the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jul 2021
| 16 Jul 2021
Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical
Rongrong Wu
Luc Vereecken
Epameinondas Tsiligiannis
Sungah Kang
Sascha R. Albrecht
Luisa Hantschke
Defeng Zhao
Anna Novelli
Hendrik Fuchs
Ralf Tillmann
Thorsten Hohaus
Philip T. M. Carlsson
Justin Shenolikar
François Bernard
John N. Crowley
Juliane L. Fry
Bellamy Brownwood
Joel A. Thornton
Steven S. Brown
Astrid Kiendler-Scharr
Andreas Wahner
Mattias Hallquist
Isoprene oxidation by nitrate radical (NO3) is a potentially important source of secondary organic aerosol (SOA). It is suggested that the second or later-generation products are the more substantial contributors to SOA. However, there are few studies investigating the multi-generation chemistry of isoprene-NO3 reaction, and information about the volatility of different isoprene nitrates, which is essential to evaluate their potential to form SOA and determine their atmospheric fate, is rare. In this work, we studied the reaction between isoprene and NO3 in the SAPHIR chamber (Jülich) under near-atmospheric conditions. Various oxidation products were measured by a high-resolution time-of-flight chemical ionization mass spectrometer using Br− as the reagent ion. Most of the products detected are organic nitrates, and they are grouped into monomers (C4 and C5 products) and dimers (C10 products) with 1–3 nitrate groups according to their chemical composition. Most of the observed products match expected termination products observed in previous studies, but some compounds such as monomers and dimers with three nitrogen atoms were rarely reported in the literature as gas-phase products from isoprene oxidation by NO3. Possible formation mechanisms for these compounds are proposed. The multi-generation chemistry of isoprene and NO3 is characterized by taking advantage of the time behavior of different products. In addition, the vapor pressures of diverse isoprene nitrates are calculated by different parametrization methods. An estimation of the vapor pressure is also derived from their condensation behavior. According to our results, isoprene monomers belong to intermediate-volatility or semi-volatile organic compounds and thus have little effect on SOA formation. In contrast, the dimers are expected to have low or extremely low volatility, indicating that they are potentially substantial contributors to SOA. However, the monomers constitute 80 % of the total explained signals on average, while the dimers contribute less than 2 %, suggesting that the contribution of isoprene NO3 oxidation to SOA by condensation should be low under atmospheric conditions. We expect a SOA mass yield of about 5 % from the wall-loss- and dilution-corrected mass concentrations, assuming that all of the isoprene dimers in the low- or extremely low-volatility organic compound (LVOC or ELVOC) range will condense completely.
- Article
(3551 KB) - Full-text XML
-
Supplement
(3279 KB) - BibTeX
- EndNote





Atmospheric submicron aerosols have an adverse effect on air quality, human health, and climate (Jimenez et al., 2009; Pöschl, 2005). Secondary organic aerosol (SOA), which is formed from oxidation of volatile organic compounds (VOCs) followed by gas-to-particle partitioning, comprises a large fraction (20 %–90 %) of the submicron aerosol mass (Jimenez et al., 2009; Zhang et al., 2007). It is confirmed that a significant proportion of SOA arises from biogenic VOC (BVOC) oxidation (Hallquist et al., 2009; Spracklen et al., 2011).
Isoprene is globally the most abundant non-methane volatile organic compound originating from vegetation, with emissions estimated to be 440–660 Tg yr−1 (Guenther et al., 2012). Due to its high abundance, as well as its high reactivity with atmospheric oxidants, isoprene plays a significant role in tropospheric chemistry, and its chemistry affects the global aerosol burden and distribution (Carlton et al., 2009; Fry et al., 2018; Ng et al., 2008, 2017; Surratt et al., 2010), although its SOA yield is much lower than those of monoterpenes and sesquiterpenes (Friedman and Farmer, 2018; Kim et al., 2015; Marais et al., 2016; McFiggans, et al., 2019; Mutzel et al., 2016; Ng et al., 2007, 2008; Surratt et al., 2010; Thornton et al., 2020). Recent model simulations suggested the isoprene-derived SOA production is 56.7 Tg C yr−1, contributing up to 41 % of global SOA (Stadtler et al., 2018). Observations in the southeastern United States suggested that isoprene-derived SOA makes up 17 %–48 % of total organic aerosol (Hu et al., 2015; Kim et al., 2015; Marais et al., 2016). As a consequence, it is essential to fully characterize the potential of isoprene to form condensable products and its contribution to SOA formation (Carlton et al., 2009).
Although the majority of isoprene emissions is emitted by plants and is light-dependent, isoprene emitted in the day can persist in the boundary layer after sunset, and its mixing ratio can remain as high as several parts per billion (ppb) (Brown et al., 2009; Starn et al., 1998; Stroud et al., 2002; Warneke et al., 2004). During the daytime, isoprene is primarily oxidized by the hydroxyl radical (OH) and somewhat by ozone (O3), but its main oxidizers shift to nitrate radical (NO3) and O3 in the nighttime (Wennberg et al., 2018). Due to the higher reactivity of NO3 with isoprene ( 6.5 × 10−13 cm3 molecules−1 s−1 and 1.28 × 10−17 cm3 molecules−1 s−1 at 298 K, respectively, IUPAC), a considerable fraction of the residual isoprene would be oxidized by NO3 at night, and therefore nocturnal nitrate radical chemistry is typically thought to be of significant importance for isoprene, especially in regions where sufficient nitrogen oxides are available (Brown et al., 2009; Fry et al., 2018; Ng et al., 2017; Wennberg et al., 2018). Although reaction with NO3 only represents ∼ 5 %–6 % of isoprene loss, it accounts for a large proportion of organic nitrates derived from isoprene oxidation (∼ 40 %–50 %) (Wennberg et al., 2018). Therefore, reaction of isoprene with NO3 is a potential source of SOA. In addition, it is found from both laboratory and chamber experiments that the SOA yield of isoprene from NO3 oxidation is higher than that from OH or O3 oxidation, which is typically less than 5 % (Carlton et al., 2009; Dommen et al., 2009; Kleindienst et al., 2007; Kroll et al., 2006). For example, Ng et al. (2008) concluded the isoprene SOA yield from NO3 was in the range of 4.3 %–23.8 %, depending on RO2 fate (higher SOA yield when the experiments were dominated by RO2+ RO2 rather than RO2+ NO3 reaction). Rollins et al. (2009) also observed a high SOA yield from isoprene (14 %) when both of its double bonds were oxidized by NO3. In an aircraft study in the southeastern United States, Fry et al. (2018) derived an isoprene-NO3 SOA yield as large as 27 % on average under high-NOx conditions, although their mass yield estimation was indirect, and based on a molar yield determination of 9 ± 5 %. In light of the relatively high SOA yield from NO3 oxidation, even though only a minor fraction of isoprene is oxidized by NO3, the SOA formed at nighttime would still probably be comparable to that produced at daytime (Brown et al., 2009; Fry et al., 2018).
However, isoprene-NO3 chemistry (Wennberg et al., 2018; Vereecken et al., 2021) has received less attention than the extensively studied OH- or O3-initiated oxidation (Barber et al., 2018; Novelli et al., 2020; Peeters et al., 2014; Wang et al., 2018; Wennberg et al., 2018; Whalley et al., 2012). It has been recognized that later-generation oxidation of isoprene by NO3 makes a more significant contribution to SOA formation (Carlton et al., 2009; Fry et al., 2018; Rollins et al., 2009). Nevertheless, although the importance of multi-generation NO3 oxidation of isoprene to SOA formation has been recognized, few studies extended the investigation beyond the first-generation oxidation, and details of isoprene-NO3 multi-generation chemistry are still lacking.
Organic compounds, especially highly oxygenated organic molecules (HOMs) that have low or extremely low volatility, contribute significantly to SOA formation by condensation, or even form new particles (Bianchi et al., 2019; Ehn et al., 2014; Kirkby et al., 2016, Tröstl et al., 2016). Previous studies have confirmed that low-volatility products from isoprene-NO3 reactions are the major precursors to SOA (Ng et al., 2008; Rollins et al., 2009; Schwantes et al., 2019). Here the low-volatility compounds refer to gas-phase products that allow fractions to exist in the particle phase and may include the groups of organic compounds with intermediate volatility (IVOC, 300 < C∗ < 3 × 106 µg m−3), semi-volatility (SVOC, 0.3 < C∗ < 300 µg m−3), low volatility (LVOC, 3 × 10−5 < C∗ < 0.3 µg m−3), and extremely low volatility (ELVOC, C∗ < 3 × 10−5 µg m−3) as proposed by Donahue et al. (2012). In general, SVOC, LVOC, and ELVOC can contribute to the SOA formation (Jimenez et al., 2009). In order to evaluate the potential of oxygenated products to form SOA, information about their vapor pressures is essential. However, due to the high degree of functionalization, low or extremely low volatility, and uncertainties in quantification and molecular structures, it is challenging to determine the exact vapor pressure of highly oxidized products. Detailed information on the volatilities of different-generation products is lacking, which impedes the assessment of their contribution to SOA formation.
In this work, we present the results of chamber experiments on isoprene oxidation by NO3 under near-atmospheric conditions, where NO3 was produced in situ by O3 reaction with NO2. Subsequent characteristics of multi-generation chemistry of isoprene with NO3 are investigated. By examining the time evolution of various gas-phase products, we propose possible reaction mechanisms that help to get the possible functionalization of the products. Saturation vapor pressures of the major gas-phase products observed by HR-ToF-CIMS are predicted by using different parameterization methods that are widely used or state of the art in the literature. In addition, we estimate the vapor pressure derived from the equilibrium partitioning coefficient according to the condensation behavior of different products in experiments with and without seed aerosols. Based on these results, the volatility of the major oxidation products stemming from isoprene-NO3 reaction and their potential to form SOA are evaluated.
2.1 Atmospheric simulation chamber SAPHIR
All the data presented here were measured in the atmospheric simulation chamber SAPHIR (Simulation of Atmospheric PHotochemical In a large Reaction Chamber) at Forschungszentrum Jülich, Germany, which is designed to investigate the oxidation processes of both biogenic and anthropogenic trace gases and formation of secondary particles and pollutants under near-atmospheric conditions. The SAPHIR chamber is a double-walled Teflon (FEP) cylinder with a volume of 270 m3 (5 m in diameter and 18 m in length). The large volume-to-surface ratio (1 m) allows experiments to be conducted under natural conditions and reduces interference from the chamber walls. The chamber is equipped with a shutter system which can be opened to admit sunlight for photochemical experiments or closed to mimic nighttime conditions. There are two fans inside the chamber to ensure good mixing of trace gases (within 2 min). The chamber is filled with synthetic air made from mixing ultrapure nitrogen and oxygen (Linde, purity ≥ 99.99990 %) and is slightly over-pressured (∼ 35 Pa) to prevent intrusion of outside air into the chamber. Due to a small amount of leakage (∼ 7 m3 h−1) and gas consumption by instrument sampling, a replenishment flow is provided by a flow control, which leads to a dilution rate of 4 %–7 % h−1. A more detailed description of the chamber setup and its characterization can be found elsewhere (Rohrer et al., 2005).
2.2 Experiment description
A series of experiments investigating the oxidation of isoprene by NO3 were conducted in the SAPHIR chamber in August 2018 (ISOPNO3 campaign) under different chemical conditions. In this work, we primarily focus on an experiment conducted on 8 August 2018 that examined the fast oxidation of isoprene by NO3 (up to ∼ 130 pptv) without seed aerosols. The experiment was performed under dry (RH < 5 %) and dark conditions and employed injections of O3 and NO2 as sources of NO3, where O3 was generated by a silent discharge ozonizer (O3onia), and high-purity NO2 was introduced from a gas bottle (Linde, purity > 99 %).
Before the experiment, the chamber was flushed overnight with a total amount of ∼ 1800 m3 synthetic air to minimize any remaining contamination. At the beginning of the experiment, the chamber air was slightly humidified (RH < 0.1 %) by flushing water vapor from boiling Milli-Q® water into the chamber. Thereafter, O3 and NO2 were added to the chamber in succession, and their concentrations in the chamber after injection were approximately 100 and 25 ppbv, respectively, as shown in Fig. 1. After that, ∼ 10 ppbv of isoprene was injected using a GC syringe, initiating the reaction with NO3. The period between the first and second injection is defined as “step I”, and so on for the other three periods. The second injection was done when isoprene from the first injection was almost completely consumed, to reach concentrations of O3, NO2, and isoprene in the chamber of ∼ 100, 30, and 10 ppbv, respectively. About 1.5 h later, the chemistry was further accelerated by a third injection of precursors, and accordingly the concentrations of O3, NO2, and isoprene reached ∼ 100, 25, and 10 ppbv, respectively. Two hours later, the fourth addition was made and the concentrations of O3 and NO2 increased to approximately 115 and 30 ppbv, respectively, aiming to promote further oxidation of early-generation products. In total the system was kept running for about 7.5 h. Calculation from measurements of isoprene, O3, OH, NO3, and dilution indicates that NO3 contributed for more than 90 % of the chemical losses of isoprene, as shown in Fig. S1, with reaction with O3 being a minor pathway in our system. The reaction of isoprene with OH was not considered as OH concentration was below the detection limit of the instrument in this study (Fig. S2). Thus, losses due to reaction with OH could not be quantified from the measurement but have been determined to contribute about 10 % of the isoprene losses according to a recently published modeling work based on the same campaign, with the contribution of the NO3 reaction accounting for up to 80 % accordingly (Vereecken et al., 2021).
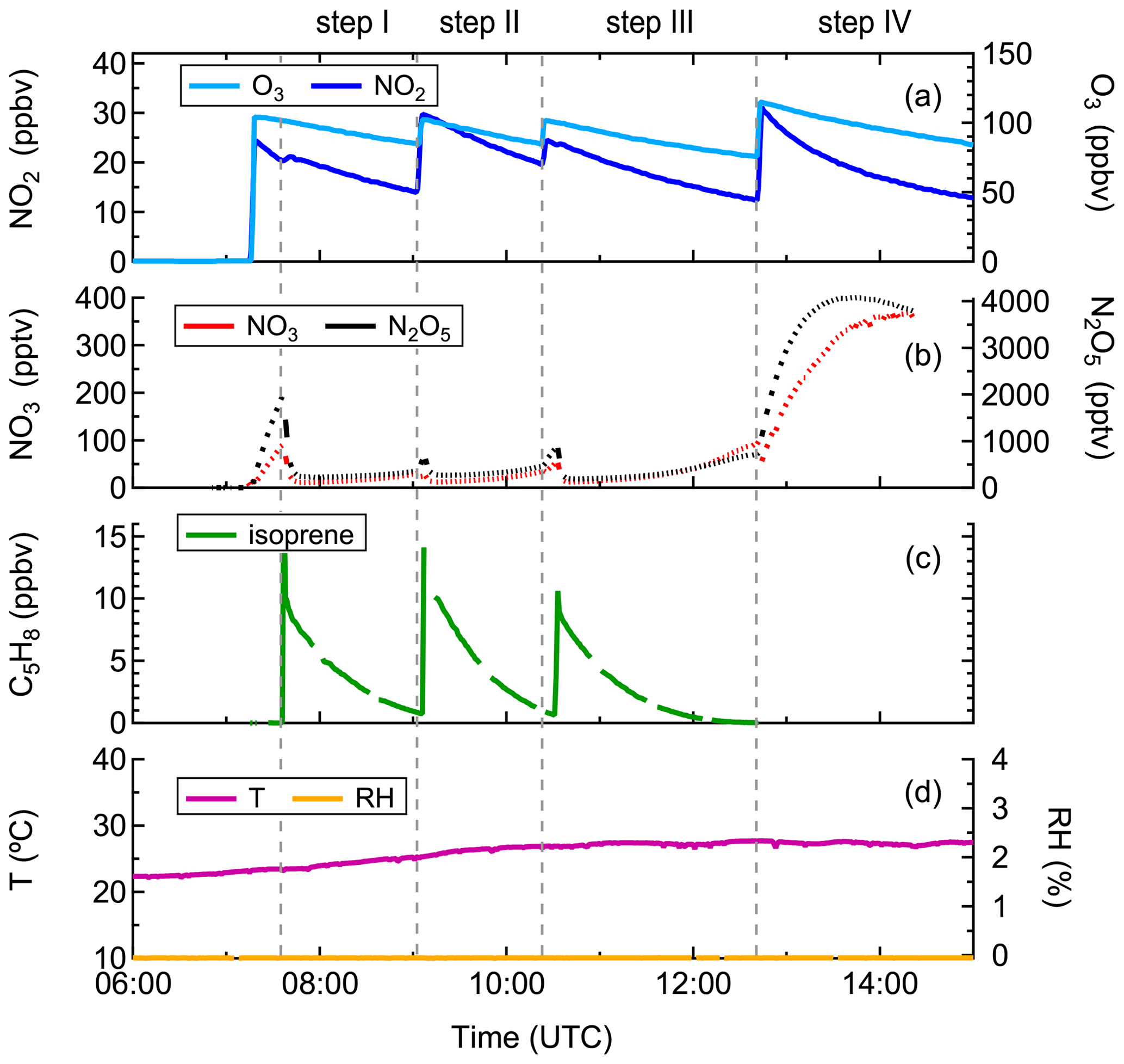
Figure 1Measurements of (a) O3 and NO2, (b) NO3 and N2O5, (c) isoprene, and (d) temperature and relative humidity in the chamber during the experiment on 8 August 2018.
