the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Feb 2020
| 26 Feb 2020
Mutual promotion between aerosol particle liquid water and particulate nitrate enhancement leads to severe nitrate-dominated particulate matter pollution and low visibility
Ying Chen
Zhijun Wu
Dongjie Shang
Yuxuan Bian
Zhuofei Du
Sebastian H. Schmitt
Rong Su
Georgios I. Gkatzelis
Patrick Schlag
Thorsten Hohaus
Aristeidis Voliotis
Keding Lu
Limin Zeng
Chunsheng Zhao
M. Rami Alfarra
Gordon McFiggans
Alfred Wiedensohler
Astrid Kiendler-Scharr
Yuanhang Zhang
As has been the case in North America and western Europe, the SO2 emissions have substantially reduced in the North China Plain (NCP) in recent years. Differential rates of reduction in SO2 and NOx concentrations result in the frequent occurrence of particulate matter pollution dominated by nitrate () over the NCP. In this study, we observed a polluted episode with the particulate nitrate mass fraction in nonrefractory PM1 (NR-PM1) being up to 44 % during wintertime in Beijing. Based on this typical -dominated haze event, the linkage between aerosol water uptake and enhancement, further impacting on visibility degradation, has been investigated based on field observations and theoretical calculations. During haze development, as ambient relative humidity (RH) increased from ∼10 % to 70 %, the aerosol particle liquid water increased from ∼1 µg m−3 at the beginning to ∼75 µg m−3 in the fully developed haze period. The aerosol liquid water further increased the aerosol surface area and volume, enhancing the condensational loss of N2O5 over particles. From the beginning to the fully developed haze, the condensational loss of N2O5 increased by a factor of 20 when only considering aerosol surface area and volume of dry particles, while increasing by a factor of 25 when considering extra surface area and volume due to water uptake. Furthermore, aerosol liquid water favored the thermodynamic equilibrium of HNO3 in the particle phase under the supersaturated HNO3 and NH3 in the atmosphere. All the above results demonstrated that is enhanced by aerosol water uptake with elevated ambient RH during haze development, in turn facilitating the aerosol take-up of water due to the hygroscopicity of particulate nitrate salt. Such mutual promotion between aerosol particle liquid water and particulate nitrate enhancement can rapidly degrade air quality and halve visibility within 1 d. Reduction of nitrogen-containing gaseous precursors, e.g., by control of traffic emissions, is essential in mitigating severe haze events in the NCP.
- Article
(4630 KB) - Full-text XML
-
Supplement
(751 KB) - BibTeX
- EndNote





Aerosol particle hygroscopicity plays an important role in air quality deterioration and cloud formation (Yu and Luo, 2009; Fitzgerald, 1973; Kreidenweis and Asa-Awuku, 2014; Wang and Chen, 2019; McFiggans et al., 2006) and can also directly influence aerosol measurements (Chen et al., 2018a). In atmospheric environments influenced by anthropogenic activities, particulate secondary inorganic compounds are often dominated by particulate sulfate and nitrate (Heintzenberg, 1989), which originate from the oxidation of sulfur dioxide (SO2) and nitrogen oxides (NOx) via multiple chemical pathways (Calvert et al., 1985; Cheng et al., 2016; Wang et al., 2016; Gen et al., 2019a, b). The abundance of secondary inorganic components is one of the most important factors determining particle hygroscopicity (Swietlicki et al., 2008), thereby governing the aerosol liquid water content under ambient moist conditions. Increased aerosol particle liquid water could accelerate secondary inorganic and organic aerosol formation by decreasing the kinetic limitation of mass transfer of gaseous precursors and providing more of a medium for multiphase reactions (Mozurkewich and Calvert, 1988; Cheng et al., 2016; Wang et al., 2016; Ervens et al., 2011; Kolb et al., 2010).
Sulfuric acid (H2SO4) is formed from the oxidation of SO2 via gaseous and multiphase reactions. H2SO4 is subsequently fully or partly neutralized by gaseous NH3 taken up on particles, resulting in the formation of (NH4)2SO4 and/or NH4HSO4. Any remaining NH3 is available to neutralize HNO3 to form particulate NH4NO3 (Seinfeld and Pandis, 2006; and further excess NH3 can neutralize any available HCl to form particulate NH4Cl). Over the past several decades, substantial efforts have reduced emissions of both SO2 and NOx, improving local and regional air quality all over the world. For example, SO2 and NOx emissions were reduced by 82 % and 54 %, respectively, in the majority of European Environment Agency member countries between 1990 and 2016 (https://www.eea.europa.eu/data-and-maps/indicators/main-anthropogenic-air-pollutant-emissions/assessment-4, last access: 15 February 2020). In consequence, an increasing trend in the molar ratio was observed in long-term measurements in Leipzig, Germany (Spindler et al., 2004), and at some other European sites from the European Monitoring and Evaluation Programme (EMEP; Putaud et al., 2004).
China also managed to reduce SO2 emissions by 75 % during 2007–2015 (C. Li et al., 2017), and they declined by ∼15.1 % yr−1 during 2013–2017 (Vu et al., 2019), whereas NOx emissions declined by only ∼10 % between 2011 and 2015 (de Foy et al., 2016) and by ∼4.3 % yr−1 during 2013–2017 (Vu et al., 2019). Strict emission control reduced the PM2.5 mass concentration and the corresponding chemical components in China significantly (Vu et al., 2019). The annual mean PM2.5 mass loading decreased by 39.6 % during 2013–2017 in the Beijing–Tianjin–Hebei region, and the and mass concentrations in the PM2.5 declined by 40 % and 34 %, respectively, during 2015–2017 in Beijing (Vu et al., 2019). However, NH3 emissions have been observed by satellites to have increased by ∼30 % from 2008 to 2016 over the North China Plain (NCP; Liu et al., 2018). The faster reduction rate of SO2 compared with NOx emissions, in conjunction with elevated NH3 level, made it reasonable to switch the dominant inorganic component in fine aerosol particles from sulfate to nitrate in recent years, similarly to European countries (Sun et al., 2015; Hu et al., 2017, 2016; Wu et al., 2018; Guo et al., 2014; Huang et al., 2014, 2010; Ge et al., 2017; Q. Xu et al., 2019; Xie et al., 2019; Li et al., 2018). Field measurements in Beijing show that the annually averaged molar ratio of NR-PM1 (nonrefractory PM1) in 2012 (1.3–1.8; Sun et al., 2015) significantly increased compared to that in 2008 (0.9–1.5; Zhang et al., 2013). Comparably, the molar ratio of PM2.5 in Beijing increased substantially, from 1.5 before 2013 to 3.33 in 2017 (Q. Xu et al., 2019).
Over the NCP region, heavy haze events are typically associated with enhanced ambient RH levels. This leads to an increased aerosol liquid water content (Wu et al., 2018), which enhances the particulate nitrate formation by increasing the reactive uptake of precursors and the thermodynamic equilibrium of ammonium nitrate (Cheng et al., 2016; Wang et al., 2016, 2017; Yun et al., 2018; Yue et al., 2019). To date, a few studies have reported aerosol liquid water content over the NCP region (Wang et al., 2018; Bian et al., 2014; Cheng et al., 2016; Wu et al., 2018; Ge et al., 2019). However, the observational and theoretical analysis of the relationship between particulate nitrate enhancement and associated liquid water during haze events in China has been infrequently reported (Wu et al., 2018).
In this study, a self-amplification effect between particulate nitrate and liquid water is demonstrated by examining a nitrate-dominated fine-particle Beijing pollution episode. The facilitation of particulate nitrate enhancement by abundant aerosol liquid water is subsequently theoretically explored through the impacts of liquid water on thermodynamic equilibrium and heterogeneous reactions. Finally, the corresponding impacts on the light extinction coefficient and visibility degradation are estimated. These results improve our quantitative understanding of the development of haze events over the NCP and help to formulate emission reduction strategies, as well as maybe also providing insights into other polluted regions.
2.1 Location and instrumentation
Measurements were conducted within the framework of the BEST-ONE (Beijing winter finE particle STudy – Oxidation, Nucleation and light Extinctions) field campaign from 1 January to 5 March 2016, at the Huairou site (40.42∘ N, 116.69∘ E), located in a rural environment, north of Beijing, China. Detailed information about the sampling site was described in Tan et al. (2018). A weather station (Met One Instruments Inc., USA) was used to measure meteorological parameters (ambient RH, temperature, wind speed, wind direction), and detailed aerosol particle physical and chemical properties were recorded using a suite of state-of-the-art instrumentation. The hygroscopic growth factor (HGF) of submicrometer aerosol particles was measured using a hygroscopicity tandem differential mobility analyzer (H-TDMA, TROPOS, Germany; Wu et al., 2011; Massling et al., 2011; Wang et al., 2018; Wu et al., 2016; Liu et al., 1978), and data retrieval followed the TDMAinv method in Gysel et al. (2009). The hygroscopicity parameter (κ) was estimated using the κ-Köhler approach (Petters and Kreidenweis, 2007; Köhler, 1936). Size-resolved NR-PM1 was recorded by an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research, Inc., USA; DeCarlo et al., 2006). Regular calibration procedures followed as reported in Jayne et al. (2000) and Jimenez et al. (2003), and composition-dependent correction followed as in Middlebrook et al. (2012). Gaseous HNO3 and NH3 were measured using a gas–aerosol collector (GAC) coupled with ion chromatography (IC; Dong et al., 2012). The mass concentration of equivalent black carbon in aerosol particles (Petzold et al., 2013) was recorded by a multiangle absorption photometer (MAAP, Model 5012, Thermo Fisher Scientific, USA) with a laser wavelength of 670 nm (Petzold and Schönlinner, 2004). Furthermore, particle number size distribution (PNSD) in the size range of 3 nm–10 µm was measured using a mobility particle size spectrometer (MPSS, Model 3776+3085 3775+3081, TSI, USA), following the recommendations described in Wiedensohler et al. (2012), and an aerodynamic particle size spectrometer (APS, Model 3021, TSI, USA; Wu et al., 2008; Pfeifer et al., 2016). Detailed descriptions of the H-TDMA, HR-ToF-AMS and GAC–IC can be found in the supporting information.
2.2 Estimation of aerosol particle liquid water
Given the absence of direct liquid water measurements, size-resolved liquid water was calculated using the corresponding HGFs measured at RH =90 % (50, 100, 150, 250, 350 nm in Stokes diameter), PNSD data (3 nm–10 µm) and meteorological parameters (RH, T), following the method proposed in Bian et al. (2014), referred to below as H-TDMA-derived liquid water. Briefly, the measured PNSD with 57 size bins was fitted using a four-mode lognormal distribution. The classification of the four modes and the fitting results are shown in Table S1 and Fig. S4 in the Supplement. Good agreement between measured values and the fitted PNSD was achieved, which indicates the reliability of the four-mode lognormal fitting method. Based on four-mode lognormal fitting results, the particle number size distribution and number fractions of each mode can be obtained. It has been assumed that particles from the same mode have constant particle hygroscopicity (κ). Under the assumption of constant particle hygroscopicity in each mode (shown in Table S1), the κ values for each mode (κ1, κ2, κ3) can be calculated by Eq. (1) from the known number fraction of the fitted four modes and the κ values of measured particle size from H-TDMA measurements.
