the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Jan 2020
| 31 Jan 2020
Peroxy radical chemistry and the volatility basis set
Meredith Schervish
Gas-phase autoxidation of organics can generate highly oxygenated organic molecules (HOMs) and thus increase secondary organic aerosol production and enable new-particle formation. Here we present a new implementation of the volatility basis set (VBS) that explicitly resolves peroxy radical (RO2) products formed via autoxidation. The model includes a strong temperature dependence for autoxidation as well as explicit termination of RO2, including reactions with NO, HO2, and other RO2. The RO2 cross-reactions can produce dimers (ROOR). We explore the temperature and NOx dependence of this chemistry, showing that temperature strongly influences the intrinsic volatility distribution and that NO can suppress autoxidation under conditions typically found in the atmosphere.
- Article
(3560 KB) - Full-text XML
-
Supplement
(277 KB) - BibTeX
- EndNote





Atmospheric particles have dangerous health effects and influence the environment both through their direct scattering of incoming radiation and their indirect effect on clouds as cloud condensation nuclei. New-particle formation (secondary particle production) constitutes the largest source of particles in the atmosphere; furthermore many if not most particles with significant health or climate effects consist largely of secondary material, having grown via condensation from a much smaller initial size. Organic vapors contribute to both nucleation and growth of new particles as well as growth of primary nanoparticles, all of which constitute secondary organic aerosol (SOA). Especially for condensation to the smallest particles, the volatility of those condensible organics is important. Therefore it is of interest to investigate and model how volatile precursors oxidize in the atmosphere to low-volatility or extremely low volatility products (LVOCs and ELVOCs) that will contribute to SOA. Furthermore, organic oxidation products alone can drive pure biogenic nucleation (Kirkby et al., 2016), and so the formation mechanisms and nucleation rates of a new class of ultra-low-volatility products (ULVOCs) that can drive nucleation are also of interest.
One important class of condensible organics is the group known as highly oxygenated organic molecules (HOMs). HOMs are organic compounds containing at least six oxygen atoms formed through peroxy radical (RO2) isomerization and subsequent addition of molecular oxygen, which preserves RO2 functionality in a recursive process known as autoxidation, followed by RO2 termination to molecular products (Crounse et al., 2013; Ehn et al., 2014; Bianchi et al., 2019). HOMs open up a rich space for gas-phase organic oxidation chemistry to strongly influence new-particle formation involving condensible organic products. Because the chemistry involves competition between internal isomerization (which typically will have a relatively high energy barrier and thus a strong temperature dependence) and radical-radical termination reactions (which typically have low barriers and thus only a modest temperature dependence), we expect a strong temperature dependence to HOM formation. Furthermore, because NOx (especially NO) can greatly shorten RO2 lifetimes, we also expect a strong NOx dependence to HOM formation. Finally, to the extent that RO2-RO2 cross-reactions between radicals from different organic precursors can be important, there may be a rich interplay among the oxidation mechanisms for a host of different organic compounds.
The volatility basis set (VBS) has been developed and used widely as a framework to represent, track, and model the oxidation of volatile organic precursors, the subsequent production of condensible products, and their evolution through multiple generations of oxidation chemistry and fragmentation (Donahue et al., 2005, 2006, 2011, 2012b, 2013; Chuang and Donahue, 2016). For numerical calculations, the 2D-VBS space is discretized into bins, with each C∗ (300 K) bin typically separated by 1 order of magnitude and each O : C bin separated by 0.1. It is not necessary to know the identity of molecules in these bins, but this can be inferred by application of composition activity relations such as SIMPOL (Pankow and Asher, 2007). In general, the perspective of the VBS has been to explicitly track the most important observable properties of organics with respect to organic aerosol formation – volatility (C∘ (T)) and the degree of oxidation (O : C) – forming the so-called two-dimensional volatility basis set (2D-VBS). Where specific products have known properties, those molecules can be located individually in the 2D-VBS. Where specific products are not known, the 2D-VBS can still track the overall carbon in a reactive system, with properties (C∘ and O : C) evolving following known chemistry during photochemical aging. In this work, we plot everything using a C∗ (300 K) x axis so as to visualize how the products of the chemistry are changing rather and inferring how this would affect condensation.
The importance of RO2 branching chemistry has so far been recognized implicitly in VBS formulations (Presto and Donahue, 2006; Henry and Donahue, 2011) but not explicitly. Instead, the product (volatility) distribution in the VBS has been based on some measure of the RO2 branching (i.e., VOC : NOx or RO2 branching in an explicit mechanism). Consequently, there have been high-NOx products and low-NOx products (volatility distributions) (Presto and Donahue, 2006), or even high-HO2 and high-RO2 products (Henry and Donahue, 2011), that can be mixed to form products at intermediate NOx (or HOx:ROx) based on this indirect measure of RO2 branching. Further, relatively little has been done to represent any temperature dependence of VOC oxidation in VBS implementations.
The purpose of this paper is to extend the 2D-VBS to explicitly represent RO2 chemistry, including RO2 oxidation and HOM formation as well as all forms of RO2 termination, and only distribute products into volatility bins after RO2 termination in an explicit gas-phase chemical mechanism. In this way we can represent the gas-phase HOM formation mechanism, its temperature dependence, and the rich array of competing RO2 termination processes in a scheme that can merge seamlessly with any gas-phase chemical mechanism (MCM, etc.) while maintaining the generality of the VBS and its ability to represent the ongoing chemistry of organic species associated with organic aerosol formation and particle growth.
HOMs and their properties have been measured in multiple laboratory and field studies. HOMs can drive pure biogenic nucleation indicating that, in the VBS context, at least some of them must be ELVOCs/ULVOCs. Stolzenburg et al. (2018) and Tröstl et al. (2016) report measurements of HOMs in the LVOC and SVOC range during experiments in the CLOUD chamber at CERN, showing that these HOMs (especially LVOCs) drive growth during new-particle formation, providing a significant reservoir of condensible species to stabilize particles after initial clustering (Stolzenburg et al., 2018; Tröstl et al., 2016). From these data, both collected under NOx-free conditions, HOM products are expected to be distributed in the SVOC, LVOC, and ELVOC range, with the largest concentration as LVOCs.
The composition and properties of HOMs produced during oxidation of a precursor such as α-pinene (as well as their quantitative yields) depend on the conditions under which they are formed. Frege et al. (2018), again at CLOUD, found that at lower temperatures fewer total HOMs are formed, with dimers seeing the sharpest falloff (Frege et al., 2018). Lehtipalo et al. (2018) showed that increasing NOx concentrations suppress the ELVOC HOMs but have less of an effect on the LVOC and SVOC HOMs (Lehtipalo et al., 2018).
There are two important elements to HOM formation and its volatility distribution. The first is autoxidation itself, which produces progressively more oxidized peroxy radicals via H-atom transfer and thus –OOH functionalization before the radicals ultimately terminate to form stable products. The second is dimerization, which is one of the termination processes, where these functionalized RO2 associate to form a covalently bonded dimer (presumably a peroxide, ROOR). The first process plays a key role in particle growth and the temperature dependence of condensible vapor yields. The second process may be rate limiting for pure biogenic nucleation.
Autoxidation involves an internal H-atom transfer, which almost certainly has a significant activation energy. Further, the process is observed to be competitive with bimolecular reactions on a timescale of 1 s or longer (Ehn et al., 2014). Unimolecular reactions have an intrinsic rate constant (A factor) given by molecular vibrational frequencies, and even loose bending modes in molecules have frequencies of THz. To slow down unimolecular reactions to 1 s−1 or so, a high activation energy is thus almost essential. Rate coefficients for competitive reactions suggest an autoxidation energy barrier in the range of 15–18 kcal mol−1 (7500–9000 K). This is also consistent with quantum chemical calculations as well as experimental data fitted to an extended MCM model (Rissanen et al., 2014; Molteni et al., 2019). This high activation energy also means that the RO2 isomerization reactions will have a strong temperature dependence, becoming very slow at low temperature.
In contrast to the strong temperature dependence of autoxidation, the two canonical RO2 loss reactions, with HO2 and NO, both have only a weak temperature dependence with similar rate coefficients near 10−11 cm3 molecules−1 s−1 largely independent of RO2 structure (Atkinson et al., 2008). HO2 levels typical in the lower atmosphere are roughly 3 pptv, or 108 molecules cm−3, giving a first-order low-NOx loss frequency of 10−3 s−1. NO in polluted areas can go much higher, increasing the high-NOx loss to 0.1 s−1 or even higher. This establishes the competitive range for RO2 first-order loss in the 0.001–1 s−1 range in the atmosphere. Because these rate coefficients are only weakly sensitive to temperature, the high activation energy for the H-atom transfer in autoxidation means we expect the reactions with HO2 and NO to be more competitive at low temperature.
Another RO2 loss is to react with other RO2. Unlike their reactions with HO2 and NO, in which the rate coefficients depend only weakly on the nature of the R group, the rate coefficients for RO2–RO2 self- and cross-reactions are extremely sensitive to the nature of the R group. This has been well established for decades for the self-reactions of different small RO2 (Madronich and Calvert, 1990; Donahue and Prinn, 1990; Wallington et al., 1992; Tyndall et al., 2001). However, because ambient RO2 concentrations rarely exceed ambient HO2 (Mihelcic et al., 2003; Tan et al., 2018), reactions with other RO2 will only be a major RO2 loss pathway in the atmosphere when the rate coefficient equals or exceeds 10−11 cm3 molecules−1 s−1. This is in contrast to low-NOx chamber experiments, where RO2 concentrations can greatly exceed HO2 concentrations, potentially biasing the chambers away from reactions important in the atmosphere.
At least for highly oxygenated peroxy radicals, association to form dimers has been found in some experimental studies to proceed with a rate coefficient near the collisional limit ( cm3 molecules−1 s−1) (Berndt et al., 2018; Molteni et al., 2019). However, Zhao et al. (2017) find that the typical rate coefficient for presumably similar RO2+RO2 association reactions is at least an order of magnitude slower (Zhao et al., 2017). These findings may or may not be directly in conflict given the wide range of peroxy radical rate coefficients. The general tendency is that electron-withdrawing groups (i.e., acyl groups in peroxy acyl radicals) increase the RO2 self-reaction rate coefficients while electron-donating groups (i.e., tert-butyl in tert-butyl peroxy radicals) lower the rate coefficients. Thus, very fast cross-reaction rate coefficients for highly oxygenated RO2 radicals are consistent with the general trend.
Autoxidation includes a unimolecular hydrogen shift reaction in which a peroxy moiety abstracts a hydrogen elsewhere on the molecule, generating an alkyl radical, which molecular oxygen will add to in turn, regenerating a peroxy radical functionality on a now-more-oxidized carbon backbone. This process is significant in its ability to rapidly generate multifunctional oxygenation products in only a single generation of chemistry (stable molecule to stable molecule). However, experimental and theoretical evidence suggests that not every peroxy radical is readily capable of undergoing autoxidation. The fraction of peroxy radicals that will autoxidize will be different depending on the precursor of interest; for α-pinene three of the four peroxy radicals from ozonolysis can autoxidize. Ehn et al. (2014) found that the yield of highly oxidized peroxy radicals from OH oxidation of α-pinene was lower than from ozonolysis, indicating that while OH-radical-derived peroxy radicals can autoxidize, the process is either slower or there are fewer that autoxidize readily (Ehn et al., 2014).