A complementary experiment was conducted on 14 August 2018 under similar conditions but with seed aerosols. Approximately 60 µg m−3 of ammonium sulfate aerosol was added at the beginning of the experiment. Thereafter, approximately 100 and 20 ppbv of O3 and NO2 was introduced to the chamber to produce NO3, followed by the addition of ∼ 10 ppbv of isoprene about 30 min later (see Fig. S3). Another 6 ppbv of NO2 and 10 ppbv of isoprene were added about 1 h later to accelerate the reaction. At the last injection, only O3 (∼ 50 ppbv) and NO2 (∼ 7 ppbv) were added, similar to for the experiment without seeds. The experiment lasted for about 8 h. The results were used to investigate the condensation behavior of various gas-phase products from isoprene oxidation, aiming to estimate equilibrium partitioning coefficients and vapor pressures.
2.3 Instrumentation
A high-resolution time-of-flight chemical ionization mass spectrometer (HR-ToF-CIMS, Aerodyne Research Inc., hereafter CIMS) was used to continuously measure the gas-phase products from isoprene oxidation by NO3. The ToF-MS was operated in “V” mode with the mass resolution power between 3000–4000 . In order to reduce the losses of HO2 radicals and HOM on the tubing, a customized inlet (Albrecht et al., 2019) was directly connected to the chamber. The CIMS was operated in negative ion mode using Br− as the reagent ion, which is selective to polar species such as acids, hydroxy, or nitrooxy carbonyls, as well as HO2 radicals (Albrecht et al., 2019; Ng et al., 2008; Rissanen et al., 2019; Riva et al., 2019).
Bromide ions were generated by passing a mixture of 10 standard cubic centimeters per minute of 0.4 % CF3Br in nitrogen and 2 standard liters of nitrogen per minute through a 370 MBq 210Po source (Type P-2021-5000, NDR Static Control LLC, USA), resulting in ∼ 105 ion counts per second (Albrecht et al., 2019). In our system, about 190 ions were identified for each mass spectrum on average, most of which were detected as adducts with Br−, while some acidic compounds (∼ 7 % of the total) like nitric acid (HNO3), glycolic acid (C2H4O3), and malonic acid (C3H4O4) were also detected as deprotonated ions. In addition, there were some ions (∼ 10 % of the total) identified as adducts with NO. The isotope distribution of 79Br and 81Br is approximately 1:1, and therefore two signals appear at MW + 79 and MW + 81 with MW being the molecular mass of the molecule that is detected as cluster with Br−. In this work, we will use Thomson (Th) as the unit for mass-to-charge (), and the of molecules discussed in following include the mass contribution from Br 79) if there is no other annotation.
In order to have an indicator of the CIMS performance, perfluoropentanoic acid (PFPA, C5F9HO2) was used as an internal standard. For calibration, five isolated peaks were used, including Br− ( 79), H2OBr− ( 97), HNO3Br− ( 142), C5F9O ( 263), and C5F9HO2Br− ( 343), covering the mass range of dominant products. The averaged accuracies of all five calibrated masses were below 5 ppm over the whole measurement period. However, due to the low signal intensity, the PFPA cluster (C10F18O4H−, 527) was not suited for mass calibration, and there were no other suitable masses with sufficient intensity and high accuracy that could be used to calibrate the higher mass range. Therefore, peak fitting in the mass range between 300 and 500 + Th might have higher uncertainties. The CIMS was optimized to gain a maximum signal of [HOBr]− isotopes, which are weakly bounded clusters. This was achieved by adjusting step by step the electrostatic field in the transfer stage to minimize fragmentation. During the campaign, the settings of CIMS were kept unchanged to keep a similar performance. However, the signal of reagent ion Br− decreased by about 65 % (from ∼ 100 000 to 34 000 counts s−1) over the campaign duration of 4 weeks. In order to minimize the effect of drift in performance, we used the normalized (by the sum of the total ion counts, norm. counts from here on) signals for analysis. The sensitivity for total carbon was calculated by determining the slope of wall-loss-corrected total carbon signals detected by CIMS (only the identified peaks were considered) versus isoprene consumed. As illustrated in Fig. S4a, the CIMS sensitivities were roughly identical in two experiments (0.026 ± 0.002 norm. counts s−1 ppbv−1 on 8 August, and 0.022 ± 0.001 norm. counts s−1 ppbv−1 on 14 August), indicating that different experimental conditions over two days had an insignificant impact on CIMS sensitivity for total carbon and thus the data from these days are comparable. In addition, an inter-comparison of measurements by Br− CIMS and I− CIMS were made. As shown in Fig. S4b, the measurements of C5H6N2O8 from the two instruments are well linearly correlated with each other at the early oxidation stages. However, the correlation coefficients of measurements from two instruments deviated from experiment to experiment. This is probably related to different experimental conditions, which might lead to different chemical processes and thus formation of isomers. Since CIMS with different reagent ions might have different sensitivities to isomers and may be selective for different compounds, the correlation coefficients of measurements from Br−, and I− CIMS may differ from day to day. Moreover, the Br− CIMS was not tuned during the campaign while the I− CIMS was optimized from time to time. In general, the performance of Br− CIMS was stable and the data taken by it are reliable.
The mass spectra data were processed using the software “Tofware” embedded in Igor as provided by Aerodyne Research Inc. (https://www.tofwerk.com/software/tofware/?cn-reloaded=1, last access: 6 December 2019). Peaks detected in the mass spectra could be isolated and identified according to their exact mass, and molecular formulas and the corresponding intensities were obtained by high-resolution peak fitting. Due to a lack of authentic standards for the products, it is difficult to quantitatively determine their individual absolute concentrations, but we have calculated the bulk sensitivity for organonitrates by using the sum of organic nitrate signals from Br− CIMS divided by measurements of the total alkyl nitrates from a thermal dissociation-cavity ring-down spectrometer during the experiment . The estimated bulk sensitivities for organonitrates are 0.016 ± 0.001 and 0.022 ± 0.001 norm. counts s−1 ppbv−1 on 8 August and 14 August, respectively, as shown in Fig. S4c, comparable to the sensitivity for total carbon, but smaller than the sensitivity for salicylic acid determined by an independent calibration (163 norm. counts µg−1 on average as shown in Fig. S4d, equal to 0.07 norm. counts s−1 ppbv−1). The bulk sensitivity for organonitrates enables estimation of the absolute concentrations of products assuming that they have identical sensitivity. In this study we use the normalized signals instead of absolute concentrations for analysis. This is sufficient here because our analysis focuses on the time evolution of signals and the relative changes of intensities, so the absolute concentrations are not necessarily needed. The sensitivity derived above is only used to convert the signals of dimers to concentrations in order to estimate the SOA yield.
Isoprene was measured by a Vocus proton transfer reaction time-of-flight mass spectrometer (Aerodyne Research Inc., hereafter Vocus), which has a higher mass-resolving power (nominal 10 000 ) and less inlet wall losses and sampling delays compared to traditional PTR-MS (Krechmer et al., 2018). The mixing ratio of O3 was monitored by an UV absorption instrument, and that of NO2 was monitored by a chemiluminescence instrument and a custom-built cavity ring-down spectrometer (CRDS). The concentrations of NO3 and N2O5 were detected by two custom-built CRDS instruments (Dubé et al., 2006; Sobanski et al., 2016). In addition, temperature and pressure inside the chamber were monitored by an ultra-sonic anemometer and a pressure sensor, respectively. The relative humidity was primarily detected as water mixing ratio by a Picarro CRDS instrument (Crosson, 2008).
The particle number concentrations and their size distributions were measured by a condensation particle counter (TSI 3783, hereafter CPC) and a scan mobility particle sizer (TSI 3081 electrostatic classifier combined with TSI 3025 CPC, hereafter SMPS). The aerosol chemical composition was identified by a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc., hereafter AMS). The ionization efficiency of AMS was determined by using the monodisperse aerosol generated from NH4NO3 and (NH4)2SO4 solutions. The collection efficiency (CE) could be estimated based on the particle mass concentration yielded from AMS and that derived from SMPS. In this study, the average CE value of 0.5 is used for correction.
2.4 Methods to estimate saturation vapor pressure
The pure liquid saturation vapor pressure is a thermodynamic metric relevant for the partitioning equilibrium of organic molecules, which determines their propensity to form SOA (Compernolle et al., 2011; O'Meara et al., 2014; Pankow and Asher, 2008). Due to their complex functionalities and low or extremely low volatility, it is challenging to determine the vapor pressures of highly oxidized molecules. As a result, theoretical and semiempirical methods are usually used for vapor pressure estimation. Commonly used semiempirical methods include composition–activity (CA), group-contribution (GC), and structure–activity (SA) methods. The CA methods are the easiest to use, as they only require information on molecular composition for estimation. They are widely applied in the context of the two-dimensional volatility basis set (2D-VBS) (Donahue et al., 2011). For GC methods, the exact functional groups are required to calculate the saturation vapor pressure. The SIMPOL.1 (Pankow and Asher, 2008), the parameterization as described by Nannoolal et al. (2008), and EVAPORATION (Compernolle et al., 2011) are three widely used GC methods. Structure–activity methods can provide more accurate estimates with sophisticated treatments of intramolecular interactions like intramolecular hydrogen bonding (Bilde et al., 2015). However, detailed molecular properties such as boiling point and evaporation enthalpy are required for estimation, which are generally obtained by complex and time-consuming quantum chemical calculations. Therefore, SA methods are not applied for vapor pressure estimation in this study.
Saturation concentration (C∗, mass based) is related to saturation vapor pressure and can be calculated following Eq. (1) (Donahue et al., 2006). The log10(C∗) is a metric used in the 2D-VBS method to evaluate the volatility of organic molecules.
where R (8.206 × 10−5 m3 atm K−1 mol−1) is the gas constant, T (K) is the temperature, Mi (g mol−1) is the molecular weight of compound i, ξi is the activity coefficient of compound i and here is assumed to be 1 (Donahue et al., 2006), and pi∘ (atm) is the pure liquid saturation vapor pressure at temperature T (298 K).
In this study, different CA methods are applied to calculate the saturation vapor pressures of various oxidation products from isoprene reaction with NO3. These include parameterizations that were constrained by chamber measurements as proposed by Donahue et al. (2011), Mohr et al. (2019), and Peräkylä et al. (2020). All of these three parameterization methods have included the effect of the presence of nitrate groups on vapor-pressure estimation. Further we test the GC methods proposed by Nannoolal et al. (2008), Pankow and Asher (2008, SIMPOL.1), and Compernolle et al. (2011, EVAPORATION). All the methods used in this study are summarized in Table 1. The calculations of EVAPORATION and the Nannool method were done via the online molecular and multiphase property prediction facility UManSysProp (http://umansysprop.seaes.manchester.ac.uk/tool/vapour_pressure, last access: 20 September 2019). For the latter the boiling point parameterization method needs to be predefined, and that from Nannoolal et al. (2004) was adopted as recommended by O'Meara et al. (2014). The information about molecular structures needed for the calculation is inferred from mechanistic information, which is described in detail in Sect. 2.5.
Table 1Summary of estimation methods of saturation vapor pressure used in this study.
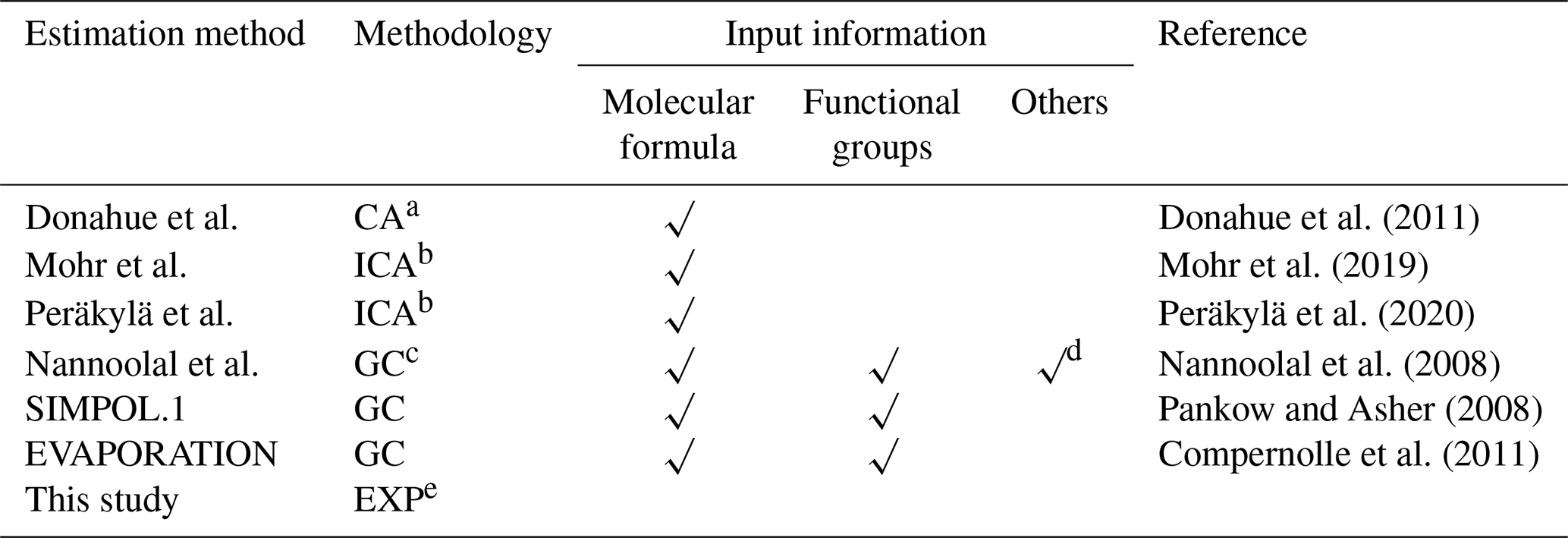
a Abbreviation of composition-activity method. b Abbreviation of improved composition-activity method, which modified the parameterization based on chamber measurements to better fit HOMs. c Abbreviation of group-contribution method. d Boiling point parameterization method is also required to be defined. e Abbreviation of experimental method.