Here, κi and fi represent the κ value and the particle number fraction of the i mode. Then, the calculated κ values for each mode and the derived number fraction of each size bin were used to obtain the κ distribution for each size bin. Figure S5 shows the comparison of the calculated size-resolved κ distribution and the κ measured by the H-TDMA; the good agreement showed the reliability of the method. Then, based on κ-Köhler theory (Petters and Kreidenweis, 2007; Köhler, 1936), the size-resolved HGFs at ambient RH were calculated. Finally, liquid water of size-resolved particles can be derived by calculating the differentials between the dry and wet PNSD of aerosol particles in Eq. (2):
where j represents the bin number of measured PNSD and Nj and Dp,j represent the number concentration and the diameter of dry particles of the jth bin, respectively, while HGF and ρw are the hygroscopic growth factor of aerosol particles and water density (1 g cm−3), respectively.
2.3 Condensation rate of trace gases
The condensation rate (k) of trace gases (dinitrogen pentoxide, N2O5, referred to as k_N2O5) was calculated by the method of Schwartz (1986), shown in Eq. (3). In order to illustrate the influences of the dry and wet PNSDs due to water uptake on the condensation rate of gases, the PNSDs of the dry and wet particles (obtained by applying the HGF estimated from the H-TDMA-derived liquid water method) were used.
where r represents the radius of the particles, and Dg represents the binary diffusion coefficient evaluated following Maitland et al. (1981) ( m2 s−1). Cg is the kinetic velocity of the gas molecules, calculated in Eq. (4). Here, R and M are the ideal gas constant (8.314 kg m2 mol−1 K−1 s−2) and molar mass of the gas, respectively, while T represents the ambient temperature. The expression dN∕dlogr is the number size distribution, and γ is the uptake coefficient of the gas.
The uptake coefficient of N2O5 was estimated following the method proposed in Chen et al. (2018b) and Chang et al. (2016) and the references therein. The influences of RH, temperature, multiple inorganic particle compositions, secondary organic aerosol (SOA) and primary organic aerosol (POA) are considered. The uptake suppression effect of N2O5 due to the presence of SOA was considered following the method in Anttila et al. (2006). Based on our source apportionment using positive matrix factorization (SoFi tool, ME2, Francesco Canonaco, PSI), two oxygenated organic aerosol (OOA) factors, usually interpreted as SOA, and three POA factors were determined. The fraction of SOA in the total organic aerosol (OA) was 60 %–90 % during the observed period, which is quite consistent with the results of a previous study in Beijing (Huang et al., 2014). Hence, 75 % was used as the ratio of SOA∕OA in our model to estimate the suppression effect of SOA on the uptake of N2O5 following the work of Anttila et al. (2006). The reaction of chloride with N2O5 was not considered in this study due to its limited mass concentration (on average 5 % of the PM1 mass concentration during the marked haze period), which could cause minor uncertainty in the k_N2O5 calculation. The detailed information regarding the estimation is given in Chen et al. (2018b), and influences of different chemical components on are summarized in Table 1 of Chen et al. (2018b).
2.4 Equilibrium of NH4NO3
The equilibrium dissociation constant of NH4NO3 (Kp) under dry conditions was calculated as a function of ambient temperature (Seinfeld and Pandis, 2006) in the following Eq. (5).
Taking into account the associated liquid water, the equilibrium vapor pressure of HNO3 and NH3 was calculated by employing the Extended Aerosol Inorganic Model (E-AIM), H+ − − − − H2O (Clegg et al., 1998), using HR-ToF-AMS data, NH3 from GAC–IC and meteorological parameters (RH, T). In this calculation, a simplified ion pairing scheme was performed to ensure the ion balance of the input chemical composition following the method in Gysel et al. (2007).
2.5 Light extinction coefficient and visibility calculation
The size-resolved chemical composition of the NR-PM1 from the HR-ToF-AMS, the mass concentration of equivalent black carbon from the MAAP, PNSD data and the H-TDMA-derived liquid water were used to calculate the light extinction coefficient (including light absorption and scattering) and visibility degradation of size-resolved particles by the Mie scattering theory described in Barnard et al. (2010). Here, the size-resolved equivalent black carbon mass concentration was inferred from the particle mass size distribution measurement by a single-particle soot photometer at Peking University Atmosphere Environment Monitoring Station (PKUERS). The method of redistribution of liquid water and HR-ToF-AMS data has been described in the supporting information (Text S1 in the Supplement, HR-ToF-AMS introductory section). Thus, with the redistributed datasets as the input into the Mie scattering theory, the light extinction coefficient for atmospheric particles in the absence and presence of liquid water with a size range of 100–2500 nm in Stokes diameter can be derived. Due to lack of measurements of aerosol particle morphology and mixing states, we assume particles are spherical as described in Barnard et al. (2010). To perform Mie calculations, the complex reflective index of each component is given in Table 1 of Barnard et al. (2010) and references therein. This method shows good agreement with measurements in Mexico City and is consistent with the regional atmospheric chemistry model WRF-Chem. Here, Ext_550nm_wet and Ext_550nm_dry represent the calculated light extinction coefficient for particles in the presence and absence of liquid water at an incident light wavelength of 550 nm. The corresponding visibility degradation (VIS) for dry and wet particles was calculated from the light extinction coefficient following the Koschmieder Eq. (6).
3.1 Nitrate-dominated fine particulate matter pollution
Figure 1 illustrates a summary of the chemical composition of NR-PM1, ambient RH, size distribution of and total aerosol particle liquid water, and size distribution of and total aerosol surface area concentration during the period of 29 February to 5 March 2016 in the BEST-ONE campaign. During this period, polluted episodes occurred under stagnant meteorological conditions with low wind speed (Fig. S6) and elevated ambient RH (Fig. 1a). As seen in the haze period shown in Fig. 1, an obvious increase in NR-PM1 was observed. The secondary inorganic components (sulfate, nitrate and ammonium) were dominant components of the NR-PM1, accounting for up to 73 % during the haze period. Particularly, nitrate was the major contributor to the secondary inorganic components and accounted for up to ∼44 % of NR-PM1 mass, while sulfate contributed for ∼12 % on average.
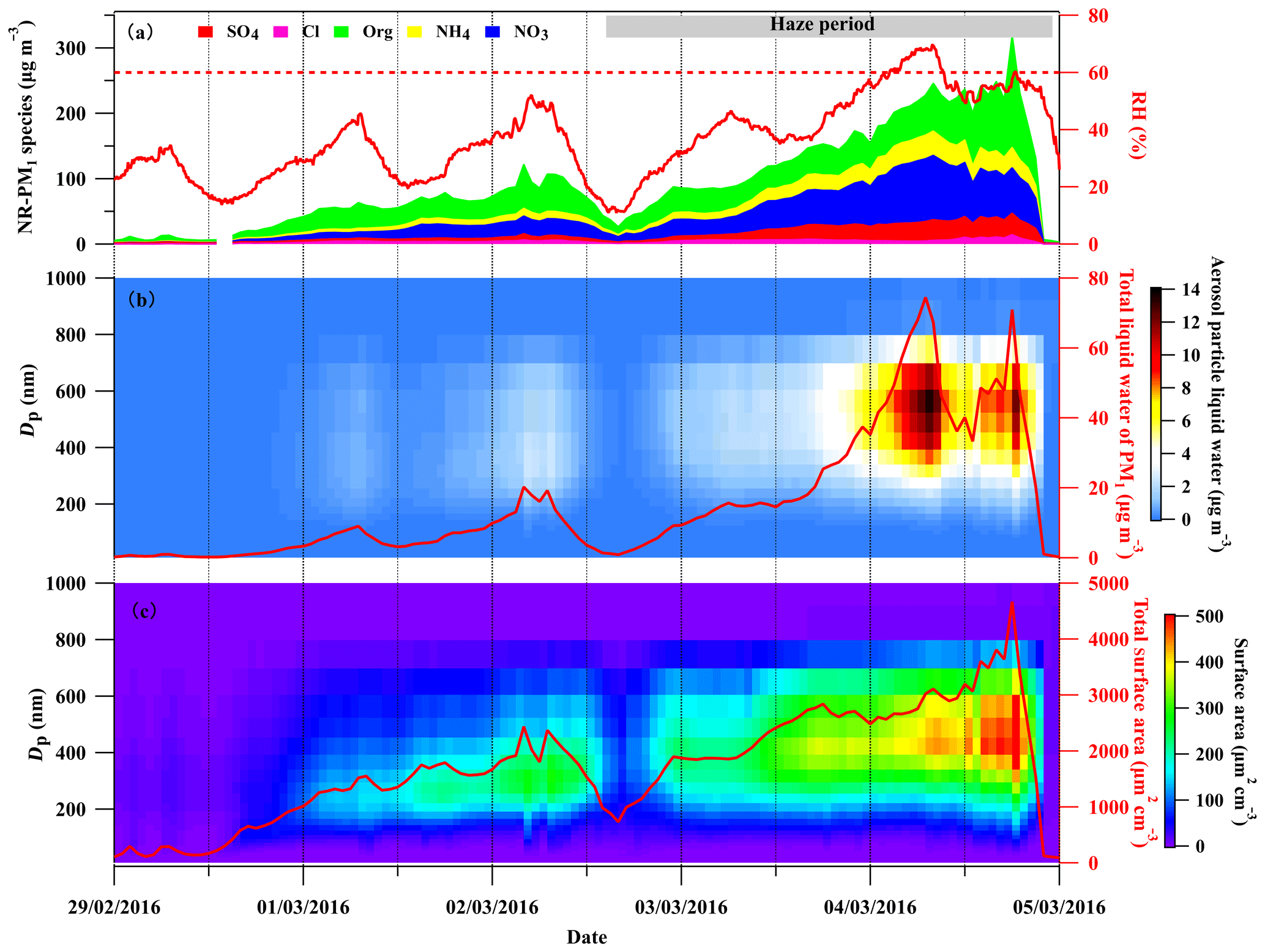
Figure 1The time series of (a) NR-PM1 chemical composition measured by the HR-ToF-AMS and ambient RH (solid red line), (b) size-segregated aerosol particle liquid water and the total mass concentration of liquid water with an aerodynamic diameter smaller than 1 µm (solid red line), and (c) size-segregated aerosol particle surface area and total aerosol particle surface area without considering particle hygroscopic growth during 29 February to 5 March 2016.
In recent decades, severe haze events with high aerosol mass loading occurred frequently in Beijing during wintertime (Hu et al., 2016, 2017; Sun et al., 2014, 2015). To mitigate the air pollution, the Beijing government implemented strict emission controls. The total mass loading of particulate matter has reduced substantially in recent years (http://sthjj.beijing.gov.cn/, last access: 15 February 2020). With decreasing PM mass concentration, the mass fraction of particulate nitrate during these haze events in Beijing was enhanced substantially. In 2014, the highest fraction of nitrate in PM1 was reported as ∼20 % and increased to ∼35 % in 2016 (W. Xu et al., 2019), which is comparable to the ratio (44 %) in this study. The particulate nitrate became more dominant in secondary inorganic compounds other than particulate sulfate with the air quality improvement over the NCP.
