the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Mar 2020
| 31 Mar 2020
Global inorganic nitrate production mechanisms: comparison of a global model with nitrate isotope observations
Tomás Sherwen
Christopher D. Holmes
Jenny A. Fisher
Qianjie Chen
Mat J. Evans
Prasad Kasibhatla
The formation of inorganic nitrate is the main sink for nitrogen oxides (NOx = NO + NO2). Due to the importance of NOx for the formation of tropospheric oxidants such as the hydroxyl radical (OH) and ozone, understanding the mechanisms and rates of nitrate formation is paramount for our ability to predict the atmospheric lifetimes of most reduced trace gases in the atmosphere. The oxygen isotopic composition of nitrate (Δ17O(nitrate)) is determined by the relative importance of NOx sinks and thus can provide an observational constraint for NOx chemistry. Until recently, the ability to utilize Δ17O(nitrate) observations for this purpose was hindered by our lack of knowledge about the oxygen isotopic composition of ozone (Δ17O(O3)). Recent and spatially widespread observations of Δ17O(O3) motivate an updated comparison of modeled and observed Δ17O(nitrate) and a reassessment of modeled nitrate formation pathways. Model updates based on recent laboratory studies of heterogeneous reactions render dinitrogen pentoxide (N2O5) hydrolysis as important as NO2 + OH (both 41 %) for global inorganic nitrate production near the surface (below 1 km altitude). All other nitrate production mechanisms individually represent less than 6 % of global nitrate production near the surface but can be dominant locally. Updated reaction rates for aerosol uptake of NO2 result in significant reduction of nitrate and nitrous acid (HONO) formed through this pathway in the model and render NO2 hydrolysis a negligible pathway for nitrate formation globally. Although photolysis of aerosol nitrate may have implications for NOx, HONO, and oxidant abundances, it does not significantly impact the relative importance of nitrate formation pathways. Modeled Δ17O(nitrate) (28.6±4.5 ‰) compares well with the average of a global compilation of observations (27.6±5.0 ‰) when assuming Δ17O(O3) = 26 ‰, giving confidence in the model's representation of the relative importance of ozone versus HOx (= OH + HO2 + RO2) in NOx cycling and nitrate formation on the global scale.
- Article
(8897 KB) - Full-text XML
-
Supplement
(1868 KB) - BibTeX
- EndNote





Nitrogen oxides (NOx = NO + NO2) are a critical ingredient for the formation of tropospheric ozone (O3). Tropospheric ozone is a greenhouse gas, is a major precursor for the hydroxyl radical (OH), and is considered an air pollutant due to its negative impacts on human health. The atmospheric lifetime of NOx is determined by its oxidation to inorganic and organic nitrate. The formation of inorganic nitrate (HNO3(g) and particulate ) is the dominant sink for NOx globally, while formation of organic nitrate may be significant in rural and remote continental locations (Browne and Cohen, 2014). Organic nitrate as a sink for NOx may be becoming more important in regions in North America and Europe where NOx emissions have declined (Zare et al., 2018). Uncertainties in the rate of oxidation of NOx to nitrate have been shown to represent a significant source of uncertainty for ozone and OH formation in models (e.g., Newsome and Evans, 2017), with implications for our understanding of the atmospheric lifetime of species such as methane, whose main sink is reaction with OH.
NOx is emitted to the atmosphere primarily as NO by fossil fuel and biomass/biofuel burning, soil microbes, and lightning. Anthropogenic sources from fossil fuel and biofuel burning and from the application of fertilizers to soil for agriculture currently dominate NOx sources to the atmosphere (Jaeglé et al., 2005). After emission, NO is rapidly oxidized to NO2 by ozone (O3), peroxy (HO2) and hydroperoxy radicals (RO2), and halogen oxides (e.g., BrO). During the daytime, NO2 is rapidly photolyzed to NO + O at wavelengths (λ) <398 nm. NOx cycling between NO and NO2 proceeds several orders of magnitude faster than oxidation of NOx to nitrate during the daytime (Michalski et al., 2003).
Formation of inorganic nitrate is dominated by oxidation of NO2 by OH during the day and by the hydrolysis of dinitrogen pentoxide (N2O5) at night (Alexander et al., 2009). Recent implementations of reactive halogen chemistry in models of tropospheric chemistry show that formation of nitrate from the hydrolysis of halogen nitrates (XNO3, where X = Br, Cl, or I) is also a sink for NOx with implications for tropospheric ozone, OH, reactive halogens, and aerosol formation (Schmidt et al., 2016; Sherwen et al., 2016; Saiz-Lopez et al., 2012; Long et al., 2014; Parrella et al., 2012; von Glasow and Crutzen, 2004; Yang et al., 2005). Other inorganic nitrate formation pathways include hydrogen abstraction of hydrocarbons by the nitrate radical (NO3), heterogeneous reaction of N2O5 with particulate chloride (Cl−), heterogeneous uptake of NO2 and NO3, direct oxidation of NO to HNO3 by HO2, and hydrolysis of organic nitrate (Atkinson, 2000). Inorganic nitrate partitions between the gas (HNO3(g)) and particle () phases, with its relative partitioning dependent upon aerosol abundance, aerosol liquid water content, aerosol chemical composition, and temperature. Inorganic nitrate is lost from the atmosphere through wet or dry deposition to the Earth's surface with a global lifetime against deposition on the order of 3–4 d (Park et al., 2004).
Formation of inorganic nitrate was thought to be a permanent sink for NOx in the troposphere due to the slow photolysis of nitrate compared to deposition. However, laboratory and field studies have shown that adsorbed on surfaces is photolyzed at rates much higher than HNO3(g) (Ye et al., 2016). For example, the photolysis of in snow grains on ice sheets has a profound impact on the oxidizing capacity of the polar atmosphere (Domine and Shepson, 2002). More recently, observations of NOx and nitrous acid (HONO) provide evidence of photolysis of aerosol in the marine (Reed et al., 2017; Ye et al., 2016) and continental (Ye et al., 2018; Chen et al., 2019) boundary layer, with implications for ozone and OH (Kasibhatla et al., 2018).
Organic nitrates form during reaction of NOx and NO3 with biogenic volatile organic compounds (BVOCs) and their oxidation products (organic peroxy radicals, RO2) (Browne and Cohen, 2014; Liang et al., 1998). Products of these reactions include peroxy nitrates (RO2NO2) and alkyl and multifunctional nitrates (RONO2) (O'Brien et al., 1995). Peroxy nitrates are thermally unstable and decompose back to NOx on the order of minutes to days at warm temperatures. Decomposition of longer-lived peroxy nitrates such as peroxyacetyl nitrate (PAN) can provide a source of NOx to remote environments (Singh et al., 1992). The fate of RONO2 is uncertain. First-generation RONO2 is oxidized to form second-generation RONO2 species with a lifetime of about a week for the first-generation species with ≥4 carbon atoms and up to several weeks for species with fewer carbon atoms (e.g., days to weeks for methyl nitrate) (Fisher et al., 2018). Subsequent photolysis and oxidation of second-generation RONO2 species can lead to the recycling of NOx (Müller et al., 2014), although recycling efficiencies are highly uncertain (Horowitz et al., 2007;Paulot et al., 2009). RONO2 can also partition to the particle phase (pRONO2) contributing to organic aerosol formation (Xu et al., 2015). pRONO2 is removed from the atmosphere by deposition to the surface or through hydrolysis to form inorganic nitrate and alcohols (Rindelaub et al., 2015; Jacobs et al., 2014).
The oxygen isotopic composition (Δ17O = δ17 − 0.52×δ18O) of nitrate is determined by the relative importance of oxidants leading to nitrate formation from the oxidation of NOx (Michalski et al., 2003). Observations of the oxygen isotopic composition of nitrate (Δ17O(nitrate)) have been used to quantify the relative importance of different nitrate formation pathways and to assess model representation of the chemistry of nitrate formation in the present day (Alexander et al., 2009; Michalski et al., 2003; Costa et al., 2011; Ishino et al., 2017; Morin et al., 2009, 2008, 2007; Savarino et al., 2007, 2013; Kunasek et al., 2008; McCabe et al., 2007; Hastings et al., 2003; Kaiser et al., 2007; Brothers et al., 2008; Ewing et al., 2007) and in the past from nitrate archived in ice cores (Sofen et al., 2014; Alexander et al., 2004; Geng et al., 2014, 2017). Ozone-influenced reactions in NOx oxidation lead to high Δ17O(nitrate) values while HOx-influenced reactions lead to Δ17O(nitrate) near zero. Oxidation by XO (where X = Br, Cl, or I) leads to Δ17O(nitrate) values similar to reactions with ozone because the oxygen atom in XO is derived from the reaction X + O3. Therefore, Δ17O(nitrate) is determined by the relative importance of O3 + XO versus HOx (= OH + HO2 + RO2) in both NOx cycling and oxidation to nitrate. Although freshly emitted NO will have Δ17O(NO) = 0 ‰, NOx achieves isotopic equilibrium during the daytime due to rapid NOx cycling, so that its Δ17O value (Δ17O(NOx)) is solely determined by the relative abundance of (O3 + XO) to (HO2 + RO2) (Michalski et al., 2003).
The Δ17O value of HOx (Δ17O(HOx)) is near zero due to isotopic exchange of OH with water vapor (Dubey et al., 1997). Previously, observations of the Δ17O value of ozone (Δ17O(O3)) showed a large range (6 ‰–54 ‰) (Johnston and Thiemens, 1997; Krankowsky et al., 1995), in contrast to laboratory and modeling studies suggesting that the range of Δ17O(O3) in the troposphere should be relatively narrow (30 ‰–46 ‰) (Morton et al., 1990; Thiemens, 1990). The large range of observed Δ17O(O3) values is thought to be due to sampling artifacts (Brenninkmeijer et al., 2003). Uncertainty in the value of Δ17O(O3) has been the largest source of uncertainty in quantification of nitrate formation pathways using observations of Δ17O(nitrate) (Alexander et al., 2009). Previous modeling studies showed good agreement with observations of Δ17O(nitrate) when assuming that the bulk oxygen isotopic composition of ozone (Δ17O(O3)) is equal to 35 ‰ (Alexander et al., 2009; Michalski et al., 2003) but varied in their assumption on terminal oxygen atom versus statistical isotopic transfer from O3 to the reactant (NO and NO2). This is an important distinction because it is now known that the 17O enrichment in O3 is contained entirely in its terminal oxygen atoms, and it is the terminal oxygen atom that is transferred from O3 (Vicars et al., 2012; Berhanu et al., 2012; Bhattacharya et al., 2008, 2014; Savarino et al., 2008; Michalski and Bhattacharya, 2009), so that the Δ17O value of the oxygen atom transferred from ozone to the product is 50 % larger than the bulk Δ17O(O3) value. Recently, much more extensive observations of Δ17O(O3) using a new technique (Vicars et al., 2012) consistently show Δ17O(O3) = 26±1 ‰ in diverse locations (Vicars et al., 2012; Ishino et al., 2017; Vicars and Savarino, 2014) and suggest that previous modeling studies are biased low in Δ17O(nitrate) (e.g., Alexander et al. 2009), which would occur if the model underestimated the relative role of ozone in NOx chemistry. These new observations of Δ17O(O3), combined with improved understanding and hence more comprehensive chemical representation of nitrate formation in models, motivate an updated comparison of observed and modeled Δ17O(nitrate) as an observational constraint for the relative importance of nitrate formation pathways in the atmosphere. Note that laboratory studies show that the magnitude of Δ17O(O3) is dependent on temperature and pressure (Heidenreich and Thiemens, 1986; Thiemens, 1990; Morton et al., 1990). The observations of Δ17O(O3) by Vicars et al. (2012, 2013) were at the surface over a large temperature range but may not reflect the value of Δ17O(O3) at higher altitudes. However, with the exception of lightning, whose emissions are presently several times smaller than NOx emissions from anthropogenic and biomass burning sources (Murray, 2016), NOx sources emit at the surface. With a NOx lifetime relative to its conversion to nitrate on the order of 1 d (Levy et al., 1999), most nitrate formation also occurs near the surface. Here, we examine the relative contribution of each nitrate formation pathway in a global chemical transport model and compare the model with surface observations of Δ17O(nitrate) from around the world.
We use the GEOS-Chem global chemical transport model version 12.0.0 driven by assimilated meteorology from the MERRA-2 reanalysis product with a native resolution of and 72 vertical levels from the surface up to the 0.01 hPa pressure level. For computational expediency, the horizontal and vertical resolution were downgraded to and 47 vertical levels. GEOS-Chem was originally described in Bey et al. (2001) and includes coupled HOx–NOx–VOC–ozone–halogen–aerosol tropospheric chemistry as described in Sherwen et al. (2016, 2017) and organic nitrate chemistry as described in Fisher et al. (2016). Aerosols interact with gas-phase chemistry through the effect of aerosol extinction on photolysis rates (Martin et al., 2003) and heterogeneous chemistry (Jacob, 2000). The model calculates deposition for both gas species and aerosols (Liu et al., 2001; Zhang et al., 2001; Wang et al., 1998).
Global anthropogenic emissions, including NOx, are from the Community Emissions Data System (CEDS) inventory from 1950 to 2014 CE (Hoesly et al., 2018). The CEDS global emissions inventory is overwritten by regional anthropogenic emissions inventories in the US (EPA/NE11), Canada (CAC), Europe (EMEP), and Asia (MIX Li et al., 2017). Global shipping emissions are from the International Comprehensive Ocean-Atmosphere Data Set (ICOADS), which was implemented into GEOS-Chem as described in Lee et al. (2011). NOx emissions from ships are processed using the PARANOX module described in Vinken et al. (2011) and Holmes et al. (2014) to account for nonlinear, in-plume ozone and HNO3 production. Lightning NOx emissions match the OTD/LIS satellite climatological observations of lightning flashes as described by Murray et al. (2012). Emissions from open fires are from the Global Fire Emissions Database (GFED4.1). Biogenic soil NOx emissions are described in Hudman et al. (2012). Aircraft emissions are from the Aviation Emissions Inventory Code (AEIC) (Stettler et al., 2011).