In addition, we take advantage of the measurements in this study to calculate the gas-particle equilibrium partitioning coefficient (K) by comparing experiments with and without seed aerosols. The partitioning coefficient K can be converted to saturation concentration C∗ by Eq. (2).
where Ci,g and Ci,p are the gas- and particle-phase concentrations (µg m−3) of species i, respectively, and COA is the organic aerosol concentration (µg m−3). In this study, Ci,g is signal of species i from CIMS in the experiment with seeds, and Ci,p is the difference of signals between experiments without and with seeds (under the same isoprene consumption condition). The COA in the experiment with seeds is in a range of 1–4 µg m−3.
2.5 Pathways to the multifunctional oxidation products
2.5.1 Basic peroxy and alkoxy radical chemistry
As mentioned before, information about molecular structures (at least functional groups) is required to calculate vapor pressures by using GC methods. Although the high-resolution ToF-CIMS allows for determining the chemical composition of the detected ions, it is unable to provide information about molecular structures, so that the constitutional or configurational isomers with the same mass cannot be distinguished without additional information. Fortunately, knowledge of detailed chemical formation mechanisms can help to infer the molecular structure information. However, the development of a comprehensive, multi-generational kinetic mechanism for NO3-initiated oxidation of isoprene is outside the scope of the current paper. Instead, in order to link the observed mass peaks to representative molecular structures, we developed a framework tracing the chemical oxidation mechanisms by taking well-known oxidation steps to predict the most likely isomeric forms of the functionalized products formed in the isoprene oxidation. For this purpose, we rely on the extensive literature on isoprene, alkylperoxy radical, and alkoxy radical chemistry (Atkinson, 2007; Atkinson and Arey, 2003; Bianchi et al., 2019; Crounse et al., 2013; Ehn et al., 2014; Jenkin et al., 2015, 2019; Kwan et al., 2012; Mentel et al., 2015; Ng et al., 2008; Novelli et al., 2021; Orlando et al., 2003; Orlando and Tyndall, 2012; Rollins et al., 2009; Schwantes et al., 2015; Vereecken and Francisco, 2012; Vereecken and Peeters, 2009, 2010; Vereecken et al., 2021; Wennberg et al., 2018; Ziemann and Atkinson, 2012). This framework is depicted in the supporting information and will be discussed in more detail in Sect. 2.5.2 and 2.5.3. They are based on the following main reactivity trends.
Generally, RO2 radicals can react with other RO2 and HO2 radicals. There are three major channels for the reaction between two RO2 radicals, leading to alkoxy radicals (RO) (Reaction R1a), as well as termination products like alcohols, aldehydes, or ketones (Reaction R1b) and accretion products (Reaction R1c). These reactions should take place with the first-generation peroxy radicals, as well as with the higher-generation RO2 radicals formed in the later oxidation steps. Hydroperoxides can be formed from the reaction of RO2 with HO2 radicals (Reaction R2a). This reaction can also yield alkoxy radicals (Reaction R2b).
In the presence of NOx, RO2 radicals can also react with NO and NO2, leading to the formation of alkoxy radicals (Reaction R3a), organic nitrates (Reaction R3b), and peroxynitrates (Reaction R4) (including peroxyacyl nitrates, PANs, if R= R′C(O)–). The channel that results in RO radicals is the major pathway for the reaction of RO2 radicals with NO (Ziemann and Atkinson, 2012). However, reactions of RO2 radicals with NO (Reactions R3a and R3b) can be neglected in this study due to the high O3 concentration, which results in rapid conversion of NO to NO2. The peroxynitrates formed from the reaction of RO2 with NO2 will undergo rapid thermal decomposition under our experimental conditions, with the exception of PANs. The reaction of RO2 with NO3 radicals mainly forms NO2 and alkoxy radicals (Reaction R5), which will continue the radical chains (Reaction R7).
In addition to bimolecular reactions, intramolecular rearrangement (H migration) is a competitive reaction pathway for RO2 radicals. RO2 radicals can undergo H migration to form a hydroperoxy functionality (–OOH) and a radical site that can subsequently recombine with an O2 molecule, leading to the formation of a new, more oxidized substituted RO2 (Reaction R6). This process is the so-called “autoxidation” path and has been confirmed as significantly important for SOA formation (Crounse et al., 2013; Ehn et al., 2014; Mentel et al., 2015; Praske et al., 2018; Rissanen et al., 2014). The rates of RO2 H migration are strongly dependent on the structure of RO2 radicals, and the most likely routes can be derived based on the structure–activity relationship proposed by Vereecken and Nozière (2020).
The RO radicals formed in the reaction of RO2+ RO2 typically have three accessible pathways, including isomerization by H migration (Reaction R7a), fragmentation (Reaction R7b), and (less important here) reaction with O2 (Reaction R7c). Like H migration in RO2, rearrangement by H shift in RO radicals leads to the formation of more oxidized RO2 radicals. Fragmentation leads to smaller carbon chains, and this becomes more important for alkoxy radicals with a higher number of (oxygen-bearing) substituents (Vereecken and Peeters, 2009, 2010).
In addition to the above general reaction pathways, we include a number of other reactions in the framework, such as fragmentation of peroxy radicals, epoxidation of β-OOH alkyl radicals, and unimolecular termination of nitrooxy or hydroperoxyl peroxy radicals. Details can be found in the supporting information.
2.5.2 Formation of first-generation products
Here “first-generation products” refers to the closed-shell compounds from the first attack of NO3 at the isoprene double bonds, while “second-generation products” follow an addition of NO3 to the remaining double bond (or any other oxidation reaction) of a first-generation product. Addition of a NO3 radical to one of isoprene double bonds and subsequent addition of O2 to the resulting (delocalized) radical sites leads to the formation of nitrooxy alkylperoxy radicals (INO2, C5H8NO3). Since isoprene contains two double bonds, NO3 can attack any of the four positions on the conjugated carbon bonds, resulting in eight possible INO2 isomers (including six constitutional and two conformational isomers), as shown in Scheme S1. However, both theoretical and experimental studies suggest that the addition occurs preferably at the primary and terminal carbons, wherein C1 addition seems to be preferred over C4 addition (Schwantes et al., 2015; Suh et al., 2001; Wennberg et al., 2018). As the GC methods have limited or no ability to distinguish between positional isomers (Kurten et al., 2016), we take exemplarily the products following the C1 addition for the vapor pressure analysis in this study.
The initial peroxy radicals (C5H8NO3) can undergo rearrangement by H shift from C–H bonds with subsequent O2 addition, yielding new –OOH functionalized peroxy radicals (Reaction R6). Repeating this process can lead to the formation of a series of peroxy radicals and termination products with a stepwise increasing number of oxygen atoms by 2, as shown in the conceptual Scheme S2 in the Supplement. This is the RO2 autoxidation channel, and the molecular formula of peroxy radicals formed via consecutive O2 additions can be represented as C5H8NO(3+2n) (n≥ 1, number of autoxidation steps). The autoxidation chain can be terminated when the H shift occurs at a carbon with an –OOH or –ONO2 group attached, leading to carbonyl formation with OH or NO2 loss (Anglada et al., 2016; Bianchi et al., 2019; Vereecken, 2008; Vereecken et al., 2004). The closed-shell products formed in these termination steps have the general molecular formula C5H7NO (OH loss channel) or C5H8O (NO2 loss channel).
The C5H8NO(3+2n) peroxy radicals can also react with HO2 radicals to form –OOH functionalized termination products with the general molecular formula C5H9NO(3+2n) (Reaction R2a), or yielding the alkoxy radicals C5H8NO (Reaction R2b). In addition, the C5H8NO(3+2n) peroxy radicals can react with other RO radicals (Reactions R1a–R1c). The reaction R1a leads to the formation of alkoxy radicals (C5H8NO) while Reaction R1b forms closed-shell products either with a carbonyl group (C5H7NO) or a hydroxyl group (C5H9NO). Alternatively, dimers can be formed following Reaction R1c, which have then two –ONO2 groups and at least eight oxygen atoms depending on the formula of RO2 radicals involved, as shown in Table S1.
The alkoxy radicals from Reactions R1a and R2b can undergo unimolecular rearrangement by H shift with subsequent O2 addition, similar to the RO2 radicals, forming new RO2 radicals with a –OH group (Reaction R7a). As mentioned above, when the H shift occurs at a carbon with an –OOH or –ONO2 group attached, the resulting intermediates tend to lose an OH group or NO2 (Bianchi et al., 2019), yielding the closed-shell carbonyl products with general formulas C5H7NO or C5H8O respectively, as shown in the conceptual scheme Scheme S3. The newly formed RO2 radicals from the alkoxy H shift channel can follow the peroxy pathways (Reactions R1–R6) like other RO2 radicals, leading to a range of compounds like hydroperoxides (Reaction R2a, C5H9NO), alcohols (Reaction R1b, C5H9NO(3+2n)), aldehydes (Reaction R1b, C5H7NO(3+2n)), and accretion products (Reaction R1c, C10H16N2Ox), as depicted in Scheme S3. Alternatively, they can also yield alkoxy radicals again following Reactions R1a and R2b and continue in that manner. Furthermore, the alkoxy radicals can break apart into two fragments according to Reaction R7b.
In general, the alkoxy reaction pathways diversify the parity of the oxygen number of the products from the reaction of isoprene with NO3, and the compounds formed via these reactions generally have one less or one more oxygen atom compared to those formed from straight peroxy reaction pathways. With the help of the mechanistic framework described above, we can infer the functionality of first-generation products. This is exemplified in Scheme S5 and S6 for the major first-generation C5 products. In addition, the reaction pathways and their corresponding structures of the first-generation C10 dimers (C10H16N2Ox) are summarized in Scheme S13.
2.5.3 Formation of second-generation products
Nitrate radicals can oxidize the first-generation products once again at the double bond remaining ( (298 K) ∼ 3–11 × 1014 cm3 molecules−1 s−1, Wennberg et al., 2018). This leads eventually to “second-generation” products that contain at least two nitrogen atoms. Addition of a NO3 radical to the remaining double bond of the first-generation products results in the formation of dinitrooxy peroxy radicals. We assume that dinitrooxy peroxy radicals can undergo unimolecular and bimolecular reactions (Reactions R1–R6) in analogy to nitrooxy peroxy radicals, which lead to secondary products containing two or more nitrogen atoms, as summarized in the conceptual scheme Scheme S4.
The reaction of first-generation nitrooxy peroxy radicals with NO2 can also yield 2N compounds (Reaction R4); however, these 2N compounds ought to be under first-generation products by definition. Such species are not discussed in detail here but will be covered to catch the diversity of the functionalities for the vapor pressure estimation. With the help of this secondary reaction framework, we can propose functional groups for the major second-generation products. Schemes S8–S10 depict the detailed (possible) reaction pathways that lead to the formation of detected C5 dinitrates, as well as their possible structures. Furthermore, the proposed formation mechanism, and their structures for C5 trinitrates are shown in Scheme S12, while those for the second-generation C10 dimers (C10H17N3Ox and C10H18N4Ox) are depicted in Scheme S13.
2.5.4 Formation of fragmentation products
In addition to the multigenerational C5 and C10 products, fragmentation products can be formed from the reaction of isoprene with NO3. As mentioned above, the alkoxy radicals can undergo C–C bond scission, producing a carbonyl compound and an alkyl fragment (Reaction R7b). As shown in Scheme S7, when the secondary nitrooxy alkoxy radicals from the further oxidation of C5 carbonyl compounds (C5H8O2 and C5H8O3 here) undergo unimolecular decomposition, C4 carbonyl products (C4H7NO5 and C4H7NO6, respectively) are formed as well as formyl radicals. Since the bond fission can occur at different positions, the generation of more reactive C2 and C3 carbonyl compounds are possible. In addition, the C4 carbonyl compounds are possibly generated through peroxy radical arrangement by 1,4 H shift and subsequent acyl radical bond scission reactions (see Scheme S7). The C4 dinitrates can be formed following similar chemistry, as depicted in Scheme S11.
2.5.5 Candidate structures for vapor pressure estimation
Among all gas-phase products detected by CIMS, we selected 32 major representative organonitrates formed from isoprene oxidation by NO3 radicals. Their structures are rationalized by the corresponding molecular formulas and proposed formation mechanisms in the reaction framework. Table S2 summarizes all the exemplified structures used for vapor pressure estimation. The functional groups covered in the selected structures include nitrate, hydroxyl, ketone, aldehyde, carboxylic acid, peroxide, hydroperoxide, hydroperoxy acid, peroxynitrate, peroxyacyl nitrate, and epoxide. The structural information allows calculation of the saturation vapor pressure by GC methods.
3.1 Chemical composition of oxidation products
Figure 2 illustrates the average mass spectra of the whole experiment measured by Br−CIMS for isoprene-NO3 reaction. Chemical sum formulas were attributed to most of the detected ions. The gas-phase products were separated into two major groups according to their chemical composition, including monomers comprising C5 compounds and dimers containing C10 compounds. There were also products from decomposition reactions with C<5, which were merged into monomers. The monomers and dimers were further classified into five subgroups as follows. Monomers consisting of compounds with one nitrogen atom (hereafter 1N-monomers) and two or three N atoms (2N- or 3N-monomers) mainly accumulate in 220–280 Th, 300–340 Th, and 350–390 Th, respectively, while dimers containing compounds with two N atoms (2N-dimers) and three N atoms (3N-dimers) appear in 370–440 Th and 450–520 Th, respectively. As shown in Fig. 2, the signal intensities decrease from 1N-monomers, 2N-monomers, and 2N-dimers to 3N-monomers and 3N-dimers. Many of the compounds detected in this work were also observed in previous isoprene-NO3 systems (Kwan et al., 2012; Ng et al., 2008; Schwantes et al., 2015). In this work, only closed-shell products are considered for analysis.
