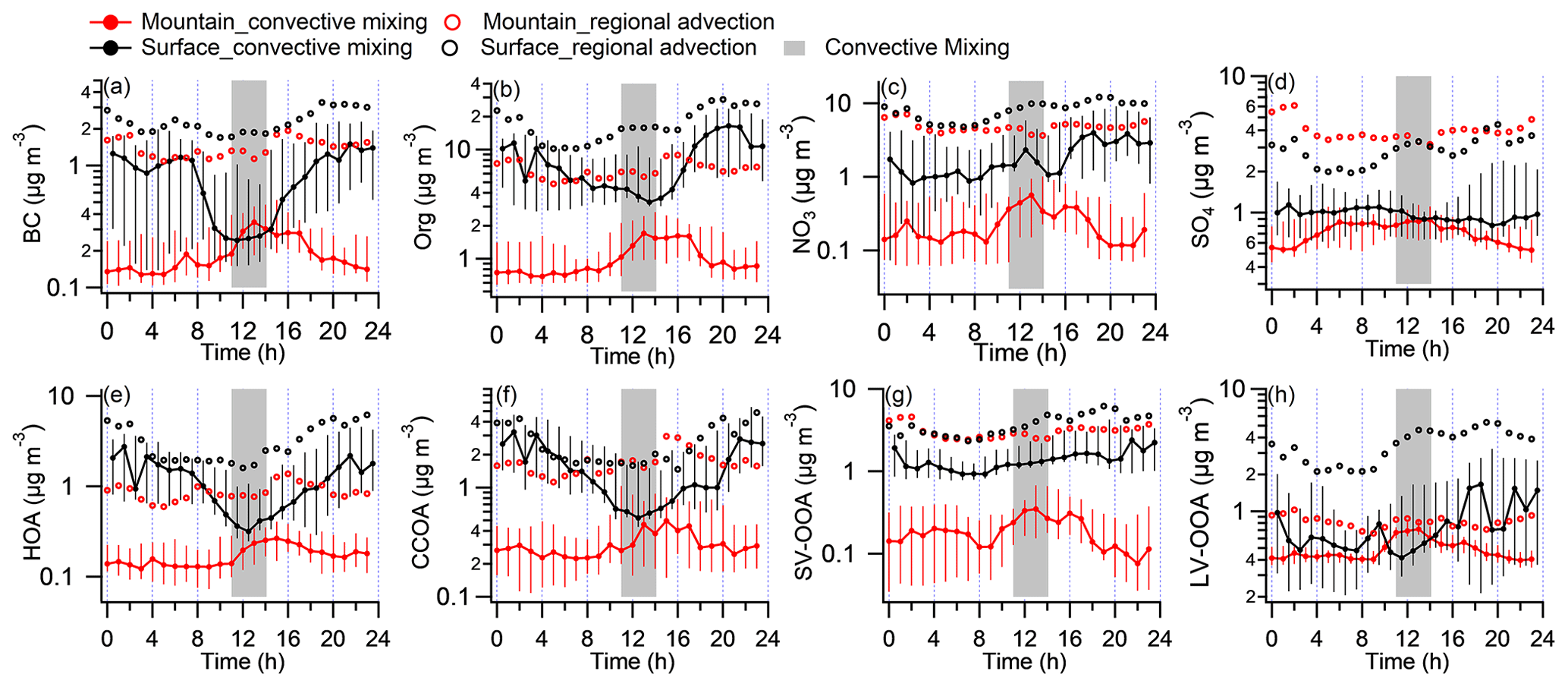
Figure 2Diurnal variations in key aerosol compositions at both sites during convective mixing (CM) and regional advection (RA) periods. (a) Black carbon (BC). (b) Organics (Org). (c) Nitrate (NO3). (d) Sulfate (SO4). (e) The sum of hydrocarbon OA (HOA) and cooking-related OA (COA). (f) Coal-combustion-related OA (CCOA). (g) Semivolatile oxygenated OA (SV-OOA). (h) Low-volatile oxygenated OA (LV-OOA). The black and red colors represent the surface and mountain site, respectively. Solid circles and error bars show the median, 75th, and 25th percentiles in the CM period. Circle markers show the mean value in RA period. The gray bar represents the time of the day (11:00–14:00) with the most developed PBL.
Figure 2 gives the statistical diurnal variations in aerosol species during CM and RA periods, respectively, with species of BC, OA, nitrate, and sulfate shown in Fig. 2a–d and PMF-derived organic components in Fig. 2e–h. During the CM period, the chemically inert species, such as BC, showed a clear diurnal pattern on the mountain, with concentrations elevated by 82 % (from 0.19 to 0.34 µg m−3) from midday to early afternoon (Fig. 2a). This pattern was highly consistent with the development of the PBLH and local air mass contribution. On the surface, the diurnal variation in BC concentration showed a minimum at the same hours, due to the dilution effect of developed PBL, but had a sharp enhancement during nighttime due to the accumulation in the shallow nighttime PBL. Notably, BC concentration at 11:00–14:00 on the mountain almost matched that on the surface, suggesting a well-mixed layer because of the daytime convective mixing. The inert gas CO was also efficiently transported without significant loss from the surface to the mountain (Fig. S6c). This means the pollutants, if without reactions, evaporation or other forms of losses, were able to be efficiently transported upwards from the surface to the mountain site through the daytime convective mixing.
