the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Aug 2019
| 01 Aug 2019
Photochemical impacts of haze pollution in an urban environment
Oliver Wild
Ting Yang
Weiqi Xu
Conghui Xie
Lisa Whalley
Eloise Slater
Dwayne Heard
Dantong Liu
Rapid economic growth in China over the past 30 years has resulted in significant increases in the concentrations of small particulates (PM2.5) over the city of Beijing. In addition to health problems, high aerosol loading can impact visibility and thus reduce photolysis rates over the city, leading to potential implications for photochemistry. Photolysis rates are highly sensitive not only to the vertical distribution of aerosols but also to their composition, as this can impact how the incoming solar radiation is scattered or absorbed. This study, for the first time, uses aerosol composition measurements and lidar optical depth to drive the Fast-JX photolysis scheme and quantify the photochemical impacts of different aerosol species during the Air Pollution and Human Health (APHH) measurement campaigns in Beijing in November–December 2016 and May–June 2017. This work demonstrates that severe haze pollution events (PM2.5 > 75 µg m−3) occur during both winter and summer, leading to reductions in O3 photolysis rates of 27 %–34 % (greatest in winter) and reductions in NO2 photolysis of 40 %–66 % (greatest in summer) at the surface. It also shows that in spite of much lower PM2.5 concentrations in the summer months, the absolute changes in photolysis rates are larger for both O3 and NO2. In the winter, absorbing species such as black carbon dominate the photolysis response to aerosols, leading to mean reductions in J[O1D] and J[NO2] in the lowest 1 km of 24 % and 23 %, respectively. In contrast, in the summer, scattering aerosol such as organic matter dominate the response, leading to mean decreases of 2 %–3 % at the surface and increases of 8 %–10 % at higher altitudes (3–4 km). During these haze events in both campaigns, the influence of aerosol on photolysis rates dominates over that from clouds. These large impacts on photochemistry can have significant implications for concentrations of important atmospheric oxidants such as the hydroxyl radical. Idealized photochemical box model studies show that such large impacts on photochemistry could lead to a 12 % reduction in surface O3 (3 % for OH) due to haze pollution. This highlights that PM2.5 mitigation strategies could have important implications for the oxidation capacity of the atmosphere both at the surface and in the free troposphere.
- Article
(3557 KB) - Full-text XML
- BibTeX
- EndNote





As a result of rapid economic growth and industrialization over the past 30 years, air pollution has become a major problem in China (Chan and Yao, 2008; Zhang et al., 2015), with an increase in the number of haze episodes emerging as a particular issue. During such haze events, concentrations of small aerosol particles (PM2.5: particles with an aerodynamic diameter of less than 2.5 µm) can climb to very high levels (higher than 75 µg m−3; Han et al., 2015; Wang et al., 2018), leading to significant reductions in visibility, health problems and potential feedbacks on atmospheric chemistry and dynamics (Cheng et al., 2011; Han et al., 2015; Lelieveld et al., 2015; Xing et al., 2017).
High aerosol loadings not only impact dynamics through their regulation of the atmospheric radiation budget (Kaiser and Qian, 2002; Bian et al., 2003; Martin et al., 2003; Hu et al., 2017) but can also have significant impacts on atmospheric oxidation capacity through altering photolysis rates (Liao et al., 1999; Lou et al., 2014; Tang et al., 2003; Li et al., 2011; Xing et al., 2017). Photolysis plays a very important role in initiating atmospheric photochemistry. Of particular importance are the photolysis of NO2 in the troposphere (J[NO2]), which is key to chemical generation of ozone (O3), and the photolysis of O3 itself (J[O1D]) to produce electronically excited oxygen (O1D). O1D may subsequently react with water vapour and is the main source of the hydroxyl radical (OH) in the atmosphere globally. In addition, in highly polluted urban environments such as Beijing, photolysis of nitrous acid (HONO) is also a major source of OH. OH is highly reactive and serves as the primary oxidation sink of many atmospheric species, whilst also playing a key role in initiating catalytic cycles that result in poor air quality (e.g. O3 formation).
Both the vertical distribution and composition of aerosols can impact how incoming solar radiation is absorbed or scattered throughout the atmospheric column, which in turn can significantly affect photolysis rates (Tang et al., 2003; Li et al., 2011). For example, Li et al. (2011) estimated that J[O1D] rates over eastern China were reduced by 53 %, 37 % and 21 % in the lower, middle and upper troposphere, respectively, during high summer aerosol loadings in 2006. This resulted in corresponding OH concentration reductions of 51 %, 40 % and 24 %, respectively. Therefore, vertical characterization of aerosols is critical to fully understanding the impact of severe haze on atmospheric photochemical processes. The key processes behind the formation and composition of severe haze events over China at ground sites (including in Beijing) have been studied extensively (Sun et al., 2013; Zhao et al., 2013; Huang et al., 2014; Ji et al., 2014; Han et al., 2015), with some studies also using aircraft and tethered balloon measurements to investigate the vertical profiles of aerosol and gaseous pollutants (Chen et al., 2009; Zhang et al., 2009; Li et al., 2015; Ran et al., 2016). Aircraft typically only permit measurements above 300 m altitude and fail to capture the full evolution of aerosol profiles, and tethered balloons are normally operated in rural settings that are not representative of urban environments. Therefore, greater understanding of the vertical distribution of aerosol loadings is still required.
Recent studies have better captured the evolution of black carbon (BC) profiles and their associated optical impacts during haze events over Beijing (Wang et al., 2018), but these are still limited to the lower boundary layer (< 260 m altitude). Lidar instruments are useful in providing vertical profiles of total aerosol extinction and have been applied over Beijing previously (Yang et al., 2010, 2017) to measure up to a height of 6 km. However, only the total extinction can be retrieved and there is no information on the contribution from different aerosol species (e.g. strong absorbers such as BC or scattering aerosol such as ammonium sulfate, (NH4)2SO4), which is required to understand how the incoming solar radiation is scattered and absorbed. As a result, atmospheric models are often used to simulate aerosol distributions and composition throughout the atmospheric column and their impacts on photolysis rates (Tang et al., 2003; Li et al., 2011). However, such model studies are often poorly constrained by observations, particularly in the upper atmosphere, and can fail to accurately predict peaks in aerosol concentrations during severe haze events (L. T. Wang et al., 2014; Y. Wang et al., 2014; Z. Wang et al., 2014; Zheng et al., 2015).
In this study, aerosol composition and lidar extinction measurements from two intensive field campaigns conducted in Beijing in winter 2016 and summer 2017 are used to derive chemically apportioned vertical profiles of aerosol extinction. These profiles are used, for the first time, to critically test an offline photolysis scheme against measurements of photolysis rates under observed aerosol loadings and to quantify the contribution of different aerosol components to changes in photolysis rates. This enables a better understanding of the impacts of severe haze episodes on pollutant photochemistry in contrasting seasons and provides insight into the photochemical implications of potential future pollution reduction strategies.
2.1 Measurement campaigns and sampling site
Two measurement campaigns were conducted at the tower site of the Institute of Atmospheric Physics, Chinese Academy of Sciences (IAP-CAS) as part of the joint UK–China Air Pollution and Human Health (APHH) programme addressing the sources, processing and impacts of air pollution in Beijing (Shi et al., 2019). The site is located in urban surroundings (39.6∘ N, 116.2∘ E) between the third and fourth ring roads in the north of Beijing, 40 m from the nearest road and around 400 m from the Jingzang Expressway (Han et al., 2015). The first campaign was conducted from 5 November to 10 December 2016 and the second campaign was conducted from 15 May to 22 June 2017, in order to monitor haze episodes during different seasons (Shi et al., 2019).
2.2 Instrumentation
Non-refractory aerosol species (including sulfate, nitrate, chloride, ammonium and organics) and black carbon (BC) were measured using an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; DeCarlo et al., 2006) and a seven-wavelength aethalometer (model AE33; Magee Scientific Corp., Drinovec et al., 2015), respectively, which provided aerosol composition data at 5 min resolution. Total aerosol extinction and aerosol absorption were measured simultaneously at 870 nm using a photoacoustic extinctiometer (PAX; Droplet Measurement Technologies, Boulder, CO, USA), with data reported for dry aerosol (approximately 40 % relative humidity) at 5 min resolution. The PAX, HR-ToF-AMS and aethalometer were all located on the roof of a three-floor laboratory building at the tower site.
Vertical profiles of aerosol extinction were obtained using a dual-wavelength (1064 and 532 nm) depolarization lidar, which was located at a height of 28 m on the roof of a building near the aerosol-monitoring equipment. The lidar provided extinction coefficients attributed to anthropogenic aerosols at 30 m vertical resolution up to an altitude of 6 km and 15 min temporal resolution for both campaign periods. Further details of the lidar instrument, calibration procedures and attribution of extinction coefficients to anthropogenic aerosol using the depolarization ratio can be found in Yang et al. (2010, 2017) and Sugimoto et al. (2002).
The spectrally resolved (≈1 nm) actinic flux was measured during both campaign periods at a temporal frequency of 1 min using a spectrometer (Ocean Optics QE65000), which was fibre-coupled to a 2pi quartz receiver optic (Meteorologie Consult GmbH). J[O1D] and J[NO2] rates were then calculated using literature values of wavelength-dependent photodissociation quantum yields and absorption cross sections (Whalley et al., 2018). The instrument provided measurements representative of a height of 3.5 m above ground level.
Finally, a single particle soot photometer (SP2) instrument was used to measure the physical properties of individual black carbon (BC) particles during each campaign period. Described in detail by Liu et al. (2010, 2014), the SP2 employs a laser at 1064 nm to detect the optical properties of BC particles, including core diameter and coating thickness for each single particle. The core size and coating information for BC covering both the winter and summer campaign periods is presented in detail in Liu et al. (2019).
2.3 Chemical apportionment of aerosol extinction
To estimate the vertical extinction profile for each species, it is necessary to chemically apportion the total aerosol extinction measured with the lidar in the 532 nm channel. The co-located PAX, HR-ToF-AMS and aethalometer data are used here to develop an empirical relationship between aerosol composition and optical properties.