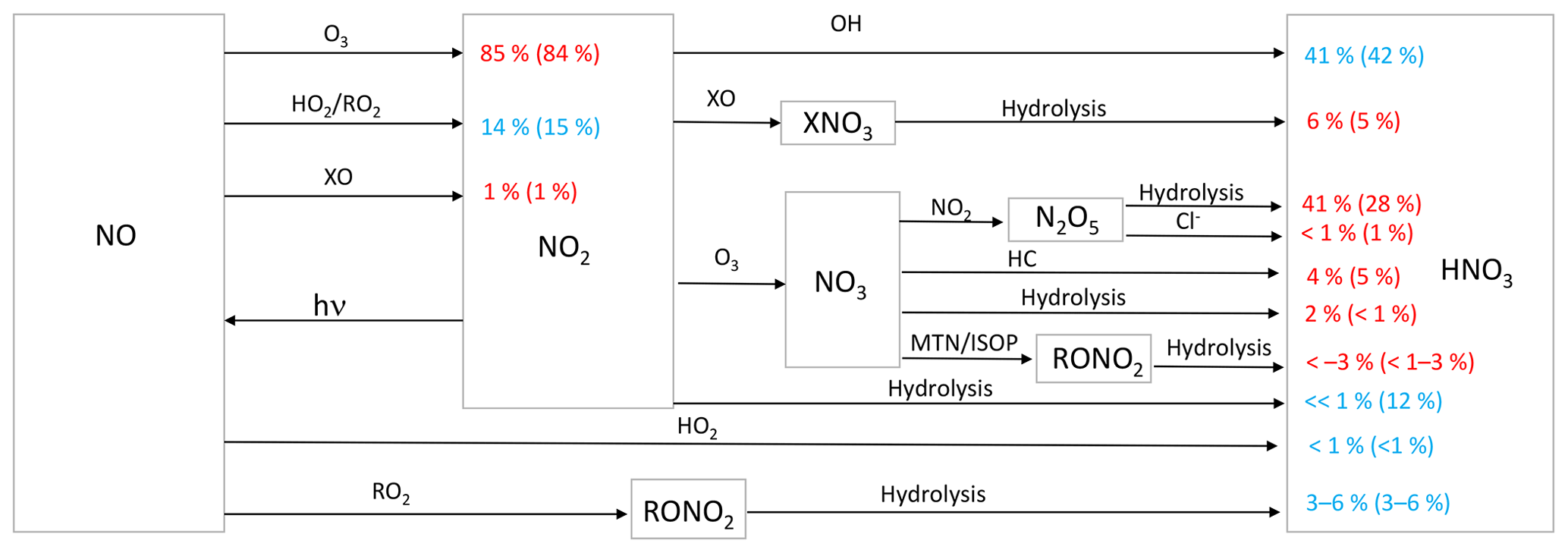
Figure 1Simplified HNO3 formation in the model. Numbers show the global annual-mean percent contribution to NO2 and HNO3 formation in the troposphere below 1 km for the cloud chemistry (standard) simulation. Red indicates reactions leading to high Δ17O values, and blue indicates reactions leading to low Δ17O values. HO2 = HO2 + RO2; X = Br + Cl + I; HC: hydrocarbons; MTN: monoterpenes; ISOP: isoprene.
Chemical processes leading to nitrate formation in GEOS-Chem have expanded since the previous work of Alexander et al. (2009). Figure 1 summarizes the formation of inorganic nitrate in the current model. In the model, NO is oxidized by O3, HO2, RO2, and halogen oxides (XO = BrO, ClO, IO, and OIO) to form NO2. The reaction of NO + HO2 can also form HNO3 directly, although the branching ratio for this pathway is <1 % (Butkovskaya et al., 2005). NO2 can form HNO3 directly from its reaction with OH and through hydrolysis on aerosol surfaces. NO2 can react with XO to form halogen nitrates (BrNO3, ClNO3, and INO3), which can then form HNO3 upon hydrolysis (as described in Sherwen et al., 2016). NO2 can also react with O3 to form NO3, which can then react with NO2, hydrocarbons (HC), and the biogenic VOC monoterpenes (MTN) and isoprene (ISOP). Reaction of NO3 with NO2 forms N2O5, which can subsequently hydrolyze or react with Cl− in aerosol to form HNO3. Reaction of NO3 with HC forms HNO3 via hydrogen abstraction. Reactions of NO3 are only important at night due to its short lifetime against photolysis. Formation of organic nitrate (RONO2) was recently updated in the model as described in Fisher et al. (2016). Reaction of NO3 with MTN and ISOP can form RONO2. RONO2 also forms from the reaction of NO with RO2 derived from OH oxidation of BVOCs. RONO2 hydrolyzes to form HNO3 on a timescale of 1 h. Inorganic nitrate partitions between the gas (HNO3(g)) and particle () phase according to local thermodynamic equilibrium as calculated in the ISORROPIA-II aerosol thermodynamic module (Fountoukis and Nenes, 2007). HNO3(g) and are mainly lost from the atmosphere via wet and dry deposition to the surface.
In the standard model, hydrolysis of N2O5, NO3 ( = ), and NO2 ( = ) occurs on aerosol surfaces only. Uptake and hydrolysis of N2O5 on aerosol surfaces depend on the chemical composition of aerosols, temperature, and humidity as described in Evans and Jacob (2005). Recently, Holmes et al. (2019) updated the reaction probabilities of the NO2 and NO3 heterogeneous reactions in the model to depend on aerosol chemical composition and relative humidity. Holmes et al. (2019) also updated the N2O5 reaction probability to additionally depend on the H2O and concentrations in aerosol (Bertram and Thornton, 2009). In addition to these updates for hydrolysis on aerosol, Holmes et al. (2019) included the uptake and hydrolysis of N2O5, NO2, and NO3 in cloud water and ice limited by cloud entrainment rates. We incorporate these updates from Holmes et al. (2019) into the cloud chemistry model to examine the impacts on global nitrate production mechanisms. We consider the cloud chemistry model as state of the science, and as such we focus on the results of this particular simulation. Additional model sensitivity studies are also performed and examined relative to the standard model simulation, which represents a more common representation of nitrate chemistry in atmospheric chemistry models. These additional sensitivity simulations are described in Sect. 4.
Δ17O(nitrate) is calculated in the model using monthly-mean, local chemical production rates rather than by treating different isotopic combinations of nitrate as separate tracers that can be transported in the model. Alexander et al. (2009) transported four nitrate tracers, one each for nitrate production by NO2 + OH, N2O5 hydrolysis, NO3 + HC, and nitrate originating from its formation in the stratosphere. Since Δ17O(NOx) was not transported in the Alexander et al. (2009) model, it was calculated using local production rates, so effectively only one-third of the Δ17O(nitrate) was transported in Alexander et al. (2009). Accurately accounting for transport of Δ17O(nitrate) in the model would require transporting all individual isotopic combinations of the primary reactant (NO), the final product (nitrate), and each reaction intermediate (e.g., N2O5), which we do not do here due to the large computational costs. Thus, the model results shown here represent Δ17O(nitrate) from local NOx cycling and nitrate production. This may lead to model biases, particularly in remote regions such as polar regions in wintertime when most nitrate is likely transported from lower latitudes or the stratosphere. This should make less of a difference in polluted regions where most nitrate is formed locally or for example in polar regions in summer when photochemical recycling of nitrate in the snowpack represents a significant local source of NOx at the surface (Domine and Shepson, 2002). Although lack of transport of the isotope tracers adds uncertainty to direct comparison of the model with observations at any particular location, this approach will reflect the full range of possible modeled Δ17O(nitrate) values for the current chemical mechanism, which can then be compared to the range of observed Δ17O(nitrate) values around the globe.
Table 1Calculated Δ17O(nitrate) in the model for each nitrate production pathway (X = Br, Cl, and I; HC: hydrocarbon; MTN: monoterpenes; ISOP: isoprene; = 39 ‰). A is defined in Eq. (1).

The Δ17O(nitrate) value of nitrate produced from each production pathway is calculated as shown in Table 1. The value of A in Table 1 represents the relative importance of the oxidation pathways of NO to NO2 where the oxygen atom transferred comes from ozone (NO + O3 and NO + XO):
In Eq. (1), k represents the local reaction rate constant for each of the four reactions, XO = BrO, ClO, IO, and OIO; and we assume Δ17O(XO) is equal to the Δ17O value of the terminal oxygen atoms of ozone, as described in more detail below. This effectively assumes that the other oxidation pathways (NO + HO2 and NO + RO2) yield Δ17O(NOx) = 0 ‰. Although HO2 may have a small 17O enrichment on the order of 1 ‰–2 ‰ (Savarino and Thiemens, 1999b), the assumption that this pathway yields Δ17O(NOx) = 0 ‰ simplifies the calculation and leads to negligible differences in calculated Δ17O(nitrate) (Michalski et al., 2003). This approach assumes that NOx cycling is in a photochemical steady state, which only occurs during the daytime. A is calculated in the model as the 24 h average NO2 production rate rather than the daytime average only. As was shown in Alexander et al. (2009), rapid daytime NOx cycling dominates the calculated 24 h averaged A value, leading to negligible differences in calculated Δ17O(nitrate) for 24 h averaged values versus daytime averaged values.
NOx formed during the day will retain its daytime Δ17O(NOx) signature throughout the night due to lack of NO2 photolysis (Morin et al., 2011), suggesting similar A values for the nighttime reactions (R2, R4, R5, R8, and R10 in Table 1). However, NO emitted at night will not undergo photochemical recycling, initially suggesting that NO will retain its emitted Δ17O(NO) value of 0 ‰ prior to sunrise. Thus, any NO emitted at night and oxidized to NO2 before sunrise will result in Δ17O(NO2) equal to one-half of the Δ17O value of the oxidant, since only one of the two oxygen atoms of NO2 will originate from the oxidant. Since HOx abundance is low at night, ozone will be the dominant oxidant. Thus, NO both emitted and oxidized to NO2 at night will lead to Anight=0.5 (half of the O atoms of NO2 originate from O3). Although isotopic exchange between NO + NO2 (Sharma et al., 1970) and NO2 and NO3 via thermal dissociation of N2O5 (Connell and Johnston, 1979) will tend to increase Δ17O(NO) above its emitted value of 0 ‰, the bulk Δ17O value of the NOx plus NO3 system will be lower at night than during the daytime due to the absence of photochemical cycling at night (Michalski et al., 2014; Morin et al., 2011). Since the atmospheric lifetime of NOx near the surface against nighttime oxidation to nitrate (R2 + R4 + R5) is typically greater than 24 h (Fig. S1 in the Supplement), most nitrate formed during the nighttime will form from NOx that reached photochemical equilibrium during the previous day. Thus, we use values of A calculated as the 24 h average NO2 production rate for calculating the Δ17O(nitrate) value of all nitrate production pathways, including those that can occur at night. Using 24 h averaged A values may lead to an overestimate of Δ17O(nitrate) in locations with more rapid nighttime nitrate formation rates such as in China and India (Fig. S1). However, even in these locations the lifetime of NOx against nighttime oxidation is greater than 12 h, suggesting that over half of nitrate formation at night occurs from the oxidation of NOx that reached photochemical equilibrium during the daytime. When comparing modeled Δ17O(nitrate) with observations, we add error bars to model values in these locations (Beijing and Mt. Lulin, Taiwan) that reflect the range of possible A values for nighttime nitrate formation, with the high end (Ahigh) reflecting 24 h average A values and the low end assuming that half of nitrate formation occurs from oxidation of NOx that reached photochemical equilibrium during the daytime (, where Anight=0.5).
Δ17O(nitrate) for total nitrate is calculated in the model according to
where fR represents the fractional importance of each nitrate production pathway (R1–R10 in Table 1) relative to total nitrate production, and Δ17O(nitrate)R is the Δ17O(nitrate) value for each reaction as described in Table 1. To calculate Δ17O(nitrate), we assume that the mean Δ17O value of the ozone molecule (Δ17O(O3)) is equal to 26 ‰ based on recent observations (Vicars et al., 2012; Ishino et al., 2017; Vicars and Savarino, 2014) and that it is the terminal oxygen atom that is transferred to the oxidation product during chemical reactions (Savarino et al., 2008; Berhanu et al., 2012). Thus, we assume that the Δ17O value of the oxygen atom transferred from O3 () = 1.5×Δ17O(O3), as in previous work (e.g., Morin et al., 2011), where represents the Δ17O value of the terminal oxygen atoms in ozone. Assuming that Δ17O(O3) = 26 ‰ based on recent observations, this leads to = 39 ‰.
Figure 1 shows the relative importance of the different oxidation pathways of NO to NO2 and nitrate formation below 1 km altitude in the model for the cloud chemistry simulation, with equivalent values for the standard simulation shown in parentheses. We focus on model results near the surface (below 1 km) because these can be compared to observations; currently only surface observations of Δ17O(nitrate) are available. We note that two observation data sets – from Bermuda (Hastings et al., 2003) and Princeton, NJ (Kaiser et al., 2007) – are rainwater samples and thus may represent nitrate formed aloft. However, since cloud water peaks on average near 1 km altitude in the MERRA2 meteorology used to drive GEOS-Chem, our model sampling strategy should capture the majority of the influence of clouds on the chemistry of nitrate formation. The dominant oxidant of NO to NO2 is O3 (84 %–85 %). Much of the remaining oxidation occurs due to the reaction with peroxy radicals (HO2 and RO2). Oxidation of NO to NO2 by XO is minor (1 %) and occurs over the oceans because the main source of tropospheric reactive halogens is from sea salt aerosol and sea water (Chen et al., 2017; Sherwen et al., 2016; Wang et al., 2019) (Fig. 2). In the model, the global annual-mean lifetime of NOx in the troposphere against oxidation to nitrate is about 1 d; about 50 % of this loss is from the reaction of NO2 + OH. NOx loss from N2O5 becomes more important near the surface where aerosol surface area is relatively high. The global annual-mean lifetime of nitrate in the troposphere against wet and dry deposition to the surface is about 3 d.

Figure 2Annual-mean fraction of NO2 formation from the oxidation of NO in the troposphere below 1 km altitude in the cloud chemistry model.
