the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Jul 2020
| 03 Jul 2020
Constraining remote oxidation capacity with ATom observations
Katherine R. Travis
Hannah M. Allen
Eric C. Apel
Stephen R. Arnold
Donald R. Blake
William H. Brune
Róisín Commane
John D. Crounse
Bruce C. Daube
Glenn S. Diskin
James W. Elkins
Mathew J. Evans
Samuel R. Hall
Eric J. Hintsa
Rebecca S. Hornbrook
Prasad S. Kasibhatla
Michelle J. Kim
Kathryn McKain
Dylan B. Millet
Fred L. Moore
Jeffrey Peischl
Thomas B. Ryerson
Tomás Sherwen
Alexander B. Thames
Kirk Ullmann
Xuan Wang
Paul O. Wennberg
Glenn M. Wolfe
Fangqun Yu
The global oxidation capacity, defined as the tropospheric mean concentration of the hydroxyl radical (OH), controls the lifetime of reactive trace gases in the atmosphere such as methane and carbon monoxide (CO). Models tend to underestimate the methane lifetime and CO concentrations throughout the troposphere, which is consistent with excessive OH. Approximately half of the oxidation of methane and non-methane volatile organic compounds (VOCs) is thought to occur over the oceans where oxidant chemistry has received little validation due to a lack of observational constraints. We use observations from the first two deployments of the NASA ATom aircraft campaign during July–August 2016 and January–February 2017 to evaluate the oxidation capacity over the remote oceans and its representation by the GEOS-Chem chemical transport model. The model successfully simulates the magnitude and vertical profile of remote OH within the measurement uncertainties. Comparisons against the drivers of OH production (water vapor, ozone, and NOy concentrations, ozone photolysis frequencies) also show minimal bias, with the exception of wintertime NOy. The severe model overestimate of NOy during this period may indicate insufficient wet scavenging and/or missing loss on sea-salt aerosols. Large uncertainties in these processes require further study to improve simulated NOy partitioning and removal in the troposphere, but preliminary tests suggest that their overall impact could marginally reduce the model bias in tropospheric OH. During the ATom-1 deployment, OH reactivity (OHR) below 3 km is significantly enhanced, and this is not captured by the sum of its measured components (cOHRobs) or by the model (cOHRmod). This enhancement could suggest missing reactive VOCs but cannot be explained by a comprehensive simulation of both biotic and abiotic ocean sources of VOCs. Additional sources of VOC reactivity in this region are difficult to reconcile with the full suite of ATom measurement constraints. The model generally reproduces the magnitude and seasonality of cOHRobs but underestimates the contribution of oxygenated VOCs, mainly acetaldehyde, which is severely underestimated throughout the troposphere despite its calculated lifetime of less than a day. Missing model acetaldehyde in previous studies was attributed to measurement uncertainties that have been largely resolved. Observations of peroxyacetic acid (PAA) provide new support for remote levels of acetaldehyde. The underestimate in both model acetaldehyde and PAA is present throughout the year in both hemispheres and peaks during Northern Hemisphere summer. The addition of ocean sources of VOCs in the model increases cOHRmod by 3 % to 9 % and improves model–measurement agreement for acetaldehyde, particularly in winter, but cannot resolve the model summertime bias. Doing so would require 100 Tg yr−1 of a long-lived unknown precursor throughout the year with significant additional emissions in the Northern Hemisphere summer. Improving the model bias for remote acetaldehyde and PAA is unlikely to fully resolve previously reported model global biases in OH and methane lifetime, suggesting that future work should examine the sources and sinks of OH over land.
- Article
(7934 KB) - Full-text XML
-
Supplement
(783 KB) - BibTeX
- EndNote




The hydroxyl radical (OH) is the main oxidant responsible for removing trace gases from the atmosphere, and its concentration defines the tropospheric oxidation capacity. OH is primarily produced by the photolysis of ozone in the presence of water vapor. The lifetimes of key atmospheric trace gases are governed by how quickly they are removed by reaction with OH. Oxidation of volatile organic compounds (VOCs) by OH produces tropospheric ozone and fine particulate matter which are detrimental to human health and vegetation and impact climate. The oxidation of VOCs, carbon monoxide (CO), and methane provides the main sink of OH in the troposphere. Oxidation of methane and VOCs accounts for over half of the global CO production (Duncan et al., 2007; Safieddine et al., 2017), resulting in a tight coupling of these compounds.
Models generally overestimate global mean tropospheric OH and its ratio in the Northern Hemisphere to Southern Hemisphere (Naik et al., 2013; Patra et al., 2014). These biases may be linked to the persistent CO underestimate in models (Shindell et al., 2006), as prescribing OH from observations improves simulated CO (Müller et al., 2018). However, constraining models with observations of ozone and water vapor cannot resolve biases in model OH (Strode et al., 2015), which is impacted by additional complex factors such as the chemical mechanism and the ozone photolysis frequency (Nicely et al., 2017). Constraining the performance of model chemical mechanisms has largely focused on regions of strong biogenic and anthropogenic activity (Emmerson and Evans, 2009; Yu et al., 2010; Marvin et al., 2017), but at least half of the oxidation of methane occurs over the ocean, where models have received little evaluation due to a lack of observational constraints.
The introduction of airborne measurements of OH reactivity (OHR) provides a method to evaluate the sink of OH across a range of altitudes and a variety of locations and chemical environments (Mao et al., 2009; Thames et al., 2020). Previous work compared surface observations of OHR at a single site to the sum of individually calculated OHR components from measurements (Di Carlo, 2004; Yoshino et al., 2006; Sinha et al., 2008, 2010; Mao et al., 2010; Dolgorouky et al., 2012; Hansen et al., 2014; Nakashima et al., 2014; Nölscher et al., 2012, 2016; Ramasamy et al., 2016; Zannoni et al., 2016, 2017) or from simple models (Ren et al., 2006; Lee et al., 2009; Lou et al., 2010; Mogensen et al., 2011; Mao et al., 2012; Edwards et al., 2013; Kaiser et al., 2016; Whalley et al., 2016). Thames et al. (2020) found evidence of missing OHR between measurements and an observationally constrained box model during the first three ATom deployments. Chen et al. (2019) compared calculated OHR from a global model to OHR determined from a suite of VOCs but did not have measurements of OHR itself. Ferracci et al. (2018) found that missing OHR estimated from surface observations could result in a small increase in the methane lifetime in a global model. Safieddine et al. (2017) and Lelieveld et al. (2016) presented the first global model simulations of OHR but with only qualitative comparison to observations. No study has quantitatively compared simulated and observed OHR in a global model in an effort to constrain the OH sink.
The ATom campaign (Wofsy et al., 2018) provides an unprecedented opportunity to test models in the remote atmosphere with a detailed suite of chemical observations. We simulate the first two deployments (ATom-1: July–Agust 2016, ATom-2: January–February 2017) using the GEOS-Chem chemical transport model (CTM) as our tool to explore potential sources of systematic errors that could explain the community-wide model overestimate in global mean OH and underestimate of the methane lifetime. We include model evaluation with measurements of OHR, a relatively new constraint available for assessing atmospheric oxidation capacity. To our knowledge, this is the first quantitative use of this measurement to evaluate a CTM.
2.1 The GEOS-Chem model
We use the GEOS-Chem global 3-D CTM in v12.3.0 (http://www.geos-chem.org, last access: 2 July 2020) driven by assimilated meteorological data from the Goddard Earth Observing System Modern-Era Retrospective analysis for Research and Applications, Version 2 (MERRA-2; Gelaro et al., 2017). The native MERRA-2 model has a horizontal resolution of and 72 vertical levels which we degrade to and 47 vertical levels for use in GEOS-Chem. The midpoint of the first model layer is 58 m. We use time steps of 20 min for chemistry and 10 min for transport as recommended by Philip et al. (2016). GEOS-Chem includes detailed treatment of HOx–NOx–VOC–halogen–aerosol chemistry, with recent improvements for isoprene (Chan Miller et al., 2017; Fisher et al., 2016; Marais et al., 2016; Travis et al., 2016), peroxyacetyl nitrate (PAN) (Fischer et al., 2014), and halogen chemistry (Sherwen et al., 2016). The production of organic aerosols is calculated using fixed yields from isoprene, monoterpenes, biomass burning, and anthropogenic fuel combustion (Pai et al., 2020). Aerosol uptake of HO2 is parameterized with a reactive uptake coefficient (γ) of 0.2 (Jacob, 2000) to produce H2O (Mao et al., 2013). Aerosol thermodynamic equilibrium is calculated by ISORROPIA II v2.0 (Pye et al., 2009). Surface methane concentrations are prescribed monthly using spatially interpolated observations from the NOAA GMD flask network. We simulate the 2016–2017 period with an 18-month initialization.
Table 1Annual emissions of CO and NOx for 2016 used in the GEOS-Chem simulations.
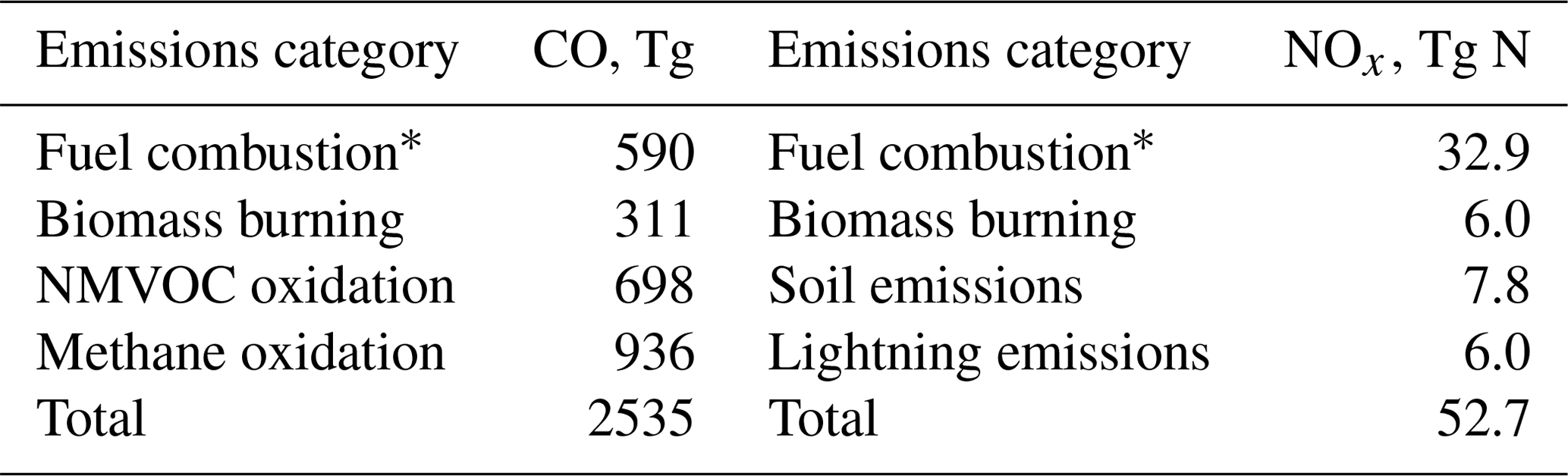
* Anthropogenic fossil fuel and biofuel combustion.
Global fire emissions at 3-hourly resolution (Mu et al., 2011) for 2016 and 2017 are from the Global Fire Emissions Database (GFED4s; van der Werf et al., 2017). The GFED4s burned area (Giglio et al., 2013) includes a parameterization of small fires (Randerson et al., 2012). Biogenic VOC emissions are from MEGANv2.1 (Guenther et al., 2012; Hu et al., 2015). Global anthropogenic emissions are from the Community Emissions Data System (CEDS) inventory (Hoesly et al., 2018), overwritten by ethanol from the POET inventory (Olivier et al., 2003; Granier et al., 2005), ethane from Tzompa-Sosa et al. (2017), and regional inventories for the United States (NEI11v1, Travis et al., 2016), Canada (CAC, https://www.canada.ca/en/services/environment/pollution-waste-management/national-pollutant-release-inventory.html, last access: 31 July 2013), Mexico (BRAVO, Kuhns et al., 2003), Europe (EMEP, http://www.emep.int/index.html, last access: 31 March 2015), Asia (MIX, Li et al., 2017), and Africa (DICE, Marais and Wiedinmyer, 2016). Lightning emissions are constrained with satellite data according to Murray et al. (2012) with a global flash rate of 280 mol NO flash−1 (Marais et al., 2018). Air–sea exchange is calculated for acetaldehyde (Millet et al., 2010), acetone (Fischer et al., 2012), and dimethyl sulfide (Breider et al., 2017). All emissions are processed by the Harvard Emissions Component (HEMCO, Keller et al., 2014). Table 1 gives the 2016 emission budget for CO and NOx.
The standard simulation includes prescribed methanol concentrations. We expand this simulation to include emissions and chemistry for methanol as well as unsaturated C2 compounds. Air–sea exchange of methanol is specified using the methodology of Millet et al. (2008) with a constant seawater concentration of 142 nM. Terrestrial biogenic methanol emissions are from MEGANv2.1, and anthropogenic and biomass burning emissions are from the inventories described above. We likewise include biomass burning and anthropogenic emissions of ethyne (C2H2) and ethene (C2H4) along with terrestrial biogenic emissions of C2H4. Oxidation of C2H2 by OH proceeds according to the Master Chemical Mechanism (MCM) v3.3.1 (Jenkin et al., 1997, 2015; Saunders et al., 2003), via http://mcm.leeds.ac.uk/MCM (last access: 8 February 2018). Simplified C2H4 chemistry is included based on Lamarque et al. (2012) with an updated OH rate constant from the MCM v3.3.1. Table S1 in the Supplement shows the reactions and species included for unsaturated C2 compounds. The standard model does not consider the OH reactivity of a subset of organic acids (RCOOH) from the oxidation of VOCs. We implement oxidation of RCOOH and evaluate the impact of excluding this species, which is minor, in Table S2 and Fig. S1 in the Supplement. The model concentration of H2 is fixed at 500 ppt, consistent with observed H2 from ATom-1 and ATom-2 (520 ppt).
The GEOS-Chem global mean tropospheric OH ([OH]GM) is calculated as an air-mass-weighted quantity below the model tropopause (see http://wiki.seas.harvard.edu/geos-chem/index.php/Mean_OH_concentration, last access: 4 May 2020, for the calculation methodology). The [OH]GM for 2016 is 11.9×105 molecules cm−3 and the corresponding methane lifetime() is 9.0 years. This result is comparable to the multi-model [OH]GM of 11.1×105 molecules cm−3 and of 9.7 years from Naik et al. (2013). The best observationally derived estimate of is 11.2±1.3 years (Prather et al., 2012), suggesting a model bias here of 20 %. We calculate the ratio of total 2016 air-mass-weighted OH in the Northern (> 0∘ N) to Southern Hemisphere (< 0∘ S) to be 1.14. This exceeds observationally derived ratios of 0.85 to 0.97 (Montzka et al., 2000; Patra et al., 2014) but is at the low end of previous model estimates ranging from 1.13 to 1.42 (Naik et al., 2013).
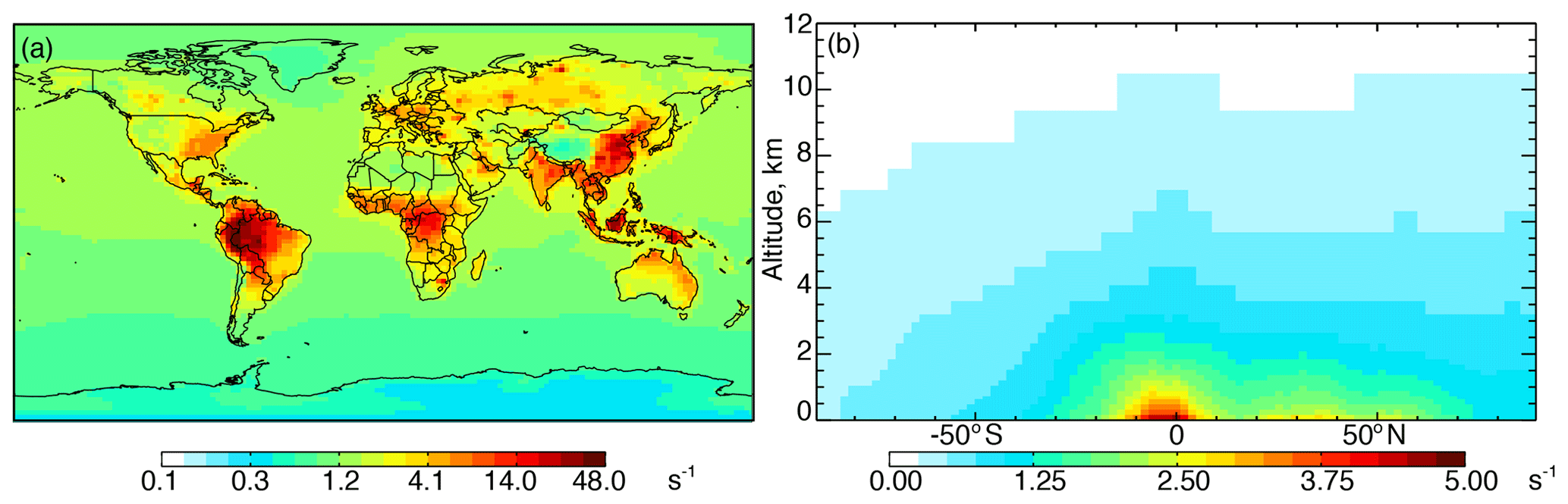
Figure 1Annual mean 2016 (a) surface (log scale) and (b) zonal mean cOHR calculated from individual model species. The GEOS-Chem species included in the calculation of cOHR are listed in Table S3.
2.2 Calculated OH reactivity
The atmosphere contains thousands of reactive organic compounds (Goldstein and Galbally, 2007). Transforming the concentrations of these compounds and reactive inorganics to calculated OH reactivity (cOHR) ranks them in order of their importance as OH sinks. The cOHR from a model (cOHRmod) can then be compared to cOHR from a suite of measurements (cOHRobs) where cOHR is defined by Eq. (1).
Figure 1a shows annual surface cOHRmod for the year 2016 based on the 90 components listed in Table S3. Figure 1b shows the zonal mean profile below 12 km. Approximately 80 % of air-mass-weighted cOHRmod resides below 3 km. The average annual surface cOHRmod is 1.8 s−1, with 40 % present over the ocean (average of 1.0 s−1). Higher cOHRmod occurs in coastal outflow regions and the lowest cOHRmod is present over the Southern Ocean. The maximum cOHRmod (48 s−1) over northern China is due to high concentrations of SO2, NOx, and CO. In the tropics, elevated cOHRmod is mainly from isoprene, other biogenic species, and CO.
Table 2Description of ATom measurements used to evaluate the model simulation.
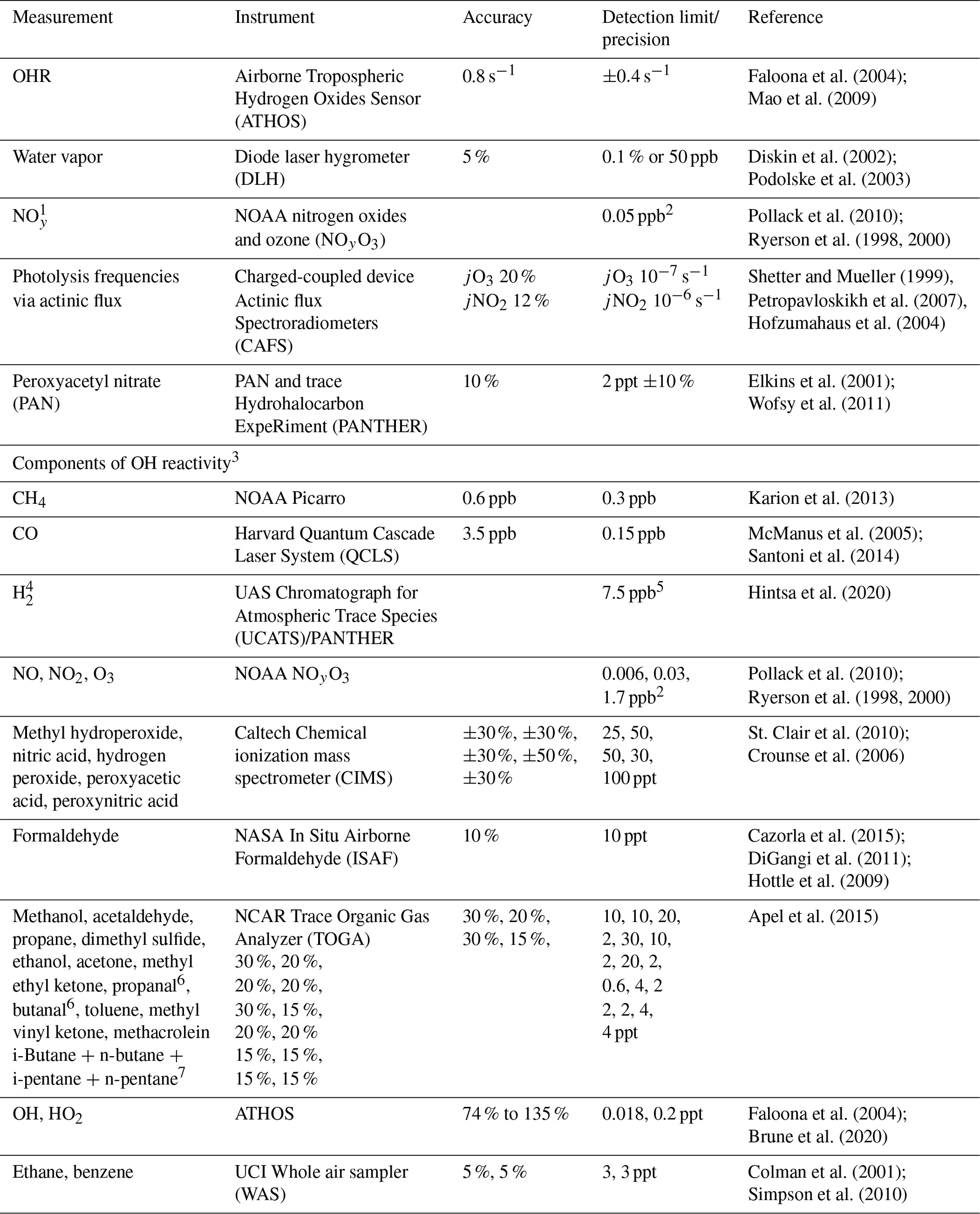
1 Model NOy is defined as ANs. 2 Average of 2σ uncertainty for each individual 1 Hz measurement for ATom-1 and ATom-2. 3 Included in cOHR are observations of species where at least 20 % of the possible available measurements below 3 km are not missing. 4 The GEOS-Chem concentration of H2 is set to a constant value of 500 ppt. 5 Average of reported error for each individual measurement for ATom-1 and ATom-2. 6 Lumped as > C4 alkanes (ALK4) in GEOS-Chem. 7 Lumped as > C3 aldehydes (RCHO) in GEOS-Chem.