Based on the assumption that aerosol particles are externally mixed and that the extinctions of individual species are independent of one another, the contribution of the non-refractory aerosol species to the total extinction coefficient (bext) is estimated at 870 nm. A differential evolution optimization algorithm is utilized on the scattering coefficient (bsct) and the respective concentrations of ammonium sulfate ((NH4)2SO4), ammonium nitrate (NH4NO3), ammonium chloride (NH4Cl) and organic aerosol (OA). The concentrations of the inorganic aerosol components are calculated from the measurements of sulfate, nitrate and chloride, assuming that these ions are neutralized by ammonium.
The scattering coefficient bsct is assumed to be the difference between bext and the absorption coefficient (babs) measured by the PAX instrument. As the algorithm is applied to lidar measurements in ambient conditions, the effects of aerosol hygroscopic growth needs to be accounted for through use of the humidity-dependent growth factor (fRH). This work uses the fRH factor from the Interagency Monitoring of Protected Visual Environments (IMPROVE) algorithm (Pitchford et al., 2007), which has previously been used to chemically apportion PM extinction over China (Shen et al., 2014). As in the IMPROVE algorithm, the effects of hygroscopic growth are only allowed to impact the inorganic ions. The differential algorithm is calibrated using an optimization approach to minimize the mean absolute error between the observed scatter (bsct) and the value estimated with the differential evolution algorithm. This allows the mass scattering efficiency (MSE) for each species to be estimated.
For the contribution of BC to overall extinction, it is assumed that all absorption from the PAX measurements can be attributed to BC, following the approach of Han et al. (2015). A simple linear regression model is fitted between the absorption coefficient (babs) and the measured BC mass concentration. The slope of this regression yields the mass absorption efficiency (MAE) for BC. Combining the calculated MSE and MAE values for each PM constituent gives the following empirical relationships for the winter (Eq. 1) and summer (Eq. 2) campaigns. Here the values in square brackets represent the concentrations of each PM constituent (µg m−3), the numbers represent the optimized MSE and MAE values (m2 g−1), and fRH represents the hygroscopic growth factor.
An optimization approach is then used to estimate the contribution of each species to the measured lidar extinction at each 30 m layer (up to a height of 6 km) given the empirical relationships derived in Eqs. 1 and 2. In this case the assumption is made that the MSE and MAE values for each species hold with height. Relative humidity is obtained for the column from the European Centre for Medium-range Weather Forecasts (ECMWF) ERA5 dataset (ECMWF, 2018) and is mapped onto the lidar height levels in order to estimate the change in the fRH with height and thus the effects of hygroscopic growth on the inorganic aerosol species. As the lidar operates at 532 nm and the PAX optical properties are measured at 870 nm, the lidar extinction is scaled to the PAX wavelength using the Angstrom exponent from a nearby Aerosol Robotic Network (AERONET) station, Beijing-CAMS (Chinese Academy of Meterological Sciences) before the optimization. Overall, this provides vertical profiles of extinction attributed to each aerosol species, which are then converted to aerosol optical depth (AOD) by integrating over each 30 m layer.
2.4 Model description and setup
Fast-JX is an interactive photolysis scheme designed to efficiently and accurately calculate photolysis rates for use in global atmospheric models at minimal computational cost (Wild et al., 2000; Bian and Prather, 2002; Neu et al., 2007; Prather, 2015). The scheme apportions light from wavelengths 177 to 850 nm into 18 bins to permit calculation of photolysis rates appropriate to both tropospheric and stratospheric chemistry. Cloud and aerosol optical depths are used along with the scattering-phase functions for appropriate particle types to solve the eight-stream multiple scattering problem (Wild et al., 2000). This allows calculation of the photolytic intensity, which can be used to determine photolysis rate coefficients for key atmospheric species (e.g. J[O1D] and J[NO2]). The scheme also utilizes a quadrature approach to average over multiple layers of overlapping clouds (Neu et al., 2007; Prather, 2015).
The Fast-JX scheme is run here in “stand-alone” mode using offline data rather than interactively within a chemical transport model (CTM) framework. This allows it to be constrained using observations where these are available. In this study, the optical depths for aerosols from the lidar chemical apportionment are used in conjunction with data on cloud-based (cloud fraction, liquid water content and ice water content) and meteorological variables (temperature and relative humidity) from the ERA5 reanalysis dataset (ECMWF, 2018). Cloud cover from each ERA5 layer is used to drive Fast-JX using the quadrature approach described above for overlapping layers. Atmospheric columns of O3 and NO2 are provided from the ERA5 and the CAMS reanalysis datasets (CAMS, 2018; Inness et al., 2018), respectively, to account for absorption from gas-phase species. Due to high concentrations of NO2 in Beijing, particularly in the boundary layer, the standard version of the Fast-JX code was modified to account for the significant attenuation that can occur when photons are absorbed by NO2. In addition, the scattering-phase function for BC was updated to account for the ageing and coating of BC particles. This is done using the BC core size and coating thickness determined from SP2 measurements to derive asymmetry parameters at each Fast-JX wavelength. These asymmetry parameters were used to estimate the first eight terms of the scattering-phase function using a Legendre expansion of the Henyey–Greenstein function (Henyey and Greenstein, 1941).
In order to critically evaluate the impact of haze pollution events on photolysis rates during the winter and summer campaign periods, a range of different scenarios are run (Table 1). These include runs where the radiative effects of each key aerosol species and clouds are switched on in isolation. This enables the contributions of clouds and aerosols to be determined and for the contribution of key aerosol species to be identified, respectively. The scenario where both cloud and aerosol effects are turned on represents the best model simulation of photolysis rate constants during the campaigns and allows critical evaluation of the model against observed values of J[O1D] and J[NO2].
Table 1Model scenarios run during both campaign periods indicating whether the radiative effects of each aerosol species or cloud is turned on in the respective scenario.
3.1 Fast-JX evaluation
Observed J[O1D] and J[NO2] are captured well by the model during both the winter and summer periods, as shown in Fig. 1. In general, when the sun is highest in the sky at local noon, Fast-JX tends to capture observed J[O1D] within 8 % (+3.7 % bias in summer and +7.8 % bias in winter, averaged over all days), marginally higher than observations in both cases. For J[NO2] the model performance is much better during the summer campaign (+5.6 % bias) than the winter (+20.4 % bias). In the summer, on clear-sky days (28 May, 1, 7, 9, 14, 15 and 16 June) the model performs slightly less well for J[O1D] (+5.5 % average bias) and slightly better for J[NO2] (+4.7 % bias). In contrast, on winter clear-sky days (19, 22, 27 November and 1 December) the model performs better for J[O1D] (+2.7 % bias) but less well for J[NO2] (+23.1 % bias). These discrepancies may be linked to uncertainties in the retrieval of AOD from the lidar instrument and, therefore, to underestimation of the background aerosol on these less polluted days. In addition, errors in the column O3 from the ERA5 reanalysis could be responsible for the positive bias of model J[O1D]. However, the ERA5 column O3 was independently validated against Brewer measurements over Beijing, which indicated that the total column was captured well by the reanalysis product (mean bias of −2.0 % for summer and +3.4 % for winter), indicating that this is likely to be a smaller source of error. There are also issues with stray light during calibration of the spectrometer instrument at shorter wavelengths (below 300 nm) which may affect the fluxes derived in this part of the spectrum and thus the measured photolysis rates, particularly for J[O1D]. Overall, however, the model is shown to perform reasonably well, capturing the magnitude of the reductions in photolysis rates observed during severe haze episodes in both winter (16–18, 20 and 29 November) and summer periods (6, 22 and 23 June). This is particularly important during the summer where photochemistry is most active and large reductions in photolysis rates can have important impacts on oxidant concentrations and the production and destruction of key pollutants (e.g. O3 and secondary organic aerosol).
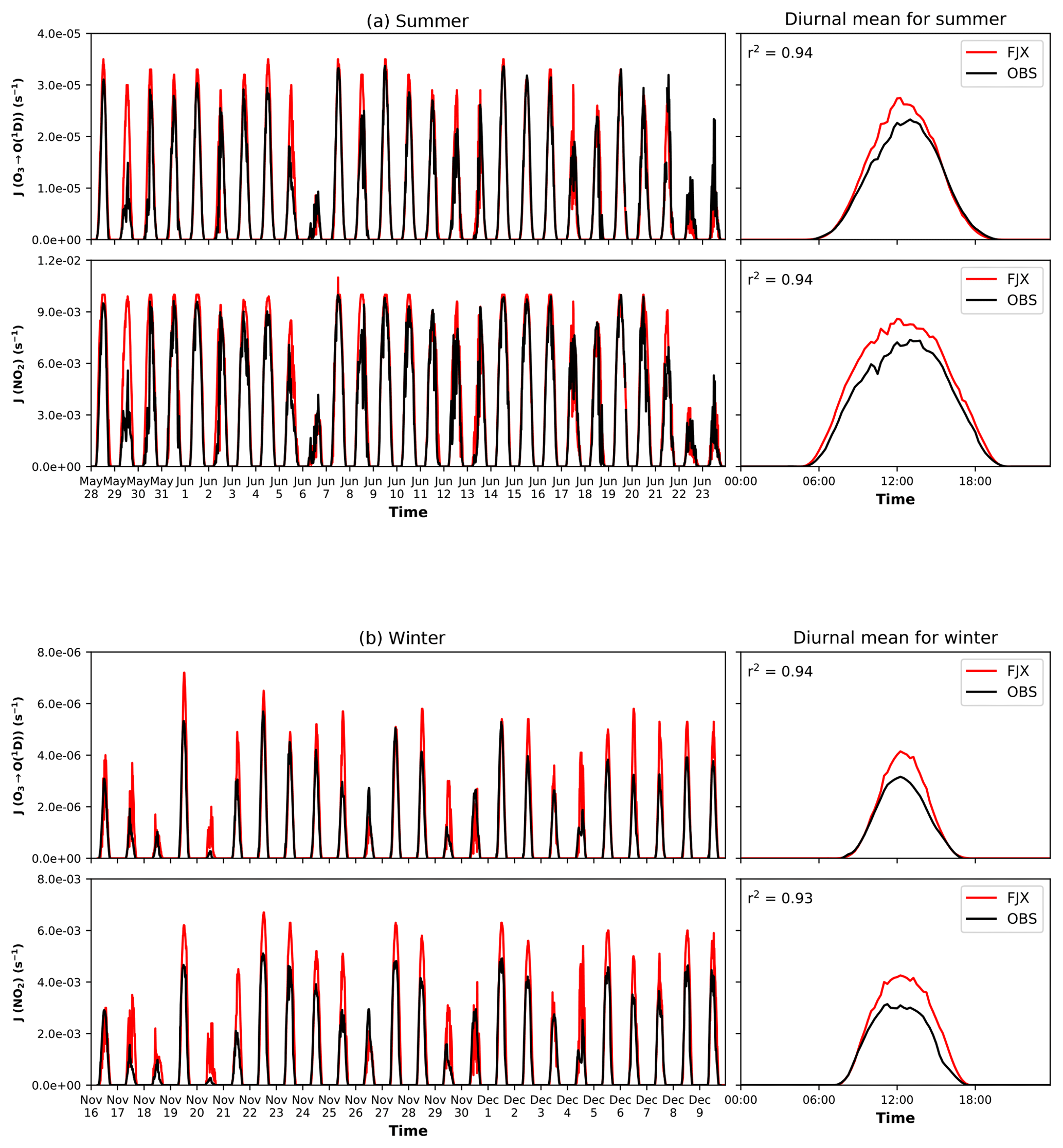
Figure 1Modelled vs. observed photolysis rate constants for the winter (a) and summer (b) campaign periods. Time series (left-hand plots) and campaign diurnal averages (right-hand plots) of J[O1D] and J[NO2] are shown for simulations with Fast-JX (FJX) including cloud and aerosol (red lines) and for observations. Note the difference in magnitude of rate constants between summer and winter. The r2 values for modelled vs. observed hourly photolysis rates are shown in the right-hand plots for reference.