For both the cloud chemistry and standard simulations, the two most important nitrate formation pathways are NO2 + OH (41 %–42 %) and N2O5 hydrolysis (28 %–41 %), the latter of which is dominant over the mid- to high northern continental latitudes during winter, where both NOx emissions and aerosol abundances are relatively large (Figs. 1 and 3). The cloud chemistry simulation results in an equal importance of nitrate formation via NO2 + OH and N2O5 hydrolysis (both 41 %) due to increases in the rate of N2O5 uptake in clouds and decreases in the importance of NO2 hydrolysis, which can compete with N2O5 formation at night. In the standard model, NO2 hydrolysis represents an important nitrate production mechanism (12 %), but it is negligible in the cloud chemistry simulation due to the reduction in the reaction probability (from = 10−4 to = 10−4 to 10−8) in the model, which is supported by laboratory studies (Burkholder et al., 2015; Crowley et al., 2010; Tan et al., 2016). The formation of HNO3 from the hydrolysis of RONO2 formed from both daytime (NO + RO2) and nighttime (NO3 + MTN/ISOP) reactions represents 6 % of total global nitrate formation (Fig. 1) and is dominant over Amazonia (Fig. 3). RONO2 hydrolysis represents up to 20 % of inorganic nitrate formation in the southeastern US (Fig. 3). This is similar to Fisher et al. (2016), who estimated that formation of RONO2 accounts for up to 20 % of NOx loss in this region during summer, with RONO2 hydrolysis representing 60 % of RONO2 loss. Globally, the formation of inorganic nitrate from the hydrolysis of RONO2 is dominated by RONO2 formation from the daytime reactions (3 %–6 %), while the formation of RONO2 from nighttime reactions represents up to 3 %. The relative importance of nighttime and daytime RONO2 formation is expressed as a range because precursors to RONO2 that formed from monoterpenes can form from both daytime and nighttime reactions, and these precursors are not separately diagnosed in the model output. HNO3 formation from NO3 + HC and the hydrolysis of XNO3 is small globally (5 %–6 %). Although XNO3 hydrolysis is the dominant nitrate formation pathway over the remote oceans (Fig. 3), its contribution to total global nitrate production is relatively small due to small local NOx sources in these regions.
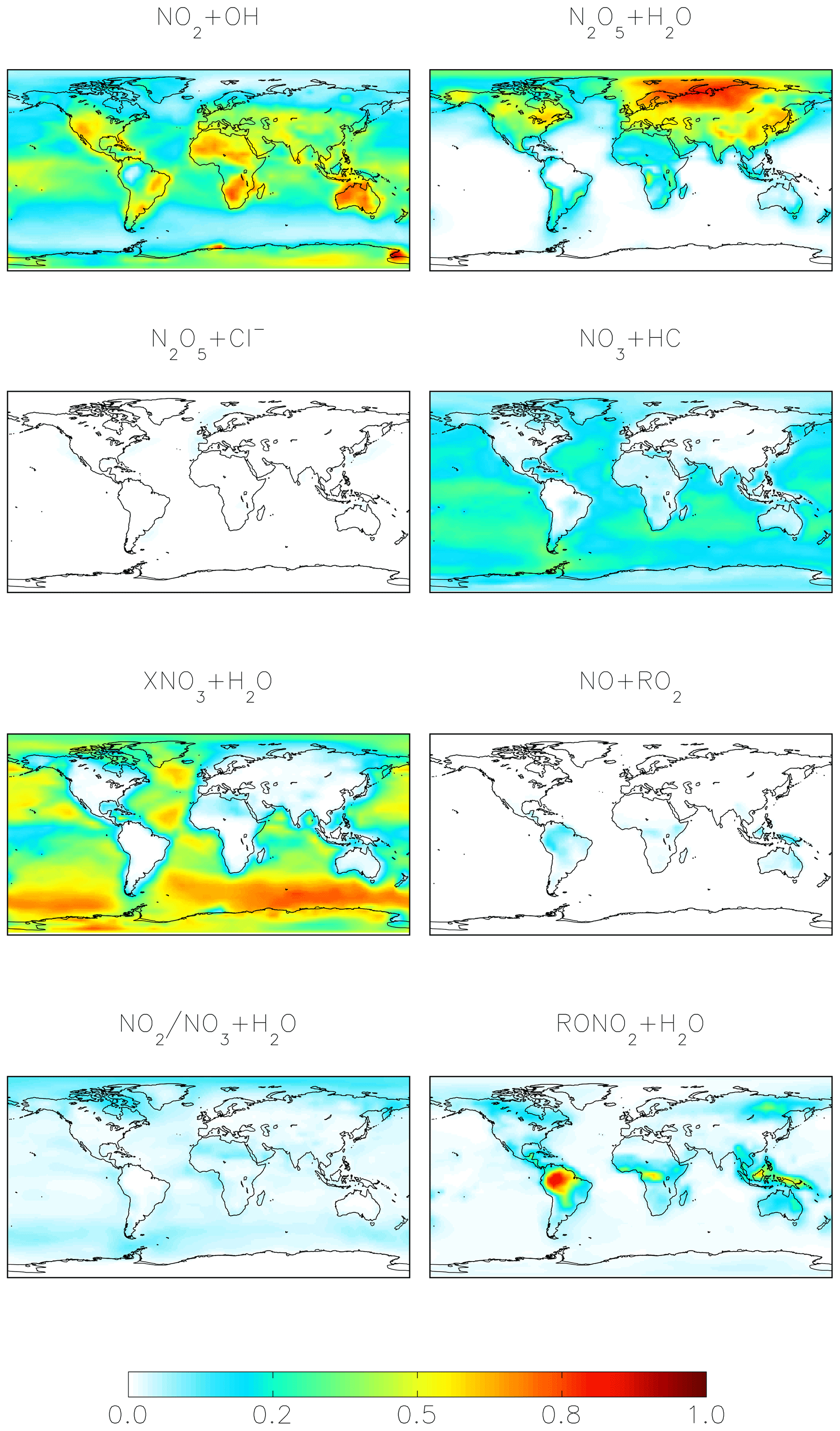
Figure 3Annual-mean fraction of HNO3 formation from the oxidation of NOx in the troposphere below 1 km altitude in the cloud chemistry model.
Figures 4–6 show modeled Δ17O(nitrate) for the cloud chemistry simulation (the standard simulation is shown in Figs. S2–S4). Figure 4 shows modeled annual-mean Δ17O(nitrate) below 1 km altitude (Δ17O(NO2) is shown in Fig. S5). The model predicts an annual-mean range of Δ17O(nitrate) = 4 ‰–33 ‰ near the surface. The lowest values are over Amazonia due to the dominance of RONO2 hydrolysis, and the highest values are over the midlatitude oceans due to the dominance of XNO3 hydrolysis (Figs. 3 and 4).

Figure 4Modeled annual-mean Δ17O(nitrate) below 1 km altitude for the cloud chemistry model.
Figure 5 compares the model with a global compilation of Δ17O(nitrate) observations from around the world. Observations included in Fig. 5 include locations where there is enough data to calculate monthly means (McCabe et al., 2006; Kunasek et al., 2008; Hastings et al., 2003; Kaiser et al., 2007; Michalski et al., 2003; Guha et al., 2017; Savarino et al., 2013, 2007; Ishino et al., 2017; Alexander et al., 2009; He et al., 2018b; Fibiger et al., 2013; Wang et al., 2014). Figure 6 compares the seasonality in modeled Δ17O(nitrate) to the observations where samples were collected over the course of approximately 1 year (McCabe et al., 2006; Kunasek et al., 2008; Kaiser et al., 2007; Michalski et al., 2003; Guha et al., 2017; Savarino et al., 2013, 2007; Ishino et al., 2017; Alexander et al., 2009). In contrast to Alexander et al. (2009), the model does not significantly underestimate the Δ17O(nitrate) observations when assuming a bulk ozone isotopic composition (Δ17O(O3)) on the order of 25 ‰ (see Fig. 2d in Alexander et al., 2009). The increase in modeled Δ17O(nitrate) is due to increased importance of O3 in NOx cycling (85 % below 1 km) compared to Alexander et al. (2009) (80 % below 1 km altitude), as well as an increase in the number and fractional importance of nitrate formation pathways that yield relatively high values of Δ17O(nitrate) (red pathways in Fig. 1). Although XO species themselves are only a minor NO oxidation pathway (1 %), the addition of reactive halogen chemistry in the model has altered the relative abundance of O3 and HOx (Sherwen et al., 2016) in such a way as to increase the modeled Δ17O(NOx). The Alexander et al. (2009) study used GEOS-Chem v8-01-01, which included tropospheric nitrate formation from the NO + OH, N2O5 + H2O, and NO3 + HC pathways only. An increased importance of N2O5 hydrolysis (R4) and additional nitrate formation pathways that yield relatively high values of Δ17O(nitrate) (R5, R6, R8, and R10) in the present study also explain the increase in modeled Δ17O(nitrate) relative to Alexander et al. (2009). An increase in the average A value from 0.80 to 0.85 would tend to increase the calculated Δ17O(nitrate) on the order of 2 ‰ (, suggesting that the increase in the relative importance of the terminal reactions R4, R5, R6, R8, and R10 explains the majority of the difference between the results presented here compared to Alexander et al. (2009). Assuming a value of 35 ‰ for bulk Δ17O(O3) in the model that did not include reactive halogen chemistry or heterogeneous reactions in cloud water produced good agreement between modeled and observed Δ17O(nitrate) in Alexander et al. (2009); however, in the current version of the model this bulk isotopic assumption leads to a model overestimate at nearly all locations (Fig. S6). The cloud chemistry model shows somewhat better agreement with the observations (R2=0.51 in Fig. 5) compared to the standard model (R2=0.48 in Fig. S3). Improved agreement with the observations occurs in the mid- to high latitudes (Figs. 6 and S4) and is due to addition of N2O5 hydrolysis in clouds (Figs. 3 and S6).
The mean Δ17O(nitrate) value of the observations (27.7±5.0 ‰) shown in Fig. 5 is not significantly different from the modeled values at the location of the observations (28.6±4.5 ‰); however, the range of Δ17O(nitrate) values of the observations (10.9 ‰–40.6 ‰) is larger than in the model (19.6 ‰–37.6 ‰). As previously noted in Savarino et al. (2007), the maximum observedΔ17O(nitrate) value (40.6 ‰) is not possible given our isotope assumption for the terminal oxygen atom of ozone ( = 39 ‰); however, it is theoretically possible given the approximately 2 ‰ uncertainty in observed . A value of Δ17O(nitrate) = 41 ‰ is possible if = 41 ‰ and all oxygen atoms of nitrate originate from ozone (A=1 and all nitrate forms from R2 and/or R5). Although this may be possible for nitrate formed locally in the Antarctic winter due to little to no sunlight, lack of local NOx sources during Antarctic winter makes it unlikely that all nitrate observed in Antarctica forms locally. Long-range transport from lower latitudes and/or the stratosphere likely contributes to nitrate observed in Antarctica during winter (Lee et al., 2014). Observed Δ17O(nitrate) > 39 ‰ (in Antarctica) has been suggested to be due to transport of nitrate from the stratosphere (Savarino et al., 2007), as stratospheric O3 is expected to have a higher Δ17O(O3) value than ozone produced in the troposphere (Krankowsky et al., 2000; Mauersberger et al., 2001; Lyons, 2001). Indeed, the model underestimates the observations at Dumont d'Urville (DDU) and the South Pole (both in Antarctica) during winter and spring (Fig. 6), where and when the stratospheric contribution is expected to be most important (Savarino et al., 2007). The model underestimate in Antarctica may also be due to model underestimates of the BrO column (Chen et al., 2017) and ozone abundance (Sherwen et al., 2016) in the southern high latitudes. The largest model overestimates occur at Mt. Lulin, Taiwan (Figs. 5 and 6). Based on nitrogen isotope observations (δ15N), nitrate at Mt. Lulin is thought to be influenced by anthropogenic nitrate emitted in polluted areas of mainland China and transported to Mt. Lulin rather than local nitrate production (Guha et al., 2017). However, observations of Δ17O(nitrate) in autumn and winter in Beijing suggest much higher values (30.6±1.8 ‰) than was measured at Mt. Lulin (15 ‰–30 ‰ in winter). A potential reason for the model overestimate of the observed values at Mt. Lulin could be qualitatively explained by transport of nitrate formed in the free troposphere to this high-altitude location, where the high-Δ17O(nitrate)-producing pathways (R4–R8) should be negligible due to minimal aerosol surface area for heterogeneous chemistry. Low Δ17O(nitrate) values from nitrate formed at higher altitudes and transported to Mt. Lulin would not be accounted for in the model since the isotopes are not transported. The model compares better to the midlatitude locations close to pollution sources (La Jolla and Princeton), although the model overestimates wintertime Δ17O(nitrate) in Princeton, NJ, USA, by up to 6 ‰ and underestimates winter time Δ17O(nitrate) in La Jolla, CA, USA, by up to 4 ‰ . The model overestimate at Princeton during winter could be due to the fact that these are precipitation samples and not ambient aerosol samples, and thus may reflect nitrate formed at altitudes higher than we are sampling in the model. The underestimate at La Jolla, CA, could be due to underestimates in reactive chlorine chemistry in the model, which would tend to increase Δ17O(nitrate) by increasing nitrate formation by the hydrolysis of halogen nitrates (R6) in this coastal location. The model underestimates the Δ17O(nitrate) observations at Cabo Verde in late summer/early autumn by up to 6 ‰ (Savarino et al., 2013). Comparison with results from the steady-state model employed in Savarino et al. (2013) suggests that the low bias could be due to an underestimate of nitrate formation via NO3 + DMS (R2). The steady-state model in Savarino et al. (2013) agreed with observations when R2 represented about one-third of total nitrate formation. The model results presented here have R2 representing about 15 % of total nitrate formation in this season. An underestimate of the relative importance of R2 could result from a model underestimate of atmospheric DMS abundances.