To summarize, three important characteristics of a strongly autoxidizing system such as O3+α-pinene are emerging. First, most of the products under atmospheric conditions are likely to be traditional oxidation products formed without an autoxidation step. Second, the amount of autoxidation is likely to be strongly temperature dependent, with more at higher temperature. Third, some termination of at least the highly oxygenated RO2 results in covalently bound (and highly oxygenated) dimers, which will have extraordinarily low vapor pressures.
Each consequence has an important implication, which we give in reverse order. First, the dimerization may be the rate-limiting step for pure organic nucleation; because it involves a cross-reaction, this means that organic nucleation may also be highly sensitive to interactions between different hydrocarbon oxidation sequences, especially at low NOx. It remains somewhat unclear how important this termination is in the real atmosphere vs. chambers, given the high RO2:HO2 in chambers; however, the characteristic dimer products are observed in the atmosphere. This strongly suggests that these association reactions are important in the atmosphere.
Second, the temperature dependence of the monomer autoxidation products may well be counterbalanced by the simple temperature dependence of vapor pressures. Both of these govern organic condensation, especially to very small particles. Consequently, particle growth rates from organic oxidation (as well as secondary organic aerosol mass yields) are likely to be less sensitive to temperature than one would otherwise expect. They are also likely to be less sensitive to interactions between different hydrocarbon oxidation sequences. Particle nucleation and growth may thus be partially decoupled, making the treatment of each separately important, especially in the “valley of death” region below 3–5 nm where highly diffusive particles are most vulnerable to coagulation loss.
Third, the large majority of organic oxidation products are likely to be oxygenated species with lower vapor pressure than the precursors that remain in the gas phase (traditional SVOCs). Later-generation chemistry of these products remains an important topic that cannot be forgotten (Donahue et al., 2005, 2012a); these subsequent steps have already been shown to be efficient sources of HOMs (Schobesberger et al., 2013; Ye et al., 2018).
For this work we will model an idealized batch reactor with oxidation of α-pinene leading to HOM products. The model is split into two pieces: a general chemical mechanism and a specific implementation for the batch reactor. Here we only simulate the gas-phase chemistry, culminating in formation (and loss) of RO2 species, with RO2 termination mapping into the 2D-VBS via a succession of kernels specific to each termination step.
3.1 Chemical mechanism
The model we use here is an explicit gas-phase photochemical box model using a custom-written Perl mechanism parser that produces Matlab code for the coupled differential equations describing an arbitrary set of input reactions. The reaction set here is relatively standard small-molecule HOx chemistry with added α-pinene. We list the full set of reactions and rate coefficients in the Supplement.
The reactions begin with oxidation of a precursor, in this case α-pinene via ozonolysis, OH, and the nitrate radical (when NOx is present). Most of the products of these oxidation reactions will be peroxy radicals, with 10 carbons each, which will undergo two types of reactions: autoxidation or radical termination. Autoxidation, as described previously, is a unimolecular reaction followed by O2 addition that generates a more oxidized peroxy radical that will have the same reaction pathways available to it again as the parent RO2. It is thus effectively recursive. The radical termination reactions available to a peroxy radical are reaction with HO2, NO, or another peroxy radical.
It is likely that peroxy radicals have a diversity of H-transfer activation energies (and A factors) and that this diversity is important to the overall system behavior. We currently lack sufficient kinetic information to constrain this, and so we adopt a simple approach. For the initial RO2 (without additional –OOH functionality), we assume that some fraction can undergo autoxidation and the remainder cannot (it is a binary choice). All of the RO2 that can autoxidize does so with a single barrier height and A factor, and this is common to all of the subsequent oxidized RO2.
A second complication is the role of alkoxy radicals (RO), which can be chain propagating (including a much more widely recognized 1–4 H shift that is broadly similar to the H shift in autoxidation) (Atkinson, 1997; Lim and Ziemann, 2009) However, for this set of simulations only we shall assume that RO radicals effectively terminate to molecular products; that is consistent with our broad objective to upgrade the 2D-VBS from simply generating (NOx-dependent) products from a precursor (i.e., α-pinene) to allow the products to be controlled by peroxy radicals. In the future we shall relax this assumption and permit regeneration of RO2 from RO.
3.1.1 α-pinene reactions
We are simulating α-pinene ozonolysis, but this chemistry will produce OH radicals and, with added NOx, NO3 as well. Consequently, we have three oxidants to consider. It has been found that OH oxidation of α-pinene produces far fewer HOMs than O3 (Kirkby et al., 2016), presumably because the OH addition does not immediately cleave the six-member ring in α-pinene. Further, oxidation of α-pinene by NO3 has been shown to produce very little first-generation SOA (Fry et al., 2014; Ng et al., 2017). However, almost all primary oxidation results in peroxy radicals (with the exception of some small fraction of stabilized Criegee intermediates), though only some of those may have rapid autoxidation pathways. To address this, we assign two first-generation RO2 radicals to each oxidation pathway; one, Ox0RO2, has the potential for autoxidation, while the second, RO2, does not.
Here we assume that , αOH=0.1, and . Further, (Presto and Donahue, 2004), yOH=0, and .
3.1.2 Autoxidation
The autoxidation process involves an internal H-atom transfer in an RO2 radical, immediately followed by O2 addition to reform a new RO2 radical with an added –OOH group. Broadly, we label these reactions
where n is the number of –OOH groups (the number of generations of autoxidation). The rate-limiting kinetic parameter is the unimolecular rate constant for H-atom transfer, which we express as an Arrhenius rate coefficient with a pre-exponential A=108 s−1 for the Ox0RO2, which decreases to and for the Ox1RO2 and the Ox2RO2, respectively, and an activation energy θa=7500 K that is consistent for all autoxidation steps in our base-case simulation. This gives an H-atom transfer rate coefficient for Ox0RO2 of 0.01 s−1 at 298 K and 10−7 s−1 at 248 K for the Ox0RO2. Raising the activation energy to K gives an H-atom transfer rate coefficient of s−1 at 298 K and s−1 at 248 K. Even though each H-atom transfer step proceeds with different rate coefficients, we are still likely missing a large amount of the diversity of barriers and rate constants. In addition it is plausible that the barriers drop with increasing extent of autoxidation.
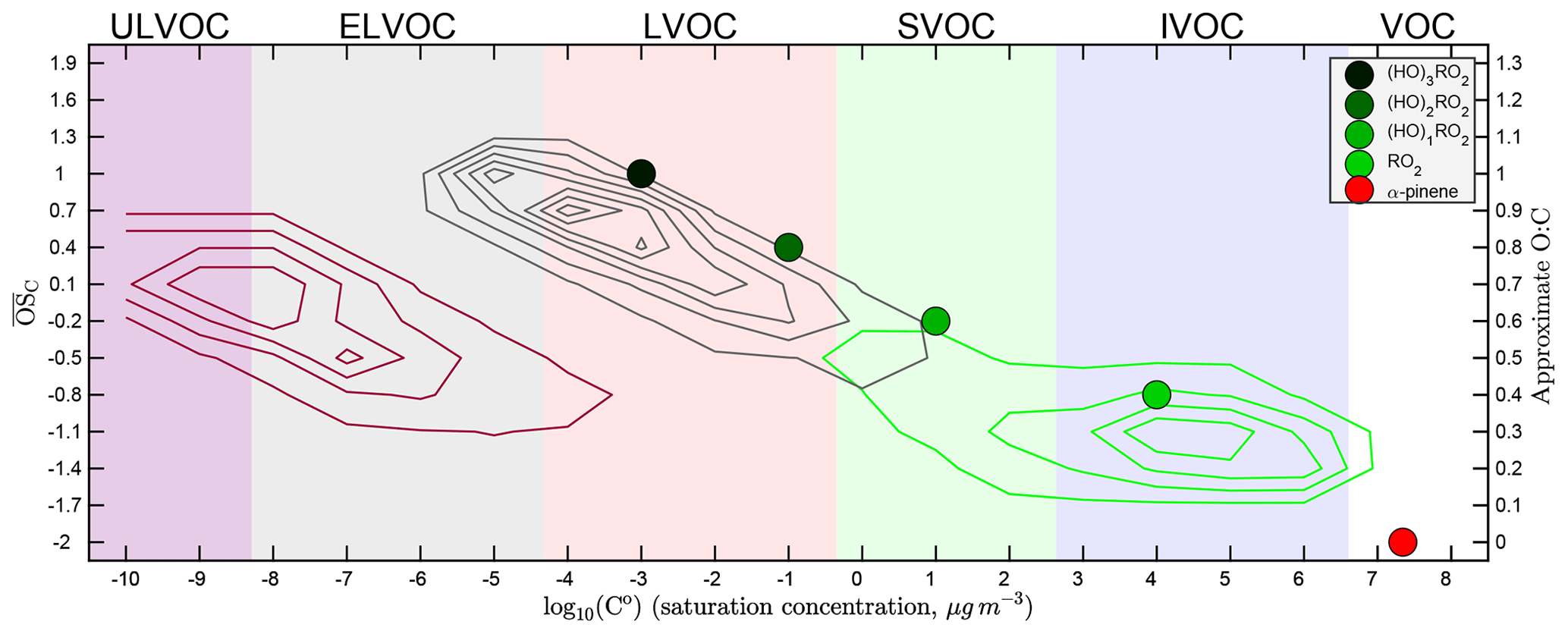
Figure 1Basic α-pinene oxidation and the two-dimensional volatility basis set (2D-VBS), including explicitly treated peroxy radicals (RO2). Axes are volatility log 10C∘(300 K) and O : C. Broad colored bands are volatility classes, indicated along the top: ULVOCs are efficient nucleators. α-Pinene (red, lower right) oxidizes to produce a succession of peroxy radicals – first a traditional radical (RO2, in light green) and then a succession of oxidized RO2 via autoxidation (OxnRO2, darker shades of green). RO2 radicals ultimately terminate into molecular products, represented here by contours depending on whether they derive from the traditional RO2 (green contours), autoxidation (C10 monomers, gray contours), or OxnRO2 dimerization (C20 ROOR, maroon contours).
There are multiple peroxy radical structures formed both initially and via every autoxidation reaction. Furthermore, HOM RO2 radicals with multiple –OOH groups can rapidly interconvert, and even the exact same peroxy radical can react with other radicals to produce different product structures. This leads to a large and complex distribution of final product structures. In order to reduce this complexity, we represent the peroxy radicals as individual surrogate molecules – points in the 2D-VBS space – with autoxidation moving the surrogate to lower volatility and higher O : C. We thus explicitly represent the volatility of the RO2 radicals, which we show in Fig. 1 as a sequence of four colored circles (along with α-pinene itself as a red circle), ranging from light green for all traditional RO2 that have not undergone autoxidation through a succession of deeper green colors for OxnRO2 that have undergone n=1–3 generations of autoxidation.