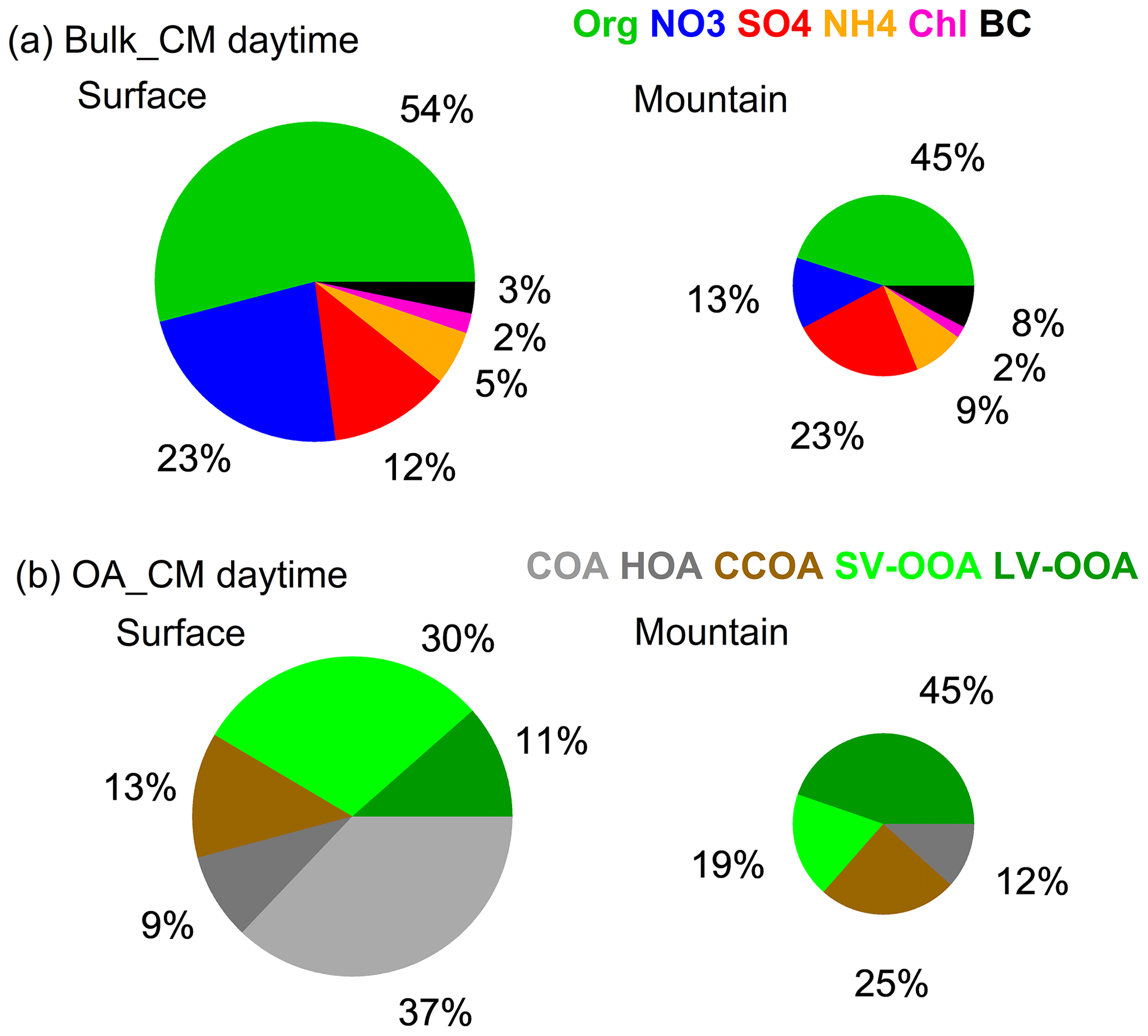
Figure 3Averaged chemical composition at both sites during the convective mixing (CM) daytime period (11:00–14:00). (a) Bulk aerosols. (b) Organic aerosols.
3.2 Loss of semivolatile particulate mass from surface to mountain
Aerosol chemical compositions showed remarkable differences between both sites, even when the PBL was fully developed (Fig. 3), i.e., nitrate (23 %) and organics (54 %) dominated at surface, while sulfate (23 %) and organics (45 %) dominated at the top of PBL. Meanwhile, the characteristics of OA varied from POA dominated (59 %) to SOA dominated (64 %). Statistical analysis in Fig. 4 highlights the difference between both sites during the periods of CM midday (11:00–14:00), CM night (23:00–02:00), and RA, where CM midday represents the period with the most efficient convective mixing. The matched concentrations of BC and CO between surface and mountain demonstrated the capability of the boundary layer in transporting pollutants upwards in terms of atmospheric dynamics. For other species, such as organics and nitrate, there was still an enhancement on the mountain peaking around midday; however, the loadings were 61 % and 74 % lower than the surface at the same hours (Figs. 2b, c, 4a), which contrasted with the efficient transport of BC. The low-volatile species of sulfate at both sites showed no apparent (or only a broad) diurnal pattern and matched the concentration at midday, consistent with its gas precursor SO2 (Fig. S6b), indicating their regional feature and the aerosol production was not sufficiently rapid to display notable diurnal variation.
