the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Sep 2021
| 14 Sep 2021
Atmospheric oxidation of α,β-unsaturated ketones: kinetics and mechanism of the OH radical reaction
Rodrigo Gastón Gibilisco
Iustinian Gabriel Bejan
Iulia Patroescu-Klotz
Peter Wiesen
The OH-radical-initiated oxidation of 3-methyl-3-penten-2-one and 4-methyl-3-penten-2-one was investigated in two atmospheric simulation chambers at 298±3 K and 990±15 mbar using long-path FTIR spectroscopy. The rate coefficients of the reactions of 3-methyl-3-penten-2-one and 4-methyl-3-penten-2-one with OH radicals were determined to be and , respectively. To enlarge the kinetics data pool the rate coefficients of the target species with Cl atoms were determined to be and , respectively. The mechanistic investigation of the OH-initiated oxidation focuses on the RO2+NO reaction. The quantified products were acetoin, acetaldehyde, biacetyl, CO2 and peroxyacetyl nitrate (PAN) for the reaction of 3-methyl-3-penten-2-one with OH radicals and acetone, methyl glyoxal, 2-hydroxy-2-methylpropanal, CO2 and peroxyacetyl nitrate (PAN) for the reaction of 4-methyl-3-penten-2-one with OH, respectively. Based on the calculated product yields an upper limit of 0.15 was determined for the yield of RONO2 derived from the OH reaction of 4-methyl-3-penten-2-one. By contrast, no RONO2 formation was observed for the OH reaction of 3-methyl-3-penten-2-one. Additionally, a simple model is presented to correct product yields for secondary processes.
- Article
(3205 KB) - Full-text XML
-
Supplement
(920 KB) - BibTeX
- EndNote





The α,β-unsaturated ketones are a particular class of oxygenated volatile organic compounds (OVOCs) emitted either from biogenic and/or anthropogenic sources or generated in the oxidation of airborne VOCs in the atmosphere. The most prominent representative among this class is methyl vinyl ketone (MVK). MVK is, on the one hand, emitted from the polymer, pharmaceutical and fungicide manufacturing industries (Siegel and Eggersdorfer, 2000). On the other hand, it is formed in the troposphere, mainly through the gas-phase oxidation of isoprene, the NMHC which is most abundantly emitted into the atmosphere, with an estimated annual emission up to 750 Tg (Calvert et al., 2000; Guenther et al., 2006). Other α,β-unsaturated ketones, like 3-methyl-3-penten-2-one (3M3P2), are used in the fragrances and food industry (Chapuis and Jacoby, 2001; Bickers et al., 2003, Wang et al., 2020). Furthermore, 4-methyl-3-penten-2-one (4M3P2), commonly known as mesityl oxide, is a precursor of methyl isobutyl ketone used extensively as solvent in the fabrication of paints and coatings. Production rates as high as 0.1 Tg yr−1 were reported in the US (Sifniades et al., 2011). α,β-Unsaturated ketones were also identified in laboratory studies on emissions from different fuels representative of biomass burning (Hatch et al., 2017).
Table 1Structures of α,β-unsaturated ketones and related literature on the corresponding OH-radical-initiated oxidation mechanism.

During daytime the main loss process of α,β-unsaturated ketones, once released into or formed in the atmosphere, is probably the oxidation initiated by OH radicals, which has a direct impact on the atmospheric ozone and secondary organic aerosol formation (Kanakidou et al., 2005; Calvert et al., 2011). However, our knowledge about the oxidation mechanisms of species presented in Table 1 is rather limited. To date, only the OH radical reaction of MVK has been intensively studied (Tuazon and Atkinson, 1989; Galloway et al., 2011; Praske et al., 2015; Fuchs et al., 2018). Under high-NO conditions, where virtually all peroxy radicals react with NO, glycolaldehyde and methyl glyoxal together with formaldehyde and PAN were identified as first-generation products. Praske et al. (2015) also found a low RONO2 yield of about 4 %. Some attention was also given to the reaction of both 3M3P2 and 4M3P2, investigated in the present study, with O3 and to the 3M3P2+Cl and 3M3P2+NO3 system (Sato et al., 2004; Canosa-Mas et al., 2005; Wang et al., 2015; Illmann et al., 2021a; Li et al., 2021). Gaona-Colmán et al. (2017) investigated the OH radical reaction of 4M3P2. However, they qualitatively identified only formaldehyde and acetone (GC-MS detection). Hence, the mechanism remains rather incomplete.
In this work, we present an in-depth investigation of the OH-radical-initiated oxidation of two di-substituted α,β-unsaturated ketones, namely 3M3P2 and 4M3P2. Besides the first determination of the rate coefficient for the reaction of OH radicals with 3M3P2, we report kinetic data for Cl atom reactions.
In order to correct the formation yields of products formed in target reactions within complex experimental systems it is quite common to use the Tuazon formalism (Tuazon et al., 1986). This is based on the assumption that reaction products are subsequently consumed in secondary processes like photolysis, wall loss and oxidation by OH radicals. Thus, their formation yields in the target reactions are underestimated when determined from plotting the formed product against the consumed compound of interest, and the yields increase when corrected. However, the formalism is difficult to apply for products with secondary sources in the reaction system. In this case, the formation yields are overestimated without proper corrections. For this purpose, we present a simple model to correct molar formation yields in the target reactions, which accounts for both consumption and secondary formation processes.
The study investigates the contribution of the OH-initiated oxidation of both 3M3P2 and 4M3P2 to the formation of NOx reservoir species, like peroxyacetyl nitrate (PAN), in the atmosphere. Apart for being an important NOx reservoir species for the atmosphere, PAN is a phytotoxic air pollutant (Vyskocil et al., 1998) with sources that are still unaccounted for (Fischer et al., 2014). PAN formation depends strongly on temperature and the levels of NO2 and NO. The study stresses the importance of determining either the ratio between PAN and CO2/HCHO or the PAN yield together with the NO2NO ratio within the experiment since these give more comprehensive information on NOx reservoir species production than the PAN yield alone. Therefore experiments were conducted under varying NO2NO ratios.
Experiments were carried out in a 1080 and a 480 L reaction chamber in 990±15 mbar of synthetic air at 298±3 K. In the following section is given an updated description of both chambers. A major improvement of both chambers is the addition of heatable injection blocks (<100 ∘C). A controlling unit allows the temperature to be adjusted for a better transfer of samples into the reaction chamber, according to the thermal stability of the investigated substances. Graphic representations of the chambers were published already by Barnes and co-workers (Barnes et al., 1993; Barnes et al., 1994).
2.1 1080 L chamber
The 1080 L chamber consists of two joint quartz-glass tubes with a total length of 6.2 m and an inner diameter of 0.47 m connected via a middle flange. It is closed at both ends by metal flanges bearing several ports for the injection of reactants, addition of bath gases and coupling with analytical devices. A total of 32 superactinic fluorescent lamps (Philips TL05 40 W: 300–460 nm, max. intensity at ca. 360 nm) and 32 low-pressure mercury vapour lamps (Philips TUV 40 W: max. intensity at 254 nm) can be used to irradiate the reaction mixture. These lamps are wired in parallel and spaced evenly around the reaction vessel. The pumping system consists of a turbo-molecular pump backed by a double-stage rotary fore pump. The chamber is cleaned between experiments by evacuating it to 10−4 mbar for at least 30 min. Cleanliness of the chamber is proved by FTIR. Homogeneity of the reaction mixtures is achieved by three magnetically coupled Teflon mixing fans, which are placed on the end and middle flanges. A White-type mirror system is installed inside the chamber to monitor reaction mixtures via FTIR spectroscopy in the spectral range 4000–700 cm−1 at a resolution of 1 cm−1. The system whose base length is (5.91±0.01) m was operated at 82 traverses which yields a total optical path length of (484.7±0.8) m. Spectra were recorded using a Nicolet iS50 instrument equipped with a liquid-nitrogen-cooled mercury–cadmium–telluride (MCT) detector.
The initial mixing ratios in the 1080 L experiments, in ppmV ( at 298 K), were 0.7–1.3 for 3-methyl-3-penten-2-one (3M3P2), 0.9–1.8 for 4-methyl-3-penten-2-one (4M3P2), 1.1 for isoprene, 0.9–1.4 for propene, 0.9–1.4 for isobutene, 0.9–1.4 for 1,3-butadiene, 0.9–1.9 for methyl nitrite, 0.9–1.9 for Cl2, 2–4 for NO and 0–2.5 for NO2.
2.2 480 L chamber
The smaller chamber in the Wuppertal laboratory consists of a borosilicate glass cylinder with a total volume of 480 L (length of 3 m and 0.45 m inner diameter). The tube is closed at both ends by aluminium flanges containing various ports for the introduction of reactants and bath gases, sampling, and instruments monitoring the physical parameters inside the chamber. To ensure homogeneous mixing of the reactants a magnetically coupled fan is mounted on the front flange inside the chamber. A total of 32 fluorescent lamps (Philips TLA 40 W, nm, Imax at 360 nm) are mounted in four boxes and spaced evenly around the chamber. The lamp housings are cooled with air and their inner surface is encased in reflective steel sheets. The lamps can be switched individually to allow a variation of the photolysis frequency and consequently the radical level during photolysis experiments. The pumping system consists of a rotary vane pump and a root pump yielding an end vacuum of up to 10−3 mbar. For a typical cleaning procedure between two experiments the chamber is completely evacuated and filled up to 200–300 mbar of synthetic air or nitrogen. This procedure is repeated until it is certain that no signals related to the previous experiment are detected. Reactants and products are monitored using in situ FTIR spectroscopy. For this purpose, a White-type mirror system (base length: 2.80±0.01 m) is installed inside the chamber and coupled to a Nicolet 6700 FTIR spectrometer (MCT detector). The system is operated at 18 traverses, which yields a total optical path length of 50.4±0.2 m. FTIR spectra are recorded in the spectral range 4000–700 cm−1 at a resolution of 1 cm−1.
The initial mixing ratios in the 480 L experiments in ppmV ( at 298 K) were 5.0–6.1 for 3-methyl-3-penten-2-one (3M3P2), 5.0–6.0 for 4-methyl-3-penten-2-one (4M3P2), 5.0–5.7 for methyl valerate, 4.2–6.3 for propene, 4.2–6.3 for isobutene, 4.2–6.3 for 1,3-butadiene, 10–16 for methyl nitrite, 13–16 for Cl2 and 20–27 for NO.
2.3 Materials
The following chemicals were used without further handling, with purities as stated by the suppliers: isobutene (Sigma Aldrich, 99 %), propene (Air Liquide, 99.95 %), 1,3–butadiene (Messer, >99 %), isoprene (Aldrich, 99 %), methyl valerate (Alfa Aesar, 99 %), 3-methyl-3-penten-2-one (Sigma Aldrich, tech. 90 %), 4-methyl-3-penten-2-one (Sigma Aldrich, tech. 90 %), acetoin (Sigma Aldrich, 96 %), biacetyl (Sigma Aldrich, 97 %), 2-methyl-3-buten-2-ol (Sigma Aldrich, 98 %), carbon monoxide (Air Liquide, 99.97 %), nitrogen monoxide (Air Liquide, 99.5 %), nitrogen dioxide (Messer Griesheim, >98 %), Cl2 (Air Liquide, 99.8 %), synthetic air (Messer, 99.9999 %), nitrogen (Messer, 99.9999 %). Methyl nitrite was synthesized by dropping sulfuric acid in an ice-cooled aqueous solution of sodium nitrite and methanol (Taylor et al., 1980). The product was collected and stored in a cooling trap at −78 ∘C. The purity of methyl nitrite was confirmed via FTIR spectroscopy.
2.4 Experimental protocol
OH radicals were generated by methyl nitrite photolysis in synthetic air at 360 nm:
NO was added to the reaction system to suppress ozone formation and hence the formation of NO3 radicals. Cl atoms were generated by photolysis of molecular chlorine in either synthetic air or nitrogen at 360 nm:
In both chambers, the target compound and the products formed during the reaction (mechanistic investigation) as well as the target and reference compound (kinetic study experiments) were monitored using FTIR spectroscopy. Typically, 50–70 interferograms were co-added per spectrum, which results in averaging period of about 80–113 s, and 15–20 spectra were recorded per experiment. In each run the first five spectra were collected in the dark, over a period of 10–20 min, to check for a potential wall loss of the unsaturated ketones and the reference compounds. Afterwards, the reaction was started by switching on the lamps. In the product study experiments, the reaction was terminated after a maximum of 10 spectra were recorded. In selected experiments additionally 5 spectra were collected in the dark, after termination, over a time interval of 10–20 min, to investigate the significance of the wall loss for the products of the oxidation reaction in the experimental system.
Usually, the housing which enfolds the transfer optics between FTIR spectrometer and chamber is flushed with purified dry air. Therefore, quantification of CO2 is, due to a slight variability in the dry air supply, unreliable under normal laboratory conditions. To be able to quantify CO2, in selected product study experiments performed in the 1080 L chamber, the transfer optics housing was flushed with ultrapure N2 evaporated from a liquid nitrogen tank.
2.5 Relative rate method
The rate coefficients for the reaction of OH radicals and Cl atoms with the α,β-unsaturated ketones were determined by relating the consumption of the ketone to the consumption of at least two reference compounds:
Both ketones and references could potentially be subject to an irreversible first-order wall loss:
Considering these processes the following equation can be derived:
where [X]t is the concentration of the species X at time t and t=0 corresponds to the time where the lamps were switched on. According to Eq. (1) a plot of against should yield a straight line with zero intercept where the slope represents the rate coefficient ratio .