2.3 ATom observations
The NASA ATom field campaign (Wofsy et al., 2018) sampled the remote troposphere with the DC-8 aircraft over the Atlantic and Pacific oceans from approximately 200 m to 12 km altitude in four seasons from 2016 to 2018 with a goal of improving the representation of trace gases and short-lived greenhouse gases in models of atmospheric chemistry and climate. We use data here from the first two deployments (ATom-1 and ATom-2), which sampled winter and summer conditions in each hemisphere. We consider only observations over the ocean (73 % of measurements). Flight tracks for ATom-1 with land crossings removed are shown in Fig. 2; ATom-2 flight tracks are nearly identical. We sample the model along the flight tracks, and both the model and observations are averaged to the model grid and time step for all the following comparisons. The aircraft carried an extensive chemical payload including observations of water vapor, methane, CO, OH, NOx, VOCs, photolysis frequencies, and OHR. Table 2 describes the observations used in this work.
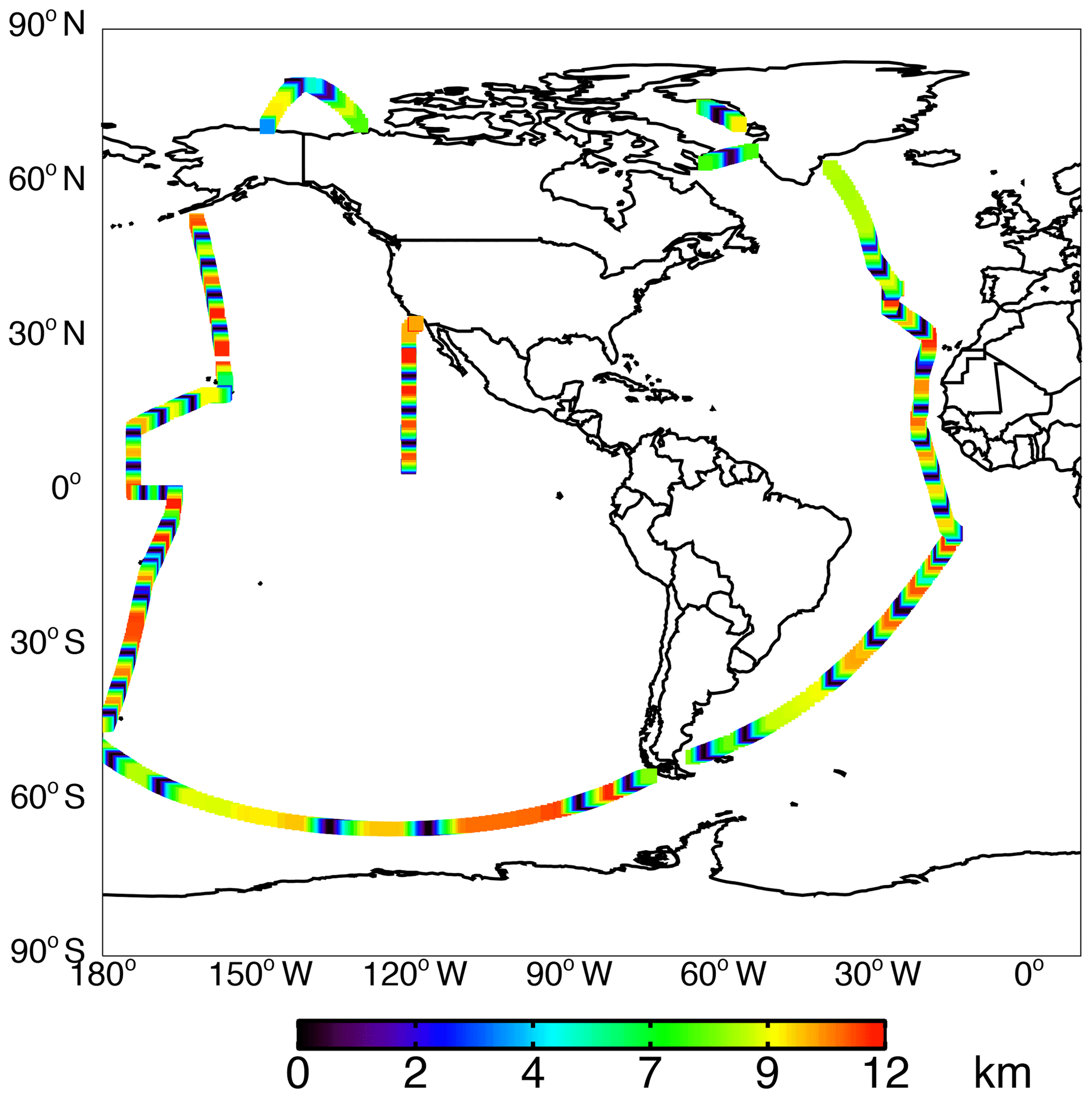
Figure 2ATom-1 ocean-only flight tracks colored by altitude.
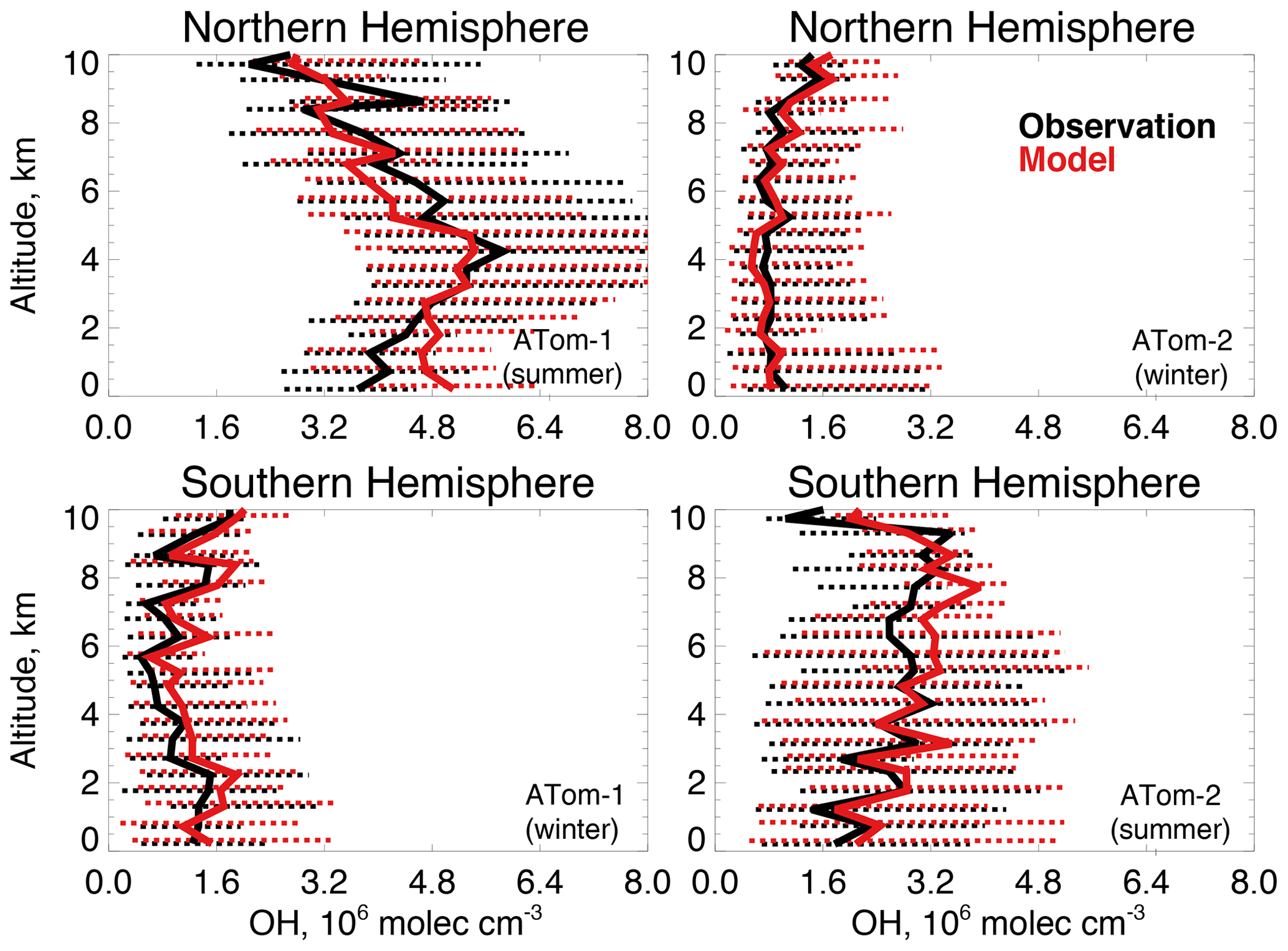
Figure 3Median OH concentrations for the Northern Hemisphere (> 0∘ N) and Southern Hemisphere (< 0∘ S) from the ATHOS instrument described in Table 2 during ATom-1 (July–August 2016) and ATom-2 (January–February 2017) compared against the GEOS-Chem model in 0.5 km altitude bins. The observations have been filtered to remove biomass burning (acetonitrile > 200 ppt) and stratospheric (O3∕CO > 1.25) influence. The dashed lines show the observed 25th–75th percentiles.
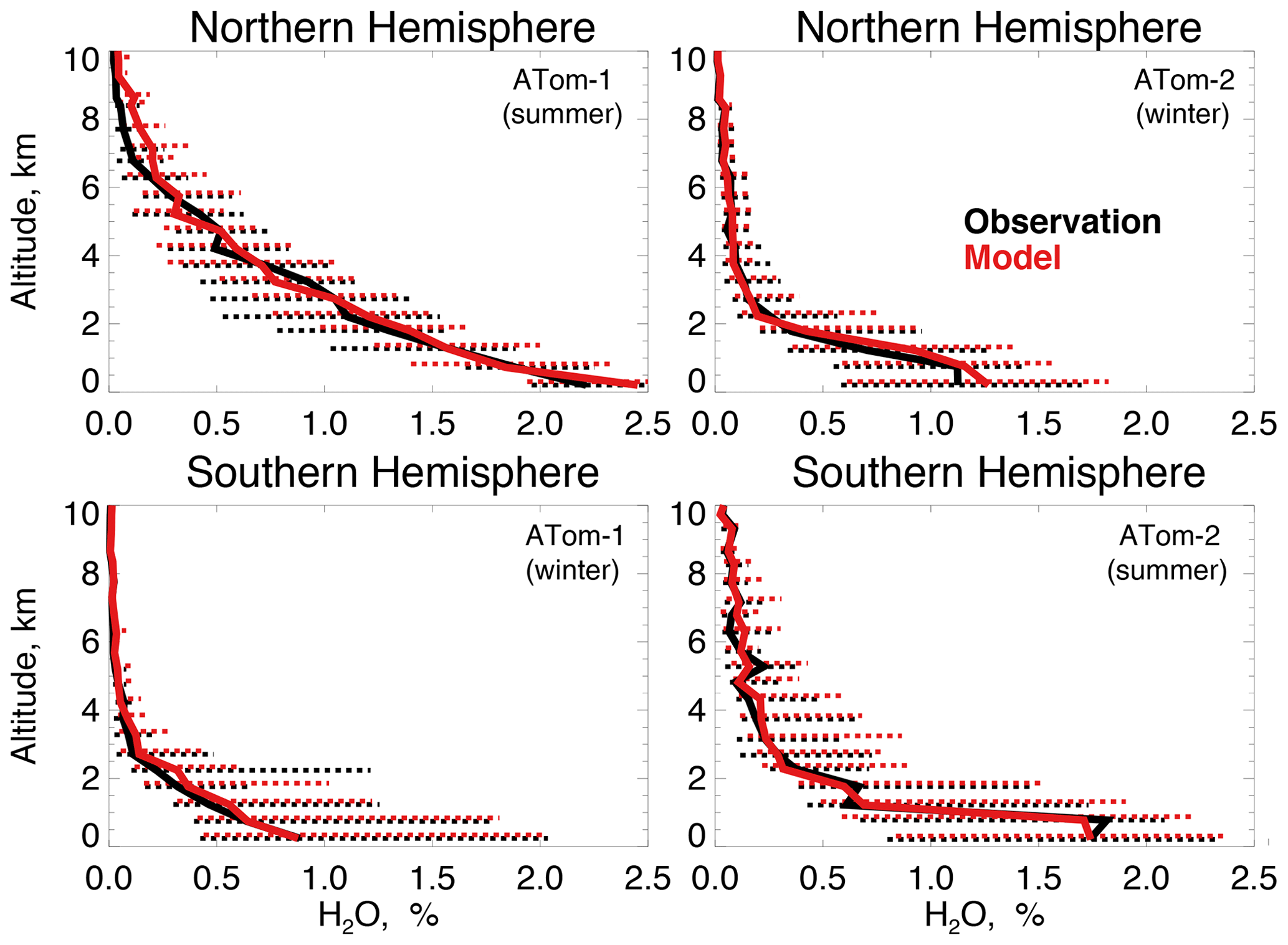
Figure 4The same as Fig. 3 for median water vapor concentrations. Water vapor mixing ratio was measured by the DLH instrument as described in Table 2.
We compare observed and simulated OH concentrations to evaluate whether differences are consistent with the bias in discussed in Sect. 2.1. Figure 3 shows modeled OH sampled along the flight tracks and compared to observed OH (Table 2) for ATom-1 (boreal summer 2016) and ATom-2 (boreal winter 2017) in each hemisphere from the lowest sampled altitude (∼200 m) to 10 km. There is no evidence of a systematic overestimate in modeled OH throughout the troposphere. Figure S2 shows similarly good agreement across the observed frequency distributions of OH concentration. A model OH overestimate is apparent in the lowest 2 km in the Northern Hemisphere summer that could indicate excessive OH production or an underestimated sink from emissions of ocean VOCs. Global models tend to overestimate OH against constraints from methyl chloroform observations (Shindell et al., 2006; Naik et al., 2013; Nicely et al., 2017), but we find here that tropospheric OH is successfully simulated within observational uncertainty (74 % to 135 %, 2σ confidence level). This result from a global CTM is consistent with good agreement between OH measurements and a box model during NASA's Pacific Exploratory Mission – Tropics (PEM-Tropic B) campaign in the clean remote Pacific (Tan et al., 2001) and a similar analysis by Brune et al. (2020) for ATom 1 through 4.
We calculate the median air-mass-weighted column average OH (OHcol) from the median OH concentrations in Fig. 3 and the total tropospheric air mass over the ocean. During ATom-1, the modeled OHcol in the Northern (Southern) Hemisphere is 4.5(1.4)×106 molecules cm−3 compared against the observations of 4.4(1.1)×106 molecules cm−3 during ATom-1. Similarly, during ATom-2, OHcol is 0.8(2.8)×106 molecules cm−3 in the model and 0.9(2.6)×106 molecules cm−3 in the observations. Median model OHcol is within 30 % of observations during both deployments, with the smallest bias in the total column during Northern Hemisphere summer when OH is at a maximum. As discussed above, model OH is overestimated in the lowest 2 km during this period, but this bias is minimized in the column average. The observed air-mass-weighted ratio of Northern to Southern Hemisphere OH, calculated in the same manner as described in Sect. 2, is 2.8 during ATom-1 and 0.2 during ATom-2, indicating a strong seasonality that the model largely reproduces (ratios of 2.3 and 0.2). This ratio is less than the ratio of OHcol because there is approximately 30 % less air mass over the ocean in the Northern Hemisphere ocean than over the Southern Hemisphere. This seasonality is masked by calculations performed on an annual mean basis. The seasonality in this ratio reported by Wolfe et al. (2019) for satellite-derived OH during ATom-1 and ATom-2 is more modest because they calculate a daily average OH that extends to the tropopause, while here, we use largely daytime aircraft observations below 10 km.
The model is in good agreement with OH measurements during ATom, but the uncertainty in the observations is similar to a recent estimate of the GEOS-Chem model uncertainty for OH of 25 % to 40 % (Christian et al., 2018). In addition, the lifetime of OH is short (seconds) and atmospheric concentrations are highly variable; thus, direct model comparison to measured OH is insufficient to demonstrate model skill in capturing the broader remote oxidation capacity. Agreement between the model and observations could also result from compensating errors in the OH source and sink. We support the model comparison in Fig. 3 with an evaluation of the key factors governing OH production and loss measured by ATom and investigate potential missing sources of VOCs from the ocean during summertime.
In the remote troposphere, OH is primarily produced from the photolysis of ozone in the presence of water vapor (Monks, 2005) and is enhanced by nitrogen oxides (NOx) from lightning and transport from continental sources. Methane, CO, and VOCs provide the main OH sinks (Murray et al., 2014). We compare the model to ATom-1 and ATom-2 observations of the drivers of the tropospheric OH source (water vapor, ozone, ozone photolysis frequency, NOx) to determine possible broader sources of model bias.
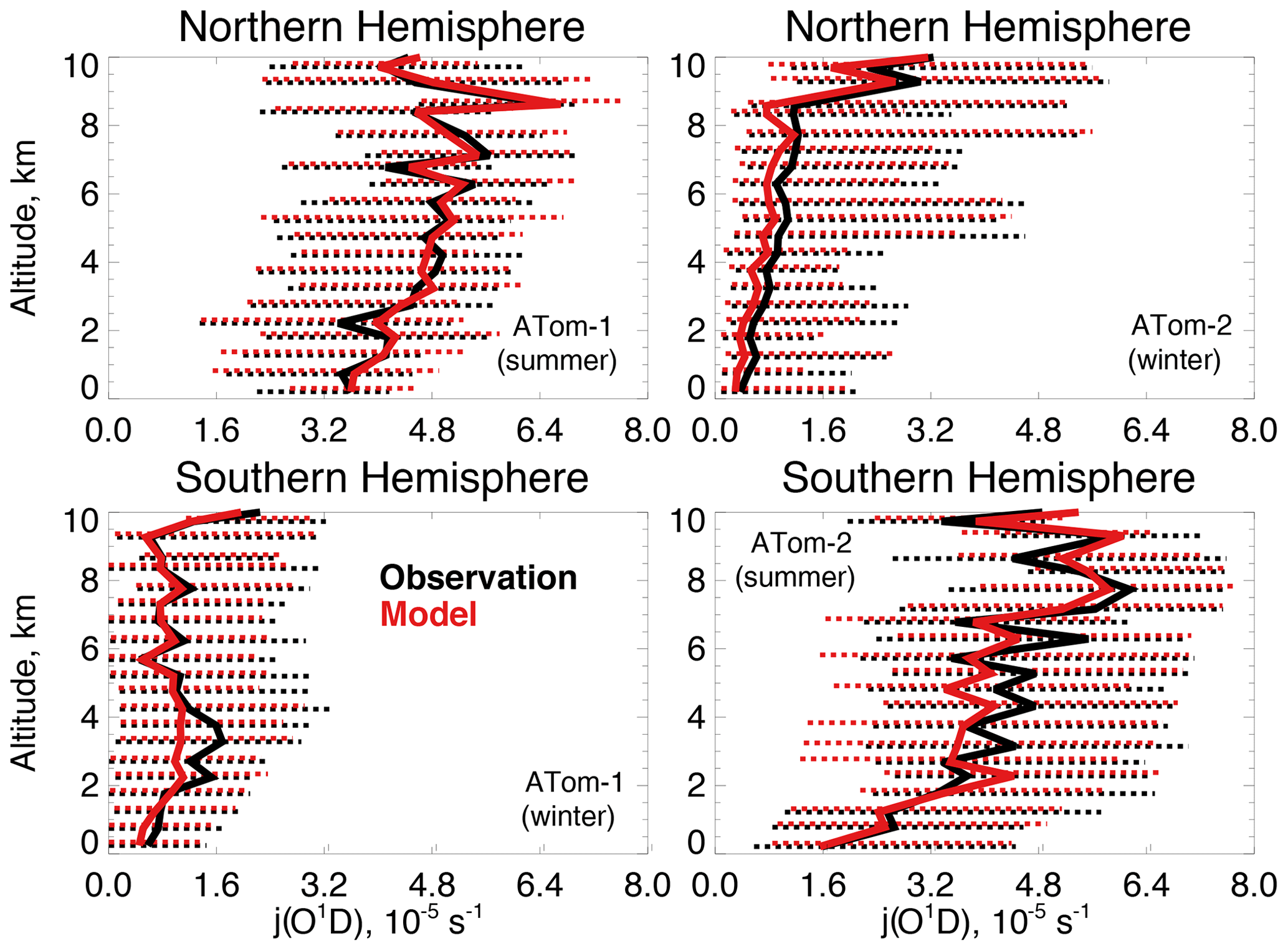
Figure 5The same as Fig. 3 for median photolysis frequencies for ozone (j(O1D)). The actinic flux measured by the CAFS instrument is used to calculate j(O1D) as described in Table 2.

Figure 6The same as Fig. 3 for median ozone concentrations. Ozone was measured by the NOAA NOyO3 instrument as described in Table 2.
Figure 4 compares observations of water vapor mixing ratios to the NASA MERRA-2 reanalysis product used by the model. MERRA-2 is generally successful at reproducing observed tropospheric water vapor (Gelaro et al., 2017), and we also find good agreement compared with ATom-1 and ATom-2 observations throughout the troposphere. We evaluate the model treatment of the incoming actinic flux and the resulting ozone photolysis frequency (j(O1D)) in Fig. 5. Hall et al. (2018) showed that GEOS-Chem actinic fluxes in both cloudy and clear skies were well simulated during the ATom-1 deployment. Figure 5 confirms the minimal model bias in j(O1D) and successful representation of the observed seasonality with median summertime values below 3 km ( s−1) approximately 4 times higher than in winter ( s−1).
The GEOS-Chem ozone simulation has been extensively tested against ozonesondes, aircraft, and satellite observations and shows no systematic overestimates (Hu et al., 2017), with the exception of continental surface concentrations (Fiore et al., 2009; Travis et al., 2016). Figure 6 shows that the highest (54–63 ppb) and lowest (14 ppb) tropospheric ozone observed during ATom-1 and ATom-2 occurs during summer in the mid to upper troposphere and marine boundary layer, respectively. Ozone is less variable in wintertime, with values between 30 and 50 ppb. The model generally reproduces the magnitude and shape of the tropospheric ozone profiles as well as the seasonality observed during both deployments. There is no evidence of the systematic Northern Hemisphere ozone bias previously seen in global model evaluations (Young et al., 2013) that was suggested as a cause of excessive OH (Naik et al., 2013). This may be reflected in the improved model interhemispheric OH ratio (Sect. 2.1) seen here over previous studies. Upper tropospheric ozone is overestimated in all cases but Northern Hemisphere summer, but this would not have a large influence on primary OH production (or the methane lifetime) at these altitudes (Brune et al., 2020).