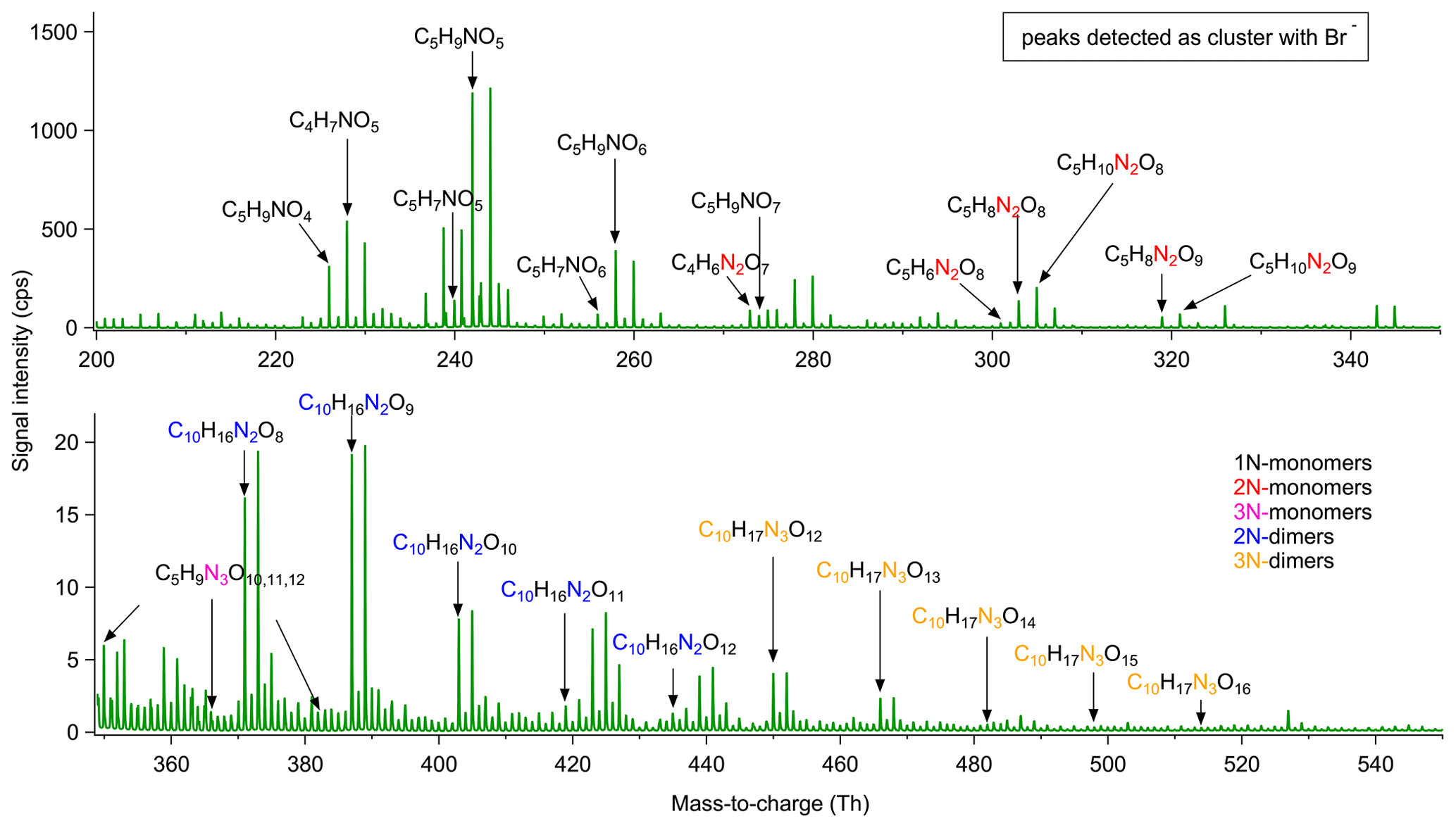
Figure 2Averaged mass spectra for isoprene-NO3 experiment on 8 August 2018. Molecular formulas were determined according to the accurate mass data provided by HR-ToF-CIMS.
The 1N-monomer C5H9NO5 at 242 is the dominant product formed from the NO3-induced isoprene oxidation in our experiment, followed by the 1N-decomposition product C4H7NO5 at 228. In addition to C5H9NO5, several analogues with molecular formulas C5H7NO4−7 and C5H9NO4 are in relatively high abundance. C5H8,10N2O8,9 and C5H9N3O10−12 are the major 2N- and 3N-monomers. Their signal intensities are 1 to 2 orders of magnitude lower than those of 1N-monomers. According to the chemical composition, the 1N-monomers are likely to be the first-generation products from NO3 oxidation of isoprene, while the 2N- and 3N-monomers probably arise from the further oxidation of 1N-monomers by NO3, which therefore should be second- or later-generation products. As mentioned before, the reaction of nitrooxy alkylperoxy radicals with NO2 can lead to the formation of peroxynitrates (for the special case peroxyacyl nitrates, PAN-like) containing two N atoms. The peroxynitrates will decompose rapidly under experimental conditions, whereas the PAN-like compounds are more stable (with lifetimes ranging from minutes to weeks at 298 K and ambient temperature). Such C5 PAN-like compounds are isomers of aforementioned 2N-monomers but ought to be first-generation products. In addition to C5-2N-monomers, we observe some C4-2N-monomers with relatively high intensity, such as C4H6N2O7 at 273 and C4H8N2O8 at 291. It is proposed that such C4 dinitrates originate from the further oxidation of C5 carbonyl compounds followed by unimolecular decomposition (Schwantes et al., 2015; Wennberg et al., 2018), as shown in Scheme S11.
2N-Dimers are C10 compounds with 8–12 oxygen atoms (C10H16N2O8−12), and their signal intensities are relatively low compared to that of monomers, approximately 3 orders of magnitude lower. They might be ROOR products from the self- or cross-reaction of two nitrooxy peroxy radicals (Berndt et al., 2018). 3N-Dimers are molecules consisting of 12–16 oxygen atoms (C10H17N3O12−16). They are probably formed from further oxidation of 2N-dimers or from the cross-reaction of a nitrooxy peroxy radical with a dinitrooxy peroxy radicals.
3.2 Multi-generation chemistry
3.2.1 Molecular composition for each step
As mentioned in Sect. 2.2, there were four injections during the experiment on 8 August (denoted as step I, II, III, IV in Fig. 3), wherein in the first three injections all components, O3, NO2, and isoprene, were added, while in the last step only O3 and NO2 were injected to promote the further oxidation of early-generation products. The extended oxidation time with reinjection of oxidants provides the opportunity to investigate the multi-generation oxidation chemistry of the isoprene-NO3 system. The mass spectra show only slow changes in the concentrations during the last period of each step, indicating weak chemical evolution. Therefore, we use integrated mass spectra over the last 10 min of each step for further analysis. Due to the similarity of the integrated mass spectra for step II and step III, the latter is omitted in Fig. 3.
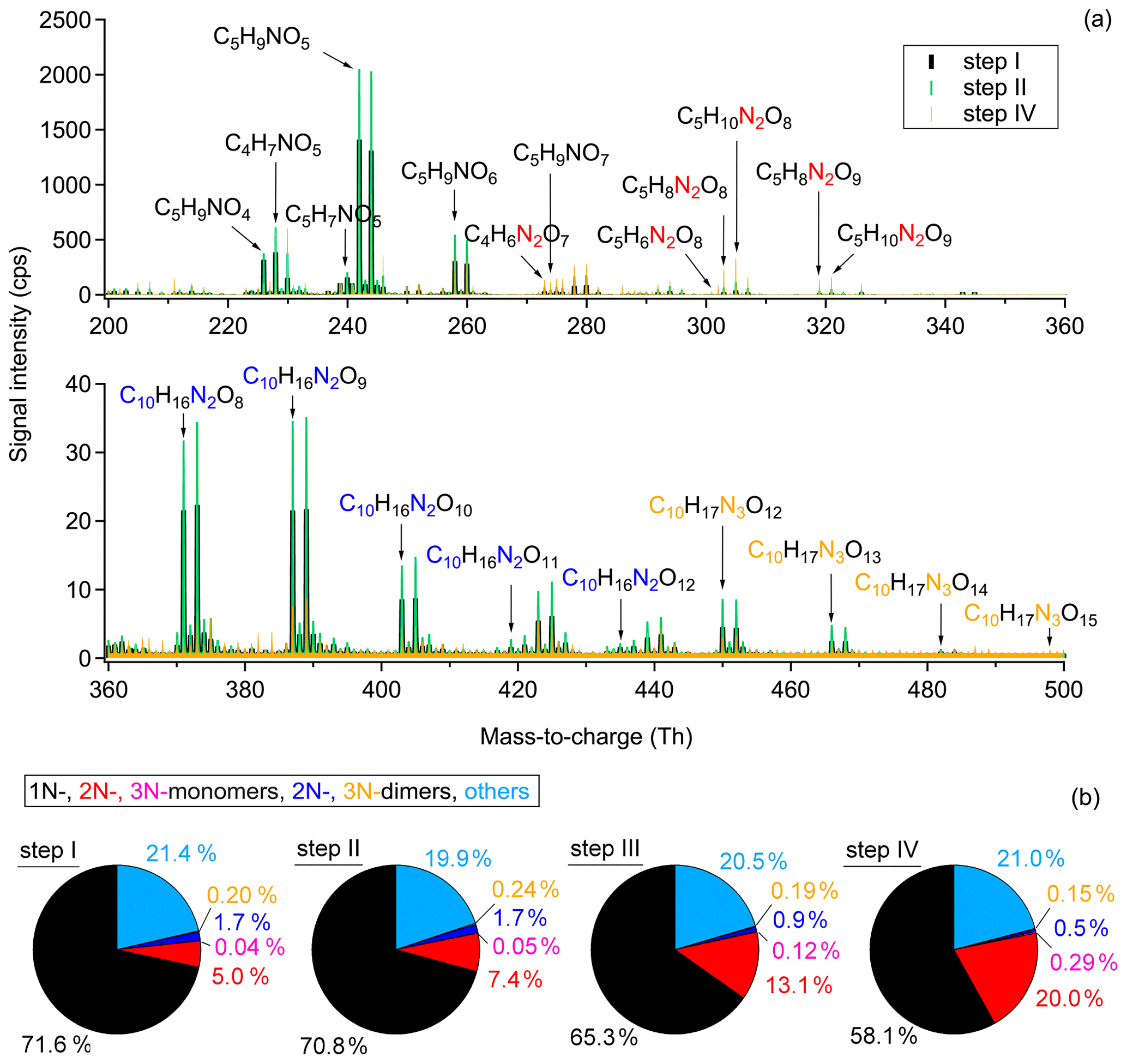
Figure 3Comparison of the chemical composition of each oxidation step. (a) Averaged mass spectra for step I, II, and IV, with the omitted spectrum of step III being very similar to that of step II. (b) Relative contribution of different chemical groups for each oxidation step. Only organic products were counted for analysis. “Others” refers to CHO compounds that do not contain nitrogen atoms (e.g., C5H8O2 and C5H8O3).
As shown in Fig. 3a, large amounts of 1N-monomers were formed from NO3 oxidation of isoprene in step I, wherein C5H9NO5, C5H9NO6, and C4H7NO5 are the most abundant compounds in signal. The 2N-monomers, which are expected from further oxidation of 1N-monomers, are much less abundant compared to 1N-monomers, accounting for 5.0 % of the total organic signals. The 3N-monomers are even less abundant (0.04 %). The low contributions of second-generation products probably results from the relatively high concentration of isoprene in step I, reducing the possibility for further oxidation of first-generation products. These results indicate that the system is dominated by first-generation chemistry at the early stage and therefore the oxidation state of products is low. In addition to monomers, some 2N- and 3N-dimers are observed. They contribute 1.7 % and 0.2 %, respectively, to the total organic signals, as shown in Fig. 3b. The low signal intensity of dimers probably results from their small yield under our experimental conditions. In this case their contribution to SOA formation might be small. However, a part of the dimers condense onto the chamber wall due to their low volatility, so only a smaller portion exists in the gas phase (compare Table S3 and Fig. S5).
In step II, the secondary chemistry was accelerated by the further addition of O3 and NO2, but the primary chemistry was also maintained by isoprene injection. As a result, more 1N-monomers (e.g., C5H9NO) were formed compared to step I, as well as dimers (e.g., C10H16N2O and C10H17N3O12,13), as shown in Fig. 3a. The signals of 2N-monomers almost double in this period compared to those in step I, and their relative contribution increases from 5.0 % to 7.4 %. This is attributed to the further oxidation of first-generation products formed in step I. The relative contributions of different chemical groups exhibited in Fig. 3b clearly show that, although NO3 produced from the second addition of NO2 and O3 still primarily reacted with newly injected isoprene, reaction of NO3 with the first-generation oxidation products retaining a double bond was inevitable, leading to more second-generation 2N or 3N products compared to step I. The visibly increasing fraction of 2N-monomers indicates that the second-generation chemistry started to play a more important role than that in the early stage. In step III, the chemical process proceeded similarly and thus is not further discussed here.
Due to the favorable conditions for further oxidation, the signals of 1N-monomers (such as C5H9NO4, C5H9NO5, and C5H9NO6), as well as 2N- and 3N-dimers, dropped dramatically in step IV, with their relative contributions decreasing to 58.1 %, 0.5 %, and 0.15 %, respectively. The decrease in signals of dimers is primarily ascribed to lack of isoprene, as there were fewer peroxy radicals under this condition, and hence fewer dimers were formed. In addition, their condensation on the wall and dilution also contributed to the decreasing signals. Furthermore, dimers with 2 or 3 nitrogen atoms possess at least one double bond in their molecular structures and can thus be further oxidized under high-NO3 conditions to form 4N- or 5N-dimers. However, only few 4N-dimers and no 5N-dimers were detected by CIMS, suggesting that the 4N- and 5N-dimers were either not formed, or if present, had lower absolute concentrations below the detection limit (approximately 5 × 107 and 5 × 105 molecules cm−3 for salicylic acid and acetic acid, for an integration time of 60 s). condensed on the wall due to their low volatilities. In contrast, 2N- and 3N-monomers increase significantly, with their relative contributions ascending to 20.0 % and 0.29 %, respectively. This indicates that 2N- and 3N-monomers might be second- or later-generation products that are formed from the further oxidation of first-generation products. Additionally, unlike the C5 monomers, the signal of C4H7NO5 increased in step IV, indicating that there is a new formation pathway for C4H7NO5 under excess-NO3 conditions. No double bond can remain in such products, as otherwise they would be oxidized and their signal should decay instead.
In summary, the above findings confirm that multi-generation chemistry happened during the NO3-initiated isoprene oxidation, and that the later generation oxidation was promoted by “excess” NO3 radicals.
3.2.2 Carbon oxidation state ()
The oxidation state of carbon () is defined as the charge a carbon atom takes with assumption that it loses completely all electrons in bonds to more electronegative atoms and vice versa (Kroll et al., 2011). This quantity is a metric for the degree of oxidation and will increase with oxidation. Moreover, together with carbon number can be used to constrain the composition of organic mixtures and provide insights into their evolutions. The carbon oxidation state of a species is determined by the relative abundances and oxidation states of non-carbon atoms in the compound. Since we observed nitrate groups in the products, is defined by Eq. (3). In this study, the group-averaged is the signal-weighted mean average carbon oxidation state of compounds with the same carbon number, and the bulk-averaged is the signal-weighted mean average carbon oxidation state of all detected compounds in the system.
wherein nO, nH, and nN are the number of the respective atoms in the molecular formula.
Figure 4 shows the distribution of gas-phase products from the isoprene-NO3 system in the oxidation state versus carbon number (OSC vs. nC) space. The bulk-averaged is −0.35 in step I, wherein the smaller molecules (C≤4) have higher oxidation states than the larger molecules. The group-averaged oxidation state of C5 compounds is relatively low (), indicating that both the oxidation and autoxidation degrees of isoprene are quite low during this period. This is consistent with the conclusion made previously from mass spectra results that at the early stage isoprene-NO3 oxidation was dominated by first-generation chemistry.
The system increases to −0.26 in step II, confirming that first-generation products were further oxidized after the second injection. During this step, the of most compound groups increases only weakly, except for that of the C5 compounds. The group-averaged of C5 compounds increases to −0.60 in step II, which is the major contributor to the increase in of the whole system. The increase in of C5 compounds is largely attributed to the formation of 2N-monomers expected from further oxidation of existing 1N-products formed in step I. This is confirmed by the detectable increase in 2N- and 3N-monomers in the mass spectra and their higher relative contributions to total signals (see Fig. 3). In addition to C5 compounds, the of C3 and C6 products increase significantly in step II.