2.6 Product identification and quantification
The quantification of identified products was done by subtraction of calibrated reference IR spectra. The corresponding concentrations of the reference spectra were calculated based on cross sections either taken from literature references, taken from the Wuppertal laboratory's database or determined within this work. For methyl glyoxal we used the cross sections determined by Talukdar et al. (2011). When employing the values reported by Profeta et al. (2011) the obtained mixing ratios of methyl glyoxal are, however, almost identical. Peroxyacetyl nitrate (PAN) has been quantified using the absorption cross section reported by Allen et al. (2005). Cross sections for acetone and acetaldehyde were taken from the Wuppertal database. The absorption cross sections of 3-methyl-3-penten-2-one, 4-methyl-3-penten-2-one, 3-hydroxy-2-butanone (acetoin) and 2,3-butanedione (biacetyl) were determined within this work by either injecting different volumes of the pure compound into the chamber, evaporating weighted solid samples (in the case of acetoin) into a flow of bath gas or injecting aliquot volumes of a solution containing the target species according to a method previously described by Etzkorn et al. (1999). Reference spectra of 2-hydroxy-2-methylpropanal (HMPr) were generated in situ by the ozonolysis of 2-methyl-3-buten-2-ol in the presence of CO to scavenge any OH radical formed in the reaction system. CO2 was quantified by integration of the absorption features in the range 2400–2349 cm−1 and a polynomial calibration function derived from the injection of various volumes of CO2 using a calibrated gas-tight syringe.
2.7 Modelling
In order to correct the experimentally determined product yields for both secondary formation and consumption a simple model was established based on the Euler–Cauchy method, which can be written down using a commercially available calculation program. In doing so, the rates for each species X are calculated for constant time intervals Δt based on the rate equation and the concentration of the species X in the previous time interval. This allows the calculation of concentration changes for each time interval, which, when added to the concentration of the previous time interval, yield the concentration of X at time t. The model assumes simplified reaction mechanisms as exemplarily shown below.
The simplest version of this approach assumes a steady-state (sst) concentration of OH radicals that can be determined from the individual loss rate of the target species A during the irradiation period of each experiment. This hypothesis is valid as long as pseudo-first-order conditions are proven by a linear correlation between and the time t. The modelled concentration time profiles are obtained by the input of [A]0, [OH]sst and the rate coefficients of all listed Reactions (R9)–(R14). The yield yx for each product of the target reaction is included as a parameter to be varied until the simulated concentration–time profile of each species matches the experimental data. However, for traceability and potential future applications, we prefer to outline in detail the stepwise procedure which turned out as best practice after several tests.
-
The rate equations for each species are written according to the simplified exemplary mechanism shown above in Reactions (R9)–(R14), where kinetic data and molar formation yields for the secondary reactions are taken from literature references. Values for the first-order rate coefficients of each species' wall loss are included if they are measured in the same experiment (thus measured before and after the irradiation of the reaction mixture). If they are not measured, values for kwall are first set to zero and included as additional variables. Molar formation yields for the target reaction are included as variable yx.
-
Time intervals Δt and the total duration t are adjusted and the initial concentration [A]0 of A is included for t=0 according to the experimental data.
-
If the experimental logarithmic decay of the concentration of A demonstrates pseudo-first-order conditions, a constant OH concentration is included based on the consumption of A during the irradiation. If pseudo-first-order conditions are not accomplished within the experiment the OH concentration has to be described as a function of t based on the consumption of A. However, including the OH concentration yields a simulated time profile for A which should reproduce the experimental data of A well.
-
Finally, the parameters yx for the target reaction are varied until the time profile of each species matches the experimental data, starting with yx corresponding to species X, which is the less affected by secondary reactions. Thus, one would start with yd and yb rather than yc in the given example. The uncorrected molar yields yx, derived from plotting the measured mixing ratio of product X against the consumed target species A, are appropriate as a starting point of the iterative process. If wall losses were not determined within the experiments one should try to match the time profile for the first data points where secondary processes are less significant. After that the wall loss parameters are varied to match the whole time profile. However, for this procedure it is mandatory to know the reasonable range of kwall for each species in the experimental set-up.
The time intervals used for the simulations were typically Δt<0.1 s. The yields errors associated with the model were found to be in the range of 0.02. If it is not possible to reproduce the time profiles with the model this will indicate either an error in the spectra evaluation, the simplified mechanism used or the experiments conducted.
All experiments were conducted at a total pressure of 990±15 mbar and 298±3 K. Irreversible first-order wall losses of 3M3P2 and 4M3P2 were found to be negligible in the 480 L chamber and in the range (1– s−1 and (1– s−1 in the 1080 L chamber, respectively. In separate control experiments, containing the target species in the bath gas only, no difference was observed for the wall loss in the dark and when the mixture was irradiated over the typical length of an experiment. Hence, an increased wall loss rate due to convection induced by heated walls could be ruled out. Dark reactions between the radical source and the target species were tested as well in separate experiments and were found to be negligible in both reaction chambers. Photolysis of the unsaturated ketones was likewise found to be negligible under the experimental conditions.
3.1 Kinetic study
Relative-rate plots according to Eq. (1) are presented in Fig. 1 for all kinetic experiments conducted. Three reference compounds have been used to determine the rate coefficient of each investigated reaction system. The relative-rate plots for individual experiments display a high linearity, with correlation coefficients from linear regression analysis being R2>0.95 and zero intercepts within a 2σ statistical error. The calculated relative ratios are summarized in Table 2. They were found to be independent of the initial concentration of the unsaturated ketone and the irradiation time. In the case of the Cl reactions, the relative ratios were independent of using either synthetic air or nitrogen as bath gas. Therefore, the obtained relative ratios likely result solely from each target reaction and any interfering process can be neglected in the present experimental set-up.
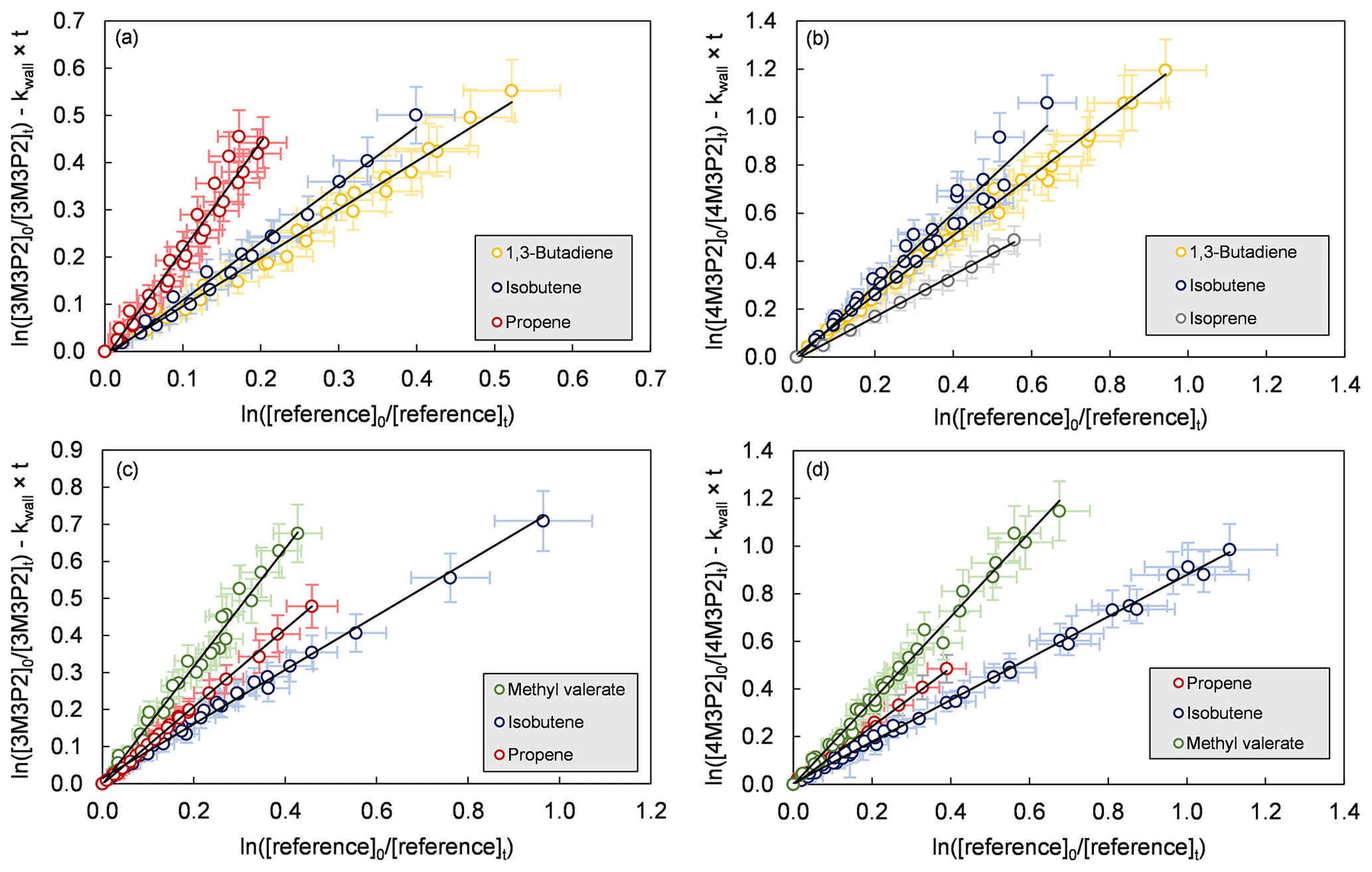
Figure 1Relative-rate plots of all experiments according to Eq. (1) for the reaction of (a) 3-methyl-3-penten-2-one+OH, (b) 4-methyl-3-penten-2-one+OH, (c) 3-methyl-3-penten-2-one+Cl and (d) 4-methyl-3-penten-2-one+Cl. The error bars consist of a systematic uncertainty and an additional 10 % relative error to cover uncertainties derived from the experimental and evaluation procedure, respectively.
Table 2Results of the kinetic study using different reference compounds.
The rate coefficients were calculated based on the determined relative ratios using the following reference data: (Mellouki et al., 2021), (Atkinson et al., 2006), (Calvert et al., 2000), (Mellouki et al., 2021), (Ezell et al., 2002), (Ezell et al., 2002), (Notario et al., 1998).
In Table 2 the calculated rate coefficients are given as an average for each reference together with the corresponding error as a combination of the relative ratio's statistical error and the error of the reference's rate coefficient. The determinations with different reference compounds agree within <4 % for the Cl reactions and <17 % for the OH reactions, respectively. The final rate coefficients for the target reactions are given as the weighted average of all experimental determinations: , , and . The quoted errors represent the 2σ statistical error of the weighted mean and an additional 10 % relative error to cover all uncertainties derived from the experimental and evaluation procedure.
Rate coefficients for the reaction of 4M3P2 with OH radicals and Cl atoms have been previously determined by Gaona-Colmán et al. (2017), using the relative-rate technique and GC-FID as the detection method. The present values are about 20 % smaller (OH reaction) and 15 % larger (Cl reaction) than those reported by Gaona-Colmán et al. (2017). However, both values are found within the uncertainties of the former study and thus are in good agreement. Wang et al. (2015) reported at 293±1 K based on a relative-rate study (GC-FID detection) employing large initial mixing ratios for the target species (100 ppm). Nevertheless, the value determined within this work is in excellent agreement with the one reported by Wang and co-workers.
3.1.1 Reactivity
It is generally accepted that OH radical and Cl atom reactions of OVOCs proceed via H atom abstraction or addition to the C=C double bond, in the case of unsaturated organic species. The AOPWIN software (US EPA, 2021; Kwok and Atkinson, 1995) estimates for the OH reaction with both investigated ketones, without discriminating, the contribution of H atom abstraction and OH addition to the olefinic bond to be and , respectively. This suggests that the OH reaction proceeds almost exclusively through the addition to the C=C double bond. This theoretical result is supported by the findings of the present product studies (see Sect. 3.3). The rate coefficients of 4M3P2+OH predicted by AOPWIN and determined here experimentally agree within 4 %. However, AOPWIN does not differentiate between certain substitution patterns. Given that there is a good agreement between the results using different references, our results show 4M3P2 to be about 1.25 times more reactive towards OH radicals than 3M3P2. Therefore, the AOPWIN prediction is less accurate in the case of 3M3P2. Estimations for the rate coefficients of the reactions of both ketones with OH radicals were given as well in an earlier study ( and ) based on linear free-energy relationships using ionization potentials (Grosjean and Williams, 1992). Being lower for both ketones, these predictions qualitatively reproduce, however, the experimentally observed rate coefficient ratio . The same applies when the estimation of the rate constants is performed according to the SAR approach by Jenkin et al. (2018).
On the other hand, if the reaction proceeds almost solely via the electrophilic addition to the C=C double bond the reactivity can be examined in terms of the electron density associated with the olefinic bond. We have recently pointed out the importance of defining the appropriate core structure when discussing gas-phase reactivity and related substituent effects in unsaturated oxygenated compounds (Illmann et al., 2021a). In the case of the α,β-unsaturated ketones this yields a comparison with structural analogue alkenes where the acetyl moiety is replaced by an H atom, thus (Z)-2-butene (for 3M3P2) and isobutene (for 4M3P2) as structural analogues. Despite the deactivating (−I) effect of carbonyl groups, the ability of the carbonyl group to form hydrogen-bonded intermediates with OH radicals (Smith and Ravishankara, 2002) lead to a presumably higher reactivity of the unsaturated ketones compared to their alkene analogues. Experimental results supporting this assumption were reported previously, although the corresponding alkenes were chosen in a different way (Blanco et al., 2012).