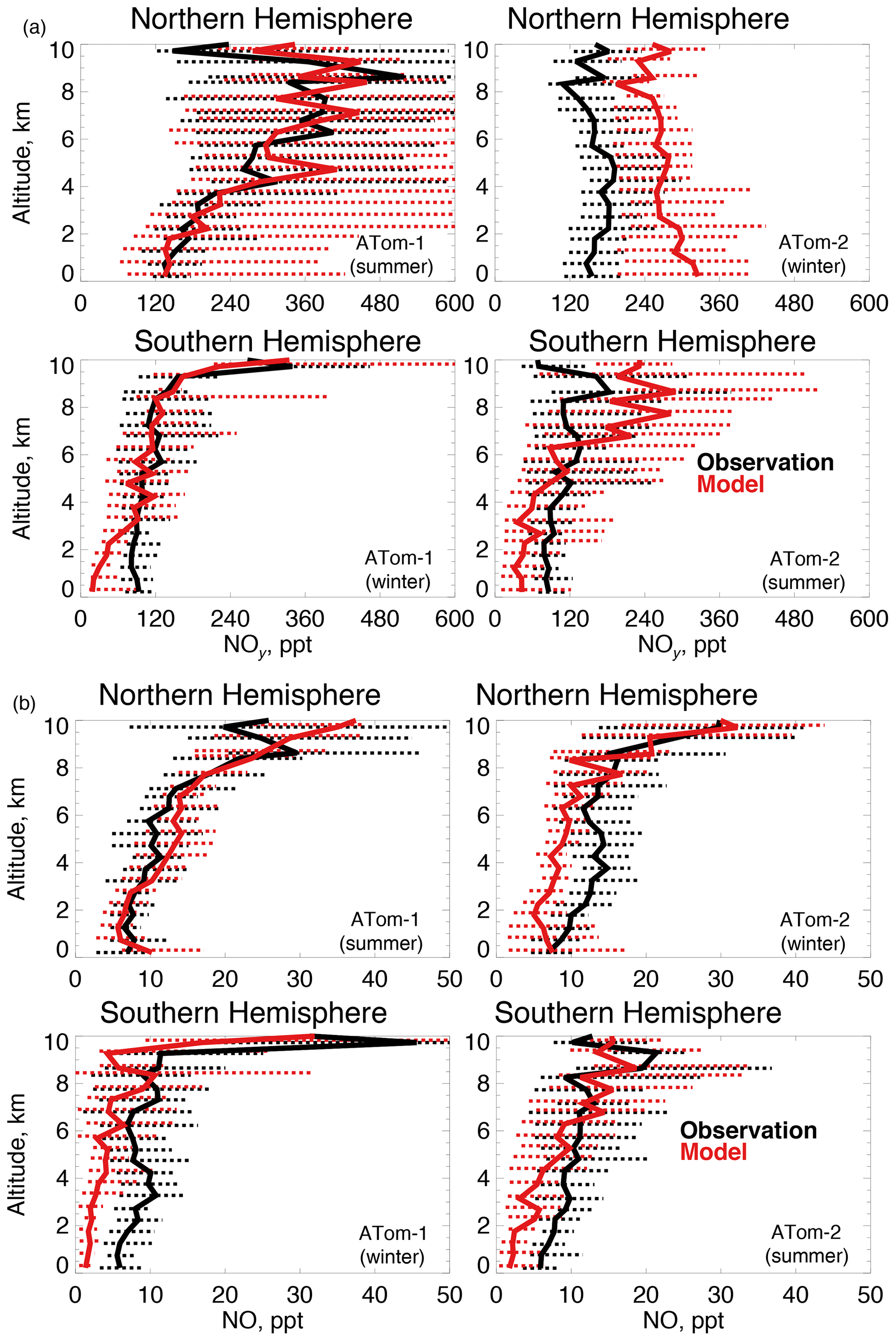
Figure 7The same as Fig. 3 for median NOy (a) and NO (b) concentrations. NOy and NO were measured by the NOAA NOyO3 instrument as described in Table 2.
OH is enhanced in the presence of NOx (≡NO+NO2). We use NOy here (Fig. 7a) as our constraint as observed NO2 was generally near the detection limit in both deployments. We also show NO (Fig. 7b) given its role in secondary OH production. The model reproduces the maximum in NOy that occurs in the Northern Hemisphere upper troposphere in summertime due to lightning (Marais et al., 2018). Observations show little variability between summer and winter NOy in the lower troposphere. Southern Hemisphere NOy is underestimated in the lowest few kilometers in both seasons, which could be due to missing ocean production of methyl nitrate (Fisher et al., 2018). The largest model discrepancy is an overestimate of approximately 70 % in the Northern Hemisphere wintertime. Observations of NO reflect the structure of NOy, with the exception of Northern Hemisphere winter.
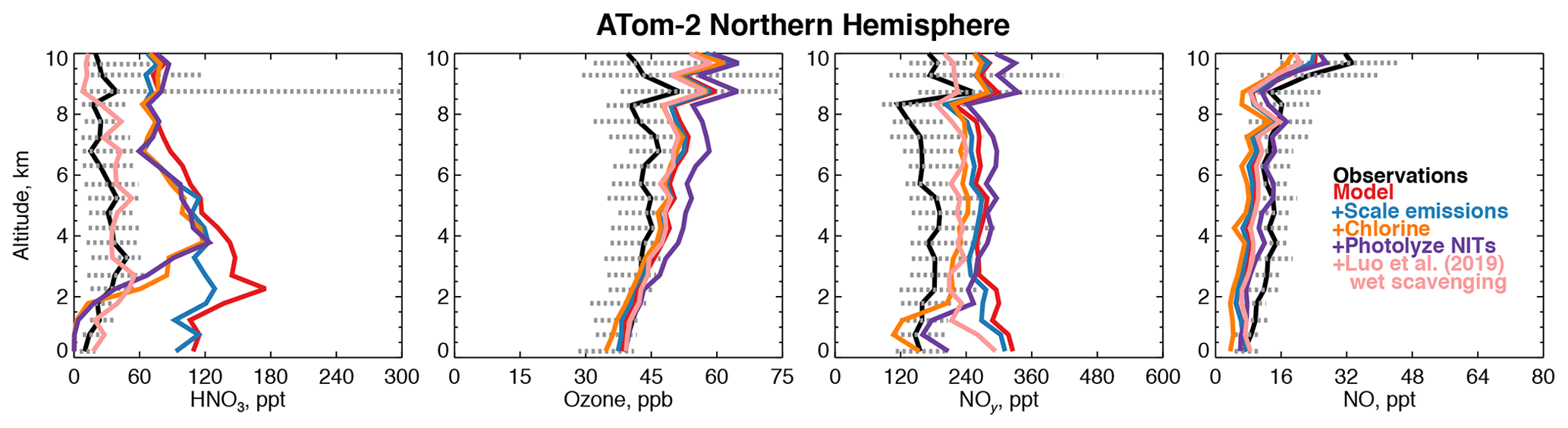
Figure 8Comparison of modeled and observed HNO3, ozone, NOy, and NO with sensitivity studies including scaling emissions from the US and Asia, improved chlorine chemistry (X. Wang et al., 2019), and the photolysis of particulate nitrate on coarse-mode sea-salt aerosols (Kasibhatla et al., 2018) as described in Sect. 4.1. HNO3 was measured by the Caltech CIMS; ozone, NOy, and NO were measured by the NOAA NOyO3 instrument (Table 2).
Causes of the remote model bias in NOy
Figure 8 shows that the model NOy overestimate in winter is primarily caused by nitric acid (HNO3). Excessive remote HNO3 is a long-standing model deficiency (Bey et al., 2001; Staudt et al., 2003; Brunner et al., 2003, 2005). The model bias identified here is unlikely to result from overestimated continental emissions due to the short lifetime of NOy against deposition (∼3 d in the Northern Hemisphere winter). Models suggest that less than 40 % of emitted NOx in the US is exported downwind (Dentener et al., 2006; Zhang et al., 2012). However, the standard model configuration here does not address the large possible bias in the US anthropogenic NOx inventory of ∼40 % (Anderson et al., 2014; Travis et al., 2016) or the downward trend in NOx emissions from Asia of ∼30 % since 2011 (Krotkov et al., 2016). As expected, scaling Asian and US NOx emissions by these percentages improves the model bias in winter by only 15 % below 3 km (Fig. 8). Recent improvements to the simulation of continental wintertime HNO3 (Jaeglé et al., 2018) would similarly be expected to have a marginal effect in our study region.
Kasibhatla et al. (2018) showed that acid displacement of chloride (Cl−) by HNO3 on sea-salt aerosols (SSA) could resolve model overestimates of gas-phase HNO3 in the marine boundary layer using the GEOS-Chem model. A more comprehensive simulation of this process was developed by X. Wang et al. (2019). Figure 8 shows sensitivity tests with the mechanism from X. Wang et al. (2019) incorporated into our simulation in the Northern Hemisphere winter. Model HNO3 decreases by approximately 100 ppt below 3 km, which would significantly improve the wintertime NOy bias in this region, but the free tropospheric bias remains. The displacement of Cl− described above generates particulate nitrate on coarse-mode SSA (NITs). Photolysis of nitrate has been proposed as a source of NOx to the marine boundary layer (Ye et al., 2016; Romer et al., 2018), which might increase HNO3. We include NITs photolysis at a frequency of 50 times that of HNO3 (Kasibhatla et al., 2018). Figure 8 shows that this mechanism is consistent with observations of NO and ozone below 3 km and does not increase HNO3 but increases the free tropospheric NOy bias due to PAN formation and exacerbates the overestimate in upper tropospheric ozone during this season.
The difficulty in resolving the bias in wintertime may be due to an overestimate of the NOy lifetime as demonstrated by our sensitivities discussed above. Luo et al. (2019) proposed a new treatment of model wet scavenging using spatially and temporally varying cloud condensation water content and an empirical description of HNO3 wet removal. This scheme drastically reduced persistent model biases in nitric acid and nitrate at the surface in the United States (Zhang et al., 2012; Heald et al., 2012). As shown in Fig. 8, the revised wet scavenging scheme could fully resolve the remote bias in HNO3 throughout the troposphere. However, this parameterization has only received testing over the surface of the continental US, and more evaluation is needed before it can be adopted widely in models.
We find that scaling NOx, implementing chlorine chemistry, and revised wet scavenging (except in Northern Hemisphere winter) have negative impacts on the modeled OHcol along the flight tracks of −1 %, −7 %, and −4 %, respectively. The addition of NITs photolysis to the chlorine chemistry simulation increases OHcol by 11 % over the base model. In Northern Hemisphere winter only, revised wet scavenging increases OHcol by 16 %, possibly due to the effect of reduced heterogeneous chemistry. Overall, the annual mean impact of revised wet scavenging from our preliminary sensitivity tests is a −3 % reduction in global mean air-mass-weighted OH and a +2 % increase in the model methane lifetime. These preliminary sensitivities suggest that resolving the model wintertime NOy bias in the Northern Hemisphere could marginally reduce the overestimates of global mean OH on an annual basis if the photolysis frequency of NITs is smaller than 50 times the rate of HNO3 photolysis. Recent work from the NASA KORUS-AQ field campaign found that a rate of 1 to 30 might be more consistent with observational constraints (Romer et al., 2018).
Overall, the main drivers of remote tropospheric OH production in our base-case simulation are in good agreement with observations from the first two ATom deployments, with the exception of an NOy overestimate in the Northern Hemisphere wintertime. Acid displacement of Cl− by HNO3 on SSA (Kasibhatla et al., 2018; X. Wang et al., 2019) may somewhat improve remote HNO3 below 3 km, but if the resulting NITs undergoes photolysis at a rate of 50 times that of HNO3 (Kasibhatla et al., 2018), the impact on remote NOy may be lessened due to the formation of PAN. Both mechanisms require significant further study as tropospheric halogen sources and chemistry and the photolysis frequency of NITs are highly uncertain. A new parameterization of wet scavenging (Luo et al., 2019) would greatly improve modeled remote HNO3 and NOy but requires further testing and evaluation of its broader impacts on atmospheric chemistry.
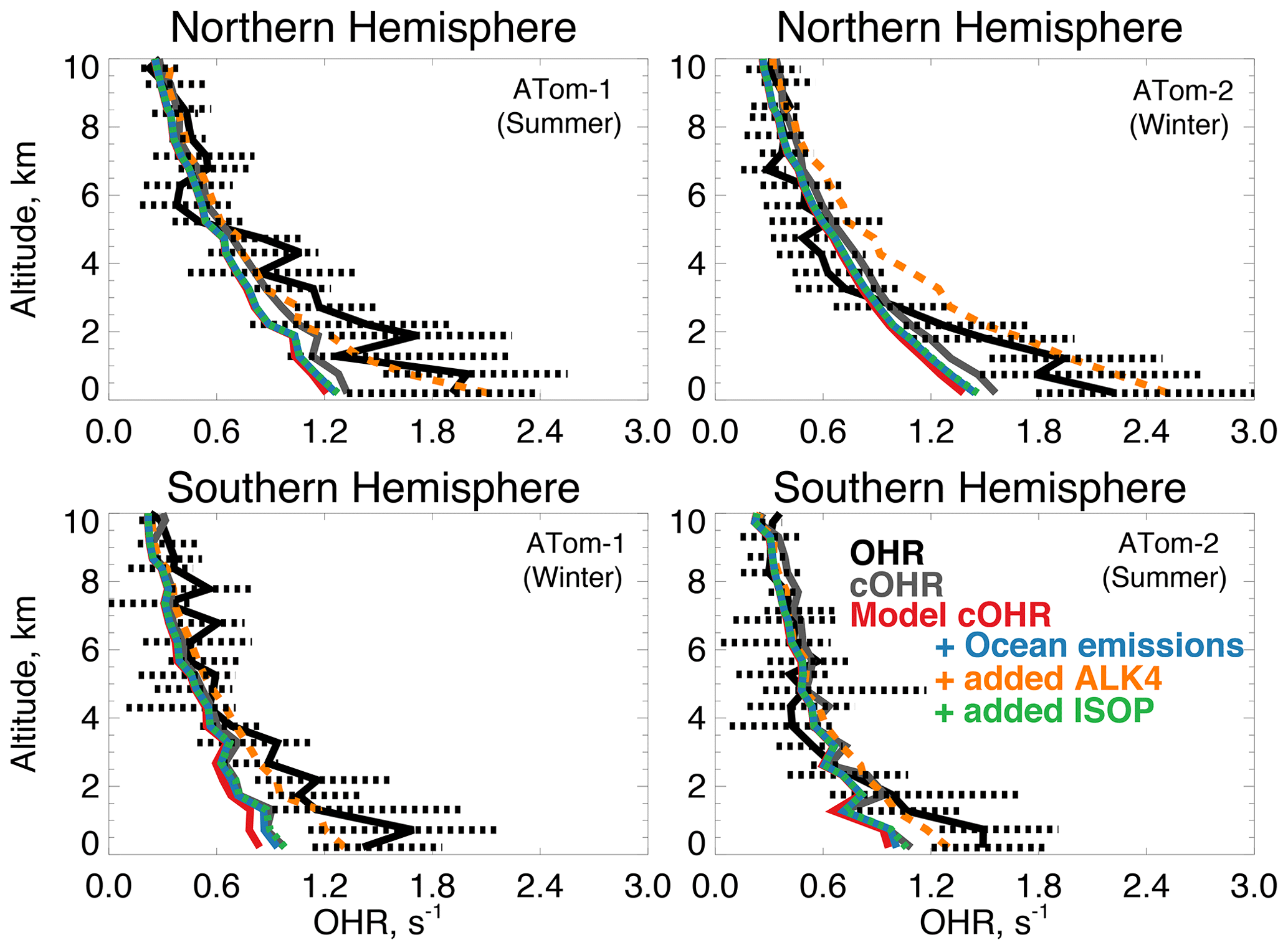
Figure 9The same as Fig. 3 for median OHR. OHR was measured by the ATHOS instrument as described in Table 2. The calculation of cOHR in the model and observations includes the species described in Table 2. In order to allow for a point-by-point comparison of cOHR in the model and observations, missing values are filled in the observational components of cOHR using linear interpolation. All calculated reactivity values are determined using the temperature and pressure of the ATHOS instrument inlet, which differ from ambient values. The sensitivity tests are described in Sect. 5.
The primary sinks of tropospheric OH are CO, methane, and VOCs; OHR measurements represent the sum effect of these species. Previous aircraft measurements of OHR provided evidence of missing reactivity in the remote atmosphere linked to unknown highly reactive VOCs (Mao et al., 2009). During ATom, Thames et al. (2020) measured OHR over the Atlantic and Pacific oceans and determined that missing OHR also correlated with oxygenated VOCs, suggesting the presence of unknown ocean emissions. We compare directly measured OHR during the ATom-1 and ATom-2 deployments to calculated OHR (cOHRobs) according to Eq. (1) from the full ATom measurement suite and from the model (cOHRmod) sampled along the flight path. Table 2 describes the observations used to calculate cOHR.
Figure 9 shows the comparison of OHR and cOHR from the model and observations. The observed cOHR is typically less than observed OHR. Along the flight tracks, cOHRobs and cOHRmod show good agreement and strong correlation (r2=0.95 for ATom-1 and ATom-2). The model underestimates cOHRobs by 10 % to 12 % in the lowest 3 km; we discuss this difference below. The measured relationship between OHR and cOHRobs is weaker (r2=0.53 for ATom-1, r2=0.56 for ATom-2) and cOHRobs is less than OHR below 3 km by 0.2 to 0.4 s−1. Thames et al. (2020) showed that median missing reactivity (between OHR and an observationally constrained box model) below 4 km during the ATom-1, ATom-2, and ATom-3 deployments was between 0.2 and 0.8 s−1. They provided statistical evidence that while near the level of the instrument accuracy, missing OHR in the marine boundary layer was statistically significant. We find that missing OHR is not associated with acetonitrile or CO (r2 < 0.06), indicating that biomass burning is not the cause. Acetaldehyde in Northern Hemisphere summer has the strongest relationship with missing OHR (r2=0.19, p-value ≪ 0.01, Fig. S3), which suggests a potential role for unmeasured reactive VOCs or their oxidation products from the ocean, as also suggested by Read et al. (2012) and Thames et al. (2020).
Table 3Biogenic ocean emissions of VOCs.
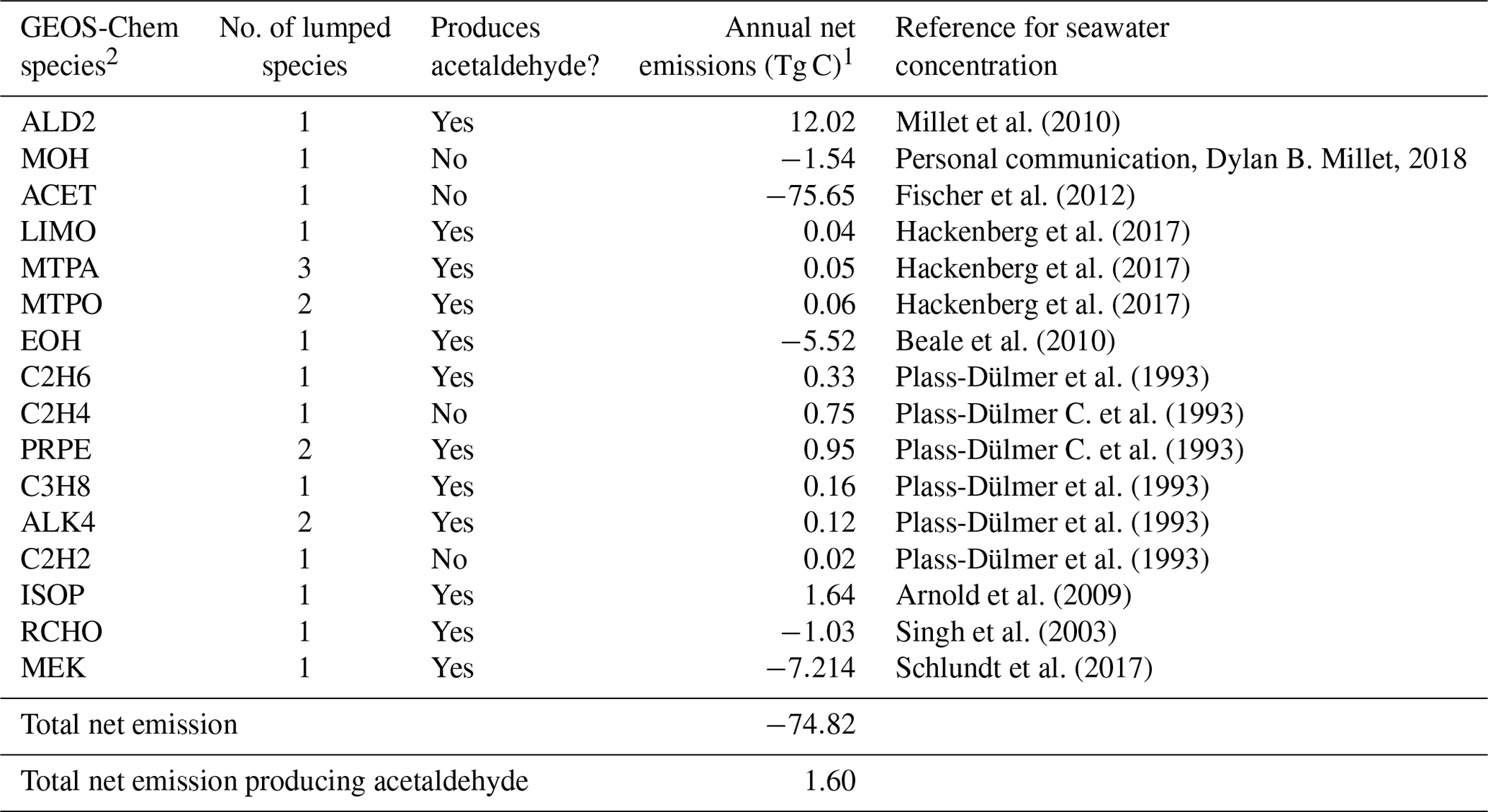
1 Net ocean emissions = upward flux out of the ocean–ocean uptake. 2 More information on the GEOS-Chem species definitions can be found here: http://wiki.seas.harvard.edu/geos-chem/index.php/Species_in_GEOS-Chem (last access: 21 May 2020).
Ocean emissions of VOCs have been suggested as a source of remote secondary organic aerosols (Gantt et al., 2010; Kim et al., 2017; Mungall et al., 2017), but their impact on remote reactivity has not been quantified. Our base simulation, described in Sect. 2.1, only includes air–sea exchange of acetone, acetaldehyde, methanol, and dimethyl sulfide. We determine whether additional compounds emitted from the ocean, but not generally included in models, could increase cOHRmod and reconcile the observed discrepancy described above. We follow the standard methodology for air–sea exchange described in Millet et al. (2008) to include emission of the species listed in Table 3 using available measured seawater concentrations, with the addition of isoprene implemented as a direct emission according to Arnold et al. (2009). As shown in Table 3, air–sea exchange represents a net sink of VOCs on an annual basis (−75 Tg C yr−1), but this is mainly due to ocean uptake of acetone, which is a negligible component of cOHR.
Interfacial photochemistry may provide an additional abiotic source of VOCs from the ocean. We model abiotic ocean emissions of VOCs according to Brüggemann et al. (2018) by applying species-specific emission factors to the monthly ocean photochemical potential derived in their study. We use the emission factor appropriate for the upper bound of this source according to Brüggemann et al. (2017) (Table S4). Table 4 provides a breakdown of these additional VOCs with a total annual emission of 28 Tg C yr−1.