3.2 Chemical apportionment of AOD
Figure 2 shows vertical profiles of extinction due to aerosol for each PM2.5 component for both campaigns, as derived from the optimization approach described in Sect. 2.3. Figure 2 also shows the contribution to total column AOD of each aerosol component. The differences between haze periods and non-haze periods are also highlighted. Haze is defined here as conditions where the PM2.5 concentration is larger than 75 µg m−3 (corresponding to an air quality index, AQI, of 100), which is the daily air quality limit for China (Shi et al., 2019). In winter, (NH4)2SO4 (39 %) and BC (31 %) provide the largest contributions to total column AOD during haze periods, with NH4NO3 and organic aerosol making the second largest contributions (13 % and 12 %, respectively) and NH4Cl making the smallest (5 %). Vertical aerosol extinction profiles show large peaks in both the boundary layer (below 1 km) and above 3 km where there is evidence of an elevated pollution layer (EPL). Elsewhere in the column extinction values are much lower; however, there are slightly elevated values at 1–2 km which are not as large as those in the boundary layer or EPL. This phenomenon has been demonstrated previously in stable conditions during haze episodes when the topography around Beijing can lead to the “mountain chimney effect”, where pollutants build up in elevated layers rather than disperse through the free troposphere (Chen et al., 2009; Liu et al., 2018). During cleaner winter periods (NH4)2SO4 (30 %) and BC (23 %) still dominate total column AOD, and NH4NO3 and organic aerosol make very similar contributions to those during haze periods (13 % and 14 %). However, in contrast to haze periods, NH4Cl makes the third biggest contribution at 20 %. As these cleaner periods are characterized by lower levels of particulates and more unstable conditions, AOD values are in general much lower than in hazy conditions and the largest values are seen within the boundary layer.
During the summer campaign, in both haze and cleaner periods, organic matter dominates the contribution to total column AOD (38 % and 44 %, respectively) with (NH4)2SO4 providing the second largest contribution (21 % and 16 %, respectively). BC is shown to contribute less to AOD during haze days (7 %) than in cleaner periods (13 %). In contrast to the winter, the highest AOD values in hazy periods are seen in an EPL, which lies between 3 and 5 km and is dominated by BC and organic matter, although all other species are also elevated. Although haze conditions occur much less frequently in the summer campaign, the extinction values for BC and organic matter at these higher levels are comparable to those seen in the winter months, with peaks of around 60 Mm−1. During the summer campaign, all species except organic matter show much lower AOD values during the cleaner periods than during haze, and there is little indication of an EPL layer. There is substantial organic matter still present in these cleaner periods, and this is maximum at 2–3 km where AOD values peak at around 20 Mm−1 (higher than during cleaner periods in the winter campaign). Finally, to check consistency, the total column optical depth from the lidar was compared to AERONET values at a nearby site (Beijing CAMS) showing good agreement for both campaigns.
3.3 Impacts of aerosol species on photolysis rates in haze conditions
As Fast-JX is run in offline mode, the effects of each aerosol species can be determined independently (Table 1) allowing the change in photolysis rates with respect to clear-sky conditions to be quantified (Fig. 3). During winter, absorption by BC has the greatest impact on both J[O1D] and J[NO2], resulting in 24 % and 23 % reductions in the lowest 1 km, respectively, compared to clear-sky conditions. The impact of BC reduces with height but continues to show the largest response up to 4 km for J[O1D] (−6 %) and 3 km for J[NO2] (−6 %). At higher altitudes the effects of scattering aerosol begin to dominate, with a general enhancement in photolysis rates compared to clear-sky conditions of 1 %–4 % for J[O1D] and 2–7 % for J[NO2]. The most pronounced effects are seen towards the top of the lidar column (6 km), where there is substantial backscattered solar radiation from the polluted boundary layer below. The largest increases are due to (NH4)2SO4 (4 % for J[O1D] and 7 % for J[NO2]), which corresponds to the presence of large amounts of (NH4)2SO4 in the EPL during haze periods in winter (Fig. 2). In the surface layer, within the elevated levels of aerosol, the scattering aerosol leads to reductions of −2 % to −4 % for J[O1D] and −3 % to −7 % for J[NO2], with OA producing the largest reduction in both cases.

Figure 2Contributions to total column AOD (a, b, c, d) and vertical profiles of extinction (e, f, g, h) for each aerosol component as derived from the optimization approach. Data are presented for haze and non-haze periods (see text for definition) in both campaigns from the surface up to 6 km altitude.
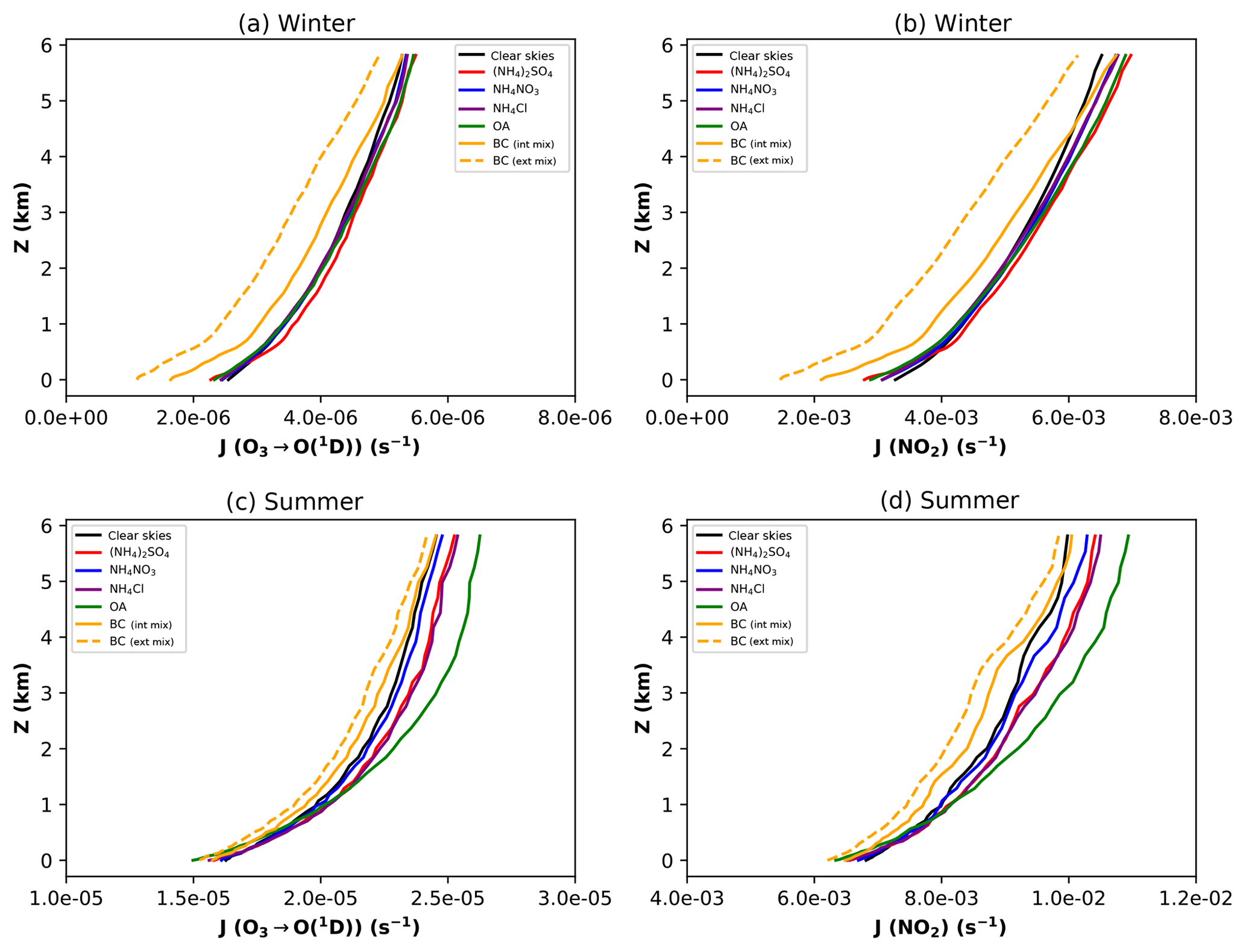
Figure 3Vertical profiles of J[O1D] (a, c) and J[NO2] (b, d) for simulations where the radiative effects of each aerosol component are included in isolation. Profiles are shown for the winter (a, b) and summer campaigns (c, d) and show impacts of OA (solid green), (NH4)2SO4 (solid red), NH4NO3 (solid blue), NH4Cl (solid purple) and coated BC (solid orange). Photolysis rate constants under clear-sky conditions (solid black line) are shown for reference. The radiative impacts of BC when it is assumed to be externally mixed rather than coated is shown for comparison (dashed orange line).
