the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 May 2019
| 29 May 2019
Non-methane hydrocarbon (C2–C8) sources and sinks around the Arabian Peninsula
Efstratios Bourtsoukidis
Lisa Ernle
John N. Crowley
Jos Lelieveld
Jean-Daniel Paris
Andrea Pozzer
David Walter
Jonathan Williams
Atmospheric non-methane hydrocarbons (NMHCs) have been extensively studied around the globe due to their importance to atmospheric chemistry and their utility in emission source and chemical sink identification. This study reports on shipborne NMHC measurements made around the Arabian Peninsula during the AQABA (Air Quality and climate change in the Arabian BAsin) ship campaign. The ship traversed the Mediterranean Sea, the Suez Canal, the Red Sea, the northern Indian Ocean, and the Arabian Gulf, before returning by the same route. The Middle East is one of the largest producers of oil and gas (O&G), yet it is among the least studied. Atmospheric mixing ratios of C2–C8 hydrocarbons ranged from a few ppt in unpolluted regions (Arabian Sea) to several ppb over the Suez Canal and Arabian Gulf (also known as the Persian Gulf), where a maximum of 166.5 ppb of alkanes was detected. The ratio between i-pentane and n-pentane was found to be 0.93±0.03 ppb ppb−1 over the Arabian Gulf, which is indicative of widespread O&G activities, while it was 1.71±0.06 ppb ppb−1 in the Suez Canal, which is a characteristic signature of ship emissions. We provide evidence that international shipping contributes to ambient C3–C8 hydrocarbon concentrations but not to ethane, which was not detected in marine traffic exhausts. NMHC relationships with propane differentiated between alkane-rich associated gas and methane-rich non-associated gas through a characteristic enrichment of ethane over propane atmospheric mixing ratios. Utilizing the variability–lifetime relationship, we show that atmospheric chemistry governs the variability of the alkanes only weakly in the source-dominated areas of the Arabian Gulf (bAG=0.16) and along the northern part of the Red Sea (bRSN=0.22), but stronger dependencies are found in unpolluted regions such as the Gulf of Aden (bGA=0.58) and the Mediterranean Sea (bMS=0.48). NMHC oxidative pair analysis indicated that OH chemistry dominates the oxidation of hydrocarbons in the region, but along the Red Sea and the Arabian Gulf the NMHC ratios occasionally provided evidence of chlorine radical chemistry. These results demonstrate the utility of NMHCs as source/sink identification tracers and provide an overview of NMHCs around the Arabian Peninsula.
- Article
(8280 KB) - Full-text XML
-
Supplement
(11644 KB) - BibTeX
- EndNote





Anthropogenic activities are estimated to be responsible for the release of 169 Mt of non-methane hydrocarbons (NMHCs) into the atmosphere each year (Huang et al., 2017). NMHCs are important in atmospheric chemistry as precursors of tropospheric ozone and particle formation, both of which have negative impacts on air quality, human health, and climate (Lelieveld et al., 2015; Seinfeld and Pandis, 2016; EEA, 2018). Ozone production is known to be efficient in Oil and Gas (O&G) basins (Edwards et al., 2014; Wei et al., 2014; Field et al., 2015) when the high NMHC concentrations react with the hydroxyl radical (OH) under low nitrogen oxide (NOx) regimes leading to carbonyls which photolyse to recycle the OH oxidant (Rohrer et al., 2014). Since ozone production is photochemical and catalysed by NOx, summertime urban pollution is frequently associated with elevated ozone concentrations (Parrish et al., 2004; Lelieveld et al., 2009; Perring et al., 2013; Helmig et al., 2014).
Globally, NMHC sources are dominated by urban centres. Road transportation (including both evaporative and combustion emissions), residential combustion, transformation industry, fuel production and transmission, and solvent use contribute about three-quarters to the estimated global total (Huang et al., 2017). Biomass burning (Andreae and Crutzen, 1997; Pozzer et al., 2010) in addition to biogenic sources (Poisson et al., 2000; Pozzer et al., 2010) and the Earth's degassing (Etiope and Ciccioli, 2009) complete the diverse and dynamic inventory of NMHC sources. The dynamic nature of the NMHC source strength and hydrocarbon composition is driven by political, socioeconomic, and environmental decisions. For example, reducing petrol- and diesel-powered vehicles can result in an overall decrease in transportation sector emissions but an increase in the share of alkanes in the summed emission NMHC (Huang et al., 2017). The general decline in fossil fuel usage had led to decreasing trends of global ethane concentrations (Aydin et al., 2011), but the demand for cleaner-than-coal burning energy resulted in an expansion of natural gas production in the US (Swarthout et al., 2013) and reversal of global atmospheric ethane and propane trends due to US O&G activities (Helmig et al., 2016).
Since most sources have distinct emission composition patterns, hydrocarbon ratios are frequently used for identification and characterization of sources. The isomeric pentane ratio (i-pentane ∕ n-pentane) has been used to characterize vehicle emissions, gasoline, O&G activities, and urban conditions in general (Gilman et al., 2013; Thompson et al., 2014; Baker et al., 2008). Correlations with propane, which is usually associated with natural gas processing and petroleum refining, can serve as an indicator of local/regional emission sources (Swarthout et al., 2013; Helmig et al., 2016), while the relative methane abundance can provide an indication of the type of gas (associated or non-associated with liquids) responsible for the emissions (Farry, 1998; Salameh et al., 2016).
Due to their relatively short atmospheric lifetimes that range from a few hours to several weeks, the variability of the NMHC mixing ratios can serve to determine to what degree sources and sinks impact the measurements on a regional scale. Based on the Junge relationship (Junge, 1974), Jobson et al. (1998) established a correlation between the natural logarithm of the standard deviation of the gas concentrations and the chemical lifetimes for shorter-lived gases such as NMHCs. This variability–lifetime relationship has been applied in characterizing the impact of chemistry at various locations (Jobson et al., 1999; Williams et al., 2000, 2001; Bartenbach et al., 2007; Helmig et al., 2008). In addition, the presence of specific oxidants can be detected by the use of NMHC oxidative pairs (Parrish et al., 2007; Baker et al., 2016; Young et al., 2014). This is particularly important for Cl radicals as there is no instrumentation available for direct Cl measurements, although measurements of ClNO2 indicate a Cl source to be present.
The objective of this study is to present a comprehensive assessment of the atmospheric mixing ratios of the NMHCs that were measured in the marine boundary layer across the Mediterranean Sea and around the Arabian Peninsula during the AQABA (Air Quality and climate change in the Arabian BAsin) ship campaign. NMHCs are used to characterize the various emission sources encountered (including ship, O&G, regional, and urban emissions) and to evaluate atmospheric oxidation processes (by using lifetime–variability and selected oxidative pair relationships). Specific sources and sinks are studied by analysing NMHC mixing ratios in the extremely clean and polluted conditions encountered within this rarely investigated region.
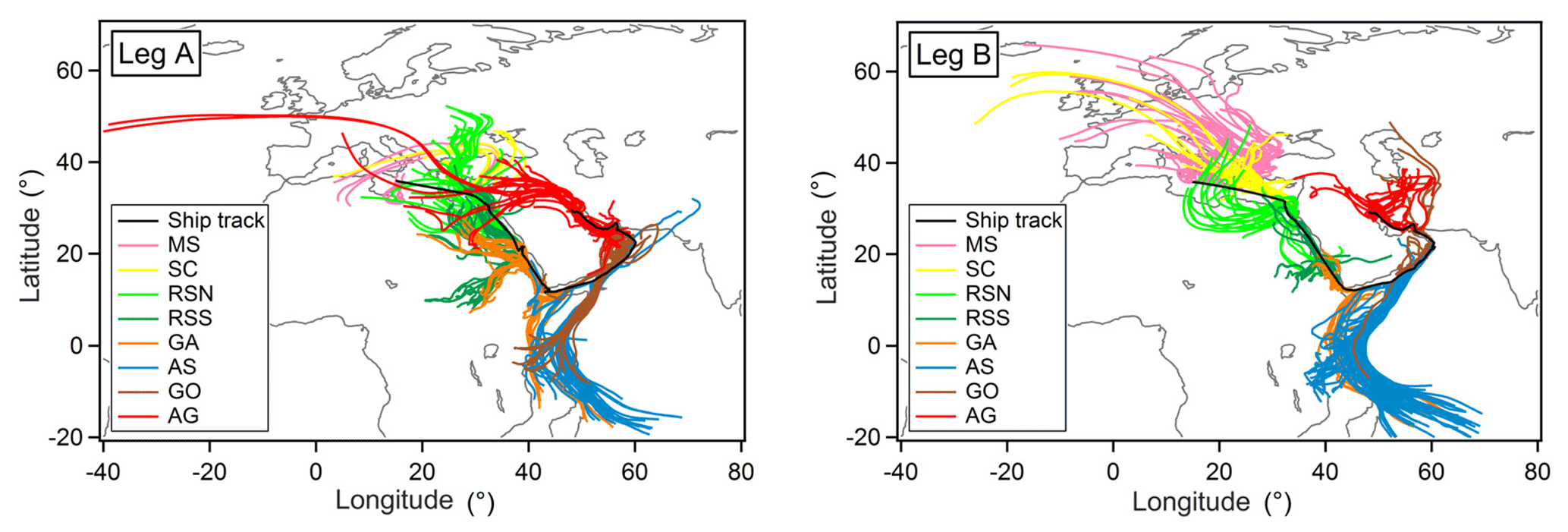
Figure 1Back-trajectory calculations along the route which start at the ship position and follow air masses back in time for 4 days on an hourly time grid.
2.1 The AQABA ship campaign
To study the Air Quality and Climate in the Arabian BAsin, AQABA, a suite of gas-phase and aerosol instruments was installed inside five air-conditioned laboratory containers and set onboard the Kommandor Iona (KI) research vessel (IMO: 8401999, flag: UK, length overall × breadth extreme: 72.55 m × 14.9 m). The shipborne measurement campaign lasted from the beginning of July to the end of August 2017, starting from the south of France, across the Mediterranean Sea, and through the Suez Canal to Kuwait and back, covering around 20 000 km at sea (Fig. 1) at an average speed of 3.4±1.8 m s−1. When possible, in order to reduce sampling of its own exhaust, the ship sailed upwind of the shipping lane and at an angle to the prevailing wind. During the campaign, the average temperature was 30.6±3.9 ∘C (range: 22.9–38.7 ∘C) and the average relative humidity was 71.4±16.3 % (range 23.5–94.5 %), while the wind speeds encountered were on average 5.4±2.8 m s−1 (range: 0.7–14 m s−1).
2.2 Gas chromatography–flame ionization detector
2.2.1 Instrumentation
Non-methane hydrocarbons were measured with two coupled GC–FID systems (GC5000VOC and GC5000BTX; AMA Instruments GmbH, Germany). The GC5000VOC was used for the quantification of light hydrocarbons (C2–C6), while the CG5000BTX was used for the heavier hydrocarbons and aromatics (C6–C8). Both systems share the same operating principle as sample collection on absorbent-filled enrichment traps (Carbosieve/Carbograph 1:1 for AMA5000VOC and Carbotrap for AMA5000BTX), subsequent thermal desorption, chromatographic separation (analytical column for GC 5000 VOC: AMA-sep Alumina, 0.32 mm ID, 50 m, 8 µm, and for GC5000BTX: AMA-sep1, 0.32 mm ID, 30 m, 1.5 µm), and detection of the eluting trace gases by flame ionization detectors. Their main difference is that the GC5000VOC system uses a dual-stage pre-concentration principle, additionally equipped with a second trap (focusing trap/Carbosieve/Carbograph 1:1) and a second chromatographic column (AMA-sep WAX, 0.32 mm ID, 30 m, 0.25 µm) in order to improve injection, peak resolution, and chromatographic separation.
To enable stand-alone combustion and carrier gas supply without the use of additional high-pressure gas cylinders, a hydrogen generator (H2PD-300-220, Parker Hannifin Corporation, USA) and zero air generator (AK6.1, Innotec GmbH & Co. KG, Germany) were operated in parallel. The zero air generator was supplied with compressed air (Compressor UA-025K/05641100, Dürr Technik GmbH & Co. KG, Germany) at 2 bar, after three-stage filtering through cartridges filled with different adsorbent materials. The first cartridge is filled with silica gel for drying, the second with charcoal for ozone and particle removal, and the third with a mixture of charcoal, molecular sieve, and soda lime for CO2 and organic trace gas removal. Inside the zero air generator, a palladium catalyst is heated to 430 ∘C to eliminate any remaining hydrocarbons and carbon monoxide. The zero air produced was used for both FID flames in addition to the Nafion drier.
2.2.2 Water and ozone interferences
The necessity of water and ozone removal in NMHC quantification has been frequently reported in the literature (Helmig and Greenberg, 1995; Plass-Dülmer et al., 2002; Slemr et al., 2002; Apel et al., 2003; Rappenglück et al., 2006). As the campaign was planned for the moist marine boundary layer and for areas that were likely to have high ambient ozone concentrations (Lelieveld et al., 2002, 2009), the effects of humidity and ozone were studied in the laboratory prior to deployment.
Moist air (relative humidity 100 %) substantially influenced ethene concentrations compared to dry air (increased by a factor of 2.2), while it had a smaller effect on propene (increased by a factor of 1.16), benzene (increased by a factor of 1.28), and all other species (differences below ±10 % between dry and moist air). The use of the Nafion drier (Perma Pure LLC) effectively eliminated these discrepancies as it proved capable of drying the ingoing sample air without producing chromatographic artefacts for the NMHCs investigated.
Stepwise addition of O3 up to mixing ratios of 200 ppb showed no significant impact of ozone on the alkane measurements. The most affected species were the alkenes and in particular isoprene, which decreased in mixing ratio by 25 % per 100 ppb of O3. Therefore two ozone scrubbers were tested. The potassium iodine scrubbers (LpDNPH, Supelco Analytical, USA) showed limited O3 removal capacity (ca. 45 L at 70 ppb O3) and induced chromatographic artefacts such as noisy background and ghost peaks. In contrast, Na2S2O3-impregnated quartz filters removed O3 with much larger capacity (>200 L at 70 ppb O3) without affecting the quality of the chromatograms. We therefore used the Na2S2O3-infused quartz filters for ozone removal in the NMHC sampling.
2.2.3 Sampling
The GC–FID system was installed inside one of the five air-conditioned laboratory containers. A 5.5 m tall (3 m above the container), 0.2 m diameter, high-flow (≈10 m3 min−1) stack was used as the common inlet for all gas-phase measurements. For NMHC sampling, a sub-flow of 2.5 L (stp) min−1 (Lpm) was drawn through a PTFE filter (5 µm pore size, Sartorius Corporate Administration GmbH, Germany) for particle removal and then through a heated (ca. 40 ∘C) and insulated PFA-Teflon line (12 m length, OD = 0.635 cm) with a residence time of approximately 9 s. The PTFE filter was exchanged every 2–5 days depending on the aerosol and sea salt load encountered. Inside the container, the GC–FID systems sampled air from the main 2.5 L min−1 stream at a rate of 90 sccm (2×45 cm3 (stp) min−1 (sccm)), through an ozone scrubber (Na2S2O3-infused quartz filters) and a Nafion dryer (500 sccm counter-flow) that were used to eliminate the effects of ozone and humidity in sample collection. Similar to ambient samples, the calibration gas was passed through the ozone scrubber and Nafion dryer to ensure identical calibration conditions through the complete campaign (Fig. S1 in the Supplement).
The broad range of NMHC concentrations encountered along the ship track dictated in situ adjustments in the sampling volume. It is worth noting that in addition to ambient concentrations, high waves that cause intense yaw, pitch, and roll movements of the ship can disturb flow controllers and influence the flame stability and signal baseline and hence detection limits. Therefore, depending on ambient NMHC concentrations and wave conditions, three sampling volumes were used throughout the campaign. In polluted conditions, such as the Arabian Gulf and the Suez Canal, short sampling times (10 min) and volumes (450 mL) allowed higher time resolution (50 min per measurement), while under clean conditions, such as found in the Arabian Sea, longer sampling times and volumes (30 min, 1350 mL, time resolution = 1 h) improved detection limits. For most of the route, the sampling time was 20 min, the sampling volume 900 mL, and the time resolution 50 min per measurement.
Temperature control of the traps is done by Peltier elements which permit rapid heating and cooling rates along with high temperature reproducibility. Sample collection was carried out at 30 ∘C for all traps, while desorption temperature was 230 ∘C. The typical container temperature was about 25 ∘C and condensation was avoided with the use of heated lines and the Nafion drier. A different oven temperature programme of a 38 min cycle was selected for the two GC–FID systems. The initial oven temperatures were set to 50 ∘C and 60 ∘C, while the final oven temperatures were 160 ∘C and 200 ∘C for the GC5000VOC and GC5000BTX systems respectively. The detailed temperature programme for each GC–FID system can be seen in Fig. S2.
2.2.4 Calibrations
Frequent calibration and blank samples ensured optimum and stable performance of the GC–FID systems. A multicomponent reference gas mixture (National Physical Laboratory, UK, 2015) was diluted with synthetic air (6.0, Westfalen AG, Germany) and passed through both the ozone scrubber and the Nafion drier to provide sampling conditions similar to ambient air. In total, 79 calibration and 20 blank samples were obtained throughout the campaign. Prior to ambient samples, a six-step calibration curve (6×3) was obtained for 450 and 900 mL sampling volumes, confirming the high sensitivity and excellent linearity of FID detectors as the R2 of the linear fit for all species was always higher than 0.98. During the campaign, single calibration points and blanks were frequently obtained to monitor the sensitivity while minimizing data loss. Analysis of the initial calibrations revealed volume-dependent sensitivity for ethane, propane, ethene, and propene. Therefore, another set of linear calibrations with 1350 mL sampling volume was performed towards the end of the campaign. For the aforementioned species, the volume-dependent calibration curve was applied for deriving the ambient mixing ratios.