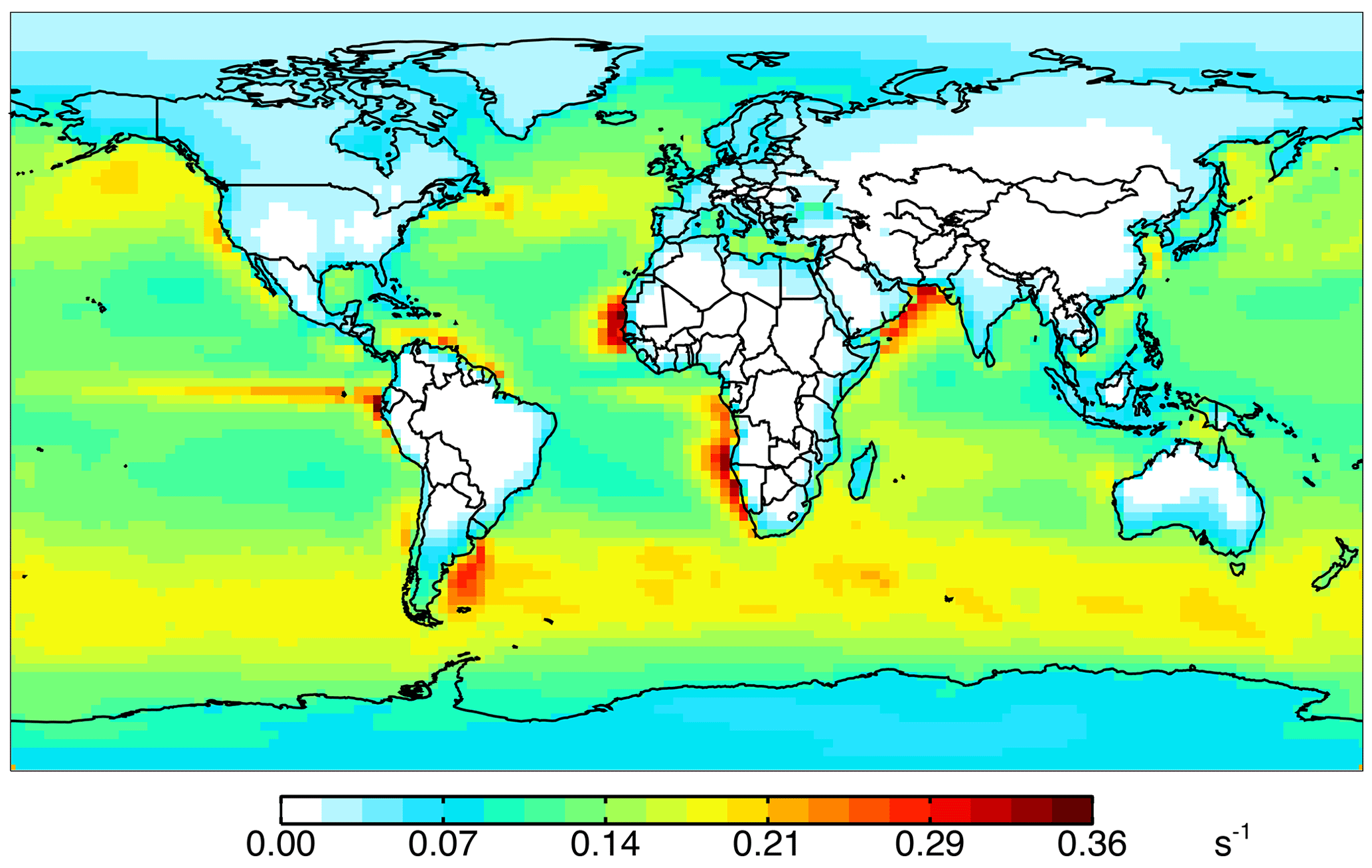
Figure 10Impact of all ocean emissions (Tables 3 and 4) on annual simulated 2016 surface cOHR as described in the text.
Figure 10 shows the annual mean impact of all ocean emissions described in Tables 3 and 4 (including an adjustment to the acetaldehyde seawater concentration described below in Sect. 5.1) on cOHRmod by turning off those ocean sources in a 1-year simulation. Average annual surface cOHRmod over the ocean increases by 6 % over the base simulation and 12 % over the simulation with no ocean emissions. The largest increases occur in regions of higher biogenic activity along coastlines and in the Southern Ocean due to the adjustment to acetaldehyde emissions discussed in Sect. 5.1. The incremental impact of the additional ocean emissions over the base simulation is shown in Fig. S4. Without any ocean emissions, global mean OH would be 2 % greater than in the case with comprehensive treatment of ocean VOCs. Figure 9 shows that along the flight tracks, cOHRmod increases below 3 km by 3 % to 9 %, which reduces the model bias against cOHRobs. However, the majority of the added species (Tables 3 and 4) were measured during ATom, would therefore contribute to cOHRobs, and cannot explain the gap in OHR.
Table 4Abiotic ocean emissions of VOCs according to Brüggemann et al. (2018)1.
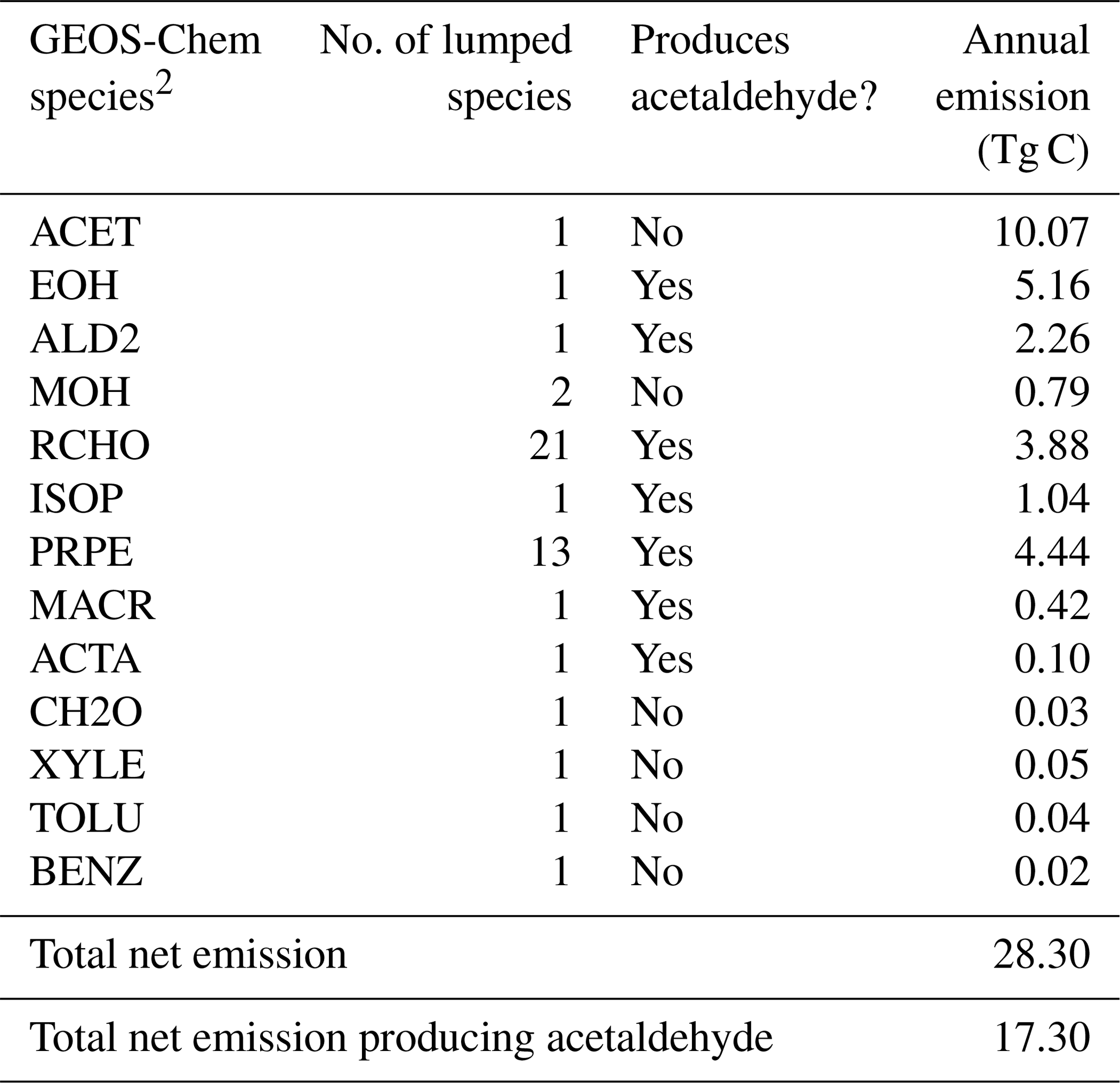
1 Table S2 shows the emission factor assumed for each species and the lumping methodology for Table 4. 2 More information on the GEOS-Chem species definitions can be found here: http://wiki.seas.harvard.edu/geos-chem/index.php/Species_in_GEOS-Chem (last access: 21 May 2020).
We evaluate the impact of further expanding the oceanic source of reactive VOCs to reconcile the discrepancy between cOHRobs and OHR in a similar manner to Mao et al. (2009). Here, we test a source of alkanes as previously suggested by Read et al. (2012), using the model species ALK4 (> C4 alkanes) that has a calculated lifetime of less than 2 d in the Northern Hemisphere summer ( cm3 molecules−1 s−1 at 298 K). Known alkanes have been measured in seawater (Plass-Dülmer et al., 1993), but the implied source is small. Consequently, we use ALK4 for testing only. Generating the missing OHR in this way requires an implausibly large oceanic ALK4 source of approximately 340 Tg C yr−1 compared against all other sources of VOCs in the model (Tables 3 and 4). A sensitivity test with this source, shown in Fig. 9, largely closes the gap between cOHRmod and OHR but would result in a 20 % to 50 % reduction in OH below 3 km, biasing the model OH simulation (Fig. 3) and degrading model NOy (Fig. 7) due to increased PAN formation.
Thames et al. (2020) found that a partial recycling of OH would be required to maintain consistency with observed OH and HO2 during ATom when adding an unknown source of reactivity. If the unknown VOC we suggest includes some OH recycling in its oxidation mechanism and does not produce PAN, the model bias in OH could be mitigated. We use isoprene as our test of a more reactive VOC that includes OH recycling by scaling the ALK4 emission source by the reaction rate of isoprene with OH to obtain a more reasonable emission source of approximately 9 Tg C yr−1. Figure 9 shows that this source actually has a minimal impact on cOHRmod of no more than 0.1 s−1. Only one-third in summer and two-thirds in winter of the additional cOHRmod from the ocean source of ALK4 are attributable to ALK4; the rest is due to CO, acetaldehyde, and other aldehydes from both increased chemical production and longer lifetimes from suppressed OH. Therefore, a larger source of even a reactive VOC like isoprene is required to close the gap in missing OHR. Reconciling cOHRmod and OHR is therefore difficult using the existing suite of ATom measurement constraints and possible known precursors; further investigation of the accuracy of the OHR measurements in challenging remote conditions may be needed.
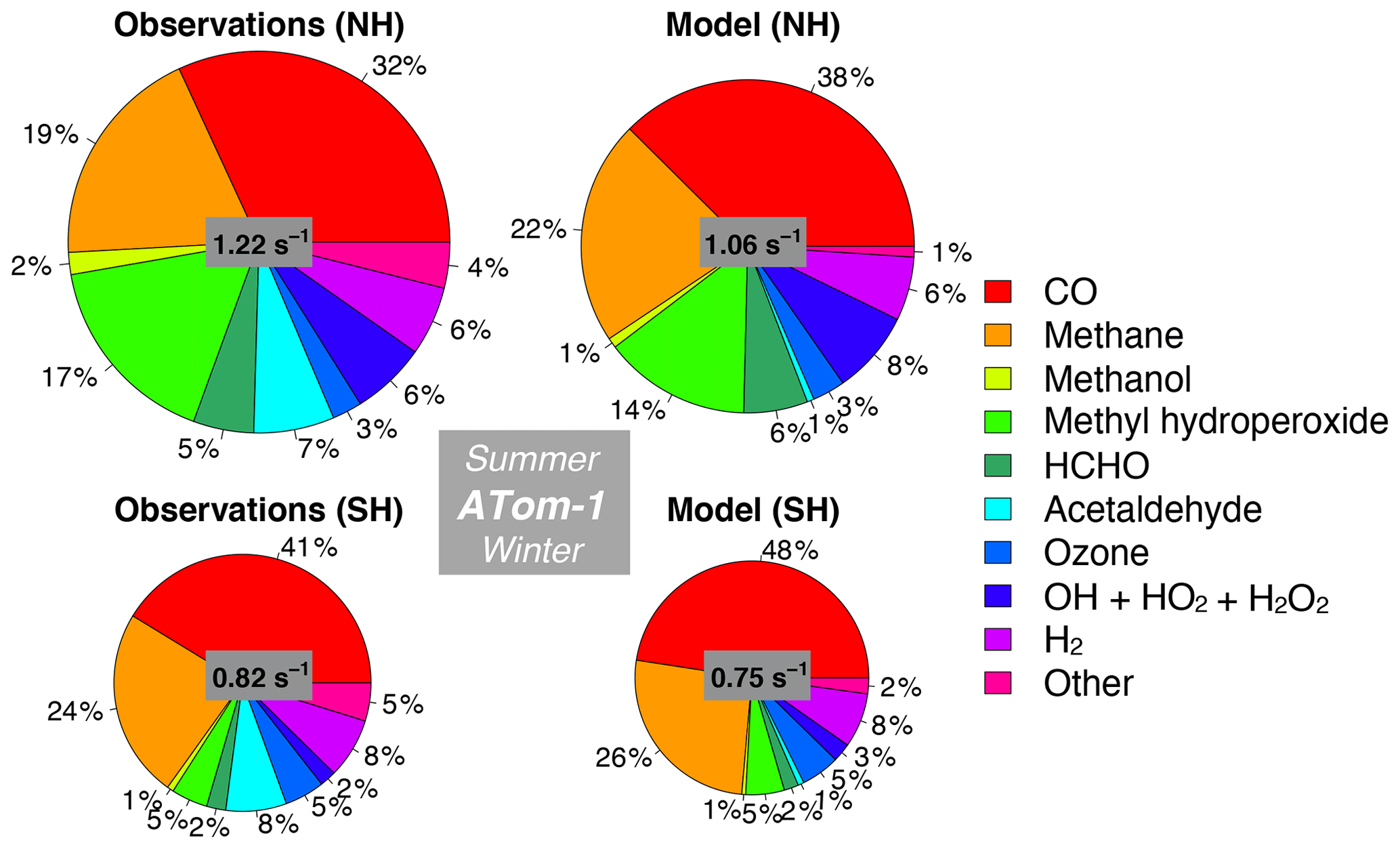
Figure 11Median observed and modeled OHR and cOHR (see text) below 3 km in the Northern Hemisphere (> 0∘ N) and Southern Hemisphere (< 0∘ S) during ATom-1. The “Other” category is the following species as described in Table 2: ethanol, propane, ethane, acetone, > C3 aldehydes, methyl ethyl ketone, methyl vinyl ketone, methacrolein, benzene, toluene, > C4 alkanes, peroxyacetic acid, peroxynitric acid, dimethyl sulfide, nitric acid, NO, and NO2. The diameter of each pie chart is scaled relative to the maximum cOHR for ATom-1.
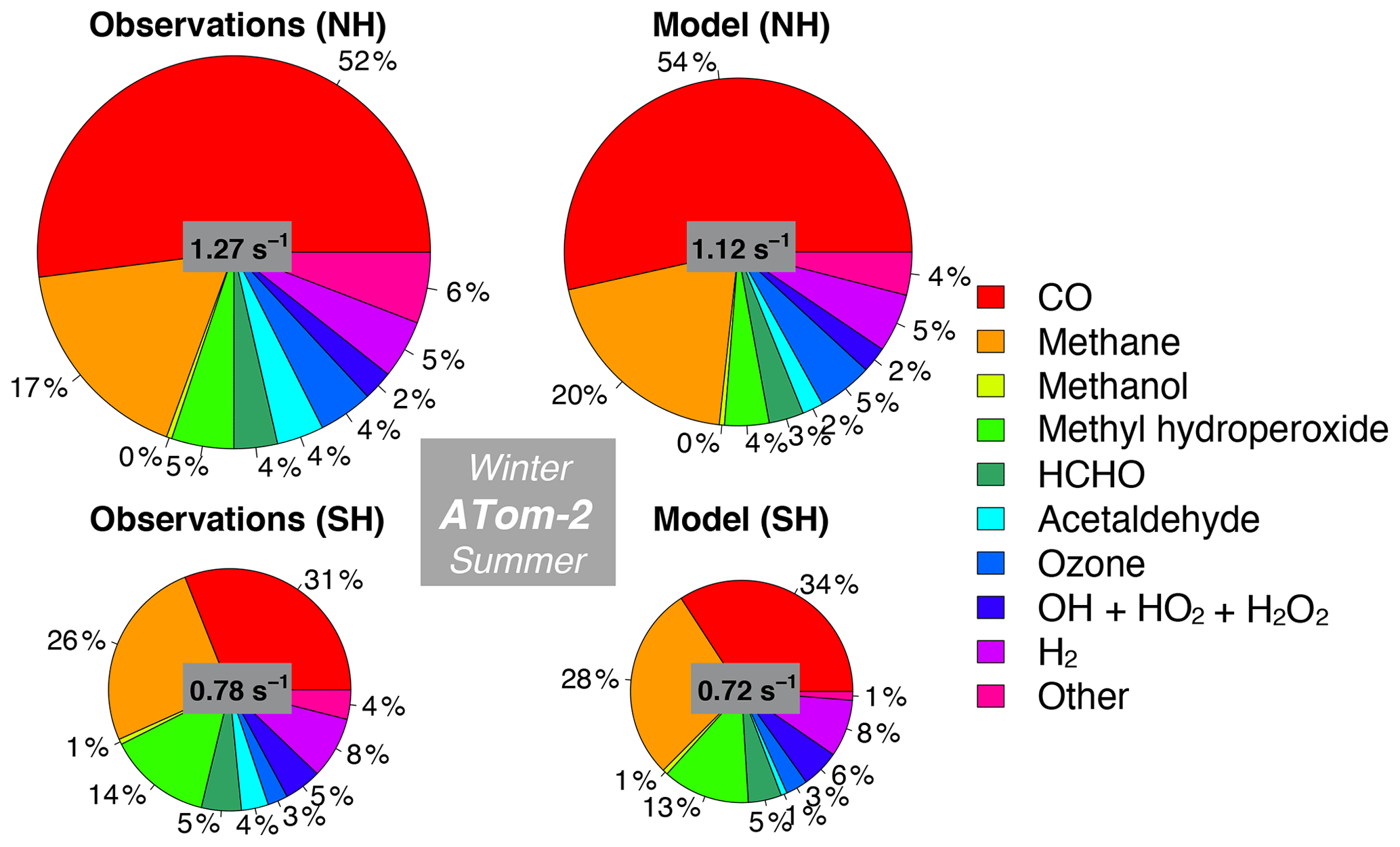
Figure 12Same as Fig. 10 but for ATom-2. The diameter of each pie chart is scaled relative to the maximum cOHR for ATom-2.
We also assess whether the model accurately represents the components of cOHRobs and explore potential additional sources of missing cOHRmod. Figures 11 and 12 show the components of median cOHR in the base simulation below 3 km for each deployment. The composition of cOHRmod is generally consistent with cOHRobs. CO and methane make up half or greater of both cOHRobs and cOHRmod. There is no systematic underestimate in CO reactivity as might be expected from the general model bias described by Shindell et al. (2006), with the exception of a 9 % underestimate during Northern Hemisphere winter when the lifetime of CO is longer and biases in continental sources could have a larger impact. During the ATom-1 deployment, cOHRobs is 50 % higher in the Northern Hemisphere (summer) than in the Southern Hemisphere (winter) primarily due to the increase in methyl hydroperoxide (MHP) concentrations. During the ATom-2 deployment, cOHRobs is 60 % higher in the Northern Hemisphere (winter) than in the Southern Hemisphere (summer) due to the large contribution of CO in Northern Hemisphere wintertime. The model successfully represents the observed seasonality during both deployments but underestimates cOHRobs by 12 % in the Northern Hemisphere and 9 % in the Southern Hemisphere.
The difference between measured and simulated cOHR is mainly due to differences between measured and simulated OVOCs. These compounds contribute on average 25 % to cOHRobs but 17 % to cOHRmod. The largest difference in reactivity is from acetaldehyde. Differences between simulated and measured MHP (Fig. S5) are also important and could reflect an error in the MHP lifetime (Müller et al., 2016). However, interferences in the MHP measurement in the boundary layer (Supplement, Sect. S6) have yet to be resolved, and therefore we do not further evaluate causes of underestimated MHP here. We do consider potential missing sources of model acetaldehyde constrained by the ATom measurements over the ocean and assess their impact on simulated OH and CO in Sect. 5.1.
Evaluation of the remote sources of acetaldehyde
Inability to reconcile remote acetaldehyde observations with models is a long-standing problem (Singh et al., 2001., 2003; Millet et al., 2010; Nicely et al., 2016). Singh et al. (2001) proposed that a large, diffuse, and as-yet unknown source of OVOCs such as acetaldehyde must exist in the troposphere to solve this discrepancy. Read et al. (2012) determined that missing cOHRmod from OVOCs (mainly acetaldehyde) in the marine tropical atmosphere, possibly from terrestrial or ocean sources of alkanes, could cause up to an 8 % underestimation of the methane lifetime. Nicely et al. (2016) showed that constraining a box model with observed acetaldehyde reduced tropospheric column OH by 9 % and that this acetaldehyde bias was present across eight different CTMs. Therefore, understanding the source of missing acetaldehyde may be part of the cause of the multi-model bias in the methane lifetime and global mean OH.