In contrast, during the summer, scattering by OA dominates the response of both J[O1D] and J[NO2]. At the surface, OA leads to a 3 % reduction in J[O1D] and a 2 % reduction in J[NO2]. Higher up the column, within and above the EPL layer, scattering aerosol leads to increases in photolysis for both species, with the effects of OA producing the dominant response (4 %–8 % for J[O1D] and 4 %–10 % for J[NO2]). This represents the high backscatter from the level of maximum OA, which occurs at a height of approximately 4 km (Fig. 2). Overall, the relative impacts of the scattering aerosol compared with clear-sky conditions are similar in winter and summer, but due to the higher rates of photolysis in summer the absolute impacts are much larger. Absorption by BC is still evident during the summer, leading to reductions in photolysis of 0 %–2 % for J[O1D] and 1 %–4 % for J[NO2]. However, in contrast to winter, the impacts of BC are lower than those of OA in all layers with the exception of J[NO2] in the surface layer.
3.4 Diurnal impacts of aerosols on photolysis rates
Figure 4 shows the impacts of all aerosol on the vertical profiles of J[O1D] and J[NO2] for each daylight hour averaged over each campaign period. During the winter, the strong effect of absorption by BC is evident for both J[O1D] and J[NO2], with reductions in excess of 20 % in the lowest 500 m and reaching maximum reductions of 34 % ( s−1) for J[O1D] and 40 % ( s−1) for J[NO2]. Reductions in J[NO2] show a stronger diurnal pattern over the day, with the greatest effects around sunrise, where reductions of at least s−1 are seen up to an altitude of 5 km. This is when the atmospheric path is at its longest and absorbing species such as BC have the greatest effect. Approaching noon, the effects of scattering aerosol start to dominate down through the column and this results in increases in J[NO2] to a maximum of 10.5 % ( s−1) at 6 km altitude. For J[O1D] the effects of scattering are less pronounced and absorption continues to dominate below 3 km. The effects of scattering are more evident above this level, and J[O1D] is enhanced by around 6 % ( s−1) in the middle of the day.
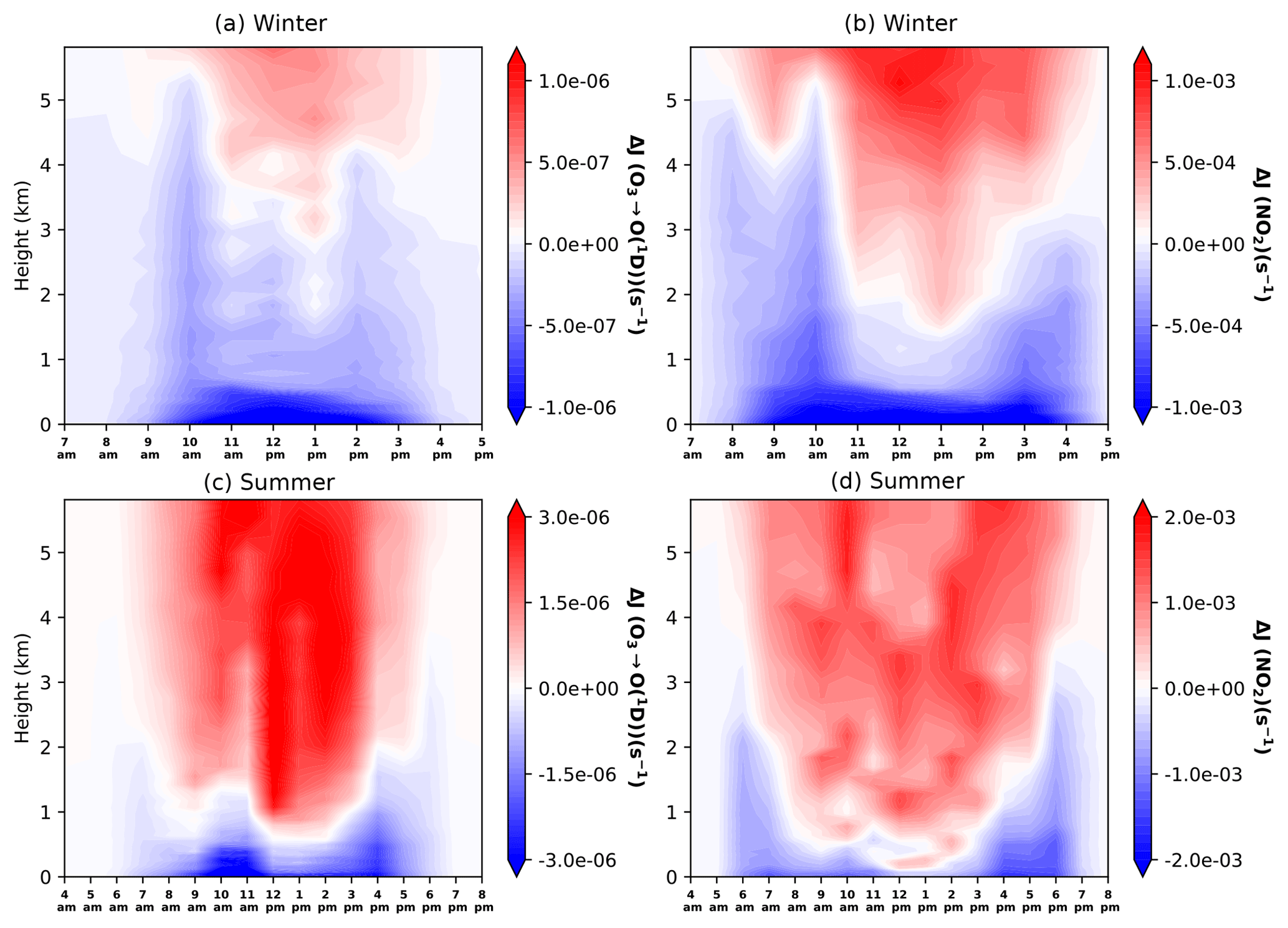
Figure 4Diurnal profiles of aerosol impacts on J[O1D] (a, c) and J[NO2] (b, d) for winter (a, b) and summer (c, d) campaign periods. Plots show the mean absolute difference with respect to clear-sky conditions, with blue representing a reduction due to aerosols and red representing an increase. Note the difference in timescales between seasons, which reflects longer daylight hours in summer.
During the summer, the responses of J[O1D] and J[NO2] are similar, with the absorption effect of BC evident in a very shallow surface layer about 200–300 m deep. The exception to this is around sunrise and sunset where reductions in photolysis rates extend to altitudes of 2–3 km. In this layer, reductions in photolysis rates for both species are in excess of 20 %, and these are most pronounced for J[NO2], where reductions reach 66 % ( s−1). The effects of scattering aerosol are more pronounced during the summer than the winter and are seen much lower in the column, particularly during the middle of the day. The impacts on J[NO2] are slightly greater than for J[O1D], with increases of 10–15 % (1.0 to s−1) compared with 5–10 % (1.5 to s−1) at 1–6 km altitude. Although these increases are not as large as the reductions seen in the boundary layer, they have the potential to have significant impacts on chemical processes through their influence on oxidant concentrations. This can have further impacts on lifetimes of other pollutants in the free troposphere. These findings allow quantification of the full impacts of haze pollution throughout the lower troposphere and highlight the non-local impacts of aerosols. Therefore, aerosols, whose origins are mainly from the surface (within the boundary layer), not only impact visibility and photolysis at the surface but can also have significant impacts at altitude. This extends findings of previous studies that largely focus on the impacts of haze pollution on surface photolysis rates and not those throughout the free troposphere (Li et al., 2005; Xing et al., 2017).
3.5 Cloud vs. aerosol impacts during campaign periods
The average impacts of clouds and aerosol on J[O1D] and J[NO2] for haze days are shown for both campaigns in Fig. 5. During the winter, aerosols produce reductions in both photolysis rates of more than 30 % at the surface. The impact of clouds in this lowest layer is much smaller, with reductions of around 7 % for both species. At higher altitudes, the effects of aerosol are less dominant and backscatter from clouds is more evident, leading to increases in photolysis rates for both species. The largest increases are seen in the 1–2 km layer where photolysis rates increase by 46 %–58 % with the impacts for J[NO2] slightly larger than for J[O1D]. The effects of cloud backscatter are smaller higher up the column, and increases of 17 %–32 % are seen in the 5–6 km layer, with largest impacts again seen for J[NO2]. This pattern is reflected in the combined impact of cloud and aerosol, which shows 33 %–34 % reductions in the surface layer, 32 %–42 % increases in the 1–2 km layer and smaller 20 %–39 % increases in the 5–6 km layer.