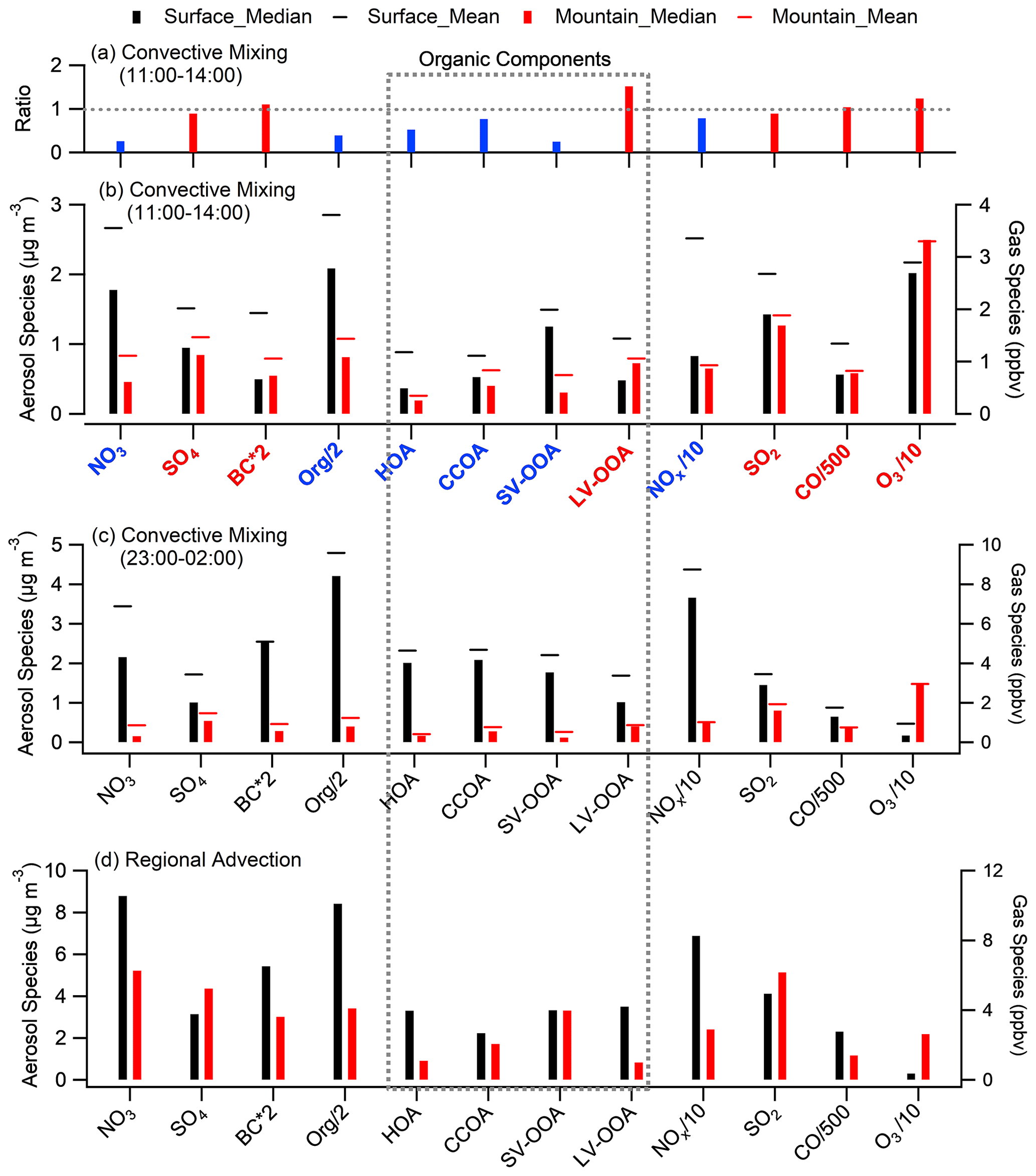
Figure 4Statistical analysis of chemical components at both sites. (a) The mountain over surface ratio of key species during convective mixing (CM) daytime period (11:00–14:00). (b) Median concentrations of key species during CM daytime, with black and red bars denoting the surface and mountain, respectively, and the cap showing the mean. (c–d) The same as panel (b) but for the periods of CM night and the RA period. Species with a mountain/surface ratio above and below 0.8 (the transport efficiency for BC and CO) are marked in red and blue, respectively.
The diurnal patterns of all PMF-resolved organic components on the mountain presented a midday peak feature at different amplitude (Fig. 2e–h), suggesting there was no additional source around the site, and the upward transport processes for these components was affected by various factors. Previous studies found that POA (e.g., HOA and CCOA) has substantial semivolatile materials and presents relatively high volatility (Cao et al., 2018). The comparison between the two sites showed significantly decreased semivolatile species (i.e., HOA, CCOA, and SV-OOA); their concentrations on the mountain were significantly lower than that at surface by 48 %, 24 %, and 76 % in the CM midday, respectively (Fig. 4a–b). The LV-OOA is a typical SOA which predominantly exists in aged air mass (Zhang et al., 2011). In contrast with semivolatile species, the low-volatile LV-OOA on the mountain showed a poorly defined diurnal pattern, and its concentrations in the midday were higher than that at surface by 52 %, which may be partially caused by further oxidation of relative fresh species in vertical transport process.
The results above demonstrated that, during vertical transport, the loss of particulate masses only occurred for semivolatile substances (nitrate, POA, and SV-OOA) but not for low-volatile species (BC, LV-OOA, and sulfate) or inert gas (CO). Meanwhile, these losses had occurred in a relatively dry condition without notable wet scavenging (Fig. S7b). Due to few anthropogenic emissions on the mountain, the concentrations of gaseous precursors, such as ammonia, nitric acid vapor, etc., should be significantly lower than that in urban environment. This suggested that the evaporation process may have played an important role in repartitioning the condensed phase rich in semivolatile species to the gas phase, which occurred when the activity of semivolatile species in the condensed phase (molar fraction multiplied by activity coefficient) was higher than the partial vapor pressure (relative to equilibrium vapor pressure of pure substance under certain temperature; Pankow, 1994, 1987). Given the winter time period of the experiment, the mean temperature shifted from −5.5 to 3.8 ∘C from surface to mountain in the CM midday (Fig. S7a), which was already sufficiently low for even the most volatile species (i.e., ammonium nitrate) to be in the condensed phase (Salo et al., 2011) at both sites. Therefore, the temperature decrease within this low range had not driven an appreciable condensation process, but the reduction in ambient concentrations at the mountain (given no additional sources but only contributed by surface emission) was the main reason leading to the evaporation. A previous study based on aircraft measurements in this region showed that aerosol chemical composition had a significant variation from the surface to the top of the PBL under high RH conditions (surface RH < 60 %), which caused by secondary formation through the enhanced aqueous/heterogeneous processes in vertical direction (Liu et al., 2020). However, the RH was quite low at both sites (most of time, RH > 40 %; Fig. S7b) and below the deliquescence RH for most substances (Cruz and Pandis, 2000); water vapor may, thus, have not played an important role in the phase transformation or chemical reactions during vertical transport. In addition, the bulk equilibrium between the gas and condensed phases may be significantly hindered under conditions of lower temperature and lower RH due to the kinetically limited diffusion rate at the aerosol surface (Koop et al., 2011). Therefore, the viscosity of aerosols may be enhanced, and OA may be present as semisolid or glassy state under these conditions. This means the evaporation process may eventually be depressed when aerosols are transported from the lower and moister boundary layer to the upper level with lower temperature and moisture; thus, aerosols may be more solid-like and resistant to the evaporation. This evaporation-dominated variation on aerosol composition from the surface to the top of the PBL tends to only occur under cold and dry conditions.