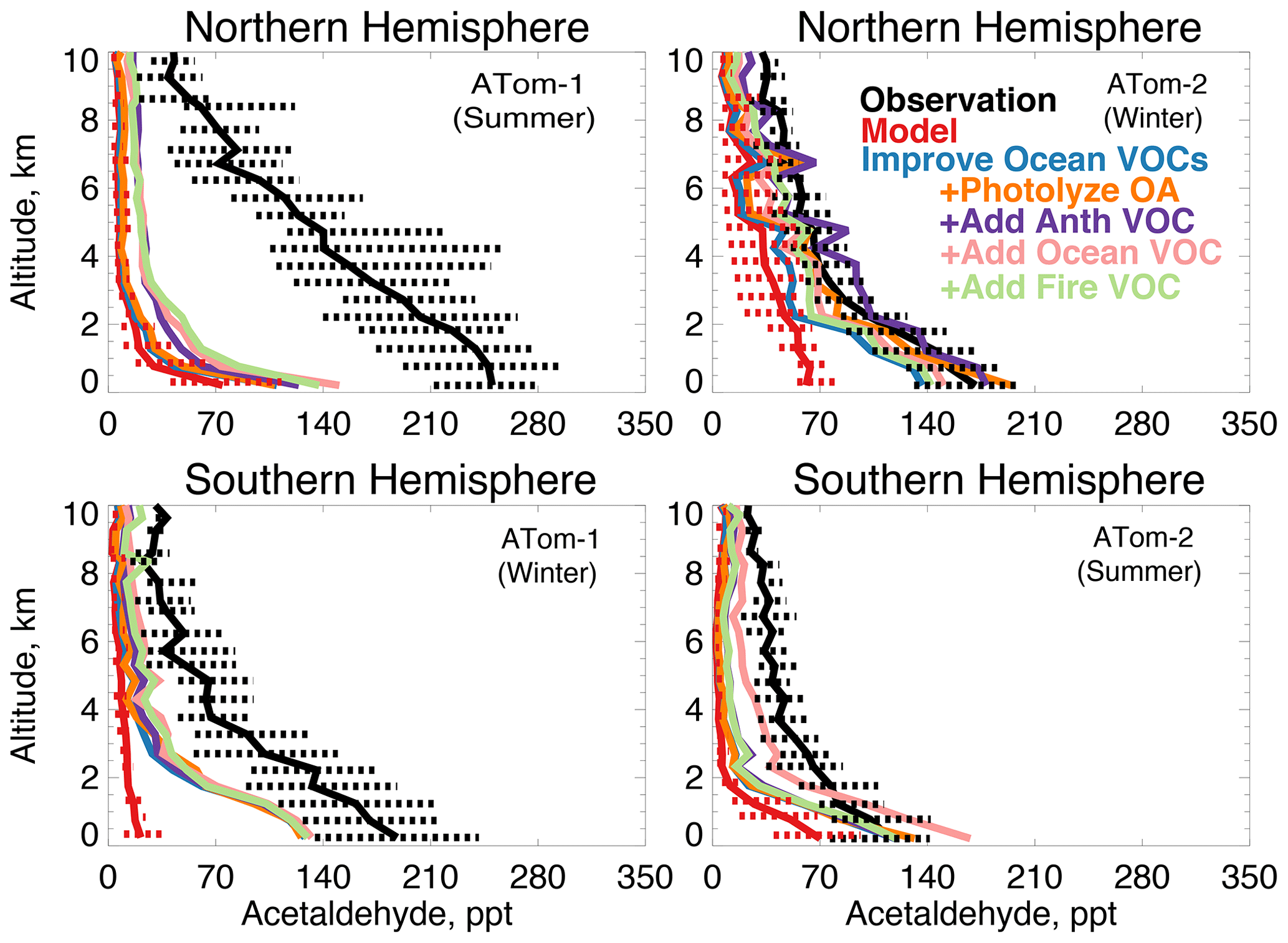
Figure 13The same as Fig. 3 for median acetaldehyde profiles. Acetaldehyde was measured by the TOGA instrument as described in Table 2. The sensitivity studies are described in Sect. 5.1.
Figure 13 compares the model simulation of acetaldehyde against observations. Average observed concentrations peak in the Northern Hemisphere during ATom-1 with an average mixing ratio of 230 ppt below 3 km and 100 ppt above 3 km despite a lifetime of only several hours in summer. The maximum model underestimate occurs during this period. Observed concentrations are at a minimum during the ATom-2 deployment, indicating a strong seasonality in the source. In each deployment, concentrations remain as high as 70 to 100 ppt as far south as 60∘ S (Fig. S6), which the model does not reproduce. There is no apparent difference in model bias between observations over the Atlantic or Pacific Ocean (Fig. S7). The model underestimates acetaldehyde on average by 60 to 90 % (50 to 200 ppt) below 3 km and does not capture the observed elevated levels throughout the troposphere.
In earlier studies, measurement uncertainties prevented interpretation of model–measurement disagreements in the remote atmosphere, including difficulties in background subtraction (Apel et al., 2008), with uncertainties as high as 70 ppt (Apel et al., 2003), which hindered analysis of clean conditions. The ATom measurement uncertainty is reduced to 10 ppt/20 % (Table 2) and does not have the biases present in previous campaigns (S. Wang et al., 2019). Studies have also disputed whether observed acetaldehyde was compatible with observed PAN due to the significant role of acetaldehyde as a PAN precursor through production of the peroxyacetyl (PA) radical (Singh et al., 2001, 2003; Millet et al., 2010). Global simulations estimate that acetaldehyde is responsible for approximately 40 % of PA radical production (Fischer et al., 2014), which would be even larger if acetaldehyde is severely underestimated by models. Reaction of the PA radical with HO2 is more prevalent in remote environments and produces peroxyacetic acid (PAA) preferentially over PAN, making PAA a more useful constraint for the conditions sampled by ATom. Figure 14 shows the average model underestimate of PAA below 3 km of 70 % to 90 % (60 to 250 ppt). The model biases for PAA and acetaldehyde both peak with similar magnitude during Northern Hemisphere summer. Figure 15 shows the model comparison with PAN, which is generally well simulated during this period.
S. Wang et al. (2019) used an observationally constrained box model to show that the levels of acetaldehyde observed during ATom are required to explain the observed PAA. The reaction rate of PAA + OH may be 3 times larger (Wu et al., 2017) than the maximum value used by S. Wang et al. (2019), which could result in even better agreement between PAA and acetaldehyde in the marine boundary layer. We evaluate the standard GEOS-Chem acetaldehyde budget, described in detail by Millet et al. (2010), against available ATom observations. The 2016 model budget for the base simulation is provided in Table 5. Acetaldehyde is produced from oxidation of VOCs (ethane, propane, ≥ C4 alkanes, ≥ C3 alkenes, isoprene, ethanol) and is directly emitted from the ocean, terrestrial plant growth, biomass burning, and anthropogenic activities. The model parameterization of acetaldehyde ocean emissions is dependent on satellite-based observations of colored dissolved organic matter (CDOM) (Millet et al., 2010).
Table 5Model sources of acetaldehyde in 2016.
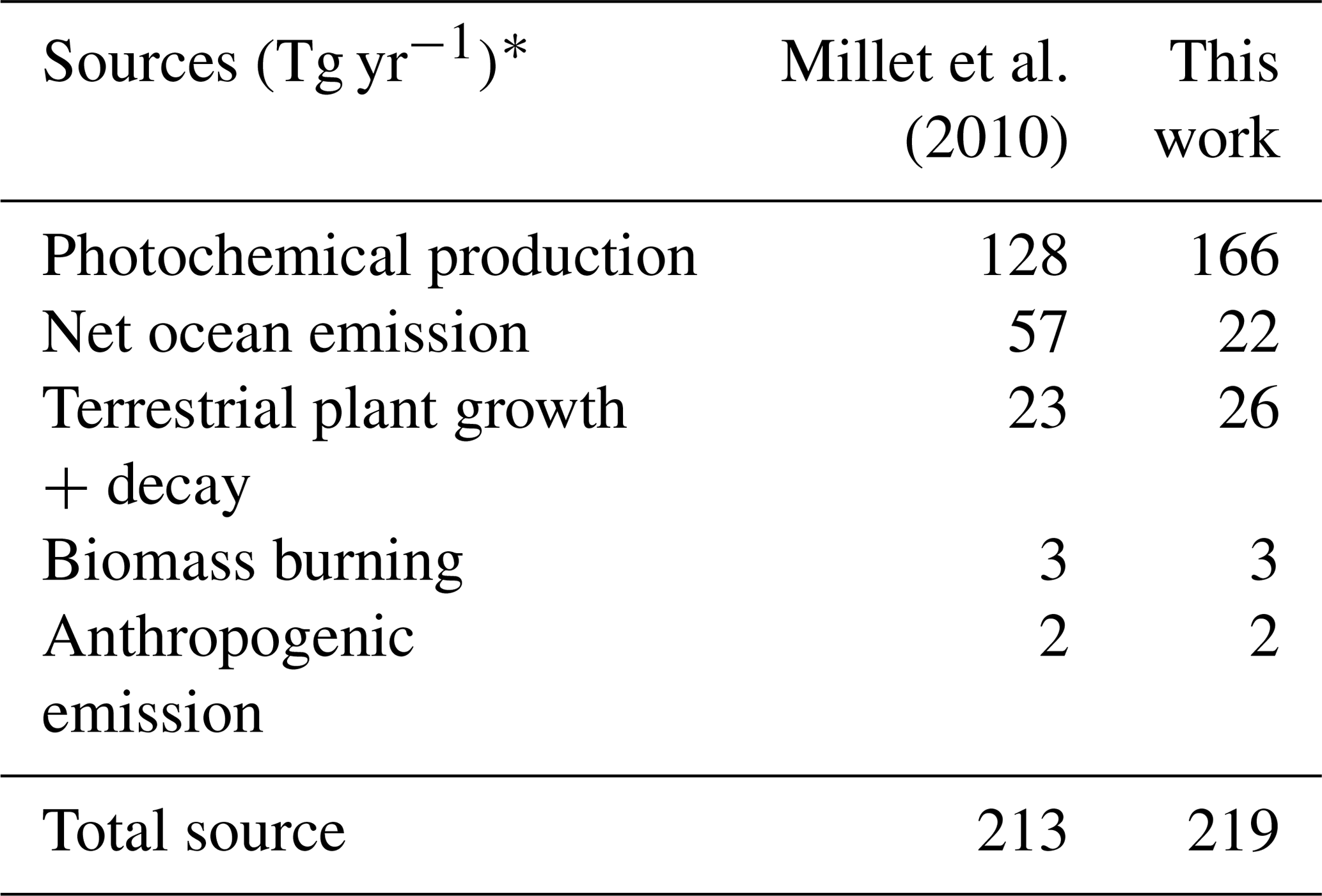
* Emissions are given in Tg of acetaldehyde per year for comparison to Millet et al. (2010). These totals are for the baseline model simulation described in Sect. 2.1.
The model free tropospheric bias suggests that long-lived oxidation of VOCs must be underestimated due to the short lifetime of acetaldehyde (< 1 d). The longest-lived precursor VOCs in the model are ethane (2 months) and propane (2 weeks). Ethane has the highest concentration of any measured non-methane VOC during ATom, with an average of 1.5 ppb below 3 km during the Northern Hemisphere winter. The model underestimates average ethane and propane below 10 km by approximately 25 % and 60 %, respectively (Figs. S8 and S9), which could be due to underestimated natural geologic and fossil fuel emissions (Dalsøren et al., 2018). However, the oxidation of these species is too slow to provide the missing model acetaldehyde and would only marginally increase remote background levels even if it was produced at higher yield at low NOx (model yields are ∼50 % for ethane and ∼20 % for propane, Millet et al., 2010). The chemical mechanism used for these species is provided in Table S5. One or more precursors able to resolve the model acetaldehyde bias must therefore be present at higher cumulative concentrations than ethane or propane. Modeled ALK4, parameterized as a butane–pentane mixture, maintains a high acetaldehyde yield at low NOx and has a shorter lifetime (∼5 d), contributing to a larger perturbation to atmospheric acetaldehyde levels than ethane or propane for a given concentration change. The sensitivity test adding substantial ALK4 emissions from the ocean described in Sect. 4 would not resolve the free tropospheric bias in the Northern Hemisphere but would result in a 40 % overestimate below 1 km. Furthermore, ALK4 is too short-lived to substantially perturb the remote atmosphere from a continental source; thus, the potential missing acetaldehyde precursors (from either a marine or terrestrial source) must have a longer lifetime.
As shown in Table 5, primary ocean emissions of acetaldehyde in the base simulation (22 Tg yr−1) are lower than previous work (57 Tg yr−1), likely due to updates to the model parameterization of the water transfer velocity (Johnson, 2010). Additional independent estimates of the ocean source are also much larger (34 to 42 Tg yr−1, Read et al., 2012; S. Wang et al., 2019). However, an increased primary ocean source would not address the bias in the free troposphere or in winter when biogenic activity from CDOM is zero in the model at high latitudes. Ship-borne measurements generally measure non-zero acetaldehyde seawater concentrations of approximately 5 nM (Read et al., 2012), and a recent trans-Atlantic campaign found that acetaldehyde concentrations from 47∘ S to 50∘ N did not always correlate with levels of CDOM (Yang et al., 2014). Therefore, we set a minimum seawater concentration of 5 nM in the model parameterization regardless of CDOM level. This change adds 2 Tg C yr−1 in emissions and increases concentrations over the remote ocean in winter by up to 50 ppt.
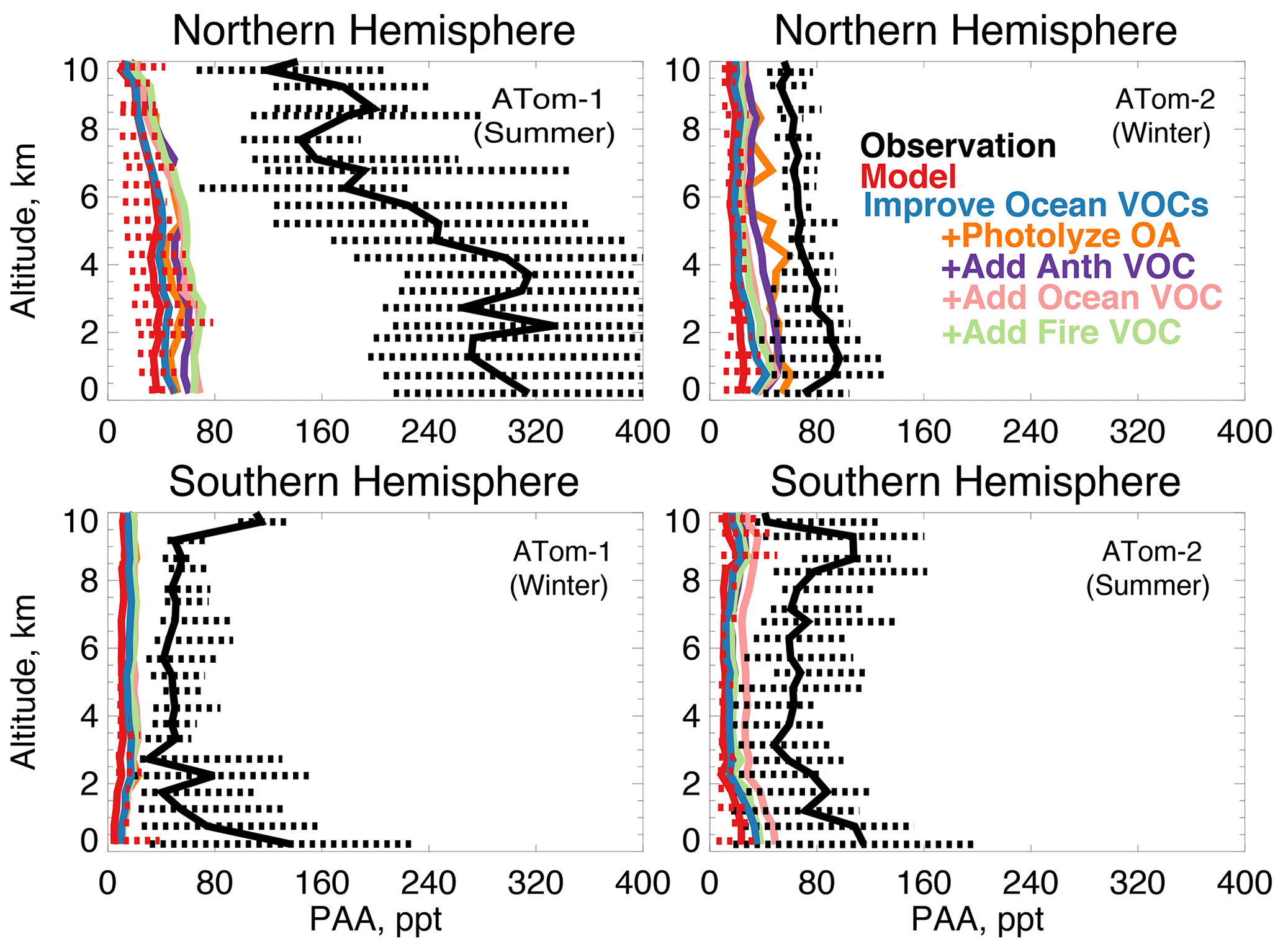
Figure 14The same as Fig. 3 for median peroxyacetic acid (PAA) profiles. PAA was measured by the Caltech CIMS instrument as described in Table 2. The sensitivity studies are described in Sect. 5.1.
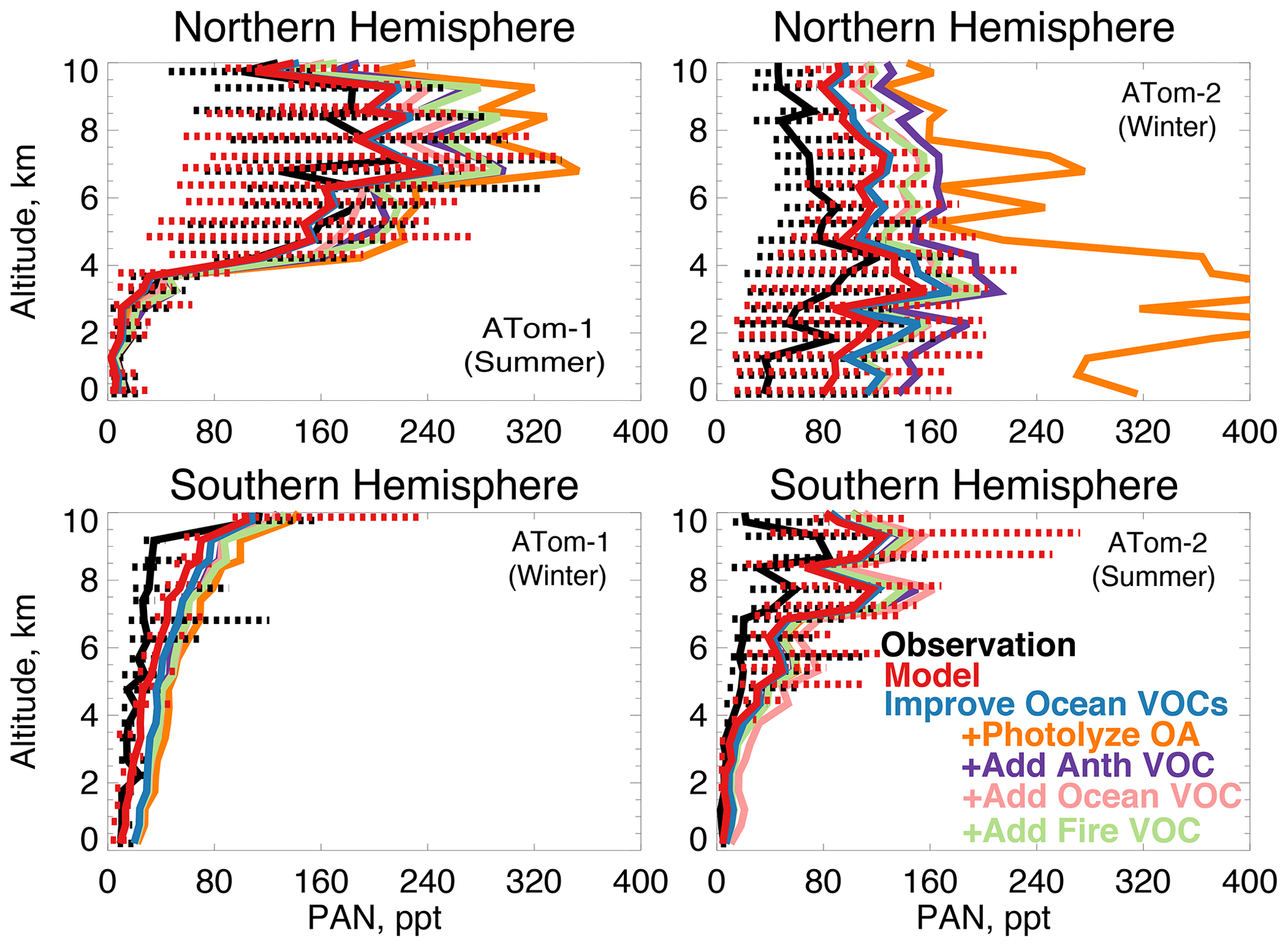
Figure 15The same as Fig. 3 for median peroxyacetyl nitrate (PAN) profiles. PAN was measured by the PANTHER instrument as described in Table 2. The sensitivity studies are described in Sect. 5.1.
Figure 13 shows the combined effect of adding new ocean VOCs in Sect. 5 and improving the seawater parameterization described above on modeled acetaldehyde (labeled “Improve Ocean VOCs”). Although the direct ocean source in this work is lower than previous estimates as described above, the secondary source from precursor VOCs is enhanced. Of the additional marine VOCs described in Sect. 5, 19 Tg C yr−1 produce acetaldehyde as an oxidation product (Tables 3 and 4). This is compared to 12 Tg C yr−1 of direct emissions in the base model. These sources substantially increase average modeled acetaldehyde below 3 km, with the largest improvement during winter (40 to 60 ppt) when atmospheric lifetimes are longer and the influence of the ocean can extend aloft. In summer, the average model increase below 3 km is only 10 to 20 ppt due to higher OH concentrations. Recent work over North America suggested that free tropospheric VOCs may be underestimated due to errors in model vertical mixing (Chen et al. 2019), but in Northern Hemisphere summer slower mixing would not be expected to compensate for the short lifetime of acetaldehyde in this region (∼5 h). Thus, the pervasive model bias in the free troposphere cannot be explained by an increase in known direct or indirect ocean sources.
Photodegradation of organic aerosols (OA) is another potential source of oxygenated VOCs such as acetaldehyde to the troposphere (Kwan et al., 2006; Epstein et al., 2014; Wong et al., 2015; S. Wang et al., 2019). The source of secondary organic aerosols (SOA) is uncertain and has been suggested to be up to 4 times larger than current estimates given an implied underestimate of the photochemical loss term (Hodzic et al., 2016). We test the potential impact of the maximum possible source of acetaldehyde from photochemical loss of OA by increasing the overall model production of SOA by a factor of 4 to maximize the impact of Reaction (R2) below. We apply a photolysis frequency for OA of (Hodzic et al., 2015) to Reactions (R1) and (R2) as an upper limit and describe the formulation of Reactions (R1) and (R2) below.
The model species OCPI and SOAS represent the majority of simulated OA in the remote atmosphere. OCPI is aged (hydrophilic) organic carbon (12 g C mol−1) and SOAS is SOA from all emissions categories (150 g mol−1). Both are assumed for the purposes of the sensitivity tests here to have an OA∕OC ratio of 2.1. In Reaction (R1), one molecule of carbon (0.5 ALD2) is produced per reaction. In Reaction (R2), one acetaldehyde molecule (ALD2) is produced per reaction. The resulting impact on acetaldehyde is only appreciable in the Northern Hemisphere winter (Fig. 13), when modeled aerosol amounts are highest and the lifetime of acetaldehyde is long. Given that this test represents an upper limit, we conclude that photolysis of organic aerosols cannot provide a sufficient source of acetaldehyde to reconcile the model with observations.
We consider whether an entirely unknown VOC with moderate lifetime and a high yield of acetaldehyde at low NOx could resolve the free tropospheric model bias. We emit such a species with a lifetime of approximately 1 month against oxidation by OH, emissions of 100 Tg yr−1 from either anthropogenic, biomass burning, or ocean sources, and a yield of one acetaldehyde molecule per reaction with OH. We do not test a terrestrial biogenic source here but expect the results would be similar to the biomass burning case. These simulations result in average tropospheric concentrations of 1 to 5 ppb. The effect of the unknown VOC is compatible with the model simulation of OH (unlike the addition of oceanic ALK4 needed to reconcile OHR observations as described in Sect. 5). The maximum cOHRmod of this species is small (< 0.03 s−1). The impact on modeled acetaldehyde (Fig. 13) is generally similar across all three source categories due to the long lifetime of this precursor. As shown in Figs. 13 and 14, the addition of this unknown VOC modestly improves the simulation of acetaldehyde and PAA everywhere, but a large residual underestimate in Northern Hemisphere summer remains. The impact on PAN is minor, with the exception of Northern Hemisphere winter (Fig. 15), but this is likely driven by the model overestimate in NOy (Fig. 7, Sect. 4.1).