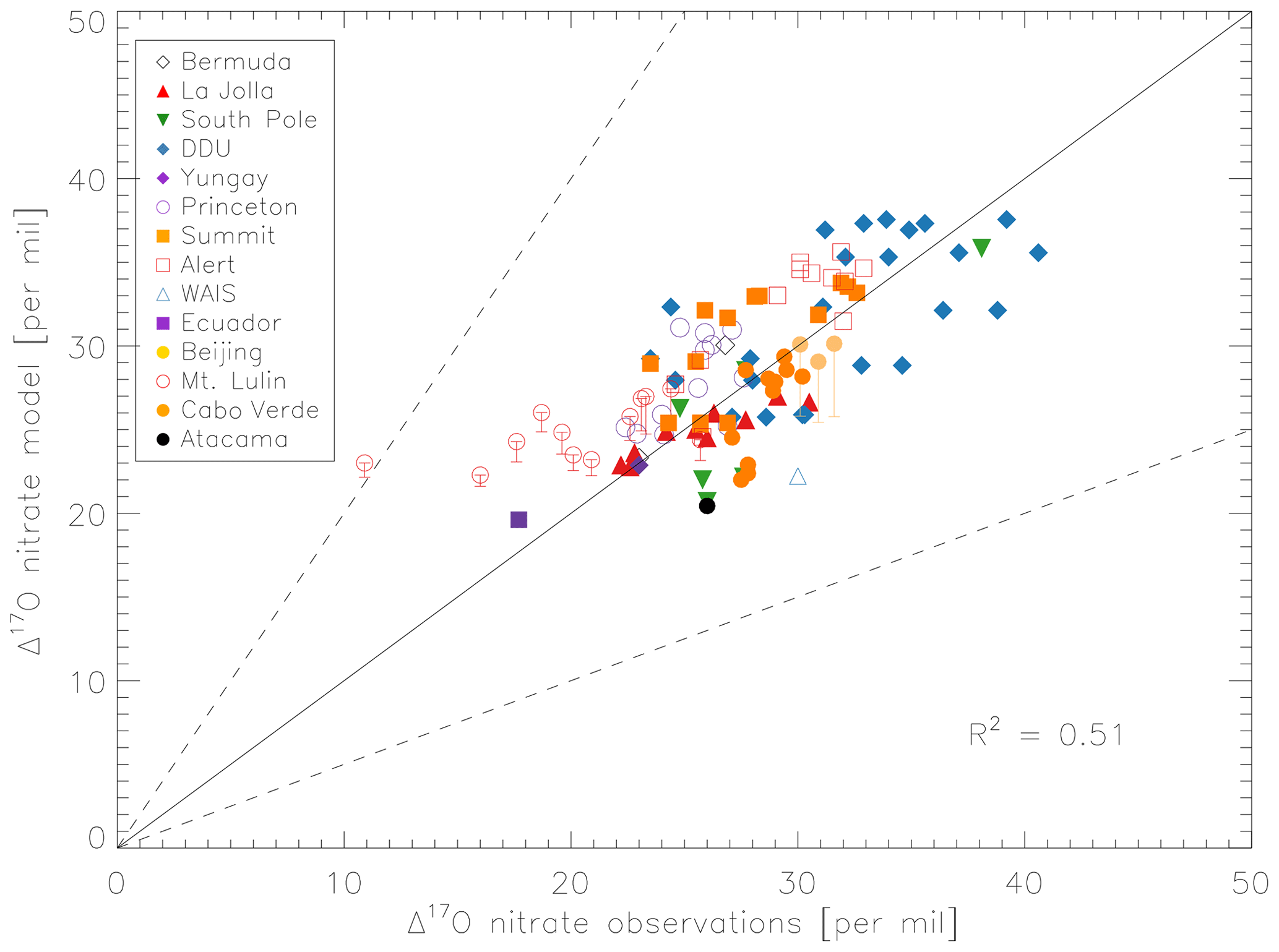
Figure 5Comparison of monthly-mean modeled (cloud chemistry) and observed Δ17O(nitrate) at locations where there are enough observations to calculate a monthly mean. References for the observations are in the text. The error bars represent different assumptions for calculated modeled A values for nighttime reactions as described in the text. Error bars for Beijing and Mt. Lulin reflect the range of possible modeled A values for nighttime reactions as described in the text. The y=x (solid line) and y=2x and y=0.5x (dashed) are shown.
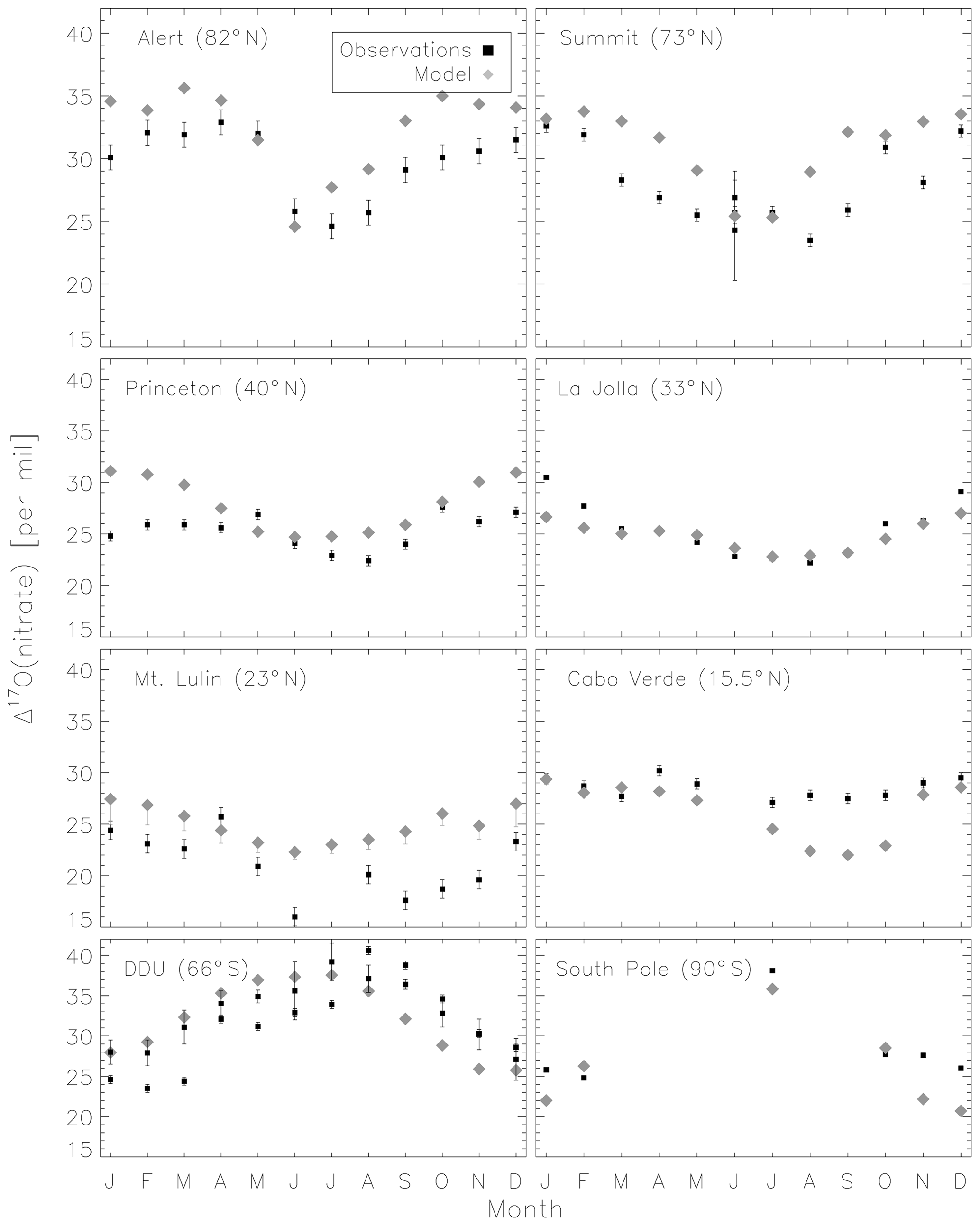
Figure 6Comparison of monthly-mean modeled (cloud chemistry) and observed Δ17O(nitrate). Error bars for model results from Mt. Lulin reflect the range of possible modeled A values for nighttime reactions as described in the text. Error bars for the observations reflect the analytical uncertainty in the measurements, except for two data points in June for Summit which reflect the standard deviation of Δ17O(nitrate) from multiple measurements during that month.
The uncertainty in the two most important nitrate formation pathways, NO2 + OH and N2O5 hydrolysis, and their impacts on NOx and oxidant budgets have been examined and discussed elsewhere (Macintyre and Evans, 2010; Newsome and Evans, 2017; Holmes et al., 2019). The impacts of the formation and hydrolysis of halogen nitrates on global NOx and oxidant budgets have also been previously examined (Sherwen et al., 2016). Here we focus on three additional processes using a set of model sensitivity studies. First, we examine the importance of the third most important nitrate production pathway on the global scale as predicted by the standard model, NO2 aerosol uptake and hydrolysis, and its implications for the global NOx, nitrate, and oxidant budgets. Second, we examine the role of changing anthropogenic NOx emissions over a 15-year period (2000 to 2015) on the relative importance of the formation of inorganic nitrate from the hydrolysis of organic nitrates. Finally, we examine the role of aerosol nitrate photolysis on the relative importance of different nitrate formation pathways. The impact of aerosol nitrate photolysis on NOx and oxidant budgets has been examined in detail elsewhere (Kasibhatla et al., 2018).
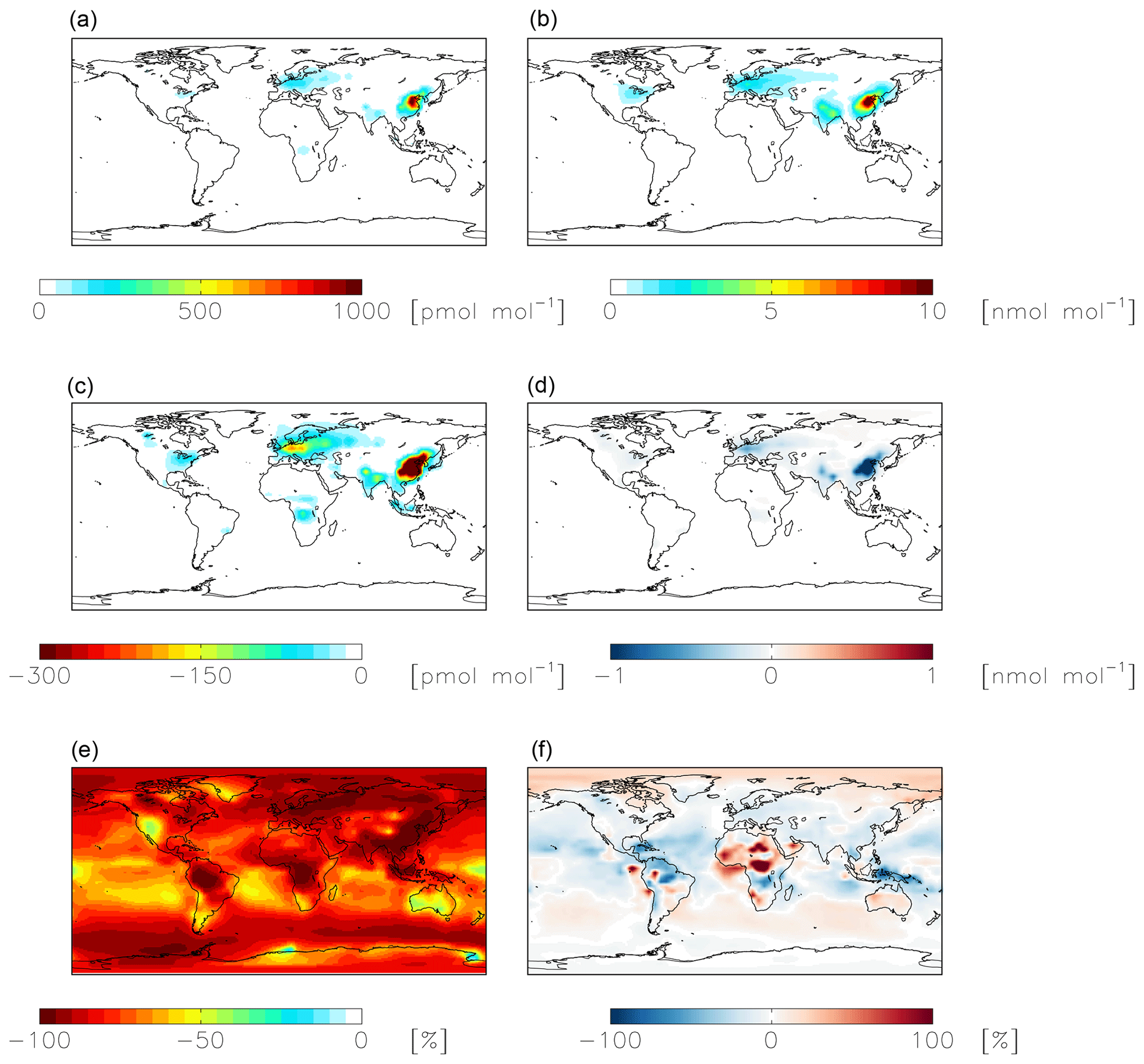
Figure 7Modeled annual-mean HONO (a, c, e) and fine-mode nitrate (b, d, f) concentrations below 1 km altitude in the standard simulation (a, b) with = 10−4 for NO2 hydrolysis. Absolute (c, d) and relative (e, f) change in concentrations below 1 km altitude between the standard model and the model simulation with = 10−7. Negative numbers represent a decrease relative to the standard simulation.
4.1 Heterogeneous uptake and hydrolysis of NO2
Heterogeneous uptake of NO2 to form HNO3 and HONO is the third most important nitrate formation pathway in the standard model on the global scale (Fig. 1). The reaction probability () measured in laboratory studies ranges between 10−8 and 10−4 depending on aerosol chemical composition (Lee and Tang, 1988; Crowley et al., 2010; Gutzwiller et al., 2002; Yabushita et al., 2009; Abbatt and Waschewsky, 1998; Burkholder et al., 2015; Bröske et al., 2003; Li et al., 2018a; Xu et al., 2018). A value of = 10−4 is used in the standard model, which is at the high end of the reported range. A molar yield of 0.5 for both HNO3 and HONO formation is assumed in the model based on laboratory studies and hypothesized reaction mechanisms (Finlayson-Pitts et al., 2003; Jenkin et al., 1988; Ramazan et al., 2004; Yabushita et al., 2009). However, both the reaction rate and mechanism of this reaction and its dependence on chemical composition and pH are still not well understood (Spataro and Ianniello, 2014).