This evaporation process tended to occur along the path of vertical transport, with a higher loss rate when there is a larger gradient of concentration between the condensed phase and ambient air (Donahue et al., 2006; Shrivastava et al., 2006; Robinson et al., 2007). Previous studies indicated the dilution could be particularly important for biomass burning emissions (Li et al., 2021). The high concentrations of the condensed phase could be an important contribution to the gaseous precursors and, under certain conditions, could form secondary aerosol. In addition, these low-volatile aerosols transported to the free troposphere may have a longer lifetime and be transported for a longer distance (Liu et al., 2020a). Here we only observed the resultant compositions after being transported to the top of PBL, but at which atmospheric layer this process had mostly occurred remained inconclusive. There may be some production process, e.g., photochemical oxidation around midday, which contributed to some increase in the SV-OOA concentration around midday (Fig. 2g). The overall reduced SV-OOA suggested its net loss, which was when evaporation prevailed in production processes, during upwards transport.
During CM night, pollutants (except O3) were mostly accumulated at the surface (Fig. 4c) due to local emissions and secondary formation processes occurring in a shallow nighttime PBL and the interruption of vertical transport to high layers. Note that, the diurnal variation in surface O3 showed a significant reduction at night due to rapid consumption by strong NO titration, which in contrast to a maintainable high O3 concentration on the mountain without an apparent diurnal pattern (Fig. S6d). For the RA period, due to the additional input of pollutants from a wider regional region besides the local influence, the diurnal variation at the mountain was diminished but showed enhanced concentrations at all hours (Fig. 2), indicating that a robust regional advection process also significantly affects aerosol concentration and variation at high layers. Because the surface site was more influenced by local sources, most species had higher concentrations at the surface than at the mountain, apart from sulfur compounds (SO2 and sulfate; in Fig. 4d). This implies the possible high-level sulfuric sources, e.g., emissions from industrial stack, directly emitting into a higher level in the PBL (Wu et al., 2018; Xu et al., 2014).
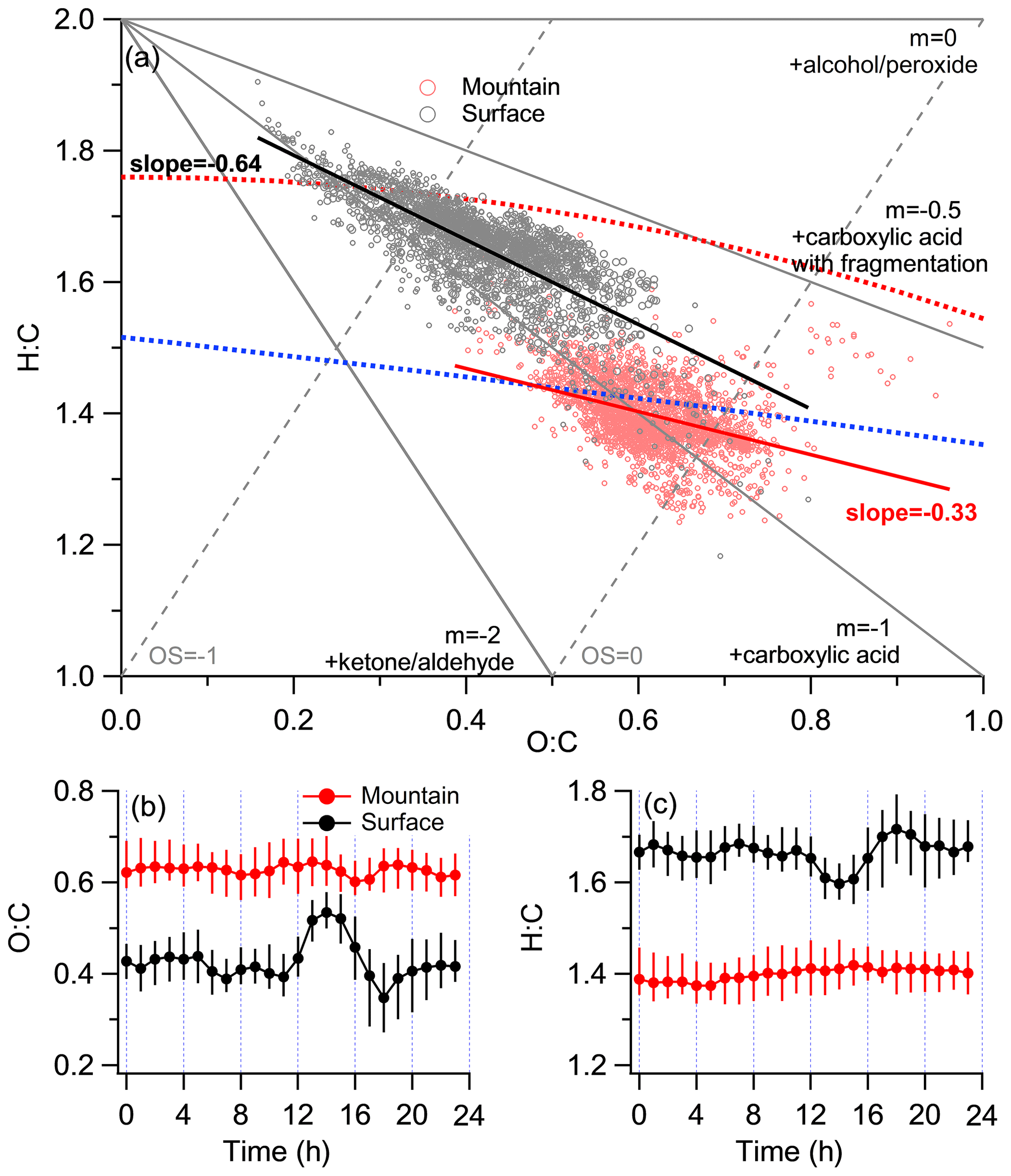
Figure 5Characterization of elemental ratios at both surface and mountain sites. (a) The standard Van Krevelen triangle diagram of H:C versus O:C. (b) Diurnal variations in O:C. (c) Diurnal variations in H:C. The gray and red circles denote the data points from the surface and mountain sites, respectively.