Table 1Detection limits and total uncertainty of measured NMHCs. The average detection limits (DLs) for each compound are reported in the first column. DLs varied during the campaign as a result of different sampling volumes and wave height. The average total uncertainty was derived by error interpolation and is reported in the third column.
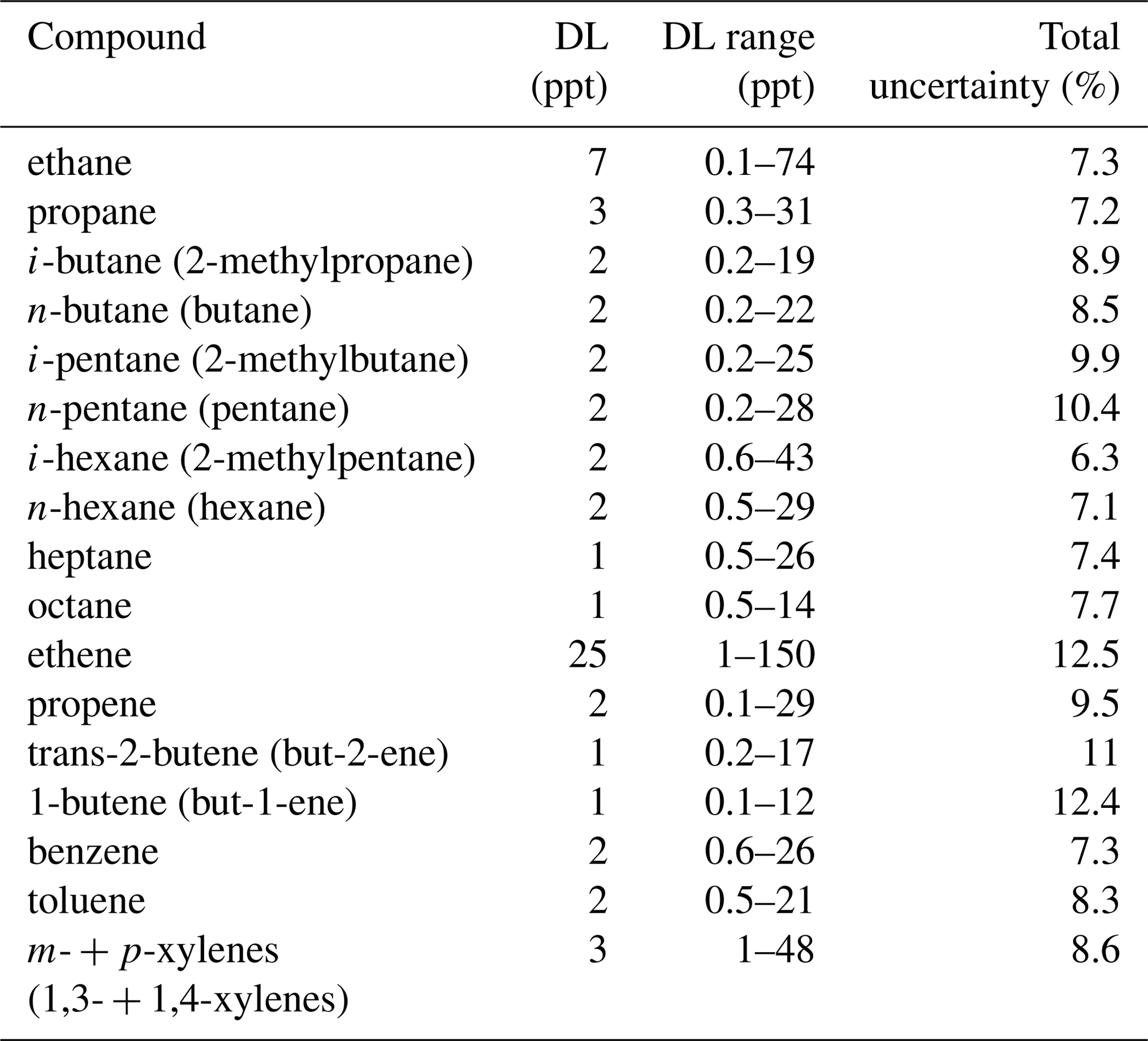
Due to varying baseline noise, dependent on the ship motion, condition-specific detection limits (DLs) and total uncertainties have been calculated (Table 1). The limit of detection was calculated from the baseline noise, as this was determined by IAU Chrom integration software (Sala et al., 2014). Peaks that were higher than 3 times the noise signal were considered to be above DL. With this approach DLs for each calibrated chromatograph were derived. Similarly, a total uncertainty for each sample was derived. For the total uncertainty we propagated 5 % uncertainty in the measured flows combined with the % inherent reference gas mixture uncertainty and the varying peak uncertainty that was calculated as the percentage signal-to-noise ratio for each chromatographic peak.
2.2.5 Ship exhaust filter
During the complete ship campaign 1393 samples were collected by the GC5000VOC system and 1244 samples by the GC5000BTX system, resulting in 26 668 integrated peaks. For the purpose of this study, all samples that have been obtained in ports or under stationary conditions have been excluded from the analysis, with the only exception being the pentane mixing ratios measured in the Jeddah port (JP). Similarly, samples influenced by the KI ship exhaust were considered only for the NMHC composition characterization and excluded from the ambient air assessments. To identify which samples were affected by our own ship exhaust, a combination of wind direction relative to the ship movement and measured NO2, O3, and SO2 was used to create a flag for affected time frames. Since ethene mixing ratios were exceptionally high inside the exhaust plume, as has been reported previously (Eyring et al., 2005), all flagged samples were individually inspected for ethene, resulting in a filter that excluded all samples that were contaminated by the emissions of the KI ship exhaust. Ethane was clearly not emitted by the KI ship exhaust, and therefore the filter was not applied for this particular hydrocarbon. The number of samples quantified per measured species can be seen in Table 2.
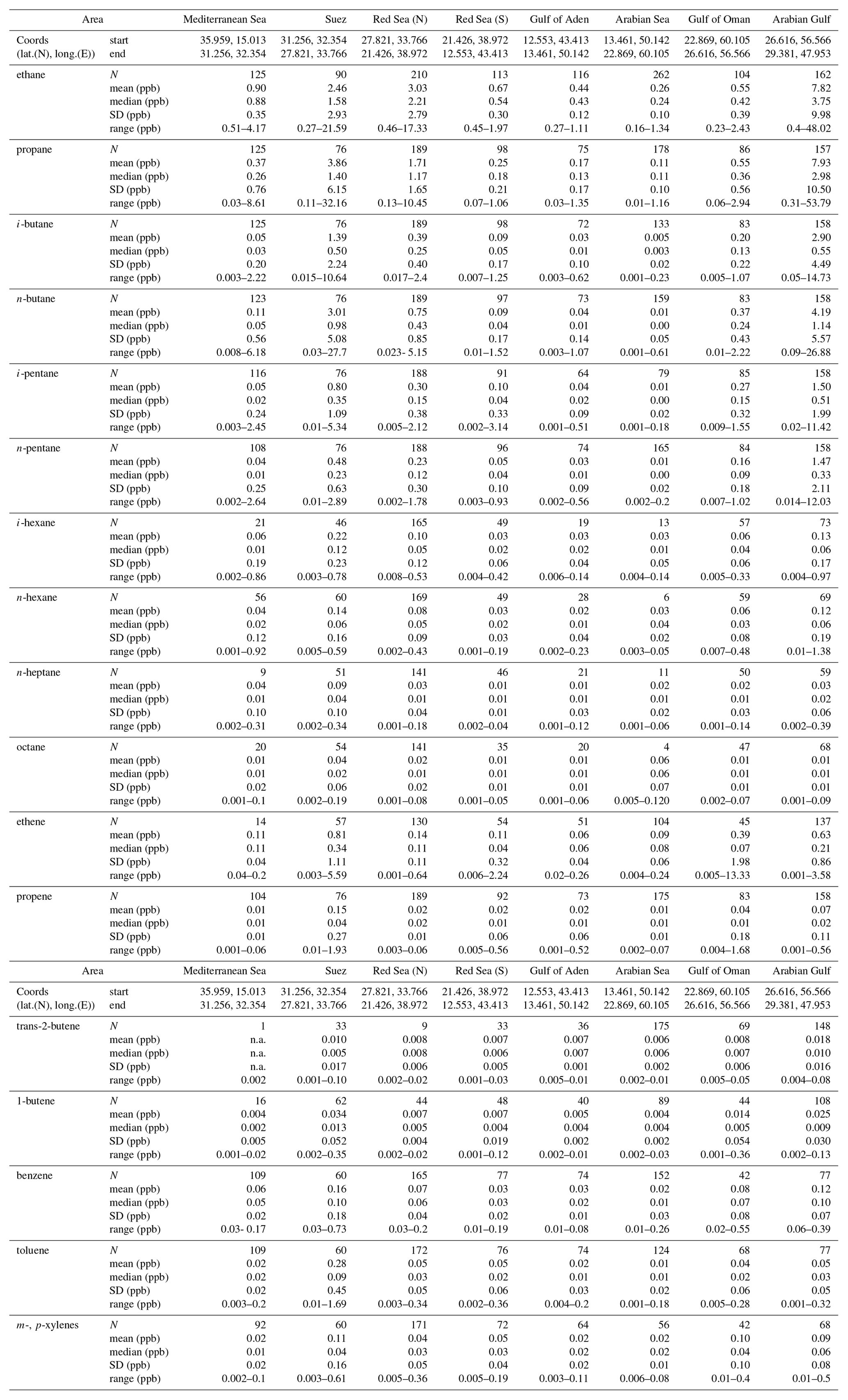
Table 2Spatial volume mixing ratio means, medians, standard deviations (SDs) and ranges of measured NMHCs.
2.3 Methane measurements
The CH4 molar mixing ratio was measured using a cavity ring-down spectroscopy analyser (Picarro G2401). Four different calibration gases traceable to WMO X2004A scale and bracketing typical ambient concentrations were injected into the analyser at ports and every 15 days for calibration purposes. The injection sequence consists of four 15 min injections of each of the four gases. An additional target gas was injected daily to assess measurement accuracy. The data have been documented and processed following ICOS (Integrated Carbon Observing System) standard procedure (Hazan et al., 2016), including the propagation of the calibration and threshold-based filters. The mean drift of measured concentrations from calibration cylinders between two sequences is 0.5 nmol mol−1, significantly below the drifts typically observed at fixed observatories (Hazan et al., 2016).
2.4 ECHAM5/MESSy Atmospheric Chemistry (EMAC) model
The ECHAM/MESSy Atmospheric Chemistry (EMAC) model is a numerical chemistry and climate simulation system that includes sub-models describing tropospheric and middle atmosphere processes and their interaction with oceans, land, and human influences (Jöckel et al., 2010). It uses the second version of the Modular Earth Submodel System (MESSy2) to link multi-institutional computer codes. The core atmospheric model is the 5th generation European Centre Hamburg general circulation model (ECHAM5, Roeckner et al., 2006). For the present study we applied EMAC (ECHAM5 version 5.3.02, MESSy version 2.53.0) in the T106L31 resolution, i.e. with a spherical truncation of T106 (corresponding to a quadratic Gaussian grid of approx. 1.1 by 1.1∘ in latitude and longitude) with 31 vertical hybrid pressure levels. A complex organic chemistry (MOM, Mainz Organic Mechanism) is integrated into the model as described in Sander et al. (2018). The simulation set-up is analogous to the one of Lelieveld et al. (2017).
2.5 Hybrid Single Particle Lagrangian Integrated Trajectory (HYSPLIT) model
Back-trajectories of air parcels encountered have been calculated with the Hybrid Single-Particle Lagrangian Integrated Trajectory model (HYSPLIT, version 4, 2014), which is a hybrid between a Lagrangian and an Eulerian model for tracing a small imaginary air parcel forward or back in time. The model can be accessed under https://ready.arl.noaa.gov/HYSPLIT.php (last access: 20 May 2019), while further details are provided in Draxler and Hess (1998). For the purposes of this study, back-trajectories with a start height of 200 m a.s.l. have been calculated, starting at the ship position and going back in time on an hourly time grid (Fig. 1).
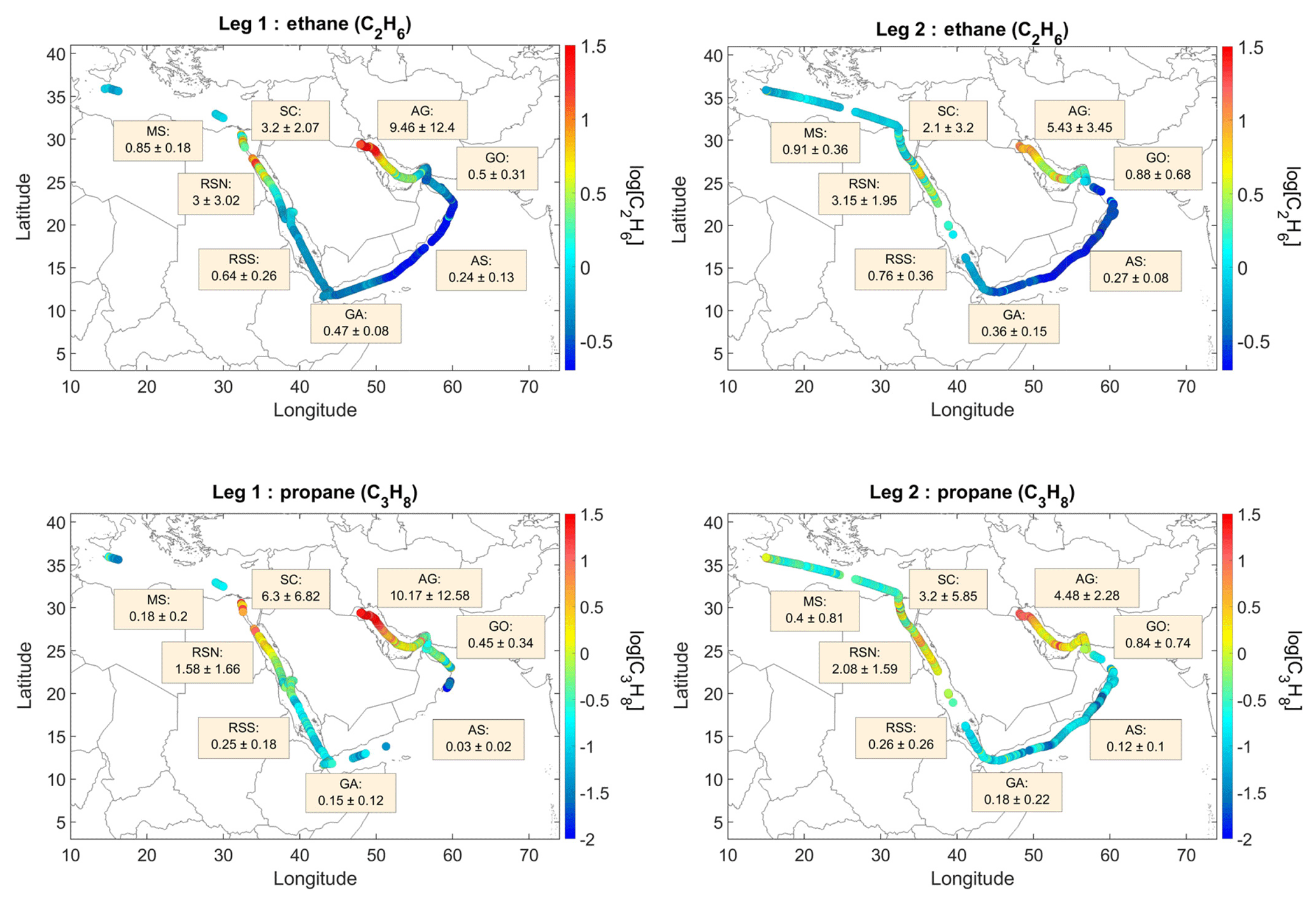
Figure 2Ethane and propane volume mixing ratios during AQABA. Average mixing ratios ± standard deviation (in ppb) for each area and leg. More detailed statistics are provided in the Supplement (Figs. S3–S4) and in Table 2.