An attempt to quantify the substituent effects in oxygenated compounds was previously performed by defining a non-dimensional reactivity factor (Illmann et al., 2021a). By using the latest IUPAC recommendations (Mellouki et al., 2021) for the OH radical reactions of isobutene and (Z)-2-butene, respectively, and the results of the present study we obtained for both ketones xr>1. This is in complete agreement with the expected enhancement of reactivity towards OH radicals. On the other hand, xr is <1 for both ketones in the case of the Cl atom reaction. This is not surprising as here the formation of an activating hydrogen-bonded intermediate is not possible. Therefore, the reactivity towards Cl atoms is determined by the deactivating inductive effect of the carbonyl moiety upon the olefinic bond. However, an in-depth analysis of these effects is more related to fundamentals of physical chemistry and will be included in a separate publication.
3.2 Infrared cross sections
Integrated absorption cross sections σ have been determined based on the Beer–Lambert law,
by plotting the integrated absorption band between υ1 and υ2 against the concentration c. These values are summarized in Table 3 together with literature data where available. Plots used to determine the cross sections are shown in Figs. S1 and S2 in the Supplement. To the best of our knowledge, there are no previous reports on the IR cross sections for 3M3P2, 4M3P2 and acetoin in the literature. Profeta and co-workers (2011) reported band intensities for the various absorptions of biacetyl in the gas phase. The integrated cross section of the carbonyl absorption (1690–1769 cm−1) determined within this work is almost identical with their value. The other cross sections agree within <10 %, where the largest discrepancies are observed for the least intense absorption features (between 870–994 and 2905–3053 cm−1).
Table 3Integrated absorption cross sections determined within this work (resolution: 1 cm−1, apodization: Happ-Genzel, phase correction: Merck, zero-filling: 0) together with available literature references.
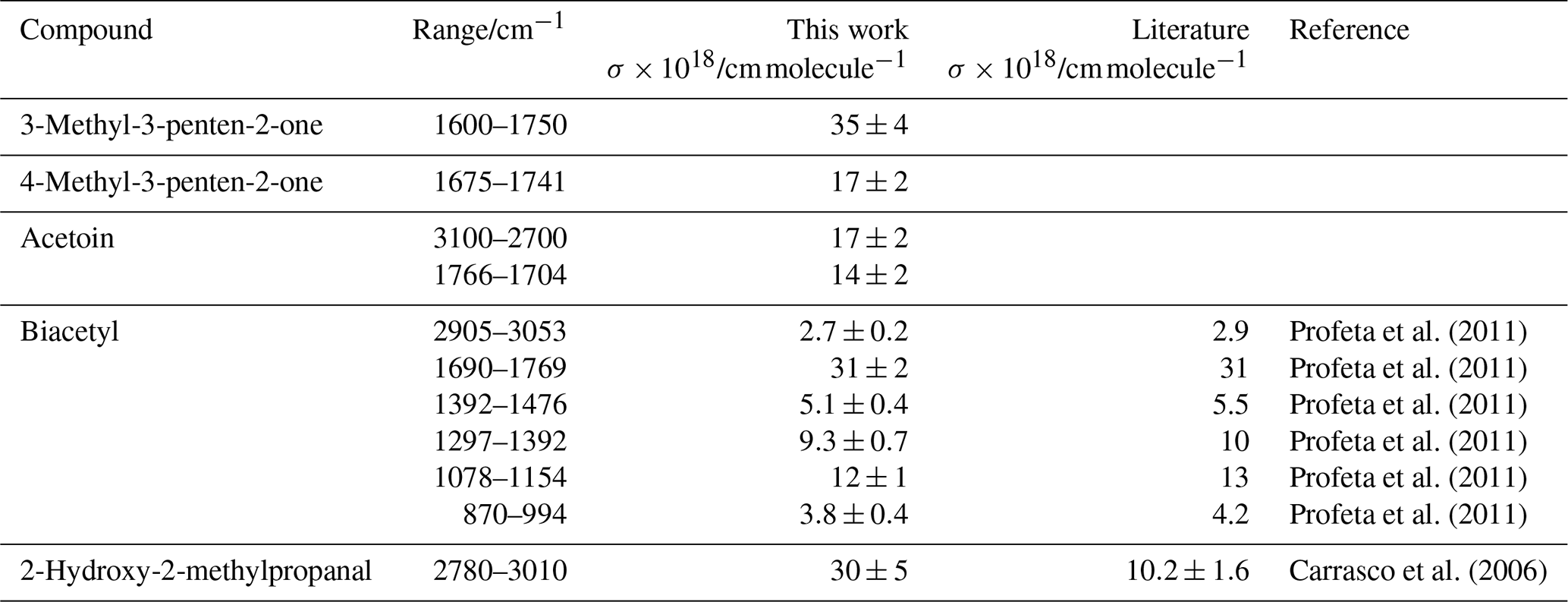
We recommend using freshly prepared or purchased samples when working with biacetyl. The investigation of an older sample stored for several months at temperatures <8 ∘C yielded cross sections 30 %–40 % lower than those reported by Profeta et al. (2011) while the gas-phase IR spectra were identical with the new sample. Thus, it seems likely that degradation takes place even in a cooled sample. However, the absence of foreign absorption features in the FTIR spectra of the older sample remains unexplained.
In order to determine its integrated absorption cross section, 2-hydroxy-2-methylpropanal (HMPr) was generated in situ through the ozonolysis of 2-methyl-3-buten-2-ol in the presence of sufficient amounts of CO to scavenge any OH radical formed during the reaction. According to the well-established gas-phase ozonolysis mechanism, the initially formed trioxolane will decompose in two possible ways to form either one or the other primary carbonyl, namely formaldehyde (HCHO) and HMPr, and the remaining Criegee intermediate. A secondary formation of both carbonyls resulting from further reactions of the Criegee intermediates is not likely based on the known mechanism. Moreover, by comparison with FTIR spectra of commercially available epoxides, we do not find any hint for epoxide formation in the gas-phase ozonolysis of 2-methyl-3-buten-2-ol, which is in agreement with previous studies (Carrasco et al., 2007, and references therein). Therefore, the sum of the molar yields of HCHO and HMPr should yield 100 %. The concentrations of HMPr in each spectrum are thus obtained based on the consumption of the HMPr-precursor 2-methyl-3-buten-2-ol and the molar formation yield of HMPr (YHMPr),
where YHCHO is the experimentally determined molar formation yield of HCHO. However, in doing so the determined cross section is cm molecule−1, which is about 300 % larger than the only available literature reference (Carrasco et al., 2006). If the literature reference is taken as the true value this would either indicate a fundamental underestimation of the HMPr concentration or an erroneous determination of the 2-methyl-3-buten-2-ol concentration in our experiments. The unsaturated alcohol is highly volatile and does not pose quantification problems. Moreover, no wall loss was measured in any of the chambers either for the alcohol or for the HMPr (see Sect. 3.3). A falsely determined cross section of the HMPr precursor by a factor up to 3 is therefore very unlikely. On the other hand, using the literature cross section would result in molar yields of about 190 % for HMPr in our experiments.
Carrasco et al. (2006) synthesized and purified HMPr in the liquid phase and evaporated different amounts of the sample. The concentrations were calculated according to the ideal gas law and subtracting the amounts of formic acid and formaldehyde still present in the liquid sample despite purification. Although the authors stated that HMPr has also been synthesized in situ, similarly to the present study, the cross section seems to be calculated based solely on the liquid sample evaporation. However, the listed integrated cross section for HCHO, based on the natural logarithm as stated by the authors, differs about a factor of 2 from other literature references (Nakanaga et al., 1982; Gratien et al., 2007). As well as this, the used value is about 2.3 smaller than the cited literature reference (Picquet-Varrault et al., 2002). All this suggests a systematic conversion error in the previous study. Therefore, we prefer to use the HMPr cross section estimated in our group for the following product study.
3.3 Product study of the OH reactions
In the following subsections the terms “α and β position” are used with respect to their position related to the carbonyl group. Thus, the Cα refers to the carbon atom adjacent to the carbonyl group. Consequently, the α-RO2 radical names the RO2 where the molecular oxygen added in α position.
3.3.1 3-Methyl-3-penten-2-one+OH
Figure 2 shows details obtained by evaluating IR spectra recorded during a 3M3P2+OH experiment and the references used to identify the reaction products. The range 3600–2600 cm−1 was chosen solely for clarity reasons. More spectra focussing on other spectral ranges can be found in Fig. S3 in the Supplement. Figure S4 in the Supplement shows reference spectra for the unsaturated ketones. The remaining absorption features present in the residual spectra after subtraction of 3M3P2, methyl nitrite, methyl nitrate, HNO3, HONO, HCHO, NO and NO2 can be unambiguously attributed to acetoin, biacetyl, acetaldehyde and peroxyacetyl nitrate (PAN), respectively. CO2 formation was clearly observed by examination of its absorption features in the range 2400–2250 cm−1 during the irradiation period. After subtraction of all clearly assigned spectral features the residual spectrum contains only weak absorption bands centred on 1654, 1364, 1297 and 856 cm−1. Although a possible indication for the formation of organic nitrates, they are too weak to be reasonably interpreted.

Figure 2Exemplary FTIR spectra of a product study experiment of 3M3P2+OH: (a) reaction mixture before irradiation; (b) reaction mixture at the end of the irradiation period; (c) residual spectrum after subtraction of methyl nitrite, methyl nitrate, HNO3, HONO, NO, NO2 and HCHO from (b); (d) reference spectrum of 3-hydroxy-2-butanone (acetoin); (e) residual spectrum after subtraction of 3M3P2 and acetoin from (c); (f) reference spectrum of 2,3-butanedione (biacetyl); and (g) reference spectrum of acetaldehyde. The spectra are shifted and scaled individually for a better overview.
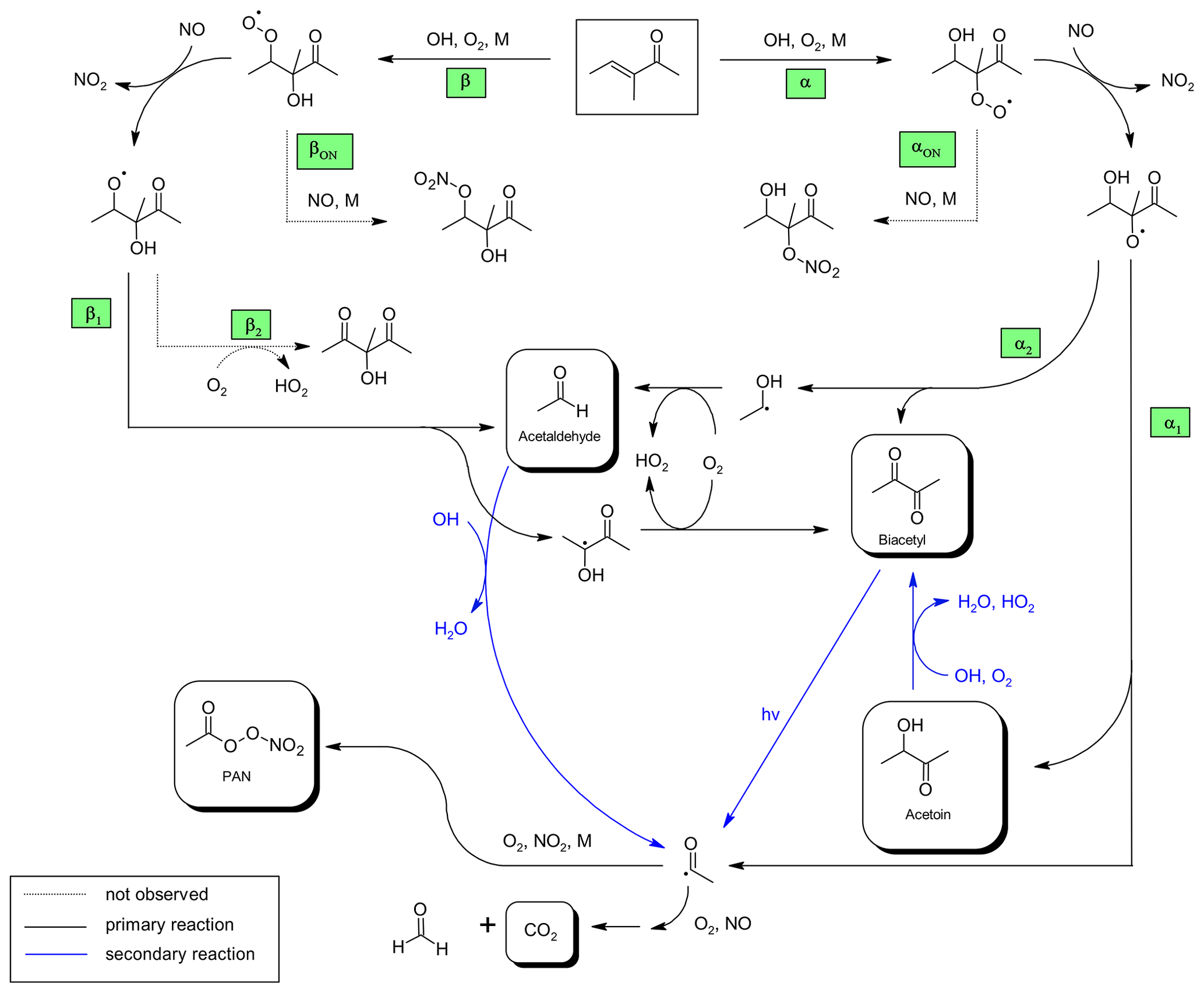
Figure 3Proposed mechanism for the OH-radical-initiated oxidation of 3-methyl-3-penten-2-one and further oxidation of the first-generation products relevant under the experimental conditions.