3.1.3 Unimolecular RO2 termination
Peroxy radicals may undergo unimolecular termination at any point in the autoxidation chain, but exact barrier height and prefactors for these reactions remain unknown. For example, radical termination may occur via OH loss following a H abstraction from a carbon with an –OOH group attached. Assuming that the termination involves a bond scission and potentially an H-atom transfer between neighboring groups, this termination is likely to have a higher A factor but a higher activation energy than the internal isomerization (which has a long cyclic transition state and thus a relatively low A factor). Here we represent this process with a prefactor of 1015 s−1 and a barrier of 13 000 K. This gives a termination rate coefficient of 10−4 s−1 at 298 K and s−1 at 248 K. As with autoxidation, there is every reason to believe that different functional groups on the peroxy radical would hinder or enhance the unimolecular termination pathway, but we use a consistent prefactor and barrier for every peroxy radical to represent a complicated reaction pathway more simply.
In our framework, unimolecular termination products are treated as monomer products previously discussed. They may or may not be HOMs depending on whether the peroxy radical that produced them underwent autoxidation to any extent.
3.1.4 RO2 cross-reaction kinetics
Based on the available evidence that RO2 self-reaction rate constants accelerate with the presence of electron-withdrawing functional groups near the –OO moiety, we assume that the self-reaction rate constants for the OxnRO2 are given as shown in Table 1.
Table 1Self-reaction rate constants for RO2 and OxnRO2 are shown. As peroxy radicals become more oxidized, the assumption is that their association rate constants increases.

Given the enormous number of possible cross-reactions (and paucity of kinetic data), Madronich (Madronich and Calvert, 1990) proposed a simple parameterization to estimate the cross-reaction rate coefficient for two dissimilar RO2 radicals as the geometric mean of their self-reaction rate constants, . The factor of 2 is due to the different symmetry of the self- and cross-reactions.
3.1.5 RO2 dimerization
The mechanism for RO2 dimerization is unknown. It is a theoretical puzzle because RO2 reactions are thought to proceed through a weakly bound tetroxide intermediate, ROO–OOR, which is a singlet closed-shell species (Lee et al., 2016). If by whatever (presumably multiple step) process this singlet species eliminates molecular oxygen, O2(3Σ), the singlet peroxide ROOR is spin forbidden. In the more common radical pathway, which produces , the two RO radicals are spin entangled in a triplet state. They cannot re-combine into a peroxide (ROOR) without first crossing to the singlet state.
Here we model the RO2 cross-reactions as
with products being treated as stable molecules within the volatility basis set. A more general treatment would also include both the radical (RO) and molecular (alcohol + carbonyl) pathways, which are treated with a branching ratio β (Tyndall et al., 2001); here we assume that β=1 for all of these peroxy radicals, consistent with findings that the radical pathway is favored for peroxy radicals with electron-withdrawing functional groups near the –OO moiety.
It may be that the ability to form dimers is directly dependent on additional functional groups in the RO2 molecules or it may be an indirect consequence, for example related to volatility and possibly cluster lifetime when two RO2 radicals interact. Because we are explicitly tracking the volatility of the RO2 radicals produced via autoxidation (and because some have very low volatility), here we shall explore the possibility that dimerization is somehow tied to the volatility of the reacting RO2 radicals. Specifically, we hypothesize that when low-volatility RO2 species collide they will form a short-lived (radical) cluster, both before and after reacting. We further assume that ROOR dimer formation in this cluster follows the more traditional radical (RO) step and that the product cluster must be long enough lived for collisions with air molecules to induce a spin flip in the initially triplet entangled system. As a rough measure of cluster lifetime we will use the geometric mean volatility of two interacting RO2 species (i.e., the average of the log C∘ values).
If dimerization is volatility dependent, the probability of dimerization occurring when two peroxy radicals interact may be approximated by
where is the geometric mean of the effective saturation concentrations of the two reacting peroxy radicals and is a temperature-dependent reference saturation concentration representing where we expect half of the reacting peroxy radicals to dimerize. The value of used in this work is 10−2 at 298 K and moves 1 order of magnitude lower in volatility per 10 K reduction in temperature. In our base case we assume a rate coefficient for highly oxidized α-pinene peroxy radical reactions near the collisional limit, but given the branching ratio in Eq. (1) the dimerization product yield can be much lower (depending on RO2 volatility).
3.2 RO2 termination kernels and the VBS
Following α-pinene oxidation, the initial, traditional RO2 is a dicarbonyl peroxy radical (C10H15O4). Each carbonyl functional group reduces the volatility by about 1 order of magnitude (Pankow and Asher, 2007), and we assume that the –OO moiety decreases the volatility by roughly another 1.5 orders of magnitude; consequently, this RO2 has a µg m−3. Each successive OxnRO2 differs from its predecessor by having an additional –OOH functional group. These nominally decrease volatility by 2.5 orders of magnitude (Pankow and Asher, 2007); however, there is evidence that multifunctional molecules with opportunities for internal hydrogen bonding have higher volatilities than simple composition activity relations suggest (Kurtén et al., 2016). To crudely represent this we assign µg m−3 for Ox1RO2 but assume that each successive generation of autoxidation decreases volatility by only 2 orders of magnitude, as shown in Fig. 1. It is interesting to note that Ox2RO2 reactions up on the autoxidation sequence have a low enough volatility that they are likely to remain in particles if they collide with them.
Figure 1 also shows the broad XVOC ranges as colored bands. This includes the new ULVOCs in purple, which have a sufficiently low volatility ( µg m−3) to nucleate efficiently under typical conditions (it is supersaturation that ultimately drives this). The ELVOCs, in contrast, will stick to any particle of any size they hit but may not contribute significantly to nucleation itself (in practice it may be the geometric mean of C∘(T) for two colliding vapors that govern nucleation, just as we hypothesize for ROOR formation).
We represent RO2 termination reactions to products as kernels anchored to the peroxy radical point that produced them. These kernels allow us to represent a wide variety of stabilization pathways producing a wide variety of different products through one surrogate species; the variety of species that the surrogate represents are instead mapped to a distribution of products within the 2D-VBS defined by a transformation relative to the surrogate RO2 volatility and O : C. In Fig. 1 we show the sum of these kernels (weighted by branching) for the three broad classes of products defined earlier: traditional oxidation products, monomers involving at least one generation of autoxidation, and ROOR dimers. We provide the individual kernels (relative to the C∘ and O : C values for each RO2) in the Supplement. The net effect is a concentration of traditional products with O : C modes in the SVOC and IVOC range, which in earlier VBS parameterizations of α-pinene SOA have constituted all of the products (Presto and Donahue, 2006; Jimenez et al., 2009; Donahue et al., 2012b), augmented by a tongue of HOM monomer products extending through the LVOC range and up to O : C >1, and finally with a parallel shoal of HOM dimer products spanning the ELVOC range and extending into the ULVOC range (causing pure biogenic nucleation; Kirkby et al., 2016).
We fully resolve the reaction products and then take the final concentration of every surrogate species (for example “Ox2RONO2” represents all organonitrates derived from second-generation autoxidized peroxy radicals) and map those to a distribution of products in the 2D-VBS using the appropriate kernel. This can readily be adapted to form a module (operator) within a larger framework representing particle microphysics, transport, wall loss, etc.
3.3 Ideal reactor
Here we model an ideal batch reactor with an initial input of precursor α-pinene that is oxidized over time, leaving only stable oxidation products, with essentially no precursor remaining at the end of the simulation. There is no wall loss, ventilation, additional vapors added after the initialization, or particle condensation or nucleation; our goal here is to just probe the chemistry of interest without interference of the physics of typical chamber experiments. We present results varying the temperature, NOx, and barrier height dependence of the yields of α-pinene ozonolysis. We simulate 600 ppt of α-pinene (1.5×1010 molecules cm−3) reacting with 40 ppb of ozone, which corresponds to a lifetime of approximately 4 h (1.4×104 s) at 298 K. When we simulate NOx chemistry we add the NOx to the system as NO, after which it undergoes reaction to NO2 and reaches an equilibrium ratio of . The value we report as the concentration of NOx is the amount of NO initially added into the system. All the simulations here are run in light so that photochemistry is able to occur.
We present results showing the final products of the chemistry described above and how they resolve within the 2D-VBS. We make qualitative comparisons with previous experimental results to show the validity of our approaches.
4.1 Base-case run
We will start with a base-case run at 298 K without NOx to demonstrate the model and its output. As the α-pinene reacts, the peroxy radicals quickly build up to a steady state and then decay in consort with the α-pinene as they autoxidize and form stable products. As Fig. 2 shows, each of the successively more oxidized peroxy radicals reaches a maximum concentration at a similar time but to progressively lower maximum values. The stable products are formed via the various peroxy radical termination processes, and in this simulation they have no sinks and so simply accumulate. Their yields are thus simply their final concentrations divided by the initial α-pinene concentration.
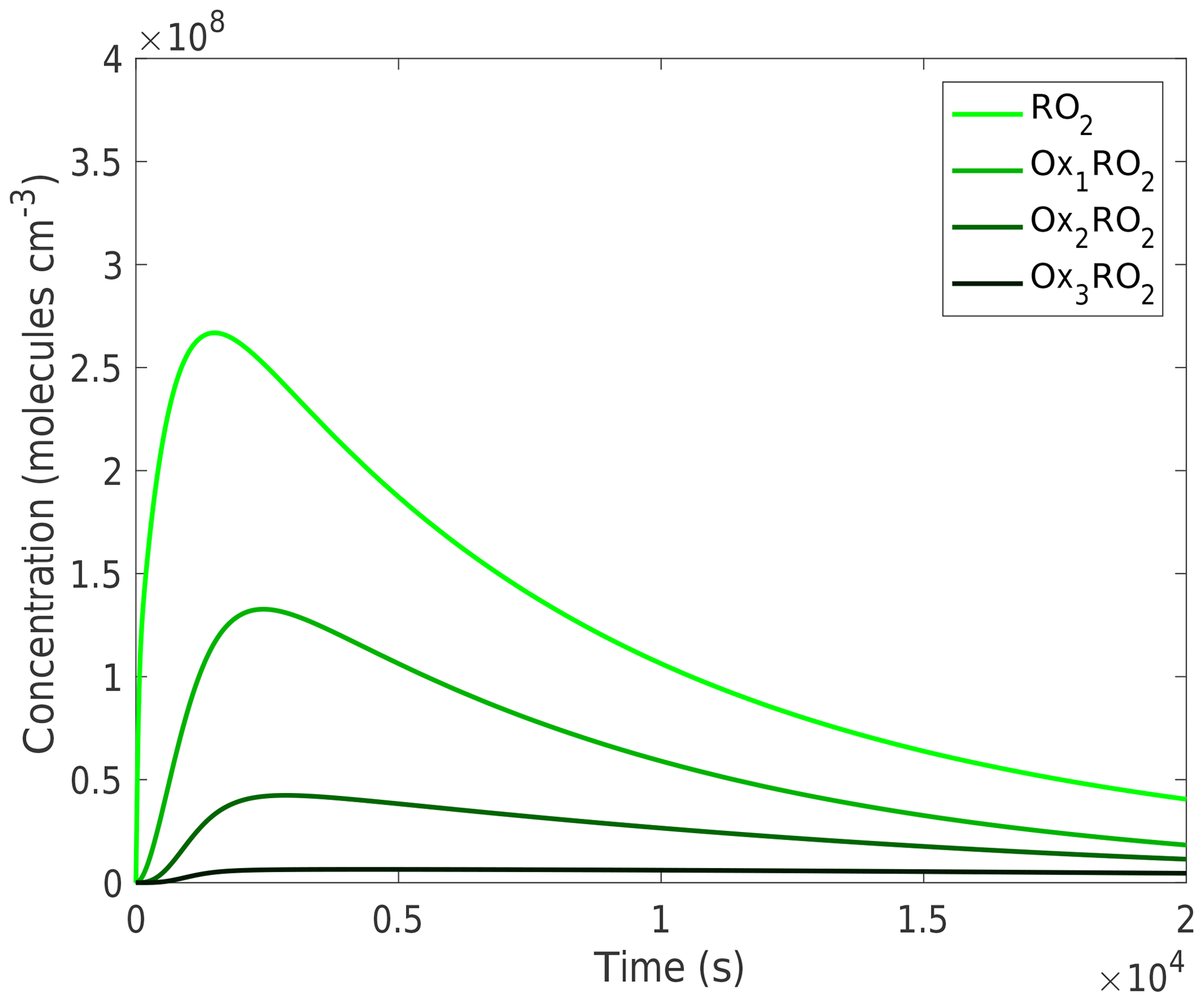
Figure 2Concentration time series of peroxy radicals (RO2). The peroxy radical concentrations over the course of a simulation peak early as they are produced via α-pinene oxidation and decrease as α-pinene decays and the RO2 react away. This simulation is run at 298 K and with no NOx, although the trends are similar across the temperatures and NOx concentrations investigated here, with the difference being in the absolute concentrations, especially those of the more oxidized peroxy radicals.