3.1 Overview of atmospheric mixing ratios
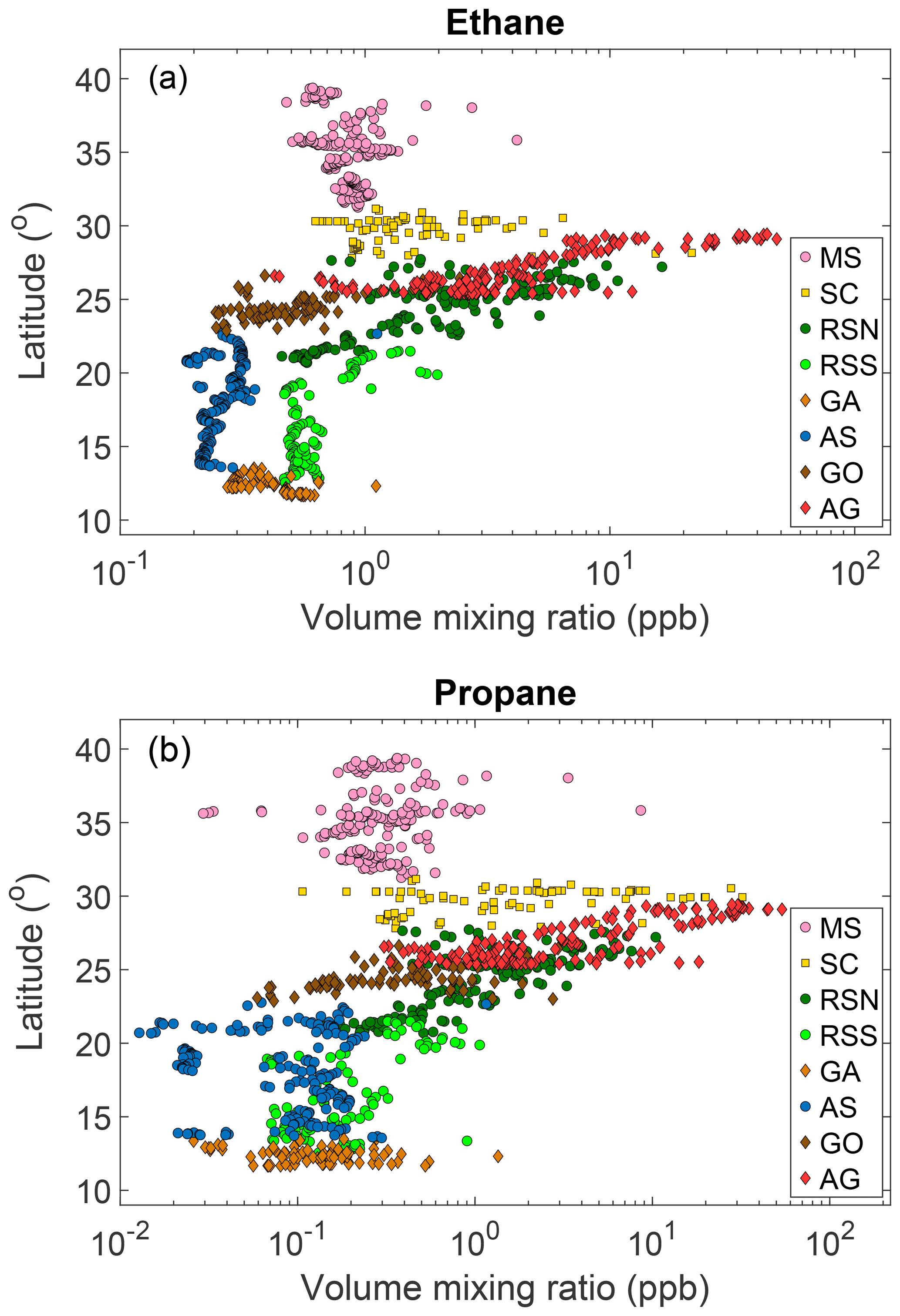
Widely varying mixing ratios of alkanes, alkenes, and aromatics were quantified along both legs of the ship campaign (Figs. 2, S3–S19). To investigate regional variations in atmospheric chemistry and abundance of NMHCs, the track was subdivided into eight regions: Mediterranean Sea (MS), Suez Canal (SC; including Great Bitter Lake and the Gulf of Suez), Red Sea North (RSN), Red Sea South (RSS), Gulf of Aden (GA), Arabian Sea (AS), Gulf of Oman (GO), and Arabian Gulf (AG) (for coordinates, see Table 2). Despite the leg-to-leg differences in the origin of the air masses (Fig. 1), ethane mixing ratios (Fig. 2) match well with this geographical demarcation that has resulted in eight different regions.
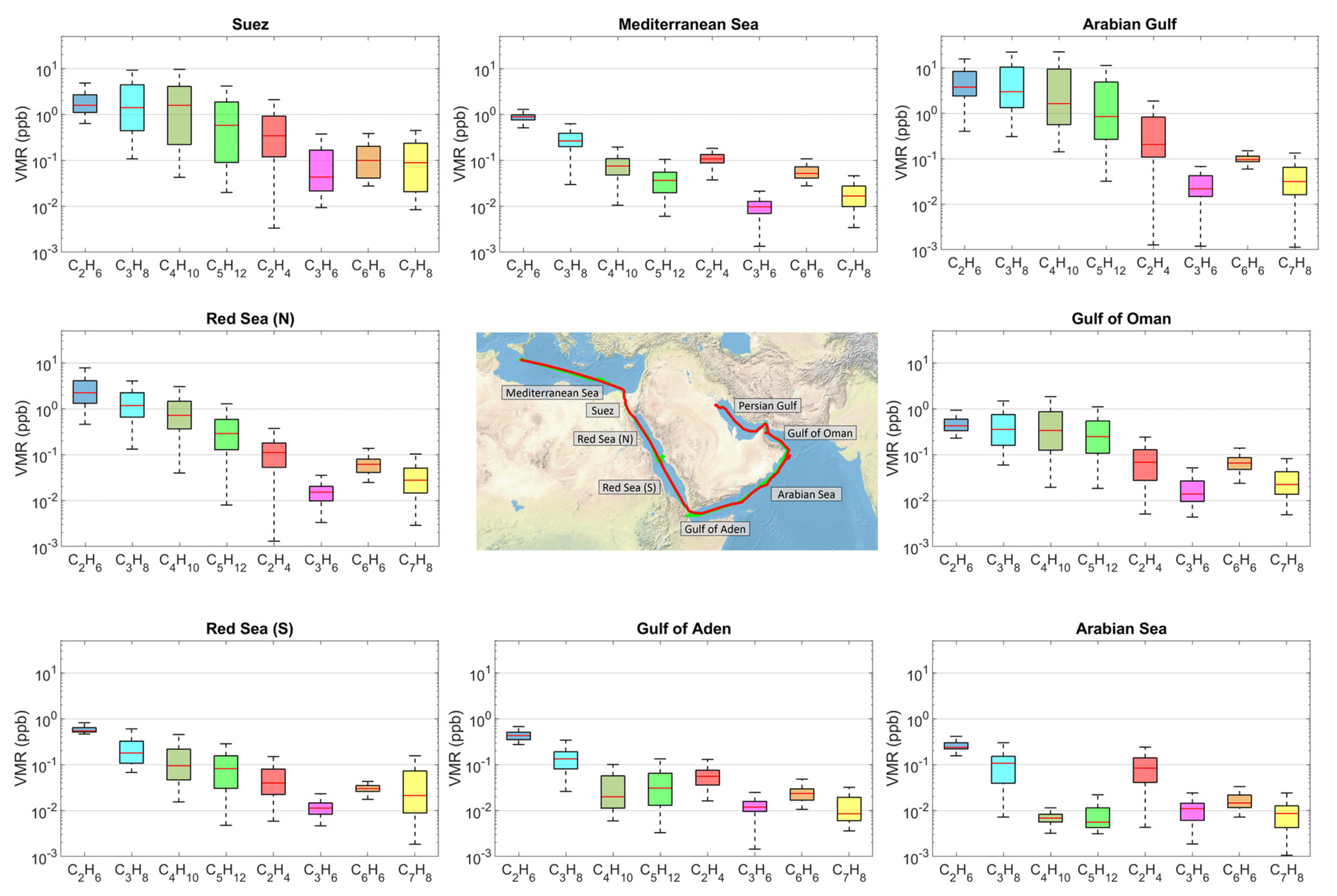
Figure 3Volume mixing ratios of selected NMHC species over the eight regions. For each box, the central red line indicates the median mixing ratio for both campaign legs. The bottom and top edges of the box indicate the 25th (q1) and 75th (q3) percentiles respectively. The boxplot draws points as outliers if they are greater than q3 + w × (q3–q1) or less than q1–w × (q3–q1). The whiskers correspond to ±2.7σ and 99.3 % coverage if the data are normally distributed. The ship track of the first leg is shown in the map with the green line, the second leg with the red line.
The cleanest air masses were measured over the AS where the majority of NMHCs were below or close to the detection limit (Table 2). HYSPLIT back-trajectories (Fig. 1) show that these air masses sampled originated from the Indian Ocean and were transported to the sampling point in the prevailing large-scale anticyclonic system. Land influence was confined to a small part of the eastern Somali desert, where NMHC sources are negligible. Mean ethane mixing ratios over the AS were 0.26±0.1 ppb on average, with the lowest recorded value being 0.16 ppb, an indicative value of the regional tropospheric background. Despite the small variability in comparison to the majority of the NMHCs measured (Fig. 3), ethene was found to be relatively high on average (0.09±0.06 ppb), with a maximum recorded value of 0.24 ppt. As ethene is highly reactive towards atmospheric oxidants and therefore has a short atmospheric lifetime (several hours), high mixing ratios are indicative of a local source. Ship traffic is unlikely to be the primary source of ethene in the AS region because of the low abundance of other ship-emission-related NMHCs.
Similar conditions were met over the MS, where ethene was 0.11±0.04 ppb on average and with a 0.2 ppb maximum. Nonetheless, average NMHC mixing ratios over the MS were generally higher compared to the AS, presumably as upwind European sources were closer to the sampling point over the MS than South-East Asian sources over the AS. The variability–lifetime dependency (Jobson et al., 1998; Williams et al., 2000) was more pronounced for the MS (see Sect. 3.3), suggesting more biogenic input in the AS. The rest of the alkenes and higher alkanes were frequently below the detection limit in the MS region and the average values presented in Table 2 are mainly driven by the elevated mixing ratios measured close to southern Italy.
Mixing ratios were markedly different in the Suez Canal and the Gulf area (SC). The increased marine traffic intensity in combination with the proximity to populated areas along the Suez Canal resulted in wide-ranging NMHC mixing ratios (Fig. 3). The most abundant hydrocarbon was propane (3.9±6.15 ppb), followed by n-butane (3±0.56 ppb), ethane (2.64±2.93), and i-butane (1.39±2.24 ppb) (average values). Accounting for both isomers, butane was the dominant NMHC for about 50 % of the samples that were collected inside the SC (Fig. S20). Alkenes were dominated by ethene (0.81±1.11 ppb) with the maximum recorded value of 5.59 ppb, but propene, trans-2-butene, and 1-butene were also present in substantial amounts. The area of the Suez had in general the highest aromatic hydrocarbons measured throughout the campaign since benzene (160±183 ppt), toluene (0.28±0.45 ppb), and m-, p-xylenes (0.11±0.16 ppb) were measured as high as 0.73, 1.69, and 0.61 ppb respectively.
Interestingly, the northern part of the Red Sea (RSN) displayed contrasting NMHC signatures compared to its southern counterpart (RSS). The air that was encountered in the RSN originated from the north-eastern part of Africa (Egypt, Libya), with vestigial influences from the south-eastern European continent. Alkane mixing ratios were dominated by ethane and propane, followed by butanes and pentanes, generally displaying a decrease in mixing ratio with higher carbon numbers. Depending on the wind direction, local sources delivered high mixing ratios of ethane (max = 17.33 ppb) and propane (max = 10.45 ppb), but, in relation to the SC, lower butanes and pentanes. For instance, the SC area had an average propane-to-butane ratio of 0.88, while the respective ratio in the RSN was 1.5 due to the different types of sources (see Sect. 3.2).
The air sampled in the southern part of the Red Sea (RSS) was mainly influenced by central Africa, with 4-day back-trajectories extending towards the desert areas of Sudan and Chad. Nonetheless, a substantial number of samples originated from along the eastern coasts of Sudan and Egypt, with influences from the RSN region. Data distribution plots (Fig. S20) show that the benzene-to-toluene ratio was higher than unity for about 60 % of the samples and below one for the rest, indicating influences by fresh ship emissions and possibly biomass burning (Andreae and Merlet 2001; Andreae, 2019). Ethane mixing ratios varied relatively little (0.67±0.3 ppb) due to the lack of O&G activities in the RS area, in addition to the negligible input from marine traffic. In contrast, marine traffic associated gases (Fig. 6) were detected in significant quantities, with the majority of NMHCs ranging from a few ppt to orders of magnitude higher mixing ratios.
The GA region showed similar atmospheric mixing ratios to both the RSS and the AS, and the back-trajectories suggest common influences with the aforementioned areas. The majority of the samples had mixing ratios in the sub-ppb range, with the isomeric sum of butanes (0.08±0.22 ppb) and pentanes (0.09±0.18 ppb) being very similar (Fig. S20) and always lower than ethane (0.44±0.12 ppb) and propane (0.17±0.17 ppb).
In contrast, the GO region was dominated by C2–C5 alkanes that were all measured to be in approximately the same concentration range. The most abundant NMHC measured was ethene (13.3 ppb), which in combination with the increased ethane (0.55±0.39 ppb), propane (0.55±56 ppb), butanes (0.57±0.64 ppb), and pentanes (0.44±0.48 ppb) indicated indirect influences from O&G production (i.e. dehydrogenation of ethane) in addition to urban pollution that includes vehicle exhausts and of marine traffic. As the air masses encountered in the GO displayed similar back-trajectories to those from the AS, the increased concentrations can be mainly attributed to anthropogenic emissions from Oman that include O&G production.
Over the AG, an area of intense O&G activity, C2–C5 alkanes were on average higher by an order of magnitude (AG ppb, AG ppb, AG ppb, AG ppb), again displaying wide-ranging mixing ratios (Figs. 2, 3, S20). Similarly, alkenes and aromatic hydrocarbons were highly abundant (AG ppb, AG ppb, AG ppb, AG ppb, AG ppb) but on average lower than the mixing ratios encountered in the SC. Due to instrumental problems the GC5000BTX was not operating in the northern part of the AG, the area with the highest observed C2–C5 mixing ratios. The high variability observed in this area is attributed to the diverse influence of local strong O&G sources. Fresh emissions from oil fields were sampled over the northern part of the AG and contained large amounts of alkanes (AGethane,max=48.02 ppb, AGpropane,max=53.79 ppb, AGbutanes,max=41.26 ppb, AGpentanes,max=23.45 ppb), but occasionally the sampled NMHCs (in particular during the second leg of the campaign) were already oxidized, in relatively clean and dry air originating from the Iranian desert (Fig. 1b). Ethane and propane were measured as low as 0.4 and 0.31 ppb respectively, at the time, while alkenes were in the sub-ppt level. Despite the variability in air mass origin between the two legs, the samples provide a distinct signature of the area which is further investigated in detail in Sect. 3.2.
Overall, the background latitudinal gradient of ethane and propane mixing ratios (Blake and Rowland, 1986; Simpson et al., 2012) was only apparent when considering the minimum mixing ratios in each region (Fig. 4). The Middle East is a global hot-spot of ethane and propane emissions as fossil fuel emissions are highly concentrated due to regional petrochemical activities. Hence, the mixing ratios that were measured along the SC, RSN, and AG are characteristic of the local emission sources, and they cannot be used to derive a latitudinal gradient. Nonetheless, ethane mixing ratios along the AS can be considered to be unaffected by anthropogenic activities, displaying a decline of 4.6 ppt per latitudinal degree. Concerning propane, the situation is different, as marine traffic emissions influence regional atmospheric mixing ratios, and hence higher variability in their abundance was observed in the AS and in most of the investigated areas.
3.2 Source identification through NMHCs
3.2.1 Pentane isomers
Typical anthropogenic sources (e.g. O&G operations, gasoline vapours, vehicle emissions, urban areas) have distinct NMHC signatures and their mixing ratios can be used for identification and characterization of the respective emission source. The relationship between pentane isomers had been frequently used for emission-source identification as natural gas (Gilman et al., 2013; Swarthout et al., 2013; Thompson et al., 2014), gasoline vapours (Gentner et al., 2009), vehicle emissions (Harley et al., 1992; Broderick and Marnane, 2002), and in general urban environments (Warneke et al., 2007; Baker et al., 2008; Von Schneidemesser et al., 2010; Barletta et al., 2017; Panopoulou et al., 2018) display a distinct relationship between i-pentane and n-pentane. Higher i-pentane is associated with fuel evaporation (McGaughey et al., 2004) and vehicle emissions (Jobson et al., 2004), while n-pentane is more abundant in natural gas (Gilman et al., 2013). Since the i- and n-pentane isomers have approximately the same reaction rate with the hydroxyl radical (Atkinson, 1986), their atmospheric oxidation proceeds at equivalent rates and their ratio is a reliable marker for identifying emission sources independent of their proximity to the sampling point.

Figure 5Relationship between pentane isomers. Linear fits of the data obtained in the Arabian Gulf, Suez Canal, and Jeddah port are compared to enhancement ratio slopes (ERs) from the literature with values for raw and natural gas (a Gilman et al., 2013), urban areas (b Baker et al., 2008), gasoline vapours (c Gentner et al., 2009), vehicle emissions (d Broderick and Marnane, 2002), and the Texas ship channel (e Blake et al., 2014). Complete regression statistics: AG: , R2=0.97; SC: , R2=0.98; JP: , R2=0.96; KI ship exhaust: , R2=0.99.
The relationship between pentane isomers is commonly expressed with the use of an enhancement ratio (ER) which is defined as the slope term in a linear regression between n-pentane and i-pentane. Figure 5 compares the values between pentane isomers for three sampled regions (JP, SC, AG) together with enhancement ratio slopes (ERs) that are reported in the literature. The samples that were collected in the Jeddah port show the highest ratio of i- to n-pentane (ERJP=2.9 ppb ppb−1, R2=0.96), with the slope being in the upper range of urban conditions and almost identical to the slope that is representative of vehicle emissions. During the 2-day stop in the Jeddah port, the ship was anchored in a part of the port with considerable activity from land vehicles inside the port, possibly explaining the observed ER. In addition, the prevailing northerly/north-easterly winds advected air from the nearby city of Jeddah, an urban centre with a population of ca. 4 million habitants. Barletta et al. (2017) reported a ratio of 2.66 ppb ppb−1 for the city of Jeddah, which agrees with our observations.
