the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Jun 2020
| 30 Jun 2020
Strong anthropogenic control of secondary organic aerosol formation from isoprene in Beijing
Daniel J. Bryant
William J. Dixon
James R. Hopkins
Rachel E. Dunmore
Kelly L. Pereira
Marvin Shaw
Freya A. Squires
Thomas J. Bannan
Archit Mehra
Stephen D. Worrall
Asan Bacak
Carl J. Percival
Lisa K. Whalley
Dwayne E. Heard
Eloise J. Slater
Bin Ouyang
Tianqu Cui
Jason D. Surratt
Di Liu
Zongbo Shi
Roy Harrison
Yele Sun
Weiqi Xu
Alastair C. Lewis
James D. Lee
Andrew R. Rickard
Jacqueline F. Hamilton
Isoprene-derived secondary organic aerosol (iSOA) is a significant contributor to organic carbon (OC) in some forested regions, such as tropical rainforests and the Southeastern US. However, its contribution to organic aerosol in urban areas that have high levels of anthropogenic pollutants is poorly understood. In this study, we examined the formation of anthropogenically influenced iSOA during summer in Beijing, China. Local isoprene emissions and high levels of anthropogenic pollutants, in particular NOx and particulate , led to the formation of iSOA under both high- and low-NO oxidation conditions, with significant heterogeneous transformations of isoprene-derived oxidation products to particulate organosulfates (OSs) and nitrooxy-organosulfates (NOSs). Ultra-high-performance liquid chromatography coupled to high-resolution mass spectrometry was combined with a rapid automated data processing technique to quantify 31 proposed iSOA tracers in offline PM2.5 filter extracts. The co-elution of the inorganic ions in the extracts caused matrix effects that impacted two authentic standards differently. The average concentration of iSOA OSs and NOSs was 82.5 ng m−3, which was around 3 times higher than the observed concentrations of their oxygenated precursors (2-methyltetrols and 2-methylglyceric acid). OS formation was dependant on both photochemistry and the sulfate available for reactive uptake, as shown by a strong correlation with the product of ozone (O3) and particulate sulfate (). A greater proportion of high-NO OS products were observed in Beijing compared with previous studies in less polluted environments. The iSOA-derived OSs and NOSs represented 0.62 % of the oxidized organic aerosol measured by aerosol mass spectrometry on average, but this increased to ∼3 % on certain days. These results indicate for the first time that iSOA formation in urban Beijing is strongly controlled by anthropogenic emissions and results in extensive conversion to OS products from heterogenous reactions.
- Article
(798 KB) - Full-text XML
-
Supplement
(394 KB) - BibTeX
- EndNote





Rapidly developing countries such as China often experience very poor air quality. Beijing regularly experiences periods of very high particle pollution, with annual and 24 hr levels well above the World Health Organization guidelines (Chan et al., 2008; Hu et al., 2014). Premature mortality, as a result of respiratory illness, cardiovascular disease and cancer, has been associated with exposure to poor air quality (Dockery et al., 1993; Pope et al., 2000; Pope and Dockery, 2006; Jerrett et al., 2009; Beelen et al., 2014; Laurent et al., 2014; Ostro et al., 2015). Lelieveld et al. (2015) estimated that 1.36 million premature deaths in China in 2010 were a result of exposure to outdoor air pollution. By far the most dangerous pollutant to health in China is particulate matter less than 2.5 µm in diameter, known as PM2.5, with a recent study suggesting that a 50 % reduction in excess mortality requires a 62 % reduction in PM2.5 in the Beijing–Tianjin–Hebei (BTH) region (Hu et al., 2017a).
Previous measurements using aerosol mass spectrometry (AMS) indicate that PM1 in Beijing is mainly composed of sulfate, nitrate, ammonium and organics (Hu et al., 2016; Zhang et al., 2013). Positive matrix factorization of AMS measurements indicates that oxidized or secondary organic aerosol (SOA) can make up a substantial fraction of the PM1 mass (> 25 %), even in urban areas, but the sources of this material are still poorly understood (Q. Zhang et al., 2011; Sun et al., 2018). Hu et al. (2017a) estimated that exposure to SOA was responsible for 0.14 million deaths in China in 2013 based on mass contribution alone, ranging from < 1 % to 23 % source contributions to PM2.5 depending on location. Zhang et al. (2017a) used 14C measurements to determine that non-fossil fuel emissions are generally a dominant contributor of secondary organic carbon (SOC) in Beijing, with a larger contribution in summer as a result of increased biogenic volatile organic compound (VOC) emissions.
Hu et al. (2017b) updated the Community Multiscale Air Quality (CMAQ) model with revised SOA yields and a more detailed description of SOA formation from isoprene oxidation. Removing all anthropogenic pollutants from the model resulted in a huge drop in isoprene SOA concentrations, indicating that controlling anthropogenic emissions would result in a reduction of both anthropogenic and biogenic SOA. The predicted SOA was dominated by isoprene in summer across China and in four cities (Beijing, Guangzhou, Shanghai and Chengdu) with concentrations of up to 30 µg m−3 in Beijing. However, there is currently very little observational evidence to support such high SOA mass concentrations from isoprene oxidation in these Chinese cities. The widely used SOA tracer method (Kleindienst et al., 2007) has been extensively utilized to estimate the fraction of isoprene-derived SOA (iSOA) across China. Ding et al. (2014) studied SOA at 14 Chinese sites and found that iSOA dominated the apportioned SOA mass (46±14 %), contributing between 0.4 and 2.17 µg m−3 with an average of 1.59 µg m−3 in Beijing. However, only a very limited subset of VOC precursors were included, and this method fails to account for heterogeneous formation processes. To overcome some of these limitations, Q. Wang et al. (2017) used tracer-based source apportionment of PM2.5 with positive matrix factorization in the Pearl River Delta region during summer. They identified an iSOA factor that contributed up to 4 µg m−3 in Guangzhou and up to 11 % of the total SOC.
A multitude of studies have examined iSOA formation (Pandis et al., 1991; Edney et al., 2005; Kroll et al., 2006; Dommen et al., 2006; Kleindienst et al., 2006; Ng et al., 2006, 2008; Surratt et al., 2007a, b, 2008, 2010; Paulot et al., 2009; Chan et al., 2010; Chhabra et al., 2010; T. B. Nguyen et al., 2011, 2014, 2015; H. Zhang et al., 2011, 2012; Lin et al., 2013; Xu et al., 2014; Krechmer et al., 2015; Clark et al., 2016; Riva et al., 2016a, b); however, the magnitude of iSOA formed can be vastly different from study to study (Carlton et al., 2009). Furthermore, there have been limited field measurements undertaken to establish if these estimates are representative of urban environments (Wang et al., 2018; Li et al., 2018; Glasius et al., 2018; Le Breton et al., 2018; Hettiyadura et al., 2018, 2019; Rattanavaraha et al., 2016; Budisulistiorini et al., 2013).
iSOA formation during the daytime is dominated by reaction with hydroxyl radicals (OH), with the concentrations of NO having a strong influence on the reaction products (Wennberg et al., 2018, and references therein). Under low-NO conditions, isoprene peroxy radicals (RO2) can react with hydroperoxy radicals (HO2) to form isoprene hydroxyhydroperoxides (ISOPOOH). The ISOPOOH isomers can react further with OH to form isoprene epoxydiol isomers (β- or δ-IEPOX; Paulot et al., 2009), which can undergo uptake into acidified sulfate particles to form 2-methyltetrol organosulfates (2-MT-OS; Surratt et al., 2010; Lin et al., 2012; T. B. Nguyen, 2014). Under high-NO conditions, isoprene RO2 can react with NO to form alkoxy radicals (RO), producing methacrolein (MACR) and methyl vinyl ketone (MVK) as the main reaction products. The reaction of MACR with OH and the subsequent addition of NO2 leads to methylacrloylperoxynitrate (MPAN), which reacts with OH to produce hydroxymethyl-methyl-α-lactone (HMML; Nguyen et al., 2015) or methacrylic acid epoxide (MAE; Lin et al., 2013). HMML is thought to be the more abundant product compared with MAE (Nguyen et al., 2015). The subsequent uptake of HMML into wet sulfate aerosols is proposed to lead to either 2-methylglyceric acid (2-MG) or its organosulfate derivative (2-MG-OS), as well as their dimers and higher order oligomers (Surratt et al., 2006, 2010; Lin et al., 2013; Nguyen et al., 2015). Recently, Schwantes et al. (2019) showed that under ambient conditions the formation of SOA from low-volatility nitrates and dinitrates, formed via reactions of isoprene-derived RO2 with NO, is also important. Chamber-derived SOA yields for OH chemistry are variable depending on the experimental conditions, but they are generally low (< 10 %). The addition of acidified sulfate aerosol, accounting for wall losses and using more atmospherically relevant radical lifetimes can lead to significantly higher SOA yields in chamber studies (Surratt et al., 2010; Lin et al., 2012; Gaston et al., 2014). However, recent work has revealed that isoprene SOA formation can be suppressed when viscous organic coatings are present on acidified sulfate aerosol, as they impede the multiphase chemistry of IEPOX that yields additional SOA (Riva et al., 2016b, 2019; Zhang et al., 2018).
Observations using aerosol mass spectrometry (AMS) indicate that IEPOX-derived SOA can make up a significant fraction of organic aerosol in isoprene-rich environments, such as Borneo (23 %; Robinson et al., 2011), the Amazon (34 %; Chen et al., 2015) and the Southeastern US (33 %–40 %; Budisulistiorini et al., 2013, 2016; Rattanvaraha et al., 2017). Hu et al. (2015) compared previous AMS studies and found a magnitude lower average IEPOX-SOA signal in urban studies ( of 0.17 %) compared with those in isoprene-rich regions ( of 2.2 %). The average IEPOX-SOA concentration measured in Nanjing, a polluted city in eastern China, in August 2013 was 0.33 µg m−3 (Zhang et al., 2017b). This represented only 3.8 % of the total OA, indicating that there is limited formation of IEPOX under high-NOx conditions (average NOx of 21 ppb). He et al. (2018) found higher concentrations of low-NO isoprene SOA tracers (average of 121 ng m−3) than high-NO iSOA tracers (average of 9 ng m−3) at a regional background site (Wanqingsha) situated within the heavily polluted Pearl River Delta region. Only two high-NO iSOA tracers were measured (2-MG and 2-MG-OS), which could lead to a significant underestimate of the strength of the high-NOx pathway. Wang et al. (2018) measured a range of OSs at a regional site 38 km northeast of Beijing during May and June 2016. Isoprene-derived OSs ranged from 0.9 to 20 ng m−3 with a mean isoprene-derived OS concentration of 14.8 ng m−3. In both of these studies, the ratio of the average concentration of the commonly used OS tracers from the low-NO versus the high-NO pathways was close to 1.5 (2-MT-OS:2-MG-OS; Beijing was 1.47 and Wanqingsha was 1.57), indicating that low-NO oxidation chemistry can even play a significant role in iSOA formation in polluted environments.
SOAs formed from anthropogenic and other biogenic (monoterpenes and sesquiterpenes) sources have also been studied. Thousands of organic species including hundreds of OS and nitroxy OS (NOS) species have been identified in studies from a range of precursors using ultra-high-performance liquid chromatography coupled to ultra-high-resolution mass spectrometry equipped with electrospray ionization (UHPLC/ESI-HR-MS) from ambient aerosol samples (X. Wang et al., 2016, 2017). Brüggemann et al. (2019) quantified both monoterpene OS (MT-OS) and sesquiterpene OS (SQT-OS) species in Melpitz, Germany, and Wangdu, China, with authentic standards. They found median daytime concentrations of 52 MT-OS species of 12.15 and 38.19 ng m−3 for Melpitz and Wangdu respectively. For the five SQT-OS species, the median concentrations were 0.3 and 3.90 ng m−3 for daytime concentrations in Melpitz and Wangdu respectively, which are much lower values than the iSOA OS species quantified in this study. Riva et al. (2016c) identified OS species from the photooxidation of C10–C12 alkanes, which were then characterized in ambient aerosol samples collected in Lahore, Pakistan, and Pasadena, CA, USA. High concentrations of OS species were identified in Lahore, with the largest observed concentration arising from a cyclodecane OS species (C10H16O7S) with a concentration of 35.93 ng m−3.
The lack of molecular-level measurements of iSOA in highly polluted urban areas makes it difficult to determine the role of isoprene in summer haze episodes in Beijing. To investigate the formation of iSOA in Beijing, offline PM2.5 filter samples were collected during summer 2017 as part of the Atmospheric Pollution and Human Health programme (Shi et al., 2019). The filters were extracted and then screened using a sensitive and selective high throughput method based on UHPLC/ESI-HR-MS. High time resolution filter sampling allowed for the formation and evolution of iSOA to be studied, with observed concentrations strongly controlled by levels of anthropogenic pollutants.
2.1 PM2.5 filter sampling and extraction
Aerosol samples were collected between 18 May and 24 June 2017 at the Institute of Atmospheric Physics (IAP) in Beijing, China. This sampling was part of the Sources and Emissions of Air Pollutants in Beijing (AIRPOLL-Beijing) project, as part of the wider Atmospheric Pollution and Human Health in a Chinese Megacity (APHH-Beijing) programme (Shi et al., 2019). PM2.5 filter samples were collected using an Ecotech HiVol 3000 (Ecotech, Australia) high-volume air sampler with a selective PM2.5 inlet and a flow rate of 1.33 m3 min−1. Filters were baked at 500 ∘C for 5 h before use. After collection, samples were wrapped in foil, stored at −20 ∘C and then shipped to the laboratory for offline analysis. Samples were collected at a height of 8 m on top of a building in the IAP complex. Samples were collected every 3 h during the day (between approximately 08:30 and 17:30 LT) and then one sample was collected overnight (between approximately 17:30 and 08:30 LT). Hourly samples were also taken on certain high-pollution days towards the end of the sampling period. A number of 24 h samples were also collected using a Digitel high-volume PM2.5 sampler at the same location.