As one of the main hydrophilic compounds in atmospheric aerosol particles, the ability of water uptake is comparable between deliquescent (NH4)2SO4 and NH4NO3 particles with the same sizes and ambient RH (Kreidenweis and Asa-Awuku, 2014; Wu et al., 2016; http://umansysprop.seaes.manchester.ac.uk/, last access: 15 February 2020). However, compared to (NH4)2SO4, NH4NO3 particles have a lower deliquescence RH (62 %, 298 K) than (NH4)2SO4 (80 %, 298 K; Kreidenweis and Asa-Awuku, 2014) and easily liquefy (Y. J. Li et al., 2017). In addition, NH4NO3 particles are semivolatile; the co-condensation of semivolatile compounds and water (Topping et al., 2013; Hu et al., 2018) could be significant. Therefore, the switching from sulfate-dominated to nitrate-dominated aerosol chemistry may impact on aerosol water uptake. The interaction between aerosol particle liquid water and particulate nitrate formation and visibility degradation should be reconsidered.
3.2 Mutual promotion between liquid water and particulate nitrate enhancement
Lu et al. (2019) conducted a box model to calculate the potential particulate nitrate formation during the same investigated period of the BEST-ONE project. They found out that HNO3 from daytime photooxidation of NO2 was the major source of the particulate nitrate (>75 %), whereas the contribution of the N2O5 pathway was lower than 25 % (Lu et al., 2019). In the following discussion, the enhancement of particulate nitrate during the haze period is elucidated by theoretical calculations of the condensational loss rate of N2O5 and the thermodynamic equilibrium of NH4NO3 and HNO3. In particular, the role of aerosol water uptake in particulate nitrate formation is comprehensively investigated.
N2O5 is an important gaseous precursor for particulate nitrate formation via its hydrolysis to form HNO3 during the nighttime (Brown et al., 2006). Liquid water can enhance aerosol surface areas and volumes, thereby increasing the available heterogeneous reacting medium. Across the development of the haze period, the estimated liquid water increased from ∼1 µg m−3 at the beginning (2 March, 14:00–18:00 LT) to ∼75 µg m−3 when the haze was fully developed (4 March, 04:00–08:00 LT). The total surface area and volume concentrations of particles were increased by the liquid water by 2 %–3 % at the beginning and by up to ∼25 and ∼40 % in the fully developed haze compared to the dry values, respectively (see Figs. S7 and S8). Additionally, from the beginning to the fully developed haze, the uptake coefficient of N2O5 was enhanced by a factor of 9 from 0.002 to 0.018, and the k_N2O5 increased by a factor of 20 (dry particles), while, considering the increased particle surface area and volume due to water uptake, the respective value of enhanced k_N2O5 increased by a factor of 25 (Fig. 2a). Apart from providing an extra reacting medium, the abundant liquid water can liquefy the aerosol particles and may reduce any kinetic limitation of mass transfer for reactive gases (Koop et al., 2011; Shiraiwa et al., 2011) and impact the thermodynamic equilibrium of semivolatile compounds (Kulmala et al., 1993; Topping et al., 2013) to contribute to secondary aerosol formation. Our previous study provided the observational evidence that particles may have transitioned from the solid phase to the liquid phase as RH increased from 20 % to 60 % during wintertime in Beijing (Liu et al., 2017). In this study, the ambient RH increased from ∼10 % up to 70 % during the haze period, suggesting a likely transition of particles from the solid to liquid phase. Such phase transition may facilitate particulate nitrate formation by increasing the diffusion coefficients of dissolved precursors.
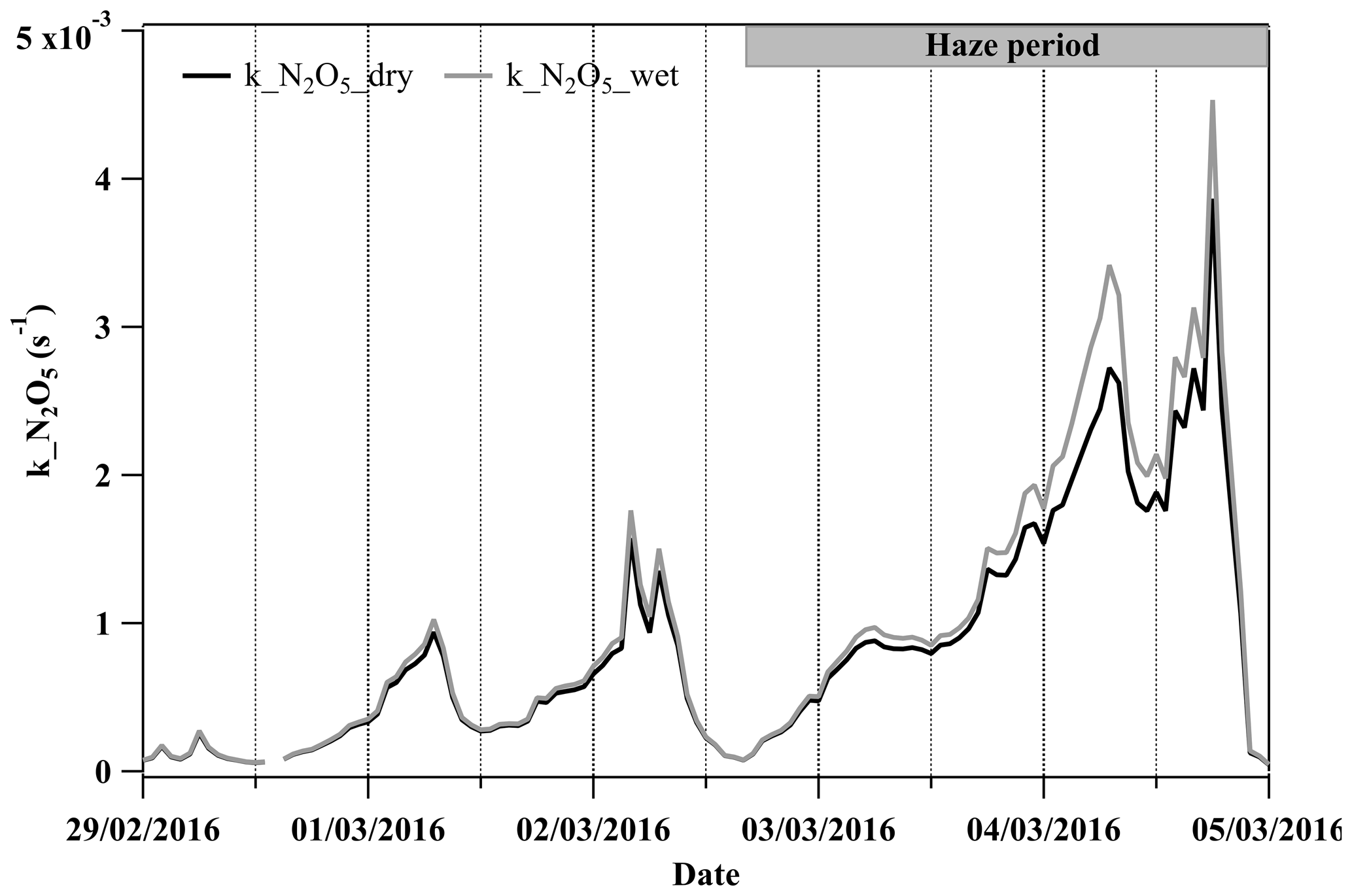
Figure 2The time series of the condensational loss rate of N2O5 (k_N2O5) with the calculation of the dry particle number size distribution (PNSD) and wet PNSD during 29 February to 5 March 2016.

Figure 3The comparison of the calculated temperature-dependent dissociation constant of NH4NO3 (Kp; Seinfeld and Pandis, 2006) in the absence of liquid water, the product of the equilibrium vapor pressure of gaseous NH3 and HNO3 from E-AIM (AIM_pNH3pHNO3), and the product of mixing ratios of gaseous NH3 and HNO3 measured by GAC–IC (M_pNH3pHNO3). Here, Kp is colored by the ambient temperature ranging 265–293 K during 29 February to 5 March 2016.
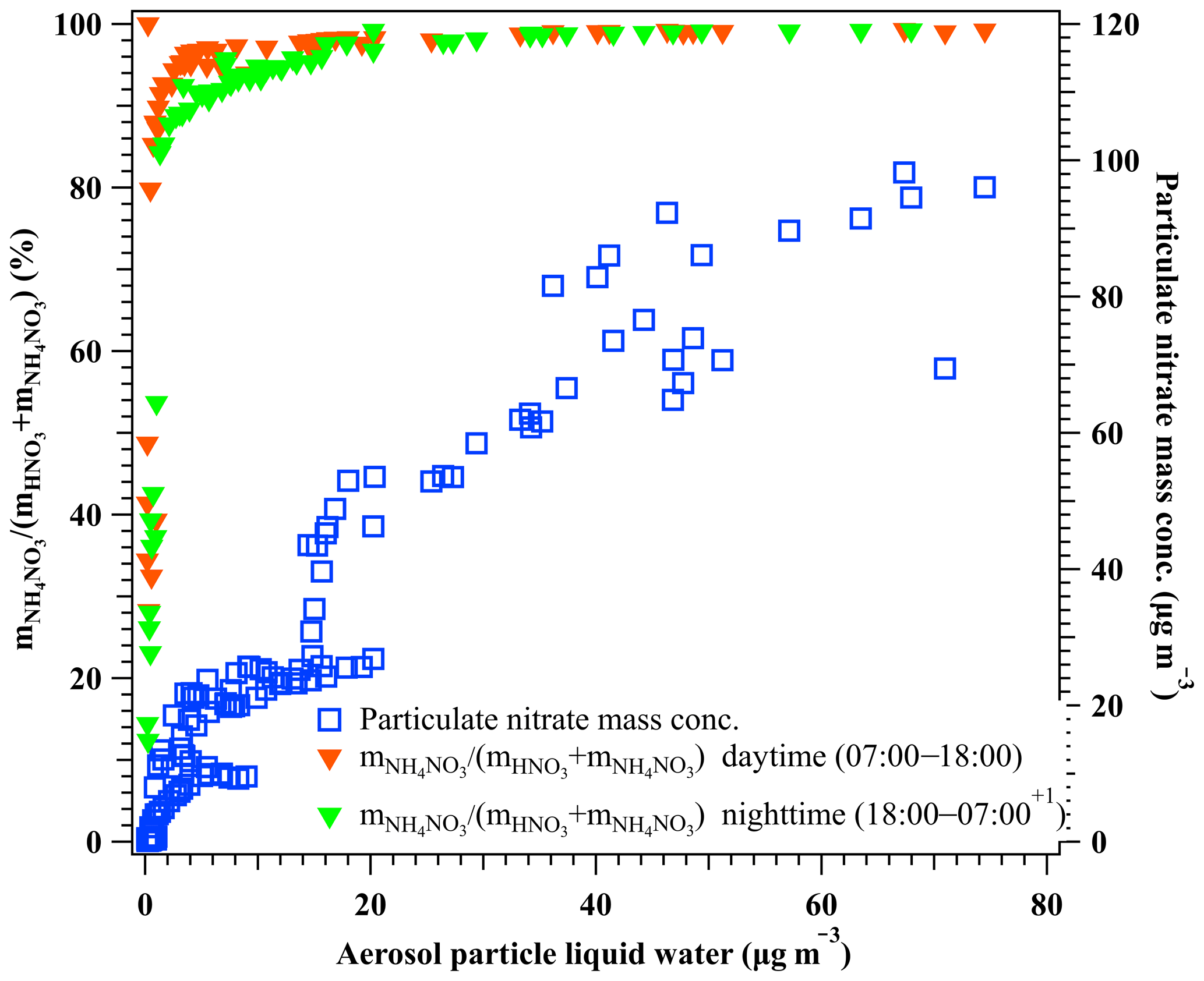
Figure 4The relationship between aerosol particle liquid water and the molar ratio of particulate nitrate in the total nitrate, (left y axis), during the nighttime 18:00–07:00+1 d (solid green triangle) and the daytime at 07:00–18:00 (solid red triangle), and the mass concentration of particulate nitrate as a function of aerosol liquid water (right y axis) during the period of 29 February to 5 March 2016. Here, particulate nitrate was measured by the HR-ToF-AMS, and the HNO3 in the gas phase was measured by GAC–IC. Aerosol liquid water was calculated using the H-TDMA-derived method.