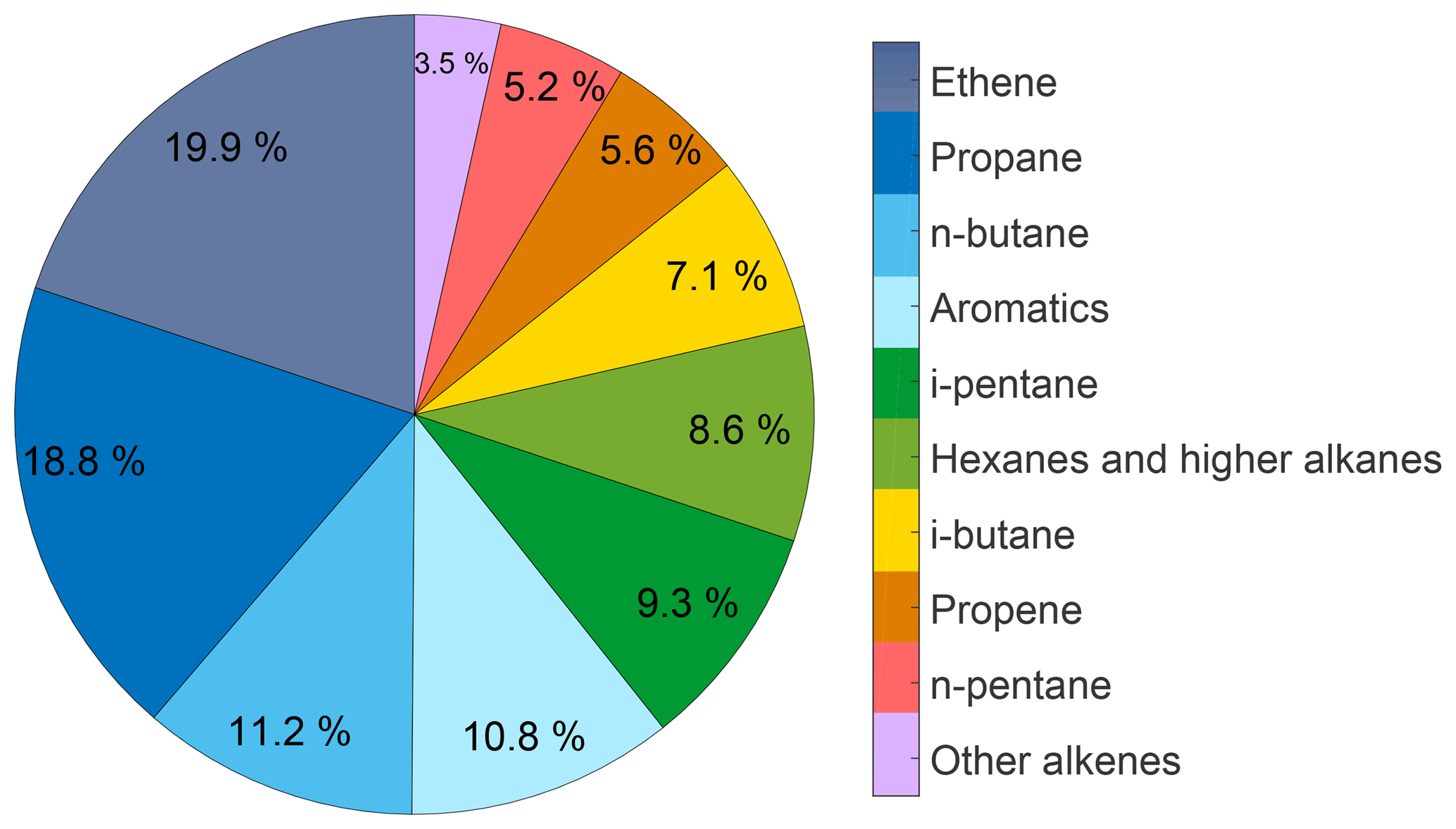
Figure 6Composition of a typical KI ship exhaust sample. Aromatics include benzene (2.8 %), toluene (4.9 %), and m-, p- xylenes (3.1 %), hexanes and higher alkanes include i-hexane (2.6 %), n-hexane (2.7 %), n-heptane (2.2 %), and octane (1.2 %), and other alkanes include trans-2-butene (0.8 %), 1-butene (1.5 %), and 1-pentene (1.2 %). The sample was measured over the Arabian Sea and background ambient mixing ratios were subtracted prior to relative composition calculation. Ethane mixing ratios were 28 ppt lower inside the exhaust sample, being within the uncertainty range of the measurement.
Along the Suez, the pentane enhancement ratio slope was ERSC=1.71 ppb ppb−1 (R2=0.98). SC air was predominantly influenced by ship traffic and potentially by urban emissions from the cities that are distributed along the channel. In addition, O&G operations along the Great Bitter Lake and Gulf of Suez, with occasional oil rigs along the route, comprise a mixture of emission sources that could affect the isomeric pentane abundance. However, the slope is identical to the one that was observed in the Texas ship channel (1.7 ppb ppb−1; Blake et al., 2014). By using all filtered samples that were directly impacted by KI exhaust, we derive an enhancement ratio of 1.59 ppb ppb−1 (R2=0.99). While this ratio is affected by regional ambient concentrations and the presence of a KI exhaust sampled volume, its values, the high linearity of fit, and the ratio slopes obtained in the SC and reported for the Texas ship channel indicate this is a characteristic signature of ship emissions. Interestingly, C2–C5 alkanes are not considered to be emitted by international shipping (Eyring et al., 2005). While we did not observe any ethane emissions from the KI exhaust, propane, butanes, and pentanes constituted about 50 % of the composition of a typical KI exhaust sample (Fig. 6). Given the obvious similarities in pentane isomer relationships when comparing KI exhaust and SC samples, it is evident that ship emissions strongly impact the hydrocarbon mixture in the SC.
The intense O&G operations in the AG area could be discerned by another characteristic relationship between pentane isomers. The enhancement ratio (ERAG=0.93 ppb ppb−1, R2=0.97) is very similar to the natural gas signature (ER = 0.86 ppb ppb−1; Gilman et al., 2013), with almost all data points falling within the O&G range (±20 %). Comparable values have been reported for the oil- and gas-rich north-eastern Colorado, including Boulder (ER = 1.1 ppb ppb−1; Gilman et al., 2013), Fort Collins (0.81 ppb ppb−1; Gilman et al., 2013), Erie/Longmont wells (ER = 0.97 ppb ppb−1; Thomson et al., 2014), and Boulder Atmospheric Observatory (ER = 0.89 ppb ppb−1; Gilman et al., 2013, ER = 1 ppb ppb−1; Swarthout et al., 2013), as well as for the natural gas activities on Kola Peninsula in Russia (ER = 1.1 ppb ppb−1; Gilman et al., 2010).
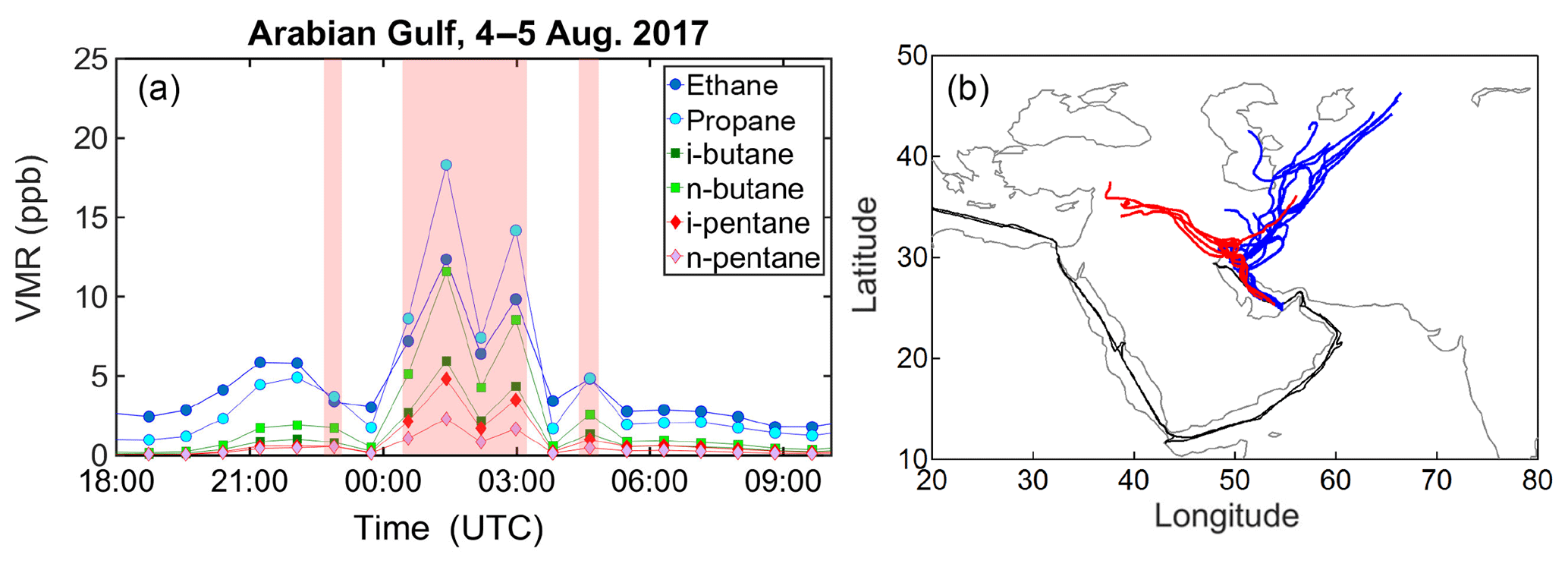
Figure 7Time series of C2–C5 alkane volume mixing ratios (a) and back-trajectories (a) of samples collected in the AG on 4–5 August 2017. In (b) the red trajectories correspond to the shaded red data points of (a), while the blue trajectories correspond to all other samples. The back-trajectories start at the ship position and follow the air masses back in time for 6 days on an hourly time grid.
Despite the high correlation between pentane isomers in the AG, several samples and in particular five samples with i-pentane >1 ppb deviate substantially from the derived slope (Fig. 5), displaying a higher i- ∕ n-pentane ratio. Six-day HYSPLIT back-trajectories show that these particular samples were influenced by the large-scale oil fields (US-CIA, 2007) and gas flaring areas (https://skytruth.org/viirs/, last access: 20 May 2019) in addition to major cities in Iraq (Fig. 7). Before and after encountering the plume of high i- to n-pentane ratios, the back-trajectories extend to the gas fields of Turkmenistan before crossing Iran and reaching the sampling point. Apart from the distinctly high i- ∕ n-pentane ratio, the common feature of all samples that originated from the Iraqi oil fields is the high n-butane and propane mixing ratios that reached values of up to 27 and 54 ppb respectively.
3.2.2 Correlation with propane mixing ratios
Elevated propane mixing ratios are usually associated with natural gas processing and petroleum refining. Light alkanes, such as ethane, n-butane and n-pentane, are co-emitted by O&G activities and, due to their short lifetimes, a tight correlation with propane indicates common sources and little photochemical processing. Therefore, propane is frequently regarded as a sensitive indicator of local/regional emission sources (Helmig et al., 2016).
Weak propane–NMHC correlations were generally observed over the MS, RSS, GA, AS, and GO despite occasional co-variation related to specific sources. In these areas, propane mixing ratios were relatively low, with median values ranging from 107 ppt over the AS to 355 ppt at the GO (Table 2). The higher mixing ratios that were measured over the GO are associated with influences by O&G activities in Oman, but the number of samples with elevated propane mixing ratios was outnumbered by the samples originating from the Arabian Sea. Considering the complete propane dataset, the aforementioned regions displayed only moderate correlations with the longer-lived ethane () and poor correlations with the rest of the alkanes, alkenes, and aromatics.
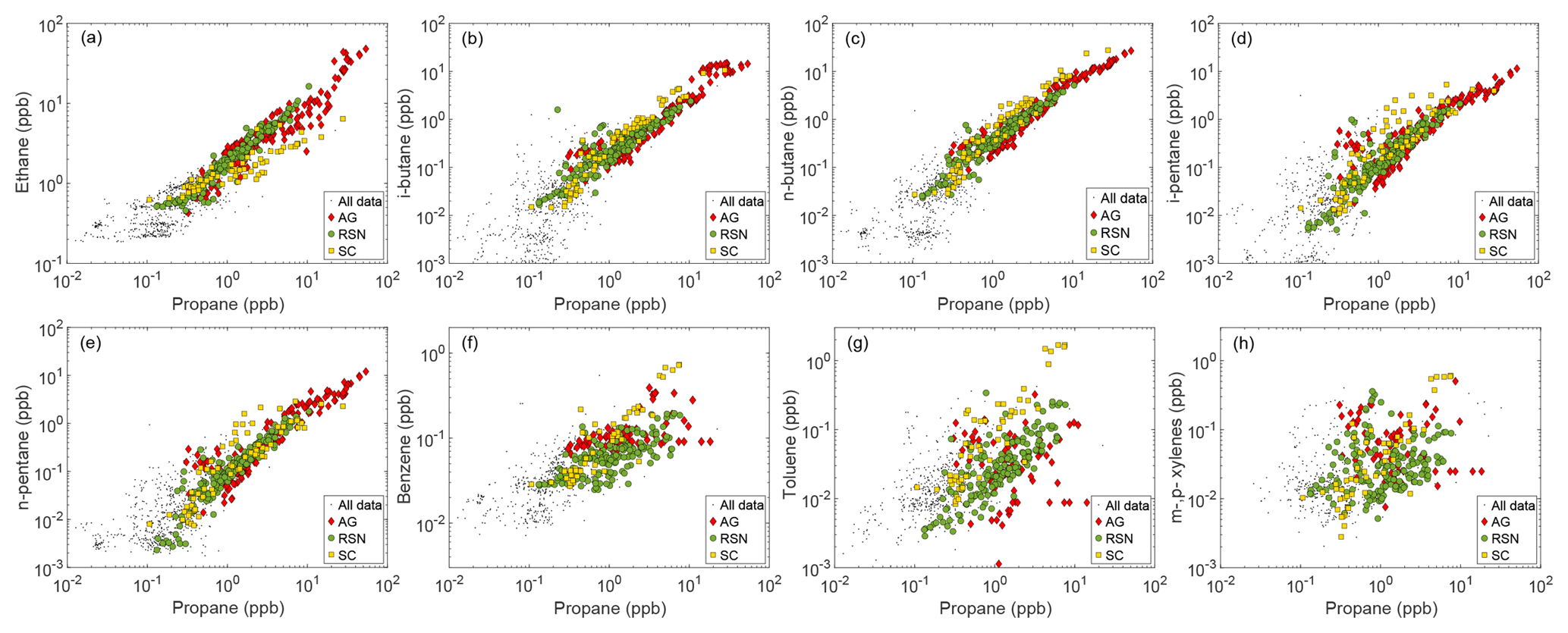
Figure 8Correlations between propane and selected NMHCs for the areas of the Arabian Gulf (AG, red diamonds), Red Sea North (RSN, green circles) and Suez Canal and Gulf (SC, yellow squares).
Table 3Correlation matrix of enhancement volume mixing ratio slopes for selected alkanes and aromatics with propane and methane mixing ratios. Bold values indicate correlations with R2>0.7.

The three regions that showed a high correlation between propane and NMHCs were the SC, RSN, and AG (Fig. 8, Table 3). Among them, the propane–NMHC relationship was markedly different in the SC as propane mixing ratios were correlating only with butanes, alkenes, and aromatic hydrocarbons. N-butane in particular was the only hydrocarbon that was emitted more strongly than propane (ER = 1.1 ppb ppb−1, R2=0.91), while the known tight relationship with its isomer i-butane resulted in an enhancement mixing ratio slope of 0.43 ppb ppb−1 (R2=0.89) for i-butane. The SC was the only region that had high propane correlations with alkenes () and aromatic hydrocarbons (), with ethene and toluene displaying the highest enhancement ratios towards propane, with values of 0.22 and 0.24 ppb ppb−1 respectively. The co-emission with combustion tracers, such as aromatic hydrocarbons and the highly reactive alkenes, provide further evidence that fresh marine traffic emissions dominate NMHC abundance in this area. Finally, the poor relationship between propane and ethane in the SC is in agreement with our ship exhaust observations, showing that ethane is not emitted by ships.
In contrast to the SC, RSN and AG measurements displayed high correlation coefficients with ethane that was found at even higher mixing ratios than propane in the RSN (ER = 1.3 ppb ppb−1, R2=0.95). A similar enhancement ratio (1.5 ppb ppb−1) has been reported by Swarthout et al. (2013) from the Boulder Atmospheric Observatory (BAO) within a wind sector representative of natural gas sources. Likewise, butane and pentane isomers showed high correlations with propane and with enhancement ratio slopes (Table 3) that are very similar to those observed in BAO (Pétron et al., 2012, 2014; Gilman et al., 2013; Thompson et al., 2014; Swarthout et al., 2013). Benzene and toluene also correlated with propane, but with moderate correlation coefficients, as the fit is mainly affected by the larger variability of these species in the southern part of the RSN. Overall, the relationships between propane and the rest of the hydrocarbons measured provide indications of a natural gas source in the region that is not documented in O&G cartographic maps (US-CIA, 2007). Etiope and Ciccioli (2009) have shown that the RSN has a substantial coverage of geothermal and volcanic spots that could potentially explain the high mixing ratios of NMHCs measured in this region.