In Fig. 3a we show one such set of terminal products, labeled Oxn, to indicate the number of autoxidation steps. These products form from both RO2+NO and RO2+RO2 reactions and so scale with the RO2 radicals themselves, but because they are not lost in the batch reaction (here we do not simulate aging with the 2D-VBS) their concentrations accumulate over time.
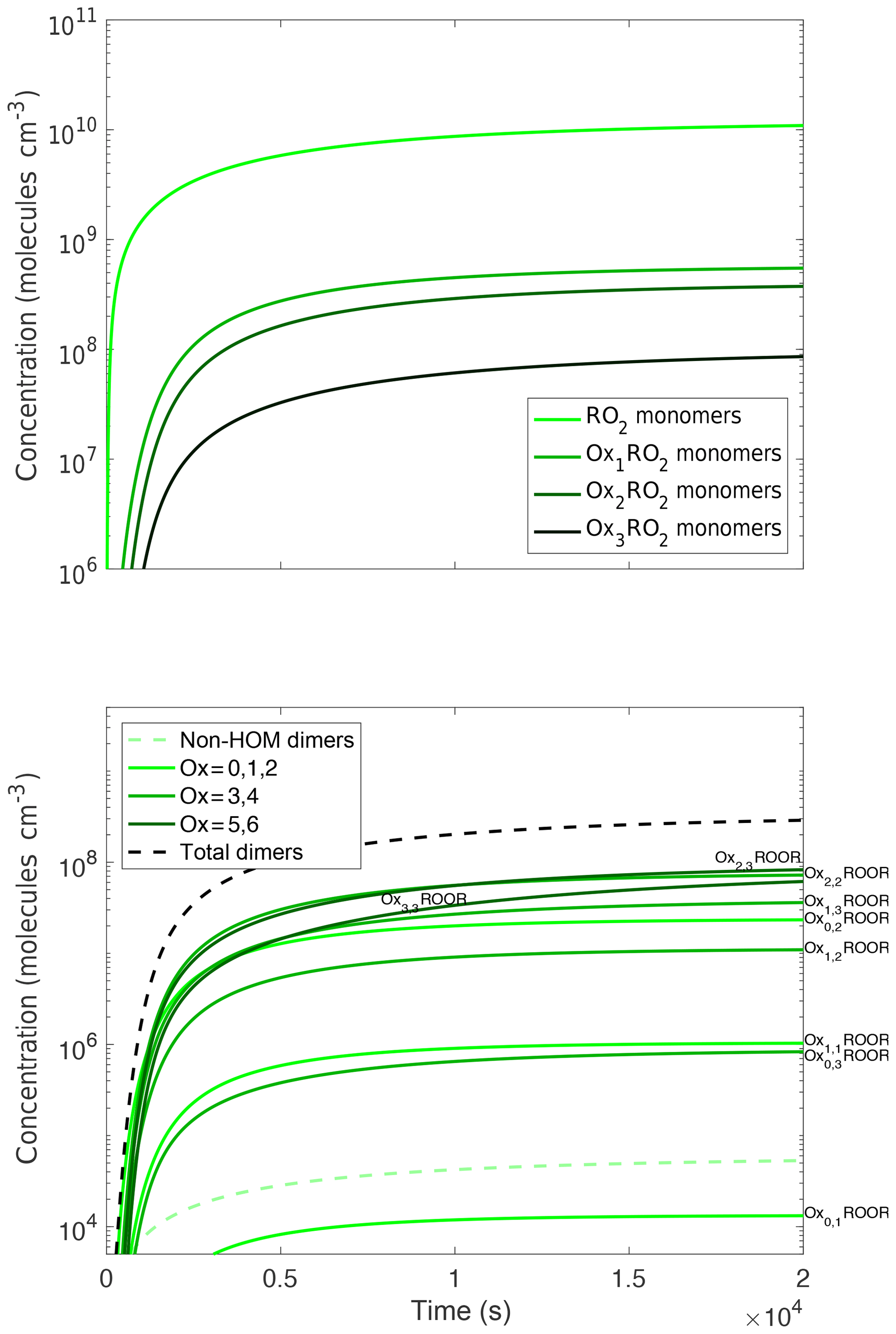
Figure 3Concentration time series of monomer and dimer products. Oxn indicates the number of autoxidation steps. All of these products are formed via peroxy radical chemistry, but the products are still predominantly non-HOM. This trend persists for hydroperoxides and organonitrates when NOx is present. The dimers do not follow the same trend of forming more from the less oxidized peroxy radical as the other species do. The dimer plot is colored by the sum of how oxidized the two peroxy radicals that reacted to form the dimer are (i.e., the dimerization of an Ox1RO2 and an Ox2RO2 would be considered the sum of Ox = 3). However, they also do not follow the exact opposite trend. This is due to the branching ratio to dimers being larger for low-volatility peroxy radicals but those peroxy radicals being less common, leading to midrange oxidized peroxy radicals forming the most dimers.
In Fig. 3b we show the corresponding dimer (ROOR) concentrations. We identify the dimers by a number pair (n,m) indicating the autoxidation extent of the associated OxnRO2. Here the situation is more nuanced because the dimers are formed exclusively through cross-reactions, and we assume (Eq. 1) that the dimer yields depend on the RO2 volatility. Consequently, even though the most oxidized RO2 have lower concentrations, they produce dimers with higher yields. The 2–2 and 3–3 symmetric dimers, along with the 2–3 cross dimer, are the most abundant.
The ultimate product distribution is dependent on the competition between autoxidation and termination via other radical species. Thus it is important to fully implement HOx and NOx chemistry, and it is informative to see how the concentrations of these radicals vary throughout the simulation. We show HO2 and NO for various levels of added NOx in Fig. 4, along with the sum of oxidized RO2 (∑OxnRO2). Characteristically for chamber experiments, HO2 remains at a low level (≲107 molecules cm−3) compared to the atmosphere due to the relative scarcity of species such as CO and CH2O that directly convert OH into HO2 and also photolyze to produce HO2. Thus HO2 remains far rarer than even the OxRO2 while most α-pinene is oxidized, with as shown in Fig. 4. Including the RO2 that was not oxidized, shown in Fig. 2, then we increase that ratio, all . Because in our simulation we add NOx as NO, at first the NO levels drop rapidly via reaction with HO2 and RO2 but then reach a steady state as NO2 photolysis replenishes NO. In general for batch chamber experiments, NOx is not added at the steady-state NO:NO2, and it is common to add NO as we simulate here; the rapidly evolving NO concentrations complicate interpretation of the results as the conditions can move from high NO early in the experiment to low NO later on. However, in this simulation, for 100 pptv of added NOx the NO stabilizes near 3 pptv. This is sufficient to slightly suppress the HO2, to more significantly reduce the oxidized RO2, and to dominate RO2 termination.

Figure 4Concentration time series of HO2, NO, and oxidized RO2 at various NOx levels. The numbers along the right-hand y axis refer to the NOx concentration for each simulation, which is also indicated by the shading of each curve, going from light at low NOx to dark at high NOx. HO2 remains reasonably stable near 107 molecules cm−3 (0.3 pptv). NOx is introduced as NO and drops to a steady state as it reacts with HO2 and RO2 to form NO2, which in turn photolyzes. By 1000 pptv of added NOx the NO greatly exceeds the HO2 and somewhat suppresses HO2 concentrations. The oxidized RO2 (∑OxnRO2) reaches a peak just over 108 molecules cm−3 at zero NOx before gradually decaying as a function of time.
The OxRO2 curves in Fig. 4 allow one to assess RO2 termination in general, remembering that the rate coefficients for HO2 and NO termination are similar and (in our model) the OxRO2 self-reaction rate coefficients are up to 10 times faster. This means that HO2 is never an important RO2 sink under these conditions, while NO becomes competitive between 50 and 100 ppt added NOx (the NO concentration must be 10 times higher than the OxRO2 to compete). Increasing NO significantly suppresses OxRO2, and so by 1000 ppt of NOx the NO pathway is dominant at all times.
4.2 Temperature dependence
The temperature dependence of the HOM formation chemistry is a key diagnostic with great atmospheric significance. Experimental data indicate that HOM yields decrease with decreasing temperature and that HOM dimers especially follow this trend. This does not mean that condensible LVOC products necessarily decrease as products with a higher C∘(300 K) will condense when it is cold; the XVOC color patches in the 2D-VBS figures show this volatility temperature dependence. Each XVOC region shifts to higher C∘(300 K) at lower temperatures, meaning that a compound with a would be in the LVOC range at 298 K, the ELVOC range at 278 K, and the ULVOC range at 248 K. However, since essentially all of the (room temperature) ULVOCs and ELVOCs are dimers, their yields will follow the dimer trends.