The formation of the identified species can be well explained as first-generation products of the OH-initiated oxidation of 3M3P2, according to the proposed mechanism depicted in Fig. 3. The OH radical is expected to add predominantly to the C=C double bond in either α or β position followed by the addition of O2 to form the corresponding α- or β-RO2 radical (=hydroxyperoxy radical). Their formation and further oxidation pathways (Fig. 3) are consecutively defined as αn and βn pathways, respectively. Due to the presence of NO in excess, under these experimental conditions it is certain that virtually all RO2 radicals will react with NO to mainly form the corresponding RO radicals (=hydroxyalkoxy radicals). The β-RO radical can undergo a bond scission (pathway β1, Fig. 3) between the α and β carbon (Cα and Cβ) to form acetaldehyde and a hydroxyalkyl radical. This, in turn, reacts with O2 to produce biacetyl. In principle, the H atom abstraction from the β-RO by O2 might lead to the formation of a 2-hydroxy-1,3-dicarbonyl species (pathway β2, Fig. 3). However, after subtraction of all identified species there are no remaining IR absorption bands to support the occurrence of this pathway. On the other hand, the reaction of RO2 with NO is known to be exothermic, resulting in chemically activated RO radicals potentially prone to “prompt” decomposition (Orlando et al., 2003). This pathway was shown to be important for RO radicals where energy barriers to decomposition are low, as expected for the β-RO radical according to available estimation methods (Atkinson, 2007; Vereecken and Peeters, 2009). As well as this, Atkinson (2007) concluded that decomposition will dominate compared to the reaction with O2 in the case of 1,2-hydroxyalkoxy radicals. This also applies for the thermalized hydroxyalkoxy radicals. All this suggests that either pathway β2 does not exist if the precursor β-RO radical is formed through RO2+NO or its branching fraction is very low.
The addition of the OH radical in β position followed by the addition of O2 and reaction with NO yields eventually acetoin (pathways α and α1, Fig. 3). The bond scission in the α-RO radical between Cα and the carbon atom of the carbonyl moiety (pathway α1, Fig. 3) yields further acetyl radicals which, after addition of O2, can either form peroxyacetyl nitrate (through addition of NO2) or react with NO to finally form CO2 and HCHO, under the experimental conditions. The yield of HCHO related to 3M3P2 oxidation cannot be experimentally determined because it is also produced in the methyl nitrite photolysis itself. On the other hand, the α-RO radical can also form biacetyl if the bond scission occurs between Cα and Cβ (pathway α2, Fig. 3). In this case acetaldehyde evolves from the simultaneously formed hydroxyalkyl radical due to H atom abstraction through molecular oxygen.
3.3.2 4-Methyl-3-penten-2-one+OH
Figure 4 depicts evaluation details of IR spectra recorded during a product study experiment of 4M3P2 and references. The spectral range 3200–2600 cm−1 was chosen as mentioned above. Acetone, methyl glyoxal and peroxyacetyl nitrate are clearly identifiable in all residual spectra after subtraction of 4M3P2, methyl nitrite, methyl nitrate, HNO3, HONO, HCHO, NO and NO2. As in the 3M3P2 product studies, the CO2 formation during the irradiation period was unambiguously observed in the range 2400–2250 cm−1 (Fig. S5 in the Supplement). Trace (f) in Fig. 4 shows an exemplary residual spectrum after subtraction of methyl glyoxal, acetone and PAN references from the spectrum in trace (c). The spectral features are in excellent agreement, in the presented range, with a reference IR spectrum of 2-hydroxy-2-methylpropanal (see Sect. 3.2). This proves its formation in the OH-radical-initiated oxidation of 4M3P2. After subtraction of all assigned absorptions the remaining residual spectra, in both 480 and 1080 L product study experiments, contain weak but well-defined absorption bands centred around 3478, 1722, 1654, 1297 and 856 cm−1 (Fig. 5). The characteristic absorption pattern of the latter three bands is a strong indication for organic nitrate formation in this reaction system.
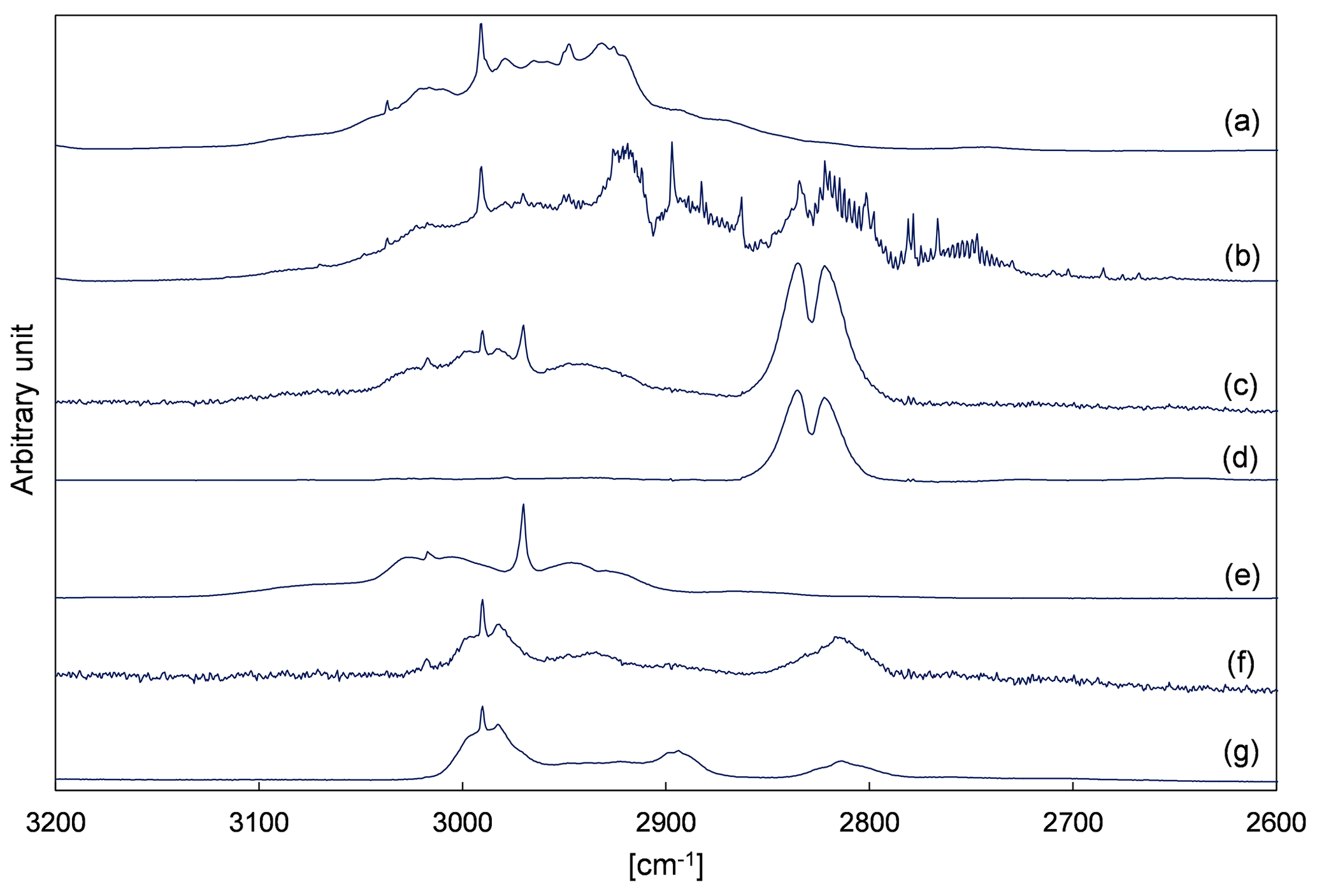
Figure 4Exemplary FTIR spectra of a product study experiment of 4M3P2+OH: (a) reaction mixture before irradiation; (b) reaction mixture at the end of the irradiation period; (c) residual spectrum after subtraction of 4M3P2, methyl nitrite, methyl nitrate, HONO, HNO3, NO, NO2 and HCHO from (b); (d) reference spectrum of methyl glyoxal; (e) reference spectrum of acetone; (f) residual spectrum after subtracting methyl gyloxal, acetone and PAN from (c); and (g) reference spectrum of 2-hydroxy-2-methylpropanal generated in situ. The spectra are shifted and scaled individually for a better overview.
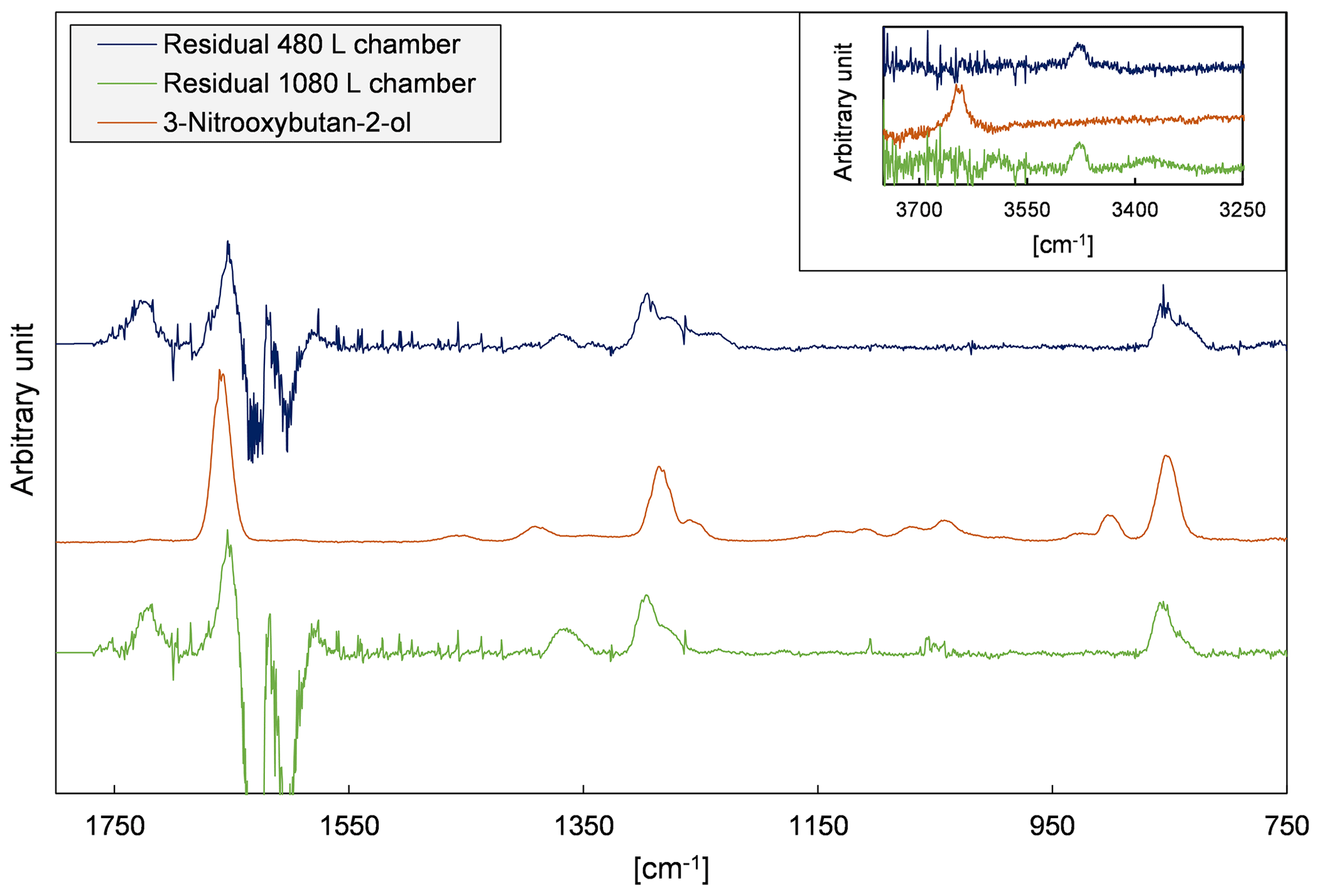
Figure 5Residual spectra of 4M3P2+OH product study experiments performed in both chambers after subtraction of all identified species and an exemplary reference spectrum of an organic nitrate recorded in our laboratory (Spittler, 2001).