The Arabian Gulf is a global hot-spot for O&G activities. The correlations of alkanes with ambient propane mixing ratios were very similar to the ones from the RSN, with the main difference being that ethane was emitted at lower rates (ER = 0.91 ppb ppb−1, R2=0.9) and i-butane at higher rates (ER = 0.52 ppb ppb−1, R2=0.83). Aromatic hydrocarbons did not correlate well with propane in the AG, where the aromatic hydrocarbon measurements are in the low range due to missing measurements along the northern, more polluted part of the AG (see Figs. S17–S19). Despite the generally high correlation between propane and the rest of the alkanes in the AG, the highest propane mixing ratios ([C3H8] ppb) had slightly different interrelationships with all hydrocarbons, which is attributed to a different air-mass origin (and therefore source), as seen for example in the case study of Fig. 7.
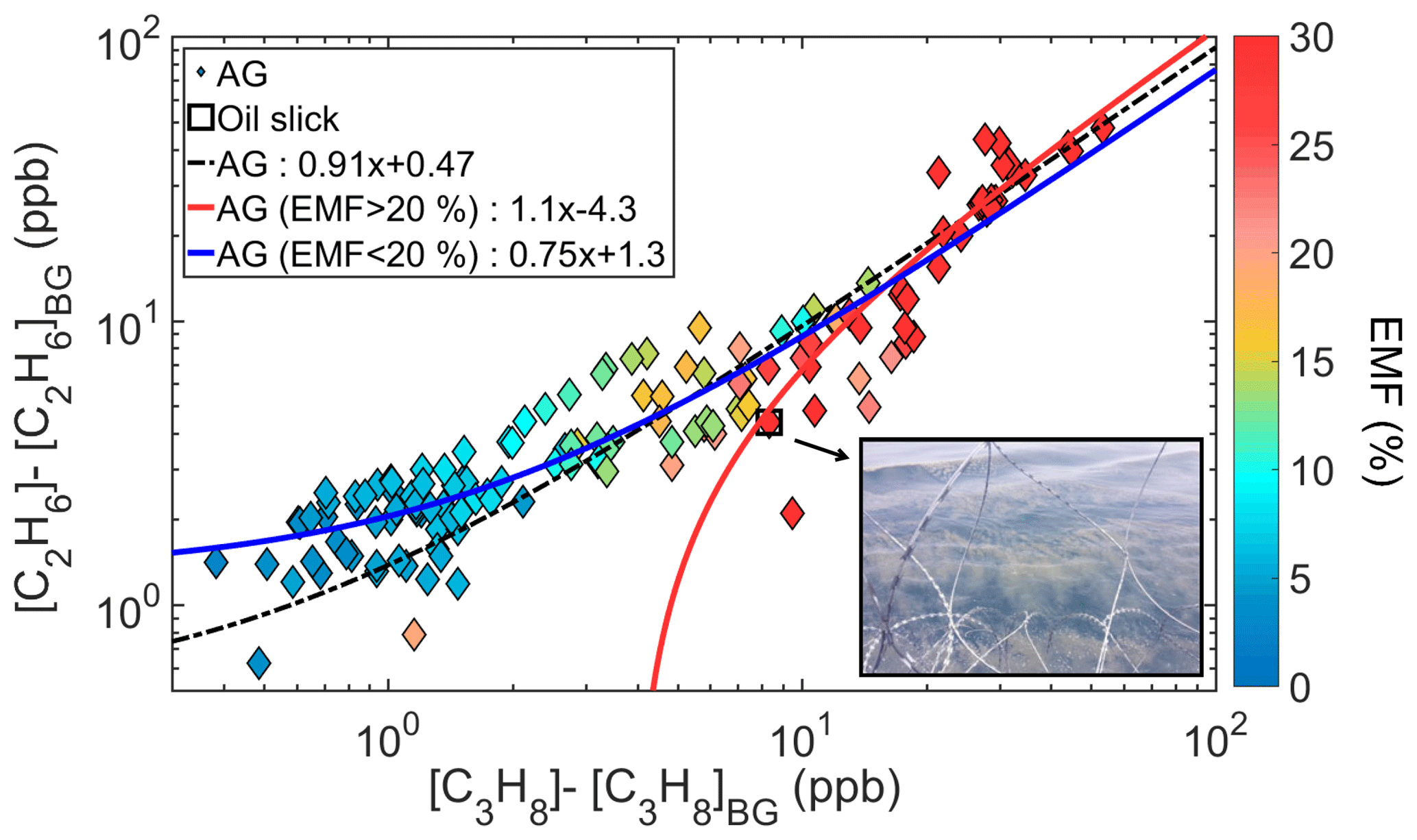
Figure 9Excess mixing ratios of ethane and propane coloured by the excess NMHC mole fraction (EMF) of each sample for the area of the AG. The photo shows the oil slick as seen from the deck during the particular sample.
3.2.3 Excess mole fraction
To further investigate the different interrelationships observed under high propane mixing ratios in the AG, and to generally provide a quantitative characterization of hydrocarbons in each region, their relative abundance was calculated. We define as “excess NMHC mole fraction” (EMF) the relative abundance of non-methane hydrocarbons to the sum of methane and non-methane mixing ratios, whereby the background mixing ratios (defined as the lowest 1 % of regional samples) of each area were subtracted from the total mixing ratios, resulting in the following equation:
While methane on the global scale is primarily emitted by wetlands, agriculture, and waste sources, natural gas and petroleum systems contribute up to a quarter of global anthropogenic emission and 10 % of the global source (Warneck and Williams, 2012; Kirschke et al., 2013; Saunois et al., 2016). Non-associated natural gas (produced by reservoirs without liquid) composition is dominated by methane, while associated gas contains smaller fractions of methane and higher fractions of alkanes (Farry, 1998; Matar and Hatch, 2001; Anosike et al., 2016). Figure 9 shows the propane versus ethane mixing ratios in the AG, coloured by the excess NMHC mole fraction. Propane and ethane display different relationships under high EMFs, where ethane is higher than propane, with an enhancement ratio slope of 1.1 ppb ppb−1 when considering EMF >20 %. Similar ratios have been obtained through the use of PMF (positive matrix factorization) analysis for oil refineries in Houston (Kim et al., 2005; Buzcu and Fraser, 2006) and with measurements over an oil well (Colombo et al., 2013).
On 29 July 2017 at 12:00 UTC, KI crossed an oil slick in the AG over which a 10 min sample was collected. This particular sample contained 30.1 ppb of C2–C8 hydrocarbons, consisting mainly of alkanes and aromatics, while the mixing ratios of alkenes remained at similar levels to the samples before and after the oil slick. Its excess mole fraction (21 %) and position on the respective linear fit indicate that associated gas contains a larger fraction of NMHCs. The composition of this sample (see Fig. S21) indicates a high fraction of light alkanes, but considering that the most volatile species tend to evaporate first, we cannot establish the representativeness of the actual composition of an oil slick.
Higher EMFs were assigned to back-trajectories originating from the oil fields and refineries of Iran, while the lower EMFs were associated with trajectories originating from the gas fields of Turkmenistan (see notably the case study of Fig. 7). Our analysis supports the notion that associated gas contains more non-methane hydrocarbons than non-associated gas, and that EMF can therefore be used to distinguish between emissions related to oil versus gas exploitation. We find also that the relationship between ethane and propane can be used to identify the type of emission source. Nonetheless, this observation is valid only for relatively fresh emissions as the shorter propane lifetime will modify the ethane-to-propane ratio in chemically processed air masses.
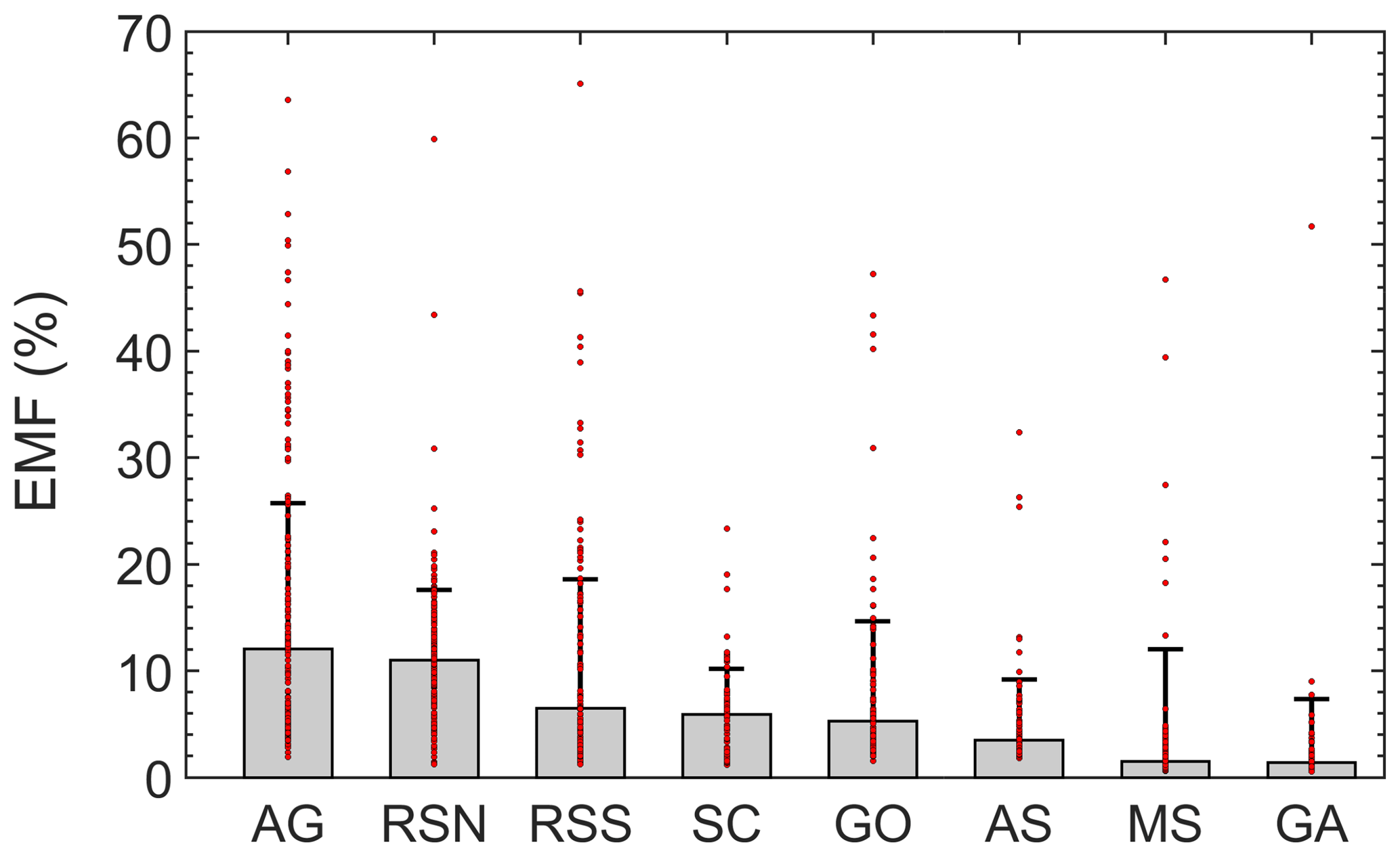
Figure 10Spatial excess NMHC mole fractions. Bars represent the regional median and error bars the standard deviation. Individual samples are shown with red dots.
All regions occasionally yielded samples with relatively high EMFs (Fig. 10) that were possibly related to oil slicks or oil production activity sources. A prominent example is the GO. Besides the AG (12±13.7 %), the northern part of Red Sea had, with a median EMF value of 11±6.6 %, the smallest regional standard deviation calculated. This observation is indicative of a widespread and relatively homogenous source of NMHCs in the area as the sum of alkanes was on average the second highest encountered along the sampled regions. Pentane isomers (Sect. 3.2.1) and relationships with propane (Sect. 3.2.2) indicated that ship emissions dominated the NMHC abundance in the SC. The high correlation coefficients between methane and the majority of NMHCs in combination with the poor correlation with ethane in this area (Table 3) confirm that the EMF in the SC is primarily driven by marine traffic, a known source of both methane and non-methane hydrocarbons (Eyring et al., 2005) but with varying relationships depending on the ship type and fuel used (Anderson et al., 2015). The areas of the GA and AS were dominated by CH4 (EMFs <5 %) which was frequently close to global background levels. In the absence of regional emission sources, the more reactive NMHCs are removed by atmospheric radicals, and the longer-lived methane, which has a large oceanic source (e.g. Bange et al., 1998; Born et al., 2017), dominates the mole fraction. Therefore, these areas are more appropriate for investigation of atmospheric oxidation processes than the identification and characterization of emissions.
3.3 Sink characterization through NMHCs
3.3.1 Variability–lifetime relationship
The relationship between the variability of atmospheric mixing ratios and their chemical lifetimes can be used to evaluate the relative importance of chemical reactions and source proximity in the observed variability of trace gases (Junge, 1974; Jobson et al., 1998, 1999; Williams et al., 2000, 2001; Pszenny et al., 2007; Helmig et al., 2008). The conceptual approach has also been extended to particles (Williams et al., 2002). Jobson et al. (1998) introduced the standard deviation of the natural logarithms of the mixing ratios as a useful measure for the variability for short-lived species. The relationship with chemical lifetime follows a power-law distribution of the form
where
Here, S is the standard deviation of the natural logarithm of hydrocarbon X with a calculated chemical lifetime (τ) with respect to reaction with the hydroxyl [OH], chlorine atoms [Cl], and the nitrate [NO3] radicals. A and b are fitting parameters that are determined by the regression. The term A has been interpreted (although interpretations vary) as related to the standard deviation of the air mass transport times, while b is a robust indicator of the dependence of the measured variability on atmospheric chemistry (as opposed to source proximity) and hence remoteness from sources (Jobson et al., 1998). The relationship has also been exploited in the past to derive gas and particle lifetimes, to determine data quality, and to estimate radical abundances (Williams et al., 2000). In general, higher variability is expected for the most reactive species (Junge, 1974). The parameter b ranges from 0 (indicating close-by sources and no dependence on chemistry) to around 0.6 (a value that is typical of remote locations in the troposphere).

The chemical lifetime depends on the respective reaction rate coefficients and on the OH, O3, NO3, and Cl concentrations. For our calculations we use the average regional radical concentrations as they have been calculated by the EMAC-MOM chemistry model (Table 4), combined with the reaction rates shown in Table 5. We chose to investigate the relationship solely with C2–C5 alkanes as these species were above the detection limit for all areas and therefore provide a robust measure for comparing different regions. They also do not react rapidly with ozone, so this can be neglected. In addition, we found that reactions with the nitrate radical (NO3) are negligible (i.e. do not affect the calculated lifetimes; <1 % changes) due to their low rate coefficients with C2–C5 alkanes (Atkinson, 1991; http://iupac.pole-ether.fr/, last access: 20 May 2019).
Figure 11 shows that the smallest b value was derived for the AG (0.16±0.09), a region where adjacent O&G activities dominated the emissions, resulting in increased and highly variable alkane mixing ratios (25.44±33.33 ppb). Due to the source proximity, C2–C5 hydrocarbon variability was not significantly related to the chemical reactions. Similarly, the small bRSN (0.22±0.11) is also indicative of nearby sources, so that wind direction rather than oxidation time determines atmospheric variability. The RSN displayed smaller standard deviations and higher atmospheric lifetimes for all species. Both bAG and bRSN fall in the lowest range of b values that are consistent with nearby emission sources.
The SC and GO displayed very similar b factors from the variability–lifetime relationships, and were both higher than found in the AG. The highest variability along the route was encountered in the SC, which is also the area with the modelled highest OH and Cl concentrations, resulting in shorter lifetimes. Therefore, the bSC (0.33±0.27) represents a combination of regional sources and oxidation processes. Similarly, the GO () had high OH but low Cl concentrations according to the model and mixing ratio variability. In this case the standard deviation of the most reactive pentanes drives the fit. Considering the AS, the poor regression fit (R2=0.33) does not allow interpretation of the derived parameters, but it is indicative of local biogenic sources as the variability–lifetime relationship is not as clear as expected for remote sources.
The areas with the highest dependence of variability on chemistry were found to be the GA, MS, and RSS. In these regions, the variability of the measured mixing ratios was more affected by chemistry than the other regions, indicating that these areas are more remote from the sources, and the air had longer chemical processing times indicated by the high b factor (, , ). Similar values have been reported for unpolluted coastal sites (0.56 at Nova Scotia, Jobson et al., 1998), marine-influenced air masses (0.54 at Appledore island, Pszenny et al., 2007; 0.64 at Surinam, Williams et al., 2000), and the eastern Mediterranean Sea (0.49, Arsene et al., 2007), validating our indications of the importance of alkane–radical reactions in these regions.
3.3.2 Characterization of radical chemistry
The atmospheric ratio of i-butane to n-butane has been frequently reported to be between 0.3 and 0.8 for a wide range of environmental conditions (e.g. Baker et al., 2016; Rossabi and Helmig, 2018). For the AQABA campaign, the isomeric ratio was on average 0.63±0.44 (median = 0.56), with 82.4 % of the samples between 0.3 and 0.8. The almost identical reaction rate coefficient of the butane isomers (i- and n-) to the OH radical means that their measured ratio will remain relatively constant and equal to the emission ratio independent of transport times, provided only OH reacts with the species and in-mixing is negligible. Deviation from this range would imply a local source with a different emission ratio or oxidation with another radical with which the isomers react at different rates. Since the reaction rate coefficient of n-butane is about 40 % faster than i-butane with Cl, these ratios can be used to investigate the presence of Cl radicals.

Figure 12(a) Relationship between butane isomers and (b) distribution of the i-butane ∕ n-butane ratio considering all data (grey bars) and the case study data (blue and red bars) as they have been selected from (a).