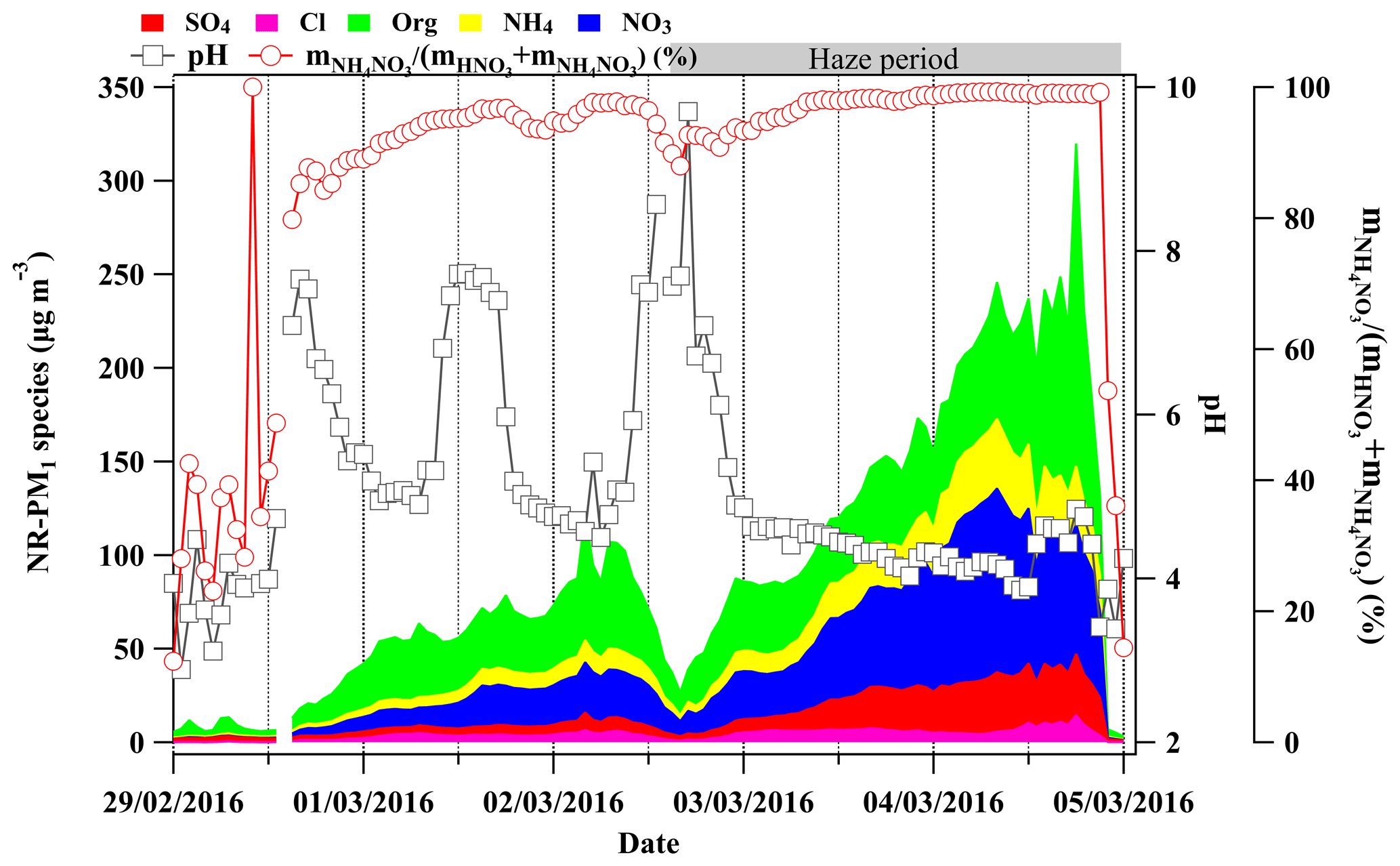
Figure 5The time series of chemical composition measured by the HR-ToF-AMS (left y axis), calculated aerosol pH by ISORROPIA II (inner right y axis) and the molar ratio of particulate nitrate in the total nitrate (gas + particle phase) shown on the outer right y axis during 29 February to 5 March 2016.

Figure 6The pH of the fine aerosol particles (left y axis) and the molar ratio of particulate nitrate in the total nitrate (gas + particle phase; right y axis) as a function of NR-PM1 mass concentrations.
To illustrate the facilitation of particulate nitrate enhancement from HNO3 in the presence of liquid water, we performed the theoretical calculation of equilibrium between particulate NH4NO3 and gaseous NH3 and HNO3 under dry and ambient conditions, respectively. The dissociation constant of NH4NO3 (Kp) in dry conditions was calculated using Eq. (5) without considering the influence of the liquid water. As shown in Fig. 3, the equilibrium Kp in the dry conditions ranged from 0.06 to 4.61 ppb2 (275.3 to 291.5 K) during the haze period. Taking account of the aerosol liquid water, the equilibrium vapor pressure of HNO3 and NH3 over particles was calculated by the E-AIM Model II (http://www.aim.env.uea.ac.uk, last access: 15 February 2020). Note that this calculation assumes negligible interaction between dissolved organic components and the activity of . In the presence of aerosol-associated water, the product of the equilibrium vapor pressure of NH3 and HNO3 calculated according to the E-AIM was 10 %–60 % lower than the equilibrium Kp in the dry conditions during the marked haze period. This means the presence of aerosol liquid water changed the equilibrium and would have favored the particulate nitrate enhancement. However, the aerosol particles did not reach the equilibrium between particulate NH4NO3 and the gases (NH3 + HNO3) during the investigated period, as the measured product of the NH3 and HNO3 partial pressure (2.55–9.63 ppb2) was supersaturated compared to the equilibrium values in both dry and deliquescent particles. In this case, the partitioning of gaseous NH3 and HNO3 in the atmosphere into the particle phase could be accelerated and leads to particulate nitrate enhancement with increasing ambient RH. Owing to its highly hydrophilic nature, the increased ammonium nitrate mass fraction leads to further water uptake. Such a mutual promotion of particulate nitrate and aerosol liquid water enhancement becomes more pronounced with the increasing pollution throughout the haze event owing to the simultaneously increasing ambient RH. Consistently, a significant co-increase of particulate nitrate and aerosol liquid water was observed during haze development as shown in Fig. 4. At first, a steep increase in particulate nitrate in the total nitrate mass ratio (from ∼12 % to ∼98 %) was observed as the aerosol liquid water was enhanced up to ∼20 µg m−3. And then, the particulate nitrate mass kept increasing with further increases in aerosol liquid water. We observed that ∼98 % of nitrate was present in the particle phase when aerosol liquid water was higher than ∼20 µg m−3. The function between the particulate nitrate fraction in the total nitrate and the aerosol particle liquid water is given in Fig. 4. It is worth noting that N2O5 hydrolysis during the nighttime can contribute extra HNO3 in the wet denuding method within the GAC–IC system. This effect explains the slight underestimation of the particulate fraction during the nighttime when aerosol liquid water is less than 10 µg m−3 (Fig. 4). However, the general consistency of this function between daytime and nighttime (Fig. 4) suggests a negligible influence of N2O5 interference on our analysis during the investigated period.
Apart from aerosol liquid water, aerosol pH is also an important factor in the particulate nitrate formation; a higher pH is favorable for the equilibrium of HNO3 into the particle phase (Nah et al., 2018). The pH of the fine aerosol particles was calculated by ISORROPIA II (Fountoukis and Nenes, 2007) during the investigated period. The model was running in forward mode with the chemical composition of NR-PM1 (, , Cl−, ) and gas precursors (HNO3, HCl, NH3) by GAC–IC as inputs. And the model was running in metastable mode when assuming no solid existed in the system. Generally, the fine aerosol particles became more acidic with pH dropping from ∼8 down to ∼4 when NR-PM1 mass concentration increased from ∼12 up to >300 µg m−3 as shown in Figs. 5 and 6. This declining trend of pH is not favorable for the HNO3 partitioning into the particle phase (Nah et al., 2018). However, a clear enhanced trend of the molar ratio of particulate nitrate in the total nitrate as a function of NR-PM1 mass concentration was observed correspondingly (as shown in Figs. 5 and 6). Therefore, in this case the increase in aerosol liquid water is more likely to be the driving factor of particulate nitrate formation compared to the influence of pH.