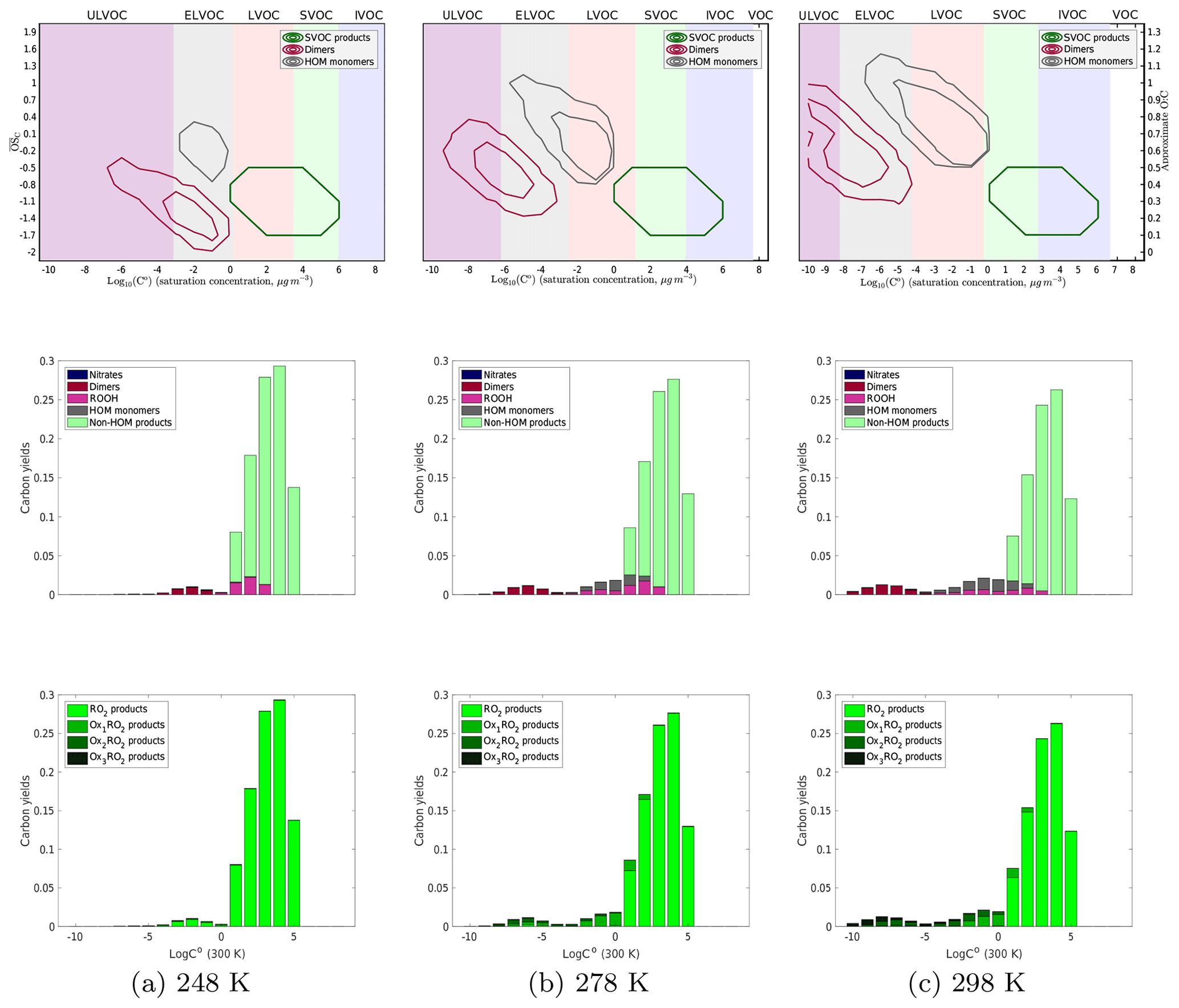
Figure 5Yields of α-pinene oxidation products without any NOx present at 248, 278, and 298 K. As temperature decreases, in the top, contour plots the dimer products shift to higher volatilities and lower O : C, even to the point where the dimers have approximately the same C∗ (300 K) as the monomer products, which is supported by experimental data. In addition, the diversity of both monomer and dimer products decreases with decreasing temperature. In the middle row of plots, where we color the yields by what type of product they are, we again see that shift of the products to higher volatilities. In addition, we see fewer dimers formed at lower temperatures. It is also of note that the formation of hydroperoxides is not a significant fraction of the yield at any temperature due to the low concentration of HO2. The bottom plots, colored by the extent of autoxidation the products underwent before terminating, show that as we decrease temperature, the extent of autoxidation that the peroxy radicals undergo decreases. At 248 K, we see almost no products that have undergone any autoxidation.
In Fig. 5 we show the temperature-dependent results for our base-case NOx-free simulation. Each column is a different temperature (248, 278, and 298 K). The top row is a 2D-VBS contour plot of the products, recapitulating Fig. 1 with the volatility classes (defined by C∘(T)) shifting with temperature following Stolzenburg (Stolzenburg et al., 2018). The middle row is a 1D-VBS plot (summing the top plots over O : C) with histograms of carbon yields in each volatility bin, colored by the RO2 termination process (NO, dimerization, HO2, etc.). The bottom row is color coded by the generation of autoxidation, with darker colors corresponding to products of RO2 that have undergone more autoxidation steps.
In the bottom row of Fig. 5 we can clearly see that the amount of products formed from autoxidized peroxy radicals decreases with decreasing temperature to the point where at 248 K there is no significant contribution of autoxidation to the yields at all. This is expected from the temperature dependence of the autoxidation rate coefficient, and the trend is supported with experimental data. In the middle plots, we see a reduction in HOM monomers at lower temperatures, but because the volatility classes shift toward higher C∘(300 K) at lower temperature, there are still relatively high LVOC yields, which is consistent with previous growth rate measurements indicating large contributions from LVOCs to growth even at low temperature. We also see that at lower temperatures dimer yields are greatly reduced. This makes sense based on the hypothesis that when two peroxy radicals react the probability that they formed a dimer is dependent on their volatility and thus dependent on the extent of autoxidation. However, at 248 K, we see an emergence of non-HOM dimers, dimers formed via reaction between two non-autoxidized peroxy radicals. These are non-HOM dimers as the definition of “HOMs” requires there to be an autoxidation step in the formation process. They are a consequence of our hypothesis that dimer formation is related to RO2 complex lifetime, as expressed in Eq. (1); any temperature dependence of dimer yields (and their O : C) is thus a test of this hypothesis.
In addition, we see almost no formation of products from an RO2+HO2 pathway. This is not a major pathway for peroxy radical termination at any temperature, given our base-case assumption of a fast RO2+RO2 reaction and the high RO2:HO2 in low-NOx chamber conditions. There is, however, a slight temperature dependence of hydroperoxide formation as at low temperatures (with no NOx) there is a larger buildup of the Ox0RO2, which have a relatively slow self-reaction rate constant and thus do terminate with HO2.
4.3 NOx dependence
Experimental data indicate that ULVOC/ELVOC yields are strongly reduced with increasing NOx while LVOC and SVOC yields are less affected. In addition, at intermediate NOx concentrations, HOM organonitrates will be formed, contributing HOMs in the SVOC to LVOC range not found under low-NOx conditions.
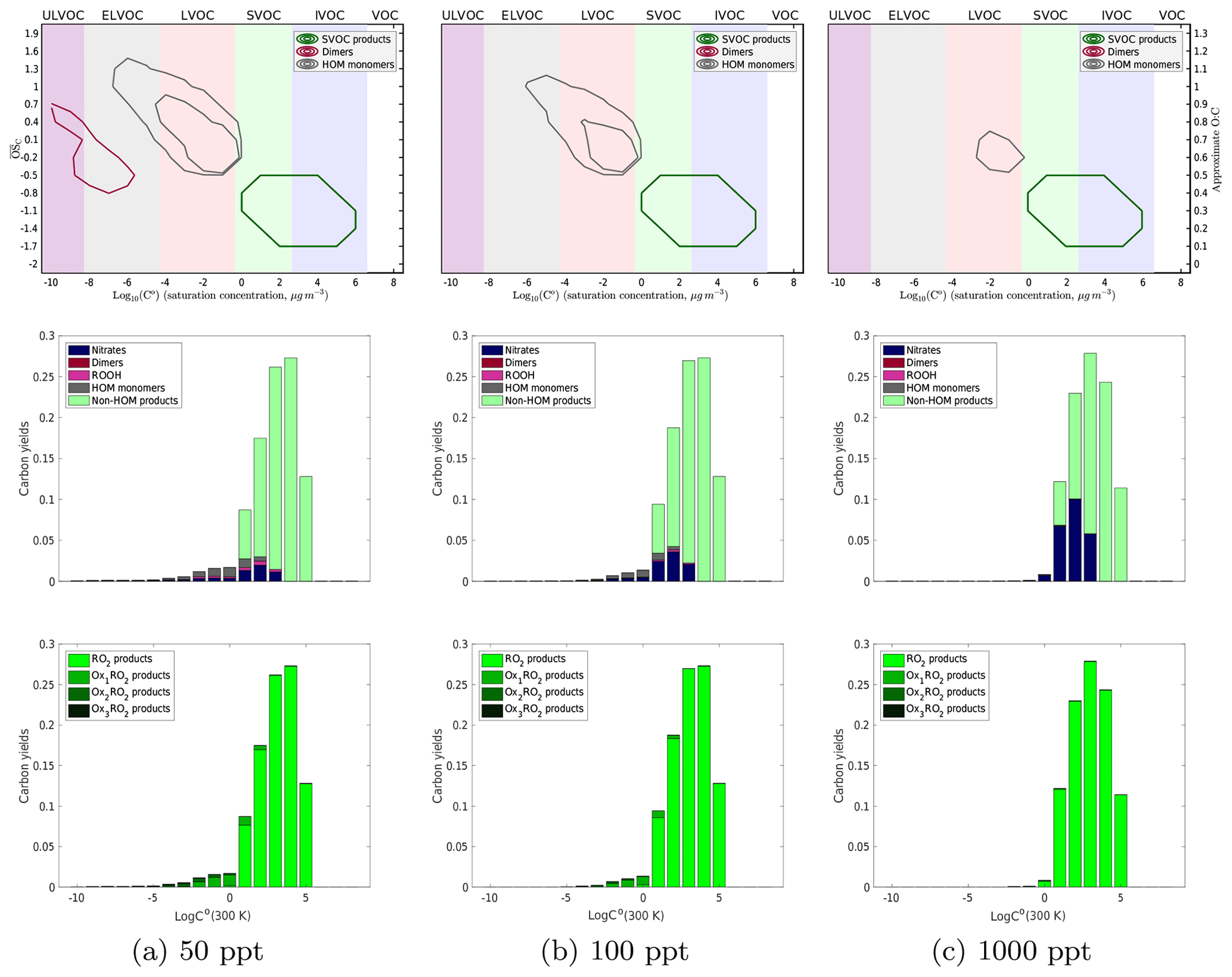
Figure 6Yields of α-pinene oxidation products at 298 K with 50, 100, and 1000 ppt of NO introduced into the system. There are similar trends seen in the contour plot with an increase in the volatility of both HOM monomer and dimer products as well as a decrease in the diversity of both with increasing NOx. In the second row of plots, we see an increase in the amount of nitrates formed and the depletion of dimers formed with increasing NOx. Similarly to the temperature trend, as we increase NOx the products shift to higher volatilities. However, once we reach a high enough NOx concentration to produce essentially no dimers, we still see some LVOC products that include nitrates. This is consistent with experimental evidence that NOx suppresses nucleation but not necessarily growth. From the last plots, we can clearly see fewer autoxidation products at higher NOx concentrations with a nearly complete suppression of autoxidation between 500 and 1000 ppt of NOx.
