the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Jul 2019
| 19 Jul 2019
On the contribution of nocturnal heterogeneous reactive nitrogen chemistry to particulate matter formation during wintertime pollution events in Northern Utah
Erin E. McDuffie
Caroline C. Womack
Dorothy L. Fibiger
William P. Dube
Alessandro Franchin
Ann M. Middlebrook
Lexie Goldberger
Ben H. Lee
Joel A. Thornton
Alexander Moravek
Jennifer G. Murphy
Munkhbayar Baasandorj
Mountain basins in Northern Utah, including the Salt Lake Valley (SLV), suffer from wintertime air pollution events associated with stagnant atmospheric conditions. During these events, fine particulate matter concentrations (PM2.5) can exceed national ambient air quality standards. Previous studies in the SLV have found that PM2.5 is primarily composed of ammonium nitrate (NH4NO3), formed from the condensation of gas-phase ammonia (NH3) and nitric acid (HNO3). Additional studies in several western basins, including the SLV, have suggested that production of HNO3 from nocturnal heterogeneous N2O5 uptake is the dominant source of NH4NO3 during winter. The rate of this process, however, remains poorly quantified, in part due to limited vertical measurements above the surface, where this chemistry is most active. The 2017 Utah Winter Fine Particulate Study (UWFPS) provided the first aircraft measurements of detailed chemical composition during wintertime pollution events in the SLV. Coupled with ground-based observations, analyses of day- and nighttime research flights confirm that PM2.5 during wintertime pollution events is principally composed of NH4NO3, limited by HNO3. Here, observations and box model analyses assess the contribution of N2O5 uptake to nitrate aerosol during pollution events using the production rate, N2O5 heterogeneous uptake coefficient (γ(N2O5)), and production yield of ClNO2 (φ(ClNO2)), which had medians of 1.6 µg m−3 h−1, 0.076, and 0.220, respectively. While fit values of γ(N2O5) may be biased high by a potential under-measurement in aerosol surface area, other fit quantities are unaffected. Lastly, additional model simulations suggest nocturnal N2O5 uptake produces between 2.4 and 3.9 µg m−3 of nitrate per day when considering the possible effects of dilution. This nocturnal production is sufficient to account for 52 %–85 % of the daily observed surface-level buildup of aerosol nitrate, though accurate quantification is dependent on modeled dilution, mixing processes, and photochemistry.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(5528 KB)
- Corrigendum
-
Supplement
(1246 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(5528 KB) - Full-text XML
- Corrigendum
-
Supplement
(1246 KB) - BibTeX
- EndNote





Over 80 % of Utah's population lives in counties that experience periods of elevated fine particulate matter (PM2.5 < 2.5 µm in diameter) during the winter season (U.S. Census Bureau, 2018; Whiteman et al., 2014). In these counties, the highest levels have been limited to three northern valleys along the Wasatch Mountains, shown in Fig. 1 (north to south: Cache Valley (Logan nonattainment area, NAA), Salt Lake Valley (Salt Lake NAA), and Utah Valley (Provo NAA)). These valleys were designated by the United States Environmental Protection Agency (EPA) as “moderate” nonattainment areas (NAA) in December 2009, with the Salt Lake and Provo areas reclassified from moderate to “serious” in May 2017 (Utah Department of Environmental Quality, 2019). Elevated PM2.5 concentrations in these regions impact public health and are associated with increases in emergency room visits for asthma (Beard et al., 2012). Short-term exposure to PM2.5 has also been shown to increase the chance of triggering acute ischemic heart disease events by 4.5 %–6 % per 10 µg m−3 in sensitive populations living in the Wasatch region (Pope et al., 2006, 2015).
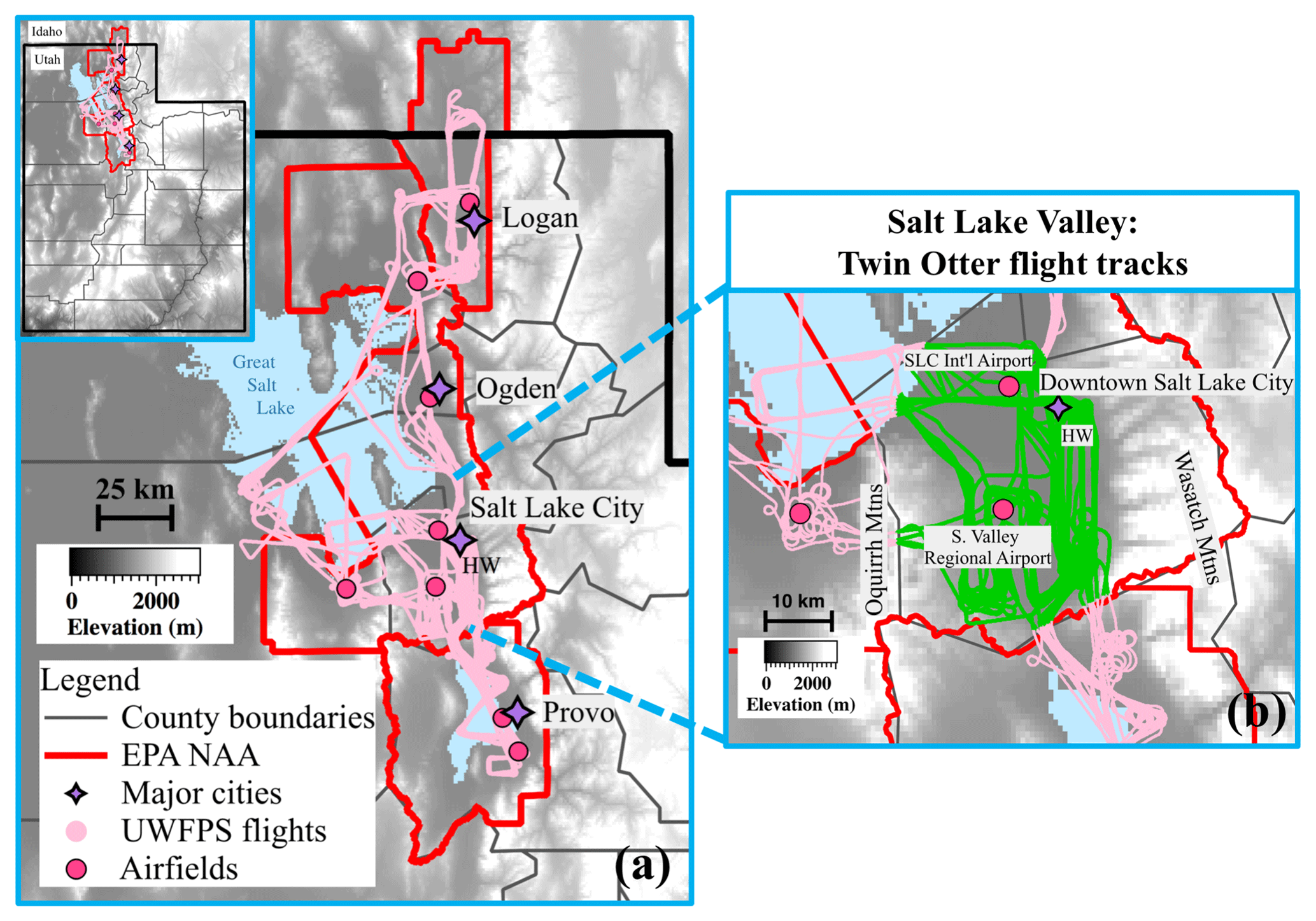
Figure 1(a) Elevation map of Utah's Wasatch region (Utah State in insert), with the Great Salt Lake (north) and Utah Lake (south) shown in blue and county borders in gray. The US-EPA-designated nonattainment areas (NAA) for PM2.5 are shown by red boundaries. From north to south these NAAs include the Logan NAA: moderate status, Salt Lake City NAA: serious status, and Provo NAA: serious status. UWFPS Twin Otter flight tracks are shown in pink. Purple markers indicate the locations of major cities, including Logan in the Cache Valley, Ogden and Salt Lake City in the SLV, and Provo in the Utah Valley. The locations of missed approaches conducted with the aircraft are shown by dark pink circles. The Hawthorne (HW) measurement site in the SLV is labeled. (b) Expanded view of the SLV, with analyzed flight tracks highlighted in green.
Elevated wintertime PM2.5 concentrations in these valleys typically correspond to multiday events of high atmospheric stability (e.g., Whiteman et al., 2014; Silcox et al., 2012; Gillies et al., 2010; Wang et al., 2012; Green et al., 2015; Silva et al., 2007; Baasandorj et al., 2017), associated with large, synoptic-scale high-pressure systems that transit from west to east, simultaneously impacting multiple basins across the intermountain western US (e.g., Reeves and Stensrud, 2009). Warm temperatures aloft cause boundary layer stratification that reduces mixing and traps cold air and emissions near the surface, illustrated in Fig. 2 and discussed further below. These events, termed persistent cold air pools (PCAPs), typically mix out after 1–5 d but have been observed to persist for as long as 18 d (Whiteman et al., 2014). Similar meteorological patterns have been linked to wintertime PM2.5 accumulation in basins across the western US (e.g., Chen et al., 2012; Green et al., 2015). During past PCAP and pollution events in Utah, data from ground-based measurements in the Salt Lake Valley (SLV) have reported day-to-day buildup rates of total PM2.5 mass in the range of ∼6–10 µg m−3 d−1 (Baasandorj et al., 2017; Silcox et al., 2012; Whiteman et al., 2014) before plateauing after ∼6 d into an event (Baasandorj et al., 2017). Average 24 h concentrations reported during PCAP events between 2001 and 2016 have been as large as 40–80 µg m−3 in Salt Lake (Baasandorj et al., 2017; Silcox et al., 2012) and Utah valleys (Malek et al., 2006) and up to 132.5 µg m−3 in Logan, Utah (Cache Valley) (Malek et al., 2006).
Previous ground-based studies have identified ammonium nitrate (NH4NO3) as the main component of PM2.5 (70 %–80 % by mass) during PCAP events in all three Northern Utah valleys (Silva et al., 2007; Hansen et al., 2010; Kuprov et al., 2014; Kelly et al., 2013; Long et al., 2003, 2005a, b; Baasandorj et al., 2017). Ammonium nitrate formation is thermodynamically favorable under cold wintertime conditions from the equilibrium between gas-phase ammonia (NH3) and nitric acid (HNO3), shown in Reaction (R1) in Fig. 2 (e.g., Kuprov et al., 2014; Nowak et al., 2012; Mozurkewich, 1993). PM2.5 mitigation strategies that are based on control of these gas-phase species are expected to be more effective if the limiting reagent and its sources can be identified. Both observationally and model-informed ground-based analyses have suggested that NH4NO3 formation in Cache and Salt Lake valleys is limited by the production of HNO3 (Kuprov et al., 2014; Mangelson et al., 1997; Martin, 2006; Utah Division of Air Quality, 2014a, b, c; Franchin et al., 2018), though uncertainties remain in how this limitation may be impacted by temporal and spatial variations.
While NH3 is directly emitted from agricultural sources, industrial processes, waste disposal, and automobile emissions (Behera et al., 2013; Livingston et al., 2009), HNO3 forms chemically in the atmosphere from the oxidation of NOx (), which in turn arises mainly from combustion emissions. There are two mechanisms by which this formation occurs, illustrated by Reactions (R2)–(R6) in Fig. 2. The first is through daytime NO2 oxidation by the hydroxyl radical (OH) (Fig. 2, Reaction R2) and the second is through the nocturnal heterogeneous uptake of dinitrogen pentoxide (N2O5) (Reaction R6), which itself is a product of nocturnal NOx oxidation (Reactions R3–R5). The former is relatively more important during the summer (Brown et al., 2004), whereas the latter, the focus of this study, may be relatively more important in winter (e.g., Wagner et al., 2013) due to reduced OH concentrations, colder temperatures that favor N2O5 in its equilibrium with NO3 (Reaction R5), and longer nights that allow more time for nocturnal reactions to occur. The nocturnal heterogeneous production of HNO3 is also expected to be largest in the residual layer (RL), due to the near-surface accumulation of NO, which titrates O3 (Reaction R3) and reacts with NO3 (Reaction R7), the precursor to N2O5 (e.g., Brown and Stutz, 2012).
The role of this nocturnal reactive nitrogen chemistry in the formation of PM2.5 has been considered in previous wintertime studies, though nocturnal, vertically resolved measurements have been relatively limited. Previous studies using ground- and tower-based observations, as well as midmorning aircraft vertical profiles, have identified heterogeneous chemistry and subsequent morning transport from aloft as a major source of surface-level NH4NO3 in California's San Joaquin Valley (e.g., Brown et al., 2006; Prabhakar et al., 2017; Pusede et al., 2016; Watson and Chow, 2002). Similarly, a box model analysis of tower- and ground-based observations in Beijing, China, also identified these processes as important contributors to surface-level particulate nitrate the following day (Wang et al., 2018). In Northern Utah specifically, nocturnal heterogeneous chemistry has been considered a source for PM2.5 (Baasandorj et al., 2017; Kuprov et al., 2014), though vertically resolved measurements have been limited to ground-based observations at different elevations along the Wasatch Mountains (Baasandorj et al., 2017). In an analysis of ground-based HNO3 and PM2.5 observations in the SLV, Kuprov et al. (2014) suggested that daytime HNO3 formation was dominant over the contribution from nocturnal heterogeneous chemistry. Baasandorj et al. (2017), however, noted that ground-based measurements in this region may not capture the extent of heterogeneous chemistry aloft in the RL, which is expected to be distinct from the surface composition (e.g., Brown et al., 2007; Brown and Stutz, 2012; Stutz et al., 2004). Therefore, vertical gradients in NOx and oxidants could promote efficient HNO3 and NH4NO3 formation aloft, which could contribute to enhanced surface-level PM2.5 concentrations the following day. Regardless of altitude, the absolute contribution at all altitudes will depend on (1) the rate of NO3 and N2O5 production, (2) the efficiency of N2O5 uptake onto aerosol (γ(N2O5)), and (3) the heterogeneous production yield of HNO3 relative to ClNO2 (φ(ClNO2)) (Osthoff et al., 2008; Behnke et al., 1997). Net accumulation of NH4NO3 at the surface, however, also depends on mixing and dilution associated with growth of the convective boundary layer and mixing of the RL down to the surface the following day. Quantification of these processes is key in designing effective mitigation strategies for Utah's wintertime air pollution and requires vertically resolved observations of chemical composition at night.
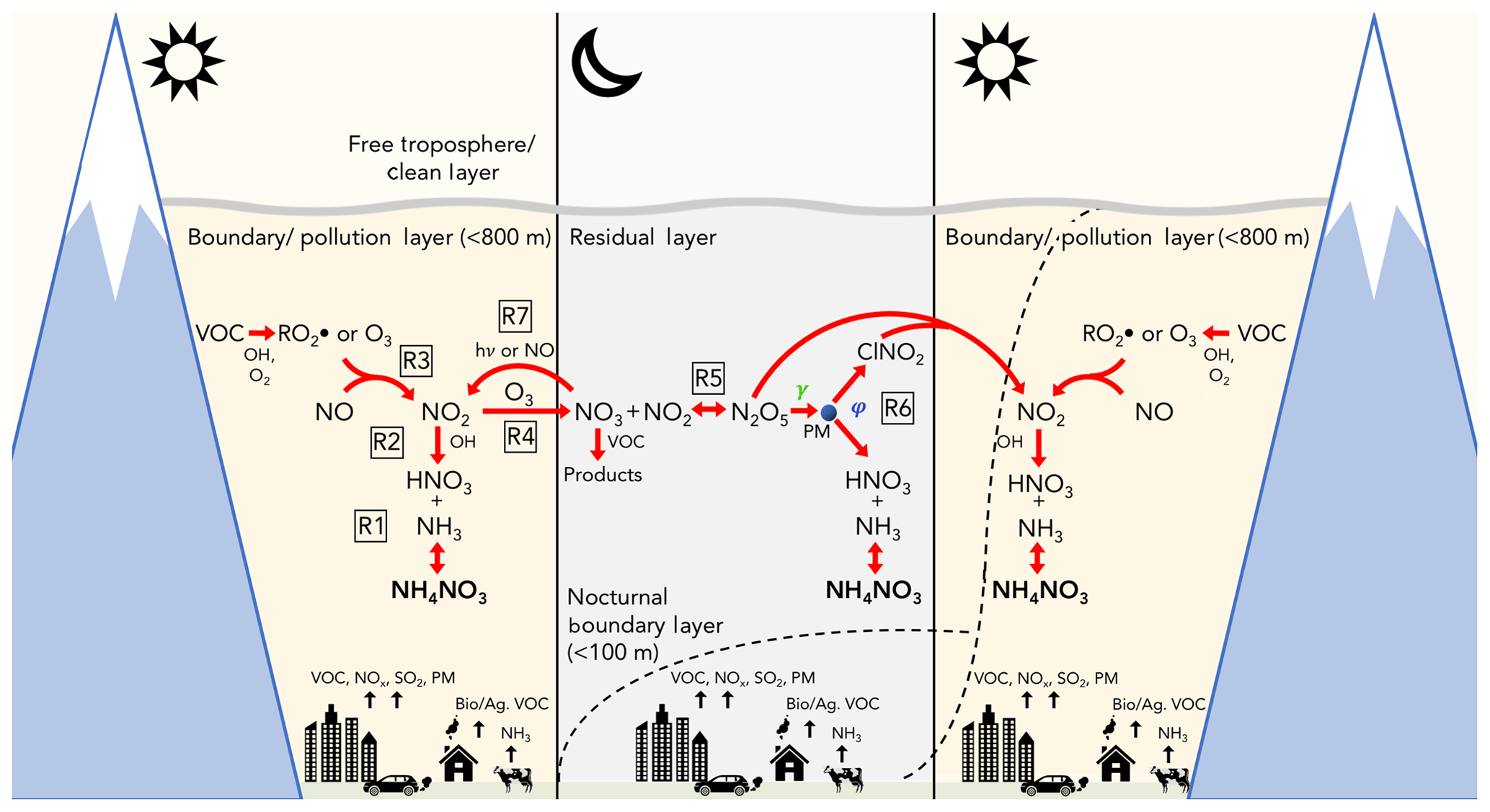
Figure 2Illustration of the day–night dynamics and chemical cycles of reactive nitrogen oxides, O3, and NH4NO3 during PCAP conditions in the SLV. The development of the nocturnal boundary layer and morning growth and mix-out are illustrated by the dashed lines. Figure is not to scale. Reaction (R6) represents the reaction .