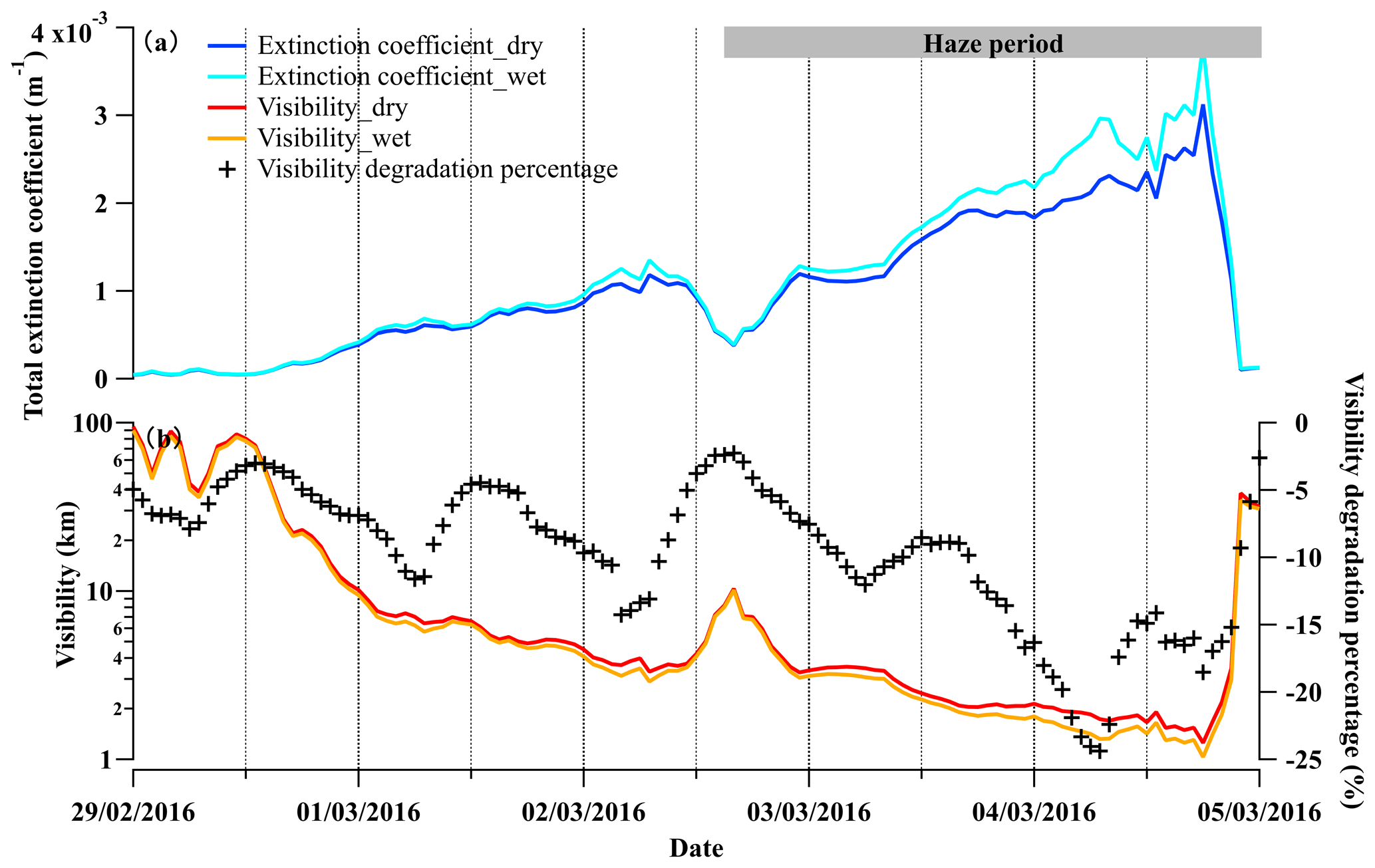
Figure 7The time series of (a) the calculated total extinction coefficient at a wavelength of 550 nm with the consideration of dry and wet PNSD, referred to as Extinction coefficient_dry and Extinction coefficient_wet and (b) calculated visibility with the consideration of dry and wet PNSD, referred to as Visibility_dry and Visibility_wet, respectively, during 29 February to 5 March 2016. The visibility degradation percentage is (Visibility_wet−Visibility_dry)∕Visibility_dry, representing the visibility degradation in the presence of liquid water.
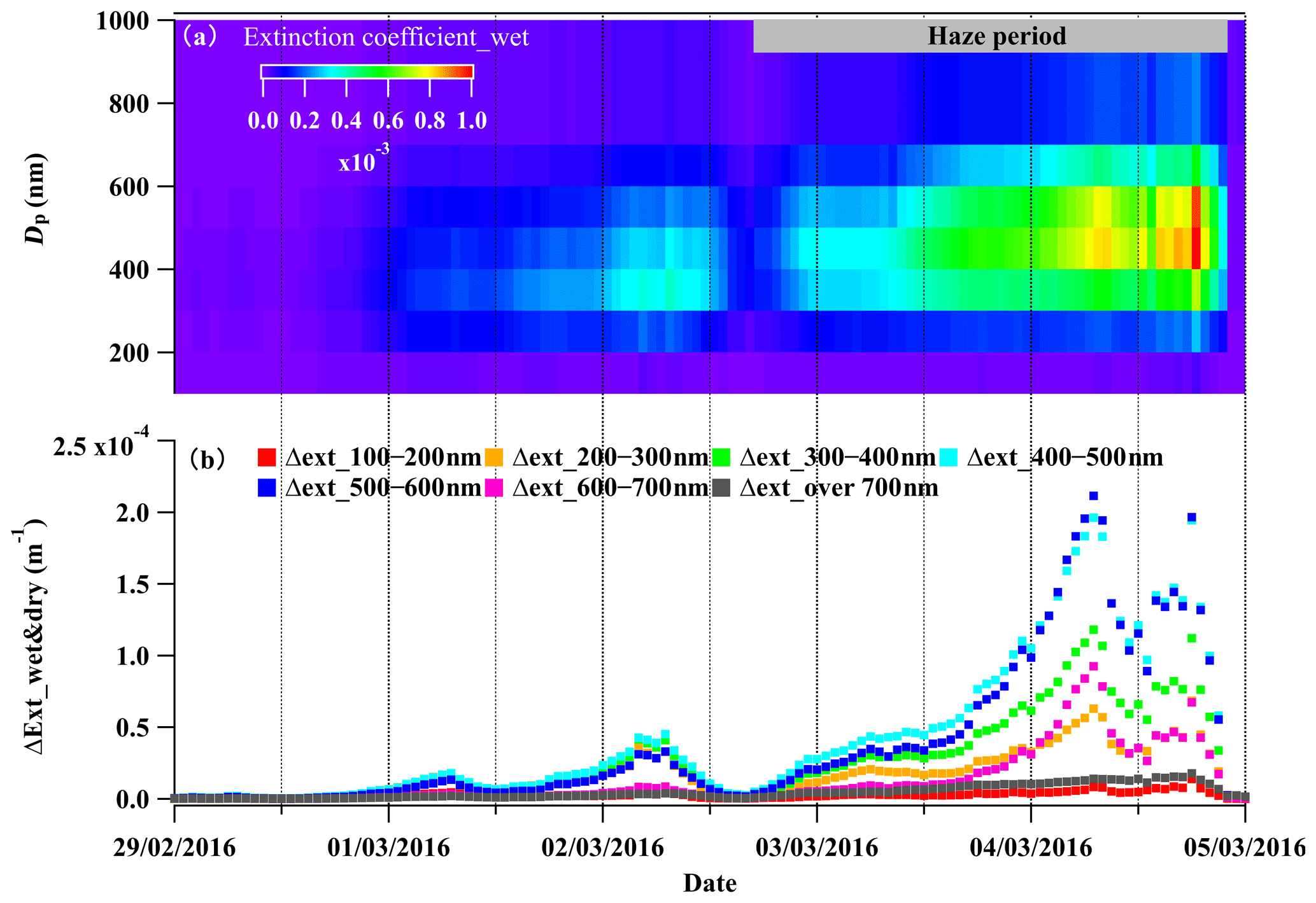
Figure 8(a) The size-segregated light extinction coefficient at a wavelength of 550 nm for wet particles (Extinction coefficient_wet) and (b) the size-segregated difference between Extinction coefficient_wet and Extinction coefficient_dry, representing the light extinction coefficient difference with and without considering liquid water during 29 February to 5 March 2016.
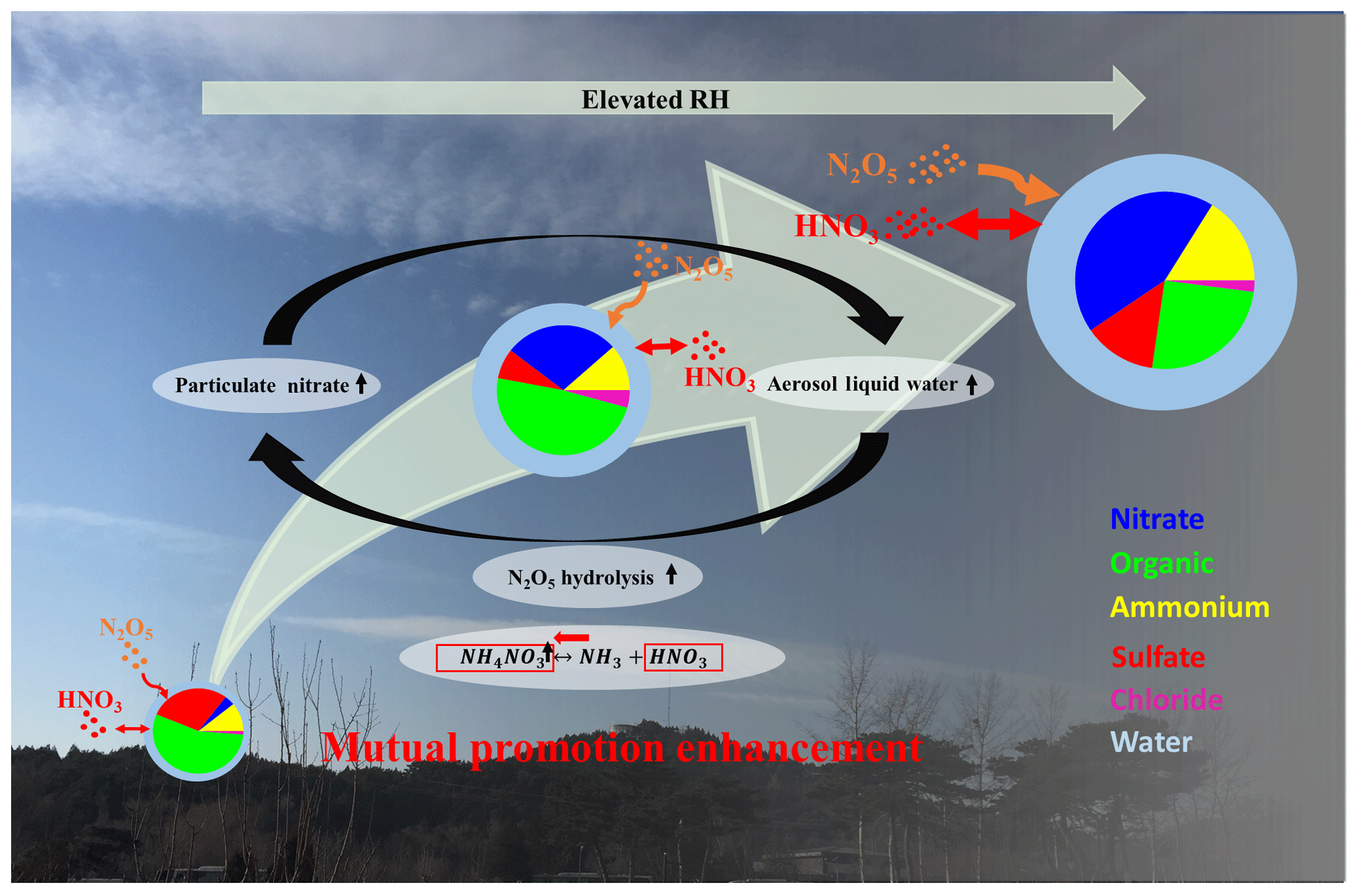
Figure 9The scheme of the mutual-promotion effect between aerosol liquid water and particulate nitrate.