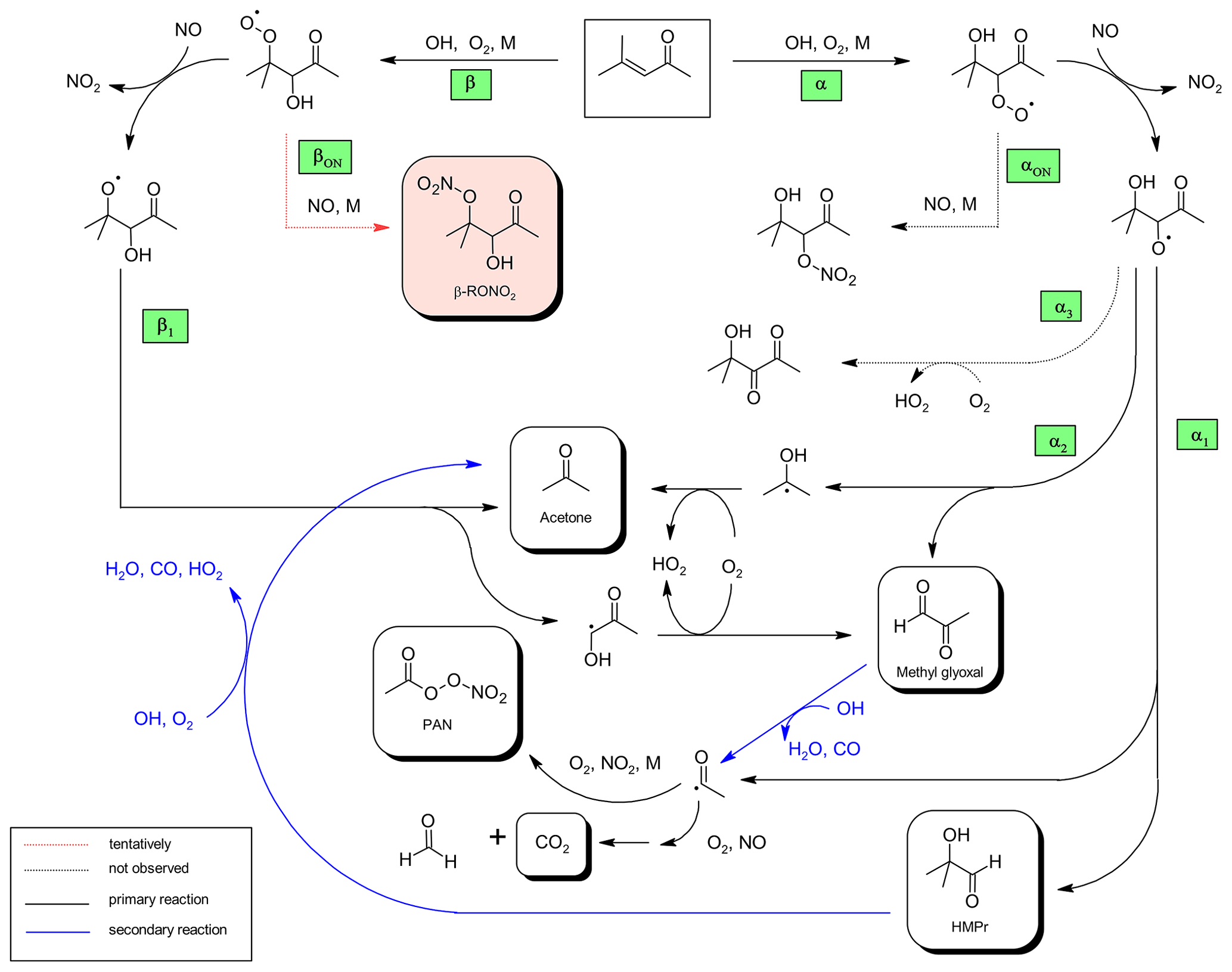
Figure 6Proposed mechanism for the OH-radical-initiated oxidation of 4-methyl-3-penten-2-one and further oxidation of the first-generation products relevant under the experimental conditions.
Based on the experimental results obtained here a mechanism for the reaction of OH radicals with 4M3P2 was drawn (Fig. 6). The OH radical will add predominantly to either the α or β carbon. The subsequent O2 addition will yield the corresponding β- or α-RO2 radical, respectively. As for 3M3P2, all 4M3P2 product studies were conducted under conditions where RO2 radicals are expected to react solely with NO. Acetone can be formed from the conversion of the β-RO2 with NO into the corresponding β-RO followed by the scission of the C−C bond between Cα and Cβ (pathway β1, Fig. 6). The synchronously generated hydroxyalkyl radical might react with O2 to yield methyl glyoxal. HMPr formation can be explained by a bond scission between Cα and the carbon atom of the carbonyl group (pathway α1, Fig. 6). In the present experimental system, the co-generated acetyl radicals yield either PAN or CO2 and HCHO, as discussed above. Methyl glyoxal and acetone can also be formed according to pathway α2 (Fig. 6) if the bond scission in the α-RO occurs between Cα and Cβ.
Hypothetically, the α-RO radical could also produce a 3-hydroxy-1,2-dicarbonyl species (pathway α3, Fig. 6) if H atom abstraction via molecular oxygen would occur rather than a C−C bond scission. However, the structure of the weak carbonyl absorption in the residual spectra seems more likely to belong to a single C=O bond rather than a vicinal diketone. There is thus no indication for the existence of this reaction pathway. Besides, the reaction RO+O2 is not expected to be competitive to the decomposition channels of the multifunctional RO radical, as discussed above.
Organic nitrate formation, which is indicated by the residual spectra, is expected to proceed through the reaction of the α-RO2 or β-RO2 radical with NO (pathway αON or βON, Fig. 6) followed by the isomerization of the nascent ROONO adduct (Calvert et al., 2015). RONO2 formation has also been observed through RO+NO2 reactions of simple alkoxy radicals (Frost and Smith, 1990; Mund et al., 1998). However, high-pressure rate coefficients for these types of reactions are about (1– (Atkinson et al., 2006), whereas for the reactions of RO with O2, k×[O2] is about 4×104 s−1 according to a recommendation provided by Atkinson (2007). Given that RO+O2 seems even not to compete with unimolecular decomposition for β-hydroxyalkoxy radicals we do not expect RO+NO2 to be experimentally and atmospherically relevant. The absorption features observed in the residual spectra, additionally to the characteristic nitrate absorptions, indicate the presence of a carbonyl (1722 cm−1) and an OH group (3478 cm−1) which can be assigned to multifunctional hydroxycarbonyl nitrates as presented in Fig. 6. In the case of the RONO2 species resulting from the α-RO radical, one would expect that intramolecular hydrogen bonding between the OH- and the carbonyl group stabilize the structure. This would cause, on the one hand, a broader and weaker OH absorption band and, on the other hand, a shift of the carbonyl absorption towards lower wavenumbers. Based on that, it is more likely that the residual absorptions to the hydroxycarbonyl nitrate result from the β-RO2 radical. The formation of organic nitrates will be further discussed in Sect. 3.3.5.
3.3.3 Product yields correction and further oxidation processes
Product yields were obtained by plotting their mixing ratio versus the mixing ratio of consumed unsaturated ketone. The data are corrected only for the wall loss of the unsaturated ketones. These plots are shown in Figs. S6 and S7 in the Supplement and exhibit a high linearity for all identified products except for PAN, CO2, or the sum of PAN and CO2. The latter was used to determine the molar formation yield of acetyl radicals. The non-linearity is a strong indication for further oxidation and secondary processes in the investigated reaction systems leading to acetyl radicals and their further oxidation products. However, this can be well explained by the oxidation of the initially formed reaction products in the OH-radical-initiated oxidation of the unsaturated ketones. On the other hand, the linearity, observed for the other oxidation products, does not necessarily indicate the absence of secondary processes. Either secondary formation compensates for loss processes or the scattering of the combined data is larger than the precise non-linearity.
According to Aschmann et al. (2000) the OH-initiated oxidation of acetoin proceeds predominantly through alkyl H atom abstraction at the −CH(OH) entity to eventually yield biacetyl, with a formation yield of about 80 %. This is also expected to be the main loss process under atmospheric conditions. Therefore, the further oxidation of acetoin is an additional source of biacetyl in the 3M3P2 experimental system. Biacetyl itself is mainly subject to photolysis (R15), under both the present experimental and atmospheric conditions yielding acetyl radicals.
Acetaldehyde also contributes to the formation of acetyl radicals since the aldehydic H atom abstraction (Reaction R16) was shown to account for about 95 % of the OH reaction (Calvert et al., 2011).
Acetone, formed in the oxidation of 4M3P2, will be mainly oxidized by OH radicals through H atom abstraction yielding acetonoxy radicals which readily decompose to HCHO and acetyl radicals (Orlando et al., 2000).
However, while being a source of HCHO and acetyl radicals under atmospheric conditions, this reaction cannot play any role in the present experimental set-up, considering the lifetime of acetone with respect to OH. By contrast, HMPr was shown to display a much higher reactivity towards OH, the reaction producing acetone with a yield of unity (Carrasco et al., 2006). Thus, this is a secondary source of acetone in our experiments. Finally, the OH reaction of methyl glyoxal will exclusively proceed via the abstraction of the aldehydic H atom. The initially formed CH3C(O)CO radical will readily dissociate into carbon monoxide and CH3C(O) radicals as well (Green et al., 1990). Based on the lamp spectrum we calculated the photolysis frequency of methyl glyoxal for all experimental conditions. The ratio between and J(CH3C(O)CHO) was found to be typically >7. Hence, while photolysis of methyl glyoxal is the main loss process under most atmospheric daytime conditions the OH reaction dominates in the present experimental system.
All these processes interfere in the determination of the intrinsic yield of CH3C(O) radicals in the reaction of the unsaturated ketones with OH. As discussed above, the further chemistry of acetyl radicals in our experiments may evolve either into PAN or formation. However, in the atmosphere the readily formed acetyl peroxy radical may also react with HO2 radicals to form peroxyacetic acid (CH3C(O)OOH), acetic acid (CH3C(O)OH), O3 and OH radicals (Winiberg et al., 2016). Therefore, in order to evaluate the atmospheric importance of the studied ketones an estimation of the acetyl radicals yield is needed.
Table 4Uncorrected and corrected molar yields of the 3M3P2+OH system.

Table 5Uncorrected and corrected molar yields of the 4M3P2+OH system.
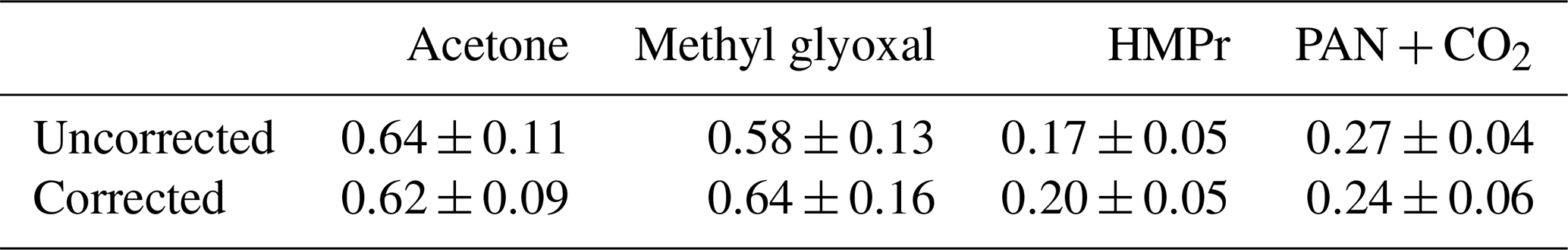
The product yields corrected for the secondary reactions mentioned above and wall losses in the simulation chambers were obtained using the model outlined in Sect. 2.7 and the kinetic parameters listed in Tables S1 and S2 in the Supplement. The molar yields for both OH reactions are summarized in Tables 4 and 5. The sum of PAN and CO2 is equal to the molar yield of CH3C(O) radicals. The errors represent the 2σ statistical error resulting from the average of all experiments and an additional 10 % relative error to cover further uncertainties derived from the evaluation procedure. Exemplary experimental and simulated time profiles of the ketones and the products are shown in Figs. 7 and 8 for both investigated reaction systems.