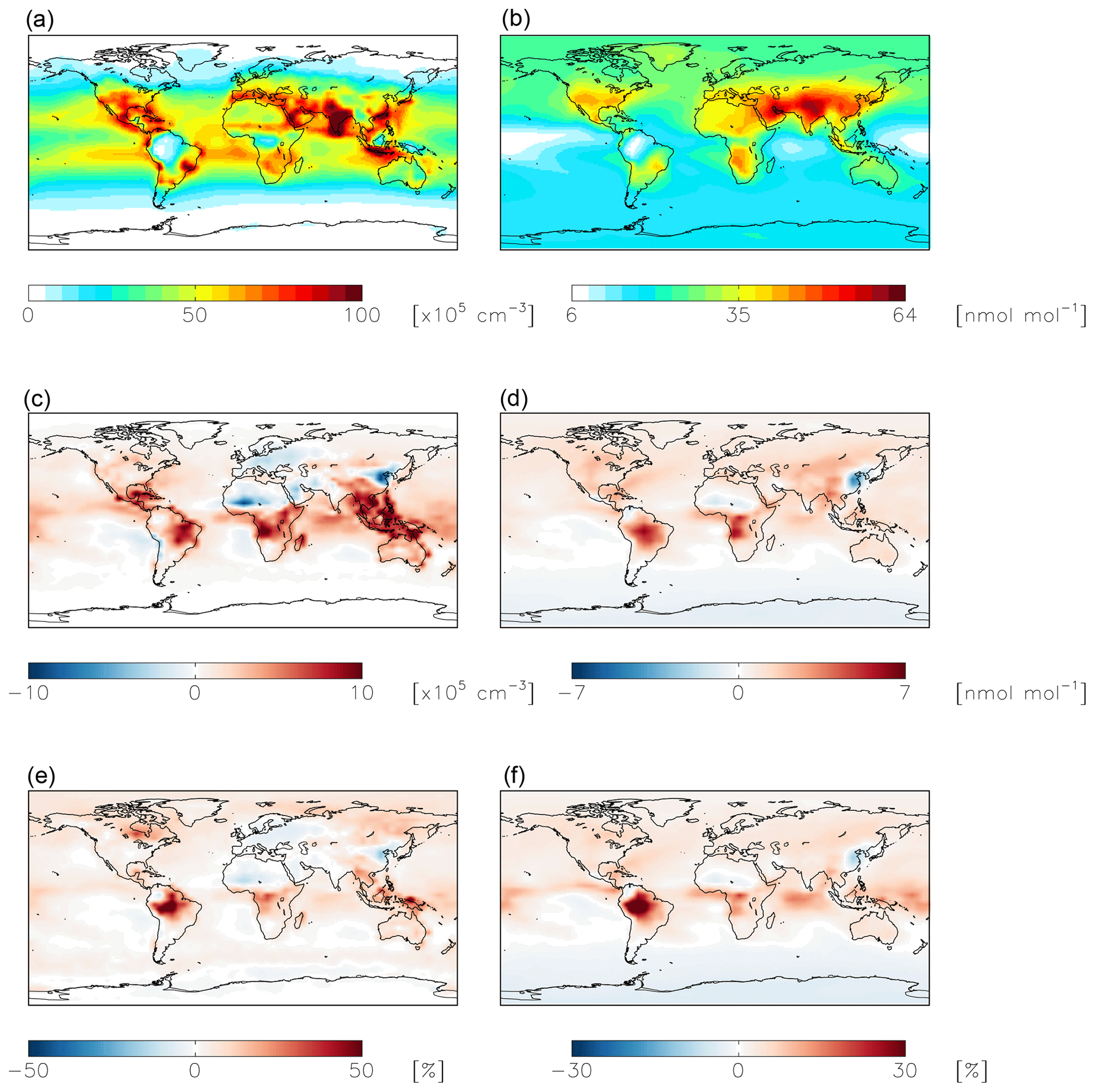
Figure 8Same as Fig. 7 except for OH (a, c, e) and ozone (b, d, f).
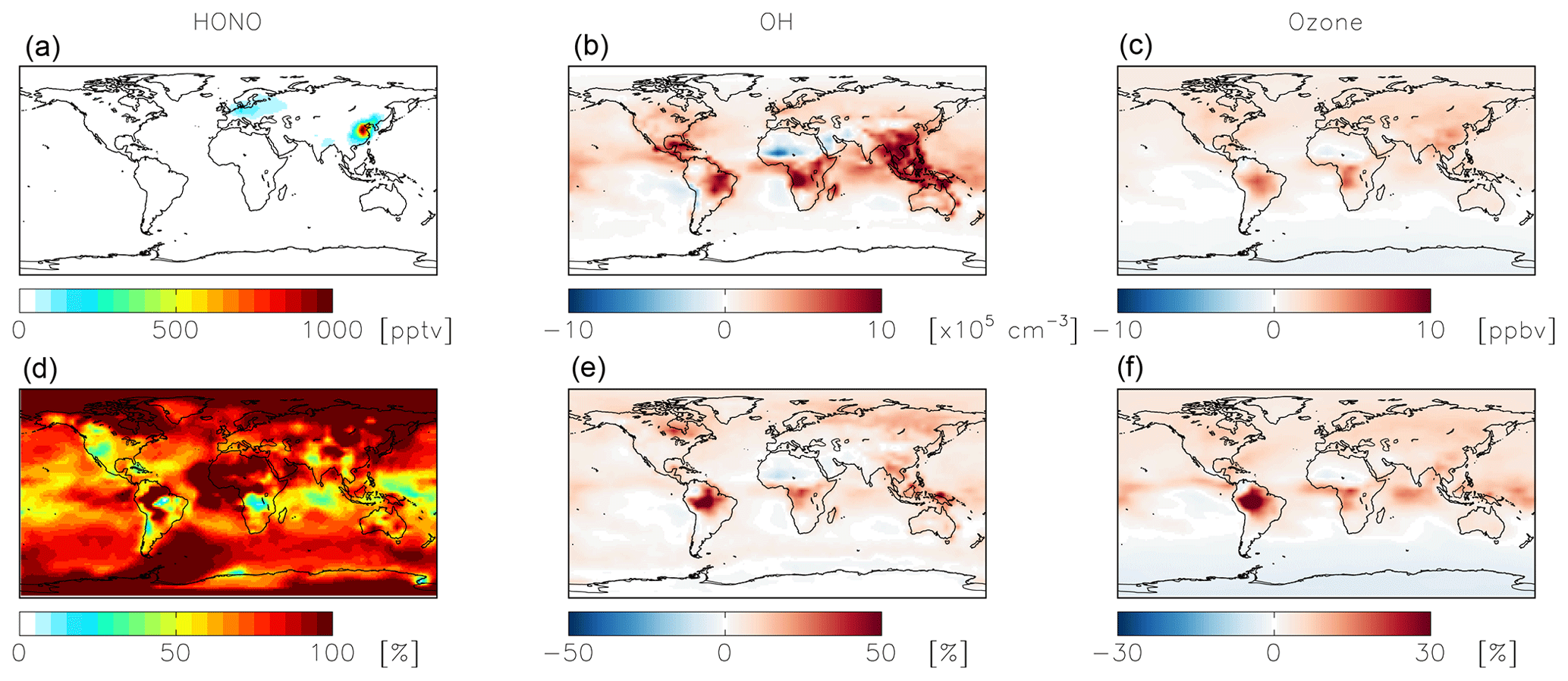
Figure 9Absolute (a, b, c) and relative (d, e, f) change in HONO (a, d), OH (b, e), and ozone (c, f) concentrations below 1 km altitude between the standard model and the model simulation with an acidity-dependent yield from NO2 hydrolysis. Positive numbers represent an increase relative to the standard simulation.
The cloud chemistry simulation uses a reaction probability formulation for aerosol uptake of NO2 ( that depends on aerosol chemical composition, ranging from ( = 10−8 for dust to () = 10−4 for black carbon based on recent laboratory studies (Holmes et al., 2019). The updated NO2 reaction probability results in a negligible (<1 %) importance of this reaction for nitrate formation, compared to a 12 % contribution in the standard model. The cloud chemistry simulation significantly increases the fractional importance of N2O5 hydrolysis (from 28 % to 41 %, globally below 1 km altitude) compared to the standard simulation, in part due to decreased competition from NO2 hydrolysis and in part due to increased N2O5 hydrolysis in clouds. To evaluate the relative importance of competition from NO2 hydrolysis and the addition of N2O5 hydrolysis in clouds, we perform a model sensitivity study that is the same as the standard simulation but decreases the reaction probability of NO2 hydrolysis on aerosol (N2O5 = 10−7), without adding N2O5 hydrolysis in clouds. Similar to the cloud chemistry simulation, using N2O5 = 10−7 renders NO2 hydrolysis a negligible nitrate formation pathway and increases the relative importance of N2O5 hydrolysis from 28 % to 37 %. This suggests that reduced competition from NO2 hydrolysis is the main reason for the increased importance of N2O5 hydrolysis in the cloud chemistry simulation, though the addition of heterogeneous reactions on clouds also plays a role.
NO2 hydrolysis represents a significant source of HONO in the standard model simulation; the reduced NO2 reaction probability from N2O5 = 10−4 to N2O5 = 10−7 results in a reduction of HONO below 1 km altitude by up to 100 % over the continents, with relatively small (up to 1 ppb) changes in nitrate concentrations (Fig. 7). The reduction in the rate of heterogeneous NO2 uptake leads to reductions in OH where this reaction was most important in the model (over China and Europe) due to reductions in HONO but leads to increases in OH elsewhere due to increases in ozone (by up to a few ppb) resulting from small increases in the NOx lifetime due to a reduction in the NOx sink (Fig. 8). Similar changes in HONO are seen when comparing the standard and cloud chemistry simulation (not shown). Increased importance of N2O5 hydrolysis in both the cloud chemistry simulation and the simulation without cloud chemistry but with a reduced reaction probability for NO2 hydrolysis increases modeled annual-mean Δ17O(nitrate) by up to 3 ‰ in China, where this reaction is most important. This improves model agreement with monthly-mean observations of Δ17O(nitrate) in Beijing (He et al., 2018a) (Figs. 5 and S3).

Figure 10Modeled annual-mean difference in the fractional production rate of HNO3 from the hydrolysis of organic nitrate below 1 km attitude in the year 2015 relative to 2000 (2015–2000).
The product yields of NO2 hydrolysis are also uncertain. Jenkin et al. (1988) proposed the formation of a water complex, NO2⋅H2O, leading to the production of HONO and HNO3. Finlayson-Pitts et al. (2003) and Ramazan et al. (2004) proposed the formation of the dimer N2O4 on the surface, followed by isomerization to form . Reaction of with H2O results in the formation of HONO and HNO3. Laboratory experiments by Yabushita et al. (2009) suggested that dissolved anions catalyzed the dissolution of NO2 to form a radical intermediate (where X = Cl, Br, or I) at the surface followed by reaction with NO2(g) to form HONO and . These experiments described above were performed at NO2 concentrations much higher than exist in the atmosphere (10–100 ppm) (Yabushita et al., 2009; Finlayson-Pitts et al., 2003; Ramazan et al., 2004). A laboratory study utilizing isotopically labeled water to investigate the reaction mechanism suggested that the formation of HONO resulted from the reaction between adsorbed NO2 and H+, while the formation of HNO3 resulted from the reaction between adsorbed NO2 and OH−, and did not involve the N2O4 intermediate (Gustafsson et al., 2009). Results from Gustafsson et al. (2009) suggest an acidity-dependent yield of HONO and HNO3, favoring HONO at low pH values. A recent study in the northeastern US during winter found that modeled nitrate abundance was overestimated using a molar yield of 0.5 for HONO and HNO3, and the model better matched the observations of NO2 and nitrate when assuming a molar yield of 1.0 for HONO (Jaeglé et al., 2018). Particles were acidic (pH<2) during this measurement campaign (Guo et al., 2017; Shah et al., 2018), which may favor HONO production over HNO3.
We examine the potential importance of this acidity-dependent yield by implementing a pH-dependent product yield in two separate sensitivity simulations, first using an NO2 aerosol uptake reaction probability of γ = 10−4 as in the standard simulation and second with = 10−7. The acidity-dependent yield for HONO and HNO3 formation is based on the laboratory study by Gustafsson et al. (2009). We use aerosol pH calculated from ISORROPIA II (Fountoukis and Nenes, 2007) to calculate the concentration of H+ and OH− in aerosol water. The yield of HONO (ΥHONO) from heterogeneous uptake of NO2 on aerosol surfaces is calculated according to Eq. (3):
where [H+] and [OH−] are in units of M. The yield of HNO3 from this reaction is equal to (1−ΥHONO). Eq. (3) yields values of ΥHONO near unity for aerosol pH values less than 6, decreasing rapidly to zero between pH values of 6 and 8 (Fig. S8). Calculated aerosol pH values are typically <6 in the model except in remote regions far from NOx sources (Fig. S9), favoring the product HONO.
The acidity-dependent yield implemented in the standard simulation with = 10−4 increases HONO concentrations by up to 1 ppbv in China, where this reaction is most important (Fig. 9). Fractional increases in HONO exceed 100 % in remote locations (Fig. 9). Increased HONO leads to increases in OH on the order of 10 %–20 % in most locations below 1 km altitude, while ozone concentrations increase in most locations by up to several ppbv (Fig. 9). The exception is the southern high latitudes, likely due to decreased formation and thus transport of nitrate to remote locations. The impact on NOx and nitrate budgets is relatively minor. The global annual-mean NOx burden near the surface (below 1 km) increases slightly (+2 %) as a result of the decreased rate of conversion of NO2 to nitrate; the change to the global tropospheric burden is negligible. Annual-mean surface nitrate concentrations show small decreases up to 1 ppbv in China, where this reaction is most important in the model; impacts on nitrate concentrations over a shorter time period may be more significant (Jaeglé et al., 2018). The fraction of HNO3 formed from NO2+OH (49 %) increases due to increases in OH from the HONO source. The fraction of HNO3 formation from the uptake and hydrolysis of N2O5 also increases (from 28 % to 32 %) due to reductions in the nighttime source of nitrate from NO2 hydrolysis. The calculated mean Δ17O(nitrate) at the location of the observations shown in Fig. 5 (27.9±5.0 ‰) is not significantly impacted due to compensating effects from changes in both high- and low-producing Δ17O(nitrate) values. Modeled monthly-mean Δ17O(nitrate) in China, where NO2 hydrolysis is most important, decreases by 0.9 ‰–1.9 ‰ and is biased low by 1.8 ‰–3.4 ‰ .