It is worth noting that a similar co-condensation effect between water vapor and semivolatile organic components (Topping and McFiggans, 2012; Topping et al., 2013; Hu et al., 2018) could promote the haze formation as well, for which there may be some evidence in the current case. Such a co-condensation effect will lead to the enhancement of semivolatile organic and inorganic (e.g., nitrate) material with the increasing RH in a developing haze. The associated water will favor partitioning of both HNO3 and semivolatile organic materials into the particle phase depending on the organic solubility, providing a linkage between the development of increasing organic and inorganic particle mass.
3.3 The key role of liquid water on visibility degradation
Aerosol particles grow in size as ambient RH increases, further enhancing their extinction coefficient and impacting visibility (Zhao et al., 2019; Kuang et al., 2016). In this section, the size-resolved extinction coefficient of aerosol particles was estimated, and the influences of liquid water on the extinction coefficient and visibility were quantitatively evaluated. As shown in Fig. 7a, the total light extinction coefficient of dry and wet aerosol particles were enhanced by a factor of 4.3 and 5.4, respectively, from the beginning to a fully developed haze. Correspondingly, the calculated visibility without considering liquid water degraded significantly from ∼10 km to less than 2 km within 48 h during the marked haze period. The contribution of aerosol-associated water to visibility impairment was negligible in the beginning (2 %), while it was significant (up to 24 %) in the fully developed haze (Fig. 7b). This indicates that liquid water facilitated visibility degradation during haze development.
The influences of liquid water on visibility degradation varied with aerosol particle size. The size-resolved chemical composition data showed that the inorganic species, mainly particulate nitrate, were dominant components in the aerosol particles within the size range of 300–700 nm (Fig. S3). Correspondingly, the particles in this size range contained most of the liquid water (50 %–80 % of the total aerosol liquid water content of PM1). According to the discussion in Sect. 3.2, the mutual-promotion effect between liquid water and particulate nitrate can promote their mass loading enhancement. Aerosol particles in this size range experienced the most significant enhancement of light extinction due to water uptake (Fig. 8a and b) and contributed 70 %–88 % of the total extinction coefficient of the total NR-PM1 (Fig. S9). In conclusion, the rapid particulate nitrate enhancement enhanced the aerosol extinction coefficient during haze development, while the aerosol water uptake further enhanced the visibility degradation by increasing the extinction coefficient and promoting particulate nitrate enhancement.
It is worth noting that the enhanced dimming effect will further decrease the planetary boundary layer (PBL), which, in turn, depresses the dilution of water vapor and particulate matter in the atmosphere, hence leading to a higher RH and higher aerosol particle mass loading (Tie et al., 2017). Such an effect is beyond the scope of this study.
In this study, we observed a particulate-nitrate-dominated (up to 44 % of nonrefractory PM1 mass concentration) particulate matter pollution episode, which is typical during winter haze in Beijing, China. A clear co-increase in aerosol particle liquid water and particulate nitrate was observed, demonstrating the mutual promotion between them via observation-based theoretical calculations.
As shown in Fig. 9, the water uptake by hygroscopic aerosols increased the aerosol surface area and volume, enhancing the condensational loss of N2O5 across particles and favoring the thermodynamic equilibrium of HNO3 into the particle phase under the supersaturated ambient HNO3 and NH3. The enhanced particulate nitrate from the above pathways increased the mass fraction of particulate nitrate, which had a lower deliquescence RH than sulfate and resulted in more water uptake at lower ambient RH (Kreidenweis and Asa-Awuku, 2014). Hence, the increased aerosol particle surface area and volume concentrations due to water uptake in turn facilitate particulate nitrate enhancement. Hence, a feedback loop between liquid water and particulate nitrate enhancement is built up. Therefore the enhanced particulate nitrate components can accelerate the feedback compared with sulfate-rich pollution over the NCP region in the past (Hu et al., 2016). This self-amplification can rapidly degrade air quality and halve visibility within 1 d. Our results highlight the importance of reducing the particulate nitrate and its precursors (e.g., NOx) for the mitigation of haze episodes in the NCP region.
The observational dataset of the BEST-ONE campaign can be accessed through the corresponding author Zhijun Wu (zhijunwu@pku.edu.cn).
The E-AIM model can be accessed via: http://www.aim.env.uea.ac.uk/aim/aim.php (Clegg et al., 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-2161-2020-supplement.
ZW, YW and YC conceived the study. YZ, MH and AKS developed the BEST-ONE field campaign program. YW, ZW, DS, ZD, SHS, RS, GIG, PS, TH, KL, LZ, CZ, AKS, YZ and MH participated in this campaign and collected the dataset. YW conducted aerosol particle liquid water calculations under the guidance of YB and the thermodynamic equilibrium of particulate ammonium nitrate under the guidance of GM. YC calculated the uptake coefficient of N2O5, optical properties and visibility. YW and YC cowrote the paper with inputs from all coauthors. ZW, GM, AKS, SHS, GIG, PS, TH, AV and AW proofread and helped improve earlier versions of the manuscript. All authors discussed the results.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Multiphase chemistry of secondary aerosol formation under severe haze”. It does not belong to a conference.
We thank Paul I. Williams for valuable advice on the reaction constant of HNO3 and N2O5.
This research has been supported by the National Natural Science Foundation of China (grant nos. 41571130021 and 41875149), the Ministry of Science and Technology of the People's Republic of China (grant no. 2016YFC0202801), the German Federal Ministry of Education and Research (ID-CLAR), and the China Scholarship Council – University of Manchester (PhD, full scholarship).
This paper was edited by Jingkun Jiang and reviewed by two anonymous referees.
Anttila, T., Kiendler-Scharr, A., Tillmann, R., and Mentel, T. F.: On the Reactive Uptake of Gaseous Compounds by Organic-Coated Aqueous Aerosols: Theoretical Analysis and Application to the Heterogeneous Hydrolysis of N2O5, J. Phys. Chem. A, 110, 10435–10443, https://doi.org/10.1021/jp062403c, 2006.
Barnard, J. C., Fast, J. D., Paredes-Miranda, G., Arnott, W. P., and Laskin, A.: Technical Note: Evaluation of the WRF-Chem “Aerosol Chemical to Aerosol Optical Properties” Module using data from the MILAGRO campaign, Atmos. Chem. Phys., 10, 7325–7340, https://doi.org/10.5194/acp-10-7325-2010, 2010.
Bian, Y. X., Zhao, C. S., Ma, N., Chen, J., and Xu, W. Y.: A study of aerosol liquid water content based on hygroscopicity measurements at high relative humidity in the North China Plain, Atmos. Chem. Phys., 14, 6417–6426, https://doi.org/10.5194/acp-14-6417-2014, 2014.
Brown, S. S., Ryerson, T. B., Wollny, A. G., Brock, C. A., Peltier, R., Sullivan, A. P., Weber, R. J., Dubé, W. P., Trainer, M., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. R.: Variability in Nocturnal Nitrogen Oxide Processing and Its Role in Regional Air Quality, Science, 311, 67–70, https://doi.org/10.1126/science.1120120, 2006.
Calvert, J. G., Lazrus, A., Kok, G. L., Heikes, B. G., Walega, J. G., Lind, J., and Cantrell, C. A.: Chemical mechanisms of acid generation in the troposphere, Nature, 317, 27–35, https://doi.org/10.1038/317027a0, 1985.
Chang, W. L., Brown, S. S., Stutz, J., Middlebrook, A. M., Bahreini, R., Wagner, N. L., Dubé, W. P., Pollack, I. B., Ryerson, T. B., and Riemer, N.: Evaluating N2O5 heterogeneous hydrolysis parameterizations for CalNex 2010, J. Geophys. Res.-Atmos., 121, 5051–5070, https://doi.org/10.1002/2015JD024737, 2016.
Chen, Y., Wild, O., Wang, Y., Ran, L., Teich, M., Größ, J., Wang, L., Spindler, G., Herrmann, H., van Pinxteren, D., McFiggans, G., and Wiedensohler, A.: The influence of impactor size cut-off shift caused by hygroscopic growth on particulate matter loading and composition measurements, Atmos. Environ., 195, 141–148, https://doi.org/10.1016/j.atmosenv.2018.09.049, 2018a.
Chen, Y., Wolke, R., Ran, L., Birmili, W., Spindler, G., Schröder, W., Su, H., Cheng, Y., Tegen, I., and Wiedensohler, A.: A parameterization of the heterogeneous hydrolysis of N2O5 for mass-based aerosol models: improvement of particulate nitrate prediction, Atmos. Chem. Phys., 18, 673–689, https://doi.org/10.5194/acp-18-673-2018, 2018b.
Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Gao, M., Zhang, Q., He, K., Carmichael, G., Pöschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, 12, https://doi.org/10.1126/sciadv.1601530, 2016.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic Model of the System at Tropospheric Temperatures, J. Phys. Chem. A, 102, 2137–2154, https://doi.org/10.1021/jp973042r, 1998.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: E-AIM model, available at: http://www.aim.env.uea.ac.uk/aim/aim.php, last access: 15 February 2020.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
de Foy, B., Lu, Z., and Streets, D. G.: Satellite NO2 retrievals suggest China has exceeded its NOx reduction goals from the twelfth Five-Year Plan, Sci. Rep., 6, 35912, https://doi.org/10.1038/srep35912, 2016.
Dong, H.-B., Zeng, L.-M., Hu, M., Wu, Y.-S., Zhang, Y.-H., Slanina, J., Zheng, M., Wang, Z.-F., and Jansen, R.: Technical Note: The application of an improved gas and aerosol collector for ambient air pollutants in China, Atmos. Chem. Phys., 12, 10519–10533, https://doi.org/10.5194/acp-12-10519-2012, 2012.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Fitzgerald, J. W.: Dependence of the Supersaturation Spectrum of CCN on Aerosol Size Distribution and Composition, J. Atmos. Scie., 30, 628–634, https://doi.org/10.1175/1520-0469(1973)030<0628:dotsso>2.0.co;2, 1973.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+––Na+–––Cl−–H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Ge, B., Xu, X., Ma, Z., Pan, X., Wang, Z., Lin, W., Ouyang, B., Xu, D., Lee, J., Zheng, M., Ji, D., Sun, Y., Dong, H., Squires, F. A., Fu, P., and Wang, Z.: Role of Ammonia on the Feedback Between AWC and Inorganic Aerosol Formation During Heavy Pollution in the North China Plain, Earth and Space Science, 6, 1675–1693, https://doi.org/10.1029/2019ea000799, 2019.
Ge, X., He, Y., Sun, Y., Xu, J., Wang, J., Shen, Y., and Chen, M.: Characteristics and Formation Mechanisms of Fine Particulate Nitrate in Typical Urban Areas in China, Atmosphere, 8, 62, https://doi.org/10.3390/atmos8030062, 2017.