In step IV, the secondary oxidation was largely accelerated by reinjection of O3 and NO2, and hence the system oxidation degree increases, with the bulk-averaged growing substantially to 0.09. Similarly, the significant increase in system is mainly attributed to the C5 compounds, with their group-averaged increasing to −0.31. In addition, the of C10 compounds increased evidently despite their decreasing signals, suggesting C10 dimers were further oxidized as well in step IV. It is worth noting that the average carbon number decreases step by step with increasing . This is the case because of fewer C10 products, but more fragments were formed with the reaction proceeding, as shown in Fig. 4 by the decreasing peak areas of larger molecules but a converse trend for smaller molecules. One conceivable explanation for the decreasing dimers but increasing fragments with the increasing is that, with more highly oxidized RO2 formed under high-NO3 conditions, the prevailing fate of RO2 changes from dimerization to forming alkoxy radicals, which would undergo unimolecular decomposition rapidly, especially when there is a neighboring oxygen-containing functional group (Molteni et al., 2019).
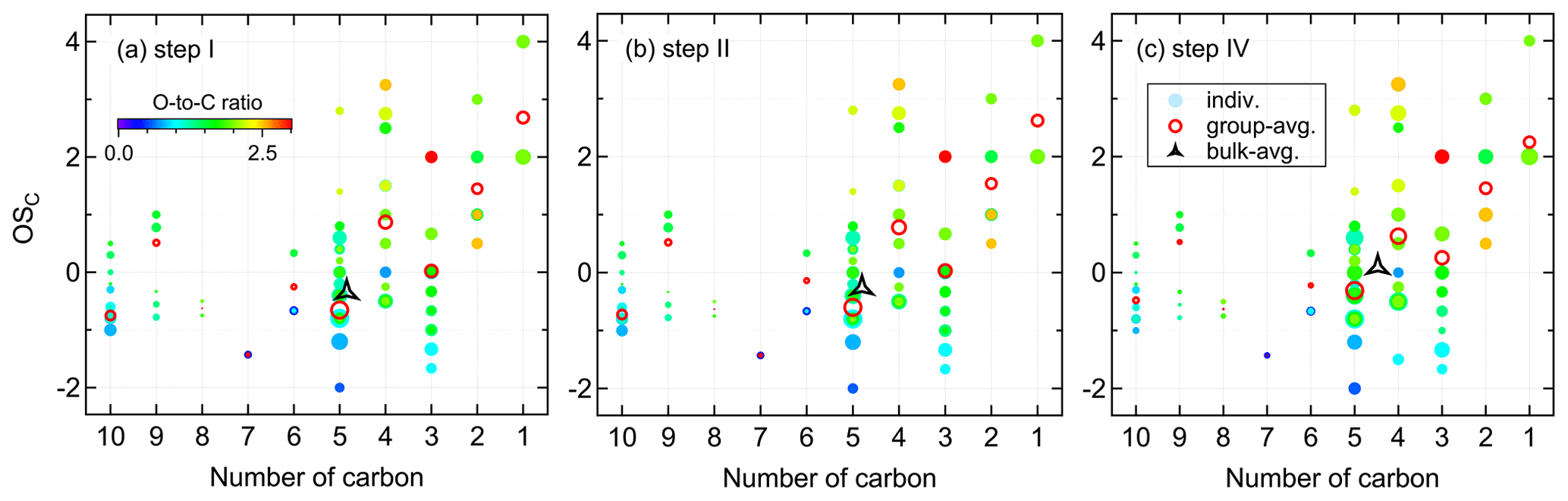
Figure 4Distribution of gas-phase products from isoprene oxidation by NO3 in the carbon oxidation state (OSC) versus carbon number (nC) space. Markers are colored by oxygen-to-carbon molar ratio and sized by the logarithm of peak areas. The group-averaged and bulk-averaged are the signal-weighted mean average carbon oxidation state of compounds with the same carbon number and of all detected compounds, respectively.
In the oxidation system, the increase in is attributed to the formation of bonds between carbon and oxygen as well as other electronegative atoms, and/or the breaking of bonds between carbon and hydrogen and other electropositive atoms (Kroll et al., 2011). The –ONO2 group has an oxidation state of −1, which means that addition of a –ONO2 group to isoprene will increase its OSC by 0.2. According to our estimates, the values of system increased by 1.25 (step I), 0.09 (step II), and 0.35 (step IV), indicating that the increases in are not only due to addition of –ONO2 group(s) but also to other oxygen-containing functionalities. In addition to functionalization, it is possible that other reactions such as fragmentation and oligomerization which can increase or reduce the oxidation state were involved during the reaction.
As mentioned above, the average carbon oxidation state of a mixture of molecules largely depends on its chemical composition. Therefore, for different oxidation systems, their may differ due to different precursors and oxidation conditions. In our study, the of a NO3-initiated isoprene oxidation system increased from −0.35 to 0.09 with further oxidation. For OH- and O3-initiated systems, the average oxidation state of laboratory-generated isoprene SOA is reported to range from −1.3 to −0.2, as listed in Table S4. It seems that the SOA generated from chloride-initiated oxidation of isoprene is more oxidized compared to other isoprene oxidation systems, for which the can be as high as +1.8 according to limited studies (Wang and Ruiz, 2017). With regard to ambient measurements, the calculated values of organic aerosol and aerosol fractions fell into a wider range between −2 and +2, depending on the site position and the corresponding oxidation environment of that site (Table S4).
In summary, isoprene and its products undergo further oxidation by NO3, leading to an increase in the degree of oxidation of products as the reaction proceeds. The increasing bulk is largely governed by the highly oxidized C5 compounds. In addition, more fragments but fewer dimers are formed as the increases, which can probably be explained by the change of RO2 fate from prevailing dimerization to fragmentation through the alkoxy radical channel.
3.2.3 Characteristics of different-generation products
1N-monomers
To illustrate the multi-generation chemistry involved in the isoprene-NO3 reaction system, Fig. 5 shows the time evolution of the major gas-phase products. The signal of the most abundant compounds, C5H9NO5, increases rapidly as soon as the reaction is initiated, reaching a maximum when its chemical production rate matches its loss rate (including chemical destruction, wall loss, dilution, etc.), and decreases slowly thereafter. Its time behavior in the first three steps is similar. In step IV, however, the injection of O3 and NO2 resulted in a strong decay of C5H9NO5, owing to the occurrence of further oxidation by NO3. The time behavior suggests that the C5H9NO5 signal is dominated by first-generation oxidation products, and the same conclusion can be made for C5H9NO4 and C5H9NO6. According to the mechanistic framework developed above, the C5H9NO4, C5H9NO5, and C5H9NO6 compounds most likely correspond to hydroxyl nitrates, nitrooxy hydroperoxides, and hydroxy hydroperoxy nitrates, respectively, but other constitutional isomers are possible. They were already observed in previous studies and were proposed to form through reactions of INO2 radicals with RO2, HO2, and unimolecular rearrangement, as shown in Scheme S5 (Ng et al., 2008; Kwan et al., 2012; Schwantes et al., 2015; Wennberg et al., 2018).

Figure 5Time evolution of selected gas-phase compounds measured during the isoprene – NO3 experiment on 8 August 2018. (a) Time series of O3, NO2, NO3, and isoprene. (b)–(f) Time evolution of major 1N-monomers (C5H9NO4−7 and C4H7NO5), 2N-monomers (C4H6N2O7, C5H6N2O8, and C5H8,10N2O8,9), 3N-monomers (C5H9N3O10−12), 2N-dimers (C10H16N2O8−12), and 3N-dimers (C10H17N3O12−16).
As shown in Fig. 5b, the temporal evolution of C5H9NO7 ( 274) is different to C5H9NO4−6 compounds, suggesting that it has a completely different formation pathway. Specifically, the formation rate of C5H9NO7 is initially much slower than that of C5H9NO4−6 but accelerates to become comparable to them later as the experiment proceeds, i.e., when a multitude of first-generation products are accumulated. This implies that C5H9NO7 is produced from the further oxidation of first-generation products, and its signal is dominated by second-generation products. Based on its molecular composition, C5H9NO7 could be the dihydroperoxy nitrate as shown in Scheme S5, but its formation through the reaction of HO2 with nitrooxy hydroperoxy radical from INO2 autoxidation suggests it should be first-generation products, not in accordance with the time behavior we actually observe. Consequently, we can conclude that it is not the major formation pathway that contributed to C5H9NO7 observed in this study. As shown in Scheme S7, the first-generation C5 hydroxy carbonyl (C5H8O2, 179) can be further oxidized by NO3 and the resulting alkyl radical would rapidly recombine with O2, producing a new peroxy radical, which then reacts with HO2 radicals to form C5H9NO7 (hydroxy hydroperoxy carbonyl nitrate). Similarly, the C5 hydroperoxy carbonyl (C5H8O3, 195) can also lead to the formation of such C5H9NO7 (isomer of that formed through C5H8O2 channel) through further oxidation (see Scheme S7). According to the above two mechanisms, C5H9NO7 formed following such reaction pathways should be second-generation products, which is consistent with its time behavior.
Considering its similar time behavior to C5H9NO7, the observed C4H7NO5 ( 228) signal is likewise thought to be dominated by second-generation products. Schwantes et al. (2015) proposed such a C4 product based on OH-initiated chemistry, but as the OH concentration in our system was below the detection limit during the experiment (see Fig. S2), this formation pathway cannot apply in our situation. Instead, we suggest that C4H7NO5 is formed through the unimolecular decomposition of the C5 alkoxy or acyl radicals, which result from further oxidation of the C5 hydroxy carbonyl (C5H8O2, 179), as shown in Scheme S7. It should be pointed out here that there may be reaction pathways forming C4H7NO5 as first-generation products that are not considered here, whereas there is no doubt that the second-generation chemistry played a dominant role in C4H7NO5 formation according to its time evolution measured by CIMS.
Although C4H7NO5 and C5H9NO7 show similar time behaviors in the first three steps, it seems that they followed fairly different reaction pathways when the concentration of NO3 in the chamber increased dramatically in step IV. As shown in Fig. 5b, the signal of C4H7NO5 drops immediately after the injection of O3 and NO2, while that of C5H9NO7 continues to increase, although its formation rate becomes slightly lower with increasing NO3 concentration. The decay of the C4H7NO5 signal can be explained by more chemical destruction or less production under high-NO3 conditions, wherein the latter seems more sensible in terms of its structure (no double bond remaining). As shown in Scheme S7, the second-generation C4H7NO5 and C5H9NO7 compounds share the same precursor in the C5H8O2 channel. Consequently, the production of C5H9NO7 through this pathway would be interrupted immediately after the injection of O3 and NO2 like C4H7NO5. In reality, its signal might decay even faster due to the larger reaction rate of RO2 H shift (leading to the formation of C4H7NO5) than that of RO2 reacting with HO2 (leading to the formation of C5H9NO7). As presented by Vereecken and Nozière (2020), the rate coefficient of aldehydic H shift is ≥ 0.5 s−1 (298 K), while the pseudo first-order rate coefficient of RO2 reacting with HO2 is ∼ 10−3 s−1 (k (298 K) = 5 × 10−12 cm3 molecules−1 s−1 (Atkinson, 2007), and [HO2] ∼ 4 × 108 molecules cm−3), about 2 orders of magnitude smaller. This result implies that the increasing C5H9NO7 observed is contributed to by other formation pathways. As mentioned before, C5H9NO7 can also be produced by C5H8O3 oxidation. We find that the signal of C4H7NO6 ( 244), which results from C5H8O3 oxidation as well, continues increasing after the injection of O3 and NO2. This tentatively confirms that the production of C5H9NO7 in step IV is mainly from the C5H8O3 oxidation channel. More experimental or theoretical studies are needed to provide insights into these differences.
2N- and 3N-monomers
As shown in Fig. 5c, 2N-monomers formed much slower than 1N-monomers in the early stage, but their formation rates were accelerated in step II and step III, probably due to the accumulation of first-generation products. According to our mechanistic framework, 2N-monomers are second-generation products resulting from the further oxidation of 1N-monomers by NO3, which is consistent with their time behaviors detected by CIMS.
Like C4H7NO5 and C5H9NO7, different 2N-monomers have similar behavior in the first three steps, but they are obviously different in step IV when the concentration of NO3 increased drastically in the chamber. For instance, the signals of C5H8N2O8, C5H8N2O9, and C5H10N2O8 continue to increase after the injection of O3 and NO2, while that of C5H10N2O9 drops immediately. This is related to their detailed formation mechanisms, which are outside the scope of this study. Furthermore, C5H8N2O9 and C5H10N2O9 decay a little bit faster than C5H8N2O8 and C5H10N2O8, which might be related to their volatility and will be further discussed in next section.
Different from other 2N-monomers, the signals of C5H6N2O8 ( 301) increase continuously under high-NO3 conditions, although its net formation rate is almost zero at the end of step IV. The characteristics of C5H6N2O8 under high-NO3 conditions reflects its different formation pathways from other dinitrates, and without having a comprehensive knowledge of its chemical mechanism, we are unable to tell what exactly leads to the differences. In the Master Chemical Mechanism (MCM v3.3.1), C5H6N2O8 is proposed to be a PAN-like compound stemming from the C5 nitrooxy carbonyl (C5H7NO4) (http://mcm.york.ac.uk/browse.htt?species=NC4CHO, last access: 18 November 2019). Such a C5H6N2O8 compound would react with NO3 radicals due to the remaining double bond, and hence this cannot be the predominant formation pathway of the C5H6N2O8 observed in this study. Based on the formation mechanism of dinitrooxyepoxides (C5H8N2O7) proposed by Kwan et al. (2012), we suggest that C5H6N2O8 can also be a dinitrooxyepoxide resulting from cyclization of specific hydroperoxy alkyl radicals, as shown in Scheme S10. Alternatively, the C5 hydroxy nitrate (C5H9NO4) can be oxidized by NO3 and then react with NO3 radicals again, forming C5H6N2O8 with two aldehyde groups ultimately (see Scheme S10). According to the proposed mechanisms above, C5H6N2O8 formed through the first two pathways are second-generation products, while those from the third channel are third-generation products, in accordance with its time behavior measured by CIMS.
In addition to C5-2N-monomers, we observe some C4 dinitrates such as C4H6N2O7 ( 273) and C4H8N2O8 ( 291), and the signal intensity of C4H6N2O7 is comparable to the major C5-2N-monomers. C4 dinitrates have rarely been mentioned in previous isoprene-NO3 studies. As shown in Fig. 5c, C4H6N2O7 has similar time behavior to C5-2N-monomers and hence is thought to be a second-generation product. Wennberg et al. (2018) proposed that such a C4 dinitrate was generated from OH-initiated further oxidation of C5H7NO4. However, this is not applicable here due to a lack of OH radicals in our system. Instead, we propose that the C4H6N2O7 observed in this study is a dinitrooxy carbonyl compound resulting from NO3 oxidation of C5H7NO4 with subsequent unimolecular decomposition (see Scheme S11 for details).