We show the NOx-dependent results for our 298 K simulation in Fig. 6, which follows the temperature-dependent results in Fig. 5 in form. Each column is a different NOx concentration (50, 100, and 1000 ppt), but the results show the same qualitative pattern as those in Fig. 5, with high NOx taking the place of low temperature. NO suppresses autoxidation and HOM formation just as low temperature does, but normal hydroperoxides (formed via HO2 termination) are replaced by organonitrates. Further, because the temperature is not reduced, the high-NOx products remain largely in the SVOC class, so high NOx will suppress nucleation.
At very low NOx (<20 ppt), we see relatively little effect on yields, either type or autoxidation, compared to the zero-NOx 298 K results in Fig. 5. There is, however, some suppression of dimers. In our simulation, NOx becomes competitive with autoxidation around 500 ppt and completely overpowers autoxidation around 1000 ppt. This is based on an RO2+NO rate coefficient of 10−11 cm3 molecules−1 s−1 and an activation barrier to autoxidation producing a rate coefficient of about 10−3 s−1 at 298 K. In this simulation we do not incorporate any variation in kinetics with the RO2 classes (e.g., the isomerization activation energy) other than the dependence of dimerization on the geometric mean of RO2 volatility expressed in Eq. (1) and the small change in the prefactor for autoxidation discussed in Sect. 3.1.2. Therefore, we caution against making quantitative comparisons to experimental results such as Zhao et al., who found that competition between autoxidation and NOx termination begins to favor the NOx pathway at about 20 ppbv of NOx. It is highly likely that the RO2 kinetics vary considerably depending on the exact RO2 structure, and it is very possible that more of this will need to be represented to obtain quantitative agreement with experimental observations.
In Fig. 7 we show the NOx effects on HOM monomers and dimers at different temperatures. For the NOx range we simulate here, the major effect of NO is to suppress the total HOMs uniformly by roughly a factor of 10. In this plot we see that while temperature has a relatively uniform effect of suppressing HOM monomers and dimers, NOx suppresses dimer yields more aggressively than it does HOM monomer yields. This can be explained due to the emergence of HOM nitrates, which are included in the monomer yields, as well as the consequence of NO reducing total OxRO2 and thus the rate of peroxy radical association reactions.
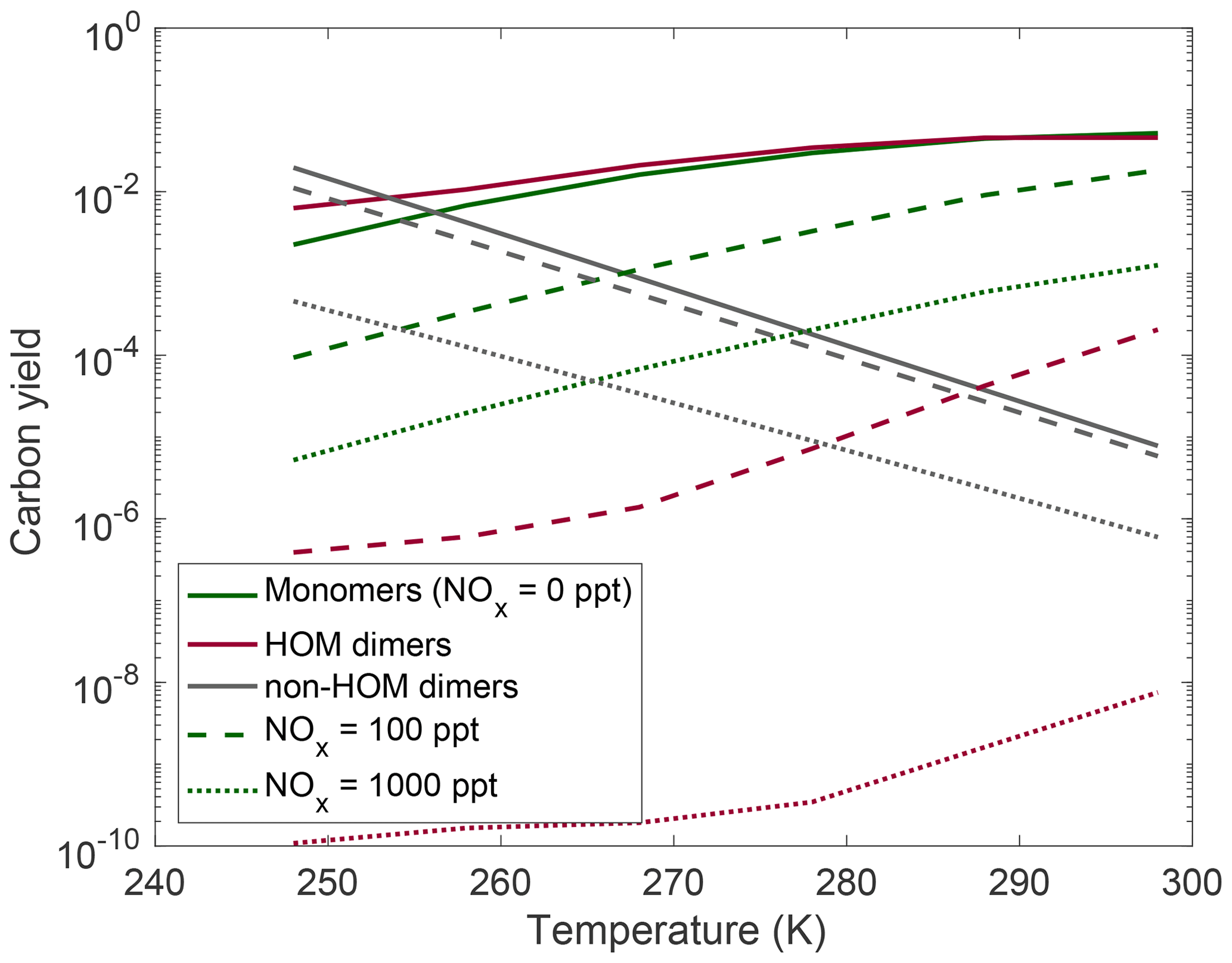
Figure 7Yields at different temperatures and various NOx levels: no NOx present (solid curves), 100 ppt of NOx present (dashed curves), and 1000 ppt of NOx present (dotted curves). Very low NOx concentrations have little effect on HOM yields; however, NOx eventually becomes competitive with HOM-producing pathways, and there is a significant reduction in both dimer and HOM monomer yields accompanied by an increase in non-HOM dimers. HOM dimers are more strongly affected by suppression of autoxidation than HOM monomers.
It is important to note that the relatively low HO2 concentrations in these runs may be enhancing the importance of NOx chemistry here. With higher HO2 concentrations, the RO2+HO2 reaction will compete with the RO2+NO and reduce the nitrate yields. However, the HO2 concentrations simulated here line up with what is often seen in very clean chamber experiments, and thus our NOx conclusions hold for those cases.
4.4 Sensitivity studies
As stated, the energy barrier to autoxidation is an important unknown in this model. It is constrained experimentally by the competition with the RO2+NO reaction, but the coefficient associated with autoxidation could span orders of magnitude for different peroxy radicals. In all the simulations described so far, we used a single barrier for autoxidation for every peroxy radical and reduced the prefactor as n, the extent of autoxidation, increased.
We employ two methods to investigate the barrier height dependence of the yields: fixing the rate coefficient at 298 K and varying its activation energy (in a Clausius–Clapeyron-like expression) and allowing the rate coefficient to vary within an order of magnitude of what was used in the base-case scenario over the whole temperature range. This is to see the effect of the autoxidation barrier height on the HOM and dimer yields as well as to provide a context for the error associated with our assumption regarding the barrier height. The effect of these methods on the autoxidation rate constant is shown in the Supplement.
As shown in Fig. 8, changing the barrier height strongly affects the calculated amount of autoxidation, especially at low temperatures. There is little effect near 298 K because that is where we prescribe the rate coefficient. In Fig. 9, we show results from allowing the rate coefficient to vary an order of magnitude over the entire temperature range, producing dimer and monomer yields that span orders of magnitude. The solid curves indicate the base-case scenario described above. This allows us to put some bounds on the rate coefficient of autoxidation as experimentally dimer and monomer yields fall well within the shaded region of the plot.
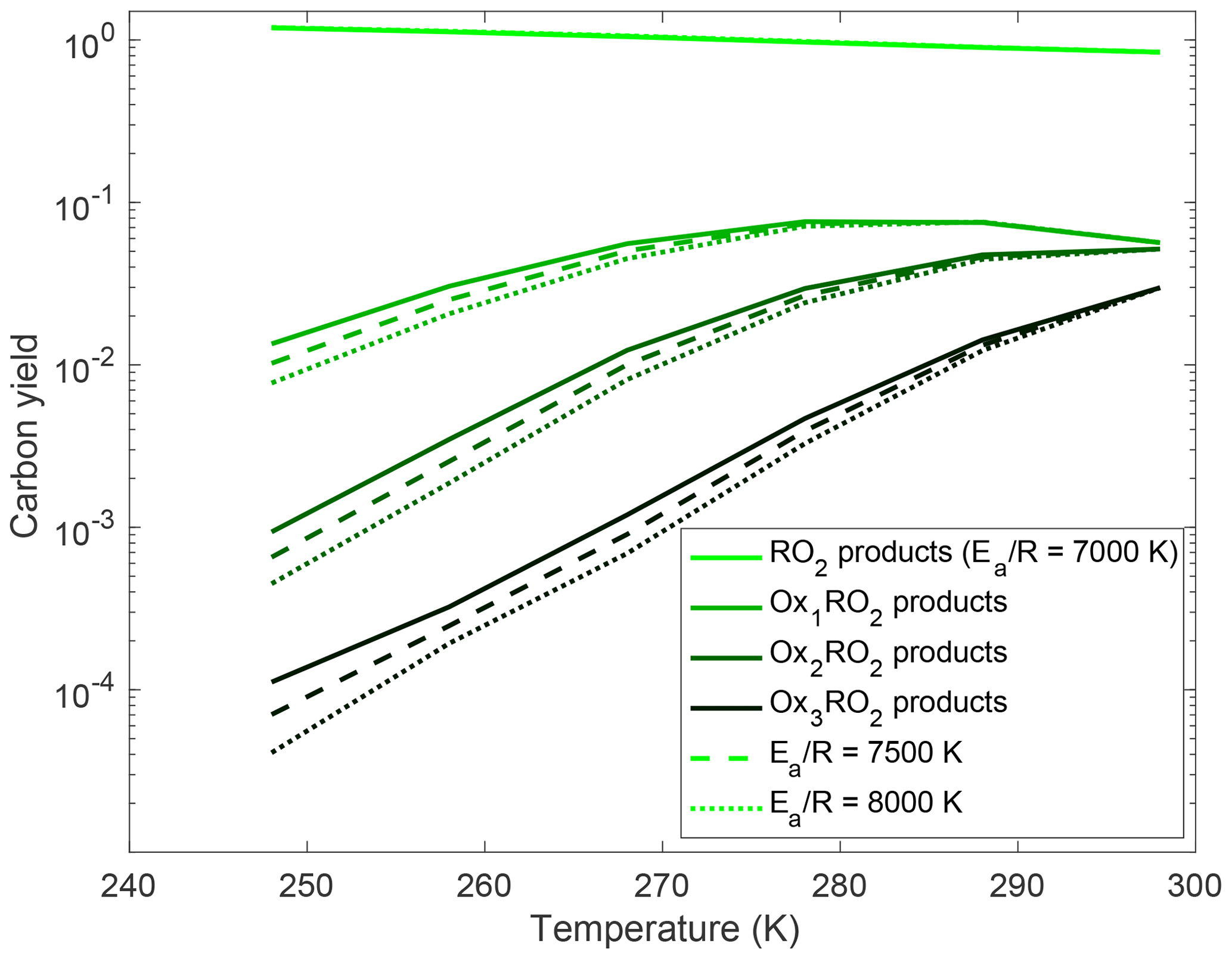
Figure 8Variation in carbon yields of the OxRO2s with temperature for different autoxidation barrier heights. Here the autoxidation rate coefficient at 298 K is fixed at 0.01 s−1, and we vary the activation energy. The dashed curve shows the results for the rate coefficient in our base-case simulation. The solid curve has a lower activation energy and thus a weaker temperature dependence, while the dotted curve has a higher activation anergy and thus a stronger temperature dependence. As expected autoxidation is suppressed more quickly as temperature drops when the activation energy is higher; this translates to lower HOM yields.