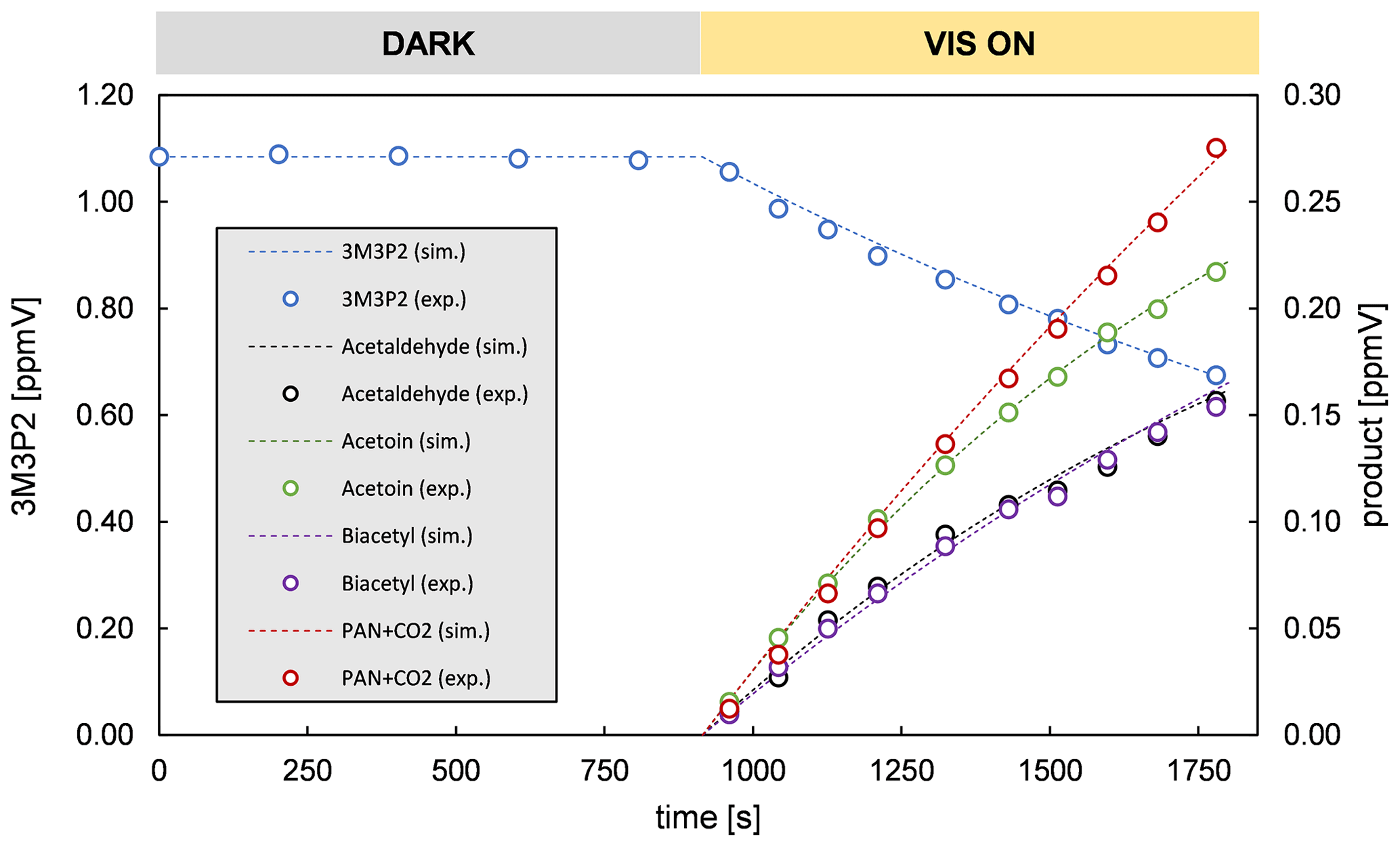
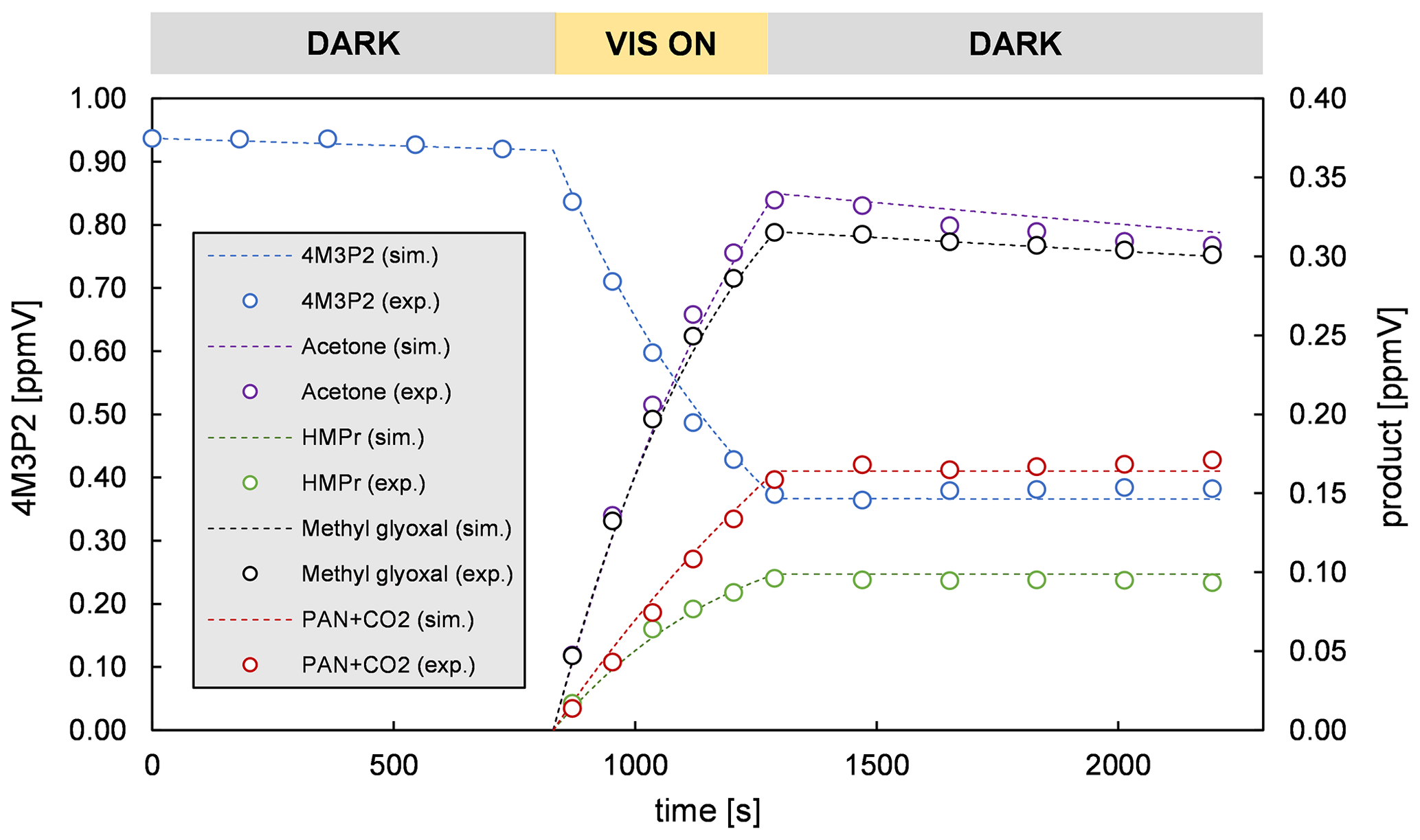
Acetoin and acetaldehyde are only affected by secondary consumption. Hence, the model estimates an increase for both yields compared to the experimental values of the 3M3P2 oxidation. By contrast, the biacetyl yields exhibit no difference indicating that the loss by photolysis is nearly compensated for by the formation through acetoin oxidation. Both acetaldehyde and biacetyl undergo further oxidation eventually forming PAN and CO2. In compliance, the experimental concentration–time profile for acetyl radicals is only reproduced when introducing significantly lower PAN+CO2 formation yields in the model. In the 4M3P2 system the modelled acetone yield decreases slightly due to the further oxidation of HMPr. Since methyl glyoxal and HMPr are influenced only by consumption processes, their corrected yields are higher than those experimentally determined. PAN+CO2 correction follows the same pattern as in the case of 3M3P2+OH.
In all cases the averaged molar yields are in excellent agreement for products expected to be formed in the same reaction channel. Thus, the molar yields of acetaldehyde/biacetyl (pathway β1 and α2, Fig. 3), acetoin/(PAN+CO2) (pathway α1, Fig. 3) and acetone/methyl glyoxal (pathway β1 and α2, Fig. 6) are nearly the same. In the case of 4M3P2 + OH, the yields of HMPr/(PAN+CO2) formed according to pathway α1 (Fig. 6) are still in good agreement within the uncertainties. This supports the absorption cross section of HMPr determined in our study. On the other hand, the results prove that the formation of acetyl radicals can be well quantified by determining the sum of PAN and CO2 in the experimental set-up.
Among α,β-unsaturated ketones of atmospheric importance only the OH-radical-initiated oxidation of methyl vinyl ketone (MVK) has been investigated in depth (Tuazon and Atkinson, 1989; Galloway et al., 2011; Praske et al., 2015; Fuchs et al., 2018). Tuazon and Atkinson (1989) quantified methyl glyoxal and glycolaldehyde as the main oxidation products and concluded that addition of the OH radical to the internal and terminal carbon atom accounts for 28±9 % and 72±21 %, respectively. This calculation is based on the assumption that the corresponding α-RO radical will favour a bond scission between the carbonyl carbon atom and Cα (according to pathway α1 in Figs. 3 and 6) due to the much lower predicted energy barrier to decomposition following this pathway (Tuazon and Atkinson, 1989). This has been confirmed by calculations performed by Praske et al. (2015) and is consistent with the SAR provided by Vereecken and Peeters (2009). Assuming α1≫α2 (Figs. 3 and 6) the addition of OH according to the α and β pathways consequently accounts for 60±18 % and 40±12 % for 3M3P2 and 26±8 % and 74±22 % in the case of 4M3P2, respectively, when referenced to the corresponding overall yield. However, at least for 4M3P2 the branching fraction α2 may be important since the estimated energy barrier is lower than for α1 according to the SAR of Vereecken and Peeters (2009). Hence, one should note that the fraction given for the addition to Cβ (α pathways) represents a lower limit and an upper limit for the addition of OH to Cα (β pathways), respectively. In the limiting case (α1≫α2) this indicates the relationship between the branching ratios of the main channels to be α>β for MVK, α≈β for 3M3P2 and α<β for 4M3P2 (see Figs. 3 and 6). Due to hyperconjugation the formation of the higher substituted alkyl radical should be favoured. The addition of OH to Cα leads to a primary alkyl radical in the case of MVK, a secondary alkyl radical for 3M3P2 and a tertiary alkyl radical in the case of 4M3P2. Therefore, while hydrogen bonding should yield a preference of the addition to the β position for all α,β-unsaturated ketones, as discussed previously with respect to the reactivity, the observed trend in the branching ratios (in the limiting case α1≫α2) is possibly related to the stability of the initially formed alkyl radicals. In the case of 4M3P2 the addition to the β position could also be sterically hindered.
However, one should emphasize that it is not possible to derive the exact branching ratios for α and β without deciphering the branching ratios α1 and α2. An attempt to obtain the corresponding rate coefficients for each decomposition channel according to Vereecken and Peeters (2009) failed since the calculated branching ratios for α1 and α2 are about 0.1 and 0.9, respectively, in the case of 4M3P2 which is contradicted by the observed first-generation yields of 2HMPr and PAN + CO2. Vereecken and Peeters (2009) stated the accuracy of the predicted rate coefficients to be within a factor of 5–10. Therefore, it is not possible to derive any further statement on the ratio α1:α2.
3.3.4 ROONO2 formation
To further elucidate the mechanism, experiments were conducted over a wider range of NO2NO ratios by adding different amounts of NO2 to the reaction mixture. In all experiments the amount of added NO was sufficient to suppress any ozone formation.
Given that CH3C(O) radicals instantaneously react with oxygen, under the employed experimental conditions, to form the corresponding RO2 radical, their fate should be described by (a) the reaction with NO, (b) the reaction with NO2 to yield peroxyacetyl nitrate (PAN), and (c) the thermal dissociation of PAN to re-generate the RO2 radical.
Based on the Reactions (R20)–(R23) the ratio PANCO2 should only depend on the NO2NO ratio during the reaction under constant pressure and temperature conditions. Therefore, PANCO2 ratios were simulated for various NO2NO ratios in the experimental temperature range 295–301 K using the IUPAC recommendations (Atkinson et al., 2006) for the Reactions (R20), (R22) and (R23) (Fig. 9). Experimental PANCO2 formation ratios were derived from plotting the generated PAN against the formed CO2 during the irradiation period. The corresponding average NO2NO ratios were determined by averaging the measured mixing ratios of NO and NO2, respectively, over the same time interval. As can be seen in Fig. 9, these data are qualitatively in quite good agreement with the expected ratios based on the model calculations. Hence, the fate of the CH3C(O) radicals is well-described by the Reactions sequence (R19)–(R23) in our experiments.
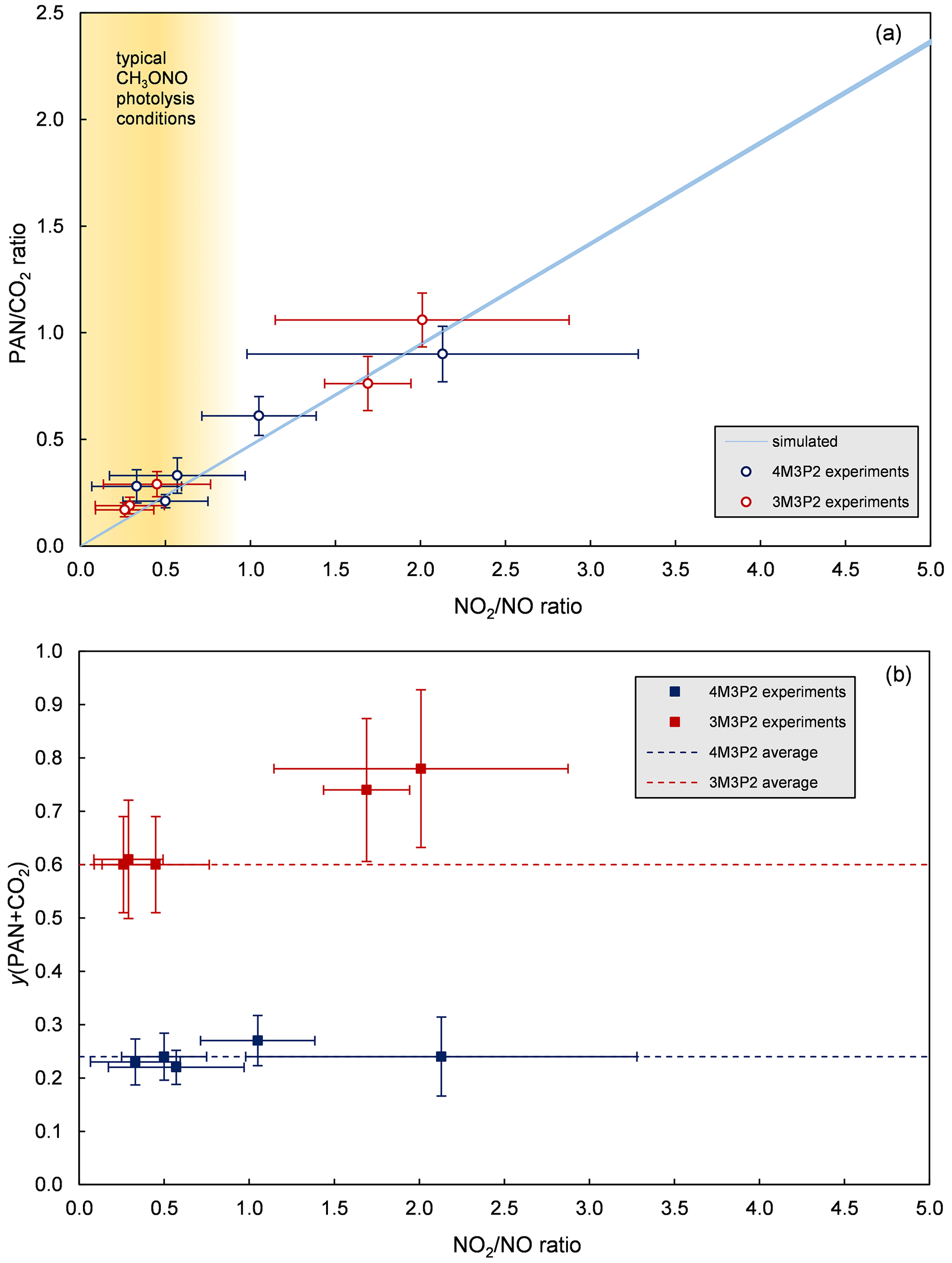
Figure 9(a) Experimentally determined and simulated formation ratio as a function of the NO2NO ratio. (b) Determined molar yields for acetyl radicals (as the sum of PAN and CO2) as a function of the NO2NO ratio.