As shown in Fig. 5d, 3N-monomers are generated more slowly than 1N-monomers, but their signals grow gradually as the experiment proceeds, with a significant increase especially for C5H9N3O10 in the last step. Furthermore, we can see from Fig. 5c and d that the signals of C5 trinitrates in step IV appear anticorrelated to that of C5H10N2O8 and C5H10N2O8. The gas-phase 3N-monomers have rarely been reported in the previous literature. Ng et al. (2008) observed a C5H9N3O10 compound in the particle phase and assumed that it was produced from NO3 oxidation of the C5 hydroxy nitrate (C5H9NO4). Similarly, C5H9N3O11 and C5H9N3O12 can be formed through NO3 reacting with dinitrooxy peroxy radicals, which result from corresponding first-generation nitrooxy compound (C5 hydroperoxy nitrate, C5H9NO5, or C5 hydroxy hydroperoxy nitrate, C5H9NO6) oxidation by NO3 radicals, as shown in Scheme S12. 3N-Monomers formed following such pathways are second-generation products by definition. Regarding the rising signals of 3N-monomers in step IV, one explanation is that although the reaction of dinitrooxy peroxy radicals with NO3 is not an oxidation process, their formation can be significantly facilitated by increasing NO3 concentration. It is also possible that 3N-monomers are formed through H abstraction of 2N-monomers. NO3 radicals can abstract the hydrogen of dihydroxy dinitrate (C5H10N2O8) or hydroxyl hydroperoxy dinitrate (C5H10N2O9) from the carbon with an –OH, –OOH or –ONO2 group attached, leading to alkyl radicals that can subsequently recombine with O2 and then react with NO2 or NO3, yielding trinitrates or peroxynitrates containing three nitrogen atoms. 3N-Monomers stemming from such reactions ought to be third-generation products. However, we should point out that 3N-monomers formed following H-abstraction pathways are less likely because abstracting hydrogen from the hydroxyl, hydroperoxy, or nitrooxy carbon would lead to fragmentation in most cases (Bianchi et al., 2019).
In addition, it is interesting to note that the signal of C5H9N3O10 increases continuously throughout step IV, whereas that of C5H9N3O11and C5H9N3O12 drop after a short period of growth. Meanwhile, the production of C5H9N3O10 is facilitated by the increasing NO3 concentration compared to that of C5H9N3O12 and C5H9N3O11. Currently, we cannot explain what exactly causes these differences, but we suspect that there may be different chemical pathways forming different 3N-monomers that are not covered here and may also be related to their different physical properties, such as vapor pressures.
2N- and 3N-dimers
As shown in Fig. 5e, 2N-dimers (except for C10H16N2O11) display very similar time behavior to 1N-monomers, which form rapidly after each injection, indicating that the signals of 2N-dimers are dominated by first-generation products like most 1N-monomers. It is noted that the time behavior of C10H16N2O11 ( 419) is completely different from that of other 2N-dimers. As illustrated in Fig. 5e, the production rate of C10H16N2O11 is initially much slower compared to other dimers. Besides, its signal increases monotonically in the first two oxidation stages, whereas that of the others always increases first, approaching the maximum as its chemical production competes against the losses, and decreases gradually thereafter. The special time behavior of C10H16N2O11 suggests that it has a different formation pathway from other 2N-dimers, and its signal is most likely dominated by secondary products. In addition, we find that the signal of C10H16N2O12 always starts to decay earlier than that of C10H16N2O8 and C10H16N2O9. If we assume that their production rates have the same order of magnitude (confirming by their formation rates after each injection), then it can be concluded that C10H16N2O12 had additional chemical destruction, or its volatility is much lower than C10H16N2O8 and C10H16N2O9 and hence has more rapid loss on the wall. It seems the second hypothesis is more likely when comparing its signal with and without dilution and wall-loss corrections (see Fig. S5). More detailed discussion about volatilities of different isoprene organonitrates will be provided in the next section.
It is proposed that dimers (ROOR′) are likely formed through the self- or cross-reaction of two peroxy radicals (Berndt et al., 2018). Consequently, the generation number of dimers depends only on how the involved peroxy radicals are formed. Table S1 summarizes the possible permutation scheme of 2N-dimers from RO2+ RO reactions, and their structural information can be found in Scheme S13. For example, self-reaction of two C5 nitrooxy peroxy radicals (C5H8NO5) leads to the formation of a C10H16N2O8 compound, while recombination of two C5 nitrooxy hydroxyl peroxy radicals (C5H8NO6) or a C5 nitrooxy peroxy radical (C5H8NO5) with a C5 nitrooxy hydroperoxy peroxy radical (C5H8NO7) results in C10H16N2O10 compound. According to their time behavior, 2N-dimers (except for C10H16N2O11) are thought to be first-generation products, and from this fact we can infer that the peroxy radicals contributing to dimer formation are dominated by first-generation intermediates. With regard to C10H16N2O11, we conclude that it is most likely a secondary product considering its typical second-generation behavior. In other words, at least one of the two C5 nitrooxy peroxy radicals involved in formation of C10H16N2O11 must be a secondary intermediate. As listed in Table S1, C10H16N2O11 can be formed through C5H8NO6+ C5H8NO7 or C5H8NO6 + C5H8NO7 reactions, wherein C5H8NO7 and C5H8NO8 would be secondary peroxy radicals if they are formed through further NO3 oxidation of the C5 hydroxy carbonyl compounds (C5H8O2 or C5H8O3), as shown in Scheme S7. In addition, it is possible that C10H16N2O11 is formed from a C5 hydroxy peroxy radical C5H9O3 reacting with a C5 dinitrooxy hydroxy carbonyl peroxy radical C5H7N2O10 (from C5H7NO5 oxidation by NO3), as we observe high abundant C5H10O3 during the experiment, although C5H10O3 is assumed to be the major product of the OH-initiated chemistry.
Apart from 2N-dimers, we observe detectable signals at 450, 466, 482, 498, and 514, which are identified as 3N-dimers with molecular formulas C10H17N3O12−16. C10H17N3O12 and C10H17N3O13 were detected in the particle phase in a previous study, suggesting that they have low volatility and can contribute to SOA formation (Ng et al., 2008). As shown in Fig. 5f, 3N-dimers form much slower than 2N-dimers, but their productions are accelerated as the experiment proceeds. This is similar to the characteristics of second-generation 2N- and 3N-monomers to some degree, suggesting that the signals of 3N-dimers we observed are most likely dominated by secondary or even later-generation compounds.
It is worth noting that C10H17N3O12−14 and C10H17N3O15,16 have two completely different types of time behavior. The signals of C10H17N3O12, C10H17N3O13, and C10H17N3O14 more or less increase in the first three oxidation steps and start to decline late in step III with increasing NO3 concentration. As depicted in Scheme S13, 3N-dimers can result from further oxidation of 2N-dimers or the cross-reaction of a first-generation nitrooxy peroxy radical with a secondary dinitrooxy peroxy radical. Accordingly, such 3N-dimers are thought to be second-generation products, and they would further react with NO3 due to the remaining double bond in their molecular structure, leading to severe chemical destruction of these compounds under high-NO3 conditions. This is consistent with the time behavior of C10H17N3O12, C10H17N3O13, and C10H17N3O14. In contrast, C10H17N3O15 and C10H17N3O16 are formed even more slowly, and their production in the first 4 h is close to zero. However, their signals start to climb late in step III, during which those of C10H17N3O12, C10H17N3O13, and C10H17N3O14 decline. This suggests that C10H17N3O15 and C10H17N3O16 formed under high-NO3 conditions probably result from further reactions of C10H17N3O12−14. However, this assumption is highly uncertain, and more experimental and theoretical studies are needed to substantiate it. In terms of their time behavior, C10H17N3O15 and C10H17N3O16 are thought to be third- or even later-generation products.
3.3 Volatility distribution of isoprene nitrates
3.3.1 C∗ estimated by experimental methods
Detailed information about the volatility of organic molecules is essential to evaluate their potential to form SOA. In order to investigate the potential contribution of various isoprene oxidation products to SOA formation, we use our (limited) experimental data to estimate the vapor pressure of different isoprene organonitrates on the basis of their condensation behavior. Figure 6 shows how the signals of gas-phase products change in experiments with and without seed aerosols (ammonium sulfate). Please note that while the two experiments were conducted under similar conditions, the procedures could not be kept fully identical as aerosol seeding required specific measures and the oxidation chemistry might be slightly altered (e.g., due to initiation of heterogeneous reactions).
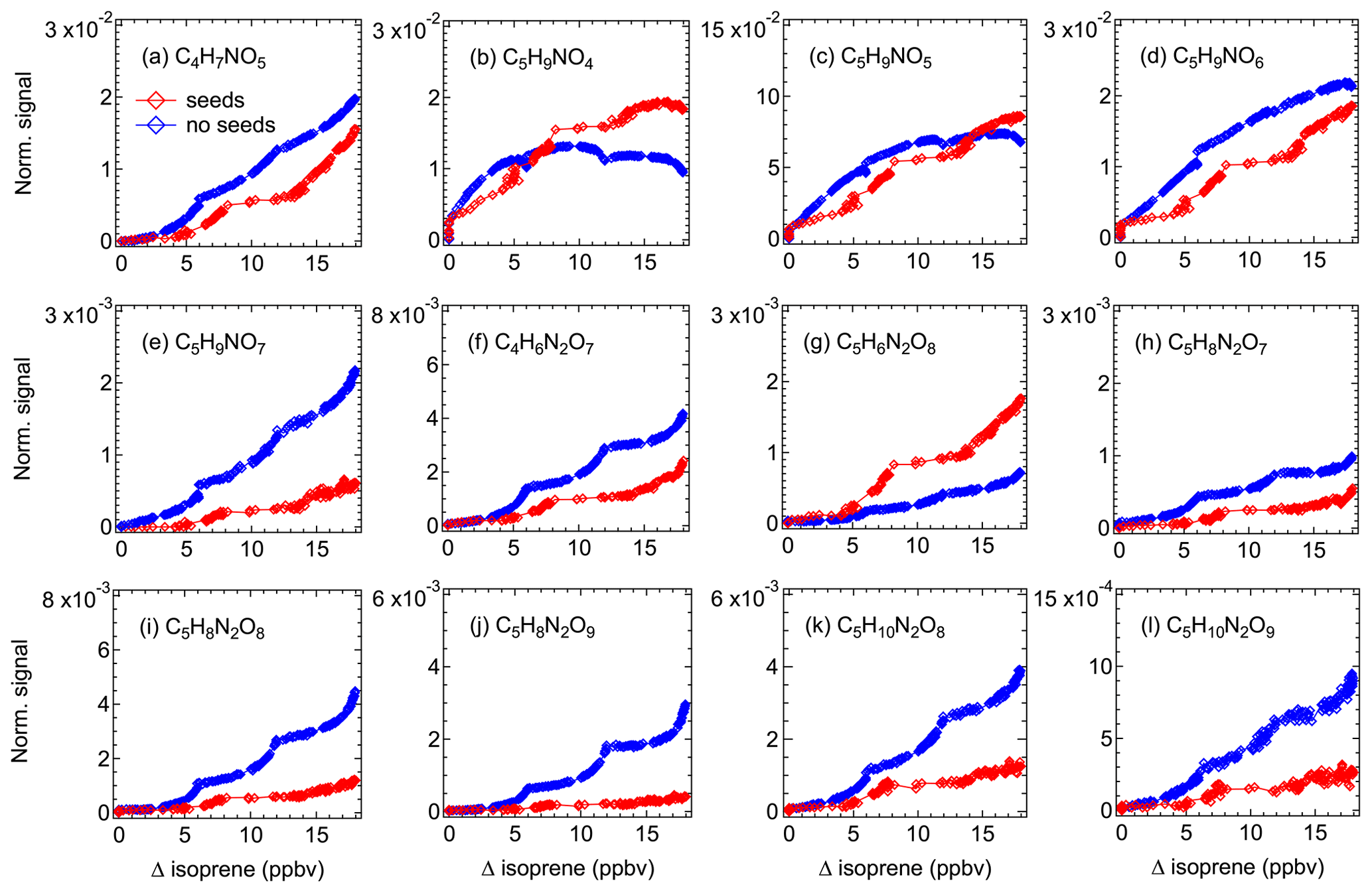
Figure 6Time evolution of selected major gas-phase products during experiments with (red) and without (blue) seed aerosols (ammonium sulfate). Signals have been corrected for dilution.
As shown in Fig. 6, the signals of most of the selected compounds decline when there are seed aerosols in the chamber, indicating that part of the condensable vapors is partitioned to the particle phase due to the introduction of condensation sinks. The decrease in signal differs for different products, mostly depending on their vapor pressures. As expected, the lower volatility of a compound, the higher the fraction that condenses. For instance, the signal of C5H9NO7 decreases by more than 70 % in an experiment with seed aerosols, compared to less than 40 % on average for other less-oxidized 1N-monomers. In some cases (e.g., C5H9NO4 and C5H9NO5), however, the product signals in an experiment with seed aerosols are higher than that without seeds after the consumed isoprene exceeding a certain level. In addition, the signal of C5H6N2O8 in the experiment with seeds is always higher compared to that without seeds. One explanation for this phenomenon is the effect of heterogeneous reactions. It is likely that some condensed compound (denoted as A) can react on the particle surface to form new products with the molecular composition of compound B, or alternatively forming a precursor of B. When they evaporate back to the gas phase, it can result in an increase in the signal of compound B. That is why a higher signal was observed for such compounds in an experiment with seeds than that without seeds, as observed for C5H6N2O8 in this case.
Based on the observed condensation behavior of different products, we can derive their vapor pressures from the gas-particle equilibrium partitioning coefficients by Eq. (2). As depicted in Fig. 7, the saturation concentrations of different organonitrates show a decreasing tendency from 1N-, 2N-monomer and 3N-monomers to 2N- and 3N-dimers, suggesting that dimers have a higher propensity of condensation and contribute to SOA formation. This is partly related to their molecular weight, as larger molecules generally have lower vapor pressures. However, it cannot explain all the features of the volatility distribution. For example, C5H9NO6 (corresponding to no. 8 in Fig. 7) has higher mass than C5H9NO5 (corresponding to no. 7 in Fig. 7) but is predicted to have higher vapor pressure. In general, chemical composition and functionalities have significant effects on vapor pressure. For instance, the 2D-VBS composition-activity relationship suggests that each carbon and oxygen decreases C∗ by 0.475 and 1.75 decades, respectively (Donahue et al., 2011). Different functional groups also have a very different effect on volatility. For example, each hydroxyl group (–OH) or hydroperoxy group (–OOH) typically reduces the volatility by 2.4 to 2.5 decades, while the less polar carbonyl group (= O) reduces the volatility by 1 decade (Pankow and Asher, 2008; Donahue et al., 2011). The nitrooxy group (–ONO2) has a similar reductive effect on vapor pressure, which typically reduces C∗ by 2.5 orders of magnitude (Pankow and Asher, 2008). Here, the irregularly high vapor pressure of C5H9NO6 is most likely attributed to the functional groups it contains. As listed in Table S2, C5H9NO6 is proposed to be a nitrooxy hydroxy hydroperoxyl compound, which consists of two highly polar functional groups –OH and –OOH, contributing to formation of intramolecular H bonding that can significantly increase the vapor pressure (Bilde et al., 2015; Kurten et al., 2016), while C5H9NO5 only contains a –OOH group and hence cannot form intramolecular H bonding. This explanation is also valid for C5H9N3O10 and C5H9N3O12. In summary, these findings underline that the constitutional and configurational information of a molecule is critical for vapor-pressure estimation.