As shown in Fig. 12, three clusters of data substantially deviate from the slope, displaying different i-butane to n-butane mixing ratios. The first cluster (termed case study CS1AG, N=20; black circles) corresponds to the high mixing ratios encountered in the AG with an isomeric ratio of 1.09±0.18. Considering the small standard deviation of the ratio along with the small geographical coverage, it is more likely that this cluster corresponds to stronger i-butane emissions rather than n-butane depletion due to reaction with Cl radicals. I-butane is more abundant in gas flares (Emam, 2015) which typically contain 45 %–70 % methane (Umukoro and Ismail, 2017). EMF values were on average 35.8±9.5 %, indicating that these particular samples had a very high NMHC content, including the gas-flaring product 1-butene which was 3 times higher compared to the AG average. In addition, these samples cover 15 h, including both day and night times during which Cl concentrations vary substantially. Therefore, Cl reactions are unlikely to be the main reason for the observed anomalous ratio cluster, and it is more likely that gas-flaring emissions are responsible for this observation. Another example is the cluster of data that belongs to the AS but falls below the commonly observed emission ratio range (CS2AS, N=43; cyan dots). These are indicative of oceanic emissions of butanes, which are estimated to be emitted at a rate of 0.11 Tg yr−1, exceeded in the alkane series only by ethane (0.54 Tg yr−1) and propane (0.35 Tg yr−1) (Pozzer et al., 2010). Broadgate et al. (1997) reported that n-butane emissions from the North Sea are ∼4 times stronger that its isomer, possibly explaining the small ratio (0.34±0.1) we observed in the unpolluted region of the AS. Finally, the third cluster of measurements (CS3, N=38, blue squares) was mainly made over the Red Sea in the regions RSS (N=15) and RSN (N=11). In addition, 12 samples from the GO, MS, and SC exhibit a higher i- ∕ n-butane ratio and were clustered together in a group that had an average isomeric ratio of 1.84±1.66. The higher and highly variable isomeric butane ratios, combined with their comparatively low concentration of both i- and n-butane, suggest Cl-influenced oxidative air masses, but nearby ship emissions could also explain this observation.
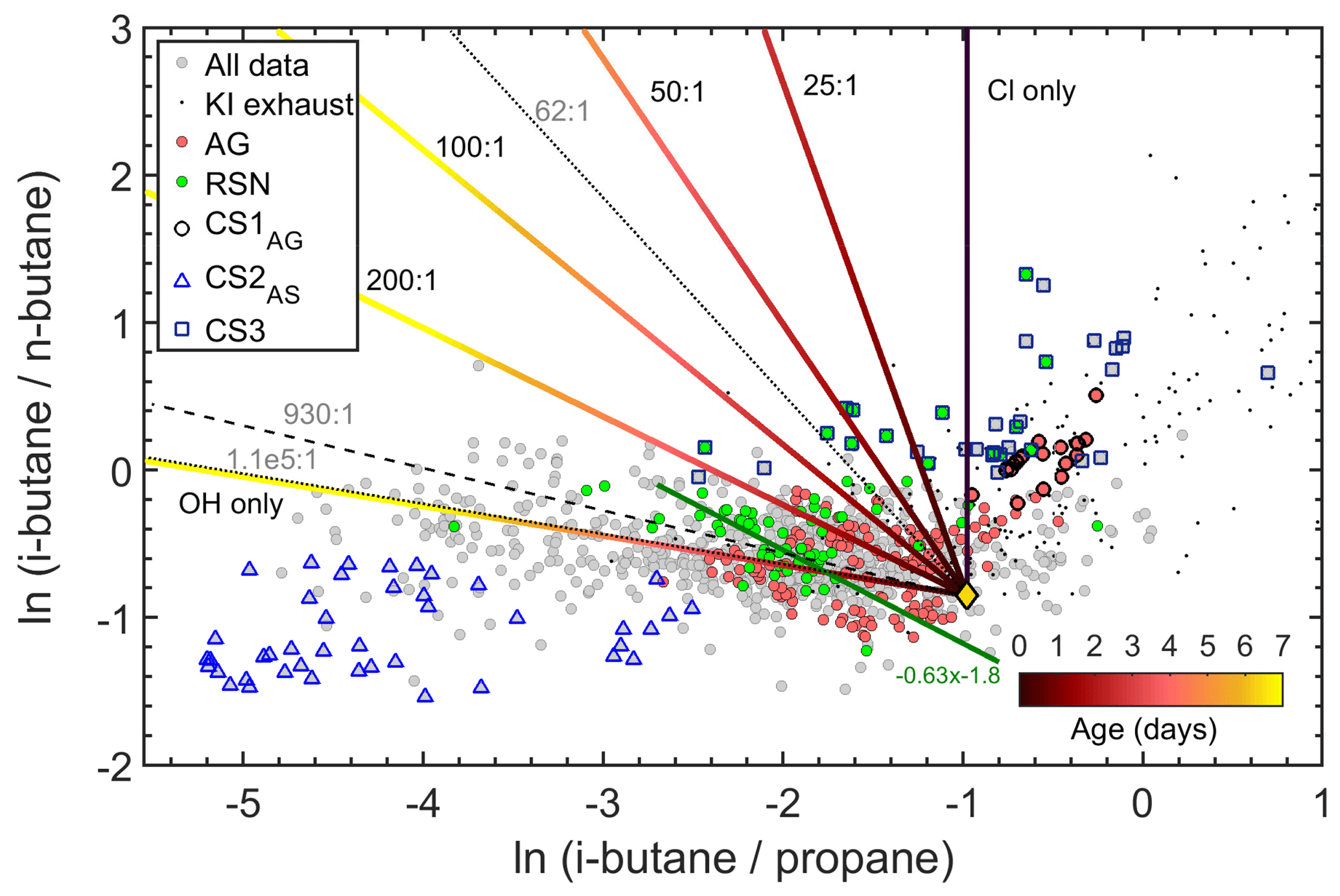
Figure 13Observations of selected VOC tracer ratios (i-butane, n-butane, propane) together with their expected evolution for OH and Cl oxidation from global emission ratios (yellow diamond). The dashed lines indicate the lowest (62:1), highest (), and average (930:1) OH-to-Cl ratios derived from the EMAC model.
Table 4Spatial average OH and Cl concentrations simulated by MOM.
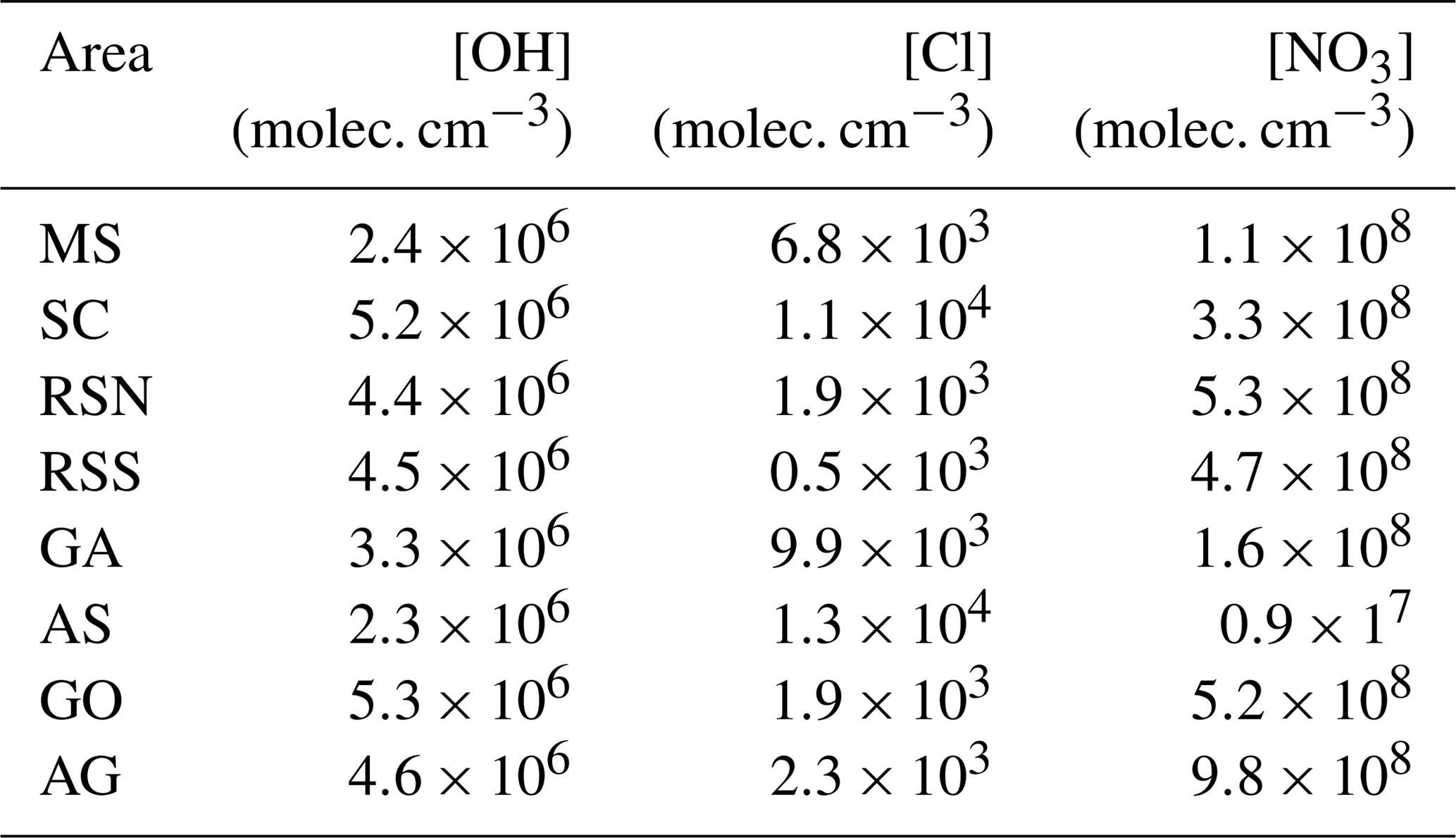
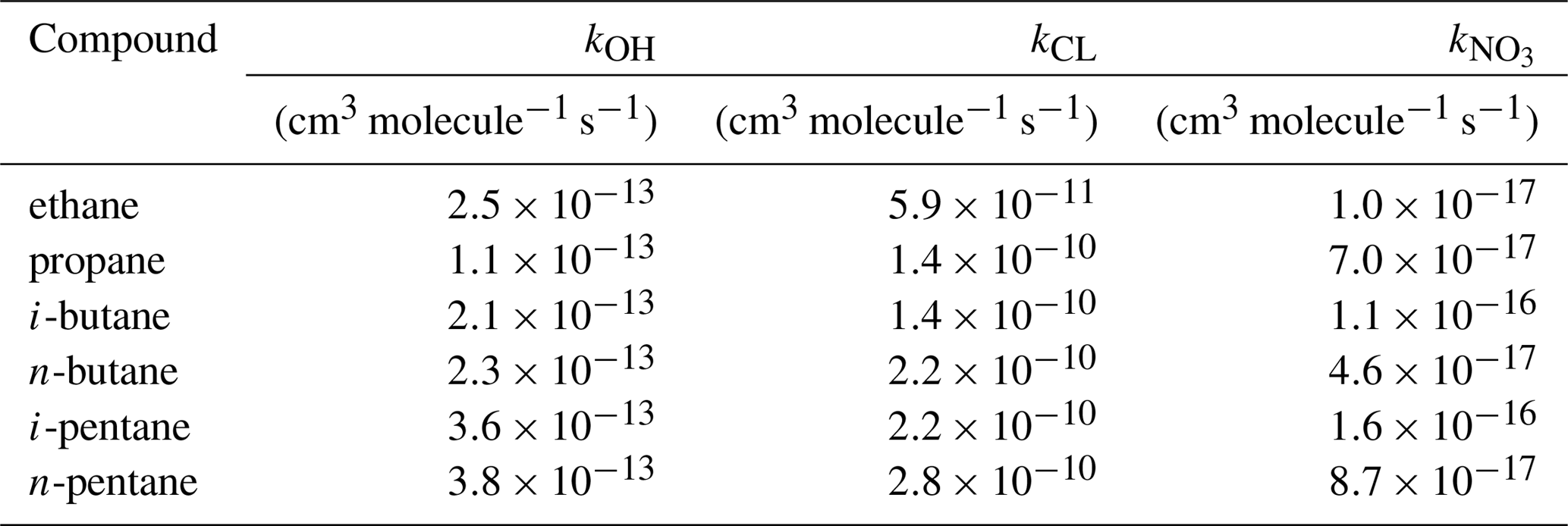
In the absence of measurement techniques that could quantify Cl radical concentrations directly, the most commonly used method to determine whether Cl chemistry has occurred is the examination of VOC ratios and inter-relationships (Jobson et al., 1994; Rudolf et al., 1997; Baker et al., 2016). Figure 13 shows the correlation between an OH-reactive pair (i-butane to propane) and a Cl-reactive pair (i-butane to n-butane). If only OH chemistry acts on the air parcel, the i-butane to n-butane ratio will remain approximately unchanged, while the i-butane to propane ratio will decrease with processing time since the OH+ i-butane rate coefficient is twice that of OH+ propane (Table 5). If only Cl chemistry acts on the air parcel, the opposite is expected: the i-butane to propane ratio will remain unchanged, while the isomeric ratio will increase with processing since n-butane reacts faster than its isomer with Cl radicals. Figure 13 shows that the majority of the samples display an unchanged isomeric butane ratio over 2 orders of magnitude of the i-butane to propane ratio, indicating that sources with similar emissions ratios and OH chemistry dominated the air parcels that were encountered along the route. However, a significant portion of the data display isomeric ratios which indicate a mixture of OH and Cl chemical processing.
To evaluate the contribution of OH and Cl radical reactions in the atmospheric processing, kinetic simulations were used to illustrate the expected tracer ratio evolution (i-butane, n-butane, and propane) starting from average global emission ratios (yellow diamond in Fig. 13, Pozzer et al., 2010). The total OH radical concentration was held at 5×106 molec. cm−3 and the OH ∕ Cl ratios were adjusted to simulate conditions that range between OH reactions only and Cl reactions only. In order to demonstrate plausible atmospheric ranges for the OH ∕ Cl ratio, the lowest (62:1), highest (), and average (930:1) ratios were generated in the EMAC model run for the campaign, and these have been implemented in Fig. 13. In the model the Cl radical is generated from HCl oxidation, the former being released from sea spray via acid displacement (Erickson et al., 1999; Keene et al., 1999; Knipping et al., 2000; Young et al., 2014). Modelled Cl concentrations are therefore expected to be more abundant over the marine boundary layer and coastal areas. In the atmosphere, ClNO2 represents a further form of reactive chlorine, which is formed in heterogeneous reactions of N2O5 with sea salt at night-time and which has been detected at significant concentrations in both coastal and continental air masses (Thornton et al. 2010; Phillips et al., 2012). ClNO2 can photolyse to produce Cl radicals (Osthoff et al., 2008; Lawler et al., 2009; Riedel et al., 2012; Young et al., 2012) and field observations have shown that ClNO2 can be responsible for more than 50 % of near-surface Cl production during early morning (Young et al., 2012; Mielke et al., 2013; Young et al., 2014). As the EMAC model does not include the formation of ClNO2, we may assume that ambient OH ∕ Cl ratios may in fact be lower than the minimum 62:1 ratio generated by the model.
Based on the kinetic model results, the OH ∕ Cl ratio can be expressed as a function of the slope (σ) of the tracer ratio evolution of these particular hydrocarbons as
where σ corresponds to the slope of ln(i-butane ∕ propane) to ln(i-butane ∕ n-butane) with evolution times illustrated in the colour scale of Fig. 13. Equation (4) is independent of the initial emission ratios and of the absolute radical concentrations, but it is sensitive to reaction rate coefficients. Therefore, it is only applicable in this form in this region, assuming that the reaction rate coefficients that are presented in Table 5 are applicable. A more general expression for the relationship of the rate coefficients and OH ∕ Cl ratio under large temperature variations (e.g. free troposphere) was presented in Rudolf et al. (1997).
Figure 13 shows that some data points lie on positive slopes which define the unrealistic case in which Cl radicals were the only oxidant acting on the hydrocarbons. This includes the clusters of points discussed previously (CS1 and CS3), reaffirming that they are unusual emission signatures rather than the result of Cl radical chemistry. It is interesting to note that many of the points in this part of the graph have been affected by our own ship exhaust. Therefore our own ship and possibly other vessels emit propane, i-butane, and n-butane in ratios that are distinctly different from those generated from any combination of OH and Cl radical chemistry acting on the gases when emitted at the average emission ratio (see the yellow marker in Fig. 13). Nonetheless, the majority of the samples collected over the RSN and over the AG lie on trend lines that match the theoretical slope of an OH ∕ Cl ratio of . As an example, Fig. 13 illustrates the regression statistics for the majority of the samples collected in the RSN (green line, ). Utilizing Eq. (4), the resulting radical ratio is 186:1. Our analysis demonstrates that oxidative pairs can be used to provide an indication of the presence of Cl atoms and provide a rough estimate of atmospheric OH-to-Cl ratios.
An overview of the atmospheric mixing ratios measured around the Arabian Peninsula during the AQABA ship campaign was provided in terms of regional abundance. The highest mixing ratios were observed over the Arabian Gulf, the Suez Canal, and the northern parts of the Red Sea, and the lowest over the Arabian Sea, Gulf of Aden, and Mediterranean Sea. The diverse environmental conditions resulted in wide-ranging (several orders of magnitude) concentrations of NMHCs, the characteristics of which were investigated in relation to regional sources and sinks and air mass ageing/oxidation.