In this study, we present results from the Utah Winter Fine Particulate Study (UWFPS), which consisted of aircraft and ground-based observations throughout the Cache, Salt Lake, and Utah valleys during January and February 2017. This analysis focuses on data from 16 aircraft flights (5 at night) during two pollution events between 16 January and 1 February 2017. These flights were carried out in the SLV, the most populated of the three Utah nonattainment areas. The first section presents an overview of PM2.5 during winter 2016–2017. In the second section, ambient mixing ratios of total (gas and particle phase) oxidized and reduced nitrogen are used to identify the limiting reagent to NH4NO3 aerosol formation, as well as its spatial and temporal trends. The final section presents upper-limit NH4NO3 production rate estimates and results from an observationally fit chemical box model to calculate γ(N2O5), φ(ClNO2), and an estimated contribution of nocturnal heterogeneous chemistry to NH4NO3 formation in the SLV. The contribution of nocturnal production relative to photochemically driven NO2 oxidation will have consequences for the development of effective mitigation strategies as day- and nighttime production processes may have different sensitivities to NOx emissions and VOC radical sources (Pusede et al., 2016; Womack et al., 2019), such that net sensitivities will be determined by the dominant formation mechanism.
2.1 UWFPS campaign overview and instrumentation
The UWFPS campaign included both aircraft and ground-based measurements throughout the Salt Lake, Cache, and Utah valleys during January and February 2017 (Fig. 1). A total of 23 research flights were conducted during both day and night with the NOAA Twin Otter (TO) aircraft. The TO was equipped with aerosol- and gas-phase instrumentation (summarized in Table 1) to probe the regional sources and formation mechanisms of PM2.5. While flights were conducted over three valleys, the focus of this analysis will be on the more densely populated SLV, with relevant flight tracks highlighted in the right panel of Fig. 1.
Table 1Aircraft measurements used in this analysis.
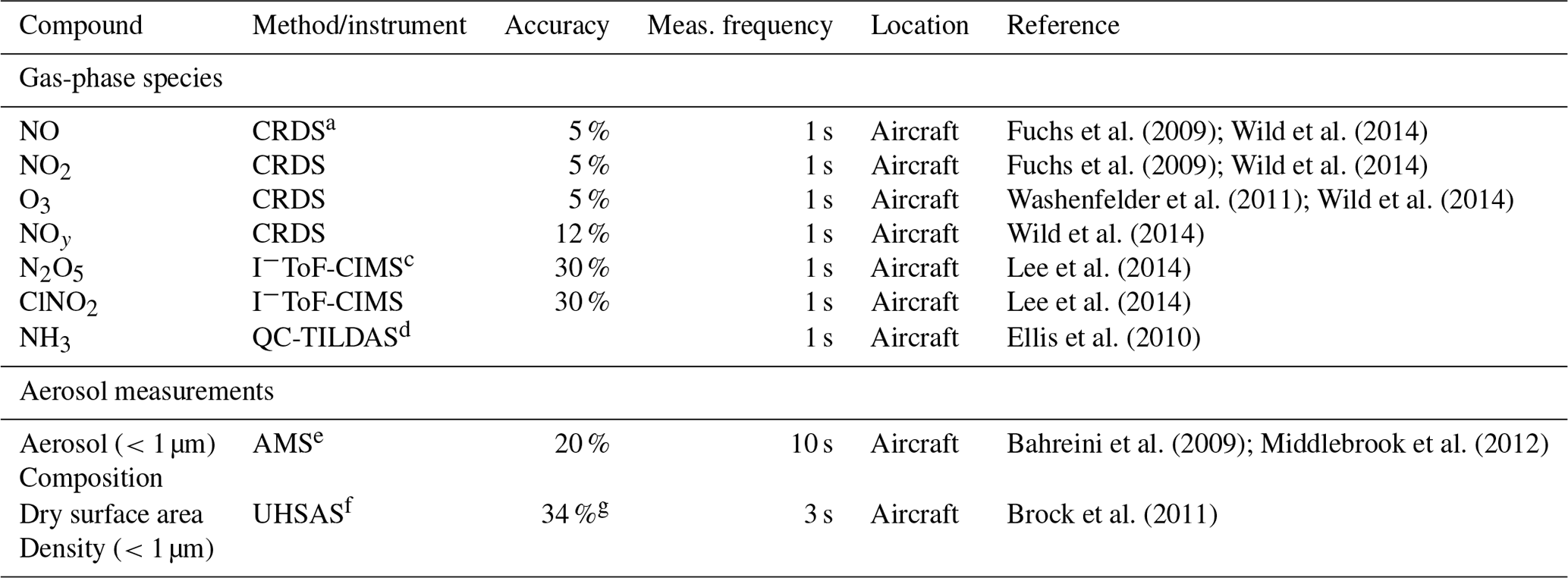
a NOAA, cavity ring-down spectrometer (CRDS, NOxCaRD). b Hawthorne. c University of Washington, iodide time-of-flight chemical ionization mass spectrometer. d University of Toronto, quantum cascade tunable infrared laser differential absorption spectrometer (see Moravek et al., 2019, for details on instrument accuracy). e NOAA, aerosol mass spectrometer. f Droplet Measurement Techniques, ultra-high-sensitivity aerosol spectrometer. g Estimated according to the performance of a different UHSAS in the WINTER campaign.
Briefly, the TO payload included gas-phase measurements of NOx, NO2, NOy, and O3 (1 Hz sample frequency) from a NOAA cavity ring-down spectrometer (NOxCaRD) (Wild et al., 2014); NH3 (1 Hz sample frequency) measurements from an Aerodyne midinfrared absorption instrument (QC-TILDAS) from the University of Toronto (Ellis et al., 2010); and N2O5, HNO3, and ClNO2 (1 Hz sample frequency) measurements from an iodide time-of-flight chemical ionization mass spectrometer (I−ToF-CIMS) from the University of Washington (Lee et al., 2014, 2018). Accuracies for NOx, NO2, and O3 were 5 % and 12 % for NOy, with stated detection limits of 60 pptv (2σ) (Wagner et al., 2011; Wild et al., 2014) in the boundary layer. Gas-phase NH3 was measured with a detection limit of 450 pptv (1 s 3σ), as described in further detail by Moravek et al. (2019). Accuracy and detection limits for N2O5, ClNO2, and HNO3 were similar to those reported from the same instrument deployed during the Wintertime Investigation of Transport, Emissions, and Reactivity (WINTER) campaign (≤0.6 pptv (1 s 1σ), 30 %) (Lee et al., 2018). Non-refractory submicron aerosol composition (sampled every ∼10 s) was measured with the NOAA aerosol mass spectrometer (AMS) (Bahreini et al., 2009; Middlebrook et al., 2012) and aerosol size (sample every ∼3 s) with a commercial ultra-high-sensitivity aerosol spectrometer (UHSAS) (Brock et al., 2011). Average detection limits for AMS aerosol composition were 0.04, 0.09, 0.33, 0.03, and 0.07 µg sm−3 (sm−3 refers to m3 under standard conditions: 1 atm and 273.15 K) for particulate nitrate, ammonium, organics, sulfate, and chloride, respectively. Uncertainties were ∼20 % for all species (Franchin et al., 2018). Ambient temperature and pressure (1 Hz sample frequency) were measured with a commercial (Avantech) meteorological probe. The accuracy of the commercial UHSAS instrument was also expected to be similar to that used during WINTER (dry surface area density: ∼34 %).
Additional ground-based measurements used in this analysis include hourly PM2.5, NO2, O3, and temperature from the Utah Department of Air Quality (UDAQ) instrumentation at the Hawthorne (HW) monitoring site (Fig. 1). Total PM2.5 mass was measured with a Thermo Scientific 1405-DF dichotomous ambient air monitor, NO2 with a Teledyne API T200U chemiluminescence detector, and O3 with a Teledyne API T400 UV absorption spectrometer, all in accordance with EPA guidelines (Environmental Protection Agency, 2018). Select volatile organic compounds (VOCs) were also measured at the University of Utah (UU) ground site by a proton-transfer-reaction mass spectrometer. Further information about the UWFPS campaign and aircraft and ground-based instrumentation can be found in additional publications (Franchin et al., 2018; UWFPS Science Team, 2018; Womack et al., 2019; Moravek et al., 2019).
2.2 Box model
2.2.1 Description
A zero-dimension chemical box model has been developed to simulate the nocturnal chemical evolution of an air parcel from sunset until the time of aircraft measurement (assuming constant temperature and relative humidity). Extensive model details have been previously discussed in McDuffie et al. (2018b). Briefly, the model forward integrates the chemical mechanism (13 reactions, Table S1 in the Supplement) starting 1.3 h prior to sunset (see below), iteratively adjusting the initial concentrations of O3 and NO2 until the model-predicted concentrations are both within 0.5 % of the aircraft observations. Holding these initial concentrations constant, the model next adjusts the total heterogeneous loss rate constant of N2O5 () until the model output reproduces ambient nighttime observations of N2O5 to within 1 %. As described in McDuffie et al. (2018b), the model iterates these steps, readjusting initial concentrations of O3 and NO2 and values of until aircraft observations of NO2, O3, and N2O5 are simultaneously reproduced by the model. The final step holds these values constant while iteratively adjusting the production rate of ClNO2 () until the modeled mixing ratios of ClNO2 are within 1 % of the nighttime ClNO2 observations. The N2O5 uptake coefficients (γ(N2O5)) and ClNO2 production yields (φ(ClNO2)) are then calculated following Eqs. (1) and (2), where c is the mean molecular speed and SA is the ambient wet PM1 surface area density (described below). The model repeats this entire process every 10 s for all flights conducted at night, as determined by time and aircraft GPS altitude.
Holding the derived and values constant, the model can further simulate the total nitrate produced overnight by forward integrating the model until the time of sunrise, as shown for a representative SLV point in Fig. 3. Here, total nitrate (gas + particulate phase) is represented as HNO3 only, as this model does not include aerosol thermodynamics that partition nitrate between the gas and particle phases. Modeled gas-phase HNO3 is assumed to partition to the particle phase with 100 % efficiency, following observations presented in Franchin et al. (2018) that show >90 % of total nitrate is in the particle phase during wintertime pollution events in the SLV. As modeled nitrate is initialized with a concentration of 0 µg m−3, concentrations predicted at sunrise represent the total amount of nitrate produced from nocturnal chemistry over the course of a single night (i.e., nocturnal nitrate production rate). These base case values assume no overnight loss from dilution and constant values of γ(N2O5) and φ(ClNO2), as discussed further in Sect. 3.3.3.
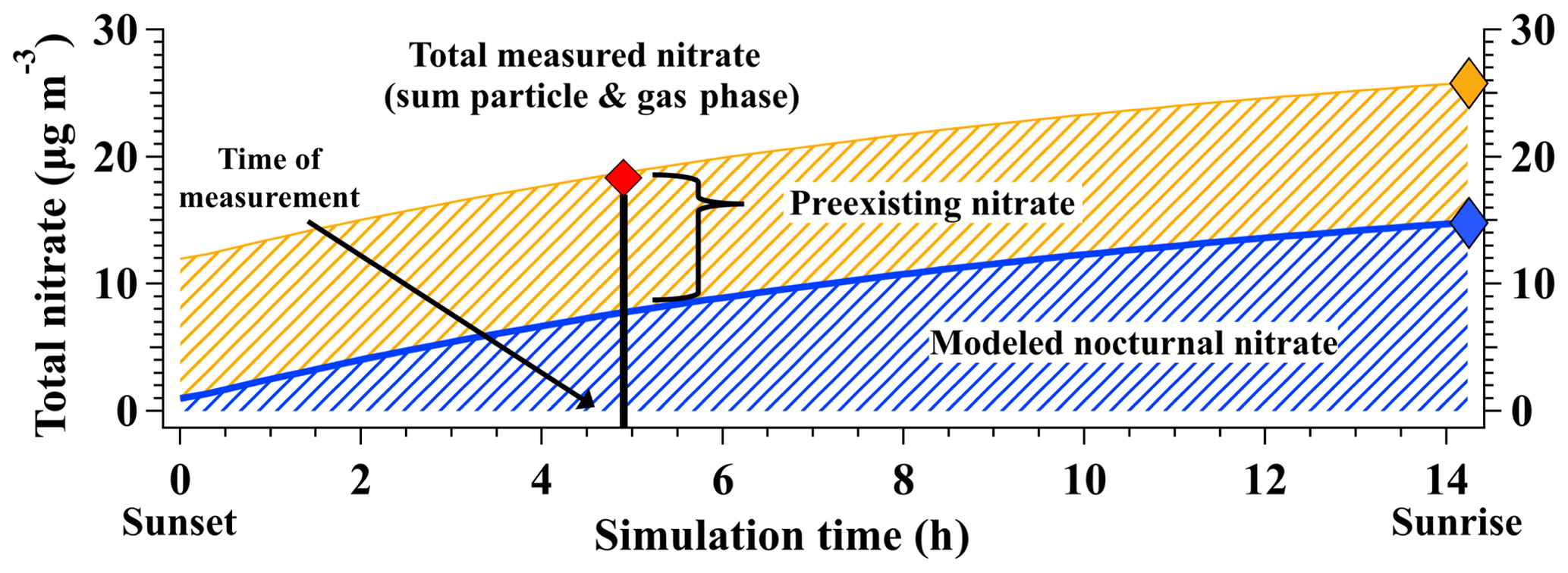
Figure 3Example simulation of total nitrate production from sunset to sunrise for an air parcel sampled over the SLV on 28 January 2017. Model-derived γ(N2O5) and φ(ClNO2) values were 0.05 and 0.21, respectively. Modeled nocturnal nitrate (blue) is the total nitrate produced by heterogeneous chemistry in the box model, with the nocturnal production rate (µg m−3 per night) represented by the blue diamond. Preexisting nitrate (yellow) represents the nitrate present at sunset and is calculated as the difference between total measured nitrate from the aircraft (red diamond) and the model-predicted nitrate at the time of aircraft measurement (vertical black line). Assuming preexisting nitrate is constant overnight (i.e., no deposition or dilution) and constant values of γ(N2O5) and φ(ClNO2), total nitrate expected at sunrise is represented by the gold diamond.
2.2.2 Model simplifications and uncertainties
For the UWFPS campaign, the box model was run in a similar manner to that described previously in McDuffie et al. (2018b), for nocturnal aircraft observations collected in the RL over the eastern US coast during the 2015 WINTER campaign. Due to more limited instrumentation during UWFPS than WINTER, a larger number of box model assumptions and simplifications were required, which are summarized below.
First, the wet SA density for the base case simulations was calculated by applying a hygroscopic growth curve as a function of RH (Fig. S2 in the Supplement) to the dry PM1 SA measured by the UHSAS (details in Sect. S1.3 in the Supplement). The growth curve was derived with the Extended AIM Aerosol Thermodynamic Model (Wexler and Clegg, 2002), assuming pure NH4NO3 particles. Alternatively, estimating the growth factor from AMS measurements and estimates of aerosol organic density and the organic hygroscopicity constant (κOrg) (described in Sect. S1.3; Jimenez et al., 2009; Mei et al., 2013; Cerully et al., 2015; e.g., Kuwata et al., 2012; Brock et al., 2016; Shingler et al., 2016) resulted in only a ∼3 % change in the total wet SA for night flights during UWFPS (Fig. S2). For the 1031 measurement periods (10 s each) with simultaneous values of γ(N2O5) and φ(ClNO2), the median dry aerosol SA was 151.9 µm2 cm−3, which increased to 353.1 µm2 cm−3 when accounting for hygroscopic growth (Fig. S2). Additional uncertainties associated with hygroscopic growth and assumptions of constant SA are discussed below in Sect. 3.3.2.
Second, loss of the nitrate radical (NO3) from its reaction with volatile organic compounds (VOCs) was assumed to occur with a single first-order rate constant (), calculated for each flight from a combination of historical ground-based VOC measurements and select VOC measurements from a PTR-MS at the UU site (see Sect. S1.2 for details; Atkinson and Arey, 2003). At night, NO3 serves as one of the primary tropospheric oxidants for VOCs and can react with RO2 and HO2 radicals to contribute to nocturnal NOx recycling (Vaughan et al., 2006). In this analysis, NO3–VOC reactions were lumped and treated as a net NOx sink with values of the first-order loss rate constant, , ranging from to s−1 (NO3 lifetime ∼ 100–1000 s). These rate constants are slightly larger than average values measured during the WINTER campaign ( to s−1) (McDuffie et al., 2018b) and within the range previously reported ( to s−1) during winter 2012 at a ground site in Colorado (Wagner et al., 2013). Additional NOx regeneration from reactions of NO3 with HO2 and RO2 radicals was not included in this analysis due to a lack of radical measurements. An underprediction in from these uncertainties would cause both an overprediction in the loss rate constant of N2O5 and the subsequent production of nitrate. While uncertainties in can lead to large model uncertainties during summertime conditions (e.g., Phillips et al., 2016), NO3–VOC reactivity is largely reduced during the winter season as a result of lower biogenic emissions and colder temperatures that favor N2O5 in its equilibrium with NO3. Sensitivity studies discussed below showed 0.2 % changes in the median model-predicted nocturnal nitrate production rate associated with ±50 % changes in (Table S4). The possibility of varying VOC reactivity with time was also investigated (Sect. S1.4.5) but resulted in a minimal (<0.1 %) impact on nitrate production results presented below. The potential for other rate constants to vary with time may additionally lead to increased variability in the results presented in Sect. 3.3.