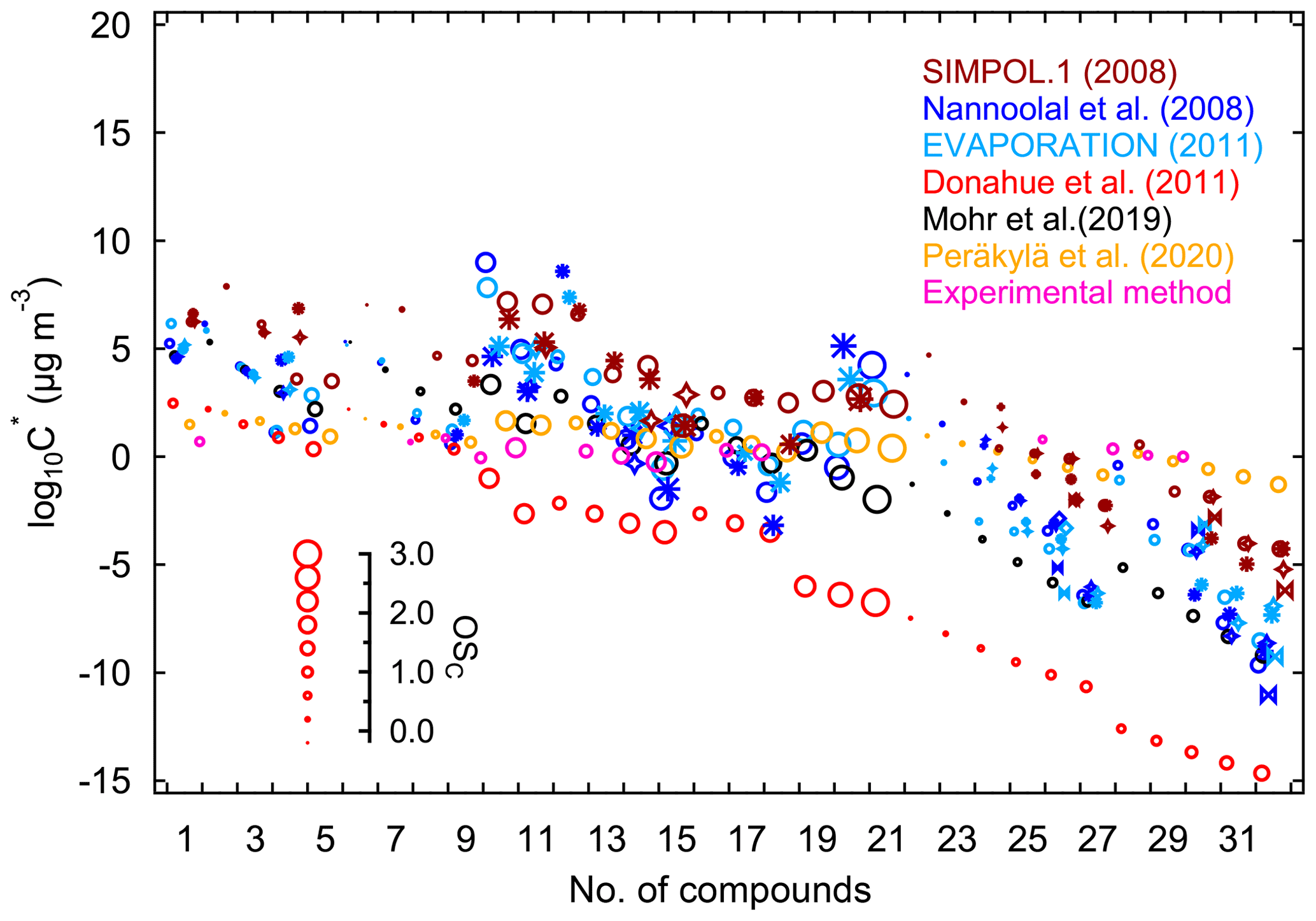
Figure 7Saturation concentrations (in µg m−3, at 298.15 K) of isoprene organonitrates estimated by using experimental and parameterization methods. The numbers correspond with the compound numbers of given in Table S2 (nos. 1–9, 10–18, 19–21, 22–27, and 28–32 corresponding to 1N-monomers, 2N-monomers, 3N-monomers, 2N-dimers, and 3N-dimers, respectively). Marker shapes indicate different isomers, with their size scaled by carbon oxidation state (OSC).
3.3.2 C∗ estimated by different parametrization methods
For comparison, we also adopt different parameterization methods to estimate the saturation vapor pressures of isoprene oxidation products based on their molecular composition and the proposed structures, with the results depicted in Fig. 7. In general, the saturation concentrations calculated by different parameterization methods show a similar volatility distribution to that calculated by the experimental method, with C∗ of 1N-, 2N-, and 3N-monomers and 2N- and 3N-dimers decreasing in turn. However, different parameterization methods lead to the predicted vapor concentrations with a variability of several orders of magnitude for the same compound, and the discrepancies become larger and larger with more complicated molecules. In addition, C∗ of structural isomers calculated by the same method could span several decades.
As shown in Fig. 7, the Donahue et al. parameterization mostly provides lower C∗ compared to the three GC methods, with a maximum discrepancy up to 12 orders of magnitude for dimers. With regard to smaller and less oxidized 1N-monomers, predicted C∗ values from different methods are in relatively good agreement with each other, whereas the disagreement increases to 11 orders of magnitude for 2N- and 3N-monomers. This is mainly the case because the organic molecules were regarded as a mixture of carbonyl (= O) and –OH functional groups in the Donahue et al. parameterization, and their relative abundance was assumed to be 1:1 (Donahue et al., 2011). In consequence, the –OOH functional group in peroxides is treated as two –OH groups when adapting the method proposed by Donahue et al. (2011). However, it is demonstrated that the extra oxygen in peroxy moieties has little contribution to reduce vapor pressure (Pankow and Asher et al., 2008); hence treating –OOH equivalently to two –OH functional groups would underestimate the vapor pressures of hydroperoxyl compounds. Furthermore, organic compounds consisting of multiple polar functional groups (such as hydroperoxy, peroxy acid, and peroxide functional groups) tend to form intramolecular H bonding, which would increase the vapor pressure (Bilde et al., 2015; Kurten et al., 2016). All these issues contribute to an underestimation of the vapor pressures of multifunctional products when using the Donahue et al. parameterization. Mohr et al. (2019) improved the parameterization for vapor-pressure estimation by taking the presence of –OOH functional groups in HOM explicitly into consideration and revising the parameters to reduce the effect of –OOH on depressing C∗. Consequently, the Mohr et al. parameterization effectively reduces the discrepancy between its estimates and those predicted by the GC methods, with the differences within 6 orders of magnitude. Nevertheless, there is a slight tendency to underestimate the vapor pressures of 3N-monomers and both 2N- and 3N-dimers. The Peräkylä et al. parameterization method, which was derived from measurements of the condensation behavior of HOM produced from α-pinene ozonolysis, predicts similar C∗ to the Donahue et al. method for 1N-monomers, but higher C∗ for 2N- and 3N-monomers like the Mohr et al. method. As for dimers, especially for the 3N-dimers containing more multifunctional groups, the Peräkylä et al. method even predicts higher C∗ than the GC methods in most cases.
Three GC methods predict similar saturation vapor pressures for different isoprene nitrates in this work, with the differences within 5 orders of magnitudes. Generally, the SIMPOL.1 method always provides higher C∗ compared to another two methods, and the disagreement between methods becomes larger for molecules containing multifunctional groups. For instance, the vapor-pressure discrepancy between SIMPOL.1 and another two GC methods are both 2 orders of magnitude for C5H9NO4,5 and C10H17N3O12−14, but it increased up to 4 and 5 orders of magnitude, respectively, for C5H9NO6,7 and C10H17N3O15,16.
It is worth noting that the Nannoolal et al. method is able to distinguish between positional isomers (e.g., the estimated C∗ for two C5H10N2O9 isomers are 0.858 and 0.333 µg m−3, respectively), whereas such capacity of EVAPORATION method is limited (e.g., it is able to distinguish between the position isomers of C5H10N2O9, but it predicts identical C∗ for C10H16N2O11 isomers). In this respect, the SIMPOL.1 method cannot distinguish between positional isomers at all. Moreover, the SIMPOL.1 method predicts smaller differences between functional group isomers for 1N-monomers and 3N-dimers compared to the Nannoolal et al. method and EVAPORATION, but there is no such regular pattern for 2N-monomers and 2N-dimers.
By comparing the results calculated by the experimental method with those by different parameterization methods, we can see that the GC methods predict higher-saturation concentrations for 1N-monomers than the experimental method, while the Donahue et al. and Peräkylä et al. method provide similar C∗ values. With regard to 2N-monomers, the GC methods predict higher vapor pressures compared to the experimental method, but the discrepancy decreases with decreasing saturation concentration. The disagreement of C∗ for 2N-monomers estimated by the experimental method and the Mohr et al. or Peräkylä et al. method are within 2 orders of magnitude. In terms of low-volatility dimers, however, the vapor pressures calculated by the experimental method were 1–3 orders of magnitude larger than that predicted by the parameterization methods except for the Peräkylä et al. method. The Peräkylä et al. method provides the most similar predictions to the experimental method for isoprene oxidation products in the full volatility range, with the disagreement within 1 order of magnitude.
In general, the vapor pressures estimated experimentally in this study are very close to those calculated by the Peräkylä et al. method for which the estimation parameters were also derived experimentally. The discrepancy between the experimental and the GC methods span several orders of magnitude depending on different compounds, with the GC methods predicting higher C∗ for less-functionalized 1N-monomers and approximate C∗ for 2N-monomers, but lower C∗ for highly functionalized dimers. It is difficult to tell which method is more reliable without any measured saturation vapor pressure data on such multifunctional organic nitrates. However, considering the fact that the existing GC methods tend to underestimate saturation vapor pressures of the highly functionalized organic molecules due to their limited capability to deal with intramolecular interactions (e.g., the intramolecular hydrogen bonding formed among polar functional groups), and the consistent results of two experimentally derived methods, we suggest that the experimental method might be a good choice to determine the volatility of highly oxidized compounds accurately.
3.3.3 Volatility distribution of isoprene nitrates and expected SOA yields
Although the vapor pressures calculated by different methods show a variability of several orders of magnitude, the predicted volatility distributions of different organic groups are consistent. To eliminate the discrepancy caused by methods and get an average trend of the volatility distribution of various isoprene nitrates, we use the median value of C∗ calculated by different methods as the estimator of the vapor pressure for each nitrate compound.
The average carbon oxidation state is plotted against Log10(C∗) in Fig. 8 to describe the volatility distribution of organic nitrates formed from isoprene oxidation by NO3. Generally, the volatility of measured gas-phase products spans a wide range from IVOCs to ELVOCs, wherein all of the 1N-monomers fall in the IVOC or SVOC range, suggesting that 1N-monomers have low potential to form SOA by simple condensation as long as the organic aerosol load is less than 200 µg m−3. The addition of a second or third –NO3 functional group decreases C∗ of most 2N- and 3N-monomers by 2–3 decades compared with 1N-monomers, and most of them belong to SVOCs. They will start to condense in significant fractions if the organic aerosol load is in a range of 1–10 µg m−3, which means 2N- and 3N-monomers with OSC > −0.8 may contribute to SOA formation under atmospheric conditions. With regard to dimers, all 3N-dimers and 2N-dimers (except for C10H16N2O8,9) are in LVOC or even ELVOC range, indicating isoprene dimers had high propensity to form SOA even at organic aerosol loads ≪ 1 µg m−3. However, we would like to emphasize here that the signals of 2N- and 3N-dimers only account for less than 2 % on average of the total assigned signals, as shown in Fig. S6. This suggests that the SOA yield of isoprene from NO3 oxidation by condensation should be low under atmospheric conditions.

Figure 8Volatility distribution of different organonitrates formed from NO3-initiated isoprene oxidation. The volatility classes are indicated along the top with corresponding colors in the plot. The position of potential SOA contributors is determined depending on the exact functionalities of molecules adapted from Bianchi et al. (2019).
The fate of RO2 determines the product distribution directly and hence could substantially affect SOA yields and aerosol physicochemical properties (Boyd et al., 2015; Fry et al., 2018; Ng et al., 2008; Schwantes et al., 2015; Ziemann and Atkinson, 2012). Consequently, it would be helpful to provide SOA yields together with the fate of RO2. In our experiment, reactions with HO2 and NO3 are the dominant loss channels for the initially formed RO2 from isoprene oxidation by NO3, contributing for ∼ 53 % and ∼ 30 % of overall RO2 loss; RO2+ RO2 reactions contributed a minor fraction (∼ 13 %) followed by unimolecular reactions with a contribution of ∼ 5 %, according to modeling results (Brownwood et al., 2021). More details about the modeling and the results can be found elsewhere (Brownwood et al., 2021; Vereecken et al., 2021).
In polluted urban regions, the fate of RO2 is typically dominated by RO2+ NO3, while in the more pristine environment, the RO2 + HO2 reaction will dominate RO2 fate (Bianchi et al., 2019; Boyd et al., 2015; Brown and Stutz, 2012). RO2+ HO2 was more important in the chamber than that in ambient conditions, and enhanced RO2+ HO2 would potentially lead to less dimer formation by RO2+ RO2 reactions and hence reduced SOA yields. However, a recent work from Brownwood et al. (2021) based on the same campaign as this study pointed out that the bulk aerosol composition and SOA yields were largely independent of RO2 fate. Similarly, Boyd et al. (2015) found for the β-pinene-NO3 system that RO2 fate (“RO2+ NO3 dominant” vs. “RO2+ HO2 dominant”) had only few effects on SOA formation. Therefore, the SOA yield estimated in this study is expected to be comparable to that in the atmosphere.
Assuming that the dimers in the LVOC or ELVOC range will condense onto particles, we estimated a SOA mass yield for condensation of isoprene organic nitrates of about 5 % ± 2 %. This value is based on an averaged bulk organonitrate sensitivity of 0.019 norm. counts s−1 ppbv−1 and has been corrected for wall loss and dilution (see Fig. S7, with uncorrected SOA mass yield of about 2 %). The estimated SOA mass yield is within the range of those reported in the literature, but at the lower end (4.3 % to 23.8 % depending on RO2 fate, Ng et al., 2008; 0.7 % for first generation oxidation and 14 % after oxidation of both double bonds, Rollins et al., 2009; 27 % on average for ambient measurements, Fry et al., 2018). The SOA yield will probably become somewhat higher if taking the contribution of SVOCs (including C10H16N2O8, C10H16N2O9, and some other monomers, as shown in Fig. 8) into consideration. Our finding is commensurable with the SOA yield for isoprene organic nitrates of 2 %–6 % derived from HR-AMS measurements in the same campaign (Brownwood et al., 2021).
In addition, Br− adduct ionization CIMS is selective for HO2 and less oxidized organic compounds (Albrecht et al., 2019; Rissanen et al., 2019), so it is reasonable to assume that there were more highly oxidized products that were not detected by Br− CIMS. This assumption is confirmed by measurements with a NO CIMS performed in another isoprene-NO3 experiment in SAPHIR (Zhao et al., 2021). Zhao et al. (2021) observed a higher fraction of dimers and more highly oxidized monomers and dimers, as well as trimers (C15 compounds). As a consequence, the SOA yields derived from NO CIMS measurements is slightly higher.
From these points of view our yield is more a lower limit. However, even if we assume an error of a factor of 2, the SOA yield of isoprene organic nitrates by condensation is more likely in a range of about 10 % or less than in the higher range of 20 %–30 % published in the literature. Of course, by our method we cannot cover any liquid phase processes that would lead to additional SOA beyond the condensation of the target compounds.