3.3 Modification on the oxidation state of OA
The Van Krevelen triangle (VK) diagram (Fig. 5a) could indicate the likely oxidation pathway of OA by investigating different degrees of changes on the oxygen or hydrogen over the carbon element ratio, depending on the manner of adding functional groups (Van Krevelen, 1950). For example, the replacement of a hydrogen atom with an alcohol/peroxide group (−OH and −OOH) results in a slope of 0, while the replacement of a hydrogen atom with a carboxylic acid group (−COOH) results in a slope of −1 without fragmentation (C-C bond breaking) and −0.5 with fragmentation (Ng et al., 2010). Results mapping on the H:C versus O:C atomic ratio showed discernible regimes between both sites (Fig. 5a). The surface O:C varied at 0.16–0.75, with an average of 0.42 ± 0.09 (0.33 ± 0.08 with the A-A method), generally consistent with those (0.29–0.41; A-A method) previously reported in Beijing (Xu et al., 2015; Zhang et al., 2014; Sun et al., 2016). Mountain OA showed significantly higher O:C (varied at 0.39 to 0.96, with an average of 0.62 ± 0.07) and lower H:C (1.23–1.64) than surface, similar with the regime of LV-OOA-dominated periods observed in Rocky Mountain National Park (∼ 2740 m a.s.l.; Schurman et al., 2015). Here the surface (slope = −0.64) showed a steeper slope than mountain (slope = −0.33). The surface thus tended to be dominated by the oxidation pathway of −COOH addition (without fragmentation), which showed an apparent decrease in . However, the OA at mountain tended to be oxidized in the mix with alcohol/peroxide and −COOH addition (with fragmentation) pathway, thus there was less decrease in .
Differences in the spectra for each PMF factor between the surface and mountain was analyzed to further investigate the chemical modification of OA from each source (Fig. 6). The mountain CO fraction in the spectra of all PMF factors enhanced at various extents, indicating the oxidation by adding carboxyl groups (Ng et al., 2011). For HOA and SV-OOA, a range of hydrocarbon fragments decreased at the mountain (Fig. 6a, c), which tended to be consistent with the evaporation mechanism proposed in Sect. 3.2 that semivolatile species may have been repartitioned to the gas phase when transported to the mountain, and these evaporated species may contain significant fractions of more volatile hydrocarbons (Cappa and Jimenez, 2010). Note that the diurnal variation in mountain O:C, characterized by a slight fluctuation at a high-value region, is in contrast with a distinct peak in the early afternoon that appears in the diurnal variation in surface O:C (Fig. 5b). This implied there were no additional primary emissions on the mountain to contribute either particles or volatile organic compounds (VOCs). Meanwhile, mountain O3 concentration is maintained at a high level without apparent diurnal pattern, which in contrast to the prominent diurnal variation in surface O3 (Fig. S6d). The maintainable high O3 concentration on the mountain meant a high oxidation capability. Thus, the repartitioned gas species during upwards transport, in addition to the directly transported VOCs, may be further oxidized by the high level of O3 throughout the day and night, only to recondense in the particle phase (Robinson et al., 2007). The continuous oxidation of these vapors potentially contributed to the production of SOA, which may partly explain the higher levels of O:C at the mountain than the surface throughout all hours. The mass spectra of mountain HOA and SV-OOA had substantial reductions mainly at ion series CxH (Fig. 6a, c), suggesting the O3 oxidation may occur on unsaturated bonds (e.g., alkenes; Paulson and Orlando, 1996), thus following a less steep slope on the VK space, which is consistent with the above.
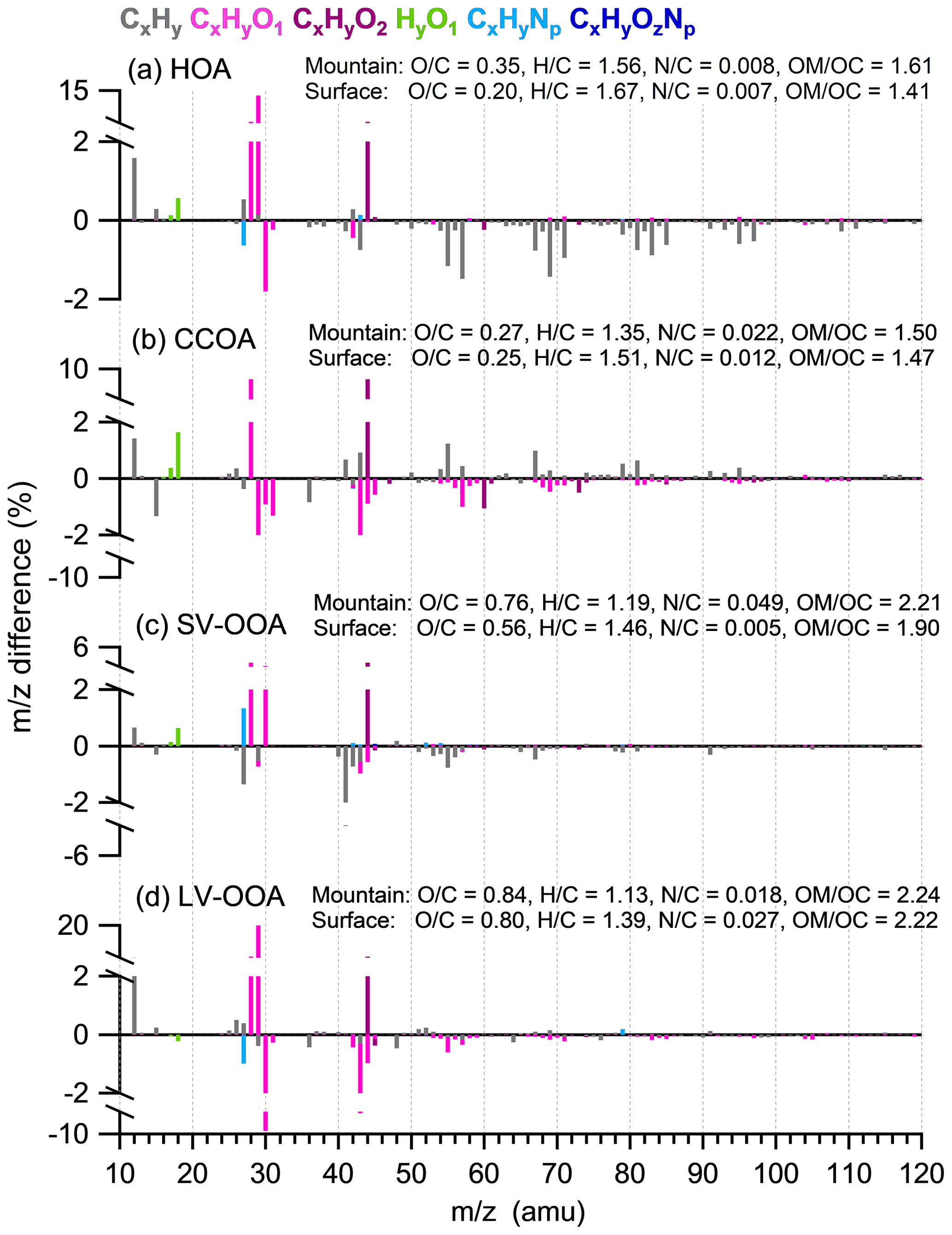
Figure 6Difference in mass spectra between the mountain and surface sites (mountain minus surface) for (a) HOA, (b) CCOA, (c) SV-OOA, and (d) LV-OOA.