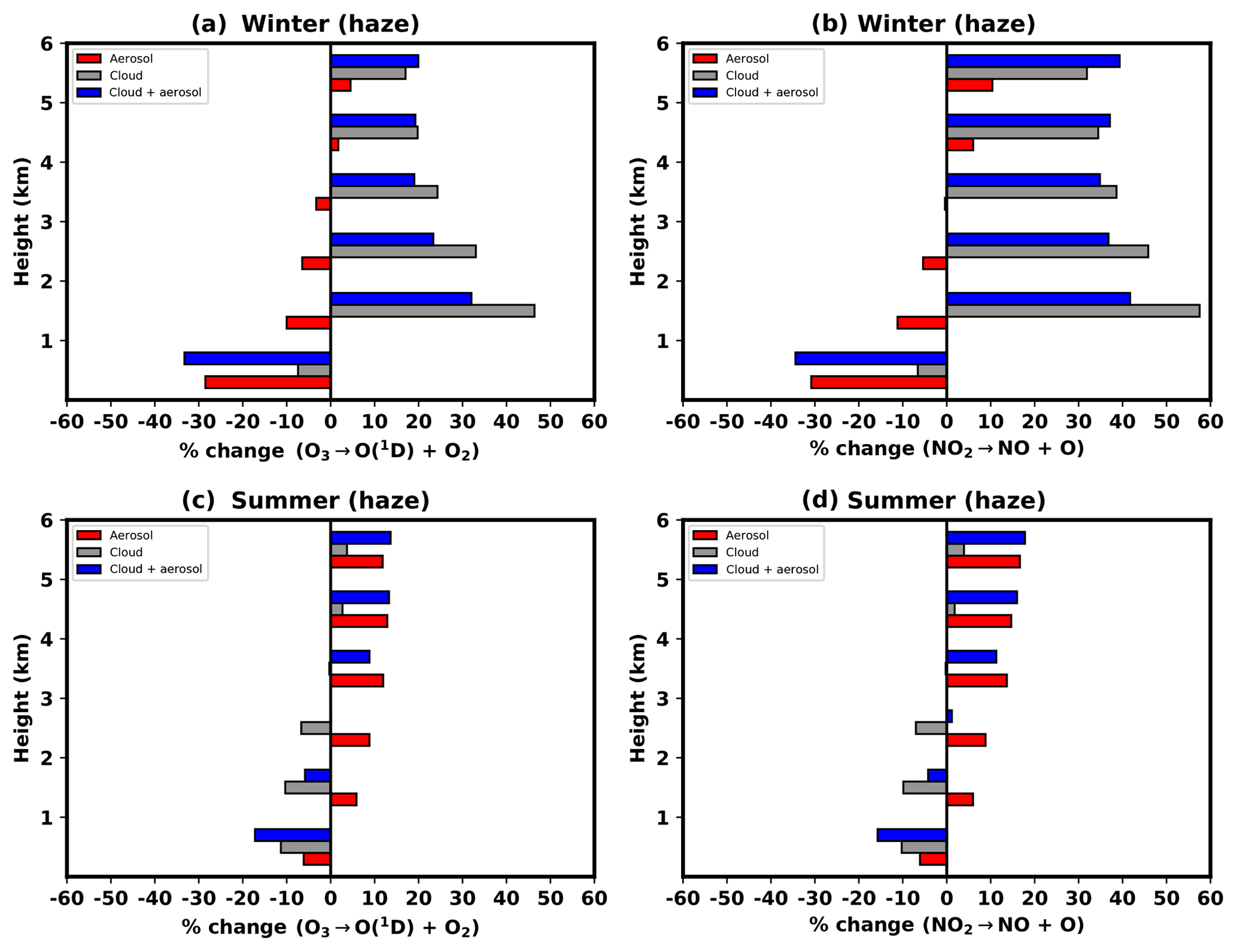
Figure 5Mean relative impacts of aerosols (red bars), clouds (grey bars), and combined cloud and aerosol (blue bars) on J[O1D] (a, c) and J[NO2] (b, d) during the winter (a, b) and summer (c, d) campaign periods. The relative differences are with respect to clear-sky conditions and are calculated as the average for periods classified as haze in 1 km layers from the surface to 6 km.
In summer, during haze conditions, clouds produce the largest impacts on photolysis rates in the surface layer (reductions of 10 %–11 %), approximately double that attributed to aerosol (≈6 %). The effects of high levels of scattering aerosol (particularly OA) during these conditions are evident higher in the column, where increases in photolysis rates due to aerosol are much larger than those from clouds. The combined effects of cloud and aerosol are reductions in the lowest 3 km (0 %–17 % for J[O1D] and 1 %–16 % J[NO2]), which are dominated by the influence of clouds. Between 3 and 6 km, the effects of backscatter from aerosol are greater than those from clouds giving net increases in photolysis rates (9 %–14 % for J[O1D] and 11 %–18 % J[NO2]).
When the impacts are averaged over all days in each campaign period (not shown), an interesting picture emerges. In the winter, aerosol produce the largest impacts in the surface layer (−16 % for J[O1D] and −17 % J[NO2]), with the effects of cloud more dominant at higher altitudes (15 %–26 % for J[O1D] and 26 %–35 % J[NO2]). This shows that background levels of aerosol during the winter, even at levels below that classified as haze, are sufficiently high to produce a greater impact on photolysis than clouds. As shown in Fig. 2, a polluted layer consisting mainly of BC and (NH4)2SO4 is present in the lowest 1 km even on non-haze days, and this most likely contributes to the dominant effect of aerosol at the surface. For the summer campaign, averaged over all days, cloud impacts dominate over aerosol impacts throughout the column for both species, consistent with the findings of Hall et al. (2018). However, the presence of an elevated layer of OA (Fig. 2) at 2–3 km is evident on the non-haze days where aerosol and clouds produce comparable increases in photolysis rates (5 % for J[O1D] and 7 % for J[NO2]).
This study presents an in-depth investigation into the impacts of haze pollution on photolysis rates during two intensive field campaigns in Beijing. For the first time, an observation-driven approach is used to quantify how different aerosol species contribute to changes in photolysis rates and to explore how they influence photochemistry during haze events in a megacity.
On haze days, in the winter and summer campaign periods, aerosols show distinct and contrasting impacts on J[O1D] and J[NO2]. The effects of absorbing species such as BC dominate during the winter, leading to large reductions in J[O1D] and J[NO2] in the lowest 1 km of 24 % and 23 %, respectively. During the summer, scattering aerosol dominates, with OA producing the largest response throughout the column, leading to reductions of around 3 % in the lowest 1 km and increases of around 10 % at 3–4 km. These differences largely reflect the different pollution sources during the campaign periods with high levels of coal burning during the winter season, leading to large emissions of soot (BC) and high production of (NH4)2SO4 particles. During the summer, emissions of biogenic volatile organic compounds (BVOCs) are much larger, leading to formation of secondary organic aerosol (Riipinen et al., 2011; Mentel et al., 2013), which can account for the dominance of OA in this season.
To test the sensitivity of the results to assumptions about the mixing state of aerosol, an additional simulation was performed treating BC as externally mixed rather than internally mixed, following Liao et al. (1999). With externally mixed BC, the effects of the absorbing species still dominate the response of photolysis rates during the winter campaign; see Fig. 3. However, the magnitudes of reductions in the lowest 1 km for both J[O1D] and J[NO2] is larger (44 % and 45 %, respectively) than when the BC is assumed to be coated (24 % and 23 %, respectively). In the summer, the effect of externally mixed BC leads to a slightly larger contribution from BC to reductions in J[O1D] and J[NO2] throughout the column, although OA still provides the dominant response for both species (Fig. 3).
It is also demonstrated that despite particulate levels being lower on average during the summer months, haze events where the AQI is higher than 100 still occur. On haze days, the mean relative impact of aerosols on photolysis rates is lower in summer than in winter, with reductions in J[O1D] of just 6 % in the surface layer compared to 29 % in winter. However, summer J[O1D] values in this layer are an order of magnitude higher than in winter ( s−1 compared to s−1) and so the absolute changes are higher in summer ( s−1 compared to s−1). This is important, as incoming solar radiation is at its highest during the summer months, which results in higher rates of O3 formation (Tie and Cao, 2009), as discussed in Sect. 4.1.
The diurnal pattern of haze impacts on photolysis is shown to differ between winter and summer (Fig. 4), with the reductions in J[O1D] and J[NO2] extending further up through the column during winter. This reflects the much deeper haze layers at the surface throughout the winter (Fig. 2). In the summer months, during the middle part of the day, significant reductions in photolysis occur near the surface but at higher altitudes aerosols result in increases of between 6 % and 16 % depending on the species. Therefore, any reductions in aerosol concentrations during the summer months, and thus reduction in this scattering effect (particularly from OA), would reduce J[O1D] and J[NO2] higher up in the column. As discussed above, lower values of J[O1D] and J[NO2] could reduce O3 and OH concentrations and thus potentially have important implications for the oxidation capacity in the free troposphere.
Previous work has shown that in air masses influenced by urban pollution, the impacts of aerosol on photolysis rates are greater than the influence of cloud cover (Tang et al., 2003). This study shows that during moderate to severe haze events in Beijing, which occur in both summer and winter, the response of photolysis to aerosol often dominates over cloud. In the winter, absorbing aerosol dominates over cloud at the surface, leading to 29 %–31 % reductions in photolysis rates compared to just 7 % reductions from the presence of clouds. In the summer, cloud impacts dominate near the surface with the scattering effect from an elevated layer of predominantly OA (Fig. 2) taking over at around 3–4 km altitude, where photolysis rates are increased by 12 %–14 % compared to very minor decreases from cloud (0.2 %–0.3 %). These patterns are in agreement with previous studies, which also demonstrate dominant responses to aerosol in urban air plumes and that the presence of a cloud layer acts to accentuate the reduction in surface photolysis rates for the two reactions considered here (Liao et al., 1999; Tang et al., 2003). Furthermore, in winter, when averaged over all days in the campaign period, the aerosol effects (16 %–17 % reductions) are roughly double the cloud impacts (8 % reductions) in the surface layer. This highlights that despite the very episodic nature of haze events in Beijing (Fig. 1), background aerosol concentrations are sufficient at the surface to produce strong reductions in photolysis rate constants. This is likely due to the high AOD values attributed to BC and (NH4)2SO4 on the cleaner days (Fig. 2). It should be noted that this response is representative for the winter of 2016 only and these effects of cloud and aerosol species are likely to vary greatly between different winter seasons. The results presented in this study, however, emphasize the large impact particulate pollution can have on photochemistry.
The results presented here are at the lower end of previous estimates of the impacts of aerosol on photolysis rates (Liao et al., 1999; Tang et al., 2003; Li et al., 2011). For example, Tang et al. (2003) estimate 29 %–39 % reductions in J[O1D] in the lowest 3 km, whereas Li et al. (2011) present reductions of 37 %–53 %. This study estimates 5 %–31 % reductions in the winter and 6 % reductions to 9 % increases in the lowest 3 km in the summer. These studies either use limited aircraft data AOD (Tang et al., 2003) or total column AOD data from a combination of satellite and ground-based instruments (Li et al., 2011) to verify the aerosol fields, and thus were unable to fully verify the accuracy of the vertical distributions of different aerosol species throughout the column. The results presented in this work greatly improve on these earlier estimates by using a combined statistical and observation-driven approach to derive chemically apportioned aerosol extinction from lidar data. This allows more accurate constraint of the vertical distribution of aerosol to be applied, including which species provide the largest contribution to extinction throughout the column. As discussed above, this results in layers of haze near to the surface (≈3 km in winter and ≈1 km in summer) dominated by absorbing species in the winter (BC) and scattering species in the summer (OA). This leads to large reductions in J[O1D] and J[NO2] at the surface and significant increases at altitude during both campaigns. These increases are greater during the summer due to the backscatter from the layer of OA present at the surface (Fig. 2). This accounts for the difference in patterns observed between this study and the previous estimates by Tang et al. (2003) and Li et al. (2011) and highlights the sensitivity of the responses in photolysis rates not only to the vertical distribution of aerosol but also the chemical speciation of the particulates. The observation-constrained approach presented here provides the best possible estimate of both, allowing more accurate quantification of the impacts of haze pollution on photolysis rates. Running the Fast-JX code in “offline” mode also enables the relative impacts of clouds and different aerosol species to be quantified independently. This allows a critical evaluation of whether clouds or a particular aerosol species are causing the greatest photochemical impact during a specific pollution episode, providing insight into where to target particulate matter control strategies.