Third, there are uncertainties in the assumptions regarding the start time and duration of each simulation. All simulations were initialized at 1.3 h prior to sunset, assuming no initial concentrations of N2O5 or ClNO2. The presunset time of 1.3 h was derived for the WINTER campaign, based on the time when predicted daytime N2O5 concentrations (described in Sect. S1.4.4 and Brown et al., 2005) diverged from ambient observations when approaching sunset. This value was not recalculated for UWFPS simulations as daytime N2O5 calculations require measurements of j(NO3) photolysis rates, which were not available during UWFPS. The median nocturnal nitrate production rate, however, changed by <0.3 % when this presunset time was varied between 0 and 2 h. Photolysis rates during this time were also calculated from those measured during the WINTER campaign (Sect. S1.4.3; Shetter and Müller, 1999). While WINTER photolysis rates may have been larger than those during Utah PCAP events, the median modeled nocturnal nitrate production rate showed a small sensitivity (<2.8 %) to ±40 % changes in these values (Sect. S1.4.3). Additional uncertainties in air age (i.e., simulation start time and duration), however, may still serve to overpredict N2O5 loss rates and nocturnal nitrate based on previous sensitivity studies (McDuffie et al., 2018b). A combination of these assumptions will lead to a greater uncertainty in model results near sunset, as discussed in Sect. 3.3.2.
Fourth, air parcel mixing and deposition of gas-phase nitric acid were not included in base case simulations. Additional simulations, described in Sect. S1.4.2, included deposition using a first-order nitric acid loss constant of s−1, calculated from a boundary layer height of 800 m; deposition velocity of 2.7 cm s−1 (Zhang et al., 2012); and gas/particle nitrate fraction of 8 % from Franchin et al. (2018). The median nocturnal nitrate production rate increased by <8 % when this depositional loss of HNO3 was included. In contrast, modeled nitrate production was most sensitive (−42.2 % reduction) to the addition of a first-order loss process, meant to simulate air parcel dilution and O3 entrainment from vertical mixing between the RL and free troposphere (Table S4). Based on a previous analysis by Womack et al. (2019), the dilution rate constant was estimated to be s−1 in the RL, with a possible range of 1.2 to 2.5 × 10−5 s−1 (described in Sect. S1.4.1). Results from simulations that include dilution are discussed further in the final section.
Finally, the absolute uncertainty associated with each individual nocturnal nitrate production rate was calculated from the quadrature addition of the uncertainties associated with sensitivity tests described above and the NO2, O3, N2O5, and ClNO2 measurements used as model fit parameters (<6 % for all tests). Production rates derived from model fits to observations as well as the absolute uncertainties associated with all 17 sensitivity tests are shown as a time series in Fig. S3. Dilution contributes 92 % of the total uncertainty (light blue in Fig. S3), on average. Both the base case results (black dots) and those from simulations including the effects of air parcel dilution are discussed in Sect. 3.3.3.
3.1 PM2.5 in Salt Lake Valley – winter 2017
To provide an overview of wintertime pollution events in the SLV, Fig. 4 shows a time series of total PM2.5 mass (1 and 24 h averages) measured at the UDAQ Hawthorne (HW) site (Fig. 1) from 1 December 2016 to 22 February 2017. Additional time series of ground-based PM2.5 measurements for all three Utah NAAs are provided in Franchin et al. (2018). The SLV data in Fig. 4 show four pollution events that exceeded the NAAQS during the 2016–2017 winter. Calculated from 24 h measurements, the four largest pollution events during December 2016 and January 2017 had daily PM2.5 buildup rates that ranged from 3.7 to 15.6 µg m−3 d−1 (see Fig. 4), encompassing the daily rates reported previously in the same valley (Whiteman et al., 2014; Silcox et al., 2012; Baasandorj et al., 2017). The last two major pollution events (10–22 January, event no. 3; and 25 January–5 February, event no. 4) overlapped with flights during UWFPS, shown by the gray shading in Fig. 4. Average non-refractory (NR) PM1 aerosol mass fractions measured during these periods by the TO AMS showed that the aerosol was primarily composed of NH4NO3 (Fig. 4 pie charts). The sum of and contributed to 76.6 % and 74.0 % of the total PM1 mass measured during the last two pollution episodes (74 % average; Franchin et al., 2018), which agree with previous ground-based observations (e.g., Baasandorj et al., 2017) of past seasons. Nitrate alone contributed to an average 57 % and 58 % of the total aerosol mass during pollution episodes no. 3 and no. 4, respectively. During the relatively clean period sampled between 8 and 12 February 2017, the combined fraction decreased to an average of 57 %, with a larger relative contribution from aerosol organics. The remaining analyses here will focus on aircraft flights during the two late January pollution events (no. 3 and no. 4) to evaluate the contribution of nocturnal RL heterogeneous nitrogen chemistry to observed surface-level nitrate during pollution events.
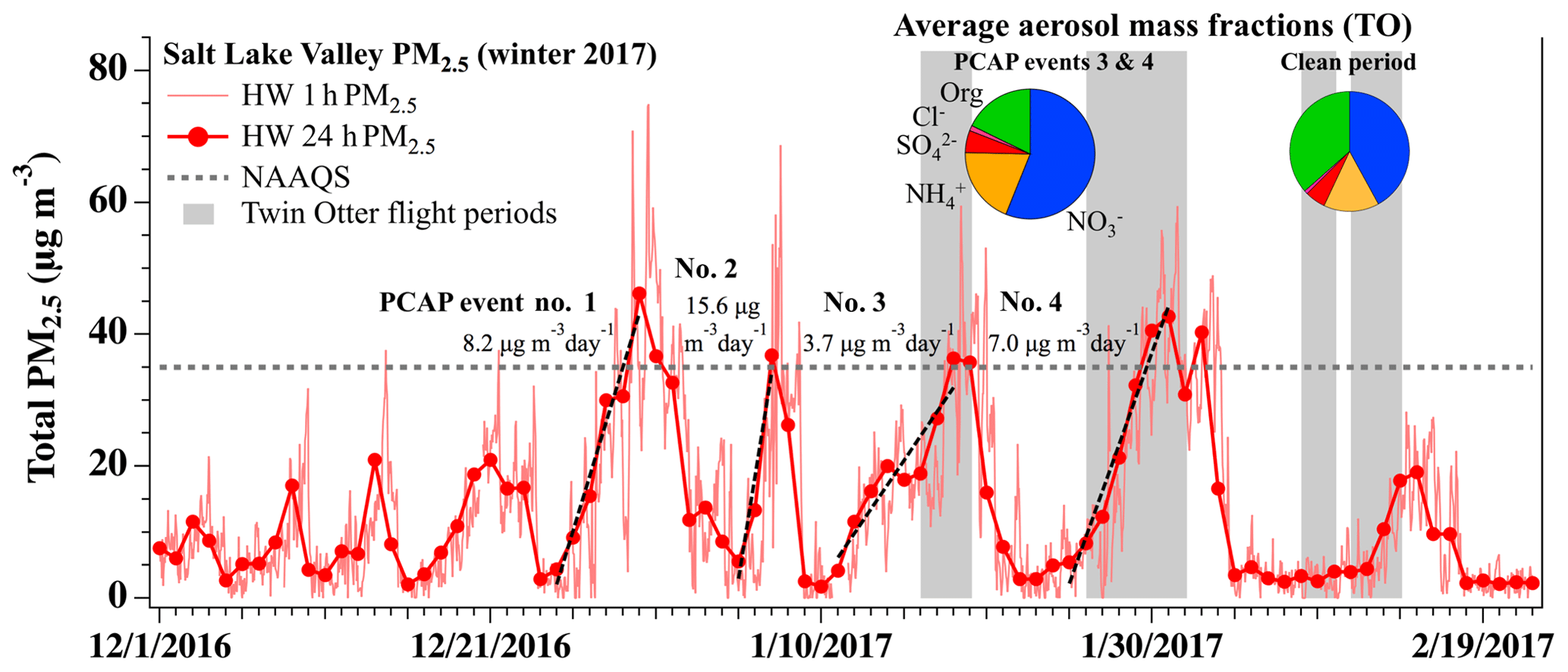
Figure 4Time series of total PM2.5 mass (µg m−3) (1 and 24 h averages) for the 2016–2017 winter, measured at the Hawthorne (HW) UDAQ site in the SLV. Dashed black lines are daily PM2.5 accumulation rates (rates given in Figure). The 24 h EPA national ambient air quality standard (NAAQS) for PM2.5 (35 µg m−3) is shown by the dashed gray line. Gray shading indicates days when the TO aircraft was flying during UWFPS. Average aerosol mass fractions measured by the AMS aboard the TO are given in pie charts for polluted and clean conditions. Aerosol components are colored by nitrate (blue), ammonium (gold), sulfate (red), non-refractory chloride (pink), and organics (green).
3.2 Limiting and excess reagents for NH4NO3 aerosol
As NH4NO3 was the principal component of PM2.5 during pollution events in the SLV (Fig. 4), the contribution from heterogeneous reactive nitrogen processes is dependent on whether NH4NO3 formation is limited by the availability of gas-phase NH3 or HNO3. Under ambient conditions, gas-phase NH3 and HNO3 are assumed to be in a thermodynamic equilibrium with their particulate equivalents ( and ). The limiting reagent can therefore be inferred from the ratio of total oxidized () to total reduced nitrogen (), shown in Eq. (3). This ratio does not account for other aerosol components such as (NH4)2SO4, NH4HSO4, and NH4Cl but should generally represent the NH4NO3 aerosol system when particulate concentrations of sulfate and inorganic chloride are low, as was observed during UWFPS 2017 (Fig. 4 and Franchin et al., 2018). A nitrogen ratio greater than 1 indicates that oxidized nitrogen is in excess and NH4NO3 particle formation is limited by the presence of NH3. Conversely, a ratio smaller than 1 indicates that formation is limited by the presence of HNO3, which itself is limited by the oxidation rate of NOx. In a HNO3-limited system, NH4NO3 formation will be sensitive to changes in HNO3 concentrations resulting from both day- and nighttime NOx oxidation processes. Daytime NOx oxidation rates during winter will depend on specific conditions but are generally slower, such that nighttime oxidation may play a dominant role (e.g., Wood et al., 2005; Kenagy et al., 2018).
A time series of nitrogen ratios in the SLV between 17 January and 1 February is shown in Fig. 5a, calculated from 10 s averaged (AMS frequency) measurements of gas- and particle-phase compounds aboard the TO aircraft. Figure 5a shows that NH4NO3 particle formation in the SLV during pollution episodes was largely limited by HNO3 (median ratio 0.77) but highly variable (range of 0.1–1.9) and time dependent, with the frequency of NH3-limited conditions increasing throughout both late January pollution events. The color scale in Fig. 5a and the vertical profiles of average and 10th–90th percentile nitrogen ratios in Fig. 5b further show that the lowest nitrogen ratios corresponded to the lowest altitudes. This evidence of HNO3 limitation near the ground is consistent with all previous ground-based observations that show exclusive HNO3 limitation in the SLV (Kelly et al., 2013; Utah Division of Air Quality, 2014c). The increased frequency of NH3-limited points throughout both pollution episodes (Fig. 5a), however, is opposite the trend predicted by Baasandorj et al. (2017), who suggested that observed surface-level oxidant depletion should lead to more HNO3-limited conditions over time. Events of NH3 limitation (excess HNO3) during 2017, however, only occurred at the highest altitudes (panel b), and their increasing frequency with time (panel a) is consistent with these events reflecting negative NH3 gradients away from the surface and/or the production of HNO3 aloft from nocturnal N2O5 chemistry. The rate of HNO3 production from nocturnal heterogeneous chemistry is expected to be maximized at higher altitudes, removed from NO emissions and O3 titration at the surface (Fig. 2). Results here are also consistent with aerosol thermodynamic modeling studies by Franchin et al. (2018), who found that simulations of total PM1 mass during UWFPS flights over the SLV were proportionally sensitive to 50 % reductions in total nitrate. Additional simulations by Franchin et al. (2018), however, also showed near 50 % PM1 reductions with 50 % reductions in total ammonium (), indicating that 50 % ammonium reductions may be enough to shift the SLV from the HNO3 to NH3-limited regime. This is consistent with nitrogen ratios in Fig. 5 approaching and exceeding values of 1.
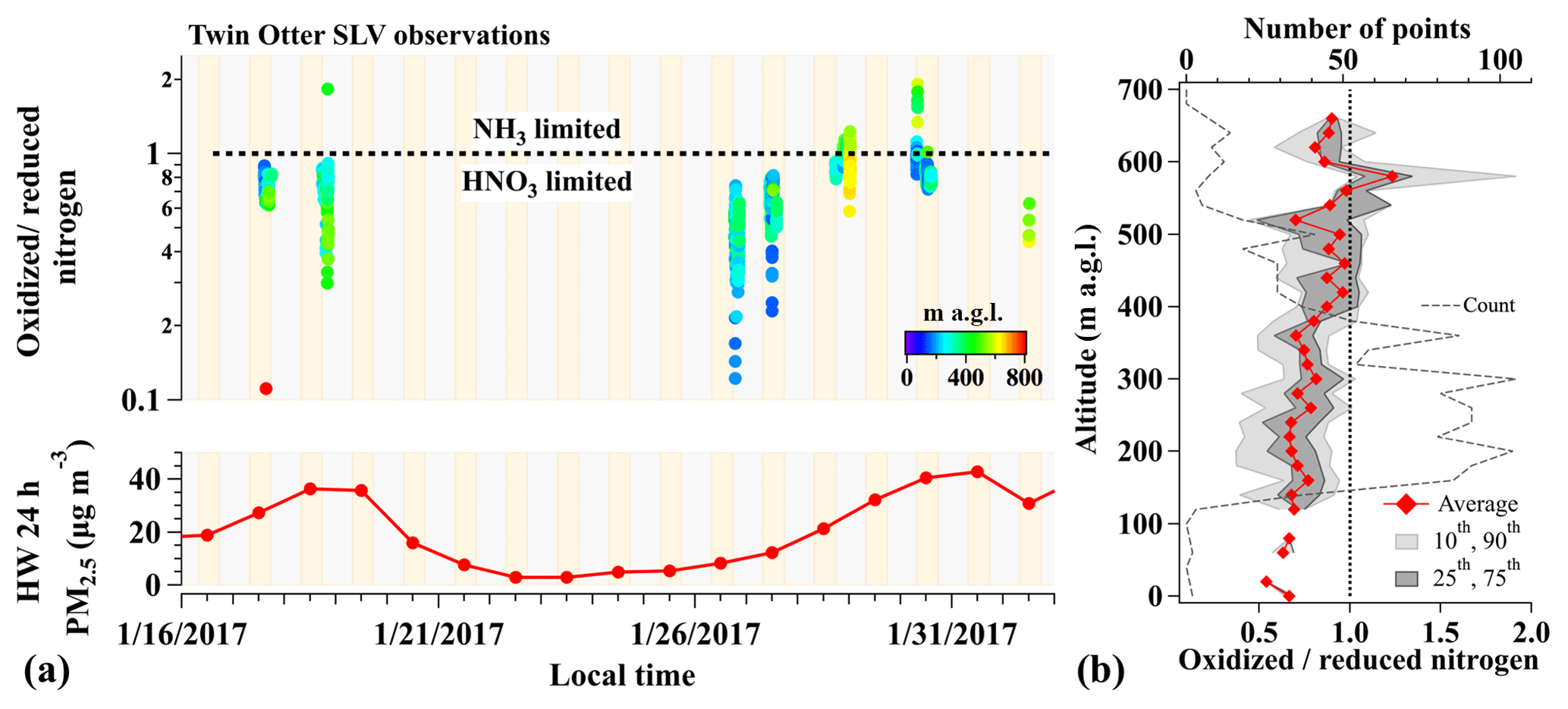
Figure 5(a, top) Time series of the ratio of total oxidized () to reduced () nitrogen between 16 January and 1 February 2017 (10 s averages), calculated from TO observations over the SLV. Individual nitrogen ratios are colored by aircraft altitude (m a.g.l.). Yellow and gray shading indicate times of day and night, respectively. (a, bottom) PM2.5 mass (24 h average) measured at the HW ground site. (b) Vertical profile of oxidized-to-reduced nitrogen ratios from panel (a). Diamonds represent the average values in each altitude bin and gray shading shows the 10th–90th (light gray) and 25th–75th (dark gray) percentiles. The number of points in each bin is shown by the gray dashed line. The vertical black line illustrates a nitrogen ratio of 1.
3.3 Nitrate production via heterogeneous reactive nitrogen chemistry
The absolute amount of nitrate that is chemically produced from heterogeneous chemistry will depend on the production rates of the nitrate radical and gas-phase N2O5 (Sect. 3.3.1), the N2O5 aerosol uptake efficiency (Sect. 3.3.2), and the yields of ClNO2 and HNO3 (Sect. 3.3.2), which are quantified below. The final section (Sect. 3.3.3) presents forward-integrated box model simulations to further quantify the nocturnal nitrate production rate and estimate the contribution of this chemistry to NH4NO3 formation during January 2017 in the SLV.