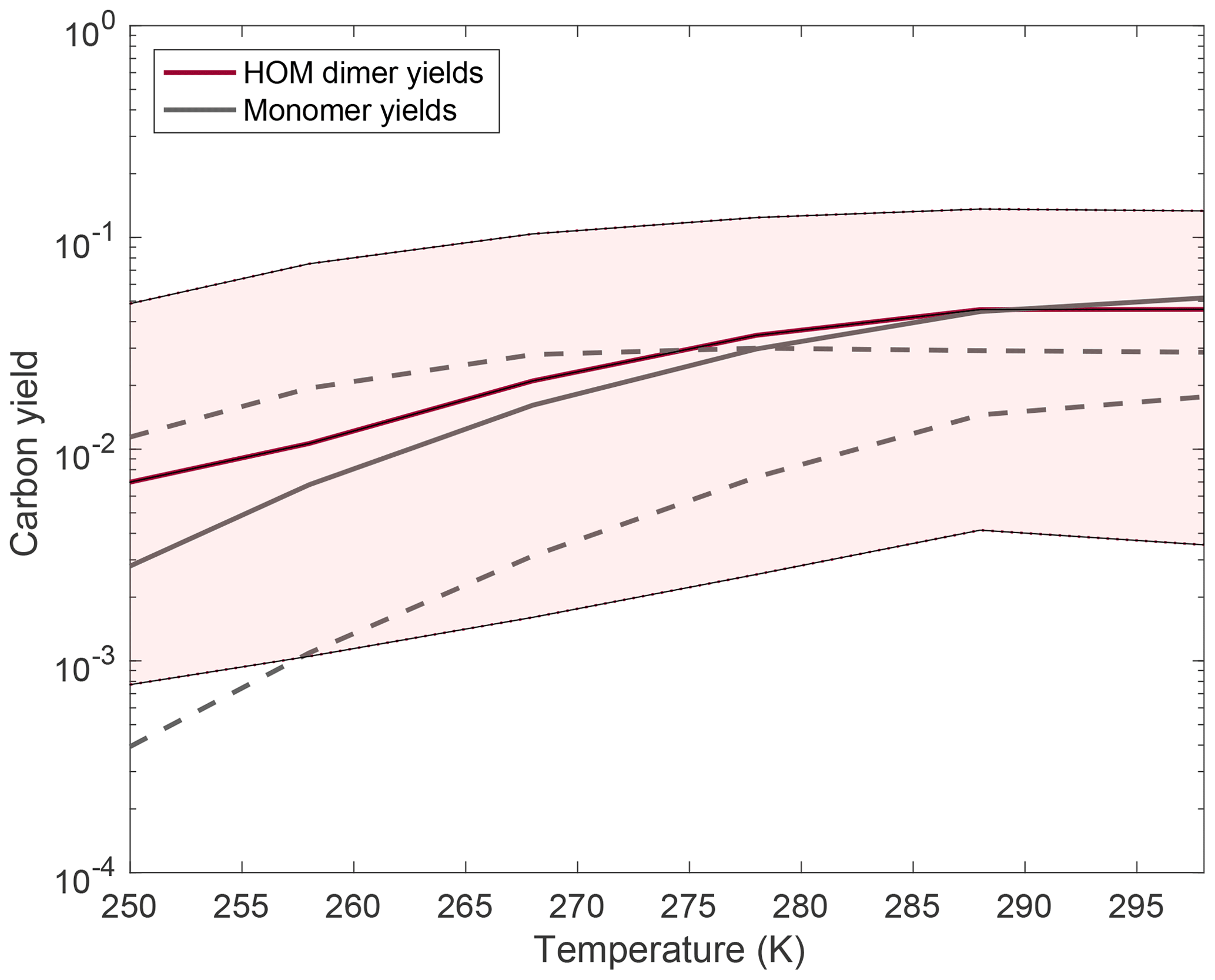
Figure 9Changes to HOM monomer and dimer yields with an order of magnitude variation in the rate coefficient at 298 K by changing the autoxidation A factor. The rate coefficient at temperatures other than 298 K is calculated in the same manner as described above. Almost all of the yields at a particular temperature are increased relative to the traditional case when we assume the rate coefficient at 298 K is higher than the traditional case, and the yields decrease at a particular temperature when we assume the rate coefficient is lower than the traditional case. The monomer yields decrease with a faster autoxidation rate constant at high temperatures because with more autoxidation more association reactions successfully form dimers, thus reducing the monomer yields. Quantitatively, as we vary the rate coefficient of autoxidation by an order of magnitude, we are seeing a corresponding increase or decrease in the HOM product also by about an order of magnitude. The monomer yields are shown in dashed lines and show a more complicated trend, with the low barrier monomer yields being higher than the base-case yields at lower temperatures but lower at high temperatures.
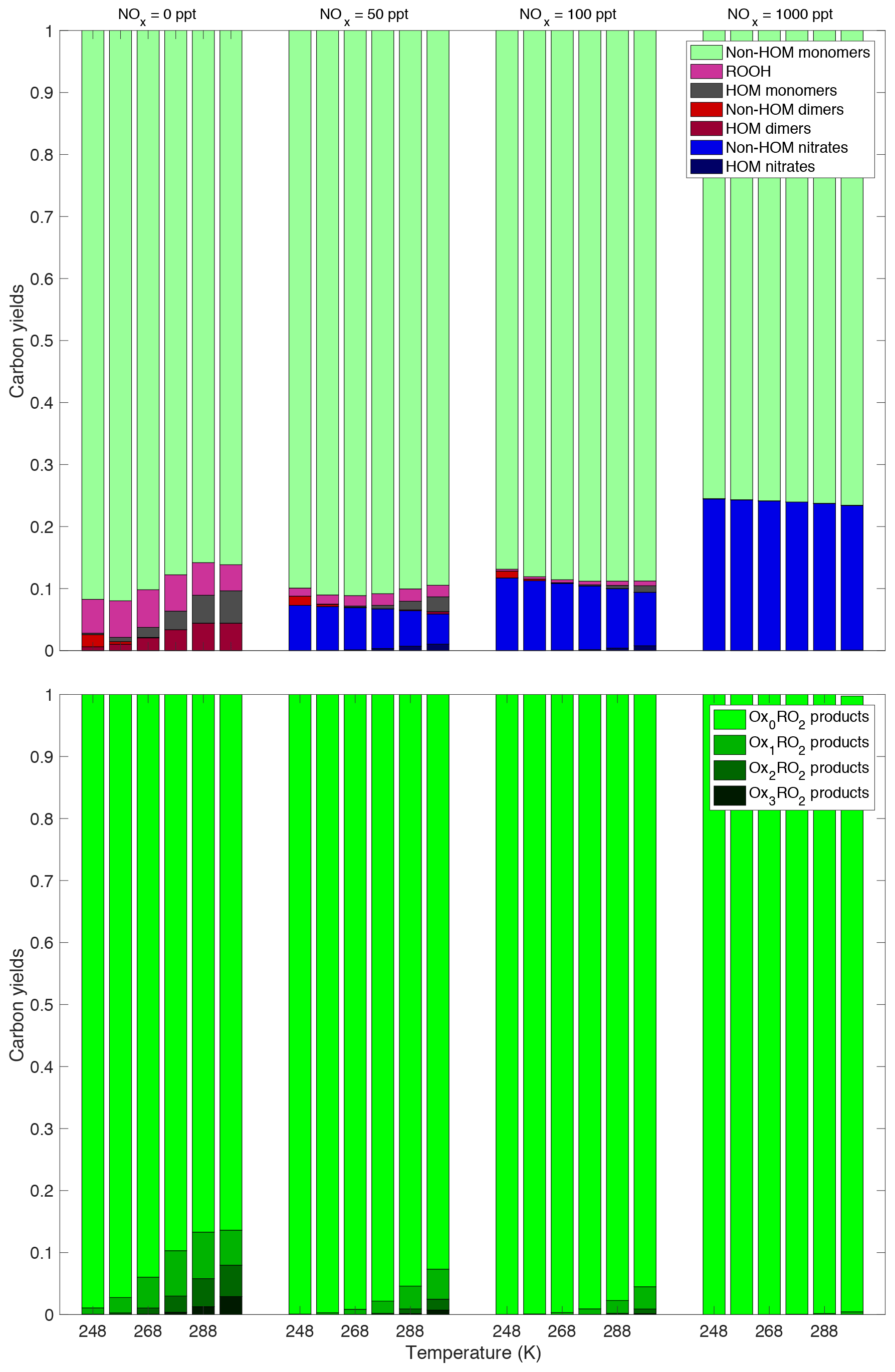
Figure 10This plot shows the synergistic effect of the NOx and temperature on the product yields. The same trend persists at every NOx concentration, specifically the decrease in the amount of HOMs – both monomers and dimers with decreasing temperature. Of note is that at the highest NOx concentration shown here (1000 ppt NO added), the HOM production is close to 0 at every temperature. Thus complete suppression of HOMs occurs around this NOx concentration. This plot shows both the temperature dependence and the NOx dependence again, but colors are based on how oxidized the peroxy radical that produced the products was. The products represented by the darker colors underwent more autoxidation before terminating than those represented by the lighter colors. We can see that autoxidation is suppressed both by temperature and by NOx. Once again, we see that at a high enough NOx concentration autoxidation is effectively suppressed at all the temperatures investigated here.