Figure 6Diurnal profiles of aerosol impacts on O3 (a, c) and OH (b, d) concentrations for winter (a, b) and summer (c, d) campaign periods. These changes are calculated using a simple chemical box model, and plots show the mean relative difference with respect to clear-sky conditions, with blue representing a reduction due to aerosols and red representing an increase.
4.1 Photochemical impacts of haze pollution and implications for pollution control strategies
The large reductions in surface J[O1D] and J[NO2] during haze events could be contributing to lower O3 than in cleaner periods, particularly in the summer months. Therefore, reduction in aerosols alone could enhance surface O3 pollution. As demonstrated by Li et al. (2011), the probability of O3 peaks greater than 120 ppbv increased dramatically with the removal of aerosol from their simulations, and the response of OH concentrations was shown to be roughly proportional to changes in J[O1D]. Therefore, under the recent reductions in PM2.5 in Beijing through emissions controls implemented as part of China’s clean-air plans (Wang et al., 2015; Liu et al., 2016), the resultant increases in J[O1D] could lead to enhanced O3 concentrations, where summer levels are already very high (Wang et al., 2006; Xue et al., 2014; Ni et al., 2018). Enhanced near-surface photolysis rates would also increase NO2 photolysis and enhance levels of NO, which subsequently react with RO2, leading to a rise in O3 production. This rise in O3 will be partially balanced by the reaction of NO with O3 itself. Furthermore, the enhancement of J[O1D] would increase OH concentrations, which would subsequently increase HO2 and RO2 and lead to a net rise in O3 concentrations. In the winter, a similar response would be expected but would be smaller in magnitude due to lower photolysis rates and much lower O3 concentrations.
To evaluate the potential photochemical impact on oxidants, a simple experiment was performed using a photochemical box model, incorporating the generic reaction set for volatile organic compound oxidation (Topping et al., 2018). The response of O3 concentrations to the presence of aerosols is similar to that of J[O1D] in both seasons with the biggest reductions (12 % or larger) seen in the lowest 500 m; see Fig. 6. These reductions are more pronounced throughout the day in the winter than in the summer months. The effect of scattering aerosol can be seen further up the column, where enhanced photolysis rates due to aerosol result in increases in O3 concentrations of 3 %–6 %. As with the photolysis rates, the largest impacts are seen in the middle of the day and lower down the column during the summer. The impacts on OH largely match those on O3, but the responses are much smaller, with 0 %–3 % decrease at the surface in winter and a similar magnitude increase higher up the column in the summer.
This suggests that if aerosols were not present surface O3 and OH concentrations would be around 12 % and 3 % higher, respectively. In contrast, removal of aerosols would see reductions in O3 higher up the column, particularly in the summer, due to the removal of scattering aerosol and the resultant reductions in J[O1D] and J[NO2]. In the winter, when O3 is much lower, this may have little impact, but during the summer months when daytime concentrations can exceed 100 ppbv, this could see a shift towards more frequent O3 pollution episodes. It should be noted that the photolysis of HONO is not included in the simple box model used here. HONO photolysis occurs at similar wavelengths to that of NO2 and is an important source of OH in urban environments. Therefore, chemical impacts simulated here, particularly for OH, may be underestimated. A more detailed treatment of gas-phase and heterogeneous chemistry in a box model or regional air quality model is needed to fully quantify the impact of these photolysis rate changes on urban oxidant concentrations under haze conditions.
This study presents, for the first time, the application of aerosol composition data and lidar extinction profiles to drive the Fast-JX photolysis model in order to quantify the effects of severe haze pollution on J[O1D] and J[NO2] over Beijing during two intensive measurement campaign periods (winter 2016 and summer 2017). The model is shown to capture observed J[O1D] and J[NO2] well during both campaigns, particularly for the reductions that occur during haze events (AQI >100). Such episodes occur during both campaign periods and lead to reductions in surface J[O1D] of 27 % (summer) to 34 % (winter) and reductions of 40 % (winter) to 66 % (summer) in J[NO2]. Despite much lower particulate concentrations in the summer campaign, the absolute changes in photolysis rates are shown to be larger than in winter for both O3 and NO2.
In the winter, absorbing aerosols such as BC dominate the photolysis response to aerosol, leading to mean reductions of 24 % and 23 %, respectively, for J[O1D] and J[NO2] in the lowest 1 km. In contrast, in the summer, scattering aerosols such as OA dominate the response, leading to mean decreases of 2 %–4 % at the surface and increases of 8 %–10 % at higher altitudes (3–4 km). During haze episodes, these effects often dominate over those attributed to cloud cover. This emphasizes that in heavily polluted urban environments such as Beijing, aerosols are present in such large concentrations that they produce greater impacts on photolysis than naturally occurring phenomena such as clouds and are likely to have other important chemical and dynamical impacts on the urban environment.
The observation-driven approach to deriving the aerosol vertical distribution allows a more accurate constraint to be placed on the estimated impacts of haze pollution on photochemistry and, more critically, allows species-specific impacts to be highlighted. This allows the identification of source sectors to target particulate control strategies on. For example, during the APHH winter campaign, sources of BC (e.g. power generation, residential heating) contribute the most to photochemical impacts during haze events. Such strategies would not only reduce particulate matter concentrations but also reduce their impacts on photolysis rates and thus potentially increase surface ozone concentrations. Furthermore, the non local impact of potential emission and pollution control strategies is emphasized. Emissions cuts implemented at the surface not only produce large impacts in surface oxidant concentrations but also result in significant impacts in the free troposphere.
Using an idealized photochemical box model, it is shown that complete removal of particulates would enhance surface O3 concentrations by 12 % and OH concentrations by 3 % during the APHH campaigns. In contrast, this would reduce O3 by 3 %–6 % in the free troposphere. However, any control policy is also likely to impact concentrations of other pollutants such as NOx and volatile organic compounds (VOCs) and, therefore, due to the complex non-linearities involved, the response of atmospheric oxidants is likely to vary dependent on the magnitude of the emissions changes. To fully quantify the effects of pollutant haze on the urban atmospheric oxidation capacity, a more detailed air quality model study is needed that fully incorporates the optical properties of urban aerosol and their impacts on photolysis rates demonstrated here, along with treatment of gas-phase and heterogeneous photochemistry and urban meteorology.
The data generated in this study are available through the CEDA data archive at https://catalogue.ceda.ac.uk/uuid/4a1d547929d44698b91e0d75d417220b (last access: 30 July 2019) (Hollaway and Wild, 2019). The processed single particle soot photometer (SP2) data used in this study are available through the CEDA data archive (https://catalogue.ceda.ac.uk/uuid/af3ccea589f9439e9e1f88c85d130965, last access: 8 July 2019) (Liu, 2017). The processed photolysis observations used in this study are available through the CEDA archive (https://catalogue.ceda.ac.uk/uuid/76b4ad364d71465d8f8b61e302eb2c4c, last access: 8 July 2019) (Whalley and Slater, 2018).
MH and OW conceived this study. MH performed the data analysis and ran model simulations. TY collected the lidar data at the IAP tower site. YS, WX and CX collected the aerosol composition data and optical properties measured at the tower site. LW, ES and DH provided the observed photolysis rates. DL collected the optical properties for black carbon with the SP2 instrument. MH and OW wrote the manuscript with input from all authors.
The authors declare that they have no conflict of interest.
This article is part of the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
The authors acknowledge funding from AIRPRO (An Integrated Study of Air Pollution PROcesses in Beijing), a Newton Innovation Fund project funded by the UK Natural Environment Research Council (grant nos. NE/N006925/1, NE/N006895/1 and NE/N007123/1), the Strategic Priority Research Program of the Chinese Academy of Sciences “Beautiful China” project (grant no. XDA19040203) and the Natural Science Foundation of China (grant no. 41571130034). ES acknowledges funding from the SPHERES NERC Doctoral Training Programme.
This research has been supported by the Natural Environment Research Council (grant nos. NE/N006925/1, NE/N006895/1 and NE/N007123/1), the Chinese Academy of Sciences (grant no. XDA19040203) and the Natural Science Foundation of China (grant no. 41571130034).
This paper was edited by Frank Keutsch and reviewed by two anonymous referees.