However, in typical methyl nitrite photolysis experiments the NO2NO ratios are <1 if NO is added to suppress ozone formation. Thus, PAN accounts for less than one-third of the CH3C(O) radical's fate (Fig. 9). If the experimental set-up does not allow HCHO or CO2 to be quantified, this leads to a fundamental underestimation of the acetyl radical reaction channels. In this case, addition of NO2 could be useful to favour PAN formation and the determination of the NO2NO ratio in the experiment could yield an estimation of the PANCO2 ratio.
On the other hand, Fig. 9 clearly shows the invariance of the PAN+CO2 yield for 4M3P2 under all experimental conditions, within the uncertainties. There is thus likely no ROONO2 formation from the initially formed α- and β-RO2 radicals since they are either not formed or their thermal dissociation is too large to play any role. This meets one's expectations as, according to literature references (Calvert et al., 2015), the lifetimes of alkylperoxy nitrates are of the order of seconds at 298 K and 1 atm and so they become relevant as a NOx reservoir when formed at lower temperatures, encountered in the upper troposphere. However, assuming an average OH concentration of 1×106 cm−3 (Bloss et al., 2005) the atmospheric lifetime (with respect to OH) is about 3.4 h, thus too short for 4M3P2 to enter higher altitudes. Therefore, except for PAN, ROONO2 formation does not play any role for 4M3P2.
In the case of 3M3P2 significantly higher yields for PAN+CO2 were observed for higher NO2NO ratios while being quite consistent at (Fig. 9). This is actually contradictory as additional ROONO2 formation from the initial α- and β-RO2 radicals should lower the overall yields of the main products. However, the molar yields of biacetyl and acetaldehyde are essentially the same as in the other experiments. Thus, the carbon balance exceeds 100 % in the higher NO2NO ratio experiments. Unfortunately, these two experiments were the last in a series and it is quite probable that due to wall loading the wall acted as a source of CH3C(O) radicals during the irradiation. The yield for PAN+CO2 was therefore only given as an average of the experiments with .
Table 6Estimated tropospheric lifetimes for the studied α,β-unsaturated ketones. The lifetimes were calculated using the following concentrations: a global mean of 1×106 molecules cm−3 (Bloss et al., 2005), b 12 h average of 3×104 molecules cm−3 (Wingenter et al., 1996), c 24 h average of 7×1011 molecules cm−3 (Logan, 1985), and d 12 h average of 5×108 molecules cm−3 (Atkinson, 1991). Rate coefficients were taken from: e this work, f Illmann et al. (2021a), and g Canosa-Mas et al. (2005).

3.3.5 RONO2 formation
Given that the carbon balance is about one for 3M3P2 and about 0.85 for 4M3P2, respectively, this results in an upper limit of the overall RONO2 yield of 0.15 for 4M3P2 while almost no RONO2 formation occurs in the case of 3M3P2. This is qualitatively in good agreement with the residual spectra, clearly indicating the presence of organic nitrates only in the case of 4M3P2. Noda et al. (2000) determined an average absorption cross section of cm molecule−1 (base 10) for the nitrooxy group absorption in the range 800–900 cm−1 by averaging available absorption cross sections of organic nitrates. Following this approach, an estimated RONO2 yield of 0.06±0.03 is determined, which should be regarded as lower limit. While the determined wall loss is of the order of s−1 the potentially formed RONO2 species could also be subject of photolysis and oxidation by OH radicals. SAR methods for the prediction of OH rate coefficients were shown to fail at carbonyl nitrates (Suarez-Bertoa et al., 2012). Therefore, Suarez-Bertoa et al. (2012) proposed alternative substituent factors optimized for carbonyl nitrates in which the factor for the −ONO2 group is less deactivating than in other SAR approaches. Applying this factor to the SAR of Kwok and Atkinson (1995) yields predicted rate coefficients of and for the α-RONO2 and β-RONO2, respectively, which would correspond to loss rates of about and s−1, respectively, due to the OH reaction. For α- and β-carbonyl nitrates it was shown that photolysis dominates over the OH-initiated oxidation (Suarez-Bertoa et al., 2012; Picquet-Varrault et al., 2020). Hence, by comparison with available data (Suarez-Bertoa et al., 2012; Picquet-Varrault et al., 2020) larger loss rates likely result from photolysis of the RONO2 species in our experiments. However, we believe that any further statement would be highly speculative. A RONO2 yield of about 0.11±0.03 has been reported for MVK based on model-assisted isoprene photooxidation experiments (Paulot et al., 2009). On the other hand, an overall RONO2 yield of about 0.040±0.006 has been determined in MVK oxidation experiments (Praske et al., 2015). Thus, the limits (0.06–0.15) reported here for the overall organic nitrate yield are consistent with the data dispersion found in the literature.
If the branching fractions for the pathways αON and βON in the 4M3P2 oxidation (Fig. 6) were the same this would correspond to a much larger formation yield of the multifunctional β-RONO2 than α-RONO2 simply due to the predominant addition of the OH radical to Cα. This would be consistent with the previous assignment of the residual's spectral features to the β-RONO2. Former studies on alkenes have shown a structure-dependent RONO2 yield derived from β-hydroxyperoxy radicals. Accordingly, the nitrate yields resulting from tertiary RO2 were larger than those produced from secondary RO2, which, in turn, were larger when compared to nitrate yields obtained from primary RO2 (Matsunaga and Ziemann, 2010). This would indicate βON>αON in the 4M3P2 oxidation (Fig. 6). Besides, Praske et al. (2015) found a 2-times-larger branching ratio for the β-RONO2 species resulting from MVK+OH than for the α-RONO2. They interpreted this finding in terms of a larger destabilizing effect on the initially formed ROONO complex caused by the closer carbonyl group in the case of the α-RONO2. Both results further support the assignment of the residual absorptions to the β-RONO2 species.
By contrast, both potentially formed RONO2 species in the 3M3P2 oxidation would contain a quaternary Cα atom surrounded by bulky substituents. Thus, the almost negligible nitrate formation in the case of 3M3P2 is possibly attributed to the steric hindrance of the hypothetically resulting RONO2 species.
Within this work we determined the rate coefficients for the OH radical and Cl atom-initiated oxidation of 3M3P2 and 4M3P2. This adds to the kinetic information concerning the reaction of these two compounds with NO3 and O3 reported previously (Sato et al., 2004; Canosa-Mas et al., 2005; Illmann et al., 2021a; Li et al., 2021) in an effort to complete the gaps in the knowledge needed for modelling chemistry in the atmosphere. The yields for the identified products formed in both target reactions were found to be independent of the chamber used, the mixing ratios and the light intensity, thus giving confidence in the experimental results.
Using the kinetic data together with reasonable average concentrations for the respective oxidants (Logan, 1985; Atkinson, 1991; Wingenter et al., 1996; Bloss et al., 2005) allows tropospheric lifetimes to be estimated as presented in Table 6, where τ(X) is the lifetime with respect to the oxidant X calculated according to . Both unsaturated ketones did not show measurable photolysis rates in the present experimental set-up. In the atmosphere, their photodissociation lifetimes are expected to have a range of the order of days (Mellouki et al., 2015). Consequently, compared to τ(OH), the photodissociation of both unsaturated ketones is evidently not an important process in the troposphere. The values in Table 6 indicate the OH radical as the dominant sink during daytime whereas the NO3 radical plays a similar role at night. However, for 3M3P2 the O3 reaction appears quite competitive during both day and night (Table 6). These estimated lifetimes indicate an oxidative degradation near the emission sources. Nevertheless, a further part of this work indicates that both ketones potentially impact atmospheric processes on a larger scale due to their huge potential of forming NOx reservoir species like PAN. In this respect, we have shown that the reaction of OH with both 3M3P2 and 4M3P2 yields CH3C(O) radicals by prompt decomposition of primary formed RO radicals. In the present work the sum of PAN and CO2 could be used successfully to determine the formation yield of CH3C(O) radicals in both reaction systems. Moreover, nearly all other identified oxidation products like methyl glyoxal, acetone, acetaldehyde, acetoin and biacetyl are known to generate CH3C(O) radicals in their further oxidation processes – simulations based on field data collected worldwide estimate that acetaldehyde, methyl glyoxal and acetone within the troposphere account for about 81 % of the global source for PAN formation (Fischer et al., 2014). Among the oxidation products of both 3M3P2 and 4M3P2 at least methyl glyoxal is a well-known source of secondary organic aerosol in atmosphere (Fu et al., 2008).
On the other hand, future work is needed to identify clearly the missing products of the OH-radical-initiated oxidation of 4M3P2. The absorptions of the residual spectra could be tentatively assigned to the β-RONO2 species. However, other detection methods are necessary for an unambiguous identification and quantification of this species.
Data can be provided upon request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-13667-2021-supplement.
NI, RGG and IPK conducted the experiments and processed the data. NI developed the model and performed the calculations. NI prepared the paper with contributions from all co-authors.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
The authors gratefully acknowledge support from the EU Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity (grant agreement no. 730997) and the Deutsche Forschungsgemeinschaft (DFG) through the grant agreement WI 958/18-1. Rodrigo Gastón Gibilisco wish to acknowledges the Alexander von Humboldt Foundation for providing a Georg Forster Research Fellowship.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant no. WI 958/18-1) and the European Commission Horizon 2020 Framework Programme (grant no. EUROCHAMP-2020 (730997)).
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Allen, G., Remedios, J. J., Newnham, D. A., Smith, K. M., and Monks, P. S.: Improved mid-infrared cross-sections for peroxyacetyl nitrate (PAN) vapour, Atmos. Chem. Phys., 5, 47–56, https://doi.org/10.5194/acp-5-47-2005, 2005.
Aschmann, S. M., Arey, J., and Atkinson, R.: Atmospheric Chemistry of Selected Hydroxycarbonyls, J. Phys. Chem. A, 104, 3998–4003, https://doi.org/10.1021/jp9939874, 2000.
Atkinson, R: Kinetics and Mechanisms of the Gas-Phase Reactions of the NO3 Radical with Organic Compounds, J. Phys. Chem. Ref. Data, 20, 459–507, https://doi.org/10.1063/1.555887, 1991.
Atkinson, R: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Barnes, I., Becker, K. H., and Zhu, T.: Near UV Absorption Spectra and Photolysis Products of Difunctional Organic Nitrates: Possible Importance as NOx Reservoirs, J. Atmos. Chem., 17, 353–373, https://doi.org/10.1007/BF00696854, 1993.
Barnes, I., Becker, K. H., and Mihalopoulos, N.: An FTIR Product Study of the Photooxidation of Dimethyl Disulfide, J. Atmos. Chem., 18, 267–289, https://doi.org/10.1007/BF00696783, 1994.
Bickers, D. R., Calow, P., Greim, H. A., Hanifin, J. M., Rogers, A. E., Saurat, J.-H., Sipes, I. G., Smith, R. L., and Tagami, H.: The safety assessment of fragrance materials, Regul. Toxicol. Pharm., 37, 218–273, https://doi.org/10.1016/S0273-2300(03)00003-5, 2003.
Blanco, M. B., Barnes, I., and Wiesen, P.: Kinetic Investigation of the OH Radical and Cl Atom Initiated Degradation of Unsaturated Ketones at Atmospheric Pressure and 298 K, J. Phys. Chem. A, 116, 6033–6040, https://doi.org/10.1021/jp2109972, 2012.
Bloss, W. J., Evans, M. J., Lee, J. D., Sommariva, R., Heard, D. E., and Piling, M. J.: The oxidative capacity of the troposphere: Coupling of field measurements of OH and a global chemistry transport model, Faraday Discuss., 130, 425–436, https://doi.org/10.1039/B419090D, 2005.
Calvert, J. G., Atkinson, R., Kerr, J. A., Madronich, S., Moortgat, G. K., Wallington, T. J., and Yarwood, G.: The mechanisms of atmospheric oxidation of the alkenes, Oxford University Press, New York, 2000.
Calvert, J. G., Mellouki, A., Orlando, J. J., Pilling, M. J., and Wallington, T. J.: The mechanisms of atmospheric oxidation of the oxygenates, Oxford University Press, New York, 2011.
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, New York, 2015.
Canosa-Mas, C. E., Flugge, M. L., King, M. D., and Wayne, R. P.: An experimental study of the gas-phase reaction of the NO3 radical with α,β-unsaturated carbonyl compounds, Phys. Chem. Chem. Phys., 7, 643–650, https://doi.org/10.1039/B416574H, 2005.