For CCOA and LV-OOA, H:C had a substantial decrease but O:C only increased slightly (Fig. 6b, d). The difference in CCOA shows substantial decrease in CxHyO+ fragments at the mountain, which may compensate for some of the O:C increase. Some CxH fragments showed increases at the mountain, which tended to be consistent with the view that the CCOA is largely contributed by non-local sources (Li et al., 2019). This factor may not have experienced significant vertical transport from the surface, thus showing some fresher signatures (the CxH fragments) at the mountain. LV-OOA had a large enhancement of CHO+ and reduction in CH2O+, which may have been caused by some transformation of alcohols to carbonyl compounds (Grosjean et al., 1993).
The increase in the oxidation state could be caused by the evaporation process losing the less oxidized and more volatile species, and the evaporated gases could be further oxidized to partition to a more oxidized phase. The evaporative loss occurred in a relatively short timescale, i.e., a few hours of vertical transport induced by daytime convective mixing of boundary layer, as reflected by the 76 % decrease in semivolatile organic masses and 74 % of nitrate. These losses occurred in a relatively dry condition without notable wet scavenging; therefore, the evaporative loss tended to dominate. For the other period rather than midday on the mountain, there were additional primary sources contributing neither particles nor VOCs; thus, most of the vapors repartitioned from the condensed phase in the midday may stay and be subjected to oxidation at the mountain. These continuous inputs of gases to the top of boundary layers on a daily basis serve as a source of precursors to be oxidized and contribute to an important fraction of highly oxidized SOA.
All of these processes lead to a consistent manner of enhancing the oxidation state of OA at the top of PBL, which may modify the hygroscopicity and viscosity of OA (Koop et al., 2011). Combining with the more efficiently transported and less-volatile species, these processes consistently led to the overall decreased volatility of aerosols at the top of the PBL, where cloud formation is initialized, influencing the activities of both cloud condensation nuclei and ice nuclei. These lower-volatile aerosols could be transported to a longer distance in the free troposphere, thus having a longer lifetime. This also implies that the aerosol characteristics at the surface may not represent those at upper levels, and the evolution during transport should be considered when evaluating the contribution of surface emissions to cloud particle nucleation and their atmospheric lifetime.
All data in this paper are available from the authors upon request (liuquan620@126.com).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-14749-2021-supplement.
DD, MH, and HH led and designed the study. QL, DL, YW, KB, WG, PT, DZ, SL, CY, GT, YW, KH, SD, QG, FW, and SK were involved in collecting, processing, and analyzing of surface and mountain data. QL and DL carried out the data analysis and wrote the paper. QL and all authors contributed to the discussions.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors would like to kindly thank the dedicated efforts of many people from IAP and BJWMO at the observation sites. Moreover, we thank the two anonymous reviewers for the helpful comments and discussions.
This research has been supported by the National Key Research and Development Program of China (grant no. 2016YFA0602001), the National Natural Science Foundation of China (grant nos. 41975177, 41875167, 41875044, and 41775138), and the Beijing Municipal Natural Science Foundation (grant nos. 8192021 and 8194065).
This paper was edited by Andreas Petzold and reviewed by two anonymous referees.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: and Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cao, L.-M., Huang, X.-F., Li, Y.-Y., Hu, M., and He, L.-Y.: Volatility measurement of atmospheric submicron aerosols in an urban atmosphere in southern China, Atmos. Chem. Phys., 18, 1729–1743, https://doi.org/10.5194/acp-18-1729-2018, 2018.
Cappa, C. D. and Jimenez, J. L.: Quantitative estimates of the volatility of ambient organic aerosol, Atmos. Chem. Phys., 10, 5409–5424, https://doi.org/10.5194/acp-10-5409-2010, 2010.
Cruz, C. and Pandis, S.: Deliquescence and Hygroscopic Growth of Mixed Inorganic−Organic Atmospheric Aerosol, Environ. Sci. Technol., 34, 4313–4319, https://doi.org/10.1021/es9907109, 2000.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
D'Andrea, S. D., Häkkinen, S. A. K., Westervelt, D. M., Kuang, C., Levin, E. J. T., Kanawade, V. P., Leaitch, W. R., Spracklen, D. V., Riipinen, I., and Pierce, J. R.: Understanding global secondary organic aerosol amount and size-resolved condensational behavior, Atmos. Chem. Phys., 13, 11519–11534, https://doi.org/10.5194/acp-13-11519-2013, 2013.
Donahue, N., Robinson, A., Stanier, C., and Pandis, S.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, 2006.
Garratt, J. R.: Review: the atmospheric boundary layer, Earth Sci. Rev., 37, 89–134, 1994.