Emission inventories of VOCs are known to be incomplete, for example neglecting emissions from volatile consumer products (McDonald et al., 2018) or failing to identify as much as half of emitted VOCs from biomass burning (Akagi et al. 2011), both of which peak in summer. The average emission factor for unidentified VOCs from biomass burning roughly corresponds to 75 Tg yr−1, similar to our sensitivity tests of 100 Tg yr−1 described above. However, recent attempts to quantify these unidentified VOCs (Stockwell et al., 2015; Koss et al., 2018) find that newly identified compounds tend to be too reactive to impact the remote atmosphere, as needed here; however, this work is ongoing and future efforts should investigate potential precursors of acetaldehyde that could be transported to the remote atmosphere. The missing source of precursor VOCs would need to have substantial additional summertime emissions above and beyond the sensitivity tests shown in Fig. 13 to address the Northern Hemisphere summertime bias. The required magnitude of this perturbation is difficult to reconcile within known measurement and emission uncertainty constraints.
The detailed set of chemical information available from the ATom field campaign provides the most comprehensive dataset ever collected to evaluate models in the remote atmosphere. The sampling strategy of collecting observations throughout the troposphere in multiple seasons is ideally suited for improving our understanding of tropospheric chemistry in a poorly observed region of the atmosphere. We use the first two deployments of the ATom field campaign during July–August 2016 and January–February 2017 to investigate sources of bias in model simulations of OH. Global models such as the GEOS-Chem CTM used here tend to overestimate the loss of methane by OH and underestimate CO, which provides the main tropospheric sink of OH. Comparisons of the model with observations from the first two ATom deployments do not show systematic bias in the simulation of OH or the drivers of remote OH production (water vapor, photolysis of ozone, ozone, and NOy), with the exception of wintertime NOy, which is overestimated by 70 %.
The model overestimate of wintertime NOy is largely attributable to nitric acid. This bias is not due to an anthropogenic inventory overestimate but may reflect insufficient wet scavenging as well as loss to sea-salt aerosols by nitric acid, although the former mechanism may be counteracted by photolysis of the resulting nitrate aerosols. The impact of resolving this wintertime NOy bias is uncertain but could marginally reduce the model overestimate of OH. Future work should improve constraints on these mechanisms, which have all received only preliminary validation, and carefully examine their impact in the context of broader atmospheric chemistry, particularly NOy partitioning throughout the troposphere.
We present the first comparison of measured OH reactivity (OHR) from aircraft with a global model to evaluate the tropospheric sink of OH. We calculate OH reactivity (cOHRobs) from relevant species observed during ATom and compare this to cOHR from the model (cOHRmod). Measured OHR is higher than cOHRobs by approximately 0.2 to 0.4 s−1 below 3 km. This missing OHR correlates with acetaldehyde during summer, indicating a potential source of missing reactive VOCs, similar to the findings of Mao et al. (2009) and S. Wang et al. (2020). The addition of a comprehensive set of ocean emissions of VOCs increases global mean cOHR by 6 % but cannot reproduce the observed OHR enhancement during ATom-1. Adding sufficient alkanes to the model to resolve this bias requires an improbably large ocean source (340 Tg C yr−1) and would degrade the model simulation of OH and NOy. Only one-third of the increase in cOHR in summer in this test is due to the alkanes; the rest is from oxidation products and changes in OH. Therefore, a more reactive VOC would still need to be emitted in large amounts.
The model successfully simulates the seasonality and hemispheric gradient in cOHR but has a persistent underestimate of up to 12 % in the lowest 3 km, primarily due to missing model acetaldehyde. The model does not underestimate CO, with the exception of Northern Hemisphere winter, which has been previously recognized by Kopacz et al. (2010) and attributed to underestimated fossil fuel emissions. The inability to reproduce observations of remote acetaldehyde was first observed during the PEM-Tropics campaign (Singh et al., 2001, 2003; Millet et al., 2010), but the measurement was uncertain. Improvements in measurement precision and the accompanying measurement of PAA during ATom (S. Wang et al., 2019) strengthen the conclusion that there is a large amount of acetaldehyde present in the atmosphere that cannot be explained by current models. We investigate possible underestimates in known sources of acetaldehyde, including emissions of VOCs from anthropogenic, biomass, or oceanic sources or production from the photolysis of organic aerosols. No known source can fully resolve the bias in acetaldehyde throughout the troposphere, and particularly in the Northern Hemisphere summer. We consider the possibility that there is a large, diffuse source of unknown VOCs by implementing 100 Tg yr−1 of such a compound from ocean, biomass burning, or anthropogenic sources. This hypothetical source modestly reduces the model acetaldehyde bias and is compatible with the simulation of OH and cOHR; however, an additional source is required to resolve the largest bias in the Northern Hemisphere summer. Errors or omissions in the oxidation mechanism of known VOCs could be another source of bias. For example, significant uncertainties exist in peroxy radical (RO2) chemistry for large RO2 molecules (Praske et al., 2017), although the flux of carbon through a minor pathway would have to be large, restricting the possible known sources. Further laboratory and field observations are needed to understand which precursors and sources could lead to the sustained production of acetaldehyde observed during ATom and prior campaigns.
This study demonstrates that long-standing model biases in global mean OH are unlikely to be due to errors in simulating tropospheric chemistry over the ocean. This implies that a large bias must be present in OH production or loss over land and future work should focus on evaluating continental OH sources and sinks. Errors in modeled OH were recently investigated by Strode et al. (2015), and when overestimates related to production terms were corrected, model OH remained too high in the Northern Hemisphere, suggesting that future studies should focus on errors in OH loss.
The ATom-1 and ATom-2 data (Wofsy et al., 2018) are available here: https://doi.org/10.3334/ORNLDAAC/1581.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-7753-2020-supplement.
CLH and KRT designed the study and wrote the paper with input from the co-authors. KRT modified the code, performed the simulations, and led the analysis. HMA, ECA, DRB, WHB, RC, JDC, BCD, GSD, JWE, SRH, EJH, SRH, MJK, KM, FLM, JP, TBR, ABT, KU, POW, and GMW provided ATom measurements used in the analysis. XW provided the model code for the sensitivity runs including acid displacement of chloride on coarse-mode sea-salt aerosols. TS, ME, and PSK provided the model code for the photolysis of particulate nitrate. GL and FY were responsible for the code for the revised treatment of wet scavenging in the model. DBM and XC provided the methanol seawater concentration and assisted in the ocean budget analysis. SRA provided the biogenic ocean isoprene emissions.
The authors declare that they have no conflict of interest.
We are grateful for helpful conversations and advice from Andrea Molod, Rachel Silvern, Eloïse Marais, Sarah Safieddine, Martin Brüggemann, Christian George, and James Crawford. We acknowledge Tom Hanisco and Jason St. Clair for the use of their formaldehyde observations from ATom and Barbara Barletta and Simone Meinardi for their contribution to the UCI WAS measurements.
This research has been supported by the National Science Foundation (grant no. AGS-1564495), the National Center for Atmospheric Research (grant no. 1852977), the National Oceanic and Atmospheric Administration (grant no. NA18OAR4310110), and the National Aeronautics and Space Administration (grant no. NNX14AP89G, grant no. IAT NNH15AB12I, grant no. NNX17AG35G, grant no. NNX15AG61A, grant no. NNX15AG71A).
This paper was edited by Yafang Cheng and reviewed by three anonymous referees.
Akagi, S. K., Yokelson, R. J., Wiedinmyer, C., Alvarado, M. J., Reid, J. S., Karl, T., Crounse, J. D., and Wennberg, P. O.: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 11, 4039–4072, https://doi.org/10.5194/acp-11-4039-2011, 2011.
Anderson, D. C., Loughner, C. P., Diskin, G., Weinheimer, A., Canty, T. P., Salawitch, R. J., Worden, H. M., Fried, A., Mikoviny, T., Wisthaler, A., and Dickerson, R. R.: Measured and modeled CO and NOy in DISCOVER-AQ: An evaluation of emissions and chemistry over the eastern US, Atmos. Environ., 96, 78–87, https://doi.org/10.1016/j.atmosenv.2014.07.004, 2014.
Apel, E. C., Hills, A. J., Lueb, R., Zindel, S., Eisele, S., and Riemer, D. D.: A fast-GC/MS system to measure C2 to C4 carbonyls and methanol aboard aircraft, J. Geophys. Res.-Atmos., 108, 8794, https://doi.org/10.1029/2002JD003199, 2003.
Apel, E. C., Brauers, T., Koppmann, R., Bandowe, B., Boßmeyer, J., Holzke, C., Tillmann, R., Wahner, A., Wegener, R., Brunner, A., Jocher, M., Ruuskanen, T., Spirig, C., Steigner, D., Steinbrecher, R., Gomez Alvarez, E., Müller, K., Burrows, J. P., Schade, G., Solomon, S. J., Ladstätter-Weißenmayer, A., Simmonds, P., Young, D., Hopkins, J. R., Lewis, A. C., Legreid, G., Reimann, S., Hansel, A., Wisthaler, A., Blake, R. S., Ellis, A. M., Monks, P. S., and Wyche, K. P.: Intercomparison of oxygenated volatile organic compound measurements at the SAPHIR atmosphere simulation chamber, J. Geophys. Res.-Atmos., 113, D20307, https://doi.org/10.1029/2008JD009865, 2008.
Apel, E. C., Hornbrook, R. S., Hills, A. J., Blake, N. J., Barth, M. C., Weinheimer, A., Cantrell, C., Rutledge, S. A., Basarab, B., Crawford, J., Diskin, G., Homeyer, C. R., Campos, T., Flocke, F., Fried, A., Blake, D. R., Brune, W., Pollack, I., Peischl, J., Ryerson, T., Wennberg, P. O., Crounse, J. D., Wisthaler, A., Mikoviny, T., Huey, G., Heikes, B., O’Sullivan, D., and Riemer, D. D.: Upper tropospheric ozone production from lightning NOx-impacted convection: Smoke ingestion case study from the DC3 campaign, J. Geophys. Res.-Atmos., 120, 2505–2523, https://doi.org/10.1002/2014JD022121, 2015.
Arnold, S. R., Spracklen, D. V., Williams, J., Yassaa, N., Sciare, J., Bonsang, B., Gros, V., Peeken, I., Lewis, A. C., Alvain, S., and Moulin, C.: Evaluation of the global oceanic isoprene source and its impacts on marine organic carbon aerosol, Atmos. Chem. Phys., 9, 1253–1262, https://doi.org/10.5194/acp-9-1253-2009, 2009.
Beale R., Liss P. S., and Nightingale P. D.: First oceanic measurements of ethanol and propanol, Geophys. Res. Lett., 37, L24607, https://doi.org/10.1029/2010GL045534, 2010.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res., 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001.
Breider, T. J., Mickley, L. J., Jacob, D. J., Ge, C., Wang, J., Sulprizio, M. P., Croft, B., Ridley, D. A., McConnell, J. R., Sharma, S., Husain, L., Dutkiewicz, V. A., Eleftheriadis, K., Skov, H., and Hopke, P. K.: Multi-decadal trends in aerosol radiative forcing over the Arctic: contribution of changes in anthropogenic aerosol to Arctic warming since 1980, J. Geophys. Res., 122, 3573–3594, https://doi.org/10.1002/2016JD025321, 2017.
Brüggemann, M., Hayeck, N., Bonnineau, C., Pesce, S., A. Alpert, P., Perrier, S., Zuth, C., Hoffmann, T., Chen, J., and George, C.: Interfacial photochemistry of biogenic surfactants: a major source of abiotic volatile organic compounds, Faraday Discuss., 200, 59–74, https://doi.org/10.1039/C7FD00022G, 2017.
Brüggemann, M., Hayeck, N., and George, C.: Interfacial photochemistry at the ocean surface is a global source of organic vapors and aerosols, Nat. Commun., 9, 2101, https://doi.org/10.1038/s41467-018-04528-7, 2018.
Brune, W. H., Miller, D. O., Thames, A. B., Allen, H. M., Apel, E. C., Blake, D. R., Bui, T. P., Commane, R., Crounse, J. D., Daube, B. C., Diskin, G. S., DiGangi, J. P., Elkins, J. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hintsa, E. J., Hornbrook, R. S., Kim, M. J., McKain, K., Moore, F. L., Neuman, J. A., Nicely, J. M., Peischl, J., Ryerson, T. B., St. Clair, J. M., Sweeney, C., Teng, A. P., Thompson, C., Ullmann, K., Veres, P. R., Wennberg, P. O., and Wolfe, G. M.: Exploring Oxidation in the Remote Free Troposphere: Insights From Atmospheric Tomography (ATom), J. Geophys. Res. Atmos., 125, e2019JD031685, https://doi.org/10.1029/2019JD031685, 2020.
Brunner, D., Staehelin, J., Rogers, H. L., Köhler, M. O., Pyle, J. A., Hauglustaine, D., Jourdain, L., Berntsen, T. K., Gauss, M., Isaksen, I. S. A., Meijer, E., van Velthoven, P., Pitari, G., Mancini, E., Grewe, G., and Sausen, R.: An evaluation of the performance of chemistry transport models by comparison with research aircraft observations. Part 1: Concepts and overall model performance, Atmos. Chem. Phys., 3, 1609–1631, https://doi.org/10.5194/acp-3-1609-2003, 2003.
Brunner, D., Staehelin, J., Rogers, H. L., Köhler, M. O., Pyle, J. A., Hauglustaine, D. A., Jourdain, L., Berntsen, T. K., Gauss, M., Isaksen, I. S. A., Meijer, E., van Velthoven, P., Pitari, G., Mancini, E., Grewe, V., and Sausen, R.: An evaluation of the performance of chemistry transport models - Part 2: Detailed comparison with two selected campaigns, Atmos. Chem. Phys., 5, 107–129, https://doi.org/10.5194/acp-5-107-2005, 2005.
Cazorla, M., Wolfe, G. M., Bailey, S. A., Swanson, A. K., Arkinson, H. L., and Hanisco, T. F.: A new airborne laser-induced fluorescence instrument for in situ detection of formaldehyde throughout the troposphere and lower stratosphere, Atmos. Meas. Tech., 8, 541–552, https://doi.org/10.5194/amt-8-541-2015, 2015.
Chan Miller, C., Jacob, D. J., Marais, E. A., Yu, K., Travis, K. R., Kim, P. S., Fisher, J. A., Zhu, L., Wolfe, G. M., Hanisco, T. F., Keutsch, F. N., Kaiser, J., Min, K.-E., Brown, S. S., Washenfelder, R. A., González Abad, G., and Chance, K.: Glyoxal yield from isoprene oxidation and relation to formaldehyde: chemical mechanism, constraints from SENEX aircraft observations, and interpretation of OMI satellite data, Atmos. Chem. Phys., 17, 8725–8738, https://doi.org/10.5194/acp-17-8725-2017, 2017.
Chen, X., Millet, D. B., Singh, H. B., Wisthaler, A., Apel, E. C., Atlas, E. L., Blake, D. R., Bourgeois, I., Brown, S. S., Crounse, J. D., de Gouw, J. A., Flocke, F. M., Fried, A., Heikes, B. G., Hornbrook, R. S., Mikoviny, T., Min, K.-E., Müller, M., Neuman, J. A., O'Sullivan, D. W., Peischl, J., Pfister, G. G., Richter, D., Roberts, J. M., Ryerson, T. B., Shertz, S. R., Thompson, C. R., Treadaway, V., Veres, P. R., Walega, J., Warneke, C., Washenfelder, R. A., Weibring, P., and Yuan, B.: On the sources and sinks of atmospheric VOCs: an integrated analysis of recent aircraft campaigns over North America, Atmos. Chem. Phys., 19, 9097–9123, https://doi.org/10.5194/acp-19-9097-2019, 2019.
Christian, K. E., Brune, W. H., Mao, J., and Ren, X.: Global sensitivity analysis of GEOS-Chem modeled ozone and hydrogen oxides during the INTEX campaigns, Atmos. Chem. Phys., 18, 2443–2460, https://doi.org/10.5194/acp-18-2443-2018, 2018.
Colman, J., Swanson, A., Meinardi, S., Sive B., Blake, D. R., and Rowland, F. S.: Description of the analysis of a wide range of volatile organic compounds in whole air samples collected during PEM-Tropics A and B, Anal. Chem., 73, 3723–3731, https://doi.org/10.1021/ac010027g, 2001.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 78, 6726–6732, 2006.
Dalsøren, S. B., Myhre, G., Hodnebrog, Ø., Myhre, C. L., Stohl, A., Pisso, I., Schwietzke, S., Höglund-Isaksson, L., Helmig, D., Reimann, S., Sauvage, S., Schmidbauer, N., Read, K. A., Carpenter, L. J., Lewis, A. C., Punjabi, S., and Wallasch, M.: Discrepancy between simulated and observed ethane and propane levels explained by underestimated fossil emissions, Nat. Geosci., 11, 178–184, https://doi.org/10.1038/s41561-018-0073-0, 2018.
Dentener, F., Drevet, J., Lamarque, J. F., Bey, I., Eickhout, B., Fiore, A. M., Hauglustaine, D., Horowitz, L. W., Krol, M., Kulshrestha, U. C., Lawrence, M., Galy-Lacaux, C., Rast, S., Shindell, D., Stevenson, D., Noije, T. V., Atherton, C., Bell, N., Bergman, D., Butler, T., Cofala, J., Collins, B., Doherty, R., Ellingsen, K., Galloway, J., Gauss, M., Montanaro, V., Müller, J. F., Pitari, G., Rodriguez, J., Sanderson, M., Solmon, F., Strahan, S., Schultz, M., Sudo, K., Szopa, S., and Wild, O.: Nitrogen and sulfur deposition on regional and global scales: A multimodel evaluation, Global Biogeochem. Cy., 20, GB4003, https://doi.org/10.1029/2005GB002672, 2006.
Di Carlo, P.: Missing OH Reactivity in a Forest: Evidence for Unknown Reactive Biogenic VOCs, Science, 304, 722–725, https://doi.org/10.1126/science.1094392, 2004.
DiGangi, J. P., Boyle, E. S., Karl, T., Harley, P., Turnipseed, A., Kim, S., Cantrell, C., Maudlin III, R. L., Zheng, W., Flocke, F., Hall, S. R., Ullmann, K., Nakashima, Y., Paul, J. B., Wolfe, G. M., Desai, A. R., Kajii, Y., Guenther, A., and Keutsch, F. N.: First direct measurements of formaldehyde flux via eddy covariance: implications for missing in-canopy formaldehyde sources, Atmos. Chem. Phys., 11, 10565–10578, https://doi.org/10.5194/acp-11-10565-2011, 2011.
Diskin, G. S., Podolske, J. R., Sachse, G. W., and Slate, T. A.: Open-Path Airborne Tunable Diode Laser Hygrometer, in: Diode Lasers and Applications in Atmospheric Sensing, SPIE Proceedings, 4817, edited by: Fried, A., 196–204, 2002.
Dolgorouky, C., Gros, V., Sarda-Esteve, R., Sinha, V., Williams, J., Marchand, N., Sauvage, S., Poulain, L., Sciare, J., and Bonsang, B.: Total OH reactivity measurements in Paris during the 2010 MEGAPOLI winter campaign, Atmos. Chem. Phys., 12, 9593–9612, https://doi.org/10.5194/acp-12-9593-2012, 2012.
Duncan, B. N., Logan, J. A., Bey, I., Megretskaia, I. A., Yantosca, R. M., Novelli, P. C., Jones, N. B., and Rinsland, C. P.: Global budget of CO, 1988–1997: Source estimates and validation with a global model, J. Geophys. Res., 112, D22301, https://doi.org/10.1029/2007JD008459, 2007.
Edwards, P. M., Evans, M. J., Furneaux, K. L., Hopkins, J., Ingham, T., Jones, C., Lee, J. D., Lewis, A. C., Moller, S. J., Stone, D., Whalley, L. K., and Heard, D. E.: OH reactivity in a South East Asian tropical rainforest during the Oxidant and Particle Photochemical Processes (OP3) project, Atmos. Chem. Phys., 13, 9497–9514, https://doi.org/10.5194/acp-13-9497-2013, 2013.
Elkins, J. W., Moorem, F., and Kline, E. S.: Next Generation Airborne Gas chromatograph for NASA Airborne Platforms, Earth Science Technology Conference 2001, 28–30 August 2001, College Park, MD, 2001.
Emmerson, K. M. and Evans, M. J.: Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 9, 1831–1845, https://doi.org/10.5194/acp-9-1831-2009, 2009.
Epstein, S. A., Blair, S. L., and Nizkorodov, S. A.: Direct Photolysis of α-Pinene Ozonolysis Secondary Organic Aerosol: Effect on Particle Mass and Peroxide Content, Environ. Sci. Technol., 48, 11251–11258, https://doi.org/10.1021/es502350u, 2014.
Faloona, I. C., Tan, D., Lesher, R. L., Hazen, N. L., Frame, C. L., Simpas, J. B., Harder, H., Martinez, M., Di Carlo, P., Ren, X., and Brune, W. H.: A Laser-induced Fluorescence Instrument for Detecting Tropospheric OH and HO 2 : Characteristics and Calibration, J. Atmos. Chem., 47, 139–167, https://doi.org/10.1023/B:JOCH.0000021036.53185.0e, 2004.
Ferracci, V., Heimann, I., Abraham, N. L., Pyle, J. A., and Archibald, A. T.: Global modelling of the total OH reactivity: investigations on the “missing” OH sink and its atmospheric implications, Atmos. Chem. Phys., 18, 7109–7129, https://doi.org/10.5194/acp-18-7109-2018, 2018.
Fiore, A. M., Dentener, F. J., Wild, O., Cuvelier, C., Schultz, M. G., Hess, P., Textor, C., Schulz, M., Doherty, R. M., Horowitz, L. W., MacKenzie, I. A., Sanderson, M. G., Shindell, D. T., Stevenson, D. S., Szopa, S., Van Dingenen, R., Zeng, G., Atherton, C., Bergmann, D., Bey, I., Carmichael, G., Collins, W. J., Duncan, B. N., Faluvegi, G., Folberth, G., Gauss, M., Gong, S., Hauglustaine, D., Holloway, T., Isaksen, I. S. A., Jacob, D. J., Jonson, J. E., Kaminski, J. W., Keating, T. J., Lupu, A., Marmer, E., Montanaro, V., Park, R. J., Pitari, G., Pringle, K. J., Pyle, J. A., Schroeder, S., Vivanco, M. G., Wind, P., Wojcik, G., Wu, S., and Zuber, A.: Multimodel estimates of intercontinental source-receptor relationships for ozone pollution, J. Geophys. Res., 114, D04301, https://doi.org/10.1029/2008JD010816, 2009.
Fischer, E. V., Jacob, D. J., Millet, D. B., Yantosca, R. M., and Mao, J.: The role of the ocean in the global atmospheric budget of acetone, Geophys. Res. Lett., 39, L01807, https://doi.org/10.1029/2011GL050086, 2012.