The extraction of the organic aerosol from the filter samples was based on the method in Hamilton et al. (2008). Initially, an eighth of the filter was cut up into roughly 1 cm2 pieces and stored in a vial. Next, 4 mL of LC-MS-grade H2O was added to the sample and left for 2 h. The samples were then sonicated for 30 min. A small subset (three) of the filter samples were also extracted via orbital shaker, and no appreciable difference was found in the concentrations of the iSOA tracers compared with sonication. Using a 2 mL syringe, the water extract was then pushed through a 0.22 µm filter (Millipore) into another sample vial. An additional 1 mL of water was added to the filter sample and then extracted through the filter to give a combined aqueous extract. This extract was reduced to dryness using a vacuum solvent evaporator (Biotage, Sweden). The dry sample was then reconstituted in 1 mL of 50:50 MeOH:H2O solution for offline chemical analysis.
2.2 Ultra-high-performance liquid chromatography tandem mass spectrometry (UHPLC-MS2)
The water-soluble fractions of the filter samples were analysed using UHPLC full-scan ddMS2 via an Ultimate 3000 UHPLC (Thermo Scientific, USA) coupled to a Q Exactive Orbitrap MS (Thermo Fisher Scientific, USA) with heated electrospray ionization (HESI). The UHPLC method uses a reversed-phase 5 µm, 4.6 mm ×100 mm, Accucore column (Thermo Scientific, UK) held at 40 ∘C. The mobile phase consists of LC-MS-grade H2O and 100 % MeOH (Fisher Chemical, USA). The H2O was acidified using 0.1 % formic acid to improve peak resolution. The injection volume was 2 µL. The solvent gradient was held for 1 min at 90:10 H2O:MeOH; changed linearly to 10:90 H2O:MeOH over 9 min; held for 2 min at this gradient before returning it to 90:10 H2O:MeOH over 2 min; and then held at 90:10 for the remaining 2 min, with a flow rate of 300 µL min−1. Due to the wide range of compounds studied, poor retention was observed for some species (retention time, RT, of less than 0.8 min). These species closely eluted to the dead time of the column where inorganic sulfate ions eluted (0.67 min). To check for ionization artefacts, an aqueous solution containing 20 ppm ammonium sulfate, 1 ppm 2-methyltetrol and 1ppm 2-methylglyceric acid was run under the same conditions as the filter samples to check for organosulfate formation (2-MT-OS and 2-MG-OS respectively). No MG-OS formation was observed, and less than 0.5 % conversion was seen for 2-MT. Therefore, this ruled out adduct formation for the two most important iSOA species, 2-MT-OS and 2-MG-OS; however, due to the lack of authentic standards and the complexity of the samples, adduct formation throughout the entire chromatogram could still be occurring. At this stage, there is not enough evidence to definitively say if adducts are forming or not. The mass spectrometer was operated in negative mode using full-scan ddMS2. The scan range was set between 50 and 750 m∕z, with a resolution of 70 000. The ESI voltage was 4 kV, with capillary and auxiliary gas temperatures of 320 ∘C. The number of the most abundant precursors for MS2 fragmentation per scan was set to 10. The samples were run in batches of 70, in a repeating sequence of 5 samples followed by one blank, and each filter sample was run only once. The calibrations were run separately after the samples were finished; the following sequence was used for the calibration runs: (3× same concentration) multiplied by the number of standards in the calibration curve from the lowest concentration to the highest followed by two blanks. The quantification method will be discussed in Sect. 3.3.
2.3 Construction of accurate mass library
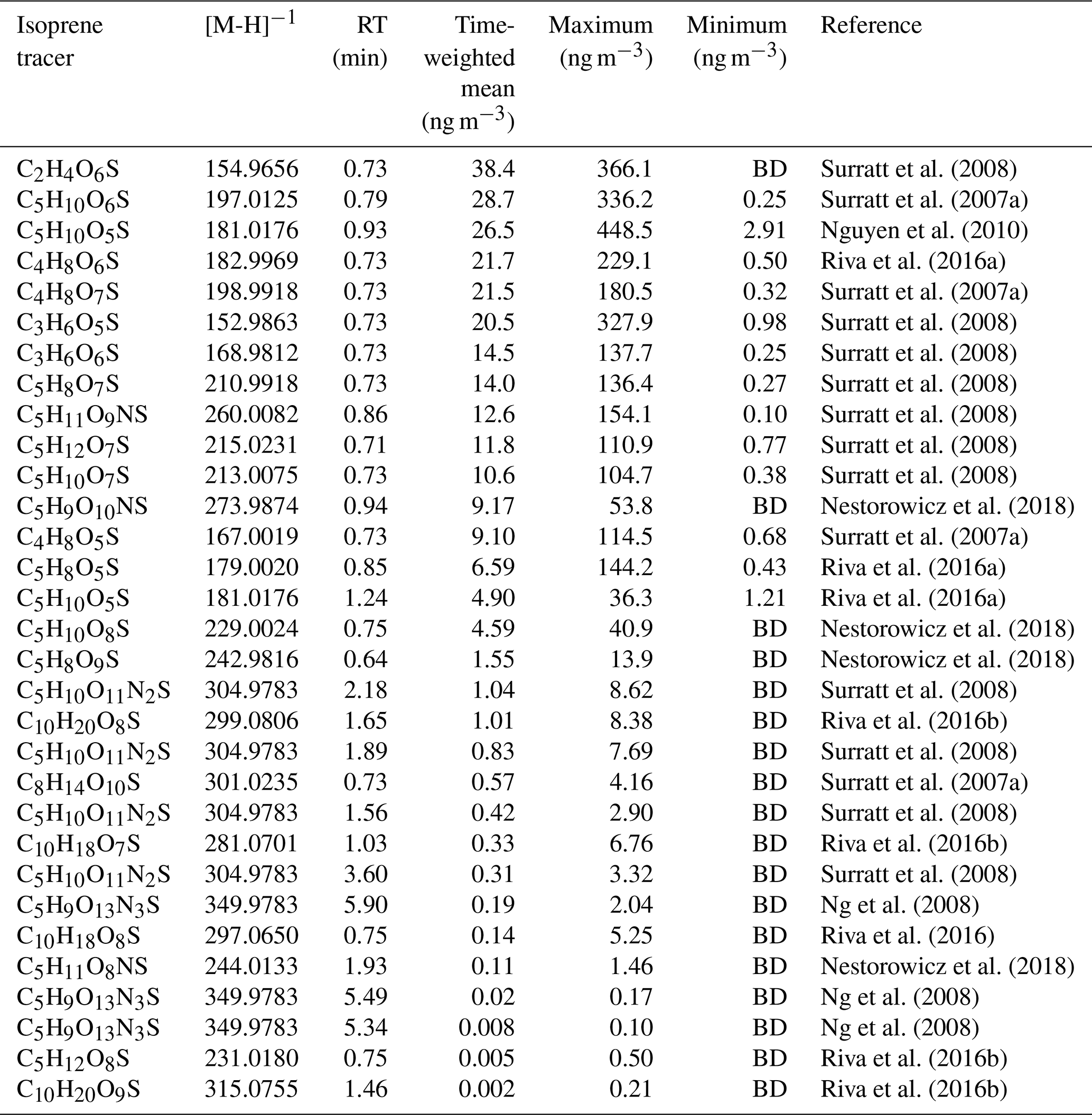
A mass spectral library was built using the compound database function in TraceFinder™ 4.1 General Quan software (Thermo Fisher Scientific, USA). Each compound was input into the compound library in the generic form: CcHhOoNnSs (where c, h, o, n and s represent the number of carbon, hydrogen, oxygen, nitrogen and sulfur atoms respectively). From the literature, species were identified, searched for in the ambient samples according to their accurate mass, and then the retention time (RT) of each isomer was obtained. Using previously observed iSOA products from the literature, extracted ion chromatograms were plotted for each m∕z value from a small subset of ambient samples, and the RT of the observed species/isomer was obtained. For most of the OS species in this study, the separation was not good enough to see individual isomers and only one peak was observed, which was added to the library. For the NOS species, individual isomers could be resolved, and each isomer was added to the library based on its RT. The accurate masses, RT and literature references for iSOA tracers are shown in Table 2.
2.4 Automated method for SOA tracer analysis
The UHPLC/ESI-HR-MS data for each ambient sample and standard were analysed using TraceFinder™ software. TraceFinder™ extracted the OS/NOS tracer peak areas from each ambient sample chromatogram using the library based on the RT and accurate mass. The mass tolerance of the method was set to 2 ppm, and the RT window was set to 30 s, although for species with multiple isomers present, the integration was checked to make sure the same peaks were not being integrated twice, and the window changed accordingly. The peak tailing factor was set to 2.0 to reduce the integration of the peak tails. The minimum signal-to-noise ratio (S/N) for a positive identification was set to 3.0. Using the output from TraceFinder™, an in-house R code script was developed to combine the identified species and peak areas with the correct filter sampling date/time midpoint and volume of air sampled. Calibration curves from the standards were then obtained (as discussed in Sect. 3.3), and the intercept and gradient were inputted to quantify the iSOA tracer concentrations in the extract. These quantified values were then converted to the mass on the whole filter, divided by the volume of air sampled for that filter sampling period and converted to units of nanograms per cubic metre (ng m−3). Higher time resolution data were averaged to the filter sampling times. It should be noted that MS2 was used to check that the iSOA species fragmented to give typical OS fragment ions.
2.5 Hydrophilic liquid interaction chromatography (HILIC)
A subset of filters (n=15) were also analysed at the University of North Carolina (UNC) using a newly developed HILIC method interfaced to high-resolution quadrupole time-of-flight mass spectrometry equipped with ESI (i.e. HILIC/ESI-HR-QTOFMS; Cui et al., 2018). Briefly, filters were extracted with 22 mL of LC/MS-grade methanol by 45 min of sonication; the samples were first extracted for 23 min, the water bath was replaced with cool water, and then the samples were extracted again for 22 min. This was done to make sure that the water bath contained within the sonicator did not exceed 30 ∘C. Extracts were filtered through polypropylene membrane syringe filters in order to remove insoluble filter fibres and soot particles. The extracts were dried under a gentle stream of nitrogen gas. Dried methanol extracts were reconstituted with 150 µL of 95:5 (v∕v) LC/MS-grade acetonitrile/Milli-Q water. Operating details of the HILIC/ESI-HR-QTOFMS used for these samples are also summarized by Cui et al. (2018).
2.6 Gas chromatography–mass spectrometry
Details of the measurement procedure used can be found elsewhere (Fu et al., 2010). Briefly, filter samples were extracted with dichloromethane/methanol (2:1 v∕v), filtered through quartz wool packed into a Pasteur pipette, concentrated using a rotary evaporator under vacuum and blown down to dryness with pure nitrogen gas. The extracts were derivatized and diluted with n-hexane containing the internal standard prior to GC-MS analysis. Separation was performed on a fused silica capillary column (DB-5MS: µm). The MS detection was conducted in electron ionization (EI) mode at 70 eV, scanning from 50 to 650 Da. Individual compounds were identified via comparison of the mass spectra with those of authentic standards or literature data. 2-Methylglyceric acid, C5-alkene triols (the sum of cis-2-methyl-1,3,4-trihydroxy-1-butene, trans-2-methyl-1,3,4-trihydroxy-1-butene and 3-methyl-2,3,4-trihydroxy-1-butene) and 2-methyltetrols (the sum of 2-methylthreitol and 2-methylerythritol) were quantified using the response factor of meso-erythritol. Field blank filters were treated as the real samples for quality assurance. Target compounds were not detected in the blanks.
2.7 High-resolution aerosol mass spectrometry measurements
The size-resolved non-refractory submicron aerosol species at the same site were measured by an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) at a time resolution of 5 min. The elemental ratios of hydrogen to carbon (H:C) and oxygen to carbon (O:C) of OAs were determined, and the sources of OAs were analysed with positive matrix factorization. Seven OA factors were identified in summer, including two primary factors; hydrocarbon-like OA (HOA); cooking OA (COA); and three oxidized OA factors with increasing degrees of oxidation, OOA1 (), OOA2 () and OOA3 ().
2.8 Iodide CIMS
A time-of-flight chemical ionization mass spectrometer (ToF-CIMS; Lee et al., 2014; Priestley et al., 2018) using an iodide ionization system coupled to a filter inlet for gases and aerosols (FIGAERO) was deployed here to make near-simultaneous, real-time measurements of both the gas- and particle-phase chemical composition. The instrument was originally developed by Lopez-Hilfiker et al. (2014) and is described and characterized in more detail by Bannan et al. (2019). The experimental set-up employed by the University of Manchester ToF-CIMS is described in Zhou et al. (2018). Only gas-phase data are presented herein.
Field calibrations were regularly carried out using a known concentration of formic acid in gas mixtures produced in a custom-made gas-phase manifold. A range of other species were calibrated for after the campaign, and relative calibration factors were derived using the measured formic acid sensitivity during the in situ calibrations (Bannan et al., 2015). Offline calibrations after the field work campaign were performed specific to the isoprene oxidation species observed here. IEPOX (C5H10O3) synthesized by the University of North Carolina, Department of Environmental Sciences and Engineering, was specifically calibrated for. Known concentrations were deposited on the FIGAERO filter in various amounts and thermally desorbed using a known continuous flow of nitrogen over the filter. For the isoprene nitrate, C5H9NO4, there was no direct calibration source available, and concentrations using the calibration factor of C5H10O3 are presented here.