Using a combination of both the low reaction probability (γ = 10−7) and the acidity-dependent yield gives similar results to using γ = 10−7 and assuming a molar yield of 0.5 for HONO and HNO3 (not shown). In other words, including a pH-dependent product yield rather than a yield of 0.5 for HONO and nitrate results in negligible differences for oxidants, NOx and nitrate abundances when the reaction probability ( is low.
4.2 Hydrolysis of organic nitrates (RONO2)
Anthropogenic NOx emissions have been increasing in China and decreasing in the US and Europe (Richter et al., 2005; Hoesly et al., 2018), with implications for the relative importance of inorganic and organic nitrate formation as a sink for NOx (Zare et al., 2018). To examine the impacts of recent changes in anthropogenic NOx emissions for nitrate formation pathways, we run the standard model using the year 2000 emissions and meteorology after a 1-year model spin-up and compare the results to the standard model simulation run in the year 2015. This time period encompasses significant changes in anthropogenic NOx emissions in the US, Europe, and China and encompasses most of the time period of the observations shown in Figs. 5 and 6. Total global anthropogenic emissions of NOx are slightly lower in the year 2000 simulation (30 Tg N yr−1) compared to the year 2015 simulation (31 Tg N yr−1) due to decreases in North America and Europe, counteracted by increases in Asia (Fig. S10). This leads to increases of less than 10 % in the annual-mean, fractional importance of the source of nitrate from the hydrolysis of organic nitrates in the US and corresponding decreases of less than 10 % over China (Fig. 10). Relatively small changes (<10 %) in nitrate formation pathways yield small changes (<2 ‰) in modeled annual-mean Δ17O(nitrate) between the year 2000 and 2015; differences in Δ17O(nitrate) over shorter time periods may be larger. Changes in the formation of nitrate from the hydrolysis of RONO2 remain unchanged globally, as increases in the US and Europe and decreases in China counteract one another.
4.3 Photolysis of aerosol nitrate
Observations have demonstrated that aerosol nitrate can be photolyzed at rates much faster than HNO3(g) (Reed et al., 2017; Ye et al., 2016); however, the magnitude of the photolytic rate constant is uncertain. We examine the implications of this process for global nitrate formation pathways by implementing the photolysis of aerosol nitrate as described in Kasibhatla et al. (2018) into the standard model simulation, scaling the photolytic rate constant for both fine- and coarse-mode aerosol nitrate to a factor of 25 times higher than that for HNO3(g) (Kasibhatla et al., 2018; Romer et al., 2018), with a molar yield of 0.67 for HONO and 0.33 for NOx production. The global annual-mean NOx burden near the surface (below 1 km) increases slightly (+2 %) as a result of the photolytic recycling of nitrate to NOx, similar to Kasibhatla et al. (2018). Aerosol nitrate photolysis results in only small impacts on the relative importance of nitrate formation pathways (<2 %), likely due to simultaneous increases in O3 and OH (Kasibhatla et al., 2018), which in turn yields small impacts on calculated Δ17O(nitrate) at the location of the observations shown in Fig. 5 (27.9±5.0 ‰). Nitrate photolysis itself has minimal impact on Δ17O(nitrate) because it is a mass-dependent process (McCabe et al., 2005).
Observations of Δ17O(nitrate) can be used to help quantify the relative importance of different nitrate formation pathways. Interpretation of Δ17O(nitrate) requires knowledge of Δ17O(O3). Previous modeling studies showed good agreement between observed and modeled Δ17O(nitrate) when assuming a bulk oxygen isotopic composition of ozone (Δ17O(O3)) of 35 ‰ based on laboratory and modeling studies (Morton et al., 1990; Thiemens, 1990; Lyons, 2001). However, recent and spatially widespread observations of Δ17O(O3) have consistently shown Δ17O(O3) = 26±1 ‰, suggesting that models are underestimating the role of ozone relative to HOx in NOx chemistry. We utilize a global compilation of observations of Δ17O(nitrate) to assess the representation of nitrate formation in a global chemical transport model (GEOS-Chem), assuming that the bulk oxygen isotopic composition of ozone (Δ17O(O3) = 26 ‰. The modeled Δ17O(nitrate) is roughly consistent with observations, with a mean modeled and observed Δ17O(nitrate) of (28.6±4.5 ‰) and (27.6±5.0 ‰), respectively, at the locations of the observations. Improved agreement between modeled and observed Δ17O(nitrate) is due to increased importance of ozone versus HO2 and RO2 in NOx cycling and an increase in the number and importance of nitrate production pathways that yield high Δ17O(nitrate) values. The former may be due to implementation of tropospheric reactive halogen chemistry in the model, which impacts ozone and HOx abundances. The latter is due mainly to increases in the relative importance of N2O5 hydrolysis, with the hydrolysis of halogen nitrates also playing an important role in remote regions.
The main nitrate formation pathways in the model below 1 km altitude are from NO2+OH and N2O5 hydrolysis (both 41 %). The relative importance of global nitrate formation from the hydrolysis of halogen nitrates and hydrogen-abstraction reactions involving the nitrate radical (NO3) is of similar magnitude (∼5 %). The formation of nitrate from the hydrolysis of organic nitrate has increased slightly in the US and decreased in China (changes <10 %) due to changing NOx emissions from the year 2000 to 2015, although the global mean fractional importance (6 %) remains unchanged as the regional changes counteract one another. Nitrate formation via heterogeneous NO2 and NO3 uptake and NO2 + HO2 are negligible (<2 %). Although aerosol nitrate photolysis has important implications for O3 and OH, the impacts on nitrate formation pathways are small.
The model parameterization for heterogeneous uptake of NO2 has significant impacts on HONO and oxidants (OH and ozone) in the model. HONO production from this reaction has been suggested to be an important source of OH in Chinese haze due to high NOx and aerosol abundances (Hendrick et al., 2014; Tong et al., 2016; Wang et al., 2017), with implications for the gas-phase formation of sulfate aerosol from the oxidation of sulfur dioxide by OH (Shao et al., 2019; Li et al., 2018b). More recent laboratory studies suggest that the reaction probability of NO2 on aerosols is lower than that previously used in the model. Using an NO2 reaction probability formulation that depends on the chemical composition of aerosols as described in Holmes et al. (2019) renders this reaction negligible for nitrate formation and has significant implications for modeled HONO, ozone, and OH. Although uncertainty also exists in the relative yield of nitrate and HONO from this reaction, the impacts of this assumption are negligible when we use these updated NO2 reaction probabilities. Observations of Δ17O(nitrate) in Chinese haze events during winter (He et al., 2018b) may help to quantify the importance of this nitrate production pathway in a region where the model predicts it is significant.
The GEOS-Chem model is available at: https://doi.org/10.5281/zenodo.1343547 (last access: 10 August 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-3859-2020-supplement.
BA designed the study and performed the model simulations and calculations. All other authors provided model code and contributed to writing and analysis.
The authors declare that they have no conflict of interest.
Becky Alexander acknowledges helpful discussions with Joel Savarino and Ron Cohen.
This research has been supported by the NSF AGS (grant nos. 1644998 and 1702266), the NASA New Investigator Program (grant no. NNX16AI57G), and the Australian Research Council (grant no. DP160101598).
This paper was edited by Jan Kaiser and reviewed by Greg Michalski and two anonymous referees.
Abbatt, J. P. D. and Waschewsky, G. C. G.: Heterogeneous interactions of OHBr, HNO3, O3, and NO2 with deliquescent NaCl aerosols at room temperature, J. Phys. Chem. A, 102, 3719–3725, 1998.
Alexander, B., Savarino, J., Kreutz, K. J., and Thiemens, M. H.: Impact of preindustrial biomass-burning emissions on the oxidation pathways of tropospheric sulfur and nitrogen, J. Geophys. Res., 109, D08303, https://doi.org/10.1029/2003JD004218, 2004.
Alexander, B., Hastings, M. G., Allman, D. J., Dachs, J., Thornton, J. A., and Kunasek, S. A.: Quantifying atmospheric nitrate formation pathways based on a global model of the oxygen isotopic composition (Δ17O) of atmospheric nitrate, Atmos. Chem. Phys., 9, 5043–5056, https://doi.org/10.5194/acp-9-5043-2009, 2009.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Berhanu, T. A., Savarino, J., Bhattacharya, S. K., and Vicars, W. C.: 17O excess transfer during the NO2 + O3–> NO3 + O2 reaction, J. Chem. Phys., 136, 044311, https://doi.org/10.1063/1.3666852, 2012.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res., 106, 23073–23095, 2001.
Bhattacharya, S. K., Pandey, A., and Savarino, J.: Determination of intramolecular isotope distribution of ozone by oxidation reaction with silver metal, J. Geophys. Res, 113, D03303, https://doi.org/10.1029/2006JF008309, 2008.
Bhattacharya, S. K., Savarino, J., Michalski, G., and Liang, M.-C.: A new feature in the internal heavy isotope distribution in ozone, J. Chem. Phys., 141, 134301, https://doi.org/10.1063/1.4895614, 2014.
Brenninkmeijer, C. A. M., Janssen, C., Kaiser, J., Rockmann, T., Rhee, T. S., and Assonov, S. S.: Isotope effects in the chemistry of atmospheric trace compounds, Chem. Rev., 102, 5125–5161, 2003.
Bröske, R., Kleffmann, J., and Wiesen, P.: Heterogeneous conversion of NO2 on secondary organic aerosol surfaces: A possible source of nitrous acid (HONO) in the atmosphere?, Atmos. Chem. Phys., 3, 469–474, https://doi.org/10.5194/acp-3-469-2003, 2003.
Brothers, L. A., Dominguez, G., Fabian, P., and Thiemens, M. H.: Using multi-isotope tracer methods to understand the sources of nitrate in aerosols, fog and river water in Podocarpus National Forest, Ecuador, Eos Trans. AGU, 89, Abstract A11C-0136, 2008.
Browne, E. C. and Cohen, R. C.: Effects of biogenic nitrate chemistry on the NOx lifetime in remote continental regions, Atmos. Chem. Phys., 12, 11917–11932, https://doi.org/10.5194/acp-12-11917-2012, 2012.
Burkholder, J. B., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies: evaluation number 18, Jet Propulsion Laboratory, Pasadena, CA, 1392 pp., 2015.
Butkovskaya, N. I., Kukui, A., Pouvesle, N., and Le Bras, G.: Formation of Nitric Acid in the Gas-Phase HO2 + NO Reaction: Effects of Temperature and Water Vapor, J. Phys. Chem. A, 109, 6509–6520, https://doi.org/10.1021/jp051534v, 2005.
Chen, Q., Schmidt, J. A., Shah, V., Jaeglé, L., Sherwen, T., and Alexander, B.: Sulfate production by reactive bromine: Implications for the global sulfur and reactive bromine budgets, Geophys. Res. Lett., 44, 7069–7078, https://doi.org/10.1002/2017GL073812, 2017.
Chen, Q., Edebeli, J., McNamara, S. M., Kulju, K., May, N. W., Bertman, S. P., Thanekar, S., Fuentes, J. D., and Pratt, K. A.: HONO, Particulate Nitrite, and Snow Nitrite at a Midlatitude Urban Site during Wintertime, ACS Earth Space Chem., 3, 811–822, https://doi.org/10.1021/acsearthspacechem.9b00023, 2019.
Connell, P. and Johnston, H. S.: Thermal dissociation of N2O5 in N2, Geophys. Res. Lett., 6, 553–556, 1979.
Costa, A. W., Michalski, G., Schauer, A. J., Alexander, B., Steig, E. J., and Shepson, P. B.: Analysis of atmospheric inputs of nitrate to a temperate forest ecosystem from Δ17O isotope ratio measurements, Geophys. Res. Lett., 38, L15805, https://doi.org/10.1029/2011GL047539, 2011.
Crowley, J. N., Ammann, M., Cox, R. A., Hynes, R. G., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V – heterogeneous reactions on solid substrates, Atmos. Chem. Phys., 10, 9059–9223, https://doi.org/10.5194/acp-10-9059-2010, 2010.
Domine, F. and Shepson, P. B.: Air-Snow Interactions and Atmospheric Chemistry, Science, 297, 1506–1510, https://doi.org/10.1126/science.1074610, 2002.
Dubey, M. K., Mohrschladt, R., Donahue, N. M., and Anderson, J. G.: Isotope-specific kinetics of hydroxyl radical (OH) with water (H2O): Testing models of reactivity and atmospheric fractionation, J. Phys. Chem. A, 101, 1494–1500, 1997.
Evans, M. J. and Jacob, D. J.: Impact of new laboratory studies of N2O5 hydrolysis on global model budgets of tropospheric nitrogen oxides, ozone, and OH, Geophys. Res. Lett., 32, L09813, https://doi.org/10.1029/2005GL022469, 2005.
Ewing, S. A., Michalski, G., Thiemens, M., Quinn, R. C., Macalady, J. L., Kohl, S., Wankel, S. D., Kendall, C., McKay, C. P., and Amundson, R.: Rainfall limit of the N cycle on Earth, Global Biogeochem. Cy., 21, GB3009, https://doi.org/10.1029/2006gb002838, 2007.
Fibiger, D. L., Hastings, M. G., Dibb, J. E., and Huey, L. G.: The preservations of atmospheric nitrate in snow at Summit, Greenland, Geophys. Res. Lett., 40, 3484–3489, https://doi.org/10.1002/grl.50659, 2013.