Gen, M., Zhang, R., Huang, D. D., Li, Y., and Chan, C. K.: Heterogeneous SO2 Oxidation in Sulfate Formation by Photolysis of Particulate Nitrate, Environ. Sci. Tech. Let., 6, 86–91, https://doi.org/10.1021/acs.estlett.8b00681, 2019a.
Gen, M., Zhang, R., Huang, D. D., Li, Y., and Chan, C. K.: Heterogeneous Oxidation of SO2 in Sulfate Production during Nitrate Photolysis at 300 nm: Effect of pH, Relative Humidity, Irradiation Intensity, and the Presence of Organic Compounds, Environ. Sci. Technol., 53, 8757–8766, https://doi.org/10.1021/acs.est.9b01623, 2019b.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Gysel, M., Crosier, J., Topping, D. O., Whitehead, J. D., Bower, K. N., Cubison, M. J., Williams, P. I., Flynn, M. J., McFiggans, G. B., and Coe, H.: Closure study between chemical composition and hygroscopic growth of aerosol particles during TORCH2, Atmos. Chem. Phys., 7, 6131–6144, https://doi.org/10.5194/acp-7-6131-2007, 2007.
Gysel, M., McFiggans, G. B., and Coe, H.: Inversion of tandem differential mobility analyser (TDMA) measurements, J. Aerosol Sci., 40, 134–151, https://doi.org/10.1016/j.jaerosci.2008.07.013, 2009.
Heintzenberg, J.: Fine particles in the global troposphere A review, Tellus B, 41, 149–160, https://doi.org/10.3402/tellusb.v41i2.15064, 1989.
Hu, D., Topping, D., and McFiggans, G.: Measured particle water uptake enhanced by co-condensing vapours, Atmos. Chem. Phys., 18, 14925–14937, https://doi.org/10.5194/acp-18-14925-2018, 2018.
Hu, W., Hu, M., Hu, W., Jimenez, J. L., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Zeng, L., and Shao, M.: Chemical composition, sources, and aging process of submicron aerosols in Beijing: Contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, https://doi.org/10.1002/2015JD024020, 2016.
Hu, W., Hu, M., Hu, W.-W., Zheng, J., Chen, C., Wu, Y., and Guo, S.: Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 17, 9979–10000, https://doi.org/10.5194/acp-17-9979-2017, 2017.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Huang, X.-F., He, L.-Y., Hu, M., Canagaratna, M. R., Sun, Y., Zhang, Q., Zhu, T., Xue, L., Zeng, L.-W., Liu, X.-G., Zhang, Y.-H., Jayne, J. T., Ng, N. L., and Worsnop, D. R.: Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer, Atmos. Chem. Phys., 10, 8933–8945, https://doi.org/10.5194/acp-10-8933-2010, 2010.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001JD001213, 2003.
Köhler, H.: The Nucleus in and the Growth of Hygroscopic Droplets, T. Faraday Soc., 32, 1152–1161, 1936.
Kolb, C. E., Cox, R. A., Abbatt, J. P. D., Ammann, M., Davis, E. J., Donaldson, D. J., Garrett, B. C., George, C., Griffiths, P. T., Hanson, D. R., Kulmala, M., McFiggans, G., Pöschl, U., Riipinen, I., Rossi, M. J., Rudich, Y., Wagner, P. E., Winkler, P. M., Worsnop, D. R., and O' Dowd, C. D.: An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds, Atmos. Chem. Phys., 10, 10561–10605, https://doi.org/10.5194/acp-10-10561-2010, 2010.
Koop, T., Bookhold, J., Shiraiwa, M., and Pöschl, U.: Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere, Phys. Chem. Chem. Phys., 13, 19238–19255, https://doi.org/10.1039/C1CP22617G, 2011.
Kreidenweis, S. M. and Asa-Awuku, A.: 5.13 – Aerosol Hygroscopicity: Particle Water Content and Its Role in Atmospheric Processes A2, in: Treatise on Geochemistry, 2nd Edn., edited by: Turekian, H. D. and Holland, K. K, Elsevier, Oxford, 331–361, 2014.
Kuang, Y., Zhao, C. S., Ma, N., Liu, H. J., Bian, Y. X., Tao, J. C., and Hu, M.: Deliquescent phenomena of ambient aerosols on the North China Plain, Geophys. Res. Lett. 43, 8744–8750, https://doi.org/10.1002/2016GL070273, 2016.
Kulmala, M., Laaksonen, A., Korhonen, P., Vesala, T., Ahonen, T., and Barrett, J. C.: The effect of atmospheric nitric acid vapor on cloud condensation nucleus activation, J. Geophys. Res.-Atmos., 98, 22949–22958, https://doi.org/10.1029/93JD02070, 1993.
Li, C., McLinden, C., Fioletov, V., Krotkov, N., Carn, S., Joiner, J., Streets, D., He, H., Ren, X., Li, Z., and Dickerson, R. R.: India Is Overtaking China as the World's Largest Emitter of Anthropogenic Sulfur Dioxide, Sci. Rep., 7, 14304, https://doi.org/10.1038/s41598-017-14639-8, 2017.
Li, H., Zhang, Q., Zheng, B., Chen, C., Wu, N., Guo, H., Zhang, Y., Zheng, Y., Li, X., and He, K.: Nitrate-driven urban haze pollution during summertime over the North China Plain, Atmos. Chem. Phys., 18, 5293–5306, https://doi.org/10.5194/acp-18-5293-2018, 2018.
Li, Y. J., Liu, P. F., Bergoend, C., Bateman, A. P., and Martin, S. T.: Rebounding hygroscopic inorganic aerosol particles: Liquids, gels, and hydrates, Aerosol Sci. Tech., 51, 388–396, https://doi.org/10.1080/02786826.2016.1263384, 2017.
Liu, B. Y. H., Pui, D. Y. H., Whitby, K. T., Kittelson, D. B., Kousaka, Y., and McKenzie, R. L.: The aerosol mobility chromatograph: A new detector for sulfuric acid aerosols, Atmos. Environ., 12, 99–104, https://doi.org/10.1016/0004-6981(78)90192-0, 1978.
Liu, M., Huang, X., Song, Y., Xu, T., Wang, S., Wu, Z., Hu, M., Zhang, L., Zhang, Q., Pan, Y., Liu, X., and Zhu, T.: Rapid SO2 emission reductions significantly increase tropospheric ammonia concentrations over the North China Plain, Atmos. Chem. Phys., 18, 17933–17943, https://doi.org/10.5194/acp-18-17933-2018, 2018.
Liu, Y., Wu, Z., Wang, Y., Xiao, Y., Gu, F., Zheng, J., Tan, T., Shang, D., Wu, Y., Zeng, L., Hu, M., Bateman, A. P., and Martin, S. T.: Submicrometer Particles Are in the Liquid State during Heavy Haze Episodes in the Urban Atmosphere of Beijing, China, Environ. Sci. Tech. Let., 4, 427–432, https://doi.org/10.1021/acs.estlett.7b00352, 2017.
Lu, K., Fuchs, H., Hofzumahaus, A., Tan, Z., Wang, H., Zhang, L., Schmitt, S. H., Rohrer, F., Bohn, B., Broch, S., Dong, H., Gkatzelis, G. I., Hohaus, T., Holland, F., Li, X., Liu, Y., Liu, Y., Ma, X., Novelli, A., Schlag, P., Shao, M., Wu, Y., Wu, Z., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Fast Photochemistry in Wintertime Haze: Consequences for Pollution Mitigation Strategies, Environ. Sci. Technol., 53, 10676–10684, https://doi.org/10.1021/acs.est.9b02422, 2019.
Maitland, G. C., Rigby, M., Smith, E. B., and Wakeham, W. A.: Intermolecular forces: their origin and determination, International series of monographs on chemistry 3, Oxford University Press, 1981.
Massling, A., Niedermeier, N., Hennig, T., Fors, E. O., Swietlicki, E., Ehn, M., Hämeri, K., Villani, P., Laj, P., Good, N., McFiggans, G., and Wiedensohler, A.: Results and recommendations from an intercomparison of six Hygroscopicity-TDMA systems, Atmos. Meas. Tech., 4, 485–497, https://doi.org/10.5194/amt-4-485-2011, 2011.
McFiggans, G., Artaxo, P., Baltensperger, U., Coe, H., Facchini, M. C., Feingold, G., Fuzzi, S., Gysel, M., Laaksonen, A., Lohmann, U., Mentel, T. F., Murphy, D. M., O'Dowd, C. D., Snider, J. R., and Weingartner, E.: The effect of physical and chemical aerosol properties on warm cloud droplet activation, Atmos. Chem. Phys., 6, 2593–2649, https://doi.org/10.5194/acp-6-2593-2006, 2006.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Mozurkewich, M. and Calvert, J. G.: Reaction probability of N2O5 on aqueous aerosols, J. Geophys. Res.-Atmos., 93, 15889–15896, https://doi.org/10.1029/JD093iD12p15889, 1988.
Nah, T., Guo, H., Sullivan, A. P., Chen, Y., Tanner, D. J., Nenes, A., Russell, A., Ng, N. L., Huey, L. G., and Weber, R. J.: Characterization of aerosol composition, aerosol acidity, and organic acid partitioning at an agriculturally intensive rural southeastern US site, Atmos. Chem. Phys., 18, 11471–11491, https://doi.org/10.5194/acp-18-11471-2018, 2018.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Petzold, A. and Schönlinner, M.: Multi-angle absorption photometry – a new method for the measurement of aerosol light absorption and atmospheric black carbon, J. Aerosol Sci., 35, 421–441, https://doi.org/10.1016/j.jaerosci.2003.09.005, 2004.
Petzold, A., Ogren, J. A., Fiebig, M., Laj, P., Li, S.-M., Baltensperger, U., Holzer-Popp, T., Kinne, S., Pappalardo, G., Sugimoto, N., Wehrli, C., Wiedensohler, A., and Zhang, X.-Y.: Recommendations for reporting “black carbon” measurements, Atmos. Chem. Phys., 13, 8365–8379, https://doi.org/10.5194/acp-13-8365-2013, 2013.
Pfeifer, S., Müller, T., Weinhold, K., Zikova, N., Martins dos Santos, S., Marinoni, A., Bischof, O. F., Kykal, C., Ries, L., Meinhardt, F., Aalto, P., Mihalopoulos, N., and Wiedensohler, A.: Intercomparison of 15 aerodynamic particle size spectrometers (APS 3321): uncertainties in particle sizing and number size distribution, Atmos. Meas. Tech., 9, 1545–1551, https://doi.org/10.5194/amt-9-1545-2016, 2016.
Putaud, J.-P., Raes, F., Van Dingenen, R., Brüggemann, E., Facchini, M. C., Decesari, S., Fuzzi, S., Gehrig, R., Hüglin, C., Laj, P., Lorbeer, G., Maenhaut, W., Mihalopoulos, N., Müller, K., Querol, X., Rodriguez, S., Schneider, J., Spindler, G., Brink, H. T., Tørseth, K., and Wiedensohler, A.: A European aerosol phenomenology – 2: chemical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe, Atmos. Environ., 38, 2579–2595, https://doi.org/10.1016/j.atmosenv.2004.01.041, 2004.