3.3.1 Maximum instantaneous nitrate production rates
An upper-limit estimate of the instantaneous aerosol nitrate production rate from heterogeneous N2O5 chemistry is defined here as . This rate can be calculated as 2 times the gas-phase production rate of the NO3 radical (), given that the reaction between NO2 and O3 (Eqs. 4–6), rather than N2O5 uptake, is the rate-limiting step for nitrate formation (discussed below). In Eq. (4), is calculated in units of molecules per cubic centimeter per second (molec. cm−3 s−1) but is typically reported in units of ppbv h−1 as shown here. The reaction kinetics in Eq. (5) between NO2 and O3 are from the 2008 IUPAC recommendation (IUPAC, 2008), ND is the number density of ambient air, and in Eq. (6) is calculated after has been converted to units of micrograms per cubic meter per hour (µg m−3 h−1), as detailed in Sect. S2. This calculation estimates a maximum contribution of N2O5 heterogeneous chemistry to nitrate production as it assumes (1) N2O5 is produced quantitatively from NO3 (i.e., no competing reaction of NO3+VOC), (2) N2O5 is produced at the rate of NO3 production (valid under cold conditions that shift the NO3–N2O5 equilibrium to favor of N2O5), (3) N2O5 is efficiently taken up onto aerosol, and (4) aqueous-phase reactions form two molecules of HNO3 for every molecule of N2O5 (i.e., φ(ClNO2)=0).
The value of is expected to vary with altitude due to boundary layer dynamics and surface NOx emissions that can deplete O3 at night near the surface, as described previously in Baasandorj et al. (2017). The time series in Fig. 6a illustrates that the O3 measured at HW was frequently absent at night during the third and fourth pollution events in January 2017. As surface-level O3 was titrated overnight, ground-site data cannot provide direct information about aloft in the RL. In the absence of vertical observations during pollution events in 2016, a previous analysis by Baasandorj et al. (2017) used late-afternoon measurements at the HW ground site to predict NO3 production rates () in the RL that varied from 0 up to ∼2 ppbv h−1 (∼0–5 µg m−3 h−1), but with values frequently <1 ppbv h−1. These values correspond to instantaneous nitrate production rates () of ∼0–10 µg m−3 h−1, with typical values closer to 5 µg m−3 h−1. Late-afternoon estimates from the same site during 2017 (dashed lines in Fig. 6, from Eq. 6) suggest smaller rates in 2017 than in 2016, with values between 1 and 5 µg m−3 h−1 during UWFPS pollution events (Fig. 6a).
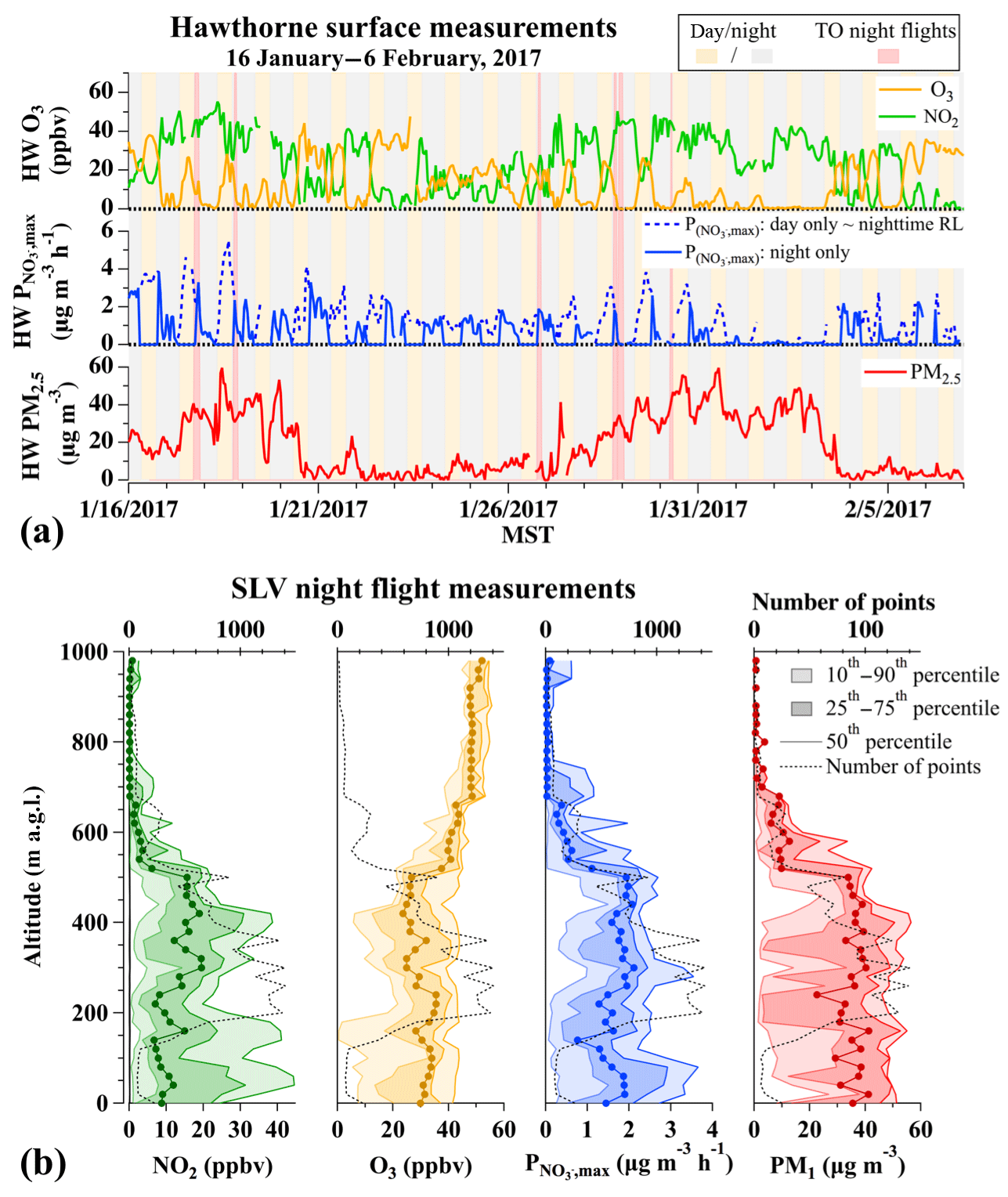
Figure 6(a) Time series of NO2, O3 (top), (middle; see text for definition), and PM2.5 (bottom) measured at the HW ground site during 16 January–6 February 2017. O3 data during the middle January pollution event were corrected to account for a 4.5 ppbv offset in the HW measurements, as shown in Fig. S4. Aircraft flight times are shown by red shading. Dashed blue line shows the calculated rates that would occur during the day if this mechanism were operative. Solid blue line assumes nitrate production from this mechanism during the day is zero. Late-afternoon at the surface (dashed line) is roughly equivalent to the expected in the RL at night. (b) Vertical profiles of O3, NO2, (1 s data), and PM1 (10 s data) measured from the aircraft on all night flights over the SLV. In each panel, light shaded regions show the 10th–90th percentile ranges, dark shaded regions are the 25th–75th percentile ranges, and the solid lines are the 50th percentile. Dashed black lines show the number of points at each altitude.
The bottom panels of Fig. 6b show the binned, vertical profiles of median, 25th, and 75th percentile instantaneous values, along with aircraft observations of O3, NO2, and PM1 for all UWFPS night flights (red shaded regions in Fig. 6a). The vertical profiles show a relatively uniform distribution of with altitude through the lowest 600 m. The dashed black lines also show that the number of points in each altitude bin was weighted toward the 100–500 m altitude range. The median instantaneous value in this polluted layer (0–600 m a.g.l., meters above ground level) was 1.6 µg m−3 h−1 (N = 21 666). This value is at the low range of estimates of 1.6–5 µg m−3 h−1 that are predicted from late-afternoon ground-based observations on each flight day (dashed line in the middle panel of Fig. 6a), following the method of Baasandorj et al. (2017).
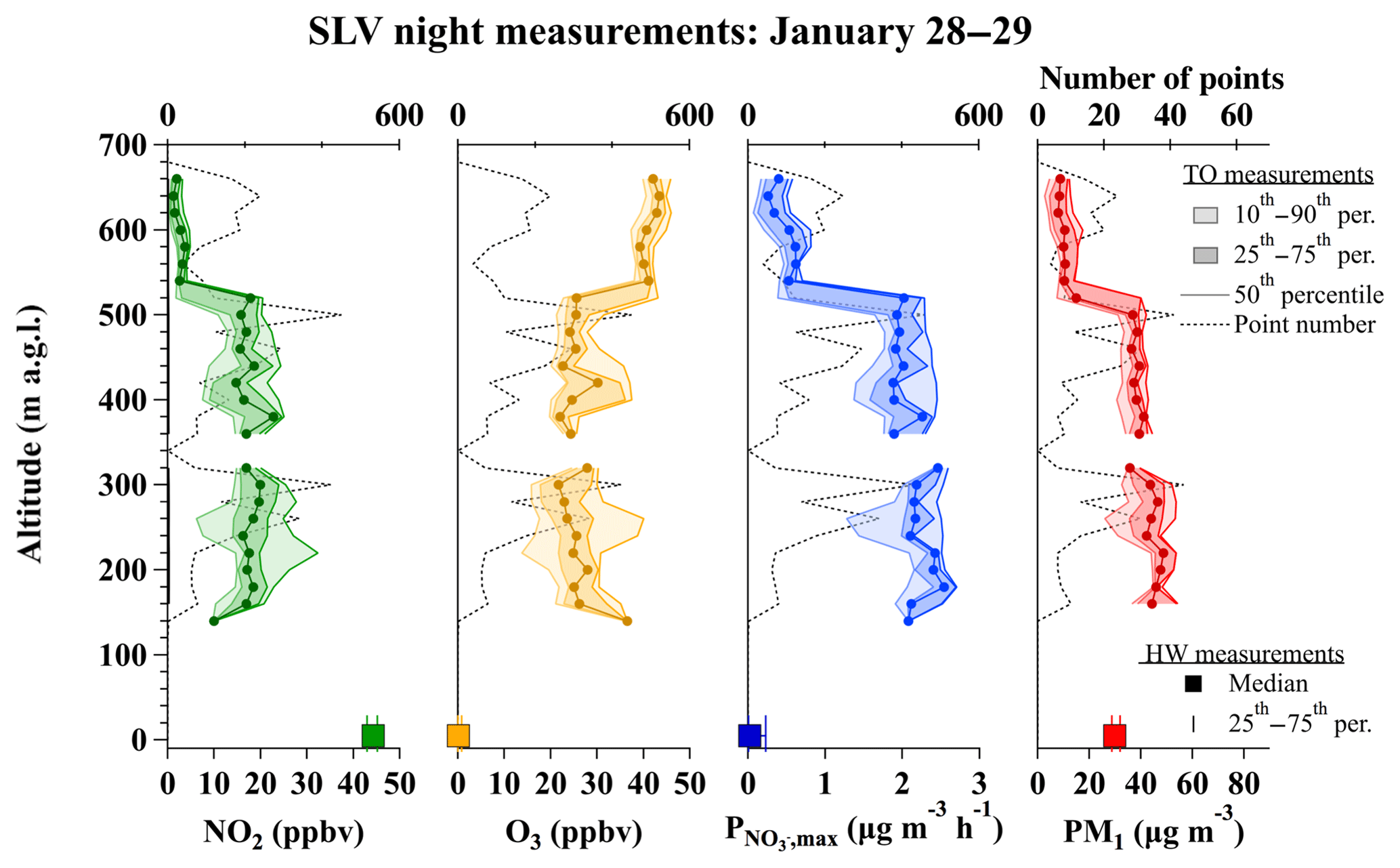
Figure 7Vertical profiles of NO2, O3, (1 s data), and PM1 (10 s data) measured from the TO aircraft during five box patterns, flown over the SLV urban core between 21:20 and 00:30 MST on 28 and 29 January. Percentiles and number of points at each altitude are shown as in Fig. 6. Square markers and error bars represent the median and 25th–75th percentile range of NO2, O3, , and PM2.5 measured concurrently at the HW ground site.
Vertical profiles in Fig. 6b do not show evidence for a reduction in or O3 near the surface, as is expected for O3 titration near the ground level (shown in panel a). The distribution in panel b, however, is affected by the location of the missed approaches/landings in the SLV (Salt Lake International and South Valley Regional airfields), which are further from the urban center of Salt Lake City than the HW ground site (see Fig. 1). Vertical profiles to the surface over urban Salt Lake City were not possible due to a lack of airfields for missed approaches. Instead, the SLV flights often executed box patterns over the eastern Salt Lake basin at several altitudes. Figure 7 shows the vertical distribution of values from these boxes on 28–29 January between 21:20 and 00:30 local time, compared to measured at the surface during the same interval. At 300 and 500 m a.g.l., the median (and interquartile range) was 2.2 (2.1 to 2.4) and 1.9 (1.8 to 2.1) µg m−3 h−1, while at 650 m, slightly above the most concentrated pollution layer, it was 0.5 (0.3 to 0.7) µg m−3 h−1. The median value at the HW ground site, directly below the aircraft, was 0.02 (0.01 to 0.2) µg m−3. These plots demonstrate that is typically low or zero at night near the surface within the urban area of Salt Lake City but large within the RL. Away from the urban area, the vertical distributions of are also likely more uniform (Fig. 6b) due to the lack of O3 titration within the nocturnal boundary layer. In the final section below, nightly integration of these instantaneous values are compared to box model predictions of total nitrate.
3.3.2 Modeled uptake coefficients and production yields
Both the aerosol uptake efficiency of N2O5 (γ(N2O5)) and the production yield of ClNO2 (φ(ClNO2)) are highly variable, dependent on aerosol composition, and can impact the absolute amount of nitrate formed from nocturnal heterogeneous nitrogen chemistry. The nighttime formation of nitrate, however, is only limited by these processes when N2O5 uptake is inefficient and is instead limited by the oxidation rate of NO2 (Reaction R4) (discussed above) at sufficiently large values of γ(N2O5).
As described in Sect. 2.2, an iterative box model was fit to observations of NO2, O3, N2O5, and ClNO2 to quantify γ(N2O5) and φ(ClNO2) during pollution events. For the SLV alone (N=1031), the distribution in Fig. 8 shows that γ(N2O5) values ranged 4 orders of magnitude from to >1 with two modes centered near 0.01 and 0.08. Values approaching or exceeding 1 are unphysical and suggest artifacts in the γ(N2O5) determinations for UWFPS (see below), at least for the largest values. Values of φ(ClNO2) encompassed the entire possible range of 0 to 1 (Fig. 8). The medians for this subset were 0.076 and 0.220 for γ(N2O5) and φ(ClNO2), respectively. For all UWFPS flights between 16 January and 1 February 2017, the median γ(N2O5) and φ(ClNO2) values in the RL (N=2195) were 0.049 and 0.256, respectively, derived from box model fits to observations. These values are compared to multiple derivation methods further below.
Compared to previous studies, the median γ(N2O5) over the SLV was twice as large as the mode derived with a similar model using data from the Nitrogen, Aerosol Composition, and Halogens on a Tall Tower (NACHTT) campaign near Denver, Colorado, in winter 2011 (Wagner et al., 2013). Similarly, the median was over 5 times larger than the median calculated using the same model from the 2015 WINTER campaign (McDuffie et al., 2018b). The largest values during UWFPS exceeded those from both WINTER and NACHTT studies, while the smallest values were also larger than either of the respective minimums. The two most common suppression mechanisms that lead to reductions in γ(N2O5) are associated with the presence of organic material and nitrate in the aerosol phase. Insoluble aerosol organics have been shown to suppress N2O5 uptake in previous laboratory studies (e.g., Griffiths et al., 2009; Thornton et al., 2003; McNeill et al., 2006; Thornton and Abbatt, 2005; Cosman et al., 2008; Badger et al., 2006; Folkers et al., 2003), and large organic mass fractions have been associated with γ(N2O5) reductions in past field studies (Bertram et al., 2009; McDuffie et al., 2018b). The average dry mass fraction of aerosol organics (i.e., organic mass/total dry aerosol mass) during the SLV pollution events was less than half of the average during the WINTER campaign (∼18 % vs. 40 %) and 40 % lower than the average during NACHTT (27 %, Wagner et al., 2013). Aerosol nitrate can also suppress uptake as soluble nitrate facilitates the reformation of gas-phase N2O5 (Bertram and Thornton, 2009; Griffiths et al., 2009), and nitrate mass fractions have been negatively correlated with γ(N2O5) in previous field studies (Wagner et al., 2013; Morgan et al., 2015; Riedel et al., 2012; Bertram et al., 2009; McDuffie et al., 2018b). The presence of sufficient aerosol water, however, can offset this nitrate suppression by promoting N2O5 aqueous solvation and reaction (e.g., Bertram and Thornton, 2009; Griffiths et al., 2009; Mentel et al., 1999; Wahner et al., 1998), resulting in increases in γ(N2O5) with the ratio of water to nitrate (McDuffie et al., 2018b). The average dry mass fraction of aerosol nitrate was much larger during UWFPS (60 %) than during NACHTT (30 %, Wagner et al., 2013) or WINTER (15 %, McDuffie et al., 2018b). High-humidity conditions during UWFPS (77 % average RH during pollution events) resulted in average aerosol water mass fractions (i.e., water mass/aerosol dry mass + water mass) near 70 %, as calculated with an aerosol thermodynamic model, described in Franchin et al. (2018). This higher RH likely contributed to efficient N2O5 uptake during UWFPS despite the presence of aerosol nitrate. In fact, the largest 25 % of UWFPS γ(N2O5) values exceed the largest value (0.175) that has been reported from recent field studies (Fig. 4 in McDuffie et al., 2018b).