Carrasco, N., Doussin, J.-F., Picquet-Varrault, B., and Carlier, P.: Tropospheric degradation of 2-hydroxy-2-methylpropanal, a photo-oxidation product of 2-methyl-3-buten-2-ol: Kinetic and mechanistic study of its photolysis and its reaction with OH radicals, Atmos. Environ., 40, 2011–2019, https://doi.org/10.1016/j.atmosenv.2005.11.042, 2006.
Carrasco, N, Doussin, J. F., O'Connor, M., Wenger, J. C., Picquet-Varrault, B., Durand-Jolibois, R., and Carlier, P: Simulation Chamber Studies of the Atmospheric Oxidation of 2-Methyl-3-buten-2-ol: Reaction with Hydroxyl Radicals and Ozone Under a Variety of Conditions, J. Atmos. Chem., 56, 33–55, https://doi.org/10.1007/s10874-006-9041-y, 2007.
Chapuis, C. and Jacoby, D.: Catalysis in the preparation of fragrances and flavours, Appl. Catal. A-Gen., 221, 93–117, https://doi.org/10.1016/S0926-860X(01)00798-0, 2001.
Etzkorn, T., Klotz, B., Sørensen, S., Patroescu, I. V., Barnes, I., Becker, K. H., and Platt, U.: Gas-phase absorption cross sections of 24 monocyclic hydrocarbons in the UV and IR spectral ranges, Atmos. Environ., 33, 525–540, https://doi.org/10.1016/S1352-2310(98)00289-1, 1999.
Ezell, M. J., Wang, W., Ezell, A. A., Soskin, G., and Finlayson-Pitts, B. J.: Kinetics of reactions of chlorine atoms with a series of alkenes at 1 atm and 298 K: structure and reactivity, Phys. Chem. Chem. Phys., 4, 5813–5820, https://doi.org/10.1039/B207529F, 2002.
Fischer, E. V., Jacob, D. J., Yantosca, R. M., Sulprizio, M. P., Millet, D. B., Mao, J., Paulot, F., Singh, H. B., Roiger, A., Ries, L., Talbot, R. W., Dzepina, K., and Pandey Deolal, S.: Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 14, 2679–2698, https://doi.org/10.5194/acp-14-2679-2014, 2014.
Frost, M. J. and Smith, I. W. M.: Rate Constants for the Reactions of CH3O and C2H5O with NO2 over a Range of Temperature and Total Pressure, J. Chem. Soc. Faraday T., 86, 1751–1756, https://doi.org/10.1039/FT9908601751, 1990.
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res. Atmos., 113, 1–17, https://doi.org/10.1029/2007JD009505, 2008.
Fuchs, H., Albrecht, S., Acir, I., Bohn, B., Breitenlechner, M., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Kaminski, M., Keutsch, F. N., Novelli, A., Reimer, D., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., Zaytsev, A., Kiendler-Scharr, A., and Wahner, A.: Investigation of the oxidation of methyl vinyl ketone (MVK) by OH radicals in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 8001–8016, https://doi.org/10.5194/acp-18-8001-2018, 2018.
Galloway, M. M., Huisman, A. J., Yee, L. D., Chan, A. W. H., Loza, C. L., Seinfeld, J. H., and Keutsch, F. N.: Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high−NOx conditions, Atmos. Chem. Phys., 11, 10779–10790, https://doi.org/10.5194/acp-11-10779-2011, 2011.
Gaona-Colmán, E., Blanco, M. B., and Teruel, M. A.: Kinetics and product identification of the reactions of (E)-2-hexenyl acetate and 4-methyl-3-penten-2-one with OH radicals and Cl atoms at 298 K and atmospheric pressure, Atmos. Environ, 161, 155–166, https://doi.org/10.1016/j.atmosenv.2017.04.033, 2017.
Gratien, A., Nilsson, E., Doussin, J.-F., Johnson, M. S., Nielsen, C. J., Stenstrøm, Y., and Picquet-Varrault, B.: UV and IR Absorption Cross-sections of HCHO, HCDO, and DCDO, J. Phys. Chem. A, 111, 11506–11513, https://doi.org/10.1021/jp074288r, 2007.
Green, M., Yarwood, G., and Niki, H.: FTIR Study of the Cl-Atom Initiated Oxidation of Methylglyoxal, Int. J. Chem. Kinet., 22, 689–699, https://doi.org/10.1002/kin.550220705, 1990.
Grosjean, D. and Williams II, E. L.: Environmental persistence of organic compounds estimated from structure-reactivity and linear free-energy relationships. Unsaturated Aliphatics, Atmos. Environ. A-Gen., 26, 1395–1405, https://doi.org/10.1016/0960-1686(92)90124-4, 1992.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hatch, L. E., Yokelson, R. J., Stockwell, C. E., Veres, P. R., Simpson, I. J., Blake, D. R., Orlando, J. J., and Barsanti, K. C.: Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 17, 1471–1489, https://doi.org/10.5194/acp-17-1471-2017, 2017.
Illmann, J. N., Patroescu-Klotz, I., and Wiesen, P.: Gas-phase reactivity of acyclic α,β-unsaturated carbonyls towards ozone, Phys. Chem. Chem. Phys., 23, 3455–3466, https://doi.org/10.1039/D0CP05881E, 2021a.
Illmann, N., Patroescu-Klotz, I., and Wiesen, P.: Biomass burning plume chemistry: OH radical initiated oxidation of 3-penten-2-one and its main oxidation product 2-hydroxypropanal, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-575, in review, 2021b.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-B, 1995.
Li, W., Dan. G., Chen, M., Wang, Z., Zhao, Y., Wang, F., Li, F., Tong, S., and Ge, M.: The gas-phase reaction kinetics of different structure of unsaturated alcohols and ketones with O3, Atmos. Environ., 254, 118394, https://doi.org/10.1016/j.atmosenv.2021.118394, 2021.
Logan, J. A.: Tropospheric Ozone: Seasonal Behavior, Trends, and Anthropogenic Influence, J. Geophys. Res., 90, 10463–10482, https://doi.org/10.1029/JD090iD06p10463, 1985.
Matsunaga, A. and Ziemann, P. J.: Yields of β-hydroxynitrates, dihydroxynitrates, and trihydroxynitrates formed from OH radical-initiated reactions of 2-methyl-1-alkenes, P. Natl. Acad. Sci. USA, 107, 6664–6669, https://doi.org/10.1073/pnas.0910585107, 2010.
Mellouki, A., Wallington, T. J., and Chen, J.: Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 115, 3984–4014, https://doi.org/10.1021/cr500549n, 2015.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021.
Mund, C., Fockenberg, C., and Zellner, R.: LIF Spectra of n-Propoxy Radicals and Kinetics of their Reactions with O2 and NO2, Ber. Bunsenges. Phys. Chem., 102, 709–715, https://doi.org/10.1002/bbpc.19981020502, 1998.
Nakanaga, T., Kondo, S., and Saëki, S.: Infrared band intensities of formaldehyde and formaldehyde-d2, J. Chem. Phys., 76, 3860–3865, https://doi.org/10.1063/1.443527, 1982.
Noda, J., Hallquist, M., Langer, S., and Ljungström, E.: Products from the gas-phase reaction of some unsaturated alcohols with nitrate radicals, Phys. Chem. Chem. Phys., 2, 2555–2564, https://doi.org/10.1039/B000251H, 2000.
Notario, A., Le Bras, G., and Mellouki, A.: Absolute Rate Constants for the Reactions of Cl Atoms with a Series of Esters, J. Phys. Chem. A., 102, 3112–3117, https://doi.org/10.1021/jp980416n, 1998.
Orlando, J. J., Tyndall, G. S., Vereecken, L., and Peeters, J.: The Atmospheric Chemistry of the Acetonoxy Radical, J. Phys. Chem. A, 104, 11578–11588, https://doi.org/10.1021/jp0026991, 2000.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4689, https://doi.org/10.1021/cr020527p, 2003.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, https://doi.org/10.5194/acp-9-1479-2009, 2009.
Picquet-Varrault, B., Doussin, J.-F., Durand-Jolibois, R., Pirali, O., and Carlier, P.: Kinetic and Mechanistic Study of the Atmospheric Oxidation by OH Radicals of Allyl Acetate, Environ. Sci. Technol., 36, 4081–4086, https://doi.org/10.1021/es0200138, 2002.
Picquet-Varrault, B., Suarez-Bertoa, R., Duncianu, M., Cazaunau, M., Pangui, E., David, M., and Doussin, J.-F.: Photolysis and oxidation by OH radicals of two carbonyl nitrates: 4-nitrooxy-2-butanone and 5-nitrooxy-2-pentanone, Atmos. Chem. Phys., 20, 487–498, https://doi.org/10.5194/acp-20-487-2020, 2020.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Profeta, L. T. M., Sams, R. L., and Johnson, T. J.: Quantitative Infrared Intensity Studies of Vapor-Phase Glyoxal, Methylglyoxal, and 2,3-Butanedione (Diacetyl), with Vibrational Assignments, J. Phys. Chem. A, 115, 9886–9900, https://doi.org/10.1021/jp204532x, 2011.
Sato, K., Klotz, B., Taketsuga, T., and Takaynagi, T.: Kinetic measurments for the reactions of ozone with crotonaldehyde and its methyl derivatives and calculations of transition-state theory, Phys. Chem. Chem. Phys., 6, 3696–3976, https://doi.org/10.1039/B402496F, 2004.
Siegel, H. and Eggersdorfer, M.: Ketones, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, https://doi.org/10.1002/14356007.a15_077, 2000.
Sifniades, S., Levy, A. B., and Bahl, H.: Acetone, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, https://doi.org/10.1002/14356007.a01_079.pub3, 2011.
Smith, I. W. M. and Ravishankara, A. R.: Role of Hydrogen-Bonded Intermediates in the Bimolecular Reactions of the Hydroxyl Radical, J. Phys. Chem. A, 106, 4798–4807, https://doi.org/10.1021/jp014234w, 2002.
Spittler, M.: Untersuchungen zur troposphärischen Oxidation von Limonen: Produktanalysen, Aerosolbildung und Photolyse von Produkten, PhD thesis, Bergische Universität Wuppertal, Germany, 2001.
Suarez-Bertoa, R., Picquet-Varrault, B., Tamas, W., Pangui, E., and Doussin, J.-F.: Atmospheric Fate of a Series of Carbonyl Nitrates: Photolysis Frequencies and OH-Oxidation Rate Constants, Environ. Sci. Technol., 46, 12502–12509, https://doi.org/10.1021/es302613x, 2012.
Talukdar, R. K., Zhu, L., Feierabend, K. J., and Burkholder, J. B.: Rate coefficients for the reaction of methylglyoxal (CH3COCHO) with OH and NO3 and glyoxal (HCO)2 with NO3, Atmos. Chem. Phys., 11, 10837–10851, https://doi.org/10.5194/acp-11-10837-2011, 2011.
Taylor, W. D., Allston, T. D., Moscato, M. J., Fazekas, G. B., Kozlowski, R., and Takacs, G. A.: Atmospheric photodissociation lifetimes for nitromethane, methyl nitrite, and methyl nitrate, Int. J. Chem. Kinet., 12, 231–240, https://doi.org/10.1002/kin.550120404, 1980.
Tuazon, E. C. and Atkinson, R.: A Product Study of the Gas-Phase Reaction of Methyl Vinyl Ketone with the OH Radical in the Presence of NOX, Int. J. Chem. Kinet., 21, 1141–1152, https://doi.org/10.1002/kin.550211207, 1989.
Tuazon, E. C., MacLeod, H., Atkinson R., and Carter, W. P. L.: α-Dicarbonyl yields from the NOx-air photooxidation of a series of aromatic hydrocarbons in air, Environ. Sci. Technol., 20, 383–387, https://doi.org/10.1021/es00146a010, 1986.
US EPA: Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11. United States Environmental Protection Agency, Washington, DC, USA, 2021.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure-activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/B909712K, 2009.
Vyskocil, A., Viau, C., and Lamy, S.: Peroxyacetyl nitrate: review of toxicity, Hum. Exp. Toxicol., 17, 212–220, https://doi.org/10.1177/096032719801700403, 1998.
Wang, J., Zhou, L., Wang, W., and Ge, M.: Gas-phase reaction of two unsaturated ketones with atomic Cl and O3: kinetics and products, Phys. Chem. Chem. Phys., 17, 12000–12012, https://doi.org/10.1039/C4CP05461J, 2015.
Wang, X., Hong, P., Kiss, A. A., Wang, Q., Li, L., Wang, H., and Qiu, T.: From Batch to Continuos Sustainable Production of 3-Methyl-3-penten-2-one for Synthetic Ketone Fragrances, ACS Sustain. Chem. Eng., 8, 17201–17214, https://doi.org/10.1021/acssuschemeng.0c05908, 2020.
Wingenter, O. W., Kubo, M. K., Blake, N. J., Smith Jr., T. W., Blake, D. R., and Rowland, F. S.: Hydrocarbon and halocarbon measurements as photochemical and dynamical indicators of atmospheric hydroxyl, atomic chlorine, and vertical mixing obtained during Lagrangian flights, J. Geophys. Res., 101, 4331–4340, https://doi.org/10.1029/95JD02457, 1996.
Winiberg, F. A. F., Dillon, T. J., Orr, S. C., Groß, C. B. M., Bejan, I., Brumby, C. A., Evans, M. J., Smith, S. C., Heard, D. E., and Seakins, P. W.: Direct measurements of OH and other product yields from the HO2+CH3C(O)O2 reaction, Atmos. Chem. Phys., 16, 4023–4042, https://doi.org/10.5194/acp-16-4023-2016, 2016.