There is ample evidence in the kinetic literature for a high degree of variability in the autoxidation rate. Therefore, it is possible that to adequately describe autoxidation; its effects on new-particle formation as well as SOA formation; and the sensitivity of both to NO, T, HO2, etc., we will need to treat fast- and slow-reacting RO2. While it is possible for us to put bounds on autoxidation with the assumptions made here, it is very likely that the autoxidation of specific peroxy radicals lies outside these bounds. For example, the low barrier shown in Fig. 9 is likely outside these bounds as at the highest temperatures the monomer yields are lower than the dimer yields, which contradicts experimental evidence. However, with relatively minor changes to other parameters (e.g., the dimerization rate coefficient, the branching ratio to dimers γ, the unimolecular termination rate coefficient), this barrier height would be completely reasonable and therefore still represents a good lower bound for the autoxidation barrier height. The OxnRO2 represents a variety of peroxy radicals, and therefore these bounds are representative of the general behavior of these peroxy radicals rather than being bounds for any specific peroxy radical species.
4.4.1 NOx dependence of temperature dependence
We also want to investigate how the similar trends of decreasing temperature and increasing NOx work together. The bottom plot of Fig. 10 clearly shows this synergistic effect on autoxidation, as products that have undergone any autoxidation are almost completely suppressed at the highest NOx concentration and the lowest temperature investigated here. The top plot of Fig. 10 shows once again that decreasing temperature and increasing NOx work together to suppress both HOM monomers and HOM dimers as well as a few other trends. As NOx is increased, the yield of nitrates increases, but at any specific NOx concentration the amount of nitrate formed is effectively temperature independent. However, HOM nitrates are only formed at the highest temperature and at lower NOx concentrations; obviously, to form nitrates some NOx needs to be present, but if too much is present, the peroxy radicals will terminate before they can autoxidize to form HOM nitrates. At low NOx concentrations the HOM formation is dependent on temperature as discussed previously, but at high NOx concentrations the yields are completely dominated by termination with NO. We can also see at low NOx and low temperatures the appearance of non-HOM dimers; formation of these less oxidized peroxides is a consequence of our assumption that a long-lived RO2 molecular cluster is required to allow the products to cross to the singlet spin surface. This plot also shows the temperature and NOx dependence of yields from the RO2+HO2 reaction. While never a large portion of the total yield, products of this reaction increase under low-temperature and low-NOx conditions. When the temperature is low, most of the peroxy radicals present are Ox0RO2, which, based on our association rate constants, will react the slowest with each other. This allows for more of the peroxy radicals to react with HO2, even at the relatively low HO2 concentrations present. At high NOx, the RO2+NO reactions dominate everything including the RO2+HO2 reaction and very few to no ROOH products are formed.
Here we present a model that represents peroxy radical chemistry semiexplicitly and maps the products of that chemistry onto the 2D-VBS. We investigate the dependence of the product yields on temperature, NOx, and the energy barrier to autoxidation. Ultimately both HOM and dimers are suppressed under conditions when autoxidation is suppressed via the competition with radical termination processes. Competition with bimolecular termination processes is enhanced when the RO2 lifetime with respect to bimolecular radical termination is shortened (higher radical concentrations) or when autoxidation is slower (higher energy barrier). Therefore, HOM and dimer production is highest under high-temperature, low-NOx conditions and if we assume a lower energy barrier to autoxidation. These simulations were conducted assuming relatively fast dimerization rate constants. Therefore should some or all peroxy radicals dimerize slower, another important competition, which is not discussed at length here due to its relatively low impact, the RO2+HO2 reaction, could be much more important.
Overall, our simulation results are consistent with emerging experimental observations and strongly suggest that HOM formation will be strongly temperature dependent under atmospheric conditions, as well as highly sensitive to NO. The new radical VBS allows us to explicitly simulate the RO2 termination without making ad hoc assumptions about high- or low-NO conditions and also enables general consideration of RO2 termination chemistry, including cross-reactions among RO2 derived from many hydrocarbon precursors. It is thus well suited to describe these rich chemical systems.
The reactions used and their rate constants as well as the kernels can be found in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-1183-2020-supplement.
MS conducted the model runs and wrote the paper; NMD designed the study, advised research progress, and commented extensively on the paper drafts.
The authors declare that they have no conflict of interest.
This research has been supported by the National Science Foundation (grant no. AGS 1801897).
This paper was edited by Dwayne Heard and reviewed by two anonymous referees.
Atkinson, R.: Gas phase tropospheric chemistry of organic compounds, J. Phys. Chem. Ref. Data, 26, 215–290, https://doi.org/10.1063/1.556012, 1997. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species, Atmos. Chem. Phys., 8, 4141–4496, https://doi.org/10.5194/acp-8-4141-2008, 2008. a
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M., Pontus, R., Berndt, T., Crounse, J., Wennberg, P., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D., Thornton, J., Donahue, N. M., Kjaergaard, H. G., and Ehn, M.: Highly-oxygenated organic molecules (HOM) from gas-phase autoxidation of organic peroxy radicals: A key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a
Chuang, W. K. and Donahue, N. M.: A two-dimensional volatility basis set – Part 3: Prognostic modeling and NOx dependence, Atmos. Chem. Phys., 16, 123–134, https://doi.org/10.5194/acp-16-123-2016, 2016. a
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a
Donahue, N. M. and Prinn, R. G.: Nonmethane Hydrocarbon Chemistry in the Remote Marine Boundary Layer, J. Geophys. Res.-Atmos., 95, 18387–18411, 1990. a
Donahue, N. M., Huff Hartz, K. E., Chuong, B., Presto, A. A., Stanier, C. O., Rosenørn, T., Robinson, A. L., and Pandis, S. N.: Critical Factors Determining the Variation in SOA Yields from Terpene Ozonolysis: A Combined Experimental and Computational Study, Faraday Discuss., 130, 295–309, 2005. a, b
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006. a
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011. a
Donahue, N. M., Henry, K. M., Mentel, T. F., Scharr, A. K., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K. H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salow, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of Biogenic Secondary Organic Aerosol via Gas-Phase OH Radical Reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012a. a
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012b. a, b
Donahue, N. M., Chuang, W. K., Epstein, S. A., Kroll, J. H., Worsnop, D. R., Robinson, A. L., Adams, P. J., and Pandis, S. N.: Why Do Organic Aerosols Exist? Understanding Aerosol Lifetimes using the Two-Dimensional Volatility Basis Set, Environ. Chem., 10, 151–157, https://doi.org/10.1071/EN13022, 2013. a
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, 2014. a, b, c, d
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018. a, b
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014. a
Henry, K. M. and Donahue, N. M.: Effect of the OH radical scavenger hydrogen peroxide on secondary organic aerosol formation from α-pinene ozonolysis, Aerosol Sci. Tech., 45, 686–690, https://doi.org/10.1080/02786826.2011.552926, 2011. a, b
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prévôt, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. D., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009. a
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 530, 521–526, https://doi.org/10.1038/nature17953, 2016. a, b, c
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., and Donahue, N. M.: Saturation Vapor Pressures of α-pinene Autoxidation Products May Be Severely Underpredicted by Group Contribution Methods, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196,, 2016. a
Lee, R., Gryn'ova, G., Ingold, K. U., and Coote, M. L.: Why are sec-alkylperoxyl bimolecular self-reactions orders of magnitude faster than the analogous reactions of tert-alkylperoxyls? The unanticipated role of CH hydrogen bond donation, Phys. Chem. Chem. Phys., 18, 23673–23679, https://doi.org/10.1039/C6CP04670C, 2016. a
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buckholz, A., Mazon, S. B., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fisher, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X. C., Heikkinen, L., Heinrizi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V.-M., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai1, H., Mathot, S., Mauldin III, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela1, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Sengupta, K., Simon, M., Tauber, C., Tomé, A., Tröst, J., Väisänen, O., Voge, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K., Curtius, J., Donahue, N., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multi-component new particle formation from sulfuric acid, ammonia and biogenic vapors, Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018. a, b
Lim, Y. B. and Ziemann, P. J.: Effects of Molecular Structure on Aerosol Yields from OH Radical-Initiated Reactions of Linear, Branched, and Cyclic Alkanes in the Presence of NOx, Environ. Sci. Technol., 43, 2328–2334, 2009. a
Madronich, S. and Calvert, J. G.: Permutation reactions of organic peroxy radicals in the troposphere, J. Geophys. Res.-Atmos., 95, 5697–5715, https://doi.org/10.1029/JD095iD05p05697, 1990. a, b
Mihelcic, D., Holland, F., Hofzumahaus, A., Hoppe, L., Konrad, S., Müsgen, P., Pätz, H.-W., Schäfer, H.-J., Schmitz, T., Volz-Thomas, A., Bächmann, K., Schlomski, S., Platt, U., Geyer, A., Alicke, B., and Moortgat, G.: Peroxy radicals during BERLIOZ at Pabstthum: Measurements, radical budgets and ozone production, J. Geophys. Res.-Atmos., 108, 8254, https://doi.org/10.1029/2001JD001014, 2003. a
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of highly-oxygenated organic molecules from α-pinene ozonolysis: chemical characteristics, mechanism and kinetic model development, Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019. a, b
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017. a
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008. a, b, c
Presto, A. A. and Donahue, N. M.: Ozonolysis Fragment Quenching by Nitrate Formation: The Pressure Dependence of Prompt OH Radical Formation, J. Phys. Chem. A, 108, 9096–9104, 2004. a
Presto, A. A. and Donahue, N. M.: Investigation of α-pinene + ozone Secondary Organic Aerosol Formation at Low Total Aerosol Mass, Environ. Sci. Technol., 40, 3536–3543, https://doi.org/10.1021/es052203z, 2006. a, b, c
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014. a
Schobesberger, S., Junninen, H., Bianchi, F., Lönn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kürten, A., Kurtén, T., Laaksonen, A., Mathot, S., Onnela, A., Rondo, A. P. P. L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Vehkamäki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petäjä, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci, USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013. a
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Buenrostro Mazon, S., Chen, D., Dias, A., Draper, D. C., Duplissy, J., El Haddad, I., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018. a, b, c
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018. a
Tröstl, J., Chuang, W. K., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Gordon, H., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 530, 527–531, https://doi.org/10.1038/nature18271, 2016. a, b
Tyndall, G. S., Cox, R. A., Granier, C., Lesclaux, R., Moortgat, G. K., Pilling, M. J., Ravishankara, A. R., and Wallington, T. J.: Atmospheric Chemistry of Small Organic Peroxy Radicals, J. Geophys. Res., 106, 12157–12182, 2001. a, b
Wallington, T. J., Daugaut, P., and Kurylo, M. J.: Ultraviolet Absorption Cross Sections and Reaction Kinetics and Mechanisms for Peroxy Radicals in the Gas Phase, Chem. Rev., 92, 667–710, 1992. a
Ye, P., Zhao, Y., Chuang, W. K., Robinson, A. L., and Donahue, N. M.: Secondary organic aerosol production from pinanediol, a semi-volatile surrogate for first-generation oxidation products of monoterpenes, Atmos. Chem. Phys., 18, 6171–6186, https://doi.org/10.5194/acp-18-6171-2018, 2018. a
Zhao, Y., Saleh, R., Saliba, G., Presto, A. A., Gordon, T. D., Drozd, G. T., Goldstein, A. H., Donahue, N. M., and Robinson, A. L.: Reducing secondary organic aerosol formation from gasoline vehicle exhaust, P. Natl. Acad. Sci. USA, 114, 6984–6989, https://doi.org/10.1073/pnas.1620911114,, 2017. a, b