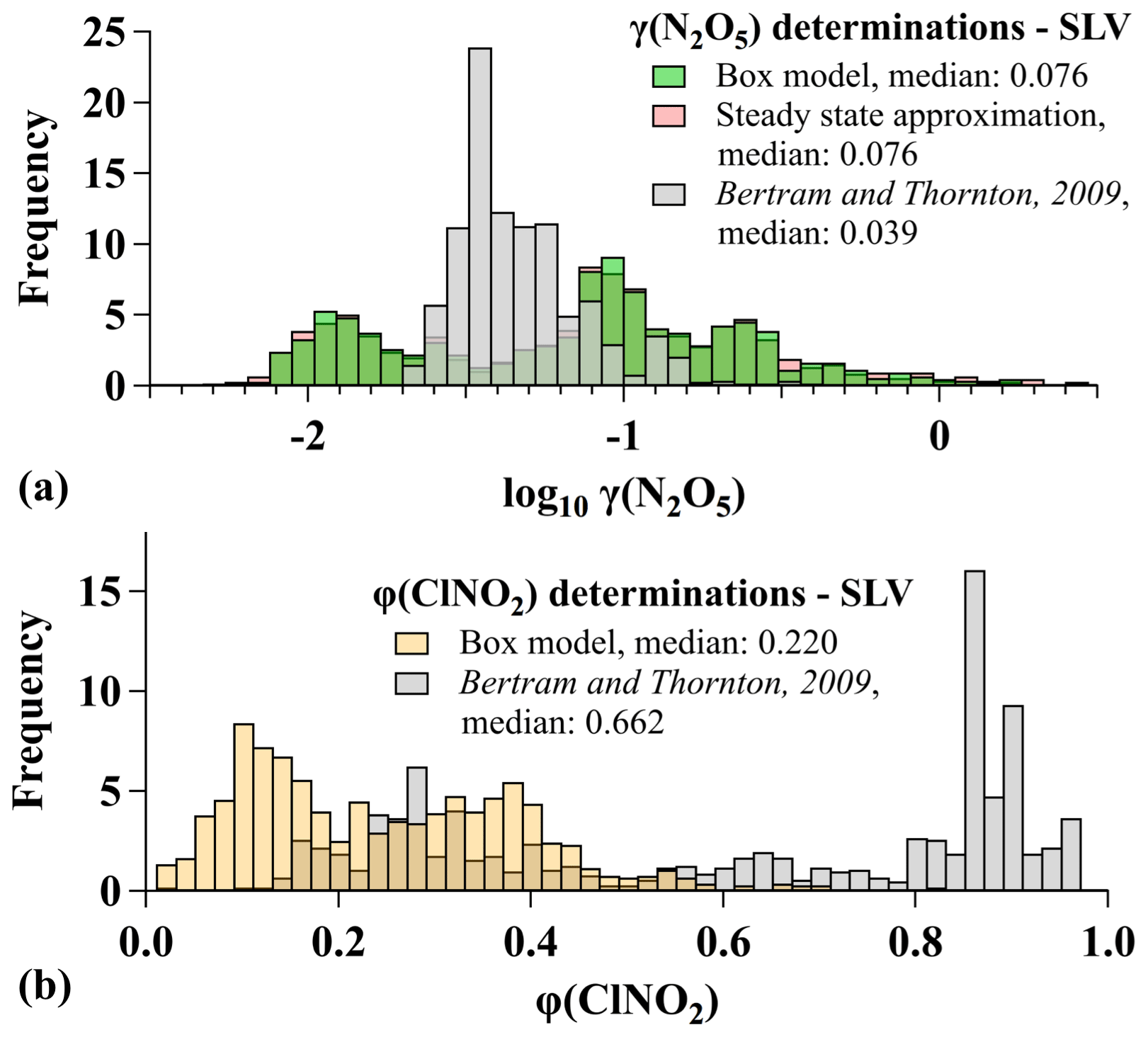
Figure 8(a) Histograms of γ(N2O5) determinations from the SLV during pollution events, calculated with the box model (green), steady-state approximation (pink), and parameterization from Bertram and Thornton (2009). (b) Histograms of φ(ClNO2) determinations from the SLV during pollution events calculated with the box model (gold) and parameterization from Bertram and Thornton (2009) (gray).
The median φ(ClNO2) value of 0.220 during the SLV pollution events was 4 times larger than during the NACHTT campaign (Riedel et al., 2013; Wagner et al., 2013) but within a factor of 2 larger than the median derived during WINTER over the US east coast (McDuffie et al., 2018a). The SLV median was also similar to medians reported from previous ground-based studies across North America (Mielke et al., 2016, 2011, 2013; Wagner et al., 2012; Thornton et al., 2010). Heterogeneous ClNO2 production requires aerosol chloride (Reaction R6) (e.g., Behnke et al., 1997), and though a consistent geographic pattern in φ(ClNO2) has not emerged from past studies (Fig. 2 in McDuffie et al., 2018a), heterogeneous chemistry in the vicinity of the Great Salt Lake appears to produce ClNO2 with the same efficiency as comparable measurements near North American ocean coastlines. ClNO2 production yields, however, remain smaller than those predicted based on measured aerosol composition, as discussed below.
While large γ(N2O5) and moderate φ(ClNO2) values indicate efficient nitrate production from heterogeneous chemistry during UWFPS, these values may be upper and lower limits, respectively. As discussed in Sect. 2.1, limited observations of VOC and photolysis rates, as well as uncertainties in air age, and dilution may cause the and values (and subsequent γ(N2O5) and φ(ClNO2)) to be over- and underpredicted, respectively. This is more likely near sunset where the model has an increased sensitivity to assumptions in simulation start time (McDuffie et al., 2018b). Uncertainties in gas-phase measurements may also contribute to uncertainties in the model predictions, though the level of uncertainty associated with these parameters is small (Table S4). Additional uncertainties in and may arise from model assumptions of constant temperature and RH (i.e., rate constants and surface area) overnight. While model sensitivities to these uncertainties cannot be directly quantified, the percent growth in SA from nitrate accumulation is estimated to be less than the uncertainty in the dry SA measurement (34 %). As modeled values are also consistent with those derived from observations (discussed below), this source of uncertainty is not discussed further. Overall, while the box model has a large number of uncertainties and assumptions, predictions of nocturnal nitrate production, which are subject to uncertainties in both and , are not highly sensitive to sources other than dilution (Table S4).
Independent of the model fits of and , unphysically large γ(N2O5) values (>0.1 in Fig. 8) may alternatively be an artifact arising from an under-measurement of ambient aerosol SA. Low aerosol SA would bias high the γ(N2O5) calculation in Eq. (1) without influencing the model derivations of and . In this study, wet aerosol SA was calculated as described above by applying an RH-dependent growth factor curve to the measured dry PM1 SA. Despite large concentrations of total dry SA (Fig. S2), an underprediction in the wet SA could arise from uncertainties in the hygroscopic growth curve or additional unmeasured SA from large particles (>1 µm). Both factors would be exacerbated by the high-humidity conditions encountered during UWFPS since large, hydrated particles would not be sampled efficiently by the aerosol inlet and hygroscopic growth curves are highly uncertain above ∼95 % RH (corresponding to 6.7 % of the SLV data). A third possible cause of under-measured SA is the presence of fog under high-humidity conditions. Fog is well known to promote rapid heterogeneous processes (Lelieveld and Crutzen, 1990) and is associated with surface areas that can be orders of magnitude larger than accumulation mode aerosol. For example, fog has been demonstrated to lead to rapid N2O5 loss at a ground site in Hong Kong, during November–December 2013 (Brown et al., 2016). It is therefore possible that unmeasured SA under high-humidity conditions could bias the calculated γ(N2O5) values high relative to values reported in previous literature. Any bias caused by aerosol SA, however, would not impact the model-derived and values that are used to calculate nocturnal nitrate production rates in the final analysis below.
To further evaluate the UWFPS γ(N2O5) and φ(ClNO2) values, box model determinations are compared to two other derivation methods in Figs. 8 and S5. The first method calculates γ(N2O5) from observations of temperature, SA, NO2, O3, and N2O5, based on the steady-state approximation (γ(N2O5)ss), described by Brown et al. (2003) and defined in Sect. S4.1. This method shows excellent agreement with box model results (Figs. 8 and S5). The steady-state method has been shown in previous analyses to overpredict γ(N2O5) values under cold, high-NOx conditions, but only if the first-order rate constants for NO3 and N2O5 loss ( and ) are modest (Brown et al., 2003). Both the steady-state and box model γ(N2O5) values are consistent with a rapid first-order loss constant of N2O5 (median s−1), suggesting the steady-state approach is valid for SLV conditions. The corresponding median lifetime () of 14 min is, for example, much shorter than the lifetimes of 2–18 h calculated from a previous steady-state analysis of aircraft measurements over Texas in fall 2006 (Brown et al., 2009). Nevertheless, the color scale in Fig. S5 shows that the largest γ(N2O5) values (≥0.1) were exclusively derived for air sampled within 3 h of sunset (4.3 h simulation time), where previous analysis has shown the steady-state approximation to be least reliable. As Fig. S5 shows large values of γ(N2O5) from both the box model and the steady-state analysis during this time, there may be a common bias between the methods if these values are indeed too large.
The second method calculates both γ(N2O5) and φ(ClNO2) using laboratory-derived parameterizations from Bertram and Thornton (2009) (BT09), based on the aerosol volume-to-surface-area ratio; N2O5 solubility (Fried et al., 1994); aerosol molarities of water, nitrate, and chloride; and laboratory-derived reaction rate constant ratios. Further details of each parameterization are provided in Sect. S4.2. These parameterizations have had mixed success in reproducing previous field-derived values (e.g., Bertram et al., 2009; Riedel et al., 2012; McDuffie et al., 2018a, b) but are commonly used to predict N2O5 uptake and ClNO2 production on internally mixed inorganic aerosol when N2O5 chemistry is included in global models (e.g., Sarwar et al., 2014; Shah et al., 2018).
Results in Fig. S5 show that the median γ(N2O5) value predicted by the BT09 parameterization is within a factor of 2 of the box model median but that this parameterization does not reproduce the observed variability (Fig. 8). For φ(ClNO2), the BT09 parameterization largely overpredicts model-derived values with a median of 0.66 relative to the model median of 0.22 (Fig. S5). This overprediction is consistent with all previous studies to compare parameterized and field-derived φ(ClNO2) results (Wagner et al., 2013; Wang et al., 2017a, b; Ryder et al., 2015; Thornton et al., 2010; Riedel et al., 2013; Tham et al., 2018; McDuffie et al., 2018a). These results also suggest that the parameterization would need to be reduced by 68 % for agreement with the box model median, similar to the 74 %–85 % reduction required for agreement of this parameterization with the WINTER campaign median (McDuffie et al., 2018a). The possible presence of additional, refractory-phase chloride (i.e., NaCl, CaCl2, and KCl) in the accumulation mode would increase the predicted γ(N2O5) and improve agreement with the box model but would further degrade the agreement of φ(ClNO2).
Lastly, the empirical γ(N2O5) parameterization from McDuffie et al. (2018b) was applied to UWFPS data, though only an estimated range for the campaign median is presented due to uncertainties in the aerosol O:C ratio and aerosol organic density, both required for this calculation (discussed in Sect. S4.2). This parameterization models N2O5 uptake onto an aqueous inorganic particle with a resistive organic coating, with the coating thickness determined by the volume ratio of inorganic to total aerosol components (McDuffie et al., 2018b; Riemer et al., 2009; Anttila et al., 2006). By estimating a range of O:C ratios using the improved-ambient O:C ratio method from Canagaratna et al. (2015) and AMS organic m∕z 44 fraction (Fig. 6, Franchin et al., 2018), assuming an organic density of 1.3 g cm−3 (e.g., Kuwata et al., 2012) to estimate the organic-associated volume, and applying additional constants described in Sect. S4.2, this parameterization estimated a median γ(N2O5) between 60 % and 85 % lower than the box model. Though there are large uncertainties in the required parameters, these results suggest that during pollution events (1) aerosol organics are not surface active, (2) aerosol organics are not resistive toward N2O5, or (3) box model γ(N2O5) values are overpredicted due to missing SA (e.g., fog, Sect. 3.3.2) or other simplifying assumptions (e.g., dilution) discussed above.
Despite disagreement between the box model and parameterizations, the γ(N2O5) values predicted by all three methods are large enough, in combination with the large measured aerosol SA, to fall within the range where models of nighttime chemistry are insensitive to variation in uptake efficiency (e.g., Macintyre and Evans, 2010; Riemer et al., 2003; Wang et al., 2018). Under these conditions, the NO2 gas-phase oxidation rate (i.e., ), rather than N2O5 uptake, becomes the limiting factor to HNO3 formation. As further evidence of this limitation, the median lifetime of NO2 with respect to O3 ( was 9 h while the lifetime of N2O5 () was just 14 min, resulting in low N2O5 mixing ratios (median = 0.03 ppbv) during the SLV pollution events (Fig. S6). In addition, explicit box modeling of day- and nighttime chemical processes during UWFPS by Womack et al. (2019) also showed that the production of Ox,total () was insensitive (<1.5 %) to order-of-magnitude changes in γ(N2O5). Short lifetimes of N2O5 relative to NO2 and nitrate insensitivity to γ(N2O5) both indicate that nocturnal heterogeneous chemistry contributes to NH4NO3 formation but that absolute production is limited by gas-phase kinetics rather than aerosol composition and γ(N2O5). This insensitivity to γ(N2O5) provides confidence in the ability of the box model to predict the magnitude of nocturnal nitrate production in the SLV, regardless of uncertainties in γ(N2O5).
3.3.3 Modeled nocturnal nitrate production rates and contribution of heterogeneous chemistry to total NH4NO3 aerosol accumulation rates
As described in Sect. 2.2 and shown in Fig. 3, the box model simulates the amount of total nitrate () produced from heterogeneous chemistry over the course of a single night. This amount of nitrate, in units of µg m−3 per night, is in addition to any nitrate present at sunset from the previous day (e.g., Fig. 3). Figure 9 shows the distribution of nightly nitrate production predicted by base case simulations (N=1031), ranging from ∼0 to 31 µg m−3 nitrate per night, with a median of 9.9 µg m−3 nitrate per night.
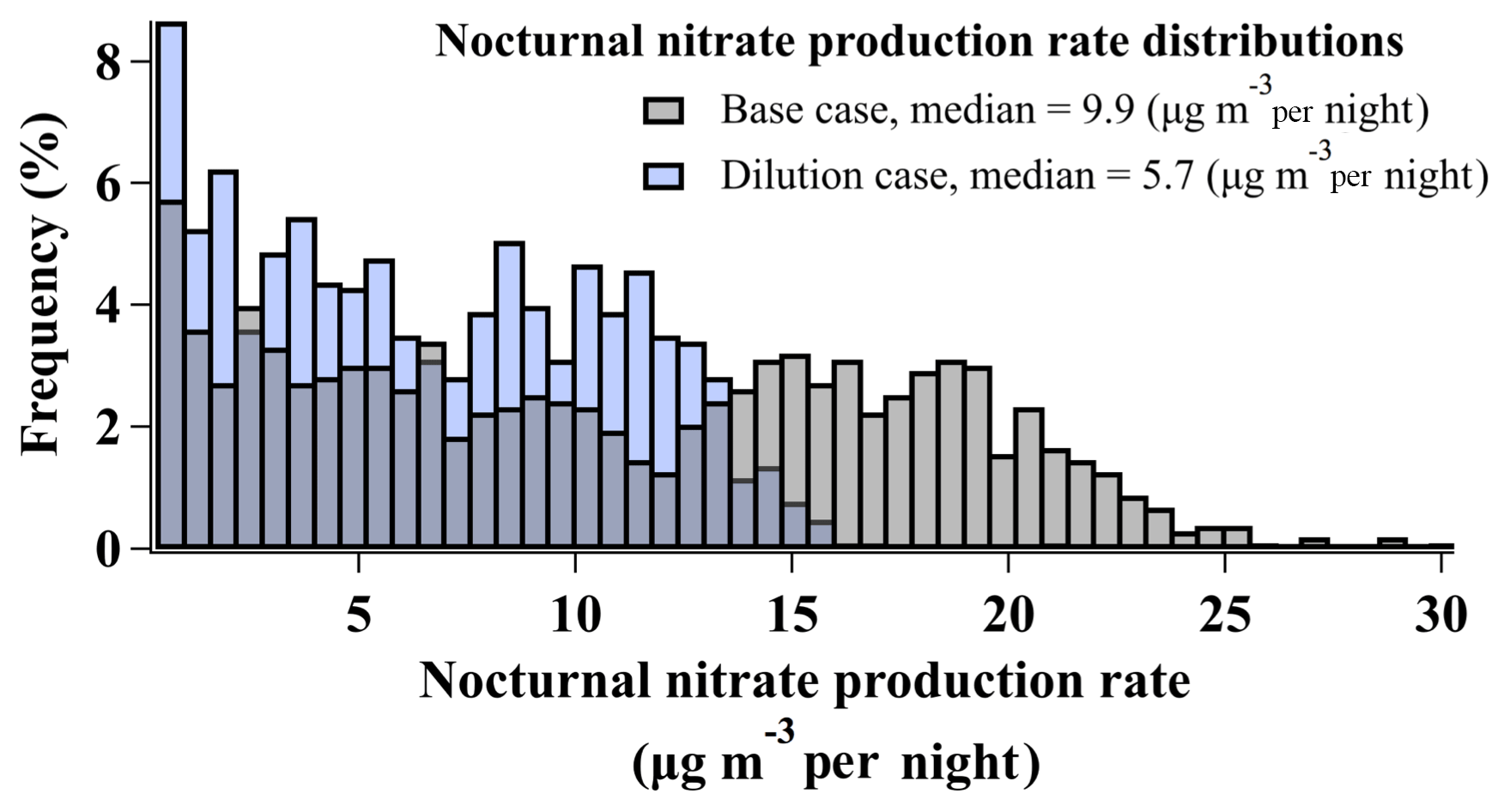
Figure 9Histograms of nocturnal nitrate production rates (µg m−3 per night) predicted by base case simulations and simulations incorporating a first-order dilution loss process with rate constant s−1.