Finlayson-Pitts, B. J., Wingen, L. M., Sumner, A. L., Syomin, D., and Ramazan, K. A.: The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: An integrated mechanism, Phys. Chem. Chem. Phys., 5, 223–242, 2003.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fisher, J. A., Atlas, E. L., Barletta, B., Meinardi, S., Blake, D. R., Thompson, C. R., Ryerson, T. B., Peischl, J., Tzompa-Sosa, Z. A., and Murray, L. T.: Methyl, Ethyl, and Propyl Nitrates: Global Distribution and Impacts on Reactive Nitrogen in Remote Marine Environments, J. Geophys. Res., 123, 12429–12451, https://doi.org/10.1029/2018JD029046, 2018.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-NH-Na+-SO--Cl−-H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Geng, L., Cole-Dai, J., Alexander, B., Erbland, J., Savarino, J., Schauer, A. J., Steig, E. J., Lin, P., Fu, Q., and Zatko, M. C.: On the origin of the occasional spring nitrate peak in Greenland snow, Atmos. Chem. Phys., 14, 13361–13376, https://doi.org/10.5194/acp-14-13361-2014, 2014.
Geng, L., Murray, L. T., Mickley, L. J., Lin, P., Fu, Q., Schauer, A. J., and Alexander, B.: Isotopic evidence of multiple controls on atmospheric oxidants over climate transitions, Nature, 546, 133–136, https://doi.org/10.1038/nature22340, 2017.
Guha, T., Lin, C. T., Bhattacharya, S. K., Mahajan, A. S., Ou-Yang, C.-F., Lan, Y.-P., Hsu, S. C., and Liang, M.-C.: Isotopic ratios of nitrate in aerosol samples from Mt. Lulin, a high-altitude station in Central Taiwan, Atmos. Environ., 154, 53–69, https://doi.org/10.1016/j.atmosenv.2017.01.036, 2017.
Guo, H., Weber, R. J., and Nenes, A.: High levels of ammonia do not raise fine particle pH sufficiently to yield nitrogen oxide-dominated sulfate production, Sci. Rep., 7, 1–7, https://doi.org/10.1038/s41598-017-11704-0, 2017.
Gustafsson, R. J., Kyiakou, G., and Lambert, R. M.: The molecular methanism of tropospheric nitrous acid production on mineral dust surfaces, Chem. Phys. Chem., 9, 1390–1393, https://doi.org/10.1002/cphc.200800259, 2009.
Gutzwiller, L., George, C., Rossler, E., and Ammann, J.: Reaction Kinetics of NO2 with Resorcinol and 2,7-Naphthalenediol in the Aqueous Phase at Different pH, J. Phys. Chem. A, 106, 12045–12050, https://doi.org/10.1021/jp026240d, 2002.
Hastings, M. G., Sigman, D. M., and Lipschultz, F.: Isotopic evidence for source changes of nitrate in rain at Bermuda, J. Geophys. Res., 108, 4790, https://doi.org/10.1029/2003JD003789, 2003.
He, P., Alexander, B., Geng, L., Chi, X., Fan, S., Zhan, H., Kang, H., Zheng, G., Cheng, Y., Su, H., Liu, C., and Xie, Z.: Isotopic constraints on heterogeneous sulfate production in Beijing haze, Atmos. Chem. Phys., 18, 5515–5528, https://doi.org/10.5194/acp-18-5515-2018, 2018a.
He, P., Xie, Z., Chi, X., Yu, X., Fan, S., Kang, H., Liu, C., and Zhan, H.: Atmospheric Δ17O() reveals nocturnal chemistry dominates nitrate production in Beijing haze, Atmos. Chem. Phys., 18, 14465–14476, https://doi.org/10.5194/acp-18-14465-2018, 2018b.
Heidenreich, J. E. and Thiemens, M. H.: A non-mass dependent oxygen isotope effect in the production of ozone from molecular oxygen: The role of molecular symmetry in isotope chemistry, J. Chem. Phys., 84, 2129–2136, 1986.
Hendrick, F., Müller, J.-F., Clémer, K., Wang, P., De Mazière, M., Fayt, C., Gielen, C., Hermans, C., Ma, J. Z., Pinardi, G., Stavrakou, T., Vlemmix, T., and Van Roozendael, M.: Four years of ground-based MAX-DOAS observations of HONO and NO2 in the Beijing area, Atmos. Chem. Phys., 14, 765–781, https://doi.org/10.5194/acp-14-765-2014, 2014.
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-I., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018.
Holmes, C. D., Prather, M. J., and Vinken, G. C. M.: The climate impact of ship NOx emissions: an improved estimate accounting for plume chemistry, Atmos. Chem. Phys., 14, 6801–6812, https://doi.org/10.5194/acp-14-6801-2014, 2014.
Holmes, C. D., Bertram, T. H., Confer, K. L., Graham, K. A., Ronan, A. C., Wirks, C. K., and Shah, V.: The role of clouds in the tropospheric NOx cycle: a new modeling approach for cloud chemistry and its global implications, Geophys. Res. Lett., 46, GL081990, https://doi.org/10.1029/2019GL081990, 2019.
Horowitz, L. W., Fiore, A. M., Milly, G. P., Cohen, R. C., Perring, A., Wooldridge, P. J., Hess, P. G., Emmons, L. K., and Lamarque, J.-F.: Observational constraints on the chemistry of isoprene nitrates over the eastern United States, J. Geophys. Res, 112, D12S08, https://doi.org/10.1029/2006JD007747, 2007.
Hudman, R. C., Moore, N. E., Mebust, A. K., Martin, R. V., Russell, A. R., Valin, L. C., and Cohen, R. C.: Steps towards a mechanistic model of global soil nitric oxide emissions: implementation and space based-constraints, Atmos. Chem. Phys., 12, 7779–7795, https://doi.org/10.5194/acp-12-7779-2012, 2012.
Ishino, S., Hattori, S., Savarino, J., Jourdain, B., Preunkert, S., Legrand, M., Caillon, N., Barbero, A., Kuribayashi, K., and Yoshida, N.: Seasonal variations of triple oxygen isotopic compositions of atmospheric sulfate, nitrate, and ozone at Dumont d'Urville, coastal Antarctica, Atmos. Chem. Phys., 17, 3713–3727, https://doi.org/10.5194/acp-17-3713-2017, 2017.
Jacob, D. J.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, 2000.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jaeglé, L., Steinberger, L., Martin, R. V., and Chance, K.: Global partitioning of NOx sources using satellite observations: Relative roles of fossil fuel combustion, biomass burning and soil emissions, Faraday Discuss., 130, 407–423, https://doi.org/10.1039/b502128f, 2005.
Jaeglé, L., Shah, V., Thornton, J. A., Lopez-Hilfiker, F. D., Lee, B. H., McDuffie, E. E., Fibiger, D., Brown, S. S., Veres, P., Sparks, T. L., Ebben, C. J., Wooldridge, P. J., Kenagy, H. S., Cohen, R. C., Weinheimer, A. J., Campos, T. L., Montzka, D. D., Digangi, J. P., Wolfe, G. M., Hanisco, T., Schroder, J. C., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Sullivan, A. P., Guo, H., and Weber, R. J.: Nitrogen oxides emissions, chemistry, deposition, and export over the Northeast United States during the WINTER aircraft campaign, J. Geophys. Res., 123, 12368–12393, doi.org/10.1029/2018JD029133, 2018.
Jenkin, M. E., Cox, R. A., and Williams, D. J.: Laboratory studies of the kinetics of formation of nitrous acid from the thermal reaction of nitrogen dioxide and water vapor, Atmos. Environ., 22, 487–498, 1988.
Johnston, J. C. and Thiemens, M. H.: The isotopic composition of tropospheric ozone in three environments, J. Geophys. Res., 102, 25395–25404, 1997.
Kaiser, J., Hastings, M. G., Houlton, B. Z., Rockmann, T., and Sigman, D. M.: Triple Oxygen Isotope Analysis of Nitrate Using the Denitrifier Method and Thermal Decomposition of N2O, Anal. Chem., 79, 599–607, 2007.
Kasibhatla, P., Sherwen, T., Evans, M. J., Carpenter, L. J., Reed, C., Alexander, B., Chen, Q., Sulprizio, M. P., Lee, J. D., Read, K. A., Bloss, W., Crilley, L. R., Keene, W. C., Pszenny, A. A. P., and Hodzic, A.: Global impact of nitrate photolysis in sea-salt aerosol on NOx, OH, and O3 in the marine boundary layer, Atmos. Chem. Phys., 18, 11185–11203, https://doi.org/10.5194/acp-18-11185-2018, 2018.
Krankowsky, D., Bartecki, F., Klees, G. G., Mauersberger, K., Schellenback, K., and Stehr, J.: Measurement of heavy isotope enrichment in tropospheric ozone, Geophys. Res. Lett., 22, 1713–1716, 1995.
Krankowsky, D., Lammerzahl, P., and Mauersberger, K.: Isotopic measurements of stratospheric ozone, Geophys. Res. Lett., 27, 2593–2595, 2000.
Kunasek, S. A., Alexander, B., Hastings, M. G., Steig, E. J., Gleason, D. J., and Jarvis, J. C.: Measurements and modeling of Δ17O of nitrate in a snowpit from Summit, Greenland, J. Geophys. Res., 113, D24302, https://doi.org/10.1029/2008JD010103, 2008.
Lee, C., Martin, R. V., van Donkelaar, A., Lee, H., Dickerson, R. R., Hains, J. C., Krotkov, N., Richter, A., innikov, K., and Schwab, J. J.: SO2 emissions and lifetimes: Estimates from inverse modeling using in situ and global, space-based (SCIAMACHY and OMI) observations, J. Geophys. Res., 116, D06304, https://doi.org/10.1029/2010JD014758, 2011.
Lee, H.-M., Henze, D. K., Alexander, B., and Murray, L. T.: Investigating the sensitivity of surface-level nitrate seasonality in Antarctica to primary sources using a global model, Atmos. Environ., 89, 757–767, https://doi.org/10.1016/j.atmosenv.2014.03.003, 2014.
Lee, J. H. and Tang, I. N.: Accommodation coefficient of gaseous NO2 on water surfaces, Atmos. Environ., 22, 1147–1151, 1988.
Levy, H., Moxim, W. J., Klonecki, A. A., and Kasibhatla, P. S.: Simulated tropospheric NOx: Its evaluation, global distribution and individual source contributions, J. Geophys. Res, 104, 26279–226306, 1999.
Li, L., Duan, Z., Li, H., Zhu, C., Henkelman, G., Francisco, J. S., and Zeng, X. C.: Formation of HONO from the NH3-promoted hydrolysis of NO2 dimers in the atmosphere, P. Natl. Acad. Sci. USA, 115, 7236–7241, https://doi.org/10.1073/pnas.1807719115, 2018a.
Li, L., Hoffmann, M. R., and Colussi, A. J.: Role of Nitrogen Dioxide in the Production of Sulfate during Chinese Haze-Aerosol Episodes, Environ. Sci. Technol., 52, 2686–2693, https://doi.org/10.1021/acs.est.7b05222, 2018b.
Li, M., Zhang, Q., Kurokawa, J.-I., Woo, J.-H., He, K., Lu, Z., Ohara, T., Song, Y., Streets, D. G., Carmichael, G. R., Cheng, Y., Hong, C., Huo, H., Jiang, X., Kang, S., Liu, F., Su, H., and Zheng, B.: MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP, Atmos. Chem. Phys., 17, 935–963, https://doi.org/10.5194/acp-17-935-2017, 2017.
Liang, J., Horowitz, L. W., Jacob, D. J., Wang, Y., Fiore, A. M., Logan, J. A., Gardner, G. M., and Munger, J. W.: Seasonal budgets of reactive nitrogen species and ozone over the United States, and export fluxes to the global atmosphere, J. Geophys. Res., 103, 13435–13450,, 1998.
Liu, H., Jacob, D. J., Bey, I., and Yantosca, R. M.: Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res., 106, 12109–12128, 2001.
Long, M. S., Keene, W. C., Easter, R. C., Sander, R., Liu, X., Kerkweg, A., and Erickson, D.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry-global climate system: halogen distributions, aerosol composition, and sensitivity of climate-relevant gases, Atmos. Chem. Phys., 14, 3397–3425, https://doi.org/10.5194/acp-14-3397-2014, 2014.
Lyons, J. R.: Transfer of mass-independent fractioation on ozone to other oxygen-containing molecules in the atmosphere, Geophys. Res. Lett., 28, 3231–3234, 2001.
Macintyre, H. L. and Evans, M. J.: Sensitivity of a global model to the uptake of N2O5 by tropospheric aerosol, Atmos. Chem. Phys., 10, 7409–7414, https://doi.org/10.5194/acp-10-7409-2010, 2010.
Martin, R. V., Jacob, D. J., Yantosca, R. M., Chin, M., and Ginoux, P.: Global and regional decreases in tropospheric oxidants form photochemical effects of aerosols, J. Geophys. Res., 108, 4097, doi:4010.1029/2002JD002622, 2003.
Mauersberger, K., Lämmerzahl, P., and Krankowsky, D.: Stratospheric Ozone Isotope Enrichments – Revisited, Geophys. Res. Lett., 28, 3155–3158, 2001.
McCabe, J. R., Boxe, C. S., Colussi, A. J., Hoffmann, M. R., and Thiemens, M. H.: Oxygen isotopic fractionation in the photochemistry of nitrate in water and ice, J. Geophys. Res., 110, D15310, https://doi.org/10.1029/2004JD005484, 2005.
McCabe, J. R., Savarino, J., Alexander, B., Gong, S., and Thiemens, M. H.: Isotopic constraints on non-photochemical sulfate production in the Arctic winter, Geophys. Res. Lett., 33, L05810, https://doi.org/10.1029/2005GL025164, 2006.
McCabe, J. R., Thiemens, M. H., and Savarino, J.: A record of ozone variability in South Pole Antarctic snow: Role of nitrate oxygen isotopes, J. Geophys. Res, 112, D12303, https://doi.org/10.1029/2006JD007822, 2007.
Michalski, G. and Bhattacharya, S. K.: The role of symmetry in the mass independent isotope effect in ozone, P. Natl. Acad. Sci. USA, 106, 5493–5496, 2009.
Michalski, G., Bhattacharya, S. K., and Girsch, G.: NOx cycle and the tropospheric ozone isotope anomaly: an experimental investigation, Atmos. Chem. Phys., 14, 4935–4953, https://doi.org/10.5194/acp-14-4935-2014, 2014.