Fischer, E. V., Jacob, D. J., Yantosca, R. M., Sulprizio, M. P., Millet, D. B., Mao, J., Paulot, F., Singh, H. B., Roiger, A., Ries, L., Talbot, R. W., Dzepina, K., and Pandey Deolal, S.: Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 14, 2679–2698, https://doi.org/10.5194/acp-14-2679-2014, 2014.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fisher, J. A., Atlas, E. L., Barletta, B., Meinardi, S., Blake, D. R., Thompson, C. R., Ryerson, T. B., Peischl, J., Tzompa-Sosa, Z. A., and Murray, L. T.: Methyl, ethyl, and propyl nitrates: global distribution and impacts on reactive nitrogen in remote marine environments, J. Geophys. Res., 123, 12429–12451, https://doi.org/10.1029/2018JD029046, 2018.
Gantt, B., Meskhidze, N., Zhang, Y., and Xu, J.: The effect of marine isoprene emissions on secondary organic aerosol and ozone formation in the coastal United States, Atmos. Environ., 44, 115–121, https://doi.org/10.1016/j.atmosenv.2009.08.027, 2010.
Gelaro, R., McCarty, W., Suárez, M. J., Todling, R., Molod, A., Takacs, L., Randles, C. A., Darmenov, A., Bosilovich, M. G., Reichle, R., Wargan, K., Coy, L., Cullather, R., Draper, C., Akella, S., Buchard, V., Conaty, A., da Silva, A. M., Gu, W., Kim, G.-K., Koster, R., Lucchesi, R., Merkova, D., Nielsen, J. E., Partyka, G., Pawson, S., Putman, W., Rienecker, M., Schubert, S. D., Sienkiewicz, M., and Zhao, B.: The Modern-Era Retrospective Analysis for Research and Applications, Version 2 (MERRA-2), J. Climate, 30, 5419–5454, https://doi.org/10.1175/JCLI-D-16-0758.1, 2017.
Giglio, L., Randerson, J. T., and van der Werf, G. R.: Analysis of daily, monthly, and annual burned area using the fourth- generation global fire emissions database (GFED4): ANALYSIS OF BURNED AREA, J. Geophys. Res.-Biogeo., 118, 317–328, https://doi.org/10.1002/jgrg.20042, 2013.
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Granier, C., Lamarque, J. F., Mieville, A., Muller, J. F., Olivier, J., Orlando, J., Peters, J., Petron, G., Tyndall, G., and Wallens, S.: POET, a database of surface emissions of ozone precursors, available at: http://www.aero.jussieu.fr/projet/ACCENT/POET.php (last access: 2 July 2020), 2005.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Mod. Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hackenberg, S. C., Andrews, S. J., Airs, R. L., Arnold, S. R., Bouman, H. A., Cummings, D., Lewis, A. C., Minaeian, J. K., Reifel, K. M., Small, A., Tarran, G. A., Tilstone, G. H., and Carpenter, L. J.: Basin-Scale Observations of Monoterpenes in the Arctic and Atlantic Oceans, Environ. Sci. Technol., 51, 10449–10458, https://doi.org/10.1021/acs.est.7b02240, 2017.
Hall, S. R., Ullmann, K., Prather, M. J., Flynn, C. M., Murray, L. T., Fiore, A. M., Correa, G., Strode, S. A., Steenrod, S. D., Lamarque, J.-F., Guth, J., Josse, B., Flemming, J., Huijnen, V., Abraham, N. L., and Archibald, A. T.: Cloud impacts on photochemistry: building a climatology of photolysis rates from the Atmospheric Tomography mission, Atmos. Chem. Phys., 18, 16809–16828, https://doi.org/10.5194/acp-18-16809-2018, 2018.
Hansen, R. F., Griffith, S. M., Dusanter, S., Rickly, P. S., Stevens, P. S., Bertman, S. B., Carroll, M. A., Erickson, M. H., Flynn, J. H., Grossberg, N., Jobson, B. T., Lefer, B. L., and Wallace, H. W.: Measurements of total hydroxyl radical reactivity during CABINEX 2009 – Part 1: field measurements, Atmos. Chem. Phys., 14, 2923–2937, https://doi.org/10.5194/acp-14-2923-2014, 2014.
Heald, C. L., Collett Jr., J. L., Lee, T., Benedict, K. B., Schwandner, F. M., Li, Y., Clarisse, L., Hurtmans, D. R., Van Damme, M., Clerbaux, C., Coheur, P.-F., Philip, S., Martin, R. V., and Pye, H. O. T.: Atmospheric ammonia and particulate inorganic nitrogen over the United States, Atmos. Chem. Phys., 12, 10295–10312, https://doi.org/10.5194/acp-12-10295-2012, 2012.
Hintsa, E. J., Moore, F. L., Hurst, D. F., Dutton, G. S., Hall, B. D., Nance, J. D., Patrick, L., McClure-Begley, A., Elkins, J. W., Hall, E. G., Jordan, A. F., Gao, R.-S., Rollins, A. W., Thornberry, T. D., Watts, L. A., Thompson, C., Peischl, J., Bourgeois, I., Ryerson, T., Daube, B. C., Pittman, J. V., Wofsy, S. C., Diskin, G., Bui, T. P., Montzka, S., and Miller, B.: UAS Chromatograph for Atmospheric Trace Species (UCATS) – a versatile instrument for trace gas measurements on different airborne platforms, Atmos. Meas. Tech., in preparation, 2020.
Hodzic, A., Madronich, S., Kasibhatla, P. S., Tyndall, G., Aumont, B., Jimenez, J. L., Lee-Taylor, J., and Orlando, J.: Organic photolysis reactions in tropospheric aerosols: effect on secondary organic aerosol formation and lifetime, Atmos. Chem. Phys., 15, 9253–9269, https://doi.org/10.5194/acp-15-9253-2015, 2015.
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016.
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018.
Hofzumahaus, A., Lefer, B. L., Monks, P. S., Hall, S. R., Kylling, A., Mayer, B., Shetter, R. E., Junkermann, W., Bais, A., Calvert, J. G., Cantrell, C. A., Madronich, S., Edwards, G. D., and Kraus, A.: Photolysis frequency of O3 to O(1D): Measurements and modeling during the International Photolysis Frequency Measurement and Modeling Intercomparison (IPMMI), J. Geophys. Res.-Atmos., 109, D08S90, https://doi.org/10.1029/2003JD004333, 2004.
Hottle, J. R., Huisman, A. J., DiGangi, J. P., Kammrath, A., Galloway, M. M., Coens, K. L., and Keutsch, F. N.: A Laser Induced Fluorescence-Based Instrument for In-Situ Measurements of Atmospheric Formaldehyde, Environ. Sci. Technol., 43, 790–795, https://doi.org/10.1021/es801621f, 2009.
Hu, L., Millet, D. B., Baasandorj, M., Griffis, T. J., Turner, P., Helmig, D., Curtis, A. J., and Hueber, J.: Isoprene emissions and impacts over an ecological transition region in the U.S. Upper Midwest inferred from tall tower measurements: Isoprene emissions in US Upper Midwest, J. Geophys. Res.-Atmos., 120, 3553–3571, https://doi.org/10.1002/2014JD022732, 2015.
Hu, L., Jacob, D. J., Liu, X., Zhang, Y., Zhang, L., Kim, P. S., Sulprizio, M. P., and Yantosca, R. M.: Global budget of tropospheric ozone: Evaluating recent model advances with satellite (OMI), aircraft (IAGOS), and ozonesonde observations, Atmos. Environ., 167, 323–334, https://doi.org/10.1016/j.atmosenv.2017.08.036, 2017.
Jacob, D. J.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, 2000.
Jaeglé, L., Shah, V., Thornton, J. A., Lopez-Hilfiker, F. D., Lee, B. H., McDuffie, E. E., Fibiger, D., Brown, S. S., Veres, P., Sparks, T. L., Ebben, C. J., Wooldridge, P. J., Kenagy, H. S., Cohen, R. C., Weinheimer, A. J., Campos, T. L., Montzka, D. D., Digangi, J. P., Wolfe, G. M., Hanisco, T., Schroder, J. C., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Sullivan, A. P., Guo, H., and Weber, R. J.: Nitrogen Oxides Emissions, Chemistry, Deposition, and Export Over the Northeast United States During the WINTER Aircraft Campaign, J. Geophys. Res.-Atmos., 123, 12368–12393, https://doi.org/10.1029/2018JD029133, 2018.
Jenkin, M. E., Saunders, S. M., and Pilling, M. E.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Johnson, M. T.: A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas, Ocean Sci., 6, 913–932, https://doi.org/10.5194/os-6-913-2010, 2010.
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016.
Karion, A., Sweeney, C., Wolter, S., Newberger, T., Chen, H., Andrews, A., Kofler, J., Neff, D., and Tans, P.: Long-term greenhouse gas measurements from aircraft, Atmos. Meas. Tech., 6, 511–526, https://doi.org/10.5194/amt-6-511-2013, 2013.
Kasibhatla, P., Sherwen, T., Evans, M. J., Carpenter, L. J., Reed, C., Alexander, B., Chen, Q., Sulprizio, M. P., Lee, J. D., Read, K. A., Bloss, W., Crilley, L. R., Keene, W. C., Pszenny, A. A. P., and Hodzic, A.: Global impact of nitrate photolysis in sea-salt aerosol on NOx, OH, and O3 in the marine boundary layer, Atmos. Chem. Phys., 18, 11185–11203, https://doi.org/10.5194/acp-18-11185-2018, 2018.
Keller, C. A., Long, M. S., Yantosca, R. M., Da Silva, A. M., Pawson, S., and Jacob, D. J.: HEMCO v1.0: a versatile, ESMF-compliant component for calculating emissions in atmospheric models, Geosci. Model Dev., 7, 1409–1417, https://doi.org/10.5194/gmd-7-1409-2014, 2014.
Kim, M. J., Novak, G. A., Zoerb, M. C., Yang, M., Blomquist, B. W., Huebert, B. J., Cappa, C. D., and Bertram, T. H.: Air-Sea exchange of biogenic volatile organic compounds and the impact on aerosol particle size distributions, Geophys. Res. Lett., 44, 3887–3896, https://doi.org/10.1002/2017GL072975, 2017.
Kopacz, M., Jacob, D. J., Fisher, J. A., Logan, J. A., Zhang, L., Megretskaia, I. A., Yantosca, R. M., Singh, K., Henze, D. K., Burrows, J. P., Buchwitz, M., Khlystova, I., McMillan, W. W., Gille, J. C., Edwards, D. P., Eldering, A., Thouret, V., and Nedelec, P.: Global estimates of CO sources with high resolution by adjoint inversion of multiple satellite datasets (MOPITT, AIRS, SCIAMACHY, TES), Atmos. Chem. Phys., 10, 855–876, https://doi.org/10.5194/acp-10-855-2010, 2010.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Krotkov, N. A., McLinden, C. A., Li, C., Lamsal, L. N., Celarier, E. A., Marchenko, S. V., Swartz, W. H., Bucsela, E. J., Joiner, J., Duncan, B. N., Boersma, K. F., Veefkind, J. P., Levelt, P. F., Fioletov, V. E., Dickerson, R. R., He, H., Lu, Z., and Streets, D. G.: Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015, Atmos. Chem. Phys., 16, 4605–4629, https://doi.org/10.5194/acp-16-4605-2016, 2016.
Kuhns, H., Green, M., and Etyemezian, V.: Big Bend Regional Aerosol and Visibility Observational (BRAVO) Study Emissions Inventory, Desert Research Institute, Las Vegas, NV, 2003.
Kwan, A. J., Crounse, J. D., Clarke, A. D., Shinozuka, Y., Anderson, B. E., Crawford, J. H., Avery, M. A., McNaughton, C. S., Brune, W. H., Singh, H. B., and Wennberg, P. O.: On the flux of oxygenated volatile organic compounds from organic aerosol oxidation, Geophys. Res. Lett, 33, https://doi.org/10.1029/2006GL026144, 2006.
Lamarque, J.-F., Emmons, L. K., Hess, P. G., Kinnison, D. E., Tilmes, S., Vitt, F., Heald, C. L., Holland, E. A., Lauritzen, P. H., Neu, J., Orlando, J. J., Rasch, P. J., and Tyndall, G. K.: CAM-chem: description and evaluation of interactive atmospheric chemistry in the Community Earth System Model, Geosci. Model Dev., 5, 369–411, https://doi.org/10.5194/gmd-5-369-2012, 2012.
Lee, J. D., Young, J. C., Read, K. A., Hamilton, J. F., Hopkins, J. R., Lewis, A. C., Bandy, B. J., Davey, J., Edwards, P., Ingham, T., Self, D. E., Smith, S. C., Pilling, M. J., and Heard, D. E.: Measurement and calculation of OH reactivity at a United Kingdom coastal site, J. Atmos. Chem., 64, 53–76, https://doi.org/10.1007/s10874-010-9171-0, 2009.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, M., Zhang, Q., Kurokawa, J.-I., Woo, J.-H., He, K., Lu, Z., Ohara, T., Song, Y., Streets, D. G., Carmichael, G. R., Cheng, Y., Hong, C., Huo, H., Jiang, X., Kang, S., Liu, F., Su, H., and Zheng, B.: MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP, Atmos. Chem. Phys., 17, 935–963, https://doi.org/10.5194/acp-17-935-2017, 2017.
Li, S. M., Leithead, A., Moussa, S. G., Liggio, J., Moran, M. D., Wang, D., Hayden, K., Darlington, A., Gordon, M., Staebler, R., Makar, P. A., Stroud, C. A., McLaren, R., Liu, P. S. K., O'Brien, J., Mittermeier, R. L., Zhang, J., Marson, G., Cober, S. G., Wolde, M., and Wentzell, J. J. B.: Differences between measured and reported volatile organic compound emissions from oil sands facilities in Alberta, Canada, P. Natl. Acad. Sci. USA, 114, E3756–E3765, https://doi.org/10.1073/pnas.1617862114, 2017.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Luo, G., Yu, F., and Schwab, J.: Revised treatment of wet scavenging processes dramatically improves GEOS-Chem 12.0.0 simulations of surface nitric acid, nitrate, and ammonium over the United States, Geosci. Model Dev., 12, 3439–3447, https://doi.org/10.5194/gmd-12-3439-2019, 2019.
Mao, J., Ren, X., Brune, W. H., Olson, J. R., Crawford, J. H., Fried, A., Huey, L. G., Cohen, R. C., Heikes, B., Singh, H. B., Blake, D. R., Sachse, G. W., Diskin, G. S., Hall, S. R., and Shetter, R. E.: Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 9, 163–173, https://doi.org/10.5194/acp-9-163-2009, 2009.
Mao, J., Ren, X., Chen, S., Brune, W. H., Chen, Z., Martinez, M., Harder, H., Lefer, B., Rappenglück, B., Flynn, J., and Leuchner, M.: Atmospheric oxidation capacity in the summer of Houston 2006: Comparison with summer measurements in other metropolitan studies, Atmos. Environ., 44, 4107–4115, https://doi.org/10.1016/j.atmosenv.2009.01.013, 2010.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Mao, J., Fan, S., Jacob, D. J., and Travis, K. R.: Radical loss in the atmosphere from Cu-Fe redox coupling in aerosols, Atmos. Chem. Phys., 13, 509–519, https://doi.org/10.5194/acp-13-509-2013, 2013.
Marais, E. A. and Wiedinmyer, C.: Air Quality Impact of Diffuse and Inefficient Combustion Emissions in Africa (DICE-Africa), Environ. Sci. Technol., 50, 10739–10745, https://doi.org/10.1021/acs.est.6b02602, 2016.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
Marais, E. A., Jacob, D. J., Choi, S., Joiner, J., Belmonte-Rivas, M., Cohen, R. C., Beirle, S., Murray, L. T., Schiferl, L. D., Shah, V., and Jaeglé, L.: Nitrogen oxides in the global upper troposphere: interpreting cloud-sliced NO2 observations from the OMI satellite instrument, Atmos. Chem. Phys., 18, 17017–17027, https://doi.org/10.5194/acp-18-17017-2018, 2018.
Marvin, M. R., Wolfe, G. M., Salawitch, R. J., Canty, T. P., Roberts, S. J., Travis, K. R., Aikin, K. C., de Gouw, J. A., Graus, M., Hanisco, T. F., Holloway, J. S., Hübler, G., Kaiser, J., Keutsch, F. N., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., Veres, P. R., and Warneke, C.: Impact of evolving isoprene mechanisms on simulated formaldehyde: An inter-comparison supported by in situ observations from SENEX, Atmos. Environ., 164, 325–336, https://doi.org/10.1016/j.atmosenv.2017.05.049, 2017.
McDonald, B. C., Gouw, J. A. de, Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S.-W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
McManus, J. B., Nelson, D. D., Shorter, J. H., Jimenez, R., Herndon, S., Saleska, S., and Zahniser, M.: A high precision pulsed quantum cascade laser spectrometer for measurements of stable isotopes of carbon dioxide, J. Mod. Opt., 52, 2309–2321, https://doi.org/10.1080/09500340500303710, 2005.
Millet, D. B., Jacob, D. J., Custer, T. G., de Gouw, J. A., Goldstein, A. H., Karl, T., Singh, H. B., Sive, B. C., Talbot, R. W., Warneke, C., and Williams, J.: New constraints on terrestrial and oceanic sources of atmospheric methanol, Atmos. Chem. Phys., 8, 6887–6905, https://doi.org/10.5194/acp-8-6887-2008, 2008.
Millet, D. B., Guenther, A., Siegel, D. A., Nelson, N. B., Singh, H. B., de Gouw, J. A., Warneke, C., Williams, J., Eerdekens, G., Sinha, V., Karl, T., Flocke, F., Apel, E., Riemer, D. D., Palmer, P. I., and Barkley, M.: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 10, 3405–3425, https://doi.org/10.5194/acp-10-3405-2010, 2010.
Mogensen, D., Smolander, S., Sogachev, A., Zhou, L., Sinha, V., Guenther, A., Williams, J., Nieminen, T., Kajos, M. K., Rinne, J., Kulmala, M., and Boy, M.: Modelling atmospheric OH-reactivity in a boreal forest ecosystem, Atmos. Chem. Phys., 11, 9709–9719, https://doi.org/10.5194/acp-11-9709-2011, 2011.
Monks, P. S.: Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 34, 376–395, https://doi.org/10.1039/b307982c, 2005.
Montzka, S. A., Spivakovsky, C. M., Butler, J. H., Elkins, J. W., Lock, L. T., and Mondeel, D. J.: New observational constraints for atmospheric hydroxyl on global and hemispheric scales, Science, 288, 5465, https://doi.org/10.1126/science.288.5465.500, 2000.
Mu, M., Randerson, J. T., van der Werf, G. R., Giglio, L., Kasibhatla, P., Morton, D., Collatz, G. J., DeFries, R. S., Hyer, E. J., Prins, E. M., Griffith, D. W. T., Wunch, D., Toon, G. C., Sherlock, V., and Wennberg, P. O.: Daily and 3-hourly variability in global fire emissions and consequences for atmospheric model predictions of carbon monoxide, J. Geophys. Res.-Atmos., 116, D24303, https://doi.org/10.1029/2011JD016245, 2011.
Müller, J.-F., Liu, Z., Nguyen, V. S., Stavrakou, T., Harvey, J. N., and Peeters, J.: The reaction of methyl peroxy and hydroxyl radicals as a major source of atmospheric methanol, Nat. Commun., 7, 13213, https://doi.org/10.1038/ncomms13213, 2016.
Müller, J.-F., Stavrakou, T., Bauwens, M., George, M., Hurtmans, D., Coheur, P.-F., Clerbaux, C., and Sweeney, C.: Top-Down CO Emissions Based On IASI Observations and Hemispheric Constraints on OH Levels, Geophys. Res. Lett., 45, 2017GL076697, https://doi.org/10.1002/2017GL076697, 2018.
Mungall, E. L., Abbatt, J. P. D., Wentzell, J. J. B., Lee, A. K. Y., Thomas, J. L., Blais, M., Gosselin, M., Miller, L. A., Papakyriakou, T., Willis, M. D., and Liggio, J.: Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer, P. Natl. Acad. Sci. USA, 114, 6203–6208, https://doi.org/10.1073/pnas.1620571114, 2017.
Murray, L. T., Jacob, D. J., Logan, J. A., Hudman, R. C., and Koshak, W. J.: Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data, J. Geophys. Res.-Atmos., 117, D20307, https://doi.org/10.1029/2012JD017934, 2012.
Murray, L. T., Mickley, L. J., Kaplan, J. O., Sofen, E. D., Pfeiffer, M., and Alexander, B.: Factors controlling variability in the oxidative capacity of the troposphere since the Last Glacial Maximum, Atmos. Chem. Phys., 14, 3589–3622, https://doi.org/10.5194/acp-14-3589-2014, 2014.
Naik, V., Voulgarakis, A., Fiore, A. M., Horowitz, L. W., Lamarque, J.-F., Lin, M., Prather, M. J., Young, P. J., Bergmann, D., Cameron-Smith, P. J., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R., Eyring, V., Faluvegi, G., Folberth, G. A., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., van Noije, T. P. C., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R., Shindell, D. T., Stevenson, D. S., Strode, S., Sudo, K., Szopa, S., and Zeng, G.: Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 5277–5298, https://doi.org/10.5194/acp-13-5277-2013, 2013.
Nakashima, Y., Kato, S., Greenberg, J., Harley, P., Karl, T., Turnipseed, A., Apel, E., Guenther, A., Smith, J., and Kajii, Y.: Total OH reactivity measurements in ambient air in a southern Rocky mountain ponderosa pine forest during BEACHON-SRM08 summer campaign, Atmos. Environ., 85, 1–8, https://doi.org/10.1016/j.atmosenv.2013.11.042, 2014.
Nicely, J. M., Anderson, D. C., Canty, T. P., Salawitch, R. J., Wolfe, G. M., Apel, E. C., Arnold, S. R., Atlas, E. L., Blake, N. J., Bresch, J. F., Campos, T. L., Dickerson, R. R., Duncan, B., Emmons, L. K., Evans, M. J., Fernandez, R. P., Flemming, J., Hall, S. R., Hanisco, T. F., Honomichl, S. B., Hornbrook, R. S., Huijnen, V., Kaser, L., Kinnison, D. E., Lamarque, J.-F., Mao, J., Monks, S. A., Montzka, D. D., Pan, L. L., Riemer, D. D., Saiz-Lopez, A., Steenrod, S. D., Stell, M. H., Tilmes, S., Turquety, S., Ullmann, K., and Weinheimer, A. J.: An observationally constrained evaluation of the oxidative capacity in the tropical western Pacific troposphere: Observationally Constrained OH in TWP, J. Geophys. Res.-Atmos., 121, 7461–7488, https://doi.org/10.1002/2016JD025067, 2016.
Nicely, J. M., Salawitch, R. J., Canty, T., Anderson, D. C., Arnold, S. R., Chipperfield, M. P., Emmons, L. K., Flemming, J., Huijnen, V., Kinnison, D. E., Lamarque, J.-F., Mao, J., Monks, S. A., Steenrod, S. D., Tilmes, S., and Turquety, S.: Quantifying the causes of differences in tropospheric OH within global models, J. Geophys. Res.-Atmos., 122, 1983–2007, https://doi.org/10.1002/2016JD026239, 2017.