In addition to evidence from the previous section, comparisons between the base case results and integrated values from Sect. 3.3.1 also suggest that nocturnal nitrate production is limited by the rate of NO2 oxidation rather than the efficiency of N2O5 aerosol uptake. Based on the calculations in Sect. 3.1, upper-limit values, integrated over an average 14 h night and reduced to account for a φ(ClNO2) value of 0.2, ranged from <0.5 to >40 µg m−3 per night, with a median of 20.2 µg m−3 per night (N = 21 666). To more directly compare with box model results, the subset of points with simultaneous γ(N2O5) determinations had a median of 10.6 µg m−3 per night, which is slightly larger but agrees well with the box-model-predicted median of 9.9 µg m−3 per night. As described in Sect. 3.3.1, the calculation assumes efficient N2O5 uptake and only considers nitrate production to be limited by gas-phase kinetics. Observed agreement between the integrated values and box-model-predicted production rates therefore suggests that nitrate production may be largely limited by gas-phase oxidation rather than multiphase processes. As a result, the large variability in predicted nitrate production rates is reflective of the variability in the observed NO3 radical production rates (Fig. 6).
Uncertainties associated with base case production rates are discussed in Sect. 2.2.2 and shown as a time series in Fig. S3. Air parcel dilution associated with the vertical entrainment of air from the free troposphere (Sect. S1.4.1) was the largest source of uncertainty (Table S4, Fig. S3). This process was not included in base case simulations, though mixing/dilution has been observed and predicted in analyses of WINTER nighttime flights (Kenagy et al., 2018; McDuffie et al., 2018b). Estimating the impact of dilution by including a single first-order dilution rate constant (kdilution) of s−1 reduced the median nocturnal nitrate production rate by 42 % to 5.7 µg m−3 per night and resulted in a smaller range of production rates (∼0 to 16 µg m−3 per night) relative to base case simulations in Fig. 9. As described in Womack et al. (2019) (and in Sect. S1.4.1), a single first-order dilution rate constant of s−1 was derived for pollution event no. 4 in the SLV by fitting a box model to best reproduce the day-to-day buildup of observed Ox,total between 28 January and 1 February at the UU ground site. In the model described by Womack et al. (2019), this rate constant was then scaled up by 40 % when simulating the nocturnal RL in order to maintain constant dilution and account for the reduced volume relative to the mixed daytime boundary layer. While dilution/entrainment rates may vary from day to night, the method of Womack et al. (2019) represents the single number that would best represent the average rate. The same procedure is followed here with a resulting kdilution value of s−1, which is ∼60 % lower than kdilution from the WINTER campaign, derived from observations of NOy () overnight in a single RL air parcel over the eastern US coast (McDuffie et al., 2018b). As processes relevant to RL dilution were not directly measured during UWFPS, there are uncertainties associated with this kdilution estimation. For instance, based on the modeled surface albedo in Womack et al. (2019), kdilution could have reproduced observed Ox,total mixing ratios with scaled values ranging between and s−1 (Fig. S10, Womack et al., 2019). This particular range of loss rate constants predicts median nitrate production rates in the SLV between 3.6 and 6.1 µg m−3 per night.
Modeled nitrate production rates are further compared in Fig. 10 to the average daily accumulation of surface-level nitrate aerosol during pollution event no. 4 at the HW ground site. This ground-based accumulation rate (red diamond in Fig. 10a) was taken as the slope of the 24 h average PM2.5 observations at HW (scaled by 0.58; average fraction from Fig. 4) during the first 6 d of event no. 4, before it began to degrade on 1 February 2017 (Fig. 10b). Only data from event no. 4 are assessed here as this was the only PCAP sampled with the aircraft on multiple nights. Figure 10a shows this average, 24 h surface accumulation rate of 4.6 µg m−3 d−1 (red diamond) compared to the 10th–90th percentile distributions, medians, and averages of the nocturnal production rates predicted by base case box model simulations (gray) and simulations including the effects of 24 h dilution (blue), described below.
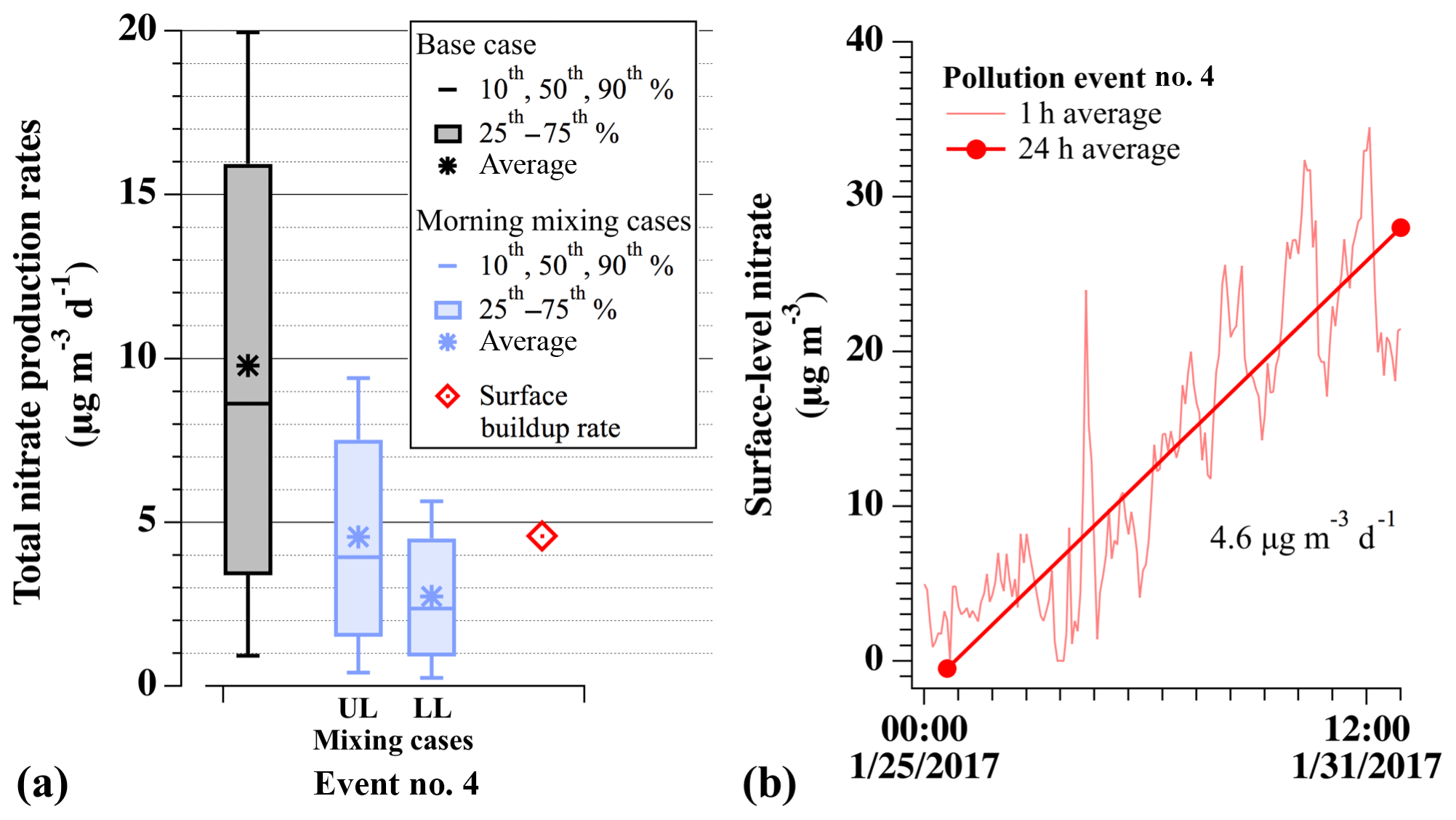
Figure 10(a) For pollution event no. 4, comparison of model-predicted nocturnal nitrate production (µg m−3 d−1) for base case simulations (gray), simulations with 24 h of dilution (blue), and the average daily nitrate buildup observed at HW (red). Dilution cases are for simulations that incorporate nocturnal dilution rate constants of (L), (M), and (H) s−1, scaled by 60 % during the day. Box-and-whisker plots show the 10th–90th percentile distributions of each set. Upper-limit (UL) values assume morning mixing between equivalent nitrate concentrations produced in the RL and NBL. Lower-limit (LL) values assume morning mixing with no nitrate production in the NBL. The red diamond shows the ground-based buildup rate, calculated from 24 h averaged data at HW in panel (b). (b) Observed concentrations and average daily buildup rate of nitrate aerosol mass (total mass ⋅ 0.58) at HW during event no. 4.
Comparing modeled RL chemical nitrate production to the observed ground-based accumulation rate can provide an estimate for the fractional contribution of N2O5 uptake to total particulate nitrate production in the SLV. Direct comparison is difficult, however, as the 24 h ground-based accumulation rate includes contributions from photochemistry and nocturnal formation in the RL and nocturnal boundary layer (NBL) and is impacted by dilution and mixing processes. For example, the amount of nocturnally produced nitrate at the surface will depend on mixing between the NBL and RL during morning boundary layer expansion (Fig. 2). In Fig. 10a, the median base case prediction without dilution or mixing (gray, 8.6 µg m−3 per night) was nearly twice as large as the 24 h average accumulation rate observed at the surface during the same event (4.6 µg m−3 per night, red). Therefore, to more directly compare box model predictions and ground-based observations, Fig. 10a also shows the results from two simulations that include upper- and lower-limit estimates of loss from nocturnal and daytime dilution. For both scenarios, the nighttime (0–14 h) kdilution value of s−1 (blue) was applied to all modeled species as described above. At sunrise, morning mixing between the NBL (taken as 40 % by volume) and RL (taken as 60 % by volume) was then estimated using the assumed volume ratio between the two layers and assuming either equivalent nocturnal nitrate production in both layers (upper limit) or no production in the NBL (lower limit). Nocturnal production in the NBL is expected to be suppressed relative to the RL due to O3 titration (e.g., Fig. 6 here and Fig. S6 in Womack et al., 2019), making the assumption of equivalence an upper-limit estimate to nocturnally produced nitrate at the surface after morning mixing. The upper-limit case required no reduction of the model-predicted nitrate concentrations at sunrise (e.g., Fig. 9), whereas these concentrations were instantaneously reduced by 40 % for the lower-limit case. To account for daytime dilution in the remaining ∼10 h, morning concentrations for both cases were further diluted with the daytime boundary dilution rate constant from Womack et al. (2019) ( s−1), described above and in Sect. S1.4.1. For a single 24 h period, this resulted in a net median of 2.4 to 3.9 µg m−3 nitrate produced from nocturnal heterogeneous N2O5 uptake for the lower and upper-limit cases, respectively. When considering the entire possible range of dilution rate constants from Womack et al. (2019), the median values from both cases were between 1.1 and 4.2 µg m−3 d−1, as shown in Fig. S7.
Comparison of modeled rates to the observed surface buildup of 4.6 µg m−3 d−1 suggests that, on average, nitrate produced from heterogenous chemistry can account for at least 50 % of the nitrate accumulation observed at the surface. This result is qualitatively consistent with an observational analysis by Pusede et al. (2016), who determined that nocturnal heterogeneous chemistry was the main source of regional aerosol nitrate during wintertime pollution events in the San Joaquin Valley. The lower-limit estimate, however, is also similar to a box model analysis of this same event by Womack et al. (2019), who found roughly equal contributions between photochemical and nocturnal nitrate production pathways, highlighting that photochemical nitrate production is also occurring during these events. Therefore, while results in Fig. 10a (including dilution) predict a median nocturnal fractional contribution of 52 %–85 % (ranging between 24 % and 91 %; Fig. S7), confirmation and further quantification of this result will require additional, vertically resolved measurements of aerosol composition, gas-phase precursors, and physical parameters, as well as more sophisticated modeling of these multiday pollution accumulation events with 3-D chemical transport models.
Aerosol- and gas-phase measurements collected during the 2017 UWFPS campaign showed multiple pollution events that exceeded PM2.5 standards in the SLV, the most populated region in the state of Utah. During these events, aerosol particles were largely composed of NH4NO3, which forms from the reaction between gas-phase NH3 and HNO3. While NH3 is emitted from surface sources, HNO3 is chemically formed from the oxidation of NOx emissions. This oxidation can occur through daytime reactions with the photochemical OH radical or through nocturnal heterogeneous reactions involving NO3 and N2O5. The contribution of nocturnal chemistry to PM2.5 formation in the SLV is dependent on whether NH4NO3 formation is NH3 or HNO3 limited, as well as the NO3 production rate, N2O5 uptake efficiency, ClNO2 and HNO3 production yields, and loss processes such as air parcel dilution.
Vertically resolved measurements of gas- and particulate-phase oxidized and reduced nitrogen in the SLV showed that NH4NO3 formation during pollution events was nearly always HNO3 limited but that oxidized and reduced nitrogen approached equivalence as pollution events progressed. This reagent balance analysis is consistent with aerosol thermodynamic modeling presented in Franchin et al. (2018), which predicted that all three major valleys in the Wasatch region were sensitive to nitrate reductions and that the SLV was also sensitive to NH3 reductions. Both observation- and modeling-based analyses agreed that NH4NO3 formation in the RL was largely HNO3 limited during pollution events, providing the possibility of a large contribution from nocturnal heterogeneous chemistry to HNO3 and PM2.5 mass.
Analysis of vertically resolved, calculated nitrate production rates (an upper-limit estimate due to heterogeneous HNO3 formation, ) and results from an observationally constrained chemical box model suggest that nocturnal chemistry is an efficient mechanism for PM2.5 production in the SLV during pollution events. Nitrate production rates had a median of 1.6 µg m−3 h−1, while values of γ(N2O5) and φ(ClNO2) had medians of 0.076 and 0.220, respectively, during pollution events. Values of γ(N2O5) were larger than previous field-based determinations (e.g., McDuffie et al., 2018b) and those predicted from the Bertram and Thornton (2009) parameterization but were in agreement with values derived using the N2O5 steady-state approach. The median φ(ClNO2) value was larger than that derived from aircraft observations over the eastern US coast but was simultaneously overpredicted by 68 % by the Bertram and Thornton (2009) parameterization, which uses measurements of aerosol chloride and aerosol water estimations.
While the box model has uncertainties associated with limited available measurements and model assumptions, the large measured aerosol SA, efficient N2O5 uptake coefficients, and moderate ClNO2 yields resulted in nightly modeled nitrate production rates that were largely insensitive to specific values of derived parameters. Agreement between base case modeled nightly nitrate production (9.9 µg m−3 per night) and that calculated from values (10.6 µg m−3 per night) alternatively suggests that nitrate production is more sensitive to gas-phase NO2 oxidation rates than γ(N2O5), providing confidence in the model's predictions of nocturnal nitrate. Of the parameters tested, the model was most sensitive to loss through air parcel dilution, with a 42 % reduction to 5.7 µg m−3 nitrate per night when including a nocturnal kdilution rate constant of s−1. When considering the possible effects of 24 h dilution, model simulations predicted a reduced median of 2.4–3.9 µg m−3 nitrate d−1, corresponding to 52 %–85 % (median) of the net aerosol nitrate accumulation that was observed at a SLV ground site. Due to model uncertainties and sensitivities to dilution, further quantification of this result will require additional vertically resolved measurements and photochemical/3-D modeling analyses. These results, however, highlight the importance of nocturnal chemistry in the formation of PM2.5 in the SLV and can provide constraints for regulatory models of PM2.5 that are used to assess control strategies in this populated nonattainment area.
Data from the UWFPS campaign can be found at the NOAA website: https://www.esrl.noaa.gov/csd/field.html (last access: March 2019). Code written in IGOR Pro for the iterative box model can be found at https://esrl.noaa.gov/csd/groups/csd7/measurements/2015winter/pubs/ (last access: March 2019). All referenced supplemental text, figures, and tables can be found in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-9287-2019-supplement.
During the UWFPS campaign, EEM, CCW, DLF, and WPD were responsible for the CRD gas-phase measurements; AF and AM for the AMS particle measurements; LG, BHL, and JAT for the I−ToF-CIMS measurements; and AM and JM for the NH3 instrument. MB and SSB organized the UWFPS campaign with technical support from WPD. EEM developed the box model code and preformed the analyses with support from CCW and SSB. EEM prepared the manuscript with contributions from coauthors.
The authors declare that they have no conflict of interest.
The authors would like to thank NOAA Aircraft Operations, staff, and pilots deployed as part of the UWFPS campaign, Jason Clark, Rob Mitchell, and Rob Militec.
NOAA acknowledges support for Twin Otter flights from the Utah Division of Air Quality under agreement number 16-049696.
This paper was edited by Yafang Cheng and reviewed by two anonymous referees.
Anttila, T., Kiendler-Scharr, A., Tillmann, R., and Mentel, T. F.: On the reactive uptake of gaseous compounds by organic-coated aqueous aerosols: Theoretical analysis and application to the heterogeneous hydrolysis of N2O5, J. Phys. Chem. A, 110, 10435–10443, https://doi.org/10.1021/jp062403c, 2006.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Baasandorj, M., Hoch, S. W., Bares, R., Lin, J. C., Brown, S. S., Millet, D. B., Martin, R., Kelly, K., Zarzana, K. J., Whiteman, C. D., Dube, W. P., Tonnesen, G., Jaramillo, I. C., and Sohl, J.: Coupling between chemical and meteorological processes under persistent cold-air pool conditions: Evolution of wintertime PM2.5 pollution events and N2O5 observations in Utah's Salt Lake Valley, Environ. Sci. Technol., 51, 5941–5950, https://doi.org/10.1021/acs.est.6b06603, 2017.