Schwartz, S. E.: Mass-Transport Considerations Pertinent to Aqueous Phase Reactions of Gases in Liquid-Water Clouds, Chemistry of Multiphase Atmospheric Systems, 6, Springer, Berlin, Heidelberg, 1986.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: from air pollution to climate change, John wiley & Sons, INC, 2006.
Shiraiwa, M., Ammann, M., Koop, T., and Pöschl, U.: Gas uptake and chemical aging of semisolid organic aerosol particles, P. Natl. Acad. Sci. USA, 108, 11003–11008, https://doi.org/10.1073/pnas.1103045108, 2011.
Spindler, G., Müller, K., Brüggemann, E., Gnauk, T., and Herrmann, H.: Long-term size-segregated characterization of PM10, PM2.5, and PM1 at the IfT research station Melpitz downwind of Leipzig (Germany) using high and low-volume filter samplers, Atmos. Environ., 38, 5333–5347, https://doi.org/10.1016/j.atmosenv.2003.12.047, 2004.
Sun, Y., Jiang, Q., Wang, Z., Fu, P., Li, J., Yang, T., and Yin, Y.: Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013, J. Geophys. Res.-Atmos., 119, 4380–4398, https://doi.org/10.1002/2014jd021641, 2014.
Sun, Y. L., Wang, Z. F., Du, W., Zhang, Q., Wang, Q. Q., Fu, P. Q., Pan, X. L., Li, J., Jayne, J., and Worsnop, D. R.: Long-term real-time measurements of aerosol particle composition in Beijing, China: seasonal variations, meteorological effects, and source analysis, Atmos. Chem. Phys., 15, 10149–10165, https://doi.org/10.5194/acp-15-10149-2015, 2015.
Swietlicki, E., Hansson, H. C., Hämeri, K., Svenningsson, B., Massling, A., McFiggans, G., McMurry, P. H., Petäjä, T., Tunved, P., Gysel, M., Topping, D., Weingartner, E., Baltensperger, U., Rissler, J., Wiedensohler, A., and Kulmala, M.: Hygroscopic properties of submicrometer atmospheric aerosol particles measured with H-TDMA instruments in various environments – a review, Tellus B, 60, 432–469, https://doi.org/10.1111/j.1600-0889.2008.00350.x, 2008.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tie, X., Huang, R.-J., Cao, J., Zhang, Q., Cheng, Y., Su, H., Chang, D., Pöschl, U., Hoffmann, T., Dusek, U., Li, G., Worsnop, D. R., and O'Dowd, C. D.: Severe Pollution in China Amplified by Atmospheric Moisture, Sci. Rep., 7, 15760, https://doi.org/10.1038/s41598-017-15909-1, 2017.
Topping, D., Connolly, P., and McFiggans, G.: Cloud droplet number enhanced by co-condensation of organic vapours, Nat. Geosci., 6, 443–446, https://doi.org/10.1038/ngeo1809, 2013.
Topping, D. O. and McFiggans, G.: Tight coupling of particle size, number and composition in atmospheric cloud droplet activation, Atmos. Chem. Phys., 12, 3253–3260, https://doi.org/10.5194/acp-12-3253-2012, 2012.
Vu, T. V., Shi, Z., Cheng, J., Zhang, Q., He, K., Wang, S., and Harrison, R. M.: Assessing the impact of clean air action on air quality trends in Beijing using a machine learning technique, Atmos. Chem. Phys., 19, 11303–11314, https://doi.org/10.5194/acp-19-11303-2019, 2019.
Wang, G., Zhang, R., Gomez, M. E., Yang, L., Levy Zamora, M., Hu, M., Lin, Y., Peng, J., Guo, S., Meng, J., Li, J., Cheng, C., Hu, T., Ren, Y., Wang, Y., Gao, J., Cao, J., An, Z., Zhou, W., Li, G., Wang, J., Tian, P., Marrero-Ortiz, W., Secrest, J., Du, Z., Zheng, J., Shang, D., Zeng, L., Shao, M., Wang, W., Huang, Y., Wang, Y., Zhu, Y., Li, Y., Hu, J., Pan, B., Cai, L., Cheng, Y., Ji, Y., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, https://doi.org/10.1073/pnas.1616540113, 2016.
Wang, H., Lu, K., Chen, X., Zhu, Q., Chen, Q., Guo, S., Jiang, M., Li, X., Shang, D., Tan, Z., Wu, Y., Wu, Z., Zou, Q., Zheng, Y., Zeng, L., Zhu, T., Hu, M., and Zhang, Y.: High N2O5 Concentrations Observed in Urban Beijing: Implications of a Large Nitrate Formation Pathway, Environ. Sci. Tech. Let., 4, 416–420, https://doi.org/10.1021/acs.estlett.7b00341, 2017.
Wang, Y. and Chen, Y.: Significant Climate Impact of Highly Hygroscopic Atmospheric Aerosols in Delhi, India, Geophys. Res. Lett., 46, 5535–5545, https://doi.org/10.1029/2019gl082339, 2019.
Wang, Y., Wu, Z., Ma, N., Wu, Y., Zeng, L., Zhao, C., and Wiedensohler, A.: Statistical analysis and parameterization of the hygroscopic growth of the sub-micrometer urban background aerosol in Beijing, Atmos. Environ., 175, 184–191, https://doi.org/10.1016/j.atmosenv.2017.12.003, 2018.
Wiedensohler, A., Birmili, W., Nowak, A., Sonntag, A., Weinhold, K., Merkel, M., Wehner, B., Tuch, T., Pfeifer, S., Fiebig, M., Fjäraa, A. M., Asmi, E., Sellegri, K., Depuy, R., Venzac, H., Villani, P., Laj, P., Aalto, P., Ogren, J. A., Swietlicki, E., Williams, P., Roldin, P., Quincey, P., Hüglin, C., Fierz-Schmidhauser, R., Gysel, M., Weingartner, E., Riccobono, F., Santos, S., Grüning, C., Faloon, K., Beddows, D., Harrison, R., Monahan, C., Jennings, S. G., O'Dowd, C. D., Marinoni, A., Horn, H.-G., Keck, L., Jiang, J., Scheckman, J., McMurry, P. H., Deng, Z., Zhao, C. S., Moerman, M., Henzing, B., de Leeuw, G., Löschau, G., and Bastian, S.: Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions, Atmos. Meas. Tech., 5, 657–685, https://doi.org/10.5194/amt-5-657-2012, 2012.
Wu, Z., Hu, M., Lin, P., Liu, S., Wehner, B., and Wiedensohler, A.: Particle number size distribution in the urban atmosphere of Beijing, China, Atmos. Environ., 42, 7967–7980, https://doi.org/10.1016/j.atmosenv.2008.06.022, 2008.
Wu, Z., Wang, Y., Tan, T., Zhu, Y., Li, M., Shang, D., Wang, H., Lu, K., Guo, S., Zeng, L., and Zhang, Y.: Aerosol Liquid Water Driven by Anthropogenic Inorganic Salts: Implying Its Key Role in Haze Formation over the North China Plain, Environ. Sci. Tech. Let., 5, 160–166, https://doi.org/10.1021/acs.estlett.8b00021, 2018.
Wu, Z. J., Nowak, A., Poulain, L., Herrmann, H., and Wiedensohler, A.: Hygroscopic behavior of atmospherically relevant water-soluble carboxylic salts and their influence on the water uptake of ammonium sulfate, Atmos. Chem. Phys., 11, 12617–12626, https://doi.org/10.5194/acp-11-12617-2011, 2011.
Wu, Z. J., Zheng, J., Shang, D. J., Du, Z. F., Wu, Y. S., Zeng, L. M., Wiedensohler, A., and Hu, M.: Particle hygroscopicity and its link to chemical composition in the urban atmosphere of Beijing, China, during summertime, Atmos. Chem. Phys., 16, 1123–1138, https://doi.org/10.5194/acp-16-1123-2016, 2016.
Xie, Y., Wang, G., Wang, X., Chen, J., Chen, Y., Tang, G., Wang, L., Ge, S., Xue, G., Wang, Y., and Gao, J.: Observation of nitrate dominant PM2.5 and particle pH elevation in urban Beijing during the winter of 2017, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-541, in review, 2019.
Xu, Q., Wang, S., Jiang, J., Bhattarai, N., Li, X., Chang, X., Qiu, X., Zheng, M., Hua, Y., and Hao, J.: Nitrate dominates the chemical composition of PM2.5 during haze event in Beijing, China, Sci. Total Environ., 689, 1293–1303, https://doi.org/10.1016/j.scitotenv.2019.06.294, 2019.
Xu, W., Sun, Y., Wang, Q., Zhao, J., Wang, J., Ge, X., Xie, C., Zhou, W., Du, W., Li, J., Fu, P., Wang, Z., Worsnop, D. R., and Coe, H.: Changes in Aerosol Chemistry From 2014 to 2016 in Winter in Beijing: Insights From High-Resolution Aerosol Mass Spectrometry, J. Geophys. Res.-Atmos., 124, 1132–1147, https://doi.org/10.1029/2018jd029245, 2019.
Yu, F. and Luo, G.: Simulation of particle size distribution with a global aerosol model: contribution of nucleation to aerosol and CCN number concentrations, Atmos. Chem. Phys., 9, 7691–7710, https://doi.org/10.5194/acp-9-7691-2009, 2009.
Yue, F., Xie, Z., Zhang, P., Song, S., He, P., Liu, C., Wang, L., Yu, X., and Kang, H.: The role of sulfate and its corresponding S(IV)+NO2 formation pathway during the evolution of haze in Beijing, Sci. Total Environ., 687, 741–751, https://doi.org/10.1016/j.scitotenv.2019.06.096, 2019.
Yun, H., Wang, W., Wang, T., Xia, M., Yu, C., Wang, Z., Poon, S. C. N., Yue, D., and Zhou, Y.: Nitrate formation from heterogeneous uptake of dinitrogen pentoxide during a severe winter haze in southern China, Atmos. Chem. Phys., 18, 17515–17527, https://doi.org/10.5194/acp-18-17515-2018, 2018.
Zhang, R., Jing, J., Tao, J., Hsu, S.-C., Wang, G., Cao, J., Lee, C. S. L., Zhu, L., Chen, Z., Zhao, Y., and Shen, Z.: Chemical characterization and source apportionment of PM2.5 in Beijing: seasonal perspective, Atmos. Chem. Phys., 13, 7053–7074, https://doi.org/10.5194/acp-13-7053-2013, 2013.
Zhao, C., Yu, Y., Kuang, Y., Tao, J., and Zhao, G.: Recent Progress of Aerosol Light-scattering Enhancement Factor Studies in China, Adv. Atmos. Sci., 36, 1015–1026, https://doi.org/10.1007/s00376-019-8248-1, 2019.