Enhancement ratios between pentane isomers identify the broad O&G activities in the Arabian Gulf by displaying a characteristic enhancement of i-pentane over n-pentane of the order of 0.93±0.03 ppb ppb−1. Similarly, the enhancement ratio of 1.71 ppb ppb−1 that was observed over the Suez Canal and Arabian Gulf points towards a characteristic value for ship emissions, found to be similar to our ship exhaust emission ratios and identical to the ratio slope observed in the Texas ship channel (Blake et al., 2014). While the exhaust from the Kommandor Iona could differ in hydrocarbon composition from other vessels, a typical ship exhaust sample in this region was rich in ethene, propane, and n-butane, which comprised ∼50 % of emitted NMHCs. The highest ethene mixing ratios were observed in the SC (0.81±1.11 ppb), where n-butane and propane were highly abundant and correlated with each other. We can therefore conclude that international shipping contributes to atmospheric NMHC abundance, which should be accounted for in emission inventories. However, atmospheric mixing ratios of ethane were consistently unaffected by marine traffic, suggesting that it can be disregarded in emission inventories. Finally, the i- ∕ n-pentane isomeric ratio characterized a mixture of vehicle emissions and urban pollution in the Jeddah port, with the value of 2.9±0.14 ppb ppb−1 being in line with previously reported observations at this urban centre (Barletta et al., 2017).
Since the atmospheric abundance of propane is influenced by nearby natural gas processing and petroleum-refining activities, its atmospheric mixing ratios were investigated in terms of co-emission with the rest of the NMHCs. Besides the ship-dominated emissions in the Suez Canal, where propane did not correlate with ethane, higher correlations with other alkanes were observed in the Arabian Gulf and the northern part of the Red Sea. By calculating the excess mole fraction of NMHCs, we showed that the ethane–propane interrelationship is different in hydrocarbon-rich associated gas compared to the methane-rich non-associated gas which contained higher fractions of propane over ethane, in line with previous observations that demonstrated the propane dominance in gas leak emissions in the region (Salameh et al., 2016). The atmospheric abundance of ethane and propane over the Arabian Gulf was inconsistent with methane mixing ratios when considering the complete dataset of this region since the different emission ratios are dependent on the source type and characteristics along with the O&G processing method and techniques.
By utilizing the variability–lifetime relationship, we confirmed the extent of remoteness from NMHC sources. The variability of measurements in the Arabian Gulf and northern Red Sea was mostly determined by the sources. These two regions, together with the Gulf of Oman and Suez Canal, showed the smallest dependency on chemical lifetimes. On the other hand, lifetime–variability regression analysis resulted in increased dependencies with the chemical lifetimes over the Mediterranean Sea and Gulf of Aden. We can therefore conclude that these regions are relatively most remote from the NMHC sources. Interestingly, measurements in the Arabian Sea area, which would have been expected to show the most remote character, showed that the relatively low mixing ratios measured there were from local emissions.
To gain insight into the radical-induced oxidation of hydrocarbons around the Arabian Peninsula, we used i-butane ∕ propane and i-butane ∕ n-butane as oxidative pairs. We found that OH chemistry dominates hydrocarbon removal in the regions of the Middle East sampled in this campaign, although indications of sporadic Cl chemistry were found along the Red Sea and Arabian Gulf.
The data are available upon request to all scientists agreeing to the AQABA protocol from August 2019 (https://doi.org/10.5281/zenodo.3050041), Bourtsoukidis et al., 2019.
| General | |
| NMHCs | Non-methane hydrocarbons |
| AQABAAir | Quality and climate in the Arabian BAsin |
| KI | Kommandor Iona (ship vessel) |
| O&G | Oil and Gas |
| Regions | |
| MS | Mediterranean Sea |
| SC | Suez Canal and Gulf |
| RSN | Red Sea North |
| RSS | Red Sea South |
| GA | Gulf of Aden |
| AS | Arabian Sea |
| GO | Gulf of Oman |
| AG | Arabian Gulf |
| JP | Jeddah port |
| Methods | |
| GC–FID | Gas chromatography–flame ionization detector |
| VOCs | Volatile organic compounds |
| HYSPLIT | Hybrid Single Particle Lagrangian Integrated Trajectory model |
| ECHAM5 | 5th generation European Centre Hamburg general circulation model |
| MESSy | Modular Earth Submodel System |
| EMAC | ECHAM/MESSy Atmospheric Chemistry model |
| stp | standard temperature (273.15 K) and pressure (1013.25 hPa) |
| sccm | Standard cubic centimetres per minute (i.e. volume flux for the case of stp) |
| Lpm | Litre per minute at standard temperature and pressure (STP) |
| PTFE | Polytetrafluoroethylene |
| DL | Detection limit |
| Results | |
| ER | enhancement ratio slopes, i.e. the slope term in a linear regression between n-pentane and i-pentane. |
| EMF | Excess mole fraction (definition in Sect. 3.2.3) |
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7209-2019-supplement.
EB performed the measurements, analysed the data, and drafted the article. LE performed the measurements. DW analysed the trajectories. JDP is responsible for the methane data, JC for the kinetic calculations, and AP for the modelled OH, Cl, and NO3 concentrations. JL conceived and realized the project. JW supervised the study. All the authors contributed to editing the article.
The authors declare that they have no conflict of interest.
We acknowledge the fruitful collaborations with the Cyprus Institute (CyI), the King Abdullah University of Science and Technology (KAUST), and the Kuwait Institute for Scientific Research (KISR). We are grateful to Hays Ships Ltd, the ship's captain Pavel Kirzner, and the ship's crew for providing the best possible working conditions onboard Kommandor Iona. We thank all the participants of the AQABA ship campaign and in particular Harwig Harder for the fruitful discussions and day-to-day organization of the campaign, and Marcel Dorf, Claus Koeppel, Thomas Klüpfel, and Rolf Hoffmann for logistics organization and assistance during the setup phase. Finally, we acknowledge Ivan Tadic and Philipp Eger for the use of a preliminary KI contamination flag. Marc Delmotte, Laurence Vialettes, and Olivier Laurent helped with setting up the methane measurements.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Jacqui Hamilton and reviewed by two anonymous referees.
Anderson, M., Salo, K., and Fridell, E.: Particle-and gaseous emissions from an LNG powered ship, Environ. Sci. Technol., 49, 12568–12575, 2015.
Andreae, M. O.: Emission of trace gases and aerosols from biomass burning – An updated assessment, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-303, in review, 2019.
Andreae, M. O. and Crutzen, P. J.: Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry, Science, 276, 1052–1058, https://doi.org/10.1126/science.276.5315.1052, 1997.
Andreae, M. O. and Merlet, P.: Emission of trace gases andaerosols from biomass burning, Glob. Biogeochem. Cy., 15, 955–966, 2001.
Anosike, N., El-Suleiman, A., and Pilidis, P.: Associated Gas Utilization Using Gas Turbine Engine, Performance Implication–Nigerian Case Study, Adv. Mater Res.-Switz., 8, 137–145, http://dx.doi.org/10.4236/epe.2016.83012, 2016.
Apel, E. C., Calvert, J. G., Gilpin, T. M., Fehsenfeld, F., and Lonneman, W. A.: Nonmethane Hydrocarbon Intercomparison Experiment (NOMHICE): Task 4, ambient air, J. Geophys. Res.-Atmos., 108, 4300, https://doi.org/10.1029/2002JD002936, 2003.
Arsene, C., Bougiatioti, A., and Mihalopoulos, N.: Non-Methane Hydrocarbons (NMHCs) Variability in the Eastern Mediterranean, in: Regional Climate Variability and its Impacts in The Mediterranean Area, Dordrecht, 197–206, 2007.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions, Chem. Rev., 86, 69–201, 1986.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the NO3 radical with organic compounds, J. Phys. Chem. Ref. Data, 20, 459–507, 1991.
Aydin, M., Verhulst, K. R., Saltzman, E. S., Battle, M. O., Montzka, S. A., Blake, D. R., Tang, Q., and Prather, M. J.: Recent decreases in fossil-fuel emissions of ethane and methane derived from firn air, Nature, 476, 198–201, https://doi.org/10.1038/nature10352, 2011.
Baker, A. K., Beyersdorf, A. J., Doezema, L. A., Katzenstein, A., Meinardi, S., Simpson, I. J., Blake, D. R., and Rowland, F. S.: Measurements of nonmethane hydrocarbons in 28 United States cities, Atmos. Environ., 42, 170–182, 2008.
Baker, A. K., Sauvage, C., Thorenz, U. R., Van Velthoven, P., Oram, D. E., Zahn, A., Brenninkmeijer, C. A., and Williams, J.: Evidence for strong, widespread chlorine radical chemistry associated with pollution outflow from continental Asia, Sci. Rep.-UK, 6, 36821, https://doi.org/10.1038/srep36821, 2016.
Bange, H. W., Ramesh, R., Rapsomanikis, S., and Andreae, M.: Methane in surface waters of the Arabian Sea, Geophys. Res. Lett., 25, 3547–3550, 1998.
Barletta, B., Simpson, I. J., Blake, N. J., Meinardi, S., Emmons, L. K., Aburizaiza, O. S., Siddique, A., Zeb, J., Yu, L. E., Khwaja, H. A., Farrukh, M. A., and Blake, D. R.: Characterization of carbon monoxide, methane and nonmethane hydrocarbons in emerging cities of Saudi Arabia and Pakistan and in Singapore, J. Atmos. Chem., 74, 87–113, https://doi.org/10.1007/s10874-016-9343-7, 2017.
Bartenbach, S., Williams, J., Plass-Dülmer, C., Berresheim, H., and Lelieveld, J.: In-situ measurement of reactive hydrocarbons at Hohenpeissenberg with comprehensive two-dimensional gas chromatography (GC × GC-FID): use in estimating HO and NO3, Atmos. Chem. Phys., 7, 1–14, https://doi.org/10.5194/acp-7-1-2007, 2007.
Blake, D. R. and Rowland, F. S.: Global atmospheric concentrations and source strength of ethane, Nature, 321, 231, https://doi.org/10.1038/321231a0, 1986.
Blake, N. J., Barletta, B., Simpson, I. J., Schroeder, J., Hughes, S., Marrero, J. E., Meinardi, S., Blake, D. R., Apel, E. C., Hornbrook, R. S., and Emmons, L. K.: Spatial distributions and source characterization of trace organic gases during SEAC4RS and comparison to DC3, American Geophysical Union, Fall Meeting 2014, A33C-3197, 2014.
Born, D. A., Ulrich, E. C., Ju, K.-S., Peck, S. C., Van Der Donk, W. A., and Drennan, C. L.: Structural basis for methylphosphonate biosynthesis, Science, 358, 1336–1339, 2017.
Bourtsoukidis, E., Ernle, L., and Williams, J.: Non-methane hydrocarbon (C2–C8) sources and sinks around the Arabian Peninsula dataset, https://doi.org/10.5281/zenodo.3050041, last access: 29 May 2019.
Broadgate, W. J., Liss, P. S., and Penkett, S. A.: Seasonal emissions of isoprene and other reactive hydrocarbon gases from the ocean, Geophys. Res. Lett., 24, 2675–2678, 1997.
Broderick, B. and Marnane, I.: A comparison of the C2-C9 hydrocarbon compositions of vehicle fuels and urban air in Dublin, Ireland, Atmos. Environ., 36, 975–986, 2002.
Buzcu, B. and Fraser, M. P.: Source identification and apportionment of volatile organic compounds in Houston, TX, Atmos. Environ., 40, 2385–2400, 2006.
Colombo, E., Rocco, M., Gardumi, F., Cassetti, G., and Barbieri, J.: Exergy as a measure of oil well potential, Proceedings of 31st UIT Heat Transfer Conference, 25–27 June 2013, Milan, Italy, 2013.
Draxler, R. R. and Hess, G.: An overview of the HYSPLIT_4 modelling system for trajectories, Aust. Meteorol. Mag., 47, 295–308, 1998.
EEA: Air quality in Europe – 2018 report, European Environment Agency, EEA report no. 12/2018, ISSN 1977-8449, available at: https://www.eea.europa.eu//publications/air-quality-in-europe-2018 (last access: 20 May 2019), 2018.
Edwards, P. M., Brown, S. S., Roberts, J. M., Ahmadov, R., Banta, R. M., Dubé, W. P., Field, R. A., Flynn, J. H., Gilman, J. B., and Graus, M.: High winter ozone pollution from carbonyl photolysis in an oil and gas basin, Nature, 514, 351–354, 2014.
Emam, E. A.: Gas flaring in industry: an overview, Petroleum & Coal, 57, 532–555, 2015.
Erickson, D. J., Seuzaret, C., Keene, W. C., and Gong, S. L.: A general circulation model based calculation of HCl and ClNO2 production from sea salt dechlorination: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8347–8372, https://doi.org/10.1029/98JD01384, 1999.
Etiope, G. and Ciccioli, P.: Earth's degassing: a missing ethane and propane source, Science, 323, 478–478, 2009.
Eyring, V., Köhler, H., Van Aardenne, J., and Lauer, A.: Emissions from international shipping: 1. The last 50 years, J. Geophys. Res.-Atmos., 110, https://doi.org/10.1029/2004JD005619, 2005.
Farry, M.: Ethane from associated gas still the most economical, Oil and Gas Journal, 96, 115–117, 1998.
Field, R. A., Soltis, J., McCarthy, M. C., Murphy, S., and Montague, D. C.: Influence of oil and gas field operations on spatial and temporal distributions of atmospheric non-methane hydrocarbons and their effect on ozone formation in winter, Atmos. Chem. Phys., 15, 3527–3542, https://doi.org/10.5194/acp-15-3527-2015, 2015.
Gentner, D. R., Harley, R. A., Miller, A. M., and Goldstein, A. H.: Diurnal and seasonal variability of gasoline-related volatile organic compound emissions in Riverside, California, Environ. Sci. Technol., 43, 4247–4252, 2009.
Gilman, J. B., Burkhart, J. F., Lerner, B. M., Williams, E. J., Kuster, W. C., Goldan, P. D., Murphy, P. C., Warneke, C., Fowler, C., Montzka, S. A., Miller, B. R., Miller, L., Oltmans, S. J., Ryerson, T. B., Cooper, O. R., Stohl, A., and de Gouw, J. A.: Ozone variability and halogen oxidation within the Arctic and sub-Arctic springtime boundary layer, Atmos. Chem. Phys., 10, 10223–10236, https://doi.org/10.5194/acp-10-10223-2010, 2010.
Gilman, J. B., Lerner, B. M., Kuster, W. C., and de Gouw, J. A.: Source Signature of Volatile Organic Compounds from Oil and Natural Gas Operations in Northeastern Colorado, Environ. Sci. Technol., 47, 1297-1305, 10.1021/es304119a, 2013.
Harley, R. A., Hannigan, M. P., and Cass, G. R.: Respeciation of organic gas emissions and the detection of excess unburned gasoline in the atmosphere, Environ. Sci. Technol., 26, 2395–2408, 1992.
Hazan, L., Tarniewicz, J., Ramonet, M., Laurent, O., and Abbaris, A.: Automatic processing of atmospheric CO2 and CH4 mole fractions at the ICOS Atmosphere Thematic Centre, Atmos. Meas. Tech., 9, 4719–4736, https://doi.org/10.5194/amt-9-4719-2016, 2016.
Helmig, D. and Greenberg, J.: Artifact formation from the use of potassium-iodide-based ozone traps during atmospheric sampling of trace organic gases, J. High Res. Chromatog., 18, 15–18, https://doi.org/10.1002/jhrc.1240180105, 1995.
Helmig, D., Tanner, D., Honrath, R., Owen, R., and Parrish, D.: Nonmethane hydrocarbons at Pico Mountain, Azores: 1. Oxidation chemistry in the North Atlantic region, J. Geophys. Res.-Atmos., 113, https://doi.org/10.1029/2007JD008930, 2008.
Helmig, D., Thompson, C. R., Evans, J., Boylan, P., Hueber, J., and Park, J. H.: Highly Elevated Atmospheric Levels of Volatile Organic Compounds in the Uintah Basin, Utah, Environ. Sci. Technol., 48, 4707–4715, https://doi.org/10.1021/es405046r, 2014.
Helmig, D., Rossabi, S., Hueber, J., Tans, P., Montzka, S. A., Masarie, K., Thoning, K., Plass-Duelmer, C., Claude, A., and Carpenter, L. J.: Reversal of global atmospheric ethane and propane trends largely due to US oil and natural gas production, Nat. Geosci., 9, 490–495, https://doi.org/10.1038/ngeo2721, 2016.
Huang, G., Brook, R., Crippa, M., Janssens-Maenhout, G., Schieberle, C., Dore, C., Guizzardi, D., Muntean, M., Schaaf, E., and Friedrich, R.: Speciation of anthropogenic emissions of non-methane volatile organic compounds: a global gridded data set for 1970–2012, Atmos. Chem. Phys., 17, 7683–7701, https://doi.org/10.5194/acp-17-7683-2017, 2017.
Jobson, B. T., Niki, H., Yokouchi, Y., Bottenheim, J., Hopper, F., and Leaitch, R.: Measurements of C2-C6 hydrocarbons during the Polar Sunrise 1992 Experiment: Evidence for Cl atom and Br atom chemistry, J. Geophys. Res.-Atmos., 99, 25355–25368, 1994.
Jobson, B. T., Parrish, D. D., Goldan, P., Kuster, W., Fehsenfeld, F. C., Blake, D. R., Blake, N. J., and Niki, H.: Spatial and temporal variability of nonmethane hydrocarbon mixing ratios and their relation to photochemical lifetime, J. Geophys. Res.-Atmos., 103, 13557–13567, https://doi.org/10.1029/97JD01715, 1998.