Badger, C. L., Griffiths, P. T., George, I., Abbatt, J. P. D., and Cox, R. A.: Reactive uptake of N2O5 by aerosol particles containing mixtures of humic acid and ammonium sulfate, J. Phys. Chem. A, 110, 6986–6994, https://doi.org/10.1021/jp0562678, 2006.
Bahreini, R., Ervens, B., Middlebrook, A. M., Warneke, C., de Gouw, J. A., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., Neuman, J. A., Ryerson, T. B., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J. S., Peischl, J., Richter, D., Walega, J., Weibring, P., Wollny, A. G., and Fehsenfeld, F. C.: Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas, J. Geophys. Res.-Atmos., 114, D00F16, https://doi.org/10.1029/2008JD011493, 2009.
Beard, J. D., Beck, C., Graham, R., Packham, S. C., Traphagan, M., Giles, R. T., and Morgan, J. G.: Winter temperature inversions and emergency department visits for asthma in Salt Lake County, Utah, 2003–2008, Environ. Health Persp., 120, 1385–1390, https://doi.org/10.1289/ehp.1104349, 2012.
Behera, S. N., Sharma, M., Aneja, V. P., and Balasubramanian, R.: Ammonia in the atmosphere: a review on emission sources, atmospheric chemistry and deposition on terrestrial bodies, Environ. Sci. Pollut. R., 20, 8092–8131, https://doi.org/10.1007/s11356-013-2051-9, 2013.
Behnke, W., George, C., Scheer, V., and Zetzsch, C.: Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments, J. Geophys. Res.-Atmos., 102, 3795–3804, https://doi.org/10.1029/96JD03057, 1997.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Bertram, T. H., Thornton, J. A., Riedel, T. P., Middlebrook, A. M., Bahreini, R., Bates, T. S., Quinn, P. K., and Coffman, D. J.: Direct observations of N2O5 reactivity on ambient aerosol particles, Geophys. Res. Lett., 36, L19803, https://doi.org/10.1029/2009GL040248, 2009.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Brock, C. A., Wagner, N. L., Anderson, B. E., Attwood, A. R., Beyersdorf, A., Campuzano-Jost, P., Carlton, A. G., Day, D. A., Diskin, G. S., Gordon, T. D., Jimenez, J. L., Lack, D. A., Liao, J., Markovic, M. Z., Middlebrook, A. M., Ng, N. L., Perring, A. E., Richardson, M. S., Schwarz, J. P., Washenfelder, R. A., Welti, A., Xu, L., Ziemba, L. D., and Murphy, D. M.: Aerosol optical properties in the southeastern United States in summer – Part 1: Hygroscopic growth, Atmos. Chem. Phys., 16, 4987–5007, https://doi.org/10.5194/acp-16-4987-2016, 2016.
Brown, S. G., Hyslop, N. P., Roberts, P. T., McCarthy, M. C., and Lurmann, F. W.: Wintertime Vertical Variations in Particulate Matter (PM) and Precursor Concentrations in the San Joaquin Valley during the California Regional Coarse PM/Fine PM Air Quality Study, J. Air Waste Manage., 56, 1267–1277, https://doi.org/10.1080/10473289.2006.10464583, 2006.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Brown, S. S., Stark, H., and Ravishankara, A. R.: Applicability of the steady state approximation to the interpretation of atmospheric observations of NO3 and N2O5, J. Geophys. Res., 108, 4539, https://doi.org/10.1029/2003jd003407, 2003.
Brown, S. S., Dibb, J. E., Stark, H., Aldener, M., Vozella, M., Whitlow, S., Williams, E. J., Lerner, B. M., Jakoubek, R., Middlebrook, A. M., DeGouw, J. A., Warneke, C., Goldan, P. D., Kuster, W. C., Angevine, W. M., Sueper, D. T., Quinn, P. K., Bates, T. S., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. R.: Nighttime removal of NOx in the summer marine boundary layer, Geophys. Res. Lett., 31, L07108, https://doi.org/10.1029/2004gl019412, 2004.
Brown, S. S., Osthoff, H. D., Stark, H., Dubé, W. P., Ryerson, T. B., Warneke, C., de Gouw, J. A., Wollny, A. G., Parrish, D. D., Fehsenfeld, F. C., and Ravishankara, A. R.: Aircraft observations of daytime NO3 and N2O5 and their implications for tropospheric chemistry, J. Photoch. Photobio. A, 176, 270–278, https://doi.org/10.1016/j.jphotochem.2005.10.004, 2005.
Brown, S. S., Dubé, W. P., Osthoff, H. D., Stutz, J., Ryerson, T. B., Wollny, A. G., Brock, C. A., Warneke, C., de Gouw, J. A., Atlas, E., Neuman, J. A., Holloway, J. S., Lerner, B. M., Williams, E. J., Kuster, W. C., Goldan, P. D., Angevine, W. M., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Vertical profiles in NO3 and N2O5 measured from an aircraft: Results from the NOAA P-3 and surface platforms during the New England Air Quality Study 2004, J. Geophys. Res., 112, D22304, https://doi.org/10.1029/2007jd008883, 2007.
Brown, S. S., Dubé, W. P., Fuchs, H., Ryerson, T. B., Wollny, A. G., Brock, C. A., Bahreini, R., Middlebrook, A. M., Neuman, J. A., Atlas, E., Roberts, J. M., Osthoff, H. D., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: Comparison to current model parameterizations, J. Geophys. Res.-Atmos., 114, D00F10, https://doi.org/10.1029/2008JD011679, 2009.
Brown, S. S., Dubé, W. P., Tham, Y. J., Zha, Q., Xue, L., Poon, S., Wang, Z., Blake, D. R., Tsui, W., Parrish, D. D., and Wang, T.: Nighttime chemistry at a high altitude site above Hong Kong, J. Geophys. Res.-Atmos., 121, 2457–2475, https://doi.org/10.1002/2015JD024566, 2016.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cerully, K. M., Bougiatioti, A., Hite Jr., J. R., Guo, H., Xu, L., Ng, N. L., Weber, R., and Nenes, A.: On the link between hygroscopicity, volatility, and oxidation state of ambient and water-soluble aerosols in the southeastern United States, Atmos. Chem. Phys., 15, 8679–8694, https://doi.org/10.5194/acp-15-8679-2015, 2015.
Chen, L.-W. A., Watson, J. G., Chow, J. C., Green, M. C., Inouye, D., and Dick, K.: Wintertime particulate pollution episodes in an urban valley of the Western US: a case study, Atmos. Chem. Phys., 12, 10051–10064, https://doi.org/10.5194/acp-12-10051-2012, 2012.
Cosman, L. M., Knopf, D. A., and Bertram, A. K.: N2O5 reactive uptake on aqueous sulfuric acid solutions coated with branched and straight-chain insoluble organic surfactants, J. Phys. Chem. A, 112, 2386–2396, https://doi.org/10.1021/jp710685r, 2008.
Ellis, R. A., Murphy, J. G., Pattey, E., van Haarlem, R., O'Brien, J. M., and Herndon, S. C.: Characterizing a Quantum Cascade Tunable Infrared Laser Differential Absorption Spectrometer (QC-TILDAS) for measurements of atmospheric ammonia, Atmos. Meas. Tech., 3, 397–406, https://doi.org/10.5194/amt-3-397-2010, 2010.
Environmental Protection Agency: Ambinet Monitoring Technology Information Center, available at: https://www.epa.gov/amtic, last access: September 2018.
Folkers, M., Mentel, T. F., and Wahner, A.: Influence of an organic coating on the reactivity of aqueous aerosols probed by the heterogeneous hydrolysis of N2O5, Geophys. Res. Lett., 30, 1644, https://doi.org/10.1029/2003GL017168, 2003.
Franchin, A., Fibiger, D. L., Goldberger, L., McDuffie, E. E., Moravek, A., Womack, C. C., Crosman, E. T., Docherty, K. S., Dube, W. P., Hoch, S. W., Lee, B. H., Long, R., Murphy, J. G., Thornton, J. A., Brown, S. S., Baasandorj, M., and Middlebrook, A. M.: Airborne and ground-based observations of ammonium-nitrate-dominated aerosols in a shallow boundary layer during intense winter pollution episodes in northern Utah, Atmos. Chem. Phys., 18, 17259–17276, https://doi.org/10.5194/acp-18-17259-2018, 2018.
Fried, A., Henry, B. E., Calvert, J. G., and Mozurkewich, M.: The reaction probability of N2O5 with sulfuric acid aerosols at stratospheric temperatures and compositions, J. Geophys. Res.-Atmos., 99, 3517–3532, https://doi.org/10.1029/93JD01907, 1994.
Fuchs, H., Dube, W. P., Lerner, B. M., Wagner, N. L., Williams, E. J., and Brown, S. S.: A sensitive and versatile detector for atmospheric NO2 and NOx based on blue diode laser cavity ring-down spectroscopy, Environ. Sci. Technol., 43, 7831–7836, https://doi.org/10.1021/es902067h, 2009.
Gillies, R. R., Wang, S.-Y., and Booth, M. R.: Atmospheric Scale Interaction on Wintertime Intermountain West Low-Level Inversions, Weather Forecast., 25, 1196–1210, https://doi.org/10.1175/2010WAF2222380.1, 2010.
Green, M. C., Chow, J. C., Watson, J. G., Dick, K., and Inouye, D.: Effects of Snow Cover and Atmospheric Stability on Winter PM2.5 Concentrations in Western U.S. Valleys, J. Appl. Meteorol. Clim., 54, 1191–1201, https://doi.org/10.1175/JAMC-D-14-0191.1, 2015.
Griffiths, P. T., Badger, C. L., Cox, R. A., Folkers, M., Henk, H. H., and Mentel, T. F.: Reactive uptake of N2O5 by aerosols containing dicarboxylic acids. Effect of particle phase, composition, and nitrate content, J. Phys. Chem. A, 113, 5082–5090, https://doi.org/10.1021/jp8096814, 2009.
Hansen, J. C., Woolwine Iii, W. R., Bates, B. L., Clark, J. M., Kuprov, R. Y., Mukherjee, P., Murray, J. A., Simmons, M. A., Waite, M. F., Eatough, N. L., Eatough, D. J., Long, R., and Grover, B. D.: Semicontinuous PM2.5 and PM10 Mass and Composition Measurements in Lindon, Utah, during Winter 2007, J. Air Waste Manage., 60, 346–355, https://doi.org/10.3155/1047-3289.60.3.346, 2010.
IUPAC: Data Sheet NOx28, available at: http://iupac.pole-ether.fr/htdocs/datasheets/pdf/NOx28_NO2_O3.pdf (last access: March 2018), 2008.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525, https://doi.org/10.1126/science.1180353, 2009.
Kelly, K. E., Kotchenruther, R., Kuprov, R., and Silcox, G. D.: Receptor model source attributions for Utah's Salt Lake City airshed and the impacts of wintertime secondary ammonium nitrate and ammonium chloride aerosol, J. Air Waste Manage., 63, 575–590, https://doi.org/10.1080/10962247.2013.774819, 2013.
Kenagy, H. S., Sparks, T. L., Ebben, C. J., Wooldridge, P. J., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., McDuffie, E. E., Fibiger, D. L., Brown, S. S., Montzka, D. D., Weinheimer, A. J., Schroder, J. C., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Dibb, J., Apel, E., Campos, T., Shah, V., Jaegle, L., and Cohen, R. C.: NOx Lifetime and NOy Partitioning During WINTER, J. Geophys. Res.-Atmos., 123, 9813–9827, https://doi.org/10.1029/2018JD028736, 2018.
Kuprov, R., Eatough, D. J., Cruickshank, T., Olson, N., Cropper, P. M., and Hansen, J. C.: Composition and secondary formation of fine particulate matter in the Salt Lake Valley: Winter 2009, J. Air Waste Manage., 64, 957–969, https://doi.org/10.1080/10962247.2014.903878, 2014.
Kuwata, M., Zorn, S. R., and Martin, S. T.: Using Elemental Ratios to Predict the Density of Organic Material Composed of Carbon, Hydrogen, and Oxygen, Environ. Sci. Technol., 46, 787–794, https://doi.org/10.1021/es202525q, 2012.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurtén, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017JD028082, 2018.
Lelieveld, J. and Crutzen, P. J.: Influences of cloud photochemical processes on tropospheric ozone, Nature, 343, 227–233, 1990.
Livingston, C., Rieger, P., and Winer, A.: Ammonia emissions from a representative in-use fleet of light and medium-duty vehicles in the California South Coast Air Basin, Atmos. Environ., 43, 3326–3333, https://doi.org/10.1016/j.atmosenv.2009.04.009, 2009.
Long, R. W., Eatough, N. L., Mangelson, N. F., Thompson, W., Fiet, K., Smith, S., Smith, R., Eatough, D. J., Pope, C. A., and Wilson, W. E.: The measurement of PM2.5, including semi-volatile components, in the EMPACT program: results from the Salt Lake City Study, Atmos. Environ., 37, 4407–4417, https://doi.org/10.1016/S1352-2310(03)00585-5, 2003.
Long, R. W., Eatough, N. L., Eatough, D. J., Meyer, M. B., and Wilson, W. E.: Continuous Determination of Fine Particulate Matter Mass in the Salt Lake City Environmental Monitoring Project: A Comparison of Real-Time and Conventional TEOM Monitor Results, J. Air Waste Manage., 55, 1782–1796, https://doi.org/10.1080/10473289.2005.10464776, 2005a.
Long, R. W., Modey, W. K., Smith, P. S., Smith, R., Merrill, C., Pratt, J., Stubbs, A., Eatough, N. L., Eatough, D. J., Malm, W. C., and Wilson, W. E.: One- and Three-Hour PM2.5 Characterization, Speciation, and Source Apportionment Using Continuous and Integrated Samplers, Aerosol Sci. Tech., 39, 238–248, https://doi.org/10.1080/027868290925633, 2005b.
Macintyre, H. L. and Evans, M. J.: Sensitivity of a global model to the uptake of N2O5 by tropospheric aerosol, Atmos. Chem. Phys., 10, 7409–7414, https://doi.org/10.5194/acp-10-7409-2010, 2010.
Malek, E., Davis, T., Martin, R. S., and Silva, P. J.: Meteorological and environmental aspects of one of the worst national air pollution episodes (January, 2004) in Logan, Cache Valley, Utah, USA, Atmos. Res., 79, 108–122, https://doi.org/10.1016/j.atmosres.2005.05.003, 2006.
Mangelson, N. F., Lewis, L., Joseph, J. M., Cui, W., Machir, J., Eatough, D. J., Rees, L. B., Wilkerson, T., and Jensen, D. T.: The contribution of sulfate and nitrate to atmospheric fine particles during winter inversion fogs in Cache Valley, Utah, J. Air Waste Manage., 47, 167–175, https://doi.org/10.1080/10473289.1997.10464429, 1997.
Martin, R.: Cache Valley Air Quality Studies, available at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.127.8179&rep=rep1&type=pdf (last access: April 2018), 2006.
McDuffie, E. E., Fibiger, D. L., Dubé, W. P., Lopez Hilfiker, F., Lee, B. H., Jaeglé, L., Guo, H., Weber, R. J., Reeves, J. M., Weinheimer, A. J., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Dibb, J. E., Veres, P., Ebben, C., Sparks, T. L., Wooldridge, P. J., Cohen, R. C., Campos, T., Hall, S. R., Ullmann, K., Roberts, J. M., Thornton, J. A., and Brown, S. S.: ClNO2 Yields From Aircraft Measurements During the 2015 WINTER Campaign and Critical Evaluation of the Current Parameterization, J. Geophys. Res.-Atmos., 123, 12994–13015, https://doi.org/10.1029/2018JD029358, 2018a.
McDuffie, E. E., Fibiger, D. L., Dubé, W. P., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., Shah, V., Jaegle, L., Guo, H., Weber, R. J., Reeves, J. M., Weinheimer, A. J., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Dibb, J. E., Veres, P., Ebben, C., Sparks, T. L., Wooldridge, P. J., Cohen, R. C., Hornbrook, R. S., Apel, E. C., Campos, T., Hall, S. R., Ullmann, K., and Brown, S. S.: Heterogeneous N2O5 uptake during winter: Aircraft measurements during the 2015 WINTER campaign and critical evaluation of current parameterizations, J. Geophys. Res.-Atmos., 123, 4345–4372, https://doi.org/10.1002/2018JD028336, 2018b.
McNeill, V. F., Patterson, J., Wolfe, G. M., and Thornton, J. A.: The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol, Atmos. Chem. Phys., 6, 1635–1644, https://doi.org/10.5194/acp-6-1635-2006, 2006.
Mei, F., Hayes, P. L., Ortega, A., Taylor, J. W., Allan, J. D., Gilman, J., Kuster, W., de Gouw, J., Jimenez, J. L., and Wang, J.: Droplet activation properties of organic aerosols observed at an urban site during CalNex-LA, J. Geophys. Res.-Atmos., 118, 2903–2917, https://doi.org/10.1002/jgrd.50285, 2013.