Bian, H. and Prather, M. J.: Fast-J2: Accurate Simulation of Stratospheric Photolysis in Global Chemical Models, J. Atmos. Chem., 41, 281–296, https://doi.org/10.1023/A:1014980619462, 2002. a
Bian, H., Prather, M. J., and Takemura, T.: Tropospheric aerosol impacts on trace gas budgets through photolysis, J. Geophys. Res.-Atmos., 108, 4242, https://doi.org/10.1029/2002JD002743, 2003. a
CAMS: CAMS Reanalysis data documentation, Copernicus Atmosphere Modelling Service (CAMS), available at: https://confluence.ecmwf.int/display/CKB/CAMS+Reanalysis+data+documentation, last access: 16 August 2018. a
Chan, C. K. and Yao, X.: Air pollution in mega cities in China, Atmos. Environ., 42, 1–42, https://doi.org/10.1016/j.atmosenv.2007.09.003, 2008. a
Chen, Y., Zhao, C., Zhang, Q., Deng, Z., Huang, M., and Ma, X.: Aircraft study of Mountain Chimney Effect of Beijing, China, J. Geophys. Res.-Atmos., 114, https://doi.org/10.1029/2008JD010610, 2009. a, b
Cheng, S. H., Yang, L. X., Zhou, X. H., Xue, L. K., Gao, X. M., Zhou, Y., and Wang, W. X.: Size-fractionated water-soluble ions, situ pH and water content in aerosol on hazy days and the influences on visibility impairment in Jinan, China, Atmos. Environ., 45, 4631–4640, https://doi.org/10.1016/j.atmosenv.2011.05.057, 2011. a
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006. a, b
Drinovec, L., Močnik, G., Zotter, P., Prévôt, A. S. H., Ruckstuhl, C., Coz, E., Rupakheti, M., Sciare, J., Müller, T., Wiedensohler, A., and Hansen, A. D. A.: The “dual-spot” Aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation, Atmos. Meas. Tech., 8, 1965–1979, https://doi.org/10.5194/amt-8-1965-2015, 2015. a, b
ECMWF: ERA5 data documentation. European Centre for Medium-range Weather Forecast (ECMWF), available at: https://confluence.ecmwf.int/display/CKB/ERA5+data+documentation, last access: 16 August 2018. a, b
Hall, S. R., Ullmann, K., Prather, M. J., Flynn, C. M., Murray, L. T., Fiore, A. M., Correa, G., Strode, S. A., Steenrod, S. D., Lamarque, J.-F., Guth, J., Josse, B., Flemming, J., Huijnen, V., Abraham, N. L., and Archibald, A. T.: Cloud impacts on photochemistry: building a climatology of photolysis rates from the Atmospheric Tomography mission, Atmos. Chem. Phys., 18, 16809–16828, https://doi.org/10.5194/acp-18-16809-2018, 2018. a
Han, T., Xu, W., Chen, C., Liu, X., Wang, Q., Li, J., Zhao, X., Du, W., Wang, Z., and Sun, Y.: Chemical apportionment of aerosol optical properties during the Asia-Pacific Economic Cooperation summit in Beijing, China, J. Geophys. Res.-Atmos., 120, 12281–12295, https://doi.org/10.1002/2015JD023918, 2015. a, b, c, d, e
Henyey, L. G. and Greenstein, J. L.: Diffuse radiation in the Galaxy, APJ, 93, 70–83, https://doi.org/10.1086/144246, 1941. a
Hollaway, M. and Wild, O.: APHH: Simulated photolysis rates using the Fast-JX model at the IAP-Beijing site during the winter and summer campaigns, Centre for Environmental Data Analysis, available at: http://catalogue.ceda.ac.uk/uuid/4a1d547929d44698b91e0d75d417220b, last access: 30 July 2019.
Hu, B., Zhao, X., Liu, H., Liu, Z., Song, T., Wang, Y., Tang, L., Xia, X., Tang, G., Ji, D., Wen, T., Wang, L., Sun, Y., and Xin, J.: Quantification of the impact of aerosol on broadband solar radiation in North China, Sci. Rep.-UK, 7, 44851, https://doi.org/10.1038/srep44851, 2017. a
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014. a
Inness, A., Ades, M., Agustí-Panareda, A., Barré, J., Benedictow, A., Blechschmidt, A.-M., Dominguez, J. J., Engelen, R., Eskes, H., Flemming, J., Huijnen, V., Jones, L., Kipling, Z., Massart, S., Parrington, M., Peuch, V.-H., Razinger, M., Remy, S., Schulz, M., and Suttie, M.: The CAMS reanalysis of atmospheric composition, Atmos. Chem. Phys., 19, 3515–3556, https://doi.org/10.5194/acp-19-3515-2019, 2019. a
Ji, D., Li, L., Wang, Y., Zhang, J., Cheng, M., Sun, Y., Liu, Z., Wang, L., Tang, G., Hu, B., Chao, N., Wen, T., and Miao, H.: The heaviest particulate air-pollution episodes occurred in northern China in January, 2013: Insights gained from observation, Atmos. Environ., 92, 546–556, https://doi.org/10.1016/j.atmosenv.2014.04.048, 2014. a
Kaiser, D. P. and Qian, Y.: Decreasing trends in sunshine duration over China for 1954–1998: Indication of increased haze pollution?, Geophys. Res. Lett., 29, 38–1–38–4, https://doi.org/10.1029/2002GL016057, 2002. a
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015. a
Li, G., Zhang, R., Fan, J., and Tie, X.: Impacts of black carbon aerosol on photolysis and ozone, J. Geophys. Res.-Atmos., 110, D23206, https://doi.org/10.1029/2005JD005898, 2005. a
Li, J., Wang, Z., Wang, X., Yamaji, K., Takigawa, M., Kanaya, Y., Pochanart, P., Liu, Y., Irie, H., Hu, B., Tanimoto, H., and Akimoto, H.: Impacts of aerosols on summertime tropospheric photolysis frequencies and photochemistry over Central Eastern China, Atmos. Environ., 45, 1817–1829, https://doi.org/10.1016/j.atmosenv.2011.01.016, 2011. a, b, c, d, e, f, g, h, i
Li, J., Fu, Q., Huo, J., Wang, D., Yang, W., Bian, Q., Duan, Y., Zhang, Y., Pan, J., Lin, Y., Huang, K., Bai, Z., Wang, S.-H., Fu, J. S., and Louie, P. K.: Tethered balloon-based black carbon profiles within the lower troposphere of Shanghai in the 2013 East China smog, Atmos. Environ., 123, 327–338, https://doi.org/10.1016/j.atmosenv.2015.08.096, 2015. a
Liao, H., Yung, Y. L., and Seinfeld, J. H.: Effects of aerosols on tropospheric photolysis rates in clear and cloudy atmospheres, J. Geophys. Res.-Atmos., 104, 23697–23707, https://doi.org/10.1029/1999JD900409, 1999. a, b, c, d
Liu, D.: APHH: Single Particle Soot Photometer measurements made at the IAP-Beijing site during the summer and winter campaigns, Centre for Environmental Data Analysis, available at: http://catalogue.ceda.ac.uk/uuid/af3ccea589f9439e9e1f88c85d130965 (last access: 8 July 2019), 2017.
Liu, D., Flynn, M., Gysel, M., Targino, A., Crawford, I., Bower, K., Choularton, T., Jurányi, Z., Steinbacher, M., Hüglin, C., Curtius, J., Kampus, M., Petzold, A., Weingartner, E., Baltensperger, U., and Coe, H.: Single particle characterization of black carbon aerosols at a tropospheric alpine site in Switzerland, Atmos. Chem. Phys., 10, 7389–7407, https://doi.org/10.5194/acp-10-7389-2010, 2010. a
Liu, D., Allan, J. D., Young, D. E., Coe, H., Beddows, D., Fleming, Z. L., Flynn, M. J., Gallagher, M. W., Harrison, R. M., Lee, J., Prevot, A. S. H., Taylor, J. W., Yin, J., Williams, P. I., and Zotter, P.: Size distribution, mixing state and source apportionment of black carbon aerosol in London during wintertime, Atmos. Chem. Phys., 14, 10061–10084, https://doi.org/10.5194/acp-14-10061-2014, 2014. a
Liu, D., Joshi, R., Wang, J., Yu, C., Allan, J. D., Coe, H., Flynn, M. J., Xie, C., Lee, J., Squires, F., Kotthaus, S., Grimmond, S., Ge, X., Sun, Y., and Fu, P.: Contrasting physical properties of black carbon in urban Beijing between winter and summer, Atmos. Chem. Phys., 19, 6749–6769, https://doi.org/10.5194/acp-19-6749-2019, 2019. a
Liu, F., Zhang, Q., van der A, R. J., Zheng, B., Tong, D., Yan, L., Zheng, Y., and He, K.: Recent reduction in NO x emissions over China: synthesis of satellite observations and emission inventories, Environmental Research Letters, 11, 114002, https://doi.org/10.1088/1748-9326/11/11/114002, 2016. a
Liu, Q., Ding, D., Huang, M., Tian, P., Zhao, D., Wang, F., Li, X., Bi, K., Sheng, J., Zhou, W., Liu, D., Huang, R., and Zhao, C.: A study of elevated pollution layer over the North China Plain using aircraft measurements, Atmos. Environ., 190, 188–194, https://doi.org/10.1016/j.atmosenv.2018.07.024, 2018. a
Lou, S., Liao, H., and Zhu, B.: Impacts of aerosols on surface-layer ozone concentrations in China through heterogeneous reactions and changes in photolysis rates, Atmos. Environ., 85, 123–138, https://doi.org/10.1016/j.atmosenv.2013.12.004, 2014. a
Martin, R. V., Jacob, D. J., Yantosca, R. M., Chin, M., and Ginoux, P.: Global and regional decreases in tropospheric oxidants from photochemical effects of aerosols, J. Geophys. Res.-Atmos., 108, 4097, https://doi.org/10.1029/2002JD002622, 2003. a
Mentel, Th. F., Kleist, E., Andres, S., Dal Maso, M., Hohaus, T., Kiendler-Scharr, A., Rudich, Y., Springer, M., Tillmann, R., Uerlings, R., Wahner, A., and Wildt, J.: Secondary aerosol formation from stress-induced biogenic emissions and possible climate feedbacks, Atmos. Chem. Phys., 13, 8755–8770, https://doi.org/10.5194/acp-13-8755-2013, 2013. a
Neu, J. L., Prather, M. J., and Penner, J. E.: Global atmospheric chemistry: Integrating over fractional cloud cover, J. Geophys. Res.-Atmos., 112, D11306, https://doi.org/10.1029/2006JD008007, 2007. a, b
Ni, R., Lin, J., Yan, Y., and Lin, W.: Foreign and domestic contributions to springtime ozone over China, Atmos. Chem. Phys., 18, 11447–11469, https://doi.org/10.5194/acp-18-11447-2018, 2018. a