Michalski, G. M., Scott, Z., Kabiling, M., and Thiemens, M. H.: First measurements and modeling of Δ17O in atmospheric nitrate, Geophys. Res. Lett., 30, 1870, doi:1810.1029/2003GL017015, 2003.
Morin, S., Savarino, J., Bekki, S., Gong, S., and Bottenheim, J. W.: Signature of Arctic surface ozone depletion events in the isotope anomaly (Δ17O) of atmospheric nitrate, Atmos. Chem. Phys., 7, 1451–1469, https://doi.org/10.5194/acp-7-1451-2007, 2007.
Morin, S., Savarino, J., Frey, M. M., Yan, N., Bekki, S., Bottenheim, J. W., and Martins, J. M. F.: Tracing the Origin and Fate of NOx in the Arctic Atmosphere Using Stable Isotopes in Nitrate, Science, 322, 730–732, https://doi.org/10.1126/science.1161910, 2008.
Morin, S., Savarino, J., Frey, M. M., Dominé, F., Jacobi, H.-W., Kaleschke, L., and Martins, J. M. F.: Comprehensive isotopic composition of atmospheric nitrate in the Atlantic Ocean boundary layer from 65∘ S to 79∘ N, J. Geophys. Res, 114, D05303, https://doi.org/10.1029/2008JD010696, 2009.
Morin, S., Sander, R., and Savarino, J.: Simulation of the diurnal variations of the oxygen isotope anomaly (Δ17O) of reactive atmospheric species, Atmos. Chem. Phys., 11, 3653–3671, https://doi.org/10.5194/acp-11-3653-2011, 2011.
Morton, J., Barnes, J., Schueler, B., and Mauersberger, K.: Laboratory studies of heavy ozone, J. Geophys. Res., 95, 901–907, 1990.
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014.
Murray, L. T., Jacob, D. J., Logan, J. A., Hudman, R. C., and Koshak, W. J.: Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data, J. Geophys. Res., 117, D20307, https://doi.org/10.1029/2012JD017934, 2012.
Murray, L. T.: Lightning NOx and Impacts on Air Quality, Curr. Pollution Rep., 2, 115–133, https://doi.org/10.1007/s40726-016-0038-0, 2016.
Newsome, B. and Evans, M.: Impact of uncertainties in inorganic chemical rate constants on tropospheric composition and ozone radiative forcing, Atmos. Chem. Phys., 17, 14333–14352, https://doi.org/10.5194/acp-17-14333-2017, 2017.
O'Brien, J., Shepson, P., Muthuramu, K., Hao, C., Niki, H., Hastie, D., Taylor, R., and Roussel, P.: Measurements of alkyl and multifunctional organic nitrates at a rural site in Ontario, J. Geophys. Res., 100, 22795–22804, 1995.
Park, R. J., Jacob, D. J., Field, B. D., Yantosca, R. M., and Chin, M.: Natural and transboundary pollution influences on sulfate-nitrate-ammonium aerosols in the United States: implications for policy, J. Geophys. Res., 109, D15204, https://doi.org/10.1029/2003JD004473, 2004.
Parrella, J. P., Jacob, D. J., Liang, Q., Zhang, Y., Mickley, L. J., Miller, B., Evans, M. J., Yang, X., Pyle, J. A., Theys, N., and Van Roozendael, M.: Tropospheric bromine chemistry: implications for present and pre-industrial ozone and mercury, Atmos. Chem. Phys., 12, 6723–6740, https://doi.org/10.5194/acp-12-6723-2012, 2012.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kroll, J. H., Seinfeld, J. H., and Wennberg, P. O.: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 9, 1479–1501, 2009.
Ramazan, K. A., Syomin, D., and Finlayson-Pitts, B. J.: The photochemical production of HONO during the heterogeneous hydrolysis of NO2, Phys. Chem. Chem. Phys., 6, 3836–3843, https://doi.org/10.1039/b402195a, 2004.
Reed, C., Evans, M. J., Crilley, L. R., Bloss, W. J., Sherwen, T., Read, K. A., Lee, J. D., and Carpenter, L. J.: Evidence for renoxification in the tropical marine boundary layer, Atmos. Chem. Phys., 17, 4081–4092, https://doi.org/10.5194/acp-17-4081-2017, 2017.
Richter, A., Borrows, J. P., Nub, H., Granier, C., and Niemier, U.: Increase in tropospheric nitrogen dioxide over China observed from space, Nature, 437, 129–132, https://doi.org/10.1038/nature04092, 2005.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis,, Atmos. Environ., 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Romer, P. S., Wooldridge, P. J., Crounse, J. D., Kim, M. J., Wennberg, P. O., Dibb, J. E., Scheuer, E., Blake, D. R., Meinardi, S., Brosius, A. L., Thames, A. B., Miller, D. O., Brune, W. H., Hall, S. R., Ryerson, T. B., and Cohen, R. C.: Constraints on Aerosol Nitrate Photolysis as a Potential Source of HONO and NOx, Environ. Sci. Technol., 52, 13738–13746, https://doi.org/10.1021/acs.est.8b03861, 2018.
Saiz-Lopez, A., Lamarque, J.-F., Kinnison, D. E., Tilmes, S., Ordóñez, C., Orlando, J. J., Conley, A. J., Plane, J. M. C., Mahajan, A. S., Sousa Santos, G., Atlas, E. L., Blake, D. R., Sander, S. P., Schauffler, S., Thompson, A. M., and Brasseur, G.: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 12, 3939–3949, https://doi.org/10.5194/acp-12-3939-2012, 2012.
Savarino, J. and Thiemens, M. H.: Analytical procedure to determine both δ18O and δ17O of H2O2 in natural water and first measurements, Atmos. Environ., 33, 3683–3690, 1999b.
Savarino, J., Kaiser, J., Morin, S., Sigman, D. M., and Thiemens, M. H.: Nitrogen and oxygen isotopic constraints on the origin of atmospheric nitrate in coastal Antarctica, Atmos. Chem. Phys., 7, 1925–1945, https://doi.org/10.5194/acp-7-1925-2007, 2007.
Savarino, J., Bhattacharya, S. K., Morin, S., Baroni, M., and Doussin, J.-F.: The NO + O3 reaction: A triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly, J. Chem. Phys., 128, 194303, https://doi.org/10.1063/1.2917581, 2008.
Savarino, J., Morin, S., Erbland, J., Grannec, F., Patey, M., Vicars, W., Alexander, B., and Achterberg, E. P.: Isotopic composition of atmospheric nitrate in a tropical marine boundary layer, P. Natl. Acad. Sci. USA, 110, 17668–17673, https://doi.org/10.1073/pnas.1216639110, 2013.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Sulieman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res., 121, 11819–11835, https://doi.org/10.1002/2015JD024229, 2016.
Shah, V., Jaeglé, L., Thornton, J. A., Lopez-Hilfiker, F. D., Lee, B. H., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Sullivan, A. P., Weber, R. J., Green, J. R., Fiddler, M. N., Bililign, S., Campos, T. L., Stell, M., Weinheimer, A. J., Montzka, D. D., and Brown, S. S.: Chemical feedbacks weaken the wintertime response of particulate sulfate and nitrate to emissions reductions over the eastern United States, P. Natl. Acad. Sci. USA, 115, 8110–8115, https://doi.org/10.1073/pnas.1803295115, 2018.
Shao, J., Chen, Q., Wang, Y., Lu, X., He, P., Sun, Y., Shah, V., Martin, R. V., Philip, S., Song, S., Zhao, Y., Xie, Z., Zhang, L., and Alexander, B.: Heterogeneous sulfate aerosol formation mechanisms during wintertime Chinese haze events: air quality model assessment using observations of sulfate oxygen isotopes in Beijing, Atmos. Chem. Phys., 19, 6107–6123, https://doi.org/10.5194/acp-19-6107-2019, 2019.
Sharma, H. D., Jervis, R. E., and Wing, K. Y.: Isotopic exchange reactions in nitrogen oxides, J. Phys. Chem., 74, 923–933, 1970.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016.
Sherwen, T., Evans, M. J., Sommariva, R., Hollis, L. D. J., Ball, S. M., Monks, P. S., Reed, C., Carpenter, L. J., Lee, J. D., Forster, G., Bandy, B., Reeves, C. E., and Bloss, W. J.: Effects of halogens on European air-quality, Faraday Discuss., 200, 75–100, https://doi.org/10.1039/C7FD00026J, 2017.
Singh, H. B., Herlth, D., O'Hara, D., Zahnle, K., Bradshaw, J. D., Sandholm, S. T., Talbot, R., Crutzen, P. J., and Kanakidou, M.: Relationship of Peroxyacetyl nitrate to active and total odd nitrogen at northern high latitudes: Influence of reservoir species on NOx and O3, J. Geophys. Res, 97, 16523–16530, 1992.
Sofen, E. D., Alexander, B., Steig, E. J., Thiemens, M. H., Kunasek, S. A., Amos, H. M., Schauer, A. J., Hastings, M. G., Bautista, J., Jackson, T. L., Vogel, L. E., McConnell, J. R., Pasteris, D. R., and Saltzman, E. S.: WAIS Divide ice core suggests sustained changes in the atmospheric formation pathways of sulfate and nitrate since the 19th century in the extratropical Southern Hemisphere, Atmos. Chem. Phys., 14, 5749–5769, https://doi.org/10.5194/acp-14-5749-2014, 2014.
Spataro, F. and Ianniello, A.: Sources of atmospheric nitrous acid: State of the science, current research needs, and future prospects, J. Air Waste Manage., 64, 1232–1250, https://doi.org/10.1080/10962247.2014.952846, 2014.
Stettler, M. E. J., Eastham, S., and Barrett, S. R. H.: Air quality and public health impacts of UK airports, Part I: Emissions, Atmos. Environ., 45, 5415–5424, https://doi.org/10.1016/j.atmosenv.2011.07.012, 2011.
Tan, F., Tong, S., Jing, B., Hou, S., Liu, Q., Li, K., Zhang, Y., and Ge, M.: Heterogeneous reactions of NO2 with CaCO3–(NH4)2SO4 mixtures at different relative humidities, Atmos. Chem. Phys., 16, 8081–8093, https://doi.org/10.5194/acp-16-8081-2016, 2016.
The International GEOS-Chem User Community: GEOS-Chem 12.0.0, https://doi.org/10.5281/zenodo.1343547, 2018.
Thiemens, M. H. and Jackson, T.: Pressure dependency for heavy isotope enhancement in ozone formation, Geophys. Res. Lett., 17, 717–719, 1990.
Tong, S. R., Hou, S. Q., Zhang, Y., Chu, B. W., Liu, Y. C., He, H., Zhao, P. S., and Ge, M. F.: Exploring the nitrous acid (HONO) formation mechanism in winter Beijing: direct emissions and heterogeneous production in urban and suburban areas, Faraday Discuss., 189, 213–230, https://doi.org/10.1039/c5fd00163c, 2016.
Vicars, W. and Savarino, J.: Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: Ambient measurements from 50∘ N to 50∘ S using the nitrite-coated filter technique, Geochem. Cosmochem. Ac., 135, 270–287, https://doi.org/10.1016/j.gca.2014.03.023, 2014.
Vicars, W. C., Bhattacharya, S. K., Erbland, J., and Savarino, J.: Measurement of the 17O-excess (Δ17O) of tropospheric ozone using a nitrite-coated filter, Rapid Commun. Mass Spec., 26, 1219–1231, https://doi.org/10.1002/rcm.6218, 2012.
Vinken, G. C. M., Boersma, K. F., Jacob, D. J., and Meijer, E. W.: Accounting for non-linear chemistry of ship plumes in the GEOS-Chem global chemistry transport model, Atmos. Chem. Phys., 11, 11707–11722, https://doi.org/10.5194/acp-11-11707-2011, 2011.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wang, F., Michalski, G., Seo, J., and Ge, W.: Geochemical, isotopic, and mineralogical constraints on atmospheric deposition in the hyper-arid Atacama Desert, Chile, Geochem. Cosmochem. Ac., 135, 29–48, https://doi.org/10.1016/j.gca.2014.03.017, 2014.
Wang, J. Q., Zhang, X. S., Guo, J., Wang, Z. W., and Zhang, M. G.: Observation of nitrous acid (HONO) in Beijing, China: Seasonal variation, nocturnal formation and daytime budget, Sci. Total Environ., 587, https://doi.org/10.1016/j.scitotenv.2017.02.159, 2017.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wang, Y. H., Jacob, D. J., and Logan, J. A.: Global simulation of tropospheric O3-NOx hydrocarbon chemistry, 1. Model formulation, J. Geophys. Res., 103, 10713–10725, 1998.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Xu, W., Kuang, Y., Zhao, C., Tao, J., Zhao, G., Bian, Y., Yang, W., Yu, Y., Shen, C., Liang, L., Zhang, G., Lin, W., and Xu, X.: NH3-promoted hydrolysis of NO2 induces explosive growth in HONO, Atmos. Chem. Phys., 19, 10557–10570, https://doi.org/10.5194/acp-19-10557-2019, 2019.
Yabushita, A., Enami, S., Sakamoto, Y., Kawasaki, M., Hoffman, M. R., and Colussi, A. J.: Anion-Catalyzed Dissolution of NO2 on Aqueous Microdroplets, J. Phys. Chem. A, 113, 4844–4848, https://doi.org/10.1021/jp900685f, 2009.
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. C., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impact on ozone: A model study, J. Geophys. Res, 110, D2331, https://doi.org/10.1029/2005JD003244, 2005.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin Iii, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, https://doi.org/10.1038/nature17195, 2016.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin III, R. L., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J., and Ortega, J.: Tropospheric HONO distribution and chemistry in the southeastern US, Atmos. Chem. Phys., 18, 9107–9120, https://doi.org/10.5194/acp-18-9107-2018, 2018.
Zare, A., Romer, P. S., Nguyen, T., Keutsch, F. N., Skog, K., and Cohen, R. C.: A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates, Atmos. Chem. Phys., 18, 15419–15436, https://doi.org/10.5194/acp-18-15419-2018, 2018.
Zhang, L., Gong, S., Padro, J., and Barrie, L.: A size-segregated particle dry deposition scheme for an atmosopheric aerosol module, Atmos. Environ., 35, 549–560, 2001.