Grosjean, D., Grosjean, E., and Williams, E. L.: Atmospheric chemistry of unsaturated alcohols, Environ. Sci. Technol., 27, 2478–2485, https://doi.org/10.1021/es00048a026, 1993.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jones, A., Thomson, D., Hort, M., and Devenish, B.: The U.K. Met Office's Next-Generation Atmospheric Dispersion Model, NAME III, Air Pollution Modeling and Its Application XVII, Boston, MA, 580–589, 2007.
Koop, T., Bookhold, J., Shiraiwa, M., and Pöschl, U.: Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere, Phys. Chem. Chem. Phys., 13, 19238–19255, https://doi.org/10.1039/C1CP22617G, 2011.
Kuang, C., McMurry, P. H., and McCormick, A. V.: Determination of cloud condensation nuclei production from measured new particle formation events, Geophys. Res. Lett., 36, https://doi.org/10.1029/2009GL03758, 2009.
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., and Rantala, P.: Direct observations of atmospheric aerosol nucleation, Science, 339, 943–946, 2013.
Li, H., Cheng, J., Zhang, Q., Zheng, B., Zhang, Y., Zheng, G., and He, K.: Rapid transition in winter aerosol composition in Beijing from 2014 to 2017: response to clean air actions, Atmos. Chem. Phys., 19, 11485–11499, https://doi.org/10.5194/acp-19-11485-2019, 2019.
Li, J., Fu, Q., Huo, J., Wang, D., Yang, W., Bian, Q., Duan, Y., Zhang, Y., Pan, J., Lin, Y., Huang, K., Bai, Z., Wang, S.-H., Fu, J. S., and Louie, P. K. K.: Tethered balloon-based black carbon profiles within the lower troposphere of Shanghai in the 2013 East China smog, Atmos. Environ., 123, 327–338, https://doi.org/10.1016/j.atmosenv.2015.08.096, 2015.
Li, S., Liu, D., Hu, D., Kong, S., Wu, Y., Ding, S., Cheng, Y., Qiu, H., Zheng, S., Yan, Q., Zheng, H., Hu, K., Zhang, J., Zhao, D., Liu, Q., Sheng, J., Ye, J., He, H., and Ding, D.: Evolution of Organic Aerosol From Wood Smoke Influenced by Burning Phase and Solar Radiation, 126, e2021JD034534, https://doi.org/10.1029/2021JD034534, 2021.
Li, Z., Guo, J., Ding, A., Liao, H., Liu, J., Sun, Y., Wang, T., Xue, H., Zhang, H., and Zhu, B.: Aerosol and boundary-layer interactions and impact on air quality, Natl. Sci. Rev., 4, 810–833, https://doi.org/10.1093/nsr/nwx117, 2017.
Liu, D., Hu, K., Zhao, D., Ding, S., Wu, Y., Zhou, C., Yu, C., Tian, P., Liu, Q., Bi, K., Wu, Y., Hu, B., Ji, D., Kong, S., Ouyang, B., He, H., Huang, M., and Ding, D.: Efficient Vertical Transport of Black Carbon in the Planetary Boundary Layer, Geophys. Res. Lett., 47, e2020GL088858, https://doi.org/10.1029/2020gl088858, 2020a.
Liu, Q., Liu, D., Gao, Q., Tian, P., Wang, F., Zhao, D., Bi, K., Wu, Y., Ding, S., Hu, K., Zhang, J., Ding, D., and Zhao, C.: Vertical characteristics of aerosol hygroscopicity and impacts on optical properties over the North China Plain during winter, Atmos. Chem. Phys., 20, 3931–3944, https://doi.org/10.5194/acp-20-3931-2020, 2020b.
May, A. A., Lee, T., McMeeking, G. R., Akagi, S., Sullivan, A. P., Urbanski, S., Yokelson, R. J., and Kreidenweis, S. M.: Observations and analysis of organic aerosol evolution in some prescribed fire smoke plumes, Atmos. Chem. Phys., 15, 6323–6335, https://doi.org/10.5194/acp-15-6323-2015, 2015.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Pankow, J. F.: Review and comparative analysis of the theories on partitioning between the gas and aerosol particulate phases in the atmosphere, Atmos. Environ., 21, 2275–2283, 1987.
Pankow, J. F.: An absorption model of gas/particle partitioning of organic compounds in the atmosphere, Atmos. Environ., 28, 185–188, 1994.
Paulson, S. E. and Orlando, J. J.: The reactions of ozone with alkenes: An important source of HOx in the boundary layer, Geophys. Res. Lett., 23, 3727–3730, https://doi.org/10.1029/96GL03477, 1996.
Ran, L., Deng, Z., Xu, X., Yan, P., Lin, W., Wang, Y., Tian, P., Wang, P., Pan, W., and Lu, D.: Vertical profiles of black carbon measured by a micro-aethalometer in summer in the North China Plain, Atmos. Chem. Phys., 16, 10441–10454, https://doi.org/10.5194/acp-16-10441-2016, 2016.
Riipinen, I., Pierce, J. R., Yli-Juuti, T., Nieminen, T., Häkkinen, S., Ehn, M., Junninen, H., Lehtipalo, K., Petäjä, T., Slowik, J., Chang, R., Shantz, N. C., Abbatt, J., Leaitch, W. R., Kerminen, V.-M., Worsnop, D. R., Pandis, S. N., Donahue, N. M., and Kulmala, M.: Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 11, 3865–3878, https://doi.org/10.5194/acp-11-3865-2011, 2011.
Riipinen, I., Yli-Juuti, T., Pierce, J. R., Petäjä, T., Worsnop, D. R., Kulmala, M., and Donahue, N. M.: The contribution of organics to atmospheric nanoparticle growth, Nat. Geosci., 5, 453–458, https://doi.org/10.1038/ngeo1499, 2012.
Robinson, A., Donahue, N., Shrivastava, M., Weitkamp, E., Sage, M., Grieshop, A., Lane, T., Pierce, J., and Pandis, S.: Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Salo, K., Westerlund, J., Andersson, P. U., Nielsen, C., D'Anna, B., and Hallquist, M.: Thermal Characterization of Aminium Nitrate Nanoparticles, J. Phys. Chem. A, 115, 11671–11677, https://doi.org/10.1021/jp204957k, 2011.