2.9 Gas-phase measurements
Additional gas-phase measurements were collected at the site from an elevated inlet at 8 m. Data included nitrogen oxide (NO) measured by chemiluminescence with a Thermo Scientific Model 42i NOx analyser and nitrogen dioxide (NO2) measured using a Teledyne Model T500U cavity-attenuated phase-shift (CAPS) spectrometer. The sum of the NOy species was measured using a Thermo Scientific Model 42C NOx analyser and a heated molybdenum converter at the sample inlet. The molybdenum converter reduces NOy compounds to NO and allows for measurement by chemiluminescence. Ozone (O3) was measured using a Thermo Scientific Model 49i UV photometric analyser. All instruments were calibrated throughout the measurement period using a “zero” or “background” calibration with a Sofnofil/charcoal trap. Span (high-concentration) calibrations were carried out using gas standards. Both the Thermo Scientific 42i and 42C instrument calibrations are traceable to the National Physical Laboratories (NPL) NO scale. The meteorological variables of wind speed, wind direction, relative humidity (RH) and temperature were measured at 102 m on the IAP 325 m meteorological tower.
Observations of VOCs were made using a dual-channel GC with flame ionization detection (DC-GC-FID). Air was sampled at 30 L min−1 at a height of 5 m, through a stainless-steel manifold ( internal diameter). Next, 500 mL subsamples were taken, dried using a glass condensation finger held at −40 ∘C and then pre-concentrated using a Markes Unity 2 pre-concentrator on a multi-bed O3 precursor adsorbent trap (Markes International Ltd). These samples were then transferred to the GC for analysis following the methods described by Hopkins et al. (2011).
Further details on the following additional gas-phase instrumentation can be found in the Supplement and in Shi et al. (2019). Isoprene was also measured at a height of ∼102 m using a Voice200 selected ion flow tube mass spectrometer (SIFT-MS, Syft Technologies, Christchurch, New Zealand). OH, HO2 and RO2 concentrations were measured using fluorescence assay by gas expansion (FAGE; Whalley et al., 2010, 2018), and NO3 concentrations were measured using broadband cavity enhanced absorption spectrometry (Le Breton et al., 2014).
The field campaign was conducted at the Institute of Physics, Beijing, situated between the third and fourth ring roads (Shi et al., 2019). The site is typical of central Beijing: it is surrounded by residential and commercial properties, and is near several busy roads. It is also close to several green spaces, including a tree-lined canal to the south and the Olympic forest park to the northeast, that provide sources for local isoprene emissions.
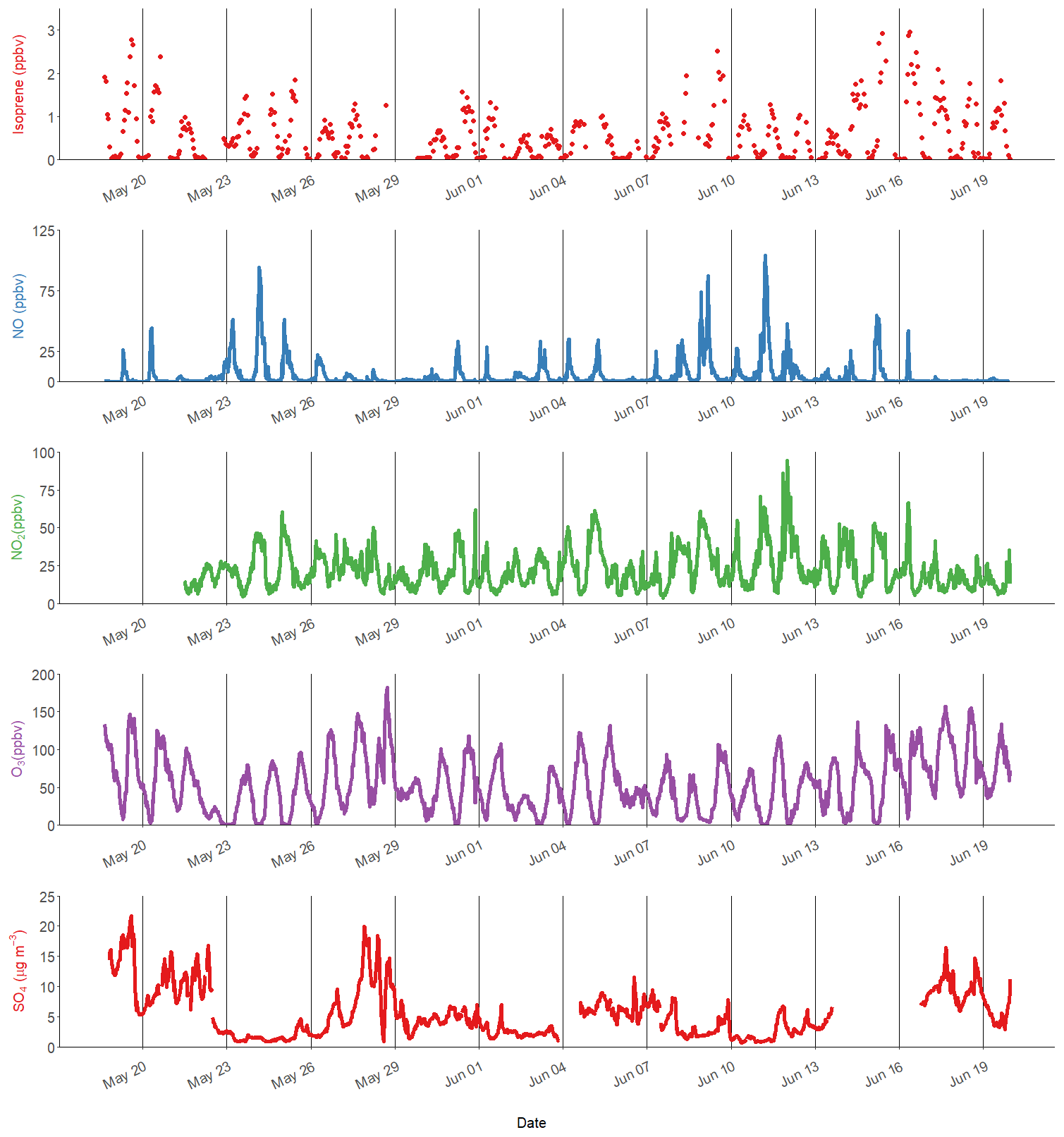
Figure 1Time series of isoprene, nitric oxide (NO), nitrogen dioxide (NO2), ozone (O3) and particulate sulfate (SO4). The black lines represent midnight every 72 h.
3.1 Isoprene gas-phase concentrations and loss processes
Isoprene was measured hourly using the DC-GC-FID between 18 May and 20 June 2017, and the observed concentrations are shown in Fig. 1 alongside NO, NO2 and O3. The mean mixing ratio of isoprene was 0.53 ppb, with a maximum of 2.9 ppb on 16 June 2017. The ambient temperature ranged from 16 to 38 ∘C. Daytime isoprene mixing ratios increased with temperature, with all isoprene mixing ratios above 1 ppb occurring when the temperature was above 25 ∘C. The average diurnal profile of isoprene in Fig. 3a shows low values overnight (< 50 ppt) with a rapid increase at 06:00 LT that reaches a maximum of around 1 ppb by the afternoon. The mixing ratio rapidly decreased after 18:00 LT and returned to very low values by around 22:00 LT. There was a strong correlation between the isoprene mixing ratio measured at 8 m using the DC-GC and at 102 m using the SIFT-MS (R2=0.77). Therefore, the SIFT-MS measurements were used to investigate the correlation with iSOA tracers when no DC-GC data were available. The slope of the linear fit between the two datasets was 0.67, indicating a loss of around 30 % of the isoprene during transport from the ground to the tower (100 m).
Using the average observed diurnal profiles of the main atmospheric oxidants, OH, O3 and NO3 (shown in Supplement Fig. S1), and isoprene (Fig. 3a), the isoprene loss rate was calculated (rate of loss =kox [oxidant][isoprene]) and is shown in Fig. 4a. The IUPAC rate constants that were used in the calculation for NO3, O3 and OH were , 1.27×1017 and 1×1010 cm3 molecule−1 s−1 respectively. The percentage contribution of each oxidant to the average diurnal isoprene loss rate is shown in Fig. 4b. During the day, OH is responsible for over 90 % of isoprene loss, with NO3 becoming relatively more important from 18:00 until around 03:00 LT, although the amount of isoprene available to react rapidly decreases during this time period. OH chemistry is still an important loss route at night (> 30 %) owing to night-time OH sources, such as the ozonolysis of alkenes (Lu et al., 2014). The loss of isoprene via ozonolysis, however, is a minor route, contributing less than 15 %. During the daytime (10:00–15:00 LT), the lifetime of isoprene was found to be around 20 min on average, increasing to a maximum of around 6 h at 03:00 LT. While the high levels of oxidants lead to a short isoprene lifetime during the day, the ambient concentrations of isoprene are still maintained at the parts per billion level. This indicates that there are significant local emissions of isoprene impacting the measurement site; therefore, there is a high potential for the formation of iSOA in this urban environment.
3.2 Anthropogenic tracers
A range of gas-phase anthropogenic tracers were measured during the campaign, as discussed in Shi et al. (2019). Figure 1 shows the time series of NO, NO2, O3 and particulate sulfate during the part of the campaign analysed in this study. Table 1 shows the average, maximum and minimum concentrations of these anthropogenic pollutants. NO mixing ratios ranged from less than 10.1 to 104 ppbv with a mean concentration of 5.1 ppb during the filter sampling period. The highest concentrations generally occurred in the morning between 04:00 and 07:00 LT and steadily decreased during the day. On some days, the mixing ratio of NO was very low in the afternoon as a result of reaction with O3 and other unknown sinks (Newland et al., 2020). The mean mixing ratio of NO2 was 22.3 ppbv, which was much higher than NO, with a range from 3.7 to 95 ppbv. NO2 peaked between 06:00 and 07:00 LT and decreased to a minima at 14:00 LT before steady increasing until about 20:00 LT. High afternoon concentrations of O3 (> 80 ppb) were found on most days, with a maximum observed mixing ratio of 182 ppbv. Night-time O3 levels were much lower due to reduced photochemistry and reaction with NO, although O3 levels were maintained above 40 ppbv on some nights, as shown in Fig. 1. Particulate sulfate concentrations measured by AMS are also shown in Fig. 1. Sulfate ranged from 0.7 to 21.7 µg m−3 with an average of 5.5 µg m−3. The time series shows a number of periods of high sulfate concentrations, and these generally matched periods of increased PM2.5 (see Fig. 9). Figure 2 shows the wind direction-dependent concentrations of particulate sulfate for the sampling period in a pollution rose plot (R, openair package). There is a strong source of sulfate from the south of the sampling site, which is enhanced under the highest wind speeds. Previous studies have shown a strong source of pollution from the southwest of Beijing, which is where many industrial factories are located (Wang et al., 2005).

Figure 2Pollution rose plot (openair) of particulate sulfate measured by AMS for the sampling period, highlighting the wind conditions under which the highest concentrations of sulfate occur.
Table 1Supplementary anthropogenic pollutants measured during the sampling period analysed in this study.
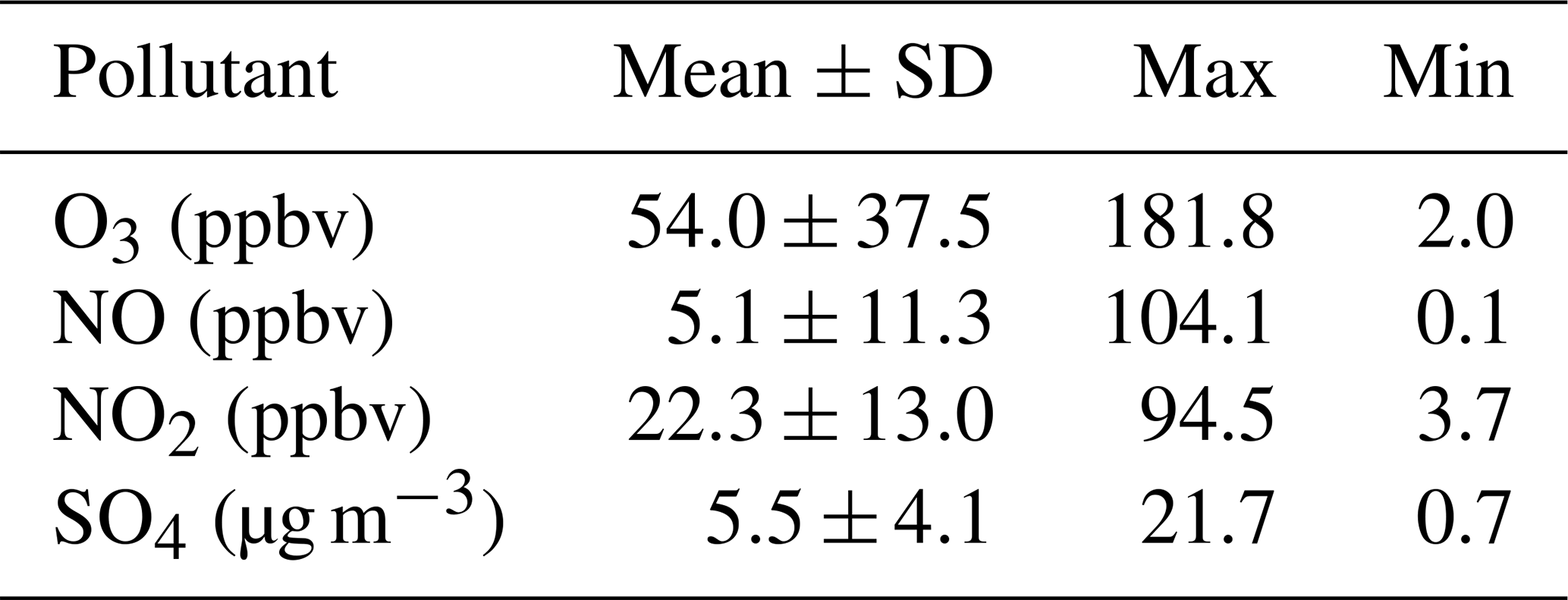
Table 2Molecular formulas, negative ion masses, retention times (RT) and time-weighted means (ng m−3) for the entire sampling period as well as the original reference where each proposed iSOA tracer was found. BD denotes “below detection”. The estimated uncertainties are 60 %, as discussed in Sect. 3.4, accounting for the use of the matrix correction factors.
3.3 Isoprene SOA in Beijing
Using the high throughput screening method described, the peak areas of 31 potential isoprene-derived OSs and NOSs, which are known iSOA tracers, were measured in 132 PM2.5 filter extracts. The full list of iSOA tracers, along with their measured m∕z values and molecular formulas, is shown in Table 2, ordered by descending average concentration (weighted by filter sampling time and reported in nanograms per cubic metre) during the campaign. The isoprene SOA tracers identified in this study are correlated towards themselves as well as common anthropogenic tracers in a correlation plot (R, openair, corPlot), which is shown in Fig. 5. The correlation plot highlights the correlations of the iSOA tracers to each other as well as the moderate to strong correlations towards some of the anthropogenic pollutants, as discussed in further sections.
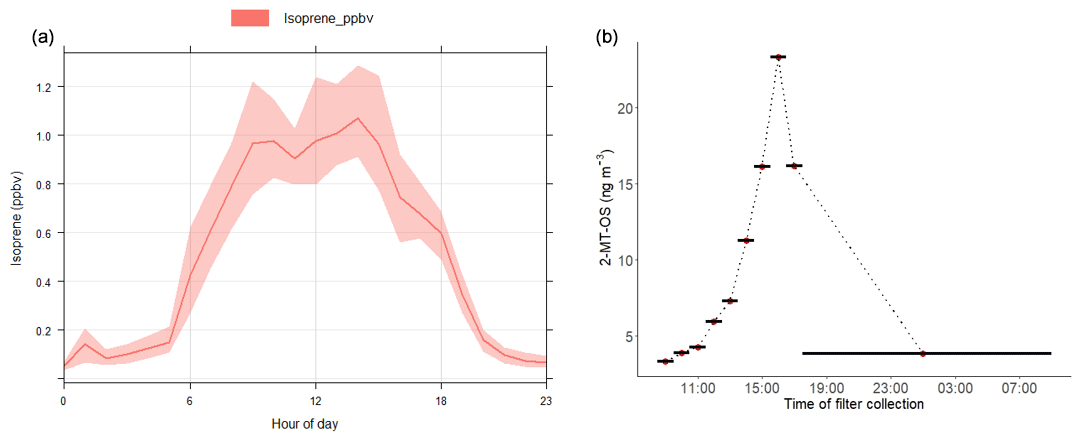
Figure 3(a) Average diurnal profile of the isoprene mixing ratio measured using DC-GC-FID. (b) Diurnal profile of 2-methyltetrol sulfate (2-MT-OS) in particulate matter (PM2.5) collected on filters hourly over the period from 11 to 12 June 2017. Black lines indicate the length of the filter sampling period.
3.4 Quantification of isoprene OS tracers
Initially, two synthesized isoprene-derived OS standards (2-MT-OS and 2-MG-OS; Cui et al., 2018; Rattanavaraha et al., 2016) were used to produce calibration curves. Both standards gave strong linear calibration curves (R2 of 0.980 and 0.996 respectively) across an appropriate range of concentrations for the peak areas in the samples. The gradient obtained for the 2-MT-OS standard was ∼4 times higher than that of the 2-MG-OS, as shown in Fig. S2. To investigate the potential for matrix effects from the large amounts of inorganic sulfate, nitrate and other particulate components that co-elute due to the poor retention of OS in reversed-phase UHPLC, standard addition calibrations were used. Five-point standard addition calibrations were run on six different filter extracts, covering both daytime and night-time samples during periods of both high and low concentrations of iSOA species. Thus, this provides a representative sample of filters for the entire sampling period. A total of 50 µL of filter sample extract and 50 µL of the calibrant solution were combined, giving a dilution factor of 2. The five-point calibration range of standard added to each sample was between 0 and 3 ppm for 2-MG-OS and between 0 and 1 ppm for 2-MT-OS. Two examples of the standard addition calibrations are shown in Figs. S3 (2-MG-OS) and S4 (2-MT-OS), with good linear fits observed (R2 of 0.997 and 0.997 respectively). A strong matrix effect was observed for 2-MT-OS: the concentration measured by standard addition calibration was 8.6–10 times higher than that measured using the external calibration, which was carried out on the same day. In contrast, 2-MG-OS showed a much lower matrix effect: the concentrations were only 1.1–1.5 times higher when using the standard addition calibration. A further comparison using camphorsulfonate, which has a longer RT (3.74 min) and, therefore, does not experience high inorganic ion concentrations in the source, showed no matrix effects when using standard addition. Tables S1 and S2 in the Supplement provide a comparison of the concentrations calculated from the standard additions and the two external calibrations. Table S1 shows the concentration of 2-MT-OS in the three filter sample extracts (144, 204 and 208) calculated via the standard addition of 2-MT-OS to the filter sample extract and via the external calibrations using both 2-MT-OS and 2-MG-OS. The ratio of the standard addition to the external calibrations then gives an estimate of the under- or overestimation that the external calibrations cause when calculating the concentration of 2-MT-OS in the samples. Both of the external calibrations would lead to an underestimation of the concentration of 2-MT-OS in the filter samples. 2-MG-OS provided a closer quantification of 2-MT-OS in the samples, with an average factor of 2.3 underestimation, whereas the 2-MT-OS external calibration gives a sample concentration that is a factor of 10 lower than the concentration determined by standard addition.
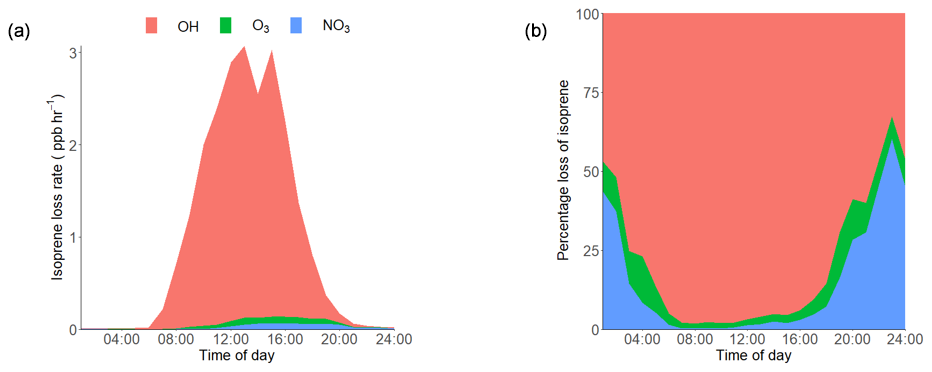
Figure 4(a) Diurnal loss rate of isoprene calculated using the measured average diurnal profiles of isoprene, OH, NO3 and O3. (b) Average diurnal percentage loss of isoprene from reactions with OH, O3 and NO3 radicals. The IUPAC rate constants used for the calculations are as follows: NO3, molecule−1 s−1; O3, 1.27×1017 molecule−1 s−1; OH, 1×1010 cm3 molecule−1 s−1 (Atkinson et al., 2006).
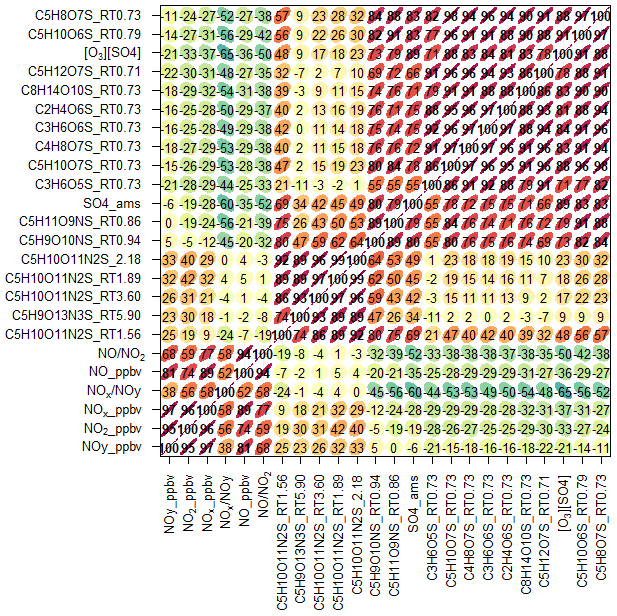
Figure 5Correlation plot (R, openair, corPlot) highlighting the correlations between known iSOA tracers and anthropogenic pollutants. The numbers represent the R correlations between the two species; the redder, more elongated circles highlight a higher correlation.
It is not realistic to carry out standard addition calibrations for all samples and all SOA tracers. When the 2-MG-OS external calibration was used to predict the 2-MT-OS concentrations during the standard addition experiments, the concentrations were within a factor of 1.5–2.5. Therefore, the 2-MG-OS external calibration was used as a proxy for all isoprene SOA tracers, with scaling factors applied to account for matrix effects (1.33 for 2-MG-OS, 2.33 for 2-MT-OS and an average of 1.83 used for all other OSs). Therefore, we estimate an uncertainty on our measured concentrations of 60 %, this uncertainty was calculated to account for the difference in the measured correction factors used when correcting for the matrix effects. The uncertainty was calculated as 2σ of the six values used to calculate the average correction factor of 1.83. The matrix effects identified in this study are likely due to the extracted samples being a complex mixture of different compounds, including a high proportion of inorganic ions that are extracted into water. This is likely to change the surface tension of the droplet produced in the ionization source and the ion distribution. Further work is needed to fully understand the reasons for this. Without these additional standard addition calibrations, the iSOA concentrations would have been largely underestimated. The dinitrate and trinitrate NOS species eluted after the sulfate peak (RT > 1.6 min). In the absence of authentic standards for these species, camphorsulfonic acid was used as a proxy for calibration. This work highlights an additional difficulty of calibration when using ESI-MS to study OSs and indicates that future studies using reversed-phase LC (RPLC) should consider the impacts of matrix effects.
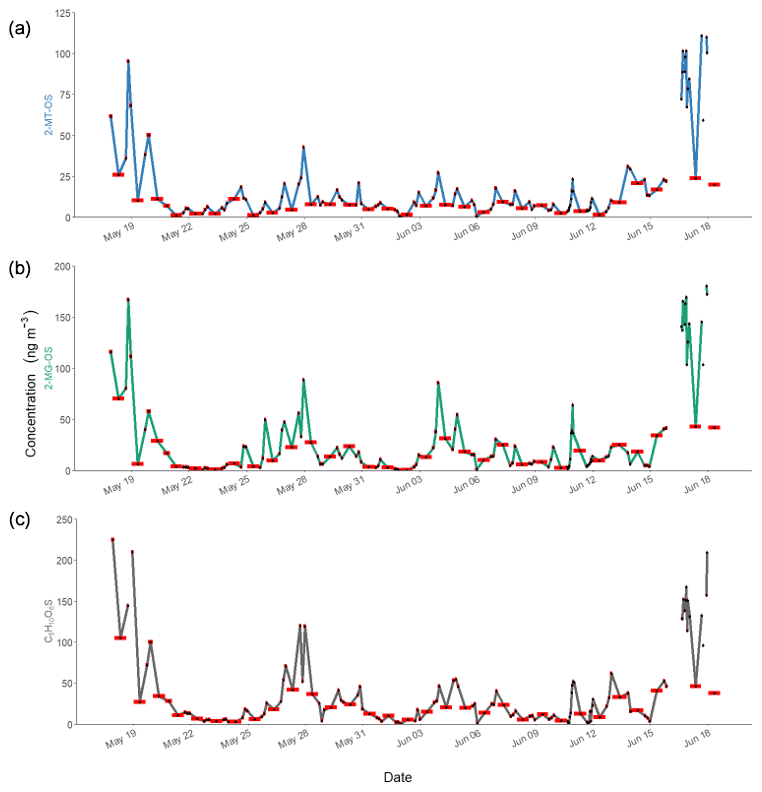
Figure 6Time series of observed concentrations of iSOA tracers in Beijing during APHH for (a) 2-MT-OS (C5H12O7S), (b) 2-MG-OS (C4H8O7S) and (c) C5H10SO6. The red bars indicate the length of the sampling period. The individual sample times will be given in the accompanying dataset (https://doi.org/10.15124/7c1a54dd-ce72-4b1a-bed1-812cf25bc407; Hamilton and Bryant, 2019).
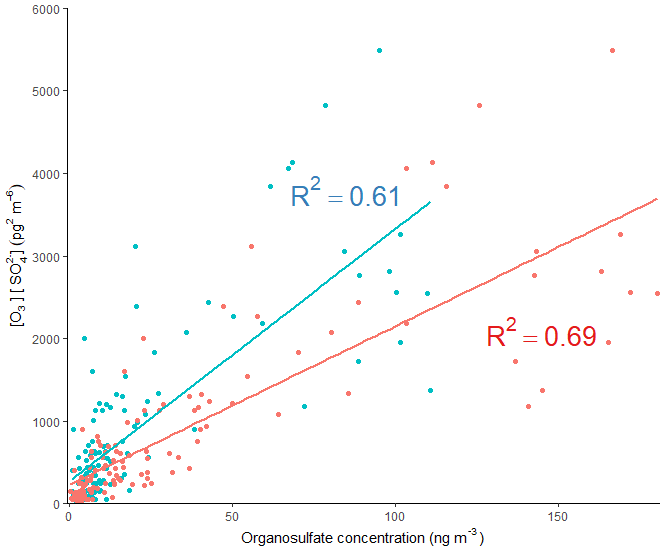
Figure 7Plot of 2-MT-OS (C5H12O7S, blue) and 2-MG-OS (C4H8O7S, red) concentrations versus [O3][SO4]. The high time resolution data (O3 and AMS ) have been averaged to the filter sampling time. The line was calculated with the “stat_smooth” function in the “ggplot2” R package, using the “lm” method.
3.5 Organosulfates
3.5.1 2-Methyltetrol OS (2-MT-OS)
The 2-MT-OS (C5H12SO7) formed from the uptake of IEPOX into the particle phase is often used as a marker of low-NO isoprene photochemistry (Wennberg et al., 2018). The time series of 2-MT-OS is shown in Fig. 6a. The particle concentration ranged from 0.7 ng m−3 to a maximum of 111 ng m−3 with a mean concentration of 11.8 ng m−3. The mean concentrations of 2-MT-OS and 2-MG-OS are compared to observations from previous studies in Table 3. The mean concentration observed in Beijing was much lower than those observed in the Amazon (Riva et al., 2019) and the Southeastern US (Budisulistiorini et al., 2015; Hettiyadura et al., 2019), but it was higher than summertime observations at polluted regional sites in China (Wang et al., 2018; He et al., 2018). The lower amounts of IEPOX-derived SOA result in an average AMS in Beijing during the APHH project of only 0.2 %, which is similar to observations in other urban studies (Hu et al., 2015).
Table 3Isoprene CHO tracer concentrations measured via GC-MS using 24 h samples between 22 May and 22 June 2017. 2-MTs is equal to the sum of 2-methylthreitol and 2-methylerythritol, and the C5-alkene triols are equal to the sum of cis-2-methyl-1,3,4-trihydroxy-1-butene, 3-methyl-2,3,4-trihydroxy-1-butene and trans-2-methyl-1,3,4-trihydroxy-1-butene.
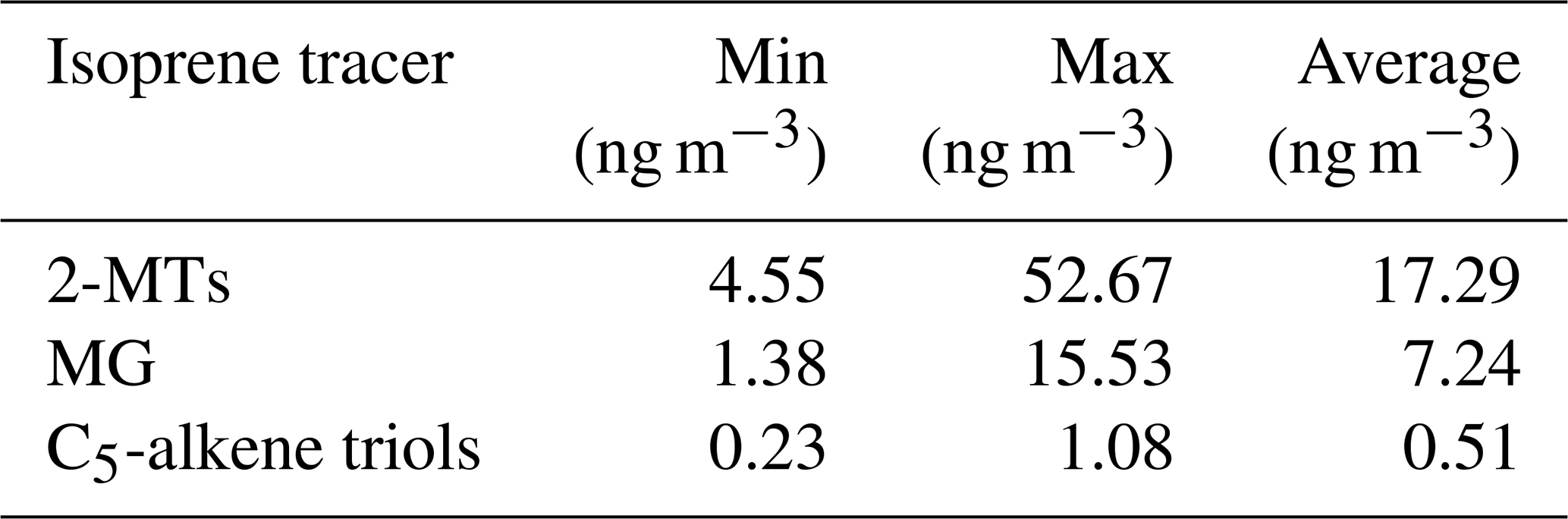
Table 4Comparison of iSOA tracer concentrations and ratios from previous studies in the Amazon, the Southeastern US and China.

* Selected sample not an average concentration.
Hourly samples were collected on selected high-pollution days and used to obtain information on the diurnal evolution of the iSOA tracers. The findings on these days are consistent with the 3-hourly data. The particulate 2-MT-OS measured by UHPLC-MS, on 11–12 June 2017, had a strong diurnal profile (Fig. 3b), peaking in the late afternoon (between 15:30 and 18:30 LT) and displaying a minimum overnight. This is consistent with the average diurnal profile of the gas-phase precursors IEPOX+ISOPOOH (C5H12O3) measured using the I-CIMS (Fig. S5). High levels of O3 were observed in the afternoon (up to 180 ppb), leading to relatively low levels of NO observed for a highly polluted environment (in some cases below 500 ppt). Thus, although the mixing ratio of NOx was high, less than 2 % was in the form of NO on most afternoons. High levels of peroxy radicals were observed, with mean afternoon concentrations of HO2 and RO2 of around 3×108 and 1.5×109 molecule cm−3 respectively. Zero-dimensional box modelling indicates that up to 35 % of the isoprene-derived RO2 radicals can react with HO2 in the afternoon on some days (Newland et al., 2020). Thus, the diurnal profile in Fig. 3b, measured in samples during the measurement period, suggests that IEPOX was formed at this urban location by the reaction of OH with local isoprene emissions, with a fraction of the RO2 radicals formed reacting with HO2 rather than NO, and subsequent uptake to aerosol forming 2-MT-OS. OH + isoprene hydroxynitrate also has a small yield of IEPOX (Jacobs et al., 2014). The average diurnal profile of isoprene hydroxynitrates (C5H9NO4) in the gas phase measured using the I-CIMS peaks at around 11:00–12:00 LT followed by a reduction during the afternoon into the evening/night (Fig. S6). This is likely to be a result of the relatively low levels of NO during the afternoon, which will reduce isoprene nitrate formation from RO2+NO reactions; thus, isoprene hydroxynitrates are unlikely to be a significant source of 2-MT-OS in Beijing.
The 2-MT-OS showed a moderate correlation with particulate sulfate (R2=0.44) and a weak anti-correlation with photochemical age, which was estimated using the NOx∕NOy ratio (R2=0.23), as shown in Fig. 5. All correlations between species are shown in Fig. 5. By taking the product of the concentration of O3, as a proxy of photochemistry, with the amount of particulate sulfate measured using AMS, [O3][pSO4], a much stronger correlation with 2-MT-OS was observed (R2=0.61), as shown in Fig. 7. This observation highlights the role of both local photochemistry and particulate sulfate mass in the formation of 2-MT-OS (Fig. 7). The correlation of [O3][pSO4] with 2-MT-OS is likely to be weaker at longer photochemical ages when the O3 concentration is not directly related to the photochemical formation of the OS. Again, this highlights the strong role of local photochemistry in the production of low-NO iSOA (2-MT-OS) in Beijing. Elevated levels of 2-MT-OS were observed at the start and end of the measurement period which were influenced by strong southwesterly winds. There were also elevated isoprene concentrations (up to 2.9 ppb) and high particulate levels. Therefore, these spikes in 2-MT-OS could be a result of either higher 2-MT-OS in regional aerosol transported to the site or a high isoprene emission source to the southwest of the site (i.e. producing IEPOX locally) that then reacts with increased regional sulfate pollution. The I-CIMS data show that the IEPOX/ISOPOOH (Fig. S5 and Newland et al., 2020) signal increases during the afternoon as the NO levels drop to below 1 ppb. The low NO levels indicate that up to 30 % of the isoprene peroxy radical from OH oxidation can react with HO2 rather than NO at this site, meaning IEPOX can be formed locally (Newland et al., 2020). There is also likely to be a regional source of IEPOX and 2-MT-OS, suggesting both local and regional anthropogenic influences.
Analysis of the 2-MT-OS isomer distribution using HILIC/ESI-HR-QTOFMS, on a subset of 15 samples, indicates that β-IEPOX is the dominant ambient IEPOX isomer, which is in line with other recent observations (Cui et al, 2018; Krechmer et al., 2016) see Fig. S7). The MT-OS derived exclusively from δ-IEPOX-OS isomers could not be observed in any of the samples. The four IEPOX-OS isomers in Fig. S7 showed similar temporal trends, although small changes in the relative proportions were observed. The sum of peak areas from the 2-MT-OS isomers measured by HILIC and the quantified 2-MT-OS (sum of isomers) measured via UHPLC/ESI-HR-MS were compared and showed a high degree of correlation (R2 of 0.84), even though the two methods used different solvents. The agreement indicates that the UHPLC/ESI-HR-MS method captures the sum of the isomers and that there is no evidence of ion source-induced artefacts.
3.5.2 2-Methyl glyceric acid OS (2-MG-OS)
The most commonly targeted SOA tracer for high-NO isoprene chemistry is 2-methylglyceric acid (2-MG) and its derivatives. As such, this tracer is the result of a direct biogenic–anthropogenic interaction. Two observed SOA tracers related to this chemistry are the OS derivatives of 2-methylglyceric acid (2-MG-OS) and the unresolved C8 dimers of 2-MG-OS (C8H14SO10) that have been identified previously in chamber-derived iSOA (Surratt et al., 2006, 2010). 2-MG-OS had an average concentration during the campaign of 21.5 ng m−3, ranging from 0.3 to 180.5 ng m−3, with the time series shown in Fig. 6b. These values are within the range of 2-MG-OS measured in other urban locations (Q. T. Nguyen et al., 2014; Rattanavaraha et al., 2016; Hettiyadura et al., 2019). However, these concentrations are considerably higher than those previously observed at two Chinese regional background sites (Wang et al., 2018; He et al., 2018). At these locations, the ratio of the low-NO to high-NO isoprene OS tracer average concentrations was close to 1.5 (2-MT-OS:2-MG-OS; Beijing was 1.47 and Wanqingsha was 1.57). However, in central Beijing, this ratio was considerably lower (2-MT-OS:2-MG-OS was 0.55), reflecting the higher proportion of RO2 radicals reacting with NO at this location compared with the regional measurements. The ratio of 2-MT-OS:2-MG-OS observed in Beijing is compared to previous studies in Table 3 and is considerably lower than measurements taken in a range of isoprene-dominated environments (Southeastern US showed a 2-MT-OS:2-MG-OS ratio of 17, Budisolistiorini et al., 2015; the Amazon showed a 2-MT-OS:2-MG-OS ratio of 13–118, Glasius et al., 2018; Atlanta showed a 2-MT-OS:2-MG-OS ratio of 33, Hettiyadura et al. 2019) reflecting the strong impact of urban NO emissions on iSOA formation. Future work will investigate how to use these ratios to quantify the effect of NO emissions on iSOA formation in different regions.
The mean concentration of the 2-MG-OS dimer (C8H14SO10) was 0.57 ng m−3. A strong linear relationship was observed between the 2-MG-OS monomer and dimer concentrations (R2=0.83) with a dimer to monomer ratio of 0.02. The formation of oligomers from reactions of 2-MG and HMML has been shown to be reduced in chamber experiments under humid conditions (Schwantes et al., 2019; Nestorowicz et al., 2018). The average RH during the afternoon of the campaign was ∼40 %, which may account for the relatively low formation of the dimer OS compared with the monomer (see Fig. S8).
The diurnal profile of the 2-MG-OS, as shown in Fig. 8, was similar to the 2-MT-OS, peaking during the early afternoon samples but with an enhanced signal at night. There was also a strong correlation between these two species (R2=0.92) during the campaign. The 2-MG-OS showed a stronger correlation with particulate sulfate (R2=0.52) than 2-MT-OS (R2=0.44), and there was also a weak anti-correlation with photochemical age (R2=0.28). A strong correlation was also observed for 2-MG-OS with [O3][pSO4] (R2=0.69), as shown in Fig. 7, highlighting that formation is dependent on both photochemistry and the sulfate aerosol availability.
3.5.3 Other isoprene-related OSs and NOSs
A total of 24 additional OSs species, with molecular formulae consistent with iSOA tracers seen in chamber experiments, were also observed in Beijing (as shown in Table 2). For C5 compounds, the most abundant species were C5H10SO6 and C5H10SO5, with mean concentrations of 28.7 and 26.5 ng m−3 respectively. The identity of the OS at m∕z 182 (C5H10SO5) is currently unknown, and the product ion MS provides little additional information other than sulfate-related fragment ions at m∕z 97 and m∕z 80. The OS at m∕z 198 (C5H10SO6) was identified as an IEPOX-related OS in chamber experiments by Nestorowicz et al. (2018), although at relatively low concentrations compared with the 2-MT-OS (1 %–4 %). This is very different to the observed ratio in Beijing, where the C5H10SO6 average concentration was more than double that of 2-MT-OS, as shown in Fig. 6c. This compound showed a strong correlation with 2-MT-OS (R2=0.77), but it is currently unclear why this compound is the most abundant C5 species. The molecular weight of this species is 18 Da (-H2O) lower than 2-MT-OS, which may indicate that it is a dehydration product enhanced under acidic aerosol conditions. In addition, this species may also be enhanced if it is formed from additional VOC precursors.
Potential low-NO iSOA tracers, seen in chamber experiments, correlated strongly with the 2-MT-OS, including unresolved isomers of cyclic hemiacetals [C5H10SO7] (R2=0.92) and lactones [C5H8SO7] (R2=0.83) (Spolnik et al., 2018). These compounds were similar in concentration to the 2-MT-OS, with lactones at MW 212 having a mean concentration of 14 ng m−3 and cyclic hemiacetals at MW 214 having a mean concentration of 10.6 ng m−3. These compounds were also observed to be the dominant type of isoprene-derived OSs in Atlanta, Georgia, although concentrations were ∼15 times lower than the observed 2-MT-OS (Hettiyadura et al., 2019).
Additional small OS compounds, previously identified during high-NO chamber experiments, were also observed in Beijing, including (in order of decreasing concentration) glycolic acid sulfate (C2H4SO6, mean of 38.4 ng m−3), hydroxyacetone sulfate (C3H6SO5, mean of 20.5 ng m−3) and lactic acid sulfate (C3H6SO6, mean of 14.5 ng m−3) (Surratt et al., 2007, 2008). These concentrations are in line with measurements made in other urban locations (Rattanavaraha et al., 2016; Huang et al., 2018; Hettiyadura et al., 2018). While all three C2–C3-OS compounds had strong correlations with the other iSOA OS tracers (R2=0.6–0.94), the relative strength of isoprene versus other VOC precursors, such as aromatics, cannot be determined. As such, they cannot be definitively assigned as iSOA tracers and are, therefore, included in the potential iSOA portion of Fig. 9. The sum of the C2 and C3 OSs had an average concentration of 73 ng m−3 with a range from 2.0 to 831 ng m−3.
In addition, nine NOS species related to isoprene were identified, as shown in Table 2 (Ng et al., 2008; Rollins et al., 2009). Some of the NOSs observed peaked in the daytime and some were enhanced at night. In total, they had a mean concentration of 24 ng m−3 during the campaign. NOS species are formed via the heterogenous uptake of isoprene nitrates (IN) into the particle phase. Nitrate radicals play a key role in the formation of IN, with nitrate radicals forming from reaction of NO2 with O3, which are both key anthropogenic pollutants. Therefore, emissions of NOx and the formation of particulate sulfate will enhance the production of isoprene NOS species.

Figure 8Diurnal profile of 2-methylglyceric acid sulfate (2-MG-OS) in particulate matter collected on filters hourly from 11 to 12 June 2017. The black lines indicate length of the sampling periods.
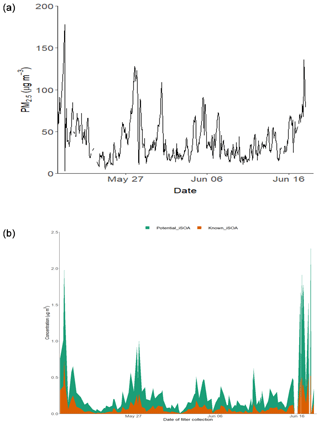
Figure 9(a) Time series of PM2.5 over the sampling period. (b) Time series of the total known isoprene SOA signal, 2-MT-OS, 2-MG-OS, C5H10SO7 (MW 214), C5H8SO7 (MW 212), C5H11NSO9 (MW 261), C5H9NSO10 (MW 275), C5H10O11N2S (MW 306) and C5H9O13N3S (MW 351); and the total signal from the other iSOA tracers quantified in this study.
3.6 Contribution of isoprene SOA in Beijing
In order to estimate the total amount of isoprene-derived OSs and NOSs, labelled here as iSOA, 13 species (including isomers) were chosen that could be confidently identified as being predominately from isoprene (2-MT-OS, 2-MG-OS, C5H10SO7, C5H8SO7, C5H11NSO9, C5H9NSO10, C5H9N2SO11 and C5H8N3SO13). Although there were a number of other compounds with formulas similar to iSOA tracers, their trends compared to previous studies and the potential for alternative sources made a confident assignment of a VOC precursor difficult. Therefore, the estimated contribution of iSOA to the observed total particulate mass determined here should be taken as a lower limit. Figure 9 shows the time series of the iSOA observed in Beijing. The average concentration was 82.5 ng m−3 during the campaign, ranging from 718 ng m−3 on 19 May 2017 (11:38–14:30 LT) to 1.9 ng m−3 on 2 June 2017 (14:36–17:28 LT). The contribution of iSOA to the OOA factors measured by the AMS was obtained by assuming that all OS and NOS species fragment in the ion source to lose the sulfate and nitrate groups. Across the whole measurement period, the iSOA tracers represented only a small fraction of the total OOA measured by AMS (0.62% of ∑[OOA1-3]). However, towards the end of the measurement period, this increased up to a maximum of 3 % on 17 June 2017 (13:32–14:23 LT).
Additional iSOA tracers containing only CHO (Table 3), including 2-methyltetrols, 2-methylglyceric acid and C5-alkene triols, were measured in separate 24 h filter samples via the commonly used derivatization GC-MS method (Claeys et al., 2004; Wang et al., 2005). The average ratio of the 2-methyltetrols to their corresponding OS (2-MT:2-MT-OS) was 1.4, indicating extensive heterogeneous conversion of isoprene oxidation products within the particles. The observed ratio is slightly higher than those measured in the Southeastern US (∼0.37–0.96 as shown in Table 3) but much lower than that measured in the Pearl River Delta region (∼40) where the 2-methyltetrols dominated. In contrast, the average ratio of the high-NO iSOA tracer (2-MG) and its corresponding organosulfate (2-MG:2-MG-OS) observed in Beijing was 0.33, indicating more extensive transformation to products from heterogeneous reactions. This ratio may also reflect the more volatile nature of 2-MG compared with 2-MT. Overall, the combined concentrations of these isoprene CHO compounds were generally low (mean of 25 ng m−3 and max of 69 ng m−3) in comparison with the heterogeneous iSOA compounds (i.e. isoprene-derived OSs and NOSs) targeted in this work. In addition, the concentrations of these CHO species may be overestimated based on recent studies demonstrating that thermal decomposition leads to these products being detected by GC-MS and FIGAERO-CIMS methods (D'Ambro et al., 2019); thus, the conversion to products from heterogenous reactions (i.e. OSs and NOSs) may in fact be larger (2MT:2MT-OS of 0.5–0.91 using the overestimates of 160 %–288 % observed in Cui et al., 2018).
The study presented here shows for the first time that OS species derived from isoprene oxidation can make a significant contribution to oxidized organic aerosol in Beijing in summer. There is significant anthropogenic control, from both NOx and sulfate aerosols, on the products and concentrations of iSOA in Beijing. The majority of the OS species showed a strong correlation towards the product of [O3][pSO4], highlighting the role of both photochemistry and the availability of particulate sulfate in heterogeneous reactions. When the observed concentrations of all of the OS and NOS species measured in this study (including the additional 19 compounds not confidently assigned to iSOA) are combined, they contribute 2.2 % to the total OOA (∑[OOA1-3]) on average, increasing to a maximum of 10.5 %, indicating the extensive heterogeneous conversion of VOC oxidation products in Beijing in summer.
Concentration data are available at https://doi.org/10.15124/7c1a54dd-ce72-4b1a-bed1-812cf25bc407 (Hamilton and Bryant, 2019). Filter collection times are shown in Table S3.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-7531-2020-supplement.
DJB analysed the aerosol samples and quantified iSOA tracers. WJD and KLP developed the UHPLC-MS method. JRH, RED and MS provided the VOC measurements. FAS and JDL collected the NO, NO2 and O3 data. TJB, AM, SDW, AB, CJP and HC collected and analysed the CIMS data. LKW, DEH and EJS provided the OH and HO2 data. BO provided the NO3 measurements. TC, JDS and WJD carried out the offline HILIC analysis. DL, ZS and RH provided the GC-MS iSOA data. YS and WX carried out the AMS measurements and the PMF analysis. ACL and RH lead the APHH projects. DJB, ARR and JFH wrote the paper with input from all co-authors.
The authors declare that they have no conflict of interest.
We acknowledge Pingqing Fu, Zifa Wang and Jie Li from IAP for their support with respect to hosting the APHH-Beijing campaign at IAP. We thank Tuan Vu and Bill Bloss from the University of Birmingham; Siyao Yue, Liangfang Wei, Hong Ren, Qiaorong Xie, Wanyu Zhao, Linjie Li, Ping Li, Shengjie Hou and Qingqing Wang from IAP; Kebin He and Xiaoting Cheng from Tsinghua University; and James Allan from the University of Manchester for providing logistic and scientific support for the field campaigns. Daniel J. Bryant, Freya A. Squires, William J. Dixon and Eloise J. Slater acknowledge NERC SPHERES PhD studentships. Marvin Shaw acknowledges Syft Technologies for his fellowship grants and scientific support. The Orbitrap MS was funded by a Natural Environment Research Council strategic capital grant (grant no. CC090). Jason D. Surratt and Tianqu Cui acknowledge support from the United States National Science Foundation (NSF) under an Atmospheric and Geospace (AGS) grant (grant no. 1703535), and they also wish to thank Avram Gold and Zhenfa Zhang from the University of North Carolina for providing the 2-MT-OS and 2-MG-OS standards.
This research has been supported by the National environment research council (NERC; grant no. NE/N006917/1).
This paper was edited by Alex Lee and reviewed by four anonymous referees.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prévôt, A. S. H., Lee, J., Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp. T., Stone, D., Heard, D. E., Fleming. Z. L., Leigh, R., Shallcross, D. E., and Percival C. J.: The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.-Atmos., 120, 5638–5657, https://doi.org/10.1002/2014JD022629, 2015.
Bannan, T. J., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Mehra, A., Hammes, J., Hallquist, M., Alfarra, M. R., Krieger, U. K., Reid, J. P., Jayne, J., Robinson, W., McFiggans, G., Coe, H., Percival, C. J., and Topping, D.: A method for extracting calibrated volatility information from the FIGAERO-HR-ToF-CIMS and its experimental application, Atmos. Meas. Tech., 12, 1429–1439, https://doi.org/10.5194/amt-12-1429-2019, 2019.
Beelen, R., Raaschou-Nielsen, O., Stafoggia, M., Andersen, Z. J., Weinmayr, G., Hoffmann, B., Wolf, K., Samoli, E., Fischer, P., Nieuwenhuijsen, M., Vineis, P., Xun, W. W., Katsouyanni, K., Dimakopoulou, K., Oudin, A., Forsberg, B., Modig, L., Havulinna, A. S., Lanki, T., Turunen, A., Oftedal, B., Nystad, W., Nafstad, P., De Faire, U., Pedersen, N. L., Ostenson, C. G., Fratiglioni, L., Penell, J., Korek, M., Pershagen, G., Eriksen, K. T., Overvad, K., Ellermann, T., Eeftens, M., Peeters, P. H., Meliefste, K., Wang, M., Bueno-de-Mesquita, B., Sugiri, D., Kramer, U., Heinrich, J., de Hoogh, K., Key, T., Peters, A., Hampel, R., Concin, H., Nagel, G., Ineichen, A., Schaffner, E., Probst-Hensch, N., Kunzli, N., Schindler, C., Schikowski, T., Adam, M., Phuleria, H., Vilier, A., Clavel-Chapelon, F., Declercq, C., Grioni, S., Krogh, V., Tsai, M. Y., Ricceri, F., Sacerdote, C., Galassi, C., Migliore, E., Ranzi, A., Cesaroni, G., Badaloni, C., Forastiere, F., Tamayo, I., Amiano, P., Dorronsoro, M., Katsoulis, M., Trichopoulou, A., Brunekreef, B., and Hoek, G.: Effects of long-term exposure to air pollution on natural-cause mortality: an analysis of 22 European cohorts within the multicentre ESCAPE project, Lancet, 383, 785–795, https://doi.org/10.1016/S0140-6736(13)62158-3, 2014.
Brüggemann, M., van Pinxteren, D., Wang, Y., Yu, J. Z., and Herrmann, H.: Quantification of known and unknown terpenoid organosulfates in PM10 using untargeted LC–HRMS/MS: contrasting summertime rural Germany and the North China Plain, Environ. Chem., 16, 333–346, https://doi.org/10.1071/EN19089, 2019.
Budisulistiorini, S. H., Canagaratna, M. R., Croteau, P. L., Marth, W. J., Baumann, K., Edgerton, E. S., Shaw, S. L., Knipping, E. M., Worsnop, D. R., Jayne, J. T., Gold, A., and Surratt, J. D.: Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in downtown Atlanta, Georgia, using aerodyne aerosol chemical speciation monitor, Environ. Sci. Technol., 47, 5686–5694, https://doi.org/10.1021/es400023n, 2013.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Budisulistiorini, S. H., Baumann, K., Edgerton, E. S., Bairai, S. T., Mueller, S., Shaw, S. L., Knipping, E. M., Gold, A., and Surratt, J. D.: Seasonal characterization of submicron aerosol chemical composition and organic aerosol sources in the southeastern United States: Atlanta, Georgia,and Look Rock, Tennessee, Atmos. Chem. Phys., 16, 5171–5189, https://doi.org/10.5194/acp-16-5171-2016, 2016.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Chan, C. K. and Yao, X.: Air pollution in mega cities in China, Atmos. Environ., 42, 1–42, https://doi.org/10.1016/j.atmosenv.2007.09.003, 2008.
Chan, M. N., Surratt, J. D., Claeys, M., Edgerton, E. S., Tanner, R. L., Shaw, S. L., Zheng, M., Knipping, E. M., Eddingsaas, N. C., Wennberg, P. O., and Seinfeld, J. H.: Characterization and quantification of isoprene-derived epoxydiols in ambient aerosol in the Southeastern United States, Environ. Sci. Technol., 44, 4590–4596, https://doi.org/10.1021/es100596b, 2010.
Chen, Q., Farmer, D. K., Rizzo, L. V., Pauliquevis, T., Kuwata, M., Karl, T. G., Guenther, A., Allan, J. D., Coe, H., Andreae, M. O., Pöschl, U., Jimenez, J. L., Artaxo, P., and Martin, S. T.: Submicron particle mass concentrations and sources in the Amazonian wet season (AMAZE-08), Atmos. Chem. Phys., 15, 3687–3701, https://doi.org/10.5194/acp-15-3687-2015, 2015.
Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Elemental analysis of chamber organic aerosol using an aerodyne high-resolution aerosol mass spectrometer, Atmos. Chem. Phys., 10, 4111–4131, https://doi.org/10.5194/acp-10-4111-2010, 2010.
Claeys, M., Graham, B., Vas, G., Wang, W., Vermeylen, R., Pashynska, Cafmeyer, J., Guyon, P., Andreae, M. O., Artaxo, P., and Maenhaut, W.: Formation of Secondary Organic Aerosols Through Photooxidation of Isoprene, Science, 303, 1173–1176, https://doi.org/10.1126/science.1092805, 2004.
Clark, C. H., Kacarab, M., Nakao, S., Asa-Awuku, A., Sato, K., and Cocker, D. R.: Temperature Effects on Secondary Organic Aerosol (SOA) from the Dark Ozonolysis and Photo-Oxidation of Isoprene, Environ. Sci. Technol., 50, 5564–5571, https://doi.org/10.1021/acs.est.5b05524, 2016.
Cui, T., Zeng, Z., dos Santos, E. O., Zhang, Z., Chen, Y., Zhang, Y., Rose, C. A., Budisulisyiorini, S. H., Collins, L. B., Bodnar, W. M., de Souza, R. A. F., Martin, S. T., Machado, C. M. D., Turpin, B. J., Gold, A., Ault, A. P., and Surratt, J. D: Development of hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci. Processes Impacts, 20, 1524–1536, https://doi.org/10.1039/c8em00308d, 2018.
D'Ambro, E. L., Schobesberger, S., Gaston, C. J., Lopez-Hilfiker, F. D., Lee, B. H., Liu, J., Zelenyuk, A., Bell, D., Cappa, C. D., Helgestad, T., Li, Z., Guenther, A., Wang, J., Wise, M., Caylor, R., Surratt, J. D., Riedel, T., Hyttinen, N., Salo, V.-T., Hasan, G., Kurtén, T., Shilling, J. E., and Thornton, J. A.: Chamber-based insights into the factors controlling epoxydiol (IEPOX) secondary organic aerosol (SOA) yield, composition, and volatility, Atmos. Chem. Phys., 19, 11253–11265, https://doi.org/10.5194/acp-19-11253-2019, 2019.
Ding, X., He, Q.-F., Shen, R.-Q., Yu, Q.-Q., and Wang, X.-M.: Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer, J. Geophys. Res.-Atmos., 119, 11877–11891, https://doi.org/10.1002/2014JD021748, 2014.
Dockery, D. W., Pope, C. A., Xu, X. P., Spengler, J. D., Ware, J. H., Fay, M. E., Ferris, B. G., and Speizer, F. E.: An Association between Air- Pollution and Mortality in 6 United-States Cities, N. Engl. J. Med., 329, 1753–1759, https://doi.org/10.1056/NEJM199312093292401, 1993.
Dommen, J., Metzger, A., Duplissy, J., Kalberer, M., Alfarra, M. R., Gascho, A., Weingartner, E., Prevot, A. S. H., Verheggen, B., and Baltensperger, B.: Laboratory observation of oligomers in the aerosol from isoprene/NOx photooxidation, Geophys. Res. Lett., 33, L13805, https://doi.org/10.1029/2006GL026523, 2016.
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowksi, M., Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005.
Fu, P., Kawamura, K., Kanaya, Y., and Wang, Z.: Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt. Tai, Central East China, Atmos. Environ., 44, 4817–4826, https://doi.org/10.1016/j.atmosenv.2010.08.040, 2010.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Glasius, M., Bering, M. S., Yee, L. D., De Sá, S. S., Isaacman-VanWertz, G., Wernis, R. A., Barbosa, H. M. J., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Shrivastava, M., Martin, S. T., and Goldstein, A. H.: Organosulfates in aerosols downwind of an urban region in central Amazon, Environ. Sci. Process. Impact., 20, 1546–1558, https://doi.org/10.1039/c8em00413g, 2018.
Hamilton, J. and Bryant, D.: Dataset for “Strong anthropogenic control of Isoprene SOA”, The York Research Database, https://doi.org/10.15124/7c1a54dd-ce72-4b1a-bed1-812cf25bc407, 2019.
Hamilton, J. F., Lewis, A. C., Carey, T. J., and Wenger, J. C.: Characterization of Polar Compounds and Oligomers in Secondary Organic Aerosol Using Liquid Chromatography Coupled to Mass Spectrometry, Anal. Chem., 80, 474–480, https://doi.org/10.1021/ac701852t, 2008.
He, Q.-F., Ding, X., Fu, X.-X., Zhang, Y.-Q., Wang, J.-Q., Liu, Y.-X., Tang, M.-J., Wang, X.-M., and Rudich, Y.: Secondary Organic Aerosol Formation From Isoprene Epoxides in the Pearl River Delta, South China: IEPOX- and HMML- Derived Tracers, J. Geophys. Res.-Atmos., 123, 6999–7012, https://doi.org/10.1029/2017JD028242, 2018.
Hettiyadura, A. P. S., Xu, L., Jayarathne, T., Skog, K., Guo, H., Weber, R. J., Nenes, A., Keutsch, F. N., Ng, N. L., and Stone, E. A.: Source apportionments of organic in Centreville, AL using organosulaftes in organic tracer-based positive matrix factorization, Atmos. Environ., 186, 74–88, https://doi.org/10.1016/j.atmosenv.2018.05.007, 2018.
Hettiyadura, A. P. S., Al-Naiema, I. M., Hughes, D. D., Fang, T., and Stone, E. A.: Organosulfates in Atlanta, Georgia: anthropogenic influences on biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 19, 3191–3206, https://doi.org/10.5194/acp-19-3191-2019, 2019.
Hopkins, J. R., Jones, C. E., and Lewis, A. C.: A dual channel gas chromatograph for atmospheric anlysis of volatile organic compounds including oxygenated and monoterpene compounds, J. Environ. Monitor., 13, 2268–2276, https://doi.org/10.1039/C1EM10050E, 2011.
Hu, J., Wang, Y., Ying, Q., and Zhang, H.: Spatial and temporal variability of PM2.5 and PM10 over the North China Plain and the Yangtze River Delta, China, Atmos. Environ., 95, 598–609, https://doi.org/10.1016/j.atmosenv.2014.07.019, 2014.
Hu, J., Huang, L., Chen, M., Liao, H., Zhang, H., Wang, S., Zhang, Q., and Ying, Q.: Premature Mortality Attributable to Particulate Matter in China: Source Contributions and Responses to Reductions, Environ. Sci. Technol., 51, 9950–9959, https://doi.org/10.1021/acs.est.7b03193, 2017a.
Hu, J., Li, X., Huang, L., Ying, Q., Zhang, Q., Zhao, B., Wang, S., and Zhang, H.: Ensemble prediction of air quality using the WRF/CMAQ model system for health effect studies in China, Atmos. Chem. Phys., 17, 13103–13118, https://doi.org/10.5194/acp-17-13103-2017, 2017b.
Hu, W., Hu, M., Hu, W., Jimenez, J. L., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Zeng, L., and Shao, M.: Chemical Composition, sources, and aging process of submicron aerosols in Beijing: Contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, https://doi.org/10.1002/2015JD024020, 2016.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Huang, R.-J., Cao, J., Chen, Y., Yang, L., Shen, J., You, Q., Wang, K., Lin, C., Xu, W., Gao, B., Li, Y., Chen, Q., Hoffmann, T., O'Dowd, C. D., Bilde, M., and Glasius, M.: Organosulfates in atmospheric aerosol: synthesis and quantitative analysis of PM2.5 from Xi'an, northwestern China, Atmos. Meas. Tech., 11, 3447–3456, https://doi.org/10.5194/amt-11-3447-2018, 2018.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jerrett, M., Burnett, R. T., Pope, C. A., Ito, K., Thurston, G., Krewski, D., Shi, Y. L., Calle, E., and Thun, M.: Long-Term Ozone Exposure and Mortality, N. Engl. J. Med., 360, 1085–1095, https://doi.org/10.1056/NEJMoa0803894, 2009.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., St.Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Krechmer, J. E., Groessl, M., Zhang, X., Junninen, H., Massoli, P., Lambe, A. T., Kimmel, J. R., Cubison, M. J., Graf, S., Lin, Y.-H., Budisulistiorini, S. H., Zhang, H., Surratt, J. D., Knochenmuss, R., Jayne, J. T., Worsnop, D. R., Jimenez, J.-L., and Canagaratna, M. R.: Ion mobility spectrometry–mass spectrometry (IMS–MS) for on- and offline analysis of atmospheric gas and aerosol species, Atmos. Meas. Tech., 9, 3245–3262, https://doi.org/10.5194/amt-9-3245-2016, 2016.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Laurent, O., Hu, J. L., Li, L. F., Cockburn, M., Escobedo, L., Kleeman, M. J., and Wu, J.: Sources and contents of air pollution affecting term low birth weight in Los Angeles County, California, 2001–2008, Environ. Res., 134, 488–495, https://doi.org/10.1016/j.envres.2014.05.003, 2014.
Le Breton, M., Bacak, A., Muller, J. B. A., Bannan, T. J., Kennedy, O., Ouyang, B., Xiao, P., Bauguitte, S. J.-B., Shallcross, D. E., Jones, R. L., Daniels, M. J. S., Ball, S. M., and Percival, C. J.: The first airbourne comparison of N2O5 measurements over the UK using a CIMS and BBCEAS during the RONOCO campaign, Anal. Methods-UK, 6, 9731–9743, https://doi.org/10.1039/C4AY02273D, 2014.
Le Breton, M., Wang, Y., Hallquist, Å. M., Pathak, R. K., Zheng, J., Yang, Y., Shang, D., Glasius, M., Bannan, T. J., Liu, Q., Chan, C. K., Percival, C. J., Zhu, W., Lou, S., Topping, D., Wang, Y., Yu, J., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 18, 10355–10371, https://doi.org/10.5194/acp-18-10355-2018, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurteìn, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Li, J., Wang, G., Wu, C., Cao, C., Ren, Y., Wang, J., Li, J., Cao, J., Zeng, L., and Zhu, T.: Characterization of isoprene-derived secondary organic aerosols at a rural site in North China Plain with implications for anthropogenic pollution effects, Sci. Rep.-UK, 8, 535, https://doi.org/10.1038/s41598-017-18983-7, 2018.
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic standards, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012.
Lin, Y.-H., Zhang, H., Pye, H. O. T., Zhang, Z., Marth, W. J., Park, S., Arashiro, M., Cui, T., Budisulistiorini, S. H., Sexton, K. G., Vizuete, W., Xie, Y., Luecken, D. J., Piletic, I. R., Edney, E. O., Bartolotti, L. J., Gold, A., and Surratt, J. D.: Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, P. Natl. Acad. Sci. USA, 110, 6718–6723, https://doi.org/10.1073/pnas.1221150110, 2013.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Brauers, T., Oebel, A., Dlugi, R., Hu, M., Li, X., Lou, S. R., Shao, M., Zhu, T., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Nighttime observation and chemistry of HOx in the Pearl River Delta and Beijing in summer 2006, Atmos. Chem. Phys., 14, 4979–4999, https://doi.org/10.5194/acp-14-4979-2014, 2014.
Nestorowicz, K., Jaoui, M., Rudzinski, K. J., Lewandowski, M., Kleindienst, T. E., Spólnik, G., Danikiewicz, W., and Szmigielski, R.: Chemical composition of isoprene SOA under acidic and non-acidic conditions: effect of relative humidity, Atmos. Chem. Phys., 18, 18101–18121, https://doi.org/10.5194/acp-18-18101-2018, 2018.
Newland, M. J., Bryant, D. J., Dunmore, R. E., Bannan, T. J., Acton, W. J. F., Langford, B., Hopkins, J. R., Squires, F. A., Dixon, W., Drysdale, W. S., Ivatt, P. D., Evans, M. J., Edwards, P. M., Whalley, L. K., Heard, D. E., Slater, E. J., Woodward-Massey, R., Ye, C., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Hewitt, C. N., Lee, J. D., Cui, T., Surratt, J. D., Wang, X., Lewis, A. C., Rickard, A. R., and Hamilton, J. F.: Rainforest-like Atmospheric Chemistry in a Polluted Megacity, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2020-35, in review, 2020.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of first-versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Nguyen, T. B., Bateman, A. P., Bones, D. L., Nizkorodov, S. A., Laskin, J., and Laskin, A.: High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 44, 1032–1042, https://doi.org/10.1016/j.atmosenv.2009.12.019, 2010.
Nguyen, Q. T., Christensen, M. K., Cozzi, F., Zare, A., Hansen, A. M. K., Kristensen, K., Tulinius, T. E., Madsen, H. H., Christensen, J. H., Brandt, J., Massling, A., Nøjgaard, J. K., and Glasius, M.: Understanding the anthropogenic influence on formation of biogenic secondary organic aerosols in Denmark via analysis of organosulfates and related oxidation products, Atmos. Chem. Phys., 14, 8961–8981, https://doi.org/10.5194/acp-14-8961-2014, 2014.
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/c5cp02001h, 2015.
Ostro, B., Hu, J., Goldberg, D., Reynolds, P., Hertz, A., Bernstein, L., and Kleeman, M. J.: Associations of Mortality with LongTerm Exposures to Fine and Ultrafine Particles, Species and Sources: Results from the California Teachers Study Cohort, Environ. Health Persp., 123, 549–556, https://doi.org/10.1289/ehp.1408565, 2015.
Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C.: Aerosol formation in the photooxidation of isoprene and β-pinene, Atmos. Environ. A.-Gen., 25, 997–1008, https://doi.org/10.1016/0960-1686(91)90141-S, 1991.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., Clair, J. M. S., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Pope, C. A.: Review: Epidemiological basis for particulate air pollution health standards, Aerosol Sci. Technol., 32, 4–14, https://doi.org/10.1080/027868200303885, 2000.
Pope, C. A. and Dockery, D. W.: Health effects of fine particulate air pollution: Lines that connect, J. Air Waste Manage., 56, 709–742, https://doi.org/10.1080/10473289.2006.10464485, 2006.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., De Vocht., F., Shallcross, B. M. A., Brazier, T., Khan, M. A., Allan, J., Shallcross, D., E., Coe, H., and Percival, C. J.: Observations of isocyanate, amide, nitrate and nitro compounds from an anthropogenic biomass burning event using a ToF-CIMS, J. Geophys. Res.-Atmos., 123, 7687–7704, https://doi.org/10.1002/2017JD027316, 2018.
Rattanavaraha, W., Chu, K., Budisulistiorini, S. H., Riva, M., Lin, Y.-H., Edgerton, E. S., Baumann, K., Shaw, S. L., Guo, H., King, L., Weber, R. J., Neff, M. E., Stone, E. A., Offenberg, J. H., Zhang, Z., Gold, A., and Surratt, J. D.: Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016, 2016.
Rattanavaraha, W., Canagaratna, M. R., Budisulistiorini, S. H., Croteau, P. L., Baumann, K., Canonaco, F., Prevot, A. S. H., Edgerton, E. S., Zhang, Z., Jayne, J. T., Worsnop, D. R., Gold, A., Shaw, S. L., and Surratt, J. D., Atmos. Environ., 167, 389–402, https://doi.org/10.1016/j.atmosenv.2017.07.055, 2017.
Riva, M., Budisulistiorini, H., Zhang, Z., Gold, A., and Surratt, J. D.: Chemical characterization of secondary organic constituents from isoprene ozonolysis in the presence of acidic aerosol, Atmos. Environ., 130, 5–13, https://doi.org/10.1016/j.atmosenv.2015.06.027, 2016a.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., canagaratna, M. R., and Surratt, J. D.: Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016b.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016.
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Riberiro, I. O., Oliveira, R. L., dos Santa, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sá, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin, S. Jr., de Souza, R. A. F., Palm, B. B., Jimenez, J.-L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing Isoprene Epoxydiol-to-inorganic Sulfate Aerosol ratio results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019.
Robinson, N. H., Hamilton, J. F., Allan, J. D., Langford, B., Oram, D. E., Chen, Q., Docherty, K., Farmer, D. K., Jimenez, J. L., Ward, M. W., Hewitt, C. N., Barley, M. H., Jenkin, M. E., Rickard, A. R., Martin, S. T., McFiggans, G., and Coe, H.: Evidence for a significant proportion of Secondary Organic Aerosol from isoprene above a maritime tropical forest, Atmos. Chem. Phys., 11, 1039–1050, https://doi.org/10.5194/acp-11-1039-2011, 2011.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Shi, Z., Vu, T., Kotthaus, S., Harrison, R. M., Grimmond, S., Yue, S., Zhu, T., Lee, J., Han, Y., Demuzere, M., Dunmore, R. E., Ren, L., Liu, D., Wang, Y., Wild, O., Allan, J., Acton, W. J., Barlow, J., Barratt, B., Beddows, D., Bloss, W. J., Calzolai, G., Carruthers, D., Carslaw, D. C., Chan, Q., Chatzidiakou, L., Chen, Y., Crilley, L., Coe, H., Dai, T., Doherty, R., Duan, F., Fu, P., Ge, B., Ge, M., Guan, D., Hamilton, J. F., He, K., Heal, M., Heard, D., Hewitt, C. N., Hollaway, M., Hu, M., Ji, D., Jiang, X., Jones, R., Kalberer, M., Kelly, F. J., Kramer, L., Langford, B., Lin, C., Lewis, A. C., Li, J., Li, W., Liu, H., Liu, J., Loh, M., Lu, K., Lucarelli, F., Mann, G., McFiggans, G., Miller, M. R., Mills, G., Monk, P., Nemitz, E., O'Connor, F., Ouyang, B., Palmer, P. I., Percival, C., Popoola, O., Reeves, C., Rickard, A. R., Shao, L., Shi, G., Spracklen, D., Stevenson, D., Sun, Y., Sun, Z., Tao, S., Tong, S., Wang, Q., Wang, W., Wang, X., Wang, X., Wang, Z., Wei, L., Whalley, L., Wu, X., Wu, Z., Xie, P., Yang, F., Zhang, Q., Zhang, Y., Zhang, Y., and Zheng, M.: Introduction to the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing)”, Atmos. Chem. Phys., 19, 7519–7546, https://doi.org/10.5194/acp-19-7519-2019, 2019.
Spolnik, G., Wach, P., Rudzinski, K. J., Skotak, K., Danikiewicz, W., and Szmigielski, R.: Improved UHPLC-MS/MS Methods for Analysis of Isoprene-Derived Organosulfates, Anal. Chem., 90, 3416–3423, https://doi.org/10.1021/acs.analchem.7b05060, 2018.
Sun, Y., Xu, W., Zhang, Q., Jiang, Q., Canonaco, F., Prévôt, A. S. H., Fu, P., Li, J., Jayne, J., Worsnop, D. R., and Wang, Z.: Source apportionment of organic aerosol from 2-year highly time-resolved measurements by an aerosol chemical speciation monitor in Beijing, China, Atmos. Chem. Phys., 18, 8469–8489, https://doi.org/10.5194/acp-18-8469-2018, 2018.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A., 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007a.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007b.
Surratt, J. D., Gómez-González, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A., 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld. J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Wang, Q., He, X., Huang, H. X. H., Griffith, S. M., Feng, Y., Zhang, T., Zhang, Q., Wu, D., and Yu, J. Z.: impact of Secondary Organic Aerosol Tracers on Tracer-Based Source Apportionment of Organic Carbon and PM2.5: A Case Study in the Pearl River Delta, China, ACS Earth Space Chem., 1, 562–571, https://doi.org/10.1021/acsearthspacechem.7b00088, 2017.
Wang, W., Kourtchev, I., Graham, B., Cafmeyer, J., Maenhaut, W., and Claeys, M.: Characterization of oxygenated derivatives of isoprene related to 2-methyltetrols in Amazonian aerosols using trimethylsilylation and gas chromatography/ion trap mass spectrometry, Rapid Commun. Mass Spectrom., 19, 1343–1351, https://doi.org/10.1002/rcm.1940, 2005.
Wang, X., Hayeck, N., Brüggemann, M., Yao, L., Chen, H., Zhang, C., Emmelin, C., Chen, J., George, C., and Wang, L.: Chemical Characteristics of Organic Aerosols in Shanghai: A Study by Ultrahigh-Performance Liquid Chromatography Coupled With Orbitrap Mass Spectrometry, J. Geophys. Res.-Atmos., 122, 11703–11722, https://doi.org/10.1002/2017JD026930, 2017.
Wang, X. K., Rossignol, S., Ma, Y., Yao, L., Wang, M. Y., Chen, J. M., George, C., and Wang, L.: Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River, Atmos. Chem. Phys., 16, 2285–2298, https://doi.org/10.5194/acp-16-2285-2016, 2016.
Wang, Y., Zhuang, G., Tang, A., Yuan, H., Sun, Y., Chen, S., and Zheng, A.: The ion chemistry and the source of PM2.5 aerosol in Beijing, Atmos. Environ., 39, 3771–3784, https://doi.org/10.1016/j.atmosenv.2005.03.013, 2005.
Wang, Y., Hu, M., Guo, S., Wang, Y., Zheng, J., Yang, Y., Zhu, W., Tang, R., Li, X., Liu, Y., Le Breton, M., Du, Z., Shang, D., Wu, Y., Wu, Z., Song, Y., Lou, S., Hallquist, M., and Yu, J.: The secondary formation of organosulfates under interactions between biogenic emissions and anthropogenic pollutants in summer in Beijing, Atmos. Chem. Phys., 18, 10693–10713, https://doi.org/10.5194/acp-18-10693-2018, 2018.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L. K., Furneaux, K. L., Goddard, A., Lee, J. D., Mahajan, A., Oetjen, H., Read, K. A., Kaaden, N., Carpenter, L. J., Lewis, A. C., Plane, J. M. C., Saltzman, E. S., Wiedensohler, A., and Heard, D. E.: The chemistry of OH and HO2 radicals in the boundary layer over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 1555–1576, https://doi.org/10.5194/acp-10-1555-2010, 2010.
Whalley, L. K., Stone, D., Dunmore, R., Hamilton, J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Williams, P., Kleffmann, J., Laufs, S., Woodward-Massey, R., and Heard, D. E.: Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 18, 2547–2571, https://doi.org/10.5194/acp-18-2547-2018, 2018.
Xu, L., Kollman, M. S., Song, C., Shiling, J. E., and Ng, N. L.: Effects of NOx on the volatility of secondary organic aerosol from isoprene photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J., and Kamens, R. M.: Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions, Atmos. Chem. Phys., 11, 6411–6424, https://doi.org/10.5194/acp-11-6411-2011, 2011.
Zhang, H., Lin, Y.-H., Zhang, Z., Zhang, X., Shaw, S. L., Knipping, E. M., Weber, R. J., Gold, A., Kamens, R. M., and Surratt, J. D.: Secondary organic aerosol formation from methacrolein photooxidation: Roles of NOx level, relative humidity and aerosol acidity, Environ. Chem., 9, 247–262, https://doi.org/10.1071/EN12004, 2012.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, Y., Sun, J., Zhang, X., Shen, X., Wang, T., and Qin, M.: Seasonal characterization of components and size distributions for submicron aerosols in Beijing, Sci. China Earth Sci., 56, 890–900, https://doi.org/10.1007/s11430-012-4515-z, 2013.
Zhang, Y., Ren, H., Sun, Y., Cao, F., Chang, Y., Liu, Shoudong, L., Lee, X., Agrios, K., Kawamura, K., Liu, D., Ren, L., Du, W., Wang, Z., Prévôt, A. S. H., Szidat, S., and Fu, P.: High Contribution of Nonfossil Sources to Submicrometer Organic Aerosols in Beijing, China, Environ. Sci. Technol., 51, 7842–7852, https://doi.org/10.1021/acs.est.7b01517, 2017a.
Zhang, Y., Tang, L., Sun, Y., Favez, O., Canonaco, F., Albinet, A., Couvidat, F., Liu, D., Jayne, J. T., Wang, Z., Croteau, P. L., Canagaratna, M. R., Zhou, H., Prévôt, A. S. H., and Worsnop, D. R.: Limited formation of isoprene epoxydiols-derived secondary organic aerosol under NOx-rich environments in Eastern China, Geophys. Res. Lett., 44, 2035–2043, https://doi.org/10.1002/2016GL072368, 2017b.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Technol. Lett., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018.
Zhou, W., Zhao, J., Ouyang, B., Mehra, A., Xu, W., Wang, Y., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Chen, Q., Xie, C., Wang, Q., Wang, J., Du, W., Zhang, Y., Ge, X., Ye, P., Lee, J. D., Fu, P., Wang, Z., Worsnop, D., Jones, R., Percival, C. J., Coe, H., and Sun, Y.: Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmos. Chem. Phys., 18, 11581–11597, https://doi.org/10.5194/acp-18-11581-2018, 2018.