Nölscher, A. C., Williams, J., Sinha, V., Custer, T., Song, W., Johnson, A. M., Axinte, R., Bozem, H., Fischer, H., Pouvesle, N., Phillips, G., Crowley, J. N., Rantala, P., Rinne, J., Kulmala, M., Gonzales, D., Valverde-Canossa, J., Vogel, A., Hoffmann, T., Ouwersloot, H. G., Vilà-Guerau de Arellano, J., and Lelieveld, J.: Summertime total OH reactivity measurements from boreal forest during HUMPPA-COPEC 2010, Atmos. Chem. Phys., 12, 8257–8270, https://doi.org/10.5194/acp-12-8257-2012, 2012.
Nölscher, A. C., Yañez-Serrano, A. M., Wolff, S., de Araujo, A. C., Lavrič, J. V., Kesselmeier, J., and Williams, J.: Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Commun., 7, 10383, https://doi.org/10.1038/ncomms10383, 2016.
Olivier, J., Peters, J., Granier, C., Petron, G., Muller, J. F., and Wallens, S.: Present and future surface emissions of atmospheric compounds, Eur. Union, Brussels, 2003.
Pai, S. J., Heald, C. L., Pierce, J. R., Farina, S. C., Marais, E. A., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Middlebrook, A. M., Coe, H., Shilling, J. E., Bahreini, R., Dingle, J. H., and Vu, K.: An evaluation of global organic aerosol schemes using airborne observations, Atmos. Chem. Phys., 20, 2637–2665, https://doi.org/10.5194/acp-20-2637-2020, 2020.
Patra, P. K., Krol, M. C., Montzka, S. A., Arnold, T., Atlas, E. L., Lintner, B. R., Stephens, B. B., Xiang, B., Elkins, J. W., Fraser, P. J., Ghosh, A., Hintsa, E. J., Hurst, D. F., Ishijima, K., Krummel, P. B., Miller, B. R., Miyazaki, K., Moore, F. L., Mühle, J., O'Doherty, S., Prinn, R. G., Steele, L. P., Takigawa, M., Wang, H. J., Weiss, R. F., Wofsy, S. C., and Young, D.: Observational evidence for interhemispheric hydroxyl-radical parity, Nature, 513, 219–223, https://doi.org/10.1038/nature13721, 2014.
Petropavlovskikh, I., Shetter, R., Hall, S., Ullmann, K., and Bhartia, P. K.: Algorithm for the charge-coupled-device scanning actinic flux spectroradiometer ozone retrieval in support of the Aura satellite validation, J. Appl. Remote Sens., 1, 013540, https://doi.org/10.1117/1.2802563, 2007.
Philip, S., Martin, R. V., and Keller, C. A.: Sensitivity of chemistry-transport model simulations to the duration of chemical and transport operators: a case study with GEOS-Chem v10-01, Geosci. Model Dev., 9, 1683–1695, https://doi.org/10.5194/gmd-9-1683-2016, 2016.
Plass-Dülmer C., Khedim A., Koppmann R., Johnen F. J., Rudolph J., and Kuosa H.: Emissions of light nonmethane hydrocarbons from the Atlantic into the atmosphere, Global Biogeochem. Cy., 7, 211–228, https://doi.org/10.1029/92GB02361, 1993.
Podolske, J. R., Sachse, G. W., and Diskin, G. S.: Calibration and data retrieval algorithms for the NASA Langley/Ames Diode Laser Hygrometer for the NASA Transport and Chemical Evolution Over the Pacific (TRACE-P) mission, J. Geophys. Res., 108, 8792, https://doi.org/10.1029/2002JD003156, 2003.
Pollack, I. B., Lerner, B. M., and Ryerson, T. B.: Evaluation of ultraviolet light-emitting diodes for detection of atmospheric NO2 by photolysis – chemiluminescence, J. Atmos. Chem., 65, 111–125, https://doi.org/10.1007/s10874-011-9184-3, 2010.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2017.
Prather, M. J., Holmes, C. D., and Hsu, J.: Reactive greenhouse gas scenarios: Systematic exploration of uncertainties and the role of atmospheric chemistry, Geophys. Res. Lett., 39, L09803, https://doi.org/10.1029/2012GL051440, 2012.
Pye, H. O. T., Liao, H., Wu, S., Mickley, L. J., Jacob, D. J., Henze, D. K., and Seinfeld, J. H.: Effect of changes in climate and emissions on future sulfate-nitrate-ammonium aerosol levels in the United States, J. Geophys. Res., 114, D01205, https://doi.org/10.1029/2008JD010701, 2009.
Ramasamy, S., Ida, A., Jones, C., Kato, S., Tsurumaru, H., Kishimoto, I., Kawasaki, S., Sadanaga, Y., Nakashima, Y., Nakayama, T., Matsumi, Y., Mochida, M., Kagami, S., Deng, Y., Ogawa, S., Kawana, K., and Kajii, Y.: Total OH reactivity measurement in a BVOC dominated temperate forest during a summer campaign, 2014, Atmos. Environ., 131, 41–54, https://doi.org/10.1016/j.atmosenv.2016.01.039, 2016.
Randerson, J. T., Chen, Y., van der Werf, G. R., Rogers, B. M., and Morton, D. C.: Global burned area and biomass burning emissions from small fires, J. Geophys. Res.-Biogeo., 117, G04012, https://doi.org/10.1029/2012JG002128, 2012.
Read, K. A., Carpenter, L. J., Arnold, S. R., Beale, R., Nightingale, P. D., Hopkins, J. R., Lewis, A. C., Lee, J. D., Mendes, L., and Pickering, S. J.: Multiannual Observations of Acetone, Methanol, and Acetaldehyde in Remote Tropical Atlantic Air: Implications for Atmospheric OVOC Budgets and Oxidative Capacity, Environ. Sci. Technol., 46, 11028–11039, https://doi.org/10.1021/es302082p, 2012.
Ren, X., Brune, W. H., Oliger, A., Metcalf, A. R., Simpas, J. B., Shirley, T., Schwab, J. J., Bai, C., Roychowdhury, U., Li, Y., Cai, C., Demerjian, K. L., He, Y., Zhou, X., Gao, H., and Hou, J.: OH, HO2, and OH reactivity during the PMTACS–NY Whiteface Mountain 2002 campaign: Observations and model comparison, J. Geophys. Res.-Atmos., 111, D10S03, https://doi.org/10.1029/2005JD006126, 2006.
Romer, P. S., Wooldridge, P. J., Crounse, J. D., Kim, M. J., Wennberg, P. O., Dibb, J. E., Scheuer, E., Blake, D. R., Meinardi, S., Brosius, A. L., Thames, A. B., Miller, D. O., Brune, W. H., Hall, S. R., Ryerson, T. B., and Cohen, R. C.: Constraints on aerosol nitrate photolysis as a potential source of HONO and NOx. Environ. Sci. Technol., 52, 13738–13746, https://doi.org/10.1021/acs.est.8b03861, 2018.
Ryerson, T. B., Buhr, M. P., Frost, G. J., Goldan, P. D., Holloway, J. S., Hübler, G., Jobson, B. T., Kuster, W. C., McKeen, S. A., Parrish, D. D., Roberts, J. M., Sueper, D. T., Trainer, M., Williams, J., and Fehsenfeld, F. C.: Emissions lifetimes and ozone formation in power plant plumes, J. Geophys. Res.-Atmos., 103, 22569–22583, https://doi.org/10.1029/98JD01620, 1998.
Ryerson, T. B., Williams, E. J., and Fehsenfeld, F. C.: An efficient photolysis system for fast-response NO2 measurements, J. Geophys. Res.-Atmos., 105, 26447–26461, https://doi.org/10.1029/2000JD900389, 2000.
Safieddine, S. A., Heald, C. L., and Henderson, B. H.: The global nonmethane reactive organic carbon budget: A modeling perspective, Geophys. Res. Lett., 44, 3897–3906, https://doi.org/10.1002/2017GL072602, 2017.
Santoni, G. W., Daube, B. C., Kort, E. A., Jiménez, R., Park, S., Pittman, J. V., Gottlieb, E., Xiang, B., Zahniser, M. S., Nelson, D. D., McManus, J. B., Peischl, J., Ryerson, T. B., Holloway, J. S., Andrews, A. E., Sweeney, C., Hall, B., Hintsa, E. J., Moore, F. L., Elkins, J. W., Hurst, D. F., Stephens, B. B., Bent, J., and Wofsy, S. C.: Evaluation of the airborne quantum cascade laser spectrometer (QCLS) measurements of the carbon and greenhouse gas suite – CO2, CH4, N2O, and CO – during the CalNex and HIPPO campaigns, Atmos. Meas. Tech., 7, 1509–1526, https://doi.org/10.5194/amt-7-1509-2014, 2014.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schlundt, C., Tegtmeier, S., Lennartz, S. T., Bracher, A., Cheah, W., Krüger, K., Quack, B., and Marandino, C. A.: Oxygenated volatile organic carbon in the western Pacific convective center: ocean cycling, air–sea gas exchange and atmospheric transport, Atmospheric Chem. Phys., 17, 10837–10854, https://doi.org/10.5194/acp-17-10837-2017, 2017.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016.
Shetter, R. E. and Muller, M.: Photolysis frequency measurements using actinic flux spectroradiometry during PEM-Tropics Mission: Instrumentation description and some results, J. Geophys. Res.-Atmos., 104, 5647–5661, https://doi.org/10.1029/98JD01381, 1999.
Shindell, D. T., Faluvegi, G., Stevenson, D. S., Krol, M. C., Emmons, L. K., Lamarque, J.-F., Pétron, G., Dentener, F. J., Ellingsen, K., Schultz, M. G., Wild, O., Amann, M., Atherton, C. S., Bergmann, D. J., Bey, I., Butler, T., Cofala, J., Collins, W. J., Derwent, R. G., Doherty, R. M., Drevet, J., Eskes, H. J., Fiore, A. M., Gauss, M., Hauglustaine, D. A., Horowitz, L. W., Isaksen, I. S. A., Lawrence, M. G., Montanaro, V., Müller, J.-F., Pitari, G., Prather, M. J., Pyle, J. A., Rast, S., Rodriguez, J. M., Sanderson, M. G., Savage, N. H., Strahan, S. E., Sudo, K., Szopa, S., Unger, N., van Noije, T. P. C., and Zeng, G.: Multimodel simulations of carbon monoxide: Comparison with observations and projected near-future changes, J. Geophys. Res., 111, D19306, https://doi.org/10.1029/2006JD007100, 2006.
Simpson, I. J., Blake, N. J., Barletta, B., Diskin, G. S., Fuelberg, H. E., Gorham, K., Huey, L. G., Meinardi, S., Rowland, F. S., Vay, S. A., Weinheimer, A. J., Yang, M., and Blake, D. R.: Characterization of trace gases measured over Alberta oil sands mining operations: 76 speciated C2–C10 volatile organic compounds (VOCs), CO2, CH4, CO, NO, NO2, NOy, O3 and SO2, Atmos. Chem. Phys., 10, 11931–11954, https://doi.org/10.5194/acp-10-11931-2010, 2010.
Singh, H., Chen, Y., Staudt, A., Jacob, D., Blake, D., Heikes, B., and Snow, J.: Evidence from the Pacific troposphere for large global sources of oxygenated organic compounds, Nature, 410, 1078, https://doi.org/10.1038/35074067, 2001.
Singh, H. B., Tabazadeh, A., Evans, M. J., Field, B. D., Jacob, D. J., Sachse, G., Crawford, J. H., Shetter, R., and Brune, W. H.: Oxygenated volatile organic chemicals in the oceans: Inferences and implications based on atmospheric observations and air-sea exchange models, Geophys. Res. Lett., 30, 1862, https://doi.org/10.1029/2003GL017933, 2003.
Sinha, V., Williams, J., Crowley, J. N., and Lelieveld, J.: The Comparative Reactivity Method – a new tool to measure total OH Reactivity in ambient air, Atmos. Chem. Phys., 8, 2213–2227, https://doi.org/10.5194/acp-8-2213-2008, 2008.
Sinha, V., Williams, J., Lelieveld, J., Ruuskanen, T. M., Kajos, M. K., Patokoski, J., Hellen, H., Hakola, H., Mogensen, D., Boy, M., Rinne, J., and Kulmala, M.: OH Reactivity Measurements within a Boreal Forest: Evidence for Unknown Reactive Emissions, Environ. Sci. Technol., 44, 6614–6620, https://doi.org/10.1021/es101780b, 2010.
Staudt, A. C., Jacob, D. J., Ravetta, F., Logan, J. A., Bachiochi, D., Krishnamurti, T. N., Sandholm, S., Ridley, B., and Singh, H. B.: Sources and chemistry of nitrogen oxides over the tropical Pacific, J. Geophys. Res., 108, 8239, https://doi.org/10.1029/2002JD002139, 2003.
St. Clair, J. M., McCabe, D. C., Crounse, J. D., Steiner, U., and Wennberg, P. O.: Chemical ionization tandem mass spectrometer for the in situ measurement of methyl hydrogen peroxide, Rev. Sci. Instrum., 81, 094102, https://doi.org/10.1063/1.3480552, 2010.
Stockwell, C. E., Veres, P. R., Williams, J., and Yokelson, R. J.: Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry, Atmos. Chem. Phys., 15, 845–865, https://doi.org/10.5194/acp-15-845-2015, 2015.
Strode, S. A., Duncan, B. N., Yegorova, E. A., Kouatchou, J., Ziemke, J. R., and Douglass, A. R.: Implications of carbon monoxide bias for methane lifetime and atmospheric composition in chemistry climate models, Atmos. Chem. Phys., 15, 11789–11805, https://doi.org/10.5194/acp-15-11789-2015, 2015.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Olson, J., Crawford, J., Avery, M., Sachse, G., Vay, S., Sandholm, S., Guan, H.-W., Vaughn, T., Mastromarino, J., Heikes, B., Snow, J., Podolske, J., and Singh, H.: OH and HO2 in the tropical Pacific: Results from PEM-Tropics B, J. Geophys. Res.-Atmos., 106, 32667–32681, https://doi.org/10.1029/2001JD900002, 2001.
Thames, A. B., Brune, W. H., Miller, D. O., Allen, H. M., Apel, E. C., Blake, D. R., Bui, T. P., Commane, R., Crounse, J. D., Daube, B. C., Diskin, G. S., DiGangi, J. P., Elkins, J. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hintsa, E., Hornbrook, R. S., Kim, M. J., McKain, K., Moore, F. L., Nicely, J. M., Peischl, J., Ryerson, T. B., St. Clair, J. M., Sweeney, C., Teng, A., Thompson, C. R., Ullmann, K., Wennberg, P. O., and Wolfe, G. M.: Missing OH reactivity in the global marine boundary layer, Atmos. Chem. Phys., 20, 4013–4029, https://doi.org/10.5194/acp-20-4013-2020, 2020.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Tzompa-Sosa Z. A., Mahieu E., Franco B., Keller C. A., Turner A. J., Helmig D., Fried A., Richter D., Weibring P., Walega J., Yacovitch T. I., Herndon S. C., Blake D. R., Hase F., Hannigan J. W., Conway S., Strong K., Schneider M., and Fischer E. V.: Revisiting global fossil fuel and biofuel emissions of ethane, J. Geophys. Res.-Atmos., 122, 2493–2512, https://doi.org/10.1002/2016JD025767, 2017.
van der Werf, G. R., Randerson, J. T., Giglio, L., van Leeuwen, T. T., Chen, Y., Rogers, B. M., Mu, M., van Marle, M. J. E., Morton, D. C., Collatz, G. J., Yokelson, R. J., and Kasibhatla, P. S.: Global fire emissions estimates during 1997–2016, Earth Syst. Sci. Data, 9, 697–720, https://doi.org/10.5194/essd-9-697-2017, 2017.
Wang, S., Hornbrook, R. S., Hills, A., Emmons, L. K., Tilmes, S., Lamarque, J.-F., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Crounse, J. D., Wennberg, P. O., Kim, M., Allen, H., Ryerson, T. B., Thompson, C. R., Peischl, J., Moore, F., Nance, D., Hall, B., Elkins, J., Tanner, D., Huey, L. G., Hall, S. R., Ullmann, K., Orlando, J. J., Tyndall, G. S., Flocke, F. M., Ray, E., Hanisco, T. F., Wolfe, G. M., Clair, J. S., Commane, R., Daube, B., Barletta, B., Blake, D. R., Weinzierl, B., Dollner, M., Conley, A., Vitt, F., Wofsy, S. C., Riemer, D. D., and Apel, E. C.: Atmospheric Acetaldehyde: Importance of Air-Sea Exchange and a Missing Source in the Remote Troposphere, Geophys. Res. Lett., 46, 5601–5613, https://doi.org/10.1029/2019GL082034, 2019.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Wofsy, S. C.: HIAPER Pole-to-Pole Observations (HIPPO): fine-grained, global-scale measurements of climatically important atmospheric gases and aerosols, Philos. T. Roy. Soc. A, 369, 2073–2086, https://doi.org/10.1098/rsta.2010.0313, 2011.
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgeois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., Chin, M., Commane, R., Correa, G., Crounse, J. D., Cullis, P. D., Daube, B. C., Day, D. A., Dean-Day, J. M., Dibb, J. E., DiGangi, J. P., Diskin, G. S., Dollner, M., Elkins, J. W., Erdesz, F., Fiore, A. M., Flynn, C. M., Froyd, K., Gesler, D. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hills, A. J., Hintsa, E. J., Hoffman, A., Hornbrook, R. S., Huey, L. G., Hughes, S., Jimenez, J. L., Johnson, B. J., Katich, J. M., Keeling, R. F., Kim, M. J., Kupc, A., Lait, L. R., Lamarque, J.-F., Liu, J., McKain, K., Mclaughlin, R. J., Meinardi, S., Miller, D. O., Montzka, S. A., Moore, F. L., Morgan, E. J., Murphy, D. M., Murray, L. T., Nault, B. A., Neuman, J. A., Newman, P. A., Nicely, J. M., Pan, X., Paplawsky, W., Peischl, J., Prather, M. J., Price, D. J., Ray, E., Reeves, J. M., Richardson, M., Rollins, A. W., Rosenlof, K. H., Ryerson, T. B., Scheuer, E., Schill, G. P., Schroder, J. C., Schwarz, J. P., St. Clair, J. M., Steenrod, S. D., Stephens, B. B., Strode, S. A., Sweeney, C., Tanner, D., Teng, A. P., Thames, A. B., Thompson, C. R., Ullmann, K., Veres, P. R., Vieznor, N., Wagner, N. L., Watt, A., Weber, R., Weinzierl, B., Wennberg, P., Williamson, C. J., Wilson, J. C., Wolfe, G. M., Woods, C. T., and Zeng, L. H.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols. ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1581, 2018.
Wolfe, G. M., Nicely, J. M., St. Clair, J. M., Hanisco, T. F., Liao, J., Oman, L. D., Brune, W. B., Miller, D., Thames, A., González Abad, G., Ryerson, T. B., Thompson, C. R., Peischl, J., McCain, K., Sweeney, C., Wennberg, P. O., Kim, M., Crounse, J. D., Hall, S. R., Ullmann, K., Diskin, G., Bui, P., Chang, C., and Dean-Day, J.: Mapping hydroxyl variability throughout the global remote troposphere via synthesis of airborne and satellite formaldehyde observations, P. Natl. Acad. Sci. USA, 116, 11171–11180, https://doi.org/10.1073/pnas.1821661116, 2019.
Wong, J. P. S., Zhou, S., and Abbatt, J. P. D.: Changes in Secondary Organic Aerosol Composition and Mass due to Photolysis: Relative Humidity Dependence, J. Phys. Chem. A, 119, 4309–4316, https://doi.org/10.1021/jp506898c, 2015.
Wu., H., Wang, Y., Li, H., Huang, L., Huang, D., Shen, H., Xing, Y., and Chen, Z.: The OH-initiated oxidation of atmospheric peroxyacetic acid: Experimental and model studies, Atmos. Environ., 164, 61–70, https://doi.org/10.1016/j.atmosenv.2017.05.038, 2017.
Yang, M., Beale, R., Liss, P., Johnson, M., Blomquist, B., and Nightingale, P.: Air–sea fluxes of oxygenated volatile organic compounds across the Atlantic Ocean, Atmos. Chem. Phys., 14, 7499–7517, https://doi.org/10.5194/acp-14-7499-2014, 2014.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, https://doi.org/10.1038/nature17195, 2016.
Young, P. J., Archibald, A. T., Bowman, K. W., Lamarque, J.-F., Naik, V., Stevenson, D. S., Tilmes, S., Voulgarakis, A., Wild, O., Bergmann, D., Cameron-Smith, P., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R. M., Eyring, V., Faluvegi, G., Horowitz, L. W., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R. B., Shindell, D. T., Strode, S. A., Sudo, K., Szopa, S., and Zeng, G.: Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 2063–2090, https://doi.org/10.5194/acp-13-2063-2013, 2013.
Yoshino, A., Sadanaga, Y., Watanabe, K., Kato, S., Miyakawa, Y., Matsumoto, J., and Kajii, Y.: Measurement of total OH reactivity by laser-induced pump and probe technique–comprehensive observations in the urban atmosphere of Tokyo, Atmos. Environ., 40, 7869–7881, https://doi.org/10.1016/j.atmosenv.2006.07.023, 2006.
Yu, S., Mathur, R., Sarwar, G., Kang, D., Tong, D., Pouliot, G., and Pleim, J.: Eta-CMAQ air quality forecasts for O3 and related species using three different photochemical mechanisms (CB4, CB05, SAPRC-99): comparisons with measurements during the 2004 ICARTT study, Atmos. Chem. Phys., 10, 3001–3025, https://doi.org/10.5194/acp-10-3001-2010, 2010.
Zannoni, N., Gros, V., Lanza, M., Sarda, R., Bonsang, B., Kalogridis, C., Preunkert, S., Legrand, M., Jambert, C., Boissard, C., and Lathiere, J.: OH reactivity and concentrations of biogenic volatile organic compounds in a Mediterranean forest of downy oak trees, Atmos. Chem. Phys., 16, 1619–1636, https://doi.org/10.5194/acp-16-1619-2016, 2016.
Zannoni, N., Gros, V., Sarda Esteve, R., Kalogridis, C., Michoud, V., Dusanter, S., Sauvage, S., Locoge, N., Colomb, A., and Bonsang, B.: Summertime OH reactivity from a receptor coastal site in the Mediterranean Basin, Atmos. Chem. Phys., 17, 12645–12658, https://doi.org/10.5194/acp-17-12645-2017, 2017.
Zhang, L., Jacob, D. J., Knipping, E. M., Kumar, N., Munger, J. W., Carouge, C. C., van Donkelaar, A., Wang, Y. X., and Chen, D.: Nitrogen deposition to the United States: distribution, sources, and processes, Atmos. Chem. Phys., 12, 4539–4554, https://doi.org/10.5194/acp-12-4539-2012, 2012.
- Abstract
- Introduction
- Description of model and observations
- Comparison of simulated and measured OH
- Constraints on the remote source of OH
- Constraints on the remote sink of OH
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Description of model and observations
- Comparison of simulated and measured OH
- Constraints on the remote source of OH
- Constraints on the remote sink of OH
- Conclusions
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement