the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Nov 2020
| 27 Nov 2020
Direct contribution of ammonia to α-pinene secondary organic aerosol formation
Liqing Hao
Eetu Kari
Ari Leskinen
Douglas R. Worsnop
Annele Virtanen
Ammonia (NH3), a gaseous compound ubiquitously present in the atmosphere, is involved in the formation of secondary organic aerosol (SOA), but the exact mechanism is still not well known. This study presents the results of SOA experiments from the photooxidation of α-pinene in the presence of NH3 in the reaction chamber. SOA was formed in in nucleation experiments and in seeded experiments with ammonium sulfate particles as seeds. The chemical composition and time series of compounds in the gas and particle phase were characterized by an online high-resolution time-of-flight proton-transfer-reaction mass spectrometer (HR-ToF-PTRMS) and a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS), respectively. Our results show that the mass concentration of ammonium (NH) was still rising even after the mass concentration of the organic component started to decrease due to aerosol wall deposition and evaporation, implying the continuous new formation of particle-phase ammonium in the process. Stoichiometric neutralization analysis of aerosol indicates that organic acids have a central role in the formation of particle-phase ammonium. Our measurements show a good correlation between the gas-phase organic mono- and dicarboxylic acids formed in the photooxidation of α-pinene and the ammonium in the particle phase, thus highlighting the contribution of gas-phase organic acids to the ammonium formation. The work shows that the gas-phase organic acids contribute to the SOA formation by forming organic ammonium salts through acid–base reaction. The changes in aerosol mass, particle size and chemical composition resulting from the NH3–SOA interaction can potentially alter the aerosol direct and indirect forcing and therefore alter its impact on climate change.
- Article
(8196 KB) - Full-text XML
-
Supplement
(495 KB) - BibTeX
- EndNote





The largest uncertainty in forward projection of global warming is related to our limited knowledge of solar radiative forcing associated with aerosols (IPCC, 2013). Formation of secondary organic aerosols (SOAs) is one of the main processes that affects the composition and properties of atmospheric aerosols. Formation of SOA occurs through two distinct mechanisms: by increasing the mass of the existing aerosol and through the formation of new particles. The estimate on the SOA formation shows its significance as a source of atmospheric organic aerosol: about 60 % of the organic aerosol mass is SOA on the global scale and regionally even more (Hallquist et al., 2009; Jimenez et al., 2009; Kanakidou et al., 2005). Hence, SOA plays an important role in the direct scattering of solar radiation, cloud formation and precipitation, and visibility reduction, and it may also have a direct impact on human health.
Ammonia (NH3) is ubiquitously present in the atmosphere as a dominant volatile base. The majority of its sources is accounted by emissions from agriculture, livestock, soil and traffic (Huang et al., 2012; Grönroos et al., 2009; Battye et al., 2003). NH3 governs the neutralization of atmospheric aerosol by reacting with inorganic acids such as sulfuric acid and nitric acid, leading to transformation of a substantial amount of ammonium sulfate (and derivatives) and ammonium nitrate (Seinfeld and Pandis, 2016). These inorganic salts play a vital role in contributing to the fine particle matter (PM2.5) and altering the chemical and physical properties of aerosol particles in the atmosphere. The mechanism between NH3 and the inorganic acids leading to the secondary inorganic aerosol has been well recognized.
A number of studies have shown that NH3 is one of the key species for the new particle formation through ternary and binary nucleation with water and sulfuric acid (e.g., Lehtipalo et al., 2018; Jokinen et al., 2018; Bianchi et al., 2016; Kirkby et al., 2011; Kurten et al., 2007; Kulmala et al., 2000). The nucleated particles are a significant source of atmospheric SOA particles, and subsequent growth to a larger size (>50 nm) allows them to serve as cloud condensation nuclei (CCN). However, the role of NH3 for the large-size (e.g., CCN size) SOA particles is still rarely studied. A study, conducted more than a decade ago, demonstrated that NH3 increased the number and volume concentrations of CCN-sized SOA particles from the α-pinene–ozone system by 15 % and 8 %, respectively (Na et al., 2007). Similar results have also been observed in the photooxidation and ozonolysis of α-pinene SOA experiments: that SOA mass yield increased by 13 % as a response to NH3 addition (Babar et al., 2017). Besides, the addition of NH3 could also promote the SOA formation from photooxidation of vehicle exhaust (Chen et al., 2019; Liu et al., 2015), anthropogenic VOCs (Wang et al., 2018; Huang et al., 2018) and acrolein (Li et al., 2019). The promotion mechanism of NH3 to SOA formation can be both through base–acid reaction (Schlag et al., 2017; Babar et al., 2017; Rubach et al., 2013; Mensah et al., 2012; Na et al., 2007) and by the NH3 uptake to the carbonyl group (Zhu et al., 2018; Liu et al., 2015). As a consequence, the changes in particle size and chemical composition could alter the CCN ability and hygroscopicity of SOA particles (Dinar et al., 2008). Moreover, the reaction of NH3 with SOA decreases the volatility of SOA particle (Paciga et al., 2014) and also results in production of light-absorbing brown carbon compounds that modify the optical properties of the aerosols (Huang et al., 2018; Updyke et al., 2012; Bones et al., 2010). Additionally, the uptake of NH3 by SOA can deplete ambient NH3 concentrations, causing indirect reductions in the amount of inorganic ammonium salts in particulate matter (Horne et al., 2018). Therefore, the interaction of NH3 and SOA could alter both direct and indirect aerosol radiative forcing and potentially alter its impact on climate change.
This work presents the results of SOA formation from photooxidation of α-pinene in the presence of NH3 in the nucleation and seeded experiments. The chemical composition of gas-phase and particle-phase compounds was characterized with a high-resolution time-of-flight proton-transfer-reaction mass spectrometer (HR-ToF-PTRMS) and a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS), respectively. Our experiments show the formation of ammonium salt (NH), and our gas-phase measurements indicate organic acids are responsible for their formation. The results will have potential applications in studying the SOA formation mechanism and the impact of SOA on climate forcing.
2.1 Reaction chamber experiments
SOA experiments were conducted in the ILMARI environmental chamber infrastructure in the University of Eastern Finland. The experimental system and experimental procedure have been described in detail in Leskinen et al. (2015) and Kari et al. (2019a), respectively. The chamber consists of a 29 m3 Teflon™ FEP film bag. Ultraviolet (UV) lights with a spectrum centered at 340 nm around the chamber enable photochemical reactions. Two sets of experiments were designed (Table 1): (1) SOA nucleation experiments in the absence of seed aerosols and (2) SOA formation experiments in the presence of ammonium sulfate seeds. Prior to each experiment, the chamber was continuously flushed overnight with laboratory clean air produced by a zero air generator (model 737-15, Aadco Instruments Inc., USA) and conditioned in a humidifier (model FC125-240-5MP-02, Perma Pure LLC., USA), aiming at 50 % relative humidity at the typical temperature of 20 ∘C during the course of each experiment. Independent on the type of experiments (nucleation and seeded), the reactants were added into the chamber in the sequence described below. The (NH4)2SO4 (ammonium sulfate, 99 %, Sigma-Aldrich) seed was injected to the chamber first. The seed was generated using an atomizer (Topas ATM226, Germany). In two experiments, nitrogen oxide (NO) was added to the chamber, and ozone (O3) was introduced into the chamber to convert NO to nitrogen dioxide (NO2) to reach the atmospherically relevant NO2-to-NO ratio of ∼3 (Kari et al., 2019a). After O3 and NOx were fed, 3 µL 9-fold butanol (butanol-d9, 98 %, Sigma-Aldrich) was injected to the chamber, from whose consumption the hydroxyl radical (OH) exposure was estimated in each experiment (Kari et al., 2019a). Next, 1 and 18 µL of α-pinene (≥99 %, Sigma-Aldrich) were added into the chamber for the seeded and nucleation SOA experiments, respectively, corresponding to concentrations of ∼5 and ∼100 ppb of α-pinene in the chamber. The α-pinene concentration in the seeded experiments was about 20 times lower than in the nucleation experiment. The relatively low α-pinene concentration was used to mimic atmospherically relevant monoterpene mixing ratios, for example, in the Hyytiälä forest area in Finland (e.g., Kourtchev et al., 2016). We aimed to study if the ammonia plays a similar role as in the high α-pinene concentration case. Last, 5 mL of hydrogen peroxide (H2O2, 30 wt % in H2O, Sigma-Aldrich) was conducted into the chamber by mixing it with purified air flowing at 10 L min−1 (liter per minute), as a precursor for OH radicals to be generated under UV radiation. The H2O2 concentration was roughly 30 ppm, estimated from the amount of its injection and the volume of the chamber. In two sets of experiments, ammonia was introduced to the chamber as an impurity. The likely sources of ammonia were the pressurized air, possible leakage of the lab air and water used for humidification. After all the compounds were introduced into the chamber, the chamber was closed and the compounds were allowed to stabilize for 15 min. Then the UV lights were switched on to initiate photochemistry.
Table 1Initial experimental parameters and results in two sets of experiments.
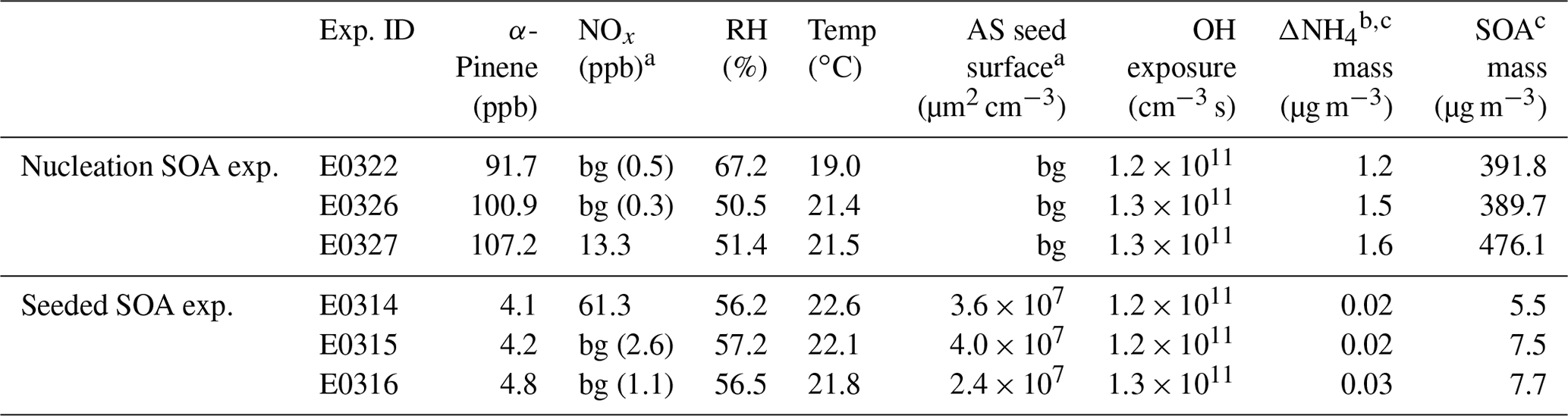
a “bg” means background concentration inside the chamber. b In the seeded SOA experiment, because of the presence of ammonium sulfate seeds, the maximum mass concentration of newly formed ammonium salt was estimated from the difference between and ; refer to the text for details. c The formed aerosol mass at maximum.
2.2 Analytical methods and instrumentation
The size-resolved chemical composition and mass concentration of aerosol particles were measured directly with an online high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, hereafter referred to as AMS) (DeCarlo et al., 2006). A detailed description of AMS operational procedure is provided in previous publications (Canagaratna et al., 2007; Jayne et al., 2000). In brief, AMS was operated in V mode in EI mode. Calibration of ionization efficiency (IE) followed the standard protocol using dried and size-selected ammonium nitrate particles. The data were analyzed using standard AMS data analysis toolkits (Squirrel V1.62D and Pika V1.22D) in Igor Pro Software (version 6.37, WaveMetrics Inc.). For determining mass concentrations, the default relative ionization efficiency (RIE) values were 1.4, 1.1 and 1.2, for organics, nitrate and sulfate, respectively. The RIE for ammonium was 2.95, as determined in the IE calibration. After a comparison to the volume concentration derived from a scanning mobility particle sizer (SMPS TSI 3081 DMA + 3775 CPC) measurement (Fig. S1 in the Supplement), a collection factor of 100 % was applied to determine the aerosol mass concentration in the reported results in this work. The positive matrix factorization (PMF) analysis was performed on the high-resolution mass spectra by using the PMF Evaluation Tool V2.08 (Paatero and Tapper, 1994; Ulbrich et al., 2009). The standard error matrices were processed following the principles of applying minimum error estimate, downweighting weak variables, removing bad variables and downweighing m∕z 44 related fragments (Ulbrich et al., 2009). The PMF was evaluated with one to six factors. Rotation (Fpeak) varied from −1 to 1 at a step of 0.1.
The concentrations of α-pinene and oxidized organic products were quantified by an online high-resolution time-of-flight proton-transfer-reaction mass spectrometer (HR-ToF-PTRMS, hereafter referred to as PTRMS) (Ionicon Analytik). A detailed description of the instrument and operation procedure has been provided in Jordan et al. (2009) and Kari et al. (2019a, b). In brief, the PTRMS instrumental settings were the same as in Kari et al. (2019a). During measurement, the mass calibration was conducted using protonated water isotope signals at m∕z 21 and internal instrumental signals of diiodobenzene and its fragment ions (protonated integer m∕z 331). The instrumental transmission calibration was conducted using a standard gas mixture containing eight different VOCs (protonated integers ranging from m∕z 79 to m∕z 181). The concentration of VOCs was determined according to the principles by Hansel et al. (1995). The selected ions that we are interested in this study are consistent with α-pinene (m∕z 136+1), formic acid (m∕z 46+1), acetic acid (m∕z 60+1), propionic acid (m∕z 74+1), pinonic acid (m∕z 184+1), butyric acid (m∕z 88+1), pentanoic acid (m∕z 102+1), malonic acid (m∕z 104+1) and succinic acid (m∕z 118+1).
For supporting information we measured the particle concentration and size distribution in a diameter range of 7–800 nm with an SMPS (TSI 3081 DMA + 3775 CPC) and the concentrations of NO, nitrogen oxides (NOx, Fig. S2), ozone (O3), and sulfur dioxide (SO2, Fig. S2), as well as the relative humidity and temperature inside the chamber. In this study, we lacked the measurement of NH3 concentration in the chamber but estimated it from our AMS measurement results, by assuming that the particulate ammonium salt (NH) was converted from the gas-phase NH3 (Fig. 1). The maximum NH concentration was in the range of 1.17–1.51 µg m−3, which corresponds to a minimum NH3 concentration level of 1.6–2.1 ppbv in our chamber. The NH3 mixing ratio over the continental range is typically between 0.1 and 10 ppb (Seinfeld and Pandis, 2016). A statistics study on the ammonia concentration in 14 Finnish office buildings shows a range of 1–49 µg m−3 and a geometric mean concentration of 14 µg m−3 (corresponds 19 ppbv) (Salonen et al., 2009). Hence, our method should provide a lowest boundary of ammonia mixing ratio in the chamber.
3.1 Time series of aerosol species
After the UV lamps were switched on, the photooxidation reaction produced oxidized gas-phase compounds and SOA particles in both nucleation and seeded experiments. The time series of mass concentrations of formed SOA (in green) and ammonium (in orange) along with sulfate and nitrate components measured by AMS are presented in Fig. 1. In the nucleation experiments (the left panels in Fig. 1), we observed a rapid increase in the SOA mass concentrations after the photooxidation reaction had started. After reaching the maximum concentration of 390–476 µg m−3, the SOA concentration declined by 27.9±9.2 % at the end of experiment because of particle deposition on the chamber wall and/or aerosol evaporation. The O : C ratio (oxygen to carbon ratio) of the SOA particles was slightly increased from the initial 0.39±0.015 to the final 0.44±0.01 because of aerosol aging. In a distinct contrast, the mass concentrations of ammonium component were still rising at the stage of decreasing SOA masses. Together taking into account the fact that aerosol wall deposition loss was present, resulting in decreasing organic mass (and decreasing sulfate mass in the seeded experiments), our results suggest new production of ammonium salts. The newly formed ammonium can be partly attributed to the cogenerated nitrate and sulfate as the photooxidized products of NO and SO2 in the chamber. However, the amount of the two inorganic species cannot fully interpret the ammonium, and we will elaborate on this in more detail in Sect. 3.2. Similar phenomena were also observed in the seeded experiments (Figs. 1 and 2). Note that as the measured mass is centered on the larger sizes of the AMS measurement range (35–1000 nm, Jayne et al., 2000; Zhang et al., 2004), we can state that the measurements represents CCN-relevant particle sizes (in general it is often assumed that atmospheric aerosol particles larger than 100 nm are able to active to cloud droplets). Additionally, the organics, nitrate and ammonium aerosols showed similar mass-based size distributions from our AMS measurement (refer to Sect. 3.2.1), and thus internally mixed aerosol can be assumed in our study, indicating that the aerosol components were lost in a similar rate to the chamber wall. Therefore, no loss correction to the aerosol wall disposition was conducted in this work.

Figure 1The mass concentrations of organics (left axis), ammonium (a, b, right axis), sulfate and nitrate (c, d) as a function of irradiation time in nucleation experiments (a, c) and in seeded SOA experiments (b, d). The irradiation time of 0 min marks the start of photooxidation reactions after UV lights were switched on.
3.2 Participation of ammonia in SOA formation
3.2.1 Reaction of ammonia with organic acids
To investigate the monotonically increasing profile of the ammonium salt in our chamber experiment, we studied the stoichiometric neutralization of formed SOA particles. The approach proposed by Zhang et al. (2007) was adopted, in which the ammonium mass concentrations measured (NH) in the particles were compared to the stoichiometric ammonium concentrations required (NH) to fully neutralize the measured concentrations of SO, NO and Cl−:
where NH, SO, NO and Cl− represent the mass concentrations (in µg m−3) of the species, and the denominators correspond to their molecular weights. The factor 18 is the molecular weight of NH.
A comparison between the predicted and measured ammonium masses is displayed in Figs. 2 and S3. In both sets of experiments, the measured ammonium mass concentration was systematically greater than the predicted value. The trend does not show a dependence on the presence of NOx in the chamber. On average, is 400±156 % and 21±11 % greater than at the end of nucleation and seeded experiments, respectively. The large discrepancy between the measured and predicted ammonium concentrations suggested that the current amount of sulfate, nitrate and chloride is insufficient to neutralize the ammonium formed in the particle phase, which indicates that the organic component must have played a role in this process. Considering the nature of NH3 as a base compound, the candidate species of organic compounds are attributed to organic acids.
Since a vast variety of molecular compositions of organic acids may be present in the photooxidation products of α-pinene, it is not possible to define the amount of individual organic acid required to neutralize the NH3. Therefore, we use the CO ion measured by AMS to represent the carboxylic functional group of organic acids. The CO is not only considered as a reliable marker of oxygenated organic aerosol (e.g., Zhang et al., 2005), but is also tightly associated with the formation of organic mono- and diacids shown in laboratory and field measurements (Yatavelli et al., 2015; Takegawa et al., 2007; Alfarra et al., 2004).
Taking into account the contribution of organic acids to ammonium salt, we reformulate Eq. (1) to
where is the predicted ammonium mass concentration after taking into account contribution of organic acids. is the mass concentration of the carboxylic function group (-CO2) representing organic acids which were required to neutralize the ammonium. The denominator 44 is the molecular weight of the carboxylic functional group.
To interpret NH, should equal to :
We can estimate the amount of by combining Eqs. (2)–(3):
So,
The time series of estimated over each experiment is shown in Fig. 3. On average, the concentration required to explain the observed ammonium concentrations was 48.6 times lower for seeded experiments than for the nucleation experiments. Figure 3 indicates that organic acids participated in reacting with NH3 much earlier in the low-NOx test (red and green mark) than in the high-NOx experiment (blue mark). Based on the time series in Fig. 3, in the high-NOx test, the time at which the organic acids started to play a role in ammonium formation was delayed by 31 and 100 min for the nucleated and seeded experiments, respectively. This observation can be associated with the formation of nitric acid (HNO3) from photooxidation of NOx compounds in the high-NOx conditions. The reaction of HNO3 and NH3 takes precedence over the reaction between organic acids and NH3. In the low-NOx test, a time lag for is also observed, especially in the seeded SOA experiments. The delay is caused by the effect of nitric acid arising from the background NOx photooxidation. In addition, the slower formation of particulate organic acids delays their reaction with ammonia in the seeded experiments compared to the nucleated cases. In general, the required accounted for the 27.0±3.1 % of total mass in the nucleation experiments and 18.7±6.0 % in the seeded SOA experiments. The amount of is assumed to be related to (1) the photooxidation and ozonolysis capacity in our chamber, which determines the amount of particulate organic acids (total CO); (2) the relative amount of low- and high-molecular-weight organic acids and their physicochemical properties; (3) the relative amount of inorganic and organic acids; and (4) the relative amount of ammonia and total acids, which needs to be thoroughly characterized in the future study.
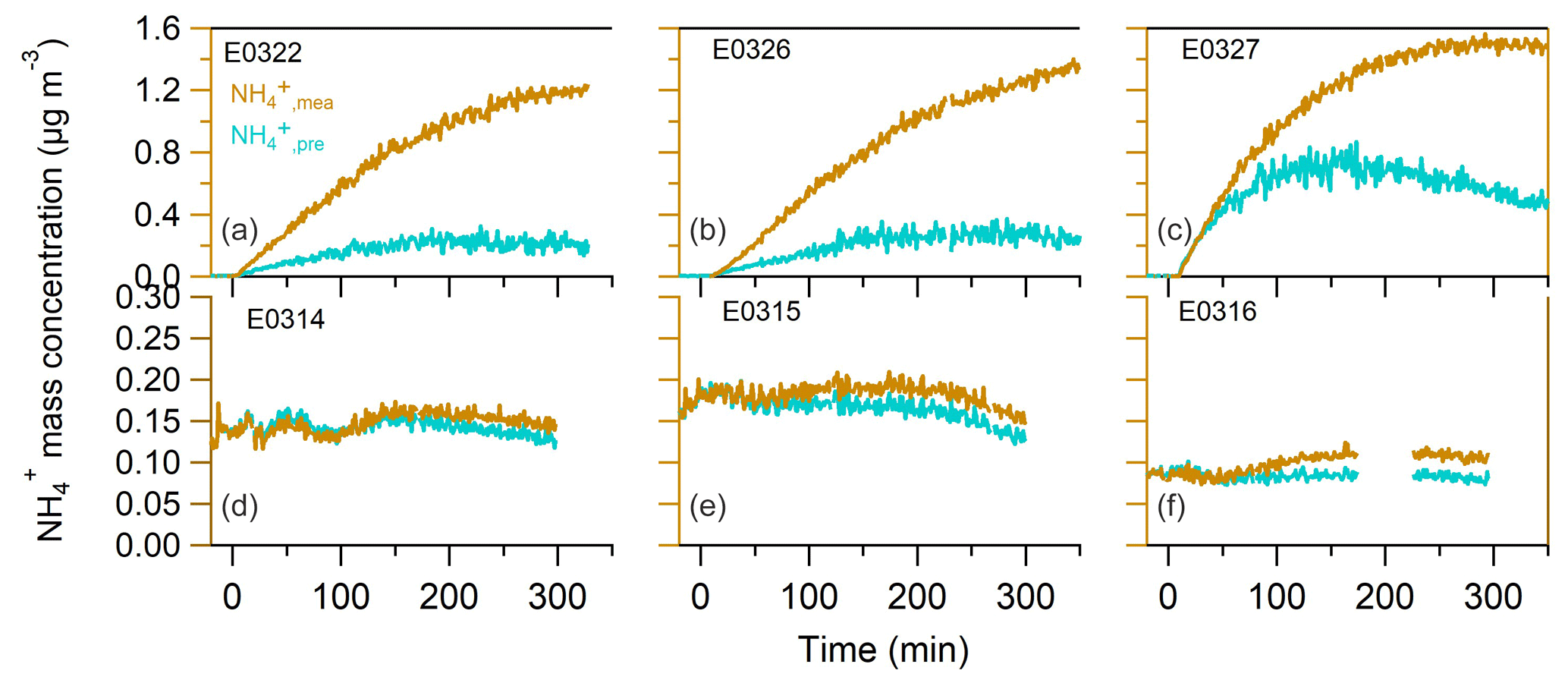
Figure 2The time series of measured and predicted ammonium in the nucleated SOA (a–c) and in the seeded SOA experiments (d–f).

Figure 3The estimated amount of organic carboxylic acid needed to fully neutralize SOA particles (). The CO group is chosen to represent organic acids. (a) Nucleation experiments. (b) Seeded experiments. The time zero marks the beginning of photooxidation reactions when UV lights were switched on.
To further verify our conclusion that organic acids are the drivers of the ammonium formation, we explored the size distribution of organic acids (represented by CO ion), ammonium and nitrate at the end of nucleation experiments (Fig. 4). The mode diameters of the three species, determined by performing lognormal fitting on the size distributions, are listed in Table S1. The mode diameter of CO ion is about 5–13 nm greater than that of ammonium and nitrate in the three individual experiments. The slight difference in two species mode diameters might be associated with the lower evaporation rate of organic CO than ammonium ions on the AMS vaporizer. Anyhow, the similarity in the mode diameters and size distributions of three chemical species suggests that they are internally mixed in the physical phase and are originated from the similar formation sources.
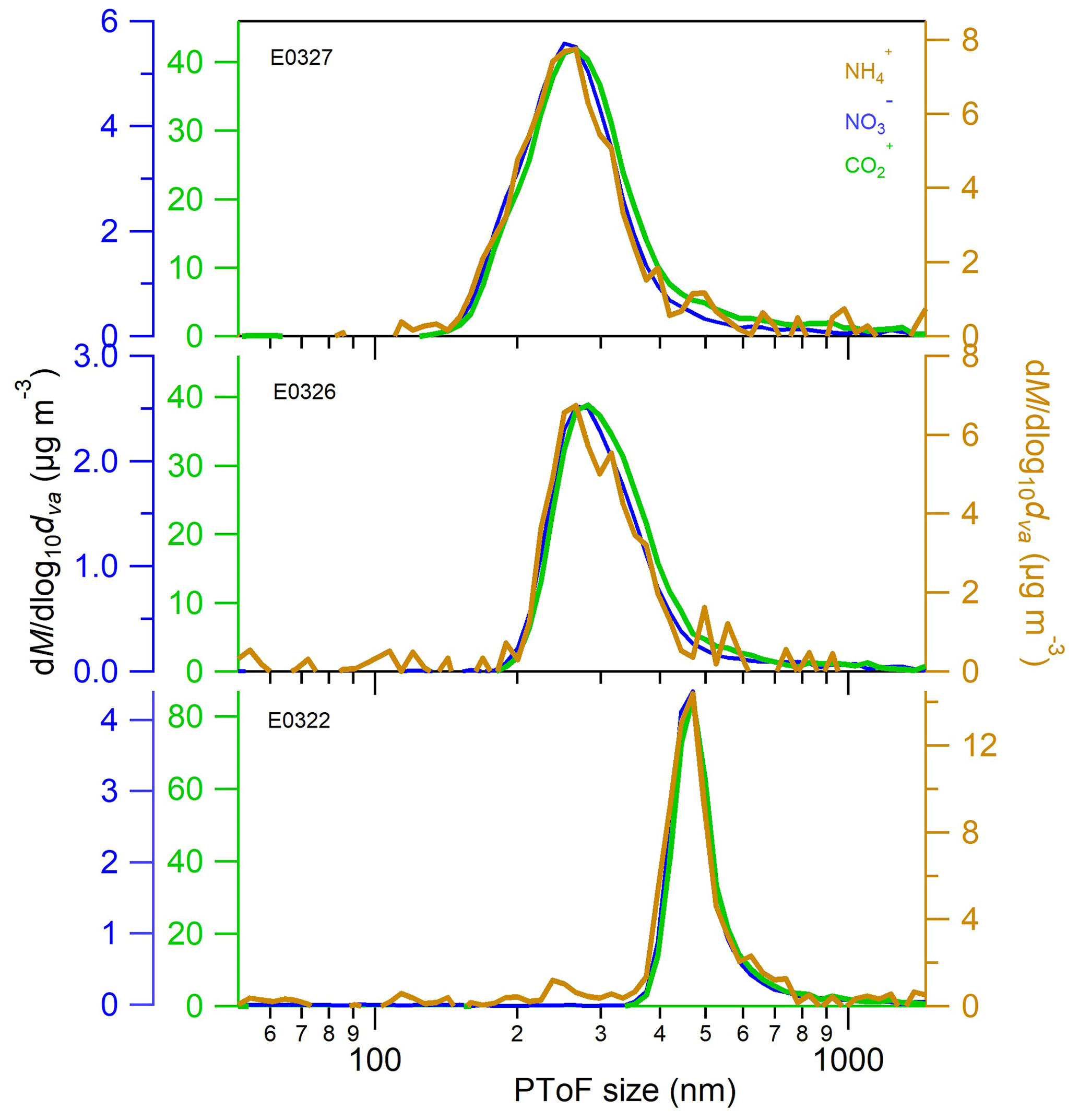
Figure 4The averaged 30 min size distribution of organic acids (represented by CO ion measured by AMS), nitrate and ammonium in the last 30 min prior to the end of the nucleated SOA experiments.
3.2.2 Connecting gas compounds by PTRMS to ammonium ion by AMS
To investigate the organic acid species which may potentially contribute to the ammonium formation in our experiments, we compared the gas-phase organic acids to the particle-phase ammonium attributed to the neutralization by organic acids (, defined as the difference between and ). We first examined the organic monocarboxylic acids. Figure 5a1 shows the correlation between the concentration of butyric acid (C4H8O2) (or its isomer) measured by PTRMS and the calculated from AMS measurement in the nucleated SOA experiments. The excellent linear correlation (coefficient R2 ≈ 0.88–0.95) of these two species implies that the formation of butyric acid is associated with the ammonium formation. In addition, we also identified a molecular ion in the AMS mass spectrum, which also shows an excellent correlation to the formed ammonium (Fig. 5c1) and thus indicates its simultaneous formation with ammonium ion. The simultaneous observation of the same ion molecules in both gas and particle phases gives us confidence to speculate that this ion is derived from gas-phase butyric acid. The observation suggests a reaction between butyric acid and NH3, which enables the production of ammonium butyrate salts (NH4C4H7O2). The formation of the salts favors the condensation of butyric acid on the particle phase, contributing to the observed ammonium ions. Although we cannot overlook the fact that ammonium butyrate is severely fragmented inside AMS, it is believed that the detected C4H8O ion signal is a residual of parental molecule. In the same way, we also explored pentanoic acid (C5H10O2) (or its isomer) (Fig. 3b1 and d1) and obtained similar results as with butyric acid.

Figure 5The comparison of measured organic monoacids in the gas phase by PTRMS and the ammonium attributed to the neutralization by organic acids () in the particle phase by AMS in the nucleated SOA (a1–d1) and seeded SOA experiments (a2–d2). (a, b) Linear correlation between butyric (C4H8O2) and pentanoic (C5H10O2) monoacids measured by PTRMS and the by AMS. (c, d) Linear correlation of two fragmental ions C4H8O and C5H10O to the by AMS. Note that the two fragmental ions have an identical ion molecular formula to those of butyric and pentanoic acids.
A similar comparison has also been made in the seeded SOA experiments (Fig. 5a2–d2). We have observed a good correlation of butyric acid to the organic acid-driven ammonium () (r2 = 0.76–0.83) and a moderate correlation of pentanoic acid to (r2 = 0.27–0.62). Compared with the nucleation experiments, the correlation relationship in the seeded SOA experiments is worse, mainly because the signals in both PTRMS and AMS measurements are weak in the seeded SOA experiments. Similar to the observation in the nucleated SOA experiments, the ion C4H8O in the AMS mass spectrum was also identified to correlate to . The ion has the same molecular formula as butyric acid. The concentration of C5H10O of the same molecular formula as pentanoic acid was in a concentration level of 10−3 µg m−3 (Fig. 5d2). Such a signal is at the same level as the background noise in AMS measurement, making it challenging to correlate with . In general, the results in the seeded SOA experiments are consistent with those in the nucleated SOA experiments, confirming the role of butyric and pentanoic acids in the formation of ammonium salt.
We also extend our study to other four types of gas-phase organic acids measured by the PTRMS. Figure 6 shows the relationship of the formic acid, acetic acid, propionic acid and pinonic acid measured in the gas phase with the ammonium ion measured by AMS. Surprisingly, the gas-phase formic acid, acetic acid and propionic acid were in good agreement with particle-phase ammonium concentration, with excellent linear correlation coefficients r2≥0.95 in the nucleated SOA experiments and r2≥0.72 in the seeded SOA experiments. The good correlation of gas-phase organic acids with the particle-phase ammonium salt suggests that theses acids played a role in the formation of ammonium. Formic acid and acetic acid are the most abundant organic monoacids in the atmosphere (Chebbi and Carlier, 1996), whose one significant source is from photooxidation of α-pinene and other alkenes and terpenes (Friedman and Farmer, 2018). Propionic and butyric acids are also other important organic acids in this process (Nah et al., 2018a, b; Chebbi and Carlier, 1996). The formation mechanisms of these low-molecular-weight organic acids are unclear in the photochemical reaction of α-pinene (e.g., Friedman and Farmer, 2018). It can be speculated that they were produced as products of stabilized Criegee intermediate associated with ozonolysis of α-pinene and double-carbon-bond-containing products (Jacob and Wofsy, 1988; Orzechowska and Paulson, 2005). The statement is verified by the fact that we have observed the highest O3 formation at the highest NOx ∕ VOC initial input in E0314 (Fig. S4). Therefore, in E0314 we have also observed higher concentrations of C1–C5 monoacids corresponding to higher ozonolysis reactivity (Fig. S5). These low-molecular-weight monoacids have a vapor pressure of 6.8–8.1 µg m−3 (log 10(C∗)) (Friedman and Farmer, 2018) and are generally considered to be too volatile to be distributed substantially to the particle phase. However, their presence in the aerosol particles is ubiquitous in various areas over the world, although the levels of these monoacids are 1 or 2 orders of magnitude lower than those in the gas phase (Nah et al., 2018a; Fisseha et al., 2004; Chebbi and Carlier, 1996). A study conducted in a forest-agriculture area in Atlanta showed that the acetic acid and formic acid are the second and third richest water-soluble organic acids in the particle phase, and their molar fractions to the total individual acid concentrations in the particulate phase were 5.8±5.0 % and 3.6±3.6 %, respectively (Nah et al., 2018a). In addition, the presence of NH3 as a strong base facilitates the shift of the equilibrium of these monocarboxylic acids and NH3 to the particle phase (Barsanti et al., 2009). Under a base environment, a higher molar fraction of formic acid and acetic acid has also been observed in the particle phase (Nah et al., 2018a). These organic acids could exist in the particle phase in chemical forms of ammonium formate, ammonium acetate and ammonium propionate salts (Barsanti et al., 2009; Smith et al., 2008; Becker and Davidson, 1963) and thus contribute to the observed ammonium ions.

Figure 6Correlations of the ammonium attributed to the neutralization by organic acids () in the particle phase by AMS to the (a) formic acid, (b) acetic acid, (c) propionic acid and (d) pinonic acid in the gas phase measured by PTRMS in the nucleated SOA experiments (left four panels) and in the seeded SOA experiments (right four panels). The light pink area in (d1) indicates a nonpositive correlation regime between two species.
In an obvious contrast to the observation of C1–C5 monoacids above, the pinonic acids did not show a linear correlation with the ammonium at the later stage of the nucleated SOA experiments (pink region, Fig. 6d1), and there was no connection in the seeded SOA experiments. Pinonic acid is a typical first-generation marker product in the photooxidation reaction of α-pinene (Hallquist et al., 2009). Na et al. (2007) reported that the addition of NH3 to the aerosolized pinonic acid in an environmental reactor led to dramatic increase in both number and volume concentrations of pinonic acid. They attributed the observation to the reaction between NH3 and pinonic acids. However, the lack of positive correlation between the pinonic acid and ammoniums can be caused by several reasons in this study. Firstly, the vapor pressure of pinonic acid is about 4–5 orders of magnitude lower than those of C1–C5 monoacids (Jimenez et al., 2009), so pinonic acid can condense on the particle phase independent of NH3. Meanwhile, the concentration of pinonic acid accounts for less than 1 % of the total monoacids based on the PTRMS results. Hence, the contribution of pinonic acid to the formed ammonium was estimated to be less than 1 %, considering that the acidity strength of pinonic acid is similar to other C1–C5 monoacids – acid dissociation constant (pKa) of pinonic acid is 4.8 (Howell and Fisher, 1958), which is close to the pKa values of formic acid (3.75), acetic acid (4.75), propionic acid (4.86), butyric acid (4.83) and pentanoic acid (4.84) (Lide, 2007). Therefore, after α-pinene was completely consumed and pinonic acid formation had ceased in the gas phase (pink region, Fig. 6), the rapid condensation of pinonic acid on the particle phase or chamber wall caused a nonlinear correlation of pinonic acid to ammonium. Secondly, the gas-phase pinonic acids could be further reacted away by OH radicals, which also contributes to the nonlinear observation. The field experiment has shown the relatively low atmospheric PM concentrations of pinonic acid measured in summer because of the consumption of pinonic acid by OH radicals (Szmigielski et al., 2007).
Analogously, the scattering plots for the gas-phase organic dicarboxylic acids and particle-phase ammonium salts are shown in Fig. 7. We chose malonic acid and succinic acid as representative organic diacids, two of the most abundant dicarboxylic acids measured in the atmospheric aerosols (Chebbi and Carlier, 1996). The nice correlations of the gas-phase malonic and succinic acids to the particle-phase ammonium study suggest that diacids contribute to the formation of ammonium in both nucleated and seeded SOA experiments. It also indicates the continuous formation of those diacids at the later stage of photooxidation in nucleated and seeded experiments. The formation mechanism of these low-molecular-weight diacids remains unclear for us and might be similar to the C1–C5 monoacids.
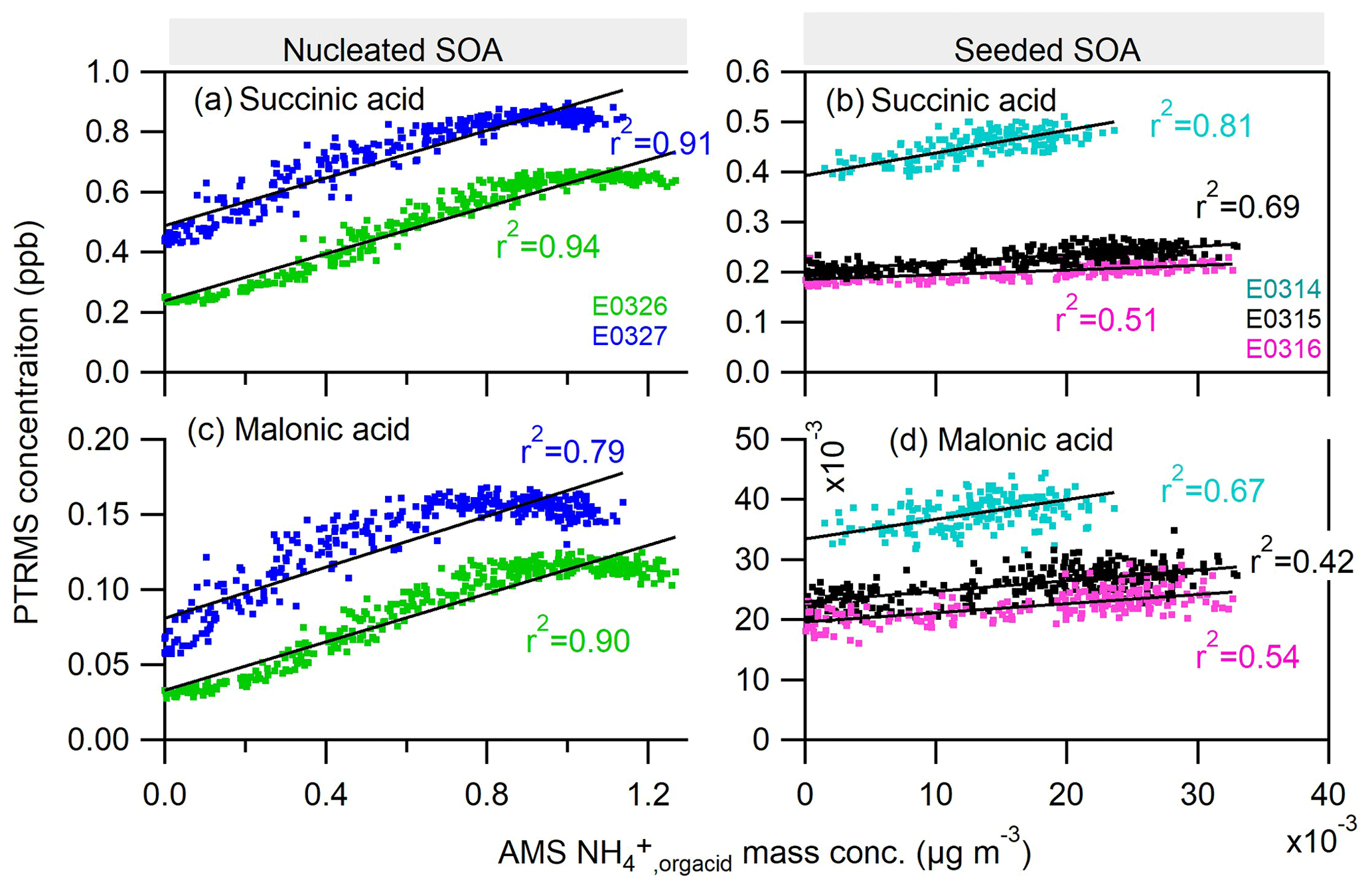
Figure 7Correlations of the ammonium attributed to the neutralization by organic acids () in the particle phase by AMS to two organic dicarboxylic acids: (a–b) succinic acid and (c–d) malonic acid in the gas phase measured by PTRMS. Neither of the diacids were observed in the experiment E0322.
Our results qualitatively demonstrate that in the photooxidation of α-pinene the presence of NH3 drives the gas-phase mono- and dicarboxylic acids to the particle phase and promotes the SOA mass concentration. Previous studies have shown that the presence of NH3 can significantly enhance SOA formation from the α-pinene–ozone–photooxidation system because of the interaction of NH3 with gas-phase organic acids (Na et al., 2007; Babar et al., 2017), which is consistent with our results. Carboxylic acid is one of the key species in determining SOA physicochemical property. Our results may prompt us to reconsider the pathway of gas-phase organic acids involved in SOA formation in the atmosphere, whether they directly participate in partitioning between the gas and particle phases or they undergo secondary conversion via reaction with NH3 before they condensate on the particle phase in the atmosphere. After SOAs are formed, the carbonyl group of chemical compounds in SOA particles can also uptake NH3 heterogeneously to form nitrogen-containing compounds (Zhu et al., 2018; Liu et al., 2015; Updyke et al., 2012; Dinar et al., 2008) and organic ammonium salts (Schlag et al., 2017). In this study the N : C ratios measured by AMS were nearly constant or slightly increasing in two sets of experiments (Fig. S6), which could suggest the formation of nitrogen-containing compounds via carbonyl-NH3 heterogeneous reaction. However, we cannot rule out that NOx also participated in the formation of SOA, resulting in an increase in the N : C ratios. The interaction of NH3 and SOA affects the cloud condensation nuclei (CCN) and hygroscopic growth of SOA particles and may have a potential impact on climate change (Dinar et al., 2008).
3.3 Ammonium relevant to PMF-solved SOA component
PMF analysis on the high-resolution organic mass spectra resolved the organic component to two factors in the nucleation experiments: MO-OOA (more oxidized oxygenated organic aerosol) and LO-OOA (less oxidized OOA). The oxidation level of MO-OOA is represented by an O : C ratio of 0.47 in this study, which is close to the value of 0.48 for MO-OOA determined in New York City in summer (Sun et al., 2012). Its time series shows a good correlation to the measured gas-phase monocarboxylic and diacids such as butyric and succinic acids. The second factor LO-OOA is featured by an O : C ratio of 0.38. Its time series is related to the gas-phase oxidant products such as pinonaldehyde. The mass spectra profiles and time series of the two factors are shown in Fig. S7. In this study, the formation of ammonium salts is consistent with MO-OOA factor and also organic mono- and diacids (Fig. 8). A higher oxidized organic factor is usually associated with the formation of organic mono- and diacids. These results further suggest that the ammonium has a close relation to the organic acids. Our observation is in agreement with the study by Schlag et al. (2017) where they showed by field data that NH4 is associated with a more oxygenated organic aerosol factor.

Figure 8The relevance of particulate ammonium salt to the resolved MO-OOA component by PMF, as well as to the gas-phase butyric monocarboxylic acid and succinic diacid.
The SOA experiments were carried out from photooxidation reaction of α-pinene in the presence of NH3 in a 29 m3 indoor simulation chamber. Experiments were designed for SOA formation in the presence of ammonium sulfate seeds and in the absence of seed aerosols. The chemical composition and time series of compounds in the gas and particle phase were characterized by an online high-resolution time-of-flight proton-transfer-reaction mass spectrometer (PTRMS) and a high-resolution time-of-flight aerosol mass spectrometer (AMS), respectively.
After the precursor α-pinene was consumed in the chamber, the mass concentration of organic aerosol was decreased because of aerosol wall deposition or evaporation, and the ammonium concentration was still rising, suggesting the continuous formation of ammonium. AMS results showed that organic acids were required to neutralize the observed ammonium salt. The CO ion was selected to represent organic acids. The amount of CO required for neutralizing ammonium accounted for the 27.0±3.1 % of total CO mass in the nucleated SOA experiments and 18.7±6.0 % in the seeded SOA experiments. The good correlation of organic monocarboxylic acids (such as formic acid, acetic acid, propionic acid, butyric acid, and pentanoic acid, or their isomers) in the gas phase to the ammonium salts further qualitatively confirms an effective role of organic acids for the ammonium formation. The same conclusion is also applied to the organic dicarboxylic acids such as malonic and succinic acids. In addition, the formed ammonium salts correlated well to the more oxidized oxygenated organic aerosol (MO-OOA), which is consistent with the conclusion that organic acids contributed to the observed particulate ammonium.
Our work firmly shows the direct contribution of NH3 to the SOA formation through the organic acid–base reaction. The increase in SOA mass and the change of chemical composition due to NH3–SOA interaction could change the hygroscopicity, CCN ability and optical property of aerosol particles, which may alter the aerosol impact on climate change and need to be studied in the future.
The data included in this paper can be obtained by contacting the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-14393-2020-supplement.
LH, EK, AL and AV designed and conducted the experiments. LH and EK performed the data analysis with contributions by DRW and AV. LH and AV wrote the paper with contributions from all coauthors.
The authors declare that they have no conflict of interest.
The authors thank Ilkka Summanen for assisting the chamber experiments.
This research has been supported by the European Union's Horizon 2020 research and innovation program through the EUROCHAMP-2020 Infrastructure Activity (grant no. 730997) and the Academy of Finland Center of Excellence program (grant no. 307331).
This paper was edited by Hang Su and reviewed by three anonymous referees.
Alfarra, R.: Insights Into Atmospheric Organic Aerosols Using An Aerosol Mass Spectrometer, PhD thesis, University of Manchester, 2004.
Babar, Z. B., Park, J., and Lim, H.: Influence of NH3 on secondary organic aerosols from the ozonolysis and photooxidation of a-pinene in a flow reactor, Atmos. Environ., 164, 71–84, 2017.
Barsanti, K. C., McMurry, P. H., and Smith, J. N.: The potential contribution of organic salts to new particle growth, Atmos. Chem. Phys., 9, 2949–2957, https://doi.org/10.5194/acp-9-2949-2009, 2009.
Battye, W., Aneja, V. P., and Roelle, P. A.: Evaluation and improvement of ammonia emission inventories, Atmos. Environ., 37, 3873–3883, 2003.
Becker, B. and Davidson, A. W.: the systems formic acid-ammonia and propionic acid-ammonia, J. Am. Chem. Soc., 85, 57–159, https://doi.org/10.1021/ja00885a010, 1963.
Bianchi, F., Trostl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kurten, A., Manninen, H. E., Munch, S., Perakyla, O., Petaja, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, 2016.
Bones, D. L., Henricksen, D. K., Mang, S. A., Sonsior, M., Bateman, A. P., Nguyen, T. B., Cooper, W. J., and Nizkorodow, S. A.: Appearance of strong absorbers and fluorophores in limonene-O3 secondary organic aerosol due to NH-mediated chemical aging over long time scales, J. Geophys. Res., 115, D05203, https://doi.org/10.1029/2009JD012864, 2010.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer, Mass Spectrom. Rev., 26, 185–222, 2007.
Chebbi, A. and Carlier, P.: Carboxylic acids in the troposphere, occurrence, sources, and sinks: a review, Atmos. Environ., 30, 4233–4249, 1996.
Chen, T., Liu, Y., Ma, Q., Chu, B., Zhang, P., Liu, C., Liu, J., and He, H.: Significant source of secondary aerosol: formation from gasoline evaporative emissions in the presence of SO2 and NH3, Atmos. Chem. Phys., 19, 8063–8081, https://doi.org/10.5194/acp-19-8063-2019, 2019.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, 2006.
Dinar, E., Anttila, T., and Rudich, Y.: CCN activity and hygroscopic growth of organic aerosols following reactive uptake of ammonia, Environ. Sci. Technol., 42, 793–799, 2008.
Fisseha, R., Dommen, J., Sax, M., Paulsen, D., Kalberer, M., Maurer, R., Höfler, F., Weingartner, E., and Baltensperger, U.: Identification of organic acids in secondary organic aerosol and the corresponding gas phase from chamber experiments, Anal. Chem., 76, 6535–6540, 2004.
Friedman, B. and Farmer, D. K.: SOA and gas phase organic acid yields from the sequential photooxidation of seven monoterpenes, Atmos. Environ., 187, 335–345, 2018.
Grönroos, J., Mattila, P., Regina, K., Nousiainen, J., Perälä, P., Saarinen, K., and Mikkola-Pusa, J.: Development of the ammonia emission inventory in Finland. Revised model for agriculture, Finnish Environment Institute, Helsinki, 2009.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansel, A., Jordan, A., Holzinger, R., Prazeller, P., Vogel, W., and Lindinger, W.: Proton-Transfer Reaction Mass-Spectrometry – Online Trace Gas-Analysis at the Ppb Level, Int. J. Mass Spectrom., 149, 609–619, https://doi.org/10.1016/0168-1176(95)04294-U, 1995.
Huang, X., Song, Y., Li, M. M., Li, J. F., Huo, Q., Cai, X. H., Zhu, T., Hu, M., and Zhang, H. S.: A high-resolution ammonia emission inventory in China, Glob. Biogeochem. Cy., 26, GB1030, https://doi.org/10.1029/2011GB004161, 2012.
Howell, H. and Fisher, G. S.: The dissociation constants of some of the terpene acids, J. Am. Chem. Soc., 80, 6316–6319, 1958.
Horne, J. R., Zhu, S., Montoya-Aguilera, J., Hinks, M. L., Wingen, L. M., Nizkorodov, S. A., and Dabdub, D.: Reactive uptake of ammonia by secondary organic aerosols: Implications for air quality, Atmos. Environ., 189, 1–8, 2018.
Huang M. Q., Xu, J., Cai, S. Y., Liu, X. Q., Zhao, W. X., Hu, C. J., Gu, X. J., Fang, L., and Zhang, W. J.: Characterization of brown carbon constituents of benzene secondary organic aerosol aged by ammonia, J. Atmos. Chem., 75, 205–218, 2018.
IPCC 2013: Climate change 2013: The physical science basis. Intergovernmental panel on Climate Change, Cambridge University Press, New York, USA, 2013.
Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 33, 49–70, 2000.
Jacob, D. J. and Wofsy, S. C.: Photochemistry of biogenic emissions over the Amazon forest, J. Geophys. Res., 93, 1477–1486, https://doi.org/10.1029/JD093iD02p01477, 1988.
Jimenez,J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, 2009.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Mark, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Mark, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, 10.1016/j.ijms.2009.07.005, 2009.
Jokinen, T., Sipila, M., Kontkanen, J., Vakkari, V., Tisler, P., Duplissy, E. M., Junninen, H., Kangasluoma, J., Manninen, H. E., Petaja, T., Kulmala, M., Worsnop, D. R., Kirkby, J., Virkkula, A., and Kerminen, V. M.: Ion-induced sulfuric acid-ammonia nucleation drives particle formation in coastal Antarctica, Sci. Adv., 4, eaat9744, https://doi.org/10.1126/sciadv.aat9744, 2018.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kari, E., Hao, L., Ylisirniö, A., Buchholz, A., Leskinen, A., Yli-Pirilä, P., Nuutinen, I., Kuuspalo, K., Jokiniemi, J., Faiola, C. L., Schobesberger, S., and Virtanen, A.: Potential dual effect of anthropogenic emissions on the formation of biogenic secondary organic aerosol (BSOA), Atmos. Chem. Phys., 19, 15651–15671, https://doi.org/10.5194/acp-19-15651-2019, 2019a.
Kari, E., Faiola, C. L., Isokääntä, S., Miettinen, P., Yli-Pirilä, P., Buchholz, A., Kivimäenpää, M., Mikkonen, S., Holopainen, J. K., and Virtanen, A.: Time-resolved characterization of biotic stress emissions from Scots pines being fed upon by the pine weevil by means of PTR-ToF-MS, Boreal Environ. Res., 24, 25–49, 2019b.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., J. H. Seinfeld, Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala,A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galacticcosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, 2011.
Kourtchev, I., Giorio, C., Manninen, A., Wilson, E., Mahon, B., Aalto, J., Kajos, M., Venables, D., Ruuskanen, T., Levula, J., Loponen, M., Connors, S., Harris, N., Zhao, D., Kiendler-Scharr, A., Mentel, T., Rudich, Y., Hullquist, M., Doussin J., Maenhaut, W., Back, J., Petaja, T., Wenger, J., Kulmala, M., and Kalberer, M.: Enhanced volatile organic compounds emissions and organic aerosol mass increase the oligomer content of atmospheric aerosols, Sci. Rep.-UK., 6, 35038, https://doi.org/10.1038/srep35038, 2016.
Kulmala, M., Pirjola, L., and Mäkelä, J. M.: Stable sulphate clusters as a source of new atmospheric particles, Nature, 404, 66–69, 2000.
Kurten, T., Torpo, L., Ding, C. G., Vehkamaki, H., Sundberg, M. R., Laasonen, K., and Kulmala, M.: A density functional study on water-sulfuric acid-ammonia clusters and implications for atmospheric cluster formation, J. Geophys. Res.-Atmos., 112, D4, https://doi.org/10.1029/2006JD007391, 2007.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V.-M., Kim, C., Kirkby, J., Kontkanen,J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipilä, M., Tauber, C., Tomé, A., Tröstl, J., Väisänen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018.
Leskinen, A., Yli-Pirilä, P., Kuuspalo, K., Sippula, O., Jalava, P., Hirvonen, M.-R., Jokiniemi, J., Virtanen, A., Komppula, M., and Lehtinen, K. E. J.: Characterization and testing of a new environmental chamber, Atmos. Meas. Tech., 8, 2267–2278, https://doi.org/10.5194/amt-8-2267-2015, 2015.
Lide, D. R.: CRC handbook of chemistry and physics, 88th Edn., CRC press/Taylor & Francis Group, Bosa Roca, United States, 2007.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Li, Z., Nizkorodov, S. A., Chen, H., Lu, X., Yang, X., and Chen, J.: Nitrogen-containing secondary organic aerosol formation by acrolein reaction with ammonia/ammonium, Atmos. Chem. Phys., 19, 1343–1356, https://doi.org/10.5194/acp-19-1343-2019, 2019.
Liu, T. Y., Wang X. M., Deng, W., Zhang, Y. L., Chu, B. W., Ding, X., Hu, Q. H., He, H., and Hao, J. M.: Role of ammonia in forming secondary aerosols from gasoline vehicle exhaust, Sci. China Chem., 58, 1377–1384, 2015.
Mensah, A. A., Holzinger, R., Otjes, R., Trimborn, A., Mentel, Th. F., ten Brink, H., Henzing, B., and Kiendler-Scharr, A.: Aerosol chemical composition at Cabauw, The Netherlands as observed in two intensive periods in May 2008 and March 2009, Atmos. Chem. Phys., 12, 4723–4742, https://doi.org/10.5194/acp-12-4723-2012, 2012.
Nah, T., Guo, H., Sullivan, A. P., Chen, Y., Tanner, D. J., Nenes, A., Russell, A., Ng, N. L., Huey, L. G., and Weber, R. J.: Characterization of aerosol composition, aerosol acidity, and organic acid partitioning at an agriculturally intensive rural southeastern US site, Atmos. Chem. Phys., 18, 11471–11491, https://doi.org/10.5194/acp-18-11471-2018, 2018a.
Nah, T., Ji, Y., Tanner, D. J., Guo, H., Sullivan, A. P., Ng, N. L., Weber, R. J., and Huey, L. G.: Real-time measurements of gas-phase organic acids using SF6− chemical ionization mass spectrometry, Atmos. Meas. Tech., 11, 5087–5104, https://doi.org/10.5194/amt-11-5087-2018, 2018b.
Na, K., Song, C., Switzer, C., and Cocker, D. R.: Effect of ammonia on secondary organic aerosol formation from α-pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, 2007.
Orzechowska, G. E. and Paulson, S. E.: Photochemical sources of organic acids. 1. Reaction of ozone with isoprene, propene, and 2-butenes under day and humid conditions using SPME, J. Phys. Chem. A, 109, 5358–5365, 2005.
Paatero, P. and Tapper, U.: Positive Matrix Factorization: a non-negative factor model with optimal utilization of error estimates of data values. Environmetrics, 5, 111–126, 1994.
Paciga, A. L., Riipinen, I., and Pandis, S. N.: Effect of ammonia on the volatility of organic diacids, Environ. Sci. Technol., 48. 13769–13775, 2014.
Rubach, F.: Aerosol processes in the planetary boundary layer: High resolution aerosol mass spectrometry on a Zeppelin NT airship, Forschungszentrum Jülich GmbH, Jülich, 2013.
Salonen, H., Pasanen, A., Lappalainen, S. K., Riuttala, H. M., Tuomi, T. M., Pasanen, P. O., Back, B. C., and Reijula, K. E.: Airborne concentrations of volatile organic compounds, formaldehyde and ammonia in Finnish office buildings with suspected indoor air problems, J. Occup. Environ. Hyg., 6, 200–209, 2009.
Schlag, P., Rubach, F., Mentel, T. F., Reimer, D., Canonaco, F., Henzing, J. S., Moerman, M., Otjes, R., Prevot, A. S. H., Rohrer, F., Rosati, B., Tillmann, R., Weingartner, E., and Kiendler-Scharr, A.: Ambient and laboratory observations of organic ammonium salts in PM1, Faraday Discuss., 200, 331–351, 2017.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, New York, USA, 2016.
Smith, J. N., Dunn, M. J., VanReken, T. M., lida, K., Stolzenburg, M. R., McMurry, P. H., and Hyey, L. G.: Chemical composition of atmospheric nanoparticles formed from nucleation in Tecamac, Mexico: Evidence for an important role for organic species in nanoparticle growth, Geophys. Res. Lett., 35, l04808, https://doi.org/10.1029/2007GL032523, 2008.
Sun, Y. L., Zhang, Q., Schwab, J. J., Yang, T., Ng, N. L., and Demerjian, K. L.: Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements, Atmos. Chem. Phys., 12, 8537–8551, https://doi.org/10.5194/acp-12-8537-2012, 2012.
Szmigielski, R., Surratt, J. D., Gomez-Gonzalez, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: an atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007GL031338, 2007.
Takegawa, N., Miyakawa, T., Kawamura, K., and Kondo, Y.: Contribution of Selected Dicarboxylic and ω-Oxocarboxylic Acids in Ambient Aerosol to the m∕z 44 Signal of an Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Technol., 41, 418–437, https://doi.org/10.1080/02786820701203215, 2007.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Updyke, K. M., Nguyen, T. B., and Nizkorodov, S. A.: Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors, Atmos. Environ., 63, 22–31, 2012.
Wang, Y., Liu, P. F., Li, Y. J., Bateman, A. P., Martin, S. T., and Hung, H. M.: The reactivity of toluene-derived secondary organic material with ammonia and influence of water vapor, J. Phys. Chem. A., 122, 7739–7747, 2018.
Yatavelli, R. L. N., Mohr, C., Stark, H., Day, D. A., Thompson, S. L., Lopez-Hilfiker, F. D., Campuzano-Jost, P., Palm, B. B., Vogel, A. L., Hoffmann, T., Heikkinen, L., Äijälä, M., Ng, N. L., Kimmel, J. R., Canagaratna, M. R., Ehn, M., Junninen, H., Cubison, M. J., Tuukka Petäjä, T., Kulmala, M., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Estimating the contribution of organic acids to northern hemispheric continental organic aerosol, Geophys. Res. Lett., 42, 6084–6090, https://doi.org/10.1002/2015GL064650, 2015.
Zhang, Q., Alfarra, M. R., Worsnop, D. R., Allan, J. D., Coe, H., Canagaratna, M. R., and Jimenez, J. L.: Deconvolution and Quantification of Hydrocarbon-Like and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry, Environ. Sci. Technol., 39, 4938–4952, https://doi.org/10.1021/es048568l, 2005.
Zhang, Q., Stanier, C. O., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Pandis, S. N., and Jimenez, J. L.: Insights into the chemistry of new particle formation and growth events in Pittsburgh based on aerosol mass spectrometry, Environ. Sci. Technol., 38, 4797–4809, 2004.
Zhang, Q., Jimenez, J. L., Worshop, D. R., and Canagaratna, M. R.: A case study of urban particle acidity and its influence on secondary organic aerosol, Environ. Sci. Technol., 41, 3213–3219, 2007.
Zhu, S., Horne, J. R., Montoya-Aguilera, J., Hinks, M. L., Nizkorodov, S. A., and Dabdub, D.: Modeling reactive ammonia uptake by secondary organic aerosol in CMAQ: application to the continental US, Atmos. Chem. Phys., 18, 3641–3657, https://doi.org/10.5194/acp-18-3641-2018, 2018.