Schurman, M. I., Lee, T., Sun, Y., Schichtel, B. A., Kreidenweis, S. M., and Collett Jr., J. L.: Investigating types and sources of organic aerosol in Rocky Mountain National Park using aerosol mass spectrometry, Atmos. Chem. Phys., 15, 737–752, https://doi.org/10.5194/acp-15-737-2015, 2015.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Shrivastava, M. K., Lipsky, E. M., Stanier, C. O., and Robinson, A. L.: Modeling Semivolatile Organic Aerosol Mass Emissions from Combustion Systems, Environ. Sci. Technol., 40, 2671–2677, https://doi.org/10.1021/es0522231, 2006.
Sun, Y., Du, W., Fu, P., Wang, Q., Li, J., Ge, X., Zhang, Q., Zhu, C., Ren, L., Xu, W., Zhao, J., Han, T., Worsnop, D. R., and Wang, Z.: Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 16, 8309–8329, https://doi.org/10.5194/acp-16-8309-2016, 2016.
Tang, G., Zhu, X., Hu, B., Xin, J., Wang, L., Münkel, C., Mao, G., and Wang, Y.: Impact of emission controls on air quality in Beijing during APEC 2014: lidar ceilometer observations, Atmos. Chem. Phys., 15, 12667–12680, https://doi.org/10.5194/acp-15-12667-2015, 2015.
Tsigaridis, K., Daskalakis, N., Kanakidou, M., Adams, P. J., Artaxo, P., Bahadur, R., Balkanski, Y., Bauer, S. E., Bellouin, N., Benedetti, A., Bergman, T., Berntsen, T. K., Beukes, J. P., Bian, H., Carslaw, K. S., Chin, M., Curci, G., Diehl, T., Easter, R. C., Ghan, S. J., Gong, S. L., Hodzic, A., Hoyle, C. R., Iversen, T., Jathar, S., Jimenez, J. L., Kaiser, J. W., Kirkevåg, A., Koch, D., Kokkola, H., Lee, Y. H., Lin, G., Liu, X., Luo, G., Ma, X., Mann, G. W., Mihalopoulos, N., Morcrette, J.-J., Müller, J.-F., Myhre, G., Myriokefalitakis, S., Ng, N. L., O'Donnell, D., Penner, J. E., Pozzoli, L., Pringle, K. J., Russell, L. M., Schulz, M., Sciare, J., Seland, Ø., Shindell, D. T., Sillman, S., Skeie, R. B., Spracklen, D., Stavrakou, T., Steenrod, S. D., Takemura, T., Tiitta, P., Tilmes, S., Tost, H., van Noije, T., van Zyl, P. G., von Salzen, K., Yu, F., Wang, Z., Wang, Z., Zaveri, R. A., Zhang, H., Zhang, K., Zhang, Q., and Zhang, X.: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 14, 10845–10895, https://doi.org/10.5194/acp-14-10845-2014, 2014.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Van Krevelen, D. W.: Graphical-statistical method for the study of structure and reaction processes of coal, Fuel, 24, 269–284, 1950.
Wang, X., Heald, C. L., Ridley, D. A., Schwarz, J. P., Spackman, J. R., Perring, A. E., Coe, H., Liu, D., and Clarke, A. D.: Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon, Atmos. Chem. Phys., 14, 10989–11010, https://doi.org/10.5194/acp-14-10989-2014, 2014.
Wu, F., Xie, P., Li, A., Mou, F., Chen, H., Zhu, Y., Zhu, T., Liu, J., and Liu, W.: Investigations of temporal and spatial distribution of precursors SO2 and NO2 vertical columns in the North China Plain using mobile DOAS, Atmos. Chem. Phys., 18, 1535–1554, https://doi.org/10.5194/acp-18-1535-2018, 2018.
Xu, W. Q., Sun, Y. L., Chen, C., Du, W., Han, T. T., Wang, Q. Q., Fu, P. Q., Wang, Z. F., Zhao, X. J., Zhou, L. B., Ji, D. S., Wang, P. C., and Worsnop, D. R.: Aerosol composition, oxidation properties, and sources in Beijing: results from the 2014 Asia-Pacific Economic Cooperation summit study, Atmos. Chem. Phys., 15, 13681–13698, https://doi.org/10.5194/acp-15-13681-2015, 2015.
Xu, W. Y., Zhao, C. S., Ran, L., Lin, W. L., Yan, P., and Xu, X. B.: SO2 noontime-peak phenomenon in the North China Plain, Atmos. Chem. Phys., 14, 7757–7768, https://doi.org/10.5194/acp-14-7757-2014, 2014.
Zhang, J. K., Sun, Y., Liu, Z. R., Ji, D. S., Hu, B., Liu, Q., and Wang, Y. S.: Characterization of submicron aerosols during a month of serious pollution in Beijing, 2013, Atmos. Chem. Phys., 14, 2887–2903, https://doi.org/10.5194/acp-14-2887-2014, 2014.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007gl029979, 2007.
Zhang, Q., Jimenez, J., Canagaratna, M., Ulbrich, I., Ng, N., Worsnop, D., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, R., Suh, I., Zhao, J., Zhang, D., Fortner, E. C., Tie, X., Molina, L. T., and Molina, M. J.: Atmospheric New Particle Formation Enhanced by Organic Acids, Science, 304, 1487–1490, https://doi.org/10.1126/science.1095139, 2004.
Zhang, X. Y., Wang, J. Z., Wang, Y. Q., Liu, H. L., Sun, J. Y., and Zhang, Y. M.: Changes in chemical components of aerosol particles in different haze regions in China from 2006 to 2013 and contribution of meteorological factors, Atmos. Chem. Phys., 15, 12935–12952, https://doi.org/10.5194/acp-15-12935-2015, 2015.
Zhao, D., Huang, M., Tian, P., He, H., Lowe, D., Zhou, W., Sheng, J., Wang, F., Bi, K., Kong, S., Yang, Y., Liu, Q., Liu, D., and Ding, D.: Vertical characteristics of black carbon physical properties over Beijing region in warm and cold seasons, Atmos. Environ., 213, 296–310, https://doi.org/10.1016/j.atmosenv.2019.06.007, 2019.