Jobson, B. T., McKeen, S., Parrish, D., Fehsenfeld, F., Blake, D., Goldstein, A., Schauffler, S., and Elkins, J.: Trace gas mixing ratio variability versus lifetime in the troposphere and stratosphere: Observations, J. Geophys. Res.-Atmos., 104, 16091–16113, 1999.
Jobson, B. T., Berkowitz, C. M., Kuster, W., Goldan, P., Williams, E., Fesenfeld, F., Apel, E., Karl, T., Lonneman, W. A., and Riemer, D.: Hydrocarbon source signatures in Houston, Texas: Influence of the petrochemical industry, J. Geophys. Res.-Atmos., 109, https://doi.org/10.1029/2004JD004887, 2004.
Jöckel, P., Kerkweg, A., Pozzer, A., Sander, R., Tost, H., Riede, H., Baumgaertner, A., Gromov, S., and Kern, B.: Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev., 3, 717–752, https://doi.org/10.5194/gmd-3-717-2010, 2010.
Junge, C. E.: Residence time and variability of tropospheric trace gases, Tellus, 26, 477–488, 1974.
Keene, W., Khalil, M. A. K., Erickson, D., McCulloch, A., Graedel, T. E., Lobert, J. M., Aucott, M. L., Gong, S. L., Harper, D. B., and Kleiman, G.: Composite global emissions of reactive chlorine from anthropogenic and natural sources: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8429–8440, 1999.
Kim, E., Brown, S. G., Hafner, H. R., and Hopke, P. K.: Characterization of non-methane volatile organic compounds sources in Houston during 2001 using positive matrix factorization, Atmos. Environ., 39, 5934–5946, 2005.
Kirschke, S., Bousquet, P., Ciais, P., Saunois, M., Canadell, J. G., Dlugokencky, E. J., Bergamaschi, P., Bergmann, D., Blake, D. R., Bruhwiler, L., Cameron-Smith, P., Castaldi, S., Chevallier, F., Feng, L., Fraser, A., Heimann, M., Hodson, E. L., Houweling, S., Josse, B., Fraser, P. J., Krummel, P. B., Lamarque, J.-F., Langenfelds, R. L., Le Quéré, C., Naik, V., O'Doherty, S., Palmer, P. I., Pison, I., Plummer, D., Poulter, B., Prinn, R. G., Rigby, M., Ringeval, B., Santini, M., Schmidt, M., Shindell, D. T., Simpson, I. J., Spahni, R., Steele, L. P., Strode, S. A., Sudo, K., Szopa, S., van der Werf, G. R., Voulgarakis, A., van Weele, M., Weiss, R. F., Williams, J. E., and Zeng, G.: Three decades of global methane sources and sinks, Nat. Geosci., 6, 813–823, 2013.
Knipping, E., Lakin, M., Foster, K., Jungwirth, P., Tobias, D., Gerber, R., Dabdub, D., and Finlayson-Pitts, B.: Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols, Science, 288, 301–306, 2000.
Lawler, M. J., Finley, B., Keene, W., Pszenny, A., Read, K., Von Glasow, R., and Saltzman, E.: Pollution-enhanced reactive chlorine chemistry in the eastern tropical Atlantic boundary layer, Geophys. Res. Lett., 36, https://doi.org/10.1029/2008GL036666, 2009.
Lelieveld, J., Berresheim, H., Borrmann, S., Crutzen, P. J., Dentener, F. J., Fischer, H., Feichter, J., Flatau, P. J., Heland, J., Holzinger, R., Korrmann, R., Lawrence, M. G., Levin, Z., Markowicz, K. M., Mihalopoulos, N., Minikin, A., Ramanathan, V., de Reus, M., Roelofs, G. J., Scheeren, H. A., Sciare, J., Schlager, H., Schultz, M., Siegmund, P., Steil, B., Stephanou, E. G., Stier, P., Traub, M., Warneke, C., Williams, J., and Ziereis, H.: Global Air Pollution Crossroads over the Mediterranean, Science, 298, 794–799, https://doi.org/10.1126/science.1075457, 2002.
Lelieveld, J., Hoor, P., Jöckel, P., Pozzer, A., Hadjinicolaou, P., Cammas, J.-P., and Beirle, S.: Severe ozone air pollution in the Persian Gulf region, Atmos. Chem. Phys., 9, 1393–1406, https://doi.org/10.5194/acp-9-1393-2009, 2009.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Matar, S. and Hatch, L. F.: Chemistry of petrochemical processes, Elsevier, 356 pp., 2001.
McGaughey, G. R., Desai, N. R., Allen, D. T., Seila, R. L., Lonneman, W. A., Fraser, M. P., Harley, R. A., Pollack, A. K., Ivy, J. M., and Price, J. H.: Analysis of motor vehicle emissions in a Houston tunnel during the Texas Air Quality Study 2000, Atmos. Environ., 38, 3363–3372, 2004.
Mielke, L., Stutz, J., Tsai, C., Hurlock, S., Roberts, J., Veres, P., Froyd, K., Hayes, P., Cubison, M., and Jimenez, J.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res.-Atmos., 118, 10638–610652, 2013.
Osthoff, H. D., Roberts, J. M., Ravishankara, A., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., and Dibb, J. E.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, 2008.
Panopoulou, A., Liakakou, E., Gros, V., Sauvage, S., Locoge, N., Bonsang, B., Psiloglou, B. E., Gerasopoulos, E., and Mihalopoulos, N.: Non-methane hydrocarbon variability in Athens during wintertime: the role of traffic and heating, Atmos. Chem. Phys., 18, 16139–16154, https://doi.org/10.5194/acp-18-16139-2018, 2018.
Parrish, D. D., Dunlea, E., Atlas, E., Schauffler, S., Donnelly, S., Stroud, V., Goldstein, A., Millet, D., McKay, M., and Jaffe, D.: Changes in the photochemical environment of the temperate North Pacific troposphere in response to increased Asian emissions, J. Geophys. Res.-Atmos., 109, D23S18, https://doi.org/10.1029/2004JD004978, 2004.
Parrish, D. D., Stohl, A., Forster, C., Atlas, E., Blake, D., Goldan, P., Kuster, W., and De Gouw, J.: Effects of mixing on evolution of hydrocarbon ratios in the troposphere, J. Geophys. Res.-Atmos., 112, D10S34, https://doi.org/10.1029/2006JD007583, 2007.
Perring, A., Pusede, S., and Cohen, R.: An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol, Chem. Rev., 113, 5848–5870, 2013.
Pétron, G., Frost, G., Miller, B. R., Hirsch, A. I., Montzka, S. A., Karion, A., Trainer, M., Sweeney, C., Andrews, A. E., and Miller, L.: Hydrocarbon emissions characterization in the Colorado Front Range: A pilot study, J. Geophys. Res.-Atmos., 117, D04304, https://doi.org/10.1029/2011JD016360, 2012 .
Pétron, G., Karion, A., Sweeney, C., Miller, B. R., Montzka, S. A., Frost, G. J., Trainer, M., Tans, P., Andrews, A., and Kofler, J.: A new look at methane and nonmethane hydrocarbon emissions from oil and natural gas operations in the Colorado Denver-Julesburg Basin, J. Geophys. Res.-Atmos., 119, 6836–6852, 2014.
Phillips, G., Tang, M., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012GL051912, 2012.
Plass-Dülmer, C., Michl, K., Ruf, R., and Berresheim, H.: C2-C8 Hydrocarbon measurement and quality control procedures at the Global Atmosphere Watch Observatory Hohenpeissenberg, J. Chromatogr. A, 953, 175–197, https://doi.org/10.1016/S0021-9673(02)00128-0, 2002.
Poisson, N., Kanakidou, M., and Crutzen, P. J.: Impact of Non-Methane Hydrocarbons on Tropospheric Chemistry and the Oxidizing Power of the Global Troposphere: 3-Dimensional Modelling Results, J. Atmos. Chem., 36, 157–230, https://doi.org/10.1023/a:1006300616544, 2000.
Pozzer, A., Pollmann, J., Taraborrelli, D., Jöckel, P., Helmig, D., Tans, P., Hueber, J., and Lelieveld, J.: Observed and simulated global distribution and budget of atmospheric C2-C5 alkanes, Atmos. Chem. Phys., 10, 4403–4422, https://doi.org/10.5194/acp-10-4403-2010, 2010.
Pszenny, A. A., Fischer, E. V., Russo, R. S., Sive, B. C., and Varner, R. K.: Estimates of Cl atom concentrations and hydrocarbon kinetic reactivity in surface air at Appledore Island, Maine (USA), during International Consortium for Atmospheric Research on Transport and Transformation/Chemistry of Halogens at the Isles of Shoals, J. Geophys. Res.-Atmos., 112, D10S13, https://doi.org/10.1029/2006JD007725, 2007.
Rappenglück, B., Apel, E., Bauerfeind, M., Bottenheim, J., Brickell, P., Čavolka, P., Cech, J., Gatti, L., Hakola, H., Honzak, J., Junek, R., Martin, D., Noone, C., Plass-Dülmer, C., Travers, D., and Wang, D.: The first VOC intercomparison exercise within the Global Atmosphere Watch (GAW), Atmos. Environ., 40, 7508–7527, https://doi.org/10.1016/j.atmosenv.2006.07.016, 2006.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S.-M., Gilman, J., De Gouw, J., and Bon, D. M.: Nitryl chloride and molecular chlorine in the coastal marine boundary layer, Environ. Sci. Technol., 46, 10463–10470, 2012.
Roeckner, E., Brokopf, R., Esch, M., Giorgetta, M., Hagemann, S., Kornblueh, L., Manzini, E., Schlese, U., and Schulzweida, U.: Sensitivity of simulated climate to horizontal and vertical resolution in the ECHAM5 atmosphere model, J. Climate, 19, 3771–3791, 2006.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Häseler, R., Holland, F., and Hu, M.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559–563, 2014.
Rossabi, S. and Helmig, D.: Changes in Atmospheric Butanes and Pentanes and Their Isomeric Ratios in the Continental United States, J. Geophys. Res.-Atmos., 123, 3772–3790, https://doi.org/10.1002/2017JD027709, 2018.
Rudolph, J., Ramacher, B., Plass-Dülmer, C., Müller, K.-P., and Koppmann, R.: The indirect determination of chlorine atom concentration in the troposphere from changes in the patterns of non-methane hydrocarbons, Tellus B, 49, 592–601, 1997.
Sala, S., Bönisch, H., Keber, T., Oram, D. E., Mills, G., and Engel, A.: Deriving an atmospheric budget of total organic bromine using airborne in situ measurements from the western Pacific area during SHIVA, Atmos. Chem. Phys., 14, 6903–6923, https://doi.org/10.5194/acp-14-6903-2014, 2014.
Salameh, T., Sauvage, S., Afif, C., Borbon, A., and Locoge, N.: Source apportionment vs. emission inventories of non-methane hydrocarbons (NMHC) in an urban area of the Middle East: local and global perspectives, Atmos. Chem. Phys., 16, 3595–3607, https://doi.org/10.5194/acp-16-3595-2016, 2016.
Sander, R., Baumgaertner, A., Cabrera-Perez, D., Frank, F., Gromov, S., Grooß, J.-U., Harder, H., Huijnen, V., Jöckel, P., Karydis, V. A., Niemeyer, K. E., Pozzer, A., Riede, H., Schultz, M. G., Taraborrelli, D., and Tauer, S.: The community atmospheric chemistry box model CAABA/MECCA-4.0, Geosci. Model Dev., 12, 1365–1385, https://doi.org/10.5194/gmd-12-1365-2019, 2019.
Saunois, M., Bousquet, P., Poulter, B., Peregon, A., Ciais, P., Canadell, J. G., Dlugokencky, E. J., Etiope, G., Bastviken, D., Houweling, S., Janssens-Maenhout, G., Tubiello, F. N., Castaldi, S., Jackson, R. B., Alexe, M., Arora, V. K., Beerling, D. J., Bergamaschi, P., Blake, D. R., Brailsford, G., Brovkin, V., Bruhwiler, L., Crevoisier, C., Crill, P., Covey, K., Curry, C., Frankenberg, C., Gedney, N., Höglund-Isaksson, L., Ishizawa, M., Ito, A., Joos, F., Kim, H.-S., Kleinen, T., Krummel, P., Lamarque, J.-F., Langenfelds, R., Locatelli, R., Machida, T., Maksyutov, S., McDonald, K. C., Marshall, J., Melton, J. R., Morino, I., Naik, V., O'Doherty, S., Parmentier, F.-J. W., Patra, P. K., Peng, C., Peng, S., Peters, G. P., Pison, I., Prigent, C., Prinn, R., Ramonet, M., Riley, W. J., Saito, M., Santini, M., Schroeder, R., Simpson, I. J., Spahni, R., Steele, P., Takizawa, A., Thornton, B. F., Tian, H., Tohjima, Y., Viovy, N., Voulgarakis, A., van Weele, M., van der Werf, G. R., Weiss, R., Wiedinmyer, C., Wilton, D. J., Wiltshire, A., Worthy, D., Wunch, D., Xu, X., Yoshida, Y., Zhang, B., Zhang, Z., and Zhu, Q.: The global methane budget 2000–2012, Earth Syst. Sci. Data, 8, 697–751, https://doi.org/10.5194/essd-8-697-2016, 2016.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, 1252 pp., 2016.
Simpson, I. J., Sulbaek Andersen, M. P., Meinardi, S., Bruhwiler, L., Blake, N. J., Helmig, D., Rowland, F. S., and Blake, D. R.: Long-term decline of global atmospheric ethane concentrations and implications for methane, Nature, 488, 490–494, 2012.
Slemr, J., Slemr, F., Partridge, R., D'Souza, H., and Schmidbauer, N.: Accurate Measurements of Hydrocarbons in the Atmosphere (AMOHA): Three European intercomparisons, J. Geophys. Res.-Atmos., 107, 4409, https://doi.org/10.1029/2001JD001357, 2002.
Swarthout, R. F., Russo, R. S., Zhou, Y., Hart, A. H., and Sive, B. C.: Volatile organic compound distributions during the NACHTT campaign at the Boulder Atmospheric Observatory: Influence of urban and natural gas sources, J. Geophys. Res.-Atmos., 118, 10614–10637, https://doi.org/10.1002/jgrd.50722, 2013.
Thompson, C. R., Hueber, J., and Helmig, D.: Influence of oil and gas emissions on ambient atmospheric non-methane hydrocarbons in residential areas of Northeastern Colorado, Elem. Sci. Anth., 3, http://doi.org/10.12952/journal.elementa.000035, 2014.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dubé, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, 2010.
Umukoro, G. E. and Ismail, O. S.: Modelling emissions from natural gas flaring, Journal of King Saud University-Engineering Sciences, 29, 178–182, 2017.
United States Central Intelligence Agency: Middle East oil and gas, Washington, D.C.: Central Intelligence Agency, available at: https://www.loc.gov/item/2007631392/ (last access: 20 May 2019), 2007.
Von Schneidemesser, E., Monks, P. S., and Plass-Duelmer, C.: Global comparison of VOC and CO observations in urban areas, Atmos. Environ., 44, 5053–5064, 2010.
Warneck, P. and Williams, J.: The atmospheric chemist's companion: Numerical data for use in the atmospheric sciences, Springer Science and Business Media B.V., Berlin, 436 pp., 2012.
Warneke, C., McKeen, S., De Gouw, J., Goldan, P., Kuster, W., Holloway, J., Williams, E., Lerner, B., Parrish, D., and Trainer, M.: Determination of urban volatile organic compound emission ratios and comparison with an emissions database, J. Geophys. Res.-Atmos., 112, https://doi.org/10.1029/2006JD007930, 2007.
Wei, W., Cheng, S., Li, G., Wang, G., and Wang, H.: Characteristics of ozone and ozone precursors (VOCs and NOx) around a petroleum refinery in Beijing, China, J. Environ. Sci., 26, 332–342, 2014.
Williams, J., Fischer, H., Harris, G. W., Crutzen, P. J., Hoor, P., Hansel, A., Holzinger, R., Warneke, C., Lindinger, W., Scheeren, B., and Lelieveld, J.: Variability-lifetime relationship for organic trace gases: A novel aid to compound identification and estimation of HO concentrations, J. Geophys. Res.-Atmos., 105, 20473–20486, https://doi.org/10.1029/2000JD900203, 2000.
Williams, J., Gros, V., Bonsang, B., and Kazan, V.: HO cycle in 1997 and 1998 over the southern Indian Ocean derived from CO, radon, and hydrocarbon measurements made at Amsterdam Island, J. Geophys. Res.-Atmos., 106, 12719–12725, 2001.
Williams, J., de Reus, M., Krejci, R., Fischer, H., and Ström, J.: Application of the variability-size relationship to atmospheric aerosol studies: estimating aerosol lifetimes and ages, Atmos. Chem. Phys., 2, 133-145, 10.5194/acp-2-133-2002, 2002.
Young, C. J., Washenfelder, R. A., Roberts, J. M., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., and Cochran, A. K.: Vertically resolved measurements of nighttime radical reservoirs in Los Angeles and their contribution to the urban radical budget, Environ. Sci. Technol., 46, 10965–10973, 2012.
Young, C. J., Washenfelder, R. A., Edwards, P. M., Parrish, D. D., Gilman, J. B., Kuster, W. C., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., Roberts, J. M., Griffith, S., Dusanter, S., Stevens, P. S., Flynn, J., Grossberg, N., Lefer, B., Holloway, J. S., Peischl, J., Ryerson, T. B., Atlas, E. L., Blake, D. R., and Brown, S. S.: Chlorine as a primary radical: evaluation of methods to understand its role in initiation of oxidative cycles, Atmos. Chem. Phys., 14, 3427–3440, https://doi.org/10.5194/acp-14-3427-2014, 2014.