In this work, a gas-phase experiment conducted in the SAPHIR chamber under near-atmospheric conditions in the dark was analyzed to primarily investigate the multi-generation chemistry of isoprene-NO3 system. The characteristics of a diversity of isoprene nitrates were measured by the CIMS using Br− as the reagent ion. Isoprene 1N-,2N-, and 3N-monomers and 2N- and 3N-dimers have different time behaviors, indicating the occurrence of multi-generation oxidation during this process. Based on their specific time behaviors as well as the general knowledge of isoprene and radical chemistry, the possible formation mechanisms of these compounds are proposed.
In order to evaluate the potential contribution of various isoprene nitrates to SOA formation, different composition-activity and group-contribution methods were used to estimate their saturation vapor pressures. We also calculated the vapor pressures of isoprene oxidation products based on the gas-particle equilibrium coefficients derived from condensation measurements. The vapor pressures estimated by different methods spans several orders of magnitude, and the discrepancies increase as the compounds become highly functionalized. It shows that existing composition-activity methods (especially the Donahue et al. method) seriously underestimate the saturation vapor pressure of multifunctional low-volatility molecules compared to the experimental methods. The group-contribution methods seem to have a better performance than the CA methods in this aspect, but they still have a tendency to underestimate the vapor pressures of multifunctional molecules. We suggest that experimental methods are a good choice to estimate the volatility of highly oxidized compounds accurately.
According to our results, 1N-monomers and most 2N- and 3N-nitrates fall in the IVOC or SVOC range. Therefore, they have, with a few exceptions, low potential to form SOA at atmospheric organic aerosol loads. In contrast, 2N- and 3N-dimers are estimated to have low or extremely low volatility, indicating that they are significant contributors to SOA formation, although dimers constitute less than 2 % of the total explained signals. In this study, no new particle formation events were observed. Assuming that the dimers in the LVOC or ELVOC range will condense onto particles completely, we estimate a SOA mass yield of about 5 % ± 2 %, which is a lower limit if one takes a possible contribution of the minor dimer products as well as SVOC species into consideration. Both the volatility distribution and calculated SOA yields indicate that isoprene dimers formed from NO3 oxidation are the major contributors to SOA formation.
All data given in figures can be displayed in tables or in digital form. This includes the data given in the Supplement. Please send all requests for data to t.mentel@fz-juelich.de and r.wu@fz-juelich.de. The data used in this work are available on the EUROCHAMP database (https://data.eurochamp.org/data-access/chamber-experiments/, EUROCHAMP, 2020) under https://doi.org/10.25326/JTYK-5V47 (Fuchs et al., 2020a) and https://doi.org/10.25326/0SPZ-BN30 (Fuchs et al., 2020b).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-10799-2021-supplement.
HF, JNC, JLF, SSB, AW, and AKS designed the study. Instrument deployment and data analysis were carried out by RW, ET, SK, SRA, LH, AN, HF, RT, TH, PTMC, JS, FB, BB, and JAT. RW, LV, ET, DZ, JAT, MH, and TFM interpreted the compiled data set. RW, TFM, and LV wrote the paper. All co-authors discussed the results and commented on the paper.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
Rongrong Wu gratefully acknowledges the fellowship from Helmholtz-OCPC (Office of China Postdoc Council) Postdoc Program for research support.
This research has been supported by the European Research Council (ERC), (SARLEP grant agreement no. 681529),
European Commission (EC) under the European Union's Horizon 2020 research and innovation program, (Eurochamp 2020 grant agreement no. 730997 and FORCeS grant agreement no. 821205),
Vetenskapsrådet (VR, grant nos. 2014-05332 and 2018-04430), and
Svenska Forskningsrådet Formas (grant nos. 2015-1537 and 2019-586).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Eliza Harris and reviewed by two anonymous referees.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019.
Anglada, J. M., Crehuet, R., and Francisco, J. S.: The Stability of α-Hydroperoxyalkyl Radicals, Chem. Eur. J., 22, 18092–18100, 2016.
Atkinson, R.: Gas-phase tropospheric chemistry of organic compounds: a review, Atmos. Environ., 41, 200–240, https://doi.org/10.1016/j.atmosenv.2007.10.068, 2007.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003.
Barber, V. P., Pandit, S., Green, A. M., Trongsiriwat, N., Walsh, P. J., Klippenstein, S. J., and Lester, M. I.: Four-carbon Criegee intermediate from isoprene ozonolysis: Methyl vinyl ketone oxide synthesis, infrared spectrum, and OH production, J. Am. Chem. Soc., 140, 10866–10880, 2018.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of alpha-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bilde, M., Barsanti, K., Booth, M., Cappa, C. D., Donahue, N. M., Emanuelsson, E. U., McFiggans, G., Krieger, U. K., Marcolli, C., Topping, D., Ziemann, P., Barley, M., Clegg, S., Dennis-Smither, B., Hallquist, M., Hallquist, A. M., Khlystov, A., Kulmala, M., Mogensen, D., Percival, C. J., Pope, F., Reid, J. P., Ribeiro da Silva, M. A., Rosenoern, T., Salo, K., Soonsin, V. P., Yli-Juuti, T., Prisle, N. L., Pagels, J., Rarey, J., Zardini, A. A., and Riipinen, I.: Saturation vapor pressures and transition enthalpies of low-volatility organic molecules of atmospheric relevance: from dicarboxylic acids to complex mixtures, Chem. Rev., 115, 4115–4156, https://doi.org/10.1021/cr5005502, 2015.
Brown, S., Degouw, J., Warneke, C., Ryerson, T., Dubé, W., Atlas, E., Weber, R., Peltier, R., Neuman, J., and Roberts, J.: Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 9, 3027–3042, https://doi.org/10.5194/acp-9-3027-2009, 2009.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, 2012.
Brownwood, B., Turdziladze, A., Hohaus, T., Wu, R., Mentel, T. F., Carlsson, P. T., Tsiligiannis, E., Hallquist, M., Andres, S., and Hantschke, L.: Gas-particle partitioning and SOA yields of organonitrate products from NO3-initiated oxidation of isoprene under varied chemical regimes, ACS Earth Space Chem., 5, 785–800, https://doi.org/10.1021/acsearthspacechem.0c003112021, 2021.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Compernolle, S., Ceulemans, K., and Müller, J. F.: EVAPORATION: a new vapour pressure estimation methodfor organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Crosson, E.: A cavity ring-down analyzer for measuring atmospheric levels of methane, carbon dioxide, and water vapor, Appl. Phys. B, 92, 403–408, https://doi.org/10.1007/s00340-008-3135-y, 2008.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dommen, J., Hellen, H., Saurer, M., Jaeggi, M., Siegwolf, R., Metzger, A., Duplissy, J., Fierz, M., and Baltensperger, U.: Determination of the aerosol yield of isoprene in the presence of an organic seed with carbon isotope analysis, Environ. Sci. Technol., 43, 6697–6702, 2009.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Dubé, W. P., Brown, S. S., Osthoff, H. D., Nunley, M. R., Ciciora, S. J., Paris, M. W., McLaughlin, R. J., and Ravishankara, A.: Aircraft instrument for simultaneous, in situ measurement of NO3 and N2O5 via pulsed cavity ring-down spectroscopy, Rev. Sci. Instrum., 77, 034101, https://doi.org/10.1063/1.2176058, 2006.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
EUROCHAMP: https://data.eurochamp.org/data-access/ (last access: 15 July 2021), 2020.
Friedman, B. and Farmer, D. K.: SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes, Atmos. Environ., 187, 335–345, https://doi.org/10.1016/j.atmosenv.2018.06.003, 2018.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J. M., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 − Gas-phase oxidation − product study [Data set], AERIS, https://doi.org/10.25326/JTYK-5V47, last access: 1 December 2020a.
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J. M., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 − Gas-phase oxidation − product study [Data set], AERIS, https://doi.org/10.25326/0SPZ-BN30, last access: 1 December 2020b.
Guenther, A., Jiang, X., Heald, C., Sakulyanontvittaya, T., Duhl, T., Emmons, L., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. .
Hallquist, M., Wenger, J., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N., George, C., and Goldstein, A.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009 .
Hu, J., Wu, L., Zheng, B., Zhang, Q., He, K., Chang, Q., Li, X., Yang, F., Ying, Q., and Zhang, H.: Source contributions and regional transport of primary particulate matter in China, Environ. Pollut., 207, 31–42, https://doi.org/10.1016/j.envpol.2015.08.037, 2015.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jenkin, M., Young, J., and Rickard, A.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jimenez, J. L., Canagaratna, M., Donahue, N., Prevot, A., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., and Ng, N.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Trostl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Perakyla, O., Piel, F., Petaja, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipila, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tome, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Jaoui, M., and Edney, E. O.: Ozone-isoprene reaction: Re-examination of the formation of secondary organic aerosol, Geophys. Res. Lett., 34, L01805, https://doi.org/10.1029/2006GL027485, 2007.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion-Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kurten, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene autoxidation products may not have extremely low saturation vapor pressures despite high O : C ratios, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016.
Kwan, A., Chan, A., Ng, N., Kjærgaard, H. G., Seinfeld, J., and Wennberg, P.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
McFiggans, G., Mentel, T. F., Wildt, J. r., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. S. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure – product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petaja, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 4442, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: Monoterpene SOA – Contribution of first-generation oxidation products to formation and chemical composition, Atmos. Environ., 130, 136–144, https://doi.org/10.1016/j.atmosenv.2015.10.080, 2016.
Nannoolal, Y., Rarey, J., Ramjugernath, D., and Cordes, W.: Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 226, 45–63, https://doi.org/10.1016/j.fluid.2004.09.001, 2004.
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 269, 117–133, https://doi.org/10.1016/j.fluid.2008.04.020, 2008.
Ng, N., Kwan, A., Surratt, J., Chan, A., Chhabra, P., Sorooshian, A., Pye, H. O., Crounse, J., Wennberg, P., and Flagan, R.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., and Rosanka, S.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020.
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/d0cp05555g, 2021.
O'Meara, S., Booth, A. M., Barley, M. H., Topping, D., and McFiggans, G.: An assessment of vapour pressure estimation methods, Phys. Chem. Chem. Phys., 16, 19453–19469, https://doi.org/10.1039/c4cp00857j, 2014.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/C2CS35166H, 2012.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The atmospheric chemistry of alkoxy radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Peeters, J., Müller, J.-F. O., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOM), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020.
Pöschl, U.: Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem. Int. Ed., 44, 7520–7540, https://doi.org/10.1002/anie.200501122, 2005.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Rissanen, M. P., Kurten, T., Sipila, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jorgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petaja, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Rissanen, M. P., Mikkilä, J., Iyer, S., and Hakala, J.: Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 12, 6635–6646, https://doi.org/10.5194/amt-12-6635-2019, 2019.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F. J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Rollins, A. W., Kiendler-Scharr, A., Fry, J., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., and Mentel, T.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Sobanski, N., Schuladen, J., Schuster, G., Lelieveld, J., and Crowley, J. N.: A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 9, 5103–5118, https://doi.org/10.5194/amt-9-5103-2016, 2016.
Spracklen, D., Jimenez, J., Carslaw, K., Worsnop, D., Evans, M., Mann, G., Zhang, Q., Canagaratna, M., Allan, J., and Coe, H.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Stadtler, S., Kühn, T., Schröder, S., Taraborrelli, D., Schultz, M. G., and Kokkola, H.: Isoprene-derived secondary organic aerosol in the global aerosol–chemistry–climate model ECHAM6.3.0–HAM2.3–MOZ1.0, Geosci. Model Dev., 11, 3235–3260, https://doi.org/10.5194/gmd-11-3235-2018, 2018.
Starn, T., Shepson, P., Bertman, S., Riemer, D., Zika, R., and Olszyna, K.: Nighttime isoprene chemistry at an urban-impacted forest site, J. Geophys. Res.-Atmos., 103, 22437–22447, 1998.
Stroud, C., Roberts, J., Williams, E., Hereid, D., Angevine, W., Fehsenfeld, F., Wisthaler, A., Hansel, A., Martinez-Harder, M., and Harder, H.: Nighttime isoprene trends at an urban forested site during the 1999 Southern Oxidant Study, J. Geophys. Res.-Atmos., 107, 4554, https://doi.org/10.1029/2001JD000959, 2002.
Suh, I., Lei, W., and Zhang, R.: Experimental and Theoretical Studies of Isoprene Reaction with NO3, J. Phys. Chem. A, 105, 6471–6478, https://doi.org/10.1021/jp0105950, 2001.
Surratt, J. D., Chan, A. W., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Thornton, J. A., Shilling, J. E., Shrivastava, M., D'Ambro, E. L., Zawadowicz, M. A., and Liu, J.: A Near-Explicit Mechanistic Evaluation of Isoprene Photochemical Secondary Organic Aerosol Formation and Evolution: Simulations of Multiple Chamber Experiments with and without Added NOx, ACS Earth Space Chem., 4, 1161–1181, https://doi.org/10.1021/acsearthspacechem.0c00118, 2020.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., and Wagner, R.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vereecken, L.: Computational study of the stability of α-nitroxy-substituted alkyl radicals, Chem. Phys. Lett., 466, 127–130, https://doi.org/10.1016/j.cplett.2008.10.042, 2008.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, https://doi.org/10.1039/C2CS35070J, 2012.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – Part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/B909712K, 2009.
Vereecken, L. and Peeters, J.: A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/C0CP00387E, 2010.
Vereecken, L., Nguyen, T. L., Hermans, I., and Peeters, J.: Computational study of the stability of α-hydroperoxyl-or α-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 393, 432–436, https://doi.org/10.1016/j.cplett.2004.06.076, 2004.
Vereecken, L., Carlsson, P., Novelli, A., Bernard, F., Brown, S. S., Cho, C., Crowley, J. N., Fuchs, H., Mellouki, W., Reimer, D., Shenolikar, Justin, Tillmann, R., Zhou, L., Kiendler-Scharr, A., and Wahner, A.: Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 23, 5496–5515, https://doi.org/10.1039/d0cp06267g, 2021.
Wang, S., Riva, M., Yan, C., Ehn, M., and Wang, L.: Primary formation of highly oxidized multifunctional products in the OH-Initiated oxidation of Isoprene: a combined theoretical and experimental study, Environ. Sci. Technol., 52, 12255–12264, https://doi.org/10.1021/acs.est.8b02783, 2018.
Warneke, C., De Gouw, J., Goldan, P., Kuster, W., Williams, E., Lerner, B., Jakoubek, R., Brown, S., Stark, H., and Aldener, M.: Comparison of daytime and nighttime oxidation of biogenic and anthropogenic VOCs along the New England coast in summer during New England Air Quality Study 2002, J. Geophys. Res.-Atmos., 109, D10309, https://doi.org/10.1029/2003JD004424, 2004.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., and Smarte, M. D.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L., Stone, D., and Heard, D.: New insights into the tropospheric oxidation of isoprene: combining field measurements, laboratory studies, chemical modelling and quantum theory, in: Atmospheric and Aerosol Chemistry, edited by: McNeill, V. F. and Ariya, P. A., Springer, Berlin, Heidelberg, Germany, 55–95, https://doi.org/10.1007/128_2012_359, 2012.
Zhang, Q., Jimenez, J. L., Canagaratna, M., Allan, J., Coe, H., Ulbrich, I., Alfarra, M., Takami, A., Middlebrook, A., and Sun, Y.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhao, D., Pullinen, I., Fuchs, H., Schrade, S., Wu, R., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Guo, Y., Kiendler-Scharr, A., Wahner, A., Kang, S., Vereecken, L., and Mentel, T. F.: Highly oxygenated organic molecules (HOM) formation in the isoprene oxidation by NO3 radical, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2020-1178, in review, 2020.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.