Pitchford, M., Malm, W., Schichtel, B., Kumar, N., Lowenthal, D., and Hand, J.: Revised Algorithm for Estimating Light Extinction from IMPROVE Particle Speciation Data, JAPCA J. Air. Waste Ma., 57, 1326–1336, https://doi.org/10.3155/1047-3289.57.11.1326, 2007. a
Prather, M. J.: Photolysis rates in correlated overlapping cloud fields: Cloud-J 7.3c, Geosci. Model Dev., 8, 2587–2595, https://doi.org/10.5194/gmd-8-2587-2015, 2015. a, b
Ran, L., Deng, Z., Xu, X., Yan, P., Lin, W., Wang, Y., Tian, P., Wang, P., Pan, W., and Lu, D.: Vertical profiles of black carbon measured by a micro-aethalometer in summer in the North China Plain, Atmos. Chem. Phys., 16, 10441–10454, https://doi.org/10.5194/acp-16-10441-2016, 2016. a
Riipinen, I., Pierce, J. R., Yli-Juuti, T., Nieminen, T., Häkkinen, S., Ehn, M., Junninen, H., Lehtipalo, K., Petäjä, T., Slowik, J., Chang, R., Shantz, N. C., Abbatt, J., Leaitch, W. R., Kerminen, V.-M., Worsnop, D. R., Pandis, S. N., Donahue, N. M., and Kulmala, M.: Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 11, 3865–3878, https://doi.org/10.5194/acp-11-3865-2011, 2011. a
Shen, G., Xue, M., Yuan, S., Zhang, J., Zhao, Q., Li, B., Wu, H., and Ding, A.: Chemical compositions and reconstructed light extinction coefficients of particulate matter in a mega-city in the western Yangtze River Delta, China, Atmos. Environ., 83, 14–20, https://doi.org/10.1016/j.atmosenv.2013.10.055, 2014. a
Shi, Z., Vu, T., Kotthaus, S., Harrison, R. M., Grimmond, S., Yue, S., Zhu, T., Lee, J., Han, Y., Demuzere, M., Dunmore, R. E., Ren, L., Liu, D., Wang, Y., Wild, O., Allan, J., Acton, W. J., Barlow, J., Barratt, B., Beddows, D., Bloss, W. J., Calzolai, G., Carruthers, D., Carslaw, D. C., Chan, Q., Chatzidiakou, L., Chen, Y., Crilley, L., Coe, H., Dai, T., Doherty, R., Duan, F., Fu, P., Ge, B., Ge, M., Guan, D., Hamilton, J. F., He, K., Heal, M., Heard, D., Hewitt, C. N., Hollaway, M., Hu, M., Ji, D., Jiang, X., Jones, R., Kalberer, M., Kelly, F. J., Kramer, L., Langford, B., Lin, C., Lewis, A. C., Li, J., Li, W., Liu, H., Liu, J., Loh, M., Lu, K., Lucarelli, F., Mann, G., McFiggans, G., Miller, M. R., Mills, G., Monk, P., Nemitz, E., O'Connor, F., Ouyang, B., Palmer, P. I., Percival, C., Popoola, O., Reeves, C., Rickard, A. R., Shao, L., Shi, G., Spracklen, D., Stevenson, D., Sun, Y., Sun, Z., Tao, S., Tong, S., Wang, Q., Wang, W., Wang, X., Wang, X., Wang, Z., Wei, L., Whalley, L., Wu, X., Wu, Z., Xie, P., Yang, F., Zhang, Q., Zhang, Y., Zhang, Y., and Zheng, M.: Introduction to the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing)”, Atmos. Chem. Phys., 19, 7519–7546, https://doi.org/10.5194/acp-19-7519-2019, 2019. a, b, c
Sugimoto, N., Matsui, I., Shimizu, A., Uno, I., Asai, K., Endoh, T., and Nakajima, T.: Observation of dust and anthropogenic aerosol plumes in the Northwest Pacific with a two-wavelength polarization lidar on board the research vessel Mirai, Geophys. Res. Lett., 29, 7-1–7-4, https://doi.org/10.1029/2002GL015112, 2002. a
Sun, Y., Song, T., Tang, G., and Wang, Y.: The vertical distribution of PM2.5 and boundary-layer structure during summer haze in Beijing, Atmos. Environ., 74, 413–421, https://doi.org/10.1016/j.atmosenv.2013.03.011, 2013. a
Tang, Y., Carmichael, G. R., Uno, I., Woo, J.-H., Kurata, G., Lefer, B., Shetter, R. E., Huang, H., Anderson, B. E., Avery, M. A., Clarke, A. D., and Blake, D. R.: Impacts of aerosols and clouds on photolysis frequencies and photochemistry during TRACE-P: 2. Three-dimensional study using a regional chemical transport model, J. Geophys. Res.-Atmos., 108, 8822, https://doi.org/10.1029/2002JD003100, 2003. a, b, c, d, e, f, g, h, i
Tie, X. and Cao, J.: Aerosol pollution in China: Present and future impact on environment, Particuology, 7, 426–431, https://doi.org/10.1016/j.partic.2009.09.003, 2009. a
Topping, D., Connolly, P., and Reid, J.: PyBox: An automated box-model generator for atmospheric chemistry and aerosol simulations., The Journal of Open Source Software, 3, 755, https://doi.org/10.21105/joss.00755, 2018. a
Wang, L. T., Wei, Z., Yang, J., Zhang, Y., Zhang, F. F., Su, J., Meng, C. C., and Zhang, Q.: The 2013 severe haze over southern Hebei, China: model evaluation, source apportionment, and policy implications, Atmos. Chem. Phys., 14, 3151-3173, https://doi.org/10.5194/acp-14-3151-2014, 2014. a
Wang, Q., Sun, Y., Xu, W., Du, W., Zhou, L., Tang, G., Chen, C., Cheng, X., Zhao, X., Ji, D., Han, T., Wang, Z., Li, J., and Wang, Z.: Vertically resolved characteristics of air pollution during two severe winter haze episodes in urban Beijing, China, Atmos. Chem. Phys., 18, 2495–2509, https://doi.org/10.5194/acp-18-2495-2018, 2018. a, b
Wang, S., Zhang, Q., Martin, R. V., Philip, S., Liu, F., Li, M., Jiang, X., and He, K.: Satellite measurements oversee China's sulfur dioxide emission reductions from coal-fired power plants, Environ. Res. Lett., 10, 114015, https://doi.org/10.1088/1748-9326/10/11/114015, 2015. a
Wang, T., Ding, A., Gao, J., and Wu, W. S.: Strong ozone production in urban plumes from Beijing, China, Geophys. Res. Lett., 33, L21806, https://doi.org/10.1029/2006GL027689, 2006. a
Wang, Y., Yao, L., Wang, L., Liu, Z., Ji, D., Tang, G., Zhang, J., Sun, Y., Hu, B., and Xin, J.: Mechanism for the formation of the January 2013 heavy haze pollution episode over central and eastern China, Sci. China Earth Sci., 57, 14–25, https://doi.org/10.1007/s11430-013-4773-4, 2014. a
Wang, Z., Li, J., Wang, Z., Yang, W., Tang, X., Ge, B., Yan, P., Zhu, L., Chen, X., Chen, H., Wand, W., Li, J., Liu, B., Wang, X., Wand, W., Zhao, Y., Lu, N., and Su, D.: Modeling study of regional severe hazes over mid-eastern China in January 2013 and its implications on pollution prevention and control, Sci. China Earth Sci., 57, 3–13, https://doi.org/10.1007/s11430-013-4793-0, 2014. a
Whalley, L. and Slater, E.: APHH: Solar actinic UV flux photolysis rates made at the IAP-Beijing site during the summer and winter campaigns, Centre for Environmental Data Analysis, available at: http://catalogue.ceda.ac.uk/uuid/76b4ad364d71465d8f8b61e302eb2c4c (last access: 8 July 2019), 2018.
Whalley, L. K., Stone, D., Dunmore, R., Hamilton, J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Williams, P., Kleffmann, J., Laufs, S., Woodward-Massey, R., and Heard, D. E.: Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 18, 2547–2571, https://doi.org/10.5194/acp-18-2547-2018, 2018. a
Wild, O., Zhu, X., and Prather, M. J.: Fast-J: Accurate Simulation of In- and Below-Cloud Photolysis in Tropospheric Chemical Models, J. Atmos. Chem., 37, 245–282, https://doi.org/10.1023/A:1006415919030, 2000. a, b
Xing, J., Wang, J., Mathur, R., Wang, S., Sarwar, G., Pleim, J., Hogrefe, C., Zhang, Y., Jiang, J., Wong, D. C., and Hao, J.: Impacts of aerosol direct effects on tropospheric ozone through changes in atmospheric dynamics and photolysis rates, Atmos. Chem. Phys., 17, 9869–9883, https://doi.org/10.5194/acp-17-9869-2017, 2017. a, b, c
Xue, L. K., Wang, T., Gao, J., Ding, A. J., Zhou, X. H., Blake, D. R., Wang, X. F., Saunders, S. M., Fan, S. J., Zuo, H. C., Zhang, Q. Z., and Wang, W. X.: Ground-level ozone in four Chinese cities: precursors, regional transport and heterogeneous processes, Atmos. Chem. Phys., 14, 13175–13188, https://doi.org/10.5194/acp-14-13175-2014, 2014. a
Yang, T., Wang, Z., Zhang, B., Wang, X., Wang, W., Gbauidi, A., and Gong, Y.: Evaluation of the effect of air pollution control during the Beijing 2008 Olympic Games using Lidar data, Chinese Sci. Bull., 55, 1311–1316, https://doi.org/10.1007/s11434-010-0081-y, 2010. a, b
Yang, T., Wang, Z., Zhang, W., Gbaguidi, A., Sugimoto, N., Wang, X., Matsui, I., and Sun, Y.: Technical note: Boundary layer height determination from lidar for improving air pollution episode modeling: development of new algorithm and evaluation, Atmos. Chem. Phys., 17, 6215–6225, https://doi.org/10.5194/acp-17-6215-2017, 2017. a, b
Zhang, Q., Ma, X., Tie, X., Huang, M., and Zhao, C.: Vertical distributions of aerosols under different weather conditions: Analysis of in-situ aircraft measurements in Beijing, China, Atmos. Environ., 43, 5526–5535, https://doi.org/10.1016/j.atmosenv.2009.05.037, 2009. a
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of Urban Fine Particulate Matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015. a
Zhao, X. J., Zhao, P. S., Xu, J., Meng,, W., Pu, W. W., Dong, F., He, D., and Shi, Q. F.: Analysis of a winter regional haze event and its formation mechanism in the North China Plain, Atmos. Chem. Phys., 13, 5685–5696, https://doi.org/10.5194/acp-13-5685-2013, 2013. a
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015. a