Mentel, T., F, Sohn, M., and Wahner, A.: Nitrate effect in the heterogeneous hydrolysis of dinitrogen pentoxide on aqueous aerosols, Phys. Chem. Chem. Phys., 1, 5451–5457, https://doi.org/10.1039/A905338G, 1999.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of ClNO2 in a Mid-Continental Urban Environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Mielke, L. H., Stutz, J., Tsai, C., Hurlock, S. C., Roberts, J. M., Veres, P. R., Froyd, K. D., Hayes, P. L., Cubison, M. J., Jimenez, J. L., Washenfelder, R. A., Young, C. J., Gilman, J. B., de Gouw, J. A., Flynn, J. H., Grossberg, N., Lefer, B. L., Liu, J., Weber, R. J., and Osthoff, H. D.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res.-Atmos., 118, 10638–10652, https://doi.org/10.1002/jgrd.50783, 2013.
Mielke, L. H., Furgeson, A., Odame-Ankrah, C. A., and Osthoff, H. D.: Ubiquity of ClNO2 in the urban boundary layer of Calgary, Alberta, Canada, Can. J. Chem., 94, 414–423, https://doi.org/10.1139/cjc-2015-0426, 2016.
Moravek, A., Murphy, J. G., Hrdina, A., Lin, J. C., Pennell, C., Franchin, A., Middlebrook, A. M., Fibiger, D. L., Womack, C. C., McDuffie, E. E., Martin, R., Moore, K., Baasandorj, M., and Brown, S. S.: Wintertime Spatial Distribution of Ammonia and its Emission Sources in the Great Salt Lake Region, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-266, in review, 2019.
Morgan, W. T., Ouyang, B., Allan, J. D., Aruffo, E., Di Carlo, P., Kennedy, O. J., Lowe, D., Flynn, M. J., Rosenberg, P. D., Williams, P. I., Jones, R., McFiggans, G. B., and Coe, H.: Influence of aerosol chemical composition on N2O5 uptake: airborne regional measurements in northwestern Europe, Atmos. Chem. Phys., 15, 973–990, https://doi.org/10.5194/acp-15-973-2015, 2015.
Mozurkewich, M.: The dissociation constant of ammonium nitrate and its dependence on temperature, relative humidity and particle size, Atmos. Environ. A-Gen., 27, 261–270, https://doi.org/10.1016/0960-1686(93)90356-4, 1993.
Nowak, J. B., Neuman, J. A., Bahreini, R., Middlebrook, A. M., Holloway, J. S., McKeen, S. A., Parrish, D. D., Ryerson, T. B., and Trainer, M.: Ammonia sources in the California South Coast Air Basin and their impact on ammonium nitrate formation, Geophys. Res. Lett., 39, L07804, https://doi.org/10.1029/2012GL051197, 2012.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Phillips, G. J., Thieser, J., Tang, M., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Pope, C. A., Muhlestein, J. B., May, H. T., Renlund, D. G., Anderson, J. L., and Horne, B. D.: Ischemic Heart Disease Events Triggered by Short-Term Exposure to Fine Particulate Air Pollution, Circulation, 114, 2443–2448, https://doi.org/10.1161/CIRCULATIONAHA.106.636977, 2006.
Pope, C. A., Muhlestein, J. B., Anderson, J. L., Cannon, J. B., Hales, N. M., Meredith, K. G., Le, V., and Horne, B. D.: Short-Term Exposure to Fine Particulate Matter Air Pollution Is Preferentially Associated With the Risk of ST-Segment Elevation Acute Coronary Events, J. Am. Heart Assoc., 4, https://doi.org/10.1161/JAHA.115.002506, 2015.
Prabhakar, G., Parworth, C. L., Zhang, X., Kim, H., Young, D. E., Beyersdorf, A. J., Ziemba, L. D., Nowak, J. B., Bertram, T. H., Faloona, I. C., Zhang, Q., and Cappa, C. D.: Observational assessment of the role of nocturnal residual-layer chemistry in determining daytime surface particulate nitrate concentrations, Atmos. Chem. Phys., 17, 14747–14770, https://doi.org/10.5194/acp-17-14747-2017, 2017.
Pusede, S. E., Duffey, K. C., Shusterman, A. A., Saleh, A., Laughner, J. L., Wooldridge, P. J., Zhang, Q., Parworth, C. L., Kim, H., Capps, S. L., Valin, L. C., Cappa, C. D., Fried, A., Walega, J., Nowak, J. B., Weinheimer, A. J., Hoff, R. M., Berkoff, T. A., Beyersdorf, A. J., Olson, J., Crawford, J. H., and Cohen, R. C.: On the effectiveness of nitrogen oxide reductions as a control over ammonium nitrate aerosol, Atmos. Chem. Phys., 16, 2575–2596, https://doi.org/10.5194/acp-16-2575-2016, 2016.
Reeves, H. D. and Stensrud, D. J.: Synoptic-Scale Flow and Valley Cold Pool Evolution in the Western United States, Weather Forecast., 24, 1625–1643, https://doi.org/10.1175/2009WAF2222234.1, 2009.
Riedel, T. P., Bertram, T. H., Ryder, O. S., Liu, S., Day, D. A., Russell, L. M., Gaston, C. J., Prather, K. A., and Thornton, J. A.: Direct N2O5 reactivity measurements at a polluted coastal site, Atmos. Chem. Phys., 12, 2959–2968, https://doi.org/10.5194/acp-12-2959-2012, 2012.
Riedel, T. P., Wagner, N. L., Dubé, W. P., Middlebrook, A. M., Young, C. J., Öztürk, F., Bahreini, R., VandenBoer, T. C., Wolfe, D. E., Williams, E. J., Roberts, J. M., Brown, S. S., and Thornton, J. A.: Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl2 measurements from a tall tower in a polluted continental setting, J. Geophys. Res.-Atmos., 118, 8702–8715, https://doi.org/10.1002/jgrd.50637, 2013.
Riemer, N., Vogel, H., Vogel, B., Schell, B., Ackermann, I., Kessler, C., and Hass, H.: Impact of the heterogeneous hydrolysis of N2O5 on chemistry and nitrate aerosol formation in the lower troposphere under photosmog conditions, J. Geophys. Res.-Atmos., 108, 4144, https://doi.org/10.1029/2002JD002436, 2003.
Riemer, N., Vogel, H., Vogel, B., Anttila, T., Kiendler-Scharr, A., and Mentel, T. F.: Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe, J. Geophys. Res.-Atmos., 114, D17307, https://doi.org/10.1029/2008JD011369, 2009.
Ryder, O. S., Campbell, N. R., Shaloski, M., Al-Mashat, H., Nathanson, G. M., and Bertram, T. H.: Role of organics in regulating ClNO2 production at the air–sea interface, J. Phys. Chem. A, 119, 8519–8526, https://doi.org/10.1021/jp5129673, 2015.
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 41, 4050–4058, https://doi.org/10.1002/2014gl059962, 2014.
Shah, V., Jaeglé, L., Thornton, J. A., Lopez-Hilfiker, F. D., Lee, B. H., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Sullivan, A. P., Weber, R. J., Green, J. R., Fiddler, M. N., Bililign, S., Campos, T. L., Stell, M., Weinheimer, A. J., Montzka, D. D., and Brown, S. S.: Chemical feedbacks weaken the wintertime response of particulate sulfate and nitrate to emissions reductions over the eastern United States, P. Natl. Acad. Sci. USA, 115, 8110–8115, https://doi.org/10.1073/pnas.1803295115, 2018.
Shetter, R. E. and Müller, M.: Photolysis frequency measurements using actinic flux spectroradiometry during the PEM-Tropics mission: Instrumentation description and some results, J. Geophys. Res.-Atmos., 104, 5647–5661, https://doi.org/10.1029/98JD01381, 1999.
Shingler, T., Crosbie, E., Ortega, A., Shiraiwa, M., Zuend, A., Beyersdorf, A., Ziemba, L., Anderson, B., Thornhill, L., Perring, A. E., Schwarz, J. P., Campazano-Jost, P., Day, D. A., Jimenez, J. L., Hair, J. W., Mikoviny, T., Wisthaler, A., and Sorooshian, A.: Airborne characterization of subsaturated aerosol hygroscopicity and dry refractive index from the surface to 6.5 km during the SEAC4RS campaign, J. Geophys. Res.-Atmos., 121, 4188–4210, https://doi.org/10.1002/2015JD024498, 2016.
Silcox, G. D., Kelly, K. E., Crosman, E. T., Whiteman, C. D., and Allen, B. L.: Wintertime PM2.5 concentrations during persistent, multi-day cold-air pools in a mountain valley, Atmos. Environ., 46, 17–24, https://doi.org/10.1016/j.atmosenv.2011.10.041, 2012.
Silva, P. J., Vawdrey, E. L., Corbett, M., and Erupe, M.: Fine particle concentrations and composition during wintertime inversions in Logan, Utah, USA, Atmos. Environ., 41, 5410–5422, https://doi.org/10.1016/j.atmosenv.2007.02.016, 2007.
Stutz, J., Alicke, B., Ackermann, R., Geyer, A., White, A., and Williams, E.: Vertical profiles of NO3, N2O5, O3, and NOx in the nocturnal boundary layer: 1. Observations during the Texas Air Quality Study 2000, J. Geophys. Res.-Atmos., 109, https://doi.org/10.1029/2003JD004209, 2004.
Tham, Y. J., Wang, Z., Li, Q., Wang, W., Wang, X., Lu, K., Ma, N., Yan, C., Kecorius, S., Wiedensohler, A., Zhang, Y., and Wang, T.: Heterogeneous N2O5 uptake coefficient and production yield of ClNO2 in polluted northern China: roles of aerosol water content and chemical composition, Atmos. Chem. Phys., 18, 13155–13171, https://doi.org/10.5194/acp-18-13155-2018, 2018.
Thornton, J. A. and Abbatt, J. P. D.: N2O5 reaction on submicron sea salt aerosol: Kinetics, products, and the effect of surface active organics, J. Phys. Chem. A, 109, 10004–10012, https://doi.org/10.1021/jp054183t, 2005.
Thornton, J. A., Braban, C. F., and Abbatt, J. P. D.: N2O5 hydrolysis on sub-micron organic aerosols: the effect of relative humidity, particle phase, and particle size, Phys. Chem. Chem. Phys., 5, 4593–4603, https://doi.org/10.1039/B307498F, 2003.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
U.S. Census Bureau: Quickfacts, available at: https://www.census.gov/quickfacts/, last access: March 2018.
Utah Department of Environmental Quality: Serious Area PM2.5 State Implementaion Plan Development, available at: https://deq.utah.gov/air-quality/pm-2-5-serious-sips-2017-2019, last access: March 2019.
Utah Division of Air Quality: Utah State Implementation: Plan Control Measures for Area and Point Sources, Fine Particulate Matter, PM2.5 SIP for the Logan, UT Nonattainment Area, https://deq.utah.gov/air-quality/pm2-5-moderate-sips-2009-2014 (last access: March 2019), 2014a.
Utah Division of Air Quality: Utah State Implementation: Plan Control Measures for Area and Point Sources, Fine Particulate Matter, PM2.5 SIP for the Provo, UT Nonattainment Area, available at: https://deq.utah.gov/air-quality/pm2-5-moderate-sips-2009-2014 (last access: March 2019), 2014b.
Utah Division of Air Quality: Utah State Implementation: Plan Control Measures for Area and Point Sources, Fine Particulate Matter, PM2.5 SIP for the Salt Lake City, UT Nonattainment Area, available at: https://deq.utah.gov/air-quality/pm2-5-moderate-sips-2009-2014 (last access: March 2019), 2014c.
UWFPS Science Team: 2017 Utah Winter Fine Particulate Study Final Report, available at: https://documents.deq.utah.gov/air-quality/planning/technical-analysis/research/northern-utah-airpollution/utah-winter-fine-particulate-study/DAQ-2018-004037.pdf (last access: March 2019), 2018.
Vaughan, S., Canosa-Mas, C. E., Pfrang, C., Shallcross, D. E., Watson, L., and Wayne, R. P.: Kinetic studies of reactions of the nitrate radical (NO3) with peroxy radicals (RO2): an indirect source of OH at night?, Phys. Chem. Chem. Phys., 8, 3749–3760, https://doi.org/10.1039/B605569A, 2006.
Wagner, N. L., Dubé, W. P., Washenfelder, R. A., Young, C. J., Pollack, I. B., Ryerson, T. B., and Brown, S. S.: Diode laser-based cavity ring-down instrument for NO3, N2O5, NO, NO2 and O3 from aircraft, Atmos. Meas. Tech., 4, 1227–1240, https://doi.org/10.5194/amt-4-1227-2011, 2011.
Wagner, N. L., Riedel, T. P., Roberts, J. M., Thornton, J. A., Angevine, W. M., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Dubé, W. P., Coffman, D. J., Bon, D. M., de Gouw, J. A., Kuster, W. C., Gilman, J. B., and Brown, S. S.: The sea breeze/land breeze circulation in Los Angeles and its influence on nitryl chloride production in this region, J. Geophys. Res.-Atmos., 117, D00V24, https://doi.org/10.1029/2012JD017810, 2012.
Wagner, N. L., Riedel, T. P., Young, C. J., Bahreini, R., Brock, C. A., Dubé, W. P., Kim, S., Middlebrook, A. M., Öztürk, F., Roberts, J. M., Russo, R., Sive, B., Swarthout, R., Thornton, J. A., VandenBoer, T. C., Zhou, Y., and Brown, S. S.: N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements, J. Geophys. Res.-Atmos., 118, 9331–9350, https://doi.org/10.1002/jgrd.50653, 2013.
Wahner, A., Mentel, T. F., Sohn, M., and Stier, J.: Heterogeneous reaction of N2O5 on sodium nitrate aerosol, J. Geophys. Res.-Atmos., 103, 31103–31112, https://doi.org/10.1029/1998JD100022, 1998.
Wang, H., Lu, K., Chen, X., Zhu, Q., Wu, Z., Wu, Y., and Sun, K.: Fast particulate nitrate formation via N2O5 uptake aloft in winter in Beijing, Atmos. Chem. Phys., 18, 10483–10495, https://doi.org/10.5194/acp-18-10483-2018, 2018.
Wang, S.-Y., Gillies, R. R., Martin, R., Davies, R. E., and Booth, M. R.: Connecting Subseasonal Movements of the Winter Mean Ridge in Western North America to Inversion Climatology in Cache Valley, Utah, J. Appl. Meteorol. Clim., 51, 617–627, https://doi.org/10.1175/JAMC-D-11-0101.1, 2012.
Wang, X., Wang, H., Xue, L., Wang, T., Wang, L., Gu, R., Wang, W., Tham, Y. J., Wang, Z., Yang, L., Chen, J., and Wang, W.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Environ., 156, 125–134, https://doi.org/10.1016/j.atmosenv.2017.02.035, 2017a.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017b.
Washenfelder, R. A., Wagner, N. L., Dube, W. P., and Brown, S. S.: Measurement of atmospheric ozone by cavity ring-down spectroscopy, Environ. Sci. Technol., 45, 2938–2944, https://doi.org/10.1021/es103340u, 2011.
Watson, J. G. and Chow, J. C.: A wintertime PM2.5 episode at the Fresno, CA, supersite, Atmos. Environ., 36, 465–475, https://doi.org/10.1016/S1352-2310(01)00309-0, 2002.
Wexler, A. S. and Clegg, S. L.: Atmospheric aerosol models for systems including the ions H+, , Na+, , , Cl−, Br−, and H2O, J. Geophys. Res.-Atmos., 107, ACH 14-11–ACH 14-14, https://doi.org/10.1029/2001JD000451, 2002.
Whiteman, C. D., Hoch, S. W., Horel, J. D., and Charland, A.: Relationship between particulate air pollution and meteorological variables in Utah's Salt Lake Valley, Atmos. Environ., 94, 742–753, https://doi.org/10.1016/j.atmosenv.2014.06.012, 2014.
Wild, R. J., Edwards, P. M., Dube, W. P., Baumann, K., Edgerton, E. S., Quinn, P. K., Roberts, J. M., Rollins, A. W., Veres, P. R., Warneke, C., Williams, E. J., Yuan, B., and Brown, S. S.: A measurement of total reactive nitrogen, NOy, together with NO2, NO, and O3 via cavity ring-down spectroscopy, Environ. Sci. Technol., 48, 9609–9615, https://doi.org/10.1021/es501896w, 2014.
Womack, C. C., McDuffie, E. E., Edwards, P. M., Bares, R., de Gouw, J. A., Docherty, K. S., Dube, W. P., Fibiger, D. L., Franchin, A., Gilman, J. B., Goldberger, L., Lee, B. H., Lin, J. C., Long, R., Middlebrook, A. M., Millet, D. B., Moravek, A., Murphy, J. G., Quinn, P. K., Riedel, T. P., Roberts, J. M., Thornton, J. A., Valin, L. C., Veres, P. R., Whitehill, A. R., Wild, R. J., Warneke, C., Yuan, B., Baasandorj, M., and Brown, S. S.: An odd oxygen framework for wintertime ammonium nitrate aerosol pollution in urban areas: NOx and VOC control as mitigation strategies, Geophys. Res. Lett., 46, 4971–4979, https://doi.org/10.1029/2019GL082028, 2019.
Wood, E. C., Bertram, T. H., Wooldridge, P. J., and Cohen, R. C.: Measurements of N2O5, NO2, and O3 east of the San Francisco Bay, Atmos. Chem. Phys., 5, 483–491, https://doi.org/10.5194/acp-5-483-2005, 2005.
Zhang, L., Jacob, D. J., Knipping, E. M., Kumar, N., Munger, J. W., Carouge, C. C., van Donkelaar, A., Wang, Y. X., and Chen, D.: Nitrogen deposition to the United States: distribution, sources, and processes, Atmos. Chem. Phys., 12, 4539–4554, https://doi.org/10.5194/acp-12-4539-2012, 2012.
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(5528 KB) - Full-text XML
- Corrigendum
-
Supplement
(1246 KB) - BibTeX
- EndNote